ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[1] Настоящее изобретение относится в целом к способам и композициям для лечения заболевания, нарушения, патологических состояний или поражения центральной нервной системы (ЦНС) путем временного снижения уровня системной иммуносупрессии в системе кровообращения.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[2] Большинство патологий центральной нервной системы (ЦНС) имеют общий нейровоспалительный компонент, который является частью проогрессирования заболевания и вносит вклад в распространение заболевания. К таким патологиям относится болезнь Альцгеймера (БА), связанное с возрастом нейродегенеративное заболевание, отличающееся прогрессирующей потерей памяти и когнитивных функций, при котором, как считается, накопление агрегатов бета-амилоидных пептидов (Аβ) играет ключевую роль в каскадном развитии воспаления ЦНС, что в конечном счете ведет к разрушению нейронов и разрушению тканей (Akiyama et al, 2000; Hardy & Selkoe, 2002; Vom Berg et al., 2012). Несмотря на хронический нейровоспалительный ответ при нейродегенеративных заболеваниях, за последнее десятилетие клинические и доклинические исследования, направленные на изучение терапии нейродегенеративных заболеваний на основе иммуносупрессии, подняли вопрос о неэффективности противовоспалительных лекарственных средств (Breitner et al, 2009; Group et al, 2007; Wyss-Coray & Rogers, 2012). Авторы изобретения обеспечивают новаторское решение для преодоления недостатков существующих способов лечения БА, а также аналогичных заболеваний и поражений ЦНС; этот способ основан на особенном понимании авторами изобретения роли различных компонентов системной и центральной иммунной системы в поддержании функционирования и восстановлении ЦНС.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[3] В одном аспекте настоящее изобретение обеспечивает фармацевтическую композицию, содержащую активный агент, который вызывает снижение уровня системной иммуносупрессии у пациента для применения при лечении заболевания, расстройства, патологического состояния или травмы ЦНС, которое не включает в себя аутоиммунное нейровоспалительное заболевание, рецидивирующе-ремиттирующий рассеянный склероз (RPMC), где указанная фармацевтическая композиция предназначена для введения с помощью режима дозирования, содержащего по меньшей мере два курса лечения, включающих последовательно курс лечения, за которым следует перерыв без лечения.
[4] В другом аспекте настоящее изобретение обеспечивает способ лечения заболевания, нарушения, патологического состояния или поражения ЦНС, которое не включает в себя аутоиммунное нейровоспалительное заболевание, рецидивирующе-ремиттирующий рассеянный склероз (RPMC), указанный способ, содержащий введение пациенту, который в этом нуждается, фармацевтической композиции, содержащей активный агент, который вызывает снижение уровня системной иммуносупрессии в соответствии с настоящим изобретением, где указанную фармацевтическую композицию вводят в режиме дозирования, содержащего по меньшей мере два курса лечения, каждый курс лечения, содержащий последовательно курс лечения, за которым следует период без лечения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКОГО МАТЕРИАЛА
[5] На фиг. 1А-В изображена активность хороидного сплетения (CP) в течение прогрессирования заболевания на трансгенных мышах 5XFAD с моделью БА (AD-Tg). (А) уровни экспрессии мРНК генов icam1, vcam1, cxcl10 и ccl2, измеренные количественной ПЦР в реальном времени (RT-qPCR), в сосудистых сплетениях (CPs), отобранных у AD-Tg мышей 1, 2, 4 и 8-месячного возраста, представленные в виде кратного изменения по сравнению с контрольными образцами дикого типа (WT) соответственно возрасту (n=6-8 на группу; т-критерий Стьюдента для каждого момента времени). (В) Репрезентативные микроскопические изображения CPs 8-месячных мышей AD-Tg и контрольных особей дикого типа (WT) соответственно возрасту, иммуноокрашенных на молекулы плотного контакта эпителия Клаудин-1, окрашивание ядер Hoechst, и лиганд интегрина, ICAM-1 (масштабная полоска, 50 мкм). На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[6] На фиг. 2А-С представлены: (А) Количественная оценка иммунореактивности ICAM-1 в CP человека (посмертно) у молодых и пожилых пациентов без расстройств ЦНС и пациентов с БА (n=5 на группу; однофакторный дисперсионный анализ (ANOVA), с последующим апостериорным анализом (post-hoc analysis) Ньюмена-Кейлса; (В) анализ методом проточной цитометрии для иммунных клеток, экспрессирующих IFN-γ (внутриклеточно окрашенных и с предварительным гейтингом по CD45) в хороидных сплетениях (CPs) 8-месячных мышей AD-Tg и у контрольных особей дикого типа по возрастным группам. Заштрихованная гистограмма отображает изотипический контроль (n=4-6 на группу; т-критерий Стьюдента); и (С) уровни экспрессии мРНК генов ifn-γ (ИФН-γ), измеренные RT-qPCR, в тканях CP, выделенных из мышей AD-Tg 4- и 8-месячного возраста, по сравнению с контролями WT по возрастным группам (n=5-8 на группу; т-критерий Стьюдента для каждого момента времени). На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[7] На фиг. 3А-В представлены (А) репрезентативные графики проточной цитометрии частот встречаемости спленоцитов CD4+Foxp3+ (с предварительным гейтингом по TCRβ) у 8-месячных мышей AD-Tg и контрольных мышей WT; и (В) количественный анализ спленоцитов 1-, 2-, 4- и 8-месячных мышей AD-Tg и контрольных мышей WT (n=6-8 на группу; т-критерий Стьюдента для каждого момента времени). На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[8] На фиг. 4 представлена стратегия гейтирования и репрезентативные графики проточной цитометрии спленоцитов мышей AD-Tg/Foxp3-DTR+/- через 1 день после последней инъекции DTx. DTx инъецировали интраперитонеально в течение 4 установленных дней, достижение ~99% истощения клеток Foxp3+.
[9] На фиг. 5A-G представлены последствия транзиторного истощения регуляторных Т-клеток у мышей AD-Tg. (A) AD-Tg/Foxp3-DTR+ (которые экспрессируют трансген DTR) и не экспрессирующих DTR однопометных животных AD-Tg (AD-Tg/Foxp3-DTR-) контрольной группы лечили DTx в течение 4 установленных дней. Уровни экспрессии мРНК генов icam1, cxcl10 и ccl2, измеряли методом RT-qPCR (количественной ПЦР в реальном времени), у леченных DTx 6-месячных мышей AD-Tg через 1 день после последней инъекции DTx (n=6-8 на группу; т-критерий Стьюдента). (B-D) Анализ паренхимы головного мозга методом проточной цитометрии (за исключением хороидного сплетения, которое иссекали отдельно), 6-месячных мышей AD-Tg и контрольных особей, леченных DTx, через 3 недели после последней инъекции DTx. Количественный анализ методом проточной цитометрии, показывающий увеличение числа D11bhigh/CD45high mo-МФ и CD4+ Т-клеток (В), и репрезентативные графики проточной цитометрии (С) и количественный анализ (D) частоты встречаемости регуляторных Т-клеток CD4+Foxp3+ в паренхиме головного мозга мышей AD-Tg/Foxp3-DTR+ и контрольных особей AD-Tg/Foxp3-DTR, леченных DTx (n=3-7 на каждую группу; т-критерий Стьюдента). (Е) уровни экспрессии мРНК foxp3 и il10 в паренхиме головного мозга, леченных DTx 6-месячных мышей AD-Tg AD-Tg/Foxp3-DTR+, и контрольных особей AD-Tg/Foxp3-DTR- через 3 недели после последней инъекции DTx (n=6-8 на группу; т-критерий Стьюдента). (F) количественный анализ иммуноокрашивания GFAP, демонстрирующий пониженный астроглиоз в срезах гиппокампа 6-месячных, леченных DTx, AD-Tg/Foxp3-DTR+ и AD-Tg/Foxp3-DTR- контрольных мышей через 3 недели после последней инъекции DTx (масштабная полоска, 50 мкм; n=3-5 на группу; т-критерий Стьюдента). (G) уровни экспрессии мРНК il-12р40 и tnf-a в паренхиме головного мозга через 3 недели после последней инъекции DTx (n=6-8 на группу; т-критерий Стьюдента). На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[10] На фиг. 6А-Е показан эффект транзиторного истощения регуляторных Т-клеток на Аβ-бляшках на функции обучения и запоминания. (А) Репрезентативные микроскопические изображения и (В) количественный анализ головного мозга, леченных DTx 5-месячных мышей AD-Tg/Foxp3-DTR+ и контрольных мышей AD-Tg/Foxp3-DTR- через 3 недели после последней инъекции DTx, иммуноокрашивание на Аβ-бляшки и окрашивание ядер Hoechst (масштабная линейка, 250 мкм). Были определены количественно средняя площадь и количество Аβ-бляшек в зубчатой извилине гиппокампа (DG) и 5-ом слое коры головного мозга (в 6 мкм срезах головного мозга; n=5 на группу; т-критерий Стьюдента). На фиг. 6С-Е представлены результаты испытаний в водном лабиринте Морриса (MWM), леченных DTx 6-месячных мышей AD-Tg/Foxp3-DTR+ и контрольных особей через 3 недели после последней инъекции DTx. После транзиторного истощения регуляторных Т-клеток мыши AD-Tg показали лучшие результаты пространственного обучения и памяти на фазах обнаружения (С), (D) изучения и (Е) разворота в WLM, по сравнению с контрольными особями AD-Tg (n=7-9 на группу; двухфакторный дисперсионный анализ с повторными измерениями ANOVA с последующими попарными апостериорными («post-hoc») сравнениями с критерием Бонферрони; *, Р<0,05 в совокупности для узнавания, зондирования и разворота). На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[11] На фиг. 7 представлены уровни экспрессии мРНК ifn-γ , измеренные методом количественной ПЦР в реальном времени (RT-qPCR), в хороидных сплетениях (CPs), выделенных у 6- и 12-месячных APP/PS1 AD-Tg мышей (мышиная модель болезни Альцгеймера (см. материалы и методы)), по сравнению с контролными особями дикого типа по возрастным группам (n=5-8 на группу; т-критерий Стьюдента). Величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05.
[12] На фиг. 8A-I представлен терапевтический эффект еженедельного введения глатирамера ацетата (GA) мышам AD-Tg. (А) Схематическое представление еженедельной схемы лечения GA. Мышам (5-месячным) подкожно инъецировали GA (100 мкг), дважды в течение первой недели (на 1 и 4 день), и после этого раз в неделю, общей продолжительностью 4 недели. У мышей исследовали когнитивные способности через 1 неделю (водный лабиринт Морриса, MWM), через 1 месяц (радиальный водный лабиринт Морриса, RAWM) и через 2 месяца (радиальный водный лабиринт Морриса, RAWM, с использованием различных экспериментальных пространственных установок) после последней инъекции, и воспаление гиппокампа. На фиг. 8B-D представлены уровни экспрессии мРНК генов в гиппокампе нелеченных мышей AD-Tg, и мышей AD-Tg, леченных еженедельно GA, в возрасте 6 месяцев, демонстрирующие (В) пониженную экспрессию провоспалительных цитокинов, таких как TNF-α, IL-1β и IL-12р40, (С) повышение противовоспалительных цитокинов IL-10 и TGF-β и (D) нейротрофических факторов, IGF-1 и BDNF, у мышей, еженедельно леченных GA (n=6-8 на группу; т-критерий Стьюдента). На фиг. 8E-G мышей AD-Tg (5-месячного возраста) лечили еженедельно либо GA, либо носителем (PBS) и сравнивали с однопометными контрольными мышами дикого типа (WT) в соответствующей возрастной группе, в возрасте 6 месяцев, в MWM-тесте. Получившие лечение мыши AD-Tg, показали лучшие результаты пространственного обучения и памяти на фазах обнаружения (Е), изучения (F) и разворота (G) в MWM, по сравнению с контрольными особями (n=6-9 на группу; двухфакторный дисперсионный анализ с повторными измерениями ANOVA с последующими попарными апостериорными («post-hoc») сравнениями с критерием Бонферрони для сравнения отдельных пар; мыши дикого типа (WT) черные круги; контрольные особи AD-Tg, белые круги; получившие лечение AD-Tg, серые круги). На фиг. 8H-I представлена когнитивная способность тех же самых мышей в RAWM-тесте через 1 месяц (Н) или 2 месяца (I) после последней инъекции GA (n=6-9 на группу; двухфакторный дисперсионный анализ с повторными измерениями ANOVA с последующими попарными апостериорными («post-hoc») сравнениями с критерием Бонферрони для сравнения отдельных пар). Данные представляют по меньшей мере три независимых эксперимента. На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[13] На фиг. 9А-Н представлены дополнительные терапевтические эффекты еженедельного введения GA мышам AD-Tg. На фиг. А-В представлены результаты на мышах 5XFAD AD-Tg, которые еженедельно получали лечение GA или носитель (PBS) и были исследованы в конце первой недели режима введения (в общей сложности после двух инъекций GA). Анализ методом проточной цитометрии на частоту встречаемости спленоцитов CD4+Foxp3+ (А) и иммунных клеток CP, экспрессирующих IFN-γ (В; внутриклеточно окрашенных и с предварительным гейтингом по CD45), у получивших лечение 6-месячных мышей AD-Tg, по сравнению с мышами WT в соответствующей возрастной группе, соответствующего возраста (n=4-6 на группу; однофакторный дисперсионный анализ ANOVA, с последующим апостериорным анализом («post-hoc») Ньюмена-Кейлса). (С) уровни экспрессии мРНК генов icam1, cxcl10 и ccl2, измеренные методом RT-qPCR, в CPs 4-месячных AD-Tg мышей, леченных еженедельно как GA, так и носителем, и исследованных как в конце 1-ой, так и 4-ой недели режима еженедельного введения GA (n=6-8 на группу; однофакторный дисперсионный анализ ANOVA, с последующим апостериорным анализом («post-hoc») Ньюмена-Кейлса). На фиг. 9D-E представлены репрезентативные изображения срезов мозга 6-месячных химер костного мозга (ВМ) AD-Tg/CX3CR1GFP/+ после недельного лечения GA. Клетки CX3CR1GFP были локализованы в CP третьего желудочка (3V; i), в прилегающем к желудочкам пространстве (ii) и в CP боковых желудочков (LV; iii) у мышей AD-Tg, еженедельно леченных GA (D; масштабная линейка, 25 мкм). Репрезентативные ортогональные проекции конфокальных изображений (стеков) по оси Z, показывающие колокализацию клеток GFP+ с миелоидным маркером, CD68, в CP 7-месячных мышей AD-Tg/CX3CR1GFP/+, леченных еженедельно GA, но не у контрольных мышей AD-Tg/CX3CR1GFP/+, леченных PBS (Е, масштабная линейка, 25 мкм). (F) Клетки CX3CR1GFP колокализованы с миелоидным маркером IBA-1 в головном мозге, леченных GA мышей AD-Tg/CX3CR1GFP/+, рядом с Аβ-бляшками и коэкспрессирующие миелоидный маркер, IBA-1 (масштабная линейка, 25 мкм). На фиг. 9G-H показаны репрезентативные графики проточной цитометрии клеток, выделенных из гиппокампа 4-месячных нелеченных мышей AD-Tg WT, и мышей AD-Tg на второй неделе еженедельного режима введения GA. CD11bhigh/CD45high mo-МФ были гейтированы (G) и определены количественно (Н; n=4-5 на группу; однофакторный дисперсионный анализ ANOVA, с последующим апостериорным анализом («post-hoc») Ньюмена-Кейлса). На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[14] На фиг. 10А-Н изображен терапевтический эффект введения ингибитора p300 (С646) мышам AD-Tg. На фиг. 10А-В мышей 18-месячного возраста лечили как p300i, так и носителем (ДМСО) в течение 1 недели и исследовали на следующий день после прекращения лечения. Репрезентативные графики проточной цитометрии показывают повышение частоты встречаемости CD4+ Т-клеток, экспрессирующих IFN-γ , в селезенке (А), и количества клеток, экспрессирующих IFN-γ , в CP (В), после лечения p300i. На фиг. 10С-Е представлены репрезентативные микроскопические изображения (С) и количественный анализ объема Аβ-бляшек в головном мозге 10-месячных AD-Tg мышей, которые получили p300i или носитель (ДМСО) в течение 1 недели и затем были исследованы еще через 3 недели. Препараты головного мозга были подвергнуты иммуноокрашиванию на Аβ-бляшки и ядра окрашивали Hoechst (n=5 на группу; масштабная линейка, 250 мкм). Были вычислены средние площадь Аβ-бляшек и количество бляшек в зубчатой извилине гиппокампа (DG) (D) и в 5-ом слое коры головного мозга (Е) (в 6 мкм срезах головного мозга; n=5-6 на группу; т-критерий Стьюдента). (F) схематическое представление режима лечения введением p300i (или ДМСО в качестве носителя) различным группам мышей AD-Tg в возрасте 7 месяцев, в течение 1 или 2 сессий. На фиг. 10G-H представлено изменение среднего значения процентного охвата коры головного мозга Аβ-бляшками (5-й слой) (G) и изменение средних уровней церебрального растворимого белка Аβ1-40 и Aβ1-42 (Н), по отношению к группе нелеченных AD-Tg (средний уровень АВ1-40 и Аβ1-42 в группе без лечения, 90,5±11,2 и 63,8±6,8 пг/мг общего количества, соответственно; n=5-6 на группу; однофакторный дисперсионный анализ ANOVA, с последующим апостериорным анализом («post-hoc») Ньюмена-Кейлса). На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[15] Как представлено на фиг. 11A-D, блокада PD-1 увеличивает процент CD4+ Т-клеток, продуцирующих IFN-γ , в селезенке, а также экспрессию IFN-γ в хороидном сплетении у мышей AD-Tg. 10-месячным мышам AD-Tg инъецировали интраперитонеально на 1 день и 4 день 250 мкг анти-PD-1 или контрольного IgG, и на 7-10-й дни исследовали эффект на системный иммунный ответ и активность СР. (А-В) Репрезентативные графики проточной цитометрии (А) и количественный анализ (В) частоты встречаемости спленоцитов CD4+IFN-γ+ (внутриклеточно окрашенных и с предварительным гейтингом по CD45 и TCR-β), у мышей AD-Tg, леченных анти-PD-1 или IgG, и нелеченных AD-Tg мышей и контрольных особей WT (n=4-6 на группу; однофакторный дисперсионный анализ ANOVA, с последующим апостериорным анализом («post-hoc») Ньюмена-Кейлса; **, Р<0,01 между указанными группами, получившими лечение; величина ошибки представляет среднее значение ± стандартная ошибка среднего). (С) уровни экспрессии мРНК ifn-g, измеренные RT-qPCR в CP мышей AD-Tg, леченных анти-PD-1, в сравнении с леченными IgG и с нелеченными контрольными AD-Tg (D) Термины генной онтологии (термины GO), обогащения в RNA-Seq (пер., RNAseq, технология РНК-секвенирования) в CPs тех же мышей (n=3-5 на группу; однофакторный дисперсионный анализ ANOVA, с последующим апостериорным анализом («post-hoc») Ньюмена-Кейлса; *, Р<0,05) (серая шкала соответствует отрицательному десятичному логарифму значения Р).
[16] Как представлено на фиг. 12А-В, блокада PD-1 уменьшает когнитивный спад у AD-Tg мышей. 10-месячным мышам AD-Tg интраперитонеально инъецировали на 1-й и 4-й день 250 мкг как анти-PD-1, так и контрольный IgG, и через 1 или 2 месяца исследовали эффект на патологию, (А) демонстрирующая результаты обучения мышей AD-Tg в RAWM-тесте после 1 сеанса лечения анти-PD-1 или контрольным IgG, и (В) демонстрирующая эффект одиночного сеанса лечения анти-PD-1, или 2 сеансов с перерывом 1 месяц на обучение. Одиночные стрелки указывают временные точки лечения, двойные стрелки указывают временные точки когнитивного тестирования. Когнитивную деятельность мышей, леченных анти-PD-1 и IgG, по сравнению с мышами AD-Tg дикого типа соответствующего возраста и нелеченными мышами, оценивали по среднему количеству ошибок в день при выполнении задач обучения и запоминания в RAWM-тесте (n=6-8 на группу; двухфакторный дисперсионный анализ с повторными измерениями ANOVA с последующими попарными апостериорными («post-hoc») сравнениями с критерием Бонферрони).
[17] На фиг. 13A-D представлены репрезентативные микроскопические изображения, показывающие, что блокада PD-1 уменьшает патологию БА (А), и количественный анализ (В, С, D) нагрузки Аβ-бляшками и астроглиоза в головном мозге AD-Tg мышей, которых лечили в возрасте 10 месяцев, как анти-PD-1 (в 1 или 2 сеансах, как показано на фиг. 12А-b), так и IgG, и затем исследовали в возрасте 12 месяцев. Препараты головного мозга подвергали иммуноокрашиванию на Аβ-бляшки (красный), GFAP (маркер астроглиоза, зеленый) и ядра окрашивали Hoechst (n=4-5 на группу; масштабная линейка, 50 мкм). Количественно выражали среднюю площадь и количество Аβ-бляшек в зубчатой извилине гиппокампа (DG) и 5-ом слое коры головного мозга, и измеряли GFAP-иммуноактивность в гиппокампе (в 6 мкм срезах головного мозга; n=5-6 на группу; т-критерий Стьюдента). На всех панелях величина ошибки представляет собой среднее ± стандартная ошибка среднего; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
[18] На фиг. 14 представлен эффект различного дозирования и частоты введения анти-PD-1-антитела на снижение когнитивных способностей у мышей AD-Tg, и иллюстрируется схема дозирования и влияние лечения анти-PD-1 антителом на пространственное обучение и память с использованием теста в радиальном рукавном водном лабиринте (RAWM) в возрасте 7 месяцев. Черные стрелки указывают моменты времени лечения, иллюстрации показывают моменты времени когнитивного тестирования.
[19] На фиг. 15А-Н представлен эффект повторного введения анти-PD-1 антитела на снижение когнитивных способностей у мышей АД-Tg. Мышей 5XFAD лечили с использованием как PD-1 специфическим антителом, так и контрольным IgG, начиная с 3-месячного возраста; лечение проводили раз в месяц до 5-месячного возраста (всего три инъекции). Схема эксперимента представлена в (А). Черные стрелки указывают моменты времени лечения, и иллюстрации показывают моменты времени когнитивного тестирования. (В) Обучение в RAWM в возрасте 5 месяцев, леченных анти-PD-1 мышей 5XFAD (n=7), мышей 5XFAD (n=9), леченных контрольным антителом (IgG); и мышей дикого типа (WT) (n=8). (С) Обучение в RAWM в возрасте 6 месяцев, леченных анти-PD-1 мышей 5XFAD (n=7), мышей 5XFAD (n=9), леченных контрольным антителом (IgG); и контрольных мышей дикого типа (WT) (n=8). (двухфакторный дисперсионный анализ с повторными измерениями ANOVA с апостериорным критерием Даннетта для множественных сравнений между двумя леченными группами 5XFAD). (D) Сравнение рекзультатов обучения в группах, леченных анти-PD-1 и IgG в возрасте 5 и 6 месяцев; значения, указывающие число ошибок для каждой мыши, взяты из последнего измерения, выполненного на второй день тестирования. (Е) Репрезентативные иммунофлуоресцентные изображения и (F) количественный анализ нейронов Neu-N+ в подставке гиппокампа мышей 5XFAD, леченных анти-PD-1 (n=9); леченных IgG мышей 5XFAD (n=10) и контрольных мышей дикого типа (WT) (n=6). (G) Репрезентативные иммунофлуоресцентные изображения нейронов гиппокампа, демонстрирующих повышенную активность каспазы-3 у леченных IgG мышей 5XFAD (n=8) по сравнению с леченными анти-PD-1 мышами 5XFAD (n=5). (Н) Количественое определение активированных каспаза-3-иммунореактивных клеток Neu-N+ в области СА3 гиппокампа (однофакторный ANOVA и точный критерий Фишера). Масштабные линейки, 100 мкм (е, g). Данные представлены как среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001
[20] На фиг. 16 представлен эффект однократного введения анти-TIM-3 анатитела на снижение когнитивных способностей у мышей AD-Tg, и иллюстрируется схема дозирования и эффект лечения анти-TIM-3 антителом на пространственное обучение и память с использованием теста в радиальном рукавном водном лабиринте (RAWM) в возрасте 7 месяцев. Черные стрелки указывают моменты времени лечения, иллюстрации показывают моменты времени когнитивного тестирования.
[21] Как показано на фиг. 17A-J, блокада PD-1 смягчает снижение когнитивных способностей у мышей AD-Tg. Мышей 5XFAD (в возрасте 7 месяцев) лечили как PD-1-специфическим антителом, PD-L1-специфическим антителом, так и изотипически сходным контрольным антителом (IgG). Схема эксперимента представлена на (А). Черные стрелки показывают моменты времени лечения, и иллюстрации показывают моменты времени оценки когнитивных функций с использованием RAWM. (В) Обучение мышей 5XFAD в тесте RAWM (мужские и женские особи использовали в равных пропорциях во всех группах, включая контрольную с IgG), леченных как 0,1 мг/мышь (n=8), 0,5 мг/мышь (n=9), так и 1,5 мг/мышь (n=9) инъекциями однократной дозы анти-PD-L1- специфического антитела, или контрольного IgG при 1,5 мг/мышь (n=10). Однопометных мышей дикого типа (WT) по возрастным группам использовали в качестве дополнительной контрольной группы (n=10). (С) Сравнение обучения в тесте RAWM мышей 5XFAD, леченных как 0,5 мг анти-PD-1-специфического антитела (n=14), так и 0,5 мг анти-PD-L1-специфического антитела (n=7); и также тестировали IgG контрольное антитело (n=15) и контроли WT (n=19) (двухфакторный дисперсионный анализ с повторными измерениями ANOVA с апостериорным критерием Даннетта для множественных сравнений между каждой группой, леченной анти-PD-1, и группой, леченной IgG). Приводятся объединенные результаты двух экспериментов. (D) Репрезентативные иммунофлуоресцентные изображения препаратоа головного мозга, иммуноокрашенных на Aβ (красный), GFAP (зеленый) и окрашивание ядер DAPI (масштабные линейки, 100 мкм), и количественный анализ (Е, F) Аβ у мышей 5XFAD, леченных анти-PD-1 (n=9), мышей 5XFAD, леченных анти-PD-L1 (n=10) и мышей 5XFAD, леченных IgG (n=9). (G) GFAP у мышей 5XFAD, леченных анти-PD-1 (n=8), леченных анти-PD-L1 (n=10), мышей 5XFAD, леченных IgG (n=15) и мышей WT (n=6), по результатам оценки через 1 месяц после лечения. Количественно определяли среднюю площадь и число бляшек (в 6 мкм срезах головного мозга) в зубчатой извилине (DG) и в коре головного мозга (слой V), и измеряли иммуноактивность к GFAP в гиппокампе (однофакторный ANOVA и точный критерий Фишера). (Н) Репрезентативные иммунофлуоресцентные изображения и (I) количественный анализ синаптофизина, оценивали через 1 месяц после лечения, в препаратах головного мозга мышей 5XFAD, леченных анти-PD-1 (n=6), мышей 5XFAD, леченных анти-PD-L1 (n=8) и мышей 5XFAD, леченных IgG (n=8). Иммунореактивность к синаптофизину измеряли в областях DG и СА3 гиппокампа (однофакторный ANOVA и точный критерий Фишера). (J) Уровни экспрессии мРНК il-12р40 относительно экспрессии il-10, измеряли RT-gPCR в ткани гиппокампа, выделенной из мышей 5XFAD через 1 месяц после лечения контрольным IgG (n=5), анти-PD-1 (n=5), или anti-PD-L1 (n=5) (однофакторный ANOVA и точный критерий Фишера). (В-С, E-G, I-J) Данные представлены как среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001
[22] На фиг. 18А-В представлено, что экспрессия PD-L1 возрастает в CP с возрастом, при этом (А) демонстрируя экспрессию PDL1 в CP молодых (левый столбец) и взрослых (правый столбец) мышей, измеренную RT-gPCR; и (В) представлено иммуногистохимическое окрашивание экспрессии PD-L1 эпителия в CP молодых (левый микроснимок) и взрослых (правый микроснимок) мышей. LV; боковой желудочек.
[23] На фиг. 19А-В представлены графики толщины внешнего ядерного слоя (ONL) по всей сетчатке, измеренной в отдельных глазах крыс RCS, получивших интраперитонеальную инъекцию анти-PD1 mAb (n=10 крыс; 20 глаз), IgG (n=10 крыс, 20 глаз), или животных, не получивших лечение (n=4 крысы, 8 глаз), посредством гистологического анализа, основанного на окрашивании гематоксилином и эозином (Н&Е), на (А) показаны все животные, и на (В) показаны только ответившие на лечение. Данные представлены как среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001
[24] На фиг. 20А-В представлены графики толщины внешнего ядерного слоя (ONL) по всей сетчатке, измеренной в отдельных глазах крыс RCS, получивших интравитреальную инъекцию анти-PD1 mAb (n=6) или IgG (n=5), или посредством гистологического анализа, основанного на окрашивании гематоксилином и эозином (Н&Е), с демонстрацией обоих глаз (инъецируемый глаз и контралатеральный глаз, который не инъецировали); на (А) показаны животные, леченные анти-PD1 mAb, и на (В) животные, леченные IgG. Данные представлены как среднее ± стандартное значение ошибки; существенные различия были определены с использованием т-критерия Стьюдента по каждой отдельной точке выборки и отмечены звездочками (*Р<0,05).
[25] Как показано на фиг. 21А-Е, блокада PD-1/PD-L1 ствола головного мозга у мышей DM-hTau повышает рекрутинг моноцитов в головной мозг.10-месячных мышей DM-hTAU, которых лечили 0,5 мг анти-PD-L1 (n=10) или IgG-совместимым контрольным антителом (IgG) (n=17), и однопометных мышей WT по возрастным группам (n=13) анализировали через 14 дней после лечения с использованием метода проточной цитометрии. (А) Стратегия гейтирования спленоцитов методом проточной цитометрии и (В) количественный анализ регуляторных Т-клеток FoxP3+, и (С) эффекторных Т-клеток (TEF) памяти CD44+CD62L-/low(TEF), а также (С) эффекторных Т-клеток (TEF) памяти CD44+CD62L-/low(TEF) по сравнению с центральными Т-клетками (TCM) памяти CD44+CD62Lhigh. (D) Стратегия гейтирования в проточной цитометрии для миелоидных клеток головного мозга CD45highCD11b+ и CD45highCD11b+. (Е) Головной мозг тех же самых мышей вырезали и анализировали на присутствие инфильтрирующих миелоидных клеток CD45highCD11b+. Количественный анализ клеток CD45highCD11b+головного мозга, показывающий повышенную частоту встречаемости инфильтрирующих миелоидных клеток, через 14 дней после блокады PD-L1 (n=10) по сравнению с леченными IgG мышами DM-hTAU (n=16) и однопометными мышами WT (n=13). Объединяли результаты двух независимых экспериментов. Данные показаны как среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001
[26] Как показано на фиг. 22A-G, блокада сигнального пути PD-1/PD-L1 ствола головного мозга у мышей DM-hTau повышает рекрутинг моноцитов в головной мозг. Мужские особи мышей, экспрессирующих ген tau человека с двумя мутациями (K257T/P301S, двойной мутант, DM-hTAU) (средний возраст в группе, 8 месяцев), лечили анти-PD-1-специфическим антителом, анти-PD-L1-специфическим антителом, или контрольным антителом соответствующего изотипа (IgG) (одна инъекция интраперитонеально по 0,5 мг/мышь); план эксперимента представлен в (А). Черная стрелка показывает момент времени лечения, и иллюстрации показывают моменты времени оценки когнитивных функций. (В) Эффект блокады PD-1/PD-L1 на пространственную память с использованием Т-лабиринта. Мыши DM-hTAU, леченные как анти-PD-1 (n=10), так и анти-PD-L1 (n=10), демонстрировали предпочтение новому рукаву, по сравнению с контрольными IgG (n=16) (В-С). Однопометных мышей WT по возрастным группам (n=19) использовали в качестве дополнительной контрольной группы. Объединяли результаты двух независимых экспериментов. (С) Репрезентативные графики теплокарт времени трех групп испытуемых, проведенного в разных рукавах. (D) Результаты выполнения задачи в Y-лабиринте мышами DM-hTAU, получавшими анти-PD-1 (n=6), анти-PD-L1 (n=4), или контрольными по изотипу IgG (n=6), а также мышами дикого типа одного помета (n=5). (Е) Репрезентативные иммунофлуоресцентные изображения и (F) количественный анализ иммунореактивности к GFAP, при оценке через 1 месяц после лечения, в гиппокампе мышей DM-TAU+IgG (n=6), DM-hTAU + анти-PD-1 (n=6), DM-hTAU + анти-PD-L1 (n=4) и нелеченных мышей однопометных мышей WT (n=5). Иммунореактивность к GFAP в головном мозге мышей DM-hTAU + IgG (n=11), DM-TAU + анти-PD-1 (n=11), DM-tau + анти-PD-L1 (n=4) и нелеченных мышей однопометных мышей WT (n=5), по сравнению с контрольными, леченными IgG (однофакторный ANOVA и точный критерий Фишера). Масштабные линейки, 100 мкм (G) уровни экспрессии мРНК tnf-a; il-6 и il-12р40 по сравнению с экспрессией il-10, измеренные RT-gPCR, в гиппокампе, выделенном из мышей DM-hTAU через 1 месяц после лечения контрольным IgG (n=5), анти-PD-1 (n=6), или анти-PD-L1 (n=4) (однофакторный ANOVA и точный критерий Фишера), (b, d, f-g) Данные представлены как среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001
[27] На фиг. 23A-G представлено, что блокада сигнального пути PD-1/PD-L1 снижает гиперфосфорилирование у мышей DM-tau. Иммуноокрашивание нейрофибриллярных клубков (NFTs) в головном мозге 8-месячных мышей DM-hTAU через 1 месяц после лечения анти-PD-1, анти-PD-L1 или контрольным антителом соответствующего изотипа, или контрольным антителом соответствующего изотипа (IgG) (A-F). (A, D) Репрезентативные иммунофлуоресцентные изображения и (В, С, Е, F) количественный анализ АТ-100 и АТ-180. Иммунореактивность АТ-100 и АТ-180 измеряли в областях гиппокампа СА1 и СА3 у мышей DM-hTAU + IgG (n=11), DM-hTAU + анти-PD-1 (n=10), DM-tau + анти-PD-L1 (n=4) (однофакторный ANOVA и точный критерий Фишера). Масштабные линейки, 100 мкм (В-С, E-F) Данные представлены как среднее ± стандартная ошибка среднего. (G) Мужские и женские особи мышей (одинаково распределенным среди всех тестируемых групп), экспрессирующих белок tau человека с двумя мутациями (K257T/P301S, двойной мутант, DM-hTAU) (средний возраст 9 месяцев), лечили как инъекциями однократной дозы анти-PD-L1-специфического антитела 0,1 мг/мышь (n=7), 0,5 мг/мышь (n=10) или 1,5 мг/мышь (n=9), так и контрольного IgG 1,5 мг/мышь (n=10). Эффект блокады PD-L1 на пространственную память определяли с использованием Т-лабиринта. Однопометных мышей WT по возрастным группам использовали в качестве дополнительной контрольной группы (n=10) (однофакторный ANOVA и точный критерий Фишера); * Р<0,05, **Р<0,01, ***Р<0,001.
[28] На фиг. 24А-В представлено, как блокада PD-1 усиливает нейрогенез в гиппокампе мышей 5XFAD, при этом (А) показаны парасагиттальные срезы головного мозга, иммунноокрашенные на нейронный маркер NeuN (зеленый), DCX (красный) и окрашивание ядер Hoechst (синий); а также (В) приведен график, количественно определяющий окрашивание у животных, леченных анти-PD-1, у иммунных контролей IgG и у контрольных особей дикого типа соответствующего возраста.
[29] На фиг. 25А-В представлено, как блокада PD-1 усиливает синаптическую пластичность гиппокампа у мышей 5XFAD, при этом (А) показаны парасагиттальные срезы головного мозга, иммунноокрашенные на VgluT1 (красный); а также (В) приведен график, количественного определяющий окрашивание у животных, леченных анти-PD-1, у иммунных контролей IgG и у контрольных особей дикого типа соответствующего возраста.
[30] На фиг. 26А-В представлено, как блокада PD-1 снижает нейронные потери в субикулуме у мышей 5XFAD, при этом (А) показаны парасагиттальные срезы головного мозга, иммунноокрашенные на нейронный маркер NeuN (зеленый); а также (В) приведен график, количественно определяющий окрашивание у животных, леченных анти-PD-1, у иммунных контролей IgG и у контрольных особей дикого типа соответствующего возраста.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[31] Механизмы контрольных точек иммунного ответа (механизмы иммунных контрольных точек), которые включают в себя внутреннюю клеточную понижающую регуляцию (даун-регуляцию, негативную регуляцию) реактивности (отвечаемости) активированных Т-клеток и эффекторную функцию при помощи ингибиторых рецепторов, поддерживают системный иммунный гомеостаз и аутоиммунную толерантность (Joller et al, 2012; Pardoll, 2012). В последние годы блокада этих иммуных контрольных точек, таких как сигнальный путь программируемой клеточной гибели-1 (PD-1) (Francisco et al, 2010), продемонстрировала заметную противоопухолевую эффективность, подчеркнув потенциал развязывания возможностей иммунной системы в борьбе с различными злокачественными новообразованиями. Недавно было показано (WO 2015/136541, Baruch et al., 2016), что введение антител к PD-1 (анти-PD-1 антител) модельному животному с болезнью Альцгеймера приводит к клиренсу Aβ, отмене снижения когнитивных способностей, и связано со снижением нейровоспалительной реакции. Таким образом, системная иммуносупрессия препятствует способности избавиться от патологии БА и путем снятия ограничений на системный иммунитет патология БА может быть смягчена.
[32] Не желая быть связанными какой-либо теорией, авторы полагают, что блокада иммунных контрольных точек, активирует каскад иммунологических событий, который начинается на периферии и завершается многочисленными активностями внутри головного мозга. Сначала иммунный ответ увеличивает доступность IFN-γ во вторичных лимфоидных органах (лимфатических узлах, селезенке и т.д.) и циркулирующих моноцитах на периферии. Этот иммунный ответ приводит к иммунной активации сосудистого (хороидного) сплетения (CP) головного мозга, эпителиального слоя в желудочках головного мозга, который формирует гематоликворный барьер (B-CSF-B) и служит селективным шлюзом для лейкоцитов, поступающих в ЦНС. Эффект блокады ингибиторных иммунных контрольных точек на активность CP-шлюза для лейкоцитов опосредуется индуцированной IFN-γ экспрессией молекул миграции лейкоцитов (молекул адгезии и хемокинов) посредством эпителия CP, который обеспечивает миграцию лейкоцитов. Эта повышенная экспрессия приводит к рекрутингу макрофагов моноцитарного происхождения и иммунорегуляторных клеток в пораженные участки внутри головного мозга. Важно, что этот рекрутинг приводит к комплексному действию на функции головного мозга, в том числе уменьшению объема бляшек, восстановлению иммунологического баланса, устранению местного воспаления, уменьшению глиоза, снижению синаптических потерь, усилению нейрогенеза, повышению защиты нейронов и повышению выживаемости нейронов, что в совокупности приводит к нейропротекции и/или ослаблению спада когнитивных способностей.
[33] Иммунные контрольные точки представляют собой молекулы иммунной системы, которые либо включают сигнал (костимуляторные молекулы), либо отключают сигнал. Четыре стимулирующие молекулы контрольной точки являются членами суперсемейства рецепторов фактора некроза опухолей (TNF) - CD27, CD40, ОХ40, GITR и CD137. Две другие стимулирующих молекулы контрольной точки принадлежат к суперсемейству B7-CD28, собственно CD28, и ICOS. Известны многие ингибиторные молекулы контрольных точек, включая, без ограничения, A2aR, В7-Н3, В7-Н4, BTLA, CTLA-4, IDO, KIR, LAG-3, PD-1, TIM-3 и VISTA.
[34] Настоящее изобретение обеспечивает способ лечения заболевания, расстройства, патологического состояния или поражения центральной нервной системы (ЦНС). В одном варианте осуществления раскрытый способ лечения болезни, расстройства, патологического состояния или поражения центральной нервной системы (ЦНС), не включает в себя аутоиммунное нейровоспалительное заболевание, рецидивирующе-ремиттирующий рассеянный склероз (РРРС). Раскрываемый способ, содержащий введение пациенту, который в этом нуждается, активного агента, который вызывает снижение уровня системной иммуносупрессии, где указанный активный агент вводят в режиме дозирования, содержащем, по меньшей мере два курса лечения, каждый курс лечения содержит последовательно сеанс лечения, за которым следует перерыв без лечения.
[35] В другом аспекте, настоящее изобретение направлено на активный агент, который вызывает снижение уровня системной иммуносупрессии у пациента, или фармацевтическую композицию, содержащую активный агент, для применения при лечении заболевания, расстройства, патологического состояния или поражения ЦНС, кроме аутоиммунного нейровоспалительного заболевания, рецидивирующе-ремиттирующего рассеянного склероза (РРРС), где указанную фармацевтическую композицию вводят в режиме дозирования, содержащем, по меньшей мере два курса лечения, при этом каждый курс лечения содержит последовательно сеанс лечения, за которым следует перерыв без лечения.
[36] В некоторых вариантах осуществления режим дозирования откалиброван таким образом, что уровень системной иммуносупрессии временно снижается.
[37] В применении здесь, термин «лечение» относится к средствам получения желаемого физиологического эффекта. Эффект может быть терапевтическим с точки зрения частичного или полного излечивания заболевания и/или симптомов, связанных с заболеванием. Термин относится к ингибированию заболевания, т.е. остановке или замедлению его развития; или смягчению заболевания, т.е. достижению регрессии заболевания.
[38] В применении здесь термин «системное присутствие» регуляторных или эффекторных Т-клеток относится к присутствию регуляторных или эффекторных Т-клеток (измеряемых их уровнем их активностью) в циркуляторной иммунной системе, то есть в крови, селезенке и лимфатических узлах. В области иммунологии хорошо известно, что профиль клеточной популяции в селезенке отражается в профиле клеточной популяции в крови (Zhao et al, 2007).
[39] Настоящее лечение применимо как к пациентам, демонстрирующим повышение системной иммуносупрессии, так и к пациентам, у которых нет такого повышения. Иногда субъект, который нуждается в лечении в соответствии с настоящим изобретением, имеет определенный уровень периферической иммуносупрессии, которая отражается повышенной частотой встречаемости или числом регуляторных Т-клеток в циркулирующей крови, и/или их повышенной функциональной активностью, и/или снижением числа лейкоцитов, продуцирующих IFNγ, и/или пониженной пролиферацией лейкоцитов в ответ на стимуляцию. Повышение частоты встречаемости или количества регуляторных Т-клеток может выражаться в общих количествах или в процентах от общего числа CD4 клеток. Например, в соответствии с настоящим изобретением было установлено, что животная модель болезни Альцгеймера имеет более высокую частоту встречаемости Foxp3 из клеток CD4 по сравнению с мышами дикого типа. Однако, даже если уровни системных регуляторных Т-клеток не повышаются, их функциональная активность не усиливается, уровень лейкоцитов, продуцирующих IFNγ, не снижается, или пролиферация лейкоцитов в ответ на стимуляцию не понижается у упомянутого субъекта, способ, предлагаемый настоящим изобретением, снижающий уровень или активность системной иммуносупрессии, является эффективным для лечения болезней, расстройств, патологических состояний или поражений ЦНС, кроме аутоиммунного нейровоспалительного заболевания RPMS. Важно, что указанная системная иммуносупрессия также может включать дополнительные типы иммунных клеток, кроме регуляторных Т-клеток, такие как супрессорные клетки миелоидного происхождения (MDSC) (Gabrilovich & Nagaraj, 2009).
[40] Уровень системной иммуносупрессии может быть обнаружен различными способами, которые хорошо известны специалисту в данной области техники. Например, уровень регуляторных Т-клеток может быть измерен методом проточной цитометрии мононуклеарных клеток или Т-лимфоцитов периферической крови, иммуноокрашенных как на маркеры клеточной поверхности, так и на ядерные внутриклеточные маркеры Treg (Chen & Oppenheim, 2011), CD45, TCR-β, или CD4 маркеры лимфоцитов, путем измерения количества антител, специфически связанных с клетками. Функциональная активность регуляторных Т-клеток может быть измерена различными способами; например, широко используется анализ включения тимидина, при котором супрессию пролиферации Т-клеток CD4+CD25- (традиционные Т-клетки), стимулированную анти-CD3 mAb, измеряют включением [3Н] тимидина или применением CFSE (5-(и 6)-карбоксифлуоресцеин диацетат сукцинимидил эфира, который способен проникать в клетки; деление клеток измеряют в виде последовательного уменьшения наполовину интенсивности флуоресценции CFSE). Количество лейкоцитов, продуцирующих IFNγ, их активность или способность к пролиферации могут быть легко оценены специалистами в данной области с использованием распространенных методов; например, уровень лейкоцитов, продуцирующих IFNγ, может быть измерен проточной цитометрией мононуклеарных клеток периферической крови, после короткой стимуляции ex-vivo, и GolgiStop, и иммуноокрашивания IFNγ-внутриклеточным красителем (например, с использованием набора для фиксации/пермеабилизации BD Biosciences Cytofix/cytoperm™), путем сбора кондиционированной среды этих клеток и количественного определения уровня секретированных цитокинов, с помощью ИФА, или сравнением соотношения различных цитокинов в кондиционированной среде, например IL2/IL10, IL2/IL4, INFγ/TGFβ, и т.д. Уровни MDSC в периферической крови человека могут быть легко определены специалистом в данной области, например, с помощью анализа методом проточной цитометрии частоты встречаемости клеток DR-/LIN-/CD11b+, DR-/LIN-/CD15+, DR-/LIN-/CD33+ и DR(-/low)/CD14+, как описано (Kotsakis et al, 2012).
[41] В организме человека периферическая/системная иммуносупрессия может считаться повышенной, когда общее количество регуляторных Т-клеток в системе кровообращения превышает 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%, или выше, чем в здоровой контрольной популяции, доля регуляторных Т-клеток от общего количества CD4+ клеток повышена на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%, или выше, чем в здоровой контрольной популяции, или же функциональная активность регуляторных Т-клеток повышена на 10, 20, 30, 40, 50, 60, 70, 80, 90, или 100%, или выше, чем в здоровой контрольной популяции. Помимо этого, периферическая/системная иммуносупрессия может считаться повышенной, когда уровень лимфоцитов, продуцирующих IFNγ, или их активность меньше, чем в здоровой контрольной популяции, на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%; или пролиферация лейкоцитов в ответ на стимуляцию снижена по сравнению со здоровой контрольной популяцией на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%.
[42] Агент может считаться агентом, который вызывает снижение уровня системной иммуносупрессии, когда при введении агента пациенту общее число регуляторных Т-клеток в системе кровообращения у данного субъекта снижается на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100% по сравнению с уровнем до введения агента, доля регуляторных Т-клеток в общем числе CD4+ клеток снижается на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100% по сравнению со здоровой контрольной популяцией, или функциональная активность регуляторных Т-клеток снижается на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100% по сравнению с уровнем до введения агента. Кроме того, агент может считаться агентом, который вызывает снижение уровня системной иммуносупрессии, когда при введении агента пациенту общее число лейкоцитов, продуцирующих IFNγ, или их активность увеличивается на 10, 20, 30, 40, 50, 60, 70, 80, 90, 100% или больше; или пролиферация лейкоцитов в ответ на стимуляцию возрастает по сравнению со здоровой контрольной популяцией на 10, 20, 30, 40, 50, 60, 70, 80, 90, 100% или более.
[43] В некоторых вариантах осуществления активный агент вызывает снижение уровня системной иммуносупрессии путем снятия ограничений, наложенных на иммунную систему одной или несколькими иммунными контрольными точками, например, блокадой одной или нескольких иммунных контрольных точек.
[44] В некоторых вариантах осуществления снижение уровня системной иммуносупрессии связано с увеличением системного присутствия или активности лейкоцитов, продуцирующих IFNγ.
[45] В некоторых вариантах осуществления активный агент вызывает снижение уровня системной иммуносупрессии и, следовательно, повышение системного присутствия или активности эффекторных Т-клеток.
[46] В некоторых вариантах осуществления снижение уровня системной иммуносупрессии связано с повышением системного присутствия или активности цитокина IFNγ.
[47] В некоторых вариантах осуществления снижение уровня системной иммуносупрессии связано со снижением системного присутствия или активности регуляторных Т-клеток.
[48] В некоторых вариантах осуществления снижение уровня системной иммуносупрессии связано со снижением системного присутствия или активности цитокина IL-10.
[49] В некоторых вариантах осуществления снижение уровня системной иммуносупрессии связано со снижением системного присутствия или активности супрессорных клеток миелоидного происхождения (MDSCs).
[50] В некоторых вариантах осуществления активный агент вызывает снижение уровня системной иммуносупрессии и, следовательно, повышение системного присутствия или активности эффекторных Т-клеток.
[51] Контрольные точки, которыми можно манипулировать для высвобождения системной иммуносупрессии, именуемые здесь как пара, состоящая из рецептора иммунной контрольной точки и его нативного лиганда, или любого из двух партнеров. Например, PD-1, который имеет два известных лиганда, здесь называется «PDL1» или «PDL2», тогда как В7Н3, лиганд который еще не идентифицирован, называется просто «В7Н3». Контрольные точки, которыми можно манипулировать для высвобождения системной иммуносупрессии по настоящему изобретению, включают в себя, без ограничения, PD-1-PDL1, PD-1-PDL2, CD28-CD80, CD28-CD86, CTLA-4-CD80, CTLA-4-CD86, ИКО-B7RP1, В7Н3, В7Н4, В7Н7, В7-CD28-подобные молекулы, BTLA-HVEM, KIR-МНС класса I или II, LAG3-MHC класса I или II, CD137-CD137L, OX40-OX40L, CD27-CD70, CD40L-CD40, TIM3-GAL9, V-доменный иммуноглобулиновый супрессор активации Т-клеток (VISTA), стимулятор генов интерферона (STING), иммуноглобулин Т-клеток и иммунорецепторный ингибиторный мотив домена на основе тирозина (TIGIT), индуцированный глюкокортикоидами ФНО-родственный белок (GITR), A2aR-аденозин и индолеамин-2,3-диоксигеназа (IDO)-L-триптофан.
[52] Агенты, способные блокировать иммунные контрольные точки, известны в данной области техники (Colombo & Piconese, 2007) и могут быть использованы в соответствии с настоящим изобретением. Каждая из приведенных ниже публикаций, и Pardoll, 2012, включены посредством ссылки, как если бы их текст был полностью приведен в настоящем документе.
[53] В некоторых вариантах осуществления активный агент, который может быть использоан по настоящему изобретению, может быть антителом. Антитело, раскрытое в данном документе, может быть поликлональнм антителом, моноклональным антителом, димером, мультимером, мультиспецифическим антителом, человеческим антителом, гуманизированным антителом, рекомбинантным антителом, химерным антителом, бифункциональным антителом, клеточно-ассоциированным антителом подобным рецептору Ig, линейным антителом, диателом, минителом или нанотелом, при условии, что фрагмент обнаруживает необходимую биологическую активность, и их одноцепочечными производными. Антитело может быть полноразмерной молекулой иммуноглобулина, содержащей VH и VL-домены, а также константный домен легкой цепи (CL) и константные домены тяжелой цепи, СН1, СН2 и СН3, или иммунологически активным фрагментом полноразмерной молекулы иммуноглобулина, такой, как, например, однодоменное антитело (sdAb), одноцепочечный вариабельный фрагмент (scFv), Fab-фрагмент, F(ab')2-фрагмент, Fc фрагмент, Fd фрагмент, Fv фрагмент. Антитело может быть получено от любых видов позвоночных (например, человека, козы, лошади, осла, мыши, крысы, кролика или курицы) и может быть любого типа (например, IgG, IgE, IgM, IgD и IgA), класса (например, IgA, IgD, IgE, IgG и IgM) или подкласса (IgG1, IgG2, IgG3, IgG4, lgA1 и lgA2). Функционально антитело, раскрытое в данном документе, может быть антителом-антагонистом, то есть антителом, которое ингибирует биологическую активность, или же антитело, описанное здесь, может быть антителом-агонистом, то есть антителом, стимулирующим биологическую активность. Аналогичным образом, антитело, описанное здесь, может быть нейтрализующим антителом, то есть антителом, которое может блокировать или нейтрализовать биологическую активность. Основные требования к раскрытию информации о структуре естественных антител, не встречающихся в природе антител и их антигенсвязывающих фрагментов, см., например: Pluckthun in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., Springer-Verlag, New York, pp. 269-315 (1994); Borrabeck, Antibody Engineering, 2d ed. (Oxford University Press 1995), оба источника во всей своей полноте включены в настоящий документ посредством ссылки.
[54] Антитело, описанное здесь, может быть, без ограничения, анти-PD-1, анти-PDL1, анти-PDL2, анти-CTLA-4, анти-CD80, анти-CD86, анти-B7RP1, анти-В7-Н3, анти-В7-Н4, анти-В7-Н7, анти-BTLA, анти-HVEM, анти-CD-27, анти-CD40, анти-CD40L, анти-CD70, анти-CD80, анти-CD86, анти-CD137, анти-CD137L, анти-ОХ40, анти-OX40L, анти-TIM-3, анти-Galectin9, анти-KIR, анти-LAG-3, анти-ICOS, анти-VISTA, анти-STING, анти-TIGIT, анти-GITR или любым их сочетанием. Описанное здесь антитело может быть введено человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[55] Белок программируемой смерти 1, также известный как PD-1 и CD279 (кластер дифференцировки 279), представляет собой рецептор на поверхности клетки, принадлежащий к суперсемейству иммуноглобулина и экспрессируемый на Т-клетках и про-В-клетках. PD-1 связывает два лиганда, PDL1 и PDL2. Выступая в качестве иммунной контрольной точки, PD-1 играет важную роль в подавлении (down-регуляции) иммунной системы, предотвращая активацию Т-клеток, что, в свою очередь, уменьшает аутоиммунитет и способствует аутотолерантности. Ингибирующее воздействие PD-1 осуществляется через двойной механизм содействия апоптозу (программируемой смерти клеток) в антигенспецифичных Т-клетках в лимфатических узлах при одновременном снижении апоптоза в регуляторных Т-клетках (супрессорных Т-лимфоцитах). По сути, соединения, которые ингибируют функционирование PD-1, такие, как ингибиторы PD-1, ингибиторы PDL1 и/или ингибиторы PDL2, служат для активации иммунной системы. Один класс ингибиторов PD-1 включает в себя антагонистические или нейтрализующие антитела к PD-1, PDL1 и PDL2. В данной области техники известно много антагонистических или нейтрализующих антител к PD-1, PDL1 и PDL2. Например, антитело к PD-1, используемое согласно настоящему изобретению, может быть выбрано из тех, что описаны в Ohaegbulam et al. (Ohaegbulam et al, 2015), все содержание которого включено здесь посредством ссылки. Примеры человеческих или гуманизированных анти-PD-1 антител включают в себя, без ограничения, CD279 (человеческое моноклональное антитело к PD1, Bio X Cell), MEDI0680 (АМР-514, анти-PD-1 гуманизированное моноклональное антитело к IgG4, AstraZeneca), Ниволумаб (BMS-936558, анти-PD1 человеческое антитело к IgG4, Bristol-Myers Squibb), Пембролизумаб (Ламбролизумаб, МК-3475, анти-PDI гуманизированное моноклональное антитело к IgG4, Merck), Пидилизумаб (СТ-011, анти-PDI гуманизированное моноклональное антитело к IgG1,Medivation) и TSR-042 (анти-PD-1 гуманизированное моноклональное антитело к IgG4, Tesaro). Примеры человеческих или гуманизированных анти-PDL1 антител включают, без ограничения, Авелумаб (MSB0010718C, анти-PDL1 моноклональное антитело к IgG1 человека, Merck-Serono), Атезолизумаб (MPDL3280A, RG7446, анти-PDL1 моноклональное антитело к IgG человека Hoffmann-La Roche), BMS-936559 (MDX-1105, анти-PDL1 моноклональное антитело к IgG4 человека, Bristol-Myers Squibb), Дурвалумаб (MEDI4736, анти-PDL1 гуманизированное моноклональное антитело к IgG1, AstraZeneca), KN035 (анти-PDL1 моноклональное антитело, 3D Medicines) и LY3300054 (анти-PDL1 моноклональное антитело, Eli Lilly). Примеры человеческих или гуманизированных анти-PDL2 антител включают, без ограничения, АМР-224 (IgG2a Fc-слитый белок PDL2, AstraZeneca). В некоторых вариантах осуществления антитело к PD-1, антитело к PDL1 и/или антитело к PDL2 можно вводить человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[56] В некоторых вариантах осуществления человеку может быть введен Пидилизумаб в дозировке 0,2-6 мг/кг или от 1,5-6 мг/кг; Пембролизумаб можно вводить человеку в дозировке 1-10 мг/кг; Ниволумаб может быть введен человеку в дозировке 0,3-20 мг/кг, 0,3-10 мг/кг, 1-10 мг/кг или 1 или 3 мг/кг; BMS-936559 может быть введен человеку в дозировке 0,3-10 мг/кг; Атезолизумаб может быть введен человеку в дозировке 1-20 мг/кг; Дурвалумаб может быть введен человеку в дозировке 0,1-15 мг/кг; и Авелумаб может быть введен человеку в дозировке 1-20 мг/кг.
[57] Т-клеточный иммуноглобулин и домен-3 муцина (TIM-3) представляет собой Th1-специфический белок на поверхности клетки, действующий в качестве иммунной контрольной точки, которая ингибирует активность лимфоцитов путем понижающей регуляции (down-регуляции) активации макрофагов и играет важную роль в истощении CD8+ Т-клеток, которое происходит при хронических патологических состояниях иммунной системы. TIM-3 действует в качестве негативного регулятора функции Th1/TC1, вызывая гибель клеток при взаимодействии с его лигандом, galectin-9 (Gal9). По сути, соединения, которые ингибируют функцию TIM-3, такие, как ингибиторы TIM-3 и/или ингибиторы PD-Gal9, служат для активации иммунной системы. Один класс ингибиторов TIM-3 включает в себя антагонистические или нейтрализующие антитела к TIM-3 и/или Gal-9. В данной области техники известно много антагонистических или нейтрализующих антител к TIM-3 и Gal-9. Примеры человеческих или гуманизированных антител к TIM-3 включают, без ограничения, AF2365 (анти-TIM-3 моноклональное антитело к IgG человека, R&D Systems), CD366 (анти-TIM-3 моноклональное антитело к IgG1 человека, BioLegend), F38-2E2 (анти-TIM-3 моноклональное антитело к IgG1, R&D Systems), L3D (анти-TIM-3 моноклональное антитело к IgG1 человека, CN 102492038 В), МАВ2365 (анти-TIM-3 моноклональное антитело к IgG2a, R&D Systems), МАВ23651 (анти-TIM-3 моноклональное антитело к IgG1 человека, R&D Systems) и TSR-022 (гуманизированное анти-TIM-3 моноклональное антитело к IgG4, Tesaro). В некоторых вариантах осуществления, анти-TIM-3 антитело и (или) анти-Gal9 антитело могут вводиться человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[58] Ассоциированный с Т-лимфоцитами цитотоксический белок 4 (CTLA-4), также известный как кластер дифференцировки 152 (CD152), является белковым рецептором, функционирующим как иммунная контрольная точка, которая отрицательно регулирует (down-регулирует) иммунную реакцию. CTLA-4 конститутивно экспрессируется в Treg-клетках, но положительно регулируется только в традиционных Т-клетках после активации. CTLA-4 действует как «выключатель», когда связан с CD80 или CD86 на поверхности антигенпредставляющих клеток. По сути, соединения, ингибирующие функцию CTLA-4, такие, как ингибиторы CTLA-4, ингибиторы CD80 и/или ингибиторы CD86, служат для активации иммунной системы. Один класс ингибиторов CTLA-4 включает в себя антагонистические или нейтрализующие антитела к CTLA-4, CD80 и/или CD86. B данной области техники известно много антагонистических или нейтрализующих антител к CTLA-4, CD80 и CD86. Примеры человеческих или гуманизированных антител к CTLA-4 включают, без ограничения, Ипилимумаб (анти-CTLA-4 моноклональное антитело к IgG1 человека, Bristol-Myers Squibb) и Тремелимумаб (анти-CTLA-4 моноклональное антитело к IgG2 человека, Pfizer). В некоторых вариантах осуществления антитело к CTLA-4, антитело к PD-CB80 и/или антитело к PD-CD86 можно вводить человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[59] Иммуноглобулин-подобные рецепторы клеток-киллеров (KIRs) представляют собой семейство трансмембранных гликопротеинов типа I, экспрессируемых на плазматической мембране естественных клеток-киллеров (NK-клеток) и меньшей части Т-клеток. Они регулируют функцию киллинга этих клеток, взаимодействуя с молекулами главного комплекса гистосовместимости класса I (МНС), которые экспрессируются на всех типах ядросодержащих клеток. Таким образом, KIRs являются ингибиторами активности лимфоцитов. По сути, соединения, которые ингибируют функцию KIR, такие, как ингибиторы KIR, служат для активации иммунной системы. Один класс ингибиторов KIR включает в себя антагонистические или нейтрализующие антитела к KIR. B данной области техники известно много антагонистических или нейтрализующих антител к KIR. Примеры человеческих или гуманизированных анти-KIR антител включают, без ограничения, Лирилумаб (BMS-986015, человеческое анти-KIR моноклональное антитело, Bristol-Myers Squibb). В некоторых вариантах осуществления, анти-KIR антитело может быть введено человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[60] Активирующий лимфоциты ген 3 (LAG-3), также известный как кластер дифференцировки 223 (CD223), представляет собой молекулу на поверхности клетки с различными биологическими эффектами на функцию Т-клеток. LAG-3 является иммунной контрольной точкой, которая ингибирует активность лимфоцитов, подавляя иммунный ответ путем воздействия на Treg-клетки, а также оказывая прямое воздействие на CD8+ Т-клетки. По сути, соединения, которые ингибируют функцию LAG-3, такие, как ингибиторы LAG-3, служат для активации иммунной системы. Один класс ингибиторов LAG-3 включает в себя антагонистические или нейтрализующие антитела к LAG-3. В данной области техники известно много антагонистических или нейтрализующих антител к LAG-3. Примеры человеческих или гуманизированных анти-LAG-3 антител включают, без ограничения, BMS-986016 (человеческое анти-LAG-3 моноклональное антитело, Bristol-Myers Squibb). В некоторых вариантах осуществления, антитело к LAG-3 может быть введено человеку в дозировке, например, 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[61] ОХ40, также известный как кластер дифференцировки 134 (CD134), является членом суперсемейства рецепторов TNFR. ОХ40 способствует экспансии эффекторных Т-клеток и Т-клеток памяти, однако он также известен своей способностью подавлять дифференциацию и активность регуляторных Т-клеток, а также регуляцией образования цитокинов. Будучи транзиентно экспрессированным после вовлечения Т-клеточного рецептора, ОХ40 положительно регулируется только на тех Т-клетках, которые позже других были активированы антигеном при воспалительных поражениях. Его лигандом является OX40L, также известный как кластер дифференцировки 252 (CD252). По сути, соединения, которые активируют или стимулируют функцию ОХ40, такие как активаторы ОХ40 и (или) активаторы OX40L, служат для активации иммунной системы. Один класс активаторов ОХ40 включает антитела-агонисты к ОХ40 и OX40L. B данной области техники известно много антител-агонистов к ОХ40 и OX40L. Примеры человеческих или гуманизированных антител против ОХ40 включают, без ограничения, GSK3174998; гуманизированное анти-ОХ40 моноклональное антитело к IgG1; GlaxoSmithKline), MEDI0562 (гуманизированное анти-ОХ40 моноклональное антитело, MedImmune) и MEDI6383 (слитый белок человека ОХ40, MedImmune). Другие антитела к ОХ40 включают, без ограничения, MEDI6469 (9 В12, моноклональное антитело к ОХ40 мыши, MedImmune). В некоторых вариантах осуществления, анти-ОХ40 антитело и (или) анти-OX40L антитело могут вводиться человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[62] Анти-GITR-антитела направлены на глюкокортикоид-индуцированный белок, связанный с рецептором фактора некроза опухолей (GITR), который регулярно экспрессируется на поверхности регуляторных Т-клеток (Treg-клеток) и на поверхности эффекторных Т-клеток после их активации. Анти-GITR антитела блокируют взаимодействие GITR, обнаруживаемого на различных типах Т-клеток, с его лигандом, тем самым индуцируя как активацию опухоле-антигенспецифических Т-эффекторных клеток, так и отмену супрессии, индуцированной неправильно активированными Т-регуляторными клетками. По сути, соединения, активирующие или стимулирующие функцию GITR, такие как активаторы GITR, служат для активации иммунной системы. Один класс активаторов GITR включает антитела-агонисты к GITR. В данной области техники известно много антител-агонистов к GITR. Примеры человеческих или гуманизированных антител против TIGR включают, без ограничения, GWN323 (гуманизированное анти-GITR моноклональное антитело, Novartis) и TRX518 (гуманизированное анти-GITR моноклональное антитело; GITR, Inc.). В некоторых вариантах осуществления анти-GITR антитело может вводиться человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[63] CD27 является членом надсемейства рецепторов фактора некроза опухоли. Активность CD27 регулируется транзиентной доступностью его лиганда, CD70, на лимфоцитах и дендритных клетках. Активация CD27 играет ключевую роль в регуляции активации В-клеток и синтеза иммуноглобулина, поддерживает антигенспецифическую экспансию наивных Т-клеток, является необходимой для генерации и долгосрочного поддержания иммунитета Т-клеток и является маркером памяти В-клеток. CD27 передает сигналы, ведущие к активации NF-κВ и MAPK8/JNK. По сути, соединения, активирующие или стимулирующие функцию CD27, такие как активаторы CD27 и (или) активаторы CD70, служат для активации иммунной системы. Один класс активаторов CD27 включает антитела-агонисты к CD27 и/или CD70. B данной области техники известно много антител-агонистов к CD27 и (или) CD70. Примеры человеческих или гуманизированных анти-CD27 антител включают, без ограничения, Варлилумаб (CDX-1127, человеческое анти-CD27 моноклональное антитело, Celldex Therapeutics). В некоторых вариантах осуществления, анти-CD27 антитело и (или) анти-CD70 антитело могут вводиться человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[64] Индуцируемый Т-клеточный костимулятор (ICOS), также известный как кластер дифференцировки 278 (CD278), представляет собой костимуляторный рецептор клеточной поверхности суперсемейства CD28, который экспрессируется на активированных Т-клетках. Он является активатором функции Т-клеток. По сути, соединения, активирующие или стимулирующие функцию ICOS, такие как активаторы ICOS и/или активаторы B7RP1, служат для активации иммунной системы. Один класс активаторов ICOS включает антитела-агонисты к ICOS и/или B7RP1. В данной области техники известно много антител-агонистов к ICOS и B7RP1. В некоторых вариантах осуществления, анти-ICOS антитело и/или анти-B7RP1 антитело могут быть введены человеку в дозировке, например, около 0,1 мг/кг - 20 мг/кг, 0,1 мг/кг - 15 мг/кг, 0,1 мг/кг - 10 мг/кг, 0,1 мг/кг - 5 мг/кг, 0,2 мг/кг - 20 мг/кг, 0,2 мг/кг - 15 мг/кг, 0,2 мг/кг - 10 мг/кг, 0,2 мг/кг - 6 мг/кг, 0,2 мг/кг - 5 мг/кг, 0,3 мг/кг - 20 мг/кг, 0,3 мг/кг - 15 мг/кг, 0,3 мг/кг - 10 мг/кг, 0,3 мг/кг - 5 мг/кг, 1 мг/кг - 20 мг/кг, 1 мг/кг - 15 мг/кг, 1 мг/кг - 10 мг/кг, 1 мг/кг - 5 мг/кг, 1,5 мг/кг - 20 мг/кг, 1,5 мг/кг - 15 мг/кг, 1,5 мг/кг - 10 мг/кг, 1,5 мг/кг - 6 мг/кг или 1,5 мг/кг - 5 мг/кг.
[65] В некоторых вариантах осуществления могут быть использовны, среди прочего, такие комбинации антител, как: СТ-011 в сочетании с Ритуксимабом (торговые названия Rituxan, MabThera и Zytux), химерное моноклональное антитело к белку CD20, например, по 3 мг/кг каждого; Ниволумаб (например, 1 мг/кг) в сочетании с Ипилимумабом; например, 3 мг/кг); или Ниволумаб (например, 1-10 мг/кг) в сочетании с ограниченной мультипептидной вакциной HLA-A*0201 (Weber et al, 2013).
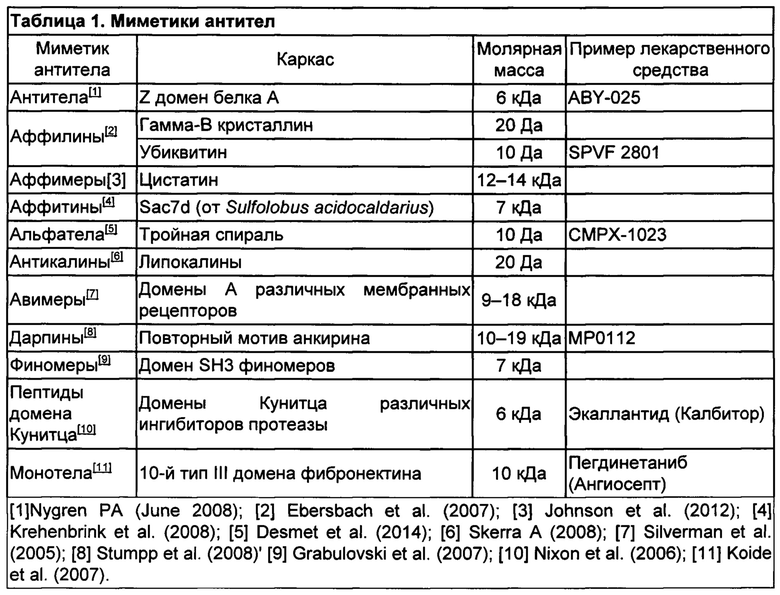
[66] В некоторых вариантах осуществления активный агент, который может быть применен по настоящему изобретению, может быть имитатором антитела. Имитаторы антител могут специфически связывать антигены, подобно антителам, но структурно они не родственны антителам. Обычно они представляют собой искусственные пептиды или белки с молярной массой от около 3 до 20 кДа. В аспектах настоящего варианта осуществления раскрываемый миметик антитела может быть молекулой аффитела; аффилином; аффимером; аффитином; альфателом; антикалином; авимером; дарпином; финомером; пептидом домена Куниц; или монотелом. Неограничивающие примеры антител-миметиков представлены в таблице 1:
[67] В некоторых вариантах осуществления активный агент, который может быть использован по настоящему изобретению, может быть аптамером. Аптамеры являются олигонуклеотидными или пептидными молекулами, которые связываются со специфической молекулой-мишенью. В аспектах настоящего варианта осуществления описанный здесь аптамер может представлять собой ДНК-аптамер, аптамер РНК, аптамер XNA или аптамер пептида.
[68] В некоторых вариантах осуществления описанное здесь антитело может быть в комбинации с адъювантом. К адъюванам относят любое вещество или смесь веществ, которые усиливают или диверсифицируют иммунный ответ. Адъювант может служить для снижения количества иммунизаций или количества антигена, необходимого для защитной иммунизации. Адъюванты включают в себя, без ограничения перечисленным, такие как, например, липосомы, масляные фазы, в том числе, без ограничения, адъюванты типа Фрейнда, такие как, например, полный адъювант Фрейнда (FCA); неполный адъювант Фрейнда (FIA); сапогениновые гликозиды, такие, как, например, сапонины; карбопол; N-ацетилмурамил-L-аланил-D-изоглутамин (общеизвестный как мурамилдипептид или «MDP»); и липополисахарид (LPS). Такие адъюванты обычно используются в виде эмульсии с водной фазой, или, чаще, с нерастворимыми в воде неорганическими солями. Эти неорганические соли включают в себя гидроксид алюминия, сульфат цинка, коллоидный гидроксид железа, фосфат кальция или хлорид кальция. В аспектах данного варианта осуществления описанное здесь антитело может быть объединено, например, с анти- CTLA-4 антителом в комбинации с анти-ОХ40 антителом и лигандом TLR9, таким как CpG (Marabelle et al, 2013).
[69] В некоторых вариантах осуществления активный агент, который может быть применен по настоящему изобретению, может быть малой молекулой. В аспектах этого варианта осуществления раскрытая здесь малая молекула может быть: (а) ингибитором p300 (Liu et al, 2013), таким, как гемцитабин (низкая доза), С646 или его аналоги, то есть соединение формулы I:
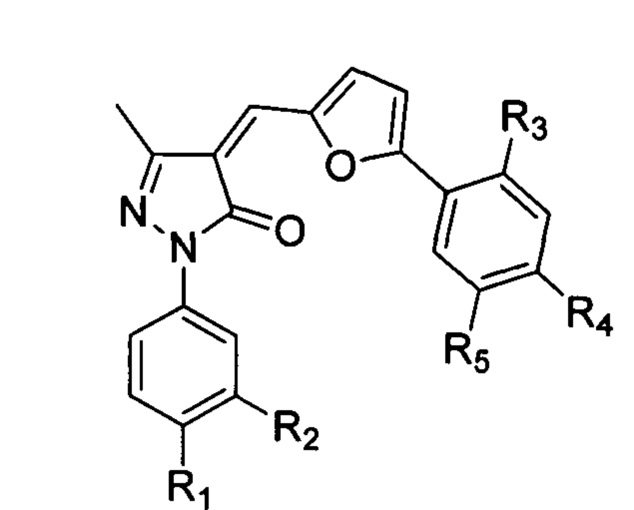
в которой выбран из Н, -CO2R6, -CONR6R7, -SO3H или -SO2NR6R7; R2 выбран из H, -CO2R6, или галогена, предпочтительно Cl; R3 выбран из галогена, предпочтительно F, -NO2, -CN, -CO2R6, предпочтительно CO2CH3 или CO2CH2CH3, или -СН2ОН; R4 и R5 каждый независимо от другого представляет собой Н или -С1-С6 алкил, предпочтительно метил; R6 представляет собой Н или -C1-C6 алкил, предпочтительно Н, метил или этил; и R7 представляет собой Н или -C1-C6 алкил, предпочтительно Н или метил; (b) Сунитиниб; (с) Полиоксомталат-1 (РОМ-1) (Ghiringhelli et al, 2012); (d) α,β-метиленаденозин 5'-дифосфат (АРСР); (е) трехокись мышьяка (As2O3); (f) GX15-070 (Обатоклакс); (g) анатагонист ретиноевой кислоты, такой как Ro 41-5253 (синтетический ретиноид и избирательный низкомолекулярный антагонист) или LE-135; (h) антагонист SIRPα (CD47), такой, как CV1-hIgG4 (вариант SIRPα) самостоятельно или в сочетании с анти-СР47-антителом; (i) антагонист CCR4, такой, как AF399/420/18025, самостоятельно или в сочетании с анти-CCR4 антителом; (j) антагонист аденозинового рецептора; (k) антагонист аденозинового рецептора А1; аденозиновый рецептор А2а; (m) антагонист аденозинового рецептора A2b; (n) антагонист аденозинового рецептора A3; (о) антагонист индолеамин-2,3-диоксигеназы; или (р) регулятор HIF-1.
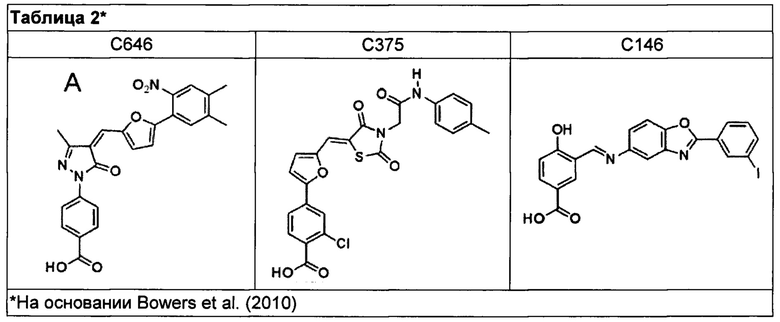
[70] В некоторых вариантах осуществления агент представляет собой ингибитор p300, формулы которого приведены в таблице 2, т.е. С646 (4-(4-((5-(4,5-диметил-2-нитрофенил)фуран-2-ил)метилен)-3-метил-5-оксо-4,5-дигидро-1Н-пиразол-1-ил)бензойная кислота), С146 (4-гидрокси-3-(((2-(3-иодфенил)бензо[д]оксазол-5-ил)имино)метил)бензойная кислота) или С375 (2-хлор-4-(5-((2,4-диоксо-3-(2-оксо-2-(п-толиламино)этил)тиазолидин-5-илиден)метил)фуран-2-ил)бензойная кислота). В частности, ингибитором p300 является С646.
[71] В некоторых вариантах осуществления антагонист аденозинового рецептора может представлять собой CGS15943 (9-Хлор-2-(2-фуранил)-[1,2,4]триазоло[1,5-с] хиназолин-5-амин); антагонистом аденозинового рецептора А1 может быть PSB 36 (1-Бутил-8-(гексагидро-2,5-метанопантенан-3а(1Н)-ил)-3,7-дигидро-3-(3-гидроксипропил)-1Н-пурин-2,6-дион); антагонистом аденозинового рецептора А2а может быть SCH58261 (5-Амино-7-(2-фенилэтил)-2-(2-фурил)-пиразоло(4,3-е)-1,2,4-триазоло(1,5-с)пиримидин), SYN115 (4-гидрокси-N-[4-метокси-7-(4-морфолинил)-2-бензотиазолил]-4-метил-1-пиперидинкарбоксамид), FSPTP (также называемый SCH58261 (5-амино-7-[2-(4-фторсульфонил)фенилэтил]-2-(2-фурил)-пиразоло[4,3-е]-1,2,4-триазоло[1,5-с]пиримидин), SCH442416 (2-(2-фуранил)-7-[3-(4-метоксифенил)пропил]-7Н-пиразоло[4,3-е][1,2,4]триазоло[1,5-с]пиримидин-5-амин), или ZM241385 (также называемый тозадентантом (4-гидрокси-N-(4-метокси-7-морфолинобензо[d] тиазол-2-ил)-4-метилпиперидин-1-карбоксамид), антагонист аденозинового рецептора A2b может представлять собой PSB 603 (8 {4-[4-(4-хлорфенил)пиперазин-1-сульфонил]фенил}-1-пропил-2,3,6,7-тетрагидро-1Н-пурин-2,6-дион (Nakatsukasa et al, 2011)), а антагонист рецептора A3 может представлять собой MRS3777 (2-фенокси-6-(циклогексиламино)пурин-гемиоксалат).
[72] В некоторых вариантах осуществления низкомолекулярным ингибитором пути индоламина-2,3-диоксигеназы может быть индоксимод (NSC-721782/NLG-9189 (1-метил-D-триптофан), NewLink Genetics), INCB024360 ((4Е)-4-[(3-хлор-4-фторанилино)нитрозометилиден]-1,2,5-оксадиазол-3-амин, Incyte) или NLG-919 (1-Циклогексил-2-(5Н-имидазо[5,1-а]изоиндол-5-ил)этанол), NewLink Genetics).
[73] HIF-1 регулятором может быть М30, (5-N-метил-N-пропаргиламинометил]-8-гидроксихинолин), описанный в Zheng et al. (Zheng et al, 2015).
[74] В некоторых вариантах осуществления активным агентом, который может быть применен по настоящему изобретению, может быть любая комбинация раскрываемого здесь антитела и раскрываемой здесь малой молекулы. В аспектах этого варианта осуществления активным агентом может быть любая комбинация описанного здесь антитела и описанной здесь малой молекулы.
[75] В некоторых вариантах осуществления активным агентом, который быть применен по настоящему изобретению, может быть белок, выбранный из группы, состоящей из: (а) гликопротеина из листьев нима (NLGP; (Roy et al, 2013)); и (или) (b) sCTLA-4 (растворимая изоформа CTLA-4) (Ward et al, 2013).
[76] В некоторых вариантах осуществления активный агент, который может быть применен по настоящему изобретению, может быть молекулой сайленсинга. В аспектах этого варианта осуществления молекула сайленсинга выбирана из группы, состоящей из антисмысловой miR-126 (Qin et al, 2013) и анти-галектина-1 (Gal-1; (Dalotto-Moreno et al, 2013)).
[77] В некоторых вариантах осуществления активным агентом, который может быть применен по настоящему изобретению, может быть OK-432 (лиофилизированный препарат Streptococcus pyogenes) (Hirayama et al, 2013).
[78] В некоторых вариантах осуществления активный агент, который может быть применен по настоящему изобретению, может быть сочетанием IL-12 и анти-CTLA-4.
[79] В некоторых вариантах осуществления агент может быть производным от широкого спектра антибиотиков, нацеленным на грамположительные и грамотрицательные бактерии, тем самым способствуя иммуномодуляции регуляторных Т-клеток, например, ванкомицин, нацеленный на грамположительные бактерии и в отношении которого было показано, что он уменьшает уровень/активность Treg-клеток.
[80] В некоторых вариантах осуществления активным агентом, который может быть применен по настоящему изобретению, может быть любая комбинация раскрытого здесь антитела, раскрытого здесь миметика антитела, раскрытого здесь аптамера, раскрытой здесь малой молекулы, раскрытого здесь гликопротеина листьев Нима, раскрытого здесь sCTLA-4, раскрытой здесь молекулы сайленсинга, раскрытого здесь OК-432 и/или раскрытая здесь комбинация IL-12 и анти-CTLA-4.
[81] Как указывалось выше, активный агент вводят в режиме дозирования, содержащем по меньшей мере два курса лечения, при этом каждый курс лечения содержит сеанс лечения, за которым следует перерыв.
[82] Термины «сеанс лечения» и «период лечения» здесь взаимозаменяемы и относятся к периоду, во время которого пациенту вводят один или несколько описанных здесь активных агентов. Как будет подробнее рассмотрено ниже, сеанс лечения может представлять собой однократное дозирование или режим с неоднократным дозированием, осуществляемый в течение определенного периода времени. Сеанс лечения заключается в том, чтобы на всем его протяжении поддерживалось терапевтически эффективное количество активного агента, описанного здесь.
[83] Термины «период без лечения», «перерыв в лечении», «период отсутствия лечения», «перерыв» здесь взаимозаменяемы и относятся к периоду, во время которого пациенту, проходящему лечение, не вводят описанный здесь активный агент. Прекращение введения активного агента во время периода без лечения приводит к снижению содержания описанного здесь активного агента до субтерапевтического уровня у пациента, проходящего лечение. Как описано здесь, «период без лечения» представляет собой не то же самое событие, что период времени между дозированиями, составляющими многократный режим дозирования, который происходит в течение сеанса лечения. Если введение одного или нескольких описанных здесь активных агентов во время сеанса лечения является повторным, период без лечения будет продолжительнее, чем промежуточный период между этими повторными приемами во время сеанса лечения.
[84] Режим дозирования может быть определен несколькими способами. Например, уровень иммуносупрессии может быть откалиброван до желаемого уровня для каждого пациента, проходящего лечение (персонализированная медицина), путем мониторинга уровня или активности лейкоцитов, продуцирующих IFN-γ , или скорости пролиферации лейкоцитов в ответ на стимуляцию индивидуально, и корректировки сеанса лечения, частоты введения и перерыва эмпирическим путем и индивидуально, как определено по результатам мониторинга.
[85] Таким образом, сеанс лечения может содержать введение активного агента или фармацевтической композиции пациенту, и сеанс лечения продолжается по меньшей мере до тех пор, пока системное присутствие или уровень лейкоцитов, продуцирующих IFN-γ , или скорость пролиферации лейкоцитов в ответ на стимуляцию поднимется выше ссылочного значения, затем введение приостанавливается на время перерыва, и перерыв продолжается до тех пор, пока этот уровень выше ссылочного значения, при этом ссылочное значение выбрано из (а) уровня системного присутствия или активности лейкоцитов, продуцирующих IFN-γ , или скорости пролиферации лейкоцитов в ответ на стимуляцию, измеренной в последнем образце крови, полученном от пациента перед упомянутым введением; или (b) уровня системного присутствия или активности лейкоцитов, продуцирующих IFN-γ , или скорости пролиферации лейкоцитов в ответ на стимуляцию, характерной для группы лиц, страдающих от заболевания, расстройства, патологического состояния или поражения ЦНС.
[86] Продолжительность сеансов лечения и сеансов без лечения или перерывов может быть определена врачами в клинических испытаниях, направленных на определенные группы пациентов, и затем последовательно применена к этой группе пациентов, без необходимости индивидуального контроля за уровнем иммуносупрессии.
[87] В некоторых вариантах осуществления сеанс лечения содержит введение активного агента пациенту, и продолжается по меньшей мере до тех пор, пока системное присутствие активного агента не достигнет терапевтического уровня, введение приостанавливается на время перерыва, а перерыв продолжается до тех пор, пока уровень остается выше 95%, 90%, 80%, 70%, 60% или 50% от упомянутого терапевтического уровня. Термин «терапевтический уровень» используется здесь по отношению к общепринятому системному уровню препаратов, используемых для блокировки иммунных контрольных точек в известных способах лечения, таких как терапия рака (см. выше).
[88] В некоторых вариантах осуществления сеанс лечения содержит введение активного агента пациенту, и продолжается по меньшей мере до тех пор, пока системное присутствие или активность активного агента не достигнет терапевтического уровня, при котором введение приостанавливается, и перерыв продолжается до тех пор, пока системное присутствие или активность активного агента остается выше порогового значения терапевтического уровня. В аспектах этого варианта осуществления пороговый терапевтический уровень является уровнем, составляющим, например, не менее 10%, не менее 20%, не менее 30%, не менее 40%, не менее 50%,не менее 60%, не менее 70%, не менее 80%, не менее 90% от терапевтического уровня. Термин «терапевтический уровень» используется здесь по отношению к общепринятому системному уровню препаратов, используемых для блокировки иммунных контрольных точек в известных методах лечения, таких как терапия рака (см. выше). В аспектах этого варианта осуществления активный агент представляет собой анти-PD-1 антитело, анти-PDL1 антитело, анти-PDL2 антитело, анти-CTLA-4 антитело, анти-B7RP1 антитело, анти-В7-Н3 антитело, анти-В7-Н4 антитело, анти-В7-Н7 антитело, анти-BTLA антитело, анти-HVEM антитело, анти-CD-27 антитело, анти-CD40 антитело, анти-CD40L антитело, анти-CD70 антитело, анти-CD80 антитело, анти-CD86 антитело, анти-CD137 антитело, анти-CD137L антитело, анти-ОХ40 антитело, анти-OX40L антитело, анти-TIM-3 антитело, анти-Galectin 9 антитело, анти-KIR антитело, анти-LAG-3 антитело, анти-ICOS антитело, анти-VISTA антитело, анти-STING, анти-TIGIT, анти-GITR или любую их комбинацию.
[89] В некоторых вариантах осуществления сеанс лечения содержит введение активного агента пациенту, и продолжается по меньшей мере до тех пор, пока системное присутствие или активность активного агента не достигнет терапевтического уровня, при котором введение приостанавливается, и перерыв продолжается до тех пор, пока положительное влияние на когнитивную деятельность остается выше уровня до начала лечения. В аспектах этого варианта осуществления поддерживается положительное влияние на когнитивную деятельность, которое показывает улучшение, например, по меньшей мере на 10%, по меньшей мере на 20%, по меньшей мере на 30%, по меньшей мере на 40%, по меньшей мере на 50%, по меньшей мере на 60%, по меньшей мере на 70%, по меньшей мере на 80%, по меньшей мере на 90% выше когнитивного уровня до начала лечения. В аспектах этого варианта осуществления активный агент представляет собой анти-PD-1 антитело, анти-PDL1 антитело, анти-PDL2 антитело, анти-CTLA-4 антитело, анти-B7RP1 антитело, анти-В7-Н3 антитело, анти-В7-Н4 антитело, анти-В7-Н7 антитело, анти-BTLA антитело, анти-HVEM антитело, анти-CD-27 антитело, анти-CD40 антитело, анти-CD40L антитело, анти-CD70 антитело, анти-CD80 антитело, анти-CD86 антитело, анти-CD137 антитело, анти-CD137L антитело, анти-ОХ40 антитело, анти-OX40L антитело, анти-TIM-3 антитело, анти-Galectin 9 антитело, анти-KIR антитело, анти-LAG-3 антитело, анти-ICOS антитело, анти-VISTA антитело, анти-STING, анти-TIGIT, анти-GITR или любую их комбинацию.
[90] В некоторых вариантах осуществления сеанс лечения содержит введение активного агента пациенту, и продолжается по меньшей мере до тех пор, пока системное присутствие или активность активного агента не достигнет терапевтического уровня, при котором введение приостанавливается, и перерыв продолжается до тех пор, пока положительное влияние на зрение остается выше уровня до начала лечения. В аспектах этого варианта осуществления поддерживается положительное влияние на зрение, которое показывает улучшение, например, по меньшей мере на 10%, по меньшей мере на 20%, по меньшей мере на 30%, по меньшей мере на 40%, по меньшей мере на 50%, по меньшей мере на 60%, по меньшей мере на 70%, по меньшей мере на 80%, по меньшей мере на 90% выше уровня зрения до начала лечения. В аспектах этого варианта осуществления активный агент представляет собой анти-PD-1 антитело, анти-PDL1 антитело, анти-PDL2 антитело, анти-CTLA-4 антитело, анти-B7RP1 антитело, анти-В7-Н3 антитело, анти-В7-Н4 антитело, анти-В7-Н7 антитело, анти-BTLA антитело, анти-HVEM антитело, анти-CD-27 антитело, анти-CD40 антитело, анти-CD40L антитело, анти-CD70 антитело, анти-CD80 антитело, анти-CD86 антитело, анти-CD137 антитело, анти-CD137L антитело, анти-ОХ40 антитело, анти-OX40L антитело, анти-TIM-3 антитело, анти-Galectin 9 антитело, анти-KIR антитело, анти-LAG-3 антитело, анти-ICOS антитело, анти-VISTA антитело, анти-STING, анти-TIGIT, анти-GITR или любую их комбинацию.
[91] В некоторых вариантах осуществления сеанс лечения может представлять собой однократное введение или содержать несколько введений, выполняющихся в течение заданного периода времени. В аспектах этого варианта осуществления сеанс лечения может представлять собой несколько введений в течение, например, от 1 дня до четырех недель, от 2 дней до четырех недель, от 3 дней до четырех недель, от 4 дней до четырех недель, от 5 дней до четырех недели, от 6 дней до четырех недель, одна неделя и четыре недели, 10 дней и четыре недели, две недели и четыре недели, 17 дней и четыре недели или три недели и четыре недели. Например, сеанс лечения может содержать два введения, оба в течение одной недели, например, если второе введение происходит на 1, 2, 3, 4, 5 или 6-й день после первого. В качестве другого примера, сеанс лечения может содержать три введения, все в течение одной недели, например, через 1, 2 или 3 дня после предшествующего введения. В качестве другого примера, сеанс лечения может содержать три введения, все в течение двух недель, например, через 1, 2, 3, 4 или 5 дней после предшествующего введения. В качестве другого примера, сеанс лечения может содержать четыре введения, все в течение двух недель, например, через 1, 2, 3, 4 или 5 дней после предшествующего введения. В качестве другого примера, сеанс лечения может содержать четыре введения, все в течение трех недель, например, через 1, 2, 3, 4, 5 или 6 дней после предшествующего введения. В качестве другого примера, сеанс лечения может содержать пять введений, все в течение трех недель, например, через 1, 2, 3, 4 или 5 дней после предшествующего введения.
[92] В некоторых вариантах осуществления перерыв без лечения может составлять от одной недели до шести месяцев, например, от 2 недель до 4 недель, от 3 недель до 4 недель, от 2 недель до 6 недель, от 3 недель до 6 недель, 4 недель до 6 недель, от 5 недель до 6 недель, от 2 недель до 2 месяцев, от 3 недель до 2 месяцев, от 4 недель до 2 месяцев, от 5 недель до 2 месяцев, от 6 недель до 2 месяцев, от 7 недель до 2 месяцев, от 2 месяцев до 3 месяцев, от 2 месяцев до 4 месяцев, от 3 месяцев до 4 месяцев, от 3 месяцев до 5 месяцев, от 3 месяцев до 5 месяцев, от 4 месяцев до 5 месяцев, от 1 недели до 6 месяцев, от 2 недель до 6 месяцев, от 3 недель до 6 месяцев, 4 недель до 6 месяцев, от 6 недель до 6 месяцев, от 2 месяцев до 6 месяцев, от 3 месяцев до 6 месяцев, от 4 месяцев до 6 месяцев, или от 5 месяцев до 6 месяцев. В некоторых вариантах осуществления перерыв, когда лечение не проводится, может составлять от 1 до 2 месяцев, от 1 до 3 месяцев или от 2 до 3 месяцев.
[93] Во время сеанса лечения введение активного агента или фармацевтической композиции может представлять собой однократное введение или повторяющееся введение, например, активное вещество или фармацевтическую композицию можно вводить только один раз, а затем немедленно следует перерыв, или его можно вводить ежедневно или по одному разу каждые два, три, четыре, пять или шесть дней, раз в неделю, раз в две недели, раз в три недели или раз в четыре недели. Эта периодичность применима к любому активному агенту, может быть основана на общепринятых практиках в данной области и может окончательно определяться врачами в клинических испытаниях. Кроме того, периодичность повторных введений во время сеанса лечения может изменяться в зависимости от характера активного агента, при этом, например, малые молекулы могут вводиться ежедневно, а антитела могут вводиться раз в 3 дня. Следует понимать, что при введении агента во время сеанса лечения с относительно малой периодичностью, например, один раз в неделю при сеансе лечения в течение одного месяца, или один раз в месяц при сеансе лечения в течение шести месяцев, за этим сеансом лечения следует перерыв, продолжительность которого больше, чем интервал между повторными введениями во время сеанса лечения (т.е. в этом примере это, соответственно, более одной недели или одного месяца). Период продолжительностью одна неделя или один месяц между введениями во время сеанса лечения в этом примере не считается перерывом в лечении.
[94] Продолжительность сеанса лечения и сеанса без лечения или перерыва может корректироваться по частоте введения, так что, например, частота введения активного агента один раз каждые 3 дня может привести к продолжительности сеанса лечения 6 или 9 дней, и соответствующему перерыву.
[95] Если сеанс лечения состоит из одного введения, режим дозирования определяется длиной перерыва без лечения, чтобы за одним введением следовал перерыв без лечения продолжительностью 7, 8, 9, 10, 14, 18, 21, 24 или 28 дней или дольше до следующего сеанса лечения с однократным введением. В частности, режим дозирования состоит из отдельных введений, чередующихся с перерывами без лечения продолжительностью 2, 3 или 4 недели. Кроме того, режим дозирования может состоять из отдельных введений, чередующихся с перерывами без лечения от 2 до 4 недель, от 2 до 3 недель или от 3 до 4 недель.
[96] Если сеанс лечения состоит из нескольких введений, режим дозирования определяется продолжительностью перерыва без лечения, так чтобы за несколькими введениями в течение одной недели следовал перерыв без лечения 7, 10, 14, 18, 21, 24 или 28 дней или дольше до следующего сеанса лечения с несколькими введениями. В частности, режим дозирования может состоять из нескольких введений в течение одной недели, чередующихся с перерывами без лечения продолжительностью 2, 3 или 4 недели. Кроме того, режим дозирования может состоять из нескольких введений в течение одной недели, чередующихся с перерывами без лечения от 2 до 4 недель, от 2 до 3 недель или от 3 до 4 недель.
[97] В качестве другого примера, режим дозирования может содержать несколько введений в течение двух недель, с последующим перерывом без лечения в 2 недели, 3 недели или 1, 2, 3 или 4 месяца или дольше, до следующего сеанса лечения с несколькими введениями. В частности, режим дозирования может состоять из нескольких введений в течение двух недель, чередующихся с перерывами без лечения продолжительностью 1, 2, 3 или 4 месяца. Кроме того, режим дозирования может состоять из нескольких введений в течение двух недель с перерывами без лечения от 1 до 2 месяцев, от 1 до 3 месяцев, от 1 до 4 месяцев, от 2 до 3 месяцев, от 2 до 4 месяцев или от 3 до 4 месяцев.
[98] В качестве другого примера, режим дозирования может содержать несколько введений в течение трех недель, с последующим перерывом без лечения в 1, 2, 3, 4, 5 или 6 месяцев или дольше, до следующего сеанса лечения с несколькими введениями. В частности, режим дозирования может состоять из нескольких введений в течение трех недель, чередующихся с перерывами без лечения продолжительностью 1, 2, 3, 4, 5 или 6 месяцев. Кроме того, режим дозирования может состоять из нескольких введений в течение трех недель с перерывами без лечения от 1 до 2 месяцев, от 1 до 3 месяцев, от 1 до 4 месяцев, от 1 до 5 месяцев, от 1 до 6 месяцев, от 2 до 3 месяцев, от 2 до 4 месяцев, от 2 до 5 месяцев, от 2 до 6 месяцев, от 3 до 4 месяцев, от 3 до 5 месяцев, от 3 до 6 месяцев, от 4 до 5 месяцев, от 4 до 6 месяцев, или от 5 до 6 месяцев.
[99] Разумеется, предусмотрен гибкий режим дозирования, который начинается с определенного режима и меняется на другой. Например, сеансы лечения, каждый из которых включает в себя 2 отдельных введения с интервалом в 3 дня, с перерывом, например, 1 неделя между сеансами лечения, может быть заменен, если это будет сочтено целесообразным, режимом дозирования, включающим сеансы лечения с однократными введениями, разделенными перерывами продолжительностью, например, 2, 3 или 4 недели. В качестве другого примера, сеансы лечения, каждый из которых включает в себя 2 отдельных введения с интервалом в 7 дней, с перерывом, например, 2 недели между сеансами лечения, может быть заменен, если это будет сочтено целесообразным, режимом дозирования, включающим сеансы лечения с однократными введениями, разделенными перерывами продолжительностью, например, 2, 3, 4, 5 или 6 недель. В качестве другого примера, сеансы лечения, каждый из которых включает в себя 3 отдельных введения с интервалом в 3 дня, с перерывом, например, 2 недели между сеансами лечения, может быть заменен, если это будет сочтено целесообразным, режимом дозирования, включающим сеансы лечения с однократными введениями, разделенными перерывами продолжительностью, например, 2, 3, 4, 5 или 6 недель.
[100] В любом случае, режим дозирования, то есть продолжительность сеанса лечения и перерыва подбирается таким образом, чтобы снижение уровня иммуносупрессии, например, согласно измерениям снижения уровня системного присутствия или активности регуляторных Т-клеток, или увеличения у пациента уровня системного присутствия или активности лейкоцитов, продуцирующих IFN-γ , являлось кратковременным.
[101] Способ, активный агент или фармацевтическая композиция в соответствии с настоящим изобретением могут использоваться для лечения заболевания, расстройства или патологического состояния ЦНС, являющегося нейродегенеративным заболеванием, расстройством или состоянием. Согласно аспектам настоящего варианта осуществления, нейродегенеративные заболевания, расстройства или патологические состояния включают в себя болезнь Альцгеймера, амиотрофический боковой склероз, болезнь Паркинсона, болезнь Хантингтона, первичный прогрессирующий рассеянный склероз; вторичный прогрессирующий рассеянный склероз, кортикобазальную дегенерацию, синдром Ретта, таупатию, дегенерацию сетчатки; переднюю ишемическую оптическую невропатию; глаукому; увеит; депрессию; связанный с травмой стресс или посттравматическое стрессовое расстройство, лобно-временную деменцию, болезнь диффузных телец Леви, умеренные когнитивные нарушения, атрофию задней коры головного мозга, первичную прогрессирующую афазию или прогрессирующий надъядерный паралич. В некоторых вариантах осуществления патологическим состоянием ЦНС является возрастная деменция. В некоторыхаспектах данного варианта осуществления к патологическим состояниям ЦНС относятся болезнь Альцгеймера, боковой амиотрофический склероз, болезнь Паркинсона, болезнь Хантингтона.
[102] Таупатии представляют собой клинически, морфологически и биохимически гетерогенный класс нейродегенеративных заболеваний, характеризующихся патологическим слипанием тау-белка в нейрофибриллярные или глиофибриллярные клубки в головном мозге человека. Тау - белок, ассоциированный с микротрубочками (MAP), связывается с микротрубочками и способствует их полимеризации. Он играет важную роль в поддержании переноса аксонов и целостности нейронов, но выполняет физиологическую роль в дендритах и экспрессируется на низких уровнях в глиальных клетках. При таупатии связки формируются путем гиперфосфорилирования тау, заставляя его агрегировать в нерастворимой форме. Примеры таупатий, без ограничения перечисленным, включают болезнь Альцгеймера, аргирофильное зерновое заболевание, хроническую травматическую энцефалопатию, кортикобазальную дегенерацию, деменцию боксеров, лобно-височную деменцию, лобно-височную дегенерацию, болезнь Галлервордена-Шпатца, болезнь Хантингтона, ганглиоглиому, ганглиоцитому, глобулярную глиальную таупатию, свинцовую энцефалопатию, липофусциноз, болезнь Литико-Бодига (комплекс болезни Паркинсона и деменции на Гуаме), менингоангиоматоз, паркинсонизм, связанный с хромосомой 17, болезнь Пика, первичную возрастную таупатию (PART), ранее известную как деменция с преобладанием нейрофибриллярных клубков (NFT-деменция), постэнцефалитический паркинсонизм, прогрессирующий надъядерный паралич, подострый склерозирующий панэнцефалит и клубневой (туберозный) склероз.
[103] Нарушения, связанные с дегенерацией сетчатки, приводят к деградации сетчатки из-за смерти фоторецепторных клеток. Существует несколько причин дегенерации сетчатки, включая окклюзию артерии или вены, диабетическую ретинопатию, ретролентальную фиброплазию/ретинопатию при недоношенности или заболевание (обычно наследственное). Симптомы включают в себя, без ограничения перечисленным, нарушение зрения, ночную слепоту, отслоение сетчатки, светочувствительность, чувствительность к бликам, туннельное зрение, потерю восприятия глубины, потерю контрастности, ночную слепоту, потерю центрального зрения, потерю периферического зрения и полную потерю зрения. Расстройства дегенерации сетчатки включают в себя, без ограничения перечисленным, возрастную дегенерацию макулы (влажную и сухую), пигментный ретинит, хороидеремию, конусно-палочковую дистрофию сетчатки, гиратную атрофию, ювенильный ретиношизис, желтушную макулярную дистрофию (болезнь Беста), абеталипопротеинемию (болезнь Бассена-Корнцвейга), синдром Барде-Бидля, синдром монохромности голубого конуса, доминантные друзы, витреоретинальную дистрофию Гольдмана-Фавра (усиленный синдром S-конуса), синдром Кернса-Сейра, синдром Лоренса-Муна, врожденный амавроз Лебера, болезнь Рефсума Лебера, болезнь Огучи, перипапиллярную (перицентральную) хороидальную дистрофию, пигментную дистрофию, макулярную дистрофию Сорсби, болезнь Штаргардта, синдром Стиклера, синдром Ашера и витреоретинальную дистрофию Вагнера.
[104] В некоторых вариантах осуществления, каждый из вышеописанных активных агентов, блокирующий одну из иммунных контрольных точек, в том числе ICOS-B7RP1, V-доменный иммуноглобулиновый супрессор активации Т-клеток (VISTA), В7-С028-подобная молекула, CD40L-CD40, CD28-CD80, CD28-CD86, В7Н3, В7Н4, В7Н7, BTLA-HVEM, CD137-CD137L, OX40L, CD27-CD70, STING, TIGIT и A2aR-аденозин, а также индоламин-2,3-диоксигеназа (IDO)-L-триптофан, такой как антитело к одному из двух компонентов иммунной контрольной точки, предназначен для лечения нейродегенеративных заболеваний, расстройств или патологических состояний, включающих в себя болезнь Альцгеймера, амиотрофический боковой склероз, болезнь Паркинсона, болезнь Хантингтона, первичный прогрессирующий рассеянный склероз; вторичный прогрессирующий рассеянный склероз, кортикобазальную дегенерацию, синдром Ретта, дегенерацию сетчатки, выбранную из группы, куда входит возрастная макулярная дегенерация и пигментная дегенерация сетчатки; переднюю ишемическую оптическую невропатию; глаукому; увеит; депрессию; связанный с травмой стресс или посттравматическое стрессовое расстройство, лобно-временную деменцию, болезнь диффузных телец Леви, умеренные когнитивные нарушения, атрофию задней коры головного мозга, первичную прогрессирующую афазию или прогрессирующий надъядерный паралич. Лечение любого из этих заболеваний с использованием любого из этих активных агентов может осуществляться согласно режиму, описанному выше.
[105] Способ, активный агент и фармацевтическая композиция в соответствии с настоящим изобретением могут дополнительно использоваться при лечении повреждений ЦНС, в том числе травмы спинного мозга, закрытой травмы головы, тупой травмы, проникающей травмы, геморрагического инсульта, ишемического инсульта, церебральной ишемии, травмы зрительного нерва, инфаркта миокарда, отравления органофосфатом и травм, вызванных удалением опухоли.
[106] В некоторых вариантах осуществления, каждый из описанных выше активных агентов блокирует одну из иммунных контрольных точек, в том числе ICOS-B7RP1, V-доменный иммуноглобулиновый супрессор активации Т-клеток (VISTA), В7-CD28-подобная молекула, CD40L-CD40, CD28-CD80, CD28-CD86, В7Н3, В7Н4, В7Н7, BTLA-HVEM, CD137-CD137L, OX40L, CD27-CD70, STING, TIGIT и A2aR-аденозин, а также индоламин-2,3-диоксигеназа (IDO)-L-триптофан, такой как антитело к одному из двух компонентов иммунной контрольной точки, предназначен для лечения травм ЦНС, в том числе травмы спинного мозга, закрытой травмы головы, тупой травмы, проникающей травмы, геморрагического инсульта, ишемического инсульта, церебральной ишемии, повреждения зрительного нерва, инфаркта миокарда, отравления органофосфатом и травм, вызванных удалением опухоли. Лечение любой из этих травм с использованием любого из этих активных агентов может осуществляться согласно режиму, описанному выше.
[107] Как указывалось выше, авторы изобретения обнаружили, что настоящее изобретение улучшает когнитивные функции у мышей с эмуляцией болезни Альцгеймера. Таким образом, данный способ, активный агент и фармацевтическая композиция могут быть применены для улучшения моторной и/или когнитивной функции ЦНС, например, для уменьшения возрастной потери когнитивной функции, которая может возникать у людей, не страдающих диагностированными заболеваниями, а также у людей, страдающих нейродегенеративными заболеваниями. Кроме того, данный способ, активный агент и фармацевтическая композиция могут быть использованы для уменьшения потери когнитивной функции в результате острого стресса или травматического происшествия. К указанным выше когнитивным функциям может относиться обучение, память или и то, и другое.
[108] Следует подчеркнуть, что улучшение когнитивных функций у мышей с эмуляцией болезни Альцгеймера (5XFAD AD-Tg мыши) наблюдалось и описывалось изобретателями в различных стадиях проявления болезни; как на ранних, так и на поздних стадиях развития болезни патологии можно уменьшить путем лечения. 5XFAD AD-Tg мыши начинают демонстрировать патологию церебральных бляшек в возрасте 2,5 месяцев, а когнитивную недостаточность в возрасте 5 месяцев (Oakley et al, 2006). Следует отметить, что, в то время как в приведенном ниже Примере 2 изобретатели описывают терапевтический эффект у 5XFAD мышей в возрасте 6 месяцев, в Примере 5 они характеризуют терапевтический эффект у 5XFAD мышей в возрасте 11 и 12 месяцев - чрезвычайно прогрессивный этап осаждения бета-амилоидных бляшек и когнитивной недостаточности в данной модели. Поэтому предполагается, что предложенное изобретение будет актуальным для пациентов на разных этапах прогрессирования заболевания, таких как Этап 1 - легкий/ранний (длится 2-4 года); Этап 2 - умеренный/средний (длится 2-10 лет); и Этап 3 - тяжелый/поздний (длится 1-3 года и дольше).
[109] В применении здесь термин «функция ЦНС» относится, среди прочего, к получению и обработке сенсорной информации, мышлению, обучению, запоминанию, восприятию, производству и пониманию языка, управлению двигательной функцией, слуховым и визуальным ответом, поддержанию баланса и равновесия, координации движения, передаче сенсорной информации и контролю таких вегетативных функций, как дыхание, сердечный ритм и пищеварение.
[110] В применении здесь термины «познание», «когнитивная функция» и «когнитивная деятельность» взаимозаменяемы и обозначают любой психический процесс или состояние, к которым относятся, без ограничения перечисленным, обучение, память, создание изображений, мышление, понимание, рассуждение, пространственная ориентация, речь и язык, приобретение языковых знаний и потенциал произвольного внимания. Познание формируется в нескольких областях головного мозга, таких как гиппокамп, кора и другие структуры головного мозга. Однако предполагается, что долговременные воспоминания хранятся, по меньшей мере, частично, в коре, а также известно, что сенсорная информация приобретается, объединяется и извлекается определенной кортикальной структурой, вкусовой корой, которая находится внутри островковой коры.
[111] У людей когнитивную функцию можно измерить при помощи любого известного метода, например, без ограничения, шкалы общего клинического впечатления (шкала CIBIC-плюс); мини-экзамена по психическому состоянию (MMSE); нейропсихиатрического опросника (NPI); клинической рейтинговой шкалы деменции (CDR); Кембриджской нейропсихологической автоматизированной батареи тестов (CANTAB) или гериатрической шкалы клинической оценки Сандоз (SCAG). Когнитивная функция также может быть измерена косвенно с использованием методов визуализации, таких как позитронная эмиссионная томография (PET), функциональная магнитно-резонансная томография (fMRI), однофотонная эмиссионная компьютерная томография (SPECT) или любая другая техника визуализации, позволяющая измерить мозговую деятельность.
[112] Улучшение одного или нескольких процессов, влияющих на познание у пациента, будет означать улучшение когнитивной функции у упомянутого пациента, поэтому в некоторых вариантах осуществления совершенствование познания включает в себя улучшение обучения, пластичности и/или долговременной памяти. Термины «совершенствование» и «повышение» могут использоваться как взаимозаменяемые.
[113] Термин «обучение» касается приобретения новых или изменения и закрепления имеющихся знаний, образцов поведения, навыков, ценностей или предпочтений.
[114] Термин «пластика» относится к синаптической пластичности, пластичности мозга или нейропластичности, связанным со способностью мозга изменяться посредством обучения и изменять уже приобретенную память. Один из поддающихся измерению параметров, отражающих пластичность, это отмирание памяти.
[115] Термин «память» относится к процессу, при котором информация кодируется, хранится и извлекается. Память имеет три различных категории: сенсорная память, кратковременная память и долговременная память.
[116] Термин «долговременная память» означает способность хранить информацию в течение длительного или неограниченного периода времени. Долговременная память состоит из двух основных отделов: эксплицитная память (декларативная память) и имплицитная память (скрытая память). Долговременная память достигается путем консолидации памяти, которая представляет собой категорию процессов, стабилизирующих след в памяти после первоначального приобретения. Консолидация делится на два конкретных процесса: синаптическая консолидация, которая происходит в течение первых нескольких часов после обучения, и системная консолидация, при которой зависимые от гиппокампа воспоминания становятся независимыми от гиппокампа в течение периода от нескольких недель до нескольких лет.
[117] Вышеизложенные варианты осуществления, описывающие различные особенности фармацевтической композиции по настоящему изобретению, также актуальны для способа по изобретению, поскольку в этом способе используется та же самая фармацевтическая композиция.
[118] В еще одном аспекте, настоящее изобретение предлагает методику снижения бремени Аβ-бляшек у пациента с диагнозом болезни Альцгеймера, включающую в себя введение упомянутому пациенту активного агента или фармацевтической композиции, как указано выше, что приводит к снижению уровня системной иммуносупрессии путем снятия ограничения, налагаемого на иммунную систему одной или несколькими иммунными контрольными точками.
[119] В еще одном аспекте, настоящее изобретение предлагает способ снижения гиппокампального глиоза у пациента с диагнозом болезни Альцгеймера, включающий в себя введение упомянутому пациенту активного агента или фармацевтической композиции, как указано выше, что приводит к снижению уровня системной иммуносупрессии путем снятия ограничения, налагаемого на иммунную систему одной или несколькими иммунными контрольными точками.
[120] Фармацевтические композиции для применения в соответствии с настоящим изобретением могут быть приготовлены обычным способом с использованием одного или нескольких физиологически приемлемых носителей или вспомогательных средств. Носитель (носители) должен быть «приемлемым» в смысле совместимости с другими ингредиентами препарата и не должен быть вредным для реципиента.
[121] Следующие примеры носителей, способов введения, лекарственных форм и т.д. перечислены как известные возможности, из которых можно выбрать носители, способы введения, лекарственные формы и т.д. для использования с настоящим изобретением. Специалистам в данной области, тем не менее, должно быть ясно, что каждую выбранную лекарственную форму и способ введения необходимо сначала испытать, чтобы определить, достигаются ли желаемые результаты.
[122] К способам введения относятся, без ограничения, парентеральный (например внутривенный, интраперитонеальный (внутрибрюшинный), внутримышечный, подкожный), слизистый (например, пероральный, интраназальный, трансбуккальный, вагинальный, ректальный, внутриглазной), подоболочечный, местный и внутрикожный пути. Введение может быть системным или местным.
[123] Термин «носитель» обозначает разбавитель, адъювант, наполнитель или проводник, с помощью которого осуществляют введение активного агента. Носители в фармацевтической композиции могут содержать связующее, такое как микрокристаллическая целлюлоза, поливинилпирролидон (поливидон или повидон), трагакантовая камедь, желатин, крахмал, лактоза или моногидрат лактозы; дезинтегрирующий агент, такой как альгиновая кислота, кукурузный крахмал и т.п.; смазывающее вещество или поверхностно-активное вещество, такое как стеарат магния или лаурилсульфат натрия; а также глидант, такой как коллоидный диоксид кремния.
[124] Для перорального введения фармацевтическая композиция может быть в жидкой форме, например растворах, сиропах или суспензиях, или может быть представлена в виде лекарственного средства для разведения водой или другой подходящей основой перед использованием. Такие жидкие препараты могут быть получены обычными способами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, производные целлюлозы или гидрированные пищевые жиры); эмульгаторы (например, лецитин или аравийская камедь); неводные носители (например, миндальное масло, масляные сложные эфиры или фракционированные растительные масла); а также консерванты (например, метил, пропил-п-гидроксибензоаты или сорбиновая кислота). Фармацевтические композиции могут принимать форму, например, таблеток или капсул, полученных обычными способами, с фармацевтически приемлемыми вспомогательными средствами, такими как связывающие агенты (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или гидрофосфат кальция); смазывающие вещества (например, стеарат магния, тальк или диоксид кремния); разрыхлители (например, картофельный крахмал или натрия гликолят крахмала); или смачивающие агенты (например, лаурилсульфат натрия). Таблетки могут быть покрыты методами, хорошо известными в уровне техники.
[125] Формулу препаратов для перорального введения можно соответствующим образом подобрать для получения контролируемого высвобождения активного компонента.
[126] Для трансбуккального (щечного) введения композиции могут принимать форму таблеток или леденцов, с обычной формулой состава.
[127] Композиции могут быть разработаны для парентерального введения путем инъекции, например, путем болюсной инъекции или непрерывной инфузии. Составы для инъекций могут быть представлены в стандартной дозированной форме, например, в ампулах или в контейнерах с множественными дозами, с добавлением консерванта. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных основах, и могут содержать формообразующие агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Кроме этого, активный ингредиент может быть в виде порошка для соединения с подходящим носителем, например, стерильной апирогенной водой, перед использованием.
[128] Композиции также могут производиться в форме ректальных составов, таких как суппозитории или удерживающие клизмы, например, с содержанием обычных основ для суппозиториев, таких как масло какао или других глицеридов.
[129] Для введения путем ингаляции композиции для применения в соответствии с настоящим изобретением доставляются в удобной форме в виде аэрозоля в упаковках под давлением или распылителя с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением дозировочная единица может определяться путем установки клапана для введения отмеренного количества. Капсулы и картриджи, например, желатин для использования в ингаляторе или инсуффляторе, могут быть составлены с использованием порошковой смеси препарата и подходящей порошкообразной основы, такой как лактоза или крахмал.
[130] Определение доз активного ингредиента, используемого при лечении людей, основано на общепринятых практиках в данной области техники и должно окончательно определяться врачами в клинических испытаниях. Ожидаемая приблизительная эквивалентная доза для введения человеку может быть рассчитана на основе экспериментальных данных in vivo, описанных здесь ниже, с использованием известных формул (например, Reagan-Show et al. (2007) Dosetranslationfromanimaltohumanstudiesrevisited. TheFASEBJournal 22:659-661). Согласно этой парадигме взрослая эквивалентная человеческая доза (мг/кг массы тела) равна дозе, рассчитанной для мыши (мг/кг массы тела), умноженной на 0,081.
[131] Аспекты настоящего описания изобретения также можно представить следующим образом:
1. Способ лечения болезни, расстройства, патологического состояния или поражения центральной нервной системы у индивидуума, который нуждается в этом, способ содержащий введение индивидууму композиции, содержащей активный агент, который вызывает снижение уровня системной иммуносупрессии путем снятия ограничения, налагаемого на иммунную систему одной или несколькими иммунными контрольными точками, где композицию вводят с помощью режима дозирования, содержащего по меньшей мере один курс лечения, причем каждый курс лечения содержит последовательно сеанс лечения, когда композицию вводят индивидууму, за которым следует сеанс без лечения, когда композицию не вводят индивидууму, причем период без лечения продолжительнее, чем сеанс лечения; при котором, если введение композиции во время сеанса лечения является повторным, период без лечения будет продолжительнее, чем период между повторными введениями во время сеанса лечения; при котором введение композиции временно снижает уровни системной иммуносупрессии и повышает "шлюзовую" активность хороидного сплетения в облегчении селективного рекрутинга иммунных клеток в центральную нервную систему, тем самым обеспечивая лечение индивидуума.
2. Активный агент или фармацевтическая композиция, содержащая активный агент, для применения при лечении болезни, расстройства, патологического состояния или поражения центральной нервной системы, где активный агент вызывает снижение уровня системной иммуносупрессии путем снятия ограничения, налагаемого на иммунную систему одной или несколькими иммунными контрольными точками, при котором активный агент или фармацевтическую композицию вводят с помощью режима дозирования, содержащего по меньшей мере один курс лечения, причем каждый курс лечения содержит последовательно сеанс лечения, когда композиция вводят индивидууму, за которым следует сеанс без лечения, когда композицию не вводят индивидууму, причем период без лечения продолжительнее, чем сеанс лечения; где, если введение композиции во время сеанса лечения является повторным, период без лечения будет продолжительнее, чем период между повторными введениями во время сеанса лечения; где введение композиции временно снижает уровни системной иммуносупрессии и повышает "шлюзовую" активность хороидного сплетения в облегчении селективного рекрутинга иммунных клеток в центральную нервную систему, тем самым обеспечивая лечение индивидуума.
3. Применение активного агента, снижающего уровень системной иммуносупрессии путем снятия ограничений, наложенных на иммунную систему одной или несколькими иммунными контрольными точками, для лечения заболевания, расстройства, патологического состояния или поражения центральной нервной системы.
4. Применение активного агента, снижающего уровень системной иммуносупрессии путем снятия ограничений, наложенных на иммунную систему одной или несколькими иммунными контрольными точками, при производстве лекарственного средства для лечения заболевания, расстройства, патологического состояния или поражения центральной нервной системы.
5. Способ по варианту осуществления 1, активный агент или фармацевтическая композиция по варианту осуществления 2 или применение в соответствии с вариантами осуществления 3 или 4, где активный агент представляет собой антитело, имитатор антитела, аптамер, малую молекулу, гликопротеин листвы нима, sCTLA-4, молекулу сайленсинга, OK-432, комбинацию IL-12 и анти-CTLA-4 или любую комбинацию перечисленного.
6. Способ по варианту осуществления 5, активный агент или фармацевтическая композиция по варианту осуществления 5, или применение по варианту осуществления 5, где антитело представляет собой поликлональное антитело или моноклональное антитело.
7. Способ по вариантам осуществления 5 или 6, активный агент или фармацевтическая композиция по вариантам осуществления 5 или 6, или применение по вариантам осуществления 5 или 6, где антитело представляет собой димер, мультимер, мультиспецифическое антитело, рекомбинантное антитело, химерное антитело, бифункциональное антитело, ассоциированное с клеткой антитело, такое, как Ig-рецептор, линейное антитело, диатело, минитело или нанотело.
8. Способ по любому из вариантов осуществления 5-7, активный агент или фармацевтическая композиция по вариантам осуществления 5-7, или применение по варианту осуществления 5-7, где антитело представляет собой человеческое или гуманизированное антитело.
9. Способ по любому из вариантов осуществления 5-8, активный агент или фармацевтическая композиция по вариантам осуществления 5-8, или применение по варианту осуществления 5-8, где антитело представляет собой антитело-антагонист или антитело-агонист.
10. Способ по любому из вариантов осуществления 5-9, активный агент или фармацевтическая композиция по вариантам осуществления 5-9, или применение по варианту осуществления 5-9, где антитело представляет собой нейтрализующее антитело.
11. Способ по любому из вариантов осуществления 5-10, активный агент или фармацевтическая композиция по вариантам осуществления 5-10, или применение по варианту осуществления 5-10, где антитело представляет собой полноразмерную молекулу иммуноглобулина или иммунологически активный фрагмент.
12. Способ по варианту осуществления 11, активный агент или фармацевтическая композиция по варианту осуществления 11, или применение в соответствии с вариантом осуществления 11, где иммунологически активный фрагмент представляет собой однодоменное антитело (sdAb), одноцепочечный вариабельный фрагмент (scFv), Fab-фрагмент, F(ab')2-фрагмент, Fc фрагмент, Fd фрагмент, Fv фрагмент.
13. Способ по варианту осуществления 5-12, активный агент или фармацевтическая композиция по варианту осуществления 3 или 4, или применение в соответствии с любым из вариантов осуществления 4-11, где антитело является анти-PD-1, анти-PDL1, анти-PDL2, анти-CTLA-3, анти-CD80, анти-CD86, анти-B7RP1, анти-В7-Н3, анти-В7-Н4, анти-В7-Н7, анти-BTLA, анти-HVEM, анти-CD-4, анти-CD40, анти-CD40L, анти-CD70, анти-CD80, анти-CD86, анти-CD137, анти-CDI 37L, анти-ОХ40, анти-OX40L анти-TIM-4-11, анти-Galectin9, анти-KIR, анти-LAG-1, анти-ICOS, анти-VISTA, анти-STING, анти-TIGIT, анти-GITR антителом или любой их комбинацией.
14. Способ по варианту осуществления 5, активный агент или фармацевтическая композиция по варианту осуществления 5, или применение в соответствии с вариантом осуществления 5, где миметик антитела представляет собой аффинную молекулу, аффилин, аффимер, аффитин, альфа-группу, антикалин, авимер, дарпин, финомер, пептид домена Куниц или монотело.
15. Способ по варианту осуществления 5, активный агент или фармацевтическая композиция по варианту осуществления 5, или применение по варианту осуществления 4, где аптамер представляет собой ДНК-аптамер, РНК-аптамер, КсНК-аптамер или аптамер пептида.
16. Способ по варианту осуществления 5, активный агент или фармацевтическая композиция по варианту осуществления 5 или применение в соответствии с вариантом осуществления 5, где малая молекула представляет собой ингибитор p300, сунитиниб, полиоксометалат-1, α,β-метиленаденозин 5'-дифосфат, триоксид мышьяка, GX15-070, антагонист ретиноевой кислоты, антагонист CCR4, антагонист аденозинового рецептора,
антагонист аденозинового рецептора А1; аденозин-А2а-рецептор, антагонист аденозинового рецептора A2b, антагонист рецептора A3, антагонист индоламина-2,3-диоксигеназы или регулятор HIF-1.
17. Способ по любому из вариантов осуществления 1-16, активный агент или фармацевтическая композиция по вариантам осуществления 2-16, или применение по варианту осуществления 3-16, где введение композиции во время сеанса лечения представляет собой однократное введение.
18. Способ по любому из вариантов осуществления 1-16, активный агент или фармацевтическая композиция по вариантам осуществления 2-16, или применение по варианту осуществления 3-16, где введение композиции во время сеанса лечения представляет собой повторяющееся введение.
19. Способ по варианту осуществления 18, активный агент или фармацевтическая композиция согласно варианту осуществления 18 или применение по варианту осуществления 18, где повторное введение выполняется по одному разу каждый день, раз в два дня, раз в три дня, раз в четыре дня, раз в пять дней или раз в шесть дней.
20. Способ по варианту осуществления 18, активный агент или фармацевтическая композиция по варианту осуществления 18 или применение по варианту осуществления 18, где повторное введение выполняется по одному разу каждую неделю, раз в две недели, раз в три недели или раз в четыре недели.
21. Способ по любому из вариантов осуществления 1-20, активный агент или фармацевтическая композиция по вариантам осуществления 2-20, или применение по варианту осуществления 3-20, где сеанс лечения занимает от 1 дня до четырех недель.
22. Способ по варианту осуществления 21, активный агент или фармацевтическая композиция по вариантам осуществления 21, или применение по варианту осуществления 21, где сеанс лечения занимает от 3 дней до четырех недель.
23. Способ по варианту осуществления 22, активный агент или фармацевтическая композиция по вариантам осуществления 22, или применение по варианту осуществления 22, где сеанс лечения занимает от одной недели до четырех недель.
24. Способ по любому из вариантов осуществления 1-23, активный агент или фармацевтическая композиция по вариантам осуществления 2-23, или применение по варианту осуществления 3-23, где период отсутствия лечения занимает от одной недели до шести месяцев.
25. Способ по варианту осуществления 24, активный агент или фармацевтическая композиция по варианту осуществления 24, или применение по варианту осуществления 24, где период отсутствия лечения занимает от двух недель до шести месяцев.
26. Способ по варианту осуществления 25, активный агент или фармацевтическая композиция по варианту осуществления 25, или применение по варианту осуществления 25, где период отсутствия лечения занимает от трех недель до шести месяцев.
27. Способ по варианту осуществления 26, активный агент или фармацевтическая композиция по варианту осуществления 26, или применение по варианту осуществления 26, где период отсутствия лечения занимает от одного до трех месяцев.
28. Способ по варианту осуществления 27, активный агент или фармацевтическая композиция по варианту осуществления 27, или применение по варианту осуществления 27, где период отсутствия лечения занимает от одного до двух месяцев.
29. Способ по любому из вариантов осуществления 1-28, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-28 или применение в соответствии с любым из вариантов осуществления 3-28, где снижение уровня системной иммуносупрессии связано с увеличением системного присутствия или активности продуцирующих IFNγ лейкоцитов и/или увеличением системного присутствия или активности цитокина IFNγ.
30. Способ по любому из вариантов осуществления 1-29, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-29 или применение в соответствии с любым из вариантов осуществления 3-29, где временное снижение уровня системной иммуносупрессии связано с увеличением системного присутствия или активности эффекторных Т-клеток.
31. Способ по любому из вариантов осуществления 1-30, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-30 или применение в соответствии с любым из вариантов осуществления 3-30, где временное снижение уровня системной иммуносупрессии связано с уменьшением системного присутствия или активности регуляторных Т-клеток и/или снижением системного присутствия цитокина IL-10.
32. Способ по любому из вариантов осуществления 1-31, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-31 или применение в соответствии с любым из вариантов осуществления 3-31, где временное снижение уровня системной иммуносупрессии связано с уменьшением системного присутствия супрессорных клеток миелоидного происхождения (MDSC).
33. Способ по любому из вариантов осуществления 1-32, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-32 или применение в соответствии с любым из вариантов осуществления 3-32, где временное снижение уровня системной иммуносупрессии происходит путем снятия ограничения, наложенного на иммунную систему одной или несколькими иммунными контрольными точками.
34. Способ по варианту осуществления 33, активный агент или фармацевтическая композиция в соответствии с вариантом осуществления 33 или применение по варианту осуществления 33, где введение композиции блокирует одну или несколько контрольных точек иммунной системы, тем самым вызывая временное снижение уровня системной иммуносупрессии.
35. Способ по варианту осуществления 34, активный агент или фармацевтическая композиция по варианту осуществления 34 или применение по варианту осуществления 34, где одна или несколько иммунных контрольных точек включают в себя PD-1-PDL1, PD-1-PDL2, CD28-CD80, CD28-CD86, CTLA-4-CD80, CTLA-4-CD86, ICOS-B7RP1, В7Н3, В7Н4, В7Н7, В7-CD28-подобную молекулу, BTLA -HVEM, KIR-МНС класса I или II, LAG3-МНС класса I или II, CD137-CD137L, OX40-OX40L, CD27-CD70, CD40L-CD40, TIM3-GAL9, V-доменный иммуноглобулиновый супрессор активации Т-клеток (VISTA), стимулятор генов интерферона (STING), иммуноглобулин Т-клеток и иммунорецепторный ингибиторный мотив домена на основе тирозина (TIGIT), индуцированный глюкокортикоидами белок, связанный с фактором некроза опухолей (GITR), A2aR-аденозин или индоламин-2,3-диоксигеназа (IDO)-L-триптофан.
36. Способ по любому из вариантов осуществления 1-35, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-35 или применение по любому из вариантов осуществления 3-35, где введение композиции во время сеанса лечения продолжается, по меньшей мере, до тех пор, пока системное присутствие или активность лейкоцитов, продуцирующих IFNγ, и/или цитокина IFNγ не станет выше контрольного, после чего введение прекращается, а период без лечения продолжается до тех пор, пока системное присутствие или активность лейкоцитов, продуцирующих IFNγ, и/или цитокина IFNγ остается выше контрольного, при этом к контролю относятся: а) уровень системного присутствия или активности лейкоцитов, продуцирующих IFNγ, и/или цитокина IFNγ, измеренного в последнем полученном образце крови от индивидуума перед введением; или b) уровень системного присутствия или активности лейкоцитов, продуцирующих IFNγ, и/или цитокин IFNγ, характерный для группы лиц, страдающих от заболевания, расстройства, патологического состояния или поражения центральной нервной системы.
37. Способ по любому из вариантов осуществления 1-36, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-36 или применение по любому из вариантов осуществления 3-36, по которому у индивидуума снижается уровень растворимого амилоидного бета-пептида в головном мозге, снижается или снижается нагрузка бета-амилоидными бляшками (Аβ) головного мозга, снижается гиппокампальный глиоз, снижается уровень провоспалительного цитокина в головном мозге, уменьшается воспаление головного мозга и/или улучшается когнитивная функция.
38. Способ по варианту осуществления 37, активный агент или фармацевтическая композиция по варианту осуществления 37 или применение по варианту осуществления 37, где улучшенная когнитивная функция представляет собой обучение, память, создание изображений, пластичность, мышление, осознание, логический ход мыслей, способность к пространственному восприятию, навыки речи и языка, приобретение языка, способность оценивать мнение или любую их комбинацию.
39. Способ по любому из вариантов осуществления 1-38, активный агент или фармацевтическая композиция по вариантам осуществления 2-38, или применение по варианту осуществления 3-38, где иммунные клетки включают в себя моноциты, макрофаги или Т-клетки.
40. Способ по варианту осуществления 39, активный агент или фармацевтическая композиция по варианту осуществления 39, или применение по варианту осуществления 39, где Т-клетки включают в себя регуляторные Т-клетки.
41. Способ по любому из вариантов осуществления 1-40, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-40 или применение по любому из вариантов осуществления 3-40, где заболевания, расстройства, патологические состояния или поражения центральной нервной системы включают в себя болезнь Альцгеймера, амиотрофический боковой склероз, болезнь Паркинсона, болезнь Хантингтона, первичный прогрессирующий рассеянный склероз; вторичный прогрессирующий рассеянный склероз, кортикобазальную дегенерацию, синдром Ретта, переднюю ишемическую оптическую невропатию; глаукому; увеит; депрессию; связанный с травмой стресс или посттравматическое стрессовое расстройство, лобно-временную деменцию, болезнь диффузных телец Леви, умеренные когнитивные нарушения, атрофию задней коры головного мозга, первичную прогрессирующую афазию или прогрессирующий надъядерный паралич.
42. Способ по любому из вариантов осуществления 1-40, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-40 или применение по любому из вариантов осуществления 1-40, по которому заболевание, расстройство, патологическое состояние или поражение центральной нервной системы является таупатией.
43. Способ по варианту осуществления 42, активный агент или фармацевтическая композиция по варианту осуществления 42, или применение по варианту осуществления 42, где таупатия представляет собой болезнь Альцгеймера, аргирофильное зерновое заболевание, хроническую травматическую энцефалопатию, кортикобазальную дегенерацию, деменцию боксеров, лобно-височную деменцию, лобно-височную дегенерацию, болезнь Галлервордена-Шпатца, болезнь Хантингтона, ганглиоглиому, ганглиоцитому, глобулярную глиальную таупатию, свинцовую энцефалопатию, липофусциноз, болезнь Литико-Бодига (комплекс болезни Паркинсона и деменции на Гуаме), менингоангиоматоз, паркинсонизм, связанный с хромосомой 17, болезнь Пика, первичную возрастную таупатию (PART), ранее известную как деменция с преобладанием нейрофибриллярных клубков (NFT-деменция), постэнцефалитический паркинсонизм, прогрессирующий надъядерный паралич, подострый склерозирующий панэнцефалит или клубневой (туберозный) склероз.
44. Способ по любому из вариантов осуществления 1-40, активный агент или фармацевтическая композиция по любому из вариантов осуществления 2-40 или применение по любому из вариантов осуществления 3-40, где заболевание, расстройство, патологическое состояние или поражение центральной нервной системы представляет собой дегенерацию сетчатки.
45. Способ по варианту осуществления 44, активный агент или фармацевтическая композиция по варианту осуществления 44, или применение по варианту осуществления 44, где дегенерация сетчатки представляет собой влажную возрастную дегенерацию макулы, сухую возрастную дегенерацию макулы, пигментный ретинит, хороидеремию, конусно-палочковую дистрофию сетчатки, гиратную атрофию, ювенильный ретиношизис, желтушную макулярную дистрофию (болезнь Беста), абеталипопротеинемию (болезнь Бассена-Корнцвейга), синдром Барде-Бидля, синдром монохромности голубого конуса, доминантные друзы, витреоретинальную дистрофию Гольдмана-Фавра (усиленный синдром S-конуса), синдром Кернса-Сейра, синдром Лоренса-Муна, врожденный амавроз Лебера, болезнь Рефсума Лебера, болезнь Огучи, перипапиллярную (перицентральную) хороидальную дистрофию, пигментную дистрофию, макулярную дистрофию Сорсби, болезнь Штаргардта, синдром Стиклера, синдром Ашера или витреоретинальную дистрофию Вагнера.
ПРИМЕРЫ
[132] Следующие примеры, без ограничения перечисленным, представлены в иллюстративных целях только для того, чтобы способствовать более полному пониманию рассматриваемых репрезентативных вариантов осуществления. Эти примеры не должны толковаться как ограничивающие любые варианты осуществления, представленные в настоящем описании, включая те, которые относятся к активным агентам, фармацевтическим композициям или способам и применениям, раскрываемым в настоящем документе.
Материалы и методы
[133] Животные. 5XFAD трансгенные мыши (Tg6799) со сверхэкспрессией семейных мутантных форм БА человеческого АРР (шведская мутация, K670N/M671L; флоридская мутация, I716V; и лондонская мутация, V717I), PS1 (M146L/L286V) трансгены при транскрипционном контроле специфического для нейрона катализатора для мышей Thy-1 (Oakley et al, 2006) и двойные трансгенные мыши с БА В6. Cg-Tg (APPswe, PSEN1dE9) 85Dbo/J (Borchelt et al, 1997) были приобретены в лаборатории Джексона. Генотипирование проведено методом ПЦР-анализа хвостовой ДНК, как описано выше (Oakley et al, 2006). Гетерозиготные мутанты мышей cx3cr1GFP/+ (Jung et al, 2000) (B6.129P-cx3cr1tm1Litt/J, где одна из CX3CR1 аллелей хемокинового рецептора заменена геном, кодирующим GFP), были использованы в качестве доноров костномозговых химер. Мыши Foxp3.LuciDTR были выведены из 5XFAD мышей для активации условного истощения Foxp3+ Treg клеток. Животные были выведены и содержались Центром по разведению животных Института Вейцмана. Все эксперименты, описанные в настоящем документе, проведены с соблюдением правил, сформулированных Комитетом по уходу за животными и их использованию в учреждениях (IACUC) Института Вейцмана.
[134] Очистка РНК, синтез кДНК и количественный анализ ПЦР в реальном времени. Общая РНК зубчатой извилины гиппокампа (ЗИ) была экстрагирована TRI Reagent (Молекулярный исследовательский центр) и очищена от лизатов с использованием комплекта RNeasy (Qiagen). Тотальную РНК хороидного сплетения получали с помощью комплекта MicroPrep РНК (Zymo Research). мРНК (1 мкг) первращали в кДНК, с использованием высокопроизводительного комплекта для обратной транскриптазы кДНК (Applied Biosystems). Экспрессию специфических мРНК анализировали с использованием флуоресцентной количественной ПЦР в реальном времени (RT-qPCR). Реакции RT-qPCR проводили с использованием смеси Fast-SYBR PCM Master Mix (Applied Biosystems). Реакции количественной оценки проводили в трех повторностях для каждого образца с использованием метода стандартной кривой. В качестве ссылочного гена был выбран ген пептидил-пролил-изомеразы А (ppia). Циклы амплификации составляли 95°С в течение 5 с, 60°С в течение 20 с и 72°С в течение 15 с. В конце анализа была построена кривая плавления для оценки специфичности реакции. Для анализа генов ifn-γ и ppia кДНК была преамплифицирована за 14 циклов ПЦР с неслучайными праймерами ПЦР, тем самым была увеличена чувствительность последующего анализа ПЦР в реальном времени, согласно протоколу производителя (PreAmp Master Mix Kit; Applied Biosystems). Экспрессию мРНК определяли с помощью TaqMan RT-qPCR, согласно инструкциям производителя (Applied Biosystems). Все реакции RT-qPCR были проведены и проанализированы с помощью программного обеспечения StepOne V2.2.2 (Applied Biosystems). Были использованы следующие зонды TaqMan Assays-on-Demand™: Mm02342430_g1 (ppia) и Mm01168134_m1 (ifn-γ).
[135] Для всех других исследованных генов использовались следующие праймеры:
[136] ppia прямой 5'-AGCATACAGGTCCTGGCATCTTGT-3' (Идентиф. номер: 33) и обратный 5'-CAAAGACCACATGCTTGCCATCCA-3' (Идентиф. номер: 34);
[137] icam1 прямой 5'-AGATCACATTCACGGTGCTGGCTA-3' (Идентиф. номер: 35) и обратный 5'-AGCTTTGGGATGGTAGCTGGAAGA-3' (Идентиф. номер: 36);
[138] vcam1 прямой 5'-TGTGAAGGGATTAACGAGGCTGGA-3' (Идентиф. номер: 37) и обратный 5'-CCATGTTTCGGGCACATTTCCACA-3' (Идентиф. номер: 38);
[139] cxcl10 прямой 5'-AACTGCATCCATATCGATGAC-3' (Идентиф. номер: 39) и обратный 5'-GTGGCAATGATCTCAACAC-3' (Идентиф. номер: 40);
[140] ccl2 прямой 5'-CATCCACGTGTTGGCTCA-3' (Идентиф. номер: 41) и обратный 5'-GATCATCTTGCTGGTGAATGAGT-3' (Идентиф. номер: 42);
[141] tnf-γ прямой 5'-GCCTCTTCTCATTCCTGCTT-3' (Идентиф. номер: 43) обратный CTCCTCCACTTGGTGGTTTG-3' (Идентиф. номер: 44);
[142] il-1β прямой 5'-CCAAAAGATGAAGGGCTGCTT-3' (Идентиф. номер: 45) и обратный 5'-TGCTGCTGCGAGATTTGAAG-3' (Идентиф. номер: 46);
[143] il-12p40 прямой 5'-GAAGTTCAACATCAAGAGCA-3' (Идентиф. номер: 47) и обратный 5'-CATAGTCCCTTTGGTCCAG-3' (Идентиф. номер: 48);
[144] il-10 прямой 5'-TGAATTCCCTGGGTGAGAAGCTGA-3' (Идентиф. номер: 49) и обратный 5'-TGGCCTTGTAGACACCTTGGTCTT-3' (Идентиф. номер: 50);
[145] tgfβ2 прямой 5'-AATTGCTGCCTTCGCCCTCTTTAC-3' (Идентиф. номер: 51) и обратный 5'-TGTACAGGCTGAGGACTTTGGTGT-3' (Идентиф. номер: 52);
[146] igf-1 прямой 5'-CCGGACCAGAGACCCTTTG (Идентиф. номер: 53) и обратный 5'-CCTGTGGGCTTGTTGAAGTAAAA-3' (Идентиф. номер: 54);
[147] bdnf прямой 5'-GATGCTCAGCAGTCAAGTGCCTTT-3' (Идентиф. номер: 55) и обратный 5'-GACATGTTTGCGGCATCCAGGTAA-3' (Идентиф. номер: 56);
[148] Иммуногистохимия. Обработка тканей и иммуногистохимия были выполнены на парафиновом срезе мозга мыши (толщиной 6 мкм) и человека (толщиной 10 мкм). Для окрашивания человеческого ICAM-1 использовали первичное антитело к ICAM мышей (1:20 Abcam, ab2213). Слайды инкубировали в течение 10 мин в 3% H2O2, и использовали вторичное биотин-конъюгированное антимышиное антитело с последующей биотин/авидиновой амплификацией с помощью набора Vectastain ABC (Vector Laboratories). Затем применяли 3,3'-диаминобензидин (субстрат ДАБ) (набор Zytomed); слайды дегидратировались и устанавливались с помощью крепежного раствора на основе ксилола. Для окрашивания тканей мышей подвергали транскардиальной перфузии PBS до вырезания ткани и фиксации. Ткани СС были отделены под препаровальной лупой (Stemi DV4; Zeiss) от боковых, третьего и четвертого желудочка головного мозга. При окрашивании цельного соединения СС ткани фиксировали 2,5% параформальдегидом (ПФА) в течение 1 часа при 4°С, а затем переносили в PBS, содержащий 0,05% азида натрия. Перед окрашиванием рассеченные ткани промывали PBS и блокировали (20% лошадиной сывороткой, 0,3% Triton Х-100 и PBS) в течение 1 часа при комнатной температуре. В течение 1 часа при комнатной температуре проводили окрашивание с полным первичным антителом (в PBS, содержащем 2% лошадиной сыворотки и 0,3% Triton Х-100), или со вторичными антителами. После каждого шага следовало три промывания в PBS. Ткани были нанесены на слайды, смонтированы с помощью Immu-mount (9990402, Thermo Scientific) и запечатаны с помощью обложки. Для окрашивания срезов мозга были применены два различных протокола подготовки тканей (внедренный парафин или микротомные свободные срезы), как описано ранее (Baruch et al, 2013; Kunis et al, 2013). Были использованы следующие первичные антитела: мышиное анти-Аβ (1:300, Covance, #SIG-39320); кроличье анти-GFP (1:100, MBL, #598); анти-CD68 крысы (1:300, eBioscience, #14-0681); анти-ICAM-1 крысы (1:200, Abcam, #АВ2213); козье анти-GFP (1:100, Abcam, #ab6658); кроличье анти-IBA-1 (1:300, Wako, #019-19741); козье анти-IL-10 (1:20, R&D systems, #AF519); крысы анти-Foxp3 (1:20, eBioscience, #13-5773-80); кроличье анти-CD3 (1:500, Dako, #IS503); мышиное анти-ZO-1, мышиное анти-E-Cahedrin и кроличье анти-Клаудин-1 (все 1:100, Invitrogen, #33-9100, #33-4000, #51-9000); кроличье анти-ГФКБ (1:200, Dako, #Z0334). Вторичные антитела включали в себя: ослиные анти-мышиные/козьи/кроличьи/крысыные антитела, конъюгированные Су2/Су3/Су5 (1:200, все от Jackson Immunoresearch). Слайды были подвергнуты ядерному окрашиванию Хехст (1 4000; Invitrogen Probes) в течение 1 мин. В процедурах иммуноокрашивания регулярно применяли два отрицательных контроля, проводилось окрашивание изотипическим контрольным антителом с последующим окрашиванием вторичным антителом, или окрашиванием только вторичным антителом. Для внутриклеточного окрашивания Foxp3 извлечение антигена из парафиновых слайдов выполняли с использованием набора Retreivagen Kit (№550524, №550527, BD Pharmingen™). Микроскопический анализ проводили с использованием флуоресцентного микроскопа (Е800, Nikon) или лазерного сканирующего конфокального микроскопа [Carl Zeiss, Inc.]). Флуоресцентный микроскоп был оснащен цифровой камерой (DXM 1200F, Nikon) и объективом 20× NA 0,50 или 40× NA 0,75 (Plan Fluor, Nikon). Конфокальный микроскоп был оснащен устройством лазерного сканирования LSM 510 (три лазера: Ar 488, HeNe 543 и HeNe 633). Записи были сделаны на вторично фиксированных тканях с использованием программного обеспечения для захвата (NIS-Elements, F3 [Nikon] или LSM [Carl Zeiss, Inc.]). Для количественной оценки интенсивности окрашивания, общее окрашивание клеток и фона измеряли с использованием программного обеспечения ImageJ (NIH), а интенсивность специфического окрашивания рассчитывали, как описано ранее (Burgess et al, 2010). Изображения были обрезаны, объединены и оптимизированы с помощью Photoshop CS6 13.0 (Adobe), и организованы с помощью Illustrator CS5 15.1 (Adobe).
[149] Парафиновые срезы СС человека. Посмертные срезы головного мозга молодых и пожилых пациентов, скончавшихся не от заболеваний ЦНС, а также пациентов с БА были получены из Оксфордского мозгового банка (ранее известного как Оксфордская коллекция головного мозга имени Томаса Уиллиса (TWOBC)) с соответствующего согласия и одобрения Комитета по этике (TW220). Эксперименты с применением этих срезов были одобрены Комитетом по биоэтике Института Вейцмана.
[150] Проточная цитометрия, подготовка и анализ проб. Мышей мышей подвергали транскардиальной перфузии PBS, и ткани обрабатывали, как описано выше (Baruch et al, 2013). Мозги были рассечены, различные участки мозга были удалены под препаровальной лупой (Stemi DV4; Zeiss) в PBS, ткани были отделены с помощью диссоциатора gentleMACS™ (Miltenyi Biotec). Ткани хороидного сплетения выделяли из боковых, третьего и четвертого желудочков головного мозга, инкубировали при 37°С в течение 45 минут в PBS (с Са2+/Mg2+), содержащем 400 ЕД/мл коллагеназы типа IV (Worthington Biochemical Corporation), а затем вручную гомогенизировали пипетированием. Селезенки раздавливали плунжером шприца и обрабатывали лизирующим буфером ACK (аммоний-хлорид-калий) для удаления эритроцитов. Во всех случаях пробы окрашивали согласно протоколам производителей. Все образцы фильтровали через нейлоновую сетку с размером пор 70 мкм и блокировали при помощи анти-Fc CD16/32 (1:100; BD Biosciences). Для внутриклеточного окрашивания IFN-γ клетки инкубировали с параметоксиамфетамином (10 нг/мл, Sigma-Aldrich) и иономицином (250 нг/мл, Sigma-Aldrich) в течение 6 ч, на последние 4 часа добавлялся Брефельдин-А (10 мкг/мл, Sigma-Aldrich). Внутриклеточное мечение цитокинов проводили с помощью набора BD Cytofix/Cytoperm™ Plus для фиксации/пермеализации (кат. №555028). Для окрашивания регуляторных Т-клеток использовали набор для окрашивания буфера eBioscience FoxP3 (кат. №00-5523-00). Следующие моноклональные антитела, меченные флуорохромом, приобретали у BD Pharmingen, BioLegend, R&D Systems или eBiosciences, и использовли в соответствии с протоколами производителей: РЕ или Alexa Fluor 450-конъюгированное анти-CD4; РЕ-конъюгированное анти-CD25; PerCP-Су5.5-конъюгированное анти-CD45; FITC-конъюгированное анти-TCRβ; АРС-конъюгированное анти-IFN-γ; АРС-конъюгированное анти-FoxP3; ярко-фиолетовое конъюгированное анти-CD45. Клетки анализировались на цитометре LSRII (BD Biosciences) с использованием программного обеспечения FlowJo. В каждом эксперименте использовались соответствующие отрицательные контрольные группы, положительные контроли и одиночные окрашенные пробы для каждой ткани для идентификации представляющих интерес популяций, и для исключения других популяций.
[151] Получение костномозговых (ВМ) химер. Костномозговые химеры получали как описано выше (Shechter et al, 2009; Shechter et al, 2013). Вкратце, мыши реципиентов с учетом пола подвергали летальному облучению всего тела (950 рад) при защите головы (Shechter et al, 2009). Мышам затем вводили внутривенно 5×106 клеток ВМ от доноров CX3CR1GFP/+. Мышей оставляли на 8-10 недель после трансплантации клеток ВМ, чтобы обеспечить восстановление гематопоэтической линии до их использования в экспериментах. Процент химеризма определяли с помощью FACS-анализа образцов крови согласно процентной доле клеток, экспрессирующих GFP, из числа циркулирующих моноцитов (CD11b+). В этой модели с защитой головного мозга было достигнуто в среднем 60% химеризма, и CNS-инфильтрирующие GFP+ миелоидные клетки были подтверждены как CD45high/CD11bhigh, представляющие собой макрофаги, полученные из моноцитов, а не микроглии (Shechter et al, 2013).
[152] Водный лабиринт Морриса. Испытания на мышах проводили три раза в день в течение 4 дней подряд, чтобы они научились находить скрытую платформу, расположенную на 1,5 см ниже поверхности воды в бассейне (1,1 м в диаметре). Температуру воды поддерживали на уровне 21-22°С. Вода была сделана непрозрачной при помощи молочного порошка. В помещении для испытаний мышам для поиска погруженной платформы были предоставлены только отдаленная визуальная форма и объекты-подсказки. Латентность спасения, т.е. время, необходимое для поиска платформы и подъема на нее, регистрировалась до 60 с. Каждой мыши разрешалось остаться на платформе в течение 15 с, затем ее забирали из лабиринта и возвращали в клетку. Если мышь не находила платформы в течение 60 с, ее вручную сажали на платформу и через 15 с возвращали в клетку. Интервал между испытаниями для каждой мыши составлял 10 мин. На 5-й день платформу убирали, и мышей подвергали одному испытанию продолжительностью 60 с без возможности найти выход. В дни 6 и 7 платформу помещали в секторе напротив первоначального сектора обучения, и мышь переобучали по три сессии каждый день. Данные записывали с помощью автоматизированной системы слежения EthoVision V7.1 (Noldus Information Technology). Статистический анализ проводили с помощью дисперсионного анализа (ANOVA) и апостериорного анализа (post-hoc) Бонферрони. Все испытания в ВЛМ проводились с 10 утра до 5 вечера во время фазы выключенного освещения.
[153] Радиальный рукавный водный лабиринт. Для тестирования пространственного обучения и памяти, как было подробно описано ранее, использовался радиальный рукавный водный лабиринт (РРВЛ) (Alamed et al, 2006). Вкратце, шесть вставок из нержавеющей стали помещались в резервуар, образуя шесть плавающих рукавов, выходящих из открытой центральной зоны. Платформа для выхода была расположена в конце одного из рукавов (целевой рукав), на 1,5 см ниже поверхности воды в бассейне диаметром 1,1 м. Температура воды поддерживалась на уровне 21-22°С. Вода была сделана непрозрачной при помощи молочного порошка. В помещении для испытаний мышам для поиска погруженной платформы были предоставлены только отдаленная визуальная форма и объекты-подсказки. Местоположение целевого рукава оставалось неизменным для данной мыши. На 1 день с мышами проводилось 15 испытаний (в течение 3 часов), с чередованием видимой и скрытой платформы, и последние 4 испытания только со скрытыми платформами. На 2 день мышей обучали посредством 15 испытаний со скрытой платформой. Вход в неверный рукав или неспособность выбрать рукав в течение 15 с записывались как ошибка. Пространственное обучение и память измерялись путем подсчета количества ошибок входа в рукав или задержки спасения мышей в каждом испытании. Данные обучения анализировались как средние ошибки или задержки спасения по блокам из трех последовательных испытаний.
[154] Введение GA. Каждой мыши подкожно вводили GA общей дозой 100 мкг (партия № Р53640, Teva Pharmaceutical Industries, Петах-Тиква, Израиль), растворенный в 200 мкл PBS. Мышам делали инъекции в соответствии с режимом либо еженедельного введения GA (Butovsky et al, 2006), либо ежедневного введения GA (фиг. 8 и фиг. 16). Мышей умерщвляли через 1 неделю после последней инъекции GA, или спустя 1 месяц после лечения, как указано по каждому эксперименту.
[155] Условная абляция регуляторных Т-клеток. Дифтерийный токсин (DTx, 8 нг/г массы тела, Sigma) вводили интраперитонеально ежедневно в течение 4 дней подряд мышам Foxp3.LuciDTR (Suffner et al, 2010). Эффективность DTx была подтверждена проточным цитометрическим анализом иммунных клеток в крови и селезенке, было достигнуто почти полное (>99%) истощение GFP-экспрессирующих FoxP3+ CD4+ Treg клеток (фиг. 4).
[156] Ингибирование Р300. Ингибирование р300 у мышей проводили аналогично ранее описанному (Liu et al, 2013). p300i (C646; Tocris Bioscience) растворяли в ДМСО и ежедневно вводили интраперитонеально (8,9 мг кг-1 д-1) в течение 1 недели. Аналогичным образом мышам, проходившим лечение носителем, вводили ДМСО.
[157] Лечение ПТРК. Политрансретиноевая кислота (ПТРК) вводилась мышам аналогично ранее описанной процедуре (Walsh et al, 2014). ПТРК (Sigma) растворялась в ДМСО и вводилась интраперитонеально. (8 мг кг-1 д-1) через день в течение 1 недели. Аналогичным образом, мышам, проходившим лечение носителем, вводили ДМСО.
[158] Изоляция и количественная оценка растворимого белка Аβ (sAβ). Гомогенизацию ткани и экстракцию белка sAβ проводили, как описано ранее (Schmidt et al, 2005) ENREF 54 ENREF 53. Вкратце, паренхиму мозга головного мозга отделяли, замораживали и выдерживали при -80°С вплоть до гомогенизации. Белки последовательно экстрагировали из образцов для получения отдельных фракций, содержащих белки с различной растворимостью. Образцы гомогенизировали в 10 объемах ледяного буфера для гомогенизации ткани, содержащего 250 мМ сахарозы, 20 мМ Tris base, 1 мМ этилендиаминтетрауксусной кислоты (ЭДТА) и 1 мМ этиленгликолевой тетрауксусной кислоты (рН 7,4) с использованием пестика из шлифованного стекла в гомогенизаторе Даунса. После шести ударов гомогенат смешивали 1:1 с 0,4% диэтиламина (DEA) в растворе 100 мМ NaCl перед еще шестью ударами, и затем центрифугировали при 135,000 g при 4°С в течение 45 минут. Супернатант (DEA-растворимая фракция, содержащая внеклеточные и цитозольные белки) собирали и нейтрализовали 10% раствором 0,5 М трис-HCl (рН 6,8). Аβ1-40 и Аβ1-42 индивидуально измеряли с помощью иммуноферментного анализа (ELISA) из растворимой фракции с использованием коммерчески доступных наборов (Biolegend; #SIG-38954 и #SIG-38956 соответственно) в соответствии с инструкцией производителя.
[159] Количественное определение Аβ-бляшек. Из каждой пробы мозга собирали 6 мкм коронарные срезы, и по восемь срезов на мышь, с четырех различных заранее определенных глубин по всей изучаемой области (зубчатая извилина или кора головного мозга), подвергали иммуноокрашиванию. Сегментация положительно окрашенных пикселей на основе гистограммы была выполнена с использованием программного обеспечения Image-Pro Plus (Media Cybernetics, Бетесда, Мэриленд, США). Алгоритм сегментации был вручную применен к каждому изображению, в области зубчатой извилины или в 5-ом кортикальном слое, и была определена процентная доля площади, занимаемой полным иммуноокрашиванием Аβ. Количество бляшек было подсчитано на тех же 6 мкм корональных срезах мозга и представлено как среднее число бляшек на область мозга. До количественной оценки срезы были закодированы, чтобы скрыть идентификаторы экспериментальных групп, и бремя бляшек подсчитывалось наблюдателем, не способным идентифицировать группы.
[160] Статистический анализ. Конкретные тесты, используемые для анализа каждой серии экспериментов, указаны в условных обозначениях к фигурам. Данные были проанализированы с использованием двухстороннего t-критерия Стьюдента для сравнения между двумя группами, однофакторного дисперсионного анализа ANOVA для сравнения нескольких групп, с последующим апостериорным анализом (post-hoc analysis) Ньюмена-Кейлса для парного сравнения групп после отклонения нулевой гипотезы (Р<0,05). Данные поведенческих тестов анализировали с использованием двухфакторного дисперсионного анализа с повторными измерениями ANOVA, и post-hoc анализ с критерием Бонферрони для парных сравнений Бонферрони. Размеры проб выбирали с достаточной статистической мощностью, основанной на литературе и прошлом опыте, мышей выделяли в экспериментальные группы в зависимости от возраста, пола и генотипа. Исследователи во время экспериментов и при оценке результатов не могли идентифицировать группы («слепой метод»). Все критерии включения и исключения были предварительно установлены в соответствии с руководящими принципами IACUC. Результаты представлены как среднее ± стандартная ошибка среднего. На графиках ось у погрешностей представляет стандартную ошибку среднего. Статистические вычисления выполнялись с использованием программного обеспечения GraphPad Prism (GraphPad Software, Сан-Диего, Калифорния).
Пример 1. "Шлюзовая" активность хороидного (сосудистого) сплетения (CP) при прогрессировании болезни в мышиной модели БА.
[161] Сначала авторы изобретения исследовали активность CP при прогрессировании заболевания в модели БА у трансгенных мышей 5XFAD (AD-Tg); эти мыши коэкспрессируют пять мутаций, связанных с семейной БА, и у них развивается церебральная Аβ-патология и глиоз уже в возрасте 2 месяцев (Oakley et al, 2006). Авторы изобретения обнаружили, что на прогрессирующих стадиях патологии болезни, CP мышей AD-Tg, по сравнению с контрольными особями дикого типа (WT) соответствующего возраста, экспрессировали значительно более низкие уровни детерминант "хоуминга" и миграции лейкоцитов, в том числе icam1, vcam1, cxcl10 и ccl2 (фиг. 1А), демонстрируя повышающее регулирование со стороны CP в ответ на острое поражение ЦНС, и нуждались в трансэпителиальной миграции лейкоцитов (Kunis et al, 2013; Shechter et al, 2013). Иммуногистохимическое окрашивание лиганда интегрина, ICAM-1, подтвердило его пониженную экспрессию эпителием CP мышей AD-Tg (фиг. 1b). Кроме того, окрашивание на ICAM-1 в мозгах человека после смерти показало его возрастное снижение в эпителии CP в соответствии с предыдущими наблюдениями авторов изобретения (Baruch et al, 2014), а количественная оценка этого эффекта выявила дальнейшее снижение у пациентов с БА по сравнению с пожилыми людьми без заболеваний ЦНС (фиг. 2А). Поскольку индукция детерминант миграции лейкоцитов со стороны CP зависит от передачи сигнала (IFN)-γ эпителиального интерферона (Kunis et al, 2013), авторы изобретения затем проверили, отражают ли наблюдаемые эффекты потерю доступности IFN-γ в СР. Изучение CP мышей 5XFAD AD-Tg с использованием внутриклеточного окрашивания проточной цитометрии выявило значительно меньшее количество клеток, продуцирующих IFN-γ, в этом отделении (фиг. 2В), а количественный анализ ПЦР в реальном времени (RT-qPCR) подтвердил более низкий уровень экспрессии мРНК ifn-γ в CP мышей AD-Tg по сравнению с контрольными особями дикого типа соответствующего возраста (фиг. 2С).
Пример 2. Функциональные связи между Treg-опосредованной системной иммуносупрессией, "шлюзовой" активностью CP и патологией БА.
[162] Регуляторные Т-клетки (Treg) играют ключевую роль в подавлении системных эффекторных иммунных ответов (Sakaguchi et al, 2008). Авторы изобретения предположили, что Treg-опосредованная системная иммуносупрессия влияет на доступность IFN-γ в CP и, соответственно, сконцентрировались на вовлечении регуляторных Т-клеток в патологию БА. В соответствии с предыдущими сообщениями о повышенных уровнях Treg и супрессии у пациентов с БА (Rosenkranz et al, 2007; Saresella et al, 2010; Torres et al, 2013), оценка частотности Foxp3+ Treg в спленоцитах мышей 5XFAD AD-Tg по сравнению с их сопоставимыми по возрасту однопометниками дикого типа показала повышенные уровни при прогрессировании заболевания (фиг. 3А, В). Для изучения функциональных связей между Treg-опосредованной системной иммуносупрессией, шлюзовой активностью CP и БА-патологией авторы изобретения скрестили 5XFAD AD-Tg мышей с мышами Foxp3-дифтерийного рецептора токсина (DTR+), что обеспечило временное условное in vivo истощение Foxp3+ Treg клеток у AD-Tg/DTR+ мышей путем введения дифтерийного токсина (DTx) ENREF 33 (фиг. 4А). Переходное истощение регуляторных Т-клеток привело к повышенной экспрессии мРНК молекул трафика лейкоцитов со стороны CP AD-Tg/DTR+ мышей в сравнении с их AD-Tg/DTR однопометниками, получавшими DTx (фиг. 5А). Анализ долгосрочного эффекта временного истощения регуляторных Т-клеток на паренхиму головного мозга (3 недели спустя) выявил накопление иммунных клеток в мозге, включая повышенное количество CD45high/CD11high миелоидных клеток, представляющих собой проникающие mo-МФ (Shechter et al, 2013) и CD4+ Т-клетки (фиг. 5В). Кроме того, кратковременное истощение регуляторных Т-клеток привело к заметному обогащению Foxp3+ Treg клеток среди CD4+ Т-клеток, которые накопились в мозге, согласно оценке по проточной цитометрии (фиг. 5С, D). Исследование гиппокампа методом количественной ПЦР в реальном времени (RT-qPCR) показало повышенную экспрессию мРНК foxp3 и il10 (фиг. 5Е).
[163] Затем авторы изобретения изучили, приводит ли краткосрочное истощение регуляторных Т-клеток, после которого иммунорегуляторные клетки накапливаются в зонах патологии головного мозга, к долгосрочному воздействию на мозговую деятельность. Авторы изобретения наблюдали снижение глиоза гиппокампа (фиг. 5F) и снижение уровня экспрессии мРНК провоспалительных цитокинов, таких как il-12р40 и tnf-α (фиг. 5G). Кроме того, бремя церебральных Аβ-бляшек в зубчатой извилине гиппокампа, а также в коре головного мозга (5-й слой), двух областях мозга, демонстрирующих устойчивую патологию Аβ-бляшек у 5XFAD AD-Tg мышей (Oakley et al, 2006), было снижено (фиг. 6А, В). Оценка влияния на когнитивную функцию с использованием водного лабиринта Морриса (ВЛМ) показала значительное улучшение пространственного обучения и памяти у мышей AD-Tg/DTR+ после истощения регуляторных Т-клеток по сравнению с AD-Tg/DTR мышами, получавшими DTx, достигая производительности, аналогичной таковой у мышей дикого типа (фиг. 6С-Е). В совокупности эти данные продемонстрировали, что кратковременное нарушение Treg-опосредованной системной иммуносупрессии у мышей AD-Tg привело к накоплению снимающих воспаление клеток, включая mo-МФ и Treg, в мозге, после чего последовало разрешение нейровоспалительной реакции, очистка Aβ и устранение когнитивного спада.
Пример 3. Еженедельное введение сополимера-1 снижает Treg-опосредованную системную иммуносупрессию, повышает шлюзовую активность CP и смягчает патологию БА.
[164] Для дальнейшего обоснования причинно-следственной природы обратной зависимости между системной иммуносупрессией, деятельностью СС и патологией БА авторами изобретения затем использовалось иммуномодулирующее соединение, глатирамер ацетат (GF, ГА, также известный как сополимер-1 или Copaxone®), которое по результатам еженедельного введения продемонстрировало терапевтический эффект в мышиной модели БА APP/PS1 (Butovsky et al, 2006); ENREF 22 этот эффект был функционально связан с рекрутингом mo-МФ в участки мозга, пораженные заболеванием (Butovsky et al, 2007). В этом случае авторы изобретения сначала исследовали, характеризуются ли СС у мышей AD-Tg APP/PS1, аналогично наблюдению авторов изобретения у мышей 5XFAD AD-Tg, недостаточностью в плане уровня экспрессии IFN-γ. Авторы изобретения обнаружили, что у мышей AD-Tg APP/PS1 уровни IFN-γ d CP были снижены по сравнению с контрольными особями дикого типа соответствующего возраста (фиг. 7А). Эти результаты побудили авторов изобретения проверить, можно ли воспроизвести терапевтический эффект еженедельного введения ГА мышам APP/PS1 (Butovsky et al, 2006) у мышей 5XFAD AD-Tg, и если да, то повлияет ли это на системные Treg клетки и активацию СС для трафика mo-МФ. Поэтому авторы изобретения применили к 5XFAD AD-Tg мышам еженедельный режим введения ГА в течение 4 недель (далее «еженедельный ГА»; схематически изображен на фиг. 8А). Авторы изобретения обнаружили, что мыши 5XFAD AD-Tg, получавшие еженедельный ГА, продемонстрировали снижение нейровоспаления (фиг. 8B-D) и улучшенные когнитивные способности, что продолжалось до 2 месяцев после лечения (фиг. 8E-I). Изучая методом проточной цитометрии воздействие еженедельного ГА на системный иммунитет и на СС, авторы изобретения обнаружили снижение уровней Foxp3+ Treg спленоцитов (фиг. 9А) и увеличение клеток, продуцирующих IFN-γ, в СС проходивших лечение мышей 5XFAD AD-Tg, с достижением уровней, аналогичных наблюдаемым у контрольных особей дикого типа (фиг. 9В). Повышенный уровень IFN-γ-экспрессирующих клеток в СС у мышей, получавших еженедельный ГА, сопровождался повышающим регулированием эпителиальной экспрессии молекул трафика лейкоцитов (фиг. 9С).
[165] Чтобы обнаружить попадание проникающих mo-МФ в ЦНС, авторы изобретения использовали мышей 5XFAD AD-Tg/CX3CR1GFP/+ с костномозговыми химерами (подготовлены с использованием защиты головы), что позволило визуализировать циркулирующие (с маркировкой зеленым флуоресцентным белком (ЗФБ/GFP)+) миелоидные клетки (Shechter et al, 2009; Shechter et al, 2013). Авторы изобретения обнаружили увеличение самонаведения GFP+ mo-МФ в СС и в соседние желудочковые пространства после терапии с еженедельным введением ГА, по сравнению с контрольными особями AD-Tg/CX3CR1GFP/+, получавшими проводник (фиг. 9D-E). Иммуногистохимия паренхимы мозга показала наличие GFP+ mo-МФ скоплений на участках формирования мозговых бляшек (фиг. 9F), а количественная оценка проникающих миелоидных клеток путем анализа гиппокампа методом проточной цитометрии у нехимерных мышей AD-Tg показала увеличение числа CD11bhighCD45high-экспрессирующих клеток (фиг. 9G, Н). Вместе эти результаты подтверждают функциональную связь между рекрутингом mo-МФ в участках патологии БА, снижением системного уровня Treg и IFN-γ-зависимой активации СС.
Пример 4. Вмешательство в деятельность регуляторных Т-клеток с использованием низкомолекулярного ингибитора гистоновой ацетилтрансферазы.
[166] Вышеприведенные данные, предполагающие, что Treg-опосредованная системная иммуносупрессия препятствует способности бороться с патологией БА, напоминают о функции, приписываемой регуляторным Т-клеткам при иммунотерапии рака, в которой эти клетки препятствуют способности иммунной системы обеспечивать эффективную анти-опухолевую реакцию (Bos & Rudensky, 2012; Nishikawa & Sakaguchi, 2010). Поэтому авторы изобретения сделали вывод, что лечение, которое непосредственно препятствует активности клеток Foxp3+ Treg, может быть эффективным при БА. Авторы изобретения испытали p300i (С646 (Bowers et al, 2010)), непептидный ингибитор р300, гистоновую ацетилтрансферазу, регулирующую функцию Treg (Liu et al, 2013); было продемонстрировано, что этот ингибитор влияет на подавляющую деятельность Treg, оставляя защитные ответы Т-эффекторных клеток без изменений (Liu et al, 2013). Авторы изобретения обнаружили, что мыши, проходившие лечение p300i, по сравнению с мышами, получавшими носитель (ДМСО), демонстрировали повышенный уровень системных IFN-γ-экспрессирующих клеток в селезенке (фиг. 10А), а также в CP (фиг. 10В). Затем авторы изобретения лечили AD-Tg мышей либо p300i, либо носителем в течение 1 недели, и исследовали животных через 3 недели в отношении нагрузки церебральных Аβ-бляшек. Иммуногистохимический анализ показал значительное снижение нагрузки церебральных Аβ-бляшек у мышей, получавших p300i, по сравнению с AD-Tg мышами (фиг. 10С-Е). Авторы изобретения также протестировали, будет ли воздействие на патологию бляшек после одного курса лечения продолжаться дольше, чем 3 недели, и если да, внесут ли дополнительные курсы лечения свой вклад в длительное воздействие. По этой причине авторы изобретения сравнили мышей AD-Tg, которые прошли один курс лечения p300i и были обследованы 2 месяца спустя, с группой, сопоставимой по возрасту, которая в течение этого периода прошла два курса лечения с интервалом в 1 месяц между ними (схематично изображена на фиг. 10F). Авторы изобретения обнаружили, что снижение нагрузки церебральных бляшек было очевидно даже через два месяца после одного курса лечения, но было интенсивнее у мышей, которые прошли два курса лечения с интервалом в 1 месяц между ними (фиг. 10G). Поскольку нарушенная синаптическая пластичность и память при БА связаны с повышенным уровнем растворимого Аβ1-40/Аβ1-42 (sAβ) в мозге (Shankar et al, 2008), авторы изобретения также измерили уровень sAβ после одного или нескольких циклов лечения p300i. И снова авторы изобретения обнаружили, что как один, так и два курса (с интервалом в 1 месяц между ними) эффективны в снижении церебрального sAβ, однако этот эффект был сильнее после нескольких курсов в плане воздействия на sAβ1-42 (фиг. 10Н). Эти результаты показали, что, несмотря на эффективность одного краткосрочного курса лечения, серия курсов лечения способствовала бы поддержанию долговременного терапевтического эффекта, аналогичного нашим наблюдениям после еженедельного лечения ГА.
Пример 5. Терапевтический потенциал блокады иммунной контрольной точки PD-1 при болезни Альцгеймера.
[167] Сперва авторы изобретения проверили, может ли таргетинг ингибирующего пути PD-1 повлиять на IFN-γ-ассоциированный системный иммунитет у трансгенных (AD-Tg) мышей 5XFAD с БА, которые коэкспрессируют пять мутаций, связанных с семейной БА (Oakley et al, 2006). Мышам AD-Tg в возрасте 10 месяцев, когда развивается церебральная патология, двумя интраперитонеальными инъекциями вводили как блокирующие антитела, направленных на контрольные антитела PD-1 (анти-PD-1), так и IgG, в дни 1 и 4, а затем на 7-й день. Анализ методом проточной цитометрии показал, что блокада пути PD-1 приводит к повышенным частотам IFN-γ-продуцирующих спленоцитов CD4+ Т (фиг. 11А, В).
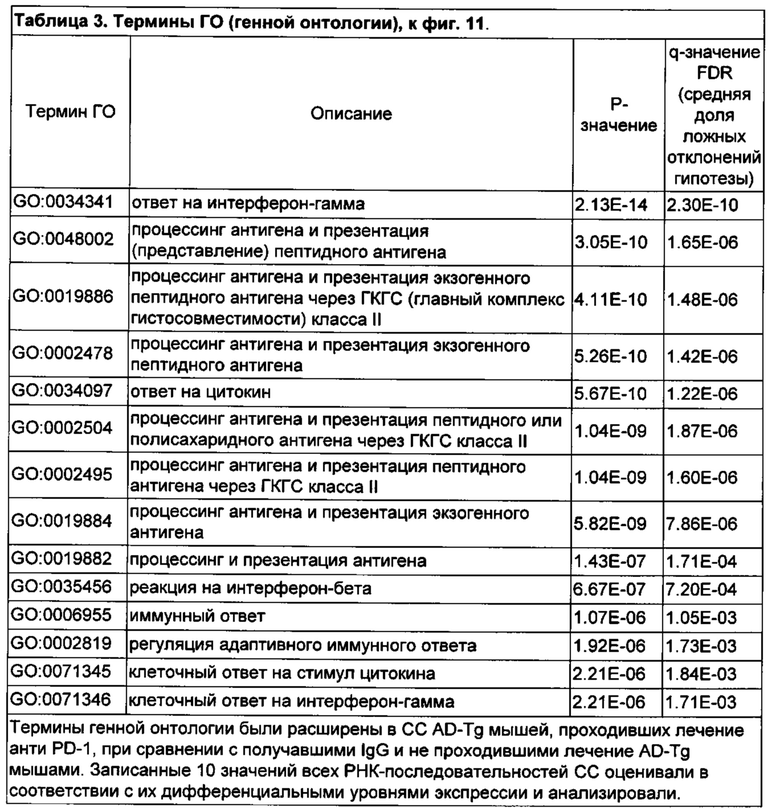
[168] Затем нами было изучено, влияет ли этот системный иммунный ответ на деятельность СС. Геномное РНК-секвенирование СС (не приведено; полный анализ будет раскрыт в отчете авторов изобретения под названием Примера 5, и может быть получен от изобретателей по запросу) показало профиль экспрессии, связанный с реакцией на IFN-γ (фиг. 11D и Таблица 3), и анализ методом количественной ПЦР в реальном времени (RT-qPCR) подтвердил повышенный уровень мРНК IFN-γ в СС, по сравнению с контрольными AD-Tg особями, получавшими IgG или не проходившими лечение (фиг. 11С). Эти выводы подтвердили системную и специфичную для тканей СС IFN-γ иммунную реакцию после блокады PD-1, и побудило нас после этого проверить воздействие на патологию заболевания.
[169] Для изучения функциональных последствий блокады PD-1 на патологию БА мы лечили 10-месячных AD-Tg мышей анти-PD-1 или контрольными антителами IgG и оценивали влияние на пространственное обучение и память с помощью радиального рукавного водного лабиринта (РРВЛ).
[170] Через месяц после лечения (две интраперинетонеальных инъекции с 3-дневным интервалом) у мышей AD-Tg, получивших анти-PD1, наблюдалось значительное улучшение когнитивной функции по сравнению с контрольными особями соответствующего возраста, получавшими IgG или не проходившими лечение, с достижением когнитивных уровней, аналогичных уровням у мышей дикого типа соответствующего возраста (фиг. 12А). Далее мы исследовали, продлится ли эффект блокады PD-1 на когнитивные функции у AD-Tg мышей дольше 1 месяца, и будут ли эффективны дополнительные терапевтические сеансы. Мы лечили AD-Tg-мышей в возрасте 10 месяцев анти-PD-1 («1 сеанс») или в возрасте 10 и 11 месяцев («2 сеанса»), и изучали результаты когнитивной деятельности в возрасте 12 месяцев (схематически изображено на фиг. 12В). В контрольные группы входили мыши дикого типа, не проходившие лечение AD-Tg мыши и AD-Tg мыши, которые прошли два сеанса лечения IgG. Мы обнаружили, что в то время как один сеанс введения анти-PD-1 оказал благотворное влияние на пространственное обучение и память через 1 месяц после лечения (фиг. 12А), никакого существенного эффекта нельзя было обнаружить у мышей, которые прошли один сеанс лечения и были исследованы через 2 месяца (фиг. 12В). Напротив, мыши AD-Tg, которые прошли два сеанса лечения анти-PD-1, с интервалом в 1 месяц, демонстрировали когнитивную деятельность, аналогичную деятельности мышей дикого типа, в конце 2-месячного периода (фиг. 12В).
[171] Мы изучили, повлияла ли блокада PD-1 на патологию БА, проявляющуюся при нагрузке церебральных Аβ-бляшек и глиозе. Мозги AD-Tg мышей, которые получали анти PD-1 или IgG в течение одного или двух сеансов, были исследованы методом иммуногистохимии на Аβ и глиальный фибриллярный кислый белок (ГФКБ). Мы обнаружили, что нагрузка церебральных Аβ-бляшек была снижена в зубчатой извилине гиппокампа (фиг. 13А, В), а также в коре головного мозга (5-й слой) (фиг. 13А, С), двух областях мозга, демонстрирующих устойчивую патологию Аβ-бляшек у 5XFAD мышей (Oakley et al, 2006). Воздействие на очистку Аβ проявилось после одного сеанса введения анти-PD-1 и стало более устойчивым после двух сеансов. Количественный анализ иммуноокрашивания ГФКБ показал сниженный гиппокампальный астроглиоз как у мышей AD-Tg, прошедших 1 сеанс, так и у особей, прошедших 2 сеанса блокады PD-1, относительно контрольных особей, получавших IgG (фиг. 13А, D). Для исследования эффекта дозирования и частоты введения, женские особи трансгенных мышей 5XFAD AD (средний возраст контингента 6 месяцев) получали либо анти-PD-1-специфическое антитело (IgG2a PD-1 против мыши, либо IgG-контроль (IgG2a крысы). Мыши, проходившие лечение анти-PD-1, получали либо 1 инъекцию 500 мкг антитела на 1-й день эксперимента, либо две инъекции 250 мкг с 3-дневным интервалом между инъекциями. Мыши дикого типа соответствующего возраста были использованы в качестве дополнительной контрольной группы. Эффект лечения на пространственное обучение и эффективность памяти мышей 5XFAD, проходивших лечение анти-PD-1-одна инъекция (n=7) или две инъекции (n=11), получавших IgG2a мышей 5XFAD (n=10) и WT (n=14) оценивался с использованием радиального рукавного водного лабиринта (РРВЛ) в возрасте 7 месяцев (фиг. 14). Черные стрелки указывают временные точки лечения, иллюстрации показывают временные точки когнитивного тестирования. Повторные измерения были проанализированы с использованием двухстороннего теста ANOVA и ретроспективного теста Даннета. Величина ошибки представляет собой среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001, проходившие лечение анти-PD-1 (1 инъекция) в сравнении с контрольными особями, проходившими лечение IgG. Через месяц после лечения (две интраперинетонеальных инъекции с 3-дневным интервалом) у мышей AD-Tg, получивших анти-PD1, наблюдалось значительное улучшение когнитивной функции по сравнению с контрольными особями соответствующего возраста, получавшими IgG или не проходившими лечение, с достижением когнитивных уровней, аналогичных уровням у мышей дикого типа соответствующего возраста (фиг. 14).
[172] Наконец, мужских особей 5XFAD AD трансгенных мышей лечили повторными сеансами, один раз в месяц, либо анти-PD-1-специфическими антителами (IgG2a анти-мышь PD-1), либо с контролем IgG (крыса IgG2a). Первая инъекция проводилась в возрасте 3 месяцев, вторая в возрасте 4 месяцев, третья в возрасте 5 месяцев. Дозировка указана в схеме эксперимента (фиг. 15А). Мыши дикого типа соответствующего возраста были использованы в качестве дополнительной контрольной группы. Влияние на пространственное обучение и память оценивалось с помощью радиального рукавного водного лабиринта (РРВЛ), в две различные временные точки - 5-месячный возраст (фиг. 15В) и возраст 6 месяцев (фиг. 15С). Черные стрелки указывают временные точки лечения, иллюстрации показывают временные точки когнитивного тестирования. Результаты в РРВЛ у 5XFAD мышей, проходивших лечение анти-PD-1 (n=7), 5XFAD мышей, проходивших лечение IgG2a (n=9) и контрольных особей дикого типа (n=8). Повторные измерения были проанализированы с использованием двухстороннего теста ANOVA и ретроспективного теста Даннета. Величина ошибки представляет собой среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001, проходившие лечение анти-PD-1 в сравнении с контрольными особями, проходившими лечение IgG. В возрасте 5 месяцев контрольные мыши, получавшие IgG, не до конца потеряли навыки пространственного обучения/памяти и, таким образом, продемонстрировали некоторое обучение во время последнего испытания на второй день (фиг. 15В), тогда как в возрасте 6 месяцев, наблюдалось прогрессирование заболевания с последующим снижением функциональных результатов (фиг. 15C, D). Фиг. 15D иллюстрирует спад у мышей, получавших IgG, в возрасте от 5 до 6 месяцев, тогда как группа, получавшая антитело к PD-1, сохранила способность к обучению. Эти данные свидетельствуют о том, что повторные сеансы лечения с блокадой PD-1 могут не только обратить вспять развитие заболевания при введении мышам 5XFAD на поздних стадиях заболевания, но и задержать начало заболевания, если лечение начинается в раннем возрасте, до когнитивного спада (фиг. 15В-D).
[173] Сообщалось, что при БА потеря нейронов и синаптическая недостаточность наиболее тесно коррелировали с ухудшением навыков пространственного обучения/памяти. Трансгенные мыши 5XFAD - одна из немногих моделей БА у животных, в которой проявляется значительная потеря нейронов, что становится очевидным у этих мышей в возрасте 6 месяцев. Поэтому мы оценивали выживаемость нейронов у мышей вслед за экспериментом, описанным выше (фиг. 15A-D), после последнего поведенческого теста. Мы проанализировали выживаемость нейронов в мозгах этих мышей, сосредоточившись на субикулуме, ранее использовавшейся для демонстрации потери нейронов в этой мышиной модели БА. Иммуногистохимический анализ выявил большее количество Neu-N+ клеток в субикулуме мышей 5XFAD, получавших антитело к PD-1, по сравнению с группой, получавшей IgG (фиг. 15Е, F).
[174] Молекулярные механизмы, лежащие в основе потери нейронов при БА, были связаны с активацией нейрональной каспазы-3. Поэтому мы оценивали, было ли спасение нейронов после лечения анти-PD-1 связано с уменьшением уровней активированной каспазы-3 в клетках Neu-N+. Мы обнаружили, что мыши, получавшие антитело к PD-1, продемонстрировали снижение иммунореактивности активированной каспазы-3 в нейронах Neu-N+ по сравнению с группой, получавшей IgG (фиг. 15G, Н), что еще раз подтверждает влияние лечения антителом к PD-1 на выживаемость нейронов.
Пример 6. Терапевтический потенциал блокады иммунной контрольной точки TIM-3 при болезни Альцгеймера.
[175] Чтобы исследовать функциональное влияние блокады TIM-3 на патологию БА, мы вводили 6-месячным самкам 5XFAD AD-Tg мышей либо анти-TIM-3-специфическое антитело (TIM-3 против мыши), либо IgG-контроль (антитело IgG2a крысы). Дозировка указана в схеме эксперимента (фиг. 16). Лечение состояло из двух интраперитонеальных инъекций антитела, по 250 мкг каждый, с 3-дневным перерывом между инъекциями. Мыши дикого типа соответствующего возраста были использованы в качестве дополнительной контрольной группы. Эффект лечения на пространственное обучение и эффективность памяти мышей 5XFAD, проходивших лечение анти-TIM-3 (n=9), мышей 5XFAD, получавших IgG (n=6), и контрольных особей дикого типа (n=7) оценивался с использованием радиального рукавного водного лабиринта (РРВЛ) в возрасте 7 месяцев (фиг. 16). Черные стрелки указывают временные точки лечения, иллюстрации показывают временные точки когнитивного тестирования. Повторные измерения были проанализированы с использованием двухстороннего теста ANOVA и ретроспективного теста Даннета. Величина ошибки представляет собой среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001, проходившие лечение анти-TIM-3 в сравнении с контрольными особями, проходившими лечение IgG. Через один месяц после лечения, мыши AD-Tg, получавшие анти-TIM-3, продемонстрировали значительное улучшение когнитивных характеристик по сравнению с 5XFAD мышами, получавшими IgG или контрольными особями дикого типа соответствующего возраста (фиг. 16).
Пример 7. Терапевтический потенциал блокады иммунной контрольной точки PDL1 при болезни Альцгеймера, и сравнение с лечением анти-PD-1.
[176] PD-1 представляет собой ингибиторный рецептор, экспрессируемый многочисленными иммунными клетками, среди которых эффекторные CD4-Т-клетки, тогда как его лиганд PDL1 экспрессируется дендритными клетками, эпителиальными клетками и регуляторными Т-клетками. В связи с этим возник вопрос, может ли блокирование PDL1 иметь эффект, аналогичный блокированию PD-1 (фиг. 17А). С этой целью 6-месячным мышам 5XFAD сначала вводили одну дозу антитела к анти-PDL1 (0,1 мг, 0,5 мг или 1,5 мг на мышь), посредством интраперитонеальной инъекции. Мышей исследовали через 1 месяц, в возрасте 7 месяцев, при помощи РРВЛ. В то время как введение 0,1 мг анти-PDL1 не оказало никакого влияния на когнитивную деятельность по сравнению с контрольным лечением изотипом IgG, обе дозы (0,5 мг и 1,5 мг) оказывали сходное положительное влияние на результаты выполнения задачи в РРВЛ (фиг. 17В). Затем мы сравнили эффект одной инъекции 0,5 мг антитела к PD-1 и 0,5 мг антитела к PDL1 на когнитивную деятельность, и, следовательно, на патологию головного мозга (фиг. 17C-J). Используя эксперимент с РРВЛ, мы обнаружили, что лечение антителом к PDL1 было столь же эффективным, как антителом к PD-1, касательно улучшения функциональных когнитивных последствий у мышей 5XFAD (фиг. 17С). Иммуногистохимический анализ мозгов этих мышей после поведенческих тестов показал аналогичное снижение бремени бляшек (измеренного по количеству бляшек и площади, покрытой бляшками) в гиппокампе (ГК) и коре, и в глиозе (измерения по иммунореактивности глиального фибриллярного кислого белка (ГФКБ)) как в группе, проходившей лечение анти-PD-1, так и в группе с анти-PDL1, по сравнению с группой, получавшей IgG (фиг. 17D). В мозгах мышей, получавших либо анти-PD-1, либо анти-PDL1 антитело, мы также наблюдали более высокий уровень иммунореактивности, характерный для синаптофизина, маркера предсинаптической активности, что указывает на лучшее сохранение синапсов (фиг. 17Н, I). Мы проанализировали методом количественной ПЦР в реальном времени уровни il-12p40 и il-10 в гиппокампе мышей 5XFAD, прошедших лечение. Результаты показали смещение к противовоспалительной активности в гиппокампе мышей, получавших либо анти-PD-1, либо анти-PDL1, что проявляется в уменьшении отношения il-12р40/il-10 по сравнению с животными 5XFAD, получавшими IgG (фиг. 17J). Следует отметить, что контрольные антитела соответствующего изотипа для анти-PD-1 и анти-PDL1 представляют собой IgG2a и IgG2b, соответственно. Однако во всех наших поведенческих исследованиях они показали аналогичные результаты, поэтому эти две контрольные группы были объединены в представленном здесь заключительном анализе.
[177] PDL1 экспрессируется активированными иммунными клетками, такими, как Т-клетки, В-клетки, макрофаги, дендритные клетки и микроглии, а также неиммунными клетками, такими, как эндотелиальные и эпителиальные клетки. Таким образом, мы предположили, что экспрессия PDL1 эпителием СС может способствовать отрицательной регуляции (down-регуляции) трафика лейкоцитов в ЦНС путем заглушения активности продуцирующих IFN-γ Т-клеток, которые экспрессируют PD-1 при общении с PDL1-экспрессирующими эпителиальными клетки внутри СС. Иммуногистохимический анализ показывает, что у взрослых мышей эпителий СС экспрессировал значительно более высокие уровни PDL1 по сравнению с молодыми мышами (фиг. 18).
Пример 8. Терапевтический потенциал блокады иммунной контрольной точки при болезни Альцгеймера.
[178] Чтобы проверить, может ли блокада иммунных контрольных точек ослабить патологию БА, мышей AD-Tg лечат в возрасте от 6 до 10 месяцев одним из следующих антител, действующих против контрольной точки: анти-ICOS, анти-B7RP1, анти-VISTA, анти-CD40, анти-CD40L, анти-CD80, анти-CD86, анти-В7-Н3, анти-В7-Н4, В7-Н7, анти-BTLA, анти-HVEM, анти-CD137, анти-CD137L, анти-OX40L, анти-CD-27, анти-CD70, анти-STING, анти-TIGIT или анти-GITR. Некоторых мышей лечат антителом к PD-1 в качестве положительного контроля, IgG в качестве отрицательного контроля или комбинацией анти-PD1 и одного из других антител к контрольной точке, упомянутых выше. Эффект лечения на пространственное обучение и память измеряется через месяц после лечения, с помощью радиального рукавного водного лабиринта (РРВЛ), бремени Аβ-бляшек с помощью иммуногистохимии на Аβ и астроглиоз гиппокампа путем иммуногистохимии на глиальный фибриллярный кислый белок (ГФКБ).
[179] Ожидается, что мыши, получавшие антитела, будут демонстрировать значительное улучшение когнитивной способности по сравнению с получавшими IgG и не проходившими лечение мышами AD-Tg, а также значительное снижение нагрузки церебральных бляшек.
Пример 9. Терапевтический потенциал блокады PD-1 в комбинации с иммунной контрольной точкой CTLA-4 при болезни Альцгеймера.
[180] В 10-месячном возрасте трансгенным (Tg) мышам 5XFAD с болезнью Альцгеймера (AD), интраперитонеально вводят либо 250 мкг анти-PD1 (RMP1-14; # ВЕ0146; Bioxcell Lifesciences Pvt. LTD.) и 250 мкг анти-CTLA-4 (InVivoMAb анти-mCD152; #ВЕ0131; Bioxcell Lifesciences Pvt. LTD.), либо IgG-контрольные (IgG2a, #BE0089 или Polyclonal Syrian Hamster IgG, #BE0087; Bioxcell Lifesciences Pvt. LTD.) антитела, на 1-й и 4-й день эксперимента, затем их исследуют через 3 недели на предмет когнитивной деятельности с использованием задач по пространственному обучению и памяти в радиальном рукавном водном лабиринте (РРВЛ), как описано выше.
[181] Некоторые мыши проходят дополнительный сеанс с перерывом в лечении продолжительностью 3 недели. Контрольные группы либо получают IgG, либо не проходят лечение, и все группы мышей исследуются на предмет когнитивной деятельности спустя 3 недели.
[182] Ожидается, что мыши, получавшие комбинацию антител, будут демонстрировать значительное улучшение когнитивной способности по сравнению с получавшими IgG и не проходившими лечение мышами AD-Tg, а также значительное снижение нагрузки церебральных бляшек.
Пример 10. Терапевтический потенциал блокады иммунной контрольной точки при патологии ПТСР.
[183] Особо стрессовые условия или хронический стресс могут привести к депрессии и посттравматическому стрессовому расстройству (ПТСР). Мы приняли ПТСР-подобную физиологическое животную модель, в которой мыши демонстрируют бессонницу, нарушение внимания, повышенный учет риска и плохой сон (Lebow et al, 2012). В этой экспериментальной модели индукции ПТСР мышей приучают за 10 дней к обратному дневному/ночному циклу, подвергают двум ударам электрическим током (травма и триггер), под названием «ПТСР индукция», и оценивают в разных временных точках после травмы. После травматического события мышам вводят упомянутый препарат, который блокирует иммунные контрольные точки. Мышей лечат по одной из следующих схем:
[184] Мышей лечат одним из следующих антител к контрольной точке: анти-ICOS, анти-B7RP1, анти-VISTA, анти-CD40, анти-CD40L анти-CD80, анти-CD86, анти-В7-Н3, анти-В7-Н4, В7-Н7, анти-BTLA, HVEM, анти-CD137, анти-CD137L, анти-OX40L, анти-CD-27, анти-CD70, анти-STING, анти-GITR или анти-TIGIT по отдельности или в сочетании с антителом анти-CTLA-4. Некоторых мышей лечат антителом к PD-1 в качестве положительного контроля, IgG в качестве отрицательного контроля или комбинацией анти-PD1 и одного из других антител к контрольной точке, упомянутых выше.
[185] Некоторые мыши проходят дополнительный сеанс лечения после соответствующего интервала.
[186] Ожидается, что мыши, получающие лечение, не проявляют тревожного поведения, связанного с ПТСР, в этой экспериментальной модели, что оценивается по времени, проведенному в исследовании и оценке риска в темном/светлом лабиринте или при выполнении других поведенческих задач, описанных в (Lebow et al, 2012).
Пример 11. Терапевтический потенциал метода блокады иммунной контрольной точки при патологии болезни Паркинсона.
[187] В этом эксперименте используются трансгенные мыши (Tg) с болезнью Паркинсона (PD, БП) или мышиные модели БП, индуцированные МФТФ. Мышей лечат на прогрессивных стадиях заболевания по одной из следующих схем:
[188] Мышей PD-Tg лечат одним из следующих антител к контрольной точке: анти-ICOS, анти-B7RP1, анти-VISTA, анти-CD40, анти-CD40L, анти-CD80, анти-CD86, анти-В7-Н3, анти-В7-Н4, В7-Н7, анти-BTLA, HVEM, анти-CD137, анти-CD137L, анти-OX40L, анти-CD-27, анти-CD70, анти-STING, анти-GITR или анти-TIGIT по отдельности или в сочетании с антителом анти-CTLA-4. Некоторых мышей лечат антителом к PD-1 в качестве положительного контроля, IgG в качестве отрицательного контроля или комбинацией анти-PD1 и одного из других антител к контрольной точке, упомянутых выше.
[189] Моторные неврологические функции оцениваются, например, с помощью теста «ротарод», который оценивает способность мышей удерживаться на вращающемся барабане.
[190] Ожидается, что мыши PD-Tg, прошедшие один сеанс лечения, будут проявлять значительно улучшенную двигательную активность по сравнению с контрольной группой, получавшей IgG или проводник, или группой, не проходившей лечение. У мышей PD-Tg, которые пройдут два сеанса лечения и которые будут исследованы после соответствующего перерыва, как ожидается, проявится долговременный терапевтический эффект. Чтобы сохранить терапевтический эффект, мышей подвергают активному сеансу лечения с соответствующим перерывом без лечения после каждого сеанса лечения.
Пример 12. Терапевтический потенциал PD-1 в комбинации с блокадой иммунной контрольной точки CTLA-4 при болезни Хантингтона.
[191] Модель, используемая в этих экспериментах, может быть тестовой системой болезни Хантингтона (HD) у R6/2 трансгенных мышей (Tg). R6/2 трансгенные мыши сверхэкспрессируют мутировавший человеческий гентингтин, что подразумевает введение множественных CAG-повторов у мышей на прогрессирующих стадиях заболевания. У этих мышей наблюдается прогрессирующая поведенческо-моторная недостаточность, начинающаяся уже в возрасте 5-6 недель и приводящая к преждевременной смерти через 10-13 недель. Симптомы включают в себя низкий вес тела, сокращение мышц, тремор и судороги.
[192] Мышей лечат по одной из следующих схем в возрасте 45 дней:
[193] Мышей лечат одним из следующих антител к контрольной точке: анти-ICOS, анти-B7RP1, анти-VISTA, анти-CD40, анти-CD40L, анти-CD80, анти-CD86, анти-В7-Н3, анти-В7-Н4, В7-Н7, анти-BTLA, HVEM, анти-CD137, анти-CD137L, анти-OX40L, анти-CD-27, анти-CD70, анти-STING, анти-GITR или анти-TIGIT по отдельности или в сочетании с антителом анти-CTLA-4. Некоторых мышей лечат антителом к PD-1 в качестве положительного контроля, IgG в качестве отрицательного контроля или комбинацией анти-PD1 и одного из других антител к контрольной точке, упомянутых выше.
[194] Моторные неврологические функции оцениваются, например, с помощью теста «ротарод», который оценивает способность мышей удерживаться на вращающемся барабане.
[195] Ожидается, что мыши HD-Tg, прошедшие один сеанс лечения, будут проявлять значительно улучшенную двигательную активность по сравнению с контрольной группой, получавшей IgG или проводник, или группой, не проходившей лечение. У мышей HD-Tg, которые пройдут два сеанса лечения и которые будут исследованы после соответствующего перерыва, как ожидается, проявится долговременный терапевтический эффект. Чтобы сохранить терапевтический эффект, мышей подвергают активному сеансу лечения с соответствующим перерывом без лечения после каждого сеанса лечения.
Пример 13. Терапевтический потенциал метода блокады иммунной контрольной точки при патологии бокового амиотрофического склероза.
[196] Модель, используемая в этом эксперименте, может представлять трансгенных мышей, сверхэкспрессирующих дефектный человеческий мутантный аллель SOD1, содержащий ген Gly93→Ala (G93A) (B6SJL-TgN (SOD1-G93A)1Gur (далее «мыши ALS»). В этой модели развивается болезнь моторных нейронов, таким образом, она является приемлемой моделью для тестирования ALS.
[197] Мышей лечат по одной из следующих схем в возрасте 75 дней:
[198] Мышей лечат одним из следующих антител к контрольной точке: анти-ICOS, анти-B7RP1, анти-VISTA, анти-CD40, анти-CD40L, анти-CD80, анти-CD86, анти-В7-Н3, анти-В7-Н4, В7-Н7, анти-BTLA, HVEM, анти-CD137, анти-CD137L анти-OX40L анти-CD-27, анти-CD70, анти-STING, анти-GITR или анти-TIGIT по отдельности или в сочетании с антителом анти-CTLA-4. Некоторых мышей лечат антителом к PD-1 в качестве положительного контроля, IgG в качестве отрицательного контроля или комбинацией анти-PD1 и одного из других антител к контрольной точке, упомянутых выше.
[199] Двигательные неврологические функции оценивают, например, с использованием теста «ротарод», при котором оценивается способность мышей оставаться на вращающемся барабане, или при этом мышам разрешается хвататься и держаться за вертикальный провод (диаметром 2 мм) с небольшой петлей на нижнем конце. Вертикальный провод позволяет мышам использовать как передние, так и нижние конечности для захвата. Провод в вертикальном положении совершает круговые движения (радиус окружности 10 см) со скоростью 24 об/мин. Время, в течение которого мышь может висеть на проводе, отсчитывается таймером.
[200] Ожидается, что мыши ALS, прошедшие один сеанс лечения, будут проявлять значительно улучшенную двигательную активность по сравнению с контрольной группой, получавшей IgG или проводник, или группой, не проходившей лечение. У мышей ALS, которые пройдут два сеанса лечения и которые будут исследованы после соответствующего перерыва, как ожидается, проявится долговременный терапевтический эффект. Чтобы сохранить терапевтический эффект, мышей подвергают активному сеансу лечения с соответствующим перерывом без лечения после каждого сеанса лечения.
Пример 14. Эксперименты «доза-эффект» для определения минимального и максимального диапазона дозы и эксперименты по определению режима лечения и его длительного терапевтического эффекта.
[201] Мы уже показали, что один сеанс лечения с использованием блокады PD-1 приводит к значительному снижению бремени бляшек и улучшенной когнитивной функции, продолжительностью как минимум 2 месяца после лечения, до последней временной точки, по которой проводилось тестирование. Здесь мы описываем исследование дозозависимого эффекта, использующего две дополнительные дозы, вводимые трансгенным мышам 5XFAD AD. В выборку информации будет включаться бремя амилоидных бляшек через один, два и три месяца после введения. Исследовательские группы будут состоять из 1) не получающих лечение мышей 5XFAD; 2) мышей 5XFAD, получающих 1 инъекцию 500 мкг контрольного анти-PD-1 (RMP1-14; # ВЕ0146; Bioxcell Lifesciences Pvt. LTD.); 3) 5XFAD мышей, получающих 1 инъекцию 250 мкг контрольного анти PD-1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.); 4) 5XFAD мышей, получающих 1 инъекцию 100 мкг контрольного анти PD-1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.); и 5) 5XFAD мышей, получающих 1 инъекцию 500 мкг контрольного анти PD-1 (IgG2a; #ВЕ0089; BioxcellLifesciencesPvt. LTD.). Всех мышей подвергают лечению в начале эксперимента, от каждой группы мышей умерщвляют и исследуют их мозг с интервалом в 1 месяц, 2 месяца и 3 месяца после начала лечения.
[202] Ожидается, что мыши, получавшие антитела к PD-1, будут демонстрировать значительное снижение бремени бета-амилоидных бляшек в головном мозге по сравнению с не проходившими лечение мышами AD-Tg или с мышами, получавшими IgG-контроль.
[203] Дополнительный сеанс лечения анти-PD-1 через месяц после первоначального лечения, как было обнаружено нами, сохраняет влияние на улучшение когнитивной способности у мышей 5XFAD AD-Tg (Пример 5). Эти данные свидетельствуют о том, что для долгосрочной эффективности необходимы повторные сеансы лечения. Здесь мы описываем исследование, в котором используются повторные инъекции для поддержания долговременного эффекта лечения.
[204] Мышам 5XFAD AD-Tg вводят инъекции препарата в дозировке, которая будет определяться по итогам предыдущего исследования. Мышам будут вводить инъекции, а их когнитивная деятельность будет контролироваться с использованием заданий в радиальном рукавном водном лабиринте во время исследования и после него. Также выполняется гистологическое исследование головного мозга на предмет бремени амилоидных бляшек.
[205] Различным группам мышей назначают многократные введения в виде одиночных инъекций (или двойных инъекций с промежутком в 3 дня, как описано в примере 5) с интервалами без лечения продолжительностью 2, 3 или 4 недели (Таблица 4). Мышей контролируют, как описано выше, через один, два или три месяца после первоначального лечения.
Пример 15. Системное введение моноклонального антитела к PD-1 у крыс RCS ослабляет дегенерацию сетчатки.
[206] Целью этого эксперимента было определить, ослабляет ли системное введение антитела к PD1 дегенерацию внешнего ядерного слоя в животной модели дегенерации сетчатки.
[207] Крысы RCS, принятая животная модель сухой возрастной дегенерации макулы, пигментного ретинита и других дегенеративных заболеваний и состояний сетчатки, содержат делеционную мутацию в гене, кодирующем белок MerTK, который приводит к дегенерации сетчатки и полной потере зрения в возрасте трех месяцев. Значительное и быстрое уменьшение толщины наружного ядерного слоя сетчатки (ONL) наблюдается в возрасте 4 недель. Таким образом, в научной литературе считается, что существует прямая корреляция между сохранением толщины ONL в данной модели и сохранением зрения.
[208] Крысам RCS вводили интраперитонеально в возрасте 4 недель либо моноклональные антитела к PD1 (всего 760 мкг на животное, n=10), либо подходящий IgG-контроль (IgG2a) в той же концентрации (n=10). Еще одна группа крыс RCS оставалась без лечения (n=4). Через 2 недели после лечения, в возрасте 6 недель, животных умерщвляли, их глаза вырезали, а толщину наружного ядерного слоя сетчатки в каждом глазу определяли гистологическим анализом с использованием окрашивания гематоксилином и эозином (Н&Е stain). Эффективность лечения определяли путем измерения толщины ONL по всей длине сетчатки и горизонтального построения данных для создания карты, которая позволяет определить результаты лечения в любой области сетчатки.
[209] Анализ средней толщины ONL всех животных, проходивших лечение, при котором каждый отдельный глаз служил независимым набором данных, показал, что в возрасте 6 недель (через 2 недели после лечения) ONL в центральной области сетчатки в непосредственной близости от головки зрительного нерва был значительно толще у животных, получавших анти-PD1, по сравнению с получавшими IgG и не проходившими лечение контрольными особями (фиг. 19А).
[210] Для оценки величины эффекта только для чувствительных к лечению животных пороговое значение для причисления животного к «положительно реагирующим животным» было установлено как значение, равное 2 стандартным значениям ошибки выше среднего значения толщины в центральной сетчатке контрольной группы IgG. Основываясь на этом установленном пороговом значении, 13 из 20 проанализированных глаз (65%) в группе анти-PD1 были охарактеризованы как положительно реагирующие. Для сравнения, только 1 из 20 (5%) анализируемых глаз был охарактеризован как положительно реагирующий в экспериментальной группе IgG; имеется значительная разница (Хи-квадрат = 15,82, Р = 0,00007). При изучении только 13 анти-PD1 положительно реагирующих глаз данной экспериментальной группы было показано, что толщина в центральной области сетчатки в 1,5-2 раза больше, чем у контрольных глаз. Кроме того, еще более широкая область центральной сетчатки была значительно толще, чем у контрольных групп (фиг. 19В). Любопытно, что, хотя антитело вводили системно и, как ожидается, оно достигло обоих глаз и воздействовало на них на одинаковом уровне, различия в реакции на лечение между двумя глазами отдельных животных наблюдались у четырех животных, получавших анти-PD-1.
Пример 16. Местное введение моноклонального антитела против PD-1 непосредственно в стекловидное тело у крыс RCS ослабляет дегенерацию сетчатки.
[211] Целью этого эксперимента было определить, ослабляет ли системное введение антитела к PD1 дегенерацию наружного ядерного слоя в животной модели дегенерации сетчатки.
[212] Крысам RCS в возрасте 4 недель вводили либо моноклональные антитела к PD1 (n=6), либо подходящий контроль IgG (50 мкг на животное). Инъекции выполняли непосредственно в стекловидное тело одного глаза каждого животного, в то время как контралатеральный глаз оставался без инъекции. Через 2 недели после лечения, в возрасте 6 недель, животных умерщвляли, их глаза вырезали, а толщину наружного ядерного слоя сетчатки в каждом глазу определяли гистологическим анализом с использованием окрашивания гематоксилином и эозином (Н&Е stain). Эффективность лечения определяли путем измерения толщины ONL по всей длине сетчатки и горизонтального построения данных для создания карты, которая позволяет определить результаты лечения в любой области сетчатки.
[213] Анализ средней толщины ONL у животных через 2 недели после лечения показал, что в экспериментальной группе с анти-PD1 ONL был значительно толще в нескольких разделах сетчатки в пролеченном глазу, по сравнению с необработанным контралатеральным глазом (фиг. 20А). Такого эффекта не было обнаружено у животных, получавших лечение IgG (фиг. 20В).
Пример 17. Местная блокада PD-1 путем интравитреальных инъекций PD-1 или PD-L1 моноклональных антител ослабляет дегенерацию сетчатки.
[214] Моноклональные антитела к PD-L1 или моноклональные антитела к PD-1, комбинация моноклональных антител к PD-L1 и анти-PD-1 или подходящие фрагменты IgG без специфической для антигена вариабельной области будут вводиться непосредственно в стекловидное тело крыс RCS в возрасте 4 недель. В течение 8 последующих недель будет проводиться оценка зрительной функции животных в ответ на зрительные стимулы. Важно отметить, что только один глаз каждого животного будет подвергаться лечению, в то время как контралатеральный глаз останется без лечения и будет служить дополнительным контролем. Кроме того, на протяжении всего эксперимента назначенные группы крыс RCS из каждой экспериментальной группы будут подвергнуты анализу, направленному на количественное и качественное определение эффекта лечения на сетчатку в плане выживаемости нейронов и локального иммунного ответа, а также влияния на сетчатку эпителиальных клеток в смысле экспрессии ими молекул трафика лейкоцитов и лигандов иммунной контрольной точки. Ожидается, что локальная блокада пути PD-1/PDL1 через интравитреальную инъекцию приведет к ослаблению дегенерации сетчатки, иммунной модуляции и сохранению зрительной функции.
Пример 18. Таргетинг пути PD-1/PDL1 в мышиной модели Тау патологии увеличивает рекрутинг макрофагов моноцитарного происхождения в паренхиме мозга.
[215] Поскольку лечение с использованием блокады иммунной контрольной точки непосредственно направлено на системные иммунные клетки, мы предположили, что его эффективность не будет ограничиваться специфическим невропатологическим признаком, связанным с БА. Чтобы проверить, может ли лечение с таргетингом пути PD-1/PD-L1 быть эффективным в других моделях БА, которые имитируют отчетливую этиологию болезни, мы использовали мышиную модель БА, которая экспрессирует две мутации гена человека-тау (K257T/P301S; двойной мутант, DM-hTAU), ассоциированные с лобно-временной деменцией. У этих мышей развивается деменция с преобладанием нейрофибриллярных клубков (NFT), характерная для широкого спектра таупатий, включая болезнь Альцгеймера (АБ) и другие нейродегенеративные заболевания. Патологии у этих мышей включают в себя когнитивный дефицит, нейровоспаление, активацию глиальных клеток и фосфорилирование белков Тау в головном мозге.
[216] Ранее мы показали обратное функциональное отношение между системной иммуносупрессией и AD-патологией у мышей 5XFAD. Кроме того, было обнаружено, что в обеих мышиных моделях AD (5XFAD и J20) прогрессирование заболевания связано с потерей доступности IFN-γ в СС. Это уменьшение IFN-γ сопровождалось повышением регуляторных Т-клеток FoxP3 у мышей 5XFAD, а обработка антителами против PD-1 приводила к восстановлению сигналов IFN-γ в СС и рекрутингу макрофагов моноцитарного происхождения в паренхиму головного мозга (Baruch et al., 2016). Здесь мы сначала проверили, влияет ли введение антитела, направленного против PD-L1, на уровни Т-клеток системной эффекторной памяти или регуляторных клеток у мышей DM-hTAU, согласно измерениям, проведенным в селезенке через 2 недели после введения антитела. Мы обнаружили, что также в этой мышиной модели при отсутствии лечения наблюдается системное повышение регуляторных Т-клеток FoxP3 и снижение системных уровней Т-клеток эффекторной памяти (CD44+CD-62Llow) по сравнению с сопоставимыми по возрасту мышами дикого типа (фиг. 21А-С). Введение антитела к PDL1 не уменьшало уровни супрессорных клеток, но увеличивало уровень эффекторных Т-клеток памяти в отношении мышей, получавших IgG (фиг. 21А-С), согласно анализу методом проточной цитометрии. Мы дополнительно проанализировали мозг тех же мышей, чтобы определить уровни макрофагов, полученных из моноцитов. Мы обнаружили значительное увеличение количества макрофагов моноцитарного происхождения в головном мозге мышей DM-hTAU, получавших антитело к PDL1, по сравнению с теми, которые получали контрольный изотип IgG (фиг. 21D, Е). Эти данные подтверждают нашу гипотезу о том, что в данной мышиной модели развивается периферическая иммуносупрессия, и что сокращение иммуносупрессии на периферии облегчает проникновение болезнетворных лейкоцитов в пораженный мозг. Эти результаты подтверждают наше утверждение, что блокада иммунной контрольной точки может быть применима к БА, характеризующейся дополнительной этиологией.
Пример 19. Блокада оси PD-1/PDL1 в мышиной модели Тау патологии смягчает когнитивные дефициты и церебральную патологию.
[217] Полученные нами данные о том, что таргетинг системных путей иммунной контрольной точки усиливает трафик моноцитов в паренхиму головного мозга, как мы наблюдали у мышей 5XFAD, побудило нас проверить влияние таргетинга PD-1 или PD-L1 на когнитивную функцию в мышиной модели Тау. Мы лечили мышей DM-hTAU в возрасте 8 месяцев с помощью анти-PD-1 или анти-PDL1, используя ту же дозу 0,5 мг, что и у мышей 5XFAD (фиг. 22А); антитела, соответствующие по изотипу, направленные на нерелевантные антигены, использовались в качестве отрицательных контролей (контроль, получавший IgG). Мыши дикого типа соответствующего возраста оценивались в качестве дополнительного контроля на предмет нормального обучения/памяти посредством испытаний, используемых в данном исследовании. В этой мышиной модели БАкратковременная пространственная память обычно оценивается с помощью испытания в Т-лабиринте и Y-лабиринте. Две группы, которые получали антитела к PD-1 или PDL1, продемонстрировали повышенное предпочтение новому рукаву при испытании в Т-лабиринте по сравнению с контрольной группой, получавшей IgG, через 1 месяц после однократной инъекции антител (фиг. 22В, С). Кроме того, обе экспериментальные группы продемонстрировали улучшенные когнитивные характеристики при испытании в Y-лабиринте по сравнению с контрольной группой, получавшей IgG (фиг. 22D).
[218] Эффект лечения на глиоз выявил снижение иммунореактивности ГФКБ в гиппокампе групп, получавших антитела к PD-1/PDL1 (фиг. 22Е, F). Далее, мы проверили, может ли положительный эффект блокады PD-1/PDL1 на глиоз быть связан с изменениями в среде воспалительных цитокинов. С этой целью мы контролировали уровни различных цитокинов с использованием метода количественной ПЦР в реальном времени (RT-qPCR) для определения того, была ли искажена среда воспаления ЦНС в результате лечения. Мы обнаружили, что и анти-PD-1, и анти-PDL1 снижают уровни экспрессии провоспалительного цитокина il-12p40 по сравнению с противовоспалительным цитокином, il-10 в гиппокампе. Кроме того, обе процедуры приводили к снижению уровней провоспалительных цитокинов, tnfa и il-6 (фиг. 22G).
[219] Показано, что нейровоспаление в животных моделях Тау патологии усиливает гиперфосфорилирование и прогрессирование заболевания. Поэтому мы проверили влияние антител к PD-1 и PDL1 на Тау гиперфосфорилирование. Иммуногистохимический анализ срезов головного мозга у мышей DM-hTAU показал снижение иммунореактивности эпитопов АТ-100 (Phospho-Tau Thr212, Ser214) и АТ-180 (Phospho-Tau Thr231) в областях СА1 и СА3 гиппокампа, после блокады анти-PD-1 и анти-PDL1, по сравнению с контрольной группой изотипа IgG (фиг. 23A-F).
[220] Наконец, поскольку при иммунотерапии рака антитело к PD-L1 используется при более высокой дозе, чем антитело к PD-1, мы протестировали, будет ли повышение дозы антитела к PDL1 иметь превосходящий эффект. Поэтому мы провели дополнительное исследование, в ходе которого тестировали влияние различных доз на краткосрочную память как у женских, так и у мужских особей (одинаково распределенных среди подгрупп) мышей DM-hTAU в возрасте 9 месяцев. Мышам вводили одну инъекцию анти-PDL1 в дозировке 0,1, 0,5 или 1,5 мг на мышь, по сравнению с одной инъекцией контрольного антитела 1,5 мг на мышь. Мышей дикого типа одного помета использовали в качестве контроля в отношении интактных когнитивных способностей. Мыши, получавшие 0,5 или 1,5 мг на мышь, показали результаты, близкие к мышам дикого типа; доза анти-PDL1 1,5 мг на мышь была несколько более эффективной, но разница между дозами была незначительной (фиг. 23G).
[221] В совокупности эти результаты указывают на то, что системная иммунная активация модифицирует ключевые процессы в головном мозге, которые связаны с патологией болезни в животной модели тау, аналогично ярко выраженному эффекту, обнаруженному в животных моделях бета-амилоидной патологии.
Пример 20. Блокада PD-1 усиливает нейрогенез гиппокампа у мышей 5XFAD.
[222] Даблкортин (DCX) представляет собой ассоциированный с микротрубочками белок, экспрессируемый клетками-предшественниками нейронов и незрелыми нейронами. В ткани зрелых нейронов DCX используется как маркер нейрогенеза, поскольку экспрессируется почти исключительно развивающимися нейронами. Самкам мышей 5XFAD (средний контингент в возрасте 6 месяцев) вводили либо специфическое антитело к PD-1 (IgG2a PD-1 против мыши; n=17), либо IgG-контроль (крысы IgG2a; n=7), спустя месяц их умерщвляли. Мышей дикого типа соответствующего возраста использовали в качестве дополнительной контрольной группы. Были подготовлены парасагиттальные срезы мозга, взятые у репрезентативных животных, и отмечен гранулированный слой зубчатой извилины (фиг. 24А). Срезы головного мозга иммунизировали на нейронный маркер-NeuN (зеленый), DCX (красный) и ядерным окрашиванием Хехст (синий). Клетки DCX+ были подсчитаны двойным слепым методом, на срезах мозга толщиной 6 мкм (фиг. 24В). Повторные измерения были проанализированы с использованием одностороннего теста ANOVA и ретроспективного теста Даннета. Величина ошибки представляет собой среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001. Результаты показывают, что системная блокада пути PD-1 / PDL1 вызывает защитную иммунную активность, которая модулирует среду мозга для поддержания нейрогенеза гиппокампа, воздействие на патологию, которое неоднократно коррелировали до благотворного влияния на поведенческую и когнитивную недостаточность.
Пример 21. Блокада PD-1 повышает синаптическую пластичность у мышей 5XFAD.
[223] Везикулярные глютаматные транспортеры 1 (VGLUT1), экспрессируемые глутаматергическими нейронами, опосредуют поглощение глутамата в синаптические везикулы и, как было показано, способствуют синаптической пластичности гиппокампа и пространственному обучению, зависящему от гиппокампа. Самкам мышей 5XFAD (средний контингент в возрасте 6 месяцев) вводили либо специфическое антитело к PD-1 (IgG2a PD-1 против мыши; n=17), либо IgG-контроль (крысы IgG2a; n=7), спустя месяц их умерщвляли. Мышей дикого типа соответствующего возраста использовали в качестве дополнительной контрольной группы. Были подготовлены парасагиттальные срезы мозга, взятые у репрезентативных животных, и отмечена зона субикулума (фиг. 25А). Срезы мозга подвергли иммуноокрашиванию на VgluT1. Интенсивность флуоресценции определяли количественно двойным слепым способом с использованием программного обеспечения ImageJ на срезах мозга толщиной 6 мкм (фиг. 25В). Повторные измерения были проанализированы с использованием одностороннего теста ANOVA и ретроспективного теста Даннета. Величина ошибки представляет собой среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001. Результаты показывают, что системная блокада пути PD-1/PDL1 вызывает защитную иммунную активность на периферии и модулирует среду мозга для поддержания синаптической пластичности и сохранения когнитивной функции.
Пример 22. Блокада PD-1 уменьшает потери нейронов в субикулуме мышей 5XFAD.
[224] Модель трансгенных мышей 5XFAD является одной из немногих амилоидных животных моделей, которая демонстрирует значительную потерю нейронов, аналогичную прогрессированию БА у людей. Потери нейронов у мышей 5XFAD характеризовались в субикулуме и в кортикальном слое 5. Самкам мышей 5XFAD (средний контингент в возрасте 6 месяцев) вводили либо специфическое антитело к PD-1 (IgG2a PD-1 против мыши; n=17), либо IgG-контроль (крысы IgG2a; n=7), спустя месяц их умерщвляли. Мышей дикого типа соответствующего возраста использовали в качестве дополнительной контрольной группы. Были подготовлены парасагиттальные срезы мозга, взятые у репрезентативных животных, и отмечена зона субикулума (фиг. 26А). Срезы мозга подвергли иммуноокрашиванию на NeuN, отмечая нейроны (зеленым цветом).
Нейроны субикулума определяли количественно двойным слепым способом с использованием программного обеспечения ImageJ на срезах мозга толщиной 6 мкм (фиг. 25В). Повторные измерения были проанализированы с использованием одностороннего теста ANOVA и ретроспективного теста Даннета. Величина ошибки представляет собой среднее ± стандартная ошибка среднего; *Р<0,05, **Р<0,01, ***Р<0,001. Результаты показывают, что системная блокада пути PD-1/PDL1 вызывает защитную иммунную активность, которая модулирует среду мозга, делая ее пермиссивной для поддержки выживаемости и спасения нейронов, в конечном итоге способствуя лучшей когнитивной деятельности.
[225] В заключение следует понимать, что, хотя аспекты настоящего описания изобретенияотмечены ссылкой на конкретные варианты осуществления, специалист в данной области техники легко поймет, что описанные варианты осуществления являются лишь иллюстрацией принципов раскрываемого здесь объекта изобретения. Поэтому следует понимать, что раскрываемый объект изобретения никоим образом не ограничивается каким-либо конкретным соединением, композицией, изделием, устройством, методологией, протоколом и/или реагентом и т.д., описанным здесь, если подобное не указано прямо. Кроме того, специалисты в данной области техники поймут, что определенные изменения, модификации, перестановки, изменения, дополнения, вычитания и их подкомбинации могут осуществляться в отношении объекта изобретения в соответствии с описанными здесь принципами без отхода от сущности настоящей спецификации. Поэтому предполагается, что следующие прилагаемая формула изобретения и ее пункты, представленные ниже, интерпретируются таким образом, чтобы включать все такие изменения, модификации, перестановки, изменения, дополнения, вычитания и подкомбинации, которые соответствуют их настоящей сути и объему.
[226] Некоторые варианты осуществления настоящего изобретения описаны здесь, включая лучший способ осуществления изобретения, известный изобретателям. Разумеется, вариации этих описанных вариантов осуществления будут очевидны для специалистов в данной области техники после прочтения вышеприведенного описания. Изобретатель ожидает, что специалисты в данной области будут использовать такие вариации согласно необходимости, и изобретатели предполагают, что настоящее изобретение будет на практике использоваться иначе, чем конкретно описано здесь. Соответственно, это изобретение включает все модификации и эквиваленты объекта, указанные в формуле изобретения, прилагаемой к настоящему документу, в пределах применимого законодательства. Более того, любая комбинация вышеописанных вариантов осуществления во всех возможных вариантах ее осуществления также охватывается изобретением, если иное не указано здесь или если контекст явно не указывает на обратное.
[227] Группирование альтернативных вариантов осуществления, элементов или этапов настоящего изобретения не следует рассматривать как ограничения. Каждый член группы может быть упомянут и заявлен отдельно или в любом сочетании с другими членами группы, встречающимися в настоящем документе. Ожидается, что один или более членов группы могут быть включены в группу или удалены из группы по причинам удобства и/или патентоспособности. При таком включении или удалении считается, что описание, как подразумевается в настоящем документе, содержит модифицированную группу, таким образом соответствуя письменному описанию всех групп Маркуша, используемых в прилагаемой формуле изобретения.
[228] Если не указано иное, все числа, выражающие характеристику, элемент, количество, параметр, свойство, период и т.д., используемые в настоящем описании и формуле изобретения, следует понимать как измененные во всех возможных случаях термином «около». Используемый здесь термин «около» означает, что характеристика, элемент, количество, параметр, свойство или период, им определяемый, включает в себя диапазон на десять процентов выше или ниже значения указанной характеристики, элемента, количества, параметра, свойства или периода. Соответственно, если не указано обратное, числовые параметры, указанные в описании и прилагаемой формуле изобретения, являются приблизительными и могут варьироваться. Например, поскольку инструменты масс-спектрометрии могут незначительно отличаться при определении массы данного аналита, термин «около» в контексте массы иона или отношения массы иона к его заряду означает +/- 0,50 единицы атомной массы. Не преследуя цель ограничить применение доктрины эквивалентов рамками формулы изобретения, каждое числовое обозначение должно, по меньшей мере, истолковываться в свете количества подтвержденных значащих цифр и с применением обычных методов округления.
[229] Использование терминов «может» или «способен» в отношении варианта осуществления или аспекта варианта осуществления также несет в себе альтернативный смысл «не может» или «не способен». Таким образом, если в настоящем описании изобретения указано, что вариант осуществления или аспект варианта осуществления могут быть или способны быть включены как часть объекта изобретения, то также явно подразумевается отрицательное ограничение или исключающее условие, означающее, что вариант осуществления или аспект варианта осуществления не могут быть или не способны быть включены как часть объекта изобретения. Аналогичным образом, использование термина «если требуется» применительно к варианту осуществления или аспекту варианта осуществления означает, что такой вариант осуществления или аспект варианта осуществления могут включаться как часть объекта изобретения или могут не включаться как часть объекта изобретения. Применимо ли такое отрицательное ограничение или исключающее условие зависит от того, указано ли отрицательное ограничение или исключающее условие в заявленном объекте.
[230] Несмотря на то, что числовые диапазоны и значения, устанавливающие широкий диапазон изобретения, являются приближениями, численные диапазоны и значения, указанные в конкретных примерах, указаны с максимально возможной точностью. Тем не менее, любой численный диапазон или значение по своей сути содержат определенные ошибки, неизбежно возникающие в результате стандартного отклонения в соответствующих тестовых измерениях. Указание здесь численных диапазонов значений предназначено только для использования в качестве сокращенного способа индивидуальной ссылки на каждое отдельное численное значение, входящее в диапазон. Если не указано иное, каждое индивидуальное значение числового диапазона включено в настоящее описание, как если бы оно было описано здесь индивидуально.
[231] Термины «некий», «какой-либо», «данный» и подобные указания, используемые в контексте описания настоящего изобретения (особенно в контексте нижеследующей формулы изобретения), должны толковаться как подразумевающие и единственное число, и множественное, если иное не указано здесь или контекст этому явно не противоречит. Кроме того, порядковые числительные, такие, как «первый», «второй», «третий» и т.д., используются с идентифицированными элементами для их различения и не указывают или не подразумевают требуемое или ограниченное число таких элементов, а также не указывают конкретное положение или порядок таких элементов, если явно не указано иное. Все описанные здесь методы могут осуществляться в любом подходящем порядке, если иное не указано здесь или контекст этому явно не противоречит. Использование всех без исключения примеров или фраз, указывающих на примеры (например, «такой, как»), здесь предназначено только для лучшего освещения настоящего изобретения и не представляет собой ограничение объема изобретения, заявленного иным образом. Никакие фразы в настоящем описании изобретения не должны толковаться как указывающие на какой-либо не заявляемый элемент, необходимый для осуществления изобретения.
[232] При использовании в формуле изобретения, независимо от того, при подаче заявки или при внесении изменений, переходный неограничивающий термин «содержащий» (и эквивалентные ему неполные фразы, такие, как включающий, охватывающий и имеющий), относится ко всем явно обозначенным элементам, ограничениям, этапам и/илипризнакам по отдельности или в сочетании с неуказанными объектами изобретения; названные элементы, ограничения и/илипризнаки имеют важное значение, но могут быть добавлены и другие еще не названные элементы, ограничения и/илипризнаки, все же образующие структуру в пределах объема формулы изобретения. Конкретные варианты осуществления, раскрываемые в настоящем документе, могут быть дополнительно ограничены в формуле изобретения с использованием закрытых переходных фраз «состоящий из» или «состоящий главным образом из» вместо или в качестве изменения термина «содержащий». При использовании в формуле изобретения, независимо от того, при подаче заявки или при внесении изменений, ограничивающая фраза «состоящий из» исключает любой элемент, ограничение, этап или признак, явно не указанные в формуле изобретения. Переходная ограничивающая фраза «состоящий главным образом из» ограничивает объем формулы изобретения до явно указанных элементов, ограничений, этапов и/или особенностей и любых других элементов, ограничений, этапов и/илипризнаков, которые не оказывают существенного влияния на основные и новые характеристики заявленного объекта изобретения. Таким образом, значение переходной неограничивающей фразы типа «содержащий» определяется как охватывающее все конкретно перечисленные элементы, ограничения, этапы и/или признаки, а также любые необязательные, дополнительные, которые не были определены. Значение переходной ограничивающей фразы «состоящий из» определяется как включающее только те элементы, ограничения, этапы и/или признаки, которые конкретно указаны в формуле изобретения, тогда как значение переходной ограничивающей фразы «состоящий главным образом из» определяется как включающее только те элементы, ограничения, этапы и/илипризнаки, которые конкретно указаны в формуле изобретения, и те элементы, ограничения, этапы и/или признаки, которые не оказывают существенного влияния на основные и новые характеристики заявленного объекта изобретения. Следовательно, переходная неограничивающая фраза«содержащий» (и эквивалентные ей переходные неограничивающие фразы) включает в своем ограничивающем значении заявляемый объект изобретения, указанный переходными ограничивающими фразами «состоящий из» или «состоящий главным образом из». Таким образом, варианты осуществления, описанные здесь или заявленные таким образом фразой «содержащий», прямо или по своей сути однозначно описаны, включены и обоснованы здесь в отношении фраз «состоящий главным образом из» и «состоящий из».
[233] Все патенты, патентные публикации и другие публикации, упомянутые и указанные настоящем описании, индивидуально и явно включены в него посредством ссылки во всей своей полноте с целью описания и раскрытия, например, композиций и способов, описанных в таких публикациях, которые могут быть использованы в связи с настоящим изобретением. Эти публикации представлены исключительно для их раскрытия до даты подачи настоящей заявки. Ничто в этом отношении не должно толковаться как признание того, что изобретатели не имеют права относить к более ранней дате такое раскрытие в силу предшествующего изобретения или по любой другой причине. Все заявления о дате или представлении в отношении содержания этих документов основаны на информации, доступной заявителям, и не являются признанием правильности дат или содержания этих документов.
[234] Наконец, используемая здесь терминология предназначена только для описания конкретных вариантов осуществления и не предназначена для ограничения объема настоящего изобретения, который определяется исключительно формулой изобретения. Соответственно, настоящее изобретение не ограничивается тем, что конкретным образом представлено и описано.
СПРАВОЧНАЯ ДОКУМЕНТАЦИЯ
• AkiyamaH, BargerS, BarnumS, BradtB, BauerJ, ColeGM, CooperNR, EikelenboomP, EmmerlingM, FiebichBL, FinchCE, FrautschyS, GriffinWS, HampelH, HullM, LandrethG, LueL, MrakR, MackenzieIR, McGeerPL,  PachterJ, PasinettiG, Plata-SalamanC, RogersJ, RydeIR, ShenY, StreitW, StrohmeyerR, Tooyomal, VanMuiswinkelFL, VeerhuisR, WalkerD, WebsterS, WegrzyniakB, WenkG, Wyss-CorayT (2000)
PachterJ, PasinettiG, Plata-SalamanC, RogersJ, RydeIR, ShenY, StreitW, StrohmeyerR, Tooyomal, VanMuiswinkelFL, VeerhuisR, WalkerD, WebsterS, WegrzyniakB, WenkG, Wyss-CorayT (2000)  . Neurobiology of aging 21:383-421
. Neurobiology of aging 21:383-421
• Alamed J, Wilcock D M, Diamond D M, Gordon M N, Morgan D (2006) Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nature protocols 1:1671-1679
• Bai A, Lu N, Guo Y, Liu Z, Chen J, Peng Z (2009) All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. Journal of leukocyte biology 86:959-969
• BaruchK, DeczkowskaA, DavidE, CastellanoJM, MillerO, KertserA, BerkutzkiT, Barnett-ltzhakiZ, BezalelD, Wyss-CorayT, Amitl, SchwartzM (2014) Aging. Aging-induced type linterferon response at the choroid plexus negatively affects brain function. Science 346:89-93
• BaruchK, KertserA, PoratZ, SchwartzM (2015) CerebralnitricoxiderepresseschoroidplexusNFkappaB-dependentgatewayactivityforleukocytetrafficking. The EMBO journal
• BaruchK, Ron-HarelN, GalH, DeczkowskaA, ShifrutE, NdifonW, Mirlas-NeisbergN, CardonM, Vakninl, CahalonL, BerkutzkiT, MattsonMP, Gomez-PinillaF, FriedmanN, SchwartzM (2013) CNS-specificimmunityatthechoroidplexusshiftstowarddestructiveTh2 inflammationinbrainaging. Proceedings of the National Academy of Sciences of the United States of America 110:2264-2269
• Baruch, A. Deczkowska, N. Rosenzweig, A. Tsitsou-Kampeli, A. M. Sharif, O. Matcovitch-Natan, A. Kertser, E. David, I. Amit, M. Schwartz (2016) PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of  disease, Nat. Med. 22, 135-137
disease, Nat. Med. 22, 135-137
• Borchelt D R, Ratovitski T, van Lare J, Lee M K, Gonzales V, Jenkins N A, Copeland N G, Price D L, Sisodia S S (1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19: 939-945
• Bos P D, Rudensky A Y (2012) Treg cells in cancer: a case of multiple personality disorder. Science translational medicine 4:164fs144
• BowersEM, YanG, MukherjeeC, OrryA, WangL, HolbertMA, CrumpNT, HazzalinCA, LiszczakG, YuanH, LaroccaC, SaldanhaSA, AbagyanR, SunY, MeyersDJ, MarmorsteinR, MahadevanLC, AlaniRM, ColePA (2010) Virtualligandscreeningofthep300/CBPhistoneacetyltransferase: identificationofaselectivesmallmoleculeinhibitor. Chemistry & biology 17:471-482
• Breitner J C, Haneuse S J, Walker R, Dublin S, Crane P K, Gray S L, Larson E В (2009) Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 72:1899-1905
• Brestoff J R, Artis D (2013) Commensal bacteria at the interface of host metabolism and the immune system. Nature immunology 14:676-684
• Burgess A, Vigneron S, Brioudes E, Labbe J C, Lorca T, Castro A (2010) Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proceedings of the National Academy of Sciences of the United States of America 107:12564-12569
• Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M (2006) Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proceedings of the National Academy of Sciences of the United States of America 103:11784-11789
• Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M (2007) Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. The European journal of neuroscience 26:413-416
• Chen X, Oppenheim J J (2011) Resolving the identity myth: key markers of functional CD4+FoxP3+ regulatory T cells. International immunopharmacology 11:1489-1496
• Colombo M P, Piconese S (2007) Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nature reviews Cancer 7:880-887
• Coyne G O, Gulley J L (2014) Adding fuel to the fire: Immunogenic intensification. Human vaccines & immunotherapeutics 10:3306-3312
• Dalotto-MorenoT, CrociDO, CerlianiJP, Martinez-AlloVC, Dergan-DylonS, Mendez-HuergoSP, StupirskiJC, MazalD, OsinagaE, ToscanoMA, SundbladV, RabinovichGA, SalatinoM (2013) Targetinggalectin-1 overcomesbreastcancerassociatedimmunosuppressionandpreventsmetastaticdisease. Cancer research 73:1107-1117
• Duraiswamy J, Freeman G J, Coukos G (2014) Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors-response. Cancer research 74:633-634; discussion 635
• Francisco L M, Sage P T, Sharpe A H (2010) The PD-1 pathway in tolerance and autoimmunity. Immunological reviews 236:219-242
• Gabrilovich D I, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nature reviews Immunology 9:162-174
• Galvin K С, Dyck L, Marshall N A, Stefanska A M, Walsh K P, Moran B, Higgins S C, Dungan L S, Mills K H (2013) Blocking retinoic acid receptor-alpha enhances the efficacy of a dendritic cell vaccine against tumours by suppressing the induction of regulatory T cells. Cancer immunology, immunotherapy: C// 62:1273-1282
• Ghiringhelli F, Bruchard M, Chalmin F, Rebe С (2012) Production of adenosine by ectonucleotidases: a key factor in tumor immunoescape. Journal of biomedicine & biotechnology 2012:473712
• Group A R, Lyketsos С G, Breitner J C, Green R C, Martin В K, Meinert С, Piantadosi S, Sabbagh M (2007) Naproxen and celecoxib do not prevent A D in early results from a randomized controlled trial. Neurology 68:1800-1808
• Hardy J, Selkoe D J (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353-356
• He F, Balling R (2013) The role of regulatory T cells in neurodegenerative diseases. Wiley interdisciplinary reviews Systems biology and medicine 5:153-180
• HirayamaM, NishikawaH, NagataY, TsujiT, KatoT, KageyamaS, UedaS, SugiyamaD, HoriS, SakaguchiS, RitterG, OldLJ, GnjaticS, ShikuH (2013) OvercomingregulatoryT-cellsuppressionbyalyophilizedpreparationof Streptococcuspyogenes. European journal of immunology 43:989-1000
• Hong J, Li N, Zhang X, Zheng B, Zhang J Z (2005) Induction of CD4+CD25+ regulatory T cells by copolymer-l through activation of transcription factor Foxp3. Proceedings of the National Academy of Sciences of the United States of America 102:6449-6454
• Joller N, Peters A, Anderson A C, Kuchroo V K (2012) Immune checkpoints in central nervous system autoimmunity. Immunological reviews 248:122-139
• JuY, ShangX, LiuZ, ZhangJ, LiY, ShenY, LiuY, LiuC, LiuB, XuL, WangY, ZhangB, ZouJ (2014) TheTim-3/galectin-9 pathwayinvolvesinthehomeostasisofhepaticTregsinamousemodelofconcanavalinA-inducedhepatitis. Molecular immunology 58:85-91
• Jung S, Aliberti J, Graemmel P, Sunshine M J, Kreutzberg G W, Sher A, Littman D R (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Molecular and cellular biology 20:4106-4114
• Kim J M, Rasmussen J P, Rudensky A Y (2007) Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nature immunology 8:191-197
• KimPS, JochemsC, Grengal, DonahueRN, TsangKY, GulleyJL, SchlomJ, FarsaciB (2014) Pan-Bcl-2 inhibitor, GX15-070 (obatoclax), decreaseshumanTregulatorylymphocyteswhilepreservingeffectorTlymphocytes: arationaleforitsuseincombinationimmunotherapy. Journal of immunology 192:2622-2633
• Kotsakis A, Harasymczuk M, Schilling B, Georgoulias V, Argiris A, Whiteside T L (2012) Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples. Journal of immunological methods 381:14-22
• Kunis G, Baruch K, Miller O, Schwartz M (2015) Immunization with a Myelin-Derived Antigen Activates the  Choroid Plexus for Recruitment of Immunoregulatory Cells to the CNS and Attenuates Disease Progression in a Mouse Model of ALS. The Journal of neuroscience: the official journal of the Society for Neuroscience 35:6381-6393
Choroid Plexus for Recruitment of Immunoregulatory Cells to the CNS and Attenuates Disease Progression in a Mouse Model of ALS. The Journal of neuroscience: the official journal of the Society for Neuroscience 35:6381-6393
• Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, Schwartz M (2013) IFN-gamma-dependent activation of the  choroid plexus for CNS immune surveillance and repair. Brain: a journal of neurology 136:3427-3440
choroid plexus for CNS immune surveillance and repair. Brain: a journal of neurology 136:3427-3440
• Lebow M, Neufeld-Cohen A, Kuperman Y, Tsoory M, Gil S, Chen A (2012) Susceptibility to PTSD-like behavior is mediated by corticotropin-releasing factor receptor type 2 levels in the bed nucleus of the stria terminalis. J Neurosci 32:6906-6916
• Lesokhin A M, Callahan M K, Postow M A, Wolchok J D (2015) On being less tolerant: enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Science translational medicine 7:280sr281
• LiuY, WangL, PredinaJ, HanR, BeierUH, WangLC, KapoorV, BhattiTR, AkimovaT, SinghalS, BrindlePK, ColePA, AlbeldaSM, HancockWW (2013) Inhibitionofp300 impairsFoxp3(+) Tregulatorycellfunctionandpromotesantitumorimmunity. Nature medicine 19:1173-1177
• MarabelleA, KohrtH, Sagiv-Barfil, AjamiB, AxtellRC, ZhouG, RajapaksaR, GreenMR, TorchiaJ, BrodyJ, LuongR, RosenblumMD, SteinmanL, LevitskyHI, TseV, LevyR (2013) Depletingtumor-specificTregsatasinglesiteeradicatesdisseminatedtumors. The Journal of clinical investigation 123:2447-2463
• Mellman I, Coukos G, Dranoff G (2011) Cancer immunotherapy comes of age. Nature 480:480-489
• Michaud J P, Bellavance M A, Prefontaine P, Rivest S (2013) Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell reports 5:646-653
• Nishikawa H, Sakaguchi S (2010) Regulatory T cells in tumor immunity. International journal of cancer Journal international du cancer Ml: 759-767
• OakleyH, ColeSL, LoganS, MausE, ShaoP, CraftJ, Guillozet-BongaartsA, OhnoM, DisterhoftJ, VanEldikL, BerryR, VassarR (2006) Intraneuronalbeta-amyloidaggregates, neurodegeneration, andneuronlossintransgenicmicewithfivefamilialAlzheimer'sdiseasemutations: potentialfactorsinamyloidplaqueformation. The Journal of neuroscience: the official journal of the Society for Neuroscience 26: 10129-10140
• Ohaegbulam K С, Assal A, Lazar-Molnar E, Yao Y, Zang X (2015) Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends in molecular medicine 21:24-33
• Pardoll D M (2012) The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer 12:252-264
• PengW, LiuC, XuC, LouY, ChenJ, YangY, YagitaH, OverwijkWW, LizeeG, RadvanyiL, HwuP (2012) PD-1 blockadeenhancesT-cellmigrationtotumorsbyelevatinglFN-gammainduciblechemokines. Cancer research 72:5209-5218
• PereH, MontierY, BayryJ, Quintin-ColonnaF, MerillonN, DransartE, BadoualC, GeyA, RavelP, MarcheteauE, BatteuxF, SandovalF, AdoteviO, ChiuC, GarciaS, TanchotC, LoneYC, FerreiraLC, NelsonBH, HanahanD, FridmanWH, JohannesL, TartourE (2011) ACCR4 antagonistcombinedwithvaccinesinducesantigen-specificCD8+ Tcellsandtumorimmunityagainstselfantigens. Blood 118:4853-4862
• Postow M A, Callahan M K, Wolchok J D (2015) Immune Checkpoint Blockade in Cancer Therapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology
• Qin A, Wen Z, Zhou Y, Li Y, Li Y, Luo J, Ren T, Xu L (2013) MicroRNA-126 regulates the induction and function of CD4(+) Foxp3(+) regulatory T cells through PI3K/AKT pathway. Journal of cellular and molecular medicine 17:252-264
• Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, Gasser T, Stoltze L (2007) Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. Journal of neuroimmunology 188:117-127
• RoyS, BarikS, BanerjeeS, BhuniyaA, PalS, BasuP, BiswasJ, GoswamiS, ChakrabortyT, BoseA, BaralR (2013) Neemleafglycoproteinovercomesindoleamine 2,3 dioxygenasemediatedtoleranceindendriticcellsbyattenuatinghyperactiveregulatoryTcellsincervic alcancerstagelll Bpatients. Human immunology 74:1015-1023
• Sakaguchi S, Yamaguchi T, Nomura T, Ono M (2008) Regulatory T cells and immune tolerance. Cell 133:775-787
• SaresellaM, CalabreseE, Marventanol, PianconeF, GattiA, CalvoMG, NemniR, ClericiM (2010) PD1 negativeandPD1 positiveCD4+  Journal of
Journal of  disease JAD 21:927-938
disease JAD 21:927-938
• Schmidt S D, Nixon R A, Mathews P M (2005) ELISA method for measurement of amyloid-beta levels. Methods in molecular biology 299:279-297
• Schreiber R D, Old L J, Smyth M J (2011) Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331:1565-1570
• Schwartz M, Baruch K (2014a) Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: boosting autoimmunity to fight-off chronic neuroinflammation. Journal of autoimmunity 54:8-14
• Schwartz M, Baruch K (2014b) The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. The EMBO journal 33:7-22
• ShankarGM, LiS, MehtaTH, Garcia-MunozA, ShepardsonNE, Smithl, BrettFM, FarrellMA, RowanMJ, LemereCA, ReganCM, WalshDM, SabatiniBL, SelkoeDJ (2008) Amyloid-  Nat ure medicine 14:837-842
Nat ure medicine 14:837-842
• Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M (2009) Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS medicine 6: e1000113
• ShechterR, MillerO, YoveIG, RosenzweigN, LondonA, RuckhJ, KimKW, KleinE, KalchenkoV, BendelP, LiraSA, JungS, SchwartzM (2013) RecruitmentofbeneficialM2 macrophagestoinjuredspinalcordisorchestratedbyremotebrainchoroidplexus. Immunity 38:555-569
• Shevchenko I, Karakhanova S, Soltek S, Link J, Bayry J, Werner J, Umansky V, Bazhin A V (2013) Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. International journal of cancer Journal international du cancer 133:98-107
• Simpson T R, Li F, Montalvo-Ortiz W, Sepulveda M A, Bergerhoff K, Arce F, Roddie C, Henry J Y, Yagita H, Wolchok J D, Peggs K S, Ravetch J V, Allison J P, Quezada S A (2013) Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. The Journal of experimental medicine 210:1695-1710
• Smith P M, Howitt M R, Panikov N, Michaud M, Gallini С A, Bohlooly Y M, Glickman J N, Garrett W S (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569-573
• Suffner J, Hochweller K, Kuhnle M C, Li X, Kroczek R A, Garbi N, Hammerling G J (2010) Dendritic cells support homeostatic expansion of Foxp3+ regulatory T cells in Foxp3.LuciDTR mice. Journal of immunology 184:1810-1820
• Terme M, Colussi O, Marcheteau E, Tanchot C, Tartour E, Taieb J (2012) Modulation of immunity by antiangiogenic molecules in cancer. Clinical & developmental immunology 2012:492920
• Thomas-Schoemann A, Batteux F, Mongaret C, Nicco C, Chereau C, Annereau M, Dauphin A, Goldwasser F, Weill B, Lemare F, Alexandre J (2012) Arsenic trioxide exerts antitumor activity through regulatory T cell depletion mediated by oxidative stress in a murine model of colon cancer. Journal of immunology 189: 5171-5177
• Torres K С, Araujo Pereira P, Lima G S, Bozzi I C, Rezende V B, Bicalho M A, Moraes E N, Miranda D M, Romano-Silva M A (2013) Increased frequency of T cells expressing IL-10 in Alzheimer disease but not in late-onset depression patients. Progress in neuro-psychopharmacology & biological psychiatry 47:40-45
• Vom Berg J, Prokop S, Miller K R, Obst J, Kalin R E, Lopategui-Cabezas I, Wegner A, Mair F, Schipke С G, Peters O, Winter Y, Becher B, Heppner F L (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nature medicine 18:1812-1819
• Voo K S, Boyer L, Harline M L, Vien L T, Facchinetti V, Arima K, Kwak L W, Liu Y J (2013) Antibodies targeting human OX40 expand effector T cells and block inducible and natural regulatory T cell function. Journal of immunology 191:3641-3650
• Walsh J T, Zheng J, Smirnov I, Lorenz U, Tung K, Kipnis J (2014) Regulatory T cells in central nervous system injury: a double-edged sword. Journal of immunology 193:5013-5022
• Ward F J, Dahal L N, Wijesekera S K, Abdul-Jawad S K, Kaewarpai T, Xu H, Vickers M A, Barker R N (2013) The soluble isoform of CTLA-4 as a regulator of T-cell responses. European journal of immunology 43:1274-1285
• Weber J S, Kudchadkar R R, Yu B, Gallenstein D, Horak С E, Inzunza H D, Zhao X, Martinez A J, Wang W, Gibney G, Kroeger J, Eysmans C, Sarnaik A A, Chen Y A (2013) Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 31:4311-4318
• Weber M S, Hohlfeld R, Zamvil S S (2007) Mechanism of action of glatiramer acetate in treatment of multiple sclerosis. Neurotherapeutics: the journal of the American Society for Experimental Neuro Therapeutics 4:647-653
• Weiskopf K, Ring A M, Schnorr P J, Volkmer J P, Volkmer A K, Weissman I L, Garcia K С (2013) Improving macrophage responses to therapeutic antibodies by molecular engineering of SIRPalpha variants. Oncoimmunology 2:e25773
• Wyss-Coray T, Rogers J (2012) Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harbor perspectives in medicine 2:a006346
• Zeng J, See A P, Phallen J, Jackson С M, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris T J, Albesiano E, Pradilla G, Ford E, Wong J, Hammers H J, Mathios D, Tyler B, Brem H, Tran P T, Pardoll D, Drake С G, Lim M (2013) Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. International journal of radiation oncology, biology, physics 86:343-349
• Zhao L, Sun L, Wang H, Ma H, Liu G, Zhao Y (2007) Changes of CD4+CD25+Foxp3+ regulatory T cells in aged Balb/c mice. Journal of leukocyte biology 81:1386-1394
• Zheng H, Fridkin M, Youdim M (2015) New approaches to treating Alzheimer's disease. Perspectives in medicinal chemistry 7:1-8
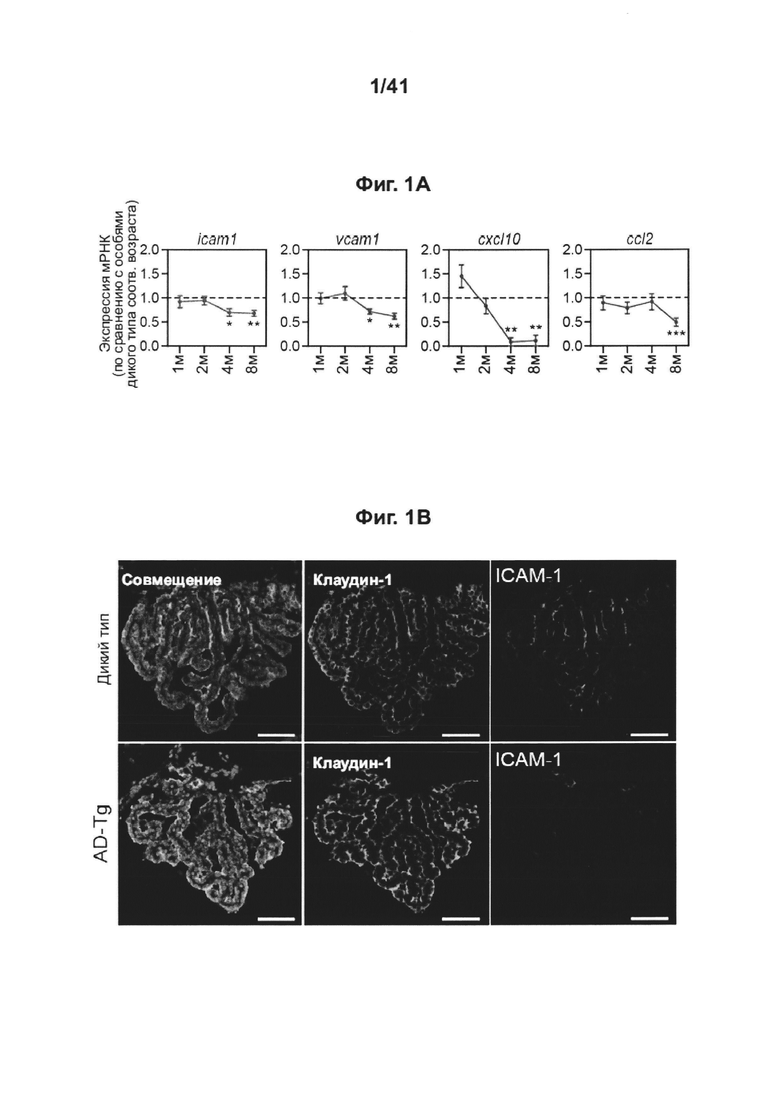
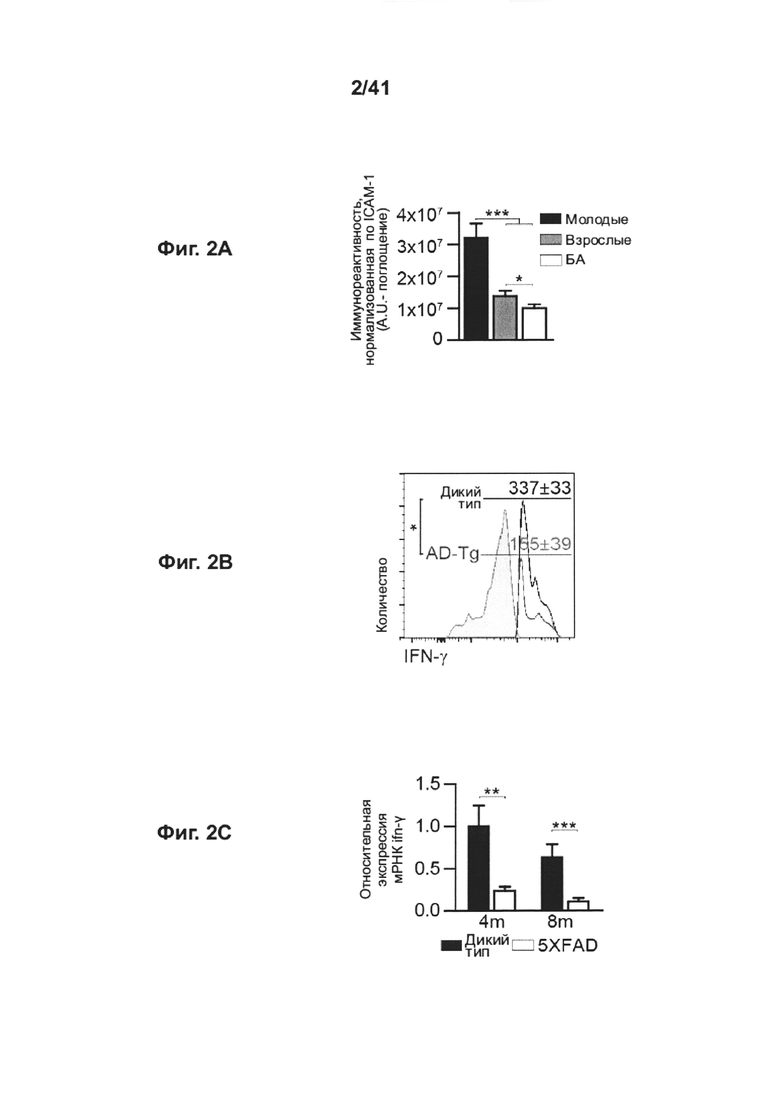
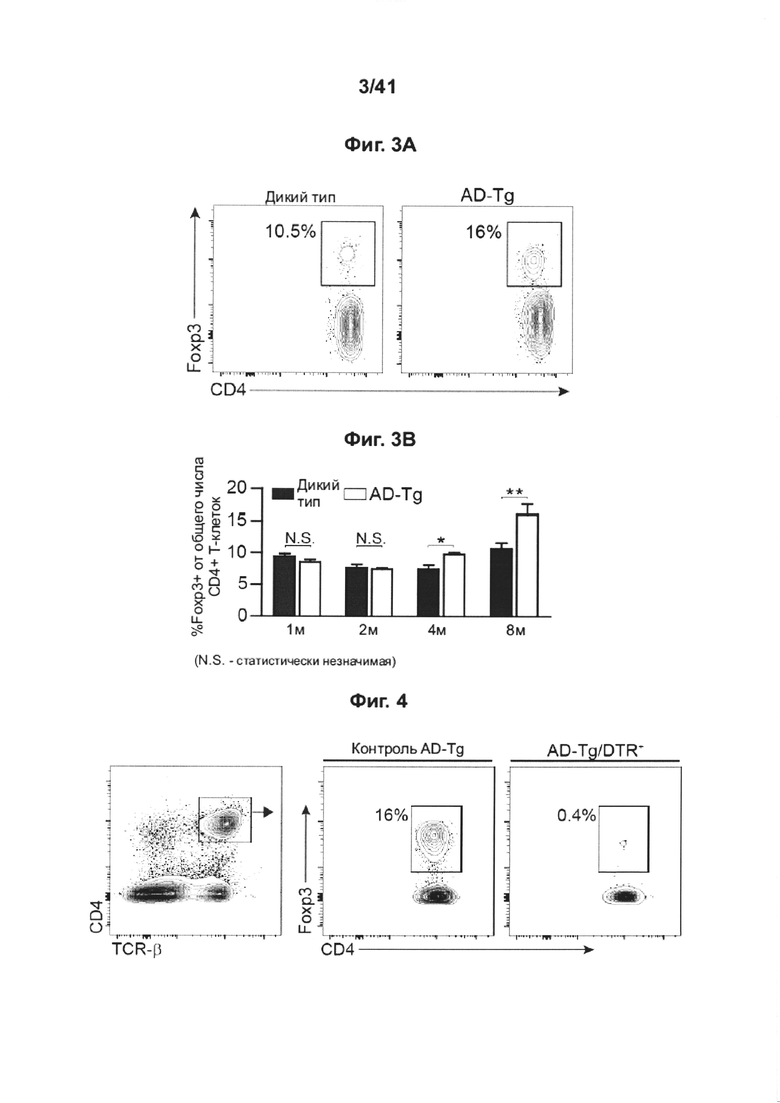
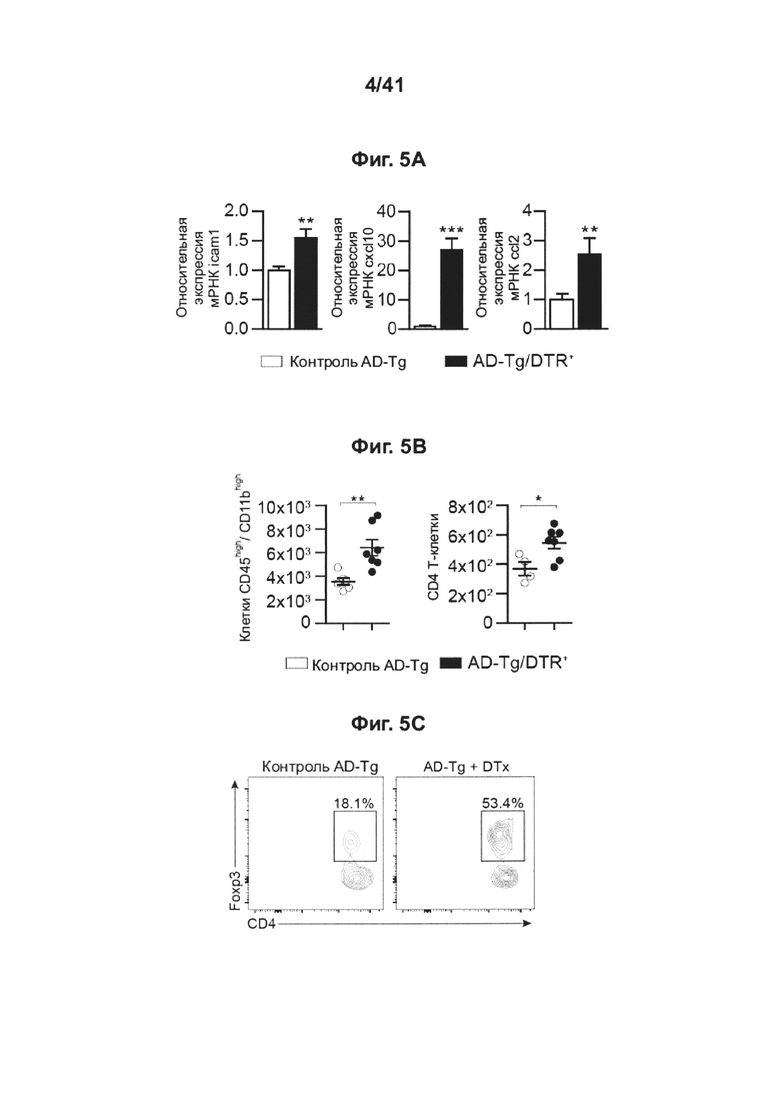
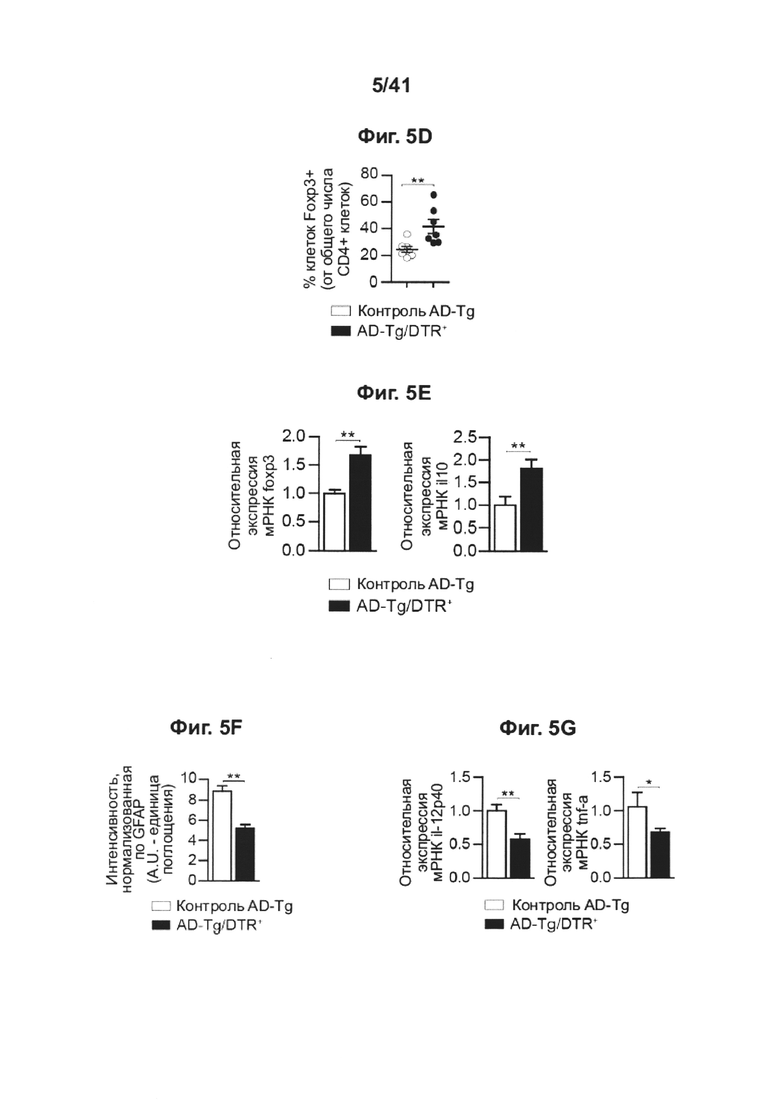
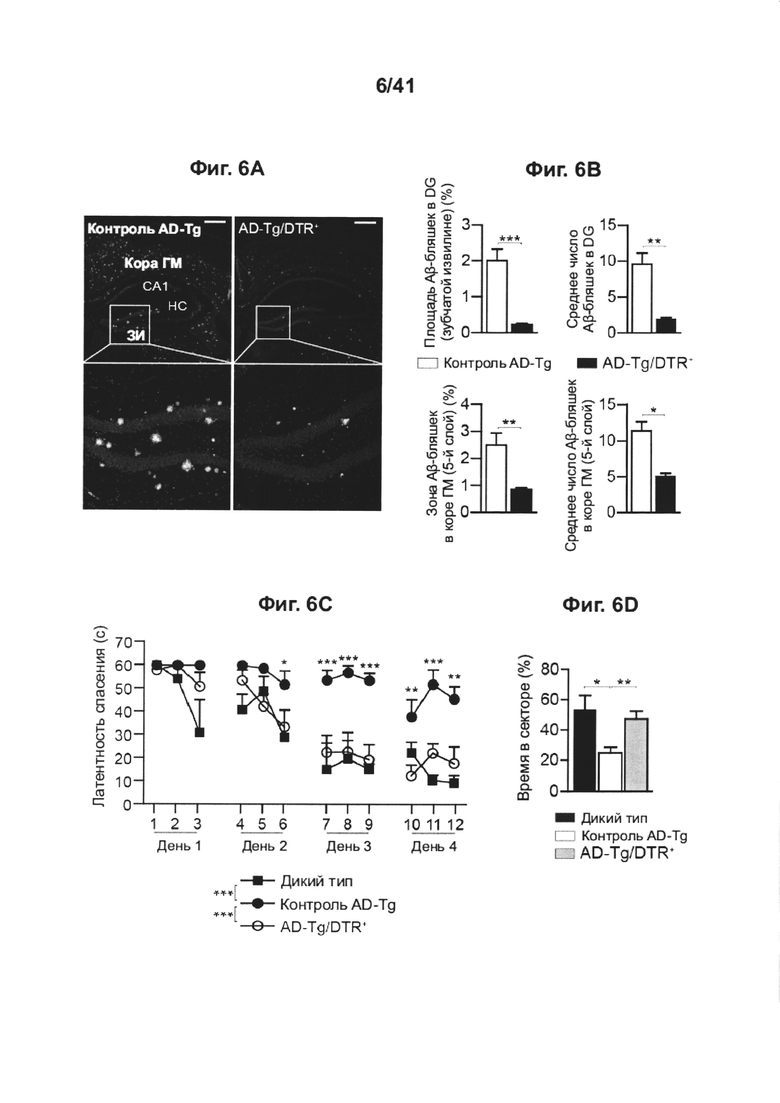
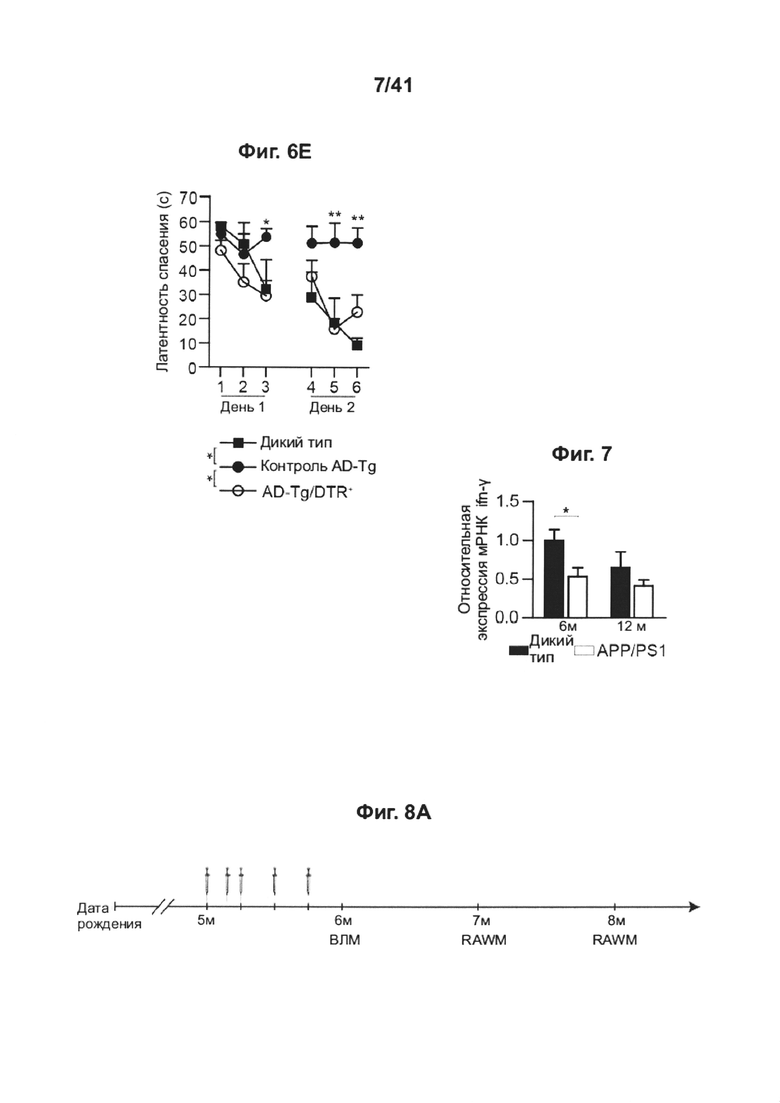
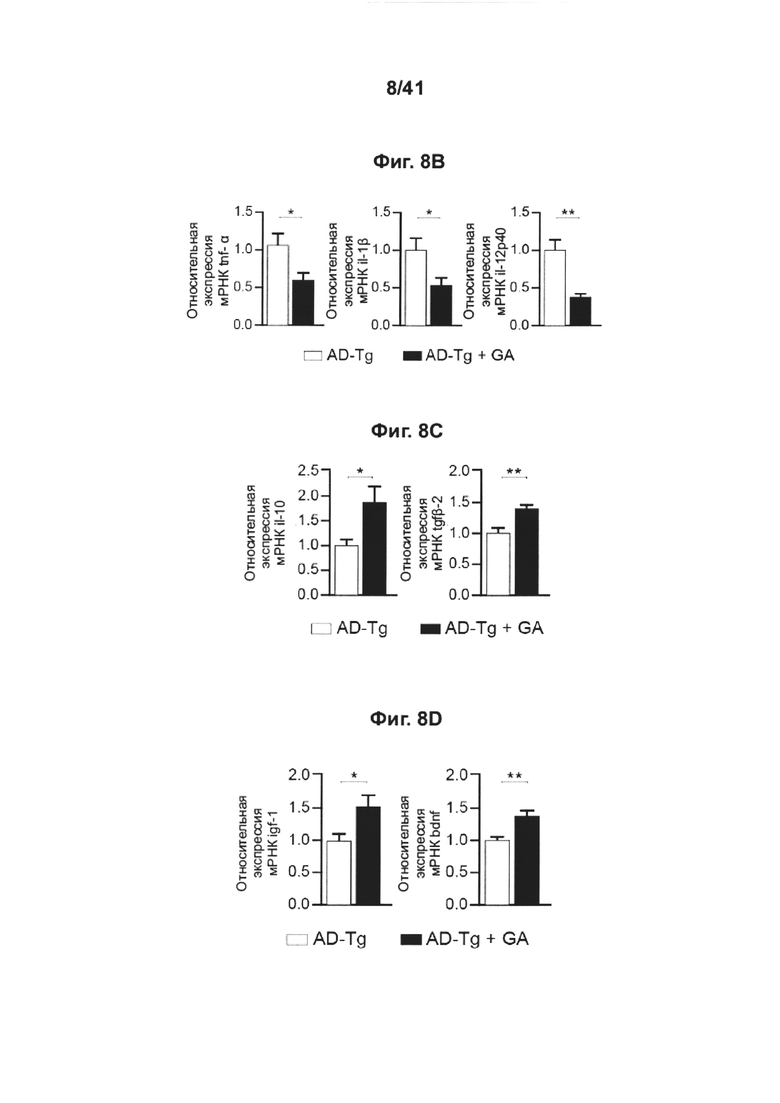
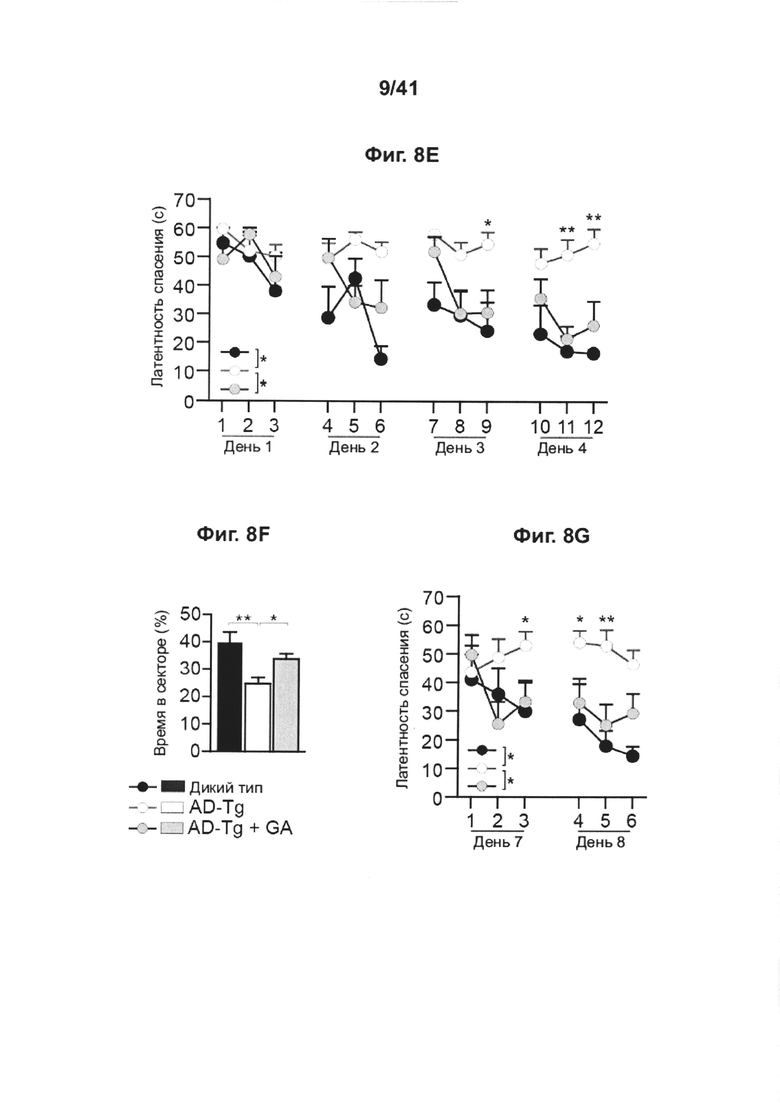
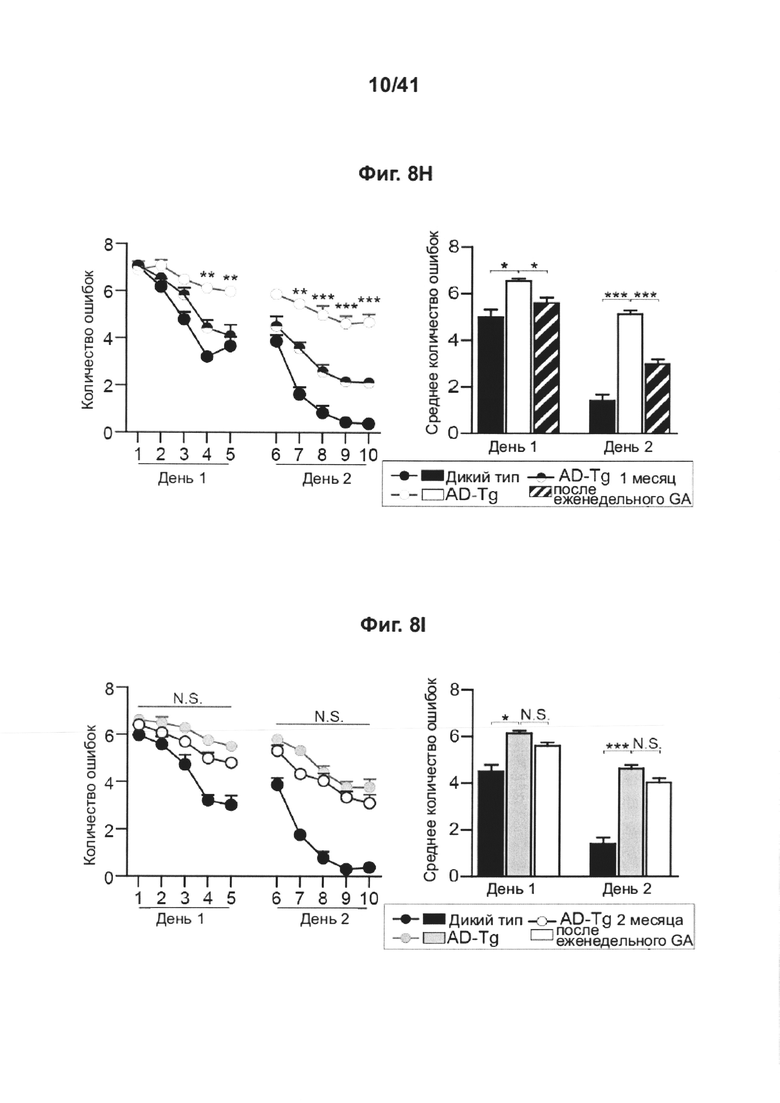
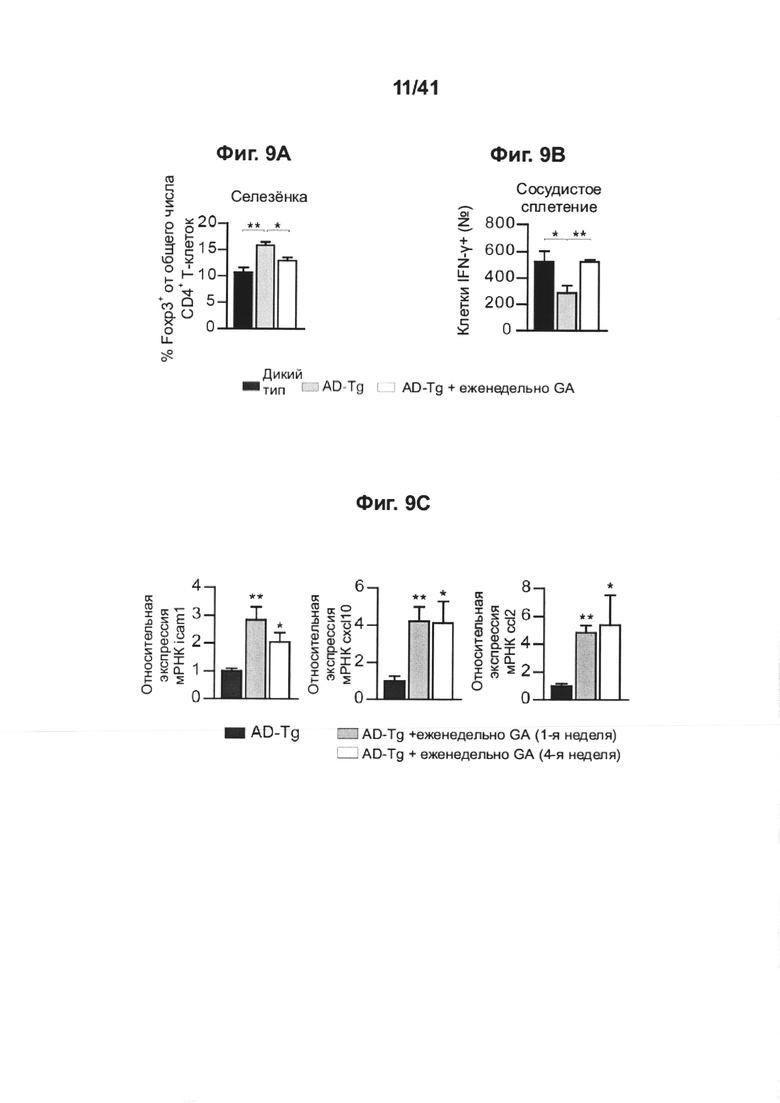
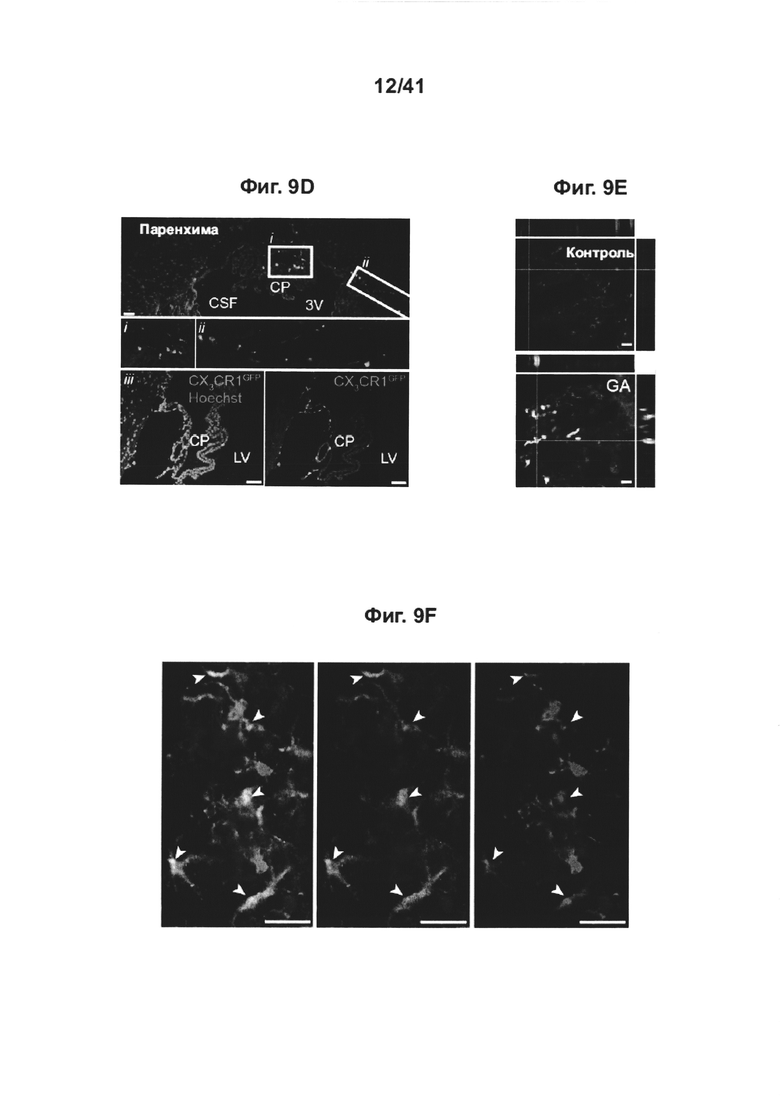
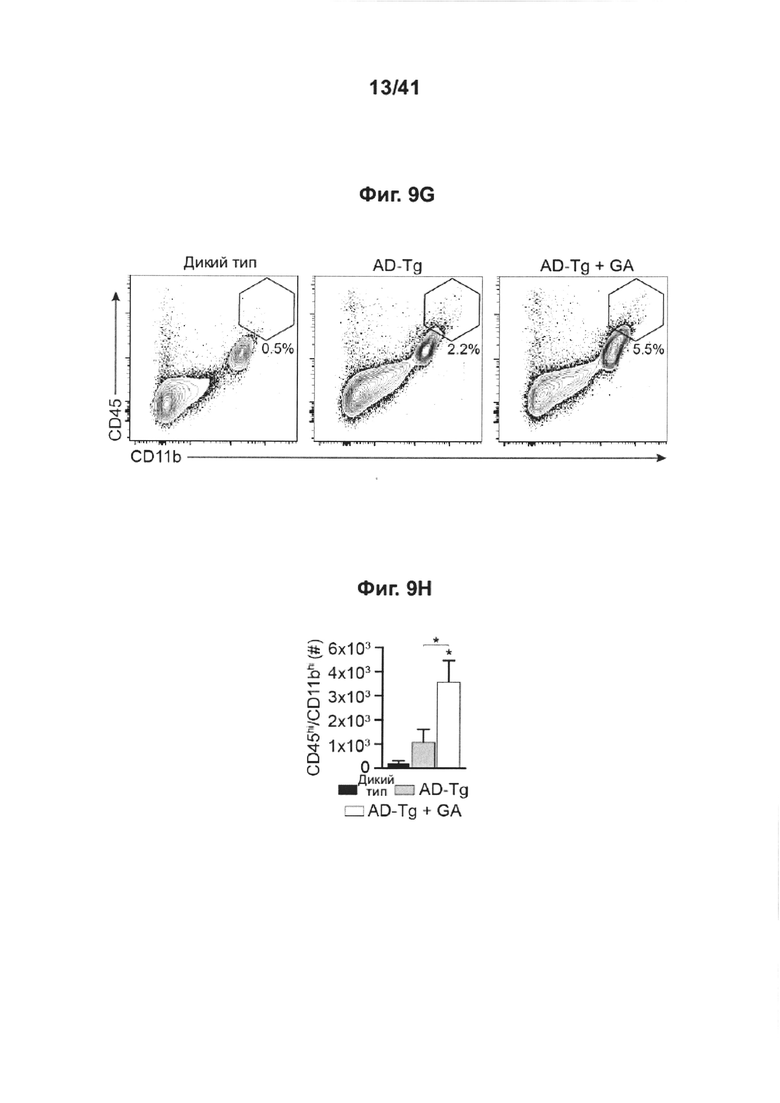
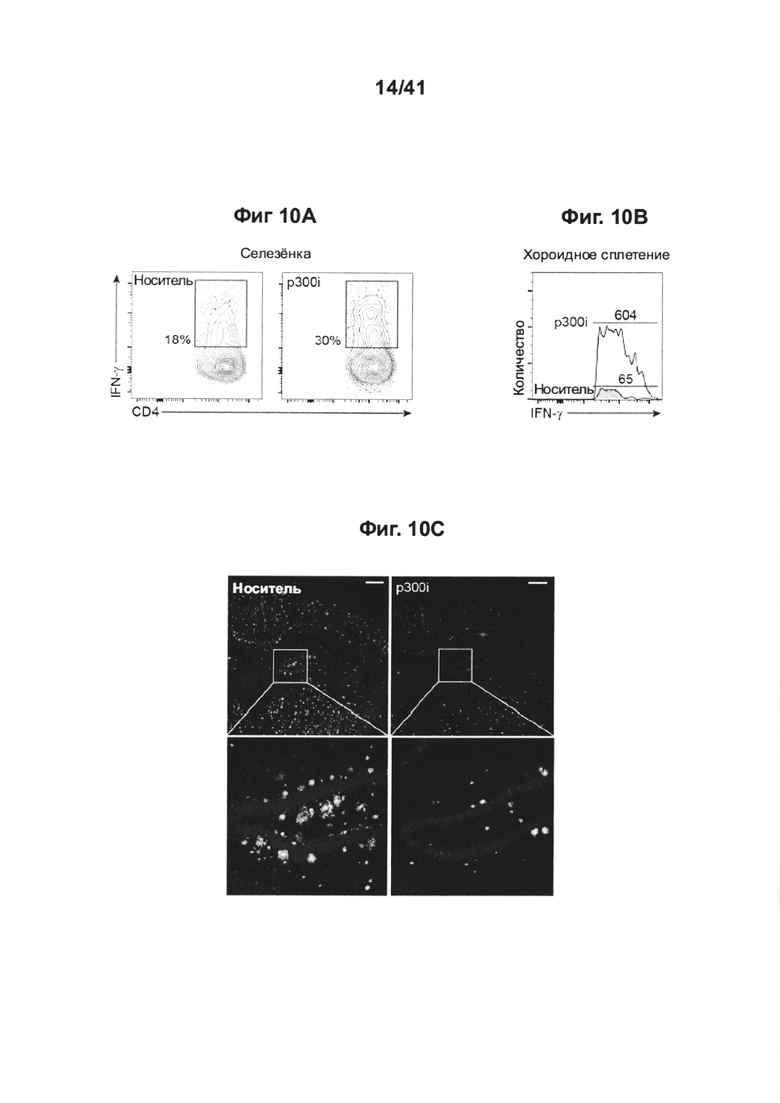
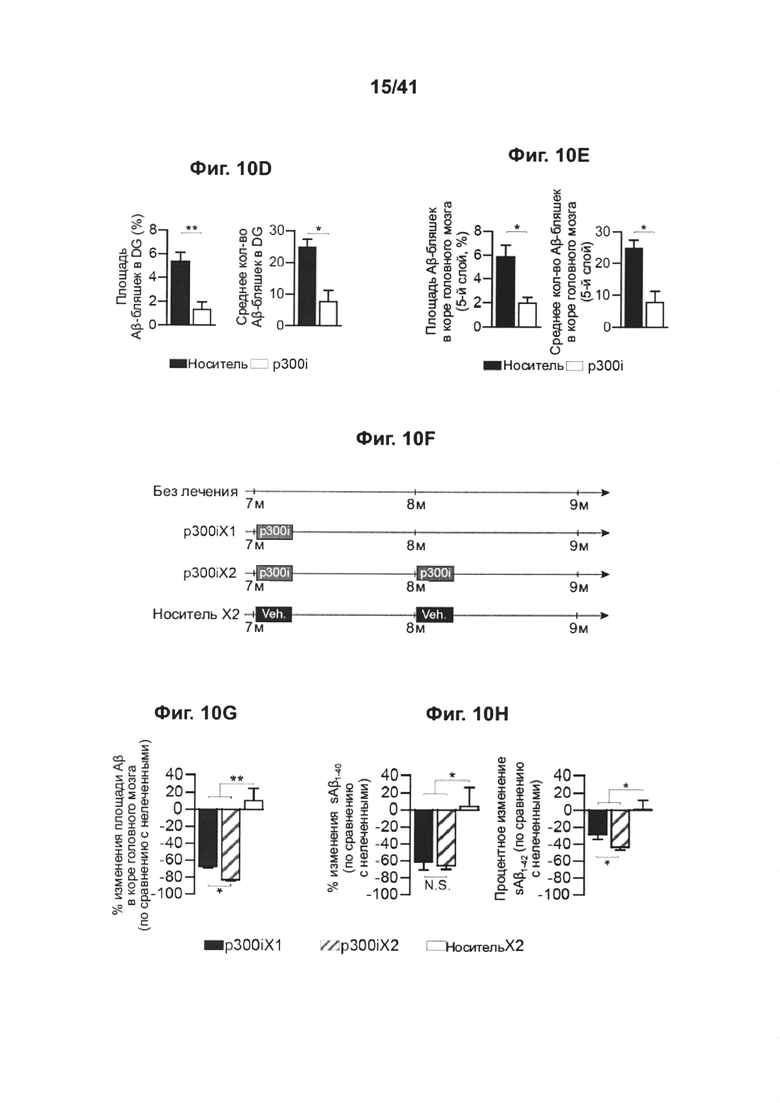
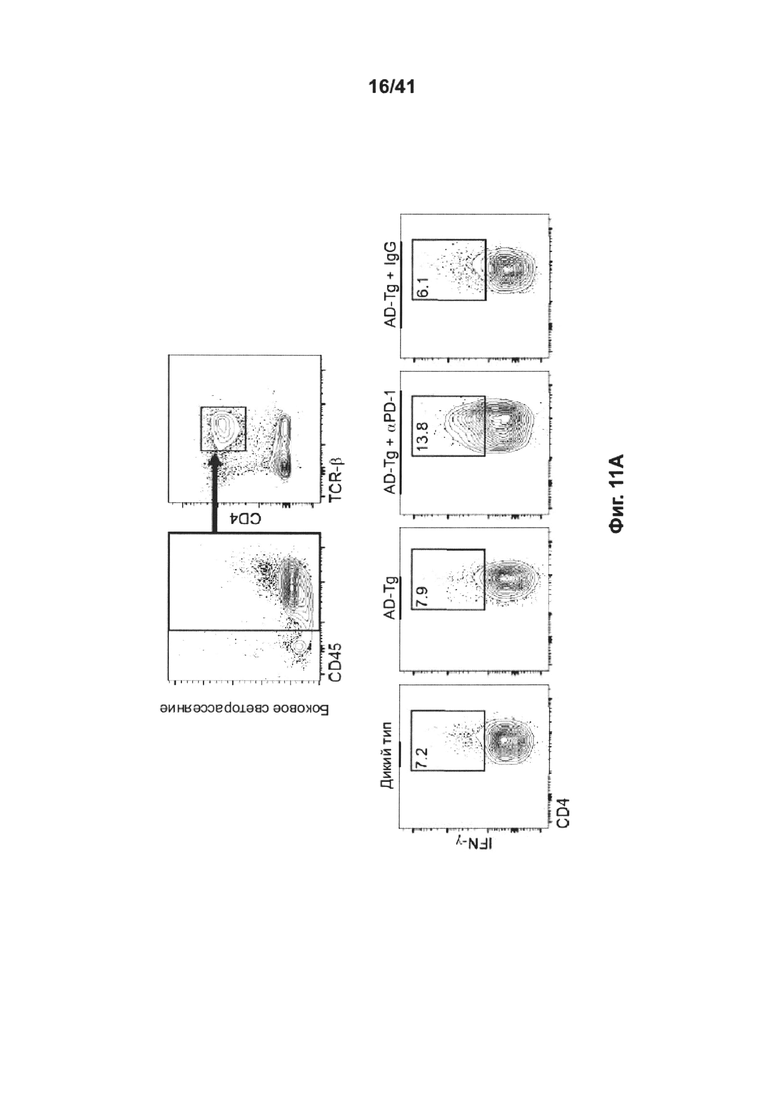
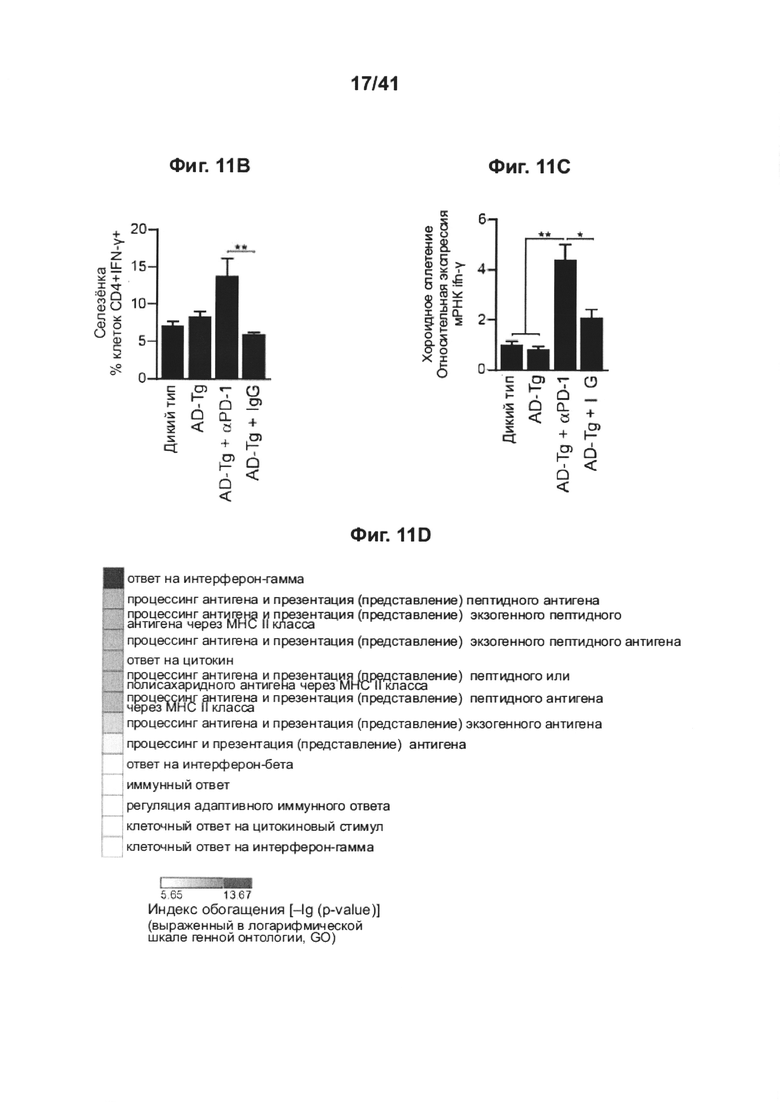
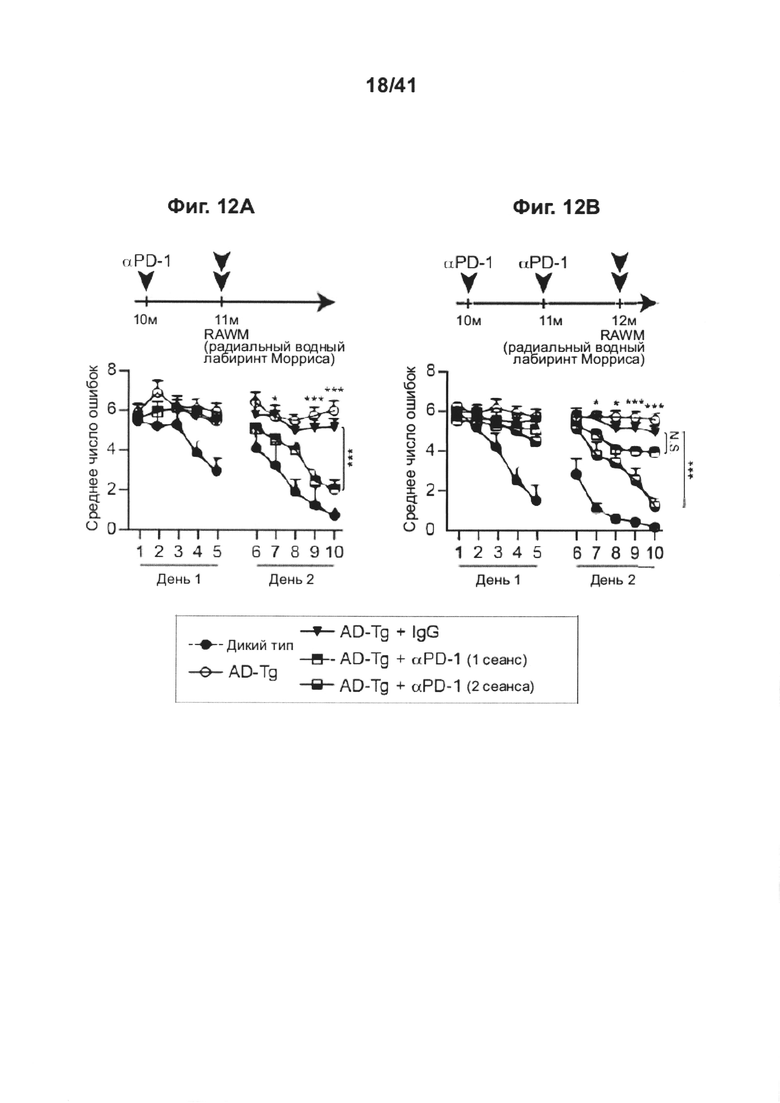
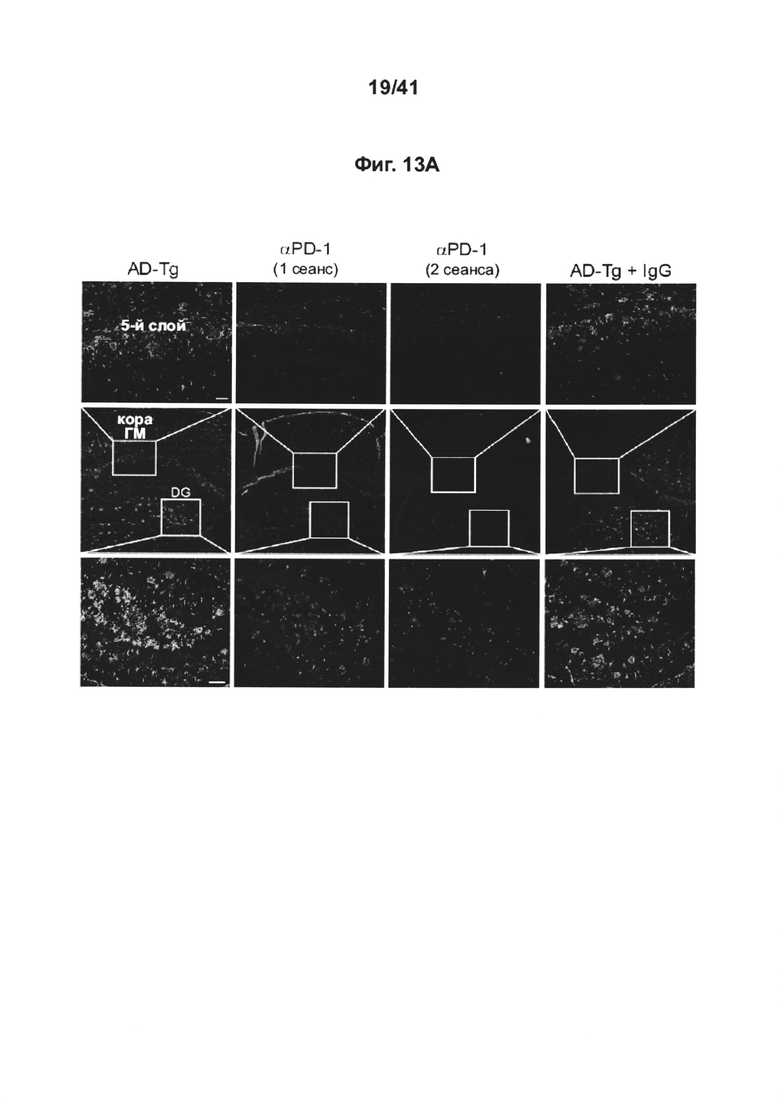
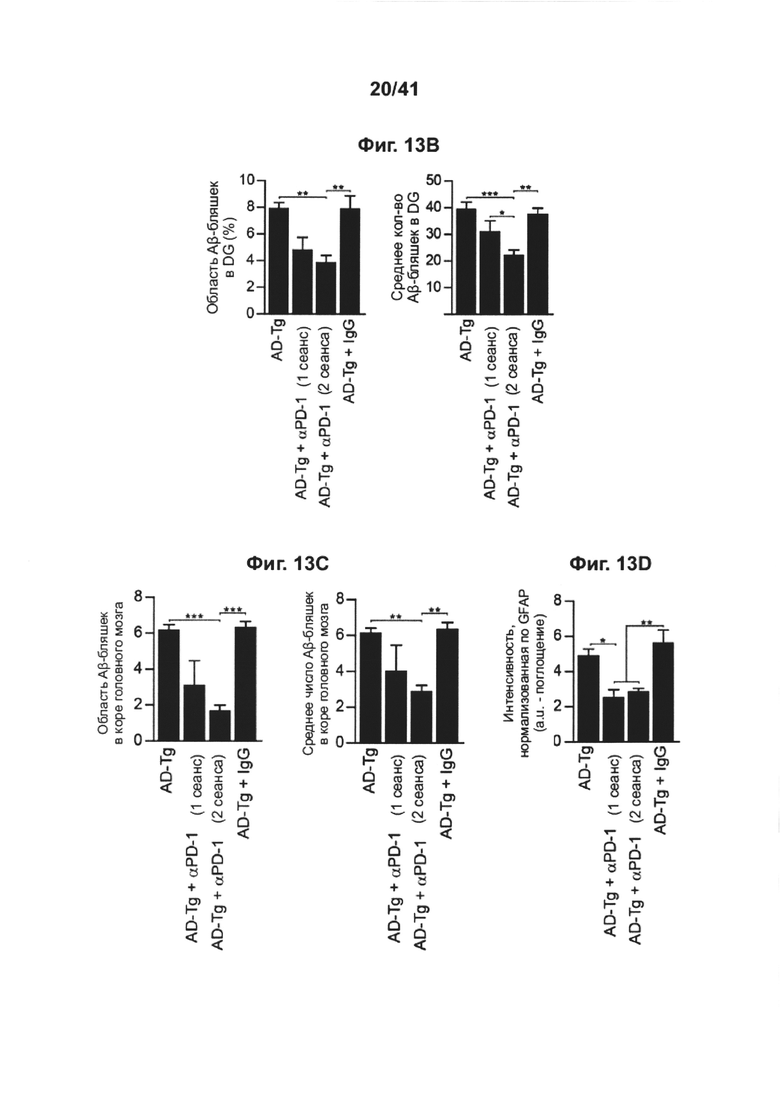
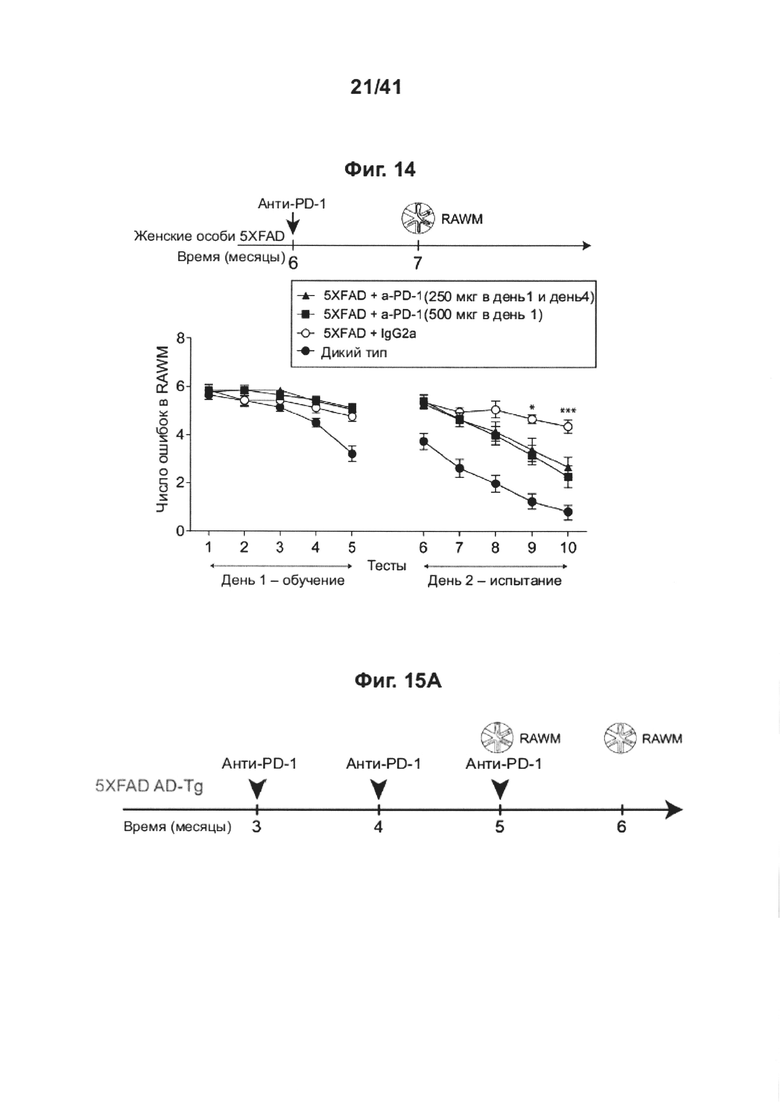
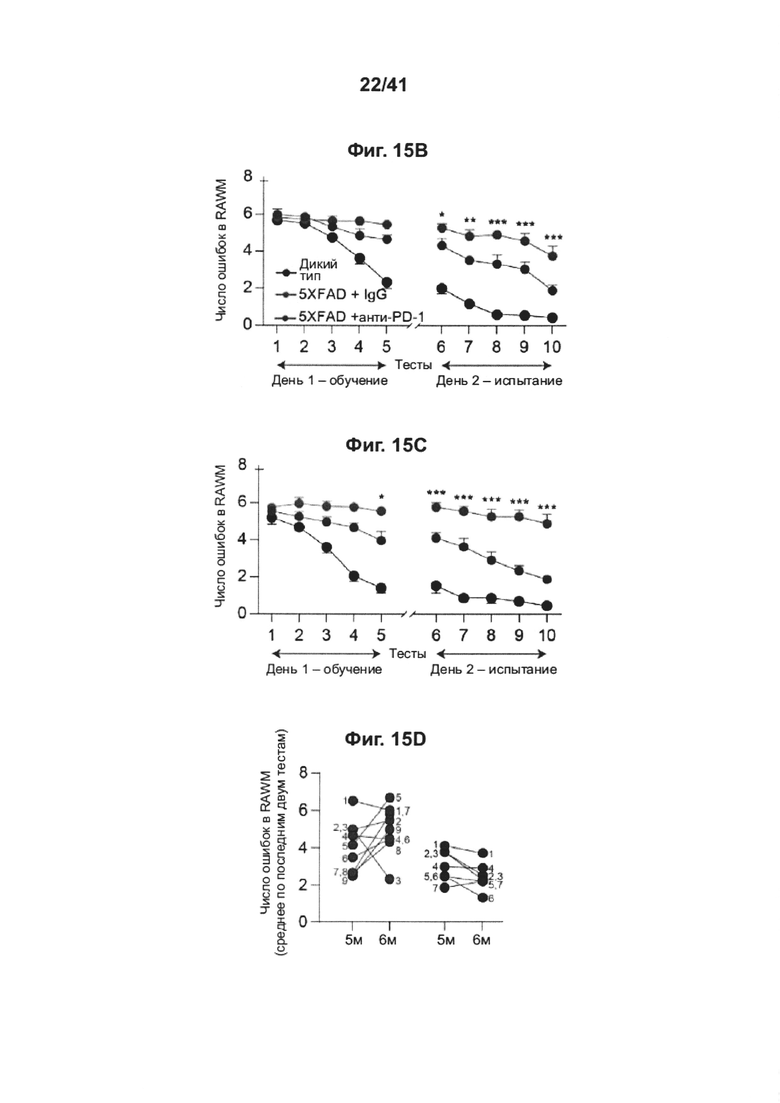
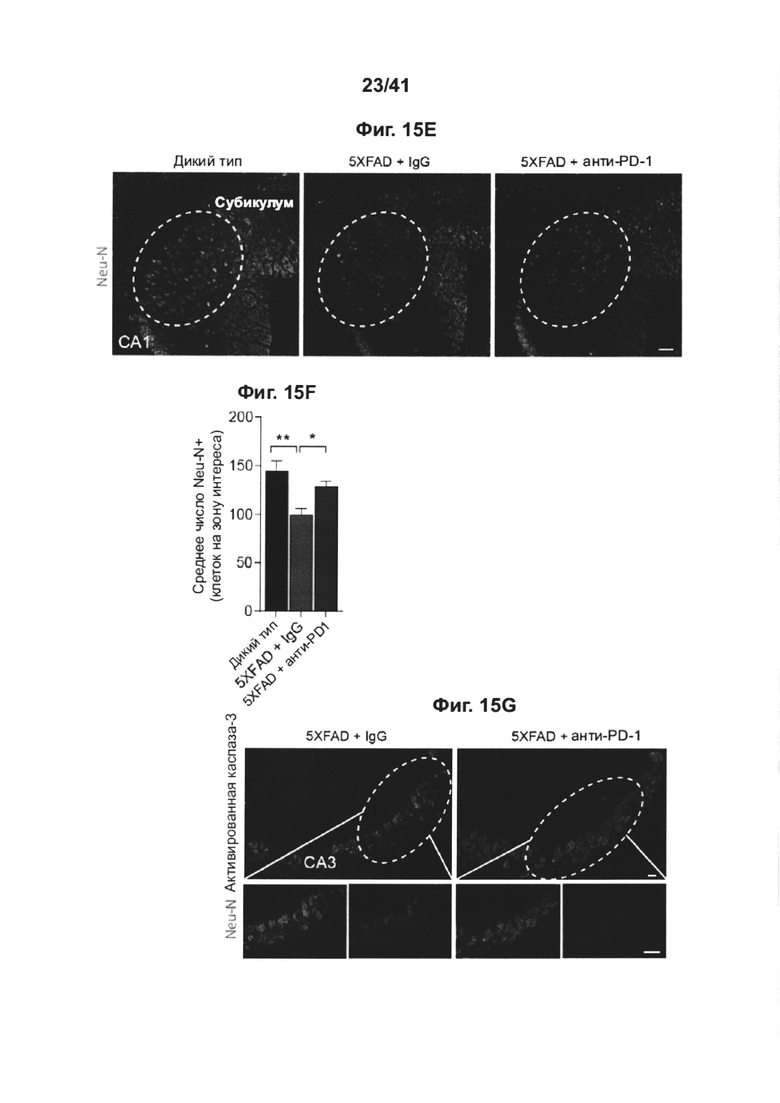
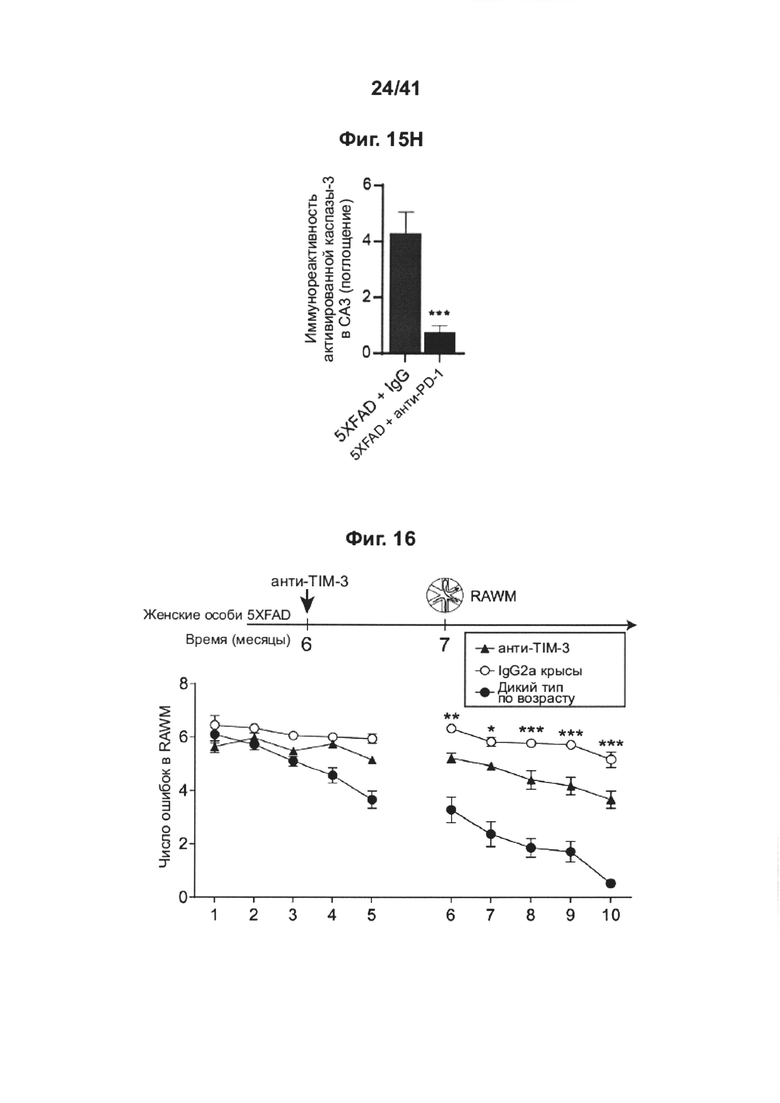
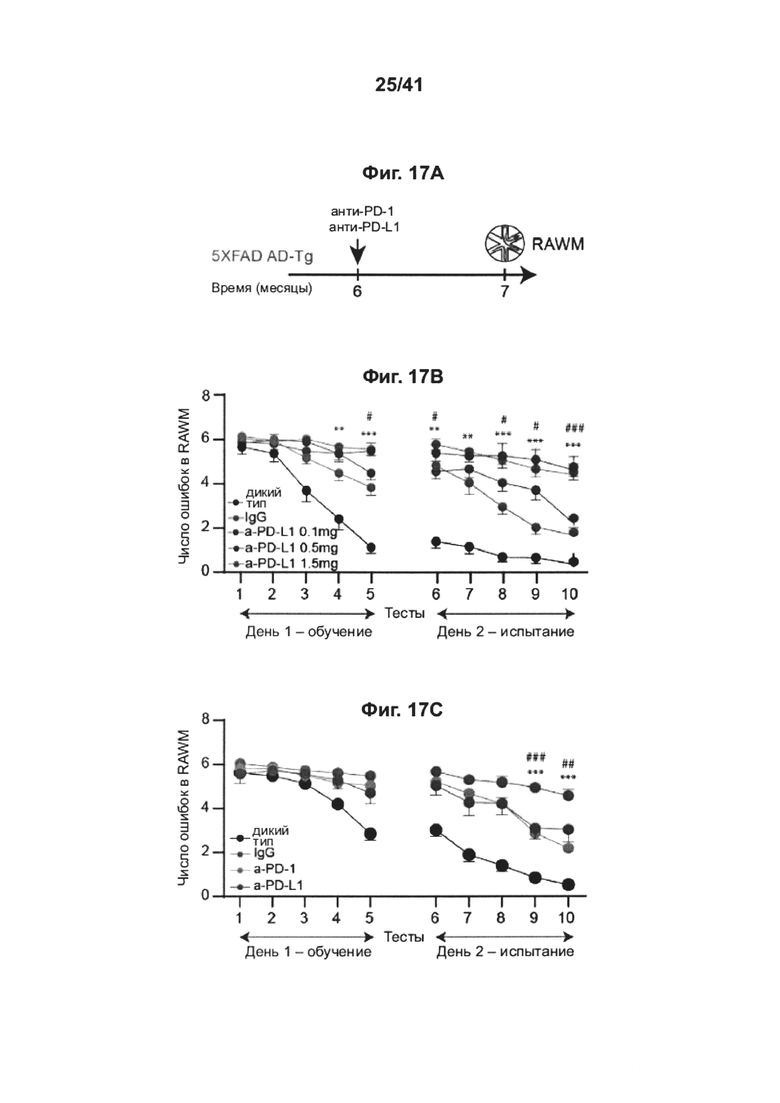
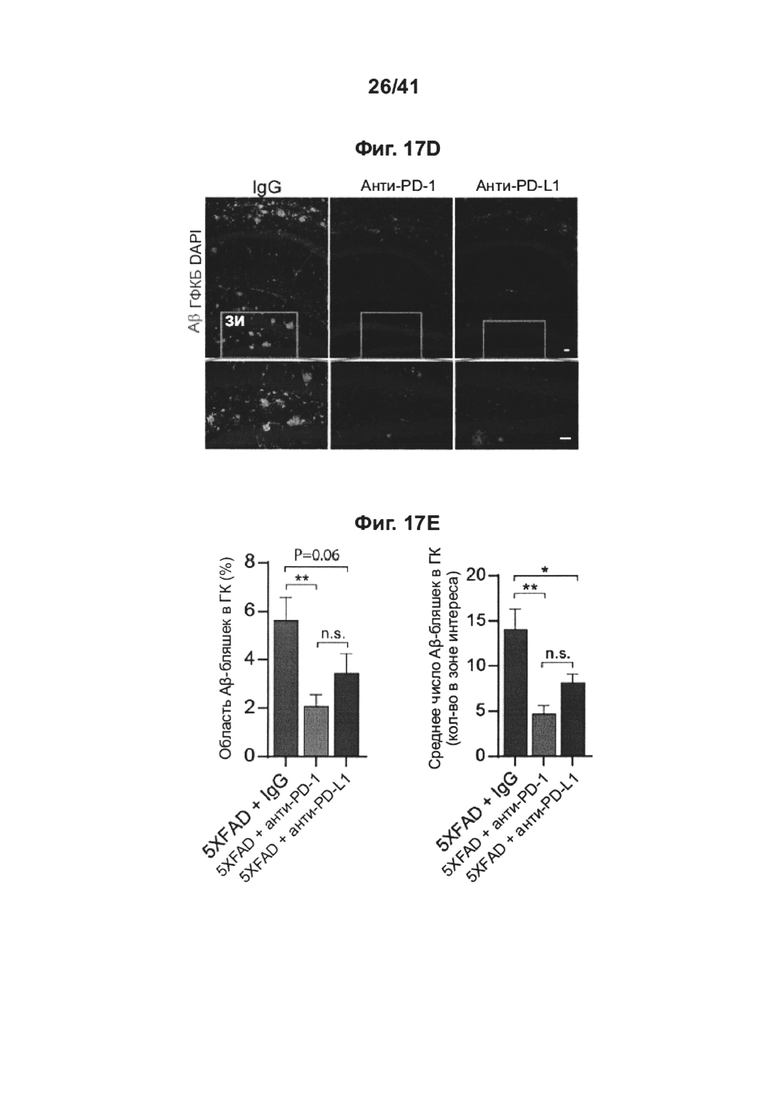
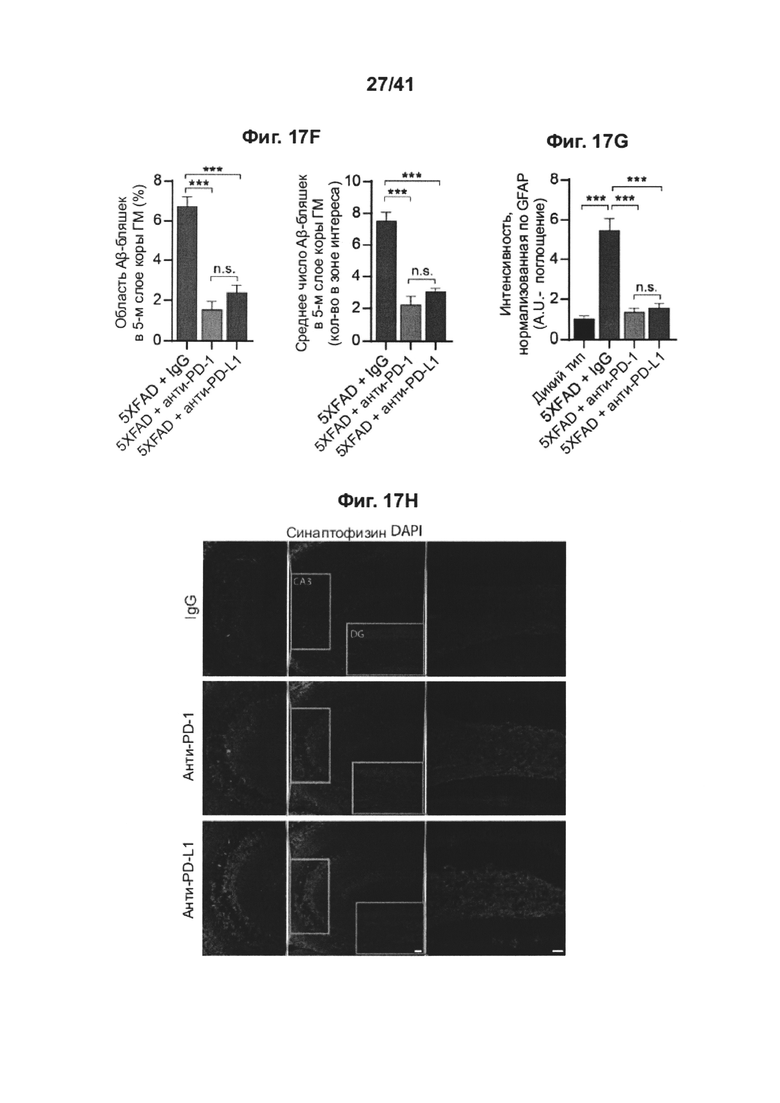
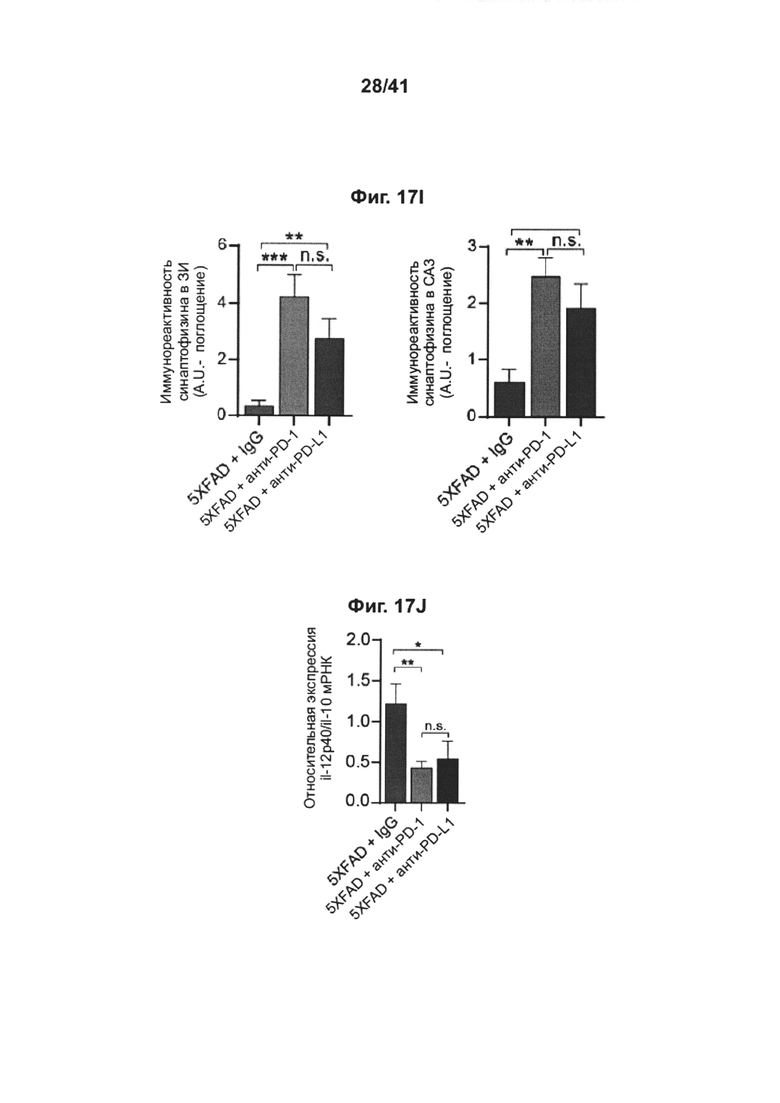
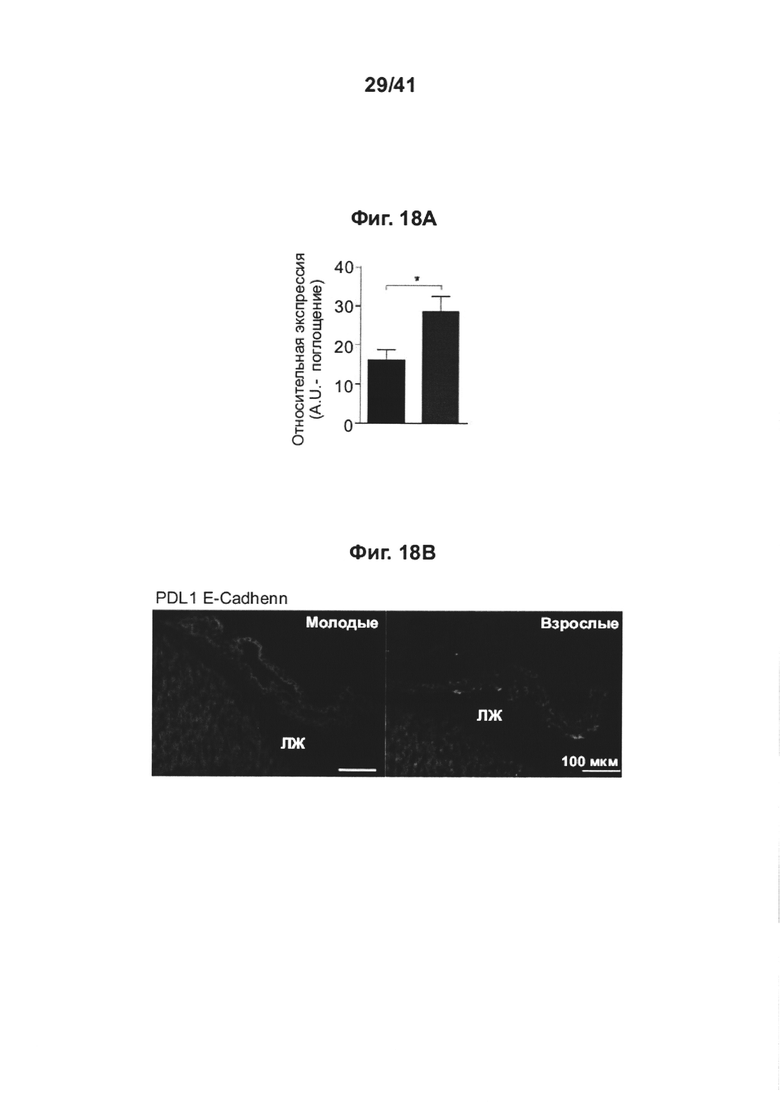
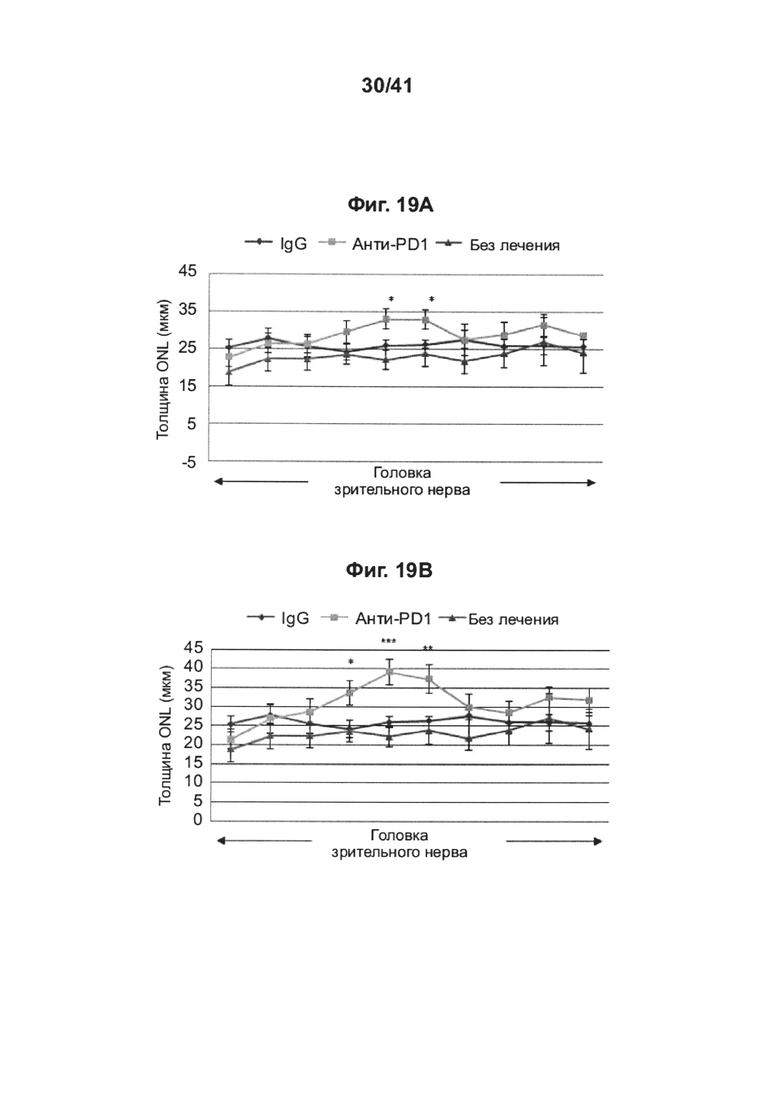
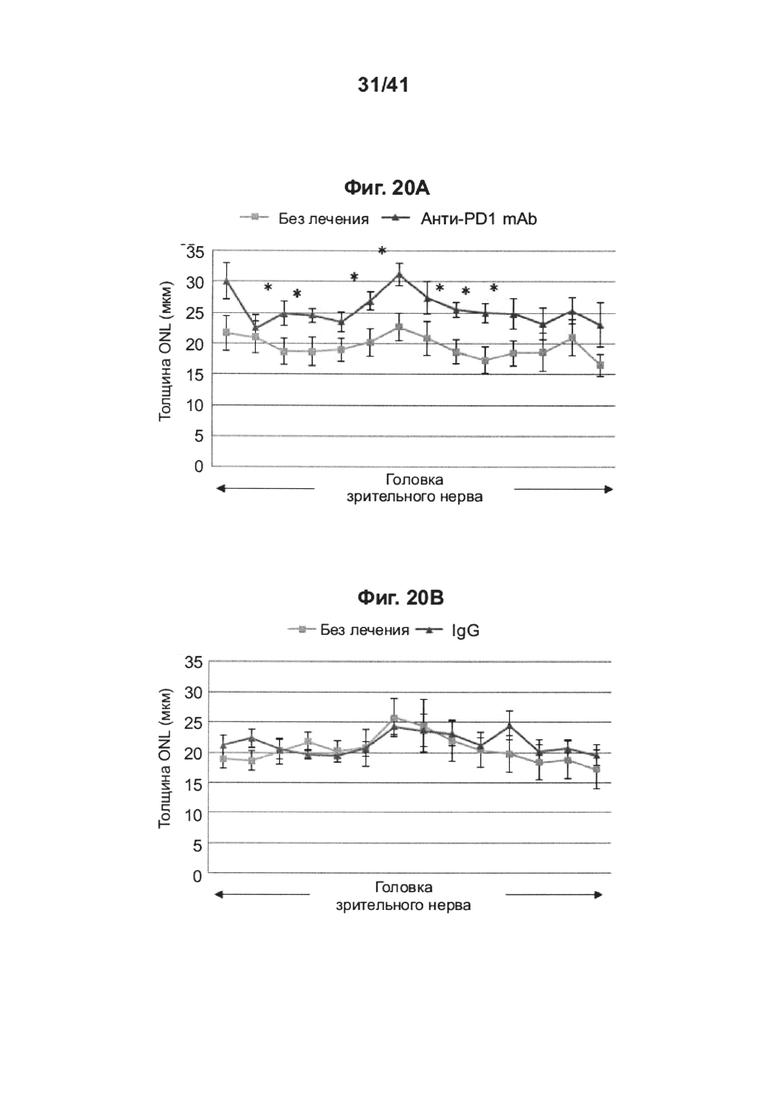
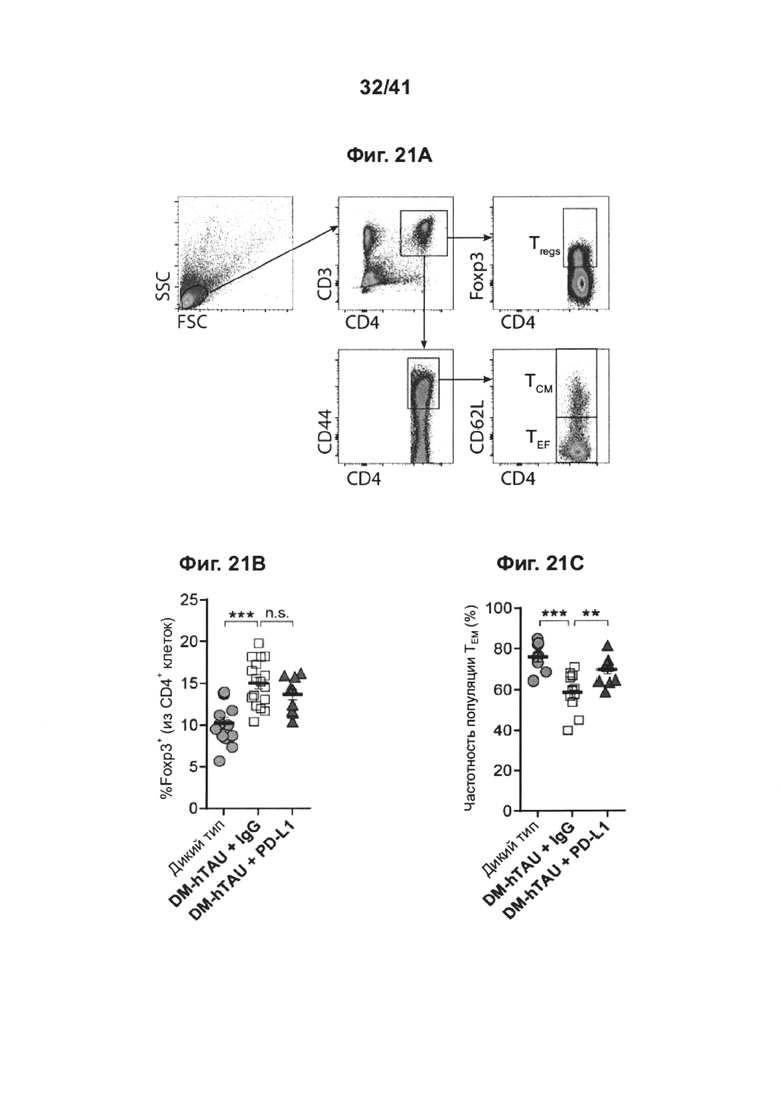
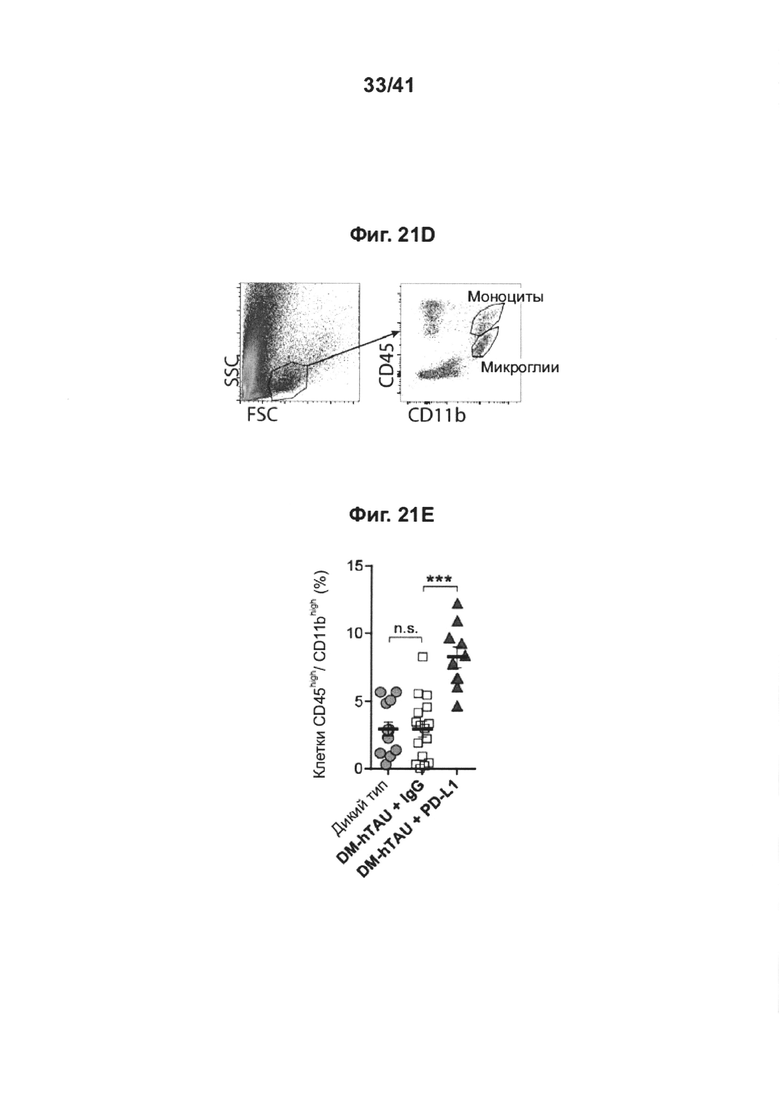
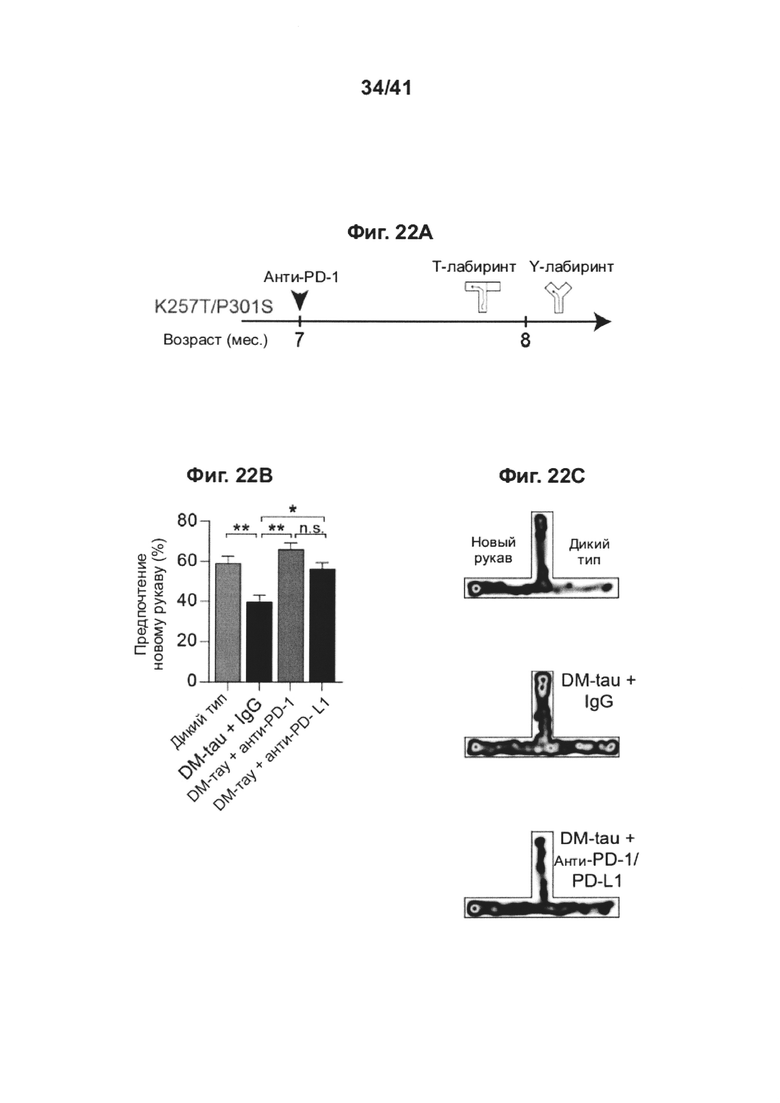
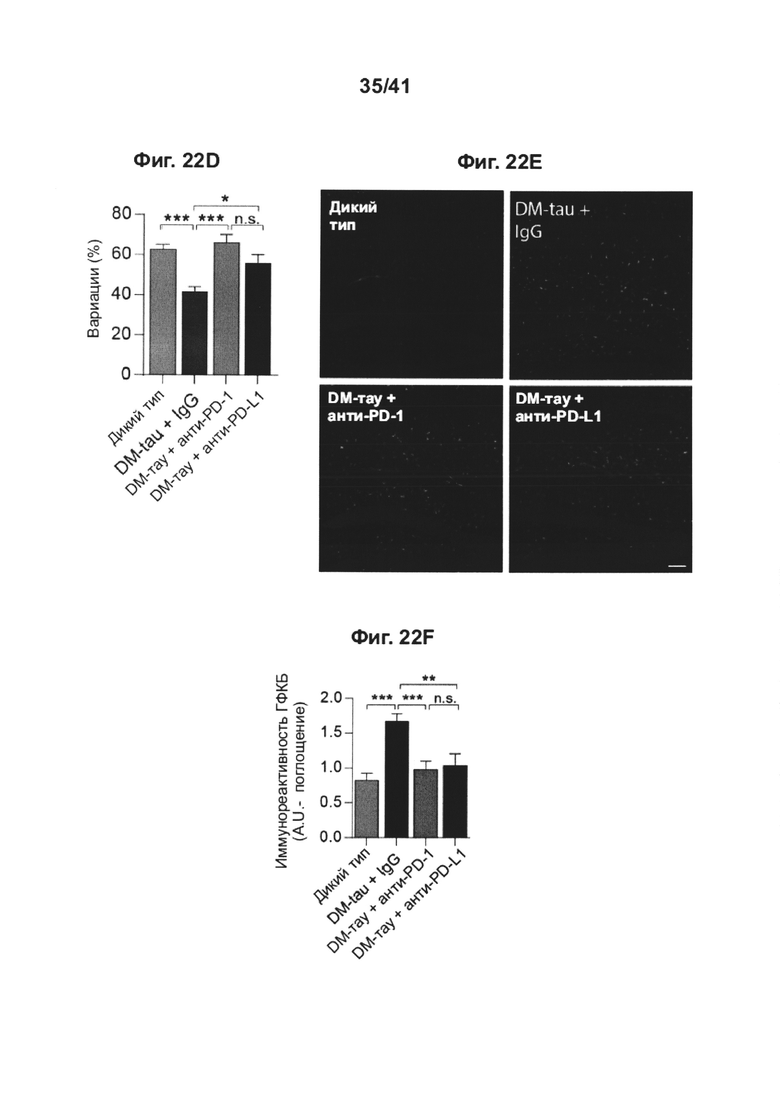
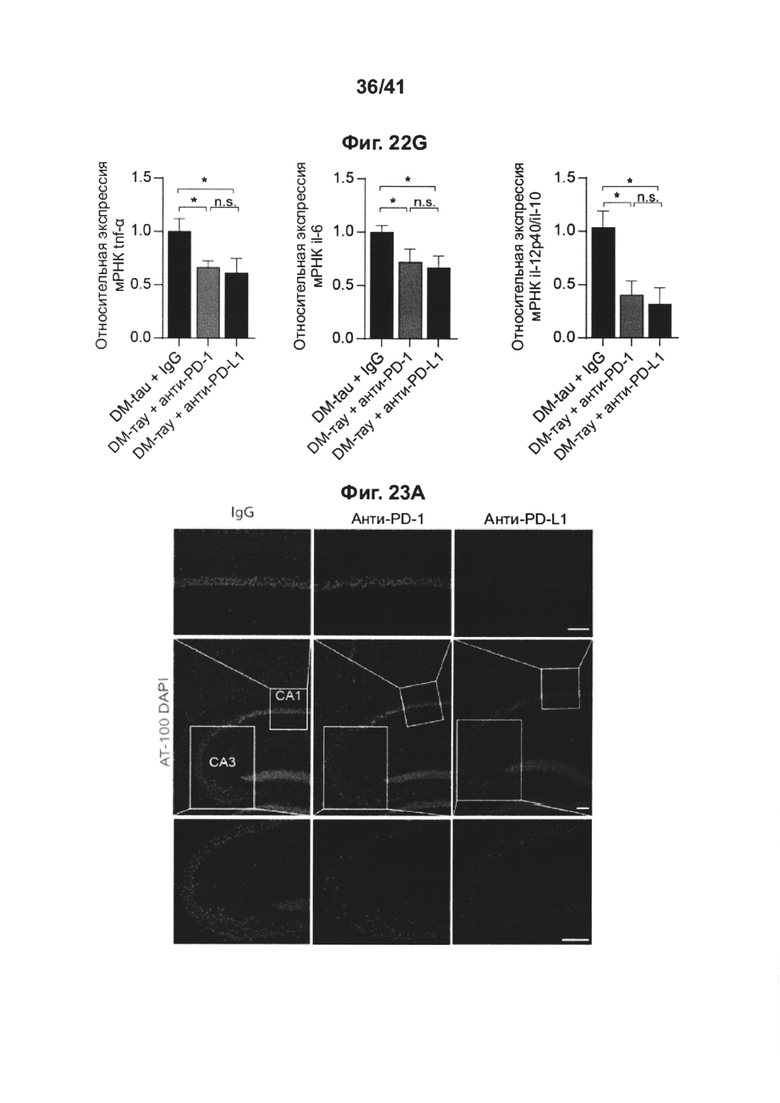
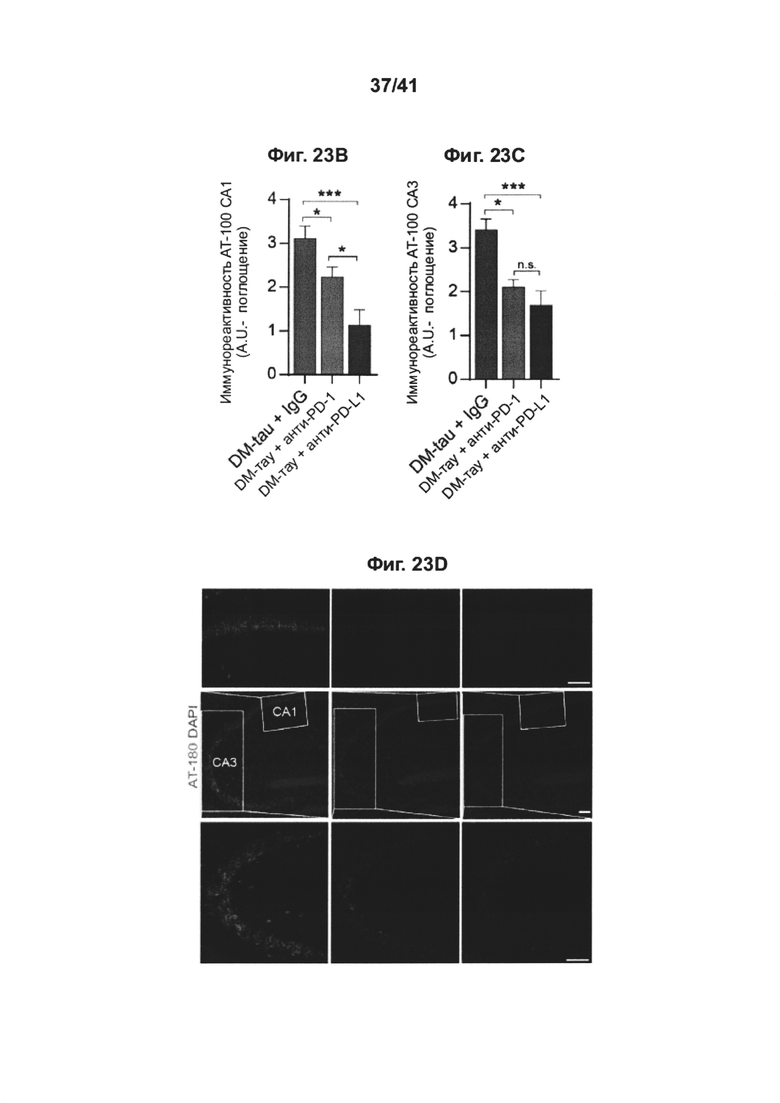
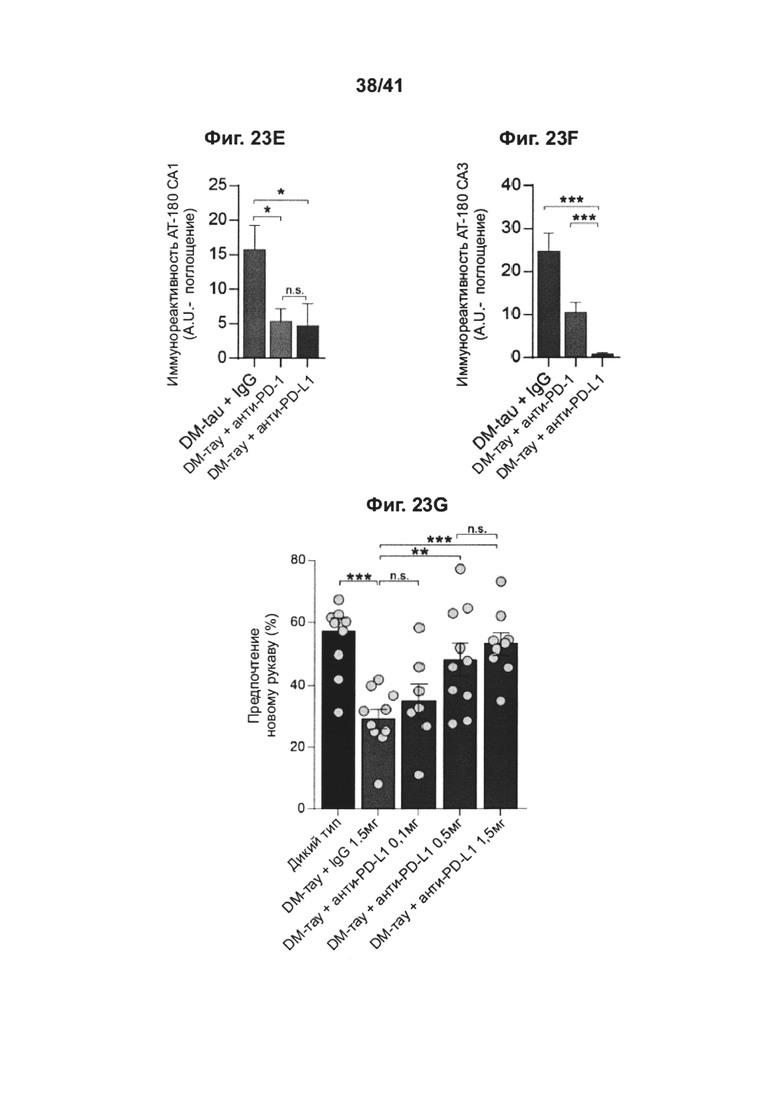
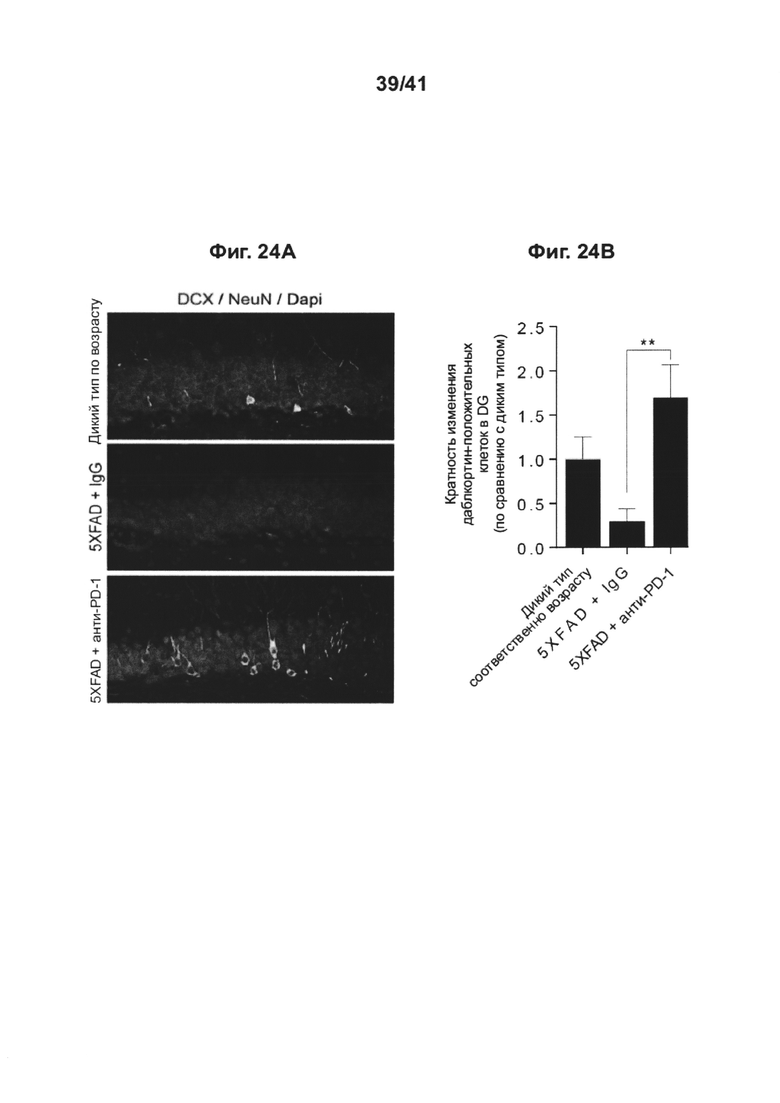
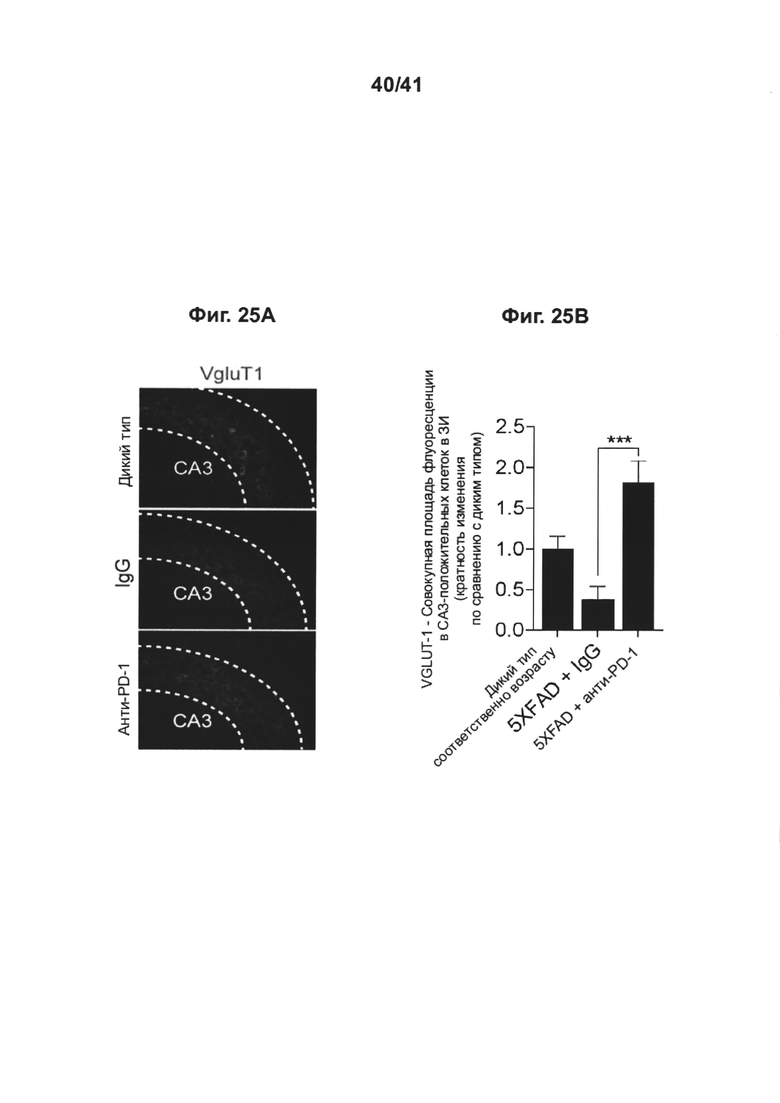
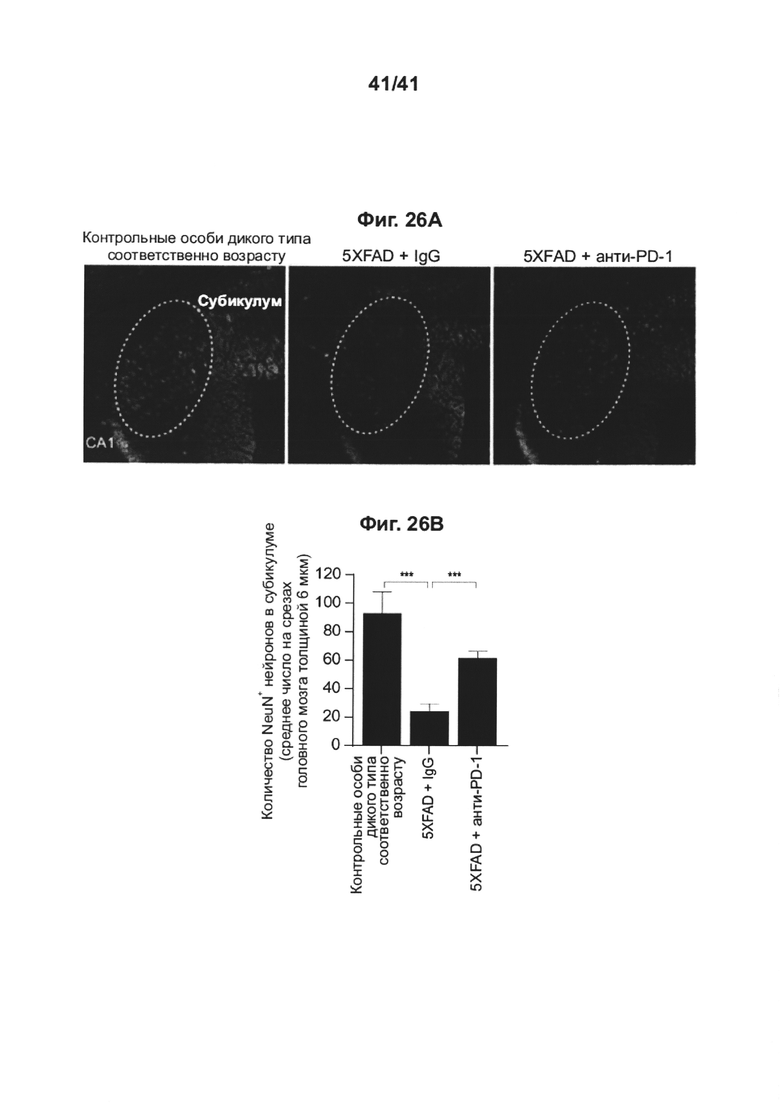
Изобретение относится к медицине, а именно к неврологии, и может быть использовано для снижения системных уровней или активности регуляторных Т-клеток для лечения заболевания и поражения ЦНС. Способ включает введение индивидууму фармацевтической композиции, содержащей анти-PD-1-антитело, анти-PDL1-антитело или анти-TIM-3-антитело. Фармацевтическую композицию вводят с помощью режима дозирования, включающего по меньшей мере два курса лечения, каждый курс лечения включает последовательно сеанс лечения, при котором фармацевтическую композицию вводят индивидууму, и последующий сеанс без лечения, когда фармацевтическую композиция не вводят индивидууму. Введение фармацевтической композиции во время сеанса лечения является однократным введением, и сеанс без лечения составляет от одного месяца до трех месяцев. Использование изобретения позволяет осуществить лечение таупатии за счет временного снижения уровня системной иммуносупрессии в системе кровообращения. 4 з.п. ф-лы, 4 табл., 107 ил., 22 пр.
1. Способ лечения таупатии у индивидуума, который нуждается в этом, содержащий введение индивидууму фармацевтической композиции, содержащей анти-PD-1-антитело, анти-PDL1-антитело или анти-TIM-3-антитело, где таупатия выбрана из болезни Пика, глобулярной глиальной таупатии, аргирофильной зерновой болезни, хронической травматической энцефалопатии и первичной возрастной таупатии (PART), ранее известной как деменция с преобладанием нейрофибриллярных клубков (NFT-деменция), где фармацевтическую композицию вводят с помощью режима дозирования, включающего по меньшей мере два курса лечения, каждый курс лечения включает последовательно сеанс лечения, при котором фармацевтическую композицию вводят индивидууму, и последующий сеанс без лечения, когда фармацевтическую композицию не вводят индивидууму, введение фармацевтической композиции во время сеанса лечения является однократным введением, и сеанс без лечения составляет от одного месяца до трех месяцев.
2. Способ по п. 1, где анти-PD-1-антитело представляет собой нейтрализующее анти-PD-1-антитело, анти-PD-L1-антитело представляет собой нейтрализующее анти-PDL1-антитело или анти-TIM-3-антитело представляет собой нейтрализующее анти-TIM-3-антитело.
3. Способ по п. 2, где анти-PD-1-антитело представляет собой нейтрализующее человеческое анти-PD-1-антитело или гуманизированное нейтрализующее анти-PD-1-антитело.
4. Способ по п. 2, где анти-PD-L1-антитело представляет собой нейтрализующее человеческое анти-PD-L1-антитело или гуманизированное нейтрализующее анти-PD-L1-антитело.
5. Способ по п. 2, где анти-TIM-3-антитело представляет собой нейтрализующее человеческое анти-TIM-3-антитело или гуманизированное нейтрализующее анти-TIM-3-антитело.
| WO 2015136541 A2, 17.09.2015 | |||
| US 9326999 B2, 03.05.2016 | |||
| ПРИМЕНЕНИЕ АНТИТЕЛА ПРОТИВ АМИЛОИДА-БЕТА ПРИ ГЛАЗНЫХ ЗАБОЛЕВАНИЯХ | 2008 |
|
RU2482876C2 |
| GRINBERG L.T | |||
| et al., Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation | |||
| Acta Neuropathol | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
