ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
1. По всему тексту данной заявки ссыпки на различные публикации приводятся в скобках с указанием имени автора и даты, или серийного номера патента или серийного номера патентной публикации. Описания данных публикаций, таким образом, включены посредством ссылки во всей своей полноте в данную заявку для того, чтобы более полно описать уровень техники, известный специалистам в данной области, на дату осуществления настоящего изобретения, описанного и заявленного в настоящем документе. Тем не менее, цитирование ссылки в настоящем описании не должно быть истолковано как признание того, что такая ссылка представляет известный уровень техники по отношению к настоящему изобретению.
Область техники изобретения
2. Данное изобретение относится к способам лечения глиомы у субъекта, включающим введение субъекту антитела против белка программируемой смерти-1 (PD-1). В некоторых вариантах осуществления данное изобретение относится к способам лечения глиомы у субъекта, включающим введение субъекту комбинации антитела против белка программируемой смерти-1 (PD-1) и другого противоракового средства, такого как антитело против антигена-4 цитотоксических Т-лимфоцитов ((cytotoxic T-Lymphocyte Antigen-4 (CTLA-4)). В некоторых вариантах осуществления глиома представляет собой глиобластому (GBM).
Предпосылки создания изобретения
3. Глиомы представляют собой новообразования или опухоли, которые возникают в головном или спинном мозге, чаще всего в головном мозге. Глиомы названы по типу клеток, с которыми они имеют общие гистопатологические признаки. GBM является наиболее частой первичной опухолью мозга у взрослых и составляет примерно от 60 до 70% всех злокачественных глиом. (Wen P.Y. et al., N Engl J Med. 359(5):492-507 (2008)). GBM связаны с особенно агрессивным клиническим течением с высокой заболеваемостью и смертностью. В одних только Соединенных Штатах ежегодно диагностируется около двенадцати-четырнадцати тысяч новых случаев множественной формы GBM.
4. Медианное выживание с GBM без лечения составляет 4 месяца. Несмотря на стандартное лечение первой линии с помощью нейрохирургии, лучевой терапии (RT) и/или темозоломида, медианная выживаемость составляет приблизительно от 12 до 15 месяцев, и почти во всех случаях происходят рецидивы. (Chang S.M. at al. Neurosurg Focus. 20(4): E4 Review (2006); Johnson, D.R. и O'Neill, B.P., J. Neurooncol. 107(2):359-64 (2012)) Авастин (бевацизумаб) представляет собой антиангиогенный терапевтический препарат, который был одобрен для лечения рецидивирующей GBM. Тем не менее, предварительные исследования показали, что, хотя лечение авастином увеличивает выживаемость без прогрессирования, оно не повышает, в сколь-либо значимой степени, общую выживаемость в случае вновь диагностированной GBM по сравнению со стандартным лечением, когда авастин используется в сочетании со стандартными схемами лечения (лучевая терапия плюс темозоломид). (Chinot et al., N Engl J Med 370:709-722 (2014); Gilbert et al., N Engl J Med 370:699-708 (2014)) Кроме того, исследования показали, что у пациентов, получавших бевацизумаб, происходило ухудшение когнитивных функций и качества жизни. (См http://www.mdanderson.org/newsroom/news-releases/2013/patients-treated-with-bevacizumab.html, последнее визит 15 декабря 2015 года) Таким образом, не существует никакого известного способа вылечивания GBM. Кроме того, GBM особенно трудно поддается лечению, поскольку некоторые препараты не могут проходить через гематоэнцефалический барьер. Таким образом, существует настоятельная потребность в новых лечебных вмешательствах для повышения выживаемости и качества жизни пациентов, страдающих от GBM.
месяца. Несмотря на стандартное лечение первой линии с помощью нейрохирургии, лучевой терапии (RT) и/или темозоломида, медианная выживаемость составляет приблизительно от 12 до 15 месяцев, и почти во всех случаях происходят рецидивы. (Chang S.M. at al. Neurosurg Focus. 20(4): E4 Review (2006); Johnson, D.R. и O'Neill, B.P., J. Neurooncol. 107(2):359-64 (2012)) Авастин (бевацизумаб) представляет собой антиангиогенный терапевтический препарат, который был одобрен для лечения рецидивирующей GBM. Тем не менее, предварительные исследования показали, что, хотя лечение авастином увеличивает выживаемость без прогрессирования, оно не повышает, в сколь-либо значимой степени, общую выживаемость в случае вновь диагностированной GBM по сравнению со стандартным лечением, когда авастин используется в сочетании со стандартными схемами лечения (лучевая терапия плюс темозоломид). (Chinot et al., N Engl J Med 370:709-722 (2014); Gilbert et al., N Engl J Med 370:699-708 (2014)) Кроме того, исследования показали, что у пациентов, получавших бевацизумаб, происходило ухудшение когнитивных функций и качества жизни. (См http://www.mdanderson.org/newsroom/news-releases/2013/patients-treated-with-bevacizumab.html, последнее визит 15 декабря 2015 года) Таким образом, не существует никакого известного способа вылечивания GBM. Кроме того, GBM особенно трудно поддается лечению, поскольку некоторые препараты не могут проходить через гематоэнцефалический барьер. Таким образом, существует настоятельная потребность в новых лечебных вмешательствах для повышения выживаемости и качества жизни пациентов, страдающих от GBM.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
5. Настоящее изобретение относится к способу лечения субъекта, пораженного глиомой, включающему введение субъекту терапевтически эффективного количества антитела или его антигенсвязывающей части, которое специфически связывается с рецептором запрограммированной смерти-1 (PD-1) и ингибирует активность PD-1 ("анти-PD-1-антитело, антитело против PD-1"). В одном варианте осуществления глиома выбрана из группы, состоящей из астроцитомы, глиомы ствола головного мозга, эпендимомы, смешанной глиомы, олигодендроглиомы, зрительной нервной глиомы или любой их комбинации. В другом варианте осуществления глиома представляет собой GBM (глиобластому).
6. В других вариантах осуществления антитело против PD-1 перекрестно конкурирует (одно препятствует связыванию или модулирующему эффекту другого) с ниволумабом за связывание с PD-1 человека. В некоторых вариантах осуществления антитело против PD-1 представляет собой химерное, гуманизированное или человеческое моноклональное антитело против PD-1. В одном варианте осуществления антитело против PD-1 представляет собой ниволумаб. В другом варианте осуществления антитело против PD-1 представляет собой пембролизумаб.
7. В некоторых вариантах осуществления субъект дополнительно получает противораковую терапию одновременно с введением, до или после введения антитела против PD-1. В некоторых вариантах осуществления противораковая терапия включает введение противоракового средства. В одном варианте осуществления противораковая терапия включает лучевую терапию. В другом варианте осуществления противораковая терапия включает хирургическое вмешательство. В следующем варианте осуществления хирургия включает MRI-управляемую лазерную абляцию.
8. В некоторых вариантах осуществления противораковое средство содержит второе антитело или его антигенсвязывающую часть. В одном варианте осуществления второе антитело или его антигенсвязывающая часть специфически связывается с цитотоксическим Т-лимфоцитарным антигеном-4 (CTLA-4) и ингибирует активность CTLA-4 ("антитело против CTLA-4, антитело против CTLA-4"). В другом варианте осуществления антитело против CTLA-4 перекрестно конкурирует с ипилимумабом за связывание с CTLA-4 человека. В других вариантах осуществления антитело против CTLA-4 представляет собой химерное, гуманизированное или человеческое моноклональное антитело. В одном варианте осуществления антитело против CTLA-4 представляет собой ипилимумаб.
9. В некоторых вариантах осуществления антитело против PD-1 вводят в виде монотерапии в дозе около 3 мг/кг массы тела. В других вариантах осуществления антитело против PD-1 вводят в дозе около 3 мг/кг массы тела в комбинации с антителом против CTLA-4, которое вводят в дозе около 1 мг/кг массы тела.
10. В определенных вариантах осуществления настоящее изобретение относится к любому раскрытому в настоящем описании способу, включающему: 1) индукционную фазу, в которой антитела против PD-1 и против CTLA-4 вводят в комбинации в 2, 4, 6, 8 или 10 дозах, причем каждая доза варьирует от по меньшей мере около 0,1 мг/кг до по меньшей мере около 10,0 мг/кг массы тела, при введении не реже одного раза приблизительно каждые 2, 3 или 4 недели; 2) с последующей поддерживающей фазой, в которой антитело против CTLA-4 не вводят и антитело против PD-1 повторно вводят в дозе в диапазоне от по меньшей мере около 0,1 мг/кг до по меньшей мере около 10 мг/кг при введении не реже одного раза приблизительно каждые 2, 3 или 4 недели.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
11. На фигуре 1 представлено схематическое изображение дизайна исследования когорты 1.
12. На фигуре 2 представлено схематическое изображение дизайна исследования когорты 1b.
13. На фигуре 3 представлено схематическое изображение дизайна исследования когорты 2.
14. На фигуре 4 показано состояние испытуемых (светлые столбики) из группы лечения N+I когорты 1 [ниволумаб (1 мг/кг) + ипилимумаб (3 мг/кг)] и состояние испытуемых (темные столбики) из группы лечения N когорты 1 [ниволумаб (3 мг/кг)]. Состояние испытуемого (субъекта) помечено как показано, включая стабильную болезнь (SD), псевдопрогрессию (PseudoPrg), прогрессирующую болезнь (PD), частичный ответ (PR) и смерть (D). Также указаны любые переходы из группы лечения N+I в группу лечения N (Переключение на N) или прекращение лечения из-за нежелательных явлений (D/C Тх АЕ).
15. На фигурах 5A-5D показаны МРТ-изображения головного мозга субъекта, отображающие псевдопрогрессию GBM после монотерапии ниволумабом. Изображения слева (фигуры 5А и 5В) были сделаны в двух разных сечениях до начала исследования. Изображения справа (фигуры 5С и 5D) были сделаны в аналогичных сечениях после 5 доз монотерапии ниволумабом. Субъект не получал одновременного лечения кортикостерои дами.
16. На фигурах 6А-6С показаны гистологические препараты резецированной опухолевой ткани, показывающие перивентрикулярную некротическую ткань (фигура 6А) и большие агрегаты иммунных клеток (фигура 6В) у субъекта, демонстрируя связанные с лечением изменения у пациента с GBM после монотерапии ниволумабом. На фигуре 6С показано МРТ-изображение головного мозга пациента.
17. На фигурах 7А-7С показаны гистологические препараты резецированной опухолевой ткани, показывающие связанные с лечением изменения, включая внутриопухолевые макрофаги (фигура 7А) и CD45-положительные клеточные агрегаты (фигура 7В), у субъекта, испытывающего псевдопрогрессирование GBM, на МРТ после монотерапии ниоблумабом. На фигуре 7С показано МРТ-изображение головного мозга пациента.
18. На фигурах 8А-8С показаны гистологические препараты резецированной опухолевой ткани, показывающие CD45-положительные (фигура 8А) и CD3-положительные клетки (фигура 8В) у субъекта, испытывающего псевдопрогрессирование GBM после монотерапии ниволумабом. МРТ-изображение головного мозга показано на фигуре 8С.
19. На фигурах 9А-9С показаны гистологические препараты резецированной опухолевой ткани, показывающие периваскулярную инфильтрацию (фигура 9А) и инфильтрирующие клетки, а также периваскулярную инфильтрацию CD45-положительных клеток (фигура 9В) у субъекта, испытывающего псевдопрогрессирование GBM после монотерапии ниоблумабом. МРТ-изображение головного мозга показано на фигуре 9С.
20. На фигурах 10А-10С показаны гистологические препараты резецированной опухолевой ткани, показывающие CD45-положительные клетки, выстилающие кровеносный сосуд опухоли (фигура 10А), а также периваскулярную инфильтрацию и инфильтрацию CD45-положительных клеток в опухоль (фигура 10 В) у субъекта, испытывающего псевдопрогрессию GBM после монотерапии ниоблумабом. МРТ-изображение головного мозга показано на фигуре 10С.
21. На фигурах 11A-11D показаны МРТ-изображения головного мозга субъекта, у которого наблюдается частичный ответ после монотерапии ниволумабом. На фигуре 11А показано исходное состояние, и на фигуре 11В показан субъект после трех доз монотерапии ниволумабом. На фигурах 11С и 11D показаны МРТ-изображения головного мозга, полученные для двух разных временных точек последующего наблюдения, в то время как субъект продолжал получать ниволумаб.
22. На фигуре 12 показан дизайн исследования клинического испытания, сравнивающего ниволумаб и темозоломид в комбинации с лучевой терапией.
23. На фигуре 13 показан график лечения, включая сбор проб.
24. Фигура 14 показывает анализ Т-клеток периферической крови и опухоль-инфильтрирующих Т-клеток при подозрении на прогрессирование заболевания у пациентов, получающих монотерапию ниволумабом.
25. Фигура 15А и 15В показывает профиль созревания CD4+ (А) и CD8+ (В) Т-лимфоцитов у пациентов, получавших ниволумаб ± ипилимумаб.
26. Фигура 16 показывает временной ход CD8+ CD28- Т-клеточной пролиферации и активации у пациента, получавшего ниволумаб + ипилимумаб.
27. Фигура 17А и 17В показывает анти-ТАА-активность TILS (А) и РВМС (В) у пациента, получавшего монотерапию ниволумабом.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
28. Настоящее изобретение относится к способам лечения глиомы или пациента с глиомой, включающим введение пациенту антитела против PD-1. В некоторых вариантах осуществления настоящее изобретение относится к способам лечения глиомы антителом против PD-1. В некоторых вариантах осуществления антитело против PD-1 вводят с другим противораковым средством.
Термины
29. Для того чтобы настоящее раскрытие было более понятным, сначала определяются определенные термины. Как используется в данной заявке, за исключением случаев, специально оговоренных в настоящем документе, каждый из следующих терминов должен иметь значение, указанное ниже. Дополнительные определения приведены по тексту всей заявки.
30. Термин "и/или", если он используется в настоящем описании, должен рассматриваться как конкретное раскрытие каждого из двух указанных признаков или компонентов как с другим, так и без него. Таким образом, предполагается, что термин "и/или", используемый во фразе, такой как "А и/или В", включает "А и В", "А или В", "А" (по отдельности) и "В" (по отдельности). Аналогично, термин "и/или", используемый во фразе, такой как "А, В и/или С", предназначен для охвата каждого из следующих аспектов: А, В и С; А, В или С; А или С; А или В; В или С; А и С; А и В; В и С; А (по отдельности); В (по отдельности); и С (по отдельности).
31. Понятно, что везде, где аспекты настоящегоизобретения описаны с помощью термина "содержащий", возможно их альтернативное аналогичное описание с использованием терминов "состоящий из" и/или "состоящий в основном из".
32. Если не указано иное, все технические и научные термины, используемые в настоящем описании, имеют то же значение, которое обычно понимается специалистом в данной области техники, к которому относится данное раскрытие. Например, Concise Dictionary of Biomedicine and Molecular Biology, Juo, Pei-Show, 2nd ed., 2002, CRC Press; The Dictionary of Cell and Molecular Biology, 3rd ed., 1999, Academic Press; и the Oxford Dictionary Of Biochemistry And Molecular Biology, Revised, 2000, Oxford University Press, предоставляют специалисту общий словарь многих терминов, используемых в данном раскрытии.
33. Единицы, префиксы и символы обозначены в их принятой форме Systeme International de Unites (SI). Числовые диапазоны включают числа, определяющие диапазон. Заголовки, приведенные в настоящем описании, не являются ограничениями различных аспектов раскрытия, которые могут быть сделаны со ссылкой на спецификацию в целом. Соответственно, термины, определенные ниже, более полно определены посредством ссылки на спецификацию в целом.
34. "Введение" относится к физическому введению терапевтического средства субъекту с использованием любого из различных способов и систем доставки, известных специалистам в данной области. Типичные пути введения для антитела против PD-1 включают внутривенный, внутримышечный, подкожный, внутрибрюшинный, спинальный или другие парентеральные пути введения, например, путем инъекции или инфузии. В некоторых вариантах осуществления путь введения направлен непосредственно в мозг или центральную нервную систему. В вариантах осуществления введение в мозг или центральную нервную систему представляет собой интрацеребровентрикулярное (ICV), внутрижелудочковое, интратекальное (ИТ), интерстициальное, эпидуральное, внутримозговое или интраназальное (обонятельный путь) введение. Используемая в настоящем описании фраза "парентеральное введение" означает способы введения, отличающиеся от энтерального и местного введения, обычно путем инъекции, и включает в себя, но без ограничения, внутривенную, внутримышечную, внутриартериальную, интратекальную, внутрилимфатическую, внутриочаговую, интракапсулярную, интраорбитальную, внутрисердечную, внутрикожную, внутрибрюшинную, транстрахеальную, подкожную, подэпидермисную, внутрисуставную, субкапсулярную, субарахноидальную, интраспинальную, эпидуральную и внутригрудинную инъекцию и инфузию, а также электропорацию in vivo. В некоторых вариантах терапевтическое средство вводят непарентеральным путем или перорально. Другие непарентеральные пути включают топический, эпидермальный или слизистый путь введения, например, интраназально, вагинально, ректально, подъязычно или топикально. Введение также может выполняться, например, один раз, множество раз и/или в течение одного или более длительных периодов времени.
35. Термин "нежелательное явление" ("adverse event" (АЕ)), при использовании в настоящем описании, означает любое неблагоприятное и обычно непредвиденное или нежелательное явление (включая отклоняющиеся от нормы лабораторные данные), симптом или заболевание, связанное с осуществлением лечения. С лечением могут быть связаны одно или несколько АЕ, и уровень тяжести отдельных АЕ может быть одинаковым или различным. Ссылка на способы, способные "изменять нежелательные явления", означает схему лечения, которая уменьшает частоту и/или тяжесть одного или нескольких АЕ, связанных с применением другой схемы лечения.
36. Термин "антитело" (Аb) включает в себя, без ограничения, гликопротеиновый иммуноглобулин, который специфически связывается с антигеном и содержит по меньшей мере две тяжелые (Н) цепи и две легкие (L) цепи, взаимосвязанные дисульфидными связями, или его антигенсвязывающую часть. Каждая Н-цепь содержит вариабельную область тяжелой цепи (сокращено называемую в настоящем описании как VH) и константную область тяжелой цепи. Константная область тяжелой цепи состоит из трех константных доменов, CH1, CH2 и СH3. Каждая легкая цепь содержит вариабельную область легкой цепи (сокращено называемую в настоящем описании как VL) и константную область легкой цепи. Константная область легкой цепи содержит один константный домен, CL. Области VH и VL могут быть дополнительно подразделены на области гипервариабельности, называемые гипервариабельными областями (CDR), перемежающиеся с областями, которые являются более консервативными, называемыми каркасными областями (FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных от амино-конца к карбокси-концу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Вариабельные области тяжелой и легкой цепей содержат домен связывания, который взаимодействует с антигеном. Константные области антител могут опосредовать связывание иммуноглобулина с тканями хозяина или факторами, включая различные клетки иммунной системы (например, эффекторные клетки) и первый компонент (C1q) классической системы комплемента.
37. Иммуноглобулин может происходить из любого из широко известных изотипов, включая, но не ограничиваясь ими, IgA, секреторный IgA, IgG и IgM. IgG-подклассы также хорошо известны специалистам в данной области техники и включают, но не ограничиваются ими, человеческие IgG1, IgG2, IgG3 и IgG4. "Изотип" относится к классу или подклассу Аb (например, IgM или IgG1), который кодируется генами константной области тяжелой цепи. Термин "антитело" включает в себя, в качестве примера, как природные, так и не встречающиеся в природе Аb; моноклональные и поликлональные Аb, химерные и гуманизированные Аb; человеческие или нечеловеческие Аb; полностью синтетические Аb; и одноцепочечные Аb. Нечеловеческое Аb можно гуманизировать рекомбинантными способами для уменьшения его иммуногенности у человека. Там, где прямо не указано, и если из контекста не следует иное, термин "антитело" также включает в себя антигенсвязывающий фрагмент или антигенсвязывающую часть любого из вышеупомянутых иммуноглобулинов и включает в себя моновалентный и двухвалентный фрагмент или часть и одноцепочечное Аb.
38. "Выделенное антитело" относится к Аb, которое по существу не содержит других Аb, имеющих отличающиеся антигенные специфичности (например, выделенное Аb, которое специфически связывается с PD-1, является по существу свободным от Аb, которые специфически связывают антигены, отличающиеся от PD-1). Однако, выделенное Аb, которое специфически связывает PD-1, может иметь перекрестную реактивность с другими антигенами, такими как молекулы PD-1 из других видов. Более того, выделенное Аb может быть по существу свободным от другого клеточного материала и/или химических веществ.
39. Термин "моноклональное антитело" ("mAb") относится к не встречающемуся в природе препарату молекул Ab единого молекулярного состава, т.е. молекул Ab, чьи первичные последовательности являются по существу идентичными, и который проявляет единственную специфичность связывания и аффинность в отношении конкретного эпитопа. МAb является примером выделенного Ab. МAb могут быть получены гибридомными, рекомбинантными, трансгенными или другими способами, известными специалистам в данной области техники.
40. "Человеческое" антитело (HuMAb) относится к Ab, имеющему вариабельные области, в которых как каркасные, так и CDR-области получены из последовательностей иммуноглобулина зародышевой линии человека. Кроме того, если Ab содержит константную область, то константная область также получена из последовательностей иммуноглобулина зародышевой линии человека. Человеческие антитела по настоящему изобретению могут включать в себя аминокислотные остатки, не кодируемые последовательностями иммуноглобулина зародышевой линии человека (например, мутации, введенные случайным или сайт-специфическим мутагенезом in vitro или путем соматической мутации in vivo). Однако, термин "человеческое антитело", при использовании в данном описании, не предназначен для включения антител, в которых CDR-последовательности, полученные из зародышевой линии другого вида млекопитающего, такого как мышь, были привиты на каркасные последовательности человека. Термины "человеческие" антитела и "полностью человеческие" антитела используются как синонимы.
41. "Гуманизированное антитело" относится к Ab, в котором некоторые, большинство или все аминокислоты вне CDR-областей нечеловеческого Ab замещены соответствующими аминокислотами, происходящими из человеческих иммуноглобулинов. В одном из вариантов осуществления гуманизированной формы Ab некоторые, большинство или все аминокислоты вне CDR-областей замещены аминокислотами из иммуноглобулинов человека, в то время как некоторые, большинство или все аминокислоты в одной или более CDR-областей остаются неизменными. Небольшие добавки, делеции, вставки, замены или модификации аминокислот допустимы до тех пор, пока они не аннулирует способность Ab связываться с конкретным антигеном. "Гуманизированное" Ab сохраняет антигенную специфичность, аналогичную таковой исходного Ab.
42. "Химерное антитело" относится к Ab, в котором вариабельные области получены из одного вида и константные области получены из другого вида, например, Ab, в котором вариабельные области получены из мышиного Ab и константные области получены из человеческого Ab.
43. "Анти-антиген"-Ab относится к Ab, которое специфически связывается с антигеном. Например, анти-PD-1-Ab специфически связывается с PD-1 и анти-СТLА-4-Ab специфически связывается с CTLA-4.
44. "Антигенсвязывающая часть" Ab (также называемая "антигенсвязывающий фрагмент") относится к одному или более фрагментам Ab, которые сохраняют способность специфически связываться с антигеном, связывающимся с целым Ab.
45. "Рак" относится к обширной группе различных заболеваний, характеризующихся неконтролируемым ростом аномальных клеток в организме. Нерегулируемое деление и рост клеток приводит к образованию злокачественных опухолей, которые вторгаются в соседние ткани, а также могут метастазировать в отдаленные части тела через лимфатическую систему или кровоток.
46. Термин "глиома" относится к опухоли, которая возникает в головном или спинном мозге, чаще всего, в головном мозге. В некоторых вариантах осуществления глиома является астроцитомой, глиомой ствола головного мозга, эпендимомой, смешанной глиомой, олигодендроглиомой или зрительной нервной глиомой. В определенных вариантах осуществления глиома включает в себя, но не ограничиваются ими, акустическую неврому, например, астроцитому: I степени - волосовидную астроцитому, II степени - фибриллярную астроцитому, III степени - анапластическую астроцитому, IV степени - глиобластому (GBM). Другие типы глиомы включают, но не ограничиваются ими, хордому, лимфому ЦНС, краниофарингиому, стволовую глиому мозга, эпендимому, смешанную глиому, оптическую нервную глиому, субэндемимому, медуллобластому, менингиому, метастатические опухоли головного мозга, олигодендроглиому, гипофизарные опухоли, примитивные нейроэктодермальные (PNET), шванному, ювенильную пилоцитарную астроцитому (JPA), опухоль шишковидной железы и/или рабдоидную опухоль.
47. В некоторых вариантах осуществления GBM представляет собой GBM с O-6-метилгуанин-ДНК-метилтрансферазой (MGMT) (MGMT GBM). В некоторых вариантах осуществления MGMT GBM представляет собой GBM с метилированным геном MGMT. В других вариантах осуществления MGMT GBM представляет собой GBM с неметилированным геном MGMT. В некоторых вариантах осуществления антитело против PD-1 для настоящих способов используют для лечения субъекта, который не прошел стандартную терапию, например, алкилирующий агент, например, темозоломид, или для лечения субъекта, который идентифицирован как имеющий тип GBM, который не реагирует на стандартную терапию, например, алкилирующий агент, например, темозоломид. Субъекты, имеющие GBM с неметилированным геном MGMT (опухолевая O-6-метилгуанин-ДНК-метилтрансфераза), представляют популяцию GBM-пациентов с самой высокой неудовлетворенной медицинской потребностью, поскольку такие субъекты менее чувствительны к алкилирующим агентам. Это связано с тем, что белок, экспрессируемый у субъектов, имеющих неметилированный ген MGMT, может восстанавливать повреждение ДНК в опухолях, вызванное алкилирующими агентами, например, темозоломидом, в то время белок, экспрессируемый у субъектов, имеющих GBM с метилированным геном MGMT, не может восстанавливать повреждение ДНК в опухоли и, таким образом, может сделать опухоль более восприимчивой к алкилирующим агентам.
48. Термин "глиобластома", "мультиформная глиобластома" или "GBM" относится к типу рака, который представляет собой подмножество астроцитом, например, астроцитому IV степени. GBM включают варианты GBM: гигантоклеточную GBM и глиосаркому, а также четыре подтипа GBM: классическую, нейральную, пронейральную и мезенхимальную. GBM опухоли могут быть первичными (de novo) или вторичными.
49. "Антиген-4 цитотоксических Т-лимфоцитов (Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4))" относится к рецептору, ингибирующему иммунную систему, принадлежащему к семейству CD28. CTLA-4 экспрессируется исключительно на Т-клетках in vivo и связывается с двумя лигандами, CD80 и CD86 (также называемыми В7-1 и В7-2, соответственно). Термин "CTLA-4", используемый в настоящем описании, включает в себя человеческий CTLA-4 (hCTLA-4), варианты, изоформы и видовые гомологи hCTLA-4 и аналоги, имеющие по меньшей мере один общий эпитоп с hCTLA-4. Полная последовательность hCTLA-4 может быть найдена под номером доступа GenBank Accession No. ААВ59385.
50. Термин "иммунотерапия" относится к лечению субъекта, пораженного заболеванием или имеющего риск заражения или страдающего рецидивом заболевания, способом, включающим индуцирование, усиление, подавление или иным образом модифицирование иммунного ответа.
51. "Лечение" или "терапия" субъекта относится к любому типу выполняемого вмешательства или процесса, или введению активного средства субъекту с целью обращения вспять, облегчения, ослабления, ингибирования, замедления или предупреждения возникновения, прогрессирования, развития, тяжести или рецидива симптома, осложнения или состояния или биохимических показателей, связанных с заболеванием.
52. "Программируемая смерть-1 (Programmed Death-1, PD-1)" относится к рецептору, ингибирующему иммунную систему, принадлежащему к семейству CD28. PD-1 экспрессируется преимущественно на ранее активированных Т-клетках in vivo и связывается с двумя лигандами, PD-L1 и PD-L2, Термин "PD-1", при использовании в данном описании, включает в себя человеческий PD-1 (hPD-1), варианты, изоформы и видовые гомологи hPD-1 и его аналоги, имеющие по меньшей мере один общий эпитоп с hPD-1. Полную последовательность hPD-1 можно найти под номером доступа GenBank Accession No. U64863. "PD-1" и "PD-1-рецептор" используются в настоящем описании взаимозаменяемо.
53. "Лиганд-1 программируемой смерти (PD-L1)" является одним из двух присутствующих на клеточной поверхности гликопротеиновых лигандов для PD-1 (другой представляет собой PD-L2), который понижающе регулирует активацию Т-клеток и секрецию цитокинов после связывания с PD-1. Термин "PD-L1", при использовании в данном описании, включает в себя человеческий PD-L1 (hPD-L1), варианты, изоформы и видовые гомологи hPD-L1 и аналоги, имеющие по меньшей мере один общий эпитоп с hPD-L1. Полную последовательность hPD-L1 можно найти под номером доступа GenBank Accession No. Q9NZQ7.
54. "Субъект" включает в себя любого человека или любого животного, не являющегося человеком. Термин " животное, не являющееся человеком" включает в себя, но не ограничиваясь ими, позвоночных, таких как приматы (не человек), овцы, собаки, и грызунов, таких как мыши, крысы и морские свинки. В некоторых вариантах осуществления субъектом является человек. Термины "субъект" и "пациент" используются в данном описании взаимозаменяемо.
55. Термин "терапевтически эффективное количество" или "терапевтически эффективная доза" лекарственного или терапевтического средства представляет собой любое количество лекарственного средства, которое, при использовании отдельно или в комбинации с другим терапевтическим средством, защищает субъекта против наступления заболевания или способствует регрессии заболевания, что подтверждается уменьшением выраженности симптомов заболевания, увеличением частоты и продолжительности бессимптомных периодов заболевания, или предотвращением ухудшения или инвалидности вследствие недуга заболевания. Способность терапевтического средства содействовать регрессии заболевания может быть оценена с использованием различных способов, известных практикующему специалисту в данной области техники, например, у людей в ходе клинических испытаний, на животных модельных системах, прогнозирующих эффективность в организме человека, или путем анализа активности данного средства в анализах in vitro.
56. При использовании в данном описании, "субтерапевтическая доза" означает дозу терапевтического соединения (например, антитела), которая ниже, чем обычно, или типичную дозу терапевтического соединения при введении по отдельности для лечения гиперпролиферативного заболевания (например, рака).
57. В качестве примера, "противораковое средство" способствует регрессии рака у субъекта. В некоторых вариантах осуществления изобретения терапевтически эффективное количество лекарственного средства способствует регрессии рака до точки устранения рака. "Содействие регрессии рака" означает, что введение эффективного количества лекарственного средства, одного или в комбинации с противоопухолевым средством, приводит к снижению роста или размера опухоли, некрозу опухоли, уменьшению степени тяжести по меньшей мере одного симптома заболевания, увеличению частоты и продолжительности бессимптомных периодов заболевания или предотвращению ухудшения или инвалидности вследствие недуга заболевания. Кроме того, термины "эффективный" и "эффективность" в отношении лечения включают в себя как фармакологическую эффективность, так и физиологическую безопасность. Фармакологическая эффективность относится к способности препарата содействовать регрессии рака у пациента. Физиологическая безопасность относится к уровню токсичности или других нежелательных физиологических эффектов на клеточном уровне, на уровне органа и/или организма (нежелательные явления) в результате введения лекарственного средства.
58. В качестве примера лечения опухолей, терапевтически эффективное количество противоракового средства может ингибировать рост клеток или рост опухоли по меньшей мере на около 20%, по меньшей мере на около 40%, по меньшей мере на около 60% или по меньшей мере на около 80% по сравнению с субъектами, не получавшими лечение.
59. В других вариантах осуществления настоящего изобретения регрессия опухоли может проводить наблюдениеся и продолжаться в течение периода, составляющего по меньшей мере около 20 дней, по меньшей мере около 40 дней или по меньшей мере около 60 дней. Несмотря на такие конечные измерения терапевтической эффективности, оценка препаратов иммунотерапии также должна делать поправку на паттерны "иммунозависимого" ответа.
60. Паттерн "иммунозависимого" ответа относится к клинической картине ответа, часто наблюдаемой у онкологических пациентов, получавших иммунотерапевтические препараты, которые производят противоопухолевое действие путем индукции специфических по отношению к раку иммунных ответов или путем модификации природных иммунных процессов. Паттерн такого ответа характеризуется благоприятным терапевтическим эффектом, который следует за первоначальным увеличением опухолевой массы или появлением новых поражений, что, при оценке традиционных химиотерапевтических средств, классифицировалось бы как прогрессирование заболевания и было бы синонимом бездействия препарата. Соответственно, надлежащая оценка иммунотерапевтических препаратов может потребовать долгосрочного мониторинга воздействия данных средств на целевую болезнь.
61. Терапевтически эффективное количество лекарственного средства включает в себя "профилактически эффективное количество", которое представляет собой любое количество лекарственного средства, которое, при введении по отдельности или в комбинации с противоопухолевым средством субъекту с повышенным риском развития рака (например, субъекту, имеющему предзлокачественное состояние) или страдающему рецидивом рака, тормозит развитие или рецидив рака. В некоторых вариантах осуществления настоящего изобретения профилактически эффективное количество предотвращает развитие или рецидив рака полностью. "Ингибирование" развития или рецидива рака означает либо уменьшение вероятности развития рака или рецидива, либо предупреждение развития или рецидива рака полностью.
62. Использование термина "фиксированная доза" (flat dose), как используется в настоящем описании, означает дозу, которую вводят пациенту без учета массы или площади поверхности тела (body surface area (BSA)) пациента. Фиксированная доза, следовательно, не предусмотрена в виде мг/кг, а скорее, как абсолютное количество средства (например, антитела против PD-1). Например, человек с массой 60 кг и человек с массой 100 кг получали бы одну и ту же дозу терапевтического средства (например, 240 мг антитела против PD-1).
63. Термин "доза, основанная на массе", как указано в настоящем описании, означает, что доза, вводимая пациенту, рассчитывается на основе массы пациента. Например, когда пациенту с массой тела 60 кг требуется 3 мг/кг антитела против PD-1, можно вычислить и использовать соответствующее количество антитела против PD-1 (т.е. 180 мг) для введения.
64. Использование термина "фиксированная (постоянная) доза" (fixed dose) в отношении способа по настоящему изобретению означает, что два или более разных антитела в одной композиции присутствуют в композиции в конкретных (фиксированных) соотношениях друг с другом. В некоторых вариантах осуществления фиксированная (постоянная) доза основана на массе (например, мг) антител. В определенных вариантах осуществления фиксированная (постоянная) доза основана на концентрации (например, мг/мл) антител. В некоторых вариантах осуществления соотношение составляет по меньшей мере около 1:1, около 1:2, около 1:3, около 1:4, около 1:5, около 1:6, около 1:7, около 1:8, около 1:9, около 1:10, около 1:15, около 1:20, около 1:30, около 1:40, около 1:50, около 1:60, около 1:70, около 1:80, около 1:90, около 1:100, около 1:120, около 1:140, около 1:160, около 1:180, около 1:200, около 200:1, около 180:1, около 160:1, около 140:1, около 120:1, около 100:1, около 90:1, около 80:1, около 70:1, около 60:1, около 50:1, около 40:1, около 30:1, около 20:1, около 15:1, около 10:1, около 9:1, около 8:1, около 7:1, около 6:1, около 5:1, около 4:1, около 3:1 или около 2:1 мг первого антитела к мг второго антитела. Например, соотношение 3:1 первого антитела и второго антитела может означать, что флакон может содержать около 240 мг первого антитела и 80 мг второго антитела или около 3 мг/мл первого антитела и 1 мг/мл второго антитела.
65. Применение альтернативы (например, "или"), как следует понимать, означает одну, обе или любую комбинацию упомянутых альтернатив. Как используется в данном описании, неопределенные артикли "а" или "an" следует понимать как относящиеся к "одному или более" из любого перечисленного или пронумерованного компонента.
66. Термины "около (приблизительно)" или "состоящий по существу из" относятся к значению или композиции, которое находится в пределах приемлемого диапазона ошибок для конкретной величины или композиции, как определено специалистом в данной области техники, которая будет зависеть отчасти от того, как данная величина или композиция измерена или определена, то есть от ограничений конкретной системы измерения. Например, "около" или "состоящий по существу из" может означать в пределах 1 или более чем 1 стандартного отклонения, согласно практике в данной области техники. Альтернативно, "около" или "состоящий по существу из" может означать диапазон до 20%. Кроме того, особенно в отношении биологических систем или процессов, данные термины могут означать вплоть до порядка величины или до 5-кратного значения величины. При указании конкретных величин в данном описании и в формуле изобретения, если не указано иное, значение "около" или "состоящий по существу из", как предполагается, должно быть в пределах приемлемого диапазона ошибок для данной конкретной величины или композиции.
67. Термины "приблизительно один раз в неделю", "приблизительно каждые две недели" или любые другие аналогичные точки интервала дозирования, используемые в настоящем описании, означают приблизительные числа. "Приблизительно один раз в неделю" может включать в себя каждые семь дней±один день, то есть, от каждых шести дней до каждых восьми дней. "Приблизительно каждые две недели" может включать в себя каждые четырнадцать дней ± три дня, то есть, от каждых одиннадцати дней до каждых семнадцати дней. Подобные приближения применяются, например, к выражению один раз приблизительно каждые три недели, один раз приблизительно каждые четыре недели, один раз приблизительно каждые пять недель, один раз приблизительно каждые шесть недель и один раз приблизительно каждые двенадцать недель. В некоторых вариантах осуществления интервал дозирования один раз приблизительно каждые шесть недель или один раз приблизительно каждые двенадцать недель означает, что первая доза может быть введена в любой день в течение первой недели, а затем следующая дозу можно вводить в любой день на шестой или двенадцатой неделе, соответственно. В других вариантах осуществления интервал дозирования один раз приблизительно каждые шесть недель или один раз приблизительно каждые двенадцать недель означает, что первая доза вводится в определенный день первой недели (например, понедельник), а затем следующую дозу вводят в тот же день из шестой или двенадцатой недели (то есть, в понедельник), соответственно.
68. Как описано в данном документе, любой диапазон концентраций, диапазон процентов, диапазон отношений или диапазон целых чисел включает в себя величину любого целого числа в пределах упомянутого диапазона и, при необходимости, его частей (например, одну десятую и одну сотую целого числа), если не указано иное.
69. Различные аспекты настоящего изобретения описаны более подробно в следующих подразделах.
Способы по настоящему изобретению
70. Настоящее изобретение относится к способу лечения глиомы или субъекта, пораженного глиомой, включающему введение субъекту терапевтически эффективного количества антагониста программированной смерти-1 (PD-1), например, антитела или его антигенсвязывающей части, которое специфически связывается с PD-1 и ингибирует активность PD-1 ("антитело против PD-1"), или антагониста лиганда программированной смерти-1 (PD-L1), например, антитела или его антигенсвязывающей части, которое специфически связывается с PD-L1 и ингибирует активность PD-L1 ("антитело против PD-L1"). В одном варианте осуществления глиома выбрана из астроцитомы, глиомы ствола головного мозга, эпендимомы, смешанной глиомы, олигодендроглиомы и глиомы зрительного нерва. В другом варианте осуществления глиома представляет собой GBM. В других вариантах осуществления глиома является глиосаркомой. В некоторых вариантах осуществления глиома представляет собой недавно диагностированную GBM или глиосаркому. В некоторых вариантах осуществления глиома является возвратной злокачественной GBM. В некоторых вариантах осуществления GBM представляет собой GBM с O-6-метилгуанин-ДНК-метилтрансферазой (MGMT) (MGMT GBM). В некоторых вариантах осуществления MGMT GBM представляет собой GBM с метилированным геном MGMT в опухоли. В других вариантах осуществления MGMT GBM представляет собой GBM с неметилированным геном MGMT в опухоли. В некоторых вариантах осуществления опухоль глиомы экспрессирует PD-L1. В других вариантах осуществления опухоль глиомы экспрессирует PD-L2. В других вариантах осуществления опухоль глиомы экспрессирует PD-L1 и PD-L2. В некоторых вариантах осуществления опухоль глиомы не экспрессирует PD-L1, PD-L2 или оба из них.
71. В некоторых вариантах осуществления настоящее изобретение включает в себя способ лечения глиомы или субъекта, пораженного глиомой, содержащий монотерапию (введение антитела против PD-1 или анти-PD-Ll-Ab).
72. В других вариантах осуществления настоящее изобретение включает в себя способ лечения глиомы или субъекта, пораженного глиомой, включающий введение анти-PD-l -антагониста или анти-PD-Ll -антагониста, или анти-PD-l-антагониста или анти-PD-Ll -антагониста в комбинации с одним или более противораковых средств, описанных в настоящем изобретении для лечения рака. В некоторых вариантах осуществления глиома представляет собой GBM. "Анти-PD-l-антагонист" или "анти-PD-Ll-антагонист", как указано в настоящем описании, включает в себя любую молекулу, которая ингибирует взаимодействие между PD-1 (рецептор) и PD-L1 (лиганд) таким образом, что сигнальный путь PD-1/PD-L1 блокируется. В одном варианте осуществления анти-PD-l-антагонист представляет собой антитело против PD-L1. В другом варианте анти-PD-l-антагонист или анти-PD-Ll-антагонист представляет собой растворимый белок PD-1 или белок PD-L1. В других вариантах анти-PD-l-антагонист или анти-PD-Ll-антагонист представляет собой слитый белок PD-1-Fc или слитый белок PD-Ll-Fc. В некоторых вариантах осуществления анти-PD-l-антагонист или анти-PD-Ll-антагонист включает в себя слитый белок анти-PD-1, слитый белок анти-PD-Ll, антисмысловую молекулу, небольшую молекулу, рибозим или нанотело, который ингибирует или предотвращает взаимодействие между PD-1 и PD-L1.
73. В некоторых вариантах осуществления настоящее изобретение включает в себя способ лечения глиомы или субъекта, пораженного глиомой, включающий комбинированную терапию. В одном варианте осуществления настоящее изобретение относится к способу лечения глиомы или субъекта, пораженного глиомой, причем данный способ включает введение субъекту комбинации терапевтически эффективных количеств: (а) антитела против PD-1 или антитела против PD-L1; и (b) другой противораковой терапии. В другом варианте осуществления настоящее изобретение относится к способу лечения глиомы или субъекта, пораженного глиомой, причем данный способ включает введение субъекту комбинации терапевтически эффективных количеств: (а) антитела против PD-1 или анти-PD-L1-Ab; и (b) стандартной терапии лечения, раскрытой в настоящем описании в другом месте.
74. В некоторых вариантах осуществления другая противораковая терапия представляет собой противораковое средство (например, химиотерапия или любая другая терапия, раскрытая в настоящем описании или известная в данной области техники), облучение, хирургическое вмешательство или второе антитело или его антигенсвязывающую часть. В некоторых вариантах осуществления изобретения противораковое средство содержит алкилирующий агент, второе антитело или его антигенсвязывающую часть, или оба средства. В некоторых вариантах осуществления алкилирующий агент представляет собой темозоломид. В некоторых вариантах осуществления изобретения противораковое средство представляет собой двухкомпонентную химиотерапию на основе препаратов платины, EGFR-нацеленный ингибитор тирозинкиназы или бевацизумаб. В некоторых вариантах осуществления второе антитело или его антигенсвязывающая часть представляет собой Ab или его антигенсвязывающую часть, которое специфически связывается с CTLA-4 и ингибирует активность CTLA-4. В некоторых вариантах осуществления второе антитело или его антигенсвязывающая часть представляет собой антитело против CTLA-4. В других вариантах осуществления субъектом является пациент-человек. В некоторых вариантах осуществления субъектом является пациент, ранее не проходивший курс химиотерапии (например, пациент, который ранее не получал какую-либо химиотерапию). В других вариантах осуществления настоящего изобретения субъект получал другую противораковую терапию (например, химиотерапию), но является резистентным или невосприимчивым к такой другой противораковой терапии. В конкретном варианте осуществления настоящее изобретение включает в себя способ лечения рецидива злокачественной глиомы у субъекта, нуждающегося в этом, включающий введение антитела против PD-1 в сочетании с МРТ-управляемой лазерной абляцией. В некоторых вариантах осуществления субъект сначала подвергается МРТ-управляемой лазерной абляции, что может вызвать разрушение перитуморального гематоэнцефалического барьера и, следовательно, позволить прохождение антитела против PD-1 или антитела против PD-L1 через гематоэнцефалический барьер.
75. В некоторых вариантах осуществления изобретения антитело против PD-1 или антитело против PD-L1 вводят в сочетании с фокальной лучевой терапией в дозе по меньшей мере около 1, около 2 или около 3 GY, около 2, около 3, около 4, около 5 или около 6 раз в неделю. В некоторых вариантах осуществления изобретения антитело против PD-1 или антитело против PD-L1 вводят в сочетании с фокальной лучевой терапией в дозе около 2 GY около 5 раз в неделю. В других вариантах осуществления лучевая терапия продолжается в течение по меньшей мере около 2, около 3, около 4, около 5, около 6, около 7, около 8, около 9 или около 10 недель. В некоторых вариантах осуществления лучевая терапия продолжается в течение около 6 недель.
76. Комбинированная терапия по настоящему изобретению включает в себя способы лечения вновь диагностированной глиомы, например, глиосаркомы или GBM, включающие введение антитела против PD-1 или его антигенсвязывающей части или антитела против PD-L1 или его антигенсвязывающей части, необязательно в сочетании с антителом против CTLA-4 или его антигенсвязывающей частью и/или противораковым средством, например, алкилирующим агентом, например, темозоломидом. В одном варианте осуществления способ дополнительно включает введение химиолучевой терапии субъекту до введения антитела против PD-1 или антитела против PD-L1, антитела против CTLA-4 или темозоломида. В другом варианте осуществление антитело против PD-1 или антитело против PD-L1, антитело против CTLA-4 и/или темозоломид вводят в течение одной недели, двух недель, трех недель, четырех недель, пяти недель, шести недель, семи недель, восьми недель или девяти недель после завершения химиолучевой терапии. В некоторых вариантах осуществления изобретения антитело против PD-1 или антитело против PD-L1 вводят в сочетании с фокальной лучевой терапией и темозоломидом. В некоторых вариантах осуществления, после введения антитела против PD-1 или антитела против PD-L1 в сочетании с фокальной лучевой терапией и темозоломидом, продолжают лечение антителом против PD-1 или антителом против PD-L1, в поддерживающих целях. В некоторых вариантах осуществления, после введения антитела против PD-1 или антитела против PD-L1 в сочетании с фокальной лучевой терапией и темозоломидом, продолжают лечение темозоломидом, в поддерживающих целях.
77. В некоторых вариантах осуществления антитело против PD-1 или его антигенсвязывающую часть, или антитело против PD-L1 или его антигенсвязывающую часть, и/или антитело против CTLA-4 или его антигенсвязывающую часть вводят в сочетании с одним или несколькими дополнительными противораковыми агентами, выбранными из группы, состоящей из антагониста белка LAG3 (белок-продукт гена активации лимфоцитов-3 (Lymphocyte activation gene 3 protein)) (например, анти-LAG-3-антитела), антагониста CD 137 (например, Urelumab, например, BMS-663513), антагониста CD27, антагониста KIR (иммуноглобулин-подобных рецепторов киллерных клеток (Killer-cell immunoglobulin-like receptors)) (например, анти-KIR-антитела, например, Lirilumab или IPH2102/BMS-986015), ингибитора IDO (индоламин-2,3-диоксигеназы), антагониста TGF бета (трансформирующего фактора роста бета), антагониста ИЛ-10 (интерлейкин-10), антагониста VEGF(R) (сосудистого эндотелиального фактора роста (Vascular endothelial growth factor)), антагониста ИЛ-21 (интерлейкин-21), антагониста ГМ-КСФ (гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF (Granulocyte-macrophage colony-stimulating factor); также известного как колониестимулирующий фактор 2 (colony stimulating factor 2 (CSF2)), агонистов TLR-9 (толл-подобного рецептора 9 (Toll-like receptor 9)) и любой их комбинации. В некоторых вариантах осуществления противораковое средство является производным от противоопухолевой вакцины. В других вариантах осуществления противораковое средство является еще одним ингибитором иммунной контрольной точки.
78. Изобретение также относится к способу улучшения или восстановления противоопухолевого иммунного ответа у субъекта, пораженного глиомой, таким образом, что улучшенные или восстановленные иммунные клетки пересекают гематоэнцефалический барьер у субъекта и, следовательно, воздействуют на глиому, включающему введение антитела против PD-1 или антитела против PD-L1, в виде монотерапии или комбинированной терапии. В других вариантах осуществления антитело против PD-1 или антитело против PD-L1 пересекает гематоэнцефалический барьер для лечения глиомы.
79. Настоящее изобретение показывает, что, когда антитело против PD-1 или антитело против PD-L1 вводят субъекту, нуждающемуся в этом, антитело против PD-1 или антитело против PD-L1 способно к индуцированию или улучшению иммунного ответа в мозге субъекта. В одном варианте осуществления введение антитела против PD-1 или антитела против PD-L1 отличается одной или более характерными особенностями: (1) увеличивает количество иммунных клеток вблизи опухоли или в опухоли, (2) увеличивает степень агрегации иммунных клеток вблизи опухоли или в опухоли, (3) индуцирует перивентрикулярную некротическую ткань вблизи опухоли или в опухоли, (4) индуцирует внутриопухолевые макрофаги или увеличивает их количество вблизи опухоли или в опухоли; (5) индуцирует CD45-положительные клетки или увеличивает их количество вблизи опухоли или в опухоли; (6) индуцирует CD3-положительные клетки или увеличивает их количество; и (7) индуцирует или увеличивает периваскулярную инфильтрацию и/или инфильтрирующие клетки вблизи опухоли или в опухоли. В других вариантах осуществления настоящие способы относятся к индукции или улучшению иммунного ответа в мозге субъекта, нуждающегося в этом, включая введение субъекту терапевтически эффективного количества антитела против PD-1 или антитела против PD-L1. Один или более типов иммунных реакций может быть выбран из от (1) до (7), как описано выше.
80. В некоторых вариантах осуществления настоящие способы обеспечивают способ индукции или улучшения иммунного ответа в мозге субъекта, нуждающегося в этом, без непосредственного введения противоракового средства (или антитела против PD-1 или антитела против PD-L1) в мозг, включающий введение внутривенно антитела против PD-1 или антитела против PD-L1 субъекту. Настоящее изобретение неожиданно показывает, что антитело против PD-1 или антитело против PD-L1 может индуцировать иммунный ответ в головном мозге субъекта без непосредственного введения в мозг. Не будучи связанными какой-либо теорией, введение антитела против PD-1 или антитела против PD-L1 субъекту, нуждающемуся в этом, индуцирует или восстанавливает иммунную реакцию (например, активацию иммунных клеток, пролиферацию иммунных клеток, увеличение в количестве иммунных клеток, рекрутинг иммунных клеток в надлежащем месте и т.д.) у субъекта, тем самым позволяя индуцированным или активированным иммунным клеткам пересечь гематоэнцефалический барьер у субъекта и устранить опухолевые клетки. Таким образом, введение антитела против PD-1 или антитела против PD-L1 может (1) увеличить количество иммунных клеток вблизи опухоли или в опухоли, (2) увеличить большую агрегацию иммунных клеток вблизи опухоли или в опухоли, (3) индуцировать перивентрикулярные некротические ткани вблизи опухоли или в опухоли, (4) индуцировать внутриопухолевые макрофаги или увеличить их количество вблизи опухоли или в опухоли; (5) индуцировать CD45-положительные клетки или увеличить их количество вблизи опухоли или в опухоли; (6) индуцировать CD3-положительные клетки или увеличить их количество; (7) индуцировать или увеличить периваскулярную инфильтрацию и/или инфильтрирующие клетки вблизи опухоли или в опухоли, или (8) осуществить любую их комбинацию.
81. В определенных вариантах осуществления способ по настоящему изобретению (например, введение антитела против PD-1 или антитела против PD-L1 или введение антитела против PD-1 или антитела против PD-L1 и другой противораковой терапии) эффективно увеличивает продолжительность жизни субъекта. Например, возрастание продолжительности выживаемости субъекта составляет по меньшей мере около 1 месяца, по меньшей мере около 2 месяцев, по меньшей мере около 3 месяцев, по меньшей мере около 4 месяцев, по меньшей мере около 5 месяцев, по меньшей мере около 6 месяцев, по меньшей мере около 7 месяцев, по меньшей мере около 8 месяцев, по меньшей мере около 9 месяцев, по меньшей мере около 10 месяцев, по меньшей мере около 11 месяцев или по меньшей мере около 1 года или больше, по сравнению с другим субъектом, получавшим либо только другую терапию (например, бевацизумаб или темозоламид), либо, в случае комбинированной терапии, только один из двух членов комбинированной терапии по отдельности (например, только антитело против PD-1), либо альтернативную комбинированную терапию (т.е. темозоламид и лучевую терапию). В некоторых вариантах осуществления продолжительность выживаемости возрастает по меньшей мере приблизительно на 2 месяца. В некоторых вариантах осуществления способ по настоящему изобретению эффективно увеличивает продолжительность безрецидивной выживаемости субъекта. Например, возрастание продолжительности безрецидивной выживаемости субъекта составляет по меньшей мере около 1 месяца, по меньшей мере около 2 месяцев, по меньшей мере около 3 месяцев, по меньшей мере около 4 месяцев, по меньшей мере около 5 месяцев, по меньшей мере около 6 месяцев, по меньшей мере около 7 месяцев, по меньшей мере около 8 месяцев, по меньшей мере около 9 месяцев, по меньшей мере около 10 месяцев, по меньшей мере около 11 месяцев или по меньшей мере около 1 года или больше, по сравнению с другим субъектом, которого лечили либо только другой терапией (например, бевацизумаб или темозоламид), либо, в случае комбинированной терапии, только одним из двух членов комбинированной терапии по отдельности (например, только антитело против PD-1), либо альтернативной комбинированной терапией (т.е. темозоламид и лучевая терапия). В некоторых вариантах осуществления безрецидивная выживаемость возрастает по меньшей мере приблизительно на 2 месяца. В определенных вариантах осуществления терапия по настоящему изобретению эффективно повышает уровень ответа в группе испытуемых. Например, уровень ответа в группе испытуемых увеличивается по меньшей мере на около 2%, по меньшей мере на около 3%, по меньшей мере на около 4%, по меньшей мере на около 5%, по меньшей мере на около 10%, по меньшей мере на около 15%, по меньшей мере на около 20%, по меньшей мере на около 25%, по меньшей мере на около 30%, по меньшей мере на около 35%, по меньшей мере на около 40%, по меньшей мере на около 45%, по меньшей мере на около 50%, по меньшей мере на около 55%, по меньшей мере на около 60%, по меньшей мере на около 70%, по меньшей мере на около 75%, по меньшей мере на около 80%, по меньшей мере на около 85%, по меньшей мере на около 90%, по меньшей мере на около 95%, по меньшей мере на около 99% или по меньшей мере на около 100% по сравнению с другой группой пациентов, получавших либо только другую терапию (например, бевацизумаб или темозоломид), либо, в случае комбинированной терапии, только один из двух членов комбинированной терапии по отдельности (например, только антитело против PD-1), либо альтернативную комбинированную терапию (т.е. темозоламид и лучевую терапию).
82. В определенных вариантах осуществления настоящее изобретение относится к способу лечения впервые диагностированной GBM с неметилированным геном MGMT у субъекта, нуждающегося в этом, включающему введение около 240 мг антитела против PD-1 (т.е. ниволумаб) приблизительно каждые две недели на протяжении 16 недель в сочетании с фокальной лучевой терапией (2 GY 5 X недель X 6 недель). В некоторых вариантах осуществления данный способ дополнительно включает введение около 480 мг ниволумаба через 4 недели после завершения первого лечения продолжительностью 16 недель.
Антитела против PD-1
83. PD-1 представляет собой ключевой рецептор иммунных контрольных точек, экспрессируемый активированными Т и В-клетками, и опосредует иммуносупрессию. PD-1 является членом семейства CD28-рецепторов, которое включает в себя CD28, CTLA-4, ICOS, PD-1 и BTLA. Было идентифицировано два присутствующих на клеточной поверхности гликопротеиновых лиганда для PD-1, лиганд-1 запрограммированной смерти (PD-L1) и лиганд-2 запрограммированной смерти (PD-L2), которые экспрессируются на антиген-презентирующих клетках, а также в случае многих онкологических заболеваний человека, и, как было показано, понижающе регулируют активацию Т-клеток и секрецию цитокинов после связывания с PD-1. Ингибирование взаимодействия PD-1/PD-L1 опосредует мощную противоопухолевую активность в доклинических моделях.
84. HuMAbs, которые специфически связываются с PD-1 с высоким сродством, были раскрыты в патенте США №8,008,449. Другие анти-PD-1-mAb были описаны, например, в патентах США №№6,808,710, 7,488,802, 8,168,757 и 8,354,509 и в публикации РСТ WO 2012/145493. Каждое из анти-PD-1-HuMAb, раскрытых в патенте США №8,008,449, как было продемонстрировано, проявляет одну или более из следующих характерных особенностей: (а) связывается с PD-1 человека с KD, равной 1×10-7 М или менее, как это определено с помощью поверхностного плазмонного резонанса с использованием системы биосенсоров Biacore; (b) по существу не связывается с человеческим CD28, CTLA-4 или ICOS; (с) увеличивает пролиферацию Т-клеток в анализе реакции лимфоцитов в смешанной культуре (Mixed Lymphocyte Reaction (MLR)); (d) увеличивает продуцирование интерферона-гамма в анализе MLR; (е) увеличивает секрецию IL-2 в анализе MLR; (f) связывается с PD-1 человека и PD-1 макак; (g) ингибирует связывание PD-L1 и/или PD-L2 с PD-1; (h) стимулирует антигенспецифические ответные реакции памяти; (i) стимулирует ответы Ab; и (j) ингибирует рост опухолевых клеток in vivo. Антитела против PD-1, применяемые в настоящем изобретении, включают mAb, которые специфически связываются с человеческим PD-1 и проявляют по меньшей мере одну, по меньшей мере две, по меньшей мере три, по меньшей мере четыре или по меньшей мере пять из предыдущих характерных особенностей.
85. В одном варианте осуществления антитело против PD-1 представляет собой ниволумаб. Ниволумаб (также известно как "OPDIVO®", ранее называемый 5С4, BMS-936558. MDX-1106 или ONO-4538) представляет собой полностью человеческое IgG4 (S228P) антитело, действующее как ингибитор рецептора иммунных контрольных точек PD-1, которое избирательно предотвращает взаимодействие с лигандами PD-1 (PD-L1 и PD-L2), тем самым блокируя понижающую регуляцию противоопухолевых функций Т-клеток (патент США №8,008,449; Wang et al, 2014 Cancer Immunol Res 2 (9): 846-56). В другом варианте осуществления антитело против PD-1 или его фрагмент перекрестного конкурирует с ниволумабом. В других вариантах осуществления антитело против PD-1 или его фрагмент связывается с тем же эпитопом, что и ниволумаб. В определенных вариантах осуществления антитело против PD-1 имеет те же последовательности CDR, что и ниволумаб.
86. В другом варианте осуществления антитело против PD-1 или его фрагмент перекрестно конкурирует с пембролизумабом. В некоторых вариантах осуществления антитело против PD-1 или его фрагмент связывается с тем же эпитопом, что и пембролизумаб. В определенных вариантах осуществления антитело против PD-1 имеет те же последовательности CDR, что и пембролизумаб. В другом варианте осуществления антитело против PD-1 представляет собой пембролизумаб. Пембролизумаб (также известно как "KEYTRUDA®", ламбролизумаб и МК-3475) представляет собой гуманизированное моноклональное IgС4-антитело, направленное против рецептора клеточной поверхности человека PD-1 (программируемой смерти-1 или программируемой клеточной смерти-1). Пембролизумаб описан, например, в патентах США №№8,354,509 и 8,900,587; см. также http://www.cancer.gov/drugdictionary?cdrid=695789 (последнее обращение: 14 декабря 2014 года). Пембролизумаб был одобрен FDA для лечения рецидивирующей или резистентной меланомы.
87. В других вариантах антитело против PD-1 или его фрагмент перекрестно конкурирует с MEDI0608. В других вариантах осуществления антитело против PD-1 или его фрагмент связывается с тем же эпитопом, что и MEDI0608. В определенных вариантах осуществления антитело против PD-1 имеет те же последовательности CDR, что и MEDI0608. В других вариантах антитело против PD-1 представляет собой MEDI0608 (ранее известное как АМР-514), который представляет собой моноклональное антитело против рецептора PD-1. MEDI0608 описан, например, в патенте США №8,609,089 В2 или в http://www.cancer.gov/drugdictionary?cdrid=756047 (последнее обращение 14 декабря 2014 года).
88. В определенных вариантах осуществления анти-PD-1-антагонист является АМР-224, который представляет собой слитый белок B7-DC-Fc. АМР-224 обсуждается в публикации США №2013/0017199 или в http://www.cancer.gov/publications/dictionaries/cancer-drug?cdrid=700595 (последнее обращение 8 июля 2015 года).
89. В других вариантах антитело против PD-1 или его фрагмент перекрестно конкурирует с BGB-A317. В некоторых вариантах осуществления антитело против PD-1 или его фрагмент связывается с тем же эпитопом, что и BGB-A317. В определенных вариантах осуществления антитело против PD-1 имеет те же последовательности CDR, что и BGB-A317. В определенных вариантах осуществления антитело против PD-1 представляет собой BGB-A317, который является гуманизированным моноклональным антителом. BGB-A317 описан в публикации США №2015/0079109.
90. Антитела против PD-1, применяемые в описанных способах, также включают выделенные антитела, которые специфически связываются с человеческим PD-1 и перекрестно конкурируют с ниволумабом за связывание с человеческим PD-1 (см., например, патент США №8008449, WO 2013/173223). Способность антител перекрестно конкурировать за связывание с антигеном указывает на то, что данные антитела связываются с одной и той же эпитопной областью антигена и стерически препятствуют связыванию других перекрестно-конкурирующих антител с областью данного конкретного эпитопа. Такие перекрестно-конкурирующие антитела должны иметь функциональные свойства, очень похожие на свойства ниволумаба в силу их связывания с той же эпитопной областью PD-1. Перекрестно-конкурирующие антитела могут быть легко идентифицированы на основе их способности к перекрестной конкуренции с ниволумабом в стандартных анализах связывания PD-1, таких как анализ Biacore, анализы ELISA или проточная цитометрия (см., например, WO 2013/173223).
91. В определенных вариантах осуществления антитела, которые перекрестно конкурируют за связывание с человеческим PD-1 или связываются с той же эпитопной областью человеческого PD-1, что и ниволумаб, являются моноклональными антителами. В целях введения человеческим субъектам такие перекрестно-конкурирующие антитела представляют собой химерные антитела или гуманизированные или человеческие антитела. Такие химерные, гуманизированные или человеческие mAb могут быть получены и выделены способами, хорошо известными в данной области техники.
92. В определенных вариантах осуществления антитело против PD-1 или его антигенсвязывающая часть содержит константную область тяжелой цепи, которая представляет собой изотип человеческого IgG1 или IgG4. В некоторых других вариантах осуществления настоящего изобретения последовательность константной области IgG4 тяжелой цепи антитела против PD-1 или его антигенсвязывающей части содержит мутацию S228P, при которой происходит замена остатка серина в шарнирной области на остаток пролина, обычно обнаруживаемый в соответствующем положении в антителах изотипа IgG1. Данная мутация, которая присутствует в ниволумабе, предотвращает обмен Fab-плеча с эндогенными IgС4-антителами, сохраняя при этом низкую аффинность к активации Fc-рецепторов, связанных с IgС4-антителами дикого типа (Wang et al, 2014). В других вариантах осуществления настоящего изобретения антитело содержит константную область легкой цепи, которая является константной областью человеческой каппа- или лямбда-цепи. В других вариантах осуществления анти-PD-1-Ab или его антигенсвязывающая часть представляет собой mAb или его антигенсвязывающую часть. В определенных вариантах осуществления любого из терапевтических способов, описанных в настоящем изобретении, включающих введение анти-PD-1-Ab, антитело против PD-1 представляет собой ниволумаб.
93. Антитела против PD-1, применяемые в способах согласно раскрытому изобретению, также включают антигенсвязывающие фрагменты вышеупомянутых антител. Было наглядно продемонстрировано, что антигенсвязывающая функция антитела может быть осуществлена фрагментами полноразмерного Ab. Примеры связывающих фрагментов, охватываемых термином "антигенсвязывающая часть" антитела, включают (i) Fab-фрагмент, моновалентный фрагмент, состоящий из доменов VL, VH, CL и CH1, (ii) F(ab')2-фрагмент, бивалентный фрагмент, содержащий два Fab-фрагмента, связанных дисульфидным мостиком в шарнирной области; (iii) Fd-фрагмент, состоящий из доменов VH и CH1, и (iv) Fv-фрагмент, состоящий из доменов VL и VH одного плеча Ab.
Антитела против PD-L1
94. В определенных вариантах осуществления настоящие способы относятся к способам лечения глиомы у субъекта, нуждающегося в этом, путем введения анти-PD-L1-антагониста (например, антитела против PD-L1) субъекту. Так, например, антитело против PD-L1 предотвращает взаимодействие между PD-1 и PD-L1, и, таким образом, оказывает аналогичное воздействие на сигнальный путь PD-1; таким образом, антитело против PD-L1 может заменить применение антитела против PD-1 в способах, описанных в настоящем изобретении.
95. В других вариантах осуществления антитело против PD-L1 представляет собой BMS-936559 (ранее известное как 12А4 или MDX-1105) (см., например, патент США №7943743; WO 2013/173223) или любое другое антитело против PD-L1, описанное в патенте США №7943743. В некоторых вариантах осуществления изобретения антитело против PD-L1 представляет собой антитело, которое перекрестно конкурирует с BMS-936559 за связывание, или антитела против PD-L1, раскрытые в патенте США №7,943,743. В других вариантах осуществления изобретения антитело против PD-L1 представляет собой антитело, которое связывается с тем же эпитопом, что и BMS-936559, или антитело против PD-L1, раскрытое в патенте США №7,943,743.
96. В других вариантах осуществления изобретения антитело против PD-L1 представляет собой MPDL3280A (также известно как RG7446) (см., например, Herbst; патент США №8217149), MEDI4736 (также называемый Durvalumab; Khleif, 2013, см. патент США 8,779,108 или заявку США 2014/0356353, поданную 6 мая 2014 года) или MSB0010718C (также называемый Avelumab; см. заявку США 2014/0341917). В некоторых вариантах осуществления изобретения антитело против PD-L1 представляет собой антитело, которое перекрестно конкурирует с MPDL3280A, MEDI4736 и/или MSB0010718C за связывание. В вариантах осуществления изобретения антитело против PD-L1 представляет собой антитело, которое связывается с тем же эпитопом, что и MPDL3280A, MEDI4736 и/или MSB0010718C.
97. В определенных вариантах осуществления антитела, которые перекрестно конкурируют за связывание с человеческим PD-L1 или связываются с той же эпитопной областью человеческого PD-L1, что и вышеупомянутые PD-L1-антитела, представляют собой моноклональные антитела (mAb). Для введения человеческим субъектам данные перекрестно-конкурирующие антитела могут быть химерными антителами или могут быть гуманизированными или человеческими антителами. Такие химерные, гуманизированные или человеческие моноклональные антитела могут быть получены и выделены способами, хорошо известными в данной области техники.
98. Антитела против PD-L1, применяемые в способах согласно раскрытому изобретению, также включают антигенсвязывающие фрагменты вышеупомянутых антител. Было наглядно продемонстрировано, что антигенсвязывающая функция антитела может быть осуществлена фрагментами полноразмерного Ab. Примеры связывающих фрагментов, охватываемых термином "антигенсвязывающая часть" Ab, включают (i) Fab-фрагмент, моновалентный фрагмент, состоящий из доменов VL, VH, CL и CH1; (ii) F(ab')2-фрагмент, бивалентный фрагмент, содержащий два Fab-фрагмента, связанных дисульфидным мостиком в шарнирной области; (III) Fd-фрагмент, состоящий из доменов VH и CH1, и (iv) Fv-фрагмент, состоящий из доменов VL и VH одного плеча Ab.
Антитела против CTLA-4
99. Антитела против CTLA-4 по настоящему изобретению связываются с человеческим CTLA-4 с тем, чтобы нарушить взаимодействие CTLA-4 с рецептором В7 человека. Поскольку взаимодействие CTLA-4 с В7 трансдуцирует сигнал, ведущий к инактивации Т-клеток, несущих рецептор CTLA-4, нарушение взаимодействия эффективно индуцирует, повышает или пролонгирует активацию таких Т-клеток, тем самым индуцируя, увеличивая или продлевая иммунный ответ.
100. HuMAb, которые специфически связываются с CTLA-4 с высокой аффинностью, описаны в патентах США №№6,984,720 и 7,605,238. Другие анти-PD-1-mAb были описаны, например, в патентах США№№5,977,318, 6,051,227, 6,682,736 и 7,034,121. Анти-PD-1-HuMAb, раскрытые в патентах США №№6,984,720 и 7,605,238, как было показано, проявляют одну или более из следующих характерных особенностей: (а) специфически связывается с человеческим CTLA-4 с аффинностью связывания, отражаемой равновесной константой ассоциации (Кa), составляющей по меньшей мере около 107 М-1, или около 109 М-1, или от около 1010 М-1 до 1011 М-1 или выше, как определено с помощью анализа Biacore; (b) кинетическая константа ассоциации (ka) составляет по меньшей мере около 103, около 104 или около 105 м-1 с-1; (с) кинетическая константа диссоциации (kd) составляет по меньшей мере около 103, около 104 или около 105 м-1 с-1; и (d) ингибирует связывание CTLA-4 с В7-1 (CD80) и В7-2 (CD86). Антитела против CTLA-4, применяемые в настоящем изобретении, включают mAb, которые специфически связываются с человеческим CTLA-4 и проявляют по меньшей мере одну, по меньшей мере две или по меньшей мере три из предыдущих характерных особенностей.
101. Примером клинического анти-СТТА-4 Ab является человеческое моноклональное антитело 10D1 (теперь известно как ипилимумаб и продается как YERVOY®), как описано в патенте США №6,984,720. Ипилимумаб представляет собой антитело против CTLA-4 для применения в способах, раскрытых в настоящем изобретении. Ипилимумаб представляет собой полностью человеческое, IgG1 моноклональное антитело, которое блокирует связывание CTLA-4 с его В7-лигандами, стимулируя тем самым активацию Т-клеток и улучшая общую выживаемость (overall survival (OS)) у пациентов с меланомой на поздней стадии.
102. Еще одно антитело против CTLA-4, применимое для настоящих способов, представляет собой тремелимумаб (также известно как СР-675,206). Тремелимумаб представляет собой человеческое IgG2 моноклональное антитело против CTLA-4. Тремелимумаб описан в WO/2012/122444, U.S. Publ. No. 2012/263677 или WO Publ. No. 2007/113648 A2.
103. Антитела против CTLA-4, применяемые в раскрытых способах, также включают выделенные антитела, которые специфически связываются с человеческим PD-1 и перекрестно конкурируют с ипилимумабом или тремелимумабом за связывание с человеческим CTLA-4 или связываются с той же эпитопной областью человеческого CTLA-4, что и ипилимумаб или тремелимумаб. В определенных вариантах осуществления антитела, которые перекрестно конкурируют за связывание с человеческим CTLA-4 или связываются с той же эпитопной областью человеческого PD-1, что и ипилимумаб или тремелимумаб, представляет собой антитела, содержащие тяжелую цепь человеческого изотипа IgG1. В целях введения больным пациентам данные перекрестно-конкурирующие антитела могут быть химерными антителами или могут быть гуманизированными или человеческими антителами. Применяемые антитела против CTLA-4 также включают антигенсвязывающие части вышеупомянутых антител, такие как Fab-, F(ab')2-, Fd- или Fv-фрагменты.
Стандартные схемы лечения GBM
104. Настоящее изобретение также включает терапию, при которой антитело против PD-1 или антитела против PD-L1 комбинируется со стандартным лечением глиомы. Субъект может получить стандартное лечение в любое время до, во время или после лечения антителом против PD-1. Стандартные схемы лечения различных видов рака хорошо известны специалистам в данной области техники. Например, Национальная Всесторонняя Онкологическая сеть (National Comprehensive Cancer Network (NCCN)), союз 21 крупных онкологических центров в США, публикует руководства по клинической практике NCCN в онкологии (NCCN Clinical Practice Guidelines in Oncology (NCCN GUIDELINES®)), которые предоставляют подробную актуальную информацию по стандартным схемам лечения для широкого спектра раковых заболеваний (см. NCCN GUIDELINES®, 2014, доступно по адресу: http://www.nccn.org/professionals/physician_gls/f_guidelines.asp, последний доступ 14 мая 2014 года.).
105. Принято считать, что GBM трудно поддается лечению, поскольку опухоли содержат много различных типов клеток и являются очень злокачественными. Лечение первой линии для GBM, как правило, представляет собой хирургическое вмешательство для того, чтобы удалить как можно больше опухоли, насколько это возможно, и варианты лечения для рецидивной GBM осложняются побочными эффектами и ограниченной эффективностью. В одном варианте осуществления стандартное лечение представляет собой МРТ-управляемую лазерную абляцию.
106. В одном варианте осуществления настоящие способы включают введение антитела против PD-1 или антитела против PD-L1 с бевацизумабом. Бевацизумаб (Авастин®), моноклональное антитело против фактора роста эндотелия сосудов (VEGF), был одобрен для лечения рецидивной GBM в 2009 году. Разрешение на применение бевацизумаба для лечение рецидивной GBM было основано на клинических испытаниях с двумя отдельными группами лечения, AVF3708g и NCI 06-С-0064Е. AVF3708g представляло собой открытое, многоцентровое, несравнительное исследование с параллельными группами, разработанное с целью оценки эффективности и безопасности монотерапии бевацизумаба и терапии бевацизумаб плюс иринотекан у ранее пролеченных пациентов с GBM. В общей сложности, 167 пациентов были включены в исследование (85 в группе лечения бевацизумабом, 82 в группе лечения бевацизумаб плюс иринотекан). Эффективность бевацизумаба была продемонстрирована с использованием оценки ответа на лечение на основе рентгенографических критериев ВОЗ. Ответы на лечение наблюдались у 25,9% (95% CI: 17,0%, 36,1%) пациентов. Средняя продолжительность ответа составила 4,2 месяцев (95% CI: 3,0, 5,7 месяцев). NCI06-C-0064E представляло собой одногрупповое, одноцентровое NCI-спонсированое исследование бевацизумаба для пациентов с ранее пролеченными глиомами. В общей сложности, 56 пациентов с глиомой высокой степени злокачественности были включены в данное исследование. Объективные ответы, наблюдаемые в данном исследовании, составляли 19,6% (95% CI; 10,9%, 31,3%), с использованием тех же критериев, что и в AVF708g. Средняя продолжительность наблюдаемого ответа составила 3,9 месяца (95% CI: 2,4, 17,4). (Friedman H.S. et al, J Clin Oncol 27:4733-40 (2009).)
107. Одиночное средство иринотекан (СРТ-11), ингибитор топоизомеразы 1, применяемый в схеме лечения при рецидиве, дает уровни ответа ≤15%. При рецидивной GBM, уровни PFS-6 обычно находятся в диапазоне от 9% до 21%, а медианная ОС составляет ≤30 недель. (Friedman H.S. et al., J Clin Oncol 27:4733-40 (2009).)
108. В случае опухолей более высокой степени злокачественности, варианты лечения дополнительно включают лучевую терапию в отдельности, комбинированную терапию, состоящую из лучевой терапиии и химиотерапии, или комбинированную терапию, состоящую из лучевой терапии и химиотерапии с последующей дополнительной химиотерапией. Терапии для рецидивной GBM включают повторную нейрохирургическую резекцию, лучевую терапию, химиотерапию и поддерживающую терапию. Повторное облучение становится более трудным, и потенциальная токсичность включает в себя некроз мозга, прогрессивный отек мозга и стойкие неврологические нарушения. (Cohen М.Е. et al., Pediatr Neurol. 7(3):157-63 (1991). Таким образом, повторное облучение мозга предлагается небольшому числу пациентов с рецидивной GBM (Chang S.M. at al. Neurosurg Focus. 20(4): E4 Review (2006))
109. Нитрозомочевины, карбоплатин, этопозид и иринотекан или комбинация данных средств являются общеизвестными спасательными средствами, вводимыми рецидивирующим пациентам с GBM. Темозоламид является одной из форм химиотерапии, которая обычно используется для лечения GBM, хотя может применяться множество препаратов. Дополнительные средства против рака, применяемые для лечения GBM, включают в себя: нитрозомочевины (например, кармустин [BCNU]), ингибиторы MGMT (например, О6-бензилгуанин), цисплатин, бевацизумаб (один или с иринотеканом) для рецидивной глиомы, ингибиторы тирозинкиназы (например, гефитиниб, эрлотиниб), а также другие экспериментальные терапии (например, генная терапия, пептидные вакцины и вакцины на основе дендритных клеток, синтетические хлортоксины, радиоактивно меченные препараты и антитела).
Фармацевтические композиции и дозировки
110. Терапевтические средства по настоящему изобретению могут быть составлены в композиции, например, фармацевтической композиции, содержащей антитело и фармацевтически приемлемый носитель. Используемый в настоящем описании термин "фармацевтически приемлемый носитель" включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, изотонические и задерживающие абсорбцию средства и т.п., которые являются физиологически совместимыми. В некоторых вариантах осуществления носитель для композиции, содержащей антитело, является подходящим для внутривенного, внутримышечного, подкожного, парентерального, спинального или эпидермального введения (например, путем инъекции или инфузии). Фармацевтическая композиция по настоящему изобретению может включать в себя одну или несколько фармацевтически приемлемых солей, антиоксидант, водные и неводные носители и/или вспомогательные вещества, такие как консерванты, смачивающие агенты, эмульгаторы и диспергирующие средства.
111. Режимы приема лекарственного препарата регулируются для обеспечения оптимального желаемого ответа, например, максимального терапевтического ответа и/или минимального количества нежелательных явлений. Для введения анти-PD-1 Ab, в том числе, в комбинации с другим противораковым средством, дозировка может варьироваться от по меньшей мере около 0,01 до по меньшей мере около 20 мг/кг, от по меньшей мере около 0,1 до по меньшей мере около 10 мг/кг массы тела субъекта. Например, дозировки могут составлять по меньшей мере около 0,1, по меньшей мере около 0,3, по меньшей мере около 1, по меньшей мере около 2, по меньшей мере около 3, по меньшей мере около 5 или по меньшей мере около 10 мг/кг массы тела, или по меньшей мере около 0,3, по меньшей мере около 1, по меньшей мере около 2, по меньшей мере около 3 или по меньшей мере около 5 мг/кг массы тела. Режим дозирования, как правило, предназначен для достижения воздействий, которые приводят к устойчивому заполнению рецепторов (receptor occupancy (RO)) на основе типичных фармакокинетических свойств Ab. Примерная схема лечения включает введение приблизительно один раз в неделю, приблизительно один раз в 2 недели, приблизительно один раз в 3 недели, приблизительно один раз в 4 недели, приблизительно один раз в месяц, приблизительно один раз каждые 3-6 месяцев или дольше. В некоторых вариантах осуществления анти-PD-1-Ab, такое как ниволумаб, вводят субъекту приблизительно один раз в 2 недели. В других вариантах осуществления настоящего изобретения антитело вводят приблизительно один раз в 3 недели. Дозирование и режим могут меняться в течение курса лечения. Например, режим дозирования анти-PD-1-монотерапии может включать в себя введение Ab: (i) каждые 2 недели в 6-недельных циклах; (ii) каждые 4 недели для шести доз, затем каждые три месяца; (iii) каждые 3 недели; (iv) от 3 до 10 мг/кг один раз, после чего 1 мг/кг каждые 2-3 недели. В некоторых вариантах осуществления схема дозирования для антитела против PD-1 по настоящему изобретению может содержать от около 0,3 до около 10 мг/кг массы тела, от около 1 до около 5 мг/кг массы тела, от около 1 до около 3 мг/кг массы тела через внутривенное введение, причем антитело вводят каждые 14-21 дней вплоть до 6-недельного или 12-недельного циклов до достижения полного ответа или подтвержденного прогрессирующего заболевания. В некоторых вариантах осуществления антитело против PD-1 вводят в дозе по массе. В некоторых вариантах осуществления изобретения антитело против PD-1 вводят в фиксированной дозе. В некоторых вариантах осуществления антитело против PD-1 вводят в дозе, составляющей по меньшей мере около 60 мг, около 80 мг, около 100 мг, около 120 мг, около 140, около 160 мг, около 180 мг, около 200 мг, около 220 мг, около 240 мг, около 260 мг, около 280 мг, около 300 мг, около 320, около 340, около 360, около 380, около 400, около 420, около 440, около 460, около 480, около 500 или около 550 мг. В некоторых вариантах осуществления изобретения антитело против PD-1 вводят в дозе 240 мг. В других вариантах осуществления изобретения антитело против PD-1 вводят в дозе 480 мг. В некоторых вариантах осуществления антитело против PD-1 вводят в фиксированной дозе (при фиксированном соотношении) с другим антителом. В некоторых вариантах осуществления соотношение составляет по меньшей мере около 1:1, около 1:2, около 1:3, около 1:4, около 1:5, около 1:6, около 1:7, около 1:8, около 1:9, около 1:10, около 1:15, около 1:20, около 1:30, около 1:40, около о 1:50, около 1:60, около 1:70, около 1:80, около 1:90, около 1:100, около 1:120, около 1:140, около 1:160, около 1:180, около 1:200, около 200:1, около 180:1, около 160:1, около 140:1, около 120:1, около 100:1, около 90:1, около 80:1, около 70:1, около 60:1, около 50:1, около 40:1, около 30:1, около 20:1, около 15:1, около 10:1, около 9:1, около 8:1, около 7:1, около 6:1, около 5:1, около 4:1, около 3:1 или около 2:1 мг PD-1-антитела к мг второго антитела.
112. При использовании в комбинациях с другими противораковыми средствами, дозировка антитела против PD-1 может быть снижена по сравнению с дозой при монотерапии. Дозировки ниволумаба, которые ниже, чем типичные 3 мг/кг, но не менее чем 0,001 мг/кг, являются субтерапевтическими дозами. Субтерапевтические дозы антитела против PD-1, используемые в способах по настоящему изобретению, выше, чем 0,001 мг/кг, и ниже, чем 3 мг/кг. В некоторых вариантах осуществления субтерапевтическая доза составляет от около 0,001 мг/кг до около 1 мг/кг, от около 0,01 мг/кг до около 1 мг/кг, от около 0,1 мг/кг до около 1 мг/кг или от около 0,001 мг/кг до около 0,1 мг/кг массы тела. В некоторых вариантах осуществления субтерапевтическая доза составляет по меньшей мере около 0,001 мг/кг, по меньшей мере около 0,005 мг/кг, по меньшей мере около 0,01 мг/кг, по меньшей мере около 0,05 мг/кг, по меньшей мере около 0,1 мг/кг, по меньшей мере около 0,5 мг/кг или по меньшей мере около 1,0 мг/кг массы тела. Данные по заполнению рецепторов от 15 субъектов, которые получали дозу ниволумаба, составлявшую от 0,3 мг/кг до 10 мг/кг, показывают, что заполнение PD-1, по-видимому, не зависит от дозы в данном диапазоне доз. При всех дозах, средний уровень заполнения составил 85% (в диапазоне от 70% до 97%), со средним плато заполнения, составившем 72% (диапазон от 59% до 81%). В некоторых вариантах осуществления дозирование 0,3 мг/кг может обеспечить достаточное воздействие для того, чтобы привести к максимальной биологической активности.
113. Ипилимумаб одобрен для лечения меланомы в дозе, составляющей по меньшей мере около 3 мг/кг, при внутривенном введении каждые 3 недели, всего 4 дозы. Таким образом, в определенных вариантах осуществления по меньшей мере около 3 мг/кг является самой высокой дозировкой ипилимумаба, используемой в комбинации с антителом против PD-1, хотя в определенных вариантах осуществления анти-СТLА-4 антитело, такое как ипилимумаб, можно дозировать в пределах диапазона по меньшей мере от около 0,3 до около 10 мг/кг массы тела приблизительно каждые две или три недели, при комбинировании с ниволумабом. Дозировка ипилимумаба, которая значительно ниже, чем утвержденная, составляющая около 3 мг/кг приблизительно каждые 3 недели, рассматривается как субтерапевтическая дозировка. Субтерапевтические дозировки антитела против CTLA-4, используемые в способах по настоящему изобретению, выше, чем 0,001 мг/кг, и ниже, чем 3 мг/кг. В некоторых вариантах осуществления субтерапевтическая доза составляет от около 0,001 мг/кг до около 1 мг/кг, от около 0,01 мг/кг до около 1 мг/кг, от около 0,1 мг/кг до около 1 мг/кг или от около 0,001 мг/кг до около 0,1 мг/кг массы тела. В некоторых вариантах осуществления субтерапевтическая доза составляет по меньшей мере около 0,001 мг/кг, по меньшей мере около 0,005 мг/кг, по меньшей мере около 0,01 мг/кг, по меньшей мере около 0,05 мг/кг, по меньшей мере около 0,1 мг/кг, по меньшей мере около 0,5 мг/кг или по меньшей мере около 1,0 мг/кг массы тела.
114. В некоторых вариантах осуществления дозы антитела против PD-1 не превышают около 3 мг/кг при комбинировании с ипилимумабом. В определенных вариантах осуществления, на основе оценок риска и пользы и PK-PD, используемая дозировка включает в себя комбинацию ниволумаб в дозе, составляющей по меньшей мере около 1 мг/кг, плюс ипилимумаб в дозе, составляющей по меньшей мере около 3 мг/кг, ниволумаб в дозе, составляющей по меньшей мере около 3 мг/кг, плюс ипилимумаб в дозе, составляющей по меньшей мере около 1 мг/кг, или применяется ниволумаб в дозе, составляющей по меньшей мере около 3 мг/кг, плюс ипилимумаб в дозе, составляющей по меньшей мере около 3 мг/кг, причем каждый препарат вводят с частотой дозирования, составляющей один раз приблизительно каждые 2-4 недели, один раз приблизительно каждые 3 недели. В некоторых других вариантах осуществления ниволумаб вводят в дозе, составляющей по меньшей мере около 0,1, около 0,3, около 2, около 3 или около 5 мг/кг, в сочетании с ипилимумабом, который вводят в дозе, составляющей по меньшей мере около 0,1, около 0,3, около 1, около 3 или около 5 мг/кг, один раз каждые 2 недели, один раз каждые 3 недели или один раз приблизительно каждые 4 недели. В конкретном варианте осуществления ниволумаб вводят в дозировке, составляющей по меньшей мере около 3 мг/кг, в сочетании с по меньшей мере около 1 мг/кг ипилимумаба при частоте дозирования, составляющей один раз приблизительно каждые каждые 3 недели в четырех дозах.
115. В определенных вариантах осуществления настоящего изобретения антитело против PD-1 в сочетании с антителом против CTLA-4 вводят внутривенно субъекту в индукционной фазе приблизительно каждые 2 или 3 недели для 2, 3 или 4 введений. В определенных вариантах осуществления ниволумаб в сочетании с ипилимумабом вводят внутривенно в индукционной фазе приблизительно каждые 3 недели для 4 введений. За индукционной фазой следует поддерживающая фаза, во время которой субъекту вводят только антитело против PD-1 в дозе, составляющей по меньшей мере около 0,1, около 0,3, около 1, около 2, около 3, около 5 или около 10 мг/кг, каждые две или три недели до тех пор, пока лечение не перестает быть эффективным, или до появления неуправляемой токсичности или наступления прогрессирования болезни. В определенных вариантах осуществления ниволумаб вводят во время поддерживающей фазы в дозе, составляющей по меньшей мере около 3 мг/кг массы тела, приблизительно каждые 2 недели.
116. Для комбинирования ниволумаба с другими противораковыми средствами данные средства можно вводить в утвержденных для них дозировках. Лечение продолжается до тех пор, пока наблюдается клиническая польза, или до появления неуправляемой токсичности или наступления прогрессирования болезни. Тем не менее, в определенных вариантах осуществления дозировки данных вводимых противораковых средств значительно ниже, чем утвержденная дозировка, т.е. субтерапевтическую дозировку средства вводят в комбинации с анти-PD-1-Ab. Антитело против PD-1 можно вводить в дозировке, которая, как было показано, демонстрирует самую высокую эффективность в качестве монотерапии в клинических испытаниях, например, около 3 мг/кг ниволумабаа, вводимые один раз приблизительно каждые три недели (Topalian et al., 2012 N Engl J Med 366:2443-54; Topalian et al., 2012 Curr Opin Immunol 24:207-12), или в значительно более низкой дозе, т.е., в субтерапевтической дозе.
117. Дозировка и частота изменяются в зависимости от периода полужизни антитела в субъекте. В общем, человеческие антитела показывают самый длинный период полужизни, за ними следуют гуманизированные антитела, химерные антитела и нечеловеческие антитела. Дозировка и частота введения могут меняться в зависимости от того, является ли лечение профилактическим или терапевтическим. При профилактическом применении относительно низкую дозу, как правило, вводят в относительно нечастых интервалах в течение длительного периода времени. Некоторые пациенты продолжают получать лечение всю оставшуюся жизнь. При терапевтическом применении иногда требуется относительно высокая доза через относительно короткие промежутки времени до тех пор, пока прогрессирование заболевания не уменьшается или не прекращается, или до тех пор, пока пациент не показывает частичное или полное облегчение симптомов заболевания. После этого пациент может быть переведен на профилактический режим.
118. Фактические уровни дозировок активных ингредиентов в фармацевтических композициях по настоящему изобретению могут быть изменены таким образом, чтобы получить количество активного ингредиента, которое эффективно для достижения желаемого терапевтического ответа для конкретного пациента, композиции и способа введения, не будучи чрезмерно токсичным по отношению к пациенту. Выбранный уровень дозировки будет зависеть от различных фармакокинетических факторов, включая активность конкретных композиций по настоящему изобретению, способа введения, времени введения, скорости выведения конкретного применяемого соединения, длительности лечения, других лекарственных средств, соединений и/или материалов, используемых в комбинации с конкретными композициями, возраста, пола, массы, состояния, общего состояния здоровья и предыдущей истории болезни пациента, подлежащего лечению, и подобных факторов, хорошо известных в области медицины. Композиция по настоящему изобретению может быть введена посредством одного или более путей введения с использованием одного или более из множества способов, хорошо известных в данной области техники. Как будет понятно специалисту в данной области, путь и/или способ введения будет изменяться в зависимости от желаемых результатов.
Антитела против PD-1 и против PD-L1, подходящие для применения в раскрытых способах
119. Антитела против PD-1, подходящие для применения в раскрытых способах, представляют собой антитела, которые связываются с PD-1 с высокой специфичностью и аффинностью, блокируют связывание PD-L1 и или PD-L2 и ингибируют иммунодепрессивное действие PD-1-сигнального пути. В любом из терапевтических способов, описанных в настоящем изобретении, "антитело" против PD-1 или против CTLA-4 включает в себя антигенсвязывающую часть или фрагмент, который связывается с рецептором PD-1 или CTLA-4, соответственно, и демонстрирует функциональные свойства, аналогичные таковым целых антител, в ингибировании связывания лиганда и положительной регуляции иммунной системы. В некоторых вариантах осуществления антитело против PD-1 или его антигенсвязывающая часть перекрестно конкурирует с ниволумабом за связывание с человеческим PD-1. В других вариантах осуществления антитело против PD-1 или его антигенсвязывающая часть представляет собой химерное, гуманизированное или человеческое моноклональное антитело или его часть. В некоторых вариантах осуществления для лечения больного пациента антитело представляет собой гуманизированное Ab. В других вариантах осуществления для лечения больного пациента антитело представляет собой человеческое Ab. Могут быть использованы Ab, имеющие изотип IgG1, IgG2, IgG3 или IgG4.
120. Поскольку антитела против PD-1 и против PD-L1 нацелены на один и тот же сигнальный путь, и, как было показано в клинических испытаниях, демонстрируют сходные уровни эффективности при различных раковых заболеваниях (см. Brahmer et al., 2012 N Engl J Med 366:2455-65; Topalian et al, 2012a N Engl J Med 366:2443-54; WO 2013/173223), антитело против PD-L1 может быть заменено на антитело против PD-1 в любом из терапевтических способов, описанных в настоящем изобретении. В некоторых вариантах осуществления изобретения антитело против PD-L1 представляет собой BMS-936559 (ранее известно как 12А4 или MDX-1105) (см., например, патент США №7943743, WO 2013/173223). В других вариантах антитело против PD-L1 представляет собой MPDL3280A (также известное как RG7446) (см., например, Herbst et al. 2013 J Clin Oncol 31(suppl):3000; патент США №8217149) или MEDI4736 (Khleif, 2013, В: Proceedings from the European Cancer Congress 2013; September 27-October 1, 2013; Amsterdam, The Netherlands. Abstract 802).
Комбинирование антитела против PD-1 или антитела против PD-L1 с антителом против CTLA-4 для лечения глиом
121. Настоящее изобретение также относится к способу лечения глиомы, включающему введение антитела против PD-1 или антитела против PD-L1 и антитела против антигена-4 цитотоксических Т-лимфоцитов ((cytotoxic T-Lymphocyte Antigen-4 (CTLA-4)), то есть антитела или его антигенсвязывающей части, которое специфически связывается с CTLA-4 и ингибирует активность CTLA-4. В настоящем изобретении (см. примеры 1, 6, 7, и 8) было продемонстрировано, что комбинирование анти-PD-1-Ab, ниволумаба, и анти-CTLA-4-Ab, ипилимумаба, производит раннюю, долговременную противоопухолевую активность у пациентов с глиобластомой. Соответственно, в определенных вариантах осуществления изобретения антитело против CTLA-4, которое используется в сочетании с антителом анти-PD-l, представляет собой ипилимумаб. В некоторых вариантах осуществления антитело против CTLA-4 представляет собой тремелимумаб. В некоторых других вариантах осуществления антитело против CTLA-4 или его антигенсвязывающая часть представляет собой химерное, гуманизированное или человеческое моноклональное антитело или его часть. В других вариантах осуществления антитело против CTLA-4 или его антигенсвязывающая часть содержит константную область тяжелой цепи, которая имеет изотип человеческого IgG1 или IgG4. В некоторых вариантах осуществления изобретения антитело против CTLA-4 содержит константную область тяжелой цепи, которая имеет изотип человеческого IgG1.
122. Для комбинации антитела против PD-1 или против PD-L1 и анти-CTLA-4-Ab, режим дозирования содержит индукционный период (также называемый в настоящем описании индукционной фазой), в течение которого пациенту вводят одну или более, две или более, три или более, или четыре или более комбинированные дозы антитела против PD-1 или против PD-L1 и анти-CTLA-4-Ab, после чего следует поддерживающий период или фаза, содержащий дозирование антитела против PD-1 или антитела против PD-L1 по отдельности, то есть, не включая антитела против CTLA-4. В определенных вариантах осуществления изобретения способ включает в себя (а) индукционную фазу, в которой антитела против PD-1 или против PD-L1 и против CTLA-4 или их антигенсвязывающие части вводят комбинированно в 2, 4, 6, 8 или 10 дозах, причем каждую дозу, варьирующую от около 0,1 до около 10,0 мг/кг массы тела, вводят по меньшей мере один раз приблизительно каждые 2 недели, один раз приблизительно каждые 3 недели или один раз приблизительно каждые 4 недели, с последующей (b) поддерживающей фазой, в которой не вводят антитело против CTLA-4 или его антигенсвязывающую часть, а антитело против PD-1 или его антигенсвязывающую часть или антитело против PD-L1 или его антигенсвязывающую часть многократно вводят в дозе, варьирующей от около 0,1 до около 10 мг/кг, по меньшей мере один раз приблизительно каждые 2 недели, один раз приблизительно каждые 3 недели или один раз приблизительно каждые 4 недели.
123. В определенных вариантах осуществления антитело против PD-1 или его антигенсвязывающую часть, или антитело против PD-L1 или его антигенсвязывающую часть вводят в субтерапевтической дозе. В некоторых других вариантах осуществления антитело против CTLA-4 или его антигенсвязывающую часть вводят в субтерапевтической дозе. В дополнительных вариантах осуществления как антитело против PD-1 или его антигенсвязывающую часть, или антитело против PD-L1 или его антигенсвязывающую часть, так и антитело против CTLA-4 или его антигенсвязывающую часть, каждое вводят в субтерапевтической дозе. В некоторых вариантах осуществления антитело против PD-1 или его антигенсвязывающую часть, или антитело против PD-L1 или его антигенсвязывающую часть, и/или антитело против CTLA-4 или его антигенсвязывающую часть вводит в терапевтической дозе.
124. В определенных вариантах осуществления (а) индукционная фаза содержит по меньшей мере 4 дозы, вводимые с 3-недельными интервалами, причем антитела против PD-1 и против CTLA-4 вводят в следующих дозах: (i) по меньшей мере около 1 мг/кг антитела против PD-1 и по меньшей мере около 3 мг/кг анти-СТLА-4-Ab; (ii) по меньшей мере около 3 мг/кг антитела против PD-1 и по меньшей мере около 3 мг/кг анти-СТLА-4-Ab; (iii) по меньшей мере около 1 мг/кг антитела против PD-1 и по меньшей мере около 3 мг/кг анти-CTLA-4-Ab; (iv) по меньшей мере около 3 мг/кг антитела против PD-1 и по меньшей мере около 3 мг/кг анти-СТLА-4-Ab; (v) по меньшей мере около 1 мг/кг антитела против PD-1 и по меньшей мере около 1 мг/кг анти-СТLА-4-Ab; (vi) по меньшей мере около 2 мг/кг антитела против PD-1 и по меньшей мере около 3 мг/кг анти-СТLА-4-Ab; (vii) по меньшей мере около 3 мг/кг антитела против PD-1 и по меньшей мере около 2 мг/кг анти-СТLА-4-Ab; (viii) по меньшей мере около 4 мг/кг антитела против PD-1 и по меньшей мере около 1 мг/кг анти-СТLА-4 Ab; (ix) по меньшей мере около 5 мг/кг антитела против PD-1 и по меньшей мере около 1 мг/кг анти-СТLА-4-Ab; (х) по меньшей мере около 5 мг/кг антитела против PD-1 и по меньшей мере около 1 мг/кг анти-СТLА-4-Ab; (xi) по меньшей мере около 6 мг/кг антитела против PD-1 и по меньшей мере около 1 мг/кг анти-СТLА-4-Ab; или (xii) по меньшей мере около 10 мг/кг антитела против PD-1 и по меньшей мере около 1 мг/кг анти-СТLА-4-Ab, и (b) поддерживающая фаза включает повторное введение антитела против PD-1 в дозе по меньшей мере около 3 мг/кг один раз каждые 2 недели.
125. Благодаря устойчивости клинического эффекта, ранее продемонстрированной с иммунотерапией путем ингибирования иммунных контрольных точек (см, например, WO 2013/173223), поддерживающая фаза может включать в себя, в альтернативных вариантах осуществления, конечное число доз, например, от около 1 до около 10 доз, или может включать дозирование с большими интервалами, например, приблизительно один раз каждые 3-6 месяцев или приблизительно один раз от 1 года до 2 лет или с более длительными интервалами. Поддерживающая фаза может продолжаться до тех пор, пока наблюдается клиническая польза или до появления неуправляемой токсичности или наступления прогрессирования заболевания.
126. Принимая во внимание неопределенность в том, привносит ли ипилимумаб, вводимый после 12-й недели, клиническую пользу при меланоме, и тот факт, что режим дозирования YERVOY®, утвержденный Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (U.S. Food and Drug Administration (FDA)) - и Европейским агентством по лекарственным средствам (European Medicines Agency (ЕМА)), представляет собой введение каждые 3 недели, в общей сложности, 4-х доз, в некоторых вариантах осуществления антитело против CTLA-4 вводят во время индукционной фазы приблизительно один раз каждые 3 недели для, в общей сложности, 4 доз. Соответственно, в определенных вариантах осуществления настоящего изобретения способ включает в себя (а) индукционную фазу, состоящую из 4 комбинированных доз, вводимых с 3-недельными интервалами, причем (i) антитело против PD-1 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 3 мг/кг массы тела, и антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 1 мг/кг массы тела; (ii) антитело против PD-1 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 1 мг/кг массы тела, и антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 3 мг/кг массы тела; (iii) антитело против PD-1 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 1 мг/кг массы тела, и антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 1 мг/кг массы тела; или (iv) антитело против PD-1 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 3 мг/кг массы тела, и антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 3 мг/кг массы тела; и (b) поддерживающая фаза включает повторное введение антитела против PD-1 или его антигенсвязывающей части в дозе по меньшей мере около 3 мг/кг приблизительно каждые 2 недели. В дополнительных вариантах осуществления данных способов поддерживающая фаза может продолжаться до тех пор, пока наблюдается клиническая польза или до появления неуправляемой токсичности или наступления прогрессирования заболевания.
127. В конкретном варианте осуществления способ включает (а) индукционную фазу, состоящую из 4 комбинированных доз, вводимых с 3-недельными интервалами, причем антитело против PD-1 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 3 мг/кг массы тела, и антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе, составляющей по меньшей мере около 1 мг/кг массы тела; и (b) поддерживающая фаза включает повторное введение антитела против PD-1 или его антигенсвязывающей части в дозе, составляющей по меньшей мере около 3 мг/кг, один раз приблизительно каждые 2 недели.
128. Как правило, антитела против PD-1 или против PD-L1 и против CTLA-4 сформулированы для внутривенного введения. В других вариантах осуществления антитело против PD-1 или антитело против PD-L1 и/или антитело против CTLA-4 сформулировано для прямого введения в центральную нервную систему. В некоторых вариантах осуществления комбинацию антитела против PD-1 или его антигенсвязывающей части, или антитела против PD-L1 или его антигенсвязывающей части и антитела против CTLA-4 или его антигенсвязывающей части вводят одновременно в виде единой композиции или в виде отдельных композиций. В некоторых вариантах осуществления антитела против PD-1 или против PD-L1 и против CTLA-4 вводят последовательно. В некоторых вариантах осуществления антитела против PD-1 или против PD-L1 и против CTLA-4 вводят последовательно во время индукционной фазы. В определенных вариантах осуществления, когда антитела против PD-1 или против PD-L1 и против CTLA-4 вводят в комбинации, их вводят с интервалом не больше 30 минут друг от друга. Одно из антител может быть введено в первую очередь, то есть, в определенных вариантах осуществления антитело против PD-1 или антитело против PD-L1 вводят перед анти-СТLА-4-Ab, в то время как в других вариантах осуществления антитело против CTLA-4 вводят перед антителом против PD-1 или антителом против PD-L1. Обычно, каждое антитело вводят внутривенным вливанием в течение 60 минут. В определенных вариантах осуществления антитела против PD-1 или против PD-L1 и против CTLA-4 вводят одновременно, либо в смеси в виде единой композиции в фармацевтически приемлемом препарате для одновременного введения, либо одновременно в виде отдельных композиций с каждым антителом в фармацевтически приемлемом препарате.
129. Определенные варианты осуществления настоящих способов включают (а) индукционную фазу, состоящую из введения ниволумаба внутривенным вливанием с последующим введением ипилимумаба внутривенным вливанием каждые 3 недели в 4 комбинированных дозах, с последующим (b) поддерживающим дозированием с ниволумабом, вводимым внутривенным вливанием каждые 2 недели, начиная через 3 недели после 4-й дозы индукционной терапии или после 113-го дня, если 4-ю дозу индукционной терапии не вводили из-за задержек в лечении.
Наборы
130. Кроме того, в пределы объема настоящего изобретения включены наборы, содержащие антитело против PD-1 и другое противораковое средство для терапевтического применения. Наборы обычно включают в себя этикетку, в которой указывается целевое назначение содержимого комплекта, и инструкции по применению. Термин этикетка включает в себя любую запись или записанный материал, поставляемый на комплекте или с комплектом, или который, в противном случае, сопровождает комплект. Соответственно, данное изобретение обеспечивает набор для лечения субъекта, пораженного глиомой, причем данный набор включает: (а) дозировку в диапазоне от по меньшей мере около 4 мг до по меньшей мере около 500 мг антитела против PD-1 или его антигенсвязывающей части; или (b) дозировку другого противоракового средства, описанного в настоящем изобретении и (с) инструкции по применению антитела против PD-1 и другого противоракового средства в любом из способов комбинированной терапии, описанных в настоящем изобретении. В определенных вариантах осуществления анти-PD-1-Ab, антитело против CTLA-4 и/или другое противораковое средство могут быть совместно упакованы в стандартной лекарственной форме. В определенных вариантах осуществления для лечения пациентов-людей набор содержит антитело против человеческого PD-1, раскрытое в настоящем изобретении, например, ниволумаб, пембролизумаб, MEDI0608 (ранее АМР-514), АМР-224 или BGB-A317. В других вариантах осуществления набор содержит антитело против человеческого CTLA-4, раскрытое в настоящем изобретении, например, ипилимумаб или тремелимумаб.
131. Настоящее изобретение далее иллюстрируется следующими примерами, которые не должны быть истолкованы как дальнейшее ограничение. Содержание всех ссылок, приведенных в данной заявке, специально включено сюда посредством ссылки.
ПРИМЕРЫ
ПРИМЕР 1
Рандомизированное открытое исследование 3-й фазы по сравнению ниволумаба с бевацизумабом и исследование безопасности ниволумаба или ниволумаба в комбинации с ипилимумабом у взрослых пациентов с рецидивной GBM
132. Цели данного исследования заключаются в следующем: 1) оценить безопасность и переносимость ниволумаба и ниволумаба в сочетании с ипилимумабом у пациентов с диагнозом рецидивной GBM в группе по первоначальной оценке безопасности (когорта 1, 1b); и 2) оценить безопасность, переносимость и эффективность ниволумаба по сравнению со стандартным лечением (бевацизумаб) у субъектов с диагнозом рецидивной GBM в рандомизированном исследовании (когорта 2).
133. Это первое исследование по изучению моноклональных антител, нацеленных на ингибиторы иммунных контрольных точек у пациентов с рецидивной GBM. Для того, чтобы гарантировать, что монотерапия ниволумабом и ниволумаб в сочетании с ипилимумабом переносимы, были инициированы когорты по первоначальной оценке безопасности (когорта 1, 1b) для оценки переносимости двух различных схем лечения до продвижения группы лечения в когорту 2. В когорте 1 исследовали, рандомизированным образом, безопасность и переносимость монотерапии ниволумабом в дозе 3 мг/кг каждые 2 недели или ниволумаба в дозе 1 мг/кг + ипилимумаба в дозе 3 мг/кг каждые 3 недели для четырех доз, а затем ниволумаб в дозе 3 мг/кг каждые 2 недели после этого. В когорте 1b исследовали, нерандомизированным образом, безопасность и переносимости ниволумаба в дозе 3 мг/кг + ипилимумаба в дозе 1 мг/кг каждые 3 недели для четырех доз, а затем ниволумаба в дозе 3 мг/кг каждые 2 недели после этого.
134. Лечение монотерапией ниволумабом в когорте 1 соответствовало заранее заданному протоколом профилю безопасности и переносимости (менее одной трети субъектов потребовалось постоянное прекращение приема вследствие связанных с лечением нежелательных явлений до получения 4 доз лечения) с тем, чтобы перейти к когорте 2, рандомизированной части исследования для сравнения монотерапии ниволумабом с бевацизумабом. Поскольку оценка режима дозирования для комбинированной терапии продолжается в когорте 1 и 1b, когорта 2 была разработана для оценки монотерапии ниволумабом в сравнении с бевацизумабом. Оценка эффективности комбинированной терапии ниволумаб + ипилимумаб будет проводиться отдельно, когда наберется достаточно данных из когорты 1 и когорты 1b, чтобы выбрать соответствующую комбинацию дозирования для продвижения в рандомизированное исследование.
135. Дозирование ниволумаба в когорте 2 основано на предварительном клиническом опыте субъектов с рецидивной GBM из когорты 1. Субъекты в группе лечения ниволумабом будут получать 3 мг/кг каждые 2 недели, и субъекты в группе лечения бевацизумабом будут получать дозу 10 мг/кг каждые 2 недели. Субъекты будут продолжать получать исследуемый препарат до подтвержденного прогрессирования опухоли, появления неприемлемой токсичности или других критериев отмены лечения, описанных в настоящем изобретении, в зависимости от того, что наступает в первую очередь. После прекращения лечения пациенты будут входить в последующую фазу сбора информации об общей выживаемости.
136. Данное исследование также будет предоставлять данные относительно выживаемости без прогрессирования заболевания, объективного ответа, биомаркеров прогрессирования заболевания, а также состояния здоровья. Субъекты в группах комбинированного лечения, прекратившие лечение ниволумаб + ипилимумаб из-за связанных с лечением нежелательных явлений до завершения четвертой дозы, могут начать прием ниволумаба в дозе 3 мг/кг каждые 2 недели с предварительного согласия спонсора.
Способы
Дизайн исследования
137. Рандомизированное, открытое, многоцентровое исследование 3-й фазы монотерапии ниволумабом по сравнению с бевацизумабом и исследование безопасности ниволумаба и ниволумаба в сочетании с ипилимумабом проводится у взрослых (>18 лет) субъектов с первым рецидивом GBM после лечения лучевой терапией и темозоломидом. Поскольку применение ниволумаба и ипилимумаба ранее не было изучено у пациентов с GBM, была образована группа по первоначальной оценке безопасности (когорта 1), состоящая из приблизительно 10 субъектов в каждой из двух групп лечения, которые зачислены и рандомизированы для лечения либо в группе лечения N (монотерапия ниволумаб), либо в группе лечения N+I (ниволумаб в сочетании с ипилимумабом). Переносимость и безопасность в группе лечения определяются после того, как все субъекты в группе лечения завершили прием четырех доз или прекратили прием доз до завершения приема четырех доз. Оценка безопасности для когорты 1 проводится для каждой группы лечения на основании критериев, описанных в настоящей заявке. Дизайн исследования для когорты 1 приведен на фигуре 1.
138. Для того чтобы лучше охарактеризовать предварительную безопасность и переносимость для комбинированной терапии, запускаются исследования дополнительной нерандомизированной когорты по оценке безопасности (когорта 1b), с целью оценить альтернативный режим дозирования для комбинированной терапии (ниволумаб в дозе 3 мг/кг + ипилимумаб в дозе 1 мг/кг каждые 3 недели в течение 4 циклов с последующим приемом ниволумаба в дозе 3 мг/кг после этого). Дизайн исследования для когорты lb приводится на фигуре 2. Оценка эффективности комбинированной терапии ниволумаб + ипилимумаб будет проводиться отдельно. Информация, полученная от пациентов, получавших комбинированную терапию в когорте 1 и когорте 1b, будет служить основой для выбора дозы для будущих исследований у субъектов с GBM.
139. Зачисление в когорту 2 планируется начать после того, как будет оценена безопасность и переносимость в группе по первоначальной оценке безопасности и принято решение относительно того, какие группы лечения (группа лечения N и/или группа лечения N+I) будут изучаться. Лечение монотерапией ниволумабом в когорте 1 соответствовало заранее заданным протоколом профилю безопасности и переносимости (менее одной трети субъектов потребовалось постоянное прекращение приема вследствие связанных с лечением нежелательных явлений до получения 4 доз лечения)с тем, чтобы перейти к когорте 2, рандомизированной части исследования для сравнения монотерапии ниволумабом с бевацизумабом. Дизайн исследования для когорты 2 приведен на фигуре 3. Поскольку оценка второго режима дозирования для комбинированной терапии продолжается, рандомизированная часть данного исследования (когорта 2) ограничена сравнением монотерапии ниволумабом с бевацизумабом. Оценка эффективности комбинированной терапии ниволумаб + ипилимумаб будет проводиться отдельно, когда наберется достаточно данных из когорты 1 и когорты 1b, чтобы выбрать соответствующую дозу для продвижения (либо ниволумаб в дозе 3 мг/кг + ипилимумаб в дозе 1 мг/кг, либо ниволумаб в дозе 1 мг/кг + ипилимумаб в дозе 3 мг/кг) в рандомизированное исследование.
140. За всеми субъектами в когорте 1, 1b и 2 будет продолжаться наблюдение в отношении безопасности и переносимости, прогрессирования опухоли и общей выживаемости. Конечные точки прогрессирования или ответа опухоли оцениваются с использованием критериев оценки эффективности лечения опухолей RANO (Radiologic Assessment in Neuro-Oncology criteria), как описано в настоящем изобретении. Лечение исследуемым препаратом продолжалось до подтвержденного прогрессирования опухоли, появления неприемлемой токсичности или других критериев прекращения лечения, как описано в настоящем изобретении, в зависимости от того, что наступало в первую очередь.
Фазы исследования
141. Данное исследование включает в себя три фазы: скрининг, лечение (когорта 1, 1b и когорта 2), и последующее наблюдение.
142. Фаза скрининга - Фаза скрининга начинается с установления первоначального права субъекта и подписания информированного согласия (informed consent form (ICF)). Субъект зачисляется с использованием системы интерактивного голосового ответа (Interactive Voice Response System (IVRS)).
143. Фаза лечения - Фаза лечения начинается с вызова для рандомизации к IVRS. Субъекты в когорте 1 и когорте 2 случайным образом распределяются в одну из групп лечения, основываясь на когорте исследования. Субъекты в когорте lb получают назначение в группу лечения N+I_1b. В течение 3-х рабочих дней с момента назначения лечения, субъект получит первую дозу исследуемого препарата. Группа лечения N (ниволумаб в качестве монотерапии) будет получать ниволумаб в дозе 3 мг/кг внутривенно (IV) каждые две недели. Группа лечения N+I (ниволумаб + ипилимумаб) будет получать ниволумаб в дозе 1 мг/кг IV в сочетании с ипилимумабом в дозе 3 мг/кг IV каждые три недели в 4 дозах, затем ниволумаб (когорта 1 только) в дозе 3 мг/кг IV каждые две недели. Группа лечения N+I_1b (ниволумаб + ипилимумаб) получит ниволумаб в дозе 3 мг/кг IV в сочетании с ипилимумабом в дозе 1 мг/кг IV каждые три недели, всего 4 дозы, затем ниволумаб (когорта 1b только) в дозе 3 мг/кг IV каждые две недели. Группа лечения В (бевацизумаб) когорты 2 будет получать бевацизумаб в дозе 10 мг/кг каждые 2 недели. Оценки нежелательных явлений документируются при каждом посещении врача на протяжении всего исследования. Оценки качества жизни с использованием EORTC QLQ-С30 и BNS20 и EQ-5D завершаются на день 1 недели 1.
144. Все лабораторные анализы и показатели жизнедеятельности собирают до начала дозирования исследуемого лекарственного средства, включая, но не ограничиваясь ими, общий анализ крови с подсчетом лейкоцитарной формулы (СВС с дифференциалом), LDH, AST, ALT, ALP, T.Bili, BUN или уровень мочевины в сыворотке крови, содержание креатинина, Са, Mg, Na, K, Cl, глюкозы, амилазы, липазы, ТСГ с рефлексивным Free Т4 и Free Т3. Также собирают образцы крови на РК, иммуногенность и биомаркеры. Дозирования исследуемого лекарственного средства задерживается в некоторых случаях из-за токсичности. Получающие лечение субъекты оцениваются исследователем в отношении ответа и в соответствии с критериями RANO. Оценки эффективности лечения опухоли выполняются, начиная с конца недели 6 (±1 неделя) после первой дозы, в конце недели 12 (±1 неделя) и продолжая каждые 8 недель (±1 неделя) до прогрессирования заболевания или прекращения лечения, в зависимости от того, что происходит позже. Получающие лечение субъекты оцениваются на неврологическое функционирование исследователем и в соответствии со шкалой NANO (когорта 2 только). Оценки выполняются, начиная с конца недели 6 (±1 неделя) после первой дозы, в конце недели 12 (±1 неделя) и продолжая каждые 8 недель (±1 неделя) до прогрессирования заболевания или прекращения лечения, в зависимости от того, что происходит позже. Получающие лечение субъекты оцениваются на неврологическое функционирование исследователем с помощью. Cogstate (когорта 2 только). Оценки проводятся при скрининге, неделя 1, неделя 13, неделя 21, неделя 45, неделя 69, и при прекращении лечения (если >4 недель после предварительной Cogstate-оценки).
145. Фаза лечения заканчивается, когда у субъекта наблюдаются подтвержденное прогрессирование опухоли, неприемлемая токсичность или другие критерии прекращения лечения, в зависимости от того, что происходит в первую очередь.
Фаза последующего наблюдения - Фаза последующего наблюдения начинается, когда принимается решение о прекращении субъектом приема исследуемой терапии (дальнейшее лечение с исследуемой терапией не происходит). Два визита последующего наблюдения включают в себя сбор образцов на РК/иммуногенность и заполнение опросников о качестве жизни. У субъектов, которые прекратили лечение по причинам, отличным от прогрессирования опухоли, продолжают проводить оценки опухоли, начиная с конца недели 6 (±1 неделя) после первой дозы, в конце недели 12 (±1 неделя), а затем каждые 8 (±1 неделя) недель до прогрессирования заболевания или отзыва согласия. Все случаи рентгенологически установленного прогрессирования болезни подтверждаются дополнительным подтверждающим МРТ-сканированием приблизительно через 12 недель после начальной оценки рентгенологического прогрессирования. Исследователи получают дополнительные МРТ-сканы для последующего наблюдения до 12 недель, насколько уместно по медицинским показаниям. За субъектами будут проводить наблюдение в отношении токсичностей, связанных с лекарственным средством до тех пор, пока данные токсичности не будут устранены, вернутся к исходному уровню или считаются необратимыми. Все нежелательные явления будут документированы в течение как минимум 100 дней после последней дозы. После завершения первых двух посещений последующего наблюдения, за субъектами будут проводить последующее наблюдение каждые 3 месяца в отношении выживания.
Популяция исследования
147. Популяция исследования включает в себя субъектов >18 лет с гистопатологически подтвержденным диагнозом злокачественной глиомы степени IV по классификации ВОЗ (GBM или глиосаркома). Популяция включает в себя субъектов, ранее получавших терапию первой линии, по меньшей мере, с лучевой терапией и темозоломидом. Субъекты, имеющие документированный первый рецидив глиобластомы по диагностической биопсии или МРТ с контрастным усилением, выполненной в течение 21 дней рандомизации по критериям RANO, выбраны в качестве исследуемой популяции.
148. Если первый рецидив GBM документирован с помощью МРТ, требуется интервал, составляющий по меньшей мере 12 недель после окончания предшествующей лучевой терапии, если нет: i) гистопатологического подтверждения рецидива опухоли или ii) нового усиления согласно данным МРТ за пределами поля лечения лучевой терапией. Все субъекты должны ждать по меньшей мере 28 дней и проявлять признаки полного восстановления (т.е., нет актуальных проблем обеспечения безопасности) после хирургической резекции перед рандомизацией. Субъекты должны ждать по меньшей мере 4 недели после последнего введения любого другого средства лечения глиобластомы. Субъекты должны иметь показатель общего состояния по шкале Карновского (Karnofsky), равный 70 или выше, и вероятную продолжительность жизни не менее 12 недель.
149. Для того, чтобы участвовать в группе 1 или 1b, субъекты должны иметь по меньшей мере одно измеряемое GBM-поражение до рандомизации, которое отвечает следующим критериям: i) усиливающие контраст и четко определенные, двумерно измеримые края, и ii) по меньшей мере два перпендикулярных друг другу диаметра, измеренные как >10 мм на >10 мм. МРТ-измерения не включают в себя хирургическую полость, кисту или зону некроза.
150. Субъектам запрещается сопутствующая терапия с любым лекарственным средством или другими экспериментальными средствами для лечения GBM (то есть, химиотерапия, гормональная терапия, иммунотерапия, лучевая терапия) и какие-либо лекарства, противопоказанные при лечении бевацизумабом.
151. Субъекты исключаются из исследования, если они имели больше одного рецидива глиобластомы, имеют внечерепное метастатическое лептоменингеальное поражение, или им был поставлен диагноз вторичной GBM (то есть, GBM, которые прогрессируют от низко дифференцированной диффузной астроцитомы или анапластической астроцитомы). Субъекты с положительным результатом теста на поверхностный антиген вируса гепатита В (hepatitis В virus surface antigen (HBV sAg)), с обнаруживаемой рибонуклеиновой кислотой вируса гепатита С (hepatitis С virus ribonucleic acid (HCV RNA)), с указанием острой или хронической инфекции или с известной историей положительного результата теста на вирус иммунодефицита человека (ВИЧ) или синдром приобретенного иммунодефицита (СПИД), также исключаются из данного исследования.
152. Кроме того, субъекты исключаются по любой из следующих причин:
а) Любое серьезное или неконтролируемое медицинское расстройство, которое, по мнению исследователя, может увеличить риск, связанный с участием в исследовании или введением препарата исследования, ухудшить способность субъекта принимать терапию по протоколу или мешать интерпретации результатов исследования.
b) Субъекты с активным, известным или подозреваемым аутоиммунным заболеванием. Субъектам с витилиго, сахарным диабетом типа I, остаточным гипотиреозом вследствие аутоиммунного состояния, требующим только замены гормонов, псориазом, не требующим хронического и системного иммуносупрессивного лечения, или с состояниями, от которых не ожидается повторения в отсутствие внешнего триггера, было разрешено зачисляться в исследование;
c) Предыдущая лучевая терапия, отличающаяся от стандартной лучевой терапии (то есть, фокально направленное облучение);
d) Субъекты, требующие эскалирования или хронических супрафизиологических доз кортикостероидов для контроля их заболевания при рандомизации, исключены;
e) Предыдущее лечение с имплантированными пластинами кармустина, за исключением случая введения в качестве терапии первой линии и по меньшей мере за 6 месяцев до рандомизации;
f) Предыдущее лечение бевацизумабом или другим VEGF, или анти-ангиогенное лечение (когорта 2 только);
g) Предыдущее лечение с PD-1- или CTLA-4-направленной терапией;
h) Свидетельство о кровоизлиянии в ЦНС >1 степени на исходном скане МРТ;
i) Недостаточно контролируемая гипертония (определяется как систолическое артериальное давление >150 мм рт.ст. и/или диастолическое артериальное давление >100 мм рт.ст.) в течение 28 дней после первого лечения в программе исследования;
j) Предшествующая история гипертонического криза, гипертонической энцефалопатии, синдром обратимой задней лейкоэнцефалопатии (reversible posterior leukoencephalopathy syndrome (RPLS));
k) Предшествующая история желудочно-кишечного дивертикулита, перфорации или абсцесса;
l) Клинически значимое (т.е., активное) сердечно-сосудистое заболевание, например, инсульты <6 месяцев до включения в исследование, инфаркт миокарда <6 месяцев до включения в исследование, нестабильная стенокардия, застойная сердечная недостаточность (congestive heart failure (CHF)) класса II или более по классификации Нью-Йоркской ассоциации сердца (NYHA), или серьезные нарушения сердечного ритма, не контролируемые с помощью лекарств или потенциально мешающие лечению по протоколу;
m) История или свидетельство о физикальном/неврологическом обследовании заболевания центральной нервной системы (например, судорога), не связанного с раком, если должным образом не контролируется с помощью лекарств или потенциально мешает лечению по протоколу;
n) Серьезные сосудистые заболевания (например, аневризма аорты, которая требует хирургического вмешательства, или недавний тромбоз артерий) в течение 6 месяцев до начала лечения в программе исследования. Любая предыдущая венозная тромбоэмболия >3-й степени по классификации NCI СТСАЕ;
о) История легочного кровотечения/кровохаркания>2-й степени (определяется как >2,5 мл ярко-красной крови за эпизод) в течение 1 месяца до рандомизации;
p) История или свидетельство наследственного геморрагического диатеза или значительной коагулопатии с риском кровотечения (то есть, при отсутствии терапевтических антикоагулянтов);
q) Текущее или недавнее (в течение 10 дней с момента включения в исследование) применение антикоагулянтов, что, по мнению следователя, подвергнет субъекта значительному риску кровотечения. Профилактическое применение антикоагулянтов было разрешено;
r) Хирургическая процедура (в том числе, открытая биопсия, хирургическая резекция, ревизия раны или любая другая крупная внутриполостная операция) или значительное травматическое повреждение в течение 28 дней до начала первого лечения в программе исследования или предвидение необходимости серьезной хирургической процедуры в ходе исследования;
s) Незначительная хирургическая процедура (например, стереотаксическая биопсия в течение 7 дней после первого лечения в программе исследования; размещение устройства для обеспечения сосудистого доступа в течение 2 дней первого лечения в программе исследования);
t) История внутричерепного абсцесса в течение 6 месяцев до рандомизации;
u) История активного желудочно-кишечного кровотечения в течение 6 месяцев до рандомизации;
v) Серьезная, незаживающая рана, активная язва или нелеченный перелом кости; и/или
w) Субъекты, неспособные (из-за существующего медицинского состояния, например, кардиостимулятор или ICD-устройство) или не желающие подвергаться МРТ головы с контрастным усилением.
Критерии прекращения
153. Лечение ниволумабом и/или ипилимумабом должно быть окончательно прекращено при наличии любого связанного с лекарственными средствами увеита 2-й степени или боли в глазах или затуманенного зрения, который не реагирует на местную терапию и не улучшается до степени тяжести 1-й степени в течение периода повторного лечения, или требуется системное лечение. Лечение также будет прекращено, если имеется любое некожное, связанное с лекарственными средствами нежелательное явление 3-й степени длительностью >7 дней, со следующими исключениями в отношении связанных с лекарственными средствами отклонений лабораторных показателей, увеита, пневмонита, бронхоспазма, диареи, колита, неврологической токсичности, реакции гиперчувствительности и реакций на инфузии:
(a) Связанные с лекарственными средствами увеит, пневмонит, бронхоспазм, диарея, колиты, неврологические токсичности, реакции гиперчувствительности или реакции на инфузии 3-й степени любой продолжительности требуют прекращения.
(b) Связанные с лекарственными средствами отклонения лабораторных показателей 3-й степени не требуют прекращения лечения, за исключением:
(i) Связанная с лекарственными средствами тромбоцитопения 3-й степени >7 дней или связанная с кровотечением требует прекращения лечения
(ii) Любое отклонение теста на функцию печени (liver function test (LFT)), связанное с лекарственными средствами, которое удовлетворяет следующим критериям, требует прекращения лечения:
AST или ALT >8 × ULN
Общий билирубин >5 × ULN
Параллельный AST или ALT >3 × ULN и общий билирубин >2 × ULN.
154. Лечение также может быть прекращено, если имеется какое-либо нежелательное явление или отклонения лабораторных показателей 4-й степени, связанные с лекарственными средствами, за исключением следующих событий, которые не требуют прекращения:
(a) Отклонения в показателях для амилазы или липазы 4-й степени, которые не связаны с симптомами или клиническими проявлениями панкреатита. Рекомендуется проконсультироваться с медицинским консультантом BMS относительно отклонений лабораторных показателей амилазы или липазы 4-й степени.
(b) Отдельные дисбалансы/отклонения электролитов 4-й степени, которые не связаны с клиническими осложнениями и скорректированы с пищевой добавкой/надлежащим управлением в течение 72 часов с момента их появления
155. Лечение будет прекращено, если имеется любое прерывание дозирования длительностью >6 недель после последней дозы, со следующими исключениями:
(a) Разрешены перерывы в дозировании, чтобы можно было с помощью длительного равномерного снижения дозировки стероидных препаратов управлять нежелательными явлениями, связанными с лекарственными средствами. До повторного начала лечения у субъекта с прерыванием дозирования длительностью >6 недель должны быть проведены консультации с медицинским консультантом BMS. Оценки опухоли следует продолжать в соответствии с протоколом, даже если дозирование прерывается.
(b) Могут быть разрешены перерывы в дозировании >6 недель после последней дозы, которые возникают по причинам, не связанным с лекарственными средствами, при условии одобрения медицинского консультанта BMS. До повторного начала лечения у субъекта с прерыванием дозирования длительностью >6 недель, должны быть проведены консультации с медицинским консультантом BMS. Оценки опухоли следует продолжать в соответствии с протоколом, даже если дозирование прерывается.
156. Лечение будет прекращено, если имеется какое-либо нежелательное явление, отклонение лабораторных показателей или интеркуррентное заболевание, которое, по мнению исследователя, представляет значительный клинический риск для субъекта при продолжении дозирования ниволумаба или ипилимумаба.
157. Кроме того, лечение бевацизумабом должно быть окончательно прекращено в соответствии с обновленным вкладышем о бевацизумабе.
158. Лечение также будет прекращено, если субъекты проявляют симптомы 3-й степени или 4-й степени (например, тяжелая реакция). Симптомы 3-й степени включают длительные симптомы (то есть, не реагирующие быстро на симптоматическое лекарство и/или кратковременное прерывание инфузии); рецидив симптомов после первоначального улучшения; или госпитализацию, указанную для других клинических осложнений (например, нарушения функции почек, легочные инфильтраты)). Степень 4 включает в себя угрожающие жизни симптомы или прессорную поддержку или вспомогательное дыхание по показаниям.
159. Исследуемое лечение должно быть прекращено после подтвержденного рентгенологического прогрессирования или клинического прогрессирования, в зависимости от того, что наступит в первую очередь. Подробная информация о рентгенологическом прогрессировании или клиническом прогрессировании дана ниже:
160. Стандартное лечение GBM (включая лучевую терапию и темозоломид) может привести к временному увеличению опухоли (псевдопрогрессирование) у части субъектов, которое, в конечном счете, спадает без какого-либо изменения в терапии. Псевдопрогрессирование можно с трудом отличить от истинного прогрессирования опухоли, и оно может иметь важные последствия для лечения пациента. Имеются также свидетельства, что у некоторых субъектов, получавших стимулирующие иммунную систему агенты, может также развиться кажущееся прогрессирование болезни (на основании обычных критериев ответа), прежде чем будут продемонстрированы клинические объективные ответы и/или стабильное заболевание. Данное явление наблюдалось приблизительно у 10% пациентов в исследовании фазы 1 ниволумаба и также отмечалось при монотерапии ипилимумабом.
161. Для того, чтобы свести к минимуму преждевременное прекращение лечения исследуемым препаратом и отличить псевдопрогрессирование от прогрессирования заболевания, субъекты, у которых первоначально наблюдаются рентгенологические критерии прогрессирования заболевания, могут продолжать прием исследуемого препарата до подтверждения прогрессирования при прохождении МРТ приблизительно через 12 недель.
162. Для того, чтобы продолжить лечение по программе исследования после оценки первоначального рентгенологической прогрессирования, должны быть выполнены следующие критерии:
(a) По мнению исследователя, у субъекта наблюдается улучшение клинических показателей.
(b) Субъект переносит исследуемый препарат.
163. Пациенты с подтвержденным прогрессированием (приблизительно через 12 недель после первоначально оцененного прогрессирования) прекратят прием исследуемого препарата и войдут в фазу последующего наблюдения/выживания исследования. Если прогрессирование подтверждается, то дата прогрессирования заболевания будет первой датой, когда субъект отвечает критериям прогрессирования.
164. В тех случаях, в которых прогрессирование, установленное рентгенологическим исследованием, нельзя отличить от псевдопрогрессирования, и, по мнению исследователя, хирургическая резекция с целью получить опухолевую ткань для гистопатологического исследования отвечает интересам субъекта, может быть проведена хирургическая резекция после консультации, проведенной с медицинским консультантом BMS. Образцы опухоли для биопсии (блоки или срезы) должны быть представлены на рассмотрение центрального невропатолога, чтобы свести к минимуму любое различие в гистопатологической оценке прогрессирования, сделанной разными наблюдателями, по сравнению с изменениями, связанными с лечением. Если патология опухоли подтверждает прогрессию, то прием исследуемого лекарственного средства данным субъектом будет прекращен в соответствии с протоколом критериев отмены лечения, и датой прогрессирования будет день, когда оно было впервые заподозрено. Если патология опухоли выявляет изменения, связанные с лечением, и не подтверждает прогрессирование заболевания, субъект может продолжить прием исследуемого препарата. МРТ после резекции требуется до продолжения лечения. У субъекта затем продолжат проведение всех оценок опухоли при последующем лечении в соответствии с графиком лечения.
165. В тех случаях, в которых прогрессирование, установленное рентгенологическим исследованием, нельзя отличить от псевдопрогрессирования, и пациент подвергается биопсии или диагностической резекции с целью получить опухолевую ткань для патоморфологического анализа, репрезентативные образцы опухолевой ткани будут рассмотрены на местном уровне и представлены на рассмотрение центральным невропатологом. Если есть несоответствие между центральным и местным нейропатологическими определениями прогрессирования по сравнению с измененениями, связанными с лечением (псевдопрогрессирование), тогда будет выполнен второй центральный нейропатологический обзор, который будет служить в качестве окончательного решения по гистопатологическому определению. Центральный обзор гистопатологии будет слепым в отношению назначения субъекта в группу лечения. Конкретные инструкции будут предоставлены в центры исследования в отношении требований к представлению опухолевой ткани для центрального нейропатологического обзора.
Информация о дозировании
166. Режим и график дозирования для группы лечения N (ниволумаб) и группы лечения N+I (ниволумаб и ипилимумаб) и группы лечения В (бевацизумаб) подробно описаны в таблице 1, таблице 2, таблице 3 и таблице 4. Когда ниволумаб и ипилимумаб вводят в тот же самый день, для каждой инфузии используются отдельные емкости и фильтры для инфузий. Ниволумаб вводят в первую очередь. Второй инфузией вводят всегда ипилимумаб, и она начинается не раньше, чем через 30 минут после завершения инфузии ниволумаба.





Окно дозирования группы лечения N+I (неделя 1-12)
167. Субъекты получают дозу с интервалом не менее 19 дней между дозами и не более 3-х дней после запланированной даты дозирования. Доза, которая дается после интервала в 3 дня, считается задержкой дозы. Допустима максимальная задержка в 42 дня между дозами. Если дозирование задерживается, оба ниволумаб и ипилимумаб задерживаются вместе. Если дозирование возобновляется после задержки, введение как ниволумаба, так и ипилимумаба возобновляется в тот же день.
Окно дозирования группы лечения N, группы лечения В и группы лечения N+I (неделя 13 и далее)
168. Субъекты получают дозу с интервалом не менее 12 дней между дозами и не более 3-х дней после запланированной даты дозирования. Доза, которая дается после интервала в 3 дня, считается задержкой дозы. Допустима максимальная задержка в 42 дней между дозами.
169. Оценки и процедуры исследования представлены в таблице 5. В частности, оценки скрининга описаны в таблице 5А; производимые во время исследования оценки групп лечения ниволумабом (N) и бевацизумабом (В) показаны в таблице 5 В; производимые во время исследования оценки группы лечения ниволумаб + ипилимумаб (N+I) описаны в таблице 5С; и оценки последующего наблюдения описаны в таблице 5D.
Оценка эффективности
МРТ головы
170. Все пациенты получали оценки эффективности по результатам МРТ головного мозга с контрастированием на 1-й день на неделе 7,1-й день на неделе 13, а затем один раз каждые 8 недель (±1 неделя). Исследования МРТ происходили в срок ±7 дней при запланированных посещениях. Исследователи получат более частые МРТ в рамках последующего наблюдения в соответствии с медицинскими показаниями.
171. Рентгенологическую оценку измерений опухоли, проводимую на месте, использовали во время исследования для клинического лечения и прогрессирования заболевания, оцениваемого исследователем. Случаи подозреваемого прогрессирования заболевания по результатам рентгенологического исследования были подтверждены МРТ, которую проводили приблизительно через 12 недель после первоначальной рентгенологической оценки прогрессирования.
Оценка безопасности
172. Исходно будет изучен медицинский анамнез с тем, чтобы оценить патологическое состояние. Основные обследования включают в себя массу, рост, показатель общего состояния по шкале Карновского, BP, HR, температуру, ЭКГ с 12 отведениями и насыщение кислородом с помощью пульсоксиметрии в покое и после нагрузки. Исходными признаками и симптомами являются те, которые оцениваются в течение 14 дней до рандомизации. Сопутствующие медицинские назначения будут собраны на протяжении 14 дней до рандомизации в течение всего периода лечения в программе исследования.
173. Исходные местные лабораторные анализы должны быть сделаны в течение 14 дней до рандомизации, чтобы включить в себя: общий анализ крови с подсчетом лейкоцитарной формулы (СВС w/дифференциал), ANC, тромбоциты, Hgb; панель химических показателей, включая ALT, AST, общий билирубин, уровень щелочной фосфатазы, BUN или уровень мочевины в сыворотке крови, креатинин, Са, Mg, Na, K, Cl, LDH, уровень глюкозы, амилазы, липазы, TSH, свободный Т4, свободный Т3 и Hep В и тестирование С (HBV sAg, антитело к ВГС или РНК ВГС). Тестирование на беременность для женщин детородного возраста (woman of childbearing potential, WOCBP, выполняется на месте) должно быть выполнено в течение 24 часов до первоначального введения исследуемого препарата на исходном уровне. Там, где это требуется местными правилами, также должны быть выполнен тест на ВИЧ.
174. Для субъектов будет оцениваться безопасность, если они получали какой-либо исследуемый медицинский препарат. Оценка токсичности будет непрерывной в течение фазы лечения и стадии последующего наблюдения за безопасностью. Побочные явления и лабораторные показатели будут классифицироваться в соответствии с критериями NCI-СТСАЕ версии 4.0.
175. В процессе исследования, масса, общее состояние по шкале Карновского и показатели жизнедеятельности будут оцениваться при каждом контрольном визите до начала дозирования. Показатели жизнедеятельности также будут сниматься в соответствии с институциональным стандартом медицинской помощи до, во время и после дозирования. Измерение насыщение кислородом с помощью пульсоксиметрии в покое и после нагрузки будет оцениваться при каждом контрольном визите в период исследования до начала дозирования. Анализ мочи (тест-полоски) будет получен в течение 72 часов до введения дозы для пациентов, рандомизированных для приема бевацизумаба. Момент времени, соответствущий началу и прекращению вливания ниволумаба и ипилимумаба и приема бевацизумаба, соотвественно, будет документирован. Физикальные обследования должны быть выполнены по клиническим показаниям. Если есть какие-либо новые клинически значимые изменения или ухудшение клинически значимых изменений с момента последнего обследования, изменения заносятся на страницу для сообщения о нежелательных явлениях различной степени серьезности.
176. Дополнительные меры, включая не требуемые для исследования лабораторные тесты, будут проводиться по клиническим показаниям или в соответствии с местными правилами. Во время лечения будут получены ЭКГ в 12 отведениях, если есть клинические указания. Выявляемые лабораторными показателями случаи токсичности (например, повышения уровней ферментов в печени, предположительно, индуцированные препаратами) будут контролироваться в течение стадии последующего наблюдения на месте/в местных лабораториях, пока все связанные с исследуемым препаратом токсичности не будут устранены, вернутся к исходному уровню или будут признаны считаются необратимыми.
177. Определение степени насыщения кислородом с помощью пульсоксиметрии будет проводиться перед введением каждой дозы ниволумаба, а также в любое время, когда у субъекта будут появляться какие-либо новые симптомы респираторных заболеваний или наступает будет наблюдаться их усиление. Считывание показателей в состоянии покоя будет осуществляться в каждый момент времени. Если у испытуемого наблюдаются изменения пульсоксиметрии или другие связанные с легочной патологией явления (например, гипоксия, лихорадка) или симптомы (например, одышка, кашель), указывающие на возможность нежелательных явлений в легких, субъект будет подвергаться немедленному осмотру с тем, чтобы исключить легочную токсичность.
178. Анализ мочи должен быть получен для всех субъектов в течение 72 часов до введения дозы. Анализ мочи будет собираться во время лечения только для пациентов, рандомизированных для введения бевацизумаба. Субъекты со значением 2+ или более для протеинурии (уровень белка в моче), полученным измерением с тест-полосками, должны пройти дополнительную оценку с 24-часовым сбором мочи перед введением дозы. Дозирование бевацизумаба должно быть приостановлено при уровне >2 г белка в моче/24 часа и может возобновиться, когда уровень белка в моче составляет <2 г/24 часа. Лечение должно быть прекращено у пациентов с нефротическим синдромом.
179. Некоторые из ранее упомянутых оценочных параметров могут не быть внесены в качестве данных в электронную историю болезни (eCRF, electronic case report form). Они предназначены для использования в качестве контроля безопасности со стороны лечащего врача. Дополнительные анализы и осмотры могут проводиться в случае клинической необходимости или по требованию институциональных или местных правил.
Оценка изображений для исследования
180. У всех субъектов проводят оценку безопасности, измерения опухоли и объемную магнитно-резонансную томографию (МРТ) головы в течение 21 дней до рандомизации. См. таблицу 5А. Для группы лечения N и группы лечения В (когорта 2) МРТ с контрастным усилением будет выполняться на 1-й день недель 7 и 13 и затем каждые 8 недель (+/- 1 неделя) недель. См. таблицу 5В. В случае группы лечения N+I, контрастную МРТ проводили на 1-й день недель 7 и 13, а затем каждые 8 недель (+/- 1 неделю). См. таблицу 5С. В отношении последующих оценок у всех субъектов, оценка опухоли требовалась для субъектов, у которых не было прогрессирования во время проведения исследуемой терапии, включая субъектов, которые начинают подпоследовательную противораковую терапию. Что касается последующих оценок, оценку опухоли получают в конце недель 6 и 12 (+/- 1), а затем каждые 8 недель (+/- 1 неделя) после этого. См. таблицу 5D.
181. Исследователи могут проводить последующие МРТ-исследования еще чаще, если имеются медицинские показания. Все рентгенологические изображения из данного исследования будут переданы в централизованную лабораторию по визуализации для хранения и могут быть рассмотрены независимым комитетом по обзору рентгенологических данных (Independent Radiology Review Committee (IRRC)), как определено спонсором. Любые побочные результаты потенциального клинического значения, которые непосредственно не связаны с целями протокола, должны быть оценены и обработаны исследователем, участвующим в исследовании, в соответствии со стандартом медицинской/клинической оценки. Центры исследования сохраняют локальный доступ к результатам визуализации в целях оценки безопасности и эффективности. Исследователь, участвующий в исследовании, будет рассматривать полученные на местах результаты МРТ как клинически приемлемые для обеспечения того, чтобы любые потенциально чрезвычайные клинические ситуации рассматривались своевременно.
182. Клинически значимые рентгенологические данные или отклонения от исходных сканирований будут кодироваться как нежелательные явления или серьезные нежелательные явления.
Оценки эффективности
183. Основной мерой эффективности исследования является общая выживаемость (OS, overall survival). OS определяется как время между датой рандомизации и датой смерти по любой причине. OS отслеживается непрерывно, пока субъекты принимают исследуемый препарат, и каждые 3 месяца при личном контакте или по телефону во время исследовательской фазы последующего наблюдения за выживанием.
184. Оцениваемый исследователем ответ опухоли основан на опубликованных критериях RANO. Оценки опухоли выполняются, как рассмотрено выше в связи с оценкой изображений для исследования. Рентгенологический ответ оценивали, сравнивая исходные сканы МРТ перед лечением и сканы МРТ во время лечения. Рентгенологическое прогрессирование определяли с помощью измерения наименьшего размера опухоли либо от исходного уровня перед лечением, либо после начала приема исследуемого препарата.
185. Требования к полному ответу: полное прекращение увеличения проявлений измеряемой и неизмеряемой болезни, устойчивое в течение по меньшей мере 4 недель; отсутствие новых опухолевых очагов; стабильные или показывающие улучшение, не характеризующиеся увеличением плотности, отслеживаемым по накоплению контрастного материала (в режиме T2/FLAIR), опухолевые очаги; пациенты не должны принимать кортикостероиды (или принимать их только в физиологические замещающих дозах); и стабильность клинического состояния или или его улучшение. Пациенты только с неизмеряемыми проявлениями болезни не могут иметь полный ответ; лучший ответ, возможно, представляет собой стабильное заболевание.
186. Требования к частичному ответу: >50%-ное снижение по сравнению с исходным уровнем в сумме произведений перпендикулярных друг другу диаметров всех измеримых усиливающих интенсивность сигнала опухолевых очагов, устойчивое в течение по меньшей мере 4 недель; нет прогрессирования неизмеряемых проявлений болезни; нет новых опухолевых очагов; стабильные или показывающие улучшение, не характеризующиеся увеличением плотности, отслеживаемым по накоплению контрастного материала (в режиме T2/FLAIR), опухолевые очаги при такой же или более низкой дозе кортикостероидов по сравнению с исходным сканированием; доза кортикостероидов на момент проведения оценки сканированием не должна превышать дозу на момент исходного сканирования; стабильность клинического состояния или или его улучшение. Пациенты только с неизмеряемыми проявлениями болезни не могут иметь частичный ответ; лучший ответ, возможно, представляет собой стабильное заболевание.
187. Требования к стабильному заболеванию: не отвечает требованиям полного ответа, частичного ответа или прогрессирования; стабильные, не характеризующиеся увеличением плотности, отслеживаемым по накоплению контрастного материала я (в режиме T2/FLAIR), опухолевые очаги при такой же или более низкой дозе кортикостероидов по сравнению с исходным сканированием. В том случае, если доза кортикостероидов увеличивается из-за новых симптомов и признаков без подтверждения прогрессирования заболевания по нейровизуализации, а последующая визуализация показывает, что данное увеличение дозы кортикостероидов было необходимо по причине прогрессирования заболевания, последним сканированием, которое может считаться показывающим стабильное заболевание, будет сканирование, проведенное, когда доза кортикостероидов была эквивалентна исходной дозе.
188. Прогрессирование определяется любым из следующих: >25%-ное увеличение суммы произведений перпендикулярных друг другу диаметров растущих опухолевых очагов по сравнению с наименьшим размером опухоли, определенным либо на исходном уровне (если нет уменьшения), либо как лучший ответ при стабильных (включая субъектов, не получающих кортикостероиды) или возрастающих дозах кортикостероидов; значительное увеличение плотности опухолевых очагов, отслеживаемое по накоплению контрастного материала в режиме T2/FLAIR, при стабильных или возрастающих дозах кортикостероидов по сравнению со исходным сканированием или лучшим ответом после начала терапии, не вызванное сопутствующими событиями (например, лучевая терапия, демиелинизация, ишемическое повреждение, инфекция, судороги, послеоперационные изменения или другие эффекты лечения); любой новый опухолевый очаг; очевидное клиническое ухудшение, не связанное с другими причинами, кроме опухоли (например, судороги, связанные с приемом лекарств нежелательные явления, осложнения терапии, нарушения мозгового кровообращения, инфекции и так далее) или изменением дозы кортикостероидов; неявка на осмотр в результате смерти или ухудшения состояния; или явное прогрессирование проявлений неизмеряемого заболевания.
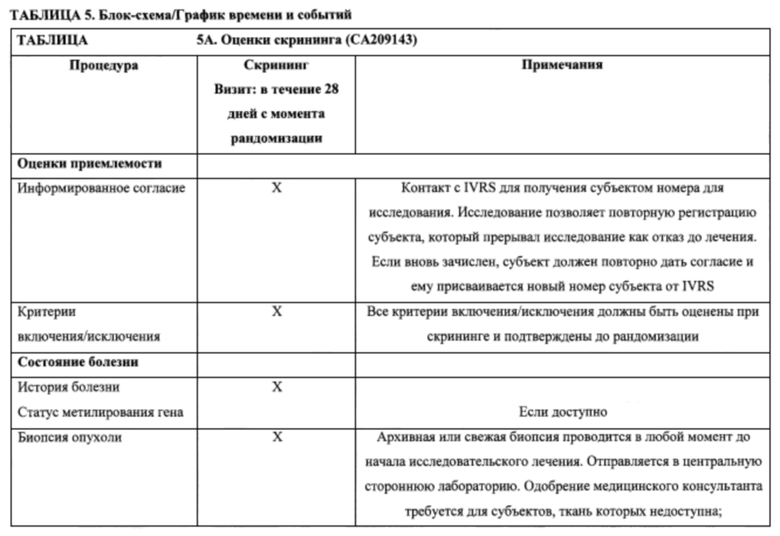


а Когда логистически осуществимо, сроки исходной МРТ должны быть перенесены как можно ближе к дате рандомизации, но не более, чем за 21 день до этого. Кроме того, данные МРТ, проведенных перед данной исходной МРТ, могут быть запрошены и получены спонсором.
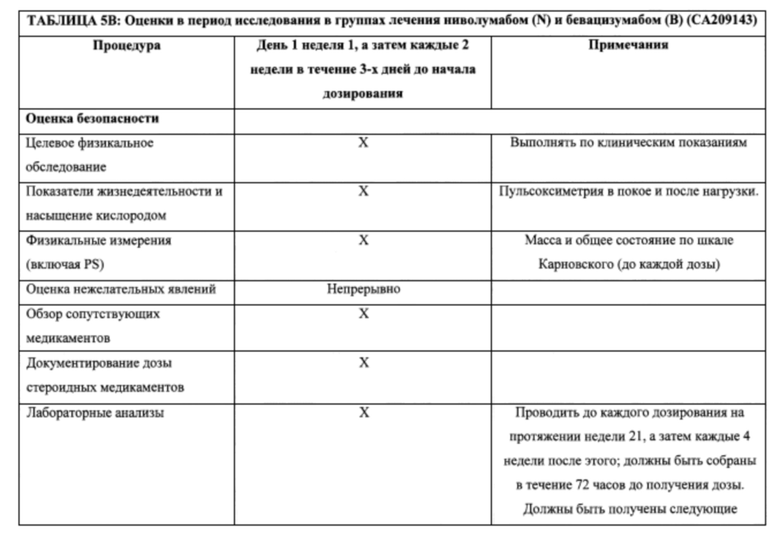

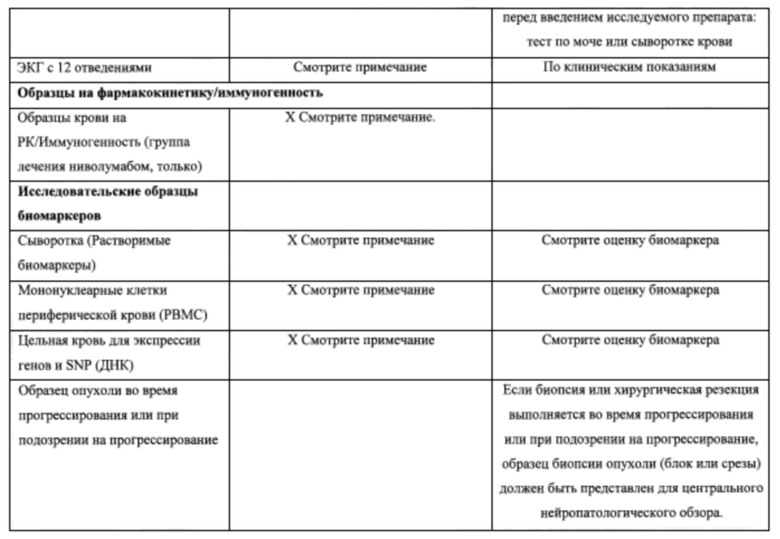
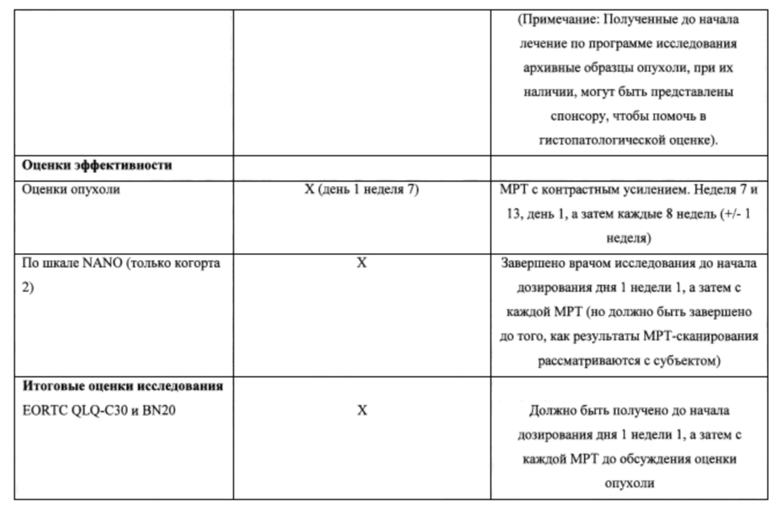

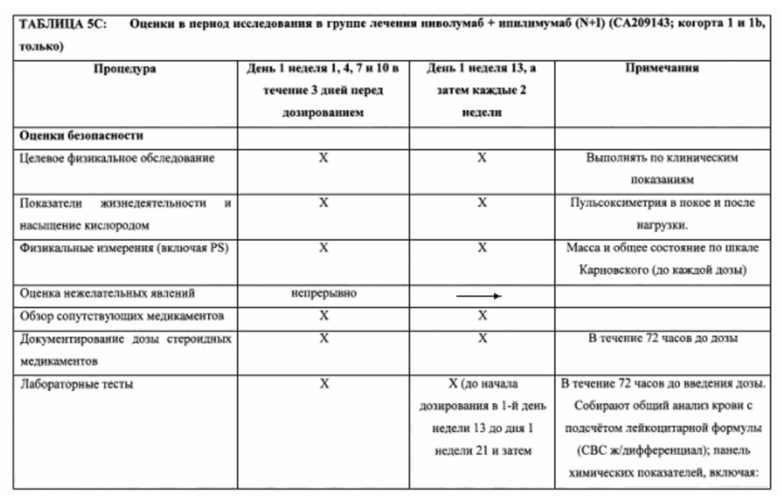
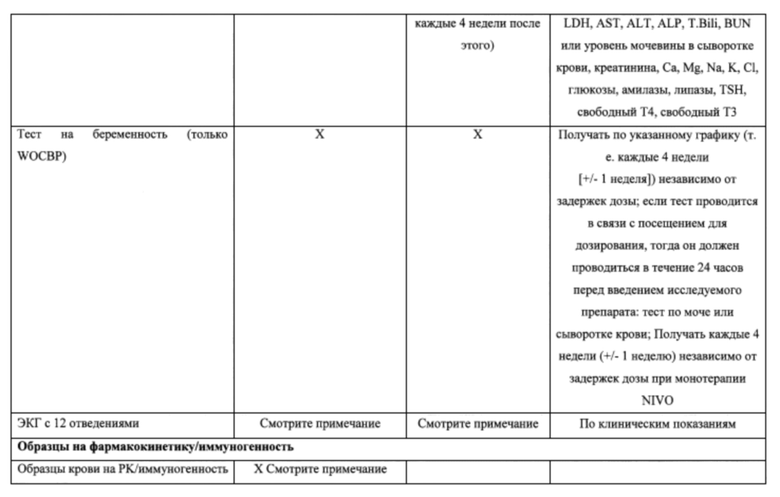
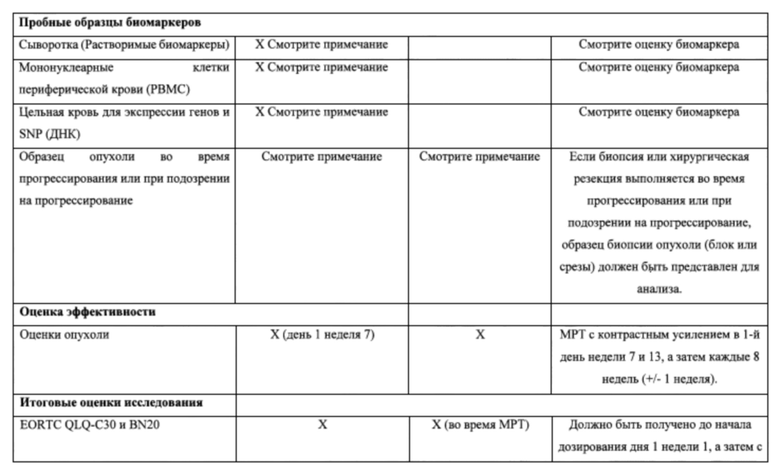
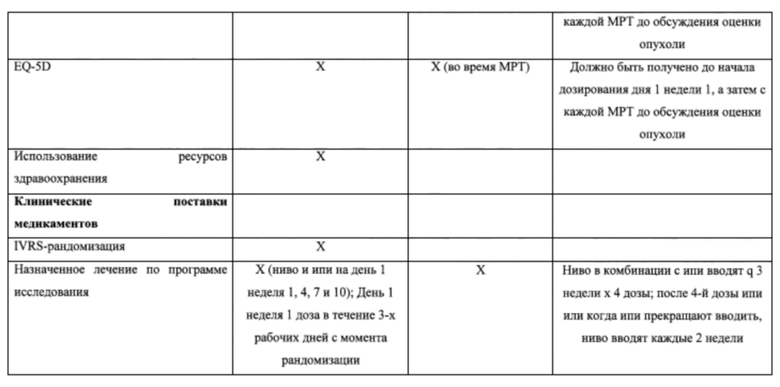
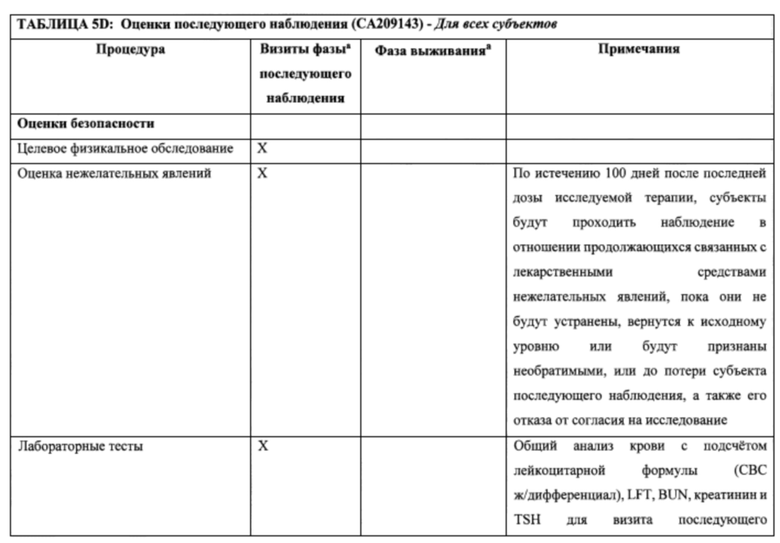


а Субъекты должны проходить наблюдение в течение не менее 100 дней после последней дозы исследуемого терапии. Последующий визит # 1 (FU1) происходит приблизительно через 35 дней (+/- 7 дней) после последней дозы или совпадает с датой прекращения (+/- 7 дней), если дата прекращения больше, чем через 35 дней после последней дозы. Последующий визит # 2 (FU2) происходит приблизительно через 80 дней (+/- 7 дней) после FU1.
a Визиты наблюдения за выживанием = каждые 3 месяца от FU2
Оценка NANO
189. Неврологическое функционирование оценивают с использованием общей шкалы неврологической оценки в нейроонкологии (Neurologic Assessment in Neuro-Oncology (NANO)), проводится исследователем, участвующего в исследовании.
190. NANO является объективной, быстрой, удобной и количественной оценкой девяти основных доменов для пациентов с опухолями головного мозга. Домены включают в себя: походку, силу, атаксию, ощущение, зрительное поле, выражение лица, язык, уровень сознания, поведение и оценку в целом. Каждый домен оценивают по шкале от 0 до 3, где 0 обозначает нормальное состояние и 3 представляет худшую степень тяжести состояния. Данный домен не оценивается, если он не может быть точно оценен из-за предшествующий состояний, коморбидных событий и/или сопутствующих медицинских препаратов. Оценка основана на непосредственном наблюдении/испытании, проведенном во время плановых посещений офиса. Оценка по шкале NANO полностью завершается исследователем или назначенным врачом исследования до начала дозирования дня 1 недели 1 (исходный уровень), а затем с каждой МРТ (но завершена до обзора результатов МРТ).
Нежелательные явления
191. Нежелательное явление (АЕ) определяется как любое новое неблагоприятное медицинское возникновение или обострение уже существующего медицинского состояния у субъекта, участвующего в клиническом исследовании, которому вводят исследуемый (лекарственный) продукт, и это не обязательно имеет причинную связь с данным лечением. Поэтому АЕ может представлять собой любой неблагоприятный и непреднамеренный признак (например, отклоняющиеся от нормальных лабораторные данные), симптом или заболевание, связанное по времени с использованием исследуемого продукта, независимо от того, относится ли оно к исследуемому продукту или нет.
192. Причинная связь с исследуемым препаратом определяется врачом и должна быть использована для оценки всех нежелательных явлений (АЕ). Случайные отношения могут быть одним из следующих:
(a) Связаны: Существует разумная причинная связь между введением исследуемого препарата и АЕ.
(b) Не связаны: Не существует разумной причинной связи между введением исследуемого препарата и АЕ.
193. Термин "разумная причинно-следственная связь" означает, что есть основания полагать наличие причинно-следственной связи. Нежелательные явления могут быть спонтанно зарегистрированы или выявлены во время открытого опроса, осмотра или оценки субъекта. (Для предотвращения предвзятости отчетности, субъекты не должны быть поставлены под сомнение в отношении конкретного случая появления одного или нескольких АЕ).
Серьезные нежелательные явления
194. Серьезное нежелательное явление (Serious Adverse Event (SAE)) является любым неблагоприятным медицинским явлением, которое при любой дозе:
(a) приводит к смерти
(b) представляет угрозу для жизни (определяется как событие, при котором субъект был подвержен риску смерти во время проведения мероприятия; оно не относится к событию, которое гипотетически могло бы вызвать смерть, если бы это было более серьезным)
(c) требует стационарной госпитализации или вызывает продлевание существующей госпитализации (Смотрите ПРИМЕЧАНИЕ ниже)
(d) приводит к стойкой или значительной инвалидности/нетрудоспособности
(e) является врожденной аномалией/врожденным дефектом
195. (f) является важным медицинским событием (определяется как медицинское событие(я), которое не может немедленно угрожать жизни или привести к смерти или госпитализации, но, на основании соответствующего медицинского и научного осмысления, может поставить под угрозу субъект или может потребовать вмешательства [например, медицинского, хирургического], чтобы предотвратить один из других серьезных результатов, перечисленных в приведенном выше определении) Примеры таких событий включают, но не ограничиваются ими, интенсивное лечение в отделении неотложной помощи или дома аллергического бронхоспазма; патологические изменения крови или конвульсия, которые не приводят к госпитализации.) Потенциальное лекарственно-индуцированное повреждение печени (drug induced liver injury (DILI)) также считается важным медицинским событием.
196. Предположительная передача инфекционного агента (например, патогенного или непатогенного) через исследуемый препарат является SAE. Хотя беременность, передозировка, рак и потенциальные лекарственно-индуцированные поражения печени (DILI) не всегда являются серьезными по регулирующему определению, данные события должны быть обработаны, как SAE. О любом компоненте конечной точки исследования, который считается связанным с изучаемой терапией (например, смерть является конечной точкой, если смерть произошла из-за анафилаксии, анафилаксия должна быть сообщена) следует сообщать как SAE.
197, Следующие госпитализации не считаются SAE в клинических исследованиях, проводимых BMS:
(a) визит в отделение неотложной помощи или другой отдел больницы продолжительностью <24 часов, что не приводит к госпитализации (если не считать важным медицинским или опасным для жизни событием)
(b) плановая операция, запланированная до подписания согласия
(c) госпитализации согласно протоколу для плановой медицинской/хирургической процедуры
(d) процедура оценки состояния здоровья, которая требует госпитализации для определения исходного/текущего состояния здоровья (например, процедура колоноскопии)
(e) медицинская/хирургическая госпитализация, не имеющая целью лечение и запланированная до включения в исследование. В данных случаях требуется соответствующая документация
(f) госпитализация из-за другого жизненного обстоятельства, которое не имеет никакого отношения к состоянию здоровья и не требует медицинского/хирургического вмешательства (например, отсутствие жилья, экономическая несостоятельность, передышка в уходе за больным, семейные обстоятельства, административные причины).
(g) госпитализация для введения противораковой терапии в отсутствии какого-либо другого SAE
Оценки биомаркеров
Обоснование и цели для оценок биомаркеров
198. Предыдущие исследования с ипилимумабом показали, что повышенная экспрессия маркеров пролиферации и поляризации в CD4+ и CD8+ клетках, снижение CCR7 и IL-7R на CD4+ Т-клетках, а также активированный Гранзим В в CD8+ Т-клетках могут служить фармакодинамическими показателями действия ипилимумаба. Повышение уровня маркеров активации ICOS и eomesodermin (EOMES) было в значительной степени связано с меньшим количеством АЕ и менее благоприятным исходом, соответственно, но исходная повышенная экспрессия EOMES, по-видимому, предсказывает благоприятную безрецидивную выживаемость в данном анализе.
199. ICOS потенциально может быть полезным маркером для ответов CD4+Т-клеток, полученных после блокады CTLA-4. Экспрессия человеческого лейкоцитарного антигена (HLA-DR) также может потенциально служить маркером Т-клеток.
200. Предыдущие исследования также показали, что экспрессия связанных с иммунитетом генов, таких как CD8a, GZMA, LCK, CXCL-9, -10, -11, и CXCR3 в опухолях на неделе 3 после лечения была связана с общей лимфоцитарной инфильтрацией, клинической эффективностью и общей выживаемостью. Предварительные результаты свидетельствуют о том, что высокая предшествующая иммунная активность способствует клиническому ответу на терапию ипилимумабом.
201. Периферическая кровь и опухолевая ткань будут собраны до терапии. Образцы периферической крови также будут собраны в выбранные моменты времени во время лечения. Если биопсия или хирургическое удаление выполняется во время прогрессирования или подозреваемого прогрессирования, образец опухоли (блок или срезы) должен быть представлен для анализа. Если образцы биомаркеров взяты, но исследуемый препарат(ы) не вводят, образцы будут сохранены. Подробное описание каждой аналитической системы представлено ниже и вымерено расписание фармакодинамических оценок. Сбор всех биомаркеров в моменты времени, когда исследуемый препарат не вводят, необязателен.
Растворимые биомаркеры
202. В данном исследовании образцы сыворотки были собраны исходно, во время лечения и после лечения от субъектов, зарегистрированных в испытании, с целью выявления потенциальных биомаркеров с прогностической и прогнозируемой ценностью для результатов (ответа, выживаемости без прогрессирования и общей выживаемости, токсичности). Предварительные результаты данного исследования будут служить информацией для оценок для исследования эффективности фазы 3. Растворимые факторы, такие как цитокины, хемокины, растворимые рецепторы и антитела к опухолевым антигенам будут охарактеризованы и определены количественно с помощью иммуноанализа в сыворотке. Анализы могут включать в себя, но не обязательно ограничиваются ими, анализ на растворимый CD25, растворимый PD-1, растворимый LAG-3 и CXCL-9. Собранные образцы сыворотки будут также использоваться для оценки опухолевых антиген-специфических ответов, вызываемых после лечения с монотерапией и комбинированной терапии, чтобы исследовать, какое из противоопухолевых антител наиболее связано с клиническим ответом. Уровни антител к тестируемым раковым антигенам будут оценены с помощью мультиплексных анализов и иммуноферментного анализа (ELISA).
Иммунофенотипирование
203. Доля специфических субпопуляций лимфоцитов и уровни экспрессии ко-стимулирующих маркеров Т-клеток в препаратах мононуклеарных клеток периферической крови (РВМС) будут количественно определяться с помощью проточной цитометрии. Анализы могут включать в себя, но не обязательно ограничиваться ими, определение соотношения Т-, В-, и NK-клеток, гранулоцитов, соотношения подмножеств клеток памяти и эффекторных Т-клеток, а также уровни экспрессии PD-1, PD-L1, других членов семейства В7, ICOS и Ki67.
Ex vivo функциональные анализы
204. Для того, чтобы выяснить, восстановит ли монотерапия и/или комбинированная терапия активацию и функцию Т-клеток, мононуклеарные клетки периферической крови (РВМС) будет изолированы и подвергнуты криоконсервации. Будут выполнены анализы функционального состояния эффекторных Т-клеток, включая, но не ограничиваясь ими, анализы на интерферон-гамма (IFN-gamma) и CD107.
205. Исследование взаимоотношений компартмента периферических Т-клеток и GBM раковых стволовых клеток также будет выполняться одним из нескольких способов. Данные способы могут включать в себя рестимуляцию периферической крови с использованием GBM раковых стволовых клеток, полученных из иммортализованных клеточных линий, с последующим измерением эффекторных цитокинов, таких как IFN-у, с помощью ELISPOT. В качестве альтернативы, раковые стволовые клетки, очищенные из биопсий субъектов, могут быть использованы в качестве ex vivo цитолитического анализа с использованием Т-клеток периферической крови.
Экспрессия генов периферической крови
206. Уровень экспрессии генов, связанных с ответом на монотерапию ниволумабом и комбинированную терапию ниволумаб/ипилимумаб, будет количественно определяться молекулярными способами, такими как микрочип компании Affymetrix, и/или анализом количественной полимеразной цепной реакции с обратной транскрипцией (ОТ-ПЦР) в образцах цельной крови. Анализ может включать в себя, но не обязательно ограничивается ими, гены, ассоциированные с иммунными путями, такими как активация Т-клеток и процессинг и презентация антигена.
Анализ Т-клеточного репертуара
207. Низкий уровень разнообразия компартмента периферических Т-клеток, как было показано, коррелирует с плохой общей выживаемостью при метастатическом раке молочной железы. Устоявшаяся теория в иммуноонкологии предполагает, что разнообразная и активированная иммунная среда обладает лучшими возможностями по ликвидации опухоли по сравнению с "перекошенным" репертуаром наивных и толерантных Т-клеток. Для того, чтобы выяснить, возможно ли прогнозировать ответ на терапию, используя разнообразный Т-клеточный репертуар, высокопропускное секвенирование ДНК нового поколения будет выполнено на ДНК, выделенной из периферической крови и ткани опухоли, для количественного определения состава Т-клеточного репертуара до и во время монотерапии и комбинированной терапии.
Опухолевые образцы
Измерения биомаркеров на основе опухолей
208. Образцы биопсии опухоли будет получены от субъектов, давших согласие, до лечения монотерапией ниволумабом, комбинированной терапией ниволумаб плюс ипилимумаб или бевацизумабом, чтобы охарактеризовать популяции иммунных клеток и экспрессию отдельных опухолевых маркеров. Блок или срезы архивных биопсий или свежая биопсия должны быть доступны для представления до рандомизации. Если опухолевая ткань отсутствует, пожалуйста, обратитесь к медицинскому консультанту, чтобы обсудить возможность включения данного субъекта в протокол. Коллекция свежих биопсий до начала терапии исследования является предпочтительной для пациентов с доступными опухолевыми очагами, но не требуется. Образцы биопсии, полученные в период лечения, не являются обязательными.
209. Свежие биопсии будут предоставлены для анализа биомаркеров, если доступны и считаются исследователем безопасными, и должны иметь приоритет над предоставлением архивного образца. Архивная биопсия, полученная до терапии, является приемлемой, если свежая биопсия не может быть получена и если архивная ткань соответствует определенным критериям, как указано ниже.
(а) Архивная биопсия (блок или срезы) должна содержать опухолевую ткань;
(b) Если архивный блок не доступен, 10 или более срезов, содержащих опухоль, доступны для исследовательского применения
210. Всякий раз, когда это возможно, архивный образец опухоли должен быть получен с момента времени после того, как субъект завершил другую системную терапию (в том числе, лечени, темозоломидом) и до рандомизации. Биопсийные образцы могут быть использованы для следующих оценок:
Характеристика опухоль-инфильтрирующих лимфоцитов (TIL) и опухолевых антигенов
211. Иммуногистохимия (ГНС) будет использоваться для оценки количества и состава иммунных инфильтратов с целью определения подмножества иммунных клеток, присутствующих в фиксированной формалином и залитой парафином (formalin-fixed, paraffin embedded (FFPE)) опухолевой ткани до и после воздействия терапии. Данные анализы IHC будут включать в себя, но не обязательно ограничиваются ими, следующие маркеры: CD4, CD8, FOXP3, PD-1, PD-L1 и PD-L2.
212. С помощью IHС также выполняется фенотипирование стволовых клеток рака, чтобы оценить фенотип, локализацию и плотность раковых стволовых клеток в биопсии.
213. В тех случаях, когда свежая опухоль доступна до терапии и/или лечения, может быть выполнено отделение и выделение TIL из опухолевых и стромальных клеток. Выделенные TIL могут быть использованы для оценки экспрессии генов, фенотипа и функции иммунных клеток. Такие способы, как секвенирование нового поколения TIL и проточная цитометрия, могут быть выполнены, чтобы охарактеризовать фенотип, функцию и специфичность.
Характеристика Т-клеточного репертуара
214. Как описано выше, секвенирование ДНК будет осуществляться с использованием опухолевой ткани, предоставленной перед лечением и после лечения, для оценки состава Т-клеточного репертуара. ДНК будет изолирована либо из любого полученного из опухоли блока, подготовленного по методике FFPE, либо из RNAlater или эквивалентных препаратов.
Профилирование экспрессии генов
215. Опухолевые биопсии, собранные в RNAlater или эквивалентном фиксаторе, будут исследованы на экспрессию гена мРНК с помощью микрочиповой технологии Affymetrix для анализа экспрессии генов и/или количественной полимеразной цепной реакции в режиме реального времени (qPCR) для выявления экспрессии выбранных генов, связанных с иммунной системой.
Генотипирование опухоли, мутационный анализ и профилирование опухолевых антигенов
216. РНК и ДНК из образцов опухолей будут проанализированы с использованием полного ЕхоМЕ и транскриптомного секвенирования, чтобы определить число мутаций, обнаруженных в пределах данного образца по отношению к нормальной ткани хозяина, такой как соседние ^трансформированные клетки или РВМС. Мутации, которые обнаруживаются, будут проанализированы на их способность связывать белки МНС I и МНС II с использованием алгоритмов предсказания, таких как NetMHC. Оценивание способности опухолевых мутаций связывать молекулы МНС предоставит доказательства того, что данные мутации служат в качестве антигенов, которые распознаются иммунной системой и являются потенциальными антигенами отторжения.
Детали сбора опухолевых образцов
Свежая биопсия:
217. Свежие биопсии исходно должны иметь приоритет над архивными образцами и настоятельно рекомендуются во время лечения.
218. Образцы опухоли, полученные из костных метастазов, не считаются приемлемыми для тестирования PD-L1, так как анализ PD-L1 не включает в себя стадию декальцинации. Для любых случаев, когда доступна только опухолевая ткань из поражения костного метастазирования, пожалуйста, дополнительно обсудите с медицинским консультантом исследования.
219. Образцы биопсий должны быть эксцизионными, послеоперационными или взяты пункционной иглой с помощью иглы калибра, который используется в учреждении. Обычно для пункционных биопсий используются иглы 16 или 18 калибра.
220. Рекомендуется, чтобы образцы быть зафиксированы в 10% нейтральном забуференном формалине в течение 24-48 часов. Образцы опухолевой ткани не должны быть погружены в формалин, так как температура и продолжительность фиксации не может контролироваться во время транспортировки.
221. Если предоставлены срезы, рекомендуемая толщина среза ткани составляет 4 мкм и срезы должны быть положительно заряженными. Срезы должны быть отгружены в холодильнике при температуре 2-8°С.
222. Отгрузка образцов должна включать заполненную реквизиционную форму, содержащую дату сбора, способ сбора, первичный/мет, сайт, условие фиксации и копию патологического отчета, если таковой имеется.
223. Если берут свежую биопсию, рекомендуется до 4-х пункционных биопсий. Оценка качества биопсии патологоанатомом настоятельно рекомендуется во время процедуры. Опухолевая ткань, которая получается из данных биопсий, будет разделена три способами в следующем порядке приоритета: в формалин для фиксации и погружения в парафин, в RNAlater для экстракции РНК/ДНК и подготовленной к выделению опухоль-инфильтрующих лимфоцитов (TIL).
224. Исследователю, в консультации с сотрудниками рентгенологического отдела, необходимо определить степень риска, связанного с процедурой, и найти его приемлемым. Биопсии могут быть сделаны с местной анестезией или седацией. Институциональные рекомендации для безопасного выполнения биопсии должны быть соблюдены. Могут быть выполнены эксцизионные биопсии, чтобы получить образцы биопсии опухоли. Инвазивные процедуры, которые требуют общей анестезии, не должны быть выполнены для получения образца биопсии. Однако, если для клинических показаний выполняется хирургическая процедура, избыток ткани опухоли может быть использован в исследовательских целях с согласия субъекта.
Архивная биопсия:
225. Минимум 1 формалин-фиксированный залитый в парафин (FFPE) блок опухолевой ткани (предпочтительно) ИЛИ не менее 10 FFPE неокрашенных срезов требуются для оценки состояния PD-L1 и оценок других биомаркеров.
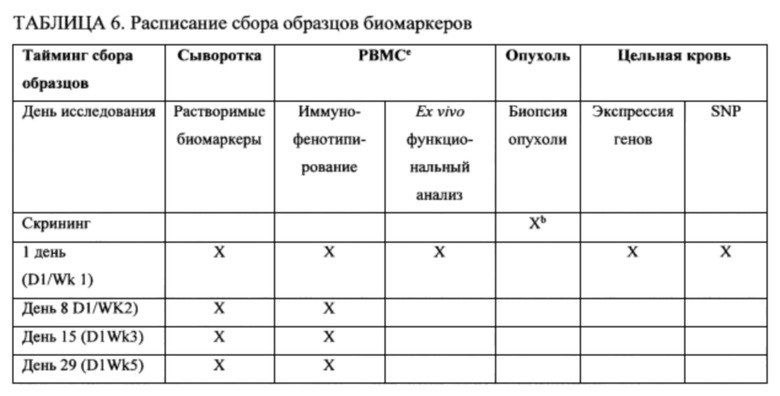
а Сбор образцов биомаркеров происходит до введения дозы исследуемого препарата и может произойти +/- 2-5 дней от запланированного времени.
b Биопсия опухоли до терапии является обязательной. Если недавняя (как определенно в протоколе) архивная биопсия не доступна при скрининге, то свежая биопсия будет принята в любой момент до начала лечения. Субъекты, для которых биопсия опухоли не доступна на момент регистрации, должны быть обсуждены с медицинским консультантом относительно возможного включения.
с Необязательными являются биопсии во время лечения и по мере прогрессирования и могут быть приняты по усмотрению исследователя
d Образцы от субъектов, которые подтвердили прогрессирование, не являются обязательными.
е Образцы биомаркеров в дни дозирования неисследуемого препарата для монотерапии или комбинированной терапии не являются обязательными.
226. Подробные инструкции получения, обработки, маркировки, обращения, хранения и отгрузки образцов будут предоставлены в отдельном процедурном руководстве на момент начала исследования.
227. Демографические данные и исходные характеристики пациентов, включенных в исследование, рассматриваются в таблице 7.
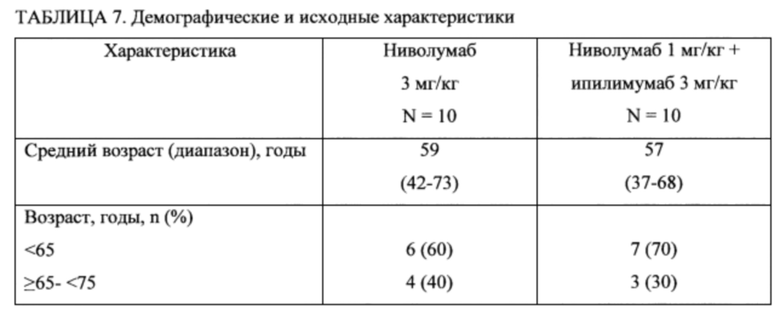

ПРИМЕР 2
Клинический обзор данных когорты 1
228. Предлагаемый режим дозирования для ниволумаба и ипилимумаба в когорте 1 и 1b был основан на данных по безопасности и переносимости от применения ниволумаба и ипилимумаба в других типах опухолей. Хотя предложенный режим дозирования должен был быть переносимым у пациентов с GBM, данное исследование сначала включало две когорты по первоначальной оценке безопасности (когорты 1 и 1b) до инициации исследования эффективности по сравнению монотерапии ниволумабом с бевацизумабом (когорты 2). Около 10 пациентов с рецидивом были рандомизированы в каждой из двух исследуемых групп лечения в группе 1 (группа лечения N и группа лечения N+I). До 20 пациентов с рецидивом будут лечиться в когорте 1b (группа лечения N+I_1b).
229. Переносимость и безопасность в группе лечения были определена после того, как все субъекты в каждой группе лечения завершили прием четырех доз или прекратили дозировки до завершения четырех доз. Переносимость за пределами четырех доз также может быть принята во внимание. Критерии переносимости, используемые для перехода к когорте 2, были основаны на связанных с лекарственными средствами нежелательных явлениях, приводящих к постоянному прекращению в когортах по первоначальной оценке безопасности и включают в себя следующее:
Если не более одной трети субъектов в данной группе лечения окончательно прекратит прием исследуемого препарата до завершения четырех доз из-за связанных с лечением нежелательных явлений, то когорта данной дозы будет считаться терпимой и прием в когорту 2 может продолжаться;
Если более чем одна треть субъектов в группе лечения окончательно прекратит прием исследуемого препарата до завершения четырех доз из-за связанных с лечением нежелательных явлений, то безопасность и переносимость данной группы лечения будет рассмотрена с DMC до рандомизации любых дополнительных субъектов. Спонсором будет принято решение о целесообразности продолжения регистрации или продвижении группы лечения в когорту 2,
230. Лечение ниволумабом в качестве монотерапии в когорте 1 отвечало заданному протоколом профилю безопасности и переносимости, подробно описанному выше, требуемому для того, чтобы перейти к рандомизированному исследованию по сравнению с бевацизумабом в когорте 2.
231. Диспозиция пациента видна в таблице 8. Ни один из пациентов, получающих монотерапию ниволумабом, не прекратил лечение из-за токсичности препарата. Не было ни одного случая смерти из-за токсичности препарата в группах лечения с монотерапией или комбинированной терапией. В момент окончания (9 сентября 2015) один пациент в группе лечения монотерапии ниволумабом продолжает получать ниволумаб после 17,5 месяцев лечения.


1 Смерть пациентов в обеих группах была обусловлена болезнью
232. Оцененные иследователем объективные ответы в соответствии с критериями RANO представлены в таблице 9. Большинство пациентов (6/10), получавших ниволумаб в качестве монотерапии, характеризовались стабильным заболеванием или улучшением состояния. Четыре пациента в группе комбинированной терапии показали лучше ответы стабильного заболевания. Исследовательский анализ общей выживаемости представлен в таблице 10. NE: не оценивается.

2 Согласно критериям RANO
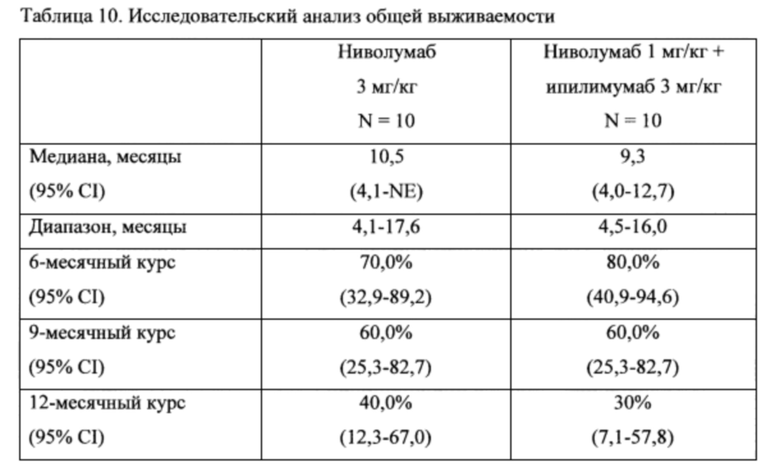
ПРИМЕР 3
Когорта 1: Пример клинического случая псевдопрогрессирования
233. 67-летней белой женщине-пациентке был первоначально поставлен диагноз правой затылочной GBM в апреле 2013 года. Пациентка подверглась правой височной резекции в апреле 2013 года с последующей одновременными химиорадиотерапией с мая по июль 2013 года и лечением эверолимусом и темозоломидом с мая по август 2013 года. Пациентка испытала первый рецидив глиобластомы в марте 2014 года. Данную пациентку рандомизировали в группу лечения ниволумабом в дозе 3 мг/кг, с началом лечения в апреле 2014 г. Пациентка получила десять доз ниволумаба в дозе 3 мг/кг.
234. Подозрение на прогрессирование глиобластомы наблюдали с помощью МРТ. На исходном уровне размер опухолевого очага GBM составил 12 мм × 10 мм. После лечения, на 43-й день, размер опухолевого очага составил 20 мм × 17 мм; и на 73-й день размер опухолевого очага составил 20 мм × 40 мм. Изображения МРТ пациентки до начала лечения (фигура 5А и 5В) и после пяти доз ниволумаба показаны на фигурах 5С и 5D. МРТ-скан после пяти доз ниволумаба (3 мг/кг Q2W) показал повышенное усиление контрастности височного опухолевого очага и новое усиление контрастности перивентрикулярной области; сообщалось о снижении зрения. МРТ-сканы на 43-й и 73-й день подтвердили наличие прогрессирования. Гистопатологический анализ резецированной опухоли показал иммунные инфильтрации в соответствии с изменениями, связанными с лечением.
235. GBM резецировали, чтобы определить, прогрессировала ли опухоль или псевдопрогрессировала. Гистопатологический анализ резецированной опухоли выявил перивентрикулярные некротические ткани и большую агрегацию иммунных клеток (фигуры 6А-С); внутриопухолевые макрофаги и агрегаты CD45-положительных клеток (фигуры 7А-С); СD45-положительные клетки и СD3-положительные клетки (фигуры 8А-С); и периваскулярную инфильтрацию и инфильтрирующие клетки (фигуры 9А-С) в ткани резецированной опухоли. Кроме того, наблюдали выстилку CD45-положительными клетками опухолевых кровеносных сосудов, как показано на фигурах 10А-С. Такая невропатология согласуется со значительными изменениями, связанными с лечением, и пациентка возобновила лечение ниволумабом в дозе 3 мг/кг.
236. Пациентка получала лечение в общей сложности 227 дней. Пациентка умерла через 397 дней после рандомизации. У пациентки было отдаленное прогрессирование заболевания в гиппокампе и правом гипоталамусе (гистопатологически доказанное на аутопсии) через 4 месяца после возобновления приема ниволумаба после операции.
ПРИМЕР 4
Когорта 1: Пример клинического случая частичного ответа
237. 42-летнему белому мужчине-пациенту был изначально поставлен диагноз левой височной GBM в октябре 2013 г. Субъекта подвергли хирургической резекции в октябре 2013 года, и по резецированной ткани было установлено, что ген MGMT метилирован и EGFRvIII положителен. Субъект прошел стандартную фокальную химиотерапию мозга с ноября по декабрь 2013 года, а затем два цикла адъювантной терапии темозоломидом в феврале 2014 года. Прогрессирующее ухудшение наблюдалось с МРТ в феврале и в начале и в конце марта 2014 года, с увеличением относительного церебрального объема крови (relative cerebral blood volume (rCBV)). Субъект испытал первый рецидив GBM 31 марта 2014 года. Субъекта затем лечили ламотриджином и алпразоламом. Данный субъект был рандомизирован в группу лечения ниволумабом в дозе 3 мг/кг.
238. Субъект отображал частичный ответ в соответствии с критериями RANO. Исходно, размер опухолевого очага GBM составил 27 мм × 10 мм. После лечения, на день 44, размер опухолевого очага GBM составил 26 мм × 9 мм; и на 85-й день размер опухолевого очага GBM составил 23 мм × 4 мм. Общее состояние испытуемого по шкале Карновского было 90 на исходном уровне и при всех последующих визитах. Изображения МРТ субъекта до начала лечения, после трех доз ниволумаба и в двух более поздних временных точках после продолжения лечения с ниволумабом показаны на фигуре 11.
ПРИМЕР 5
Когорта 1 Группа лечения N+I: Примеры клинических конкретных случаев после лечения с ниволумабом (1 мг/кг) + ипилимумаб (3 мг/кг)
239. 45-летняя женщина-пациент получила первую дозу ниволумаба (1 мг/кг) + ипилимумаба (3 мг/кг) 12 февраля 2014 года. Пациентка завершила три цикла лечения, с конечной дозой, введенной 2 апреля 2014 года. Лечение затем было прекращено из-за тяжелого диабетического кетоацидоза. Дополнительные АЕ во время лечения, в различные моменты времени, включали: пневмонит 2-й степени, повышение уровня липазы 4-й степени, повышение уровня амилазы 3-й степени, и повышения уровней AST/ALT 1-й степени. МРТ головного мозга испытуемого, проведенная 10 сентября 2014 года, показала прогрессирование заболевания. Клиническое состояние пациентки также ухудшилось. Пациентка затем начала монотерапию бевацизумабом, и в настоящее время ее клиническое состояние хорошее. Был утвержден благотворительный доступ монотерапии ниволумабом, и предстоит лечение.
240. 57-летняя женщина-пациент получила первую дозу ниволумаба (1 мг/кг) + ипилимумаба (3 мг/кг) 17 апреля 2014 года. У пациентки развилась тяжелая форма бреда 3-й степени 15 мая 2014 года (С2 D8), требующая госпитализации. Все инфекционные и метаболические обследования дали отрицательные результаты, и ЭЭГ показала только медленные волны. Пациентка восстановилась до исходного уровня после двух дней лечения высокими дозами стероидов. Пациентка была затем переведена на монотерапию ниволумабом 19 июня 2014 года. После этого пациентка получала три дозы ниволумаба в 3 мг/кг, с последней введенной дозой 17 июля 2014 года. 31 июля 2014 года, перед введением шестой дозы ниволумаба, пациентка сообщила об одышке 2-й степени, усталости и слабости в левой стороне. Кроме того, пациентка получала коническую дозу преднизолона для лечения диареи (20 мг/сут). МРТ головного мозга пациентки показала увеличение интервалов в усилении и сигнал FLAIR. Данные изменения не были измеряемы. Лечение было прекращено по желанию пациентки. Повторная МРТ головного мозга 8/19/2014 показала клиническое ухудшение с левосторонним гемипарезом и увеличенным усилением. Пациентка была подвергнута хирургической резекции 27 августа 2014 года. Анализ ткани резецированной опухоли подтвердил прогрессирование опухоли. Пациентка покинула исследование и в настоящее время проходит лечение только бевацизумабом. Пациентка отказалась от добавления CCNU. Другие токсические проявления у пациентки при данном исследовании включали: бессимптомное повышение уровня липазы 3-й степени, понос 2-й степени, бессимптомное повышение уровня ALT 2-й степени и повышение уровня AST 1-й степени.
ПРИМЕР 6
Когорта 1 Группа лечения N: Пример клинического случая после лечения монотерапией ниволумабом (3 мг/кг)
241. 67-летняя пациентка получила первую дозу ниволумаба в дозе 3 мг/кг в качестве монотерапии 24 марта 2014 года. Пациентка завершила прием десяти доз ниволумаба, с последней введенной дозой 5 ноября 2014 года. МРТ головного мозга испытуемой, выполненная 5 мая 2014 года после трех процедур, показала прогрессирование болезни в соответствии с критериями RANO, особенно вокруг хирургической полости, и наличие новой перивентрикулярной области. Пациентка согласилась с лечением при прогрессировании и получила две последующих инфузии, с четвертой процедурой 7 мая 2014 года и пятой процедурой 21 мая 2014 года. 4 июня 2014 года, до шестой инфузии, у пациентки зафиксировали сужение поля зрения. МРТ головного мозга показала дальнейший рост первичной опухоли в затылочной области и аномальности в перивентрикулярной области. Тогда лечение не назначили, и пациентку подвергли хирургической резекции 12 июня 2014 г.Патологический обзор показал жизнеспособную опухоль в образцах из затылочной области с инфильтрацией Т-лимфоцитами. В той же самой области имелся значительный некроз (радиационный некроз). Перивентрикулярная область показала только некроз и фибринозный экссудат (радиационный некроз) и крупные агрегаты лимфоцитов, без опухоли.
242. Пациентка хорошо восстановилась после операции. МРТ головного мозга 10 июля 2014 года, через четыре недели после резекции, не показала рост опухоли. Пациентка затем вновь начала ниволумаб в качестве монотерапии, получив шестую дозу 15 июля 2014 года и седьмую дозу 30 июля 2014 года. Приблизительно через одну неделю после возобновления монотерапии ниволумабом пациентка сообщила об ухудшении неврологических симптомов (степень 2), включая ухудшение в сужении поля зрения. В то время пациентка не была на стероидной терапии. МРТ головного мозга показала увеличение интервала в FLAIR-сигналах и увеличение в диффузном ободковом усилении в послеоперационной постели. Имелась устойчивая ограниченная диффузия в соответствии с изменениями лечения. В соответствии с протоколом, лечение было отложено. Пациентка начала лечение дексаметазоном в дозе 4 мг BID, и клиническое состояние пациентки улучшилось. МРТ головного мозга 28 августа 2014 показала улучшение с рассасыванием высококонтрастной зоны на краях опухоли, отмеченной при предыдущей МРТ. Пациентка вновь начала монотерапию ниволумабом 25 сентября 2014 года, и доза при лечении дексаметазоном постепенно была понижена до 3 мг/дня без существенных неврологических изменений. Состояние пациентки будет продолжать контролироваться с помощью МРТ в соответствии с протоколом.
ПРИМЕР 7
Когорта 1b Группа лечения N+I (1b): Примеры клинических случаев после лечения с ниволумабом (1 мг/кг) + ипилимумаб (3 мг/кг)
243. 46-летний мужчина-пациент получил первую дозу ниволумаба (3 мг/кг) + ипилимумаба (1 мг/кг) 27 июня 2014 года, вторую дозу 18 июля 2014 года, третью дозу 11 августа 2014 года и четвертую дозу 4 сентября 2014 года. Пациентом дополнительно завершено четыре цикла монотерапии ниволумабом, с конечной дозой, введенной 4 ноября 2014 года. Пациент переносил лечение очень хорошо, без каких-либо побочных эффектов. У пациента наблюдалось значительное клиническое улучшение в движении и когнитивной способности. МРТ головного мозга испытуемого показала уменьшение размера опухоли (стабильное в соответствии с критериями RANO), несмотря на то, что в начале была очень большая опухоль. Пациент принимает дексаметазон в дозе 4 в чередовании с 2 мг в день. Состояние пациента будет продолжать контролироваться с помощью МРТ в соответствии с протоколом.
244. 74-летний мужчина-пациент получил первую дозу ниволумаба (3 мг/кг) + ипилимумаба (1 мг/кг) 9 июля 2014 года, вторую дозу 31 июля 2014 года, третью дозу 21 августа 2014 года и четвертую дозу 11 сентября 2014 года. Пациент дополнительно завершил три цикла монотерапии ниволумабом, с конечной дозой, введенной 30 октября 2014 года. Нежелательные явления во время лечения, в разные моменты времени, включали: сыпь и понос 2-й степени, пневмонит 2-й степени, повышение уровня липазы и ALT 1-й степени, а также слабую нейропатию. Данные АЕ были улучшены лечением высокими дозами стероидов. Пациент также получиал метилпреднизолон в дозе 32 мг/день. МРТ головного мозга испытуемого 29 сентября 2014 года, после четырех циклов комбинированной терапии и до первого цикла монотерапии ниволумабом, показала прогрессирование заболевания (в соответствии с критериями RANO). Пациент продолжал лечение и последующая МРТ головного мозга была стабильной. Состояние пациента по-прежнему будет контролироваться с помощью МРТ в соответствии с протоколом.
245. 62-летний мужчина-пациент получил первую дозу ниволумаба (3 мг/кг)+ипилимумаба (1 мг/кг) 28 июля 2014 года, вторую дозу 25 августа 2014 года, третью дозу 8 сентября 2014 года и четвертую дозу 21 октября 2014 года. Нежелательные явления на протяжении лечения включали: сыпь 3-й степени и усталость 2-й степени, которые оба исчезли после лечения стероидами. МРТ головного мозга испытуемого 9/8/2014, до третьего цикла, показала прогрессирование заболевания в соответствии с критериями RANO. Пациент избрал продолжение лечение при прогрессировании. Однако пациенту потребовалось некоторое время для решения. Повторная МРТ головного мозга в момент времени 8 недель показала стабильное заболевание в сравнении с последней МРТ. Тем не менее, результатами по-прежнему считалось прогрессирование от исходного уровня, и пациент выбрал проведение хирургической резекции опухоли 11/12/2014. Пациент будет решать, следует ли продолжать лечение, на основе результатов резекции.
ПРИМЕР 8
246. Оценивали Т-клеточное созревание, активацию и функцию у пациентов с GBM, получавших ниволумаб ± ипилимумаб. И определяли частоту и функцию Tregs и супрессорных клеток миелоидного происхождения у пациентов с GBM, получавших ниволумаб ± ипилимумаб.
247. Анализы проводились на периферической крови, взятой от пациентов в заранее определенных временных точках (фигура 13), и получали образцы опухолевой ткани, если проводили резекцию во время прогрессирования или предполагаемого прогрессирования. Исходные и взятые во время лечения образцы были получены от пациентов (n=20), распределенных случайным образом 1:1 для получения либо ниволумаба в дозе 3 мг/кг через каждые 2 недели (Q2W), либо ниволумаба в дозе 1 мг/кг + ипилимумаба в дозе 3 мг/кг каждые 3 недели с последующим ниволумабом в дозе 3 мг/кг Q2W (фигура 13). Фенотипические анализы, анализы созревания, а также анализы пролиферации проводили с использованием проточной цитометрии с популяционно-специфичными панелями антител (Таблица 11). Анализы высвобождения цитокина были проведены на супернатантах из отсортированных Tregs (CD4+CD25+CD127-) ± стимуляция (РМА + иономицин). Оценивали активность против опухоль-ассоциированного антигена (tumor-associated antigen (ТАА)) мононуклеарных клеток периферической крови (peripheral blood mononuclear cells (РВМС)) и опухоль-инфильтрирующих лимфоцитов (TIL). Клетки стимулировали интерлейкином (IL) -2, IL-15 и пептидами ТАА; после 9 дней культивирования с IL-2 и IL-15, клетки повторно стимулировали IL-2, IL-15 и пептидами ТАА. Анализировали продукцию интерферона (IFN)γ/гранзима В с использованием двухцветного анализа FluoroSpot.

248. Цитометрический анализ выявил чистые популяции CD4 Т-клеток, CD8 Т-клеток и Tregs как в образцах периферической крови, так и в опухолевых образцах (фигура 14). Уникальные иммунные профили были обнаружены в иммунных инфильтратах у пациентов, перенесших прогрессирование, в сравнении с пациентами с псевдопрогрессированием, включая разительные качественные различия в соотношениях CD8.CD4 (фигура 14). У пациентов, получавших ниволумаб + ипилимумаб, произошло снижение относительной частоты CDD+CD28+Т-лимфоцитов по сравнению с пациентами, получавшими ниволумаб в качестве монотерапии (фигура 15). Лечение с ниволумаб + ипилимумаб повышало пролиферацию и активацию CD8+CD28- Т-клеток по сравнению с исходным уровнем (фигура 16). При рестимуляции, TILS и РВМС, выделенные из пациентов, получавших ниволумаб в качестве монотерапии, показали повышенную анти-ТАА активность (фигура 17).
249. Лечение ниволумабом увеличивает продукцию IFNγ с помощью TREGS, что указывает на снижение иммуносупрессивной активности. Лечение ниволумабом приводило к появлению функциональных ответов на ТАА у обоих TIL и РВМС. Иммунный профиль опухоль-инфильтрирующих Т-клеток был очень отличительным у одного пациента, испытывавшего связанные с лечением иммунные эффекты в опухоли во время лечения ниволумабом.
ПРИМЕР 9
Темозоламид в комбинации с ипилимумабом и/или ниволумабом в лечении больных с впервые диагностированной GBM или глиосаркомой
250. Данное рандомизированное испытание фазы I изучает безопасность и преимущества темозоломида, когда он дается вместе с ипилимумабом, ниволумабом или комбинацией обоих в лечении больных с впервые диагностированной GBM или глиосаркомой. Лекарственные средства, используемые в химиотерапии, такие как темозоломид, работают по-разному, чтобы остановить рост опухолевых клеток, либо убивая клетки, путем остановки их деления, либо путем остановки их распространения. Моноклональные антитела, такие как ипилимумаб и ниволумаб, могут блокировать рост опухоли по-разному, путем направленного взаимодействия с определенными клетками. Пока не известно, является ли лечение темозоламидом вместе с ипилимумабом, ниволумабом или комбинацией обоих лучшим средством для лечения глиобластомы или глиосаркомы.
251. Данное исследование проводится для того, чтобы определить оптимальную дозу однокомпонентого лечения с ипилимумабом, ниволумабом и комбинацией, когда они даются с темозоломидом во время поддерживающей терапии для вновь диагностированной глиобластомы.
252. Дополнительные цели заключаются в том, чтобы (а) собирать и записывать профили нежелательных явлений для однокомпонентого лечения с ниволумабом и комбинации ипилимумаба и ниволумаба, когда они даются с темозоломидом во время поддерживающей фазы для вновь диагностированной GBM и (b) для выполнения экспериментальных исследований иммунных клеток в образцах опухолей, например, фенотипирования опухоль-инфильтрирующих лимфоцитов (TIL) при рассмотрении опухолевых тканей от диагностических опухолевых блоков.
253. Измерения первичных результатов включают (а) иммунозависимые DLT для однокомпонентного лечения с ниволумабом [Временные рамки: до 8 недель]; или (b) иммунозависимые DLT для комбинации ипилимумаб и ниволумаб, когда они даются с темозоломидом [Временные рамки: до 8 недел ь].
254. Измерения вторичных результатов включают частоту нежелательных явлений, оцененную с использованием критериев Национального института рака общей терминологии для нежелательных явлений (National Cancer Institute Common Terminology Criteria for Adverse Events) версии 4.0 [Временные рамки: до 1 месяца после лечения]. Исследователи будут собирать и записывать профили побочных явлений для однокомпонентного лечения с ниволумабом и комбинации ипилимумаб и ниволумаб, когда они даются с темозоломидом во время поддерживающей фазы для вновь диагностированной GBM. Измерение вторичных результатов будет также включать в себя анализ биомаркеров иммунных клеток в образцах опухолей с использованием стандартного анализа [Временные рамки: до 1 месяца после лечения]. Результаты данного ряда анализов будут использоваться для получения пилотных данных и оценки возможности в получении воспроизводимых результатов при подготовке к последующему рандомизированному, статистически проведенному сравнительному клиническому исследованию.
Группы лечения, участвующие в исследовании
Экспериментальная часть: Группа лечения I (темозоламид и ниволумаб)
255. В течение 5 недель после завершения химиолучевой терапии, пациенты получают темозоломид РО в дни 1-5. Лечение повторяется каждые 28 дней до 6 курсов при отсутствии прогрессирования заболевания или неприемлемой токсичности. Пациенты также получают ниволумаб IV в течение 90 минут один раз каждые 2 недели в течение 24 недель и затем один раз каждые 2 недели в течение 42 недель в отсутствии неприемлемой токсичности.
Экспериментальная часть: Группа лечения II (темозоломид, ниволумаб, ипилимумаб)
256. В течение 5 недель после завершения химиолучевой терапии, пациенты получают темозоломид РО в дни 1-5. Лечение повторяется каждые 28 дней до 6 курсов при отсутствии прогрессирования заболевания или неприемлемой токсичности. Пациенты также получают ипилимумаб IV в течение 90 минут один раз каждые 4 недели в течение 6 курсов и ниволумаб IV в течение 90 минут каждые 2 недели в течение 48 недель при отсутствии неприемлемой токсичности.
257. После завершения лечения исследования, пациенты наблюдаются через 1 месяц, а затем каждые 3 месяца в течение 1 года, каждые 4 месяца в течение 1 года, а затем каждые 6 месяцев после этого.
258. Пациенты, имеющие право на исследование, должны быть 18 лет и старше. Критерии включения для исследования включают в себя:
(a) Гистопатологически доказанный диагноз GBM или глиосаркомы в течение 7 дней до регистрации;
(b) Опухоль должна быть однофокусной опухолью, ограниченной супратенториальным отсеком и подвергнутой общему тотальному или почти общему тотальному удалению;
(c) Блок фиксированной формалином и залитой в парафин (FFPE) опухолевой ткани должен быть доступен для отправки для ретроспективного центрального обзора патологии после регистрации;
(d) Пациенты должны быть зарегистрированы в течение 28 дней после завершения химиолучевой терапии;
(e) История/физикальное обследование в течение 7 дней до регистрации;
(f) Пациенты должны были подвергнуты обследованию с помощью магнитно-резонансной томографии (МРТ) в течение 28 дней после завершения курса облучения, а также должны быть обследованы в течение 7 дней до регистрации; МРТ не должна показывать прогрессирования опухоли
(g) общее состояние по шкале Карновского >= 70 в течение 7 дней до начала регистрации
(h) Абсолютное количество нейтрофилов >= 1500 клеток/мм^3
(i) Количество тромбоцитов >= 100000 клеток/мм^3
(j) азот мочевины крови (АМК) =< 30 мг/дл
(k) Сывороточный креатинин =< 1,7 мг/дл
(l) Общий билирубин (за исключением пациентов с синдромом Гилберта, которые имеют право на исследование, но освобождены от общего критерия приемлемости билирубина) =< 2,0 мг/дл
(m) Аланинаминотрансфераза (ALT) и аспартатаминотрансфераза (AST) =< 2,5× верхний предел нормального (ULN)
(n) Пациент должен завершить химиолучевое лечение (все когорты) в пределах стандартов медицинской помощи, установленных предварительными исследованиями групп Radiation Therapy Oncology Group (RTOG)/NRG Oncology, следующим образом:
(i) Лучевая терапия
Модальность: разрешена 3-мерная (3D) или модулированная по интенсивности лучевая терапия (IMRT) или протонная терапия;
Время инициирования: лучевая терапия должна быть начата в течение или точно через 35 дней после операции
Целевые объемы: определение целевого объема будет основываться на послеоперационной магнитно-резонансной томографии (МРТ) с повышенной контрастностью; предоперационная визуализация должна использоваться для корреляции и улучшенной идентификации, по мере необходимости
Рекомендации по дозированию: начальный целевой объем будет подвергаться воздействию до 46 Гр за 23 фракции; после 46 Гр, будет лечиться conedown или увеличенный объем, в общей сложности, до 60 Гр, с семью дополнительными фракциями в 2 Гр каждая (14 Гр повышенная доза)
(ii) Темозоламид в ходе сопутствующей лучевой терапии
Темозоламид должен непрерывно вводиться с 1 -го дня лучевой терапии до последнего дня лучевой терапии при суточной пероральной дозе 75 мг/м^2 в течение максимально 49 дней
(о) Пациент не должен принимать дозы кортикостероидов, превышающие физиологически замещающую дозу, определяемую как 30 мг кортизона в день или его эквивалента; а также
(р) Пациент должен предоставить конкретное информированное согласие на исследование до начала исследования.
259. Критерии исключения включают в себя:
(a) Окончательные клинические или рентгенологические доказательства прогрессирования заболевания;
(b) Предварительное имплантирование пластины Глиадел или местная брахитерапия;
(c) Применение иммунотерапии, такой как вакцинная терапия, вакцина на основе дендритных клеток или внутриполостная или конвекционная расширенная доставка терапии;
(d) Ранняя инвазивная злокачественность (за исключением немеланоматозного рака кожи), если без болезни в течение как минимум 3-х лет;
(e) Нестабильная стенокардия в течение последних 6 месяцев до регистрации;
(f) Трансмуральный инфаркт миокард в течение последних 6 месяцев до регистрации;
(g) Доказательства недавнего инфаркта миокарда или ишемии по результатам подъемов сегмента ST >= 2 мм с помощью анализа электрокардиограммы (ЭКГ), проведенного в течение 7 дней до регистрации;
(h) Хроническая сердечная недостаточность класса II по классификации Нью-Йоркской кардиологической ассоциации или более, требующая госпитализации в течение 12 месяцев до регистрации;
(i) История инсульта, церебральная сосудистая авария (CVA) или транзиторная ишемическая атака в течение 6 месяцев до регистрации;
(j) Серьезная и неадекватно контролируемая сердечная аритмия;
(k) Значительные сосудистые заболевания (например, аневризма аорты, история расслаивающейся аневризмы аорты) или клинически значимое заболевание периферических сосудов;
(l) Доказательства геморрагического диатеза или коагулопатии;
(m) Серьезная или незаживающая рана, язва или перелом кости или история свища брюшной полости, желудочно-кишечная перфорация, серьезная хирургическая операция из-за внутрибрюшного абсцесса, операционная биопсия или значительное травматическое повреждение в течение 28 дней до начала регистрации, за исключением трепанации для резекции опухоли;
(n) Острая бактериальная или грибковая инфекция, которая требует внутривенного введения антибиотиков на момент регистрации;
(о) Обострение хронической обструктивной болезни легких или другое респираторное заболевание, требующее госпитализации или исключающее исследуемую терапию в момент регистрации;
(р) Печеночная недостаточность, приводящая к клинической желтухе и/или дефектам свертывания крови; однако, следует отметить, что лабораторные тесты на дополнительные функции печени и параметры коагуляции не требуются для вступления в данный протокол;
(q) Синдром приобретенного иммунодефицита (СПИД), основанный на существующем определении Центров по контролю и профилактике заболеваний (Centers for Disease Control and Prevention (CDC)); однако, следует отметить, что тестирование на вирус иммунодефицита человека (ВИЧ) не требуется для вступления в данный протокол;
(r) Активные заболевания соединительной ткани, такие как волчанка или склеродермия, которые, по мнению лечащего врача, могут подвергнуть пациента высокому риску развития иммунологической токсичности;
(s) Пациенты с активным аутоиммунным заболеванием или историей аутоиммунного заболевания, которое может рецидивировать, что может повлиять на жизненно важные функции органа или требует иммуносупрессивного лечения, включая системные кортикостероиды, должно быть исключены; они включают, но не ограничиваются ими, пациентов с историей иммунносвязанного неврологического заболевания, рассеянного склероза, аутоиммунной (демиелинизирующей) невропатии, синдрома Гийена-Барре или хронической воспалительной демиелинизирующей полиневропатии (chronic inflammatory demyelinating polyneuropathy (CIDP)), миастении; системного аутоиммунного заболевания, такого как системная красная волчанка (systemic lupus erythematosus (SLE)), заболевание соединительной ткани, склеродермия, воспалительное заболевание кишечника (inflammatory bowel disease (IBD)), язвенный колит Крона, гепатит; и пациенты с историей токсического эпидермального некролиза (toxic epidermal necrolysis (TEN)), синдрома Стивенса-Джонсона, или фосфолипидного синдрома должны быть исключены;
(i) Следует отметить, что пациенты с витилиго, эндокринными недостатками, включая тиреоидит, управляемые с замещающими гормонами, включая физиологические кортикостероиды, имеют право на включение в исследование; пациенты с ревматоидным артритом и другими артропатиями, синдромом Шегрена и псориазом, который контролируется при наружном применении медицинского препарата, и пациенты с положительной серологией, такой как антинуклеарные антитела (ANA), антитиреоидные антитела, должны быть оценены на наличие участия целевого органа и потенциальной необходимости системного лечения, но в противном случае должны иметь право на включение в исследование;
(t) любые другие существенные медицинские болезни или психические нарушения, которые, по мнению исследователя, помешают введению или завершению протокола терапии;
(u) беременность или кормящие женщины; женщины детородного возраста должны иметь отрицательный серологический тест на беременность в течение 7 дней до регистрации;
(v) Пациенты, которых являются ВИЧ-инфицированными, не имеют права; отметим также, что тестирование на ВИЧ не требуется для получения права на участие в данном протоколе; однако, история ВИЧ вызывает данную неправомочность; а также
(w) История тяжелой реакции гиперчувствительности к любому моноклональному антителу.
ПРИМЕР 10
Рандомизированное открытое исследование фазы 3 ниволумаба в сравнении с темозоламидом, каждого в комбинации с лучевой терапией на недавно диагностированных взрослых пациентах с неметилированным геном MGMT (опухолевая O-6-метилгуанин-ДНК-метилтрансфераза) GBM
260. Основными целями данного исследования являются сравнение общей выживаемости (overall survival (OS)) пациентов, получающих ниволумаб плюс лучевую терапию (RT + ниволумаб) по сравнению с темозоломидом плюс лучевая терапия (RT + TMZ) у пациентов с недавно диагностированной глиобластомой и опухолями с неметилированным геном MGMT после хирургической резекции. Пациенты с неметилированным геном MGMT представляют популяцию GBM с самой высокой неудовлетворенной медицинской необходимостью. В ретроспективном исследовании впервые выявленных больных с GBM, получавших химиолучевое лечение, пациенты с опухолями GBM с неметилированным геном MGMT быстро прогрессируют и имеют медианную общую выживаемость только от 12 до 15 месяцев (по сравнению с метиловым MGMT, с медианной OS от 22 до 26 месяцев).
261. Вторичными целями данного исследования являются: 1) сравнить оцениваемую исследователем выживаемость без прогрессирования (PFS) в группе лечения RT + ниволумаб в сравнении с группой лечения RT + TMZ и 2) оценить общую выживаемость в течение 24 месяцев (OS [24] в группе лечения RT + ниволумаб по сравнению с группой лечения RT + TMZ).
262. Как можно видеть на фигуре 12 и в таблице 7, ниволумаб вводят в дозе 240 мг внутривенно (IV) в виде 30-минутной инфузии каждые 2 недели в течение 16 недель с последующим введением ниволумаба в дозе 480 мг в виде 30-минутной инфузии каждые 4 недели, начиная с 17-й недели, до прогрессирования, неприемлемой токсичности, снятия согласия или завершения исследования, в зависимости от того, что наступит в первую очередь, или темозоломид вводят в дозе 75 мг/м2 перорально ежедневно во время лучевой терапии с последующим 4-недельным перерывом, а затем 6 (28-дневных) циклов темозоломида, дозированного в дни 1-5 при 150 мг/м2 в цикле 1, с увеличением до 200 мг/м2, в зависимости от переносимости, до 6 циклов.
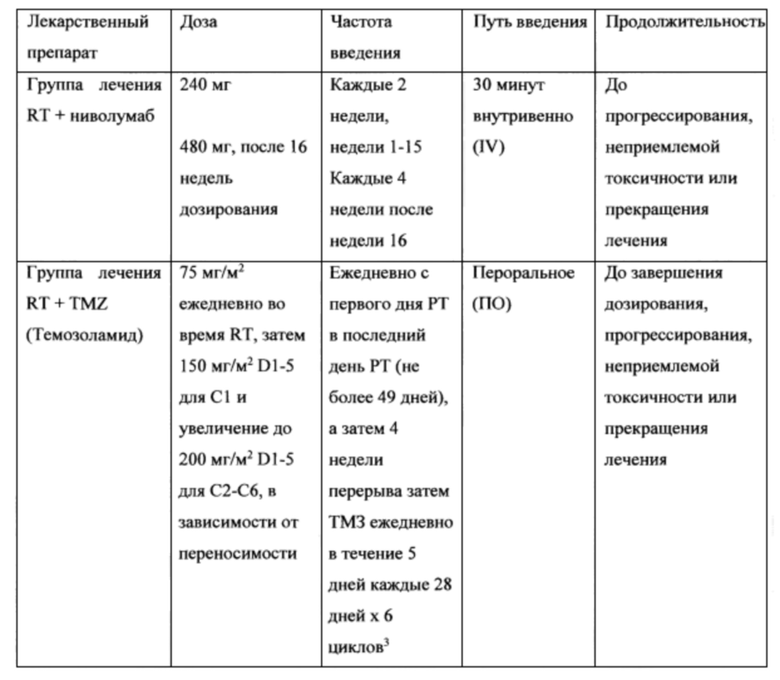
3 Дополнительные циклы темозоломида разрешены для субъектов, зачисленных в исследование в Японии.
263. Пациенты, рандомизированные в группу лечения ниволумабом должны начать лечение в течение 3-х дней после рандомизации и продолжать с дозой 240 мг в виде 30-минутной внутривенной инфузии на 1-й день каждого цикла лечения каждые 2 недели, до прогрессирования, неприемлемой токсичности, отзыва согласия или завершения исследования, в зависимости от того, что наступит в первую очередь. Пациенты, рандомизированные в группу лечения RT+ниволумаб, которые остаются на терапии ниволумабом в течение 16 недель, будет переходить к ниволумабу в дозе 480 мг в виде 30-минутной внутривенной инфузии, вводимой каждые 4 недели (q4w), начиная с 17-й недели. При необходимости для планирования клинического распорядка, пациенты могут получать дозу вплоть до 3-х дней до или после запланированной даты, т.е. с интервалом не менее 12 дней или более чем 17 дней. Доза, которая дается более чем через 3 дня после предполагаемой даты дозирования, будет считаться задержкой.
Последующее дозирование должно быть основано на фактической дате введения предыдущей дозы препарата.
264. Пациентам в группе лечения RT+TMZ будут вводить темозоламид (TMZ, TEMODAR®) ежедневно во время RT и в качестве поддерживающей терапии в течение 6 циклов. Темозоламид (TMZ) будет дозирован при 75 мг/м2 один раз в день непрерывно в течение RT, как правило, в течение 42 дней и не более 49 дней во время сопутствующего дозирования RT + TMZ. После завершения RT будет 4-недельный перерыв. Пациенты получат 6 циклов темозоломида ежедневно × 5 дней каждые 28 дней. Уровни доз для поддерживающей монотерапии TMZ приведены в таблице 13.
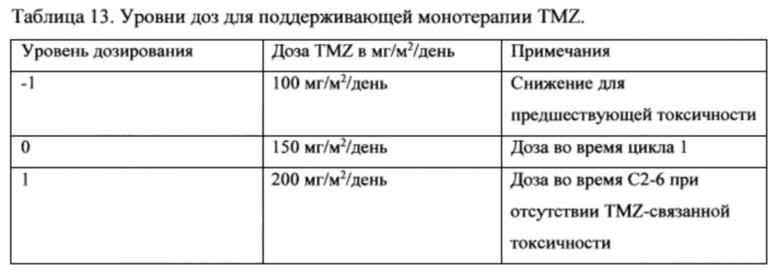
265. В первом поддерживающем цикле TMZ дают в дозе 150 мг/м2, а затем с увеличением до 200 мг/м2 в цикле 2 следующим образом: Если в ходе первого цикла все негематологические наблюдаемые АЕ были ≤2-й степени (за исключением алопеции, тошноты и рвоты) и с уровнем тромбоцитов ≥100×109/л и АНК ≥1,5×109/л, то доза темозоломида должна была эскалировать до дозы на уровне 1, и данная доза должна быть использована в качестве исходной дозы для последующих циклов. Если лечение после цикла 1 должно быть задержано из-за продолжающихся негематологических АЕ 2-й степени, то никакая эскалация не возможна. Если доза не эскалировала в цикле 2, то доза не должна эскалировать в последующих циклах на основе нежелательных явлений. Если в течение сопутствующего периода с RT было ранее достигнуто снижение дозы, то доза -1 должна быть начальной дозой для последующих циклов.
266. Поддерживающая терапия начинается после 4 недельного перерыва лечения после RT. Лечение TMZ может начаться, если анализ крови (получен в течение предыдущих 3-х дней) показывает ANC ≥1,5×109/л, количество тромбоцитов ≥100×109/л, и любое негематологическое АЕ ≥3-й степени (за исключением алопеции), должно быть уменьшено до степени ≤1. Если АЕ сохраняется, лечение должно быть отложено на 1 неделю в течение 4 недель подряд. Если, после 4 х недель задержки, все АЕ до сих пор не уменьшены до степени ≤1: то любая дальнейшая адьювантная терапия с темозоломидом должна быть прекращена.
267. Поисковыми целями исследования являются следующие: 1) оценить безопасность групп лечения RT + ниволумаб и РТА + TMZ; 2) оценить связанное со здоровьем качество жизни, используя анкету-опросник EQ-5D и модуль (QLQ-C30) Европейской организации по исследованию и лечению рака (European Organization for Research and Treatment of Care General Cancer Module) и модуль рака мозга (QLQ-BN20); 3) оценить неврологическое функционирование в группах лечения RT + ниволумаб и RT+TMZ с использованием шкалы неврологической оценки в нейроонкологии (Neurologic Assessment in Neuro-Oncology (NANO)); 4) оценить когнитивные возможности в группах лечения RT + ниволумаб и RT+TMZ с помощью набора тестов Cogstate; 5) оценить фармакодинамическую активность ниволумаба в периферической крови и опухолевой ткани, измеренную с помощью проточной цитометрии, иммуногистохимии, анализа растворимых факторов и экспрессии генов (технология микрочипов, количественная RT-PCR); 7) исследовать связь между биомаркерами в периферической крови и опухолевой ткани, например, экспресию PD-L1, с безопасностью и эффективностью для пациентов с впервые диагностированной GBM, получавших ниволумаб; 8) охарактеризовать РК ниволумаба и исследовать взаимосвязь воздействия и ответа; и 9) характеризовать иммуногенность ниволумаба в данной установке.
268. Данное исследование будет регистрировать пациентов с вновь диагностированной глиобластомой, после хирургической резекции опухоли. После получения информированного согласия, испытуемые вступят в фазу скрининга. Опухолевая ткань будет оцениваться на метилирование гена MGMT в анализе центральной лаборатории. Для того, чтобы рандомизировать 550 правомочных пациентов, ожидается, что, в общей сложности, около 1200 пациентов должны пройти скрининг опухоли, из которых около 600 пациентов, как ожидается, имеют опухоли с неметилированным геном MGMT.
269. Пациенты с полученным в центральной лаборатории результатом неметилированного гена MGMT могут продолжать проходить стадию скрининга, в которой будет задокументировано право на получение рандомизации и представлена исходная демографическая информация и информация о болезни; для деталей. Во время фазы скрининга, в течение 48 часов после операции (в течение 24 часов, предпочтительнее) должна быть получена МРТ с контрастным усилением.
270. Когда все готово для того, чтобы начать лечение по программе исследования, испытуемые будут переходить к фазе лечения данного исследования. Все пациенты, которые входят в фазу лечения, т.е. все рандомизированные пациенты, будут проводить наблюдениеся в отношении безопасности и переносимости, прогрессирования опухоли и выживаемости. МРТ с повышенным контрастом следует проводить через 4 недели после завершения лучевой терапии, а затем каждые 8 недель (±1 неделю) до прогрессирования, независимо от схемы лечения. Прогрессирование опухоли будет оцениваться с помощью критериев рентгенологической оценки в нейроонкологии (Radiologic Assessment in Neuro-Oncology criteria (RANO)). Дополнительные оценки будут выполнены для когнитивной функции, неврологической функции, биомаркеров и сообщенных пациентом результатов о качестве жизни.
271. После прекращения лечения исследования по какой-либо причине, все рандомизированные пациенты будут входить в фазу последующего наблюдения. В краткосрочной перспективе, посещения определены для отчетности о связанных с лечением нежелательных явлениях. Все пациенты, у которых не было обнаружено прогрессирование заболевания в момент времени, когда лечение по программе исследования было остановлено, будут внимательно проводить наблюдениеся в отношении прогрессирования, с проведением МРТ с контрастным усилением каждые 8 недель (±1 неделю). О последующем лечении после прогрессирования будет сообщено. Все рандомизированные пациенты должны проводить наблюдениеся в отношении выживания (первичная конечная точка).
272. Общее время, прошедшее от диагностической операции до начала RT, не может превышать 42 дней, включая максимум 7 дней от рандомизации до начала RT. Пациенты в группе лечения RT + ниволумаб должны начать получать ниволумаб в течение 3 дней после рандомизации, и могут быть предоставлены в любое время до начала RT, как клинически уместно. Пациенты, рандомизированные в группу лечения RT + TMZ, начнут комбинированную химиолучевую терапию в тот же день, до 7 дней после рандомизации, как клинически уместно.
273. Лучевая терапия должна начинаться после существенного восстановления после хирургической резекции, не более чем через 42 дней после операции. Суммарная доза RT с внешним пучком, составляющая 60 Гр, будет вводиться в суточных дозах 2 Гр, как правило, по расписанию 5 дней введение/2 выходных дня, в зависимости от обстоятельств для планирования, в течение 6-7 недель. Зона опухолевого очага RT включает предоперационной объем опухоли плюс запас в 2-3 см, по указанию радиационного онколога. Предположительное прогрессирование, которое происходит в течение 12 недель после RT, может быть "псевдопрогрессированием"; в данной ситуации, подозреваемое прогрессирование должна быть подтверждено до прекращения лечения.
274. Фаза последующего наблюдения начинается, когда принимается решение о прекращении пациентом лечения в программе исследования, включая появление нежелательных явлений, для максимального клинического эффекта (решение исследователя), по требованию пациента, для завершения TMZA-поддерживающей терапии (6 циклов), прогрессирования заболевания или по другой причине.
275. Пациенты будут оцениваться в отношении нежелательных явлений с визитами через 35 дней (±7 дней) и 115 дней (±7 дней) после задокументированной последней дозы в течение как минимум 100 дней после прекращения приема препарата, но за связанными с лекарственными средствами токсическими явлениями должно продолжаться наблюдение, пока они не исчезнут, вернутся к исходному уровню или считаются необратимыми.
276. Пациенты, которые прекращают лечение по причине, отличной от прогрессирования заболевания, будут продолжать проходить оценку опухоли и оценку по шкале NANO до прогрессирования (вторичноя конечная точка исследования). Введение противоопухолевой терапии при отсутствии прогрессирования настоятельно рекомендуется.
277. Все пациенты должны проводить наблюдениеся в отношении выживания (первичная конечная точка исследования). О последующем лечении, если таковое имеется, будет сообщено.
278. Критерии включения заключаются в следующем:
Подписано информированное письменное согласие:
Письменное информированное согласие и разрешение HIPAA (относятся к охватываемым лицам только в США), полученные от пациента/законного представителя перед выполнением каких-либо процедур, связанных с протоколом;
Пациенты должны быть готовы и в состоянии выполнить запланированные визиты, график лечения, лабораторные исследования, а также другие требования исследования.
Целевая аудитория:
Недавно диагностированная гистопатологически подтвержденная супратенториальная GBM (злокачественная глиома 4-й степени по классификации Всемирной организация здравоохранения, включая глиосаркому)
a) Нет лечения для GBM, кроме хирургического вмешательства;
b) Послеоперационная исходная МРТ в течение 48 часов (предпочтительно 24 часов) после хирургической резекции
Полное восстановление после хирургической резекции;
a) Нет каких-либо серьезных проблем, связанных с безопасностью, после операции
b) ≤20 мг преднизолона в день или ≤3 мг дексаметазона в день (или эквивалент)
Централизованное подтверждение GBM с неметилированным геном MGMT;
Оценка общего состояния по шкале Карновского ≥70;
Приемлемый для лучевой терапии, основанной на руководящих принципах NCCN.
Пациент может повторно зарегистрироваться в исследовании.
Возраст и репродуктивный статус:
Мужчины и женщины в возрасте ≥18 лет;
Женщины детородного возраста (Women of childbearing potential (WOCBP)) должны иметь отрицательный тест, выполненный на сыворотке или моче, на беременность (минимальная чувствительность 25 МЕ/л или эквивалентные единицы HCG) в течение 24 часов до начала введения исследуемого препарата;
Женщины не должны кормить грудью;
Женщины детородного возраста (WOCBP) должны согласиться следовать инструкциям для способа(ов) контрацепции в течение 30 дней (продолжительность овуляторного цикла) плюс время, необходимое для того, чтобы прошло пять периодов полувыведения исследуемого препарата. Конечный элиминационный период полувыведения ниволумаба составляет до 25 дней. Конечный элиминационный период полувыведения темозоломида составляет 1,2 часа.
WOCBP, рандомизированным для получения ниволумаба, следует использовать адекватный способ избегания беременности в течение 23 недель (30 дней плюс время, необходимое для того, чтобы прошло пять периодов полувыведения ниволумаба) после последней дозы исследуемого препарата. WOCBP, рандомизированным для получения темозоламида, следует использовать адекватный способ, чтобы избежать беременности в течение 6 недель (30 дней плюс время, необходимое для того, чтобы прошло пять периодов полувыведения темозоламида) после последней дозы темозоломида.
Мужчины, которые сексуально активны с WOCBP, должны согласиться следовать инструкциям для способа(ов) контрацепции в течение 90 дней (длительность оборота спермы) плюс время, необходимое для того, чтобы прошло пять периодов полувыведения исследуемого лекарственного средства. Конечный элиминационный период полувыведения ниволумаба составляет до 25 дней.
Мужчины, рандомизированные для получения ниволумаба, которые ведут активную половую жизнь с WOCBP, должны продолжать контрацепцию в течение 31 недель (90 дней плюс время, необходимое необходимое для того, чтобы прошло пять периодов полувыведения ниволумаба) после последней дозы исследуемого препарата. Мужчинам, рандомизированным для получения темозоламида, следует рекомендовать не зачинать ребенка вплоть до 6 месяцев после приема последней дозы и обратиться за консультацией по криоконсервации спермы до начала лечения из-за возможности необратимого бесплодия вследствие терапии с TMZ.
Азооспермические мужчины и WOCBP, которые не являются постоянно активными гетеросексуальными лицами, освобождаются от контрацептивов. Однако WOCBP должны еще пройти тестирование на беременность, как описано в данном разделе.
Исследователи должны напоминать WOCBP и пациентам мужского пола, которые сексуально активны с WOCBP, о важности профилактики беременности и последствиях неожиданной беременности. Исследователи должны консультировать WOCBP и пациентов мужского пола, которые ведут активную половую жизнь с WOCBP, по вопросам использования высокоэффективных способов контрацепции. Высокоэффективные способы контрацепции имеют частоту отказов <1% при использовании последовательно и правильно.
Как минимум, пациенты должны согласиться с использованием двух способов контрацепции, с одним из способов, который является весьма эффективным, и другим способом, который является либо весьма эффективным, либо менее эффективным, что можно найти в информированном согласии.
Результаты физикальных и лабораторных тестов:
WBC ≥2000/мкл;
Нейтрофилы ≥1500/мкл;
Тромбоциты ≥100×103/мкл;
Гемоглобин ≥9,0 г/дл;
Креатинин сыворотки крови ≤1.5 × ULN или клиренс креатинина (CrCl)≥50 мл/мин (с использованием формулы Кокрофта-Голта)
Женский CrCl = (140 - возраст в годах) × масса в кг × 0,85 72 × сывороточный креатинин в мг/дл
Мужской CrCl = (140 - возраст в годах) × масса в кг × 1,00 72 × сывороточный креатинин в мг/дл
AST ≤3,0 × ULN;
ALT ≤3,0 × ULN;
Общий билирубин ≤1,5 х ULN (за исключением пациентов с синдромом Гилберта, которые могут иметь общий билирубин <3,0 × ULN).
279. Критерии исключения являются следующими:
Исключения целевой болезни:
Предшествующее лечение GBM (кроме хирургической резекции);
Рецидив;
GMB с метилированным, частично метилированным или неопределенным геном MGMT;
Биопсии только GBM во время операции, которая определяется как <20% резекции;
Постоянное требование супрафизиологического стероида, которое определяется как >20 мг преднизолона в день или >3 мг дексаметазона в день (или эквивалент), из-за внутричерепного массового эффекта;
Кровоизлияние в ЦНС степени >1 на исходном МРТ-скане, если впоследствии не документировано, что исчезло;
Любое известное метастатическое экстракраниальное или лептоменингиальное заболевание;
Вторичная GBM (т.е. прогрессирование от предыдущей астроцитомы низкой степени злокачественности или анапластической астроцитомы); а также
Опухоль с известной мутацией в гене IDH (если доступно; тест не требуется).
История болезни и сопутствующие заболевания:
Любое серьезное или неконтролируемое медицинское расстройство, которое, по мнению исследователя, может увеличить риск, связанный с участием в исследовании или введении изучаемого препарата, ухудшить способность пациента получить терапию по протоколу или мешать интерпретации результатов исследования;
Предыдущее лечение с анти-PD-1, анти-PD-L1, анти-PD-L2, антителом против CTLA-4 или любым антителом или другим препаратом, специфически нацеленным на костимуляцию Т-клеток или пути иммунных контрольных точек;
Пациенты с активным, известным или подозреваемым аутоиммунным заболеванием. Пациентам с сахарным диабетом типа I, гипотиреозом, только требующим замены гормона, кожными заболеваниями (такими как псориаз, витилиго или алопеция), не требующими системного лечения, или состояниями, от которых не ожидается их повторения в отсутствие внешнего триггера, разрешается регистрироваться для исследования;
Пациенты с состоянием, требующим системное лечение либо с кортикостероидами (>20 мг ежедневно преднизона или >3 мг дексаметазона или эквивалента) или другими иммуносупрессивными препаратами, в течение 14 дней после рандомизации. Ингаляционные или топикальные стероиды и дозы стероидов для замены гормонов коры надпочечников >20 мг ежедневно преднизона или >3 мг дексаметазона или эквивалента разрешаются при отсутствии активного аутоиммунного заболевания;
Пациенты с интерстициальным заболеванием легких, которое является симптоматическим или может помешать обнаружению или управлению предполагаемой, связанной с лекарственными средствами токсичности для легких;
Пациенты с историей угрожающей жизни токсичности, включая реакцию гиперчувствительности, связанную с предшествующим лечением иммуноглобулином другого состояния (за исключением тех, которые, как считается, вряд ли вновь произойдут, с письменного разрешения медицинского консультанта), или любым другим компонентом изучаемого лекарственного средства;
История или свидетельство о физикальном/неврологическом обследовании другого состояния центральной нервной системы (например, судороги, абсцесс), не связанного с раком, если должным образом контролируется с помощью лекарств или считается потенциально не мешающим протоколу лечения;
Хирургическая процедура за <7 дней до начала лечения исследования, устройство сосудистого доступа никаких ограничений;
Пациенты, не способные (например, в связи с кардиостимулятором или ICD устройством) или не желающие иметь МРТ головы с контрастным усилением;
История аллергии или повышенной чувствительности к компонентам изучаемого препарата;
Невозможность проглотить пероральное лекарство или любое желудочно-кишечное заболевание или хирургическая процедура, которые могут повлиять на поглощение исследуемого препарата.
Результаты физикальных и лабораторных тестов:
Любой положительный тест на наличие вируса гепатита В или вируса гепатита С;
Известная история положительного теста на вирус иммунодефицита человека (ВИЧ) или известный синдром приобретенного иммунодефицита (СПИД). Примечание: Тестирование на ВИЧ должно проводиться в местах, санкционированных местными правилами.
Аллергии и нежелательная реакция на лекарственный препарат:
История аллергии или повышенной чувствительности к компонентам препарата исследования
Другие критерии исключения:
Заключенные или пациенты, которые находятся непроизвольно в местах лишения свободы (Примечание: при определенных обстоятельствах, человек, который был заключен в тюрьму, может быть включен или ему может быть разрешено продолжать участие в качестве пациента Применяются строгие условия и требуется одобрение.);
Пациенты, которые в обязательном порядке задержаны для лечения либо психиатрической, либо физикальной (например, инфекционные заболевания) болезни.
280. Сопутствующее лечения. Ожидается, что зачисленные пациенты будут снижать дозу приема системных кортикостероидов настолько быстро, насколько это клинически возможно, во время фазы скрининга и прекратят прием, если это возможно до рандомизации. Поддерживающий уход, обусловленный всеми связанными с заболеванием или связанными с лечением нежелательными явлениями, должен быть максимально полным для всех пациентов данного исследования.
281. Запрещенные и/или ограниченные процедуры. Параллельное противоопухолевое лечение, включая другую химиотерапию, иммунотерапию, дополнительную лучевую терапию или исследуемые агенты для лечения GBM, запрещено во время фазы лечения.
Использование любого дополнительного неинвазивного лечения GBM с использованием медицинских устройств (например, novoTTF, OPTUNE©) запрещено.
282. Иммуносупрессивные агенты, включая системные кортикостероиды, запрещены во время лечения в рамках данного исследования, если не используются для лечения нежелательного явления, связанного с лекарственными средствами. Пациенты с состоянием, требующим системного лечения либо кортикостероидами (>20 мг ежедневно преднизона или >3 мг дексаметазона в день (или эквивалент), либо другими иммуносупрессивными препаратами (включая в течение 14 дней с момента рандомизации), исключены. Пациенты, которые продолжают требовать супрафизиологических доз стероидов (преднизон >20 мг в день или >3 мг дексаметазона в день или эквивалент) не могут быть рандомизированы. Ингаляционные или топикальные стероиды и дозы стероидов для замены гормонов коры надпочечников ≤20 мг ежедневно преднизона или ≤3 мг дексаметазона в день или эквивалента разрешаются в отсутствие активного аутоиммунного заболевания.
283. Применение вальпроевой кислоты запрещено в группе лечения RT + TMZ; пациент должен быть переведен на альтернативную противосудорожную терапию.
284. Прекращение участия пациентов в любых схемах лечения с исследуемым препаратом. Пациенты должны прекратить получать исследуемый продукт (и неисследуемый продукт, на усмотрении исследователя) по любой из следующих причин
Прогрессирование заболевания, за исключением специально указанных случаев;
Максимальный клинический эффект, как определено исследователем;
Любое клиническое нежелательное явление (АЕ), отклонения от лабораторных показателей или интеркуррентные заболевания, которые, по мнению исследователя, указывают на то, что дальнейшее участие в исследовании не в интересах пациента;
Запрос пациента прекратить лечение по программе исследования;
Завершение 6 циклов поддерживающей терапии TMZ;
Прекращение исследования компанией Bristol-Myers Squibb (BMS);
Потеря способности свободно предоставлять согласие по причине тюремного заключения или непроизвольного лишения свободы для лечения либо психиатрической, либо физикальной (например, инфекционные заболевания) болезни;
Инициирование противоопухолевой терапии, отличающейся от применяемой в рандомизированных группах лечения.
285. Поскольку данное исследование - это исследование выживаемости, пациенты будут оставаться в исследовании и после прекращения лечения в рамках исследования для документирования прогрессирования болезни и смерти.
Оценки исследования
286. Оценка безопасности: Нежелательные явления будут оцениваться непрерывно в ходе исследования и в течение 100 дней после последнего лечения. Нежелательные явления будут закодированы с использованием самой последней версии MedDRA и рассмотрены в отношении потенциальной значимости и важности. Нежелательные явления будут оцениваться в соответствии с критериями NCI СТСАЕ версии 4.03. За пациентами должны проводить наблюдения, пока все связанные с лечением нежелательные явления не восстановятся до исходного уровня или будут признаны исследователем необратимыми.
287. Оценки эффективности: Исходная МРТ с повышенным контрастированием в течение 48 часов после операции (в течение 24 часов, предпочтительно) будет выполняться на установке МРТ, удовлетворяющей требованием исследования по по сбору специфических для исследования данных. Оценки по визуализации опухоли будут проводиться через 4 недели после завершения лучевой терапии, затем каждые 8 недель (+/- 1 неделю), после этого - до прогрессирования заболевания. Кроме того, оценка по шкале NANO будет завершена до дозирования дня 1 недели 1, а затем с каждой МРТ. Испытуемые будут подвергаться лечению до проявления неприемлемой токсичности или прогрессирования заболевания. После прекращения терапии будет оцениваться безопасность во время визита 2 для последующего наблюдения (Follow Up) (через ~100 дней после последней дозы). Статус выживания будет оцениваться каждые 3 месяца после того, как последующие визиты будут завершены, и может быть завершен по телефону или при посещениях человека.
Статистические соображения:
288. Размер образца: Около 550 пациентов будут рандомизированы в две группы лечения (RT + ниволумаб в сравнении с RT + TMZ) в соотношении 1:1, стратифицированных по полной или частичной резекции на исходном уровне.
289. Данное исследование требует по меньшей мере 397 событий (например, смертей), с промежуточным анализом после по меньшей мере 298 событий (75% от общего числа событий OS), чтобы обеспечить 90% мощности критерия для выявления отношения рисков (hazard ratio (HR)), равного 0,72, с общей ошибкой 1-го типа, равной 0,05 (двустороннего). Критерии досрочного завершения исследования будут определяться для промежуточных и окончательных анализов, основываясь на фактическом количестве событий OS на момент проведения анализа с использованием альфа-функции Lan DeMets с границами статистической значимости О'Брайана-Флеминга. Если промежуточные и окончательные анализы проводятся именно на планируемом количестве событий, прогнозируемый уровень альфа будет составлять 0,019 и 0,044, для промежуточных и окончательных анализов, соответственно. Это приводит к наблюдаемому HR, равному 0,762 (медианная ОС 13,0 против 17,0 месяцев) и 0,817 (медианная OS 13,0 против 15,9 месяцев) или менее, приводя к статистически значимому улучшению в промежуточном и окончательном анализе, соответственно.
290. Вторичная конечная точка только для выживания без прогрессирования (progress-free survival, PFS) будет протестирована иерархически.
291. Конечные точки: Первичной конечной точкой является общая выживаемость (OS). OS определяется как время между датой рандомизации и датой смерти по любой причине. Пациент, который не умер, будет цензурирован по последней дате, когда он был известен живым. За OS будут проводить наблюдение непрерывно, пока пациенты получают исследуемый препарат, и каждые 3 месяца лично или с помощью контакта по телефону во время фазы последующего выживания исследования.
292. Вторичная конечная точка PFS определяется как время от рандомизации до даты прогрессирования первой документированной опухоли или смерти по какой-либо причине. Пациенты, которые умирают без сообщения о прогрессировании, будут считаться имеющими прогрессирование по дате смерти. Пациенты, которые не имели прогрессирования заболевания или умирают, будут цензурированы на дату последней оценки опухоли. Пациенты, которые не имеют какой-либо оценки при исследовании опухоли и не умерли, будут цензурированы на дату рандомизации. Пациенты, которые начали любую последующую противораковую терапию без предварительного отчетного прогрессирования, будут цензурированы на дату последней оценки опухоли до начала последующей противораковой терапии. Пациенты, которые имели хирургическую резекцию после начала лечения исследования, будут цензурированы на дату последней оценки опухоли до начала хирургической резекции. PFS будет определяться ответом, сообщенным исследователем, на основании критериев RANO.
293. Вторичная конечная точка уровня выживаемости в течение 24 месяцев определяется как вероятность выживания по Каплану-Мейеру в течение 24 месяцев.
Анализы:
294. Анализы первичной конечной точки OS и вторичных конечных точек PFS и OS [24] будут основываться на всех рандомизированных пациентах. Процедура последовательного тестирования группы будет применяться к OS для контроля общей ошибки типа I из промежуточных и окончательных анализов. Если демонстрируется повышение OS, то иерархический подход проверки гипотез для вторичной конечной точки PFS будет использоваться для сохранения полученной в исследовании (study-wise) частоты ошибки I типа, равной 0.05. OS[24] будет оценена с использованием способа Каплана-Мейера.
295. Распределения OS и PFS у всех рандомизированных пациентов будут сравниваться между группами лечения с использованием двустороннего стратифицированного логрангового теста. Отношение рисков (HR) и соответствующие двусторонние 100 × (с 1-регулируемым параметром α) % доверительные интервалы (ДИ, confidence intervals (CI)) будут оцениваться в модели пропорционального риска Кокса, используя лечение как один ковариат. Стратифицировано по полной или частичной резекции на исходном уровне.
296. Кривые OS и PFS будут оценены с использованием продукт-лимит способа Каплана-Мейера. Медиана OS и PFS, наряду с соответствующими двухсторонними 95%-ными CI, будут вычислены с помощью преобразования повторным логарифмированием.
297. OS[24] и соответствующие 95%-ные CI будут вычислены с помощью преобразования повторным логарифмированием после того, как все пациенты пройдут последующее наблюдение в течение по меньшей мере 24 месяцев.
Конечные точки.
298. Первичная конечная точка(и). Первичная конечная точка представляет собой общую выживаемость (OS). OS определяется как время между датой рандомизации и датой смерти по любой причине. Пациента, который не умер, цензурируют по последней дате, когда известно, что он жив. За OS следят непрерывно, пока субъекты принимают исследуемый препарат, и каждые 3 месяца при личном контакте или по телефону во время исследовательской фазы последующего наблюдения за выживанием.
299. Вторая конечная точка(и). Первая вторичная цель (сравнение PFS между группами лечения RT + ниволумаб и RT + TMZ) будет измеряться по конечной точке PFS. PFS определяется как время от рандомизации до даты прогрессирования первой документированной опухоли или смерти из-за какой-либо причине. Пациенты, которые умирают без сообщенного прогрессирования, будут считаться имевшими прогрессирование на дату смерти. Пациенты, которые не имели прогрессирования заболевания или умирают, будут цензурированы на дату последней оценки опухоли. Пациенты, которые не имеют какой-либо оценки на исследование опухоли и не умерли, будут цензурированыы на дату рандомизации. Пациенты, которые начали любую последующую противораковую терапию без предварительного сообщенного прогрессирования, будут цензурированы при последней оценке опухоли до начала последующей противораковой терапии. Пациенты, которые имели хирургическую резекцию после начала лечения исследования, будут цензурированы на дату последней оценки опухоли до начала хирургической резекции PFS будет определяться сообщенным исследователем ответом на основании критериев RANO. 300. Исследовательские конечные точки.
Безопасность и переносимость будут измеряться по частоте нежелательных явлений, серьезных нежелательных явлений, смерти и лабораторных аномалий.
Оценка NANO будет измеряться распределением изменения от исходного уровня с оценкой функции в баллах для каждого домена.
Фармакокинетические параметры будут оценены с использованием данных зависимости сывороточной концентрации от времени. Оценка HRQoL будет измеряться изменениями от исходной линии в композитной шкале EORTC-QLQ-C30 global health status/QoL и изменениями от исходной линии в оставшихся шкалах EORTC QLQ-C30 и BN-20. Все шкалы и отдельные элементы пересчитываются на категориальные шкалы и линейно преобразуются в шкалы 0-100, где более высоким оценкам по функциональной шкале соотвествуют более высокие уровни функционирования, более высокие показатели для глобального состояния здоровья/качества жизни, представляющие более высокие уровни глобального состояния здоровья/качества жизни и более высокие баллы по шкале симптомов, представляющие более высокий уровень симптомов. Оценка HRQoL общего состояния здоровья будет измеряться изменениями от исходной линии в шкалах EQ-5D.
Когнитивная способность будет оцениваться изменениями от исходного уровня в нейрокогнитивном функционировании, когнитивных нарушениях и/или исполнительном функционировании с использованием Cogstate. Неврологическое функционирование будет оцениваться с использованием шкалы NANO.
301. Настоящая заявка испрашивает преимущество предварительной заявки США №62/092783, поданной 16 декабря 2014 и 62/261130, поданной 30 ноября 2015 года, которые включены в данное описание в качестве ссылки во всей их полноте.
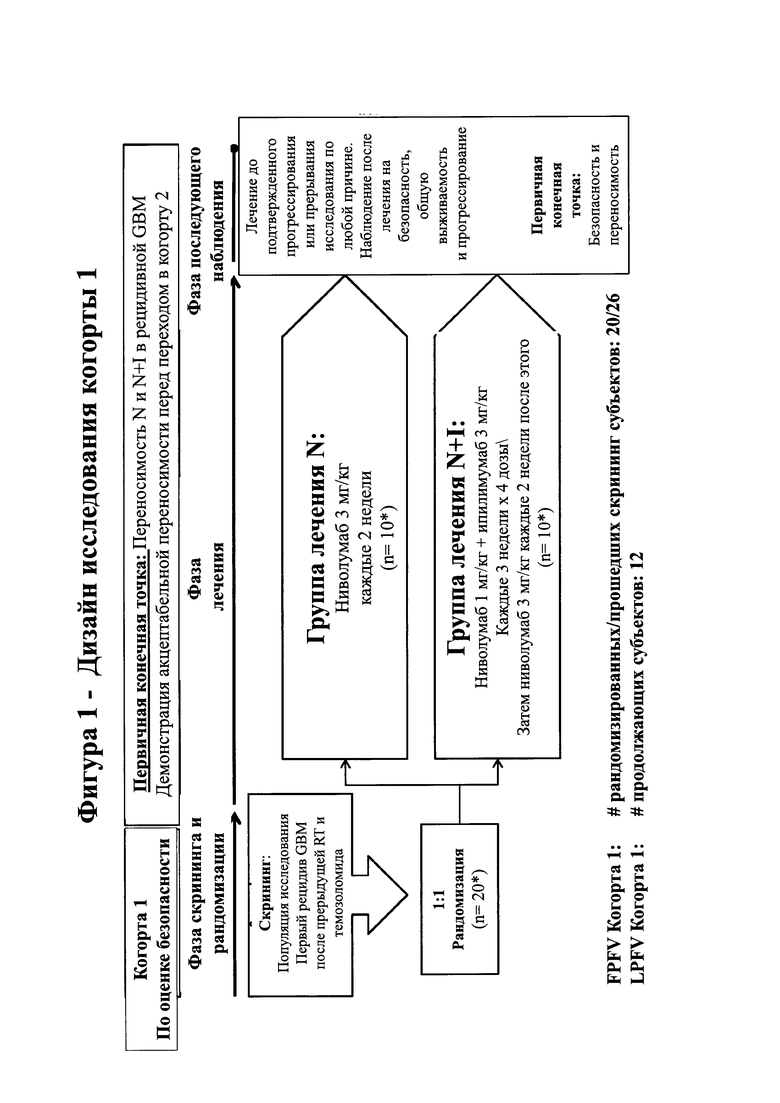


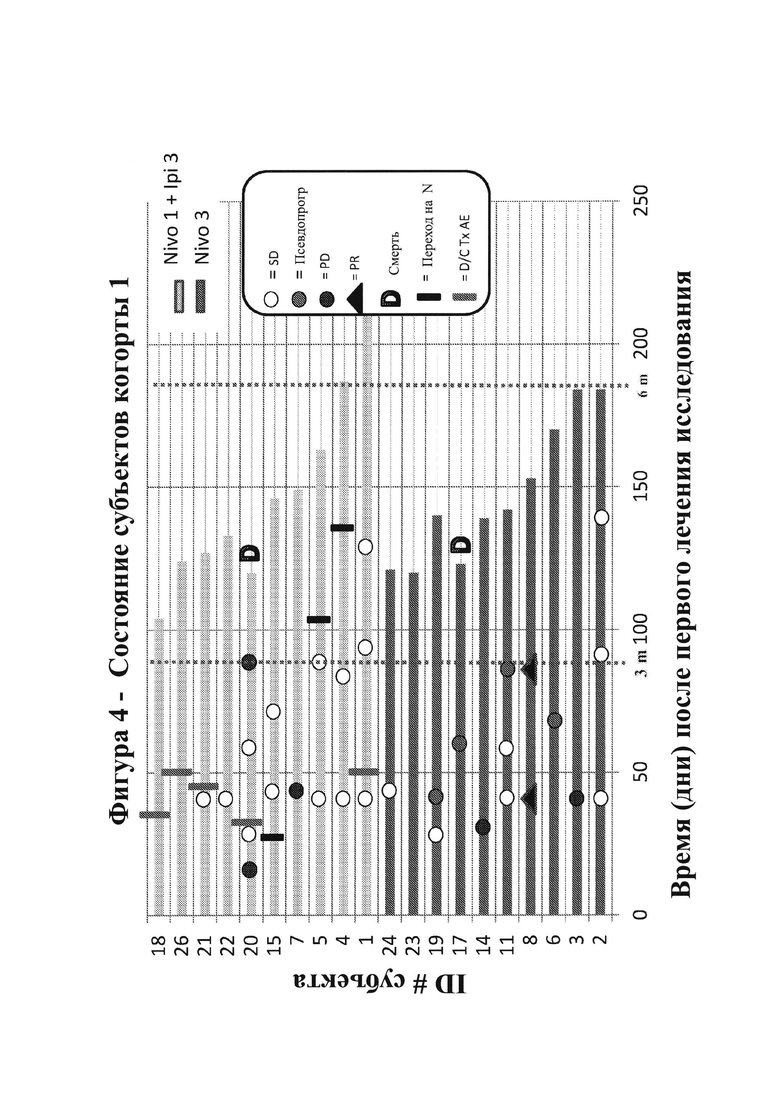

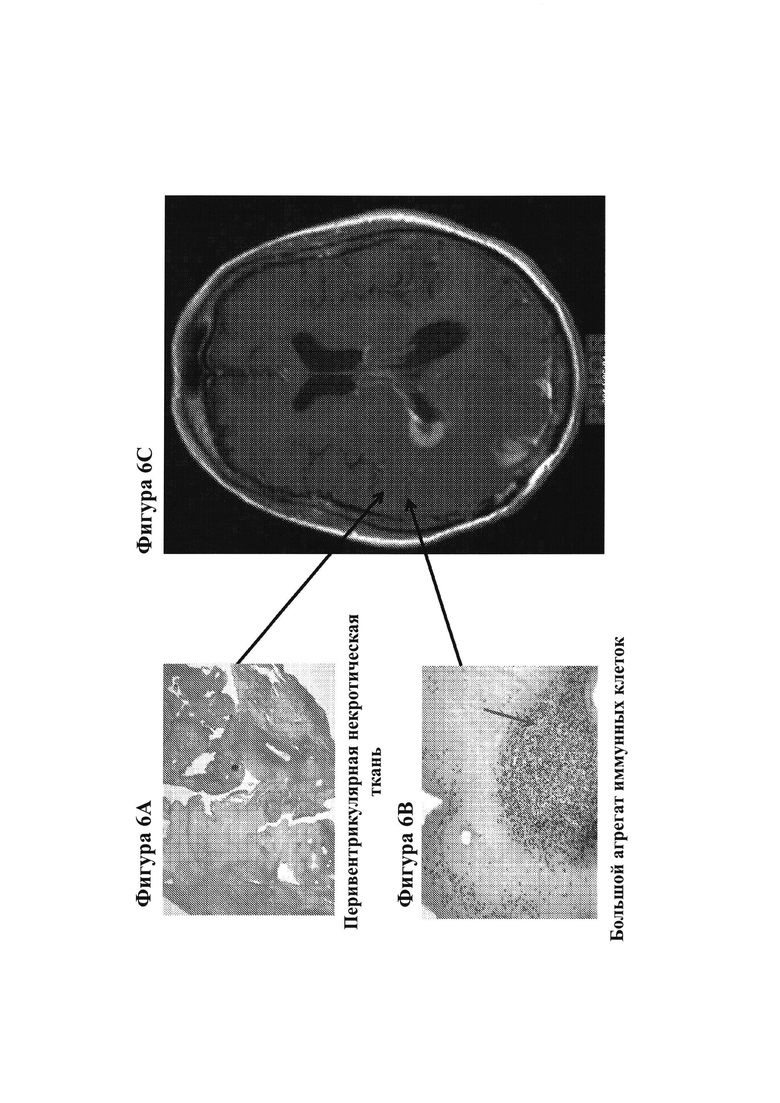


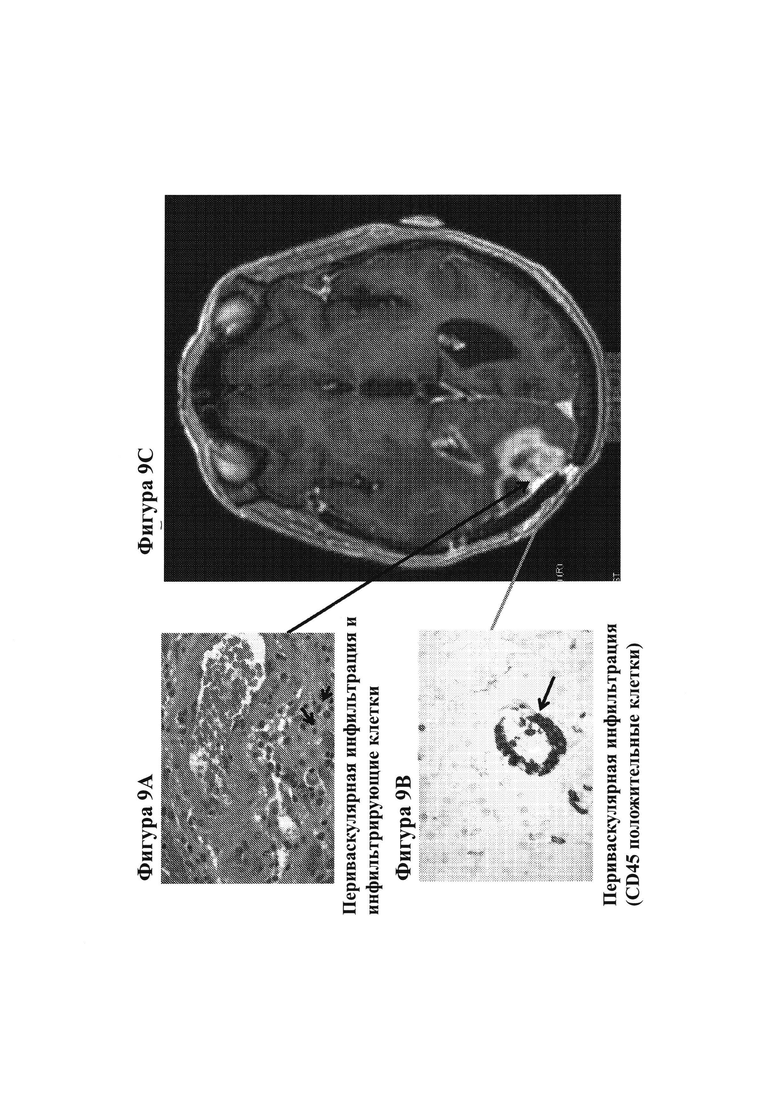
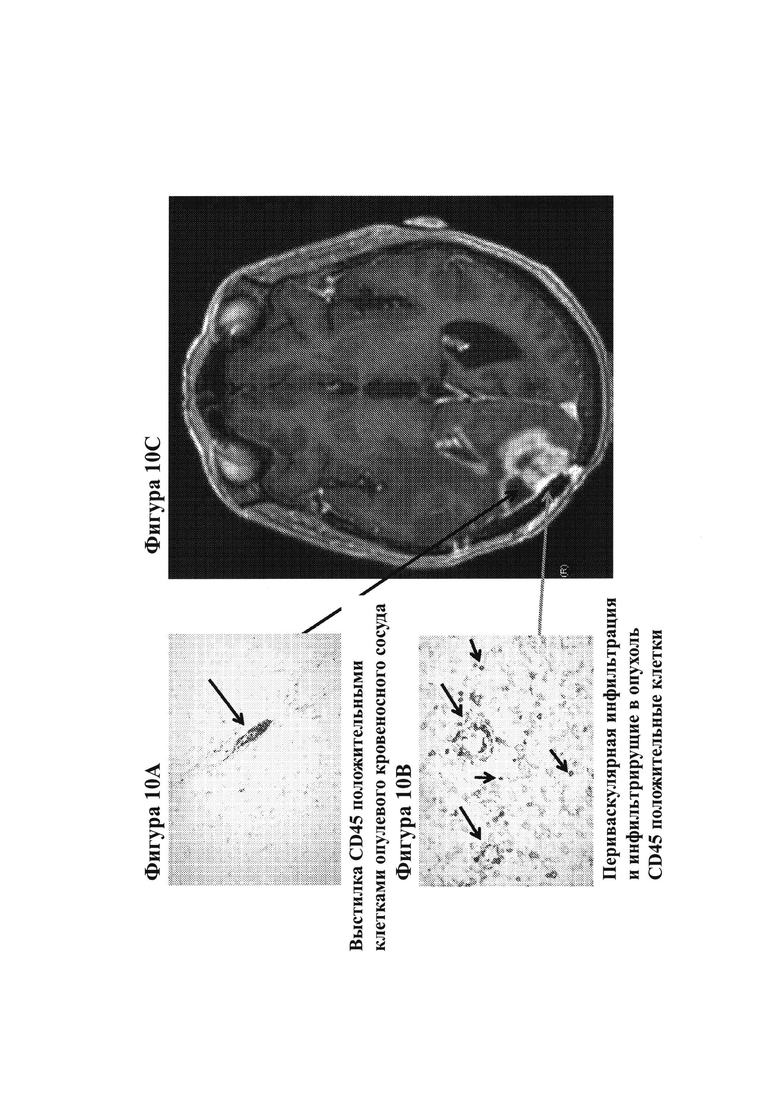
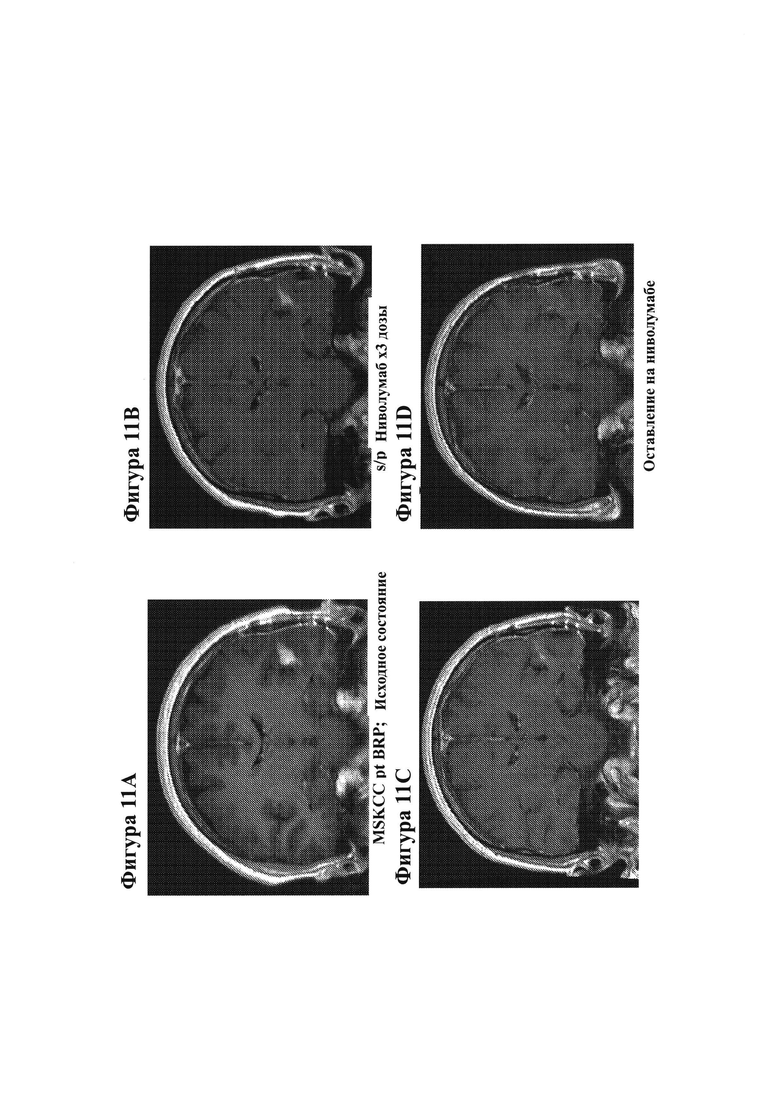
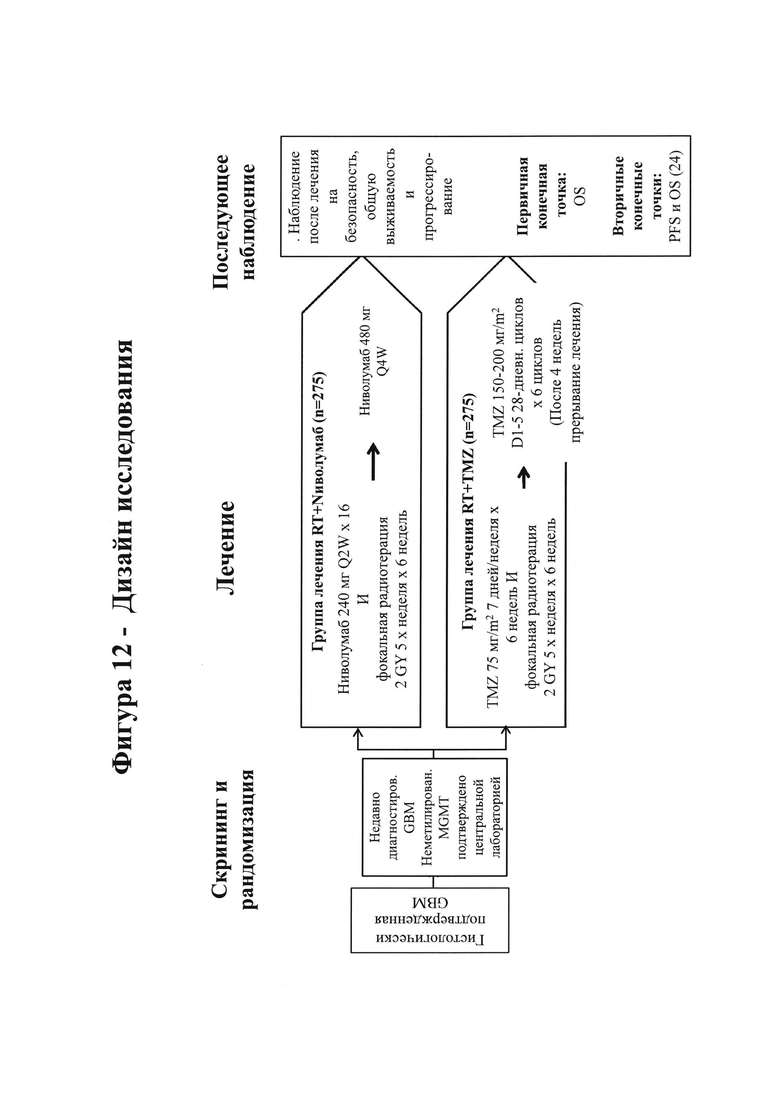
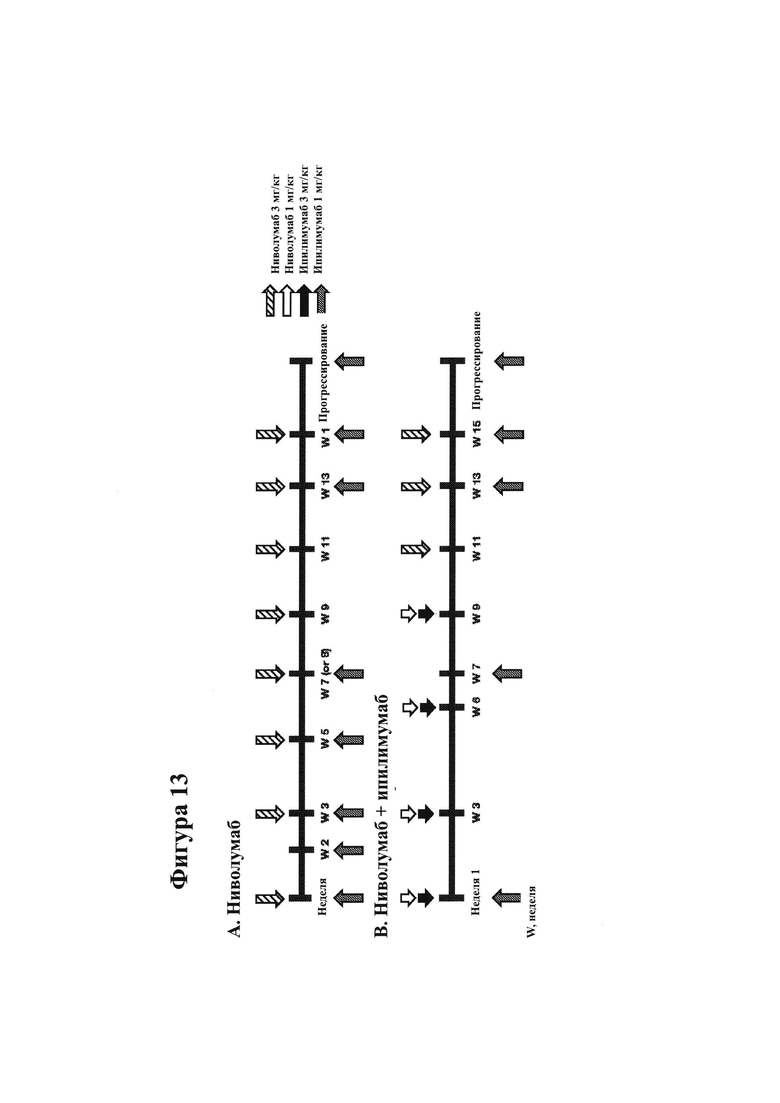


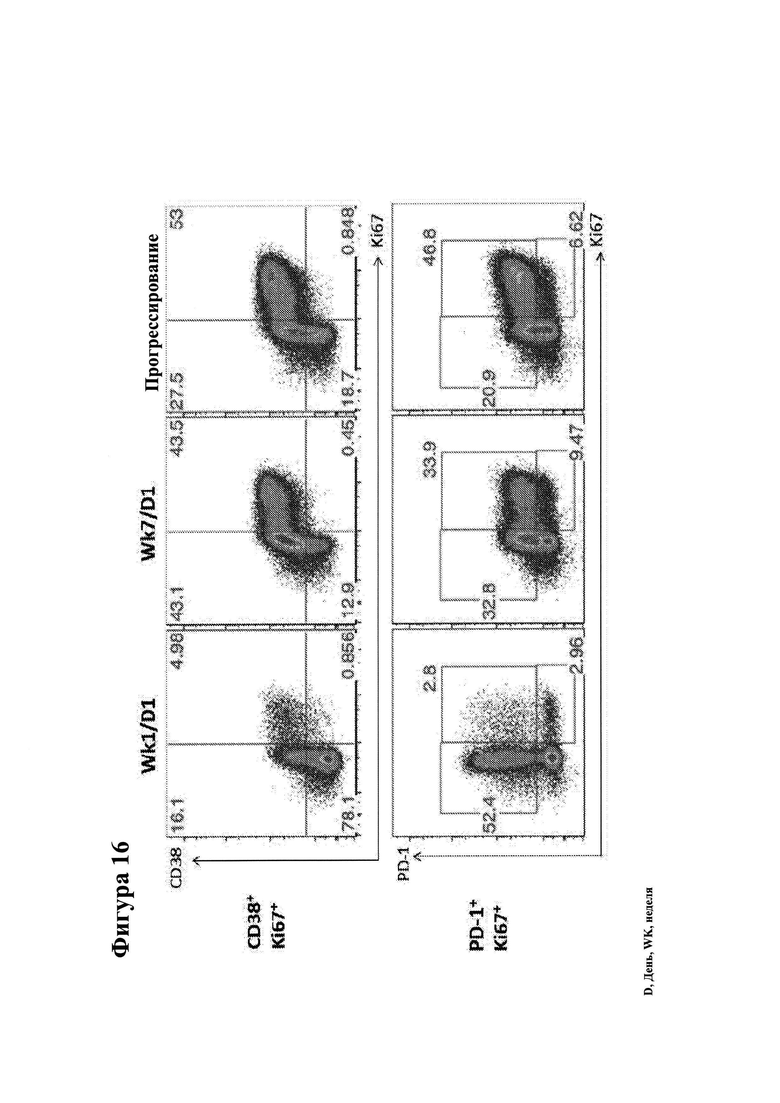

Изобретение относится к способу лечения глиобластомы у субъекта путем введения терапевтически эффективного количества антитела или его антигенсвязывающей части, которая специфически связывается с рецептором программированной смерти-1 (PD-1) и ингибирует активность PD-1 ("антитело против PD-1"), и темозоломида. Способ позволяет повысить эффективность лечения глиобластомы за счет синергитического эффекта комбинации. 52 з.п. ф-лы, 17 ил., 1 табл., 10 пр.
1. Способ лечения субъекта, страдающего глиобластомой, включающий введение субъекту (а) терапевтически эффективного количества антитела или его антигенсвязывающей части, которая специфически связывается с рецептором программированной смерти-1 (PD-1) и ингибирует активность PD-1 ("антитело против PD-1"), и (b) темозоломида.
2. Способ по п. 1, в котором глиобластома выбрана из следующих подтипов: классической, нервной, пронейральной и мезенхимальной.
3. Способ по любому из пп. 1 или 2, в котором антитело против PD-1 перекрестно конкурирует с ниволумабом за связывание с человеческим PD-1.
4. Способ по любому из пп. 1-3, в котором антитело против PD-1 содержит константную область тяжелой цепи, которая имеет человеческий IgG1 или IgG4 изотип.
5. Способ по любому из пп. 1-4, в котором антитело против PD-1 представляет собой химерное, гуманизированное или человеческое моноклональное антитело против PD-1 или его антигенсвязывающую часть.
6. Способ по любому из пп. 1-5, в котором антитело против PD-1 представляет собой ниволумаб.
7. Способ по любому из пп. 1-5, в котором антитело против PD-1 представляет собой пембролизумаб.
8. Способ по любому из пп. 1-7, в котором субъекта дополнительно лечат противораковой терапией одновременно с введением, до или после введения.
9. Способ по п. 8, в котором противораковая терапия включает введение противоракового средства.
10. Способ по п. 8, в котором противораковая терапия включает лучевую терапию.
11. Способ по п. 8, в котором противораковая терапия включает операцию.
12. Способ по п. 11, в котором операция включает в себя МРТ-управляемую лазерную абляцию.
13. Способ по п, 9, в котором противораковое средство содержит дополнительный алкилирующий агент, второе антитело или его антигенсвязывающую часть, или оба.
14. Способ по п. 9, в котором противораковое средство содержит второе антитело или его антигенсвязывающую часть.
15. Способ по п. 14, в котором второе антитело или его антигенсвязывающая часть специфически связывается с цитотоксическим Т-лимфоцитарным антигеном-4 (CTLA-4) и ингибирует активность CTLA-4 ("антитело против CTLA-4").
16. Способ по п. 15, в котором антитело против CTLA-4 или его антигенсвязывающая часть перекрестно конкурирует с ипилимумабом за связывание с человеческим CTLA-4.
17. Способ по п. 15 или 16, в котором антитело против CTLA-4 или его антигенсвязывающая часть представляет собой химерное, гуманизированное или человеческое моноклональное антитело или его антигенсвязывающую часть.
18. Способ по любому из пп. 15-17, в котором антитело против CTLA-4 или его антигенсвязывающая часть содержит константную область тяжелой цепи, которая имеет человеческий IgG1 изотип.
19. Способ по любому из пп. 15-18, в котором антитело против CTLA-4 представляет собой ипилимумаб.
20. Способ по любому из пп. 15-19, в котором комбинация антитела против PD-1 или его антигенсвязывающей части и антитела анти-CTLA-4 или его антигенсвязывающей части проявляет синергетический эффект на лечение глиомы.
21. Способ по любому из пп. 1-20, в котором антитело против PD-1 или его антигенсвязывающую часть вводят в дозе от 0,1 мг/кг до 10 мг/кг массы тела.
22. Способ по любому из пп. 1-21, в котором антитело против PD-1 или его антигенсвязывающую часть вводят в дозе, составляющей 1 мг/кг или 3 мг/кг массы тела.
23. Способ по любому из пп. 1-20, в котором антитело против PD-1 или его антигенсвязывающую часть вводят в фиксированной дозе, составляющей 240 мг или 480 мг.
24. Способ по любому из пп. 1-23, в котором антитело против PD-1 или его антигенсвязывающую часть вводят один раз каждые 1, 2, 3 или 4 недели.
25. Способ по любому из пп. 1-24, в котором антитело против PD-1 или его антигенсвязывающую часть вводят один раз каждые 2 недели.
26. Способ по любому из пп. 1-20, в котором антитело против PD-1 вводят в фиксированной дозе, составляющей 240 мг, один раз каждые 2 недели.
27. Способ по любому из пп. 1-24, в котором антитело против PD-1 или его антигенсвязывающую часть вводят один раз каждые 3 недели.
28. Способ по любому из пп. 1-28, в котором темозоломид вводят в ходе сопутствующей лучевой терапии непрерывно с 1-го дня лучевой терапии до последнего дня лучевой терапии при суточной пероральной дозе 75 мг/м2 в течение максимально 49 дней.
29. Способ по любому из пп. 1-27, в котором темозоломид вводят в дозе, составляющей 150 мг/м2.
30. Способ по любому из пп. 1-27, в котором темозоломид вводят в дозе, составляющей 200 мг/м2.
31. Способ по любому из пп. 15-30, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе от 0,1 мг/кг до 10 мг/кг массы тела.
32. Способ по любому из пп. 15-31, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе от 1 мг/кг до 5 мг/кг массы тела.
33. Способ по любому из пп. 15-32, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе, составляющей 3 мг/кг массы тела.
34. Способ по любому из пп. 15-32, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе, составляющей 1 мг/кг массы тела.
35. Способ по любому из пп. 15-34, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят один раз каждые 1, 2, 3 или 4 недели.
36. Способ по любому из пп. 15-35, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят один раз каждые 2 недели.
37. Способ по любому из пп. 15-35, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят один раз каждые 3 недели.
38. Способ по любому из пп. 15-20, в котором антитело против PD-1 или его антигенсвязывающую часть вводят в дозе, составляющей 3 мг/кг массы тела, и антитело против CTLA-4 или его антигенсвязывающую часть вводят в дозе, составляющей 1 мг/кг массы тела.
39. Способ по любому из п. 15-20, включающий:
индукционную фазу, где анти-PD-1 и антитела против CTLA-4 или их антигенсвязывающие части вводят в комбинации в 2, 4, 6, 8 или 10 дозах, причем каждую дозу в диапазоне от 0,1 мг/кг до 10,0 мг/кг массы тела вводят по меньшей мере один раз каждые 2, 3 или 4 недели;
с последующей поддерживающей фазой, в которой не вводят антитело против CTLA-4 или его антигенсвязывающую часть и антитело против PD-1 или его антигенсвязывающую часть повторно вводят в дозе в диапазоне от 0,1 мг/кг до 10 мг/кг по меньшей мере один раз каждые 2, 3 или 4 недели.
40. Способ по п. 39, в котором
a. во время индукционной фазы антитело против PD-1 или его антигенсвязывающую часть вводят в количестве 1 мг/кг массы тела и антитело против CTLA-4 или его антигенсвязывающий участок вводят в количестве 3 мг/кг массы тела один раз каждые 3 недели в 4-х дозах, и
b. во время поддерживающей фазы, антитело против PD-1 или его антигенсвязывающую часть повторно вводят в дозе 3 мг/кг массы тела по меньшей мере один раз каждые 2 недели.
41. Способ по п. 39, в котором
a. во время индукционной фазы антитело против PD-1 или его антигенсвязывающую часть вводят в количестве 3 мг/кг массы тела и антитело против CTLA-4 или его антигенсвязывающую часть вводят в количестве 1 мг/кг массы тела в 4-х дозах, один раз каждые 3 недели, и
b. во время поддерживающей фазы антитело против PD-1 или его антигенсвязывающую часть повторно вводят в дозе 3 мг/кг массы тела по меньшей мере один раз каждые 2 недели.
42. Способ по любому из пп. 39-41, в котором введение антитела против PD-1 или его антигенсвязывающей части в поддерживающей фазе продолжается до тех пор, пока наблюдаются клинический эффект, или до появления неуправляемой токсичности или прогрессирования заболевания.
43. Способ по любому из пп. 18-42, в котором анти-PD-l и антитела против CTLA-4 или их антигенсвязывающие части сформулированы для внутривенного введения.
44. Способ по любому из пп. 18-43, в котором комбинацию антитела против PD-1 или его антигенсвязывающей части и антитела анти-CTLA-4 или его антигенсвязывающей части вводят одновременно как единую композицию или в виде отдельных композиций.
45. Способ по любому из пп. 18-43, в котором комбинацию антитела против PD-1 или его антигенсвязывающей части и антитела анти-CTLA-4 или его антигенсвязывающей части вводят последовательно.
46. Способ по любому из пп. 39-43, в котором антитело против PD-1 или его антигенсвязывающую часть и антитело против CTLA-4 или его антигенсвязывающую часть вводят последовательно субъекту во время индукционной фазы.
47. Способ по любому из пп. 18-43, в котором антитело против PD-1 или его антигенсвязывающую часть и антитело против CTLA-4 или его антигенсвязывающую часть вводят с интервалом не более 30 минут.
48. Способ по любому из пп. 1-20, в котором антитело против PD-1 или его антигенсвязывающую часть вводят в субтерапевтической дозе.
49. Способ по любому из пп. 1-20, в котором антитело против PD-1 или его антигенсвязывающую часть вводят в терапевтической дозе.
50. Способ по любому из пп. 15-30, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят в субтерапевтической дозе.
51. Способ по любому из пп. 15-30, в котором антитело против CTLA-4 или его антигенсвязывающую часть вводят в терапевтической дозе.
52. Способ по любому из пп. 1 -51, в котором глиобластома является глиобластомой с неметилированным геном O-6-метилгуанина ДНК-метилтрансферазы (MGMT) в опухоли.
53. Способ по любому из пп. 1 -51, в котором глиобластома является глиобластомой с метилированным геном MGMT в опухоли.
| US 2013291136 A 1, 31.10.2013 | |||
| US 2014341920 A1, 20.11.2014 | |||
| СN 102178676 A, 31.10.2013 | |||
| СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОЙ ГЛИОМЫ ГОЛОВНОГО МОЗГА | 2009 |
|
RU2402360C1 |
| ЧИССОВ В.И | |||
| под ред | |||
| и др | |||
| Онкология | |||
| Национальное руководство// М., "ГЭОТАР-Медиа", 2008, с | |||
| ПРИБОР ДЛЯ СОЖИГАНИЯ НЕФТИ В ПЕЧНЫХ ТОПКАХ | 1923 |
|
SU1021A1 |
| CHINNAIYA P | |||
| Плавучий цепной ветро-водяной двигатель | 1923 |
|
SU913A1 |

