ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится в основном к способам и композициям для лечения заболевания, нарушения, состояния или повреждения центральной нервной системы (ЦНС) путем временного снижения уровня системной иммуносупрессии в кровообращении.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Большинство патологий центральной нервной системы (ЦНС) имеют общий нейровоспалительный компонент, который является частью прогрессирования заболевания, и вносит вклад в обострение заболевания. Среди этих патологий находится болезнь Альцгеймера (AD), связанное с возрастом нейродегенеративное заболевание, отличающееся прогрессирующей потерей памяти и когнитивных функций, при котором, как было предложено, накопление агрегатов бета-амилоидного пептида (Аβ) играет ключевую роль в воспалительном каскаде в ЦНС, в конечном счете, приводящее к повреждению нейронов и разрушению ткани (Akiyama et al, 2000; Hardy & Selkoe, 2002; Vom Berg et al, 2012). Несмотря на хронический нейровоспалительный ответ при нейродегенеративных заболеваниях, клинические и доклинические испытания в течение последнего десятилетия, исследуя способы на основе иммуносупрессивной терапии при нейродегенеративных заболеваниях, подняли вопрос о том, почему противовоспалительные лекарственные средства не оправдывают ожиданий (Breitner et al, 2009; Group et al, 2007; Wyss-Coray & Rogers, 2012). Изобретатели обеспечивают новый ответ, который преодолевает недостатки существующих способов лечения AD и подобных заболеваний и повреждений ЦНС; этот способ основан на уникальном понимании изобретателями роли различных компонентов системной и центральной иммунной системы в поддержании и восстановлении ЦНС.
КРАТКОЕ ИЗЛОЖЕНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте данное изобретение обеспечивает фармацевтическую композицию, содержащую активный агент, который вызывает снижение уровня системной иммуносупрессии у индивидуума, для применения в лечении заболевания, нарушения, состояния или повреждения ЦНС, которое не включает аутоиммунное нейровоспалительное заболевание, рецидивирующе-ремитирующий рассеянный склероз (RRMS), где указанная фармацевтическая композиция предназначена для введения в режиме дозирования, содержащем по меньшей мере два курса лечения, каждый курс лечения, содержащий последовательно сеанс лечения с последующим интервалом между сеансами, периодом без лечения.
В другом аспекте данное изобретение обеспечивает способ лечения заболевания, нарушения, состояния или повреждения центральной нервной системы (ЦНС), которое не включает аутоиммунное нейровоспалительное заболевание, рецидивирующе-ремитирующий рассеянный склероз (RRMS), указанный способ, содержащий введение индивидууму, нуждающемуся в этом, фармацевтической композиции по любому из пп. 1-24 формулы изобретения, где указанную фармацевтическую композицию вводят в режиме дозирования, содержащем по меньшей мере два курса лечения, каждый курс лечения содержащий последовательно курс лечения с последующим интервалом между сеансами, периодом без лечения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКОГО МАТЕРИАЛА
Фиг. 1А-В представляют собой активность хороидного (сосудистого) сплетения (CP) на протяжении прогрессирования заболевания у трансгенных мышей линии 5XFAD с моделью болезни Альцгеймера (AD) (AD-Tg). (А) уровни экспрессии мРНК генов icam1, vcam1, cxcl10 и ccl2, измеренные RT-qPCR в хороидных сплетениях (CPs), выделенных у 1-, 2-, 4- и 8-месячных мышей AD-Tg, показанные в виде кратного изменения, по сравнению с контрольными мышами дикого типа (WT) в соответствующей возрастной группе (n=6-8 на группу; t-критерий Стьюдента для каждого периода времени). (В) Репрезентативные микроскопические изображения CPs мышей AD-Tg 8-месячного возраста и контрольных мышей WT, соответствующей возрастной группы, иммуноокрашенные на наличие молекулы плотных контактов эпителия клаудина-1, окрашивание ядер Hoechst, и лиганда для интегринов ICAM-1 (пер., молекул межклеточной адгезии) (масштабная линейка, 50 мкм). На всех панелях черточки ошибок показывают среднее значение ± s.e.m. (пер., стандартная ошибка среднего); *, Р<0,05; **, Р<0,01; ***, Р<0,001.
Фиг. 2А-С представляют собой (А) количественную оценку иммунореактивности ICAM-1 в патоморфологическом препарате CP молодых и пожилых людей без заболеваний ЦНС, а также пациентов с AD (n=5 на группу; однофакторный дисперсионный анализ (ANOVA) с последующим использованием критерия Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений); (В) анализ методом проточной цитометрии IFN-γ-экспрессирующих клеток иммунной системы (пер., иммуноцитов) (внутриклеточно окрашенных и предварительно гейтированных (pre-gated) по CD45) в препаратах CP 8-месячных мышей AD-Tg и контролях WT по возрастным группам. Заштрихованная гистограмма обозначает изотипический контроль (n=4-6 на группу; t-критерий Стьюдента); и (С) уровни экспрессии мРНК ifn-γ, измеренные RT-qPCR, в тканях CP, выделенных у мышей AD-Tg 4- и 8-месячного возраста, по сравнению с контрольными мышами дикого типа (WT) в соответствующей возрастной группе (n=5-8 на группу; t-критерий Стьюдента для каждого периода времени). На всех панелях черточки ошибок показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
Фиг. 3А-В представляют собой (А) репрезентативные частотные гистограммы распределения спленоцитов CD4+Foxp3+ (предварительно гейтированных (pre-gated) по TCRβ) при использовании проточной цитометрии у мышей AD-Tg 8-месячного возраста и контрольных мышей WT; и (В) количественный анализ спленоцитов мышей AD-Tg 1-, 2-, 4- и 8-месячного возраста и контрольных мышей WT (n=6-8 на группу; t-критерий Стьюдента для каждого периода времени). На всех панелях черточки ошибок показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
Фиг. 4 представляет собой стратегию гейтирования и репрезентативные гистограммы распределения спленоцитов мышей AD-Tg/Foxp3-DTR+/- при использовании проточной цитометрии, через 1 день после последней инъекции DTx. DTx инъецировали интраперитонеально в течение 4 дней подряд, достигая ~99% истощения клеток Foxp3+.
Фиг. 5A-G представляют собой эффекты транзиторного истощения Tregs (пер., регуляторных Т-клеток, Treg-клеток) у мышей AD-Tg. (А) мышей AD-Tg/Foxp3-DTR+ (которые экспрессируют трансген DTR) и однопометную AD-Tg, не экспрессирующую DTR (AD-Tg/Foxp3-DTR) контрольную группу, обрабатывали DTx (дифтерийным токсином) в течение 4 конститутивных дней. Уровни экспрессии мРНК CP генов icam1, cxcl10 и ccl2 измеряли RT-qPCR у 6-месячных AD-Tg мышей, обработанных DTx, через 1 день после последней инъекции DTx (n=6-8 на группу; t-критерий Стьюдента). (B-D) Анализ методом проточной цитометрии паренхимы мозга (за исключением хороидного сплетения, которое было отдельно удалено) мышей AD-Tg 6-месячного возраста, обработанных DTx и контролей, через 3 недели после последней инъекции DTx. Количественный анализ методом проточной цитометрии, показывающий повышенное число CD11bhigh/CD45high mo-МФ и CD4+ Т-клеток (В), и репрезентативные гистограммы распределения при использовании проточной цитометрии (С) и количественный анализ (D) частоты CD4+Foxp3+ Treg (лед, клеток) в паренхиме мозга мышей AD-Tg/Foxp3-DTR+ и контрольных AD-Tg/Foxp3-DTR, обработанных DTx (n=3-7 на группу; t-критерий Стьюдента). (Е) уровни экспрессии мРНК foxp3 и il10 в паренхиме мозга 6-месячных мышей AD-Tg AD-Tg/Foxp3-DTR+, обработанных DTx, и контрольных AD-Tg/Foxp3-DTR, через 3 недели после последней инъекции DTx (n=6-8 на группу; t-критерий Стьюдента). (F) количественный анализ иммуноокрашивания GFAP (пер., глиального фибриллярного кислого белка), показывающий пониженный астроглиоз в срезах гиппокампа мышей AD-Tg/Foxp3-DTR+6-месячного возраста, обработанных DTx, и контрольных мышей AD-Tg/Foxp3-DTR", через 3 недели после последней инъекции DTx (масштабная линейка 50 мкм; n=3-5 на группу; t-критерий Стьюдента). (G) уровни экспрессии мРНК Н-12р40 и tnf-а в паренхиме мозга, через 3 недели после последней инъекции DTx (n=6-8 на группу; t-критерий Стьюдента). На всех панелях черточки ошибок показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
Фиг. 6А-Е представляют собой эффекты транзиторного истощения Tregs (пер., регуляторных Т-клеток) на Аβ-бляшки и способность к обучению/памяти. (А) Репрезентативные микроскопические изображения и (В) количественный анализ мозга 5-месячного возраста, обработанных DTx мышей AD-Tg/Foxp3-DTR+ и контрольных мышей AD-Tg/Foxp3-DTR-, через 3 недели после последней инъекции DTx, иммуноокрашенных на Аβ-бляшки, окрашивание ядер Hoechst (масштабная линейка, 250 мкм). Количественно определяли среднюю площадь и число бляшек Аβ в зубчатой извилине (DG) гиппокампа и 5-м слое коры головного мозга (в срезах мозга толщиной 6 мкм; n=5-6 на группу; t-критерий Стьюдента). Фиг. 6С-Е представляют собой результаты проведения теста водного лабиринта Морриса (MWM) на 6-месячного возраста обработанных DTx мышах AD-Tg/Foxp3-DTR+ и контрольных мышах, через 3 недели после последней инъекции DTx. После транзиентного истощения Treg-клеток, мыши AD-Tg показали лучшую способность к пространственному обучению/памяти в фазы (С) приобретения навыков, (D) проведения испытания и (Е) инверсии в MWM, по отношению к контрольным AD-Tg (n=7-9 на группу; двухфакторный дисперсионный анализ с повторениями ANOVA с последующим апостериорным анализом с поправкой Бонферрони для индивидуальных попарных сравнений; *, Р<0,05 общее для приобретения навыков, проведения испытания и инверсии). На всех панелях черточки ошибок показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
Фиг. 7 представляет собой уровни экспрессии мРНК ifn-γ, измеренные RT-qPCR, в хороидных сплетениях (CPs), выделенных у 6-ти и 12-месячного возраста мышей APP/PS1 AD-Tg (у трансгенных мышей с моделью болезни Альцгеймера (см. материалы и методы)), по сравнению с контрольными мышами дикого типа (WT) в соответствующей возрастной группе (n=5-8 на группу; t-критерий Стьюдента). Черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05.
Фиг. 8A-I представляют собой терапевтический эффект еженедельного введения глатирамера ацетата (GA) мышам AD-Tg. (А) Схематическое представление режима лечения еженедельно-GA. Мышам (5-месячного возраста) подкожно (s.c.) инъецировали GA (100 мкг), дважды в течение первой недели (на 1-й день и 4-й), и после этого один раз в неделю, в течение общего периода 4 недели. После последней инъекции мышей исследовали на когнитивную деятельность, 1 неделю (MWM), 1 месяц (RAWM, пер., радиальный водный лабиринт) и 2 месяца (RAWM, с использованием различных экспериментальных пространственных установок), и на воспаление гиппокампа. Фиг. 8B-D представляют собой уровни экспрессии мРНК генов в гиппокампе нелеченных мышей AD-Tg, и мышей AD-Tg, печенных еженедельно-GA, в возрасте 6 месяцев, демонстрирующие (В) пониженную экспрессию провоспалительных цитокинов, таких как TNF-α, IL-1β и IL-12р40, (С) повышение противовоспалительных цитокинов IL-10 и TGF-β, и (D) нейротропных факторов IGF-1 и BDNF у мышей, леченных еженедельно-GA (n=6-8 на группу; t-критерий Стьюдента). На фиг. 8E-G, мышей AD-Tg (5-месячного возраста) лечили еженедельно как GA, так и носителем (PBS, пер., фосфатно-солевым буферным раствором), и сравнивали с контрольными однопометными животными дикого типа WT в соответствующей возрастной группе в тесте MWM в возрасте 6 месяцев. Получившие лечение мыши показали лучшую способность к пространственному обучению/памяти в фазы приобретения навыков (Е), проведения испытания (F) и инверсии (G) в MWM, по отношению к контролям (n=6-9 на группу; двухфакторный дисперсионный анализ с повторениями ANOVA с последующим апостериорным анализом с поправкой Бонферрони для индивидуальных попарных сравнений). Фиг. 8H-I представляют собой когнитивную деятельность тех же самых мышей в тесте RAWM, чрез 1 месяц (Н) или 2 месяца (I) после последней инъекции GA (n=6-9 на группу; двухфакторный дисперсионный анализ с повторениями ANOVA с последующим апостериорным анализом с поправкой Бонферрони для индивидуальных попарных сравнений). Данные являются репрезентативными по меньшей мере для трех независимых экспериментов. На всех панелях черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
Фиг. 9А-Н представляют собой дополнительные терапевтические эффекты от еженедельного введения GA мышам AD-Tg. А-В представляют собой мышей AD-Tg линии 5XFAD (пер., трансгенных мышей линии 5XFAD с моделью AD), которых лечили еженедельно как GA, так и носителем (PBS), и обследовали в конце 1-й недели режима введения (после двух инъекций GA в общей сложности). Анализ методом проточной цитометрии на частоту встречаемости спленоцитов CD4+Foxp3+ (А), и IFN-γ-экспрессирующих иммуноцитов в CP (В); внутриклеточно окрашенных и предварительно гейтированных по CD45, у леченных мышей AD-Tg 6-месячного возраста, по сравнению с контрольными животными дикого типа WT в соответствующей возрастной группе (n=4-6 на группу; однофакторный ANOVA с последующим использованием критерия Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений). (С) уровни экспрессии мРНК генов icam1, cxcl10 и ccl2, измеренные RT-qPCR, в хороидных сплетениях (CPs) мышей AD-Tg 4-месячного возраста, леченных еженедельно как GA, так и носителем, и обследованных как в конце 1-й, так и 4-й недели при еженедельном режиме введения GA (n=6-8 на группу; однофакторный ANOVA с последующим использованием критерия Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений). Фиг. 9D-E представляют собой репрезентативные изображения срезов мозга 6-месячных химер ВМ AD-Tg/CX3CR1GFP/+ после еженедельного введения GA. Клетки CX3CR1GFP были локализованы в CP третьего желудочка (3V; i), прилегающих желудочковых полостей (ii), и CP боковых желудочков (LV; iii) у мышей AD-Tg, леченных еженедельно-GA (D; масштабная линейка, 25 мкм). Репрезентативные ортогональные проекции конфокальных срезов по оси z, демонстрирующие колокализацию GFP+ клеток с миелоидным маркером CD68, в хороидном сплетении (CP) 7-месячных мышей AD-Tg/CX3CR1GFP/+, леченных еженедельно-GA, но не в контрольных мышах AD-Tg/CX3CR1GFP/+, леченных PBS (Е; масштабная линейка, 25 мкм). (F) Клетки CX3CR1GFP, являются колокализованными с миелоидным маркером IBA-1 в мозге мышей AD-Tg/CX3CR1GFP/+, леченных GA, вблизи Аβ бляшек, и коэкспрессирующими миелоидный маркер IBA-1 (масштабная линейка, 25 мкм). Фиг. 9G-H представляют собой репрезентативные графики и гистограммы (пер., событий) при анализе методом проточной цитометрии клеток, выделенных из гиппокампа 4-месячных мышей WT, нелеченных AD-Tg, и леченных мышей AD-Tg, на 2-ю неделю при еженедельном режиме лечения GA. Гейтировали CD11bhigh/CD45high mo-МФ (G) и определяли количественно (Н; n=4-5 на группу; однофакторный ANOVA с последующим использованием критерия Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений). На всех панелях черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
На фиг. 10А-Н изображен терапевтический эффект введения ингибитора р300 (С646) мышам AD-Tg. На фиг. 10А-В, взрослых мышей (18-месячных) лечили как p300i, так и носителем (DMSO) в течение 1 недели и исследовали на следующий день после прекращения лечения. Репрезентативные графики и гистограммы событий при анализе методом проточной цитометрии, показывающие повышение частоты встречаемости CD4+ Т-клеток, экспрессирующих IFN-γ в селезенке (А), и число иммуноцитов, экспрессирующих IFN-γ в CP (В), после лечения p300i. Фиг. 10С-Е представляют собой репрезентативные микроскопические изображения (С), и количественный анализ объема Аβ-бляшек в препаратах мозга 10-месячных мышей AD-Tg, которые получали как p300i, так и носитель (DMSO) в течение 1 недели, и впоследствии были обследованы после 3-х дополнительных недель. Препараты мозга подвергали иммуноокрашиванию на Аβ-бляшки и окрашиванию ядер с использованием (пер., красителя) Hoechst (n=5 на группу; масштабная линейка 250 мкм). Среднюю площадь Аβ-бляшек и число бляшек определяли количественно в зубчатой извилине (DG) гиппокампа (D) и 5-м слое коры головного мозга (Е) (в 6 мкм срезах мозга; n=5-6 на группу; t-критерий Стьюдента). (F) Схематическое представление лечения с использованием p300i (или DMSO в качестве носителя) при режиме введения для различных групп мышей AD-Tg в возрасте от 7 месяцев, как в 1, так и в 2 сеанса. Фиг. 10G-H представляют собой среднее изменение процента покрытия Аβ-бляшками коры головного мозга (5-й слой) (G), и изменение средних уровней растворимого белка Аβ1-40 и Аβ1-42 в коре головного мозга (Н), по сравнению с нелеченной группой AD-Tg (средний уровень Аβ1-40 и Aβ1-42 в нелеченной группе, 90,5±11,2 и 63,8±6,8 пг/мг от общей части, соответственно; n=5-6 на группу; однофакторный ANOVA с последующим использованием критерия Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений). На всех панелях черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
На фиг. 11А-В изображен терапевтический эффект введения анти-PD1 антитела мышам AD-Tg. (А) Схематическое представление экспериментальных групп мышей, их возраст, режимы лечения (обработки) и введения, а также момент времени, в который мышей исследовали. В 10-месячном возрасте, трансгенным мышам (AD) линии 5XFAD с болезнью Альцгеймера инъецировали внутрибрюшинно (i.p.) как 250 мкг анти-PD1 (RMP1-14), так и контрольные антитела IgG (крысы), на день 1 и день 4 эксперимента, и через 3 недели исследовали их когнитивные способности путем теста на пространственное обучение и память на установке рукавный водный лабиринт (RAWM). В качестве контролей использовали нелеченных мышей дикого типа (WT) и мышей AD-Tg в соответствующей возрастной группе. (В) представляет собой когнитивные способности, в виде оцененных тестом на пространственное обучение и память на установке рукавный водный лабиринт (RAWM). Данные анализировали с использованием двухфакторного дисперсионного анализа с повторениями ANOVA и методом Бонферрони для апостериорных (post hoc) сравнений для последующего попарного сравнения. n=6-12 на группу. На всех панелях черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01;***, Р<0,001.
На фиг. 12А-В изображен системный эффект введения анти-PD1 антитела на IFN-γ+-продуцирующие Т-клетки у мышей AD-Tg. (А) Схематическое представление экспериментальных групп мышей, их возраст, режимы лечения и введения, а также момент времени, в который мышей исследовали. Мышам инъецировали внутрибрюшинно (i.p.) как 250 мкг анти-PD1 (RMP1-14), так и контрольные антитела IgG (крысы), на день 1 и день 4 эксперимента, и исследовали на 7 день. (В) Анализ методом проточной цитометрии частоты встречаемости CD4+IFN-γ+ Т-клеток спленоцитов (внутриклеточно окрашенных и предварительно отобранных (pre-gated) по CD45 и TCR-β) у мышей AD-Tg, леченных PD-1 или IgG, и нелеченных контролей AD-Tg и WT (n=4-6 на группу; однофакторный ANOVA с последующим использованием анализа Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений; **, Р<0,01 между указанными леченными группами; черточки погрешностей показывают среднее значение ± s.e.m.).
Фиг. 13А-В представляют собой влияние на CP мышей AD-Tg после лечения анти-PD1 Мышей AD-Tg 10-месячного возраста лечили как PD-1, IgG, так и оставляли без лечения. Мышам инъецировали внутрибрюшинно (i.p.) как 250 мкг анти-PD1 (RMP1-14), так и контрольные антитела IgG (крысы) на день 1 и день 4 эксперимента и исследовали на 7 день. (А) Уровни IFN-γ в CP, измеряемые количественной ПЦР в режиме реального времени (RT-qPCR), коррелировали положительно (коэффициент корреляции Пирсона r=0,6284, Р<0,05), и отрицательно с частотой встречаемости CD4+IFN-γ+ Т-клеток спленоцитов, измеряемых проточной цитометрией. Напротив, отрицательную тенденцию наблюдали у тех же самых мышей, когда сравнивали уровни IFN-γ в CP с частотой встречаемости CD4+Foxp3+CD25+ Tregs {пер., регуляторных Т-клеток) спленоцитов (n=3-4 на группу). (В) уровни экспрессии мРНК генов cxcl10 и ccl2, измеренные RT-qPCR, в CPs тех же самых мышей (n=3-4 на группу; однофакторный ANOVA с последующим использованием анализа Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений). На всех панелях черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05.
Фиг. 14А-В представляют собой терапевтический эффект введения анти-PD1 антитела мышам AD-Tg при сравнении одного против двух курсов лечения. Половина мышей в группе мышей, описанных на фиг. 11А-В, которые получили один курс лечения анти-PD1, или получили еще один курс лечения анти-PD1 после первого теста на установке RAWM, либо остались без лечения. После дополнительных 3 недель, всех мышей анализировали на установке RAWM с использованием других и новых экспериментальных установок с пространственными сигналами для когнитивного обучения и памяти. (А) Схематическое представление экспериментальных групп мышей, их возраст, режимы лечения и введения и момент времени, в который мышей исследовали. Для каждого курса лечения мышам инъецировали внутрибрюшинно (i.p.) как 250 мкг анти-PD1 (RMP1-14), так и контрольные антитела IgG (крысы). (В) представляет собой когнитивную деятельность, оцененную тестом на установке RAWM для оценки способности к пространственному обучению/памяти. Данные анализировали с использованием двухфакторного дисперсионного анализа с повторениями ANOVA, и использовали апостериорный анализ (post-hoc) Бонферрони для индивидуальных попарных сравнений. n=6-12 на группу. На всех панелях черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
Фиг. 15А-Н представляют собой отрицательное действие на патологию AD уровней системных Treg-клеток, усиленное полностью транс-ретиноевой кислотой (ATRA). Фиг. 15А-В представляют собой репрезентативные графики (пер., распределения) при анализе методом проточной цитометрии (А), и количественный анализ (В), показывающий повышение частоты встречаемости CD47Foxp3+/CD25+Treg спленоцитов мышей AD-Tg 5-месячного возраста, которые получали как ATRA, так и носитель (DMSO) в течение 1 недели (n=5 на группу; t-критерий Стьюдента). Фиг. 15C-F представляют собой репрезентативные микроскопические изображения (С) и количественный анализ (D, Е, F), объема бляшек Аβ и астроглиоз в мозге мышей AD-Tg, которых в 5-месячном возрасте лечили как ATRA, так и носителем (DMSO) в течение 1 недели, а затем исследовали через 3 дополнительных недели. Препараты мозга были иммуноокрашены на Аβ-бляшки, GFAP (маркирование астроглиозиса), и окрашивание ядер Hoechst (n=4-5 на группу; масштабная линейка, 250 мкм). Количественно определяли среднюю площадь и число бляшек Аβ в зубчатой извилине (DG) гиппокампа и 5-м слое коры головного мозга и измеряли иммунореактивность GFAP в гиппокампе (в срезах мозга толщиной бмкм; n=5-6 на группу; t-критерий Стьюдента). (G) Уровни растворимого Аβ1-40 и Аβ1-42, определенные количественно методом ИФА (ELISA), в церебральной паренхиме мозга мышей AD-Tg, которых в 5-месячном возрасте лечили как ATRA, так и носителем (DMSO) в течение 1 недели, а затем исследовали через 3 дополнительных недели (n=5-6 на группу; t-критерий Стьюдента). (Н) Когнитивную способность в тесте на установке RAWM мышей AD-Tg, которых в 5-месячном возрасте лечили как ATRA, так и носителем (DMSO) в течение 1 недели, а затем исследовали через 3 дополнительных недели (n=5 на группу; двухфакторный дисперсионный анализ с повторениями ANOVA, с последующим апостериорным анализом (post-hoc) Бонферрони для индивидуальных попарных сравнений). На всех панелях черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
Фиг. 16A-F представляют собой отрицательное действие на патологию AD уровней системных Treg-клеток, усиленное еженедельным введением GA. (А) Схематическое представление режима лечения ежедневно-GA по сравнению с режимом лечения еженедельно-GA. В группе с ежедневным лечением GA, мышам ежедневно инъецировали подкожно (s.c.) 100 мкг GA в течение 1 месяца. (В) Когнитивную способность мышей AD-Tg 7-месячного возраста, леченных ежедневно-GA и еженедельно-GA, по сравнению с контрольными мышами дикого типа (WT) в соответствующей возрастной группе и нелеченными мышами AD-Tg, оцененную средним числом ошибок за день в тесте на способность к обучению и запоминанию в тестах на установке RAWM (n=6-8 на группу; однофакторный ANOVA с последующим использованием анализа Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений). (С) Репрезентативные микроскопические изображения коры головного мозга и гиппокампа (НС), нелеченных AD-Tg и леченных ежедневно или еженедельно GA мышей AD-Tg, иммуноокрашенные на Aβ-бляшки, окрашивание ядер Hoechst (масштабная линейка, 250 мкм). Фиг. 16D-F представляют собой количественное определение размера и числа бляшек Аβ (на 6 мкм срезах) у мышей AD-Tg, леченных GA (группы GA ежедневно и GA еженедельно) и нелеченных мышей AD-Tg. Леченные еженедельно GA мыши AD-Tg показали уменьшение нагрузки бляшек Аβ как в процентах от их общей площади в зубчатой извилине гиппокампа (DG), так и среднего числа бляшек Аβ (n=6 на группу; однофакторный ANOVA с последующим использованием анализа Ньюмена-Кейлса (Newman-Keuls) для апостериорных (post hoc) сравнений). На всех панелях черточки погрешностей показывают среднее значение ± s.e.m.; *, Р<0,05; **, Р<0,01; ***, Р<0,001.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с настоящим изобретением было обнаружено, что кратковременное транзиторное истощение (снижение) Foxp3+ регуляторных Т-клеток (Tregs) на мышиной модели с болезнью Альцгеймера (мышах AD-Tg) приводит к улучшенному рекрутированию лейкоцитов в ЦНС через хороидное сплетение мозга, повышенному числу проникающих в ЦНС (пер., инфильтрирующих ЦНС) противовоспалительных макрофагов моноцитарного происхождения mo-МФ и CD4+ Т-клеток, и выраженному обогащению Foxp3+ Tregs, которые накапливаются в головном мозге. Кроме того, длительный эффект единственного сеанса лечения приводит к уменьшению глиоза гиппокампа и пониженным уровням экспрессии мРНК провоспалительных цитокинов в головном мозге. Важно отметить, что влияние на связанную с заболеванием патологию включает пониженный объем бляшек церебрального амилоида бета (Аβ) в зубчатой извилине гиппокампа, и коре головного мозга (5-м слое), двух участках мозга, демонстрирующих устойчивую патологию с Aβ-бляшками у мышей AD-Tg. Самое главное, что кратковременное транзиторное истощение Tregs сопровождается поразительным улучшением способности к пространственному обучению и запоминанию, достигая когнитивной деятельности, подобной мышам дикого типа (примеры 2 и 3). Эти, взятые вместе, полученные результаты демонстрируют, что короткий сеанс истощения Treg, за которым следует период без вмешательства, приводит к временно нарушенной Treg-опосредованной системной иммуносупрессии у мышей AD-Tg, которая обеспечивает рекрутирование в мозг клеток, разрешающих воспаление mo-МФ и Tregs, и приводит к разрешению нейровоспалительного ответа, клиренсу Аβ, и отмене снижения когнитивных способностей. Эти результаты убедительно опровергают общепринятую точку зрения в данной области исследования, согласно которой повышение системной иммуносупрессии привело бы к снижению нейровоспалительного ответа. Наоборот, результаты, полученные авторами данного изобретения показывают, что повышение системного ответа, кратковременным, кратким и транзиторным снижением Treg-опосредованной системной иммуносупрессии, необходимо для достижения накопления разрешающих воспаление иммунноцитов, в том числе самих Tregs, в головном мозге, тем самым отбивая AD патологию.
Специфичность подхода, представленного здесь изобретателями, была подтверждена применением нескольких независимых экспериментальных парадигм, как подробно описано ниже. Кратко, сначала изобретатели использовали иммуномодулирующее соединение в двух различных режимах введения, что привело к противоположным эффектам на уровнях периферических Treg, на активации CP, и на патологии заболевания; ежедневный режим введения, который увеличивает уровни периферических Treg (Weber et al, 2007), и еженедельный режим введения, который как было обнаружено авторами, снижает уровни периферических Treg (пример 3 и пример 5). Изобретатели также обеспечивают прямую функциональную связь между уровнями периферических Treg и связанной с заболеванием патологией, при демонстрации на мышах AD-Tg, как путем транзиентного генетического истощения Tregs in vivo (пример 2), так и фармакологическим ингибированием функции их Foxp3 (примеры 3 и 4), так что эти манипуляции приводят к активации CP для облегчения миграции лейкоцитов в ЦНС, накоплению разрешающих воспаление иммуноцитов в местах патологии, клиренсу церебральных Аβ-бляшек, и сдвигу иммунологической среды паренхимы мозга в направлении к разрешению воспаления.
В соответствии с данным изобретением, дополнительно было обнаружено, что нерегулярное введение универсального антигена, сополимера-1 (Copolymer-1), в течение ограниченного периода времени (представляющего один сеанс лечения) снижает опосредованную Treg системную иммуносупрессию, и улучшает избирательную инфильтрацию лейкоцитов в ЦНС, повышением активности шлюза (gateway) хороидного сплетения мозга, что приводит к поразительному благоприятному эффекту при патологии болезни Альцгеймера (пример 3), тогда как ежедневное введение сополимера-1, который усиливает опосредованную Treg иммуносупрессию (Hong et al, 2005; Weber et al, 2007), не оказало положительного эффекта, или даже некоторое незначительное вредное воздействие на патологию болезни (пример 5). Изобретатели дополнительно показывают здесь, что прямое нарушение активности Foxp3 Treg, как путем ингибирования р300 специфическим низкомолеклярным ингибитором (p300i), так и путем взаимодействия с рецептором PD-1 антитела против PD-1 (анти-PD-1), улучшает активность шлюза (gateway) хороидного сплетения у мышей AD-Tg, и смягчает патологию болезни Альцгеймера (пример 4).
Важно, что каждый из этих примеров, обеспечиваемых изобретателями, демонстрирует различное вмешательство, которое вызывает краткосрочное ослабление системной иммуносупресии: сополимер-1 действует в качестве иммуномодулирующего соединения, p300i в виде малой молекулы, которая уменьшает ацетилирование Foxp3 и функционирование Treg, и анти-PD-1 используют в качестве нейтрализующего антитела к PD-1, экспрессируемого на Treg-клетках. Эти терапевтические подходы использовали для короткого сеанса лечения, который временно усиливал иммунный ответ на периферии, преимущественно повышением уровней периферического IFN-γ и, IFN-γ-продуцирующих клеток, таким образом активируя хороидное сплетение мозга, обеспечивая селективную инфильтрацию Т-клеток и моноцитов в ЦНС, и хоуминг (homing, пер., способность клеток к миграции) этих клеток к сайтам патологии и нейровоспаления. Также здесь было обнаружено, что повторные сеансы лечения, прерванные интервалами сеансов без лечения, значительно улучшают эффективность лечения относительно единственного сеанса лечения (пример 4). Последующий интервал времени без лечения обеспечил временное повышение уровней и активности Treg в мозге, облегчая разрешение нейровоспаления, и индуцируя окружающие условия в сторону излечивания и восстановления ЦНС, что впоследствии приводит к восстановлению тканей. В каждом из этих случаев эффект на патологию головного мозга был устойчивым, связанный с разрешением нейровоспалительного ответа, очищением головного мозга мышей с AD от бляшек бета-амилоида, и отменой снижения когнитивных способностей. Специфичность данного подхода была дополнительно обоснована с использованием генетической модели транзиторного истощения Foxp3+ регуляторных Т-клеток на модели трансгенных мышей с AD (пример 2).
Таким образом, в соответствии с настоящим изобретением было обнаружено, что системная иммуносупрессия, опосредованная Foxp3+CD4+ Treg-клетками, мешает способности побороть патологию при AD, действуя, по меньшей мере частично, путем ингибирования IFN-γ-зависимой активации CP, необходимой для организации рекрутирования разрешающих воспаление лейкоцитов в ЦНС (Schwartz & Baruch, 2014b). Системные Tregs (пер., Treg-клетки) являются очень важными для поддержания аутоиммунного гомеостаза и защиты от аутоиммунных заболеваний (Kim et al, 2007). Однако, результаты полученные авторами данного изобретения, позволяют предположить, что при нейродегенеративных состояниях, когда головному мозгу необходим репаративный иммунный ответ, способности установить этот ответ препятствуют системные Tregs. Тем не менее, в соответствии с результатами авторов изобретения, Tregs необходимы в головном мозге, месте, где находятся участки невропатологии, и выполняют локальное противовоспалительное действие. Данное изобретение предоставляет уникальное и неожиданное решение для очевидных противоречивых потребностей в борьбе с прогрессирующей гибелью нейронов как при AD; временное уменьшение/ингибирование Tregs в циркулирующей крови ради повышения Tregs в пораженном болезнью мозге. Следовательно, кратковременное и преходящее (транзиторное) снижение подавления периферического иммунного ответа (лер., иммуносупрессии), которое обеспечивает рекрутирование противовоспалительных клеток, в том числе Tregs и mo-МФ, к участкам головного мозга с бляшками, что приводит к длительному эффекту на патологию. Примечательно, однако, что транзиторное снижение уровней или активности системных Treg может способствовать ослаблению заболевания при помощи дополнительных механизмов, в том числе поддержания ЦНС-специфического защитного аутоиммунного ответа (Schwartz & Baruch, 2014а), или увеличения уровней циркулирующих моноцитов, которые играют роль в клиренсе васкулярного Аβ (амилоида бета) (Michaud et al, 2013).
Хотя нейродегенеративные заболевания различной этиологии, имеют общий локальный нейровоспалительный компонент, результаты авторов изобретения убедительно опровергают упрощенную характеристику всех патологий ЦНС как заболеваний, которые постоянно приносили бы выгоду от системной противовоспалительной терапии. Таким образом, тогда как аутоиммунные воспалительные патологии головного мозга, такие как рецидивирующе-ремиттирующий рассеянный склероз (RRMS), получают положительный результат от постоянного системного введения противовоспалительных и иммуносупрессорных лекарственных средств, для достижения длительной периферической иммунной супрессии, это будет как неэффективно, так и будет оказывать вредное влияние (пример 5) на патологию при хронических нейродегенеративных заболеваниях, в том числе в случае AD, при первично-прогрессирующем рассеянном склерозе (PP-MS) и вторично-прогрессирующем рассеянном склерозе (SP-MS). Кроме того, результаты авторов изобретения проливают свет на ошибочное представление относительно роли системных Tregs против Treg-клеток, ассоциированных с тканями при этих патологиях (Не & Balling, 2013). Поскольку связь между головным мозгом и иммунной системой является частью пожизненной пластичности мозга (Baruch et al, 2014), а нейродегенеративные заболевания являются преимущественно возрастными, настоящие результаты авторов изобретения также указывают на более общий феномен, при котором системная иммуносупрессия затрагивает функцию мозга. Таким образом, кратковременные периодические курсы понижения системной иммуносупрессии могут представлять собой терапевтический или даже профилактический подход, применимый к широкому спектру патологий головного мозга, в том числе AD и старческому слабоумию (деменции).
Важно, что полученные изобретателями подходы и результаты, присутствующие здесь в мышиных моделях с AD, не нацелены непосредственно на любой специфический для AD фактор, такой как бета-амилоид или tau-патология, а скорее демонстрируют новый подход, который, как ожидается, будет клинически применим в широком спектре патологий ЦНС - транзиентном снижении опосредованной Treg системной иммуносупрессии, для усиления рекрутирования разрешающих воспаление иммуноцитов к участкам патологии в ЦНС.
Принимая во внимание описанные выше неожиданные результаты, данное изобретение обеспечивает фармацевтическую композицию, содержащую активный агент, который вызывает снижение уровня системной иммуносупрессии у индивидуума для применения в лечении заболевания, нарушения, состояния или повреждения ЦНС, которое не включает аутоиммунное нейровоспалительное заболевание, рецидивирующе-ремитирующий рассеянный склероз (RRMC), где указанная фармацевтическая композиция предназначена для введения в режиме дозирования, содержащем по меньшей мере два курса лечения, каждый курс лечения, содержащий последовательно курс лечения с последующим интервалом курса без лечения.
В определенных воплощениях режим дозирования (схему приема) калибруют таким образом, что уровень системной иммуносупрессии временно снижается.
В применении здесь термин "лечение" относится к способам получения необходимого физиологического эффекта. Эффект может быть терапевтическим с точки зрения частичного или полного излечивания заболевания и/или симптомов, связанных с заболеванием. Термин относится к подавлению заболевания, т.е. остановке или замедлению его развития; или облегчению заболевания, т.е. регрессии заболевания.
Термин "курс без лечения" используется в данном описании взаимозаменяемо с термином "период отсутствия лечения" и относится к курсу, во время которого, подлежащему лечению пациенту, активный агент не вводят.
В применении здесь термин "системное присутствие" регуляторных Т-клеток относится к присутствию регуляторных Т-клеток (измеряемых их уровнем или активностью) в циркулирующей иммунной системе, т.е. в крови, селезенке и лимфатических узлах. В области иммунологии хорошо известным является факт, что профиль клеточной популяции в селезенке отражается на профиле популяции клеток крови (Zhao et al, 2007).
Данное лечение применимо как для пациентов, которые демонстрируют повышение системной иммуносупрессии, так и для пациентов, которые не демонстрируют такое повышение. Иногда индивидуум, который нуждается в лечении в соответствии с данным изобретением, имеет определенный уровень периферической иммуносупресии, который выражается повышенной частотой или числом Tregs в циркулирующей крови и/или их повышенной функциональной активностью, и/или понижением IFNγ-продуцирующих лейкоцитов, и/или пониженной пролиферацией лейкоцитов в ответ на стимулирование. Повышение частоты или числа Tregs может быть повышением общего числа или в процентах от общего числа CD4 клеток. Например, было обнаружено в соответствии с данным изобретением, что животные модели болезни Альцгеймера имеют более высокие частоты Foxp3 из CD4-клеток по сравнению с мышами дикого типа. Однако даже если у указанного индивидуума уровни системных Treg-клеток не повышены, их функциональная активность не повышена, уровень IFNγ-продуцирующих лейкоцитов не понижен, или пролиферация лейкоцитов в ответ на стимулирование не понижена, способ данного изобретения, который снижает уровень или активность системной иммуносупрессии является эффективным для лечения заболевания, нарушения, состояния или повреждения ЦНС, которое не включает аутоиммунное нейровоспалительное заболевание RRMS. Важно, что указанная системная иммуносупрессия также может вовлекать дополнительные типы иммунных клеток кроме Tregs, такие как супрессорные клетки миелоидного происхождения (MDSCs) (Gabrilovich & Nagaraj, 2009).
Уровень системной иммуносупрессии может быть определен различными способами, хорошо известными специалисту с обычной квалификацией в данной области. Например, уровень Tregs может быть измерен анализом методом проточной цитометрии мононуклеарных клеток периферической крови или Т-лимфоцитов, путем иммуноокрашивания как на поверхностные маркеры клеток, так и на ядерные внутриклеточные маркеры Treg (Chen & Oppenheim, 2011), CD45, TCR-B, или CD4-маркеры лимфоцитов, и измерения количества антитела, специфически связанного с клетками. Функциональная активность Tregs может быть измерена различными анализами; например, в настоящее время широко используется анализ включения тимидина, в котором супрессию (подавление), стимулированной анти-СРЗ mAb (пер., моноклональным антителом к CD3) пролиферации CD4+CD25- Т-клеток (традиционных Т-клеток), измеряют включением [3Н]-тимидина или с использованием CFSE (5-(и 6)-карбоксифлуоресцеина диацетат сукцинимидил эфира, который способен проникать внутрь клеток; деление клеток измеряют в виде последовательного двукратного снижения интенсивности флуоресценции CFSE). Число IFNγ-продуцирующих лейкоцитов или их активность, или их способность к пролиферации могут быть легко оценены специалистом в данной области с использованием способов, известных в данной области; например, уровень IFNγ-продуцирующих лейкоцитов может быть измерен анализом методом проточной цитометрии мононуклеарных клеток периферической крови, после короткой стимуляции ex-vivo и golgi-stop (пер., добавления реактива golgi-stop), и иммуноокрашивания путем внутриклеточного окрашивания IFNγ (с использованием, например, набора для внутриклеточного окрашивания BD Biosciences Cytofix/cytoperm™ fixation/permeabilization kit), путем сбора кондиционированной среды от этих клеток и количественного определения уровня секретируемых цитокинов с использованием ELISA, или путем сравнения соотношения различных цитокинов в кондиционированной среде, например, IL2/IL10, IL2/IL4, INFγ/TGFβ и т.д.. Уровни MDSCs (пер., супрессорных клеток миелоидного происхождения) в периферической крови человека легко могут быть оценены специалистом в данной области, например, с использованием анализа методом проточной цитометрии частоты клеток DR-/LIN-/CD11b+, DR-/LIN-/CD15+, DR-/LIN-/CD33+ и DR(-/low)/CD14+, как описано (Kotsakis et al, 2012).
В организме человека, периферическая/системная иммуносупрессиия может считаться повышенной, когда общее количество Treg-клеток в токе крови выше, чем 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%, или выше, чем у здоровой контрольной популяции, процентное содержание Treg-клеток от общего числа клеток CD4+ повышено на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%, или выше, чем у здоровой контрольной популяции, или функциональная активность Tregs повышена на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100% или выше, чем у здоровой контрольной популяции. Альтернативно, периферическая/системная иммуносупрессиия может считаться повышенной, когда уровень IFNγ-продуцирующих лейкоцитов или их активность снижается по сравнению с таковым у здоровой контрольной популяции на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%; или пролиферация лейкоцитов в ответ на стимуляцию снижается по сравнению с таковой у здоровой контрольной популяции на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%.
Агентом может считаться агент, который вызывает снижение уровня системной иммуносупрессии когда при введении агента индивидууму, общее число Tregs в токе крови этого индивидуума снижается на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100% по сравнению с уровнем до введения агента, процент Treg-клеток от общего числа клеток CD4+ падает на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100% по сравнению с таковым у здоровой контрольной популяции или функциональная активность Tregs понижена на 30, 40, 50, 60, 70, 80, 90 или 100% по сравнению с уровнем до введения агента. Альтернативно, агент может считаться агентом, который вызывает снижение уровня системной иммуносупрессии когда при введении агента индивидууму, общее число IFNγ-продуцирующих лейкоцитов или их активность повышается на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%; или пролиферация лейкоцитов в ответ на стимуляцию повышается по сравнению с таковым у здоровой контрольной популяции на 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100%.
Агент, используемый в соответствии с данным изобретением, может быть любым агентом, который подавляет уровень или активность регуляторных Т-клеток или препятствует их активности, но альтернативно может быть ограничен группой таких агентов, за исключением агента, выбранного из группы, состоящей из: (i) дофамина (dopamine) или его фармацевтически приемлемой соли; (ii) предшественника дофамина или его фармацевтически приемлемой соли; (iii) агониста семейства дофаминовых рецепторов 1-типа (агониста D1-R) или его фармацевтически приемлемой соли; и (iv) антагониста семейства дофаминовых рецепторов 2-типа (антагониста D2-R) или его фармацевтически приемлемой соли, даже если эти агенты неизвестны для применения в соответствии с курсом лечения согласно настоящему изобретению.
В определенных воплощениях, сеанс лечения содержит введение фармацевтической композиции индивидууму и сеанс лечения поддерживают по меньшей мере до тех пор, пока уровень не опустится ниже ссылочного, введение приостанавливают во время интервала между сеансами, и интервал между сеансами поддерживают до тех пор, пока уровень не опустится ниже ссылочного. Ссылочный уровень может быть выбран из (а) уровня системного присутствия или активности регуляторных Т-клеток или супрессорных клеток миелоидного происхождения, измеренного в самой последней пробе крови, полученной от указанного индивидуума до указанного введения; или (b) уровня системного присутствия или активности регуляторных Т-клеток или супрессорных клеток миелоидного происхождения, характерного для популяции индивидуумов, страдающих от заболевания, нарушения, состояния или повреждения ЦНС.
Альтернативно, сеанс лечения содержит введение фармацевтической композиции индивидууму и сеанс лечения поддерживают по меньшей мере до тех пор, пока системное присутствие или уровень IFNγ-продуцирующих лейкоцитов, или скорость пролиферации лейкоцитов в ответ на стимуляцию не поднимется выше ссылочного, введение приостанавливают во время интервала между сеансами, и интервал между сеансами поддерживают до тех пор, пока уровень не поднимется выше указанного ссылочного, где ссылочное (значение) выбрано из (а) уровня системного присутствия или активности IFNγ-продуцирующих лейкоцитов, или скорости пролиферации лейкоцитов в ответ на стимуляцию, измеренного в самой последней пробе крови, полученной от указанного индивидуума до указанного введения; или (b) уровня системного присутствия или активности IFNγ-продуцирующих лейкоцитов, или скорости пролиферации лейкоцитов в ответ на стимуляцию, характерного для популяции индивидуумов, страдающих от заболевания, нарушения, состояния или повреждения ЦНС.
Продолжительность лечения и интервалы между сеансами могут быть определены врачами в клинических испытаниях, направленных на определенную популяцию пациентов, а затем последовательно применены к этой популяции пациентов, без необходимости контроля уровня иммуносупрессии на индивидуальной основе.
В определенных воплощениях, сеанс лечения может быть продолжительностью от 3 дней до четырех недель, например, длиной от одной до четырех недель.
В определенных воплощениях, интервал между сеансами может быть от одной недели до шести месяцев, например, продолжительностью от двух недель до шести месяцев, в частности, продолжительностью от 3 недель до шести месяцев.
В сеансе лечения введение фармацевтической композиции может быть повторным введением, например, фармацевтическая композиция может быть введена ежедневно, или один раз в два, три, четыре, пять или шесть дней, один раз в неделю, раз в две недели, один раз каждые три недели, или один раз каждые четыре недели. Эти частоты применимые для любого агента, могут быть основаны на обычно используемых способах применения в данной области, и в конечном итоге, могут быть определены врачами в клинических испытаниях. Альтернативно, частота повторного введения в сеансе лечения может быть адаптирована в соответствии с природой активного агента, при которой, например, малая молекула может быть введена ежедневно; антитело может быть введено один раз каждые 3 дня; и сополимер 1 вводят еженедельно, раз в две недели, один раз каждые три недели или один раз каждые четыре недели. Должно быть понятно, что когда агент, такой как сополимер 1, вводят во время сеанса лечения при относительно низкой частоте, например, один раз в неделю в течение сеанса лечения в один месяц, или один раз в месяц в течение сеанса лечения в шесть месяцев, этот сеанс лечения сопровождается периодом отсутствия лечения, продолжительность которого длиннее, чем период между повторными ведениями во время сеанса лечения (т.е., длиннее, чем на одну неделю или один месяц, соответственно, в этом примере). Пауза продолжительностью в одну неделю или один месяц между введениями во время сеанса лечения в этом примере не считается интервалом между курсами.
Продолжительности сеанса лечения и интервала между сеансами могут быть отрегулированы с частотой введения, так что, например, частота введения активного агента один раз через каждые три дня может привести к сеансу лечения 6 или 9 дней и интервалу между сеансами, который таким образом начинается.
В качестве альтернативы предварительно установленной общей схеме лечения, уровень иммуносупрессии может быть откалиброван до необходимого уровня для каждого, подлежащего лечению, пациента индивидуально (персонализированная медицина), путем мониторинга уровня или активности Treg-клеток (или IFN-γ-продуцирующих лейкоцитов или скорости пролиферации лейкоцитов в ответ на стимуляцию), и корректировки сеанса лечения, частоты введения и интервала между сеансами эмпирически и персонально, как определено на основе результатов мониторинга.
Таким образом, продолжительность сеанса лечения может быть определена (а) мониторингом уровня системного присутствия или активности регуляторных Т-клеток у индивидуума путем измерения уровня в пробе крови, полученной от индивидуума в течение предварительно установленного периода времени после указанного введения; (b) сравнением уровня, измеренного в (а) со ссылочным, указанным выше, и определения является ли уровень отличным от ссылочного; (с) принятием решения, на основе отношения указанного уровня, измеренного в (а), к указанному ссылочному уровню, следует ли продолжить сеанс лечения, повторяя введения, или начать следующий интервал между сеансами, воздерживаясь от повторения введения; и (d) повторением введения или началом следующего интервала между сеансами в соответствии с решением в (с). Альтернативно, уровень IFN-γ-продуцирующих лейкоцитов или скорость пролиферации лейкоцитов в ответ на стимуляцию могут быть проконтролированы и сравнены с подходящей ссылочной пробой, как указано выше.
Аналогично, продолжительность интервала между сеансами может быть определена (а) мониторингом уровня системного присутствия или активности регуляторных Т-клеток у индивидуума путем измерения уровня в пробе крови, полученной от индивидуума в течение предварительно установленного периода времени после указанного введения; (b) сравнением уровня, измеренного в (а) со ссылочным, указанным выше, и определения является ли уровень отличным от ссылочного; (с) принятием решения, на основе отношения указанного уровня, измеренного в (а), к указанному ссылочному уровню, следует ли начать новый сеанс лечения, повторяя введения и этапы (а) и (b) или продолжить интервал между сеансами, повторяя только этапы (а) и (b); и (d) повторением введения и этапов (а) и (b) или только этапов (а) и (b) в соответствии с решением в (с). Альтернативно, уровень IFN-γ-продуцирующих лейкоцитов или скорость пролиферации лейкоцитов в ответ на стимуляцию могут быть проконтролированы и сравнены с подходящей ссылочной пробой, как указано выше.
В любом случае, режим дозирования, т.е. продолжительность сеанса лечения и интервал между сеансами, отлаживают так, что снижение уровня иммуносупрессии, например, измеряемого снижением уровня системного присутствия или активности регуляторных Т-клеток у индивидуума, является временным.
В определенных воплощениях, предварительно установленный период времени, т.е. время, прошедшее между самым последним введением активного агента и этапом мониторинга, составляет от 2 дней до шести месяцев.
В определенных воплощениях, регуляторные Т-клетки, которые подвергают мониторированию, являются клетками CD4+, выбранными из FoxP3+-клеток, экспрессирующих одну или несколько CD25, CD127, GITR, CTLA-4 или PD-1; или FoxP3--клеток, экспрессирующих одну или несколько CD25, CD127, GITR, CTLA-4 или PD-1 поверхностных молекул. В частности, общий фенотип регуляторных Т-клеток представляет собой CD4+CD25+FoxP3+клетки или CD4+CD25+FoxP3- клетки.
Агенты, способные снижать уровень регуляторных Т-клеток, известны в данной области (Colombo & Piconese, 2007), и эти агенты могут быть использованы в соответствии с данным изобретением. Каждая из цитируемых ниже публикаций включена в данный документ ссылкой в полном объеме.
Таким образом, агент может быть выбран, но не обязательно ограничивается ими, из: (i) антитела, такого как: (а) анти-PD-1, (b) анти-PD-L1 (с) анти-PD-L2 (Coyne & Gulley, 2014; Duraiswamy et al, 2014; Zeng et al, 2013); (d) анти-CTLA-4 (Simpson et al, 2013; Terme et al, 2012); (e) анти-PD-1 в сочетании с интерфероном α (Terawaki et al, 2011); (f) анти-PD-1 в сочетании с анти-CTLA4; (g) анти-CD47 (Tseng et al, 2013); (h) анти-ОХ40 (Voo et al, 2013); (i) анти-VEGF-A (бевацизумаб, bevacizumab) (Terme et al, 2013); (j) анти-CD25 (Zhou et al, 2013); (k) анти-GITR (GITR-активирующего mAb (DTA-1) (Colombo & Piconese, 2007); (l) анти-CCR4; (m) анти-TIM-3/галектин9 (Ju et al, 2014); (n) анти-иммуноглобулиноподобных рецепторов клеток-киллеров (KIR); (о) анти-LAG-S; или (р) анти-4-1ВВ (ii) любой комбинации от (а) до (р); (iii) любой комбинации от (а) до (р) в комбинации с адъювантом, например, анти-CTLA-4 антитела в комбинации с анти-ОХ40 антителом и лиганда TLR9, такого как CpG (Marabelle et al, 2013); (iv) малой молекулы выбранной из: (а) ингибитора р300 (Liu et al, 2013), такого как гемцитабин (gemcitabine) (низкая доза) (Shevchenko et al, 2013), или С646 или их аналогов, т.е. соединений формулы I:

где,
R1 выбран из Н, -CO2R6, -CONR6R7, -SO3H или -SO2NR6R7;
R2 выбран из Н, -CO2R6, или галогена, предпочтительно Cl;
R3 выбран из галогена, предпочтительно F, -NO2, -CN, -CO2R6, предпочтительно CO2CH3 или CO2CH2CH3, или -СН2ОН;
R4 и R5 каждый независимо представляет собой Н или -C1-С6-алкил, предпочтительно метил;
R6 представляет собой Н или -C1-С6-алкил, предпочтительно Н, метил или этил; и
R7 представляет собой Н или -C1-C6-алкил, предпочтительно Н или метил [см., (Bowers et al, 2010)];
(b) сунитиниба (Sunitinib) (Terme et al, 2012); (с) полиоксометалата-1 (Polyoxometalate-1) (POM-1) (Ghiringhelli et al, 2012); (d) α,β-метиленаденозин 5'-дифосфата (APCP) (Ghiringhelli et al, 2012); (e) триоксида мышьяка (As2O3) (Thomas-Schoemann et al, 2012); (f) GX15-070 (обатоклакса) (Obatoclax) (Kim et al, 2014); (g) антагониста ретиноевой ксилоты, такого как Ro 41-5253 (синетического ретиноида и селективного антагониста малых молекул) (Galvin et al, 2013) или LE-135 (Bai et al, 2009); (h) антагониста SIRPα (CD47), такого как CV1-hlgG4 (вариант SIRPα) в качестве единственного агента или в комбинации с анти-CD47 антителом (Weiskopf et al, 2013); (i) антагониста CCR4, такого как AF399/420/18025 в качестве единственного агента или в комбинации с анти-CCR4 антителом (Pere et al, 2011); (j) антагониста А2В рецептора аденозина, такого как PSB603 (Nakatsukasa et al, 2011), (k) антагониста индоламин-2,3-диоксигеназы (IDO); или (l) регулятора HIF-1; (iv) белка, выбранного из: (а) гликопротеина листьев растения нима (NLGP) (Roy et al, 2013); или (b) sCTLA-4 (растворимой изоформы CTLA-4) (Ward et al, 2013); (vi) молекулы силенсинга, такой как антисмысловая miR-126 (Qin et al, 2013) и анти-галектин-1 (Gal-1) (Dalotto-Moreno et al, 2013); (vii) OK-432 (лиофилизированного препарата Streptococcus pyogenes) (Hirayama et al, 2013); (viii) комбинации IL-12 и анти-CTLA-4; (ix) Сополимера-1 или сополимера, который модулирует активность или уровень Treg; (х) антибиотического агента, такого как ванкомицин (Brestoff & Artis, 2013; Smith et al, 2013) или (xi) любой комбинации от (i) до (х).
В определенных воплощениях, агент является анти-PD-1 антителом, т.е. антителом, специфическим к PD-1.
Многие анти-PD-1 антитела известны в данной области. Например, анти-PD-1 антитело, применяемое в соответствии с данным изобретением, может быть выбрано из таковых, раскрытых у Ohaegbulam et al. (Ohaegbulam et al, 2015), которые непосредственно включены в данный документ ссылками в полном объеме, т.е. СТ-011 (пидилизумаба (pidilizumab); гуманизированного IgG1; Curetech), МК-3475 (ламбролизумаба (lambrolizumab), пембролизумаба (pembrolizumab); гуманизированного IgG4; Merck), BMS-936558 (ниволумаба (nivolumab); человеческого IgG4; Bristol-Myers Squibb), AMP-224 (слитого белка PD-L2 IgG2a; AstraZeneca), BMS-936559 (человеческого IgG4; Bristol-Myers Squibb), MEDI4736 (гуманизированного IgG; AstraZeneca), MPDL3280A (человеческого IgG; Genentech), MSB0010718C (человеческого IgG1; Merck-Serono); или антитело, применяемое в соответствии с данным изобретением, может быть MEDI0680 (АМР-514; AstraZeneca) гуманизированным IgG4 mAb.
В определенных воплощениях антитело СТ-011 может быть введено человеку в дозировке 0,2-6 мг/кг или между 1,5-6 мг/кг; MK-3475 антитело может быть введено человеку в дозировке 1-10 мг/кг; BMS-936558 может быть введено человеку в дозировке 0,3-20 мг/кг, 0,3-10 мг/кг, 1-10 мг/кг или в дозировке 1 или 3 мг/кг; BMS-936559 может быть введено человеку в дозировке 0,3-10 мг/кг; MPDL3280A может быть введено человеку в дозировке 1-20 мг/кг; MEDI4736 может быть введено человеку в дозировке 0,1-15 мг/кг и MSB0010718C может быть введено человеку в дозировке 1-20 мг/кг.
Анти-CTLA4 антитело может быть тремелимумабом (Tremelimumab) (Pfizer), полностью человеческим моноклональным антителом IgG2; или ипилимумаб (ipilimumab), полностью человеческим моноклональным антителом IgG1.
Антитело к иммуноглобулиноподобным рецепторам клеток-киллеров (KIR) может быть лирилумабом (Lirilumab) (BMS-986015; разработанным Innate Pharma и лицензированным Bristol-Myers Squibb), полностью человеческим моноклональным антителом.
Анти-LAG-3 антитело направлено против активирующего лимфоциты гена-3. Одно такое антитело, которое может быть применено в соответствии с данным изобретением, является моноклональным антителом BMS-986016 (пембролизумабом (pembrolizumab); гуманизированным IgG4; Merck).
Анти-4-1 ВВ антитело может быть PF-05082566 (Pfizer Oncology), полностью гуманизированным моноклональным антителом-агонистом IgG2; или урелумабом (Urelumab) (BMS-663513; Bristol-Myers Squibb), полностью человеческим моноклональным антителом IgG4, антителом против 4-1ВВ.
В определенных воплощениях могут быть использованы комбинации антител такие как, но не ограничиваются ими: СТ-011 в комбинации с ритуксимабом (Rituximab) (коммерческие названия ритуксан (Rituxan), мабтера (MabThera) и Zytux) химерным моноклональным антителом против белка CD20, например, каждый по 3 мг/кг; BMS-936558 (например, 1 мг/кг) в комбинации с ипилимумабом (ipilimumab); например, по 3 мг/кг); или BMS-936558 (например, 1-10 мг/кг) в комбинации с мультипептидной вакциной, рестрицированной по HLA-A* (Weber et al, 2013).
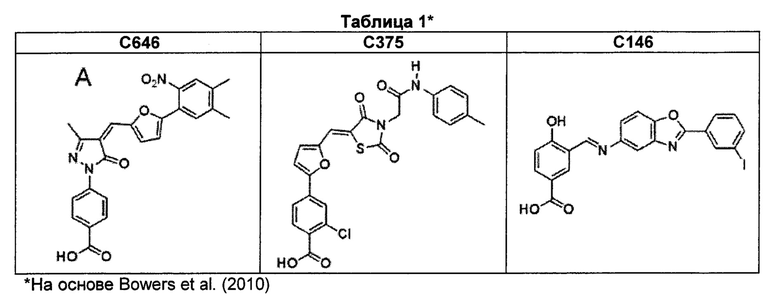
В определенных воплощениях агент является ингибитором р300, формулы которого приведены в таблице 1, т.е. С646 (4-(4-((5-(4,5-диметил-2-нитрофенил)фуран-2-ил)метилен)-3-метил-5-оксо-4,5-дигидро-1Н-пиразол-1-ил)бензойной кислотой), С146 (4-гидрокси-3-(((2-(3-йодофенил)бензо[d]оксазол-5-ил)имино)метил)бензойной кислотой) или С375 (2-хлор-4-(5-((2,4-диоксо-3-(2-оксо-2-(p-толиамино)этил)тиазолидин-5-илиден)метил)фуран-2-ил)бензойной кислотой). В частности, р300 ингибитором является С646.
В определенных воплощениях, малой молекулой-ингибитором индоламин-2,3-диоксигеназного пути может быть индоксимод (Indoximod) (NLG-9189; NewLink Genetics), INCB024360 (инцит (Incyte)) или NLG-919 (NewLink Genetics).
HIF-1-регулятором может быть M30, 5-[N-метил-N-пропаргиламинометил]-8-гидроксихинолин, описанный Zheng et al. (Zheng et al, 2015).
В определенных воплощениях агент может быть получен из широкого спектра антибиотиков, которые нацелены на грамположительные и грамотрицательные бактерии, и посредством чего облегчается иммуномодуляция Tregs, и, например, было показано, что ванкомицин, который нацелен на грамположительные бактерии, снижает уровни/активность Treg (Brestoff & Artis, 2013; Smith et al, 2013).
В определенных воплощениях агент может быть любым сополимером, который в определенном режиме приведет к понижающей регуляции (down regulation) Tregs, таким как YFAK, VYAK, VWAK, VEAK, FEAK, FAK, VAK или WAK. В применении здесь, термины "Сор-1" и "сополимер 1" ("Copolymer 1") используют здесь взаимозаменяемо.
Фармацевтическая композиция по изобретению может содержать активный агент статистический сополимер, который модулирует активность или уровень Treg, содержащая подходящее количество положительно заряженной аминокислоты, такой как лизин или аргинин, в комбинации с отрицательно заряженной аминокислотой (предпочтительно в меньшем количестве), такой как глутаминовая кислота или аспарагиновая кислота, возможно в комбинации с незаряженной нейтральной аминокислотой, такой как аланин или глицин, выступающей в качестве наполнителя, и, если требуется, с аминокислотой, адаптированной для придания иммуногенных свойств сополимеру, такой как ароматическая аминокислота, такая как тирозин или триптофан. Такие композиции могут включать любой из этих сополимеров, раскрытых в WO 00/05250, полное содержание которой включено в данный документ ссылкой.
В частности, композиция для применения по изобретению содержит по меньшей мере один сополимер, выбранный из группы, состоящей из статистических сополимеров, содержащих одну аминокислоту, выбранную из каждой, по меньшей мере из трех следующих групп: (а) лизин и аргинин; (b) глутаминовая кислота и аспарагиновая кислота; (с) аланин и глицин; и (d) тирозин и триптофан.
Сополимеры для использования в данном изобретении могут состоять из L- или D-аминокислот или их смесей. Как известно специалистам в данной области техники, L-аминокислоты встречаются в большинстве природных белков. Однако D-аминокислоты являются коммерчески доступными и могут быть использованы вместо некоторых или всех аминокислот, используемых для изготовления тройных сополимеров и других сополимеров, используемых в настоящем изобретении. Данное изобретение предполагает использование сополимеров, содержащих как D-, так и L-аминокислоты, а также сополимеры, состоящие по существу или из L-, или из D-аминокислот.
В определенных воплощениях фармацевтическая композиция изобретения содержит сополимер 1, смесь статистических полипептидов, состоящая по существу из аминокислот L-глутаминовой кислоты (Е), L-аланина (А), L-тирозина (Y) и L-лизина (K) в примерном соотношении 1,5:4,8:1:3,6, имеющая суммарный общий положительный электрический заряд и молекулярную массу от около 2 кДа до около 40 кДа. В определенных воплощениях Сор 1 имеет среднюю молекулярную массу от около 2 кДа до около 20 кДа, от около 4,7 кДа до около 13 кДа, от около 4 кДа до около 8,6 кДа, от около 5 кДа до 9 кДа, или от около 6,25 кДа до 8,4 кДА. В других воплощениях Сор 1 имеет среднюю молекулярную массу от около 13 кДа до около 20 кДа, от около 13 кДа до около 16 кДа или от около 15 кДа до около 16 кДа. Другие средние значения молекулярных масс для Сор 1, ниже чем 40 кДа, также охвачены данным изобретением. Сополимер 1 указанных диапазонов молекулярных масс может быть получен способами, известными в данной области техники, например, способами, описанными в US №5800808, полное содержание которой включено здесь в качестве ссылки в полном объеме. Сополимер 1 может быть полипептидом, содержащим в длину от около 15 до около 100, или от около 40 до около 80 аминокислот. В определенных воплощениях Сор 1 находится в форме его ацетатной соли, известной под международным непатентованным наименованием глатирамера ацетат, который был одобрен в нескольких странах для лечения рассеянного склероза (MS) под торговым названием копаксон (Copaxone®) (торговый знак Teva Pharmaceuticals Ltd., Petach Tikva, Israel). Как ожидается, активность сополимера 1 для описанной здесь фармацевтической композиции, останется, если произведена одна или несколько следующих замен: аспарагиновая кислота на глутаминовую кислоту, глицин на аланин, аргинин на лизин и триптофан на тирозин.
В определенных воплощениях изобретения сополимер, который модулирует активность или уровень Treg является сополимером трех различных аминокислот, каждая из другой одной из трех групп, групп от (а) до (d). Эти сополимеры здесь называют терполимерами.
В одном воплощении сополимер, который модулирует активность или уровень Treg, является терполимером, содержащим тирозин, аланин и лизин, далее называемым YAK, в котором средняя молярная концентрация аминокислот может изменяться: тирозин может присутствовать в молярной концентрации приблизительно 0,05-0,250; аланин в молярной концентрации приблизительно 0,3-0,6; и лизин в молярной концентрации приблизительно 0,1-0,5. Молярные соотношения тирозина, аланина и лизина могут быть около 0,10:0,54:0,35, соответственно. Можно заменить аргинин на лизин, глицин на аланин и/или триптофан на тирозин.
В определенных воплощениях сополимер, который модулирует активность или уровень Treg, является терполимером, содержащим тирозин, глутаминовую кислоту и лизин, далее называемым YEK, в котором средняя молярная концентрация аминокислот может изменяться: глутаминовая кислота может присутствовать в молярной концентрации приблизительно 0,005-0,300, тирозин может присутствовать в молярной концентрации приблизительно 0,005-0,250 и лизин может присутствовать в молярной концентрации приблизительно 0,3-0,7. Молярные соотношения глутаминовой кислоты, тирозина и лизина могут быть около 0,26:0,16:0,58, соответственно. Можно заменить аспарагиновую кислоту на глутаминовую кислоту, аргинин на лизин и/или триптофан на тирозин.
В определенных воплощениях сополимер, который модулирует активность или уровень Treg, является терполимером, содержащим лизин, глутаминовую кислоту и аланин, далее называемым KEA, в котором средняя молярная концентрация аминокислот может изменяться: глутаминовая кислота может присутствовать в молярной концентрации приблизительно 0,005-0,300, аланин в молярной концентрации приблизительно 0,005-0,600 и лизин может присутствовать в молярной концентрации приблизительно 0,2-0,7. Молярные соотношения глутаминовой кислоты, аланина и лизина могут быть около 0,15:0,48:0,36, соответственно. Можно заменить аспарагиновую кислоту на глутаминовую кислоту, глицин на аланин и/или аргинин на лизин.
В определенных воплощениях сополимер, который модулирует активность или уровень Treg, является терполимером, содержащим тирозин, глутаминовую кислоту и аланин, далее называемым YEA, в котором средняя молярная концентрация аминокислот может изменяться: тирозин может присутствовать в молярной концентрации приблизительно 0,005-0,250, глутаминовая кислота в молярной концентрации приблизительно 0,005-0,300 и аланин в молярной концентрации приблизительно 0,005-0,800. Молярные соотношения глутаминовой кислоты, аланина и тирозина могут быть около 0,21:0,65:0,14, соответственно. Можно заменить триптофан на тирозин, аспарагиновую кислоту на глутаминовую кислоту и/или глицин на аланин.
Средняя молекулярная масса терполимеров YAK, YEK, KEA и YEA может изменяться между около 2 кДа до 40 кДа, предпочтительно между около 3 кДа до 35 кДа, более предпочтительно между около 5 кДа до 25 кДа.
Сополимер 1 и другие сополимеры, которые модулируют активность или уровень Treg, могут быть получены способами, известными в данной области техники, например, в условиях конденсации с использованием необходимого молярного соотношения аминокислот в растворе, процедурами твердофазного синтеза. Условия конденсации включают правильную температуру, рН и условия растворения для конденсации карбоксильной группы одной аминокислоты с аминогруппой другой аминокислоты с образованием пептидной связи. Конденсирующие агенты, например, дициклогексилкарбодиимид, могут быть использованы для облегчения образования пептидной связи. Блокирующие группы могут быть использованы для защиты функциональных групп, таких как звенья в боковой цепи и некоторые из амино- или карбоксильных групп от нежелательных побочных реакций.
Например, сополимеры могут быть получены способом, описанным в US №3849550, где N-карбоксиангидриды тирозина, аланина, γ-бензилглутамата и N ε-трифторацетил-лизина полимеризуют при температурах окружающей среды (20°С-26°С) в безводном диоксане с диэтиламином в качестве инициатора. γ-Карбоксильная группа глутаминовой кислоты может быть деблокирована бромистым водородом в ледяной уксусной кислоте. Трифторацетильные группы удаляют из лизина 1М пиперидином. Специалист в данной области техники легко поймет, что способ может быть отрегулирован для изготовления пептидов и полипептидов, содержащих необходимые аминокислоты, то есть три из четырех аминокислот в сополимере 1, путем селективного устранения реакций, которые имеют отношение к любой из глутаминовой кислоты, аланина, тирозина или лизина.
Молекулярная масса сополимеров может быть отрегулирована во время синтеза полипептида или после того, как сополимеры были произведены. Для регулирования молекулярной массы во время синтеза полипептидов, условия синтеза или количество аминокислот регулируют таким образом, чтобы синтез прекращался при достижении необходимой приблизительной длины полипептида. После синтеза полипептиды с необходимой молекулярной массой могут быть получены с помощью любой доступной процедуры отбора по размеру, такой как хроматография полипептидов на колонке или в геле для разделения по молекулярной массе, и сбора в диапазонах необходимых молекулярных масс. Сополимеры также могут быть частично гидролизованы для удаления высокомолекулярных разновидностей, например, кислотным или ферментативным гидролизом, и затем очищены для удаления кислоты или ферментов.
В одном воплощении сополимеры с необходимой молекулярной массой могут быть получены способом, который включает взаимодействие защищенного полипептида с бромистоводородной кислотой с образованием трифторацетил-полипептида, имеющего необходимый профиль молекулярной массы. Взаимодействие выполняют в течение времени и при температуре, которая задана одной или несколькими тестовыми взаимодействиями. Во время тестового взаимодействия меняют время и температуру и определяют диапазон молекулярных масс данной партии тестируемых полипептидов. Условия испытаний, которые обеспечивают оптимальный диапазон молекулярных масс для этой партии полипептидов, используют для серии. Таким образом, трифторацетил-полипептид, имеющий необходимый профиль молекулярной массы, может быть получен способом, который включает в себя взаимодействие защищенного полипептида с бромистоводородной кислотой в течение времени и при температуре, заданной тестовым взаимодействием. Затем трифторацетил-полипептид с необходимым профилем молекулярной массы дополнительно обрабатывают водным раствором пиперидина для получения малотоксичного полипептида, имеющего необходимую молекулярную массу.
В определенных воплощениях, тестируемая проба защищенного полипептида из данной партии взаимодействует с бромистоводородной кислотой в течение приблизительно 10-50 часов при температуре приблизительно 20-28°С. Оптимальные условия для данной партии определяют путем выполнения нескольких тестовых взаимодействий. Например, в одном варианте воплощения защищенный полипептид взаимодействует с бромистоводородной кислотой в течение приблизительно 17 часов при температуре около 26°С.
Поскольку мотивы, связывающие Сор 1 с MS-ассоциированными HLA-DR-молекулами известны (Fridkis-Hareli et al, 1999), то полипептиды, имеющие установленную последовательность, могут быть легко получены и протестированы на связывание с пептидсвязывающей бороздкой молекул HLA-DR, как описано в публикации Fridkis-Hareli et al (1999). Примерами таких пептидов являются пептиды, описанные в WO 00/05249 и WO 00/05250, описания которых включены в данный документ в качестве ссылки в их полном виде, и включают пептиды SEQ ID NOs. 1-32 (таблица 2).
Можно ожидать, что такие полипептиды и другие похожие пептиды будут обладать такой же активностью, что Сор 1. Указанные пептиды и другие похожие пептиды, как полагают, также относятся к родственным Сор-1 пептидам или полипептидам, а их применение рассматривается как часть в рамках определения сополимеров, которые дают перекрестную реакцию с антигенами миелина ЦНС и их применение считается частью данного изобретения.

Определение "сополимер, который модулирует активность или уровень Treg" в соответствии с данным изобретением, предназначено для охвата других синтетических аминокислотных сополимеров, таких как, составленные случайным образом сополимеры из четырех аминокислот, описанные Fridkis-Hareli et al., 2002 и в US №8017125 (в качестве кандидатов для лечения рассеянного склероза), а именно сополимеры VFAK, содержащие аминокислоты валин (V), фенилаланин (F), аланин (А) и лизин (K); VYAK, содержащие аминокислоты валин (V), тирозин (Y), аланин (А) и лизин (K); VWAK, содержащие аминокислоты валин (V), триптофан (W), аланин (А) и лизин (K); VEAK, содержащие аминокислоты валин (V), глутаминовую кислоту (Е), аланин (А) и лизин (K); FEAK, содержащие аминокислоты фенилаланин (F), глутаминовую кислоту (Е), аланин (А) и лизин (K); FAK, содержащие аминокислоты фенилаланин (F), аланин (А) и лизин (K); VAK, содержащие аминокислоты валин (V), аланин (А) и лизин (K) и WAK, содержащие аминокислоты триптофан (W), аланин (А) и лизин (K).
Фармацевтическая композиция по изобретению может быть для лечения заболевания, нарушения или состояния ЦНС, которое является нейродегенеративным заболеванием, нарушением или состоянием, выбранным из болезни Альцгеймера, бокового амиотрофического склероза, болезни Паркинсона и болезни Хантингтона, первичного прогрессирующего рассеянного склероза; вторичного прогрессирующего рассеянного склероза, кортико-базальной дегенерации, синдрома Ретта, дегенеративного изменения сетчатки, выбранной из группы состоящей из возрастной дегенерации желтого пятна и пигментного ретинита; передней ишемической оптической нейропатии; глаукомы; увеита; депрессии; травматического стресса или посттравматического стрессового расстройства, лобно-височной деменции, деменции с тельцами Леви, умеренных когнитивных нарушений, задней кортикальной атрофии, первичной прогрессирующей афазии или прогрессирующего надъядерного паралича. В определенных воплощениях состояние ЦНС является возрастной деменцией.
В определенных воплощениях, состояние CNS является болезнью Альцгеймера, боковым амиотрофическим склерозом, болезнью Паркинсона и болезнью Хантингтона.
Фармацевтическая композиция в соответствии с настоящим изобретением дополнительно может быть для лечения повреждения ЦНС, выбранного из повреждения спинного мозга, закрытой травмы черепа, тупой травмы, проникающей травмы, геморрагического инсульта, ишемического инсульта, ишемии головного мозга, повреждения зрительного нерва, инфаркта миокарда, отравлении фосфорорганическими соединениями и повреждения, вызванного иссечением опухоли.
Как указано выше, изобретатели обнаружили, что настоящее изобретение улучшает когнитивную функцию у мышей, имитирующих болезнь Альцгеймера. Таким образом, фармацевтическая композиция может быть применима для улучшения двигательных и/или когнитивных функций ЦНС, например, для применения для облегчения, связанной с возрастом потери когнитивной функции, которая может возникнуть у индивидуумов, не имеющих диагностированного заболевания, а также у людей, страдающих от нейродегенеративного заболевания. Кроме того, фармацевтическая композиция может быть применима для облегчения потери когнитивной функции, возникающей в результате острого стресса или травматического эпизода. Когнитивные функции, упомянутые здесь выше, могут включать обучение, память или оба.
В применении здесь, термин "функция ЦНС", относится, inter alia (как таковой), к получению и обработке сенсорной информации, мышлению, обучению, запоминанию, восприятию, приему и пониманию языка, контролю двигательной функции и слуховых и зрительных реакций, сохраняя баланс и равновесие, координации движений, проведению сенсорной информации и управлению такими вегетативными функциями, как дыхание, частота сердечных сокращений и пищеварение.
Термины "познание", "когнитивная (познавательная) функция" и "когнитивная деятельность" используются здесь взаимозаменяемо и относятся к любому умственному процессу или состоянию, которое включают в себя, но не ограничивается этим, обучение, память, создание образов, мышление, осознание, способности к пространственному ориентированию, речи и языковым навыкам, приобретению языка и способности к здравомыслию. Познание формируется во множестве областей мозга, таких как гиппокамп, кора головного мозга и другие структуры мозга. Однако считается, что долговременная память хранится по меньшей мере частично в коре и известно, что сенсорная информация приобретается, объединяется и извлекается специфической кортикальной структурой, вкусовой корой, которая находится в островковой области.
У людей, когнитивная (познавательная) функция может быть измерена любым известным способом, например, и без ограничения, оценкой изменения по шкале общего клинического впечатления (шкала CIBIC-plus); кратким исследованием психического состояния (MMSE); нейропсихиатрическим опросником (NPI); по клинической рейтинговой шкале деменции (CDR); по Кембриджской автоматизированной батарее нейропсихологических тестов (CANTAB) или по гериатрической шкале клинической оценки компании Sandoz (SCAG). Когнитивная функция также может быть измерена косвенно с использованием средств воспроизведения изображений, таких как позитронно-эмиссионная томография (PET), функциональная магнитно-резонансная томография (fMRI), однофотонная эмиссионная компьютерная томография (SPECT) или с помощью любого другого средства воспроизведения изображений, которое позволит любому специалисту измерить функцию мозга.
Улучшение одного или нескольких процессов, влияющих на процесс познания у пациента, будет означать улучшение когнитивной функции у указанного пациента, таким образом, в определенных воплощениях улучшение познания содержит улучшение обучения, пластичности и/или долговременной памяти. Термины "улучшение" и "усиление" могут быть использованы взаимозаменяемо.
Термин "обучение" относится к приобретению или получению новых, или модификации и укреплению существующего знания, поведения, навыков, ценностей или предпочтений.
Термин "пластичность" относится к синаптической пластичности, пластичности мозга или нейропластичности, связанной со способностью мозга меняться с обучением и менять уже приобретенную память. Одним измеряемым параметром, отражающим пластичность, является угасание памяти.
Термин "память" относится к процессу, при котором информация кодируется, хранится и извлекается. Память имеет три различимые категории: сенсорная память, кратковременная память и долговременная память.
Термин "долговременная память" означает способность хранить информацию в течение длительного или неограниченного периода времени. Долговременная память содержит два основных деления: эксплицитная память (декларативная память) и имплицитная (недекларативная память). Долговременная память достигается за счет консолидации памяти, которая является категорией процессов, которые стабилизируют след памяти после его первоначального приобретения. Консолидация подразделяется на два конкретных процесса: синаптическиую консолидацию, которая происходит в течение первых нескольких часов после обучения, и системную консолидацию, когда гиппокамп-зависимые воспоминания становятся независимыми от гиппокампа в течение от нескольких недель до нескольких лет.
В дополнительном аспекте настоящее изобретение направлено на способ лечения заболевания, нарушения, состояния или повреждения центральной нервной системы (ЦНС), которое не включает аутоиммунное нейровоспалительное заболевание, рецидивирующе-ремитирующий рассеянный склероз (RRMS), указанный способ, содержащий введение индивидууму, нуждающемуся в этом, фармацевтической композиции по изобретению, как определено выше, где указанную фармацевтическую композицию вводят в режиме дозирования, содержащем по меньшей мере два сеанса лечения, каждый сеанс лечения, содержащий последовательно сеанс лечения, за которым следует интервал между сеансами.
В определенных воплощениях, сеанс лечения содержит введение фармацевтической композиции индивидууму и сеанс лечения поддерживают по меньшей мере до падения уровня ниже ссылочного, введение приостанавливают во время интервала между сеансами, и интервал между сеансами поддерживают до тех пор, пока уровень не опустится ниже ссылочного. Ссылочный уровень может быть выбран из (а) уровня системного присутствия или активности регуляторных Т-клеток или супрессорных клеток миелоидного происхождения, измеренных в самой последней пробе крови, полученной от указанного индивидуума до указанного введения; или (b) уровня системного присутствия или активности регуляторных Т-клеток или супрессорных клеток миелоидного происхождения, характерных для популяции индивидуумов, страдающих от заболевания, нарушения, состояния или повреждения ЦНС.
Альтернативно, сеанс лечения содержит введение фармацевтической композиции индивидууму и сеанс лечения поддерживают по меньшей мере до тех пор, пока системное присутствие или уровень IFNγ-продуцирующих лейкоцитов, или скорость пролиферации лейкоцитов в ответ на стимуляцию поднимется выше ссылочного, введение приостанавливают во время интервала между сеансами, и интервал между сеансами поддерживают до тех пор, пока уровень не поднимется выше указанного ссылочного, где ссылочное (значение) выбрано из (а) уровня системного присутствия или активности IFNγ-продуцирующих лейкоцитов, или скорости пролиферации лейкоцитов в ответ на стимуляцию, измеренных в самой последней пробе крови, полученной от указанного индивидуума до указанного введения; или (b) уровня системного присутствия или активности IFNγ-продуцирующих лейкоцитов, или скорости пролиферации лейкоцитов в ответ на стимуляцию, характерных для популяции индивидуумов, страдающих от заболевания, нарушения, состояния или повреждения ЦНС.
Вышеуказанные варианты осуществления, которые описывают различные признаки фармацевтической композиции по настоящему изобретению, актуальны также для способа изобретения, поскольку способ использует одну и ту же фармацевтическую композицию.
Еще в дополнительном аспекте, настоящее изобретение обеспечивает фармацевтическую композицию для применения в лечении заболевания, нарушения, состояния или повреждения ЦНС, которое не включает в себя аутоиммунное нейровоспалительное заболевание RRMS, указанную фармацевтическую композицию, содержащую активный агент, который вызывает снижение уровня системной иммуносупрессии у индивидуума, выбранный из: (i) антитела, специфического к: (a) CD47; (b) ОХ40; (с) VEGF-A (бевацизумабу) (bevacizumab); (d) CD25; (е) GITR (GITR-активирующему mAb (DTA-1)); (f) CCR4; или (g) TIM-3/Galectin9; (h) антителу к иммуноглобулиноподобным рецепторам клеток-киллеров; (i) анти-LAG-3; или (j) анти-4-1ВВ; (ii) любой комбинации от (а) до (j); (iii) любой комбинации от (а) до (j) в комбинации с адъювантом, таким как лиганд TLR9, например, CpG; (iv) белка, выбранного из: (а) гликопротеина листьев (растения) нима (NLGP); или (b) sCTLA-4; (v) малой молекулы, выбранной из: (а) сунитиниба (Sunitinib); (b) полиоксиметаллата-1 (Polyoxometalate-1) (РОМ-1); (с) α,β-метиленаденозин 5'-дифосфата (АРСР); (d) триоксида мышьяка (As2O3); (е) GX15-070 (обатоклакса) (Obatoclax); (f) антагониста ретиноевой кислоты, такого как Ro 41-5253 или LE-135; (g) антагониста SIRPα (CD47), такого как CV1-hlgG4 в качестве единственного агента или в комбинации с анти-СР47 антителом; (h) антагониста CCR4, такого как AF399/420/18025 в качестве единственного агента или в комбинации с анти-CCR4 антителом; или (i) антагониста А2В рецептора аденозина, такого как PSB603; (j) антагониста индоламин-2,3-диоксигеназы; (к) регулятора HIF-1; (vi) молекулы силенсинга, такой как антисмысловая miR-126 и анти-галектин-1 (Gal-1); (vii) OK-432; (viii) комбинации IL-12 и анти-CTLA-4; (ix) антибиотического агента, такого как ванкомицин; или (х) любой комбинации от (i) до (ix).
Фармацевтические композиции для применения в соответствии с настоящим изобретением могут быть приготовлены обычным способом с использованием одного или нескольких физиологически приемлемых носителей или наполнителей. Носитель(и) должен быть "приемлемым" в смысле совместимости с другими ингредиентами композиции и безвредности по отношению к их реципиенту.
Следующие иллюстративные примеры носителей, способов введения, лекарственных форм и т.п., перечислены в качестве известных возможностей, из которых носители, способы введения, лекарственные формы и т.п., могут быть выбраны для использования с настоящим изобретением. Однако, специалистам в данной области техники будет понятно, что, любая данная композиция и выбранный способ введения сначала должны быть протестированы для определения, что они достигают необходимых результатов.
Способы введения включают в себя, но не ограничиваются ими, парентеральный, например, внутривенный, внутрибрюшинный, внутримышечный, подкожный, относящийся к слизистой оболочке (например, пероральный, интраназальный, буккальный, влагалищный, ректальный, внутриглазной), интратекальный, топический и интрадермальный способы. Введение может быть системным или локальным.
Термин "носитель" относится к разбавителю, адъюванту, наполнителю или носителю, с которым активный агент вводится. Носители в фармацевтической композиции могут содержать связующее вещество, такое как микрокристаллическая целлюлоза, поливинилпирролидон (поливидон или повидон), трагакантовая камедь, желатин, крахмал, лактоза или моногидрат лактозы; агент для улучшения распадаемости таблеток, такой как альгиновая кислота, кукурузный крахмал и т.п.; смягчающий компонент или поверхностно-активное вещество, такое как магния стеарат или лаурилсульфат натрия; и вещество, способствующее скольжению, такое как коллоидная двуокись кремния.
Для перорального введения фармацевтический препарат может быть в жидкой форме, например, растворов, сиропов или суспензий, или может быть представлен в виде лекарственной формы для восстановления водой или другим подходящим носителем перед применением. Такие жидкие препараты могут быть получены традиционными способами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сорбитовый сироп, производные целлюлозы или гидрогенизированные пищевые жиры); эмульгирующие агенты (например, лецитин или камедь); неводные носители (например, миндальное масло, жирные сложные эфиры, или фракционированные растительные масла); и консерванты (например, метил- или пропил-р-гидроксибензоаты или сорбиновая кислота). Фармацевтические композиции могут принимать форму, например, таблеток или капсул, полученных традиционными способами с фармацевтически приемлемыми наполнителями, такими как связующие агенты (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или гидрофосфат кальция); смазывающие вещества (например, стеарат магния, тальк или диоксид кремния); разрыхлители (например, картофельный крахмал или натрия крахмала гликолят); или смачивающие агенты (например, лаурилсульфат натрия). Таблетки могут быть покрыты способами, хорошо известными в данной области.
Препараты для перорального введения могут быть подходящим образом приготовлены для получения контролируемого высвобождения активного соединения.
Для трансбуккального введения композиции могут принимать форму таблеток или лепешек, приготовленных традиционным способом.
Композиции могут быть приготовлены для парентерального введения путем инъекции, например, путем болюсной инъекции или непрерывной инфузии. Композиции для инъекции могут быть представлены в стандартной дозированной форме, например, в ампулах или в контейнерах для лекарственных средств для многократного приема, с добавлением консерванта. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать агенты для приготовления композиции, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. В качестве альтернативы, активный ингредиент может быть в форме порошка для соединения с подходящим носителем, например, стерильной свободной от пирогенов водой, перед применением.
Композиции могут быть также приготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такие как масло какао или другие глицериды.
Для введения путем ингаляции, композиции для применения в соответствии с настоящим изобретением традиционно поставляют в форме аэрозоля из упаковок под давлением или распылителя с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, углекислого газа или другого подходящего газа. В случае аэрозоля под давлением единица дозирования может быть определена с помощью клапана для доставки отмеренного количества. Капсулы и картриджи, например, из желатина, для применения в ингаляторе или инсуффляторе могут быть приготовлены, содержащими порошкообразную смесь соединения и подходящей порошковой основы, такой как лактоза или крахмал.
Определение доз активного ингредиента, подлежащего применению в медицинских целях основано на традиционно применяемых практических подходах в данной области техники, и будет окончательно определено врачами в клинических испытаниях. Ожидаемая приблизительная эквивалентная доза для введения человеку может быть вычислена на основе экспериментально полученных данных in vivo, раскрытых здесь ниже, с использованием известных формул (например, Reagan-Show et al. (2007) Dose translation from animal to human studies revisited. The FASEB Journal 22:659-661). Согласно этой парадигме, эквивалентная доза для взрослого человека (мг/кг массы тела) равна дозе, которая дана мыши (мг/кг массы тела), умноженной на 0,081.
Изобретение теперь будет проиллюстрировано следующими не ограничивающими примерами.
ПРИМЕРЫ
Материалы и методы
Животные. Трансгенных мышей 5XFAD (Tg6799), которые совместно сверхэкспрессируют наследственные AD мутантные формы трансгенов человеческого АРР (шведская мутация, K670N/M671L; мутация Флорида, I716V; и Лондонская мутация, V717I) и PS1 (M146L/L286V) под транскрипционным контролем нейрон-специфического мышиного промотора Thy-1 (Oakley et al, 2006), и двойных трансгенных мышей с AD B6.Cg-Tg (APPswe, PSEN1dE9) 85Dbo/J (Borchelt et al, 1997) приобретали в лаборатории Джексона (The Jackson Laboratory). Генотипирование осуществляли ПЦР-анализом хвоста ДНК, как ранее описано (Oakley et al, 2006). Гетерозиготный мутант мышей cx3cr1GFP/+(Jung et al, 2000) (B6.129P-cx3cr1tm1Litt/J, у которого один из аллелей хемокинового рецептора CX3CR1 заменяли геном, кодирующим GFP) использовали в качестве доноров для химер ВМ (пер., химер костного мозга). Мышей Foxp3.LuciDTR (Suffner et al, 2010) скрещивали с мышами 5XFAD для обеспечения условного истощения Foxp3+ Tregs. Животных размножали и содержали в Центре разведения животных Научного института Вейцмана (Animal Breeding Center of the Weizmann Institute of Science). Все подробно описанные здесь эксперименты отвечают требованиям, разработанным комитетом по содержанию и использованию лабораторных животных (Institutional Animal Care and Use Committee (IACUC) Научного института Вейцмана.
Очистка РНК, синтез кДНК и анализ количественной ПЦР в реальном времени. Тотальную РНК зубчатой извилины гиппокампа (DG) экстрагировали реагентом TRI Reagent (Molecular Research Center) и очищали из лизатов с использованием набора RNeasy Kit (Qiagen). Тотальную РНК хороидного сплетения экстрагировали с использованием набора RNA MicroPrep Kit (Zymo Research). мРНК (1 мкг) превращали в кДНК с использованием набора High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Экспрессию специфических мРНК анализировали количественной ПЦР в реальном времени (RT-qPCR) с использованием флуоресценции. Реакции RT-qPCR выполняли с использованием основной смеси Fast-SYBR PCR Master Mix (Applied Biosystems). Количественные реакции выполняли в трех повторностях для каждой пробы с использованием метода построения калибровочной кривой. Пептидилпролилизомеразу А (рр/а) выбирали в качестве ссылочного гена ("гена домашнего хозяйства"). Циклы амплификации были при 95°С в течение 5 секунд, 60°С в течение 20 секунд и 72°С в течение 15 секунд. В конце анализа строили кривую плавления для оценки специфичности реакции. Для анализа генов ifn-γ и ppia, кДНК предварительно амплифицировали в течение 14 циклов ПЦР с неслучайными праймерами для ПЦР, посредством чего увеличивали чувствительность последующего анализа ПЦР в режиме реального времени, в соответствии с протоколом фирмы-производителя (PreAmp Master Mix Kit; Applied Biosystems). Экспрессию мРНК определяли с использованием TaqMan RT-qPCR, в соответствии с инструкциями фирмы-производителя (Applied Biosystems). Все реакции RT-qPCR выполняли и анализировали с использованием программного обеспечения StepOne software V2.2.2 (Applied Biosystems). Использовали следующие зонды TaqMan Assays-on-Demand™: Mm02342430_g1 (ppia) и Mm01168134_m1 (ifn-γ).
Для всех других исследованных генов использовали следующие праймеры:
ppia прямой 5'-AGCATACAGGTCCTGGCATCTTGT-3' (SEQ ID NO: 33) и обратный 5'-CAAAGACCACATGCTTGCCATCCA-3' (SEQ ID NO: 34);
icam1 прямой 5'-AGATCACATTCACGGTGCTGGCTA-3' (SEQ ID NO: 35) и обратный 5'-AGCTTTGGGATGGTAGCTGGAAGA-3' (SEQ ID NO: 36);
vcam1 прямой 5'-TGTGAAGGGATTAACGAGGCTGGA-3' (SEQ ID NO: 37) и обратный 5'-CCATGTTTCGGGCACATTTCCACA-3' (SEQ ID NO: 38);
cxcl10 прямой 5'-AACTGCATCCATATCGATGAC-3' (SEQ ID NO: 39) и обратный 5'-GTGGCAATGATCTCAACAC-3' (SEQ ID NO: 40);
ccl2 прямой 5'-CATCCACGTGTTGGCTCA-3' (SEQ ID NO: 41) и обратный 5'-GATCATCTTGCTGGTGAATGAGT-3' (SEQ ID NO: 42);
tnf-γ прямой 5'-GCCTCTTCTCATTCCTGCTT-3' (SEQ ID NO: 43) и обратный CTCCTCCACTTGGTGGTTTG-3' (SEQ ID NO: 44);
il-1β прямой 5'-CCAAAAGATGAAGGGCTGCTT-3' (SEQ ID NO: 45) и обратный 5'-TGCTGCTGCGAGATTTGAAG-3' (SEQ ID NO: 46);
il-12p40 прямой 5'-GAAGTTCAACATCAAGAGCA-3' (SEQ ID NO: 47) и обратный 5'-CATAGTCCCTTTGGTCCAG-3' (SEQ ID NO: 48);
il-10 прямой 5'-TGAATTCCCTGGGTGAGAAGCTGA-3' (SEQ ID NO: 49) и обратный 5'-TGGCCTTGTAGACACCTTGGTCTT-3' (SEQ ID NO: 50);
tgfβ2 прямой 5'-AATTGCTGCCTTCGCCCTCTTTAC-3' (SEQ ID NO: 51) и обратный 5'-TGTACAGGCTGAGGACTTTGGTGT-3' (SEQ ID NO: 52);
igf-1 прямой 5'-CCGGACCAGAGACCCTTTG (SEQ ID NO: 53) и обратный 5'-CCTGTGGGCTTGTTGAAGTAAAA-3' (SEQ ID NO: 54);
bdnf прямой 5'-GATGCTCAGCAGTCAAGTGCCTTT-3' (SEQ ID NO: 55) и обратный 5'-GACATGTTTGCGGCATCCAGGTAA-3' (SEQ ID NO: 56);
Иммуногистохимия. Проводку тканей и иммуногистохимию выполняли на залитых в парафин срезах мозга мышей (6 мкм толщиной) и человека (10 мкм толщиной). Для окрашивания ICAM-1 (пер., молекулы межклеточной адгезии) человека использовали первичное мышиное анти-ICAM (1:20 Abeam; ab2213) антитело. Препараты инкубировали в течение 10 минут с 3% H2O2 и использовали вторичное антимышиное антитело, конъюгированное с биотином, с последующей биотин/авидин амплификацией с использованием набора Vectastain ABC kit (Vector Laboratories). Впоследствии, применяли субстрат 3,3'-диаминобензидин (substrate DAB) (Zytomed kit); предметные стекла высушивали и оснащали монтирующей средой на основе ксилола. Для окрашивания тканей, мышей вначале транскардиально перфузируют PBS до иссечения ткани и фиксации. Ткани CP выделяли под препаровальной лупой (Stemi DV4; Zeiss) из бокового, третьего и четвертого желудочков головного мозга. Для окрашивания тотального препарата ткани фиксировали 2,5% параформальдегидом (PFA) в течение 1 часа при 4°С, и впоследствии переносили в PBS, содержащий 0,05% азид натрия. Перед окрашиванием иссеченные ткани промывали PBS и блокировали (20% лошадиной сывороткой, 0,3% Triton Х-100 и PBS) в течение 1 часа при комнатной температуре. Окрашивание тотального препарата первичными антителами (в PBS, содержащем 2% лошадиную сыворотку и 0,3% Triton Х-100) или вторичными антителами выполняли в течение 1 часа при комнатной температуре. Каждый шаг сопровождали тремя промывками в PBS. Ткани наносили на предметные стекла, монтировали с использованием Immu-mount (9990402, от Thermo Scientific), и накрывали покровными стеклами. Для окрашивания срезов головного мозга применяли два различных протокола приготовления тканей (залитые в парафин или микротомные свободно-плавающие срезы) как описано ранее (Baruch et al, 2013; Kunis et al, 2013). Использовали следующие первичные антитела: мышиное анти-Aβ (1:300, Covance, #SIG-39320); кроличье анти-GFP (1:100, MBL, #598); крысиное анти-CD68 (1:300, eBioscience, #14-0681); крысиное анти-ICAM-1 (1:200, Abeam, #АВ2213); козье анти-GFP (1:100, Abeam, #ab6658); кроличье анти-IBA-1 (1:300, Wako, #019-19741); козье анти-1L-10 (1:20, R&D systems, #AF519); крысиное анти-Fox3 (1:20, eBioscience, #13-5773-80); кроличье анти-CD3 (1:500, Dako, #IS503); мышиное анти-ZO-1, мышиное анти-E-Cahedrin (анти-Е-кагедрин) и кроличье анти-Claudin-1 (анти-клаудин-1) (все 1:100, Invitrogen, #33-9100, #33-4000, #51-9000); кроличье анти-GFAP (1:200, Dako, #Z0334). Вторичные антитела включали в себя: Су2/Су3/Су5-конъюгированные ослиные анти-мышиные/козьи/кроличьи/крысиные антитела (1:200; все от Jackson Immunoresearch). Препараты на предметных стеклах подвергали действию ядерного красителя Hoechst nuclear staining (1:4000; Invitrogen Probes) в течение 1 минуты. Обычно использовали два отрицательных контроля в процедурах иммуноокрашивания, окрашивание изотипным контрольным антителом с последующим вторичным антителом, или окрашивание только вторичным антителом. Для внутриклеточного окрашивания Foxp3, извлечение антигена из залитых в парафин на стекле препаратов выполняли с использованием набора Retreivagen Kit (#550524, #550527; BD Pharmingen™). Микроскопический анализ выполняли с использованием флуоресцентного микроскопа (Е800; Nikon) или конфокального лазерного сканирующего микроскопа (Carl Zeiss, Inc.). Флуоресцентный микроскоп оснащали цифровой фотокамерой (DXM 1200F; Nikon), и объективами как 20×NA 0,50, так и 40×NA 0,75 (Plan Fluor; Nikon). Конфокальный микроскоп оснащали лазерным сканирующим модулем LSM 510 (три лазера: Ar 488, HeNe 543 и HeNe 633). Записи данных получали на тканях после фиксирования с использованием программного обеспечения для сбора данных (NIS-Elements, F3 [Nikon] или LSM [Carl Zeiss, Inc.]). Для количественного определения интенсивности окрашивания общее клеточное и фоновое окрашивание измеряли с использованием программного обеспечения ImageJ software (NIH), и рассчитывали интенсивность специфического окрашивания, как описано ранее (Burgess et al, 2010). Изображения обрезали, объединяли и оптимизировали с использованием Photoshop CS6 13.0 (Adobe), и упорядочивали с использованием Illustrator CS5 15.1 (Adobe).
Залитые парафином срезы CP человека. Посмертные срезы препаратов головного мозга человека, молодых и пожилых индивидуумов без заболеваний ЦНС, а также пациентов с AD, получали из банка мозга в Оксфорде, Oxford Brain Bank (ранее известного как коллекция образцов мозга Томаса Виллиса в Оксфорде, Thomas Willis Oxford Brain Collection (TWOBC)) с соответствующего согласия и одобрения от комитета по этике (TW220). Эксперименты с использованием этих срезов были одобрены этическим комитетом научного института Вейцмана.
Проточная цитометрия, получение проб и анализ. Мышей транскардиально перфузировали PBS и ткани обрабатывали, как описано ранее (Baruch et al, 2013). Препараты мозга рассекали и различные участки головного мозга извлекали под препаровальной лупой (Stemi DV4; Zeiss) в PBS, и ткани диссоциировали с помощью диссоциатора gentleMACS™ (Miltenyi Biotec). Ткани хороидного сплетения отделяли от бокового, третьего и четвертого желудочков головного мозга, инкубировали при 37°С в течение 45 мин. в PBS (с Са2+/Mg2+), содержащем 400 ЕД/мл коллагеназы IV типа (Worthington Biochemical Corporation), и затем вручную гомогенизировали пипетированием. Селезенки разминали поршнем шприца и обрабатывали лизирующим буфером ACK (хлорид аммония, калий) для удаления эритроцитов. Во всех случаях пробы окрашивали в соответствии с протоколами производителей. Все пробы фильтровали через 70 мкм нейлоновую сетку и блокировали анти-Fc CD16/32 (1:100; BD Biosciences). Для внутриклеточного окрашивания IFN-γ, клетки инкубировали с пара-метоксиамфетамином (10 нг/мл; Sigma-Aldrich) и индомицином (250 нг/мл; Sigma-Aldrich) в течение 6 ч, и добавляли Brefeldin-A (10 мкг/мл; Sigma-Aldrich) в течение последних 4 часов. Внутриклеточное мечение цитокинов делали с использованием набора BD Cytofix/Cytoperm™ Plus fixation/permeabilization kit (номер по каталогу 555028). Для окрашивания Treg использовали набор для окрашивания eBioscience FoxP3 staining buffer set (номер по каталогу 00-5523-00). Следующие моноклональные антитела, меченные флуорохромом, приобретали у BD Pharmingen, BioLegend, R&D Systems, или eBiosciences, и использовали в соответствии с протоколами изготовителей: анти-CD4, конъюгированные с РЕ или Alexa Fluor 450; анти-CD25, конъюгированные с РЕ; анти-CD45, конъюгированные с PerCP-Су5.5; анти-TCRβ, конъюгированные с FITC; анти-IFN-γ, конъюгированные с АРС; анти-FoxPS, конъюгированные с АРС; анти-CD45, конъюгированные с Brilliant-violet. Клетки анализировали на цитометре LSRII (BD Biosciences) с использованием программного обеспечения FlowJo. В каждом эксперименте релевантные группы отрицательных контролей, положительных контролей и одинарные окрашенные пробы для каждой ткани использовали для идентификации представляющих интерес популяций и для исключения других популяций.
Получение химер ВМ. Химеры ВМ (костного мозга) получали, как описано ранее (Shechter et al, 2009; Shechter et al, 2013). Вкратце, подобранных по полу мышей-реципиентов подвергали летальному облучению всего тела (950 рад), при экранировании головы (Shechter et al, 2009). Затем мышам внутривенно инъецировали 5×106 клеток ВМ от доноров CX3CR1GFP/+. Мышей оставляли на 8-10 недель после трансплантации ВМ для обеспечения восстановления гематопоэтической линии до их использования в экспериментах. Процент химеризма определяли в FACS анализе (пер., анализе диаграмм рассеяния возбужденной флуоресценции сортированных клеток) проб крови в соответствии с процентным содержанием экспрессирующих GFP клеток из циркулирующих моноцитов (CD11b+). В этой модели с экранированием головы достигали в среднем 60% химеризма и подтвердили, что инфильтрирующие (проникающие в) ЦНС GFP+миелоидные клетки являются CD45high/CD11bhigh, представляющие макрофаги моноцитарного происхождения, а не микроглии (Shechter et al, 2013).
Водный лабиринт Морриса. Мышам предоставляли три испытания в день в течение 4 дней подряд, чтобы они научились находить скрытую платформу, расположенную на 1,5 см ниже поверхности воды в бассейне (1,1 м в диаметре). Температуру воды поддерживали между 21-22°С. Воду делали непрозрачной с помощью сухого молока. В помещении для испытаний только отдаленные визуальные формы и признаки объектов были доступны для мышей, чтобы помочь при обнаружении местонахождения затопленной платформы. Латентность спасения, т.е. время, необходимое чтобы найти и подняться на платформу, регистрировали в течение периода длительностью до 60 секунд. Каждой мыши позволяли оставаться на платформе в течение 15 секунд, а затем удаляли из лабиринта в ее собственную клетку. Если мышь не находила платформу в течение 60 секунд, ее вручную помещали на платформу и возвращали в ее собственную клетку через 15 секунд. Интервал между испытаниями для каждой мыши составлял 10 минут. На 5-й день платформу убирали и мышам давали единственное испытание продолжительностью 60 секунд без доступного избегания. На 6-й и 7-й день платформу помещали в квадрант, находящийся напротив исходного тренировочного квадранта, и мышь переучивалась в течение трех сеансов каждый день. Данные регистрировали с помощью автоматизированной системы слежения EthoVision V7.1 (Noldus Information Technology). Статистический анализ выполняли с использованием дисперсионного анализа ANOVA и методом Бонферрони для апостериорных (post hoc) сравнений. Все испытания MWM проводили с 10 часов утра до 5 часов вечера во время фазы выключенного света.
Радиальный водный рукавный лабиринт. Радиальный водный рукавный лабиринт (RAWM) использовали для испытания способности к пространственному обучению/памяти, как это было ранее подробно описано (Alamed et al, 2006). Вкратце, шесть вставок из нержавеющей стали были помещены в бак, образуя шесть водных рукавов, расходящихся от открытого центрального участка. Платформу для побега располагали в конце одного из рукавов (целевого рукава) на 1,5 см ниже поверхности воды в бассейне (1,1 м в диаметре). Температуру воды поддерживали между 21-22°C. Воду делали непрозрачной с помощью сухого молока. В помещении для испытаний только отдаленные визуальные формы и признаки объектов были доступны для мышей, чтобы помочь при обнаружении местонахождения затопленной платформы. Местоположение целевого рукава оставалось неизменным для данной мыши. В 1 день мышей тренировали в течение 15 испытаний (разнесенных в течение 3 часов), с испытаниями, чередующимися между видимой и скрытой платформой, и последние 4 испытания только со скрытой платформой. На 2-й день мышей тренировали в течение 15 испытаний со скрытой платформой. Попадание в неправильный рукав или неспособность выбрать рукав в течение 15 секунд засчитывалось как ошибка. Пространственное обучение/память измеряли подсчетом количества ошибок попадания в рукав или латентности спасения мыши в каждом испытании. Данные тренировок анализировали в виде средних погрешностей или латентности спасения для блоков данных тренировок трех последовательных испытаний.
Введение GA. Каждой мыши подкожно (s.c.) инъецировали суммарную дозу 100 мкг GA (серия номер Р53640; Teva Pharmaceutical Industries, Petah Tiqva, Israel), растворенного в 200 мкл PBS. Мышей инъецировали как в соответствии с еженедельной схемой лечения GA (Butovsky et al, 2006), так и ежедневным введением GA (фиг. 8 и фиг. 16). Мышей умервщляли как через 1 неделю после последней инъекции GA, так и через 1 месяц после лечения, как указано для каждого эксперимента.
Условия при выполнении абляции Treg. Дифтерийный токсин (DTx; 8 нг/г массы тела; Sigma) инъецировали интраперитонеально (i.p.) ежедневно в течение 4 дней подряд мышам Foxp3.LuciDTR (Suffner et al, 2010). Эффективность DTx подтверждали анализом методом проточной цитометрии иммунных клеток в крови и селезенке, добиваясь почти полного (>99%) истощения FoxP3+ CD4+ Treg-клеток, экспрессирующих GFP (фиг. 4).
Ингибирование Р300. Ингибирование р300 у мышей выполняли аналогично описанному выше (Liu et al, 2013). p300i (C646; Tocris Bioscience) растворяли в DMSO и инъецировали интраперитонеально (i.p.) ежедневно (8,9 мг кг-1 день-1, i.p.) в течение 1 недели. Мышам, леченным носителем, аналогично инъецировали DMSO.
Лечение ATRA. Введение полностью транс-ретиноевой кислоты (ATRA) мышам выполняли аналогично описанному ранее (Walsh et al, 2014). ATRA (Sigma) растворяли в DMSO и инъецировали интраперитонеально (8 мг кг-1 день-1) через день в течение 1 недели. Мышам, леченным носителем, аналогично инъецировали DMSO.
Выделение и количественное определение растворимого белка Аβ (sAβ). Гомогенизацию тканей и экстракцию белка sAβ выполняли как описано ранее (Schmidt et al, 2005). Вкратце, церебральную мозговую паренхиму отсекали и быстро замораживали и хранили при -80°С до гомогенизации. Белки последовательно экстрагировали из проб для получения отдельных фракций, содержащих белки, отличающиеся растворимостью. Пробы гомогенизировали в 10 объемах ледяного буфера для гомогенизации тканей, содержащего 250 мМ сахарозы, 20 мМ Tris base (трис-основания), 1 мМ этилендиаминтетрауксусной кислоты (EDTA) и 1 мМ этиленгликольтетрауксусной кислоты (рН 7,4) с использованием пестика из притертого стекла в гомогенизаторе Даунса. После шести ударов гомогенат смешивали 1:1 с 0,4% диэтиламином (DEA) в 100-мМ растворе NaCl перед дополнительными шестью ударами и затем центрифугировали при 135000g при 4°С в течение 45 мин. Супернатант (фракцию, растворимую в DEA, содержащую внеклеточные и цитозольные белки) собирали и нейтрализовали 10% 0,5М Tris-HCl (рН 6,8). Каждый в отдельности Аβ1-40 и Aβ1-42 из растворимой фракции измеряли твердофазным иммуноферментным анализом (ELISA) с использованием коммерчески доступных наборов (Biolegend; #SIG-38954 и #SIG-38956, соответственно) в соответствии с инструкциями производителя.
Количественное определение Аβ-бляшек (Аβ-отложений). Из каждой пробы головного мозга собирали 6 мкм коронарные срезы, и иммуноокрашивали восемь срезов на мышь, из четырех различных предварительно установленных глубин по всей представляющей интерес области (зубчатой извилины или коры головного мозга). Сегментацию на основе гистограмм положительно окрашенных пикселей осуществляли с использованием программного обеспечения Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA). Алгоритм сегментации был вручную применен к каждому изображению, в области зубчатой извилины или в корктикальном слое V, и определен процент площади, занимаемой общим иммунноокрашиванием на Аβ. Число бляшек было определено количественно в тех же самых 6 мкм коронарных срезах мозга и представлено в виде среднего числа бляшек на область головного мозга. До количественной оценки срезы были закодированы, чтобы скрыть идентичность экспериментальных групп, и объем бляшки определялся количественно исследователем вслепую относительно идентичности групп.
Статистический анализ. Специфические тесты, использованные для анализа каждого набора экспериментов, указаны в кратком описании к фигурам. Данные анализировали с использованием двустороннего критерия Стьюдента для сравнения между двумя группами, однофакторный ANOVA использовали для сравнения нескольких групп, с последующим использованием критерия Ньюмена-Кейлса (Newman-Keuls) для апостериорной (post hoc) процедуры попарного сравнения групп, после того как нулевая гипотеза была отвергнута (Р<0,05). Данные, полученные из поведенческих тестов, анализировали с использованием двухфакторного дисперсионного анализа с повторениями ANOVA, и процедуру Bonferroni для апостериорных (post hoc) сравнений использовали для последующего попарного сравнения. Размеры выборки были выбраны с надлежащей статистической мощностью на основе публикаций и прошлого опыта, и мыши были распределены в экспериментальные группы в зависимости от возраста, пола и генотипа. Исследователи работали с обезличенной идентичностью групп во время экспериментов и оценки результатов. Все критерии включения и исключения были установлены заранее в соответствии с принципами IACUC (пер., комитет по содержанию и использованию лабораторных животных). Результаты представлены в виде среднего ± s.e.m. На графиках, планки погрешностей представляют на оси Y s.e.m. Статистические вычисления выполняли с использованием программного обеспечения GraphPad Prism (GraphPad Software, San Diego, CA).
Введение. Болезнь Альцгеймера (AD) является связанным с возрастом нейродегенеративным заболеванием, отличающимся повреждением нейронов, образованием бляшек бета-амилоида (Аβ) и хроническим воспалением центральной нервной системы (ЦНС), что приводит к постепенной потере когнитивной функции и разрушению ткани головного мозга (Akiyama et al, 2000; Hardy & Selkoe, 2002). При этих условиях циркулирующие миелоидные клетки и резидентные миелоидные клетки ЦНС, микроглия, играют основные роли в смягчении нейровоспалительного ответа (Britschgi & Wyss-Coray, 2007; Cameron & Landreth, 2010; Lai & McLaurin, 2012). В частности, тогда как микроглии, в конечном счете, не в состоянии очистить отложения Аβ, инфильтрирующие ЦНС макрофаги моноцитарного происхождения (mo-МФ) играют существенную роль в ограничении образования бляшек Аβ и отбивают патологию, подобную AD (Butovsky et al, 2007; Koronyo-Hamaoui et al, 2009; Mildner et al, 2011; Simard et al, 2006; Town et al, 2008). Хороидное сплетение головного мозга (CP), эпителиальные слои которого образуют гематоэнцефалический барьер (BCSFB), было идентифицировано в качестве селективного шлюза для проникновения лейкоцитов в ЦНС, обеспечивая возможность рекрутирования mo-МФ и Т-клеток после повреждения нервной ткани (Kunis et al, 2013; Shechter et al, 2013). В настоящем описании изобретатели выдвигают гипотезу, что при AD, близкое к оптимальному рекрутирование разрешающих воспаление иммунноцитов в пораженную заболеванием паренхиму является последствием нарушения системного иммунитета, с вовлечением нарушения функции шлюза СР.
Пример 1. Активность шлюза хороидного сплетения (CP) на всем протяжении прогрессирования заболевания в мышиной модели с AD.
Сначала изобретатели исследовали активность CP на всем протяжении прогрессирования заболевания у трансгенных мышей 5XFAD с моделью AD (AD-Tg); у этих мышей коэкспрессируется пять мутаций, связанных с наследственной AD и развивается церебральная Ар патологии и глиоз уже с 2-х месячного возраста (Oakley et al, 2006). Изобретатели обнаружили, что на протяжении стадий прогрессирования патологии заболевания в CP мышей AD-Tg, по сравнению с контрольными мышами дикого типа (WT) в соответствующей возрастной группе, экспрессировались значимо более низкие уровни детерминант хоуминга и миграции лейкоцитов, в том числе icam1, vcam1, cxcl10 и ccl2 (фиг. 1А), которые, как показано, положительно регулируются CP (хороидным сплетением) в ответ на острое повреждение ЦНС, и необходимы для трансэпителиальной миграции лейкоцитов (Kunis et al, 2013; Shechter et al, 2013). Иммуногистохимическое окрашивание на лиганд интегрина, ICAM-1, подтвердило его пониженную экспрессию эпителием CP мышей AD-Tg (фиг. 1b). Кроме того, окрашивание на ICAM-1 в посмертных препаратах головного мозга человека показало его, связанное с возрастом, снижение в эпителии CP, в соответствии с предыдущими наблюдениями авторов изобретения (Baruch et al, 2014), и количественная оценка этого эффекта выявила дальнейшее снижение (ICAM-1) у пациентов с AD по сравнению с пожилыми индивидуумами без заболевания ЦНС (фиг. 2А). Поскольку индукция детерминант миграции лейкоцитов хороидным сплетением (CP) зависит от передачи сигнала интерфероном (IFN)-γ в эпителии (Kunis et al, 2013), затем изобретатели проанализировали, могут ли наблюдаемые эффекты отражать потерю доступности IFN-γ в СР. Исследование CP мышей 5XFAD AD-Tg (пер., мышей AD-Tg линии 5XFAD) с использованием проточной цитометрии для анализа внутриклеточного окрашивания, обнаружило значимо меньшее число IFN-γ-продуцирующих клеток в этом компартменте (фиг. 2 В), и анализ количественной ПЦР в реальном времени (RT-qPCR) подтвердил более низкие уровни экспрессии мРНК ifn-γ в CP мышей AD-Tg по сравнению с контрольными животными дикого типа WT в соответствующей возрастной группе (фиг. 2С).
Пример 2. Функциональные взаимоотношения между системной иммуносупрессией, опосредованной Treg, активностью шлюза CP и патологией AD.
Регуляторные Т-клетки (Tregs) играют ключевую роль в подавлении системных эффекторов иммунных ответов (Sakaguchi et al, 2008). Изобретатели представили, что опосредованная Treg системная иммуносупрессия, влияет на доступность IFN-γ в CP, и следовательно, сосредоточились на вовлечении Tregs в патологию AD. В соответствии с предыдущими публикациями о повышенных уровнях Treg и супрессивной активности у пациентов с AD (Rosenkranz et al, 2007; Saresella et al, 2010; Torres et al, 2013), оценка частоты Foxp3+ Treg в спленоцитах мышей 5XFAD AD-Tg, по сравнению с их однопометными животными (WT) в соответствующей возрастной группе, выявила их повышенные уровни на всем протяжении прогрессирования заболевания (фиг. 3А, В). Для исследования функциональных взаимоотношений между системной иммуносупрессией, опосредованной Treg, активностью шлюза CP и патологией AD изобретатели скрещивали 5XFAD AD-Tg мышей с мышами Foxp3-рецептор дифтерийного токсина (DTR+), обеспечивая транзиторное условное истощение in vivo Foxp3+ Tregs у мышей AD-Tg/DTR+ путем введения дифтерийного токсина (DTx) (фиг. 4А). Транзиторное истощение Tregs приводит к повышенной экспрессии мРНК молекул миграции лейкоцитов хороидным сплетением (CP) мышей AD-Tg/DTR+ по сравнению с леченными DTx однопометными животными AD-Tg/DTR- (фиг. 5А). Анализ длительного эффекта транзиторного истощения Treg в паренхиме головного мозга (через 3 недели), обнаружил накопление иммунных клеток в мозге, в том числе повышенные количества миелоидных клеток CD45high/CD11bhigh, представляющих проникающие mo-МФ (Shechter et al, 2013) и CD4+ Т-клетки (фиг. 5 В). Кроме того, короткое и транзиторное истощение Tregs привело к заметному обогащению Foxp3+ Tregs среди CD4+ Т-клеток, которые накапливались в головном мозге, как определено с помощью проточной цитометрии (фиг. 5С, D). Анализ RT-qPCR гиппокампа показал повышенную экспрессию мРНК foxp3 и il10 (фиг. 5Е).
Далее изобретатели исследовали, приводило ли кратковременное истощение Tregs, которое сопровождалось накоплением иммунорегуляторных клеток в участках патологии мозга, к длительному эффекту на функционирование мозга. Изобретатели наблюдали уменьшение глиоза гиппокампа (фиг. 5F) и пониженные уровни экспрессии мРНК провоспалительных цитокинов, таких как il-12р40 и tnf-α (фиг. 5G). Кроме того, был понижен объем бляшки церебрального Аβ в зубчатой извилине гиппокампа, и коре головного мозга (5м слое), двух областях мозга, проявляющих устойчивую патологию с Аβ бляшками у мышей 5XFAD AD-Tg (Oakley et al, 2006) (фиг. 6A, В). Оценка влияния на когнитивную функцию с использованием теста водного лабиринта Морриса (MWM), показало значительное улучшение пространственного обучения и памяти у мышей AD-Tg/DTR+ после истощения Treg, по сравнению с мышами AD-Tg/DTR-, леченными DTx, в соответствующей возрастной группе, достигая функционирования сходного с таковым у мышей WT (фиг. 6С-Е). Взятые вместе, эти данные показали, что временно нарушенная Treg-опосредованная системная иммуносупрессия у мышей AD-Tg приводила к накоплению разрешающих воспаление клеток, в том числе mo-МФ и Tregs, в головном мозге, и сопровождалась разрешением воспалительного ответа, клиренсом Aβ и отмене снижения когнитивного расстройства.
Пример 3. Еженедельное введение сополимера-1 снижает системную иммуносупрессию, опосредованную Treg, улучшает активность шлюза CP (gateway activity) и смягчает патологию AD.
Для дополнительного доказательства причинной природы обратной взаимосвязи между системной иммуносупрессией, функционированием CP и патологией AD, изобретатели далее использовали иммуномодулирующее соединение, глатирамера ацетат (GA; также известное как сополимер-1 (Copolymer-1), или копаксон (Copaxone®)), который, как было обнаружено, при еженедельном режиме введения оказывает терапевтический эффект на APP/PS1 мышиной модели с AD (пер., двойной трансгенной мышиной модели болезни Альцгеймера) (Butovsky et al, 2006); этот эффект был функционально связан с рекрутированием mo-МФ в участки головного мозга с патологией заболевания (Butovsky et al, 2007). В настоящем описании изобретатели впервые исследовали, является ли CP у APP/PS1 AD-Tg мышей, аналогично наблюдениям авторов на мышах 5XFAD AD-Tg, также дефицитным в отношении уровней экспрессии IFN-γ. Изобретатели обнаружили, что у мышей APP/PS1 AD-Tg уровни IFN-γ в CP были снижены по сравнению с контрольными мышами дикого типа (WT) в соответствующей возрастной группе (фиг. 7А). Эти результаты побудили авторов изобретения проверить, может ли терапевтический эффект еженедельного введения GA на мышах APP/PS1 (Butovsky et al, 2006), быть воспроизведен на мышах 5XFAD AD-Tg, и если да, то будет ли это влиять на системные Tregs и активацию CP из-за миграции mo-МФ. Поэтому изобретатели лечили мышей 5XFAD AD-Tg по еженедельной схеме введения GA в течение 4 недель (здесь и далее, "раз в неделю-GA" ("еженедельно-GA"); схематически изображено на фиг. 8А). Изобретатели обнаружили, что мыши 5XFAD AD-Tg, леченные еженедельно-GA, показали пониженное нейровоспаление (фиг. 8B-D) и улучшенные когнитивные способности, которые сохранялись до 2 месяцев после лечения (фиг. 8E-I). Исследуя с помощью проточной цитометрии эффект еженедельного-GA на системный иммунитет и на CP, изобретатели обнаружили пониженные уровни Foxp3+ Treg в спленоцитах (фиг. 9А), и увеличение IFN-γ-продуцирующих клеток в CP леченных мышей 5XFAD AD-Tg, достигающее сходных уровней, которые наблюдали в контролях WT (фиг. 9В). Повышенный уровень IFN-γ-экспрессирующих клеток в CP у мышей, леченных ежедневно-GA, сопровождалось положительной регуляцией (upregulated) экспрессии молекул миграции лейкоцитов в эпителии (фиг. 9С).
Для обнаружения инфильтрирующих mo-МФ, которые проникают в ЦНС, изобретатели использовали 5XFAD AD-Tg/CX3CR1GFP/+ химерных мышей, полученных введением клеток костного мозга (ВМ), (полученных с помощью экранирования головы), что обеспечивает визуализацию циркулирующих миелоидных клеток (меченных зеленым флуоресцентным белком (GFP)+) (Shechter et al, 2009; Shechter et al, 2013). Авторы обнаружили повышенную миграционную способность (хоуминг) GFP+mo-МФ в CP и к прилегающим желудочковым пространствам после лечения еженедельно-GA, по сравнению с контролями AD-Tg/CX3CR1GFP/+, леченными носителем (фиг. 9D-E). Иммуногистохимия паренхимы мозга выявила наличие накопления GFP+mo-МФ в участках образования церебральных бляшек (фиг. 9F), и количественное определение инфильтрирующих миелоидных клеток методом проточной цитометрии гиппокампа нехимерных мышей AD-Tg, показало повышенные количества клеток, экспрессирующих CD11bhighCD45high (фиг. 9G, Н). Вместе взятые, эти результаты обосновали функциональную связь между рекрутированием mo-МФ в участки патологии AD, снижением уровней системных Treg и IFN-γ-зависимой активации СР.
Пример 4. Кратковременное прямое нарушение активности Treg, улучшает активность шлюза CP и смягчает патологию AD.
4.1 Нарушение активности Treg с использованием малой молекулы ингибитора гистонацетилтрансферазы. Приведенные выше результаты, которые дают основание предположить, что опосредованная Treg системная иммуносупрессия, препятствует способности бороться с патологией AD, напоминают функцию, приписываемую Tregs в иммунотерапии рака, при которой эти клетки препятствуют способности иммунной системы установить эффективный противоопухолевый ответ (Bos & Rudensky, 2012; Nishikawa & Sakaguchi, 2010). Следовательно, изобретатели считали, что лечение, которое непосредственно затрагивает активность Foxp3+ Treg клеток, может быть полезным при AD. Изобретатели тестировали p300i (С646 (Bowers et al, 2010)), неспецифический ингибитор р300 гистонацетилтрансферазы, который регулирует функцию Treg (Liu et al, 2013); было показано, что этот ингибитор влияет на супрессивную активность Treg при сохранении ответов защитных эффекторных Т-клеток неповрежденными (Liu et al, 2013). Изобретатели обнаружили, что мыши, леченные p300i, по сравнению с контрольными, леченными носителем (DMSO), показали повышенные уровни системных IFN-γ-экспрессирующих клеток в селезенке (Fig. 10А), а также в CP (Fig. 10В). Далее авторы лечили мышей AD-Tg как p300i, так и носителем в течение 1 недели, и через 3 недели исследовали у них объем церебральных Аβ-бляшек. Иммуногистохимический анализ выявил значимое снижение нагрузки Аβ-бляшек в мозге AD-Tg мышей, леченных p300i (Fig. 10С-Е). Авторы также протестировали, действительно ли эффект на патологию в виде бляшек после одного курса лечения будет длиться дольше 3 недель, и если да, то дополнительные курсы лечения вносили бы вклад в длительный эффект. Поэтому изобретатели сравнивали мышей AD-Tg, которые получили единственный курс лечения p300i и были обследованы через 2 месяца, с мышами соответствующей возрастной группы, которые получали два курса лечения во время этого периода, с 1-месячным интервалом между ними (схематически показано на фиг. 10F). Авторы обнаружили, что такое снижение нагрузки бляшек в мозге было очевидным даже через два месяца после единственного курса лечения, но было сильнее у мышей, которые получали два курса лечения с 1-месячным интервалом между ними (фиг. 10G). Поскольку нарушенная синаптическая пластичность и память при AD связана с повышенными уровнями в мозге растворимых AB1-40/Aβ1-42 (sAβ) (Shankar et al, 2008), авторы также измерили уровни sAβ после единственного или повторных курсов лечения p300i. Изобретатели снова обнаружили, что как один, так и два курса (с интервалом 1 месяц между ними) были эффективны в снижении церебрального sAβ, однако этот эффект был сильнее после повторных курсов по отношению к эффекту на sAβ1-42 (фиг. 10Н). Эти результаты показали, что тогда как единственный кратковременный курс лечения является эффективным, повторные курсы лечения были бы предпочтительнее для поддержания длительного терапевтического эффекта, сходного с наблюдениями авторов после еженедельного лечения GA.
4.2 Нарушение активности Treg с использованием анти-PD1 антитела. В 10-месячном возрасте, трансгенным (Tg) мышам линии 5XFAD с болезнью Альцгеймера (AD) инъецировали внутрибрюшинно (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и контрольные IgG антитела (IgG2a; #ВЕ0089; Bioxcell Lifesciences Pvt. LTD.), в 1-й и 4-й день эксперимента, и исследовали через 3 недели (схематически изображено на фиг. 11А) их когнитивную способность, оцененную тестом на пространственное обучение и память на установке радиальный рукавный водный лабиринт (RAWM), как было подробно описано ранее (Alamed et al, 2006). Кратко, в 1-й день теста на установке RAWM, мышей обучали в течение 15 испытаний (разнесенных в течение 3 часов), с испытаниями, чередующимися между видимой и скрытой платформой, и последние 4 испытания со скрытой платформой только. На 2-й день мышей обучали в течение 15 испытаний со скрытой платформой. На 2-й день мышей тренировали в течение 15 испытаний со скрытой платформой. Попадание в неправильный рукав или неспособность выбрать рукав в течение 15 секунд засчитывалось как ошибка. Пространственное обучение и память измеряли подсчетом количества ошибок попадания в рукав или латентности спасения мыши в каждом испытании. Нелеченные мыши WT и AD-Tg соответствующей возрастной группы использовали в качестве контролей. Изобретатели обнаружили, что мыши 5XFAD AD-Tg, леченные в течение одного сеанса лечения, n, который включал две инъекции анти-PD1 (в 1-й и 4-й день), показали, как оценено через 3 недели после лечения, существенное улучшение пространственных когнитивных способностей в RAWM (фиг. 11В).
Далее изобретатели исследовали, был ли эффект на патологию заболевания связан со снижением системной иммуносупрессии. Авторы повторили описанный выше эксперимент, на этот раз исследуя мышей в конце сеанса лечения (7-й день эксперимента; схематически изображено на фиг. 12А). Изобретатели наблюдали, что в этот момент времени ослабление системной иммуносупрессии у леченных PD-1 мышей AD-Tg, сопровождался системным эффектом повышения IFN-γ продуцирующих CD4 спленоцитов (фиг 12 В), что коррелировало с локальным эффектом в CP повышения уровней мРНК IFN-γ (фиг. 13А), и повышением экспрессии молекул миграции лейкоцитов в CP, хемокинов CCL2 и CXCL10 (фиг. 13В). Эти данные показали, что короткий сеанс лечения с помощью анти-PD-1 мышей AD-Tg был связан, как ожидалось, с системным ответом, ослабляющим иммуносупрессию, опосредованную Treg (Naidoo et al, 2014), и активацией активности шлюза CP для миграции лейкоцитов в ЦНС.
В заключение, изобретатели исследовали влияние на патологию заболевания у мышей AD-Tg и был ли дополнительный сеанс лечения полезным в своем влиянии на патологию. С этой целью 10-месячные мыши AD-Tg получали как 1 сеанс лечения анти-PD-1, как описано выше, так и дополнительное лечение с интервалом в 3 недели. Контрольные группы лечили как с помощью IgG, так оставляли без лечения, и все группы мышей анализировали через 3 недели на их когнитивную способность (схематически изображено на фиг. 14А). Изобретатели обнаружили, что у мышей AD-Tg получивших 1 сеанс лечения анти-PD-1 ("AD-Tg+PD-1 Х1") и были обследованных через 2 месяца, было обнаружено существенное когнитивное улучшение по сравнению с мышами AD-Tg, леченными IgG и нелеченными, эффект был менее устойчивым, чем когда у тех же самых мышей оценивали когнитивные способности месяцем ранее. В противоположность этому, мыши AD-Tg, которые получили второй сеанс лечения анти-PD-1 ("AD-Tg+PD-1 Х2") показали значительно лучшие способности к пространственному обучению и (пространственной) памяти в RAWM по сравнению с AD-Tg, которые получили один сеанс, а также по сравнению с леченными IgG или нелеченными мышами AD-Tg (фиг. 14 В). Эти результаты показали, что для поддержания длительного терапевтического эффекта необходимы повторные сеансы.
4.3 Нарушение активности Treg с использованием комбинации анти-PD1 антитела и анти-CTLA4 антитела.
В 10-ти месячном возрасте трансгенным (Tg) мышам 5XFAD с болезнью Альцгеймера (AD) инъецировали интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и 250 мкг анти-CTLA4 (InVivoMAb anti-mCD152; #ВЕ0131; Bioxcell Lifesciences Pvt. LTD.), или контрольные IgG (IgG2a, #BE0089 или поликлональные IgG сирийского хомяка, #BE0087; Bioxcell Lifesciences Pvt. LTD.) антитела в день 1-й и день 4-й эксперимента, и через 3 недели исследовали их когнитивные способности тестом на пространственное обучение и память на установке рукавный (с закрытыми рукавами) водный лабиринт (RAWM), как описано выше.
Некоторые мыши получали дополнительный сеанс лечения с интервалом между сеансами 3 недели. Контрольные группы лечили как IgG, так и оставляли без лечения, и через 3 недели все группы мышей анализировали на их когнитивные способности.
Предполагается, что мыши, леченные комбинацией антител обнаружат значительное когнитивное улучшение по сравнению с мышами AD-Tg, леченными IgG или без лечения, а также значительное снижение нагрузки мозга бляшками.
Пример 5. Увеличение активности Treg оказывает отрицательное действие на патологию AD.
Для обоснования отрицательной роли опосредованной Treg системной иммуносупрессии при AD, изобретатели исследовали могут ли увеличивающиеся уровни системных Treg оказывать обратное, неблагоприятное действие на патологию AD. Для проверки этого, изобретатели усилили подавляющую функцию Treg у мышей AD-Tg введением полностью транс-ретиноевой кислоты (ATRA), которая индуцирует дифференцировку Treg (Mucida et al, 2007), стабилизирует фенотип Treg (Zhou et al, 2010) и делает Tregs более подавляющими (Zhou et al, 2010). Изобретатели использовали мышей 5XFAD AD-Tg на относительно ранних стадиях прогрессирования заболевания, и лечили их как ATRA, так и носителем (DMSO). Леченные ATRA мыши AD-Tg показали значимо более высокую частоту встречаемости Foxp3+CD25+ Tregs в спленоцитах (фиг. 15А, В). Исследование мышей через 3 недели после последней инъекции ATRA, выявило относительно высокий объем бляшек церебрального Aβ и глиоз (приблизительно 2-3-кратное увеличение; фиг. 15С-Е), и оценка sAβ выявила повышенные уровни церебрального sAβ1-40 и SAβ1-42 после увеличения системных Tregs (фиг. 15F-G). Оценка когнитивной способности с использованием RAWM, показала усугубление дефицита пространственной памяти у мышей AD-Tg, леченных ATRA по сравнению с мышами AD-Tg, леченных носителем (фиг. 15Н).
Учитывая полученные авторами в настоящем изобретении результаты о негативном влиянии системных Tregs на патологию AD, вместе с известным фактом, что ежедневное введение GA индуцирует Tregs и используется в клинике для лечения рассеянного (множественного) склероза (MS) (Haas et al, 2009; Hong et al, 2005; Weber et al, 2007), изобретатели анализировали может ли GA при ежедневной схеме приема (в течение 1 месяца), в отличие от еженедельной схемы приема GA, иметь негативное влияние на патологию заболевания у мышей AD-Tg. Изобретатели сравнили влияние ежедневного введения GA против еженедельного введения GA на мышах 5XFAD AD-Tg (схематически изображено на фиг. 16А). Оценка когнитивных способностей тестом на установке RAWM выявила, что в отличие от благоприятного действия еженедельной обработки GA, либо не наблюдали благоприятное действие на пространственную память, либо наблюдали тенденцию к ухудшению эффекта у мышей AD-Tg, которые получали GA ежедневно (фиг. 16В). Дополнительно, в отличие от сильного эффекта при еженедельном введении GA на клиренс бляшек, ежедневная обработка мышей AD-Tg не показала какого бы то ни было положительного эффекта, или показала умеренное отрицательное воздействие на нагрузку бляшек (фиг. 16C-F). Эти результаты делают особое ударение на то, что MS и AD, две патологии ЦНС, связанные с нейровоспалением, могли бы быть противоположным образом и заметно затронуты одним и тем же ммуномодулирующим лечением с помощью ежедневного (введения) GA (Schwartz & Baruch, 2014а).
Пример 6. Прямое нарушение активности Treg улучшает активность шлюза (gateway) CP и предотвращает или смягчает патологию PTSD.
Чрезвычайно стрессовые состояния или хронический стресс могут привести к посттравматическому стрессу (PTSD) и депрессии. Изобретатели ранее предположили, что активность шлюза CP может быть критической для преодоления эмоционального стресса, и в случае неоптимального функционирования CP, психический травматический эпизод может привести к PTSD (Schwartz & Baruch, 2012). Дополнительно изобретатели гипотетически предположили, что своевременное системное вмешательство после травмы, которое сможет помочь модифицировать ответ CP, может предотвратить развитие хронических состояний PTSD. Выводы авторов изобретения о том, что опосредованное Treg кратковременное истощение системной иммуносупрессии, оказывает продолжительный эффект на патологию головного мозга, позволяют предположить, что это вмешательство, сразу же после травматического события, позволило бы предотвратить развитие PTSD.
Для проверки рабочей гипотезы изобретелей, что хороидное сплетение (CP) участвует в преодолении травматического стресса и что, возможно, дисфункция в случаях, когда травматический стресс приводит к развитию PTSD, изобретатели адаптировали физиологическую PTSD-подобную модель животного, в которой мыши проявляют чрезмерно бодрствующее поведение, ослабленное внимание, повышенную оценку риска и плохой сон (Lebow et al, 2012). В этой экспериментальной модели индукции PTSD, мышей в течение 10 дней приучали менять день на ночь, применяя два поражения электрическим током (травму и триггер), называемую "индукцией PTSD", и проводили оценку в различные моменты времени после травмы. После травматического события мышам инъецировали указанное соединение, которое временно ослабляет периферическую иммуносупрессию. Мыши получают лечение в соответствии с одним или несколькими следующими режимами:
- Мышам инъецируют интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и контрольные IgG (IgG2a; #ВЕ0089; Bioxcell Lifesciences Pvt. LTD.) антитела, на день 1-й и день 4-й после травматического события, и обследуют после дополнительного двухнедельного интервала между курсами;
- Мышам инъецируют интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и 250 мкг анти-CTLA4 (InVivoMAb anti-mCD152; #ВЕ0131; Bioxcell Lifesciences Pvt. LTD.), или контрольные IgG (IgG2a, #BE0089 или поликлональные сирийского хомяка IgG, #ВЕ0087; Bioxcell Lifesciences Pvt. LTD.) антитела, на день 1-й и день 4-й эксперимента, и обследуют после дополнительного двухнедельного интервала между курсами;
- Мышам инъецируют интраперитонеально (i.p.) ежедневно-GA, как описано выше, после травматического события и обследуют после дополнительного двухнедельного интервала между курсами;
- Мыши получают лечение p300i или носителем в течение курса (продолжительностью) 1 неделя после травматического события и мышей обследуют через 3 недели, как описано выше.
Некоторые мыши получают дополнительный сеанс лечения с подходящим интервалом между сеансами.
Предполагается, что мыши, которые получают лечение не демонстрируют тревожное поведение, связанное с PTSD в этой экспериментальной модели, в виде оцененного времени, затраченного на исследование и оценку риска в лабиринте темнота/свет или в других поведенческих задачах, описанных (Lebow et al, 2012).
Пример 7. Транзиторное (временное) снижение системной иммуносупрессии смягчает патологию болезни Паркинсона.
Трансгенных (Tg) мышей с болезнью Паркинсона (PD) использовали в этом эксперименте. Мыши получали лечение на прогрессирующих стадиях заболевания в соответствии с одним или несколькими следующими режимами:
- Мышам инъецируют интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и контрольные IgG (IgG2a; #ВЕ0089; Bioxcell Lifesciences Pvt. LTD.) антитела, на день 1-й и день 4-й после травматического события, и обследуют после дополнительного двухнедельного интервала между сеансами;
- Мышам инъецируют интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и 250 мкг анти-CTLA4 (InVivoMAb anti-mCD152; #ВЕ0131; Bioxcell Lifesciences Pvt. LTD.), или контрольные IgG (IgG2a, #BE0089 или поликлональные сирийского хомяка IgG, #ВЕ0087; Bioxcell Lifesciences Pvt. LTD.) антитела, на день 1-й и день 4-й эксперимента, и обследуют после дополнительного двухнедельного интервала между сеансами;
- Мышам инъецируют интраперитонеально (i.p.) еженедельно-GA, как описано выше, после травматического события и обследуют после дополнительного двухнедельного интервала между сеансами;
- Мыши получают лечение p300i или носителем в течение сеанса (продолжительностью) 1 неделя после травматического события и мышей обследуют через 3 недели, как описано выше.
Некоторые мыши получают дополнительный сеанс лечения с подходящим интервалом между сеансами (приблизительно от 3 недель до одного месяца).
Моторно-неврологические функции оценивали, например, в тесте «ротарод» для оценки способности мышей удерживаться на вращающемся стержне.
Предполагается, что мыши PD-Tg, которые проходили один сеанс лечения показывают значимо улучшенную двигательную активность по сравнению с мышами, которым вводили IgG или носитель в контрольной группе, или группе, не подвергавшейся лечению. Ожидается, что мыши PD-Tg, которые получают два сеанса лечения, и которые были обследованы после подходящего интервала между сеансами, покажут длительный терапевтический эффект. Для поддержания этого терапевтического эффекта мышей подвергают активному сеансу лечения с подходящим интервалом между каждым сеансом.
Пример 8. Транзиторное (временное) снижение системной иммуносупрессии смягчает патологию болезни Хантингтона.
Используемая в этих экспериментах модель, может быть тест-системой трансгенных мышей (Tg) R6/2 с болезнью Хантингтона (HD). Трансгенные мыши R6/2 сверхэкспрессируют мутантный ген хантингтина человека, который включает в себя инсерцию (вставку) множественных повторов CAG мыши на прогрессирующих стадиях заболевания. Эти мыши демонстрируют прогрессивный поведенческий и двигательный дефект, начиная уже с 5-6-недельного возраста, что приводит к преждевременной смерти в 10-13 недель. Симптомы включают в себя низкую массу тела, зажимы, тремор и конвульсии.
Мышей лечили в соответствии с одой или несколькими следующими схемами лечения в 45-дневном возрасте:
- Мышам инъецируют интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и контрольные IgG (IgG2a; #ВЕ0089; Bioxcell Lifesciences Pvt. LTD.) антитела, на день 1-й и день 4-й после травматического события, и обследуют после дополнительного двухнедельного интервала между сеансами;
- Мышам инъецируют интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и 250 мкг анти-СТ1_А4 (InVivoMAb anti-mCD152; #ВЕ0131; Bioxcell Lifesciences Pvt. LTD.), или контрольные IgG (IgG2a, #BE0089 или поликлональные сирийского хомяка IgG, #ВЕ0087; Bioxcell Lifesciences Pvt. LTD.) антитела, на день 1-й и день 4-й эксперимента, и обследуют после дополнительного двухнедельного интервала между сеансами.
- Мышам инъецируют интраперитонеально (i.p.) еженедельно-GA, как описано выше, после травматического события и обследуют после дополнительного двухнедельного интервала между сеансами;
- Мыши получают лечение p300i или носителем в течение сеанса (продолжительностью) 1 неделя после травматического события и мышей обследуют через 3 недели, как описано выше.
Некоторые мыши получают дополнительный сеанс лечения с подходящим интервалом между сеансами (приблизительно от 3 недель до одного месяца).
Моторно-неврологические функции оценивали, например, в тесте «ротарод» для оценки способности мышей удерживаться на вращающемся стержне.
Предполагается, что мыши HD-Tg, которые проходили один сеанс лечения показывают значимо улучшенную двигательную активность по сравнению с мышами, которым вводили IgG или носитель в контрольной группе, или группе, не подвергавшейся лечению. Ожидается, что мыши HD-Tg, которые получают два сеанса лечения, и которые были обследованы после подходящего интервала между сеансами, покажут длительный терапевтический эффект. Для поддержания этого терапевтического эффекта мышей подвергают активному сеансу лечения с подходящим интервалом между каждым сеансом.
Пример 9. Транзиторное (временное) снижение системной иммуносупрессии смягчает патологию бокового амиотрофического склероза.
Используемая в этих экспериментах модель, может быть трансгенными мышами, сверхэкспрессирующими дефектный человеческий мутантный аллель SOD1, содержащий ген Gly93→Ala (G93A) (B6SJL-TgN (SOD1-G93A)1Gur (здесь "мыши с ALS"). У этой модели развивается болезнь двигательного нейрона и таким образом она представляет собой общепринятую модель животного для тестирования ALS.
Мышей лечили в соответствии с одой или несколькими следующими схемами лечения в 75-дневном возрасте:
- Мышам инъецируют интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и контрольные IgG (IgG2a; #ВЕ0089; Bioxcell Lifesciences Pvt. LTD.) антитела, на день 1-й и день 4-й после травматического события, и обследуют после дополнительного двухнедельного интервала между сеансами;
- Мышам инъецируют интраперитонеально (i.p.) как 250 мкг анти-PD1 (RMP1-14; #ВЕ0146; Bioxcell Lifesciences Pvt. LTD.), так и 250 мкг анти-CTLA4 (InVivoMAb anti-mCD152; #ВЕ0131; Bioxcell Lifesciences Pvt. LTD.), или контрольные IgG (IgG2a, #BE0089 или поликлональные сирийского хомяка IgG, #ВЕ0087; Bioxcell Lifesciences Pvt. LTD.) антитела, на день 1-й и день 4-й эксперимента, и обследуют после дополнительного двухнедельного интервала между сеансами.
- Мышам инъецируют интраперитонеально (i.p.) еженедельно-GA, как описано выше, после травматического события и обследуют после дополнительного двухнедельного интервала между сеансами;
- Мыши получают лечение p300i или носителем в течение сеанса (продолжительностью) 1 неделя после травматического события и мышей обследуют через 3 недели, как описано выше.
Некоторые мыши получают дополнительный сеанс лечения с подходящим интервалом между сеансами (приблизительно от 3 недель до одного месяца).
Моторно-неврологические функции оценивали, например, в тесте «ротарод» для оценки способности мышей удерживаться на вращающемся стержне, или мышам позволяют захватывать и удерживаться на вертикальном проводе (диаметром 2 мм) с небольшой петлей на нижнем конце. Вертикальный провод позволяет мышам использовать как передние, так и задние конечности для захвата провода. Провод поддерживается в вертикально ориентированном круговом движении (радиус окружности составлял 10 см) при 24 оборотах в минуту. Время, в течение которого мышь способна висеть на проводе, записывается таймером.
Как и ожидается, мыши с ALS, получившие лечение одним сеансом лечения показывают значительно улучшенную двигательную активность, по сравнению с получившей IgG или получившей носитель контрольной группой, или группой, которая не подвергалась лечению. Как и ожидается, мыши с ALS, которые получают два сеанса лечения и обследованные после подходящего интервала между сеансами, показывают длительный терапевтический эффект. Для поддержания этого терапевтического эффекта мышей подвергают активному сеансу лечения с подходящим интервалом между каждым сеансом.
ССЫЛКИ
Akiyama Н, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer's disease. Neurobiology of aging 21: 383-421
Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D (2006) Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nature protocols 1: 1671-1679
Bai A, Lu N, Guo Y, Liu Z, Chen J, Peng Z (2009) All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. Journal of leukocyte biology 86: 959-969
Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-ltzhaki Z, Bezalel D, Wyss-Coray T, Amit I, Schwartz M (2014) Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346: 89-93
Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, Mirlas-Neisberg N, Cardon M, Vaknin I, Cahalon L, Berkutzki T, Mattson MP, Gomez-Pinilla F, Friedman N, Schwartz M (2013) CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proceedings of the National Academy of Sciences of the United States of America 110: 2264-2269
Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS (1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19: 939-945
Bos PD, Rudensky AY (2012) Treg cells in cancer: a case of multiple personality disorder. Science translational medicine 4: 164fs144
Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, Larocca C, Saldanha SA, Abagyan R, Sun Y, Meyers DJ, Marmorstein R, Mahadevan LC, Alani RM, Cole PA (2010) Virtual ligand screening of the р300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chemistry & biology 17: 471-482
Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, Larson EB (2009) Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 72: 1899-1905
Brestoff JR, Artis D (2013) Commensal bacteria at the interface of host metabolism and the immune system. Nature immunology 14: 676-684
Britschgi M, Wyss-Coray T (2007) Immune cells may fend off Alzheimer disease. Nature medicine 13: 408-409
Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, Castro A (2010) Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proceedings of the National Academy of Sciences of the United States of America 107: 12564-12569
Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M (2006) Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proceedings of the National Academy of Sciences of the United States of America 103: 11784-11789
Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M (2007) Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. The European journal of neuroscience 26: 413-416
Cameron B, Landreth GE (2010) Inflammation, microglia, and Alzheimer's disease. Neurobiology of disease 37: 503-509
Chen X, Oppenheim JJ (2011) Resolving the identity myth: key markers of functional CD4+FoxP3+ regulatory T cells. International immunopharmacology 11: 1489-1496
Colombo MP, Piconese S (2007) Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nature reviews Cancer 7: 880-887
Coyne GO, Gulley JL (2014) Adding fuel to the fire: Immunogenic intensification. Human vaccines & immunotherapeutics 10: 3306-3312
Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon S, Mendez-Huergo SP, Stupirski JC, Mazal D, Osinaga E, Toscano MA, Sundblad V, Rabinovich GA, Salatino M (2013) Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer research 73: 1107-1117
Duraiswamy J, Freeman GJ, Coukos G (2014) Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors-response. Cancer research 74: 633-634; discussion 635
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nature reviews Immunology 9: 162-174
Galvin KC, Dyck L, Marshall NA, Stefanska AM, Walsh KP, Moran B, Higgins SC, Dungan LS, Mills KH (2013) Blocking retinoic acid receptor-alpha enhances the efficacy of a dendritic cell vaccine against tumours by suppressing the induction of regulatory T cells. Cancer immunology, immunotherapy: Cll 62:1273-1282
Ghiringhelli F, Bruchard M, Chalmin F, Rebe С (2012) Production of adenosine by ectonucleotidases: a key factor in tumor immunoescape. Journal of biomedicine & biotechnology 2012: 473712
Group AR, Lyketsos CG, Breitner JC, Green RC, Martin BK, Meinert C, Piantadosi S, Sabbagh M (2007) Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68: 1800-1808
Haas J, Korporal M, Balint B, Fritzsching B, Schwarz A, Wildemann В (2009) Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4(+)CD25(+)FOXP3(+)CD31(+) T-cells in patients with multiple sclerosis. Journal of neuroimmunology 216:113-117
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356
He F, Balling R (2013) The role of regulatory T cells in neurodegenerative diseases. Wiley interdisciplinary reviews Systems biology and medicine 5: 153-180
Hirayama M, Nishikawa H, Nagata Y, Tsuji T, Kato T, Kageyama S, Ueda S, Sugiyama D, Hori S, Sakaguchi S, Ritter G, Old LJ, Gnjatic S, Shiku H (2013) Overcoming regulatory T-cell suppression by a lyophilized preparation of Streptococcus pyogenes. European journal of immunology 43: 989-1000
Hong J, Li N, Zhang X, Zheng B, Zhang JZ (2005) Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proceedings of the National Academy of Sciences of the United States of America 102: 6449-6454
Ju Y, Shang X, Liu Z, Zhang J, Li Y, Shen Y, Liu Y, Liu C, Liu B, Xu L, Wang Y, Zhang B, Zou J (2014) The Tim-3/galectin-9 pathway involves in the homeostasis of hepatic Tregs in a mouse model of concanavalin A-induced hepatitis. Molecular immunology 58: 85-91
Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Liftman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Molecular and cellular biology 20: 4106-4114
Kim JM, Rasmussen JP, Rudensky AY (2007) Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nature immunology 8: 191-197
Kim PS, Jochems C, Grenga I, Donahue RN, Tsang KY, Gulley JL, Schlom J, Farsaci В (2014) Pan-Bcl-2 inhibitor, GX15-070 (obatoclax), decreases human T regulatory lymphocytes while preserving effector T lymphocytes: a rationale for its use in combination immunotherapy. Journal of immunology 192: 2622-2633
Koronyo-Hamaoui M, Ko MK, Koronyo Y, Azoulay D, Seksenyan A, Kunis G, Pham M, Bakhsheshian J, Rogeri P, Black KL, Farkas DL, Schwartz M (2009) Attenuation of AD-like neuropathology by harnessing peripheral immune cells: local elevation of IL-10 and MMP-9. Journal of neurochemistry 111:1409-1424
Kotsakis A, Harasymczuk M, Schilling B, Georgoulias V, Argiris A, Whiteside TL (2012) Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples. Journal of immunological methods 381: 14-22
Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, Schwartz M (2013) IFN-gamma-dependent activation of the brain's choroid plexus for CNS immune surveillance and repair. Brain: a journal of neurology 136: 3427-3440
Lai AY, McLaurin J (2012) Clearance of amyloid-beta peptides by microglia and macrophages: the issue of what, when and where. Future neurology 7: 165-176
Lebow M, Neufeld-Cohen A, Kuperman Y, Tsoory M, Gil S, Chen A (2012) Susceptibility to PTSD-like behavior is mediated by corticotropin-releasing factor receptor type 2 levels in the bed nucleus of the stria terminalis. The Journal of neuroscience: the official journal of the Society for Neuroscience 32: 6906-6916
Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LC, Kapoor V, Bhatti TR, Akimova T, Singhal S, Brindle PK, Cole PA, Albelda SM, Hancock WW (2013) Inhibition of р300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nature medicine 19: 1173-1177
Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, Luong R, Rosenblum MD, Steinman L, Levitsky HI, Tse V, Levy R (2013) Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. The Journal of clinical investigation 123: 2447-2463
Michaud JP, Bellavance MA, Prefontaine P, Rivest S (2013) Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell reports 5: 646-653
Mildner A, Schlevogt B, Kierdorf K, Bottcher C, Erny D, Kummer MP, Quinn M, Bruck W, Bechmann I, Heneka MT, Priller J, Prinz M (2011) Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience 31: 11159-11171
Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317: 256-260
Naidoo J, Page DB, Wolchok JD (2014) Immune modulation for cancer therapy. British journal of cancer 111: 2214-2219
Nakatsukasa H, Tsukimoto M, Harada H, Kojima S (2011) Adenosine A2B receptor antagonist suppresses differentiation to regulatory T cells without suppressing activation of T cells. Biochemical and biophysical research communications 409: 114-119
Nishikawa H, Sakaguchi S (2010) Regulatory T cells in tumor immunity. International journal of cancer Journal international du cancer 127: 759-767
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. The Journal of neuroscience: the official journal of the Society for Neuroscience 26: 10129-10140
Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X (2015) Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends in molecular medicine 21: 24-33
Pere H, Montier Y, Bayry J, Quintin-Colonna F, Merillon N, Dransart E, Badoual C, Gey A, Ravel P, Marcheteau E, Batteux F, Sandoval F, Adotevi O, Chiu C, Garcia S, Tanchot C, Lone YC, Ferreira LC, Nelson BH, Hanahan D, Fridman WH, Johannes L, Tartour E (2011) A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood 118: 4853-4862
Qin A, Wen Z, Zhou Y, Li Y, Li Y, Luo J, Ren T, Xu L (2013) MicroRNA-126 regulates the induction and function of CD4(+) Foxp3(+) regulatory T cells through PI3K/AKT pathway. Journal of cellular and molecular medicine 17: 252-264
Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, Gasser T, Stoltze L (2007) Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. Journal of neuroimmunology 188: 117-127
Roy S, Barik S, Banerjee S, Bhuniya A, Pal S, Basu P, Biswas J, Goswami S, Chakraborty T, Bose A, Baral R (2013) Neem leaf glycoprotein overcomes indoleamine 2,3 dioxygenase mediated tolerance in dendritic cells by attenuating hyperactive regulatory T cells in cervical cancer stage IIIB patients. Human immunology 74: 1015-1023
Sakaguchi S, Yamaguchi T, Nomura T, Ono M (2008) Regulatory T cells and immune tolerance. Cell 133: 775-787
Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, Nemni R, Clerici M (2010) PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer's disease. Journal of Alzheimer's disease: JAD 21: 927-938
Schmidt SD, Nixon RA, Mathews PM (2005) ELISA method for measurement of amyloid-beta levels. Methods in molecular biology 299: 279-297
Schwartz M, Baruch K (2012) Vaccine for the mind: Immunity against self at the choroid plexus for erasing biochemical consequences of stressful episodes. Human vaccines & immunotherapeutics 8: 1465-1468
Schwartz M, Baruch K (2014a) Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: boosting autoimmunity to fight-off chronic neuroinflammation. Journal of autoimmunity 54: 8-14
Schwartz M, Baruch K (2014b) The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. The EMBO journal 33: 7-22
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nature medicine 14: 837-842
Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M (2009) Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS medicine 6: e1000113
Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim KW, Klein E, Kalchenko V, Bendel P, Lira SA, Jung S, Schwartz M (2013) Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38: 555-569
Shevchenko I, Karakhanova S, Soltek S, Link J, Bayry J, Werner J, Umansky V, Bazhin AV (2013) Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. International journal of cancer Journal international du cancer133: 98-107
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49: 489-502
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA (2013) Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. The Journal of experimental medicine 210: 1695-1710
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341: 569-573
Suffner J, Hochweller K, Kuhnle MC, Li X, Kroczek RA, Garbi N, Hammerling GJ (2010) Dendritic cells support homeostatic expansion of Foxp3+ regulatory T cells in Foxp3.LuciDTR mice. Journal of immunology 184: 1810-1820
Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, Honjo T (2011) IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. Journal of immunology 186: 2772-2779
Terme M, Colussi O, Marcheteau E, Tanchot C, Tartour E, Taieb J (2012) Modulation of immunity by antiangiogenic molecules in cancer. Clinical & developmental immunology 2012: 492920
Terme M, Tartour E, Taieb J (2013) VEGFA/VEGFR2-targeted therapies prevent the VEGFA-induced proliferation of regulatory T cells in cancer. Oncoimmunology 2: e25156
Thomas-Schoemann A, Batteux F, Mongaret C, Nicco C, Chereau C, Annereau M, Dauphin A, Goldwasser F, Weill B, Lemare F, Alexandre J (2012) Arsenic trioxide exerts antitumor activity through regulatory T cell depletion mediated by oxidative stress in a murine model of colon cancer. Journal of immunology 189: 5171-5177
Torres KC, Araujo Pereira P, Lima GS, Bozzi 1С, Rezende VB, Bicalho MA, Moraes EN, Miranda DM, Romano-Silva MA (2013) Increased frequency of T cells expressing IL-10 in Alzheimer disease but not in late-onset depression patients. Progress in neuro-psychopharmacology & biological psychiatry 47: 40-45
Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA (2008) Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nature medicine 14: 681-687
Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M, Weissman IL (2013) Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proceedings of the National Academy of Sciences of the United States of America 110: 11103-11108
Vom Berg J, Prokop S, Miller KR, Obst J, Kalin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nature medicine 18: 1812-1819
Voo KS, Bover L, Hariine ML, Vien LT, Facchinetti V, Arima K, Kwak LW, Liu YJ (2013) Antibodies targeting human OX40 expand effector T cells and block inducible and natural regulatory T cell function. Journal of immunology 191: 3641-3650
Walsh JT, Zheng J, Smirnov I, Lorenz U, Tung K, Kipnis J (2014) Regulatory T cells in central nervous system injury: a double-edged sword. Journal of immunology 193: 5013-5022
Ward FJ, Dahal LN, Wijesekera SK, Abdul-Jawad SK, Kaewarpai T, Xu H, Vickers MA, Barker RN (2013) The soluble isoform of CTLA-4 as a regulator of T-cell responses. European journal of immunology 43: 1274-1285
Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, Zhao X, Martinez AJ, Wang W, Gibney G, Kroeger J, Eysmans C, Sarnaik AA, Chen YA (2013) Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 31: 4311-4318
Weber MS, Hohlfeld R, Zamvil SS (2007) Mechanism of action of glatiramer acetate in treatment of multiple sclerosis. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics 4: 647-653
Weiskopf K, Ring AM, Schnorr PJ, Volkmer JP, Volkmer AK, Weissman IL, Garcia KC (2013) Improving macrophage responses to therapeutic antibodies by molecular engineering of SIRPalpha variants. Oncoimmunology 2: e25773
Wyss-Coray T, Rogers J (2012) Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harbor perspectives in medicine 2: a006346
Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, Pradilla G, Ford E, Wong J, Hammers HJ, Mathios D, Tyler B, Brem H, Tran PT, Pardoll D, Drake CG, Lim M (2013) Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. International journal of radiation oncology, biology, physics 86: 343-349
Zhao L, Sun L, Wang H, Ma H, Liu G, Zhao Y (2007) Changes of CD4+CD25+Foxp3+ regulatory T cells in aged Balb/c mice. Journal of leukocyte biology 81: 1386-1394
Zheng H, Fridkin M, Youdim M (2015) New approaches to treating Alzheimer's disease. Perspectives in medicinal chemistry 7: 1-8
Zhou S, Chen L, Qin J, Li R, Tao H, Zhen Z, Chen H, Chen G, Yang Y, Liu B, She Z,
Zhong C, Liang С (2013) Depletion of CD4+ CD25+ regulatory T cells promotes CCL21-mediated antitumor immunity. PloS one 8: e73952
Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, Brand D, Liu Z, Zheng SG (2010) Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. Journal of immunology 185: 2675-2679
| название | год | авторы | номер документа |
|---|---|---|---|
| СНИЖЕНИЕ СИСТЕМНЫХ УРОВНЕЙ ИЛИ АКТИВНОСТИ РЕГУЛЯТОРНЫХ Т-КЛЕТОК ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ И ПОРАЖЕНИЯ ЦНС | 2017 |
|
RU2815892C1 |
| СНИЖЕНИЕ СИСТЕМНОГО УРОВНЯ РЕГУЛЯТОРНЫХ Т-КЛЕТОК ИЛИ ИХ АКТИВНОСТИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ И ТРАМВ ЦНС | 2016 |
|
RU2741366C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ НЕВРОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2005 |
|
RU2387453C2 |
| МОДИФИЦИРОВАННЫЕ УГЛЕВОДАМИ ЧАСТИЦЫ И ПОРОШКООБРАЗНЫЕ КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ ИММУННОГО ОТВЕТА | 2016 |
|
RU2752620C2 |
| Пептидный толерогенный компаунд | 2021 |
|
RU2766340C1 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВОЛЧАНКИ | 2010 |
|
RU2607022C2 |
| АНТИ-ICOS АНТИТЕЛА | 2017 |
|
RU2764548C2 |
| Способ получения противовоспалительного препарата на основе пептидов селезенки млекопитающих, препарат, полученный данным способом, и его применение | 2021 |
|
RU2790158C1 |
| БИСПЕЦИФИЧЕСКИЕ АНТИТЕЛА К CD25 И Fc ГАММА-РЕЦЕПТОРУ ДЛЯ ЭЛИМИНАЦИИ ОПУХОЛЕСПЕЦИФИЧЕСКИХ КЛЕТОК | 2017 |
|
RU2759970C2 |
| МОДУЛЯТОРЫ АЛЬФА-СИНУКЛЕИНА | 2018 |
|
RU2797677C2 |













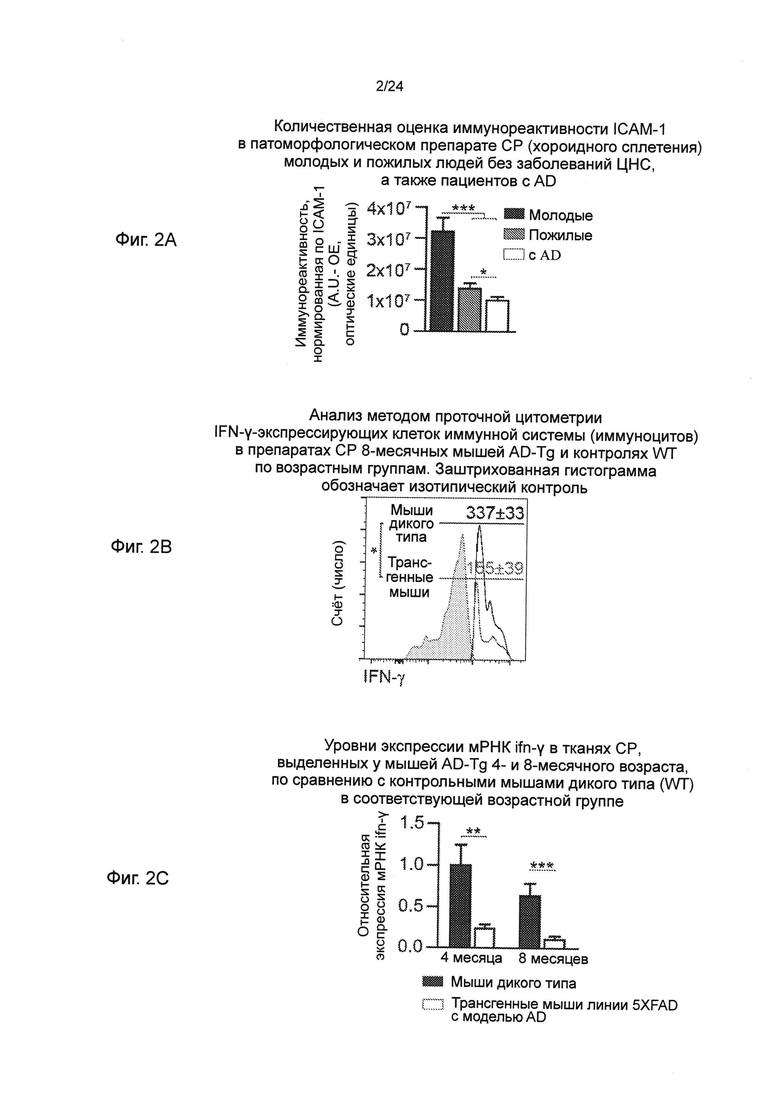

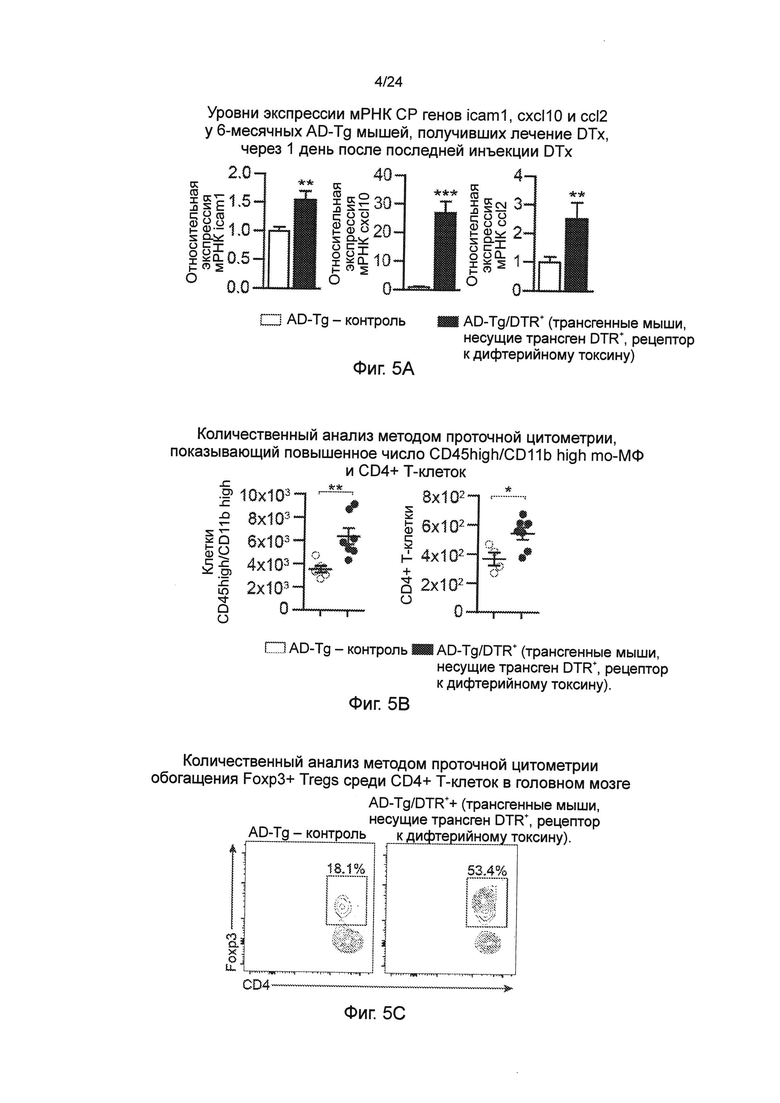



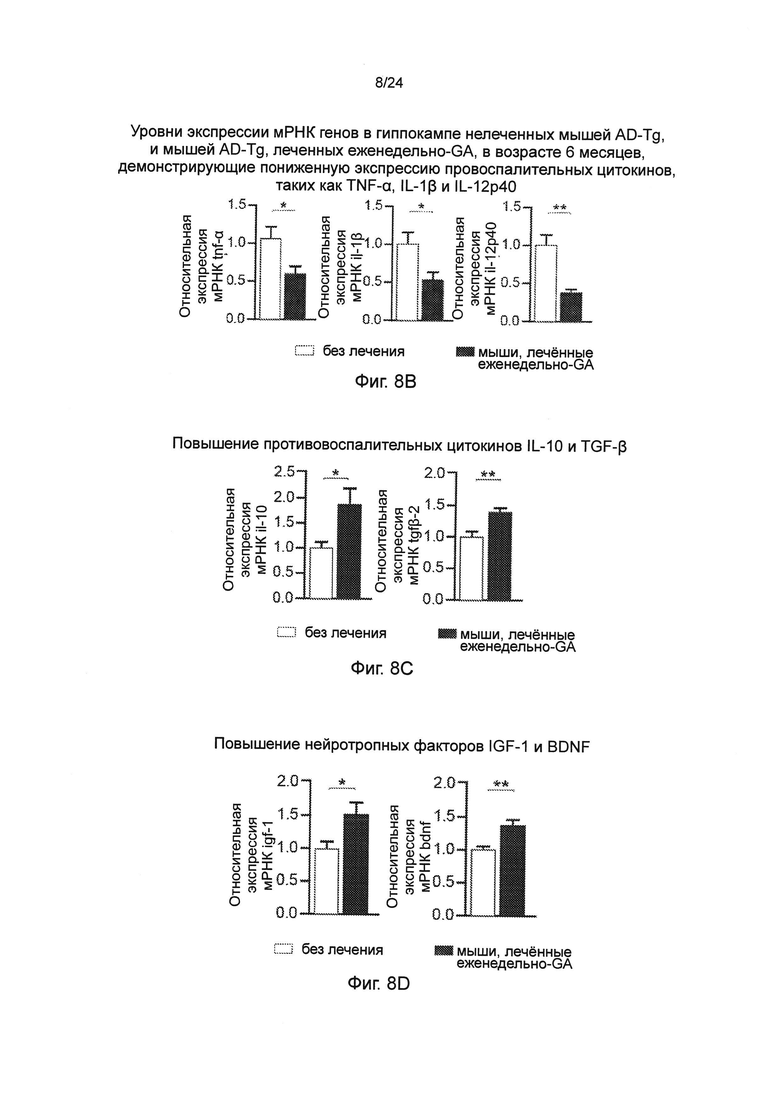
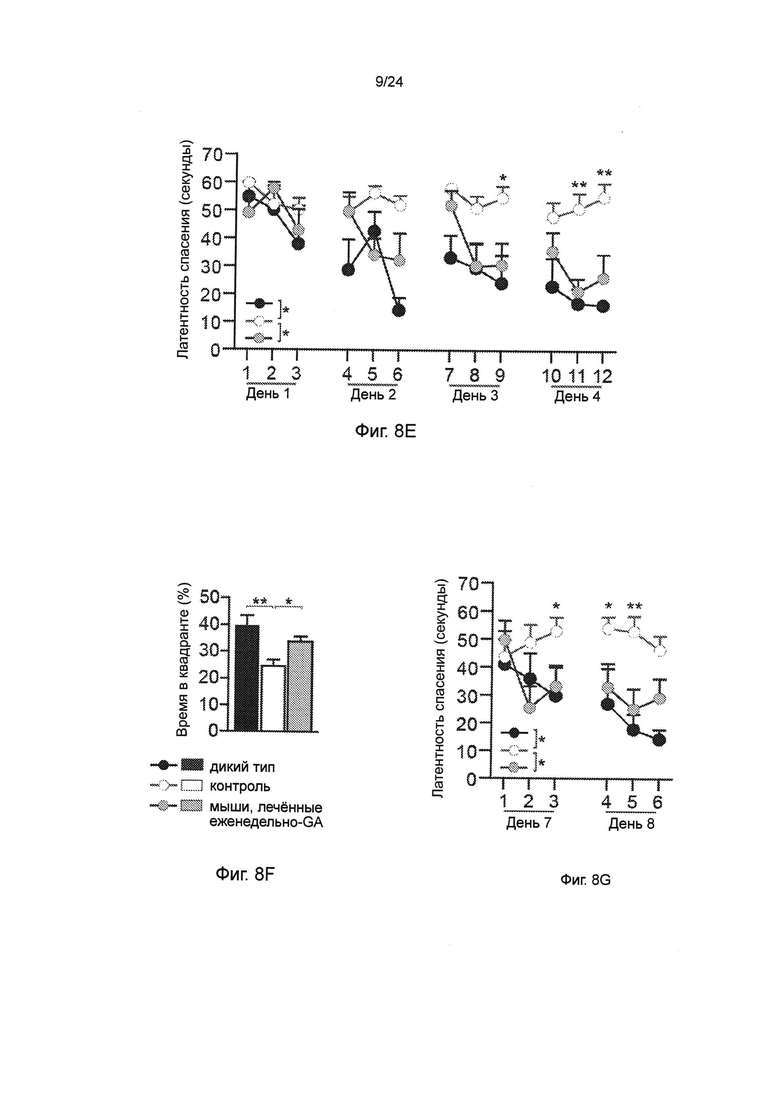
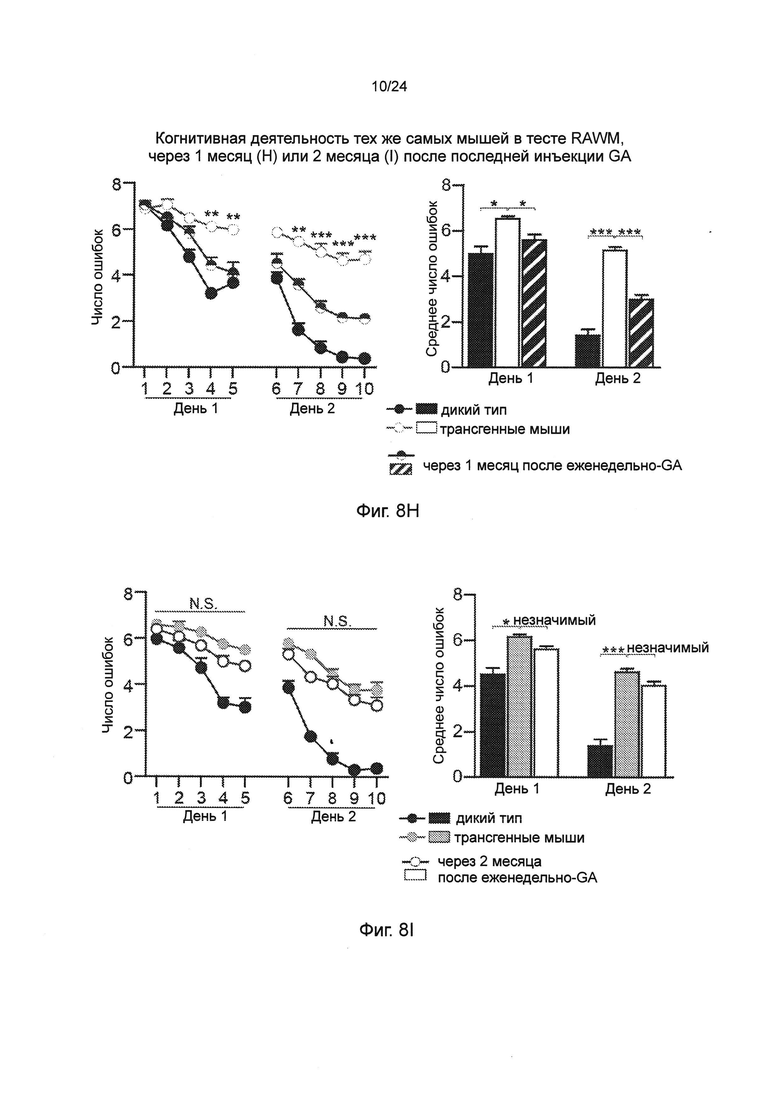
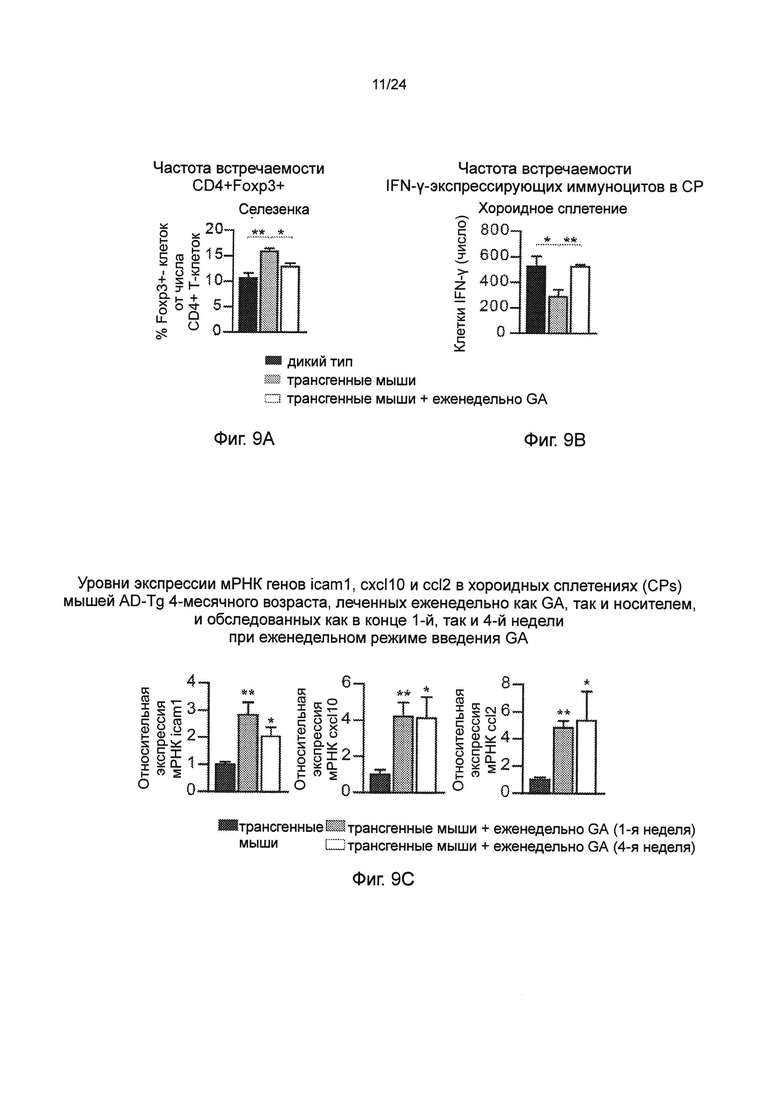
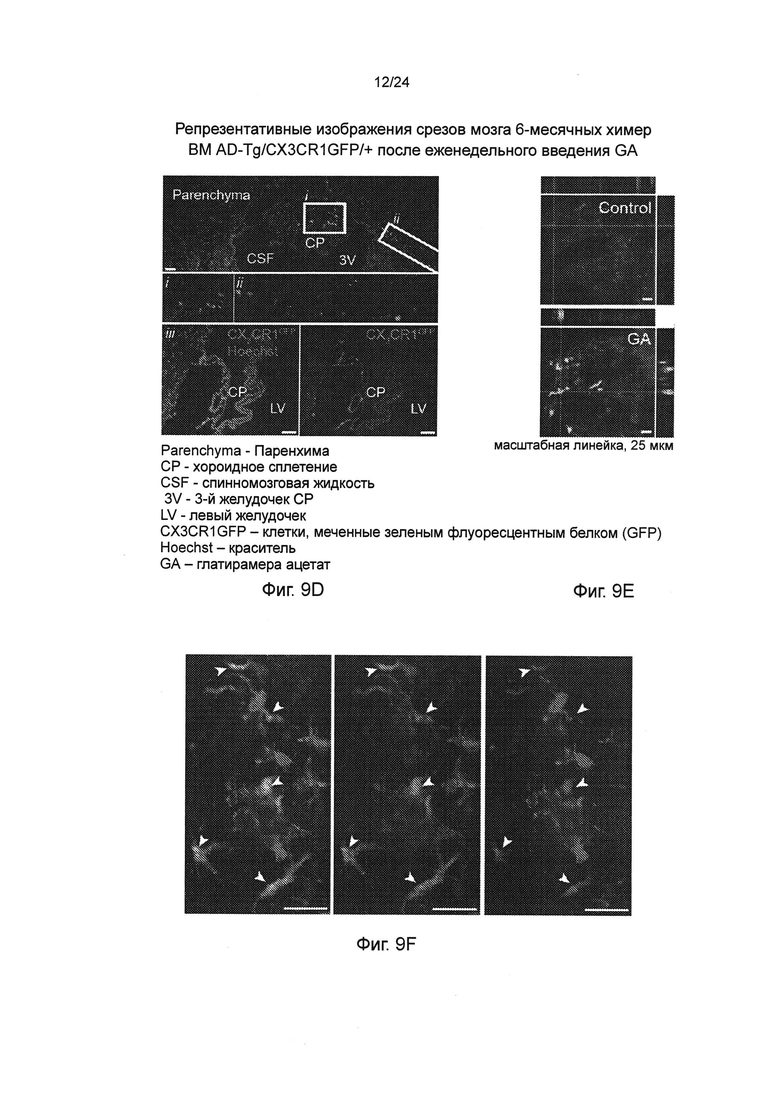
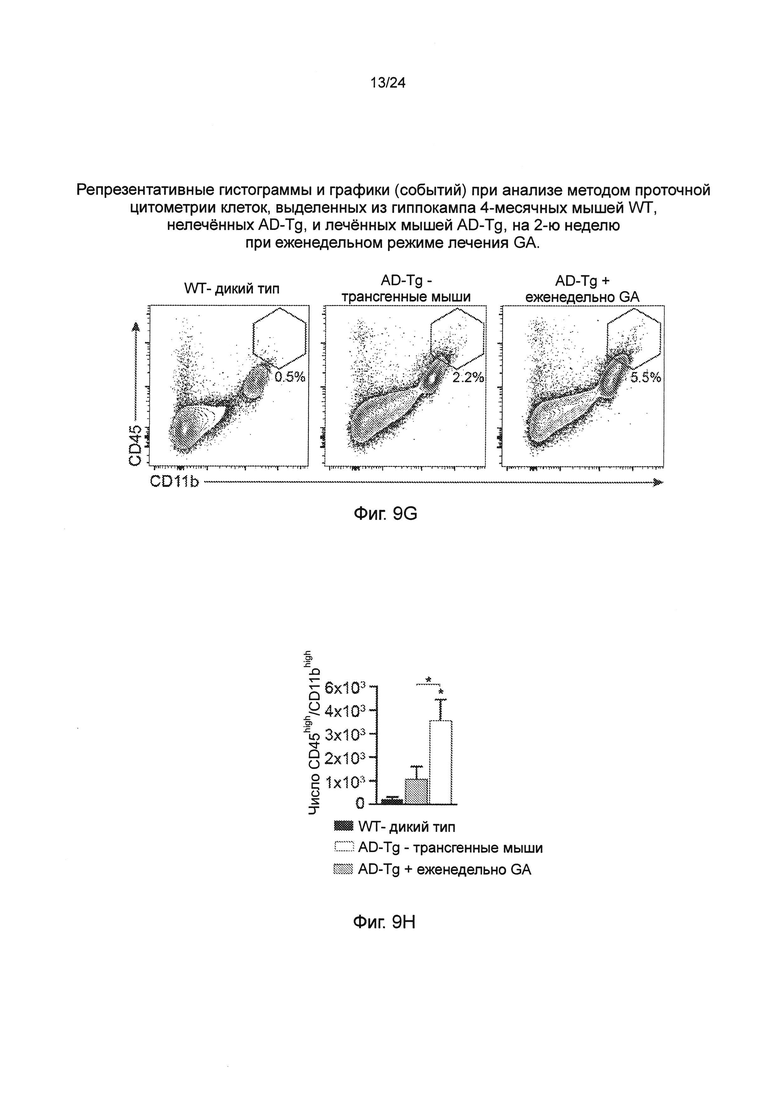

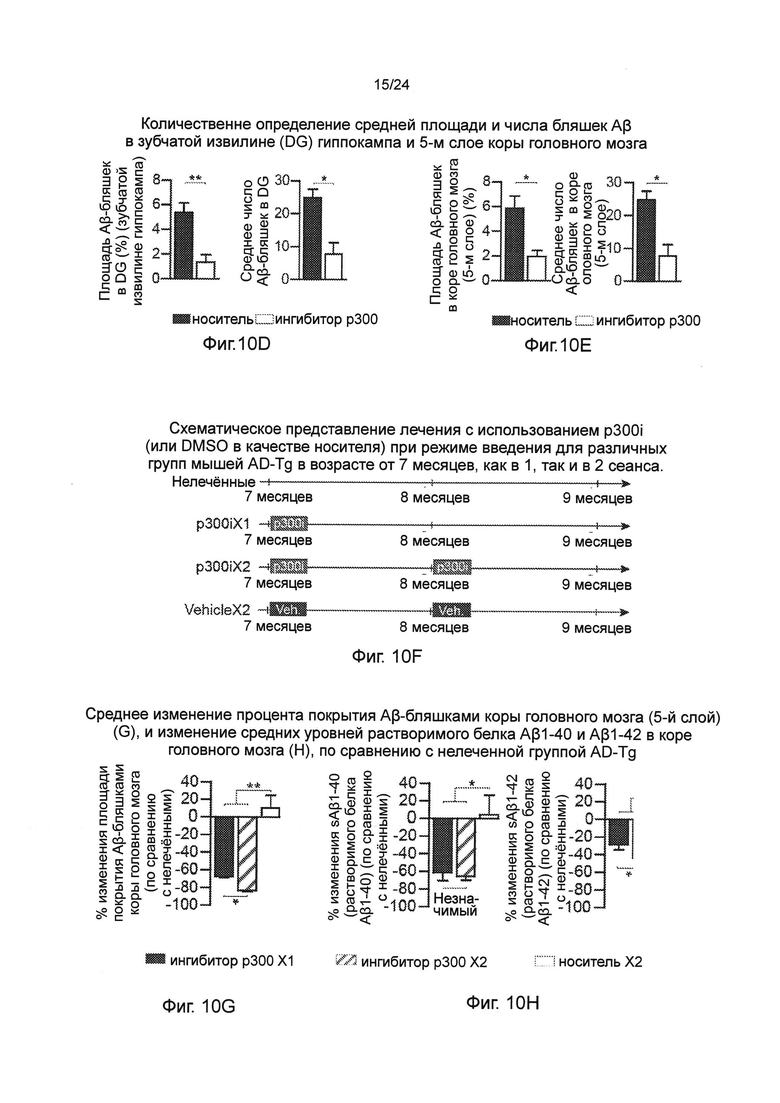

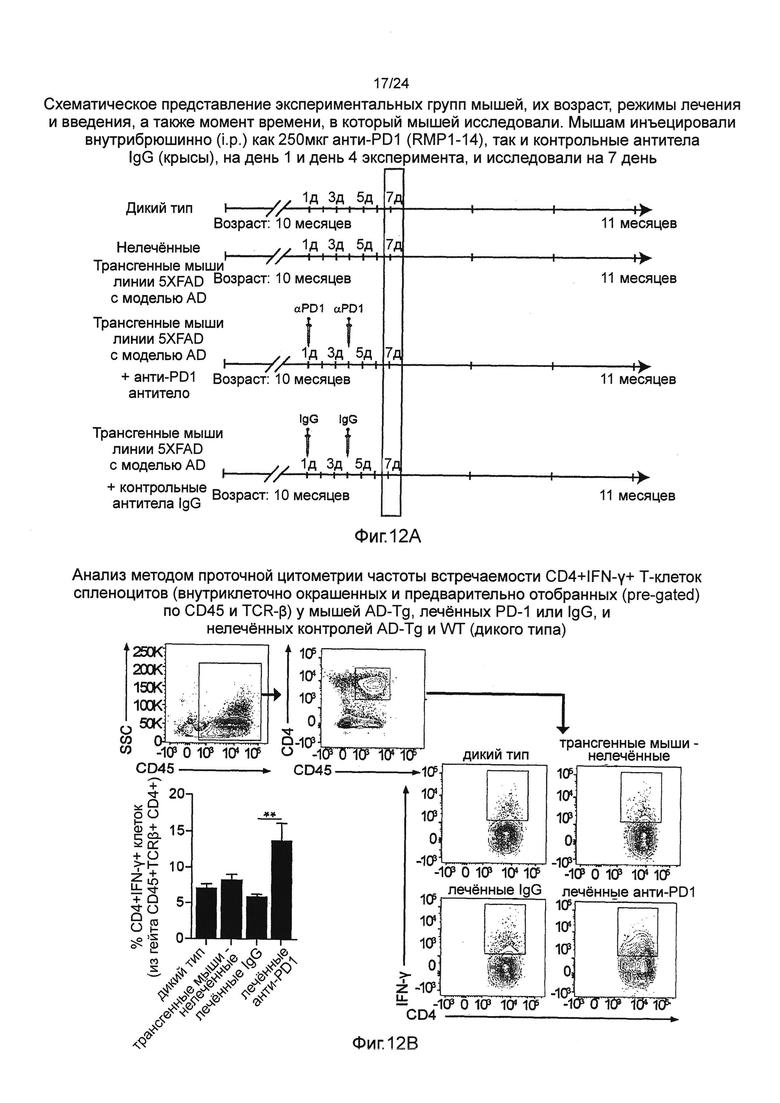
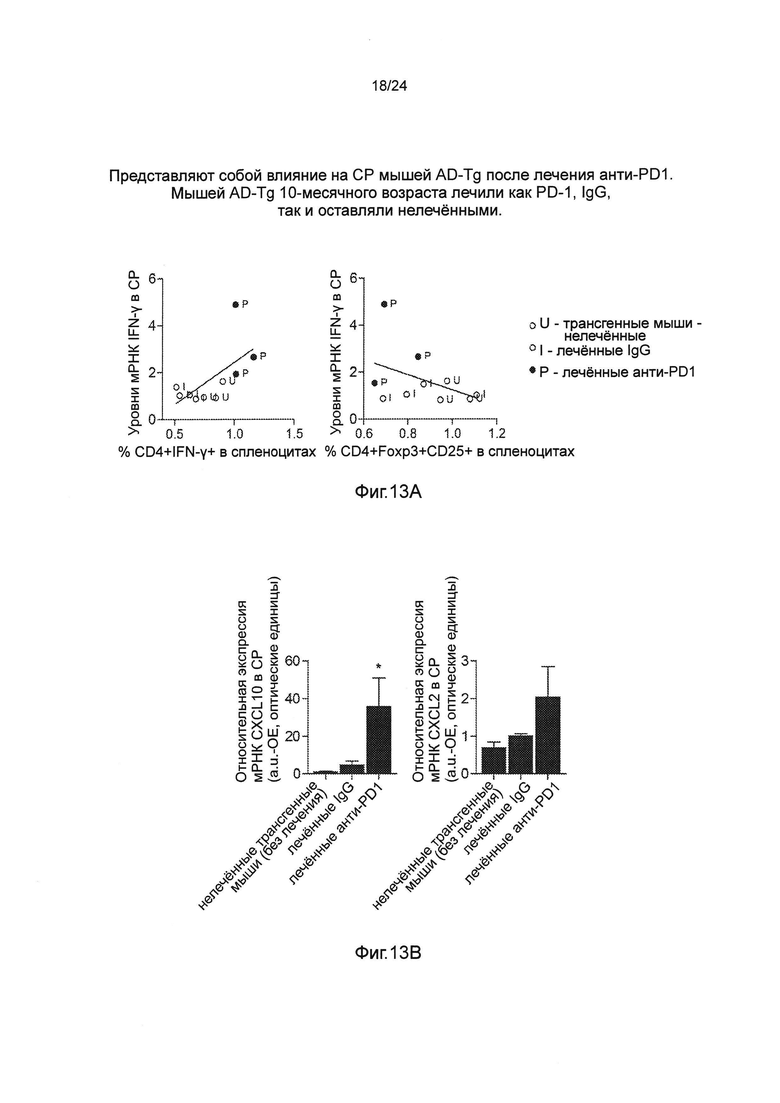

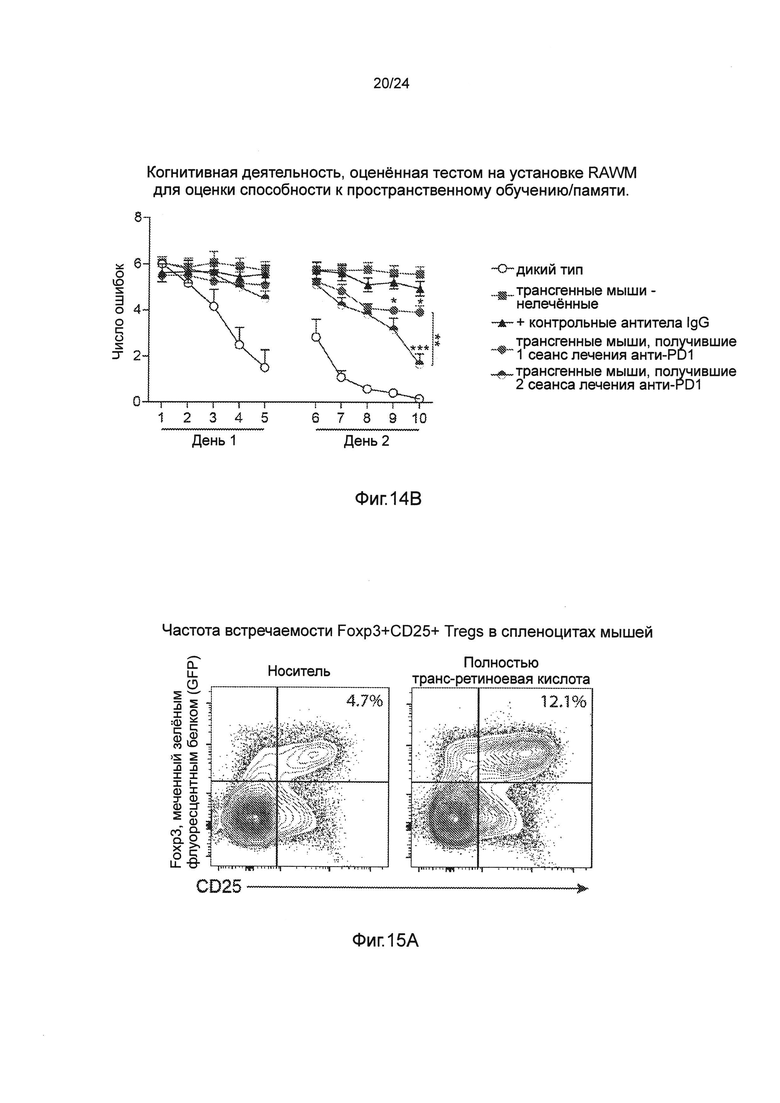
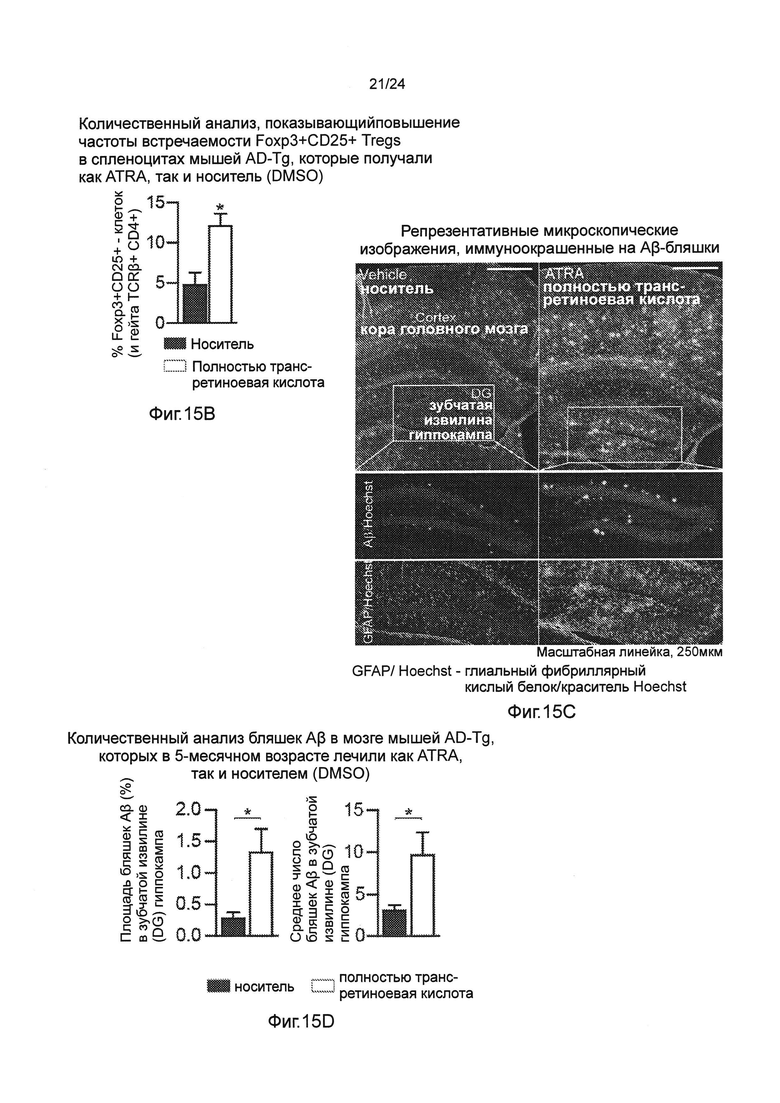

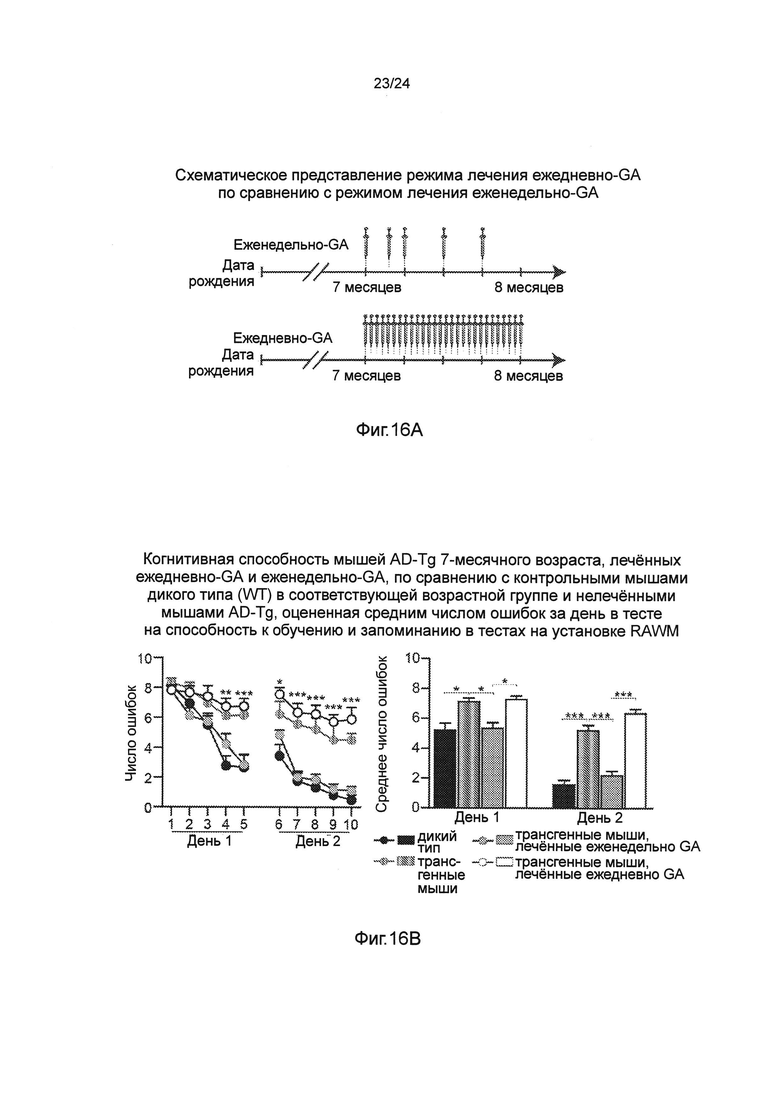
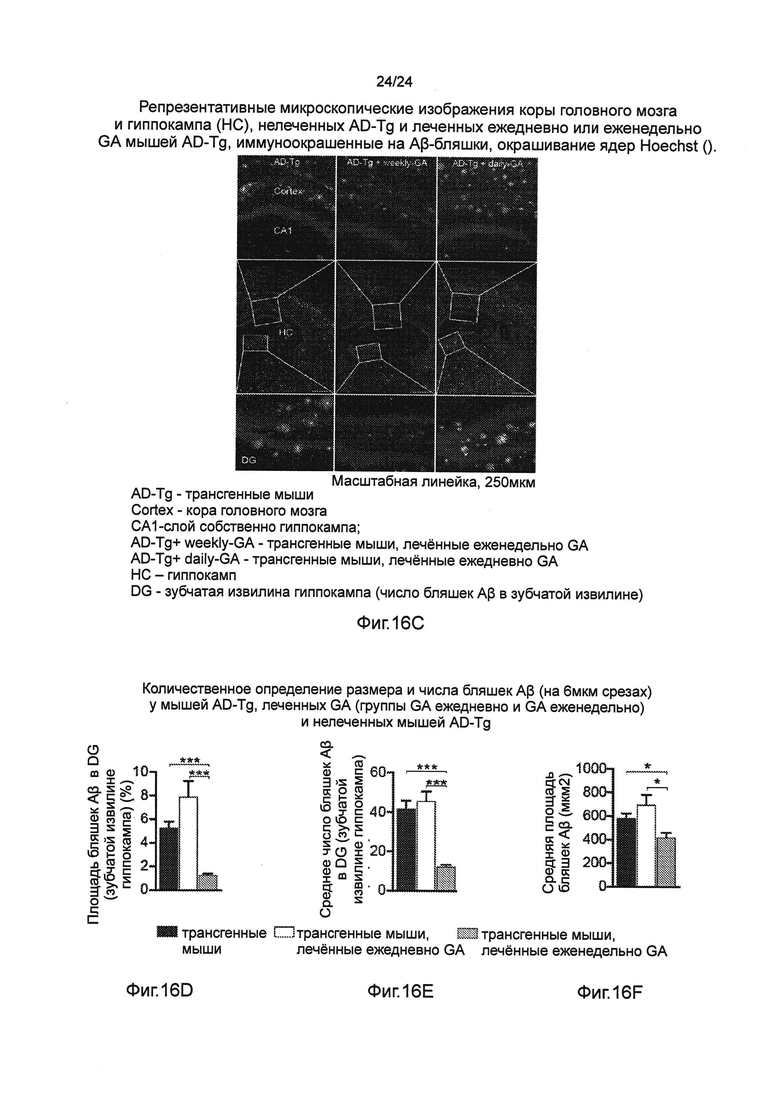
Настоящее изобретение относится к медицине. а именно к неврологии и психиатрии, и касается лечения болезни Альцгеймера. Для этого осуществляют введение фармацевтической композиции, содержащей активный агент, который вызывает снижение уровня системной иммуносупрессии. В качестве таких агентов используют антитела, нейтрализующие PD-1, PD-L1, или TIM-3. Введение осуществляют по меньшей мере двумя курсами лечения. Каждый курс лечения включает последовательность сеансов, которая содержит введение фармацевтической композиции пациенту, за которым следует интервал в лечении без введения фармацевтической композиции пациенту, и при этом интервалы без введения длиннее, чем периоды меду повторяющимися сеансами введения во время курса лечения. Такой режим введения обеспечивает транзиторное снижение уровня системной иммуносупрессии, что, в свою очередь, способствует улучшению когнитивного статуса пациентов с болезнью Альцгеймера. 13 з.п. ф-лы, 16 ил., 2 табл., 9 пр.
1. Способ лечения болезни Альцгеймера, включающий введение пациенту, нуждающемуся в этом, фармацевтической композиции, содержащей активный агент, который вызывает снижение уровня системной иммуносупрессии у индивидуума, где активный агент выбирают из нейтрализующего анти-PD-1 антитела, нейтрализующего анти-PD-L1 антитела, нейтрализующего анти-TIM-3 антитела или их комбинации и фармацевтическая композиция предназначена для введения в режиме дозирования, который вызывает транзиторное снижение уровня системной иммуносупрессии, и содержит по меньшей мере два курса лечения, при этом каждый курс лечения включает последовательность сеансов, которая содержит введение фармацевтической композиции пациенту, за которым следует интервал в лечении без введения фармацевтической композиции пациенту, и при этом интервалы без введения длиннее, чем периоды между повторяющимися сеансами введения во время курса лечения.
2. Способ лечения по п. 1, отличающийся тем, что сеанс лечения включает введение фармацевтической композиции пациенту и указанный сеанс лечения поддерживают по меньшей мере до тех пор, пока уровень не опустится ниже ссылочного, введение приостанавливают во время интервала между сеансами и указанный интервал между сеансами поддерживают до тех пор, пока указанный уровень не опустится ниже ссылочного, при этом ссылочный уровень выбран из:
(a) уровня системного присутствия или активности регуляторных Т-клеток или супрессорных клеток миелоидного происхождения, измеренного в самой последней пробе крови, полученной от указанного индивидуума до указанного введения; или
(b) уровня системного присутствия или активности регуляторных Т-клеток или супрессорных клеток миелоидного происхождения, характерных для популяции пациентов, страдающих от заболевания Альцгеймера.
3. Способ по п. 2, отличающийся тем, что ссылочный уровень означает уровень системного присутствия или активности регуляторных Т-клеток, измеренный в самой последней пробе крови, полученной от указанного пациента до введения.
4. Способ по п. 1, отличающийся тем, что сеанс лечения содержит введение фармацевтической композиции пациенту и сеанс лечения поддерживают по меньшей мере до тех пор, пока системное присутствие или уровень IFNγ-продуцирующих лейкоцитов не поднимется выше ссылочного, и введение приостанавливают во время интервала между сеансами, в котором отсутствует введение, и интервал между сеансами без введения поддерживают так долго, пока уровень не поднимется выше ссылочного, где указанный ссылочный (уровень) выбран из:
(a) уровня системного присутствия или активности IFNγ-продуцирующих лейкоцитов, измеренного в самой последней пробе крови, полученной от указанного индивидуума до указанного введения; или
(b) уровня системного присутствия или активности IFNγ-продуцирующих лейкоцитов, характерных для популяции пациентов, страдающих от заболевания Альцгеймера.
5. Способ лечения по п. 1, отличающийся тем, что сеанс введения включает повторение введения фармацевтической композиции, которое происходит один раз в два, три, четыре, пять или шесть дней, один раз в неделю, один раз в две недели, один раз каждые три недели, один раз каждые четыре недели.
6. Способ лечения по п. 1, отличающийся тем, что сеанс введения продолжается между одной и четырьмя неделями или между тремя днями и четырьмя неделями.
7. Способ лечения по п. 1, отличающийся тем, что длительность периодов без введения продолжается между одной неделей и шестью месяцами, между двумя неделями и шестью месяцами или между тремя неделями и шестью месяцами.
8. Способ лечения по п. 2, отличающийся тем, что регуляторные Т-клетки являются клетками CD4+, выбранными из FoxP3+ клеток, экспрессирующих одну или несколько из CD25, CD127, GITR, CTLA-4 или PD-1; или FoxP3- клеток, экспрессирующих одну или несколько из CD25, CD127, GITR, CTLA-4 или PD-1 поверхностных молекул.
9. Способ лечения по п. 8, отличающийся тем, что регуляторные Т-клетки являются клетками CD4+CD25+FoxP3+ или клетками CD4+CD25+FoxP3-.
10. Способ лечения по любому из пп. 1-9, отличающийся тем, что активным агентом является анти-PDL-1 антитело.
11. Способ лечения по любому из пп. 1-9, отличающийся тем, что активным агентом является нейтрализующее анти-PDL-1 антитело.
12. Способ лечения по любому из пп. 1-9, отличающийся тем, что активным агентом является анти-TIM-3 антитело.
13. Способ лечения по п. 1, отличающийся тем, что лечение болезни Альцгеймера улучшает когнитивную функцию.
14. Способ лечения по п. 13, отличающийся тем, что когнитивная функция означает обучение, память, воспроизведение изображений, мышления, восприятия, оценки, ориентации в пространстве, речи и способности к языкам, узнавания речи и/или способности к оценочному вниманию.
| BUTOVSKY O | |||
| et al | |||
| "Selective ablation og bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model" | |||
| European Jornal of Neuroscience" | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| FINNEFROCK AC | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| J Immunol | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| SHIMURA-TOMITA M | |||
| et al | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| KEIMOWITZ R.M | |||
| "Dementia improvement with cytotoxic chemotherapy" | |||
| Arch Nevrol | |||
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |

