ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №62/520430, поданной 15 июня 2017 года; предварительной заявки на патент США №62/591247, поданной 28 ноября 2017 года; предварительной заявки на патент США №62/649856, поданной 29 марта 2018 года; и предварительной заявки на патент США №62/672261, поданной 16 мая 2018; содержание каждой из которых включено в настоящую заявку во всей полноте посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к производным тетрагидропиридопиразина, которые являются модуляторами рецептора 6, сопряженного с G-белком (GPR6), к содержащим их фармацевтическим композициям и к их применению для лечения заболеваний, нарушений и состояний, связанных с GPR6.
УРОВЕНЬ ТЕХНИКИ
GPR6 является членом семейства трансмембранных рецепторов, сопряженных с G-белком (GPCR). Передача сигнала GPR6 происходит по пути G-белка (Gs). Он в высоком количестве экспрессируется в центральной нервной системе (ЦНС), в частности, в средних шипиковых нейронах (СШН) в полосатом теле, и в минимальном количестве экспрессируется в периферических тканях. Основные мишени дофаминергической иннервации полосатого тела находятся в СШН стриатопаллидарных (непрямых) и стриатонигральных (прямых) выходных путей. СШН прямых выходных путей экспрессируют рецепторы дофамина D1, при этом СШН непрямых путей экспрессируют рецепторы D2. GPR6 в большом количестве присутствует в СШН полосатого тела, экспрессирующих рецептор D2, в которых активность GPR6 повышает уровень внутриклеточного вторичного мессенджера цАМФ, который выполняет функцию, противоположную сигнальной системе рецептора D2. Антагонизм или обратный агонизм GPR6, сопряженного с Gs, понижает уровень цАМФ в СШН и, таким образом, обеспечивает функциональную альтернативу активации рецепторов D2, опосредованной дофамином.
В опубликованной международной заявке на патент WO 2015/095728A1, содержание которой включено в настоящую заявку во всей полноте посредством ссылки, описано несколько производных тетрагидропиридопиразина, которые являются модуляторами GPR6. Указанные соединения включают (S)-1-(2-(4-(2,4-дифторфенокси)-пиперидин-1-ил)-3-((тетрагидрофуран-3-ил)амино)-7,8-дигидропиридо[3,4-b]пиразин-6(5H)-ил)этан-1-он («соединение A»). Несмотря на то, что соединение A потенциально является эффективным модулятором GPR6, оно имеет неоптимальный резерв безопасности, связанный с ингибированием активности hERG (ген специфических калиевых каналов человека, англ. «human ether-a-go-go-related gene»). См., например, X. Yao et al., British Journal of Pharmacology (2008) 154:1446-56. Ингибирование hERG является одним из факторов, связанных с возможным увеличением интервала QT и сердечной аритмией.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено производное тетрагидропиридопиразина и содержащие его фармацевтические композиции. Производное тетрагидропиридопиразина является модулятором GPR6 и может применяться для лечения заболеваний, нарушений и состояний, связанных с GPR6, включая неврологические нарушения, такие как болезнь Паркинсона.
Согласно одному из аспектов изобретения предложено соединение формулы 1:
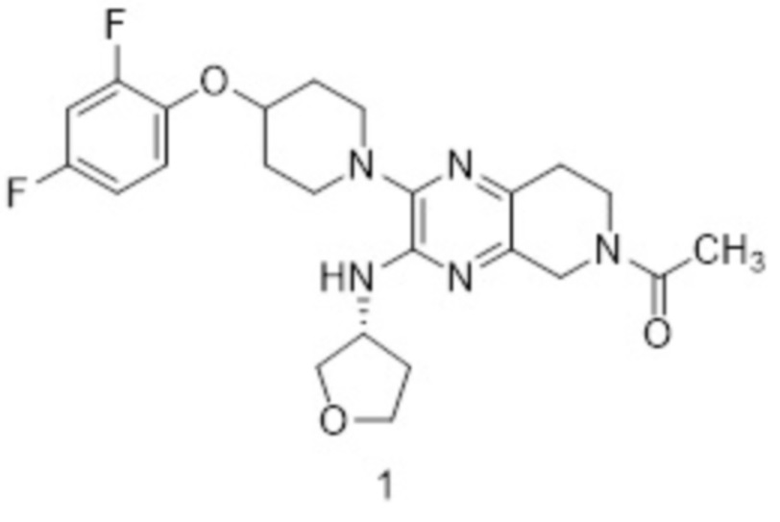
или его фармацевтически приемлемая соль. На формуле 1 изображено соединение (R)-1-(2-(4-(2,4-дифторфенокси)пиперидин-1-ил)-3-((тетрагидрофуран-3-ил)амино)-7,8-дигидропиридо[3,4-b]пиразин-6(5H)-ил)этан-1-он.
В определенных вариантах реализации соединение или его фармацевтически приемлемая соль формулы 1 имеет энантиомерную чистоту, соответствующую энантиомерному избытку (э.и.) 20 % или более, 40 % э.и., 60 % э.и., 80 % э.и., 90 % э.и., 98 % э.и., 99 % э.и. или 100 % э.и. В определенных вариантах реализации соединение присутствует в свободной форме.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, которая содержит соединение формулы 1 или его фармацевтически приемлемую соль, такие как описано в настоящем документе; и фармацевтически приемлемое вспомогательное вещество.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы 1 или его фармацевтически приемлемую соль, такие как описано в настоящем документе, для применения в качестве лекарственного средства. В определенных вариантах реализации фармацевтическая композиция предназначена для лечения болезни Паркинсона, дискинезий, вызванных леводопой, болезни Хантингтона, наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии, болезни Альцгеймера, тревоги и депрессии. В определенных вариантах реализации фармацевтическая композиция дополнительно содержит амантадин. Согласно другому аспекту изобретения предложена фармацевтическая композиция для применения в лечении заболевания, нарушения или состояния, связанного с GPR6.
В различных вариантах реализации изобретения, описанного в настоящем документе, предложена фармацевтическая композиция, содержащая соединение формулы 1 или его фармацевтически приемлемую соль, такие как описано в настоящем документе, и фармацевтически приемлемое вспомогательное вещество. В определенных вариантах реализации фармацевтическая композиция предназначена для применения в качестве лекарственного средства. В определенных вариантах реализации фармацевтическую композицию применяют для лечения заболевания, нарушения или состояния, выбранного из группы, состоящей из: болезни Паркинсона, дискинезий, вызванных леводопой, болезни Хантингтона, наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии, болезни Альцгеймера, тревоги и депрессии.
Согласно дополнительному аспекту изобретения предложено применение соединения формулы 1 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения заболевания, нарушения или состояния, связанного с GPR6. В определенных вариантах реализации заболевание, нарушение или состояние выбрано из группы, состоящей из: болезни Паркинсона, дискинезий, вызванных леводопой, болезни Хантингтона, наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии, болезни Альцгеймера, тревоги и депрессии.
Согласно дополнительному аспекту изобретения предложен способ лечения заболевания, нарушения или состояния, связанного с GPR6, включающий введение субъекту эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли, таких как описано в настоящем документе. В определенных вариантах реализации соединение формулы 1 или его фармацевтически приемлемую соль вводят перорально.
Согласно другому аспекту изобретения предложен способ лечения заболевания, нарушения или состояния у субъекта, включающий введение субъекту эффективного количества соединения формулы 1 или его фармацевтически приемлемой соли, где заболевание, нарушение или состояние выбрано из болезни Паркинсона, дискинезий, вызванных леводопой, болезни Хантингтона, наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии, болезни Альцгеймера, тревоги и депрессии.
В определенных вариантах реализации соединение формулы 1 или его фармацевтически приемлемую соль, которые вводят в способе, описанном в настоящем документе, вводят в дозе, выбранной из диапазона от примерно 0,1 мг/кг до примерно 1,0 мг/кг или от примерно 0,5 мг/кг до примерно 5,0 мг/кг. В определенных вариантах реализации соединение или его фармацевтически приемлемую соль вводят в дозе в диапазоне от примерно 40 мг/кг до примерно 60 мг/кг. В определенных вариантах реализации соединение формулы 1 или его фармацевтически приемлемую соль, которые вводят в способе, описанном в настоящем документе, вводят в дозе, выбранной из группы диапазонов, состоящей из: от примерно 5 мг/кг до примерно 15 мг/кг, от примерно 10 мг/кг до примерно 20 мг/кг, от примерно 15 мг/кг до примерно 25 мг/кг, от примерно 20 до примерно 30 мг/кг, от примерно 25 мг/кг до примерно 35 мг/кг, от примерно 30 мг/кг до примерно 40 мг/кг, от примерно 35 мг/кг до примерно 45 мг/кг и от примерно 45 мг/кг до примерно 55 мг/кг. В определенных вариантах реализации доза выбрана из группы диапазонов, состоящей из: от примерно 30 мг/кг до примерно 40 мг/кг, от примерно 35 мг/кг до примерно 45 мг/кг, от примерно 40 мг/кг до примерно 50 мг/кг, от примерно 45 мг/кг до примерно 55 мг/кг, от примерно 50 мг/кг до примерно 60 мг/кг, от примерно 55 мг/кг до примерно 65 мг/кг и от примерно 60 мг/кг до примерно 70 мг/кг. В определенных вариантах реализации доза может составлять примерно 50 мг/кг. В определенных вариантах реализации доза составляет более чем примерно 1 мг/кг. В определенных вариантах реализации доза составляет примерно 1 мг/кг. В определенных вариантах реализации доза находится в диапазоне, выбранном из группы, состоящей из: от примерно 50 мг/кг до примерно 100 мг/кг, от примерно 100 мг/кг до примерно 150 мг/кг, от примерно 150 мг/кг до примерно 200 мг/кг, от примерно 200 мг/кг до примерно 250 мг/кг, от примерно 250 мг/кг до примерно 350 мг/кг, от примерно 300 мг/кг до примерно 350 мг/кг, от примерно 350 мг/кг до примерно 400 мг/кг, от примерно 400 мг/кг до примерно 450 мг/кг, от примерно 450 мг/кг до примерно 500 мг/кг. В определенных вариантах реализации доза составляет примерно 500 мг/кг. В качестве альтернативы доза составляет менее 500 мг/кг.
В определенных вариантах реализации доза составляет 35 мг/кг. В определенных вариантах реализации доза составляет 36 мг/кг. В определенных вариантах реализации доза составляет 37 мг/кг. В определенных вариантах реализации доза составляет 38 мг/кг. В определенных вариантах реализации доза составляет 39 мг/кг. В определенных вариантах реализации доза составляет 40 мг/кг. В определенных вариантах реализации доза составляет 41 мг/кг. В определенных вариантах реализации доза составляет 42 мг/кг. В определенных вариантах реализации доза составляет 43 мг/кг. В определенных вариантах реализации доза составляет 44 мг/кг. В определенных вариантах реализации доза составляет 45 мг/кг. В определенных вариантах реализации доза составляет 46 мг/кг. В определенных вариантах реализации доза составляет 47 мг/кг. В определенных вариантах реализации доза составляет 48 мг/кг. В определенных вариантах реализации доза составляет 49 мг/кг. В определенных вариантах реализации доза составляет 50 мг/кг. В определенных вариантах реализации доза составляет 51 мг/кг. В определенных вариантах реализации доза составляет 52 мг/кг. В определенных вариантах реализации доза составляет 53 мг/кг. В определенных вариантах реализации доза составляет 54 мг/кг. В определенных вариантах реализации доза составляет 55 мг/кг. В определенных вариантах реализации доза составляет 56 мг/кг. В определенных вариантах реализации доза составляет 57 мг/кг. В определенных вариантах реализации доза составляет 58 мг/кг. В определенных вариантах реализации доза составляет 59 мг/кг. В определенных вариантах реализации доза составляет 60 мг/кг. В определенных вариантах реализации доза составляет 61 мг/кг. В определенных вариантах реализации доза составляет 62 мг/кг. В определенных вариантах реализации доза составляет 63 мг/кг. В определенных вариантах реализации доза составляет 64 мг/кг. В определенных вариантах реализации доза составляет 65 мг/кг. В определенных вариантах реализации стадию введения проводят перорально.
Согласно дополнительному аспекту изобретения предложена комбинированная терапия, включающая введение эффективного количества фармацевтической композиции; и по меньшей мере одного дополнительного фармакологически активного агента. В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: леводопы, ингибитора DOPA-декарбоксилазы, агониста дофамина, антихолинергического средства, селективного ингибитора моноаминоксидазы B и ингибитора катехол-O-метилтрансферазы. В других вариантах реализации дополнительный фармакологически активный агент представляет собой леводопу в комбинации с ингибитором DOPA-декарбоксилазы. В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: карбидопы; бенсеразида; метилдопы; α-дифторметил-DOPA; 3',4',5,7-тетрагидрокси-8-метоксиизофлавона; апоморфина гидрохлорида; бромокриптина; ротиготина; прамипексола; ропинирола; тригексилфенидила; бензтропина мезилата; сафинамида; селегилина; разагилина; энтакапона; и толкапона. В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: ингибиторов бета-секретазы, ингибиторов гамма-секретазы, ингибиторов HMG-CoA редуктазы, нестероидных противовоспалительных препаратов (НПВП). В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: апазона, аспирина, целекоксиба, диклофенака (совместно с мизопростолом или без него), дифлунизала, этодолака, фенопрофена, флурбипрофена, ибупрофена, индометацина, кетопрофена, меклофенамата натрия, мефенаминовой кислоты, мелоксикама, набуметона, напроксена, оксапрозина, фенилбутазона, пироксикама, холина и магния салицилатов, салсалата и сулиндака. В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: донепезила, ривастигмина, мемантина и галантамина. В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: седативных средств, снотворных средств, анксиолитических средств, нейролептических средств и транквилизаторов. В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: амитриптилина, амоксапина, арипипразола, азенапина, бупропиона, хлордиазепоксида, циталопрама, хлорпромазина, клозапина, дезипрамина, десвенлафаксина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флуоксетина, флуфеназина, галоперидола, илоперидона, имипрамина, изоксарбоксазида, ламотригина, левомилнаципрана, луразидона, миртазапина, нефазодона, нортриптилина, оланзапина, палиперидона, пароксетина, перфеназина, фенелзина, протриптилина, кветиапина, рисперидона, сафинамида, селегилина, сертралина, транилципромина, тразодона, тримипрамина, венлафаксина, вилазодона, вортиоксетина и зипрасидона. В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: алпразолама, хлордиазепоксида, клобазепама, клоназепама, клоразепата, диазепама, эстазолама, флуразепама, лоразепама, мидазолама, оксазепама, празепама, квазепама, темазепама и триазолама, гидроксизина, эсзопиклона, залеплона, золпидема и зопиклона и буспирона. В определенных вариантах реализации дополнительный фармакологически активный агент выбран из группы, состоящей из: ацетазоламида, карбамазепина, клобазама, клоназепама, эсликарбазепина ацетата, этосуксимида, габапентина, лакозамида, ламотригина, леветирацетама, нитразепама, окскарбазепина, перампанела, пирацетама, фенобарбитала, фенитоина, прегабалина, примидона, ретигабина, руфинамида, натрия вальпроата, стирипентола, тиагабина, топирамата, вигабатрина и зонисамида.
В различных вариантах реализации настоящего изобретения предложен способ лечения болезни Паркинсона у субъекта, включающий: введение фармацевтической композиции, такой как определено в настоящем документе, субъекту. В некоторых вариантах реализации введение фармацевтической композиции улучшает моторные симптомы у субъекта. В некоторых вариантах реализации стадию введения проводят перорально.
В различных вариантах реализации настоящего изобретения предложена лекарственная форма соединения формулы 1 или его фармацевтически приемлемой соли, предназначенная для перорального введения соединения или его фармацевтически приемлемой соли в дозе, выбранной из группы диапазонов, состоящей из: от примерно 0,1 мг/кг до примерно 1,0 мг/кг или от примерно 0,5 мг/кг до примерно 5,0 мг/кг. В определенных вариантах реализации соединение или его фармацевтически приемлемую соль вводят в дозе в диапазоне от примерно 40 мг/кг до примерно 60 мг/кг. В определенных вариантах реализации соединение формулы 1 или его фармацевтически приемлемую соль вводят в дозе, выбранной из группы диапазонов, состоящей из: от примерно 5 мг/кг до примерно 15 мг/кг, от примерно 10 мг/кг до примерно 20 мг/кг, от примерно 15 мг/кг до примерно 25 мг/кг, от примерно 20 до примерно 30 мг/кг, от примерно 25 мг/кг до примерно 35 мг/кг, от примерно 30 мг/кг до примерно 40 мг/кг, от примерно 35 мг/кг до примерно 45 мг/кг и от примерно 45 мг/кг до примерно 55 мг/кг. В определенных вариантах реализации доза выбрана из группы диапазонов, состоящей из: от примерно 30 мг/кг до примерно 40 мг/кг, от примерно 35 мг/кг до примерно 45 мг/кг, от примерно 40 мг/кг до примерно 50 мг/кг, от примерно 45 мг/кг до примерно 55 мг/кг, от примерно 50 мг/кг до примерно 60 мг/кг, от примерно 55 мг/кг до примерно 65 мг/кг и от примерно 60 мг/кг до примерно 70 мг/кг. В определенных вариантах реализации доза может составлять примерно 50 мг/кг. В определенных вариантах реализации доза составляет более чем примерно 1 мг/кг. В определенных вариантах реализации доза составляет примерно 1 мг/кг. В определенных вариантах реализации доза находится в диапазоне, выбранном из группы, состоящей из: от примерно 50 мг/кг до примерно 100 мг/кг, от примерно 100 мг/кг до примерно 150 мг/кг, от примерно 150 мг/кг до примерно 200 мг/кг, от примерно 200 мг/кг до примерно 250 мг/кг, от примерно 250 мг/кг до примерно 350 мг/кг, от примерно 300 мг/кг до примерно 350 мг/кг, от примерно 350 мг/кг до примерно 400 мг/кг, от примерно 400 мг/кг до примерно 450 мг/кг, от примерно 450 мг/кг до примерно 500 мг/кг. В определенных вариантах реализации доза составляет примерно 500 мг/кг. В качестве альтернативы доза составляет менее 500 мг/кг.
В определенных вариантах реализации доза составляет 35 мг/кг. В определенных вариантах реализации доза составляет 36 мг/кг. В определенных вариантах реализации доза составляет 37 мг/кг. В определенных вариантах реализации доза составляет 38 мг/кг. В определенных вариантах реализации доза составляет 39 мг/кг. В определенных вариантах реализации доза составляет 40 мг/кг. В определенных вариантах реализации доза составляет 41 мг/кг. В определенных вариантах реализации доза составляет 42 мг/кг. В определенных вариантах реализации доза составляет 43 мг/кг. В определенных вариантах реализации доза составляет 44 мг/кг. В определенных вариантах реализации доза составляет 45 мг/кг. В определенных вариантах реализации доза составляет 46 мг/кг. В определенных вариантах реализации доза составляет 47 мг/кг. В определенных вариантах реализации доза составляет 48 мг/кг. В определенных вариантах реализации доза составляет 49 мг/кг. В определенных вариантах реализации доза составляет 50 мг/кг. В определенных вариантах реализации доза составляет 51 мг/кг. В определенных вариантах реализации доза составляет 52 мг/кг. В определенных вариантах реализации доза составляет 53 мг/кг. В определенных вариантах реализации доза составляет 54 мг/кг. В определенных вариантах реализации доза составляет 55 мг/кг. В определенных вариантах реализации доза составляет 56 мг/кг. В определенных вариантах реализации доза составляет 57 мг/кг. В определенных вариантах реализации доза составляет 58 мг/кг. В определенных вариантах реализации доза составляет 59 мг/кг. В определенных вариантах реализации доза составляет 60 мг/кг. В определенных вариантах реализации доза составляет 61 мг/кг. В определенных вариантах реализации доза составляет 62 мг/кг. В определенных вариантах реализации доза составляет 63 мг/кг. В определенных вариантах реализации доза составляет 64 мг/кг. В определенных вариантах реализации доза составляет 65 мг/кг.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Указанные выше и другие задачи, отличительные признаки и преимущества станут понятны из последующего описания конкретных вариантов реализации изобретения, проиллюстрированных на прилагаемых чертежах. Чертежи не обязательно приведены в масштабе; напротив, основное внимание уделяется иллюстрации принципов, лежащих в основе различных вариантов реализации изобретения.
На фигуре 1 приведен график, на котором показано влияние лечения в модели болезни Паркинсона, вызванной 6-гидроксидофамином (6-OHDA).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящая заявка является родственной с публикацией WO 2015/095728, содержание которой включено в настоящую заявку во всей полноте посредством ссылки.
I. ОПРЕДЕЛЕНИЯ
Если не указано иное, то в настоящем изобретении используют определения, приведенные ниже.
«Примерно» или «приблизительно» при использовании для описания измеряемой числовой переменной относится к указанному значению переменной и ко всем значениям переменной в пределах экспериментальной ошибки для данного значения или в пределах ±10 процентов от указанного значения в зависимости от того, какая ошибка больше.
«Энантиомерный избыток» или «э.и.» относится к избытку одного энантиомера по сравнению с другим, выраженному как доля в процентах от целого, и является мерой энантиомерной (хиральной) чистоты образца, содержащего энантиомер. Например, если образец содержит в избытке R-энантиомер, то э.и. может быть определен при помощи выражения

где AR и AS представляют собой количества R- и S- энантиомеров в образце.
«Комбинированная терапия» относится к подходу для лечения заболевания, нарушения или состояния, включающему один или более агентов терапевтического воздействия. Например, агенты терапевтического воздействия могут включать более чем одну (один) фармацевтическую композицию или фармацевтически активный агент или их комбинацию. Агенты терапевтического воздействия можно вводить одновременно, последовательно или в любом порядке. Их можно вводить в разных дозировках, с разной частотой введения или разными способами, которые являются подходящими. Мольное отношение агентов терапевтического воздействия не ограничено каким-либо конкретным образом.
«Моторные симптомы» относятся к снижению активности субъекта, возникающему при заболевании, нарушении и состоянии, связанном с GPR6, таком как болезнь Паркинсона.
«По существу чистый энантиомер» и его варианты относятся к энантиомеру, который присутствует в образце с 90 % э.и. или более.
«Чистый энантиомер» и его варианты относятся к энантиомеру, который присутствует в образце с 98 % э.и. или более.
«Субъект» относится к млекопитающему, включая человека.
«Фармацевтически приемлемые» вещества относятся к веществам, которые подходят для введения субъектам.
«Лечение» относится к купированию, облегчению, подавлению прогрессирования или предупреждению заболевания, нарушения или состояния, к которому относится этот термин, или к купированию, облегчению, подавлению прогрессирования или предупреждению одного или более симптомов указанного заболевания, нарушения или состояния.
«Способ лечения» относится к осуществлению «лечения», такого как определено непосредственно выше.
«Лекарственное средство», «лекарственное вещество», «активный фармацевтический ингредиент» и т.д. относятся к соединению (например, к соединению формулы 1), которое можно применять для лечения субъекта, нуждающегося в лечении.
«Эффективное количество» лекарственного средства, «терапевтически эффективное количество» лекарственного средства и т.д. относятся к количеству лекарственного средства, которое можно применять для лечения субъекта, и могут зависеть от массы тела и возраста субъекта и способа введения, помимо прочих факторов.
«Вспомогательное вещество» относится к любому разбавителю или наполнителю для лекарственного средства.
«Фармацевтическая композиция» относится к комбинации одного или более лекарственных веществ и одного или более вспомогательных веществ.
«Лекарственный продукт», «фармацевтическая лекарственная форма», «лекарственная форма», «конечная лекарственная форма» и т.д. относятся к фармацевтической композиции, подходящей для лечения субъекта, нуждающегося в лечении, и в общем случае могут иметь форму таблеток, капсул, порционных пакетов, содержащих порошок или гранулы, жидких растворов или суспензий, пластырей, пленок и т.д.
«Состояние, связанное с GPR6» и схожие фразы относятся к заболеванию, нарушению или состоянию субъекта, при котором модуляция GPR6, включая антагонизм или обратный агонизм в отношении GPR6, может обеспечивать терапевтическое или профилактическое благоприятное действие.
В описании могут использоваться следующие сокращения: Ac (ацетил); ACN (ацетонитрил); AIBN (азо-бис-изобутиронитрил); АФИ (активный фармацевтический ингредиент); водн. (водный); BINAP (2,2′-бис(дифенилфосфино)-1,1′-бинафтил); Boc (трет-бутоксикарбонил); Cbz (карбобензилокси); dba (дибензилиденацетон); DCC (1,3-дициклогексилкарбодиимид); ДХЭ (1,1-дихлорэтан); ДХМ (дихлорметан); DIPEA (N,N-диизопропилэтиламин, основание Хюнига); ДМА (N,N-диметилацетамид); DMAP (4-диметиламинопиридин); ДМЭ (1,2-диметоксиэтан); ДМФ (N,N-диметилформамид); ДМСО (диметилсульфоксид); dppf (1,1′-бис(дифенилфосфино)ферроцен); DTT (дитиотреитол); EC50 (эффективная концентрация, обеспечивающая полумаксимальный ответ); EDA (этоксилированный додециловый спирт, BRIJ® 35); EDC (N-(3-диметиламинопропил)-N’-этилкарбодиимид); ЭДТА (этилендиаминтетрауксусная кислота); э.и. (энантиомерный избыток); экв. (эквиваленты); Et (этил); Et3N (триэтиламин); EtOAc (этилацетат); EtOH (этанол); HATU (2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилурония гексафторфосфат (V)); HEPES (4-(2-гидроксиэтил)пиперазин-1-этансульфокислота); HOAc (уксусная кислота); HOBt (1H-бензо[d][1,2,3]триазол-1-ол); IC50 (концентрация, обеспечивающая 50% ингибирование); IPA (изопропанол); iPrOAc (изопропилацетат); IPE (изопропиловый эфир); Ki (константа ингибирования; концентрация конкурентного лиганда в конкурентном исследовании, при которой он занимает 50% рецепторов в отсутствие исследуемого лиганда); LDA (диизопропиламид лития); LiHMDS (бис(триметилсилил)амид лития); мХПБК (м-хлорпероксибензойная кислота); Me (метил); MeOH (метанол); МТБЭ (метил-трет-бутиловый эфир); Тпл (температура плавления); NaOt-Bu (трет-бутоксид натрия); NMM (N-метилморфолин); NMP (N-метил-пирролидон); OTf (трифлат); ПЭ (петролейный эфир); Ph (фенил); pEC50 (-log10(EC50), где EC50 указана в единицах молярности (M)); pIC50 (-log10(IC50), где IC50 указана в единицах молярности (M)); Pr (пропил); c-Pr (циклопропил), i-Pr (изопропил); ПТФЭ (политетрафторэтилен); КТ (комнатная температура примерно от 20°C до 25°C); T3P (2,4,6-трипропил-1,3,5,2,4,6-триокситрифосфинана 2,4,6-триоксид); TCEP (трис(2-карбоксиэтил)фосфин); ТФУК (трифторуксусная кислота); ТФУКА (ангидрид 2,2,2-трифторуксусной кислоты); ТГФ (тетрагидрофуран); TMEDA (N1,N1,N2,N2-тетраметилэтан-1,2-диамин); ТМС (триметилсилил); и Tris-буфер (буфер 2-амино-2-гидроксиметилпропан-1,3-диола).
II. КОМПОЗИЦИИ СОГЛАСНО ИЗОБРЕТЕНИЮ
Как описано ниже, настоящее изобретение относится к соединению формулы 1 и его фармацевтически приемлемым солям. Настоящее изобретение также относится к материалам и способам для получения соединения формулы 1, к содержащим его фармацевтическим композициям и к применению соединения формулы 1 и его фармацевтически приемлемых солей (необязательно в комбинации с другими фармакологически активными агентами) для лечения заболеваний, нарушений или состояний ЦНС, включая болезнь Паркинсона, и других заболеваний, нарушений или состояний, связанных с GPR6.
Соединение формулы 1 может существовать в виде соли, комплекса, сольвата, гидрата и жидкого кристалла. Аналогично, соль соединения формулы 1 может существовать в виде комплекса, сольвата, гидрата или жидкого кристалла.
Соединение формулы 1 может образовывать фармацевтически приемлемую соль. Такие соли включают соли присоединения кислоты (включая двухосновные кислоты) и соли оснований. Фармацевтически приемлемые соли присоединения кислоты могут включать соли, полученные из неорганических кислот, таких как хлороводородная кислота, азотная кислота, фосфорная кислота, серная кислота, бромоводородная кислота, йодоводородная кислота, фтороводородная кислота и фосфорные кислоты, а также нетоксичные соли, полученные из органических кислот, таких как алифатические одно- и двухосновные карбоновые кислоты, фенил-замещенные алкановые кислоты, гидроксиалкановые кислоты, алкандиовые кислоты, ароматические кислоты, алифатические и ароматические сульфокислоты и т.д. Указанные соли могут включать ацетатные, адипатные, аспартатные, бензоатные, безилатные, бикарбонатные, карбонатные, бисульфатные, сульфатные, боратные, камзилатные, цитратные, цикламатные, эдизилатные, эзилатные, формиатные, фумаратные, глюцептатные, глюконатные, глюкуронатные, гексафторфосфатные, гибензатные, гидрохлоридные/хлоридные, гидробромидные/бромидные, гидроиодидные/йодидные, изетионатные, лактатные, малатные, малеатные, малонатные, мезилатные, метилсульфонатные, нафтилатные, 2-напзилатные, никотинатные, нитратные, оротатные, оксалатные, пальмитатные, памоатные, фосфатные, гидрофосфатные, дигидрофосфатные, пироглутаматные, сахаратные, стеаратные, сукцинатные, таннатные, тартратные, тозилатные, трифторацетатные и ксинофоатные соли.
Фармацевтически приемлемые соли оснований могут включать соли, полученные из оснований, включая катионы металлов, такие как катионы щелочных или щелочно-земельных металлов, а также аминов. Примеры подходящих катионов металлов могут включать натрий, калий, магний, кальций, цинк и алюминий. Примеры подходящих аминов могут включать аргинин, N,N’-дибензилэтилендиамин, хлорпрокаин, холин, диэтиламин, диэтаноламин, дициклогексиламин, этилендиамин, глицин, лизин, N-метилглюкамин, оламин, 2-амино-2-гидроксиметилпропан-1,3-диол и прокаин. Обсуждение подходящих солей присоединения кислоты и оснований см. в S. M. Berge et al., J. Pharm. Sci. (1977) 66:1-19; также см. Stahl and Wermuth, Handbook of Pharmaceutical Salts: Properties, Selection, and Use (2002); содержание обеих из которых включено в настоящую заявку во всей полноте посредством ссылок.
Фармацевтически приемлемые соли могут быть получены разными способами. Например, можно проводить реакцию соединения формулы 1 с соответствующей кислотой или основанием с получением целевой соли. В качестве альтернативы, можно проводить реакцию предшественника соединения формулы 1 с кислотой или основанием для удаления неустойчивых к кислотам или основаниям защитных групп или для раскрытия лактоновой или лактамовой группы в предшественнике. Кроме того, соль соединения формулы 1 можно превращать в другую соль (или в свободную форму) путем обработки соответствующей кислотой или основанием или приведения в контакт с ионообменной смолой. После проведения реакции можно выделять соль фильтрованием, если она осаждается из раствора, или выпариванием для выделения соли. Степень ионизации соли может изменяться от полностью ионизированной до практически неионизированной.
Соединение формулы 1 может существовать в разнообразных твердых состояниях в диапазоне от полностью аморфного до полностью кристаллического. Термин «аморфный» относится к состоянию материала, при котором отсутствует дальний порядок на молекулярном уровне и в зависимости от температуры могут проявляться физические свойства твердого вещества или жидкости. Как правило, для таких материалов не могут быть получены характеристические дифрактограммы рентгеновской дифракции, и, несмотря на то, что они имеют свойства твердого вещества, их более формально описывают как жидкость. При нагревании происходит изменение свойств твердого вещества на свойства жидкости, которое характеризуется типичным фазовым переходом второго рода («стеклование»). Термин «кристаллический» относится к твердой фазе, в которой материал имеет регулярно упорядоченную внутреннюю структуру на молекулярном уровне и для которой может быть получена характеристическая дифрактограмма рентгеновской дифракции с выраженными пиками. Указанные материалы при достаточном нагревании также обладают свойствами жидкости, но изменение формы с твердой на жидкую характеризуется типичным фазовым переходом первого рода («температура плавления»).
Соединение формулы 1 также может существовать в несольватированной и сольватированной формах. Термин «сольват» описывает молекулярный комплекс, содержащий соединение и одну или более молекул фармацевтически приемлемого растворителя (например, этанола). Термин «гидрат» представляет собой сольват, в котором растворитель представляет собой воду. Фармацевтически приемлемые сольваты включают сольваты, в которых растворитель может быть замещен изотопами (например, D2O, ацетон-d6, ДМСО-d6).
В принятой в настоящее время системе классификации сольватов и гидратов органических соединений проводят различия между сольватами и гидратамиостровного типа, канальными сольватами и гидратами, и координационными сольватами и гидратами, содержащими связи металл-ион. См., например, K. R. Morris (H. G. Brittain, ред.) Polymorphism in Pharmaceutical Solids (1995), содержание которой включено в настоящую заявку во всей полноте посредством ссылки. Сольваты и гидраты островного типа представляют собой формы, в которых молекулы растворителя (например, воды) изолированы от прямого контакта друг с другом молекулами органического соединения. В канальных сольватах молекулы растворителя расположены в каналах в кристаллической решетке, в которых они находятся по соседству с другими молекулами растворителя. В координационных сольватах молекулы растворителя связаны с ионом металла.
Если растворитель или вода связаны прочно, то комплекс имеет хорошо определенную стехиометрию независимо от влажности. Тем не менее, если растворитель или вода связаны слабо, как в случае канальных сольватов и гигроскопичных соединений, то содержание воды или растворителя зависит от влажности и условий сушки. В этих случаях, как правило, наблюдают нестехиометрические сольваты.
Соединение формулы 1 также может существовать в виде многокомпонентного комплекса (отличного от солей и сольватов), в котором соединение (лекарственное средство) и по меньшей мере один другой компонент присутствуют в стехиометрических или нестехиометрических количествах. Комплексы этого типа включают клатраты (комплексы включения лекарственное средство-хозяин) и смешанные кристаллы. Последние, как правило, определены как кристаллические комплексы нейтральных молекулярных компонентов, которые связаны друг с другом посредством нековалентных взаимодействий, но также могут представлять собой комплексы нейтральной молекулы с солью. Смешанные кристаллы могут быть получены кристаллизацией из расплава, перекристаллизацией из растворителей или физическим измельчением компонентов друг с другом. См., например, O. Almarsson and M. J. Zaworotko, Chem. Commun. (2004) 17:1889-1896, содержание которой включено в настоящую заявку во всей полноте посредством ссылки. Общий обзор многокомпонентных комплексов см. в J. K. Haleblian, J. Pharm. Sci. (1975) 64(8):1269-88, содержание которой включено в настоящую заявку во всей полноте посредством ссылки.
При воздействии подходящих условий соединение формулы 1 может существовать в мезоморфном состоянии (мезофаза или жидкий кристалл). Мезоморфное состояние находится между истинным кристаллическим состоянием и истинным жидким состоянием (расплава или раствора). Мезоморфизм, возникающий в результате изменения температуры, называют «термотропным», а мезоморфизм, возникающий в результате добавления второго компонента, такого как вода или другой растворитель, называют «лиотропным». Соединения, которые могут образовывать лиотропные мезофазы, называются «амфифильными» и включают молекулы, содержащие полярный ионный фрагмент (например, -COOˉNa+, -COOˉK+, -SO3ˉNa+) или полярный неионный фрагмент (такой как -NˉN+(CH3)3). См., например, N. H. Hartshorne and A. Stuart, Crystals and the Polarizing Microscope (4е изд., 1970), содержание которой включено в настоящую заявку во всей полноте посредством ссылки.
Соединение формулы 1 может существовать в виде полиморфов, может содержать изотопные метки, может образовываться в результате введения пролекарства или может образовывать метаболиты после введения.
«Пролекарства» относятся к соединениям, обладающим незначительной фармакологической активностью или не имеющим ее, которые могут в результате метаболизма in vivo, претерпевать конверсию в соединения, обладающие желаемой фармакологической активностью. Пролекарства могут быть получены путем замены соответствующих функциональных групп, присутствующих в фармакологически активных соединениях, на «про-фрагменты», такие как описано, например, в H. Bundgaar, Design of Prodrugs (1985), содержание которой включено в настоящую заявку во всей полноте посредством ссылки. Примеры пролекарств включают сложноэфирные, простые эфирные или амидные производные соединения формулы 1, содержащего карбоксильные, гидрокси- или амино-функциональные группы, соответственно. Дополнительное обсуждение пролекарств см., например, в T. Higuchi and V. Stella “Pro-drugs as Novel Delivery Systems,” ACS Symposium Series 14 (1975), и E. B. Roche, ред., Bioreversible Carriers in Drug Design (1987), содержание каждой из которых включено в настоящую заявку во всей полноте посредством ссылки.
«Метаболиты» относятся к соединениям, образующимся in vivo после введения фармакологически активных соединений. Примеры включают гидроксиметильные, гидрокси-, вторичные амино-, первичные амино-, фенольные и карбоксильные производные соединения формулы 1, содержащего метильные, алкокси, третичные амино-, вторичные амино-, фенильные и амидные группы, соответственно.
Соединения формулы 1 могут содержать изотопные формы, в которых по меньшей мере один атом заменен на атом, имеющий такое же атомное число, атомная масса которого отличается от атомной массы, обычно встречающейся в природе. Изотопы, подходящие для включения в соединение формулы 1, включают, например, изотопы водорода, такие как 2H и 3H; изотопы углерода, такие как 11C, 13C и 14C; изотопы азота, такие как 13N и 15N; изотопы кислорода, такие как 15O, 17O и 18O; и изотопы фтора, такие как 18F. Применение изотопных форм (например, дейтерированных, 2H) может обеспечивать определенные терапевтические преимущества, определяемые повышенной метаболической стабильностью, например, повышенный период полувыведения in vivo или пониженные требования к дозировке. Кроме того, определенные изотопные формы описанных соединений могут включать радиоактивный изотоп (например, тритий 3H, или 14C), который можно применять в исследованиях распределения лекарственного средства и/или субстрата в тканях. Замещение на позитрон-излучающие изотопы, такие как 11C, 18F, 15O и 13N, можно применять в исследованиях позитронно-эмиссионной томографии (ПЭТ) для изучения степени занятости рецепторов субстратом. Изотопно-меченные соединения могут быть получены способами, аналогичными тем, что описаны в различных разделах изобретения, с использованием соответствующего изотопно-меченного реагента вместо немеченного реагента.
III. СПОСОБЫ ПОЛУЧЕНИЯ КОМПОЗИЦИИ СОГЛАСНО ИЗОБРЕТЕНИЮ
Соединение формулы 1 может быть получено способами, описанными ниже. На некоторых схемах и в примерах могут быть опущено подробное описание традиционных реакций, включая окисление, восстановление и т.д., способов разделения (экстракция, выпаривание, осаждение, хроматография, фильтрование, порошкование, кристаллизация и т.д.) и способов анализа, которые известны квалифицированным специалистам в области органической химии. Подробное описание указанных реакций и способов можно найти в разных трудах, включая Richard Larock, Comprehensive Organic Transformations (1999), и многотомную серию под редакцией Michael B. Smith и других, Compendium of Organic Synthetic Methods (1974 и далее), содержание каждой из которых включено в настоящую заявку во всей полноте посредством ссылки. Исходные вещества и реагенты могут быть получены из коммерческих источников или описанными в литературе способами. На некоторых схемах реакций могут быть опущены побочные продукты химических превращений (например, спирт при гидролизе сложного эфира, CO2 при декарбоксилировании двухосновной кислоты и т.д.). Кроме того, в некоторых случаях промежуточные соединения, полученные в реакции, можно применять на последующих стадиях, не проводя выделение или очистку (т.е. in situ).
На некоторых схемах реакций и в приведенных ниже примерах определенные соединения могут быть получены с использованием защитных групп, которые предотвращают нежелательные химические реакции на участках, которые в ином случае могли бы вступать в реакцию. Защитные группы также можно применять для повышения растворимости или иной модификации физических свойств соединения. Обсуждение стратегии использования защитных групп, описание материалов и способов для введения и удаления защитных групп и обобщенную информацию о подходящих защитных группах для часто используемых функциональных групп, включая амины, карбоновые кислоты, спирты, кетоны, альдегиды и т.д., см. в T. W. Greene and P. G. Wuts; Protective Groups in Organic Chemistry; 3е издание; John Wiley & Sons, Inc.; New York (1999), и P. Kocienski, Protective Groups, Georg Thieme, Stuttgart (2000), содержание каждой из которых включено в настоящую заявку во всей полноте посредством ссылки.
В общем случае, химические превращения, описанные в настоящем документе, можно проводить с использованием по существу стехиометрических количеств реагентов, хотя некоторые реакции могут проходить более эффективно при использовании избытка одного или более реагентов. Кроме того, многие реакции, описанные в настоящем документе, можно проводить примерно при комнатной температуре (КТ) и при давлении окружающей среды, но в зависимости от кинетики реакции, выхода и т.д. некоторые реакции можно проводить при повышенном давлении или с использованием повышенных температур (например, в условиях обратной конденсации) или пониженных температур (например, от минус 78 °C до 0 °C). Любое упоминание в изобретении и формуле изобретения стехиометрического диапазона, температурного диапазона, диапазона pH и т.д., независимо от того, используется напрямую слово «диапазон» или нет, также включает указанные конечные значения.
Во многих химических превращениях также можно применять один или более совместимых растворителей, которые могут влиять на скорость реакции и выход. В зависимости от природы реагентов один или более растворителей могут представлять собой полярные протонные растворители (включая воду), полярные апротонные растворители, неполярные растворители или некоторые их комбинации. Типовые растворители включают насыщенные алифатические углеводороды (например, н-пентан, н-гексан, н-гептан, н-октан, циклогексан, метилциклогексан); ароматические углеводороды (например, бензол, толуол, ксилолы); галогенированные углеводороды (например, метиленхлорид, хлороформ, тетрахлорид углерода); алифатические спирты (например, метанол, этанол, пропан-1-ол, пропан-2-ол, бутан-1-ол, 2-метилпропан-1-ол, бутан-2-ол, 2-метилпропан-2-ол, пентан-1-ол, 3-метилбутан-1-ол, гексан-1-ол, 2-метоксиэтанол, 2-этоксиэтанол, 2-бутоксиэтанол, 2-(2-метоксиэтокси)этанол, 2-(2-этоксиэтокси)этанол, 2-(2-бутоксиэтокси)этанол); простые эфиры (например, диэтиловый эфир, диизопропиловый эфир, дибутиловый эфир, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, 1-метокси-2-(2-метоксиэтокси)этан, 1-этокси-2-(2-этоксиэтокси)этан, тетрагидрофуран, 1,4-диоксан); кетоны (например, ацетон, метилэтилкетон); сложные эфиры (метилацетат, этилацетат); азотсодержащие растворители (например, формамид, N,N-диметилформамид, ацетонитрил, N-метилпирролидон, пиридин, хинолин, нитробензол); серосодержащие растворители (например, дисульфид углерода, диметилсульфоксид, тетрагидротиофен-1,1-диоксид); и фосфорсодержащие растворители (например, триамид гексаметилфосфора).
IV. СОСТАВЫ И ВВЕДЕНИЕ
Необходимо проводить оценку биофармацевтических свойств соединения формулы 1, включая его фармацевтически приемлемые комплексы, соли, сольваты и гидраты, таких как растворимость и стабильность раствора при различных pH, проницаемость и т.д., для выбора соответствующей лекарственной формы и способа введения. Соединение, предназначенное для фармацевтического применения, можно вводить в виде кристаллического или аморфного продукта, и можно получать, например, в виде твердых прессованных форм, порошков или пленок, способами, такими как осаждение, кристаллизация, лиофилизация, сушка распылением, сушка выпариванием, микроволновая сушка или радиочастотная сушка.
Соединение формулы 1 можно вводить отдельно или в комбинации с одним или более фармакологически активными соединениями. В общем случае, одно или более указанных соединений вводят в виде фармацевтической композиции (состава) совместно с одним или более фармацевтически приемлемыми вспомогательными веществами. Выбор вспомогательных веществ зависит от конкретного способа введения, влияния вспомогательного вещества на растворимость и стабильность и природы лекарственной формы, помимо прочих факторов. Подходящие фармацевтические композиции и способы их получения можно найти, например, в A. R. Gennaro (ред.), Remington: The Science and Practice of Pharmacy (20е изд., 2000).
Соединение формулы 1 можно вводить перорально. Пероральное введение может включать проглатывание, и в этом случае соединение поступает в кровоток через желудочно-кишечный тракт. В качестве альтернативы или в дополнение, пероральное введение может включать чресслизистое введение (например, трансбуккальное, подъязычное, надъязычное введение), в результате чего соединение поступает в кровоток через слизистую оболочку полости рта.
Составы, подходящие для перорального введения, включая твердые, полутвердые и жидкие системы, такие как таблетки; мягкие или твердые капсулы, содержащие дисперсные материалы, состоящие из множества отдельных частиц или наночастиц, жидкости или порошки; пастилки, которые могут содержать жидкий наполнитель; жевательные леденцы; гели; быстро диспергируемые лекарственные формы; пленки; вагинальные суппозитории; распыляемые составы; и трансбуккальные или мукоадгезивные пластыри. Жидкие составы включают суспензии, растворы, сиропы и эликсиры. Указанные составы могут применяться в качестве наполнителей в мягких или твердых капсулах (полученных, например, из желатина или гидроксипропилметилцеллюлозы) и, как правило, содержат носитель (например, воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло) и один или более эмульгаторов, суспендирующих агентов или оба вида указанных агентов. Жидкие составы также могут быть получены перерастворением твердого вещества (например, из порционного пакета).
Соединение формулы 1 также можно применять в быстрорастворимых быстрораспадающихся лекарственных формах, таких как те, что описаны в Liang and Chen, Expert Opinion in Therapeutic Patents (2001) 11(6):981-986, содержание которой включено в настоящую заявку во всей полноте посредством ссылки.
В случае таблетированных лекарственных форм в зависимости от дозы активный фармацевтический ингредиент (АФИ) может составлять от примерно 1 мас.% до примерно 80 мас.% лекарственной формы или чаще от примерно 5 мас.% до примерно 60 мас.% лекарственной формы. Помимо АФИ таблетки могут включать один или более разрыхлителей, связывающих веществ, разбавителей, поверхностно-активных веществ, глидантов, смазывающих веществ, антиоксидантов, красителей, вкусоароматических добавок, консервантов и веществ, исправляющих вкус. Примеры разрыхлителей включают натрия крахмал гликолят, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, C1-6 алкил-замещенную гидроксипропилцеллюлозу, крахмал, прежелатинизированный крахмал и альгинат натрия. В общем случае, разрыхлитель составляет от примерно 1 мас.% до примерно 25 мас.% или от примерно 5 мас.% до примерно 20 мас.% лекарственной формы.
Связывающие вещества, в общем случае, используют для придания когезивных качеств таблетированному составу. Подходящие связывающие соединения включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, натуральные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки также могут содержать разбавители, такие как лактоза (моногидрат, высушенный распылением моногидрат, безводная), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и дигидрат двухосновного фосфата кальция.
Таблетки также могут включать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и глиданты, такие как диоксид кремния и тальк. Поверхностно-активные вещества, если они содержатся, могут составлять от примерно 0,2 мас.% до примерно 5 мас.% таблетки, а глиданты могут составлять от примерно 0,2 мас.% до примерно 1 мас.% таблетки.
Таблетки также могут содержать смазывающие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Смазывающие вещества могут составлять от примерно 0,25 мас.% до примерно 10 мас.% или от примерно 0,5 мас.% до примерно 3 мас.% таблетки.
Смеси для получения таблеток можно прессовать непосредственно или с использованием прессующих роликов для получения таблеток. В качестве альтернативы, смеси для получения таблеток или части смесей можно гранулировать мокрым, сухим способом или из расплава, замораживать в расплаве или экструдировать перед получением таблеток. При желании перед смешением один или более компонентов можно доводить до нужного размера просеиванием или измельчением или обоими указанными способами. Конечная лекарственная форма может содержать один или более слоев и может содержать покрытие, не содержать покрытие или быть инкапсулированной. Типовые таблетки могут содержать вплоть до примерно 80 мас.% АФИ, от примерно 10 мас.% до примерно 90 мас.% связывающего вещества, от примерно 0 мас.% до примерно 85 мас.% разбавителя, от примерно 2 мас.% до примерно 10 мас.% разрыхлителя и от примерно 0,25 мас.% до примерно 10 мас.% смазывающего вещества. Обсуждение смешения, гранулирования, измельчения, просеивания, получения таблеток, нанесения покрытий, а также описание альтернативных способов получения лекарственных продуктов см. в A. R. Gennaro (ред.), Remington: The Science and Practice of Pharmacy (20е изд., 2000); H. A. Lieberman et al. (ред.), Pharmaceutical Dosage Forms: Tablets, Vol. 1-3 (2е изд., 1990); и D. K. Parikh & C. K. Parikh, Handbook of Pharmaceutical Granulation Technology, Vol. 81 (1997), содержание каждой из которых включено в настоящую заявку во всей полноте посредством ссылки.
Принимаемые внутрь пероральные пленки для применения у человека или в ветеринарии представляют собой мягкие водорастворимые или набухающие в воде тонкие пленочные лекарственные формы, которые могут быстро растворяться или обладают мукоадгезивными свойствами. Помимо АФИ типовая пленка включает один или более пленкообразующих полимеров, связывающих веществ, растворителей, смачивающих веществ, пластификаторов, стабилизаторов или эмульгаторов, агентов, модифицирующих вязкость, и растворителей. Другие ингредиенты пленок могут включать антиоксиданты, красители, вкусоароматические добавки и усилители вкуса, консерванты, агенты, стимулирующие выработку слюны, охлаждающие средства, сорастворители (включая масла), смягчающие средства, объемообразующие агенты, противопенные агенты, поверхностно-активные вещества и вещества, исправляющие вкус. Некоторые компоненты состава могут выполнять более чем одну функцию.
Помимо требований к введению количество АФИ в пленке может зависеть от его растворимости. Если АФИ является водорастворимым, то он, как правило, составляет от примерно 1 мас.% до примерно 80 мас.% компонентов, не являющихся растворителем (растворенных веществ), в пленке или от примерно 20 мас.% до примерно 50 мас.% растворенных веществ в пленке. Менее растворимый АФИ может иметь более высокое содержание в композиции, как правило, вплоть до примерно 88 мас.% от компонентов пленки, не являющихся растворителем.
Пленкообразующий полимер может быть выбран из натуральных полисахаридов, белков или синтетических гидроколлоидов и, как правило, составляет от примерно 0,01 мас.% до примерно 99 мас.% или от примерно 30 мас.% до примерно 80 мас.% пленки.
Пленочные лекарственные формы, как правило, получают путем сушки выпариванием тонких водных пленок, нанесенных на легко отслаивающуюся подложку или бумагу, которую можно проводить в сушильной печи или туннеле (например, в комбинированном аппарате для нанесения покрытия-сушки), в оборудовании для лиофилизации или в вакуумной печи.
Подходящие твердые составы для перорального введения могут включать составы с немедленным высвобождением и составы с модифицированным высвобождением. Составы с модифицированным высвобождением включают составы с отсроченным, замедленным, пульсирующим, контролируемым, направленным и запрограммированным высвобождением. Общее описание подходящих составов с модифицированным высвобождением см. в патенте США №6106864, содержание которого включено в настоящую заявку во всей полноте посредством ссылки. Подробное описание других подходящих технологий высвобождения, таких как использованием высокоэнергетических дисперсий и осмотических и содержащих покрытие частиц, см. в Verma et al, Pharmaceutical Technology On-line (2001) 25(2):1-14, содержание которой включено в настоящую заявку во всей полноте посредством ссылки.
Соединение формулы 1 также можно вводить напрямую в кровоток, в мышцу или во внутренний орган субъекта. Подходящие способы парентерального введения включают внутривенное, внутриартериальное, интраперитонеальное, интратекальное, внутрижелудочковое, внутриуретральное, внутригрудинное, интракраниальное, внутримышечное, внутрисуставное и подкожное введение. Подходящие устройства для парентерального введения включают игольные устройства для инъекций, включая микроигольные устройства для инъекций, безыгольные устройства для инъекций и устройства для инфузии.
Парентеральные составы, как правило, представляют собой водные растворы, которые могут содержать вспомогательные вещества, такие как соли, углеводы и буферные агенты (например, с pH от примерно 3 до примерно 9). Для некоторых применений, тем не менее, соединение формулы 1 может быть более эффективно получено в виде стерильного неводного раствора или в высушенной форме для использования совместно с подходящим наполнителем, таким как стерильная апирогенная вода. Парентеральные составы могут быть легко получены в стерильных условиях (например, путем лиофилизации) стандартными фармацевтическими способами.
Растворимость соединений, которые используют при получении парентеральных растворов, может быть повышена соответствующими способами получения составов, такими как введение агентов, повышающих растворимость. Составы для парентерального введения могут быть получены с возможностью немедленного или модифицированного высвобождения. Составы с модифицированным высвобождением включают составы с отсроченным, замедленным, пульсирующим, контролируемым, направленным и запрограммированным высвобождением. Таким образом, соединение формулы 1 может быть получено в виде суспензии, твердой, полутвердой формы или тиксотропной жидкости для введения в качестве имплантируемого депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры указанных составов включают стенты с покрытием лекарственного средства и полутвердые формы и суспензии, содержащие микросферы поли(DL-молочной-гликолевой)кислоты (PGLA), включающие лекарственное средство.
Соединение формулы 1 также можно вводить местно, внутрикожно или чрескожно на кожу или в слизистую. Типовые составы для этой задачи включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, кожные пластыри, имплантируемые капсулы, имплантаты, губки, волоконные материалы, бинты и микроэмульсии. Также можно применять липосомы. Типовые носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Местные составы также могут включать усилители проникновения. См., например, Finnin and Morgan, J. Pharm. Sci. 88(10):955-958 (1999), содержание которой включено в настоящую заявку во всей полноте посредством ссылки.
Другие средства для местного введения включают доставку путем электропорации, ионтофореза, фонофореза, сонофореза и микроигольной или безыгольной (например, POWDERJECT™ и BIOJECT™) инъекции. Составы для местного введения могут быть получены с возможностью немедленного или модифицированного высвобождения, как описано выше.
Соединение формулы 1 также можно вводить интраназально или путем ингаляции, как правило, в виде сухого порошка, распыляемого аэрозоля или назальных капель. Ингалятор можно применять для введения сухого порошка, содержащего только АФИ, порошковую смесь АФИ и разбавителя, такого как лактоза, или частицы, содержащие смесь компонентов, включающую АФИ и фосфолипид, такой как фосфатидилхолин. Порошок для интраназального применения может включать биоадгезивный агент, например, хитозан или циклодекстрин. Контейнер под давлением, помпу, распылитель, аэрозольный ингалятор или небулайзер можно применять для получения распыляемого аэрозоля из раствора или суспензии, содержащего(-ей) АФИ, один или более агентов для диспергирования, растворения или продления высвобождения АФИ (например, EtOH совместно с водой или без нее), один или более растворителей (например, 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан), которые выступают в качестве вытеснителя, и необязательно поверхностно-активное вещество, такое как сорбитана триолеат, олеиновая кислота или олигомолочная кислота. Аэрозольный электрогидродинамический ингалятор можно применять для получения мелкодисперсной распыляемой взвеси.
Перед применением в составе в виде сухого порошка или суспензии лекарственный продукт, как правило, измельчают до достижения размера частиц, подходящего для доставки путем ингаляции (как правило, чтобы 90 % частиц по объему имели самый большой размер менее 5 микрон). Это может быть обеспечено любым подходящим способом измельчения, таким как измельчение на спиральной струйной мельнице, измельчение на струйной мельнице с кипящим слоем, сверхкритическая флюидная обработка, гомогенизация под высоким давлением или сушка распылением.
Капсулы, блистеры и картриджи (изготовленные, например, из желатина или гидроксипропилметилцеллюлозы) для применения в ингаляторе или инсуффляторе получают таким образом, чтобы они могли содержать порошковую смесь активного соединения, подходящую порошковую основу, такую как лактоза или крахмал, и модификатор рабочих характеристик, такой как L-лейцин, маннит или стеарат магния. Лактоза может быть безводной или представлять собой моногидрат. Другие подходящие вспомогательные вещества включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу.
Подходящий состав в виде раствора для применения в аэрозольном электрогидродинамическом ингаляторе для получения тонкодисперсной распыляемой взвеси может содержать от примерно 1 мкг до примерно 20 мг АФИ при каждой активации устройства, а высвобождаемый при активации объем может составлять от примерно 1 мкл до примерно 100 мкл. Типовой состав может содержать соединение формулы 1, пропиленгликоль, стерильную воду, EtOH и NaCl. Альтернативные растворители, которые можно применять вместо пропиленгликоля, включают глицерин и полиэтиленгликоль.
Составы для ингаляционного введения, интраназального введения или обоих указанных способов введения могут быть получены с возможностью немедленного или модифицированного высвобождения, например, с использованием PGLA. В составы, предназначенные для ингаляционного/интраназального введения, можно добавлять подходящие вкусоароматические добавки, такие как ментол и левоментол, или подсластители, такие как сахарин или сахаринат натрия.
В случае ингаляторов сухих порошков и аэрозолей стандартная доза определяется при помощи клапана, который доставляет измеренное количество. Дозирующие блоки, как правило, выполнены с возможностью введения отмеренной дозы или «пшика», содержащей(-его) от примерно 10 мкг до примерно 1000 мкг АФИ. Общая дневная доза, как правило, находится в диапазоне от примерно 1 мг/кг до примерно 500 мг/кг и может быть введена в виде одной дозы или чаще в виде отдельных доз в течение дня.
Активные соединения можно вводить ректально или внутривагинально, например, в виде суппозитория, пессария или клизмы. Масло какао является традиционной основой для суппозиториев, но при необходимости можно использовать разные альтернативные варианты. Составы для ректального или внутривагинального введения могут быть получены с возможностью немедленного или модифицированного высвобождения, как описано выше.
Соединение формулы 1 также можно вводить напрямую в глаза или уши, как правило, в виде капель измельченной суспензии или раствора в изотоническом стерильном солевом растворе с контролируемым pH. Другие составы, подходящие для глазного и ушного введения, включают мази, гели, биоразлагаемые имплантаты (например, рассасывающиеся гелевые губки, коллаген), небиоразлагаемые имплантаты (например, из силикона), имплантируемые капсулы, линзы и дисперсные или пузырьковые системы, такие как ниосомы или липосомы. Состав может включать один или более полимеров и консервант, такой как хлорид бензалкония. Типовые полимеры включают поперечно-сшитую полиакриловую кислоту, поливиниловый спирт, гиалуроновую кислоту, целлюлозные полимеры (например, гидроксипропилметилцеллюлозу, гидроксиэтилцеллюлозу, метилцеллюлозу) и гетерополисахаридные полимеры (например, геллановую камедь). Указанные составы также могут быть доставлены путем ионтофореза. Составы для глазного или ушного введения могут быть получены с возможностью немедленного или модифицированного высвобождения, как описано выше.
Для улучшения растворимости, скорости растворения, исправления вкуса, биодоступности или стабильности соединение формулы 1 можно объединять с растворимыми макромолекулярными фрагментами, включая циклодекстрин или его производные и полимеры, содержащие полиэтиленгликоль. Например, комплексы АФИ-циклодекстрин, в общем случае, подходят для большинства лекарственных форм и способов введения. Можно применять как комплексы включения, так и комплексы, не являющиеся комплексами включения. В качестве альтернативы прямому комплексообразованию с АФИ можно применять циклодекстрин в качестве вспомогательной добавки, т.е. в качестве носителя, разбавителя или вещества, повышающего растворимость. Для этих задач обычно применяют альфа-, бета- и гамма-циклодекстрины. См., например, публикации WO 91/11172, WO 94/02518 и WO 98/55148, содержание каждой из которых включено в настоящую заявку во всей полноте посредством ссылки.
Как отмечалось выше, соединение формулы 1, включая его фармацевтически приемлемые комплексы, соли, сольваты и гидраты, можно применять в комбинации с одним или более другими фармацевтически активными соединениями для лечения различных заболеваний, нарушений и состояний. В этих случаях активные соединения могут быть объединены в одной лекарственной форме, как описано выше, или могут быть обеспечены в виде набора, который подходит для совместного введения композиций. Набор содержит (1) две или более различных фармацевтических композиций, по меньшей мере одна из которых содержит соединение формулы 1; и (2) устройство для раздельного хранения двух фармацевтических композиций, такое как бутыль с отделениями или пакет из фольги с отделениями. Примером такого набора является общеизвестная блистерная упаковка, которую используют при упаковывании таблеток или капсул. Набор подходит для введения различных типов лекарственных форм (например, пероральных и парентеральных) или для введения различных фармацевтических композиций с разными интервалами введения или для подбора относительных доз различных фармацевтических композиций. Для улучшения соблюдения пациентом схемы лечения набор, как правило, содержит руководство по введению и может быть обеспечен вместе с памяткой.
Для введения пациенту-человеку общая дневная доза предложенного соединения, как правило, находится в диапазоне от примерно 0,1 мг/кг до примерно 1,0 мг/кг или от примерно 0,5 мг/кг до примерно 5,0 мг/кг. В определенных вариантах реализации соединение или его фармацевтически приемлемую соль вводят в дозе в диапазоне от примерно 40 мг/кг до примерно 60 мг/кг. В определенных вариантах реализации соединение формулы 1 или его фармацевтически приемлемую соль, которые вводят в способе, описанном в настоящем документе, вводят в дозе, выбранной из группы диапазонов, состоящей из: от примерно 5 мг/кг до примерно 15 мг/кг, от примерно 10 мг/кг до примерно 20 мг/кг, от примерно 15 мг/кг до примерно 25 мг/кг, от примерно 20 до примерно 30 мг/кг, от примерно 25 мг/кг до примерно 35 мг/кг, от примерно 30 мг/кг до примерно 40 мг/кг, от примерно 35 мг/кг до примерно 45 мг/кг и от примерно 45 мг/кг до примерно 55 мг/кг. В определенных вариантах реализации доза находится в диапазоне, выбранном из группы, состоящей из: от примерно 30 мг/кг до примерно 40 мг/кг, от примерно 35 мг/кг до примерно 45 мг/кг, от примерно 40 мг/кг до примерно 50 мг/кг, от примерно 45 мг/кг до примерно 55 мг/кг, от примерно 50 мг/кг до примерно 60 мг/кг, от примерно 55 мг/кг до примерно 65 мг/кг и от примерно 60 мг/кг до примерно 70 мг/кг. В определенных вариантах реализации доза может составлять примерно 50 мг/кг. В определенных вариантах реализации доза составляет более чем примерно 1 мг/кг. В определенных вариантах реализации доза составляет примерно 1 мг/кг. В определенных вариантах реализации доза находится в диапазоне, выбранном из группы, состоящей из: от примерно 50 мг/кг до примерно 100 мг/кг, от примерно 100 мг/кг до примерно 150 мг/кг, от примерно 150 мг/кг до примерно 200 мг/кг, от примерно 200 мг/кг до примерно 250 мг/кг, от примерно 250 мг/кг до примерно 350 мг/кг, от примерно 300 мг/кг до примерно 350 мг/кг, от примерно 350 мг/кг до примерно 400 мг/кг, от примерно 400 мг/кг до примерно 450 мг/кг, от примерно 450 мг/кг до примерно 500 мг/кг. В определенных вариантах реализации доза составляет примерно 500 мг/кг. В качестве альтернативы доза составляет менее 500 мг/кг.
В определенных вариантах реализации доза составляет 35 мг/кг. В определенных вариантах реализации доза составляет 36 мг/кг. В определенных вариантах реализации доза составляет 37 мг/кг. В определенных вариантах реализации доза составляет 38 мг/кг. В определенных вариантах реализации доза составляет 39 мг/кг. В определенных вариантах реализации доза составляет 40 мг/кг. В определенных вариантах реализации доза составляет 41 мг/кг. В определенных вариантах реализации доза составляет 42 мг/кг. В определенных вариантах реализации доза составляет 43 мг/кг. В определенных вариантах реализации доза составляет 44 мг/кг. В определенных вариантах реализации доза составляет 45 мг/кг. В определенных вариантах реализации доза составляет 46 мг/кг. В определенных вариантах реализации доза составляет 47 мг/кг. В определенных вариантах реализации доза составляет 48 мг/кг. В определенных вариантах реализации доза составляет 49 мг/кг. В определенных вариантах реализации доза составляет 50 мг/кг. В определенных вариантах реализации доза составляет 51 мг/кг. В определенных вариантах реализации доза составляет 52 мг/кг. В определенных вариантах реализации доза составляет 53 мг/кг. В определенных вариантах реализации доза составляет 54 мг/кг. В определенных вариантах реализации доза составляет 55 мг/кг. В определенных вариантах реализации доза составляет 56 мг/кг. В определенных вариантах реализации доза составляет 57 мг/кг. В определенных вариантах реализации доза составляет 58 мг/кг. В определенных вариантах реализации доза составляет 59 мг/кг. В определенных вариантах реализации доза составляет 60 мг/кг. В определенных вариантах реализации доза составляет 61 мг/кг. В определенных вариантах реализации доза составляет 62 мг/кг. В определенных вариантах реализации доза составляет 63 мг/кг. В определенных вариантах реализации доза составляет 64 мг/кг. В определенных вариантах реализации доза составляет 65 мг/кг. Общую дневную дозу можно вводить в виде одной или нескольких отдельных доз, и по решению лечащего врача она может выходить за рамки типовых диапазонов, приведенных выше. Несмотря на то, что указанные дозировки указаны для среднестатистического субъекта-человека с массой тела от примерно 60 кг до примерно 70 кг, лечащий врач сможет определить соответствующую дозу для пациента (например, пациента детского возраста), масса которого выходит за рамки указанного диапазона массы тела.
Соединение формулы 1 можно применять для лечения заболеваний, нарушений и состояний, при которых показана модуляция GPR6. Как было отмечено выше, антагонизм или обратный агонизм GPR6, сопряженного с Gs, обеспечивает функциональную альтернативу активации рецепторов D2, опосредованной дофамином. Таким образом, соединения, которые модулируют активность GPR6, могут подходить для лечения разных неврологических и психиатрических расстройств, включая нарушения подвижности, такие как болезнь Паркинсона, дискинезии, вызванные леводопой, и болезнь Хантингтона, а также наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии и депрессии. В определенных вариантах реализации соединение формулы 1 улучшает моторные симптомы субъекта для излечения болезни Паркинсона. В определенных вариантах реализации соединение формулы 1 используют в комбинированной терапии совместно с амантадином для лечения указанных нарушений.
Патологическим признаком болезни Паркинсона является утрата нервных клеток в черном веществе. Дегенерация нигростриарного пути приводит к снижению концентрации дофамина в полосатом теле, которое клинически проявляется в виде моторных и немоторных симптомов. Многих пациентов с болезнью Паркинсона лечат леводопой, пролекарством дофамина. Леводопа часто вызывает тяжелые побочные эффекты, включая дискинезию, вызванную леводопой (LID), расстройства импульсивного контроля (ICD), психотические симптомы и нарушения сна. LID представляет собой прогрессирующее заболевание, и примерно у 90 % пациентов с болезнью Паркинсона LID развивается в течение 10 лет. В мышиных моделях LID происходят необратимые изменения сигнальной системы рецептора D1 в СШН, включая пониженную десенсибилизацию, которая приводит к повышенной чувствительности в прямом пути. Генетическая инактивация рецепторов D1, но не D2, устраняет LID у мышей. Тем не менее, блокада сигнальной системы рецептора D1 не влияет на эффективность леводопы в отношении болезни Паркинсона. Модель 6-OHDA имитирует многие аспекты болезни Паркинсона, включая утрату дофаминовой нейротрансмиссии и моторные расстройства.
Предложенное соединение можно применять в комбинации с одним или более другими фармакологически активными соединениями или способами терапии для лечения одного или более заболеваний, нарушений или состояний, связанных с GPR6. Указанные комбинации могут обеспечивать значительные терапевтические преимущества, включая пониженные побочные эффекты, улучшенную эффективность при лечении малообеспеченных популяций пациентов или синергическую активность. Например, соединение формулы 1, включая его фармацевтически приемлемые комплексы, соли, сольваты и гидраты, можно вводить одновременно, последовательно или раздельно в комбинации с одним или более соединениями или способами терапии для лечения двигательных расстройств, включая болезнь Паркинсона. Указанные соединения включают леводопу; ингибиторы DOPA-декарбоксилазы, такие как карбидопа, бенсеразид, метилдопа, α-дифторметил-DOPA, и 3',4',5,7-тетрагидрокси-8-метоксиизофлавон; агонисты дофамина, такие как апоморфина гидрохлорид, бромокриптин, ротиготин, прамипексол и ропинирол; амантадин; антихолинергические средства, такие как тригексифенидил и бензтропина мезилат; селективные ингибиторы моноаминоксидазы B (MAO-B), такие как сафинамид, селегилин и разагилин; и ингибиторы катехол-O-метилтрансферазы (COMT), такие как энтакапон и толкапон.
Помимо лекарственных средств, применяемых для лечения двигательных расстройств, соединение формулы 1 можно применять в комбинации с лекарственными средствами, применяемыми для лечения болезни Альцгеймера и других заболеваний, нарушений и состояний, влияющих на когнитивные функции. Указанные лекарственные средства включают ингибиторы бета-секретазы, ингибиторы гамма-секретазы, ингибиторы HMG-CoA-редуктазы, нестероидные противовоспалительные препараты (НПВП, такие как апазон, аспирин, целекоксиб, диклофенак (совместно с мизопростолом или без него), дифлунизал, этодолак, фенопрофен, флурбипрофен, ибупрофен, индометацин, кетопрофен, меклофенамат натрия, мефенаминовая кислота, мелоксикам, набуметон, напроксен, оксапрозин, фенилбутазон, пироксикам, холина и магния салицилаты, салсалат и сулиндак), витамин Е и антитела к амилоидам. Конкретные примеры соединений, применяемых для лечения болезни Альцгеймера, включают донепезил, ривастигмин, мемантин и галантамин.
В дополнение или в качестве альтернативы, соединение формулы 1 можно применять в комбинации с седативными средствами, снотворными средствами, нейролептическими средствами, антипсихотическими средствами, транквилизаторами и другими лекарственными средствами, применяемыми в лечении неврологических или психиатрических заболеваний. Например, соединение формулы 1 можно применять в комбинации с одним или более агентами для лечения депрессии (антидепрессанты) и/или шизофрении (атипичные или типичные антипсихотики), включая амитриптилин, амоксапин, арипипразол, азенапин, бупропион, хлордиазепоксид, циталопрам, хлорпромазин, клозапин, дезипрамин, десвенлафаксин, доксепин, дулоксетин, эсциталопрам, флуоксетин, флуоксетин, флуфеназин, галоперидол, илоперидон, имипрамин, изокарбоксазид, ламотригин, левомилнаципран, луразидон, миртазапин, нефазодон, нортриптилин, оланзапин, палиперидон, пароксетин, перфеназин, фенелзин, протриптилин, кветиапин, рисперидон, селегилин, сертралин, транилципромин, тразодон, тримипрамин, венлафаксин, вилазодон и вортиоксетин и зипрасидон.
Аналогично, соединение формулы 1 можно применять в комбинации с одним или более агентами для лечения тревоги (анксиолитические средства), включая бензодиазепины (алпразолам, хлордиазепоксид, клобазепам, клоназепам, клоразепат, диазепам, эстазолам, флуразепам, лоразепам, мидазолам, оксазепам, празепам, квазепам, темазепам и триазолам), антигистаминными средствами (гидроксизин), агентами, отличными от бензодиазепинов (эсзопиклон, залеплон, золпидем и зопиклон), и буспироном.
Соединение формулы 1 также можно применять в комбинации с одним или более агентами для лечения эпилепсии (антиэпилептические или противосудорожные средства), включая ацетазоламид, карбамазепин, клобазам, клоназепам, эсликарбазепина ацетат, этосуксимид, габапентин, лакозамид, ламотригин, леветирацетам, нитразепам, окскарбазепин, перампанел, пирацетам, фенобарбитал, фенитоин, прегабалин, примидон, ретигабин, руфинамид, натрия вальпроат, стирипентол, тиагабин, топирамат, вигабатрин и зонисамид.
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
Активность соединений в качестве модуляторов GPR6 может быть определена различными способами, включая способы in vitro и in vivo.
I. Ингибирование цАМФ in vitro (EC50)
В данном клеточном исследовании измеряют способность исследуемых соединений ингибировать конститутивную активность цАМФ рецептора GPR6, экспрессируемого клетками CHO-K1. Клетки CHO устойчиво экспрессируют рецептор GPR6, экспрессия которого контролируется элементом, индуцируемым тетрациклином. Выращивали клетки в среде, содержащей F12K, 10% ЭБС, 1% пенициллина/стрептомицина, 200 мкг/мл гигромицина. Рецептор GPR6 экспрессировали в течение 20 часов с использованием 2 мкг/мл доксициклина (Sigma D9891) в питательной среде. После завершения добавления доксициклина высеивали клетки с плотностью 450-750 клеток на лунку в 96-луночные черные планшеты для тканевых культур с половинным объемом лунок (Costar) и помещали в инкубатор (37 °C, 5% CO2) на 20 часов перед анализом цАМФ.
Удаляли питательную среду из клеток и промывали клетки 50 мкл/лунка буфером Рингера (MgCl2 0,047 мг/мл, NaH2PO4 0,18 мг/мл, Na2HPO4 0,1 мг/мл, KCl 0,34 мг/мл, NaHCO3 1,26 мг/мл, D-глюкоза 1,8 мг/мл, NaCl 7 мг/мл; pH=7,4). Суспендировали исследуемые соединения в ДМСО, разбавляли буфером Рингера, содержащим 0,5% БСА, не содержащего жирные кислоты, и 300 мкМ 3-изобутил-1-метилксантина (IBMX), и инкубировали совместно с клетками в течение 45 минут при 37 °C и 5% CO2. После инкубации обрабатывали клетки в течение 10 минут при комнатной температуре раствором с меткой Eu-цАМФ из набора для исследования цАМФ PerkinElmer LANCE® Ultra (TRF0263). Затем добавляли раствор ULIGHT™-anti-cAMP из набора LANCE® и инкубировали на шейкерепри комнатной температуре в течение 1 часа, после чего детектировали в режиме гомогенной флуоресценции с временным разрешением (HTRF) на анализаторе планшетов PerkinElmer EnVision. Строили кривые для определения EC50 при помощи четырехпараметрового логистического уравнения с использованием GraphPad Prism 5.03.
II. Ингибирование цАМФ in vitro (IC50)
В данном клеточном исследовании также измеряют способность соединений ингибировать конститутивную активность цАМФ рецептора GPR6, экспрессируемого клетками CHO-K1. Клетки CHO устойчиво экспрессируют рецептор GPR6, экспрессия которого контролируется элементом, индуцируемым тетрациклином. Выращивали клетки в среде, содержащей F12K, 10% ЭБС, 1% пенициллина/стрептомицина, 200 мкг/мл гигромицина. Рецептора GPR6 экспрессировали в течение 20 часов с использованием 1 мкг/мл доксициклина (Sigma D9891) в питательной среде. После завершения добавления доксициклина высеивали клетки с плотностью 250-500 клеток на лунку в черные планшеты с половинным объемом лунок и прозрачным дном (Costar) и помещали в инкубатор (37 °C, 5% CO2) на 20 часов перед анализом цАМФ.
Удаляли питательную среду из клеток и промывали клетки 50 мкл буфером Рингера (MgCl2 0,047 мг/мл, NaH2PO4 0,18 мг/мл, Na2HPO4 0,1 мг/мл, KCl 0,34 мг/мл, NaHCO3 1,26 мг/мл, D-глюкоза 1,8 мг/мл, NaCl 7 мг/мл; pH=7,4). Разбавляли соединения, суспендированные в ДМСО, буфером Рингера, содержащим 0,5% БСА, не содержащего жирные кислоты, и инкубировали совместно с клетками в течение 45 минут при 37 °C и 5% CO2. После инкубации инкубировали клетки в течение 10 минут при комнатной температуре совместно с раствором с меткой Eu-цАМФ из набора для исследования цАМФ PerkinElmer LANCE® Ultra (TRF0264). Затем добавляли раствор ULIGHT™-anti-cAMP из набора LANCE® и инкубировали на шейкере при комнатной температуре в течение 1 часа перед детектированием в режиме HTRF на анализаторе планшетов BMG POLARSTAR® Omega. Строили кривые для определения IC50 при помощи четырехпараметрового логистического уравнения с использованием GraphPad Prism 5.03.
III. Конкурентное связывание с GPR6 in vitro (Ki)
Исследование конкурентного связывания на основе фильтрования используют для изучения характеристик связывания обратных агонистов GPR6. В способе применяют мембраны, полученные из клеток CHO-K1, экспрессирующих человеческую кДНК GPR6 под действием индуцируемого доксициклином промотора. Готовые для исследования препараты в ДМСО в 96-луночных планшетах (651201, Greiner, USA), содержащих последовательно разбавленные исследуемые соединения (1 мкл исследуемого лиганда/лунка), получали с использованием жидкостных манипуляторов (5 мкМ, конечная максимальная концентрация в исследовании). Добавляли буфер для исследования (50 мМ Tris, pH 7,4, 50 мМ NaCl, 6 мМ MgCl2, 0,1% БСА, не содержащего жирные кислоты, 1:100 коктейль ингибиторов протеиназы, Sigma USA) (39 мкл/лунка) и перемешивали планшеты на шейкере планшетов в течение 10 минут. Готовили GPR6-специфический 3H-радиолиганд в буфере для исследования и добавляли в каждую лунку (40 мкл, 2,4 нМ конечная концентрация в исследовании).
Для запуска реакций связывания добавляли 40 мкл цельных мембран, полученных из клеток, экспрессирующих человеческие рецепторы GPR6. Готовили препараты мембран в буфере для исследования и добавляли в лунки до достижения конечной концентрации в исследовании 15 мкг/лунка. Закрывали планшеты, перемешивали в течение 30 секунд при 300 об./мин и инкубировали в течение 2 часов при комнатной температуре. Затем фильтровали реакционные смеси через систему FilterMate (1450-421, FilterMate A, PerkinElmer, USA) и промывали 5 раз буфером (50 мМ Tris, pH 7,4, 50 мМ NaCl, 6 мМ MgCl2, 0,1% БСА, не содержащего жирные кислоты) при помощи инструмента Tomtec HARVERSTER96™. Сушили фильтры в микроволновой печи. На фильтрах плавили сцинтилляционные листы (1450-411, PerkinElmer, USA) и запаивали их перед количественной оценкой CPM/лунка на инструменте MICROBETA® Trilux (PerkinElmer, USA). Перед использованием вымачивали систему FilterMate в 0,5% растворе полиэтиленимина в течение 3 часов при осторожном встряхивании, после чего сушили на воздухе в течение ночи. Вычисляли значения IC50 и Ki при помощи анализа нелинейной регрессии в Prism (GraphPad, USA). Определяли значения Kd в стандартных экспериментах с насыщением радиолиганда.
IV. Модель болезни Паркинсона in vivo - каталепсия, вызванная галоперидолом
Моторные симптомы болезни Паркинсона включают акинезию, брадикинезию, оцепенелость, тремор и нарушения осанки и связаны с утратой нигральных дофаминергических клеток и снижением уровня дофамина в полосатом теле. Введение галоперидола грызунам приводит к временному состоянию, схожему с болезнью Паркинсона, которое купируется при введении леводопы и других лекарственных средств, которые были клинические одобрены для лечения болезни Паркинсона. См. Duty, S. & Jenner, P. Br. J. Pharmacol. 164:1357-1391 (2011), содержание которой включено в настоящую заявку во всей полноте посредством ссылки. Галоперидол действует как антагонист дофаминовых рецепторов D2 и в меньшей степени дофаминовых рецепторов D1 в средних шипиковых нейронах, которые включают непрямые и прямые пути моторного контура, соответственно. Возникающая в результате блокада дофаминовой трансмиссии в полосатом теле приводит к нарушенным последующим «выстрелам» нейронов в контурах базальных ганглиев, которые проявляются в виде симптомов мышечной оцепенелости и каталепсии. Было установлено, что каталепсия отражает клинические признаки болезни Паркинсона, когда пациенты отмечают неспособность к осуществлению движений.
Использовали самцов мышей C57Bl6 с массой тела 25-35 г. Каталепсию вызывали путем подкожного (п.к.) введения антагониста дофаминовых рецепторов галоперидола (0,45 мг/кг) по меньшей мере за 30 минут перед исследованием животных на вертикальной сетке. Для этого испытания крыс или мышей помещали на проволочную крышку плексигласовой клетки размером 25 см × 43 см, размещенной под углом примерно 70 градусов относительно лабораторного стола. Субъекта помещали на сетку, отводя и вытягивая все четыре лапы («поза лягушки»). Использование такой неестественной позы критически важно для специфики данного исследования каталепсии. Временной интервал от момента размещения лап крысы в такой позе до полного удаления первой лапы (время до начала спуска) измеряли не более 120 секунд. В случае мышей помещали передние лапы на расположенный горизонтально металлический стержень, поднятый на 2 дюйма (5 см) относительно платформы из плексигласа, и отмечали время, но не более 30 секунд для каждого испытания. Завершали исследование, когда животное возвращало передние лапы на платформу, или через 30 секунд. Испытание повторяли три раза и указывали среднее значение трех испытаний как индекс интенсивности каталепсии. Животных оценивали через 30 минут после введения дозы, а повторную оценку проводили через 60 или 90 минут после введения дозы галоперидола.
Эффективность модуляторов GPR6 в отношении купирования каталепсии, вызванной галоперидолом, измеряли через 30 минут, 60 минут и/или 90 минут после введения субъектам 0,45 мг/кг и.п. (интраперитонеальная инъекция) галоперидола совместно с исследуемым соединением-модулятором GPR6. Соединение формулы 1 вводили в дозе в диапазоне от 0,1 до 100 мг/кг (перорально/п.о. в 0,5% метилцеллюлозе) совместно с галоперидолом. Антагонист аденозина A2A SCH 420814 (преладенант) вводили в дозе 3 мг/кг и.п. в качестве положительного контроля.
V. Ингибирование человеческого hERG способом фиксации потенциала (patch-clamp)
Автоматизированную цельноклеточную систему фиксации потенциала (QPATCH® 16) использовали для регистрации калиевых токов, выходящих из одной клетки. В исследовании использовали клетки CHO-K1 (клетки яичников китайских хомячков), устойчиво трансфицированные кДНК человеческого hERG. Собирали клетки после обработки трипсином и выдерживали в бессывороточной среде при комнатной температуре перед анализом. Промывали клетки и повторно суспендировали во внеклеточном растворе, после чего наносили на участки для автоматизированной фиксации потенциала. Готовили исследуемые растворы в водном внеклеточном растворе (137 мМ NaCl, 4 мМ KCl, 1,8 мМ CaCl2, 1 мМ MgCl2, 10 мМ D(+)-глюкоза, 10 мМ HEPES, pH доводили до 7,4 при помощи NaOH) в день исследования фиксации потенциала. Исследуемое соединение в семи концентрациях (0,03, 0,1, 0,3, 1, 3, 10 и 30 мкМ) использовали для определения IC50. Водный внутриклеточный раствор содержал 130 мМ KCl, 10 мМ NaCl, 1 мМ MgCl2, 10 мМ ЭГТА, 5 мМ Mg-АТФ и 10 мМ HEPES (pH доводили до 7,2 при помощи KOH).
После достижения конфигурации целой клетки выдерживали клетку при -80 мВ. Доставляли -40 мВ импульсы продолжительностью 50 мс для измерения тока утечки, который вычитали из следового тока в режиме онлайн. Затем деполяризовали клетку до +20 мВ в течение 2 секунд, после чего доставляли 1-секундный -40 мВ импульс для определения следового тока hERG. Такую последовательность импульсов доставляли каждые 5 секунд для отслеживания амплитуды тока. Исследование проводили при комнатной температуре. Сначала использовали внеклеточный раствор (контроль) и стабилизировали клетку в растворе в течение 5 минут. Затем на ту же клетку наносили исследуемое соединение последовательно в концентрации от низкой до высокой. Инкубировали клетки совместно с соединением в каждой исследуемой концентрации в течение 5 минут. Одновременно с этим исследовали соединение сравнения E-4031 (N-(4-(1-(2-(6-метилпиридин-2-ил)этил)пиперидин-4-карбонил)фенил)метансульфонамид) в нескольких концентрациях для получения значения IC50. Ингибирование в процентах канала hERG вычисляли путем сравнения амплитуды следового тока до и после нанесения соединения (разницу токов нормировали по контролю).
ПРИМЕРЫ
Следующие примеры являются иллюстративными и неограничивающими и представляют конкретные варианты реализации настоящего изобретения.
I. 1H ядерный магнитный резонанс (ЯМР)
Спектры 1H ЯМР получали для многих соединений, приведенных в последующих примерах. Характеристические химические сдвиги (δ) указаны в миллионных долях относительно сигнала тетраметилсилана с использованием традиционных сокращений для обозначения основных пиков, включая s (синглет), d (дуплет), t (триплет), q (квартет), m (мультиплет) и шир. (широкий). Следующие сокращения используют для описания традиционных растворителей: CDCl3 (дейтерированный хлороформ), ДМСО-d6 (дейтерированный диметилсульфоксид), CD3OD (дейтерированный метанол), CD3CN (дейтерированный ацетонитрил) и ТГФ-d8 (дейтерированный тетрагидрофуран). Масс-спектры (m/z для [M+H]+) получали путем масс-спектрометрии с ионизацией электронным распылением (ИЭР-МС) или химической ионизацией при атмосферном давлении (ХИАД-МС).
II. Высокоэффективная жидкостная хроматография (ВЭЖХ)
В тех случаях, где это указано, продукты, полученные в определенных примерах получения и примерах, очищали на системе ВЭЖХ, активируемой в определенном диапазоне масс (например, насос: WATER™ 2525; МС: ZQ™; программное обеспечение: MASSLYNX™), путем флэш-хроматографии или препаративной тонкослойной хроматографии (ТСХ). Обращенно-фазовую хроматографию, как правило, проводили на колонке (например, Phenomenex GEMINI™ 5 мкм, C18, 30 мм × 150 мм; AXIA™, 5 мкм, 30 мм × 75 мм) в кислотных условиях («кислотный режим»), элюируя подвижными фазами на основе CH3CN и воды, содержащими 0,035% и 0,05% трифторуксусной кислоты (ТФУК), соответственно, или в основных условиях («основный режим»), элюируя подвижными фазами на основе воды и 20/80 (об./об.) смеси вода/ацетонитрил, каждая из которых содержала 10 мМ NH4HCO3. Препаративную ТСХ, как правило, проводили на пластинах силикагеля 60 F254. После выделения путем хроматографии удаляли растворитель и получали продукт путем сушки на центрифужном испарителе (например, GeneVac™), роторном испарителе, в вакуумируемой колбе и т.д. Реакции в инертной атмосфере (например, азота) или реакционноактивной атмосфере (например, H2), как правило, проводили под давлением примерно 1 атмосфера (14,7 psi).
III. Синтез
ПРИМЕР ПОЛУЧЕНИЯ 1: (R)-2-(4-(2,4-дифторфенокси)пиперидин-1-ил)-N-(тетрагидрофуран-3-ил)пиридо[3,4-b]пиразин-3-амин
В раствор 3-хлор-2-(4-(2,4-дифторфенокси)пиперидин-1-ил)пиридо[3,4-b]пиразина (10 г, 26,5 ммоль) в ДМСО (50 мл) добавляли (R)-тетрагидрофуран-3-амин (ArkPharm, AK-75910, № партии WZG082316-PB01) (5,32 мл, 61,0 ммоль). Грели раствор при 70 °C в течение 10 часов, затем разбавляли водой (300 мл) и экстрагировали iPrOAc (300 мл). Дополнительно экстрагировали водную фазу iPrOAc (100 мл). Объединяли органические слои, промывали насыщенным водным NH4Cl (300 мл) и солевым раствором (200 мл), сушили над MgSO4, концентрировали в вакууме и сушили на бытовом вакуумном насосе с получением светло-желтого твердого вещества (11,4 г). Растворяли твердое вещество в iPrOAc (55 мл) при перемешивании и нагревали до температуры обратной конденсации. Медленно по частям добавляли гептан (33 мл) при нагревании для предотвращения осаждения. Затем оставляли раствор охлаждаться до 20 °C при перемешивании (примерно 400 об./мин), за это время образовывался осадок. Медленно охлаждали смесь до температуры окружающей среды и перемешивали в течение ночи. Собирали твердое вещество путем вакуумного фильтрования, промывали ледяным 20% iPrOAc в гептане, сушили, вакуумируя осадок на фильтре, в течение по меньшей мере 30 минут и собирали с получением титульного соединения в виде светло-желтого твердого вещества (9,771 г, 86 %). 1H ЯМР (500 МГц, ДМСО-d6) δ ppm 1,84 - 1,96 (m, 2H), 2,01 - 2,16 (m, 3H), 2,20 - 2,31 (m, 1H), 3,22 - 3,31 (m, 2H), 3,64 - 3,80 (m, 4H), 3,85 - 3,93 (m, 1H), 4,00 (dd, J=9,28, 6,35 Гц, 1H), 4,55 - 4,67 (m, 2H), 6,94 (d, J=5,86 Гц, 1H), 6,99 - 7,06 (m, 1H), 7,25 - 7,37 (m, 2H), 7,46 (d, J=5,37 Гц, 1H), 8,31 (d, J=5,37 Гц, 1H), 8,79 (s, 1H); ИЭР-МС m/z [M+H]+ 428.
ПРИМЕР ПОЛУЧЕНИЯ 2: (S)-2-(4-(2,4-дифторфенокси)пиперидин-1-ил)-N-(тетрагидрофуран-3-ил)пиридо[3,4-b]пиразин-3-амин
В раствор 3-хлор-2-(4-(2,4-дифторфенокси)пиперидин-1-ил)пиридо[3,4-b]пиразина (1,0 г, 2,65 ммоль) в ДМСО (5 мл) добавляли (S)-тетрагидрофуран-3-амин (AstaTech, кат.№37021) (0,578 мл, 6,64 ммоль). Грели раствор при 70 °C в закрытой пробирке для микроволнового реактора в течение 22 часов, после чего анализ ВЭЖХ-МС указывал на завершение реакции. Разбавляли реакционную смесь (5 мл) водой (150 мл) и экстрагировали iPrOAc (150 мл). Дополнительно экстрагировали водную фазу iPrOAc (50 мл). Объединяли органические слои, промывали насыщенным водным NH4Cl (150 мл) и солевым раствором (100 мл), сушили над MgSO4 и концентрировали в вакууме на CELITE®. Очищали неочищенный продукт путем колоночной хроматографии (30 г колонка с силикагелем NH), элюируя с градиентом от 0 до 60% смесями EtOAc в гептане, с получением титульного соединения в виде белого твердого вещества (1,05 г, 93 %). 1H ЯМР (500 МГц, ДМСО-d6) δ ppm 1,90 (td, J=8,54, 3,91 Гц, 2H), 2,01 - 2,16 (m, 3H), 2,20 - 2,31 (m, 1H), 3,21 - 3,32 (m, 2H), 3,63 - 3,80 (m, 4H), 3,89 (q, J=7,49 Гц, 1H), 4,00 (dd, J=9,28, 6,35 Гц, 1H), 4,54 - 4,67 (m, 2H), 6,96 (d, J=6,35 Гц, 1H), 7,00 - 7,08 (m, 1H), 7,26 - 7,39 (m, 2H), 7,46 (d, J=5,37 Гц, 1H), 8,31 (d, J=5,37 Гц, 1H), 8,79 (s, 1H); ИЭР-МС m/z [M+H]+ 428.
ПРИМЕР 1: (R)-1-(2-(4-(2,4-дифторфенокси)пиперидин-1-ил)-3-((тетрагидрофуран-3-ил)амино)-7,8-дигидропиридо[3,4-b]пиразин-6(5H)-ил)этан-1-он
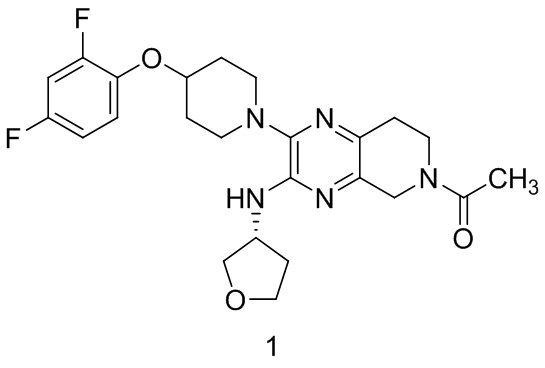
В колбу, содержащую (R)-2-(4-(2,4-дифторфенокси)пиперидин-1-ил)-N-(тетрагидрофуран-3-ил)пиридо[3,4-b]пиразин-3-амин (16 г, 37,4 ммоль) в HOAc (80 мл) и ТГФ (80 мл), добавляли ангидрид уксусной кислоты (17,66 мл, 187 ммоль) в атмосфере азота. Добавляли палладий на углеродной подложке (10%, Aldrich 205699-10G, № партии MKBZ3284V) (3,19 г, 2,99 ммоль) в атмосфере азота. Присоединяли колбу к баллону, наполненному водородом, и восемь раз вакуумировали с использованием бытового вакуумного насоса и повторно заполняли водородом. Перемешивали реакционную смесь в атмосфере водорода в течение 40 часов, а затем фильтровали через подложку CELITE®, стараясь не допускать высыхание осадка. Промывали колбу и осадок на фильтре EtOAc (48 мл), метанолом (48 мл) и EtOAc (48 мл). Концентрировали фильтрат в вакууме для удаления ТГФ, EtOAc и метанола (температура бани меньше или равна 40 °C). Разбавляли раствор гептаном (480 мл) и повторно концентрировали в вакууме для удаления HOAc с помощью азеотропной перегонки (температура бани меньше или равна 45 °C). Помещали остаток в iPrOAc (320 мл), промывали 10 мас.% водным K2CO3 (320 мл, 230 ммоль) (pH 13 до промывки, pH 10 после промывки) и солевым раствором (240 мл, pH 7 после промывки), сушили над MgSO4, концентрировали в вакууме и сушили с использованием бытового вакуумного насоса в течение по меньшей мере 1 часа с получением светло-желтого твердого вещества (16,71 г). Помещали неочищенный продукт в этанол (84 мл) и нагревали на масляной бане при перемешивании. После растворения твердых веществ оставляли раствор медленно охлаждаться на масляной бане при перемешивании, в это время начинал образовываться осадок, и раствор становился мутным. Оставляли смесь охлаждаться до температуры окружающей среды на масляной бане и перемешивали в течение ночи. Собирали белое твердое вещество после перекристаллизации путем вакуумного фильтрования, промывали ледяным этанолом и сушили в глубоком вакууме с получением титульного соединения в виде белого твердого вещества (13,34 г, 75 %). 1H ЯМР (500 МГц, ДМСО-d6) δ ppm 1,81 - 2,00 (m, 3H), 2,02 - 2,12 (m, 5H), 2,14 - 2,24 (m, 1H), 2,60 (t, J=5,61 Гц, 1H), 2,72 (t, J=5,86 Гц, 1H), 2,84 - 2,96 (m, 2H), 3,26 - 3,32 (m, 2H), 3,56 (dt, J=8,79, 5,13 Гц, 1H), 3,65 - 3,78 (m, 3H), 3,81 - 3,94 (m, 2H), 4,33 - 4,47 (m, 3H), 4,52 (tt, J=8,18, 4,03 Гц, 1H), 5,91 (dd, J=13,42, 6,10 Гц, 1H), 6,97 - 7,05 (m, 1H), 7,24 - 7,36 (m, 2H); ИЭР-МС m/z [M+H]+ 474; Тпл 150 °C (пик ДСК); хиральная чистота (хиральная колоночная хроматография) больше 98 % э.и.
СОЕДИНЕНИЕ А: (S)-1-(2-(4-(2,4-дифторфенокси)пиперидин-1-ил)-3-((тетрагидрофуран-3-ил)амино)-7,8-дигидропиридо[3,4-b]пиразин-6(5H)-ил)этан-1-он
В колбу, содержащую (S)-2-(4-(2,4-дифторфенокси)пиперидин-1-ил)-N-(тетрагидрофуран-3-ил)пиридо[3,4-b]пиразин-3-амин (1,05 г, 2,456 ммоль) в HOAc (5 мл) и ТГФ (5 мл), в атмосфере азота добавляли ангидрид уксусной кислоты (1,159 мл, 12,28 ммоль). Добавляли палладий на углеродной подложке (10%, Aldrich 205699-10G, № партии MKBZ3284V) (0,523 г, 0,491 ммоль) в атмосфере азота. Присоединяли колбу к баллону, наполненному водородом, и восемь раз вакуумировали с использованием бытового вакуумного насоса и повторно заполняли водородом. Перемешивали реакционную смесь в атмосфере водорода в течение 18 часов, а затем фильтровали через подложку Celite®, стараясь не допускать высыхание осадка. Промывали колбу и осадок на фильтре EtOAc (20 мл), метанолом (20 мл) и EtOAc (20 мл) и концентрировали фильтрат в вакууме на CELITE®. Очищали неочищенный продукт путем колоночной хроматографии (120 г колонка с силикагелем NH, размер 200), элюируя с градиентом от 0 до 60 % смесями EtOAc в гептане, с получением белого твердого вещества (1,0 г). Помещали белое твердое вещество в этанол (5 мл) и нагревали до 80 °C на масляной бане при перемешивании. После растворения твердых веществ убирали источник нагревания и оставляли раствор медленно охлаждаться на масляной бане до 20 °C при перемешивании. Перемешивали смесь в течение 3 дней при комнатной температуре. Собирали твердые вещества путем вакуумного фильтрования, промывали ледяным этанолом и сушили в глубоком вакууме с получением титульного соединения в виде белого твердого вещества (848 мг, 72,9 %). 1H ЯМР (500 МГц, ДМСО-d6) δ ppm 1,82 - 2,00 (m, 3H), 2,02 - 2,12 (m, 5H), 2,13 - 2,24 (m, 1H), 2,60 (t, J=5,86 Гц, 1H), 2,72 (t, J=5,86 Гц, 1H), 2,83 - 2,96 (m, 2H), 3,24 - 3,32 (m, 2H), 3,56 (dt, J=8,79, 5,25 Гц, 1H), 3,66 - 3,77 (m, 3H), 3,81 - 3,94 (m, 2H), 4,33 - 4,47 (m, 3H), 4,52 (tt, J=8,08, 3,87 Гц, 1H), 5,93 (dd, J=13,30, 6,22 Гц, 1H), 6,98 - 7,06 (m, 1H), 7,24 - 7,35 (m, 2H); ИЭР-МС m/z [M+H]+ 474; Тпл 149 °C (пик ДСК); хиральная чистота (хиральная колоночная хроматография) больше 98 % э.и.
IV. Исследование ингибирования цАМФ in vitro (EC50)
В таблице 1 приведены данные биологического исследования (ингибирование цАМФ in vitro) соединения формулы 1 (пример 1) и соединения A, которые исследовали в клеточном исследовании, в котором измеряли способность исследуемых соединений ингибировать конститутивную активность цАМФ рецептора GPR6, экспрессируемого клетками CHO-K1 (указана как pEC50). Исследование было описано в настоящем документе под заголовком «Ингибирование цАМФ in vitro (EC50)».
Таблица 1. Ингибирование конститутивной активности цАМФ рецептора GPR6 in vitro
Проводили дополнительный анализ для определения IC50 соединений в исследовании, описанном в настоящем документе под заголовком «Ингибирование цАМФ in vitro (IC50)».
V. Исследование ингибирования цАМФ in vitro (IC50)
В таблице 2 приведены константы ингибирования (Ki), определенные в исследовании конкурентного связывания GPR6, и значения IC50, определенные в функциональном исследовании hERG, для соединения формулы 1 (пример 1) и соединения A. Как было описано выше, Ki для каждого соединения определяли в исследовании конкурентного связывания в формате фильтрования, в котором использовали мембраны, полученные из клеток CHO-K1, экспрессирующих кДНК человеческого GPR6; IC50 для каждого соединения определяли в функциональном исследовании hERG, в котором использовали автоматизированную цельноклеточную систему фиксации потенциала и клетки CHO-K1, трансфицированные кДНК человеческого hERG.
Данные, приведенные в таблице 2, указывают на то, что соединение формулы 1 является значительно менее активным ингибитором hERG, чем соединение A. Если при максимальной концентрации свободного соединения (лекарственного средства) предполагается 50% занятость GPR6, то отношение (hERG IC50)/Ki, приведенное в таблице 2, можно рассматривать в качестве резерва безопасности для свободного лекарственного средства («SM-своб»), как определено в литературных источниках (см. X. Yao et al., British Journal of Pharmacology (2008) 154:1446-56, 50, содержание которой включено в настоящую заявку во всей полноте посредством ссылки). Если сравнивать резервы безопасности, то соединение формулы 1 имеет улучшенное значение SM-своб. для перехода на стадию исследований in vivo. Значение SM-своб. менее 300 рассматривают как подходящее для перехода на стадию исследований in vivo, согласно определению, данному в работе Яо с соавторами (Yao et al) на странице 1452 («данные позволяют предположить, что значение SM-своб. 300 или более, может обозначать, что соединение не имеет потенциала для удлинения интервала QTc»).
Таблица 2. Конкурентное связывание GPR6 (Ki) и ингибирование hERG (IC50)
Также анализировали способность соединения формулы 1 (пример 1) и соединения А купировать вызванную галоперидолом каталепсию. Соединения изучали согласно исследованию, описанному в настоящем документе под заголовком «Модель болезни Паркинсона in vivo - каталепсия, вызванная галоперидолом».
VI. Лечение в крысиной модели 6-гидроксидофамина (6-OHDA)
Соединения, которые улучшают моторную активность, рассматриваются в качестве возможных способов терапии болезни Паркинсона. Крысам в рамках модели 6-OHDA болезни Паркинсона проводили двусторонние инъекции 6-OHDA в полосатое тело с координатами по передне-задней оси (AP): медиально-латеральной оси (ML): верхней-нижней оси (DV), 1±3, -5 мм относительно брегмы под анестезией. Исследовали локомоторную активность крыс по меньшей мере через 28 дней. Животных оставляли для привыкания на 30 минут в боксах для измерения локомоторной активности перед введением соединения формулы 1 (пример 1) или носителя (контроль).
Локомоторную активность измеряли на открытой площадке, покрытой сеткой инфракрасных лучей. Проводили анализ прерывания лучей животными с использованием программного обеспечения AMLOGGER для отслеживания активности. Соединение формулы 1 (пример 1) обеспечивало зависящее от дозы улучшение локомоторной активности в обеих дозировках (5 мг/кг и 10 мг/кг) по сравнению с крысами, которым вводили носитель (контроль), через 50 минут, что показано на фигуре 1. У крыс, которым вводили 10 мг/кг дозу, наблюдали значительное увеличение активности. Полученные данные указывают на то, что соединение формулы 1 (пример 1) может эффективно облегчать симптомы у пациентов с болезнью Паркинсона.
VII. Влияние соединения формулы 1 (пример 1) на сердечно-сосудистую систему, определенное телеметрическим способом
Возможные эффекты на сердечно-сосудистую систему соединения формулы 1 (пример 1) изучали на бодрствующих собаках породы бигль (Marsahall BioResources, North Rose, NY) возрастом как минимум 8 месяцев с массой тела 7-15 кг. Биглям вживляли телеметрические устройства в передающих имплантатах DSI PHYSIOTEL® Digital L21 согласно соответствующим стандартным операционным процедурам Charles River Laboratories, Montreal, QC (TB 12-04-06). Электроды для измерения биопотенциала размещали в модифицированной II конфигурации электродов. Минимальный допустимый период акклиматизации между поступлением животного и началом хирургической имплантации телеметрических устройств составлял 6 дней, чтобы приучить животных к условиям лаборатории. Минимальный допустимый период восстановления между хирургией и началом лечения составлял 4 недели. Целевые условия содержания животных включали температуру от 17 °C до 23 °C при 30-70 % влажности и цикл 12 часов день и 12 часов ночь за исключением проведения обозначенных процедур.
Животных можно размещать группами в клетки из нержавеющей стали, оборудованные автоматическими клапанами для подачи воды, как описано в Руководстве по содержанию и уходу за лабораторными животными (8е изд., National Academies Press, 2111), содержание которой включено в настоящую заявку во всей полноте посредством ссылки, за исключением периодов введения и мониторинга, когда животных помещали в клетки по отдельности.
В течение 7 дней после хирургической имплантации цифровых передатчиков DSI PHYSIOTEL® Digital L21 животным давали кормовые добавки, которые включали 1 банку корма AID Prescription Diet совместно с 300 г международного сертифицированного корма для собак PMI NUTRITION® International Certified Canine Chow №5007 в течение 3 дней и 1 банку корма AID Prescription Diet совместно с 300 г международного сертифицированного корма для собак PMI NUTRITION® International Certified Canine Chow №5007 в течение 4 дней. В день введения доступ к корму обеспечивали по меньшей мере за 5 часов перед введением, предоставляли доступ на 1 час, а затем убирали корм по меньшей мере за 4 часа перед введением. Мясные лакомства были недоступны после удаления корма в течение 4 часов перед введением. Корм, если он оставался, возвращали после завершения отслеживания смертности/заболеваемости в день введения и оставляли на ночь. Дополнительный корм предлагали животным по мере необходимости согласно клиническим признакам или другим изменениям.
Водопроводная вода после обработки обратным осмосом и ультрафиолетовым излучением предоставлялась в свободном доступе каждому животному через автоматическую систему подачи воды за исключением проведения обозначенных процедур. При необходимости были обеспечены поилки. Периодически проводили анализ воды, и результаты этих анализов были доступны на исследуемых установках. Какие-либо примеси, которые заведомо могли бы помешать задачам исследования, в воде отмечены не были.
Перед началом введения получали данные ЭКГ, LVP, кровяного давления и температуры тела с использованием системы Data Sciences International (DSI) PONEMAH™ по меньшей мере за 24 часа для оценки параметров сердечно-сосудистой системы и качества сигналов на ЭКГ. ЭКГ снимали как минимум в течение 30 секунд и направляли кардиологу для качественной оценки. Только животных с нормальными параметрами гематологии/клинической химии и нормальными параметрами гемодинамики/ЭКГ отбирали в исследование.
Животных оставляли для привыкания к процедуре принудительного перорального введения через зонд по меньшей мере на 3 дня перед началом введения дозы состава. Воду из клетки/водопроводную воду вводили через пероральный зонд с использованием сменного катетера, присоединенного к пластиковому шприцу, в объеме 5 мл/кг. Вводимые составы перемешивали в помещении, в котором содержались животные в течение по меньшей мере 30 минут до введения и непрерывно во время введения дозы.
Через пероральный зонд вводили одну дозу носителя (контроль), который содержал 2 % лецитина и 0,5 % метилцеллюлозы в воде ULTRAPURE™ (Charles River Laboratories, Montreal, QC), или исследуемого соединения в количестве 30, 100 и 300 мг/кг. Носитель готовили для каждой серии опытов и хранили в холодильнике при 4 °C на плите смесителя без доступа света и использовали по мере необходимости. Удаляли носитель из холодильника и перемешивали по меньшей мере в течение 30 минут при комнатной температуре перед введением и непрерывно во время введения. Вводимые составы хранили в холодильнике при 4°C на плите смесителя без доступа света и использовали по мере необходимости. Удаляли вводимые составы из холодильника, перемешивали по меньшей мере в течение 30 минут при комнатной температуре перед введением и непрерывно во время введения.
Каждому из четырех кобелей вводили дозу носителя и три дозировки соединения формулы 1 (пример 1) с интервалом 7 дней между введением доз. Отслеживаемые параметры включали: частоту сердечных сокращений, определенную по кровяному давлению, давление в левом желудочке (LVP) и формы сигнала на электрокардиограмме; LVP (пиковое систолическое и конечное систолическое LVP и максимальные положительные/отрицательные значения dP/dt); электрокардиограмму (интервал PR (PR), погрешность RR (RR"), комплекс QRS (QRS), интервалы QT и QTcv, вычисленные при помощи уравнения ван де Вотера QTcv=QT-87(60/HR-1)); и температуру тела.
Маркировку сегментов ЭКГ проводили с использованием программного обеспечения для расшифровки ЭКГ ECG Pattern Recognition. Для каждого животного собирали библиотеку из типовых циклов, полученных как днем, так и ночью, и использовали во время мониторинга для обеспечения надлежащей маркировки для проведения количественной оценки, согласно соответствующим СОП Charles River Laboratories, Montreal, QC. Любые данные, которые превышали электрофизиологические нормы для этого вида, были исключены из дальнейшего анализа.
Во время каждого телеметрического исследования в дни введения дважды оценивали ЭКГ перед введением каждой дозы (с интервалом по меньшей мере 30 минут) и примерно через 1 (±5 минут), 2, 4, 6, 8, 10, 12, 15, 18 и 23 (±15 минут) часов после введения дозы. Для каждой временной точки оценку проводили как минимум 30 секунд. Проводили качественную оценку всех сигналов для обнаружения нарушений ритма или проведения импульсов или других отклонений сигнала P-QRS-T. Образцы крови собирали из яремной вены натощак после прекращения доступа к корму на ночь (для параметров клинической химии).
У биглей, которым вводили соединение формулы 1 (пример 1), не наблюдали изменения артериального кровяного давления, сократительной способности или продолжительности интервала P-R, комплекса QRS или интервала QT по сравнению с результатами, полученными при введении носителя.
ЭКВИВАЛЕНТЫ И ОБЪЕМ
Специалисты в данной области смогут распознать или выявить, не выходя за рамки обычной экспериментальной работы, множество эквивалентов конкретных вариантов реализации согласно изобретению, описанных в настоящем документе. Предполагается, что объем настоящего изобретения не ограничен приведенным выше описанием, а напротив, соответствует описанию в прилагаемой формуле изобретения.
В формуле изобретения формы единственного числа (соответствующие англ. «a», «an» и «the») могут обозначать один или более чем один, если не указано иное или если из контекста не очевидно иное. Условия в пунктах формулы изобретения или описании, которые включают «или» между одним или более элементами группы, считаются выполненными, если один, более одного или все элементы группы присутствуют, применяются или иным образом относятся к данному продукту или способу, если не указано иное или если из контекста не очевидно иное. Изобретение включает варианты реализации, в которых ровно один элемент группы присутствует, применяется или иным образом относится к данному продукту или способу. Изобретение включает варианты реализации, в которых более одного или все элементы группы присутствуют, применяются или иным образом относятся к данному продукту или способу.
Также следует отметить, что термин «содержащий» рассматривается как открытый, и он допускает, но не требует включения дополнительных элементов или стадий. Если в настоящем документе используют термин «содержащий», то при этом также включен и описан термин «состоящий из».
В описание диапазонов включены конечные значения. Кроме того, следует понимать, что если не указано иное или если иное не очевидно из контекста и на основании общих знаний, доступных специалистам в данной области, то значения, выраженные в виде диапазонов, могут подразумевать любое конкретное значение или поддиапазон в рамках указанных диапазонов в различных вариантах реализации изобретения с точностью до десятой части единицы нижнего предела диапазона, если из контекста явно не следует иное.
Кроме того, следует понимать, что любой конкретный вариант реализации настоящего изобретения, который относится к предшествующему уровню техники, может быть в явной форме исключен из любого одного или более пунктов формулы изобретения. Так как такие варианты реализации считаются известными специалистам в данной области, они могут быть исключены, даже если это исключение явным образом не выражено в настоящем документе. Любой конкретный вариант реализации композиций согласно изобретению (например, любой антибиотик, терапевтический или активный ингредиент; любой способ получения; любой способ применения; и т.д.) может быть исключен из любых одного или более пунктов формулы изобретения по любой причине независимо от того, относится ли он к предшествующему уровню техники или нет.
Следует понимать, что используемые формулировки представляют собой описательные, а не ограничивающие формулировки, и можно проводить изменения в прилагаемой формуле изобретения, не выходя за рамки истинного объема и сущности изобретения в его более общих аспектах.
Несмотря на то, что настоящее изобретение было описано довольно детально и подробно при помощи нескольких вариантов реализации, предполагается, что оно не должно быть ограничено любыми указанными конкретными деталями или вариантами реализации или любым конкретным вариантом реализации, но его следует рассматривать с учетом прилагаемой формулы изобретения, чтобы была обеспечена самая широкая возможная интерпретация указанной формулы изобретения с учетом предшествующего уровня техники и, таким образом, эффективно описан предполагаемый объем изобретения. Настоящее изобретение дополнительно проиллюстрировано неограничивающими примерами, приведенными в настоящем документе.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПРОИЗВОДНОЕ ОКСИМА ХРОМОНА И ЕГО ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ АЛЛОСТЕРИЧЕСКОГО МОДУЛЯТОРА МЕТАБОТРОПНЫХ РЕЦЕПТОРОВ ГЛУТАМАТА | 2015 |
|
RU2672569C2 |
| ПРИМЕНЕНИЕ 2-(2-НИТРО-4-ТРИФТОРМЕТИЛБЕНЗОИЛ)-1,3-ЦИКЛОГЕКСАНДИОНА ПРИ ЛЕЧЕНИИ БОЛЕЗНИ ПАРКИНСОНА | 2006 |
|
RU2420272C2 |
| МЕЗИЛАТНОЕ ПРОЛЕКАРСТВО ЛЕВОДОПЫ, ЕГО КОМПОЗИЦИИ И ПРИМЕНЕНИЕ | 2006 |
|
RU2429223C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПЕРОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ПРОЛЕКАРСТВА ЛЕВОДОПЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2537137C2 |
| СПОСОБЫ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2004 |
|
RU2342929C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2017 |
|
RU2733975C2 |
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИНА КАК ПОЗИТИВНЫЙ АЛЛОСТЕРИЧЕСКИЙ МОДУЛЯТОР D1 | 2020 |
|
RU2824581C2 |
| ПРОЛЕКАРСТВА КАРБИДОПА И L-DOPA И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2015 |
|
RU2743347C2 |
| ПРОЛЕКАРСТВА ЛЕВОДОПА, КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2365580C2 |
| СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2014 |
|
RU2677278C2 |
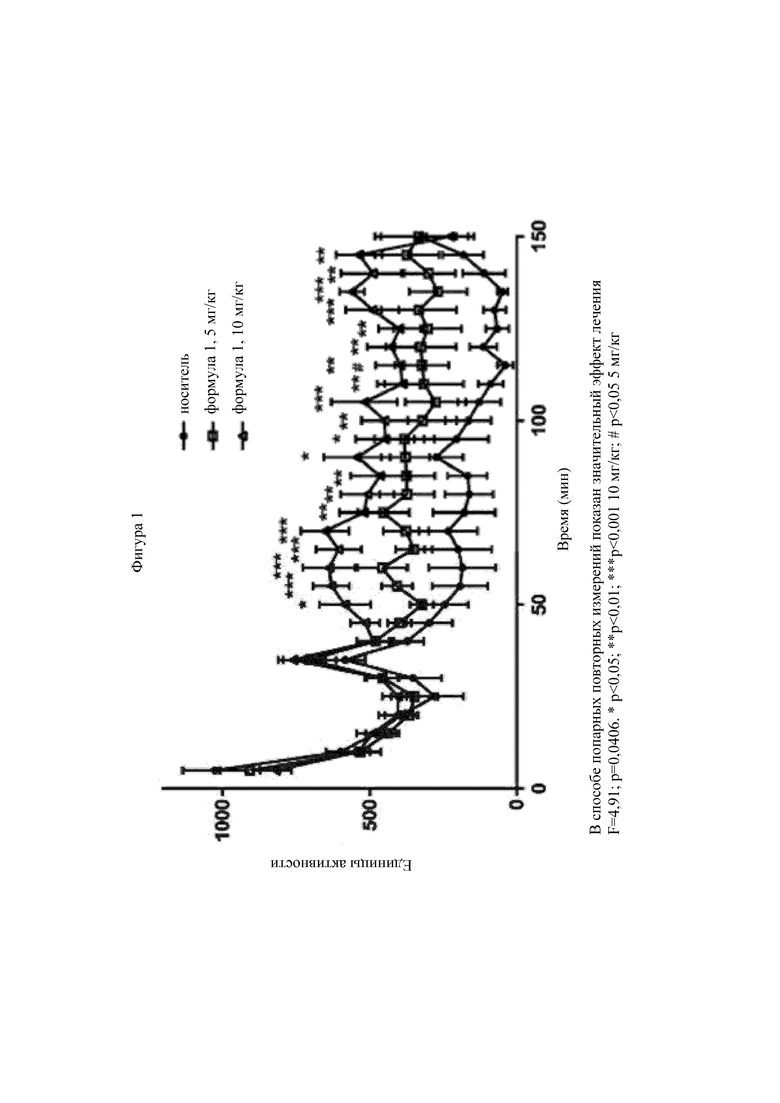
Изобретение относится к соединению формулы 1 или его фармацевтически приемлемой соли, которые являются модуляторами рецептора 6, сопряженного с G-белком (GPR6). Изобретение относится также к фармацевтической композиции, набору, лекарственной форме, содержащим указанное соединение, способу лечения с его использованием и его применению для получения лекарственного средства для лечения заболевания, нарушения или состояния, связанного с GPR6. 6 н. и 34 з.п. ф-лы, 2 табл., 1 ил., 1 пр.
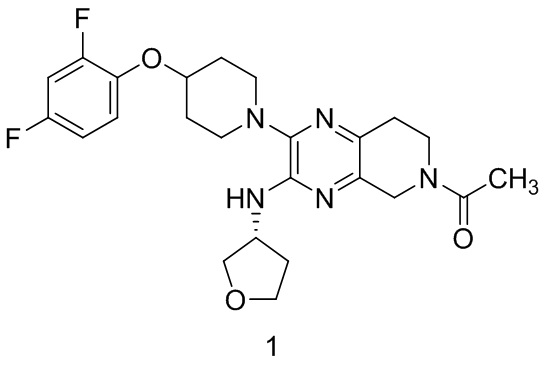
1. Соединение формулы 1
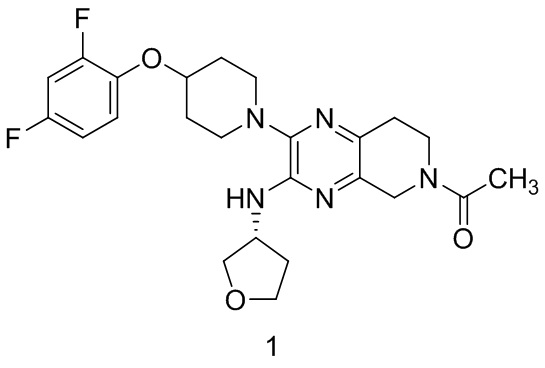
или его фармацевтически приемлемая соль.
2. Соединение или фармацевтически приемлемая соль по п. 1, имеющее энантиомерную чистоту, соответствующую энантиомерному избытку (э.и.) 20% или более.
3. Соединение или фармацевтически приемлемая соль по п. 1, имеющее энантиомерную чистоту, соответствующую э.и. 40% или более.
4. Соединение или фармацевтически приемлемая соль по п. 1, имеющее энантиомерную чистоту, соответствующую э.и. 60% или более.
5. Соединение или фармацевтически приемлемая соль по п. 1, имеющее энантиомерную чистоту, соответствующую э.и. 80% или более.
6. Соединение или фармацевтически приемлемая соль по п. 1, имеющее энантиомерную чистоту, соответствующую э.и. 90% или более.
7. Соединение или фармацевтически приемлемая соль по п. 1, имеющее энантиомерную чистоту, соответствующую э.и. 98% или более.
8. Соединение или фармацевтически приемлемая соль по п. 1, имеющее энантиомерную чистоту, соответствующую э.и. 99% или более.
9. Соединение или фармацевтически приемлемая соль по п. 1, имеющее энантиомерную чистоту, соответствующую э.и. 100%.
10. Соединение или фармацевтически приемлемая соль по любому из пп. 1-9, отличающееся тем, что указанное соединение присутствует в свободной форме.
11. Соединение или фармацевтически приемлемая соль по любому из пп. 1-10 для применения в качестве лекарственного средства.
12. Соединение или фармацевтически приемлемая соль по любому из пп. 1-11 для применения в лечении заболевания, нарушения или состояния, связанного с GPR6.
13. Соединение или фармацевтически приемлемая соль по любому из пп. 1-12 для применения в лечении:
(i) заболевания, нарушения или состояния, выбранного из группы, состоящей из болезни Паркинсона, дискинезий, вызванных леводопой, болезни Хантингтона, наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии, болезни Альцгеймера, тревоги и депрессии; и/или
(ii) болезни Паркинсона; и/или
(iii) болезни Паркинсона у субъекта,
отличающегося тем, что лечение улучшает моторные симптомы у субъекта; и/или
(iv) немоторных клинических проявлений болезни Паркинсона.
14. Соединение или фармацевтически приемлемая соль по любому из пп. 1-13, отличающееся тем, что указанное соединение или фармацевтически приемлемая соль предназначены для:
(i) введения в дозе в диапазоне от 0,5 до 5,0 мг/кг; и/или
(ii) перорального введения.
15. Фармацевтическая композиция, имеющая активность модулятора GPR6 и содержащая:
(a) эффективное количество соединения или фармацевтически приемлемой соли по любому из пп. 1-14; и
(b) фармацевтически приемлемое вспомогательное вещество.
16. Фармацевтическая композиция по п. 15 для применения в качестве лекарственного средства.
17. Фармацевтическая композиция по п. 15 или 16 для применения в лечении заболевания, нарушения или состояния, связанного с GPR6.
18. Фармацевтическая композиция по любому из пп. 15-17 для применения в лечении:
(i) заболевания, нарушения или состояния, выбранного из группы, состоящей из болезни Паркинсона, дискинезий, вызванных леводопой, болезни Хантингтона, наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии, болезни Альцгеймера, тревоги и депрессии; и/или
(ii) болезни Паркинсона; и/или
(iii) болезни Паркинсона у субъекта, отличающегося тем, что лечение улучшает моторные симптомы у субъекта; и/или
(iv) немоторных клинических проявлений болезни Паркинсона.
19. Фармацевтическая композиция по любому из пп. 15-18, отличающаяся тем, что указанное соединение или фармацевтически приемлемая соль предназначены для:
(i) введения в дозе в диапазоне от 0,5 до 5,0 мг/кг; и/или
(ii) перорального введения.
20. Фармацевтическая композиция по любому из пп. 15-19, дополнительно содержащая по меньшей мере один дополнительный фармакологически активный агент.
21. Фармацевтическая композиция по п. 20, отличающаяся тем, что указанный дополнительный фармакологически активный агент выбран из группы, состоящей из леводопы, ингибитора DOPA-декарбоксилазы, агониста дофамина, антихолинергического средства, селективного ингибитора моноаминоксидазы B и ингибитора катехол-O-метилтрансферазы.
22. Фармацевтическая композиция по п. 21, отличающаяся тем, что указанный дополнительный фармакологически активный агент выбран из:
(a) амантадина; и/или
(b) леводопы в комбинации с ингибитором DOPA-декарбоксилазы; и/или
(c) группы, состоящей из карбидопы; бенсеразида; метилдопы; α-дифторметил-DOPA; 3',4',5,7-тетрагидрокси-8-метоксиизофлавона; апоморфина гидрохлорида; бромокриптина; ротиготина; прамипексола; ропинирола; тригексилфенидила; бензтропина мезилата; сафинамида; селегилина; разагилина; энтакапона; и толкапона; и/или
(d) группы, состоящей из ингибиторов бета-секретазы, ингибиторов гамма-секретазы, ингибиторов HMG-CoA редуктазы, нестероидных противовоспалительных препаратов (НПВП); и/или
(e) группы, состоящей из апазона, аспирина, целекоксиба, диклофенака (совместно с мизопростолом или без него), дифлунизала, этодолака, фенопрофена, флурбипрофена, ибупрофена, индометацина, кетопрофена, меклофенамата натрия, мефенаминовой кислоты, мелоксикама, набуметона, напроксена, оксапрозина, фенилбутазона, пироксикама, салицилатов холина и магния, салсалата и сулиндака; и/или
(f) группы, состоящей из донепезила, ривастигмина, мемантина и галантамина; и/или
(g) группы, состоящей из седативных средств, снотворных средств, анксиолитических средств, нейролептических средств и транквилизаторов; и/или
(h) группы, состоящей из амитриптилина, амоксапина, арипипразола, азенапина, бупропиона, хлордиазепоксида, циталопрама, хлорпромазина, клозапина, дезипрамина, десвенлафаксина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флуфеназина, галоперидола, илоперидона, имипрамина, изокарбоксазида, ламотригина, левомилнаципрана, луразидона, миртазапина, нефазодона, нортриптилина, оланзапина, палиперидона, пароксетина, перфеназина, фенелзина, протриптилина, кветиапина, рисперидона, сафинамида, селегилина, сертралина, транилципромина, тразодона, тримипрамина, венлафаксина, вилазодона, вортиоксетина и зипрасидона; и/или
(i) группы, состоящей из алпразолама, хлордиазепоксида, клобазепама, клоназепама, клоразепата, диазепама, эстазолама, флуразепама, лоразепама, мидазолама, оксазепама, празепама, квазепама, темазепама и триазолама, гидроксизина, эсзопиклона, залеплона, золпидема, зопиклона и буспирона; и/или
(j) группы, состоящей из ацетазоламида, карбамазепина, клобазама, клоназепама, эсликарбазепина ацетата, этосуксимида, габапентина, лакозамида, ламотригина, леветирацетама, нитразепама, окскарбазепина, перампанела, пирацетама, фенобарбитала, фенитоина, прегабалина, примидона, ретигабина, руфинамида, натрия вальпроата, стирипентола, тиагабина, топирамата, вигабатрина и зонисамида.
23. Набор, имеющий активность модулятора GPR6, причем набор пригоден для лечения болезни Паркинсона и содержит фармацевтическую композицию по любому из пп. 15-19 и фармацевтическую композицию, содержащую по меньшей мере один дополнительный фармакологически активный агент, выбранный из группы, состоящей из леводопы, ингибитора DOPA-декарбоксилазы, агониста дофамина, антихолинергического средства, селективного ингибитора моноаминоксидазы B и ингибитора катехол-O-метилтрансферазы.
24. Набор по п. 23, отличающийся тем, что указанный набор подходит для введения фармацевтической композиции по любому из пп. 15-19 и фармацевтической композиции, содержащей по меньшей мере один дополнительный фармакологически активный агент одновременно, последовательно или раздельно.
25. Набор по п. 23 или 24, отличающийся тем, что указанный дополнительный фармакологически активный агент выбран из:
(a) амантадина; и/или
(b) леводопы в комбинации с ингибитором DOPA-декарбоксилазы; и/или
(c) группы, состоящей из карбидопы; бенсеразида; метилдопы; α-дифторметил-DOPA; 3',4',5,7-тетрагидрокси-8-метоксиизофлавона; апоморфина гидрохлорида; бромокриптина; ротиготина; прамипексола; ропинирола; тригексилфенидила; бензтропина мезилата; сафинамида; селегилина; разагилина; энтакапона; и толкапона; и/или
(d) группы, состоящей из ингибиторов бета-секретазы, ингибиторов гамма-секретазы, ингибиторов HMG-CoA редуктазы, нестероидных противовоспалительных препаратов (НПВП); и/или
(e) группы, состоящей из апазона, аспирина, целекоксиба, диклофенака (совместно с мизопростолом или без него), дифлунизала, этодолака, фенопрофена, флурбипрофена, ибупрофена, индометацина, кетопрофена, меклофенамата натрия, мефенаминовой кислоты, мелоксикама, набуметона, напроксена, оксапрозина, фенилбутазона, пироксикама, салицилатов холина и магния, салсалата и сулиндака; и/или
(f) группы, состоящей из донепезила, ривастигмина, мемантина и галантамина; и/или
(g) группы, состоящей из седативных средств, снотворных средств, анксиолитических средств, нейролептических средств и транквилизаторов; и/или
(h) группы, состоящей из амитриптилина, амоксапина, арипипразола, азенапина, бупропиона, хлордиазепоксида, циталопрама, хлорпромазина, клозапина, дезипрамина, десвенлафаксина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флуфеназина, галоперидола, илоперидона, имипрамина, изокарбоксазида, ламотригина, левомилнаципрана, луразидона, миртазапина, нефазодона, нортриптилина, оланзапина, палиперидона, пароксетина, перфеназина, фенелзина, протриптилина, кветиапина, рисперидона, сафинамида, селегилина, сертралина, транилципромина, тразодона, тримипрамина, венлафаксина, вилазодона, вортиоксетина и зипрасидона; и/или
(i) группы, состоящей из алпразолама, хлордиазепоксида, клобазепама, клоназепама, клоразепата, диазепама, эстазолама, флуразепама, лоразепама, мидазолама, оксазепама, празепама, квазепама, темазепама и триазолама, гидроксизина, эсзопиклона, залеплона, золпидема, зопиклона и буспирона; и/или
(j) группы, состоящей из ацетазоламида, карбамазепина, клобазама, клоназепама, эсликарбазепина ацетата, этосуксимида, габапентина, лакозамида, ламотригина, леветирацетама, нитразепама, окскарбазепина, перампанела, пирацетама, фенобарбитала, фенитоина, прегабалина, примидона, ретигабина, руфинамида, натрия вальпроата, стирипентола, тиагабина, топирамата, вигабатрина и зонисамида.
26. Лекарственная форма, имеющая активность модулятора GPR6 и содержащая эффективное количество соединения или фармацевтически приемлемой соли по любому из пп. 1-14 и фармацевтически приемлемое вспомогательное вещество, где лекарственная форма предназначена для перорального введения соединения или его фармацевтически приемлемой соли в дозе в диапазоне от 0,5 до 5,0 мг/кг.
27. Способ лечения заболевания, нарушения или состояния, связанного с GPR6, у субъекта, включающий введение субъекту эффективного количества соединения или фармацевтически приемлемой соли по любому из пп. 1-14, или фармацевтической композиции по любому из пп. 15-22, или набора по любому из пп. 23-25, или лекарственной формы по п. 26.
28. Способ по п. 27, отличающийся тем, что указанное заболевание, нарушение или состояние выбрано из группы, состоящей из болезни Паркинсона, дискинезий, вызванных леводопой, болезни Хантингтона, наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии, болезни Альцгеймера, тревоги и депрессии.
29. Способ по п. 27, отличающийся тем, что указанное заболевание, нарушение или состояние представляет собой болезнь Паркинсона.
30. Способ по п. 29, отличающийся тем, что лечение улучшает моторные симптомы у субъекта.
31. Способ по п. 27, отличающийся тем, что указанное заболевание, нарушение или состояние представляет собой немоторные клинические проявления болезни Паркинсона.
32. Способ по п. 27, отличающийся тем, что указанное соединение или фармацевтически приемлемая соль предназначены для введения в дозе в диапазоне от 0,5 до 5,0 мг/кг.
33. Способ по п. 27, отличающийся тем, что указанное соединение или фармацевтически приемлемая соль предназначены для перорального введения.
34. Применение соединения или фармацевтически приемлемой соли по любому из пп. 1-14 или фармацевтической композиции по любому из пп. 15-22, или набора по любому из пп. 23-25, или лекарственной формы по п. 26 для получения лекарственного средства для лечения заболевания, нарушения или состояния, связанного с GPR6.
35. Применение по п. 34, отличающееся тем, что указанное заболевание, нарушение или состояние выбрано из группы, состоящей из болезни Паркинсона, дискинезий, вызванных леводопой, болезни Хантингтона, наркотической зависимости, нарушений пищевого поведения, когнитивных нарушений, шизофрении, биполярного расстройства, эпилепсии, болезни Альцгеймера, тревоги и депрессии.
36. Применение по п. 34, отличающееся тем, что указанное заболевание, нарушение или состояние представляет собой болезнь Паркинсона.
37. Применение по п. 36, отличающееся тем, что лечение улучшает моторные симптомы у субъекта.
38. Применение по п. 34, отличающееся тем, что указанное заболевание, нарушение или состояние представляет собой немоторные клинические проявления болезни Паркинсона.
39. Применение по п. 34, отличающееся тем, что соединение или фармацевтически приемлемая соль предназначены для введения в дозе в диапазоне от 0,5 до 5,0 мг/кг.
40. Применение по п. 34, отличающееся тем, что соединение или фармацевтически приемлемая соль предназначены для перорального введения.
| Токарный резец | 1924 |
|
SU2016A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| EA 201691632 A1, 28.02.2017. | |||

