По данной заявке испрашивается приоритет согласно предварительной заявке США 60/577087, поданной 4 июня 2004, которая включена в данное описание во всей своей полноте посредством ссылки.
Варианты осуществления настоящего изобретения относятся к пролекарствам леводопа, способам получения пролекарств леводопа, способам применения пролекарств леводопа и композициям пролекарств леводопа.
Болезнь Паркинсона представляет собой инвалидизирующее прогрессирующее заболевание, которое поражает одного из 1000 человек и обычно наступает у людей в возрасте старше 50 лет. У пациентов с болезнью Паркинсона в результате повреждения нигростриарного пути, вызванного дегенерацией черной субстанции, в головном мозге наблюдается дефицит нейромедиатора допамина. Леводопа (L-допа или L-3,4-дигидроксифенилаланин), непосредственный предшественник допамина, представляет собой лекарственное средство, чаще всего прописываемое для лечения данного заболевания.
После орального введения леводопа быстро абсорбируется с помощью переносчика аминокислот, присутствующего в верхних отделах тонкой кишки. Вследствие того, что распространение такой системы переносчика ограничено, ограничен промежуток времени, доступный для абсорбции леводопа и степень абсорбции может зависеть от скорости, с которой лекарственное средство проходит через верхние отделы желудочно-кишечного тракта. После орального введения пациентам приблизительно 35% вводимой дозы достигает большого круга кровообращения в виде интактного леводопа (Sasahara, 1980, J.Pharm. Sci., 69, 261). Абсолютная биодоступность леводопа зависит от дозировки вследствие насыщения пути активного переноса. Чтобы достичь оптимальной терапевтической активности, следует тщательно подбирать уровни леводопа в плазме каждого пациента. Если концентрация леводопа в плазме (и соответственно в головном мозге) слишком низкая, пациент может ощущать возобновление симптомов болезни Паркинсона (ригидности, тремора, брадикинезии). С другой стороны, флуктуация двигательной функции может представлять собой значительный побочный эффект, если уровни лекарственного средства в плазме слишком высокие. Неконтролируемые флуктуации уровней леводопа в плазме могут весьма способствовать частоте возникновения флуктуаций по типу «on/off» (дискинезий). Наиболее эффективный контроль паркинсонизма наблюдается, когда уровни леводопа в плазме поддерживаются в узком диапазоне, например, путем постоянной интрадуоденальной инфузии.
Сразу после абсорбции леводопа под действием L-ароматической аминокислотной декарбоксилазы (AADC) быстро превращается в допамин в периферических тканях (например, в кишечнике и печени). Известно, что кишечный метаболизм леводопа является важным источником потери при первом проходе лекарственного средства. У пациентов только 1% вводимой дозы интактно достигает центральной нервной системы после переноса через гематоэнцефалический барьер с помощью переносчика нейтральных аминокислот. По указанной причине обычно леводопа вводится совместно с лекарственным средством, предназначенным для ингибирования его периферического декарбоксилирования, таким как карбидопа или бензеразид. При совместном введении со средством корбидопа количество интактного леводопа в плазме повышается и, таким образом, более значительное количество леводопа становится доступным для переноса в центральную нервную систему, где он превращается в допамин. Сами карбидопа и бензеразид не пересекают гематоэнцефалический барьер в значительной степени и, следовательно, не ингибируют необходимое превращение леводопа в допамин в головном мозге.
Биодоступность леводопа при оральном введении общепринятых препаратов леводопа/корбидопа (например, препарат Sinemet®) составляет 84-99% (Physician's Desk Reference (Настольный справочник врача)). Когда леводопа вводится в чистом виде, период полужизни леводопа в плазме пациентов составляет приблизительно 50 минут, или 1-2 часа, когда вводят вместе со средством корбидопа. По указанной причине лекарственное средство должно вводиться три или более раз в день.
Препарат леводопа/корбидопа (Sinemet® СR), предназначенный для обеспечения контролируемого высвобождения обоих лекарственных средств, коммерчески доступен. Sinemet® CR предназначен для высвобождения как леводопа, так и корбидопа в течение 4-6 часов. Однако абсорбция леводопа ограничена тонкой кишкой и результирующая биодоступность леводопа из Sinemet® CR уменьшается по сравнению с непосредственно высвобождаемым продуктом. В большинстве случаев для достижения терапевтического уровня леводопа препарат Sinemet® CR также следует давать более двух раз в день. Поскольку леводопа плохо абсорбируется в толстой кишке, препараты отсроченного и продленного высвобождения, из которых лекарственное средство высвобождается в течение приблизительно 10-24 часов и, следовательно, большая часть лекарственного средства высвобождается в толстой кишке, не эффективны для доставки леводопа. Препарат леводопа с простым энтеросолюбильным покрытием приводит к повышенным гастроинтестинальным побочным эффектам (тошноте), но не улучшает абсорбции. Описан препарат леводопа/корбидопа замедленного высвобождения, в котором использована система доставки с набухающей матрицей (Geomatrix), для задержки лекарственного средства в желудке (информация о лицензировании продукта Genta Jago, июнь 1997). Однако такой препарат предназначен стать биоэквивалентом коммерчески доступному Sinemet® CR и, следовательно, не обеспечивает необходимого режима введения дозы один или два раза в день.
Для улучшения фармокинетики лекарственного средства было предложено применение простых пролекарств леводопа на основе сложных эфиров (патенты США №№ 5017607; 4826875; 4873263; 4771073; 4663349; 4311706; японский патент № JP58024547; публикации Juncos et al., 1987, Neurology, 37: 1242; и Cooper et al., 1987, J. Pharm. Pharmacol., 39: 627-635; Dumont, публикация международной заявки РСТ WO-А-8604579; и Bundgaard et al., публикация международной заявки РСТ WO-А-8801615). Описан препарат (Levomet®, CHF 1301) для орального введения на основе сложного метилового эфира леводопа (производства Chiesi Pharmaceuticals). Сложный этиловый эфир леводопа (TV-1203) применялся при клиническом исследовании в качестве возможной терапии паркинсонизма при совместном введении с корбидопа (патент США № 5607969). Описан препарат замедленного действия с целлюлозой на основе сложного этилового эфира леводопа в смеси с гидроксипропилметилцеллюлозой, гидроксипропилцеллюлозой и карбоксивиниловым полимером (патент США № 5840756). Однако оральное введение такого препарата здоровым взрослым людям, подвергнутым предварительному действию корбидопа, приводило к тому, что конечный период полужизни леводопа в плазме составлял всего лишь 2 часа, что сопоставимо с периодом полужизни Sinemet® CR.
Описан сложный пивалоиловый эфир леводопа (NB-355) (европейский патент № 0309827). После орального введения NB-355 не наблюдалось быстрого повышения или выведения леводопа и продолжительность пребывания была длительной, в то время как уровни леводопа были низкими. Описано возможное применение сложноэфирных пролекарств леводопа для улучшения ректальной абсорбции лекарственного средства (патенты США №№ 4663349; 4771073 и 4873263). В частности, было показано, что абсорбция простых алкиловых эфиров леводопа является более значительной после ректальной абсорбции, чем после орального введения (Fix et al., Pharm. Res., 1989, 6:501-5; Fix et al., Pharm. Res., 1990, 4:384-7). Такой эффект объясняется пониженным относительным содержанием эстераз в толстой кишке по сравнению с тонкой кишкой. Следовательно, для препарата замедленного высвобождения можно было бы ожидать избирательной доставки пролекарства леводопа в толстую кишку для обеспечения более значительной биодоступности при оральном введении и пролонгированного воздействия лекарственного средства.
Описан ряд пролекарств леводопа, содержащих сложные эфиры гликолевой кислоты (Wermuth, патент США № 4134991). Также описаны липидные конъюгаты леводопа, предназначенные для облегчения поступления лекарственного средства в клетки и ткани (Yatvin, патент США № 5827819).
Период полужизни леводопа удлиняется и его биодоступность повышается путем совместного введения со средством корбидопа. Оба лекарственных средства имеют относительно короткие периоды полужизни, приблизительно менее 2 часов. Следовательно, при любом способе замедленной доставки леводопа в большой круг кровообращения для непрерывного ингибирования периферического декарбоксилирования леводопа потребовался бы достаточный уровень корбидопа. Чтобы избавиться от необходимости частого введения (более двух раз в день) леводопа и корбидопа, желательно доставлять как леводопа, так и карбидопа (или их пролекарство) замедленным способом. Предполагалось, что в качестве средства снижения метаболического клиренса леводопа можно было бы применять совместное ректальное введение ингибитора AADC, такого как карбидопа, с пролекарством леводопа на основе сложного эфира (патенты США №№ 4663349; 4771073 и 4873263). Однако исследования на крысах показали, что абсорбция корбидопа после ректального введения является недостаточной (Leppert et al., 1988, Pharm. Res., 5:587-591).
Таким образом, весьма желательным является создание пролекарств леводопа, которые могут эффективно абсорбироваться на всем протяжении желудочно-кишечного тракта, включая ободочную кишку, и уменьшать метаболизм при первом проходе леводопа.
Некоторые варианты осуществления настоящего изобретения относятся к пролекарствам леводопа, которые способны подвергаться абсорбции через кишечный эпителий путем активного и/или пассивного переноса.
Некоторые варианты осуществления настоящего изобретения относятся к пролекарствам леводопа, которые способны подвергаться абсорбции через кишечный эпителий с помощью механизмов активного переноса, и более конкретно к пролекарствам леводопа, которые представляют собой субстраты для переносчиков органических катионов, экспрессируемых на всем протяжении желудочно-кишечного тракта.
Желудочно-кишечный тракт человека включает тонкую кишку и толстую кишку. Тонкая кишка человека представляет собой извитую трубку между желудком и толстой кишкой длиной приблизительно двадцать футов. Тонкая кишка подразделяется на двенадцатиперстную кишку, тощую кишку и подвздошную кишку. Толстая кишка имеет приблизительно 5 футов в длину и соединяет подвздошную кишку и анус. Толстая кишка подразделяется на слепую кишку, ободочную кишку и прямую кишку. Ободочная кишка подразделяется на четыре части, включающих восходящую часть, поперечную часть, нисходящую часть и сигмовидный изгиб ободочной кишки. В общем случае соединение, принимаемое внутрь орально, находится приблизительно 1-6 часов в желудке, приблизительно 2-7 часов в тонкой кишке и приблизительно 8-18 часов в ободочной кишке. Таким образом, самым длительным периодом времени для замедленного высвобождения соединения оказывается период, когда соединение проходит через ободочную кишку.
Известно, что некоторые активные белки-переносчики экспрессируются на всем протяжении желудочно-кишечного тракта. Активный переносчик относится к мембраносвязанному белку, который распознает субстрат и воздействует на проникновение субстрата в клетку или на выход из клетки с помощью переноса, опосредованного переносчиками, или переноса, опосредованного рецепторами. Активный перенос включает перенос молекул через клеточные мембраны, который прямо или косвенно зависит от опосредованного энергией процесса, такого как, например, процесс, запускаемый гидролизом АТФ или ионным градиентом, который происходит под действием облегченной диффузии, опосредованной взаимодействием со специфическими белками-переносчиками, и который происходит через модулированный раствором канал. Например, переносчики органических катионов, такие как OCTN1 и OCTN2, экспрессируются в клетках эпителиальной выстилки ободочной кишки человека, а также в тонкой кишке.
Таким образом, когда соединение проходит через желудочно-кишечный тракт, пролекарства леводопа, которые действуют как субстраты для одного или нескольких переносчиков органических катионов, в течение продленного периода времени могут обладать повышенной абсорбцией, опосредованной активным переносчиком. Повышенная абсорбция и, в частности, абсорбция пролекарства леводопа в ободочной кишке, может приводить к повышенной системной биодоступности соединения в течение продленного периода времени. Под системной биодоступностью понимается скорость и степень системного воздействия лекарственного средства или его активного метаболита, которая отражается на суммарной системной концентрации в крови в течение периода времени, также именуемой как «площадь под кривой».
В некоторых вариантах осуществления пролекарства леводопа способны абсорбироваться на значительном протяжении желудочно-кишечного тракта, включая толстую кишку и, в частности ободочную кишку. Такие пролекарства можно включать в общепринятые препараты замедленного высвобождения, включая осмотические средства доставки, для обеспечения замедленного системного воздействия леводопа после орального введения пациенту. Многие из таких пролекарств можно вводить совместно с ингибитором декарбоксилазы, таким как карбидопа или бензеразид, или его пролекарством, и в некоторых вариантах осуществления также можно получать препарат в виде композиций замедленного высвобождения на основе композиций карбидопа/пролекарство леводопа или композиций бензеразид/пролекарство леводопа, тем самым обеспечивая пролонгированное воздействие леводопа при уровнях, необходимых для замедленного воздействия терапии, направленной на лечение болезни Паркинсона. Некоторые варианты осуществления включают пролекарства корбидопа, которые могут блокировать декарбоксилирование первого прохода леводопа в кишечных энтероцитах либо в виде интактного пролекарства корбидопа, либо через образование корбидопа при расщеплении пролекарства корбидопа в энтероцитах и которые можно расщеплять для обеспечения концентрации корбидопа в большом круге кровообращения. Композиции замедленного высвобождения на основе ингибитора декарбоксилазы/пролекарства леводопа или пролекарства ингибитора декарбоксилазы/пролекарства леводопа также можно вводить вместе с ингибиторами катехин-O-метилтрансферазы (COMT), такими как энтакапон или толкапон, для дополнительной блокировки периферического клиренса леводопа.
Описанные в данном описании пролекарства леводопа представляют собой пролекарства, в которых карбоксильная группировка леводопа маскируется с помощью образования сложного карбоксильного эфира, который может отщепляться in vivo для высвобождения исходного лекарственного средства (например, леводопа). Катехиновые группировки леводопа необязательно можно дополнительно маскировать предгруппировками, причем такие предгруппировки отщепляются либо до, либо после отщепления предгруппировки сложного карбоксильного эфира.
Подходящие катехин-защитные группы в вышеприведенных пролекарствах можно усовершенствовать функционализацией одной или нескольких фенольных гидроксигрупп путем ацилирования или другими подходящими способами. Соответствующие сложные эфиры, карбонаты и (полу)ацетали/(полу)кетали могут расщепляться в организме (in vivo) с восстановлением катехиновых групп исходного лекарственного средства.
Варианты осуществления настоящего изобретения относятся, по меньшей мере, к одному пролекарству леводопа формулы (I)
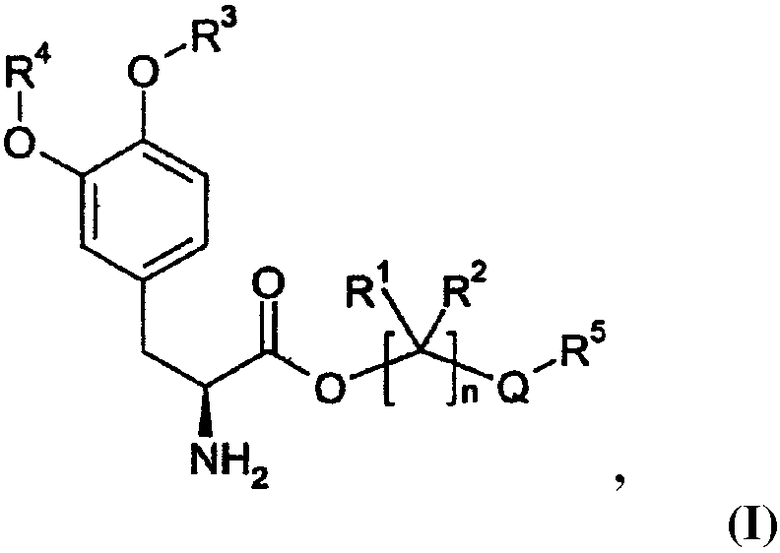
его стереоизомеру, его энантиомеру, его фармацевтически приемлемой соли, его гидрату или сольвату любого из указанных выше, где Q выбран из -X-CO- и -CO-X-; X выбран из -О- и -NR6-;
n равно целому числу от 2 до 4;
каждый R1 и R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, циклоалкила, замещенного циклоалкила, циклогетероалкила, замещенного циклогетероалкила, галогена, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила;
R3 и R4 независимо выбраны из водорода, -C(O)OR7, -C(O)R7 и -(CR8R9)OC(O)R10;
R5 выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, циклоалкила, замещенного циклоалкила, гетероалкила, замещенного гетероалкила, циклогетероалкила, замещенного циклогетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила; и когда Q представляет собой -X-CO-, R5 дополнительно выбран из алкокси, замещенного алкокси, циклоалкокси и замещенного циклоалкокси;
R6 выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила и замещенного арилалкила;
R7 выбран из алкила, замещенного алкила, циклоалкила, замещенного циклоалкила, циклогетероалкила, замещенного циклогетероалкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила;
R8 и R9 независимо выбраны из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, циклоалкила, замещенного циклоалкила, циклогетероалкила, замещенного циклогетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила, или необязательно R8 и R9 вместе с атомом углерода, к которому R8 и R9 присоединены, образуют циклоалкильное, замещенное циклоалкильное, циклогетероалкильное или замещенное циклогетероалкильное кольцо; и
R10 выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, циклоалкила, замещенного циклоалкила, гетероалкила, замещенного гетероалкила, циклогетероалкила, замещенного циклогетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила;
при условии, что соединение формулы (I) не является производным 1,3-дигексадеканоилпропан-1,2,3-триола.
Некоторые варианты осуществления настоящего изобретения относятся к композициям, содержащим, по меньшей мере, одно пролекарство леводопа. В некоторых вариантах осуществления композиции содержат, по меньшей мере, одно пролекарство леводопа или энантиомер и стереоизомер любого из вышеприведенных соединений, или его фармацевтически приемлемую соль, его гидрат или сольват любого из указанных выше и фармацевтически приемлемый разбавитель, носитель, эксципиент и/или адъювант любого из вышеприведенных соединений. Выбор разбавителя, носителя, наполнителя и/или адъюванта среди прочих факторов может зависеть от желательного способа введения.
Некоторые варианты осуществления настоящего изобретения относятся к способам лечения болезни Паркинсона. Способы включают совместное введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества, по меньшей мере, одного из следующих соединений: (i) по меньшей мере, одного пролекарства леводопа; (ii) по меньшей мере, одного пролекарства леводопа и, по меньшей мере, одного ингибитора декарбоксилазы; (iii) по меньшей мере, одного пролекарства леводопа и, по меньшей мере, одного пролекарства (предшественника) ингибитора декарбоксилазы; (iv) стереоизомера или энантиомера любого из вышеприведенных соединений; и (v) его фармацевтически приемлемой соли, его гидрата или сольвата любого из указанных выше. В некоторых вариантах осуществления композиция вводится пациенту с помощью дозированной формы замедленного высвобождения.
В некоторых вариантах осуществления, по меньшей мере, одно пролекарство леводопа может высвобождаться из дозированной формы, например дозированной формы для орального введения, в течение достаточного периода времени для обеспечения устойчивых терапевтических концентраций леводопа в крови пациента, позволяющих осуществлять введение дозированной формы на основе только одного или двух введений в день. В некоторых вариантах осуществления после орального введения пролекарства леводопа, по меньшей мере, одно пролекарство леводопа может поддерживать в крови терапевтическую или профилактическую концентрацию леводопа или пролекарства леводопа в большом круге кровообращения пациента в течение, по меньшей мере, 4 часов; в некоторых вариантах осуществления в течение, по меньшей мере, 8 часов, и в некоторых вариантах осуществления в течение, по меньшей мере, 12 часов. Аналогичным образом ингибитор декарбоксилазы (например, карбидопа, бензеразид или их пролекарство), если вводится с пролекарством леводопа, может высвобождаться из дозированной формы или лекарственного средства непосредственно после введения дозированной формы в течение периода до нескольких часов, например, в течение 16 часов после введения дозированной формы с высвобождением более 75% ингибитора декарбоксилазы, или высвобождаться коэкстенсивно с высвобождением пролекарства леводопа.
Применяемые в некоторых вариантах осуществления дозированные формы замедленного высвобождения для орального введения могут принимать любую форму при условии, что ее характеристики высвобождения и упомянутые выше фармакокинетические профили отвечают требованиям. Например, дозированные формы могут находиться в форме осмотической дозированной формы, полимера, высвобождающего пролекарство, очень маленьких пилюль с регулируемым во времени высвобождением пролекарства, липидов, высвобождающих пролекарство, восков, высвобождающих пролекарство, и/или гранул, высвобождающих пролекарство.
Некоторые варианты осуществления настоящего изобретения относятся к композициям, предназначенным для лечения болезни Паркинсона у пациента, нуждающегося в таком лечении. Композиции содержат терапевтически эффективное количество, по меньшей мере, одного из следующего: (i) пролекарство леводопа; (ii) пролекарство леводопа и ингибитор декарбоксилазы; (iii) пролекарство леводопа и пролекарство ингибитора декарбоксилазы; (iv) стереоизомер или энантиомер любого из вышеприведенных соединений; и (v) его фармацевтически приемлемую соль, его гидрат или сольват любого из указанных выше. В некоторых вариантах осуществления композиция дополнительно содержит дозированную форму замедленного высвобождения.
Некоторые варианты осуществления настоящего изобретения относятся к способам получения пролекарств леводопа, композициям, содержащим, по меньшей мере, одно пролекарство леводопа, способам применения пролекарств леводопа и способам применения композиций, содержащих, по меньшей мере, одно пролекарство леводопа, для лечения болезни Паркинсона.
Конкретные варианты осуществления
Определения
Если не указано иное, под всеми используемыми в описании и формуле изобретения числами, выражающими количества ингредиентов, условия реакции и прочее, во всех случаях следует понимать числа, определяемые термином «приблизительно». Соответственно если не указано иное, числовые параметры, изложенные в следующем описании и прилагаемой формуле изобретения, представляют собой приближенные значения, которые могут меняться в зависимости от свойств, которые требуется получить. Самое меньшее, и без попытки ограничить применение доктрины эквивалентов к объему формулы изобретения, каждый числовой параметр должен толковаться, по меньшей мере, в свете ряда опубликованных значащих цифр и с помощью применения обычных способов округления чисел.
Несмотря на то что числовые диапазоны и параметры, изложенные в широком диапазоне вариантов осуществления, представляют собой приближенные значения, изложенные в конкретных примерах числовые значения публикуются с наиболее возможной точностью. Однако любые числовые значения заведомо содержат некоторые неточности, непременно приводящие к стандартному отклонению, найденному в соответствующих экспериментальных измерениях.
Определения терминов в публикациях, патентах и патентных заявках, включенных в данное описание посредством ссылки, в некоторой степени не совсем такие, как определения, приведенные в данном описании, определения в данном описании имеют преимущественную силу для всего описания, включая формулу изобретения. Любые другие определения в публикациях, патентах и патентных заявках, включенных в данное описание посредством ссылки, которые неявно используются в данном описании, применяются только для вариантов осуществления, обсуждаемых в публикациях, патентах и патентных заявках, включенных в данное описание посредством ссылки.
Термин «соединения» относится к соединениям, которые охватываются описанными в данном описании общими формулами, любым подклассом таких общих формул и любыми конкретными соединениями в пределах таких общих формул или формул подкласса. Соединения могут представлять собой конкретный вид, подкласс или большой класс, идентифицируемый или по их химической структуре, и/или химическому названию. Кроме того, соединения также включают замены или модификации любых таких видов, подклассов или классов, которые описаны в данном описании. Когда химическая структура и химическое название вступают в противоречие, при идентификации соединения определяющей является химическая структура. Соединения могут содержать один или несколько хиральных центров и/или двойные связи и, следовательно, могут существовать в виде стереоизомеров, таких как изомеры относительно двойной связи (например, геометрические изомеры), энантиомеров или диастереомеров. Соответственно химические структуры в пределах объема описания охватывают все возможные энантиомеры и стереоизомеры иллюстрируемых соединений, включая чистую стереоизомерную форму (например, чистую относительно геометрического изомера, энантиомерно чистую или диастереомерно чистую) и смеси энантиомеров и стереоизомеров. Кроме того, когда в качестве иллюстрации приводятся частичные структуры соединений, точка присоединения частичной структуры к остальной молекуле обозначается звездочками. Смеси энантиомеров и стереоизомеров можно разделять на составляющие энантиомеры или стереоизомеры, используя методы разделения или способы хирального синтеза, хорошо известные специалисту в данной области.
Термин «алкил» относится к насыщенной или ненасыщенной одновалентной углеводородной группе с прямой или разветвленной цепью или к циклической одновалентной углеводородной группе, получаемой удалением одного атома водорода от одного атома углерода исходного алкана, алкена или алкина. Конкретные алкильные группы включают, но не ограничиваются перечисленным, метил; этилы, такие как этанил, этенил, этинил; пропилы, такие как пропан-1-ил, пропан-2-ил, циклопропан-1-ил, проп-1-ен-1-ил, проп-1-ен-2-ил, проп-2-ен-1-ил (аллил), циклопроп-1-ен-1-ил, циклопроп-2-ен-1-ил, проп-1-ин-1-ил, проп-2-ин-1-ил; бутилы, такие как бутан-1-ил, бутан-2-ил, 2-метилпропан-1-ил, 2-метилпропан-2-ил, циклобутан-1-ил, бут-1-ен-1-ил, бут-1-ен-2-ил, 2-метилпроп-1-ен-1-ил, бут-2-ен-1-ил, бут-2-ен-2-ил, бута-1,3-диен-1-ил, бута-1,3-диен-2-ил, циклобут-1-ен-1-ил, циклобут-1-ен-3-ил, циклобута-1,3-диен-1-ил, бут-1-ин-1-ил, бут-1-ин-3-ил, бут-3-ин-1-ил; и т.п.
Термин «алкил», в частности, предназначен для включения групп, содержащих любую степень или уровень насыщения, то есть групп, содержащих только одинарные углерод-углеродные связи, групп, содержащих одну или несколько двойных углерод-углеродных связей, групп, содержащих одну или несколько тройных углерод-углеродных связей, и групп, содержащих смеси одинарных, двойных и тройных углерод-углеродных связей. Если имеется в виду определенный уровень насыщения, применяются выражения «алканил», «алкенил» и «алкинил». В некоторых вариантах осуществления алкильная группа содержит от 1 до 20 атомов углерода.
Термин «алканил» относится к насыщенной алкильной группе с прямой или разветвленной цепью или к циклической алкильной группе, получаемой удалением одного атома водорода от одного атома углерода исходного алкана. Конкретные алканильные группы включают, но не ограничиваются перечисленным, метанил; этанил; пропанилы, такие как пропан-1-ил, пропан-2-ил (изопропил), циклопропан-1-ил; бутанилы, такие как бутан-1-ил, бутан-2-ил (втор-бутил), 2-метилпропан-1-ил (изобутил), 2-метилпропан-2-ил-(т-бутил), циклобутан-1-ил; и т.п.
Термин «алкенил» относится к ненасыщенной алкильной группе с прямой или разветвленной цепью или к циклической алкильной группе, содержащей, по меньшей мере, одну углерод-углеродную двойную связь, получаемой удалением одного атома водорода от одного атома углерода исходного алкена. Группа может иметь либо цис-, либо транс-конформацию относительно двойной связи (связей). Конкретные алкенильные группы включают, но не ограничиваются перечисленным, этенил; пропенилы, такие как проп-1-ен-1-ил, проп-1-ен-2-ил, проп-2-ен-1-ил (аллил), проп-2-ен-2-ил, циклопроп-1-ен-1-ил, циклопроп-2-ен-1-ил; бутенилы, такие как бут-1-ен-1-ил, бут-1-ен-2-ил, 2-метилпроп-1-ен-1-ил, бут-2-ен-1-ил, бут-2-ен-1-ил, бут-2-ен-2-ил, бута-1,3-диен-1-ил, бута-1,3-диен-2-ил, циклобут-1-ен-1-ил, циклобут-1-ен-3-ил, циклобута-1,3-диен-1-ил и т.п.
Термин «алкинил» относится к ненасыщенной алкильной группе с прямой или разветвленной цепью или к циклической алкильной группе, содержащей, по меньшей мере, одну углерод-углеродную тройную связь, получаемой удалением одного атома водорода от одного атома углерода исходного алкина. Конкретные алкинильные группы включают, но не ограничиваются перечисленным, этинил; пропинилы, такие как проп-1-ин-1-ил, проп-2-ин-1-ил; бутинилы, такие как бут-1-ин-1-ил, бут-1-ин-3-ил, бут-3-ин-1-ил и т.п.
Термин «алкилен» относится к насыщенной или ненасыщенной двухвалентной углеводородной группе с прямой или разветвленной цепью или к циклической двухвалентной углеводородной группе, получаемой удалением двух атомов водорода от исходного алкана, алкена или алкина. Конкретные алкиленовые группы включают, но не ограничиваются перечисленным, метилен, этилен, пропилен, бутилены и т.п.
Термин «ацил» относится к радикалу -C(O)R, где R представляет собой водород, алкил, циклоалкил, циклогетероалкил, арил, арилалкил, гетероалкил, гетероарил, гетероарилалкил, как определено выше. Конкретные примеры включают, но не ограничиваются перечисленным, формил, ацетил, циклогексилкарбонил, циклогексилметилкарбонил, бензоил, бензилкарбонил и т.п.
Термин «алкокси» относится к радикалу -OR, где R представляет собой алкильную или циклоалкильную группу, как определено выше. Конкретные примеры включают, но не ограничиваются перечисленным, метокси, этокси, пропокси, бутокси, циклогексилокси и т.п.
Термин «арил» относится к одновалентной ароматической углеводородной группе, получаемой удалением одного атома водорода от одного атома углерода исходной ароматической циклической системы. Конкретные арильные группы включают, но не ограничиваются перечисленным, группы, производные ацеантрилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, коронена, флуорантена, флуорена, гексацена, гексафена, гексалена, ас-индацена, с-индацена, индана, индена, нафталина, октацена, октафена, окталена, овалена, пента-2,4-диена, пентацена, пенталена, пентафена, перилена, феналена, фенантрена, пицена, плеиадена, пирена, пирантрена, рубицена, трифенилена, тринафталина и т.п. В некоторых вариантах осуществления арильная группа содержит от 6 до 20 атомов углерода.
Термин «арилен» относится к двухвалентной ароматической углеводородной группе, получаемой удалением двух атомов водорода из исходной ароматической циклической системы.
Термин «арилалкил» относится к ациклической алкильной группе, в которой один из атомов водорода, связанных с атомом углерода, обычно с концевым или sp 3-атомом углерода, заменен арильной группой. Конкретные арилалкильные группы включают, но не ограничиваются перечисленным, бензил, 2-фенилэтан-1-ил, 2-фенилэтен-1-ил, нафтилметил, 2-нафтилэтан-1-ил, 2-нафтилэтен-1-ил, нафтобензил, 2-нафтофенилэтан-1-ил и т.п. Если имеются в виду определенные алкильные группы, применяются обозначения «арилалканил», «арилалкенил» и/или «арилалкинил». В некоторых вариантах осуществления арилалкильная группа представляет собой (C6-C30)арилалкил, например алканильный, алкенильный или алкинильный фрагмент арилалкильной группы представляет собой (C1-C10), и арильный фрагмент представляет собой (C5-C20).
Термин «арилалкилен» относится к двухвалентной ациклической алкильной группе, в которой один из атомов водорода, связанных с атомом углерода, обычно с концевым или sp 3-атомом углерода, заменен арильной группой.
Термин «арилалкилокси» относится к -Oарилалкильной группе, где арилалкил является таким, как определено выше.
Термин «циано» относится к радикалу -CN.
Термин «циклоалкил» относится к насыщенной или ненасыщенной циклической алкильной группе. Если имеется в виду определенный уровень насыщения, применяются обозначения «циклоалканил» или «циклоалкенил». Конкретные циклоалкильные группы включают, но не ограничиваются перечисленным, группы, производные циклопропана, циклобутана, циклопентана, циклогексана и т.п. В некоторых вариантах осуществления циклоалкильная группа представляет собой (C3-C10)циклоалкил, или в некоторых вариантах осуществления представляет собой (C3-C6)циклоалкил.
Термин «циклогетероалкил» относится к насыщенной или ненасыщенной циклической алкильной группе, в которой один или несколько атомов углерода (и любой из связанных атомов водорода) независимо заменены одинаковыми или различными гетероатомами. Конкретные гетероатомы, заменяющие атом(ы) углерода, представляют собой, но не ограничиваются перечисленным, N, P, О, S и Si. Если имеется в виду определенный уровень насыщения, применяются обозначения «циклогетероалканил» или «циклогетероалкенил». Конкретные циклогетероалкильные группы включают, но не ограничиваются перечисленным, группы, производные эпоксидов, имидазолидина, морфолина, пиперазина, пиперидина, пиразолидина, пирролидина, хинукледина и т.п.
Выражение «соединение формулы (I), производное 1,3-дигексадеканоилпропан-1,2,3-триола» относится к группе со структурной формулой:
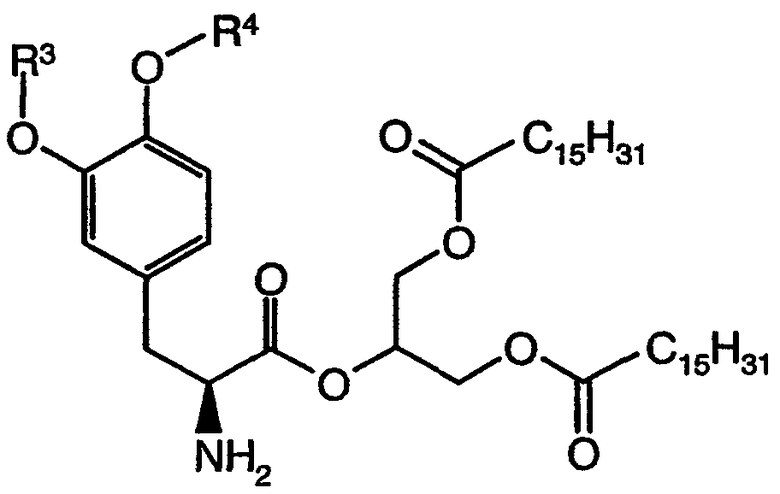
Термин «галоген» относится к фтору, хлору, брому или иоду.
Термин «гетероалкилокси» относится к -O-гетероалкильной группе, где гетероалкил является таким, как определено выше.
Термины «гетероалкил, гетероалканил, гетероалкенил, гетероалкинил» относятся соответственно к алкильной, алканильной, алкенильной и алкинильной группам, в которых один или несколько из атомов углерода (и любые связанные атомы водорода) независимо заменены одинаковыми или различными гетероатомными группами. Конкретные гетероатомные группы включают, но не ограничиваются перечисленным, -О-, -S-, -О-О-, -S-S-, -O-S-, -NR′-, =N-N=, -N=N-, -N=N-NR′-, -PH-, -P(О)2-, -O-P(O)2-, -S(O)-, -S(O)2-, -SnH2- и т.п., где R′ представляет собой водород, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, арил или замещенный арил.
Термин «гетероарил» относится к одновалентной гетероароматической группе, получаемой удалением одного атома водорода от одного атома исходной гетероароматической циклической системы. Конкретные гетероарильные группы включают, но не ограничиваются перечисленным, группы, производные акридина, арсиндола, карбазола, β-карболина, хромана, хромена, циннолина, фурана, имидазола, индазола, индола, индолина, индолизина, изобензофурана, изохромена, изоиндола, изоиндолина, изохинолина, изотиазола, изоксазола, нафтиридина, оксадиазола, оксазола, перимидина, фенантридина, фенантролина, феназина, фталазина, птеридина, пурина, пирана, пиразина, пиразола, пиридазина, пиридина, пиримидина, пиррола, пирролизина, хиназолина, хинолина, хинолизина, хиноксалина, тетразола, тиадиазола, тиазола, тиофена, триазола, ксантена и т.п. В некоторых вариантах осуществления гетероарильная группа представляет собой 5-20-членный гетероарил и в других вариантах осуществления представляет собой 5-10-членный гетероарил. В некоторых вариантах осуществления гетероарильные группы представляют собой гетероарильные группы, производные тиофена, пиррола, бензотиофена, бензофурана, индола, пиридина, хинолина, имидазола, оксазола и пиразина.
Термин «гетероарилоксикарбонил» относится к радикалу -C(O)-OR, где R представляет собой гетероарил, как определено выше.
Термин «гетероарилалкил» относится к ациклической алкильной группе, в которой один из атомов водорода, связанных с атомом углерода, обычно с концевым или sp 3 -атомом углерода, заменен гетероарильной группой. Если имеются в виду определенные алкильные группы, применяются обозначения «гетероарилалканил», «гетероарилалкенил» и/или «гетероарилалкинил». В некоторых вариантах осуществления гетероарилалкильная группа представляет собой 6-30-членный гетероарилалкил, например, алканильный, алкенильный или алкинильный фрагмент гетероарилалкила является 1-10-членным, и гетероарильный фрагмент представляет собой 5-20-членный гетероарил.
Термин «уходящая группа» имеет общепринятое значение, связанное с общепринятым значением в синтетической органической химии, то есть представляет собой атом или группу, способную заменяться нуклеофилом, и включает галоген (такой как хлор, бром и иод), ацилокси (например, ацетокси и бензоилокси), мезилокси, тозилокси, трифторметансульфонилокси, арилокси (например, 2,4-динитрофенокси), метокси, N,O-диметилгидроксиламино и т.п.
Термин «фармацевтически приемлемый» относится к фармацевтическому препарату, утвержденному или одобренному органом государственного регулирования федерального правительства или правительства штата или перечисленному в Фармакопее США, или к другому общепризнанному фармацевтическому препарату для применения на животных и более конкретно для человека.
Термин «фармацевтически приемлемая соль» относится к соли соединения, которая является фармацевтически приемлемой и которая обладает требуемой фармакологической активностью исходного соединения. Такие соли включают в себя: (1) аддитивные соли кислот, образуемые неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п.; или образуемые органическими кислотами, такими как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, оксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п.; или (2) соли, образуемые, когда кислотный протон, присутствующий в исходном соединении, либо заменяется ионом металла, например ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо образует координационную связь с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, N-метилглюкамин, дициклогексиламин и т.п.
Термин «фармацевтически приемлемое вспомогательное средство» относится к разбавителю, адъюванту, эксципиенту или носителю, с которым вводится соединение.
Термин «продленное высвобождение» относится к дозированным формам, которые обеспечивают отсроченное, медленное в течение периода времени, непрерывное, дискретное или замедленное высвобождение соединения или композиции.
Термин «пациент» включает млекопитающих и человека. Термины «человек» и «пациент» используются в данном описании взаимозаменяемо.
Термин «пролекарство» относится к производному молекулы лекарственного средства, которому требуется одна или несколько трансформаций, например метаболизм пролекарства в организме пациента, чтобы вызвать образование активного лекарственного средства. До превращения в исходное лекарственное средство пролекарства могут быть (но необязательно) фармакологически неактивными.
Термин «предгруппировка» относится к группе, которая ковалентно присоединяется к активной молекуле и которая теоретически способна легко отщепляться in vivo с помощью ферментативных или неферментативных способов. Предгруппировка, например, может представлять собой защитную группу, используемую для маскировки функциональной группы; группу, которая действует как субстрат для одного или нескольких механизмов активного или пассивного переноса; или группу, которая влияет на придание или улучшение определенного свойства молекуле, например, такого как растворимость.
Термин «защитная группа» относится к группе атомов, которая во время присоединения к химически активной группе в молекуле маскирует, уменьшает или препятствует ее химической активности. Примеры защитных групп можно обнаружить в публикациях Green et al., «Protective Groups in Organic Chemistry», (Wiley, 2 edition, 1991) и Harrison et al., «Compendium of Syntetic Organic Methods», vol. 1-8 (John Wiley and Sons, 1971-1996). Конкретные аминозащитные группы включают, но не ограничиваются перечисленным, формил, ацетил, трифторацетил, бензил, бензилоксикарбонил («CBZ»), трет-бутоксикарбонил («Boc»), триметилсилил («TMS»), 2-триметилсилилэтансульфонил («SES»), тритил и замещенные тритильные группы, аллилоксикарбонил, 9-флуоренилметилокикарбонил («FMOC»), нитровератрилоксикарбонил («NVOC») и т.п. Конкретные гидроксизащитные группы включают, но не ограничиваются перечисленным, группы, в которых гидроксигруппа либо ацилирована, либо алкилирована, такие как бензил и простые тритиловые эфиры, а также простые алкиловые эфиры, простые тетрагидропираниловые эфиры, простые триалкилсилиловые эфиры и простые аллиловые эфиры.
Термин «замещенная» относится к группе, в которой один или несколько атомов водорода независимо заменены одинаковыми или различными заместителями. Конкретные заместители включают, но не ограничиваются перечисленным, -X, -R33, -O-, =О, -OR33, -SR33, -S-, =S, -NR33R34, =NR33, -CX3, -CF3, -CN, -OCN, -SCN, -NO, -NО2, =N2, -N3, -S(O)2O-, -S(O)2OH, -S(O)2R33, -OS(O2)O-, -OS(O)2R33, -P(O)(O-)2, -P(О)(OR33)(О-), -OP(O)(OR33)(OR34), -C(О)R33, -C(S)R33, -C(O)OR33, C(O)NR33R34, -C(О)О-, -C(S)OR33, -NR35C(O)NR33R34, -NR35C(S)NR33R34, -NR35C(NR33)NR33R34 и -C(NR33)NR33R34, где каждый X независимо представляет собой галоген; каждый R33 и R34 независимо представляют собой водород, алкил, замещенный алкил, арил, замещенный арил, арилалкил, замещенный арилалкил, циклоалкил, замещенный циклоалкил, циклогетероалкил, замещенный циклогетероалкил, гетероалкил, замещенный гетероалкил, гетероарил, замещенный гетероарил, гетероарилалкил, замещенный гетероарилалкил, -NR35R36, -C(O)R35 или -S(O)2R35, или необязательно R33 и R34 вместе с атомом, к которому присоединены R33 и R34, образуют циклогетероалкильное или замещенное циклогетероалкильное кольцо; и R35 и R36 независимо представляют собой водород, алкил, замещенный алкил, арил, замещенный арил, арилалкил, замещенный арилалкил, циклоалкил, замещенный циклоалкил, циклогетероалкил, замещенный циклогетероалкил, гетероалкил, замещенный гетероалкил, гетероарил, замещенный гетероарил, гетероарилалкил или замещенный гетероарилалкил. В некоторых вариантах осуществления замещающая группа выбрана из галогена, -CN, -NО2, -OH, C1-6алкила и C1-6алкокси. В некоторых вариантах осуществления замещающая группа выбрана из галогена, -OH, C1-3алкила и C1-3алкокси.
Термин «проведение лечения» или «лечение» любого заболевания или расстройства относится к прекращению или улучшению течения заболевания или расстройства, уменьшению риска приобретения заболевания или расстройства, уменьшению развития заболевания или расстройства или, по меньшей мере, одного из клинических симптомов заболевания или расстройства, или к уменьшению риска развития заболевания или расстройства или, по меньшей мере, одного из клинических симптомов заболевания или расстройства. «Проведение лечения» или «лечение» также относится к ингибированию (замедлению) заболевания или расстройства либо в физическом смысле (например, путем стабилизации явного симптома), в физиологическом смысле (например, путем стабилизации физического параметра), либо в том и другом смысле и к ингибированию, по меньшей мере, одного физического параметра, который может быть неявным для пациента. Кроме того, «проведение лечения» или «лечение» относится к отсрочке во времени начала заболевания или расстройства. Кроме того, «проведение лечения» или «лечение» также относится к отсрочке во времени начала заболевания или расстройства, или, по меньшей мере их симптомов у пациента, который может быть подвержен или предрасположен к заболеванию или расстройству, даже несмотря на то что пациент еще не испытывает или не обнаруживает симптомов заболевания или расстройства.
Термин «терапевтически эффективное количество» относится к количеству соединения, которое при введении пациенту для проведения лечения заболевания или расстройства является достаточным для воздействия такого лечения на заболевание или расстройство. «Терапевтически эффективное количество» будет изменяться в зависимости от соединения, заболевания или расстройства и их степени тяжести, возраста и массы пациента, подвергаемого лечению.
Термин «отщепление» относится к разрыву химических связей и не ограничивается химическими или ферментативными реакциями или механизмами за исключением явно указанных в контексте.
Теперь будет приведена подробная ссылка на некоторые варианты осуществления.
Соединения
Соединения включают пролекарства леводопа, к которым присоединены предгруппировки. В вариантах осуществления соединения представляют собой производные леводопа формулы (I):
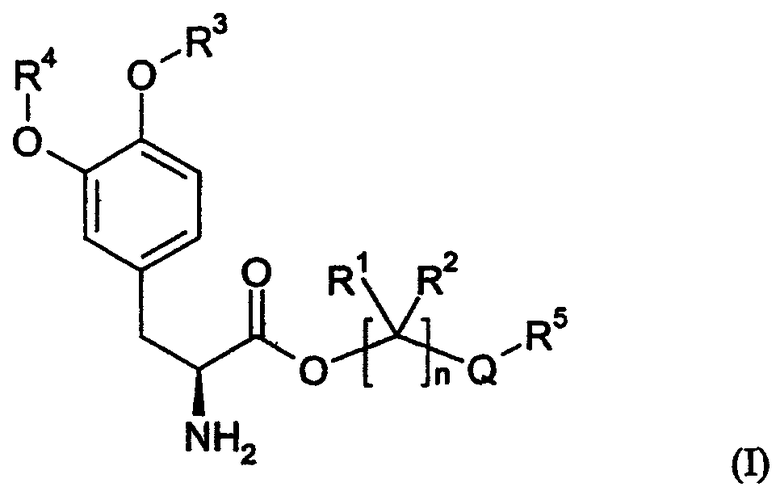
их стереоизомеры, их энантиомеры, их фармацевтически приемлемые соли, их гидраты или сольваты любого из указанных выше, где Q выбран из -X-CO- и -CO-X-; X выбран из -О- и -NR6-;
n равно целому числу от 2 до 4;
каждый R1 и R2 независимо выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, циклоалкила, замещенного циклоалкила, циклогетероалкила, замещенного циклогетероалкила, галогена, гетероалкила, замещенного гетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила;
R3 и R4 независимо выбраны из водорода, -C(O)OR7, -C(O)R7 и -(CR8R9)OC(О)R10;
R5 выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, циклоалкила, замещенного циклоалкила, гетероалкила, замещенного гетероалкила, циклогетероалкила, замещенного циклогетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила; и когда Q представляет собой -X-CO-, R5 дополнительно выбран из алкокси, замещенного алкокси, циклоалкокси и замещенного циклоалкокси;
R6 выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила и замещенного арилалкила;
R7 выбран из алкила, замещенного алкила, циклоалкила, замещенного циклоалкила, циклогетероалкила, замещенного циклогетероалкила, арила, замещенного арила, арилалкила, замещенного арилалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила;
R8 и R9 независимо выбраны из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, циклоалкила, замещенного циклоалкила, циклогетероалкила, замещенного циклогетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила, или необязательно R8 и R9 вместе с атомом углерода, к которому они присоединены, образуют циклоалкильное, замещенное циклоалкильное, циклогетероалкильное или замещенное циклогетероалкильное кольцо; и
R10 выбран из водорода, алкила, замещенного алкила, арила, замещенного арила, арилалкила, замещенного арилалкила, циклоалкила, замещенного циклоалкила, гетероалкила, замещенного гетероалкила, циклогетероалкила, замещенного циклогетероалкила, гетероарила, замещенного гетероарила, гетероарилалкила и замещенного гетероарилалкила;
при условии, что соединение формулы (I) не является производным 1,3-дигексадеканоилпропан-1,2,3-триола.
В некоторых вариантах осуществления соединений формулы I Q представляет собой -X-CO-. В некоторых вариантах осуществления соединений формулы I Q представляет собой -X-CO-, X представляет собой O. В некоторых вариантах осуществления соединений формулы I Q представляет собой -X-CO-, X представляет собой -NR6-.
В некоторых вариантах осуществления соединений формулы I Q представляет собой -CO-X-. В некоторых вариантах осуществления соединений формулы I Q представляет собой -CO-X-, X представляет собой O. В некоторых вариантах осуществления соединений формулы I Q представляет собой -CO-X, X представляет собой -NR6-.
В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 независимо выбран из водорода, -OH, C1-6алкила и замещенного C1-6алкила.
В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 независимо выбран из водорода, -OH, C1-3алкила и замещенного C1-3алкила.
В некоторых вариантах осуществления соединений формулы I R5 выбран из алканила, замещенного алканила, алкенила, замещенного алкенила, арилалканила, замещенного арилалканила, арилалкенила, замещенного арилалкенила, циклоалканила, замещенного циклоалканила, циклогетероалканила, замещенного циклогетероалканила, гетероарилалканила и замещенного гетероарилалканила. В некоторых вариантах осуществления соединений формулы I R5 выбран из метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила, трет-бутила, пентила, гексила, циклопропила, циклобутила, циклопентила, циклогексила, бензила, фенетила и стирила, где арильное кольцо бензильной или стирильной группы необязательно замещено одним или несколькими заместителями, выбранными из галогена, -CN, -NО2, -OH, C1-6алкила и C1-6алкокси.
В некоторых вариантах осуществления соединений формулы I R5 выбран из арила, замещенного арила, гетероарила и замещенного гетероарила. В некоторых вариантах осуществления соединений формулы I R5 выбран из C5-8арила и замещенного C5-8арила, замещенного одним или несколькими заместителями, выбранными из галогена, -CN, -NО2, -OH, C1-6алкила и C1-6алкокси. В некоторых вариантах осуществления соединений формулы I R5 выбран из фенила и пиридила, которые необязательно замещены галогеном, -OH, C1-3алкилом и C1-3алкокси.
В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 независимо выбран из водорода, алканила, замещенного алканила, арилалканила, замещенного арилалканила, циклоалканила, замещенного циклоалканила, циклогетероалканила, замещенного циклогетероалканила, галогена, гетероалканила, замещенного гетероалканила, гетероарилалканила и замещенного гетероарилалканила. В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 независимо выбран из водорода, метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила, трет-бутила, циклопропила, циклобутила, циклопентила, циклогексила и бензила.
В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 независимо выбран из водорода, арила, замещенного арила, гетероарила и замещенного гетероарила. В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 независимо выбран из водорода и фенила, где фенильная группа необязательно замещена одним или несколькими заместителями, выбранными из галогена, -CN, -NО2, -OH, C1-6алкила и C1-6алкокси.
В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 независимо выбран из водорода, -OH, C1-4алкила и замещенного C1-4алкила.
В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 независимо выбран из водорода, -OH, C1-3алкила и замещенного C1-3алкила.
В некоторых вариантах осуществления соединений формулы I каждый R1 и R2 представляет собой водород.
В некоторых вариантах осуществления соединений формулы I R6 выбран из водорода и C1-6алкила. В некоторых вариантах осуществления R6 представляет собой водород, и в некоторых вариантах осуществления R6 представляет собой метил.
В некоторых вариантах осуществления соединений формулы I R3 и R4 независимо выбраны из водорода, -C(O)OR7 и -C(O)R7.
В некоторых вариантах осуществления соединений формулы I R7 выбран из алканила, замещенного алканила, циклоалканила, замещенного циклоалканила, арилалканила, замещенного арилалканила, гетероарилалканила и замещенного гетероарилалканила. В некоторых вариантах осуществления R7 выбран из метила, этила, пропила, изопропила, бутила, изобутила, втор-бутил, трет-бутила, циклопропила, циклобутила, циклопентила, циклогексила и бензила, в котором арильное кольцо бензильной группы необязательно замещено одним или несколькими заместителями, выбранными из галогена, -CN, -NО2, -OH, C1-6алкила и C1-6алкокси.
В некоторых вариантах осуществления соединений формулы I R7 выбран из арила, замещенного арила, гетероарила и замещенного гетероарила. В некоторых вариантах осуществления R7 выбран из C5-8арила, замещенного C5-8арила, C6-10арилалкила и замещенного C6-10арилалкила. В некоторых вариантах осуществления R7 выбран из фенила, пиридила, фурила и тиенила, ароматические кольца которых необязательно замещены одним или несколькими заместителями, выбранными из галогена, -CN, -NО2, -OH, C1-6алкила и C1-6алкокси.
В некоторых вариантах осуществления соединений формулы I R3 и R4 независимо выбраны из водорода и -(CR8R9)OC(O)R10.
В некоторых вариантах осуществления соединений формулы I R10 выбран из водорода, C1-10алкила, замещенного C1-10алкила, C5-8арила, замещенного C5-8арила, C1-15алкокси и замещенного C1-15алкокси.
В некоторых вариантах осуществления соединений формулы I R8 и R9 независимо выбраны из водорода, C1-16алкила, замещенного C1-16алкила, C5-8арила, замещенного C5-8арила, C6-10арилалкила и замещенного C6-10арилалкила.
В некоторых других вариантах осуществления соединения включают пролекарства леводопа формулы (II):
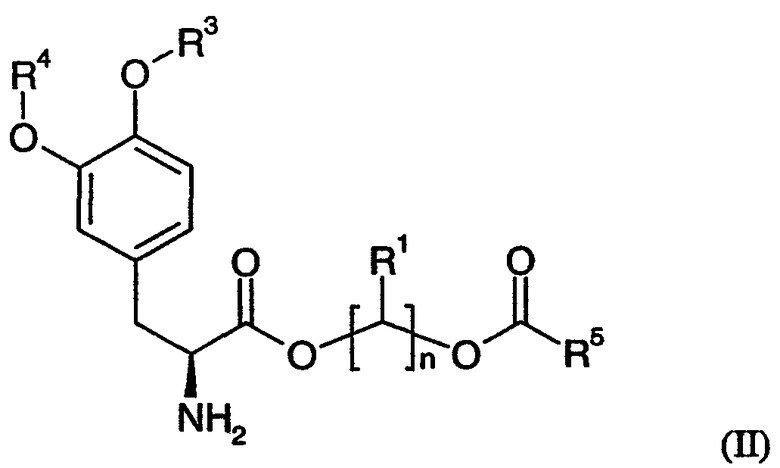
их стереоизомеры, их энантиомеры, их фармацевтически приемлемые соли, их гидраты или сольваты любого из указанных выше,
где n равно целому числу от 2 до 4, R1 выбран из водорода, C1-3алкила с прямой цепью и C1-3алкила с разветвленной цепью, и R5 выбран из фенила и замещенного фенила, в котором один или несколько заместителей выбраны из галогена, -CN, -NО2, -OH, C1-6алкила и C1-6алкокси. Некоторые варианты соединения формулы (II) содержат следующие структуры:
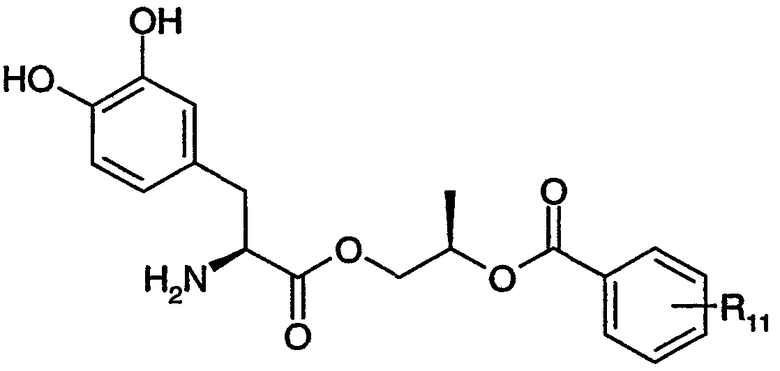 или
или
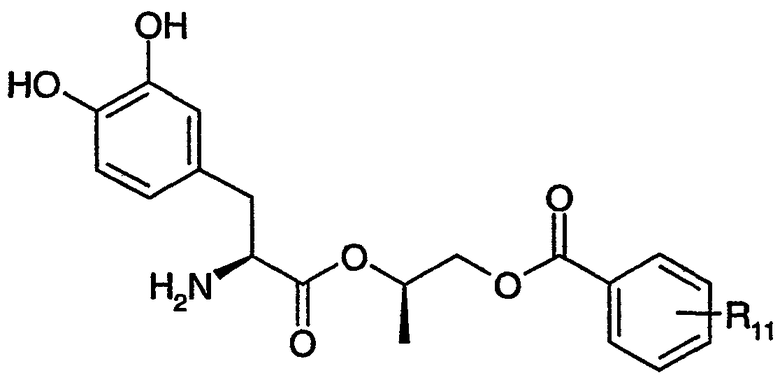
где R11 выбран из водорода, галогена, -CN, -NО2, -OH, C1-6алкила и C1-6алкокси.
В некоторых вариантах осуществления соединений формулы I соединение выбрано из:
2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-ацетилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2S)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2S)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R)-1-метил-2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S)-1-метил-2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S,2S)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R,2R)-1-метил-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S,2S)-1-метил-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-гидрокси-3-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-[(2-гидроксифенил)карбониламино]этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(R)-(3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(S)-(3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(R)-(4-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(S)-(4-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(R)-(2-этокси-3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(S)-(2-этокси-3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(R)-(2-метил-5-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(S)-(2-метил-5-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата и
их фармацевтически приемлемых солей.
В некоторых вариантах осуществления указанных выше соединений фармацевтически приемлемая соль представляет собой гидрохлорид.
Синтез некоторых соединений
Согласно вариантам осуществления пролекарства леводопа можно получить с помощью способов, хорошо известных в данной области.
В некоторых вариантах осуществления соединения можно получать из легкодоступных исходных соединений с применением следующих известных способов и методик. Следует принять во внимание, что там, где приведены конкретные или предпочтительные условия процесса (то есть температуры проведения реакции, время, мольные отношения реагентов, растворители, давления), если специально не указано иное, можно также использовать другие условия процесса. Оптимальные условия реакции можно варьировать в зависимости от используемых конкретных реагентов или растворителя, но такие условия может определить специалист в данной области с помощью установившейся практики метода оптимизации.
Дополнительно, как очевидно специалисту в данной области, чтобы не допустить вовлечения некоторых функциональных групп в нежелательные реакции, можно использовать общеизвестные защитные группы. Подходящие защитные группы для различных функциональных групп, а также подходящие условия для защиты конкретных функциональных групп и снятия защиты хорошо известны в данной области. Например, многочисленные защитные группы описаны в T.W. Greene и G.M. Wuts, Protectig Groups in Organic Syntesis и процитированных там ссылках.
Кроме того, в некоторых вариантах осуществления пролекарства леводопа могут содержать один или несколько хиральных центров. Соответственно такие соединения можно получать или выделять в виде чистых стереоизомеров, то есть в виде индивидуальных энантиомеров или диастереомеров, или в виде смесей, обогащенных стереоизомерами. Все такие стереоизомеры (и обогащенные смеси) включены в объем вариантов осуществления, если специально не указано иное. Чистые стереоизомеры (или обогащенные смеси) можно получать, например, используя оптически активные исходные вещества или стереоселективные реагенты, хорошо известные в данной области. Альтернативно, можно разделять рацемические смеси таких соединений, например, используя хиральную колоночную хроматографию, хиральные разделяющие агенты и т.п.
В некоторых вариантах осуществления пролекарства леводопа можно получать с помощью способов, хорошо известных в данной области (см. публикацию Greene et al., Protective Groups in Organic Syntesis, 3-th edition, John Wiley & Sons, 1999, и процитированные там ссылки; Larock, Comprehensive Organic Transformations, John Wiley & Sons, second edition, March 1999; Advanced Organic Chemistry, John Wiley & Sons, 4-th edition, 1992; Smith, Organic Syntesis, John Wiley & Sons, 1994; патент США № 4966915; патент США № 5462933. Описание указанных публикаций включено в данное описание посредством данной ссылки.
Некоторые препаративные методы можно обнаружить у Gallop et al., в публикации патентной заявки США № 2002/0099041 и у Gallop et al., в публикации международной заявки WO 02/28882.
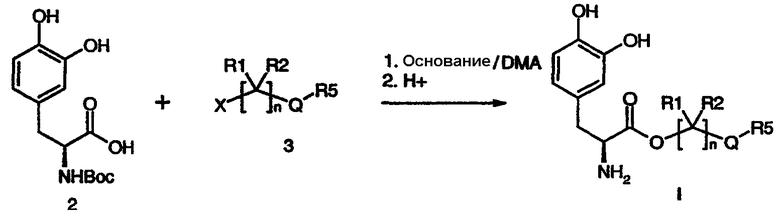
Соединение формулы I можно получать, как проиллюстрировано ниже на схеме 1. Взаимодействие леводопа, содержащего защитную Boc-группу (2), с галогенидом формулы (3) в присутствии подходящего основания, такого как бикарбонат или карбонат щелочного металла, с последующим гидролизом защитной Boc-группы в кислых условиях обеспечивает соединение формулы (I).
Схема 1
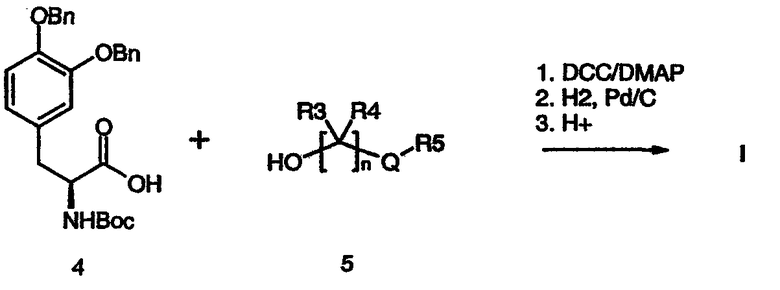
Альтернативно, при взаимодействии подходящим образом защищенного производного леводопа (4) со спиртом (5) в стандартных условиях реакции сочетания (схема 2) с последующим удалением защитных групп получают соединение формулы (I).
Схема 2
Как показано на схеме 3, эпоксид формулы (6) может взаимодействовать с кислотой формулы (7) в присутствии реагента (катализатора) межфазного переноса, такого как бромид тетрабутиламмония, в подходящем растворителе (например, ацетонитриле, толуоле и т.д.) при подходящей повышенной температуре, такой как 50°C, с получением спирта формулы (8), соединения формулы (5), где Q представляет собой -X-C(O), и X представляет собой O.
Схема 3
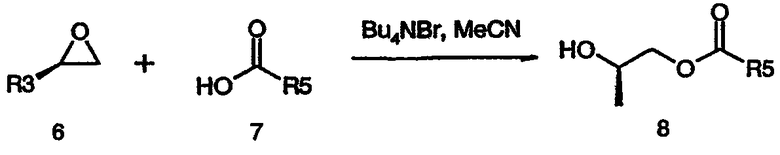
Альтернативно гидроксиламин HO(CR1R2)nNHR6 можно сочетать с кислотой формулы (7), получая соединение формулы (5), где Q представляет собой -XC(O) и X представляет собой -NR6-.
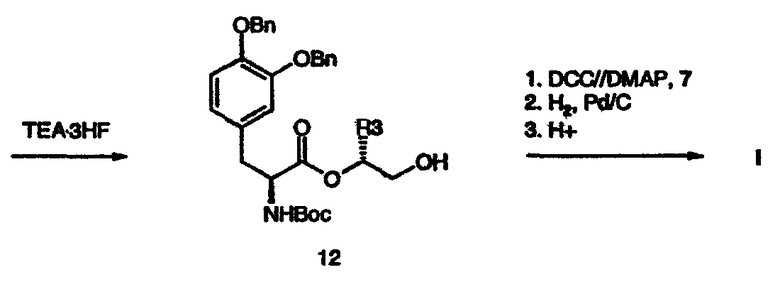
Альтернативно диол формулы (9) может быть превращен в соединение формулы (10), которое дополнительно подвергают сочетанию с производным леводопа формулы (4), получая простой силиловый эфир формулы (11). При взаимодействии простого силилового эфира формулы (11) с фторидом водорода получают спирт формулы (12). При сочетании спирта формулы (12) с кислотой формулы (7) в подходящих условиях (например, в присутствии DCC/DMAP/DCM) с последующим удалением защитных групп в описанных выше условиях получают соединение формулы I (схема 4).
Схема 4

С помощью способов, аналогичных способам, описанным выше, и в экспериментальной части с помощью подходящей операции и защиты химических функциональных групп проводят синтез остальных соединений формулы (I).
Терапевтические применения некоторых соединений
В соответствии с некоторыми вариантами осуществления пролекарства леводопа являются предшественниками допамина. Следовательно, пролекарства леводопа формулы (I) можно вводить пациенту, такому как человек, для лечения болезни Паркинсона. В некоторых вариантах осуществления, по меньшей мере, одно пролекарство леводопа можно вводить совместно с другим терапевтическим средством или лекарственным средством, таким как ингибитор декарбоксилазы или его пролекарство, которое может действовать как защитное средство для ингибирования или предотвращения преждевременного декарбоксилирования пролекарства леводопа и/или метаболита леводопа.
Пролекарства леводопа можно доставлять из той же самой дозированной формы, что и ингибитор декарбоксилазы, или из другой дозированной формы. Пролекарства леводопа можно вводить одновременно с введением ингибитора декарбоксилазы, до введения ингибитора декарбоксилазы или после введения ингибитора декарбоксилазы. Пролекарства леводопа вместе с ингибитором декарбоксилазы или пролекарством или производным ингибитора декарбоксилазы можно вводить пациенту, такому как человек, для лечения болезни Паркинсона.
Некоторые варианты осуществления соединений и композиций, содержащих, по меньшей мере, одно пролекарство леводопа вместе, по меньшей мере, с одним ингибитором декарбоксилазы или, по меньшей мере, с одним пролекарством или производным ингибитора декарбоксилазы, можно успешно применять в лекарственном средстве для человека. Как описано в данном описании, в некоторых вариантах осуществления соединения и композиции полезны для лечения болезни Паркинсона. Когда пролекарства леводопа применяются для лечения болезни Паркинсона, их можно вводить или применять в сочетании с ингибитором декарбоксилазы, таким как карбидопа и/или пролекарство корбидопа, или бензеразид и/или пролекарство бензеразида. Кроме того, терапевтическую эффективность приведенных выше комбинаций можно дополнительно увеличивать путем совместного ведения другого фармацевтически активного средства, такого как ингибитор катехол-кислород-метилтрансферазы (COMT). Кроме того, в некоторых вариантах осуществления для лечения болезни Паркинсона пролекарства леводопа можно вводить пациенту, такому как человек, вместе с (i) ингибитором декарбоксилазы, таким как карбидопа, бензеразид или их пролекарство и (ii) фармацевтически активным средством, таким как ингибитор катехол-кислород-метилтрансферазы (COMT) или его пролекарство.
Описанные в данном описании пролекарства леводопа, в частности, адаптируют для орального введения, хотя их также можно вводить любым другим удобным способом, например, таким как, инъекция, инфузия, ингаляция, трансдермальное введение, абсорбция через эпителиальные или слизистые оболочки (например, слизистую оболочку полости рта, ректальную и/или кишечную слизистую оболочку).
В некоторых вариантах осуществления соединения и/или композиции обеспечивают леводопа и пролекарства леводопа при введении пациенту in vivo. В настоящее время считается, что предгруппировка или предгруппировки пролекарств леводопа подвергаются расщеплению или химически и/или ферментативно. Один или несколько ферментов, таких как холестеразы, присутствующие в желудке, кишечном просвете, кишечной ткани, крови, печени, головном мозге или любой другой подходящей ткани млекопитающего, могут ферментативно отщеплять предгруппировку или предгруппировки соединений и/или композиций. Механизм отщепления не важен для вариантов осуществления.
В некоторых вариантах осуществления соединений и/или композиций можно запланировать, чтобы предгруппировка или предгруппировки подвергались отщеплению после абсорбции в желудочно-кишечном тракте, например в кишечной ткани, крови, печени или других подходящих тканях млекопитающего. В таком случае пролекарства леводопа могут абсорбироваться в большом круге кровообращения из тонкого и толстого кишечника либо путем активного переноса, пассивной диффузии, либо с помощью как активных, так и пассивных способов. В некоторых вариантах осуществления пролекарства леводопа активно переносятся через кишечный эндотелий с помощью переносчиков органических катионов, экспрессируемых на всем протяжении желудочно-кишечного тракта, включая тонкую кишку и толстую кишку. Некоторые соединения и/или композиции пролекарств леводопа можно вводить в виде систем замедленного высвобождения. В некоторых вариантах осуществления соединения можно доставлять путем орального введения с замедленным высвобождением. В некоторых вариантах осуществления соединения можно вводить два раза в день, в некоторых вариантах осуществления один раз в день и в некоторых вариантах осуществления - с интервалами, большими, чем один раз в день.
Некоторые пролекарства леводопа могут применяться для лечения паркинсонизма путем введения одного или нескольких пролекарств леводопа вместе с ингибитором декарбоксилазы, таким как карбидопа или пролекарство корбидопа, в некоторых вариантах осуществления - путем орального введения млекопитающему, нуждающемуся в таком лечении. Человеку массой 70 кг пролекарство леводопа можно вводить в дозировке, содержащей эквивалентную массу леводопа в диапазоне от 10 мг до 10 г в день, и в некоторых вариантах осуществления в дозировке, содержащей эквивалентную массу леводопа в диапазоне от 100 мг до 3 г в день. Дозу можно регулировать с помощью специалиста в данной области исходя из индивидуальных факторов, например массы тела и/или состояния подвергаемого лечению субъекта, вводимой дозы ингибитора декарбоксилазы или пролекарства ингибитора декарбоксилазы, степени тяжести болезни Паркинсона и частоты возникновения побочных эффектов, способа введения и решения лечащего врача. Диапазоны доз можно определить способами, известными специалисту в данной области.
Перед применением для человека пролекарства леводопа можно подвергать испытанию in vitro и in vivo для определения требуемой терапевтической или профилактической активности. Например, испытания in vitro можно применять для определения того, является ли вводимое определенное пролекарство леводопа субстратом для белка-переносчика, включая переносчики органических катионов, такие как OCTN1 и OCTN2. Примеры некоторых способов испытаний, используемых для анализа способности определенного пролекарства леводопа действовать в качестве субстрата для белка-переносчика, описаны Zerangue et al., в публикации патентной заявки США 2003/0158254. Испытания in vitro также применяются для определения того, является ли введение определенного пролекарства леводопа терапевтически эффективным. Также можно показать, что пролекарства леводопа можно эффективно и безопасно применять на системах экспериментальных моделей животных.
В некоторых вариантах осуществления терапевтически эффективная доза пролекарства леводопа может обеспечивать терапевтический эффект без оказания значительной токсичности. Токсичность пролекарств леводопа может быть определена, используя стандартные фармацевтические методики, и уточнена специалистом в данной области. Отношение между дозами, определяющими токсический и терапевтический эффект, представляет собой терапевтический индекс. Некоторые пролекарства леводопа могут показывать особенно высокие терапевтические индексы при лечении заболеваний и расстройств, таких как болезнь Паркинсона. Дозировка пролекарства леводопа может находиться в диапазоне концентраций, циркулирующих в крови, которые включают терапевтически эффективное количество пролекарства леводопа с небольшой токсичностью или без токсичности.
В добавление к применению пролекарств леводопа и композиций, содержащих пролекарства леводопа настоящего изобретения, для лечения болезни Паркинсона, в некоторых вариантах осуществления пролекарства и композиции настоящего изобретения также могут быть полезны для лечения других допаминзависимых заболеваний. Допаминзависимые заболевания можно охарактеризовать либо недостаточной, либо повышенной функциональной допаминэргической активностью в центральной нервной системе. Примеры других допаминзависимых заболеваний включают, но не ограничиваются перечисленным, аффективные расстройства, такие как депрессия и расстройство с дефицитом внимания; психотические расстройства, такие как шизофрения и маниакальная депрессия, расстройства, связанные с нарушением познавательной способности; нарушения движений, такие как синдром усталых ног, периодические нарушения движений конечности, поздняя дискинезия, гипертензия, заболевание Гентингтона и синдром Туретта, расстройства, связанные с болезненным пристрастием, застойная сердечная недостаточность и повышенная дневная сонливость. Для лечения таких заболеваний пролекарство леводопа можно вводить совместно с вспомогательным активным средством. Терапевтически эффективные дозы для лечения допаминзависимых заболеваний можно определять с помощью способов, описанных в данном описании для лечения болезни Паркинсона, и способов, известных в данной области.
Композиции некоторых соединений
В некоторых вариантах осуществления пролекарства леводопа можно включать в фармацевтические композиции, предназначенные для орального введения. Оральное введение таких фармацевтических композиций может приводить к поглощению пролекарств леводопа на всем протяжении кишечного тракта и поступлению в большой круг кровообращения. Такие композиции могут быть получены хорошо известным в фармацевтической области способом и содержат, по меньшей мере, одно пролекарство леводопа. Настоящие композиции могут включать терапевтически эффективное количество, по меньшей мере, одного пролекарства леводопа, в некоторых вариантах осуществления в чистой форме, вместе с ингибитором декарбоксилазы, таким как карбидопа, бензеразид или их пролекарство, и подходящим количеством фармацевтически приемлемого вспомогательного средства, с тем чтобы обеспечить подходящую для введения пациенту форму.
Некоторые варианты осуществления также включают композиции, которые содержат в качестве активного ингредиента, по меньшей мере, одно из пролекарств леводопа, связанное с фармацевтически приемлемыми эксципиентами, носителями, разбавителями и/или адъювантами. При образовании композиций активный ингредиент можно смешивать с эксципиентом, разбавлять разбавителем или заключать в носитель, который может находиться в форме капсулы, саше, бумаги или другого контейнера. Когда эксципиент служит в качестве разбавителя, он может представлять собой твердый, полутвердый или жидкий материал, который действует как вспомогательное средство, носитель или среда для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, пастилок, пакетов-саше, капсул, эликсиров, суспензий, эмульсий, растворов и сиропов, содержащих, например, до 90 мас.% активного соединения с применением, например, мягких и твердых желатиновых капсул.
При получении композиции может применяться измельчение активного соединения для обеспечения подходящего размера частиц перед объединением с другими ингредиентами. Например, если активное соединение в значительной степени нерастворимо, активное соединение можно измельчать до размера частиц менее 200 меш. Если активное соединение в значительной степени растворимо в воде, размер частиц активного соединения можно регулировать путем измельчения для обеспечения в значительной степени равномерного распределения в композиции, например, приблизительно до 40 меш.
Некоторые примеры подходящих эксципиентов включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, гуммиарабик, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Композиции могут дополнительно включать лубриканты, такие как тальк, стеарат магния и минеральное масло, увлажняющие вещества, эмульгаторы и суспендирующие средства, консерванты, такие как метил- и пропилгидроксибензоаты, подсластители, средства для регулирования pH и буферные вещества, средства для регулирования токсичности, ароматизаторы и т.п. Путем использования методик, известных в данной области, можно составлять композиции с тем, чтобы обеспечить быстрое, замедленное или отсроченное высвобождение активного ингредиента после введения пациенту.
Композицию можно составлять в единичной дозированной форме, причем каждая дозировка содержит эквивалентную массу леводопа в диапазоне от 10 мг до 10 г. «Единичная дозированная форма» относится к физически дискретной единице, подходящей в качестве однократной дозировки для человека и других млекопитающих, причем каждый элемент содержит предварительно определенное количество активного вещества, рассчитанное на получение требуемого терапевтического эффекта, совместно с подходящим фармацевтическим эксципиентом, разбавителем, носителем и/или адъювантом.
Пролекарство леводопа можно вводить в терапевтически эффективном количестве. Однако следует понимать, что количество фактически вводимого соединения будет определяться врачом в свете соответствующих условий, включая подлежащее лечению состояние, выбор способа введения, конкретное вводимое соединение, возраст, массу и реакцию отдельного пациента, степень тяжести симптомов у пациента и т.п.
Для получения твердых композиций, таких как таблетки, основной активный ингредиент можно смешивать с фармацевтическим эксципиентом, разбавителем, носителем и/или адъювантом для образования твердой композиции с предварительным составом, содержащей гомогенную смесь, содержащую пролекарство леводопа. Когда указанную композицию с предварительным составом обозначают как гомогенную, это означает, что пролекарство равномерно распределено по всему объему композиции так, что композицию можно легко разделить на одинаково эффективные единичные дозированные формы, такие как таблетки, пилюли и капсулы. Такой твердый предварительный состав можно затем разделять на единичные дозированные формы описанного в данном описании типа, содержащие, например, эквивалентную массу леводопа в диапазоне от 10 мг до 10 г.
Таблетки или пилюли, содержащие пролекарство леводопа, можно покрывать или другим образом обрабатывать для обеспечения дозированной формы, предусматривающей преимущество замедленного высвобождения. Например, таблетка или пилюля может содержать внутренний дозированный компонент и наружный дозированный компонент, причем последний из двух находится в форме оболочки сверху первого и/или окружающей первый компонент. Два компонента можно разделить растворимым в кишечнике слоем. Растворимый в кишечнике слой может служить для противодействия дезинтеграции в желудке и давать возможность внутреннему компоненту проходить интактно в двенадцатиперстную кишку или высвобождаться с отсрочкой. Для таких растворимых в кишечнике слоев или покрытий можно использовать различные материалы. Например, такие материалы включают ряд полимерных кислот и смеси полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы, в которых композиции, содержащие пролекарства леводопа, можно включать для введения орально или путем инъекции, включают подходящим образом ароматизированные сиропы на основе водных растворов, водные или масляные суспензии и ароматизированные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические вспомогательные средства.
Дозированные формы замедленного высвобождения для орального введения
Некоторые пролекарства леводопа можно применять на практике с рядом различных дозированных форм, которые можно адаптировать для обеспечения замедленного высвобождения пролекарства леводопа после орального введения.
В некоторых варианты осуществления дозированная форма может содержать гранулы, которые во время растворения или диффузии высвобождают пролекарство в течение продолжительного периода нескольких часов; в некоторых вариантах осуществления в течение периода, по меньшей мере, 4 часов, в некоторых вариантах осуществления в течение периода, по меньшей мере, 8 часов; в течение периода, по меньшей мере, 12 часов; в течение периода, по меньшей мере, 24 часов; и в других вариантах осуществления в течение периода более 24 часов. Гранулы, высвобождающие пролекарство, могут содержать расположенную в центре композицию или сердцевину, содержащую пролекарство и фармацевтически приемлемые вспомогательные средства, включая необязательный лубрикант, антиоксидант и буфер. Подходящие гранулы с высвобождением, приуроченным к определенному времени, описаны Lu, Int. J. Phann., 1994, 112, 117-124; Farmaceutical Sciences by Remington, 14 edition, pp. 1626-1628 (1970); Fincher, J. Pharm. Sci., 1968, 57, 1825-1835; и в патенте США № 4083949. Подходящие таблетки описаны в Farmaceutical Sciences by Remington, 17-th ed., Ch. 90, pp. 1603-1625 (1985).
В некоторых вариантах осуществления можно применять насос замедленного высвобождения для орального введения (см. Langer, 1990, Science, 249:1527-1533; Sefton, 1987, CRC Crit. Ref. Biomed. Eng., 14:201; Saudek et al., 1989, N. Engl. J. Med., 321:574).
В некоторых вариантах осуществления можно применять полимерные материалы для оральной доставки замедленного высвобождения, такие как описано, например, в «Medical Applications of Controlled Release», Langer and Wise (eds.), CRC Press, Boca Raton, Florida (1974); «Controlled Drug Bioavailability», Drug Product Design and Performance, Smolen и Ball (eds.), Wiley, New York (1984); Ranger and Peppas, 1983, J. Macromol. Sci.Rev. Macromol Chem., 23:61; Levy et al., 1985, Science, 228:190; During et al., 1989, Ann. Neurol., 25:351; и Howard et al., 1989, J. Neurosurg., 71:105.
В некоторых вариантах осуществления можно применять препараты с энтеросолюбильным покрытием для орального введения с замедленным высвобождением. В некоторых вариантах осуществления материалы с покрытием включают полимеры с растворимостью, зависящей от pH (то есть pH-контролируемого высвобождения); полимеры с замедленной или зависящей от pH скоростью набухания, растворения или вымывания (то есть контролируемого во времени высвобождения); полимеры, которые могут подвергаться разрушению под действием ферментов (то есть контролируемого под действием ферментов высвобождения); и полимеры, которые образуют прочные слои, которые могут разрушаться при повышении давления (то есть контролируемого давлением высвобождения).
В некоторых вариантах осуществления для орального введения с замедленным высвобождением можно применять липидные матрицы, высвобождающие лекарственное средство, или воски, высвобождающие пролекарство.
В некоторых вариантах осуществления систему с контролируемым высвобождением можно помещать рядом с мишенью пролекарства леводопа, соответственно требующей только определенной фракции системной дозы (см. Goodson, «Medical Applications of Controlled Release», supra, vol. 2, pp. 115-138 (1984)). Также можно применять другие системы с контролируемым высвобождением, обсуждаемые в публикации Langer, 1990, Science, 249:1527-1533.
В некоторых вариантах осуществления дозированная форма может содержать пролекарство леводопа, наносимое на полимерный субстрат. Полимер может представлять собой полимер, поддающийся или не поддающийся разложению (эрозии). Конкретные биоразлагаемые полимеры описаны, например, в Rosoff, Controlled Release of Drugs, chap. 2, pp. 53-95 (1989); и патентах США №№ 3811444; 3962414; 4066747; 4070347; 4079038 и 4093709.
В некоторых вариантах осуществления дозированная форма может содержать пролекарство леводопа, загруженное в полимер, который высвобождает пролекарство путем диффузии через полимер или путем истечения через поры или путем разрушения полимерной матрицы, как описано, например, Coleman et al., Polimers, 1990, 31, 1187-1231; Roerdink et al., Drug Carrier Systems, 1989, 9, 57-100; Leong et al., Adv.Drug Delivery Rev., 1987, 1, 199-233; Roff et al., Handbook of Common Polimers 1971, CRC Press; и патенте США № 3992518.
В некоторых вариантах осуществления для орального введения с замедленным высвобождением можно применять осмотические системы доставки (см. публикацию Verma et al., Drug Dev. Ind. Phann., 2000, 26:695-708).
Независимо от конкретного вида применяемой дозированной формы замедленного высвобождения для орального введения пролекарство леводопа может высвобождаться из дозированной формы, например, из орально вводимой дозированной формы, в течение достаточного периода времени для обеспечения устойчивых терапевтических концентраций леводопа в крови пациента, позволяя вводить дозированную форму только один раз или два раза в день. В некоторых вариантах осуществления пролекарство леводопа может поддерживать терапевтическую или профилактическую концентрацию леводопа в крови или концентрацию пролекарства леводопа в большом круге кровообращения пациента после орального введения пролекарства леводопа в течение периода времени, по меньшей мере, 4 часов; в некоторых вариантах осуществления в течение периода времени, по меньшей мере, 8 часов; и в некоторых вариантах осуществления в течение периода времени, по меньшей мере, 12 часов.
Композиции можно вводить для профилактического и/или терапевтического лечения. Терапевтическое количество представляет собой количество, достаточное для устранения болезненного состояния или симптомов, или иного способа предотвращения, замедления, задержки или обратного хода развития заболевания или любых других нежелательных симптомов тем или иным, каким угодно способом. При профилактических применениях композиции вводят пациенту, восприимчивому к определенному заболеванию или инфекции или иным образом подверженному риску определенного заболевания или инфекции. Следовательно, профилактически эффективное количество представляет собой количество, достаточное для предотвращения, замедления или задержки болезненного состояния или его симптомов. Точное количество, по меньшей мере, одного соединения, содержащегося в композиции, может зависеть от состояния здоровья и массы пациента.
Подходящую дозировку фармацевтической композиции можно определить согласно любой из нескольких хорошо известных регламентированных методик. Например, для определения подходящей дозы фармацевтического соединения можно применять исследования на животных, такие как исследования с использованием мышей или крыс. Результаты, полученные в исследованиях на животных, можно экстраполировать для определения доз, применяемых для других биологических видов, например, таких как человек.
В некоторых вариантах осуществления дозированные формы можно вводить два раза в день, в некоторых вариантах осуществления - один раз в день и в некоторых вариантах осуществления - с более длительными интервалами.
Некоторые варианты осуществления можно дополнительно осуществлять посредством отсылки к следующим примерам, которые описаны в подробном описании получения соединений и композиций, содержащих, по меньшей мере, одно пролекарство леводопа, и в описании испытаний применения соединений и композиций, содержащих, по меньшей мере, одно пролекарство леводопа. Специалисту в данной области будет очевидно, что многие модификации, относящиеся как к материалам, так и к способам, можно осуществлять на практике, не выходя за пределы вариантов осуществления.
Примеры
Следующие примеры синтеза и биологические примеры предлагаются для иллюстрации некоторых вариантов осуществления и, во всяком случае, не должны истолковываться как ограничивающие объем изобретения. Если специально не указано иное, все температуры приводятся в градусах по Цельсию. В приведенных ниже примерах следующие аббревиатуры имеют следующие значения. Если аббревиатура не обозначена, она соответствует своему общепринятому значению.
Boc = трет-бутилоксикарбонил
DCC = дициклогексилкарбодиимид
DCM = дихлорметан
DMAP =4-N,N-диметиламинопиридин
EDTA = этилендиаминтетрауксусная кислота
г = грамм
ч = час
ВЭЖХ = высокоэффективная жидкостная хроматография
л = литр
ЖХ/МС = жидкостная хроматография/масс-спектроскопия
M = молярный
мг = миллиграмм
мин = минута
мл = миллилитр
ммоль = миллимоль
Pd/C = палладий на активированном угле
ТГФ = тетрагидрофуран
мкг = микрограмм
мкл = микролитр
мкM = микромолярный
Пример 1
1(R)- и 1(S)-циклогексилоксикарбонилэтил-2(S)-амино-3-(3,4-дигидроксифенил)пропаноат
К смеси циклогексанола (10,9 г, 10,9 ммоль), пиридина (8,62 г, 10,9 ммоль) в дихлорметане добавляли 2-бромпропионилхлорид (18,53 г, 10,9 ммоль) при 0°C. Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Продукт распределяли между гексаном и 10%-ной лимонной кислотой. Органическую фазу отделяли, сушили над MgSО4 и концентрировали, получая циклогексиловый эфир 2-бромпропионовой кислоты, который использовали в следующей реакции без дополнительной очистки.
К суспензии соединения Boc-DOPA (297 мг, 1 ммоль) и гидрокарбоната цезия (194 мг, 1 ммоль) в ацетоне добавляли циклогексиловый эфир 2-бромпропионовой кислоты (235 мг, 1 ммоль) и полученную смесь перемешивали при 55°C в течение 40 часов. После удаления растворителя остаток распределяли между этилацетатом и 10%-ной лимонной кислотой. Органической фазу отделяли, сушили над MgSО4 и концентрировали. Затем полученный остаток обрабатывали 30%-ной трифторуксусной кислотой в дихлорметане при комнатной температуре в течение 30 минут. После удаления растворителя полученный остаток очищали препаративной ВЭЖХ с обращенной фазой, получая 40 мг смеси двух диастереоизомеров указанных в заголовке соединений. МС(ESI) m/z 352,73(M+H)+.
Пример 2
1(R)- и 1(S)-изопроксикарбонилэтил-2(S)-амино-3-(3,4-дигидроксифенил)пропаноат
Следуя методике, описанной в примере 1, и заменяя циклогексанол изопропанолом, получали смесь двух диастереоизомеров указанного в заголовке соединения. МС(ESI) m/z 312,70 (M+H+)+.
Пример 3
Гидрохлорид 2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Стадия A: бромэтилбензоат
К раствору бензойной кислоты (2,44 г, 20 ммоль) и 2-бромэтан-1-ола (1,42 мл, 20 ммоль) в 40 мл безводного дихлорметана медленно добавляли раствор 1,3-дициклогексилкарбодиимида (4,12 г, 20 ммоль) в дихлорметане с последующим добавлением каталитического количества 4-(диметиламино)пиридина. Полученную смесь перемешивали при комнатной температуре в течение 16 часов. После фильтрования фильтрат промывали 5%-ным NaHCО3, насыщенным раствором соли и сушили над Na2SО4. После удаления растворителя с помощью хроматографии (силикагель, 10% этилацетата в гексане) остатка получали 3,7 г (82%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): δ 3,62 (т, J=6 Гц, 2H), 4,60 (т, J=6 Гц, 2H), 7,42 (м, 2H), 7,54 (м, 2H), 8,04 (м, 2H).
Стадия B: гидрохлорид 2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Суспензию бромэтилбензоата (2,29 г, 10 ммоль), N-Boc-L-DOPACOOH (3,2 г, 11 ммоль) и бикарбоната цезия (2,1 г, 11 ммоль) в N,N-диметилацетамиде (50 мл) перемешивали при 55°C в течение 16 часов. Растворитель выпаривали в вакууме. Полученный остаток растворяли в этилацетате, промывали водой, 5%-ным NaHCО3, насыщенным раствором соли и сушили над Na2SО4. После удаления растворителя с помощью хроматографии (силикагель, 30% этилацетата в гексане) остатка получали 3,8 г белого твердого вещества. Белое твердое вещество обрабатывали 4M HCl в диоксане при комнатной температуре в течение 30 минут. После удаления растворителя полученное твердое вещество растворяли в 10 мл безводного ацетонитрила и охлаждали. Полученный белый осадок отфильтровывали, промывали простым эфиром и сушили в вакууме, получая 2,2 г (58%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CD3OD): δ 3,02 (дд, J=7,2, 14,4 Гц, 1H), 3,11 (дд, J=5,6, 14,4 Гц, 1H), 4,25 (т, J=6,4 Гц, 1H), 4,52-4,64 (м, 4H), 6,53 (дд, J=2, 8 Гц, 1H), 6,67 (д, J=2 Гц, 1H), 6,69 (д, J=8 Гц, 1H), 7,47 (т, J=7,6 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 8,02 (д, J=7,6 Гц, 2H). МС (ESI) m/z 346,17 (M+H)+ и 344,13 (M-H)-.
Пример 4
Гидрохлорид 2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 3, и заменяя бензойную кислоту 4-фторбензойной кислотой, получали указанное в заголовке соединение (62% по 2 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 3,03 (дд, J=6,8, 14,4 Гц, 1H), 3,11 (дд, J=6, 14,4 Гц, 1H), 4,26 (т, J=6,4 Гц, 1H), 4,50-4,63 (м, 4H), 6,53 (дд, J=2, 8 Гц, 1H), 6,67 (д, J=2 Гц, 1H), 6,69 (д, J=8 Гц, 1H), 7,20 (т, J=8,8 Гц, 2H), 8,05 (дд, J=5,2, 8,8 Гц, 2H). МС (ESI) m/z 363,92 (M+H)+ и 362,02 (M-H)-.
Пример 5
Гидрохлорид 3-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 3, и заменяя 2-бромэтан-1-ол 3-бромпропан-1-олом, получали указанное в заголовке соединение (61% по 2 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 2,40 (м, 2H), 3,02 (дд, J=7,2, 14,4 Гц, 1H), 3,09 (дд, J=6,4, 14,4 Гц, 1H), 4,20 (т, J=6,4 Гц, 1H), 4,35 (т, J=6,4 Гц, 1H), 4,37 (т, J=6,4 Гц, 1H), 6,53 (дд, J=2, 8 Гц, 1H), 6,66 (д, J=2 Гц, 1H), 6,73 (д, J=8 Гц, 1H), 7,48 (т, J=8 Гц, 2H), 7,61 (т, J=8,0 Гц, 1H), 8,01 (м, 2H). МС (ESI) m/z 360,13 (M+H)+ и 358,06 (M-H)-.
Пример 6
Гидрохлорид 3-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 3, и заменяя бензойную кислоту 4-фторбензойной кислотой и 2-бромэтан-1-ол 3-бромпропан-1-олом соответственно, получали указанное в заголовке соединение (55% по 2 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 2,12 (м, 2H), 3,03 (дд, J=7,2, 14,4 Гц, 1H), 3,09 (дд, J=6,4, 14,4 Гц, 1H), 4,21 (т, J=7,2 Гц, 1H), 4,35 (т, J=6,4 Гц, 2H), 4,37 (т, J=6,4 Гц, 2H), 6,54 (дд, J=2, 8 Гц, 1H), 6,67 (д, J=2 Гц, 1H), 6,73 (д, J=8 Гц, 1H), 7,21 (т, J=8,8 Гц, 2H), 8,07 (дд, J=5,6, 8,8 Гц, 2H). МС (ESI) m/z 378,27 (M+H)+ и 376,24 (M-H)-.
Пример 7
Гидрохлорид 2-ацетилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 3, и заменяя 2-бромэтил 2-бромэтилацетатом, получали указанное в заголовке соединение, которое очищали ВЭЖХ (0,05% муравьиная кислота/вода/ацетонитрил) с последующей лиофилизацией в присутствии гидрохлорида.
1H ЯМР (400 МГц, CD3OD): δ 2,50 (с, 3H), 3,03 (дд, J=6,8, 14,4 Гц, 1H), 3,11 (дд, J=6,4, 14,4 Гц, 1H), 4,24 (т, J=6,4 Гц, 1H), 4,27 (т, J=7,2 Гц, 2H), 4,44 (м, 2H), 6,55 (дд, J=2, 8 Гц, 1H), 6,67 (д, J=2 Гц, 1H), 6,73 (д, J=8 Гц, 1H). МС (ESI) m/z 284,10 (M+H)+ и 282,13 (M-H)-.
Пример 8
Гидрохлорид (2R)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Стадия A: (2R)-1-(трет-бутилдиметил-1-силилокси)пропан-2-ол
(R)-(-)-1,2-пропандиол (5 г, 65,7 ммоль) и имидазол (4,47 г, 65,7 ммоль) растворяли в безводном дихлорметане (40 мл). При 0°C добавляли раствор трет-бутилдиметилхлорсилана (9,9 г, 65,7 ммоль) в дихлорметане. Смесь перемешивали при 0°C в течение 2 часов. После фильтрования фильтрат сушили над Na2SО4 и концентрировали, получая 12,5 г (100%) указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3): δ 0 (с, 6H), 0,83 (с, 9H), 1,04 (д, J=6,4 Гц, 3H), 2,64 (с, ушир., 1H), 3,26 (дд, J=8, 9,6 Гц, 1H), 3,50 (дд, J=3,2, 9,6 Гц, 1H), 3,74 (м, 1H).
Стадия B: (1R)-1-метил-2-(трет-бутилдиметилсилилокси)этилбензоат
Бензойную кислоту (2,44 г, 20 ммоль) и (2R)-1-(трет-бутилдиметил-1-силилокси)пропан-2-ол (4,18 г, 22 ммоль) растворяли в 40 мл безводного дихлорметана. Медленно добавляли раствор 1,3-дициклогексилкарбодиимида (4,94 г, 24 ммоль) в дихлорметане с последующим добавлением каталитического количества 4-(диметиламино)пиридина. Смесь перемешивали при комнатной температуре в течение 16 часов. После фильтрования фильтрат промывали 5%-ным NaHCО3, насыщенным раствором соли и сушили над Na2SО4. После удаления растворителя с помощью хроматографии (силикагель, 10% этилацетата в гексане) остатка получали 5,8 г (98%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): δ 0 (с, 3H), 0,2 (с, 3H), 0,83 (с, 9H), 1,30 (д, J=6,4 Гц, 1H), 3,66 (дд, J=4,8, 10,8 Гц, 1H), 3,72 (дд, J=5,6, 10,8 Гц, 1H), 5,15 (м, 1H), 7,36 (т, J=8,4 Гц, 2H), 7,48 (т, J=8,4 Гц, 1H), 7,98 (д, J=8,4 Гц, 2H).
Стадия C: (1R)-2-гидроксиизопропилбензоат
(1R)-1-метил-2-(трет-бутилдиметилсилилокси)этилбензоат (5,8 г, 19,7 ммоль) растворяли в безводном тетрагидрофуране. Медленно добавляли тригидрофторид триэтиламина. Смесь перемешивали при комнатной температуре в течение 8 часов и выпаривали растворитель при пониженном давлении. С помощью хроматографии (силикагель, 30% этилацетата в гексане) остатка получали 3,2 г (90%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): δ 1,37 (д, J=6,4 Гц, 1H), 2,30 (т, J=6,4 Гц, 1H), 3,78 (м, 1H), 5,23 (м, 1H), 7,42 (т, J=7,6 Гц, 2H), 7,54 (т, J=7,6 Гц, 1H), 8,03 (д, J=7,6 Гц, 2H).
Стадия D: (1R)-2-бромизопропилбензоат
К суспензии дибромтрифенилфосфорана (9 г, 21,3 ммоль) в безводном дихлорметане медленно при 0°C добавляли раствор (1R)-2-гидроксиизопропилбензоата (3,2 г, 17,7 ммоль) в дихлорметане. Смесь перемешивали при температуре от 0°C до комнатной температуры в течение 16 часов, затем промывали водой, 5%-ным NaHCО3, насыщенным раствором соли и сушили над Na2SО4. После концентрирования к полученному остатку добавляли гексан. Ph3PO осаждался. После фильтрования и тщательной промывки гексаном фильтрат концентрировали. С помощью хроматографии остатка на силикагеле при элюировании 10% этилацетата в гексане получали 3,6 г (85%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3): δ 1,47 (д, J=6,4 Гц, 3H), 3,75 (м, 2H), 5,31 (м, 1H), 7,42 (т, J=8 Гц, 2H), 7,54 (т, J=8 Гц, 1H), 8,04 (д, J=8 Гц, 2H).
Стадия F: гидрохлорид (2R)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Суспензию (1R)-2-бромизопропилбензоата (4,98 г, 20,6 ммоль), N-Boc-L-DOPA-COOH (7,3 г, 25 ммоль) и бикарбоната цезия (4,85 г, 25 ммоль) в N,N-диметилацетамиде (100 мл) перемешивали при 55°C в течение 16 часов. Растворитель выпаривали в вакууме. К остатку добавляли этилацетат и полученный раствор промывали водой, 5%-ным NaHCО3, насыщенным раствором соли и сушили над Na2SО4. После удаления растворителя при пониженном давлении с помощью хроматографии (силикагель, 30% этилацетата в гексане) остатка получали 6,3 г (68%) белого твердого вещества. Белое твердое вещество обрабатывали 50 мл 4M HCl в диоксане при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали досуха при пониженном давлении. Полученный остаток растворяли приблизительно в 20 мл безводного ацетонитрила и 4 мл простого эфира. Раствор охлаждали, полученный белый осадок отфильтровывали, промывали простым эфиром и сушили в вакууме, получая 4,7 г (87%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CD3OD): δ 1,40 (д, J=6,4 Гц, 3H), 2,99 (дд, J=7,6, 14,4 Гц, 1H), 3,10 (дд, J=5,6, 14,4 Гц, 1H), 4,24 (дд, J=6, 8 Гц, 1H), 4,38 (дд, J=6,8, 11,6 Гц, 1H), 4,52 (дд, J=3,2, 11,6 Гц, 1H), 5,40 (м, 1H), (1H, дд, J=2, 8 Гц, 1H), 6,66 (д, J=2 Гц, 1H), 6,69 (д, J=8 Гц, 1H), 7,47 (т, J=7,6 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 8,02 (д, J=7,6 Гц, 2H). МС (ESI) m/z 360,15 (M+H)+ и 358,09 (M-H)-.
Пример 9
Гидрохлорид (2S)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 8, и заменяя (R)-(-)-1,2-дипропандиол (S)-(+)-1,2-дипропандиолом, получали указанное в заголовке соединение (32% по 5 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,37 (д, J=6,4 Гц, 3H), 2,94 (дд, J=7,2, 14,4 Гц, 1H), 3,05 (дд, J=6, 14,4 Гц, 1H), 4,23 (т, J=6,4 Гц, 1H), 4,40 (дд, J=5,2, 11,6 Гц, 1H), 4,47 (дд, J=3,6, 11,6 Гц, 1H), 5,40 (м, 1H), 6,48 (дд, J=2, 8 Гц, 1H), 6,64 (д, J=2 Гц, 1H), 6,69 (д, J=8 Гц, 1H), 7,47 (т, J=8 Гц, 2H), 7,60 (т, J=7,2 Гц, 1H), 8,00 (д, J=8 Гц, 2H). МС (ESI) m/z 360,33 (M+H)+ и 358,31 (M-H)-.
Пример 10
Гидрохлорид (2R)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 8, и заменяя бензойную кислоту 4-фторбензойной кислотой, получали указанное в заголовке соединение (23% по 5 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,38 (д, J=6,4 Гц, 3H), 3,01 (дд, J=7,2, 14,4 Гц, 1H), 3,09 (дд, J=5,6, 14,4 Гц, 1H), 4,23 (т, J=6,4 Гц, 1H), 4,37 (дд, J=6,4, 11,6 Гц, 1H), 4,49 (дд, J=3,2, 11,6 Гц, 1H), 5,36 (м, 1H), 6,53 (дд, J=2, 8 Гц, 1H), 6,67 (д, J=2 Гц, 1H), 6,69 (д, J=8 Гц, 1H), 7,20 (т, J=8,8 Гц, 2H), 8,05 (дд, J=5,6, 8,8 Гц, 2H). МС (ESI) m/z 378,11 (M+H)+ и 376,06 (M-H)-.
Пример 11
Гидрохлорид (2S)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 8, и заменяя по отдельности бензойную кислоту 4-фторбензойной кислотой и (R)-(-)-1,2-пропандиол (S)-(+)-1,2-пропандиолом, получали указанное в заголовке соединение (43% по 5 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,36 (д, J=6,4 Гц, 3H), 2,96 (дд, J=7,2, 14,4 Гц, 1H), 3,05 (дд, J=6,1, 4,4 Гц, 1H), 4,24 (дд, J=6, 6,8 Гц, 1H), 4,38 (дд, J=6,8, 11,6 Гц, 1H), 4,46 (дд, J=3,2, 11,6 Гц, 1H), 5,38 (м, 1H), 6,49 (дд, J=2, 8 Гц, 1H), 6,64 (д, J=2 Гц, 1H), 6,71 (д, J=8 Гц, 1H), 7,20 (т, J=8,8 Гц, 2H), 8,05 (дд, J=5,6, 8,8 Гц, 2H). МС (ESI) m/z 378,48 (M+H)+ и 376,34 (M-H)-.
Пример 12
Гидрохлорид (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Стадия A: (2R)-1-(трет-бутилдиметил-1-силилокси)пропан-2-ол
(R)-(-)-1,2-пропандиол (5 г, 65,7 ммоль) и имидазол (4,47 г, 65,7 ммоль) растворяли в безводном дихлорметане. При 0°C добавляли раствор хлордиметил-трет-бутилсилана (9,9 г, 65,7 ммоль) в дихлорметане. Смесь перемешивали при 0°C в течение 2 часов. После фильтрования фильтрат сушили над Na2SО4. При концентрировании получали 12,5 г (100%) (2R)-1-(трет-бутилдиметил-1-силилокси)пропан-2-ол, который использовали в следующей реакции без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3): δ 0 (с, 6H), 0,83 (с, 9H), 1,04 (д, J=6,4 Гц, 3H), 2,64 (с, ушир., 1H), 3,26 (дд, J=8, 9,6 Гц, 1H), 3,50 (дд, J=3,2, 9,6 Гц, 1H), 3,74 (м, 1H).
Стадия B: (1R)-1-метил-2-(трет-бутилдиметилсилилокси)этил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноат
N-Boc-L-DOPA-(OBn)2-COOH (3,6 г, 7,5 ммоль) растворяли в безводном дихлорметане. Добавляли триэтиламин (2,6 мл, 18,5 ммоль) и 2,4,6-трихлорбензоилхлорид (1,4 мл, 9 ммоль) и перемешивали раствор в течение 30 минут. К реакционной смеси медленно добавляли раствор (2R)-1-(трет-бутилдиметил-1-силилокси)пропан-2-ола (1,7 г, 9 ммоль) в дихлорметане с последующим добавлением каталитического количества 4-(диметиламино)пиридина. Полученную смесь перемешивали при комнатной температуре в течение 16 часов, затем промывали 10%-ной лимонной кислотой, сушили над Na2SО4 и концентрировали. С помощью хроматографии (силикагель, 10% этилацетата в гексане) получали 3,4 г (70%) (1R)-1-метил-2-(трет-бутилдиметилсилилокси)этил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата.
1H ЯМР (400 МГц, CDCl3): δ 0,08 (с, 6H), 0,88 (с, 9H), 1,12 (д, J=6,4 Гц, 3H), 1,42 (с, 9H), 2,99 (м, 2H), 3,35 (м, 1H), 3,59 (м, 1H), 3,84 (м, 1H), 4,50 (м, 1H), 4,89 (д, NH, 1H), 5,10 (с, 4H), 6,60 (д, J=8 Гц, 1H), 6,71 (с, 1H), 6,87 (д, J=8 Гц, 1H), 7,26-7,43 (м, 10H).
Стадия C: (1R)-2-гидроксиизопропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноат
(1R)-1-метил-2-(трет-бутилдиметилсилилокси)этил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноат (3,4 г, 5,2 ммоль) растворяли в безводном тетрагидрофуране. Медленно добавляли тригидрофторид триэтиламина. Смесь перемешивали при комнатной температуре в течение 4 часов и выпаривали растворитель при пониженном давлении. С помощью хроматографии (силикагель, 30% этилацетата в гексане) получали 2,5 г (90%) (1R)-2-гидроксиизопропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата.
1H ЯМР (400 МГц, CDCl3): δ 1,09 (д, J=6,4 Гц, 3H), 1,41 (с, 9H), 2,78 (с, ушир., 1H), 2,96 (м, 2H), 3,51 (м, 1H), 3,59 (м, 1H), 4,34 (м, 1H), 4,98 (м, 1H), 5,05 (д, NH, 1H), 5,10 (с, 4H), 6,66 (д, J=8 Гц, 1H), 6,77 (с, 1H), 6,83 (д, J=8 Гц, 1H), 7,26-7,43 (м, 10H).
Стадия D: (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноат
К раствору бензойной кислоты (0,57 г, 4,67 ммоль) и (1R)-2-гидроксиизопропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата (2,5 г, 4,67 ммоль), растворенному в 60 мл безводного дихлорметана, медленно добавляли раствор 1,3-дициклогексилкарбодиимида (1,15 г, 5,6 ммоль) в дихлорметане с последующим добавлением каталитического количества 4-(диметиламино)пиридина. Полученную смесь перемешивали при комнатной температуре в течение 16 часов. После фильтрования фильтрат промывали 5%-ным NaHCО3 и сушили над Na2SО4. После удаления растворителя с помощью хроматографии (силикагель, 10% этилацетата в гексане) остатка получали 2,6 г (87%) (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата.
1H ЯМР (400 МГц, CDCl3): δ 1,23 (д, J=6,4 Гц, 3H), 1,41 (с, 9H), 2,98 (м, 2H), 4,26 (м, 1H), 4,33 (м, 1H), 4,51 (м, 1H), 4,93 (д, NH, 1H), 5,10 (с, 4H), 5,24 (м, 1H), 6,65 (д, J=8 Гц, 1H), 6,76 (с, 1H), 6,81 (д, J=8 Гц, 1H), 7,25-7,45 (м, 12H), 7,54 (т, J=7,6 Гц, 1H), 8,00 (д, J=7,6 Гц, 2H).
Стадия E: (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноат
К раствору (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата (2,6 г, 4,85 ммоль) в 40 мл тетрагидрофурана добавляли 200 мг 10% Pd/C, предварительно смешанного с 10 мл метанола, в атмосфере азота. Полученную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 2 часов. После фильтрования и промывки метанолом фильтрат концентрировали с помощью хроматографии остатка (силикагель, 30% этилацетата в гексане) получали 1,87 г (100%) (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноата. МС(ESI) m/z 460,20(M+H)+ и 458,17 (M-H)-.
Стадия F: гидрохлорид (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
(1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноат (1,87 г, 4 ммоль) растворяли в 40 мл 4M HCl в диоксане. Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Диоксан полностью выпаривали при пониженном давлении. Полученное белое твердое вещество растворяли в ацетонитриле (5 мл) и добавляли простой эфир до тех пор, пока раствор не становился слегка мутным. Раствор охлаждали в течение ночи и кристаллизовали продукт. Белое кристаллическое твердое вещество собирали и сушили в вакууме, получая 1,5 г (93%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CD3OD): δ 1,35 (д, J=6,4 Гц, 3H), 3,01 (дд, J=6,8, 14,4 Гц, 1H), 3,08 (дд, J=6,4, 14,4 Гц, 1H), 4,19 (т, J=6,4 Гц, 1H), 4,34 (дд, J=6, 12,4 Гц, 1H), 4,49 (дд, J=2,8, 12,4 Гц, 1H), 5,35 (м, 1H), 6,55 (дд, J=2, 8 Гц, 1H), 6,66 (д, J=2 Гц, 1H), 6,71 (д, J=8 Гц, 1H), 7,48 (т, J=7,2 Гц, 2H), 7,61 (т, J=7,2 Гц, 1H), 8,02 (д, J=7,6 Гц, 2H). МС (ESI) m/z 360,16 (M+H)+ и 358,13 (M-H)-.
Альтернативно, (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноат (стадия D в примере 12) можно получать следующим образом:
Стадия A (способ раскрытия эпоксида): (2R)-2-гидроксипропилбензоат
Раствор (R)-(+)-пропиленоксида (10,5 мл, 150 ммоль), бензойной кислоты (12,2 г, 100 ммоль) и бромида тетрабутиламмония (3,22 г, 10 ммоль) в безводном ацетонитриле нагревали до 50°C в герметизированном сосуде под давлением в течение 24 часов. Реакционную смесь концентрировали досуха при пониженном давлении, разбавляли этилацетатом и промывали водой два раза с последующим добавлением насыщенного раствора NaHCО3 и насыщенного раствора соли. Органический слой сушили над MgSО4 и концентрировали при пониженном давлении, получая 18,0 г (100%) смеси (2R)-2-гидроксипропилбензоата и его регио-изомера (1R)-2-гидроксиизопропилбензоат в соотношении 7,2:1. Раствор смеси регио-изомеров (1,8 г, 10 ммоль) и 2,4,6-коллидина (1,1 мл, 8 ммоль) в 50 мл безводного дихлорметана охлаждали до -78°C перед тем, как добавить по каплям ацетилхлорид (0,28 мл, 4 ммоль). Реакционную смесь перемешивали при -78°C в течение 3 часов перед тем, как проводить нагревание до комнатной температуры в течение 1 часа. Реакционную смесь разбавляли дихлорметаном и трижды промывали 0,5н. HCl с последующим добавлением насыщенного раствора соли. Органический слой сушили над MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:2,5) получали 1,6 г (89%) (2R)-2-гидроксипропилбензоата.
Стадия A (способ бензоилирования диола): (2R)-2-гидроксипропилбензоат
К раствору (R)-(-)-1,2-пропандиола (6,00 г, 78,85 ммоль) и 2,4,6-коллидина (7,22 мл, 54,67 ммоль) в 100 мл безводного дихлорметана добавляли по каплям бензоилхлорид (10,98 мл, 94,62 ммоль) при -78°C. Реакционную смесь перемешивали при -78°C в течение трех часов и при комнатной температуре в течение 1 часа перед гошением водой (10 мл) в течение 15 минут. Погашенную смесь промывали 0,5н. HCl (4×50 мл) до уменьшения темной окраски и затем насыщенным раствором NaHCО3 (4×50 мл) и насыщенным раствором соли. Органический слой отделяли, сушили над Na2SО4 и концентрировали. С помощью хроматографии (силикагель 230-400 меш, этилацетат/гексан 1:9) остатка получали 8,9 г (63%) (2R)-2-гидроксипропилбензоата в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6): δ 1,13 (д, J=6,4 Гц, 3H), 3,93 (м, 1H), 4,10 (м, 2H), 4,95 (д, J=4,8 Гц, 1H), 7,51 (т, J=7,2 Гц, 2H), 7,64 (т, J=7,6 Гц, 1H), 7,99 (д, J=6,8 Гц, 2H); МС (ESI) m/z l81 (M+H)+.
Стадия B: (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-[(трет-бутокси)карбониламино]-3-[(3,4-бис-(фенилметокси)фенил]пропаноат
К раствору (2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропанoвой кислоты (15,9 г, 33,3 ммоль), (2R)-2-гидроксипропилбензоата (5,0 г, 27,7 ммоль) и 4-(диметиламино)пиридина (340 мг) в 250 мл безводного дихлорметана медленно добавляли гидрохлорид 1-(3-диметиламинопропил)3-этилкарбодиимида (EDAC) (8,0 г, 41,6 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном и дважды промывали 0,5н. HCl с последующим добавлением насыщенного раствора соли. Органический слой отделяли, сушили над рыхлым слоем MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:5, затем 1:4) остатка с последующей кристаллизацией из смеси этилацетат/гексан 1:5 получали 8,0 г (45%) указанного в заголовке соединения.
Пример 13
Гидрохлорид (1S)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 12, и заменяя (R)-(-)-1,2-пропандиол (S)-(+)-1,2-пропандиолом, получали указанное в заголовке соединение (41% по 6 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,41 (д, J=6,4 Гц, 3H), 2,92 (дд, J=8, 14,8 Гц, 1H), 3,10 (дд, J=5,2, 14,8 Гц, 1H), 4,23 (т, J=6,4 Гц, 1H), 4,38 (дд, J=7,2, 12,4 Гц, 1H), 4,47 (дд, J=3,2, 12,4 Гц, 1H), 5,42 (м, 1H), 6,51 (дд, J=2, 8 Гц, 1H), 6,65 (д, J=2 Гц, 1H), 6,67 (д, J=8 Гц, 1H), 7,47 (т, J=8,8 Гц, 2H), 7,60 (т, J=8,8 Гц, 1H), 8,02 (м, 2H). МС (ESI) m/z 360,21 (M+H)+ и 358,13 (M-H)-.
Пример 14
Гидрохлорид (1R)-1-метил-2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 12, и заменяя бензойную кислоту 4-фторбензойной кислотой, получали указанное в заголовке соединение (33% по 6 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,34 (д, J=6,4 Гц, 3H), 3,04 (дд, J=6,8, 14,4 Гц, 1H), 3,08 (дд, J=5,6, 14,4 Гц, 1H), 4,20 (т, J=6,4 Гц, 1H), 4,32 (дд, J=6, 11,6 Гц, 1H), 4,48 (дд, J=3,2, 11,6 Гц, 1H), 5,36 (м, 1H), 6,55 (дд, J=2,8 Гц, 1H), 6,68 (д, J=2 Гц, 1H), 6,74 (д, J=8 Гц, 1H), 7,22 (т, J=8,8 Гц, 2H), 8,05 (дд, J=5,6, 8,8 Гц, 2H). МС (ESI) m/z 378,11 (M+H)+ и 376,03 (M-H)-.
Пример 15
Гидрохлорид (1S)-1-метил-2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 12, и заменяя бензойную кислоту 4-фторбензойной кислотой и (R)-(-)-1,2-пропандиол (S)-(+)-1,2-пропандиолом, получали указанное в заголовке соединение (46% по 6 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,41 (д, J=6,4 Гц, 3H), 2,94 (дд, J=7,6, 14,4 Гц, 1H), 3,08 (дд, J=6, 14,4 Гц, 1H), 4,20 (т, J=7,2 Гц, 1H), 4,36 (дд, J=6,8, 11,2 Гц, 1H), 4,46 (дд, J=2,8, 11,2 Гц, 1H), 5,41 (м, 1H), 6,51 (дд, J=2, 8 Гц, 1H), 6,66 (д, J=2 Гц, 1H), 6,72 (д, J=8 Гц, 1H), 7,20 (т, J=8,8 Гц, 2H), 8,04 (дд, J=5,6, 8,8 Гц, 2H). МС (ESI) m/z 378,16 (M+H)+ и 376,10 (M-H)-.
Пример 16
Гидрохлорид (1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Стадия A: (1R,2R)-2-гидрокси-1-метилпропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноат
N-Boc-L-DOPA-(OBn)2COOH (4,3 г, 9 ммоль) растворяли в безводном дихлорметане. Добавляли триэтиламин (3 мл, 22 ммоль) и 2,4,6-трихлорбензоилхлорид (1,7 мл, 11 ммоль) и перемешивали раствор в течение 30 минут. Раствор (2R,3R-(-)-2,3-бутандиола (1,0 мл, 11 ммоль) в дихлорметане медленно добавляли к реакционной смеси с последующим добавлением каталитического количества 4-(диметиламино)пиридина. Полученную смесь перемешивали при комнатной температуре в течение 16 часов, затем промывали 10%-ной лимонной кислотой, 5%-ным NaHCО3, насыщенным раствором соли и сушили над Na2SО4. После удаления растворителя с помощью хроматографии (силикагель, градиент 20%-30% этилацетата в гексане) остатка получали 3,4 г (69%) (1R,2R)-2-гидрокси-1-метилпропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата.
1H ЯМР (400 МГц, CDCl3): δ 1,08 (д, J=6,4 Гц, 3H), 1,11 (д, J=6,4 Гц, 3H), 1,41 (с, 9H), 2,96 (м, 2H), 3,66 (с, ушир., 1H), 4,34 (м, 1H), 4,75 (м, 1H), 4,98 (м, 1H), 5,10 (с, 4H), 6,66 (д, J=8 Гц, 1H), 6,77 (с, 1H), 6,82 (д, J=8 Гц, 1H), 7,26-7,41 (м, 10H).
Стадия B: (1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-[(трет-бутокси)карбониламино]-3-(3,4-бис-(фенилметокси)фенил]пропаноат
К раствору бензойной кислоты (0,57 г, 4,8 ммоль) в безводном дихлорметане добавляли триэтиламин (1,7 мл, 12 ммоль) и добавляли 2,4,6-трихлорбензоилхлорид (0,9 мл, 5,76 ммоль). Полученную смесь перемешивали в течение 30 минут и медленно добавляли к реакционной смеси раствор (1R,2R)-2-гидрокси-1-метилпропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата (2,4 г, 4,4 ммоль) в дихлорметане с последующим добавлением каталитического количества 4-(диметиламино)пиридина. Полученную смесь перемешивали при комнатной температуре в течение 16 часов, промывали 10%-ной лимонной кислотой, сушили над Na2SО4 и концентрировали. С помощью хроматографии (силикагель, 20% этилацетата в гексане) остатка получали 2,6 г (90%) (1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата.
1H ЯМР (400 МГц, CDCl3): δ 1,26 (д, J=6,4 Гц, 6H), 1,39 (с, 9H), 2,78 (м, 1H), 2,96 (м, 1H), 4,51 (м, 1H), 4,89 (д, 1H), 5,10 (с, 4H), 5,15 (м, 1H), 6,57 (д, J=8 Гц, 1H), 6,71 (с, 1H), 6,77 (д, J=8 Гц, 1H), 7,25-7,43 (м, 12H), 7,52 (т, J=7,6 Гц, 1H), 7,99 (д, J=7,6 Гц, 2H).
Стадия C: (1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноат
К раствору (1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата (2,6 г, 3,9 ммоль) в 40 мл тетрагидрофурана в атмосфере азота добавляли 200 мг 10% Pd/C, предварительно смешанного с 10 мл метанола. Полученную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 2 часов. После фильтрования и промывки метанолом фильтрат концентрировали с помощью хроматографии (силикагель, 30% этилацетата в гексане) остатка получали 1,8 г (95%) (1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноата. МС(ESI) m/z 474,31 (M+H)+ и 472,18 (M-H)-.
Стадия D: гидрохлорид (1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
(1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноат (1,8 г, 2,7 ммоль) растворяли в 40 мл 4M HCl в диоксане. Смесь перемешивали при комнатной температуре в течение 30 минут. Диоксан полностью выпаривали при пониженном давлении. Полученное белое твердое вещество растворяли в ацетонитриле (5 мл) и добавляли простой эфир, пока раствор не становился слегка мутным. Раствор охлаждали в течение ночи и кристаллизовали продукт. Белое кристаллическое твердое вещество собирали и сушили в вакууме, получая 1,0 г (87%) указанного в заголовке соединения.
1H ЯМР (400 МГц, CD3OD): δ 1,12 (д, J=6 Гц, 3H), 1,24 (д, J=6 Гц, 3H), 2,85 (дд, J=8, 14 Гц, 1H), 3,02 (дд, J=5,2, 14 Гц, 1H), 4,15 (т, J=5,6 Гц, 1H), 5,06 (м, 2H), 6,46 (дд, J=2, 8 Гц, 1H), 6,61 (д, J=2 Гц, 1H), 6,65 (д, J=8 Гц, 1H), 7,54 (т, J=7,6 Гц, 2H), 7,65 (т, J=7,6 Гц, 1H), 7,90 (м, 2H). МС (ESI) m/z 374,11 (M+H)+ и 372,08 (M-H)-.
Пример 17
Гидрохлорид (1S,2S)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 16, и заменяя (2R,3R)-(-)-2,3-бутанолдиол (2S,3S)-(+)-2,3-бутандиолом, получали указанное в заголовке соединение (34% по 4 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,30 (д, J=6 Гц, 3H), 1,34 (д, J=6 Гц, 3H), 2,68 (дд, J=8, 14,4 Гц, 1H), 2,94 (дд, J=6, 14,4 Гц, 1H), 4,01 (дд, J=5,6, 8 Гц, 1H), 5,20 (м, 2H), 6,44 (дд, J=2, 8 Гц, 1H), 6,59 (д, J=2 Гц, 1H), 6,66 (д, J=8 Гц, 1H), 7,46 (т, J=7,6 Гц, 2H), 7,59 (т, J=7,6 Гц, 1H), 7,99 (м, 2H). МС (ESI) m/z 374,16 (M+H)+ и 372,08 (M-H)-.
Пример 18
Гидрохлорид (1R,2R)-1-метил-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 16, и заменяя бензойную кислоту 4-фторбензойной кислотой, получали указанное в заголовке соединение (42% по 4 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,24 (д, J=6,4 Гц, 3H), 1,32 (д, J=6,4 Гц, 3H), 3,06 (д, J=6,4 Гц, 2H), 4,21 (т, J=6,8 Гц, 1H), 5,19 (м, 2H), 6,57 (дд, J=2, 8 Гц, 1H), 6,71 (д, J=2 Гц, 1H), 6,74 (д, J=8 Гц, 1H), 7,24 (т, J=8,8 Гц, 2H), 8,02 (дд, J=5,2, 8,8 Гц, 2H). МС (ESI) m/z 392,20 (M+H)+ и 390,15 (M-H)-.
Пример 19
Гидрохлорид (1S,2S)-1-метил-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 16, и заменяя по отдельности бензойную кислоту 4-фторбензойной кислотой и (2R,3R)-(-)-2,3-бутандиол (2S,3S)-(+)-2,3-бутандиолом, получали указанное в заголовке соединение (47% по 4 стадиям) в виде белого твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,32 (д, J=6 Гц, 3H), 1,36 (д, J=6 Гц, 3H), 2,75 (дд, J=8, 14,4 Гц, 1H), 3,02 (дд, J=5,6, 14,4 Гц, 1H), 4,22 (дд, J=6, 8 Гц, 1H), 5,23 (м, 1H), 6,46 (дд, J=2, 8 Гц, 1H), 6,61 (д, J=2 Гц, 1H), 6,68 (д, J=8 Гц, 1H), 7,19 (т, J=8,4 Гц, 2H), 8,05 (дд, J=5,2, 1,4 Гц, 2H). МС (ESI) m/z 392,15 (M+H)+ и 390,10 (M-H)-.
Пример 20
Гидрохлорид 3-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Стадия A: 3-бромпропил-4-метоксибензоат
Гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDAC) (3,4 г, 17,7 ммоль) медленно добавляли к раствору 4-метоксибензойной кислоты (2,0 г, 13,1 ммоль), 3-бромпропан-1-ола (1,1 мл, 12,6 ммоль) и 4-(диметиламино)пиридина (100 мг) в 80 мл безводного дихлорметана. Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном и дважды промывали 0,5н. HCl с последующим добавлением насыщенного раствора NaHCО3 и насыщенного раствора соли. Органический слой сушили над MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:10) остатка получали 2,1 г (61 %) 3-бромпропил-4-метоксибензоата.
Стадия B: гидрохлорид 3-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Суспензию 3-бромпропил-4-метоксибензоата (2,1 г, 7,7 ммоль), (2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропановой кислоты (3,43 г, 11,5 ммоль) и бикарбоната цезия (2,98 г, 15,4 ммоль) в 1-метил-2-пирролидиноне (40 мл) перемешивали при 50°C в течение 3 часов. Реакционную смесь разбавляли простым эфиром и трижды промывали водой и затем насыщенным раствором соли. Органический слой отделяли, сушили над рыхлым слоем MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:1) остатка получали 3,7 г (98%) прозрачного густого масла. Масло обрабатывали 4,0M раствором HCl в 1,4-диоксане при комнатной температуре в течение 30 минут. Реакционною смесь концентрировали досуха при пониженном давлении. Полученное густое масло очищали препаративной ВЭЖХ. Фракции ВЭЖХ объединяли, обрабатывали 20 мл 0,5н. HCl и сушили лиофилизацией, получая 1,3 г (41%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,98-2,16 (м, 2H), 2,91 (дд, J=7,4, 15,0 Гц, 1H), 2,97 (дд, J=6,4, 15,2 Гц, 1H), 3,79 (с, 3H), 4,20 (т, J=6,8 Гц, 1H), 4,26 (т, J=5,8 Гц, 2H), 4,29-4,40 (м, 2H), 6,47 (дд, J=2,2, 8,2 Гц, 1H), 6,60 (д, J=2,0 Гц, 1H), 6,72 (д, J=8,0 Гц, 1H), 6,91 (д, J=8,8 Гц, 2H), 7,87 (д, J=8,8 Гц, 2H); МС (ESI) m/z 390,17 (M+H)+.
Пример 21
Гидрохлорид 3-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Стадия A: 3-бромпропил-2-(фенилметокси)бензоат
К раствору 2-(фенилметокси)бензойной кислоты (1,0 г, 4,4 ммоль), 3-бромпропан-1-ола (0,35 мл, 4,0 ммоль) и 4-(диметиламино)пиридина (50 мг) в 20 мл безводного дихлорметана медленно добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDAC) (1,3 г, 6,6 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном и трижды промывали 0,5н. HCl с последующим добавлением насыщенного раствора NaHCО3 и насыщенного раствора соли. Органический слой отделяли, сушили над рыхлым слоем MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:9) остатка получали 0,8 г (58%) 3-бромпропил-2-(фенилметокси)бензоата.
Стадия B: гидрохлорид 3-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Суспензию 3-бромпропил-2-(фенилметокси)бензоата (0,8 г, 2,3 ммоль), (2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропановой кислоты (1,0 г, 3,4 ммоль) и бикарбоната цезия (0,89 г, 4,6 ммоль) в 1-метил-2-пирролидиноне (15 мл) перемешивали при 50°C в течение 3 часов. Реакционную смесь разбавляли простым эфиром, дважды промывали водой и затем насыщенным раствором соли. Органический слой сушили над MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:1) остатка получали 1,2 г (93%) прозрачного густого масла. К раствору масла в ТГФ добавляли 300 мг 10% Pd/C. Воздух из колбы удаляли в вакууме и заменяли H2 при 1 атм. Суспензию перемешивали в атмосфере H2 при комнатной температуре в течение ночи. Реакционную смесь фильтровали через рыхлый слой целита. Растворитель удаляли в вакууме. Полученное густое масло обрабатывали 4,0M раствором HCl в 1,4-диоксане при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали досуха при пониженном давлении и очищали препаративной ВЭЖХ. Фракции ВЭЖХ объединяли, обрабатывали 10 мл 0,5н. HCl и сушили лиофилизацией, получая 545 мг (68%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,86-2,10 (м, 2H), 2,91 (дд, J=7,0, 14,6 Гц, 1H), 2,96 (дд, J=6,6, 15,0 Гц, 1H), 4,08-4,20 (м, 2H), 4,19 (т, J=6,8 Гц, 1H), 4,26 (т, J=5,8 Гц, 2H), 6,42 (дд, J=2,2, 8,2 Гц, 1H), 6,58 (д, J=2,0 Гц, 1H), 6,67 (д, J=8,4 Гц, 1H), 6,78 (т, J=7,6 Гц, 1H), 6,80 (д, J=8,0 Гц, 1H), 7,36 (дт, J=1,4, 7,2 Гц, 1H), 7,59 (дд, J=1,2, 8,0 Гц, 1H); МС (ESI) m/z 376,08 (M+H)+.
Пример 22
Гидрохлорид 3-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 21, и заменяя 2-(фенилметокси)бензойную кислоту 4-(фенилметокси)бензойной кислотой, получали указанное в заголовке соединение в виде белого твердого вещества (41% по двум стадиям).
1H ЯМР (400 МГц, D2O): δ 1,94-2,14 (м, 2H), 2,99 (дд, J=6,4, 14,8 Гц, 1H), 2,95 (дд, J=7,2, 14,4 Гц, 1H), 4,12-4,28 (м, 3H), 4,32 (т, J=5,8 Гц, 2H), 6,47 (дд, J=2,0, 8,0 Гц, 1H), 6,61 (д, J=2,0 Гц, 1H), 6,72 (д, J=8,4 Гц, 1H), 6,83 (д, J=8,8 Гц, 2H), 7,80 (д, J=8,8 Гц, 2H); МС (ESI) m/z 376,08 (M+H)+.
Пример 23
Гидрохлорид 2-гидрокси-3-фенилкарбонилоксипропил-(2S)-2-амино-3-
(3,4-дигидроксифенил)пропаноата
Стадия A: оксиран-2-илметилбензоат
Бензоилхлорид (1,2 мл, 10,0 ммоль) добавляли к раствору глицидола (0,67 мл, 10,0 ммоль) и пиридина (0,81 мл, 10,0 ммоль) в безводном дихлорметане при 0°C. Реакционную смесь дополнительно перемешивали при 0°C в течение 60 минут. Затем реакционную смесь концентрировали досуха при пониженном давлении, разбавляли этилацетатом и дважды промывали 10%-ной лимонной кислотой с последующим добавлением насыщенного раствора NaHCО3 и насыщенного раствора соли. Органическую фазу сушили над MgSО4 и концентрировали досуха, получая оксиран-2-илметилбензоат с выходом 1,8 г (100%).
Стадия B: 2-гидрокси-3-фенилкарбонилоксипропил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноат
Раствор оксиран-2-илметилбензоата (3,0 г, 16,8 ммоль), (2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропановой кислоты (6,0 г, 20,2 ммоль) и тетрабутиламмонийбромида (542 мг, 1,7 ммоль) в безводном толуоле нагревали до 90°C в течение 18 часов. Реакционную смесь концентрировали досуха при пониженном давлении, разбавляли этилацетатом и дважды промывали водой с последующим добавлением насыщенного раствора NaHCО3 и насыщенного раствора соли. Органический слоем сушили над MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:1) остатка получали 2,05 г (26%) 2-гидрокси-3-фенилкарбонилоксипропил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноата.
Стадия C: гидрохлорид 2-гидрокси-3-фенилкарбонилоксипропил (2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
2-Гидрокси-3-фенилкарбонилоксипропил-(2S)-3-(3,4-дигидроксифенил)-2-[(трет-бутокси)карбониламино]пропаноат (2,05 г, 4,3 ммоль) обрабатывали 4,0 M HCl в 1,4-диоксане при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали досуха при пониженном давлении и очищали препаративной ВЭЖХ. Фракции ВЭЖХ объединяли, обрабатывали 10 мл 0,5н. HCl и сушили лиофилизацией, получая указанное в заголовке соединение в виде белого твердого вещества с выходом 0,85 г (48%).
1H ЯМР (400 МГц, D2O): δ 3,11 (т, J=6,6 Гц, 2H), 4,38-4,44 (м, 6H), 6,60 (дд, J=2,2, 7,8 Гц, 1/2H), 6,61 (дд, J=2,4, 8,0 Гц, 1/2H), 6,70 (д, J=2,0 Гц, 1/2H), 6,71 (д, J=2,0 Гц, 1/2H), 6,77 (д, J=8,0 Гц, 1/2H), 6,78 (д, J=8,0 Гц, 1/2H), 7,46-7,52 (м, 2H), 7,60-7,68 (м, 1H), 7,96-8,02 (м, 2H); МС (ESI) m/z 376,15 (M+H)+.
Пример 24
Гидрохлорид (2R)-2-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Стадия A: (2R)-1-(трет-бутилдиметилсилокси)пропан-2-ол
Раствор трет-бутилдиметилхлорсилана (9,9 г, 65,7 ммоль) в дихлорметане добавляли по каплям к раствору (R)-(-)-1,2-пропандиола (5 г, 65,7 ммоль) и имидазола (4,47 г, 65,7 ммоль) в безводном дихлорметане при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 минут перед разбавлением дихлорметаном. Раствор трижды промывали водой с последующим добавлением насыщенного раствора соли. Органический слой отделяли, сушили над рыхлым слоем MgSО4 и концентрировали при пониженном давлении, получая (2R)-1-(трет-бутилдиметилсилокси)пропан-2-ол с выходом 12,0 г (96%).
Стадия B: (1R)-1-метил-2-(трет-бутилдиметилсилокси)этил-2-(фенилметокси)бензоат
К раствору 2-(фенилметокси)бензойной кислоты (4,0 г, 17,5 ммоль), (2R)-1-(трет-бутилдиметилсилокси)пропан-2-ола (2,78 г, 14,6 ммоль) и 4-(диметиламино)пиридина (183 мг) в 100 мл безводного дихлорметана медленно добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDAC) (4,2 г, 17,5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 48 часов. Реакционную смесь разбавляли дихлорметаном и дважды промывали 0,5н. раствором HCl с последующим добавлением насыщенного раствора NaHCО3 и насыщенного раствора соли. Органический слой отделяли, сушили над рыхлым слоем MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:12) остатка получали (1R)-1-метил-2-(трет-бутилдиметилсилокси)этил-2-(фенилметокси)бензоат с выходом 1,7 г (29%).
Стадия C: (1R)-2-гидроксиизопропил-2-(фенилметокси)бензоат
Тригидрофторид триэтиламина (1,7 мл, 10,5 ммоль) медленно добавляли к раствору (1R)-1-метил-2-(трет-бутилдиметилсилокси)этил-2-(фенилметокси)бензоата (1,7 г, 4,24 ммоль) в безводном тетрагидрофуране. Смесь перемешивали при комнатной температуре в течение 48 часов. Растворитель удаляли при пониженном давлении. Реакционную смесь разбавляли дихлорметаном и дважды промывали насыщенным раствором NaHCО3 с последующим добавлением насыщенного раствора соли. Органический слой сушили над MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:5) остатка получали (1R)-2-гидроксиизопропил-2-(фенилметокси)бензоат с выходом 1,5 г (100%).
Стадия D: (2R)-2-[2-(фенилметилокси)фенилкарбонилокси]пропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметилокси)фенил]пропаноат
Гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDAC) (1,5 г, 7,86 ммоль) медленно добавляли к раствору (2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропанoвой кислоты (2,75 г, 5,76 ммоль), (1R)-2-гидроксиизопропил-2-(фенилметокси)бензоата (1,5 г, 5,24 ммоль) и 4-(диметиламино)пиридина (64 мг) в 40 мл безводного дихлорметана. Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном и дважды промывали 0,5н. HCl с последующим добавлением насыщенного раствора NaHCО3 и насыщенного раствора соли. Органический слой отделяли, сушили над рыхлым слоем MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:3) остатка получали (2R)-2-[2-(фенилметилокси)фенилкарбонилокси]пропил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметилокси)фенил]пропаноат с выходом 3,5 г (90%).
Стадия E: гидрохлорид (2R)-2-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
К раствору (2R)-2-[2-(фенилметилокси)фенилкарбонилокси]пропил-(2S)-2-[(1,1-диметилэтилокси)карбониламино]-3-[3,4-бис-(фенилметилокси)фенил]пропаноата (3,5 г, 4,69 ммоль) в ТГФ добавляли 1,0 г 10% Pd/C. Воздух из колбы удаляли в вакууме и заменяли H2 при давлении 1 атм. Суспензию перемешивали в атмосфере H2 при комнатной температуре в течение ночи. Реакционную смесь фильтровали через рыхлый слой целита. Растворитель удаляли в вакууме. Полученное густое масло обрабатывали раствором 4,0M HCl в 1,4-диоксане при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали досуха при пониженном давлении и очищали препаративной ВЭЖХ. Фракции ВЭЖХ объединяли, обрабатывали 10 мл 0,5н. HCl и сушили лиофилизацией, получая указанное в заголовке соединение в виде белого твердого вещества с выходом 1,2 г (62%).
1H ЯМР (400 МГц, D2O): δ 1,30 (д, J=6,4 Гц, 3H), 2,97 (дд, J=6,6, 14,2 Гц, 1H), 3,02 (дд, J=6,2, 14,2 Гц, 1H), 4,27 (дд, J=6,6, 12,2 Гц, 1H), 4,30 (т, J=7,0 Гц, 1H), 4,49 (дд, J=2,8, 12,0 Гц, 1H), 5,22 (дублет пентетов, J=2,4, 6,4 Гц, 1H), 6,47 (дд, J=2,2, 8,2 Гц, 1H), 6,62 (д, J=2,0 Гц, 1H), 6,63 (д, J=8,4 Гц, 1H), 6,81 (т, J=7,6 Гц, 1H), 6,85 (д, J=8,4 Гц, 1H), 7,39 (дт, J=1,6, 7,0 Гц, 1H), 7,62 (дд, J=1,4, 7,8 Гц, 1H); МС (ESI) m/z 376,15 (M+H)+.
Пример 25
Гидрохлорид (2R)-2-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 24, и заменяя 2-(фенилметокси)бензойную кислоту 4-(фенилметокси)бензойной кислотой, получали указанное в заголовке соединение (14% по пяти стадиям).
1H ЯМР (400 МГц, D2O): δ 1,26 (д, J=6,4 Гц, 3H), 2,95 (дд, J=7,0, 14,6 Гц, 1H), 3,01 (дд, J=6,6, 14,6 Гц, 1H), 4,24 (дд, J=6,4, 12,0 Гц, 1H), 4,27 (т, J=6,6 Гц, 1H), 4,45 (дд, J=3,0, 11,8 Гц, 1H), 5,16 (дублет пентетов, J=2,4, 6,4 Гц, 1H), 6,45 (дд, J=2,0, 8,0 Гц, 1H), 6,61 (д, J=2,0 Гц, 1H), 6,63 (д, J=8,0 Гц, 1H), 6,78 (д, J=8,8 Гц, 2H), 7,70 (д, J=8,8 Гц, 2H); МС (ESI) m/z 376,15 (M+H)+.
Пример 26
Гидрохлорид (2R)-2-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Следуя методике, описанной в примере 24, и заменяя 2-(фенилметокси)бензойную кислоту 4-метоксибензойной кислотой, получали указанное в заголовке соединение (32% по пяти стадиям).
1H ЯМР (400 МГц, D2O): δ 1,26 (д, J=6,8 Гц, 3H), 2,91 (дд, J=7,4, 14,6 Гц, 1H), 2,98 (дд, J=6,2, 14,6 Гц, 1H), 3,64 (с, 3H), 4,22 (дд, J=6,4, 12,0 Гц, 1H), 4,27 (т, J=6,8 Гц, 1H), 4,47 (дд, J=2,6, 11,8 Гц, 1H), 5,17 (дублет пентетов, J=2,8, 6,4 Гц, 1H), 6,41 (дд, J=2,0, 8,4 Гц, 1H), 6,60 (д, J=2,4 Гц, 1H), 6,61 (д, J=8,0 Гц, 1H), 6,69 (д, J=8,8 Гц, 2H), 7,65 (д, J=8,8 Гц, 2H); МС (ESI) m/z 390,32 (M+H)+.
Пример 27
Гидрохлорид 2-[(2-гидроксифенил)карбониламино]этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
Стадия A: N-(2-гидроксиэтил)-[2-(фенилметокси)фенил]карбоксамид
Гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDAC) (1,3 г, 6,57 ммоль) медленно добавляли к раствору 2-(фенилметокси)бензойной кислоты (1,5 г, 6,57 ммоль), 1-гидроксибензотриазола (HOBt) (0,89 г, 6,57 ммоль) и этанoламина (0,40 мл, 6,57 ммоль) в 50 мл безводного ТГФ при 0°C. Суспензию перемешивали и медленно нагревали до комнатной температуры в течение 24 часов. Реакционную смесь концентрировали досуха при пониженном давлении, полученный остаток разбавляли дихлорметаном и дважды промывали 0,5н. HCl с последующим добавлением насыщенного раствора NaHCО3 и насыщенного раствора соли. Органический слой отделяли, сушили над рыхлым слоем MgSО4 и концентрировали при пониженном давлении, получая N-(2-гидроксиэтил)-[2-(фенилметокси)фенил]карбоксамид с выходом 1,8 г (100%).
Стадия B: 2-{[2-(фенилметокси)фенил]карбониламино}этил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноат
К раствору (2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропанoвой кислоты (3,95 г, 8,27 ммоль), N-(2-гидроксиэтил)-[2-(фенилметокси)фенил]карбоксамида (1,8 г, 6,63 ммоль) и 4-(диметиламино)пиридина (84 мг) в 40 мл безводного дихлорметана медленно добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDAC) (2,0 г, 10,34 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли дихлорметаном и дважды промывали 0,5н. HCl с последующим добавлением насыщенного раствора соли. Органический слой отделяли, сушили над MgSО4 и концентрировали при пониженном давлении. С помощью хроматографии (силикагель, этилацетат/гексан 1:2, затем 1:1,5) остатка получали 3,7 г (73%) 2-{[2-(фенилметокси)фенил]карбониламино}этил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата.
Стадия C: гидрохлорид 2-[(2-гидроксифенил)карбониламино]этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата
К раствору 2-{[2-(фенилметокси)фенил]карбониламино}этил-(2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропаноата (3,7 г, 5,06 ммоль) в ТГФ добавляли 1,0 г 10% Pd/C. Воздух из колбы удаляли в вакууме и заменяли H2 при давлении 1 атм. Суспензию перемешивали в атмосфере H2 при комнатной температуре в течение ночи. Реакционную смесь фильтровали через рыхлый слой целита. Растворитель удаляли в вакууме. Полученное густое масло обрабатывали 4,0M HCl в 1,4-диоксане при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали досуха при пониженном давлении и очищали препаративной ВЭЖХ. Фракции ВЭЖХ объединяли, обрабатывали 15 мл 0,5н. HCl и сушили лиофилизацией, получая указанное в заголовке соединение в виде белого твердого вещества с выходом 1,2 г (61 %).
1H ЯМР (400 МГц, D2O): δ 2,86 (дд, J=6,8, 14,8 Гц, 1H), 2,91 (дд, J=6,0, 14,8 Гц, 1H), 3,38-3,62 (м, 2H), 4,14-4,30 (м, 2H), 4,19 (т, J=6,6 Гц, 1H), 6,34 (дд, J=2,2, 8,2 Гц, 1H), 6,49 (д, J=2,0 Гц, 1H), 6,52 (д, J=8,0 Гц, 1H), 6,70 (дд, J=0,8, 8,4 Гц, 1H), 6,73 (дт, J=1,2, 7,8 Гц, 1H), 7,18 (ддд, J=1,6, 7,2, 7,4 Гц, 1H), 7,42 (дд, J=1,6, 8,0 Гц, 1H); МС (ESI) m/z 361,28 (M+H)+.
Пример 28
2(R)- и 2(S)-(3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноат
Стадия A: 1-бромизопропилникотинат
Хлорангидрид никотиновой кислоты (2,56 г, 20 ммоль) и 30 мг DMAP добавляли к смеси 2-бром-2-пропанола (2,8 г, 20 ммоль), триэтиламина (5,6 мл, 20 ммоль) в дихлорметане при 0°C. Полученную смесь перемешивали при комнатной температуре в течение ночи. Полученный продукт распределяли между этилацетатом и водой. Органическую фазу отделяли, сушили над MgSО4 и концентрировали, получая 1-бромизопропилникотинат, который использовали в следующей реакции без дополнительной очистки.
Стадия B: цезиевая соль (2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропионовой кислоты
Гидрокарбонат цезия (194 мг, 1 ммоль) добавляли к раствору (2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропионовой кислоты (297 мг, 1 ммоль) в 5 мл воды и 5 мл ацетонитрила. Полученную смесь перемешивали при комнатной температуре в течение 10 минут, затем замораживали и лиофилизовали, получая цезиевую соль (2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропионовой кислоты в виде белого твердого вещества, которое использовали в следующей реакции без дополнительной очистки.
Стадия C: 2(R)- и 2(S)-(3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноат
1-бромизопропилникотинат (366 мг, 1,5 ммоль) добавляли к раствору цезиевой соли (2S)-2-[(трет-бутокси)карбониламино]-3-[3,4-бис-(фенилметокси)фенил]пропионовой кислоты (432 мг, 1 ммоль) в диметилацетамиде при комнатной температуре и перемешивали смесь при 55°C в течение 16 часов. После удаления растворителя при пониженном давлении остаток распределяли между этилацетатом и водой. Органическую фазу отделяли, сушили над MgSО4 и концентрировали. Полученный остаток обрабатывали 30% трифторуксусной кислотой в дихлорметане при комнатной температуре в течение 30 минут. После удаления растворителя полученный остаток очищали препаративной ВЭЖХ с обращенной фазой, получая 180 мг указанных в заголовке соединений в виде смеси двух диастереоизомеров. МС(ESI) m/z 362,22(M+H)+.
Пример 29
2(R)- и 2(S)-(4-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноат
Следуя методике, описанной в примере 28, и заменяя хлорангидрид никотиновой кислоты хлорангидридом изоникотиновой кислоты, получали указанные в заголовке соединения в виде смеси двух диастереоизомеров. МС(ESI) m/z 362,13 (M+H)+.
Пример 30
2(R)- и 2(S)-(2-этокси-3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноат
Следуя методике, описанной в примере 28, и заменяя хлорангидрид никотиновой кислоты хлорангидридом 1′-этоксиникотиновой кислоты, получали указанные в заголовке соединения в виде смеси двух диастереоизомеров.
МС (ESI) m/z 406,15 (M+H)+.
Пример 31
2(R)- и 2(S)-(2-метил-5-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноат
Следуя методике, описанной в примере 28, и заменяя хлорангидрид никотиновой кислоты хлорангидридом 6′-метилникотиновой кислоты, получали указанные в заголовке соединения в виде смеси двух диастереоизомеров. МС(ESI) m/z 376,31(M+H)+.
Пример 32
Поглощение пролекарств леводопа после введения пролекарств леводопа и корбидопа крысам
Дозированные формы замедленного высвобождения для орального введения, которые медленно высвобождают лекарственное средство в течение периодов времени 6-24 часа, обычно высвобождают значительную часть дозы в ободочной кишке. Следовательно, лекарственные средства, подходящие для применения в таких дозированных формах, могут обладать хорошей абсорбцией в ободочной кишке. Данный эксперимент проводили для определения поглощения и фактических уровней леводопа в крови после введения пролекарств леводопа в ободочную кишку при coвместном введении со средством корбидопа (внутрь ободочной кишки, внутрибрюшинно или орально) и тем самым определяли пригодность пролекарств леводопа для применения в дозированных формах замедленного высвобождения для орального введения. Биодоступность леводопа после совместного введения пролекарств леводопа и корбидопа рассчитывали относительно орального coвместного введения леводопа и корбидопа.
Стадия A: Протокол введения
Крыс получали коммерческим путем и вводили предварительно канюли как в восходящую ободочную кишку, так и в яремную вену. Во время эксперимента животные находились в сознании. Все животные не ели в течение ночи и до 4 часов после введения дозы пролекарства леводопа. Карбидопа вводили в виде раствора в воде или цитратном буфере либо орально, либо внутрибрюшинно, либо внутрь ободочной кишки при эквивалентной дозе до 25 мг корбидопа на кг. Либо одновременно с введением корбидопа, либо спустя 1 час после введения дозы корбидопа непосредственно в ободочную кишку с помощью катетера вводили HCl-соль леводопа или HCl-соль пролекарства леводопа в виде раствора (в воде) при эквивалентной дозе до 75 мг леводопа на кг. Пробы крови (0,3 мл) получали из яремного катетера с интервалами более 8 часов и немедленно добавляли метабисульфит натрия для предотвращения окисления леводопа. Затем в пробу крови дополнительно добавляли метанол/перхлорную кислоту для предотвращения гидролиза пролекарства леводопа. Пробы крови анализировали, как описано ниже.
Стадия B: Подготовка пробы лекарственного средства, абсорбируемого в ободочной кишке
1. В 1,5 мл контрольные пробирки добавляли 300 мкл метанола/перхлорной кислоты.
2. Крысиную кровь (300 мкл) собирали через различные промежутки времени в EDTA-пробирки, содержащие 75 мкл метабисульфита натрия, и встряхивали для перемешивания. Фиксированный объем крови (100 мкл) добавляли непосредственно в пробирку Эппендорфа и встряхивали для перемешивания.
3. Для получения конечной калибровочной кривой (0,004, 0,02, 0,1, 0,5, 2,5, 10 мкг/мл) к 80 мкл контрольной крысиной крови добавляли по десять микролитров стандартного исходного раствора леводопа (0,04, 0,2, 1, 5, 25, 100 мкг/мл) и по 10 мкл 10% метабисульфита натрия. Затем в каждую пробирку добавляли по 300 мкл 50/50 метанола/перхлорной кислоты с последующим добавлением 20 мкм п-хлорфенилаланина.
4. Пробы встряхивали и центрифугировали в течение 10 минут при 14000 об/мин.
5. Супернатант анализировали ЖХ/МС/МС-анализом.
Стадия C: ЖХ/МС/МС-анализ
При проведении анализа использовали ЖХ/МС/МС-спектрометр API 4000, оборудованный бинарными насосами Agilent 1100 и автоматическим пробоотборником CTC HTS-PAL. Во время анализа использовали колонку (4,6×150 мм) Zorbax XDB C8. Подвижная фаза представляла собой 0,1% муравьиную кислоту (A) и ацетонитрил с 0,1% муравьиной кислоты (B). Градиентные условия: 5% B в течение 0,5 минут, затем увеличение до 98% B в течение 3 минут, затем поддержание на уровне 98% B в течение 2,5 минут. Подвижную фазу возвращали к 2% B в течение 2 минут. На спектрометре API 4000A использовали источник TurboIonSpray. Анализ выполняли в режиме регистрации положительных ионов (положительная мода) и MRM-переход для каждого анализируемого вещества оптимизировали с помощью стандартного раствора. Инжектировали по 5 мкл пробы. Некомпартментарный анализ выполняли, применяя WinNonlin (v.3.1 Professional Version, Pharsight Corporation, Mountain View, California) на профилях отдельных животных. Суммарную статистику в отношении оценки главных параметров проводили для Cmax (максимальная наблюдаемая концентрация после введения дозы), Tmax (время максимальной концентрации представляет собой время, при котором наблюдается максимальная концентрация), AUC(o-t) (площадь под кривой зависимости сывороточной концентрации от времени от начала отсчета времени до времени последнего сбора, оцениваемая с применением метода log-линейной трапецеидальной интерполяции), AUC(0-∞)(площадь под кривой зависимости сывороточной концентрации от времени от начала отсчета времени до бесконечности, оцениваемая с применением метода log-линейной трапецеидальной интерполяции для времени последнего сбора с экстраполяцией до бесконечности), и t1/2,z (конечный период полужизни). Максимальные концентрации леводопа в крови (значения Cmax) и значения площади под кривой зависимости сывороточной концентрации от времени (AUC) после введения дозы пролекарств леводопа со средством корбидопа в ободочную кишку были значительно выше (>2-кратно), чем те же значения, достигаемые при введении в ободочную кишку леводопа со средством корбидопа.
Совместное введение леводопа и корбидопа в ободочную кишку приводит к очень низкой относительной биодоступности леводопа (то есть только 3% при оральном coвместном введении леводопа и корбидопа). Для сравнения, coвместное введение пролекарств леводопа, перечисленных ниже, со средством корбидопа показывает улучшение относительной биодоступности леводопа, по меньшей мере, 2-кратно. Диапазон улучшения относительной биодоступности леводопа составляет от 2 до 20-кратного. Полученные данные показывают, что некоторые пролекарства леводопа можно получать в виде композиций, подходящих при оральном введении для эффективного замедленного высвобождения и поглощения леводопа из ободочной кишки.
Пролекарства леводопа, которые при введении продуцируют относительную биодоступность леводопа, по меньшей мере, 2-кратно большую, чем биодоступность леводопа, продуцируемая после введения леводопа, включают:
гидрохлорид (2R)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (2R)-2-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (2R)-2-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (2R)-2-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид 2-гидрокси-3-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид 3-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид 3-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (1R,2R)-1-метил-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (1S,2S)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (1R)-1-метил-2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (1S)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (2S)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (2R)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид (2S)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
гидрохлорид 2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата и
гидрохлорид (1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата.
| название | год | авторы | номер документа |
|---|---|---|---|
| МЕЗИЛАТНОЕ ПРОЛЕКАРСТВО ЛЕВОДОПЫ, ЕГО КОМПОЗИЦИИ И ПРИМЕНЕНИЕ | 2006 |
|
RU2429223C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПЕРОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ПРОЛЕКАРСТВА ЛЕВОДОПЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2537137C2 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
| НЕСТЕРОИДНЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИОДИДНЫХ РЕЦЕПТОРОВ ДЛЯ МЕСТНОЙ ДОСТАВКИ ЛЕКАРСТВ | 2016 |
|
RU2731618C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2014 |
|
RU2704112C2 |
| PPAR-ПОДДЕРЖИВАЮЩИЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ МЕТАБОЛИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2687490C2 |
Изобретение относится к пролекарствам левадопа, их стереоизомерам, энантиомерам или фармацевтически приемлемым солям и фармацевтической композиции на их основе для обеспечения замедленного высвобождения леводопа, а также к их вариантам их применения и способам их получения, соединения могут найти применение для лечения или профилактики заболеваний, при которых показано использование леводопа. В общей формуле (I):
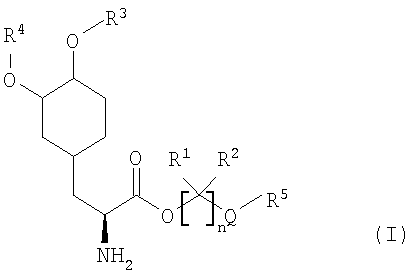
Q выбран из -Х-СО-; Х выбран из -О- и -NH-; n равно целому числу от 2 до 4; каждый R1 и R2 независимо выбран из водорода, -ОН, С1-4алкила и замещенного С1-4алкила, где замещающей группой является -ОН; R3 и R4 представляют водород; R5 выбран из водорода, С1-4алкила, фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из C1-6алкокси, C1-6алкила, галогена и -ОН; С3-8циклоалкила, пиридила, замещенного пиридила, где каждая замещающая группа независимо выбрана из C1-6алкила и C1-6алкокси; при условии, что соединение формулы (I) не является производным 1,3-дигексадеканоилпропан-1,2,3-триола. 9 н. и 39 з.п. ф-лы.
1. Соединение формулы (I)
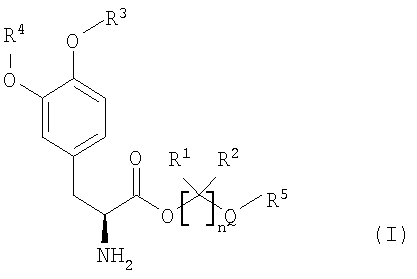
его стереоизомер, его энантиомер или его фармацевтически приемлемая соль, где
Q выбран из -Х-СО-;
Х выбран из -О- и -NH-;
n равно целому числу от 2 до 4;
каждый R1 и R2 независимо выбран из водорода, -ОН, С1-4алкила и замещенного С1-4алкила, где замещающей группой является -ОН;
R3 и R4 представляют водород;
R5 выбран из водорода, С1-4алкила, фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из C1-6алкокси, C1-6алкила, галогена и -ОН; С3-8циклоалкила, пиридила, замещенного пиридила, где каждая замещающая группа независимо выбрана из C1-6алкила и C1-6алкокси;
при условии, что соединение формулы (I) не является производным 1,3-дигексадеканоилпропан-1,2,3-триола.
2. Соединение по п.1, где Q представляет собой -Х-СО-.
3. Соединение по п.1, где каждый R1 и R2 независимо выбран из водорода, -ОН, С1-4алкила и замещенного С1-4алкила, где каждой замещающей группой является -ОН.
4. Соединение по п.1, где каждый R1 и R2 независимо выбран из водорода, -ОН, C1-3алкила и замещенного C1-3алкила, где каждой замещающей группой является -ОН.
5. Соединение по п.1, где каждый R1 и R2 представляет собой водород.
6. Соединение по п.1, где R5 выбран из С1-4алкила и С3-8циклоалкила.
7. Соединение по п.1, где R5 выбран из метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила, трет-бутила, циклопропила, циклобутила, циклопентила и циклогексила.
8. Соединение по п.1, где R5 выбран из фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из C1-6алкокси, C1-6алкила, галогена и -ОН, пиридила и замещенного пиридила, где каждая замещающая группа независимо выбрана из C1-6алкила и C1-6алкокси.
9. Соединение по п.1, где R5 выбран из фенила и замещенного фенила, замещенного одним или несколькими заместителями, выбранными из галогена, -ОН, C1-6алкила и
C1-6алкокси.
10. Соединение по п.1, где R5 выбран из фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из C1-3алкокси, C1-3алкила, галогена и -ОН; пиридила и замещенного пиридила, где каждая замещающая группа независимо выбрана из С1-3алкила и C1-3алкокси.
11. Соединение по п.2, где Х представляет собой -О-.
12. Соединение по п.2, где Х представляет собой -NH-.
13. Соединение по п.1, где соединение имеет структуру формулы (II)
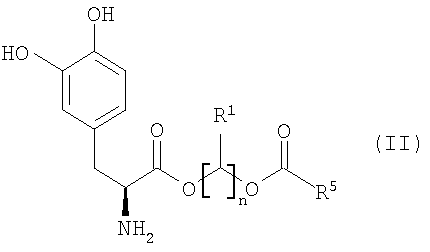
его стереоизомер, его энантиомер или его фармацевтически приемлемая соль, где n равно целому числу от 2 до 4;
каждый R1 выбран из водорода, C1-3алкила с прямой цепью и C1-3алкила с разветвленной цепью; и
R5 выбран из фенила и замещенного фенила, где одна или несколько замещающих групп выбраны из галогена, -ОН, C1-6алкила и С1-6алкокси.
14. Соединение по п.13, где соединение имеет структуру
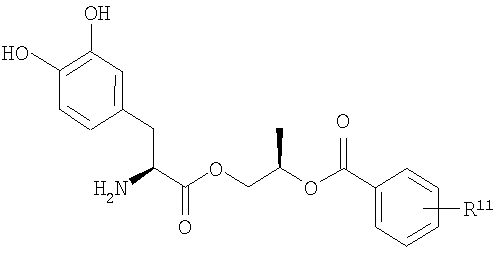
где R11 выбран из водорода, галогена, -ОН, C1-6алкила и С1-6алкокси.
15. Соединение по п.13, где соединение имеет структуру
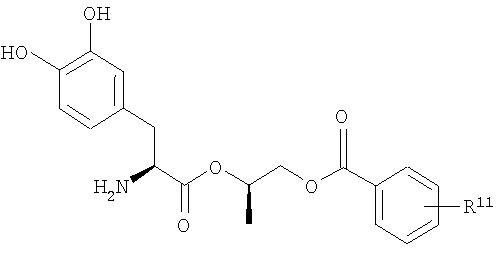
где R11 выбран из водорода, галогена, -ОН, C1-6алкила и C1-6алкокси.
16. Соединение по п.1, где соединение выбрано из:
2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-ацетилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2S)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2S)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R)-1-метил-2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S)-1-метил-2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R,2R)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S,2S)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R,2R)-1-метил-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S,2S)-1-метил-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-гидрокси-3-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-[(2-гидроксифенил)карбониламино]этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(R)-(3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(S)-(3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(R)-(4-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(S)-(4-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(R)-(2-этокси-3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(S)-(2-этокси-3-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(R)-(2-метил-5-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2(S)-(2-метил-5-пиридилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата и
их фармацевтически приемлемых солей.
17. Соединение по п.1, где соединение выбрано из:
(2R)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(2-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-гидрокси-3-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(4-гидроксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
3-(4-метоксифенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R,2R)-1-метил-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S,2S)-1-метил-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R)-1-метил-2-(4-фторфенилкарбонилокси)этил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2S)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2R)-2-(4-фторфенилкарбонилокси)пропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2S)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата и
их фармацевтически приемлемых солей.
18. Соединение по п.16 или 17, где фармацевтически приемлемая соль представляет собой гидрохлорид.
19. Фармацевтическая композиция для обеспечения замедленного высвобождения леводопа, содержащая, по меньшей мере, один фармацевтически приемлемый эксципиент и терапевтически эффективное количество, по меньшей мере, одного соединения формулы (I) по п.1.
20. Фармацевтическая композиция по п.19, дополнительно содержащая, по меньшей мере, один ингибитор декарбоксилазы.
21. Фармацевтическая композиция по п.20, где, по меньшей мере, один ингибитор декарбоксилазы выбран из корбидопа и бензеразида.
22. Фармацевтическая композиция по п.19, дополнительно содержащая, по меньшей мере, один ингибитор катехол-O-метилтрансферазы.
23. Фармацевтическая композиция по п.22, где, по меньшей мере, один ингибитор катехол-O-метилтрансферазы выбран из энтакапона и толкапона.
24. Фармацевтическая композиция по п.19, где фармацевтическая композиция представляет собой препарат для орального введения.
25. Фармацевтическая композиция по п.19, где фармацевтическая композиция представляет собой композицию замедленного высвобождения.
26. Фармацевтическая композиция по п.19, где, по меньшей мере, одно соединение присутствует в количестве, эффективном для лечения у пациента болезни Паркинсона.
27. Фармацевтическая композиция по п.19, где, по меньшей мере, одно соединение присутствует в количестве, эффективном для лечения у пациента заболевания, выбранного из депрессии, расстройства с дефицитом внимания; шизофрении, маниакальной депрессии, расстройств, связанных с нарушением познавательной способности, синдрома усталых ног, периодических нарушений движений конечностей, поздней дискинезии, заболевания Гентингтона, синдрома Туретта, гипертензии, расстройств, связанных с болезненным пристрастием, застойной сердечной недостаточности и повышенной дневной сонливости.
28. Применение, по меньшей мере, одного соединения по п.1 в терапевтически эффективном количестве для лечения заболевания у пациента, где заболевание выбрано из болезни Паркинсона, расстройства с дефицитом внимания, шизофрении, расстройств, связанных с нарушением познавательной способности, синдрома усталых ног, периодических нарушений движений конечностей, поздней дискинезии, заболевания Гентингтона и повышенной дневной сонливости.
29. Применение по п.28, где заболевание представляет собой болезнь Паркинсона.
30. Применение по п.28, где заболевание выбрано из расстройства с дефицитом внимания, шизофрении, расстройств, связанных с нарушением познавательной способности, синдрома усталых ног, периодических нарушений движений конечностей, поздней дискинезии, заболевания Гентингтона и повышенной дневной сонливости.
31. Применение, по меньшей мере, одного соединения формулы (I) по п.1 для достижения устойчивых терапевтических или профилактических концентраций в крови леводопа или пролекарства леводопа в большом круге кровообращения пациента, включающее оральное введение.
32. Применение по п.31, где терапевтическую или профилактическую концентрацию в крови поддерживают в течение, по меньшей мере, 4 ч после орального введения однократной дозы, содержащей, по меньшей мере, одно соединение формулы (I) по п.1.
33. Соединение по п.13, где соединение выбрано из:
(2R)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2S)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
фармацевтически приемлемой соли любого из вышеуказанных, и комбинации любого из вышеуказанных.
34. Соединение по п.13, где соединение выбрано из:
(1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
фармацевтически приемлемой соли любого из вышеуказанных, и комбинации любого из вышеуказанных.
35. Соединение по любому из пп.33 или 34, где фармацевтически приемлемая соль выбрана из гидрохлоридной соли.
36. Соединение по любому из пп.14 или 15, где R11 представляет собой одну замещающую группу, выбранную из хлора, брома, метила и метокси.
37. Способ получения соединения формулы (I)
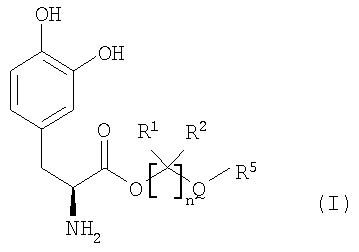
или его фармацевтически приемлемой соли, включающий:
взаимодействие соединения формулы (2)
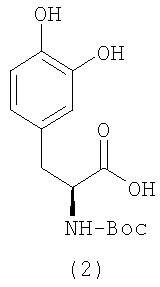
с галогенидом формулы (3)
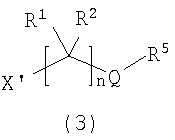
в присутствии основания; и
удаление Вос-защитной группы для получения соединения формулы (I),
где X' представляет собой галоген;
Q выбран из -Х-СО-;
Х выбран из -О- и -NH-;
n равно целому числу от 2 до 4;
каждый R1 и R2 независимо выбран из водорода, -ОН, С1-4алкила и замещенного С1-4алкила, где замещающей группой является -ОН;
R5 выбран из водорода, С1-4алкила, фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из C1-6алкокси, C1-6алкила, галогена и -ОН; С3-8циклоалкила, пиридила, замещенного пиридила, где каждая замещающая группа независимо выбрана из C1-6алкила и C1-6алкокси.
38. Способ получения соединения формулы (I)
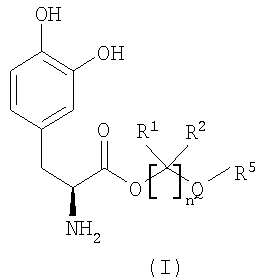
или его фармацевтически приемлемой соли, включающий:
взаимодействие соединения формулы (4)
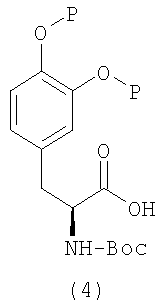
со спиртом формулы (5)
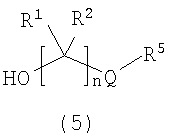
и удаление Вос- и Р-защитных групп для получения соединения формулы (I),
где Р представляет собой гидрокси-защитную группу;
Q выбран из -Х-СО-;
Х выбран из -О- и -NH-;
n равно целому числу от 2 до 4;
каждый R1 и R2 независимо выбран из водорода, -ОН, С1-4алкила и замещенного С1-4алкила, где замещающей группой является -ОН;
R5 выбран из водорода, С1-4алкила, фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из C1-6алкокси, C1-6алкила, галогена и -ОН; С3-8циклоалкила, пиридила, замещенного пиридила, где каждая замещающая группа независимо выбрана из C1-6алкила и C1-6алкокси.
39. Способ по п.37 или 38, где соединение формулы (I) имеет структуру формулы (II)
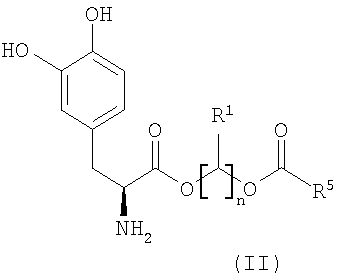
его стереоизомер, его энантиомер или его фармацевтически приемлемая соль,
где n равно целому числу от 2 до 4;
каждый R1 выбран из водорода, C1-3алкила с прямой цепью и C1-3алкила с разветвленной цепью; и
R5 выбран из фенила и замещенного фенила, где одна или несколько замещающих групп выбраны из галогена, -ОН, C1-6алкила и C1-6алкокси.
40. Способ по п.37 или 38, где соединение формулы (I) имеет структуру
или
где R11 выбран из водорода, галогена, -ОН, C1-6алкила и C1-6алкокси.
41. Способ по п.37 или 38, где соединение формулы (I) выбрано из:
(2R)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(2S)-2-фенилкарбонилоксипропил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата; и
фармацевтически приемлемой соли любого из указанных соединений.
42. Способ получения пролекарства леводопа формулы (а)
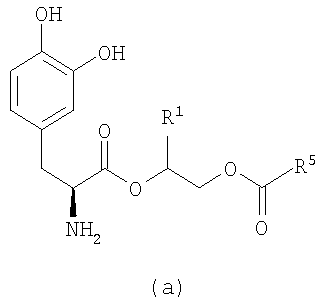
или его фармацевтически приемлемой соли, включающий:
взаимодействие простого силилового эфира формулы (11)
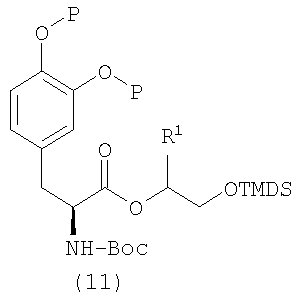
с фторидом водорода для получения соединения формулы (12)
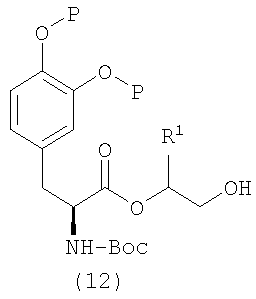
и взаимодействие соединения формулы (12) с кислотой формулы (7)
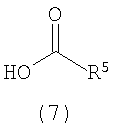 ;
;
и удаление Вос- и Р-защитных групп для получения соединения формулы (а),
где Р представляет собой гидрокси-защитную группу;
каждый R1 и R2 независимо выбран из водорода, -ОН, С1-4алкила и замещенного
С1-4алкила, где замещающей группой является -ОН;
R5 выбран из водорода, С1-4алкила, фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из C1-6алкокси, C1-6алкила, галогена и -ОН; С3-8циклоалкила, пиридила, замещенного пиридила, где каждая замещающая группа независимо выбрана из C1-6алкила и C1-6алкокси.
43. Способ по п.42, где
R1 выбран из водорода, C1-3алкила с прямой цепью и С1-3алкила с разветвленной цепью; и
R5 выбран из фенила и замещенного фенила, где одна или несколько замещающих групп выбраны из галогена, -ОН, C1-6алкила и С1-6алкокси.
44. Способ по п.42, где соединение формулы (I) выбрано из:
(1R)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата;
(1S)-1-метил-2-фенилкарбонилоксиэтил-(2S)-2-амино-3-(3,4-дигидроксифенил)пропаноата; и
фармацевтически приемлемой соли любого из указанных соединений.
45. Способ по любому из пп.37, 38 или 42, где фармацевтически приемлемая соль представляет собой гидрохлорид.
46. Способ по п.38 или 42, где Р представляет собой бензил.
47. Соединение формулы (I), полученное способом по п.37 или 38:
или его фармацевтически приемлемая соль,
где Q выбран из -Х-СО-;
Х выбран из -О- и -NH-;
n равно целому числу от 2 до 4;
каждый R1 и R2 независимо выбран из водорода, -ОН, С1-4алкила и замещенного
С1-4алкила, где замещающей группой является -ОН;
R5 выбран из водорода, С1-4алкила, фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из C1-6алкокси, C1-6алкила, галогена и -ОН; С3-8циклоалкила, пиридила, замещенного пиридила, где каждая замещающая группа независимо выбрана из C1-6алкила и C1-6алкокси.
48. Соединение формулы (а), полученное способом по п.42:
или его фармацевтически приемлемая соль, где
каждый R1 и R2 независимо выбран из водорода, -ОН, С1-4алкила и замещенного
С1-4алкила, где замещающей группой является -ОН;
R5 выбран из водорода, С1-4алкила, фенила, замещенного фенила, где каждая замещающая группа независимо выбрана из С1-6алкокси, C1-6алкила, галогена и -ОН; С3-8циклоалкила, пиридила, замещенного пиридила, где каждая замещающая группа независимо выбрана из С1-6алкила и C1-6алкокси.
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| Шланговое соединение | 0 |
|
SU88A1 |
| US 4771073 A, 13.09.1988 | |||
| US 4873263 A, 10.10.1989 | |||
| ПЕРОРАЛЬНО ВВОДИМАЯ ЛЕКАРСТВЕННАЯ ФОРМА | 1992 |
|
RU2114619C1 |
| ЗАЖИМ ДЛЯ КРЕПЛЕНИЯ ДЕТАЛЕЙ НА СТОЙКАХ | 0 |
|
SU253490A1 |

