Настоящее изобретение относится к производному тетрагидроизохинолина и к его применению в терапии. В частности, настоящее изобретение относится к фармакологически активному замещенному производному тетрагидроизохинолина.
Это соединение действует, как позитивный аллостерический модулятор D1, и поэтому полезно для применения в качестве фармацевтического средства, предназначенного для лечения заболеваний, в которых играют роль рецепторы D1.
Относящийся к моноаминам допамин воздействует на GPCR (рецепторы, связанные с G-белком) двух семейств и модулирует двигательную функцию, механизмы, связанные с поощрением, познавательные процессы и другие физиологические функции. В частности, допамин воздействует на нейроны посредством D1-подобных рецепторов, включающих допаминовые рецепторы D1 и D5, которые в основном сопряжены с G-белком Gs, и поэтому стимулируют продуцирование цАМФ (циклический аденозинмонофосфат) посредством D2-подобных рецепторов, которые включают рецепторы D2, D3 и D4, которые сопряжены с G-белками Gi/q и которые подавляют продуцирование цАМФ. Эти рецепторы широко экспрессируются в разных областях головного мозга. В частности, рецепторы D1 участвуют во многих физиологических функциях и поведенческих процессах. Так, например, рецепторы D1 участвуют в синаптической пластичности, когнитивной функции и направленных на достижение цели двигательных функциях, а также в процессах, связанных с поощрением. Вследствие их роли в нескольких физиологических/неврологических процессах рецепторы D1 участвуют в различных нарушениях, включая связанные с познавательной способностью и негативные симптомы шизофрении, нарушение познавательной способности, связанное с лечением нейролептическим средством, слабое нарушение познавательной способности (СНП), импульсивность, синдром дефицита внимания с гиперактивностью (СДВГ), болезнь Паркинсона и другие нарушения движений, дистонию, слабоумие при болезни Паркинсона, болезнь Гентингтона, слабоумие с тельцами Леви, болезнь Альцгеймера, привыкание к чрезмерному употреблению лекарственного средства или наркотика, нарушения сна, апатию, травматическое повреждение спинного мозга или невропатическую боль.
Показано, что затруднительно разработать биологически доступные при пероральном введении малые молекулы, действие которых направлено на рецепторы D1. Разработанные к настоящему времени агонисты D1 характеризуются наличием пирокатехинового фрагмента и поэтому их применение в клинической практике ограничено инвазивным лечением. Также затруднительным оказалось обеспечение достаточной селективности вследствие высокой степени гомологии центров связывания лиганда у подтипов допаминовых рецепторов (например, допаминовых рецепторов D1 и D5). Кроме того, использование агонистов D1 ограничено в связи с возможными побочными эффектами, включая дискинезию и гипотензию.
Поэтому необходимо разработать новые средства, которые могут модулировать рецепторы D1.
Проявлялся больший интерес к идентификации аллостерических модуляторов GPCR, как средств, обеспечивающих понимание механизма действия рецептора, и как возможных терапевтических средств. GPCR представляют собой самое большое семейство рецепторов клеточной поверхности и большое количество имеющихся в продаже лекарственных средств непосредственно активируют или блокируют пути передачи сигналов, опосредуемые этими рецепторами. Однако показано, что для некоторых GPCR (например, пептидных рецепторов) затруднительно разработать малые молекулы или обеспечить достаточную селективность вследствие высокой степени гомологии центров связывания лиганда у подтипов рецептора (например, допаминовых рецепторов D1 и D5 или D2 и D3). Соответственно, направление многих исследований в области лекарственных средств изменилось в сторону идентификации малых молекул, действие которых направлено на центры, отличающиеся от центров воздействия природного ортостерического агониста. Лиганды, которые связываются с этими центрами, индуцируют изменение конформации GPCR и тем самым аллостерически модулируют функцию рецептора. Аллостерические лиганды обладают самыми различными функциями, включая способность усиливать (позитивный аллостерический модулятор, ПАМ) или ослаблять (негативный аллостерический модулятор, НАМ) воздействие эндогенного лиганда, путем воздействия на его сродство и/или эффективность. Кроме селективности к подтипу аллостерические модуляторы могут обладать другими возможными преимуществами с точки зрения разработки лекарственных средств, такими как отсутствие прямого воздействия или собственной эффективности; только усиление воздействия нативного медиатора в том месте и в тот момент времени, в котором он высвобождается; ослабление склонности к индуцированию десенсибилизации, возникающей в связи с постоянным воздействием агониста, а также ослабление склонности вызывать связанные с мишенями побочные эффекты.
Соединения, предлагаемые в настоящем изобретении, усиливают воздействие агонистов D1 или эндогенного лиганда на рецепторы D1 по аллостерическому механизму и поэтому они являются позитивными аллостерическими модуляторами D1 (ПАМ D1).
Поэтому соединение, предлагаемое в настоящем изобретении, являющееся ПАМ D1, полезно для лечения и/или предупреждения заболеваний и нарушений, в которых играют роль рецепторы D1. Такие заболевания включают связанные с познавательной способностью и негативные симптомы шизофрении, нарушение познавательной способности, связанное с лечением нейролептическим средством, слабое нарушение познавательной способности (СНП), импульсивность, синдром дефицита внимания с гиперактивностью (СДВГ), болезнь Паркинсона и другие нарушения движений, дистонию, слабоумие при болезни Паркинсона, болезнь Гентингтона, слабоумие с тельцами Леви, болезнь Альцгеймера, привыкание к чрезмерному употреблению лекарственного средства или наркотика, нарушения сна, апатию, травматическое повреждение спинного мозга или невропатическую боль.
В заявке на международный патент WO 2013/051869 А1 раскрыты некоторые производные 3,4-дигидро-1Н-изохинолин-2-ила, которые являются антагонистами NK2.
В заявке на международный патент WO 2008/109336 А1 раскрыты некоторые соединения тетрагидроизохинолина, которые являются модуляторами гистаминовых рецепторов Н3.
В заявке на международный патент WO 2014/193781 А1 раскрыты некоторые производные 3,4-дигидроизохинолин-2(1Н)-ила, применимые для лечения нарушения познавательной способности, связанной с болезнью Паркинсона или шизофренией.
В заявке на международный патент WO 2016/055479 раскрыты замещенные производные 3,4-дигидроизохинолин-2(1Н)-ила и его аналоги, которые могут быть применимы для лечения заболеваний, в которых играют роль рецепторы D1.
В заявке на международный патент WO 2019/204418 раскрыты некоторые производные пиразотетрагидроизохинолинов, которые являются позитивными аллостерическими модуляторами D1, и могут быть применимы для лечения болезни Паркинсона, болезни Альцгеймера, шизофрении и синдрома дефицита внимания с гиперактивностью (СДВГ).
Однако сохраняется необходимость разработки активных позитивных аллостерических модуляторов D1, которые обладают комбинацией благоприятных фармакокинетических и фармакодинамических характеристик, и при этом обеспечивают уменьшение побочных эффектов, обычно связанных с лечением, включающим использование селективных агонистов D1, таких как, например, гипотензия или дискинезия.
Настоящее изобретение относится к 2-(3,5-дихлор-1-метилиндазол-4-ил)-1-[(1S,3R)-3-(гидроксиметил)-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1H-изохинолин-2-ил]этанону формулы (I),
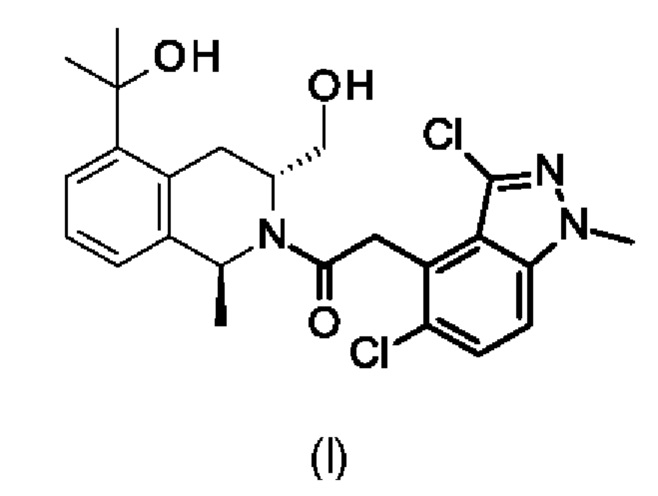
или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к соединению формулы (I), определенной выше, или его фармацевтически приемлемой соли, предназначенной для применения в терапии.
Другим объектом настоящего изобретения является соединение формулы (I), определенной выше, или его фармацевтически приемлемая соль, предназначенная для применения для лечения и заболеваний и/или нарушений, в которых играют роль рецепторы D1.
Другим объектом настоящего изобретения является соединение формулы (I), определенной выше, или его фармацевтически приемлемая соль, предназначенная для применения для лечения и/или предупреждения связанных с познавательной способностью и негативных симптомов шизофрении, нарушения познавательной способности, связанного с лечением нейролептическим средством, слабого нарушения познавательной способности (СНП), импульсивности, синдрома дефицита внимания с гиперактивностью (СДВГ), болезни Паркинсона и других нарушений движений, дистонии, слабоумия при болезни Паркинсона, болезни Гентингтона, слабоумия с тельцами Леви, болезни Альцгеймера, привыкания к чрезмерному употреблению лекарственного средства или наркотика, нарушений сна, апатии, травматического повреждения спинного мозга или невропатической боли.
В предпочтительном варианте осуществления этого объекта настоящее изобретение относится к соединению формулы (I), определенной выше, или его фармацевтически приемлемой соли, предназначенной для применения для лечения болезни Паркинсона и других нарушений движений, болезни Альцгеймера или связанных с познавательной способностью и негативных симптомов шизофрении.
Поэтому одним предпочтительным объектом настоящего изобретения является соединение формулы (I), определенной выше, или его фармацевтически приемлемая соль, предназначенная для применения для лечения болезни Паркинсона и других нарушений движений.
Другим объектом настоящего изобретения является применение соединения формулы (I), определенной выше, или его фармацевтически приемлемой соли для приготовления лекарственного средства, применимого для лечения и/или предупреждения заболеваний и/или нарушений, в которых играют роль рецепторы D1.
Еще одним объектом настоящего изобретения является применение соединения формулы (I), определенной выше, или его фармацевтически приемлемой соли для приготовления лекарственного средства, применимого для лечения и/или предупреждения связанных с познавательной способностью и негативных симптомов шизофрении, нарушения познавательной способности, связанного с лечением нейролептическим средством, слабого нарушения познавательной способности (СНП), импульсивности, синдрома дефицита внимания с гиперактивностью (СДВГ), болезни Паркинсона и других нарушений движений, дистонии, слабоумия при болезни Паркинсона, болезни Гентингтона, слабоумия с тельцами Леви, болезни Альцгеймера, привыкания к чрезмерному употреблению лекарственного средства или наркотика, нарушений сна, апатии, травматического повреждения спинного мозга или невропатической боли.
В предпочтительном варианте осуществления этого объекта настоящее изобретение относится к применению соединения формулы (I), определенной выше, или его фармацевтически приемлемой соли для приготовления лекарственного средства, применимого для лечения болезни Паркинсона и других нарушений движений, болезни Альцгеймера или связанных с познавательной способностью и негативных симптомов шизофрении.
Одним предпочтительным объектом настоящего изобретения является применение соединения формулы (I), определенной выше, или его фармацевтически приемлемой соли для приготовления лекарственного средства, применимого для лечения болезни Паркинсона и других нарушений движений.
Настоящее изобретение также относится к способу лечения и/или предупреждения нарушений, для которых показано введение позитивного аллостерического модулятора D1, который включает введение пациенту, нуждающемуся в таком лечении, соединения формулы (I), определенной выше, или его фармацевтически приемлемой соли в эффективном количестве.
Другим объектом настоящего изобретения является способ лечения и/или предупреждения связанных с познавательной способностью и негативных симптомов шизофрении, нарушения познавательной способности, связанного с лечением нейролептическим средством, слабого нарушения познавательной способности (СНП), импульсивности, синдрома дефицита внимания с гиперактивностью (СДВГ), болезни Паркинсона и других нарушений движений, дистонии, слабоумия при болезни Паркинсона, болезни Гентингтона, слабоумия с тельцами Леви, болезни Альцгеймера, привыкания к чрезмерному употреблению лекарственного средства или наркотика, нарушений сна, апатии, травматического повреждения спинного мозга или невропатической боли, который включает введение пациенту, нуждающемуся в таком лечении, соединения формулы (I), определенной выше, или его фармацевтически приемлемой соли в эффективном количестве.
В предпочтительном варианте осуществления этого объекта настоящее изобретение относится к способу лечения болезни Паркинсона и других нарушений движений, болезни Альцгеймера или связанных с познавательной способностью и негативных симптомов шизофрении, который включает введение пациенту, нуждающемуся в таком лечении, соединения формулы (I), определенной выше, или его фармацевтически приемлемой соли в эффективном количестве.
Одним предпочтительным объектом настоящего изобретения является способ лечения болезни Паркинсона и других нарушений движений, который включает введение пациенту, нуждающемуся в таком лечении, соединения формулы (I), определенной выше, или его фармацевтически приемлемой соли в эффективном количестве.
В объем настоящего изобретения входят соли соединений формулы (I), приведенной выше. Для применения в медицине соли соединений формулы (I) должны являться фармацевтически приемлемыми солями. Однако для получения соединений, применимых в настоящем изобретении, или их фармацевтически приемлемых солей можно использовать другие соли. Стандартные принципы, лежащие в основе выбора и получения фармацевтически приемлемых солей описаны, например, в публикации Handbook of Pharmaceutical Salts: Properties, Selection and Use, ed. Р.Н. Stahl & C.G. Wermuth, Wiley-VCH, 2002. Подходящие фармацевтически приемлемые соли соединения формулы (I) включают соли присоединения с кислотами, которые, например, можно приготовить путем смешивания раствора соединения формулы (I) с раствором фармацевтически приемлемой кислоты.
Следует понимать, что каждый отдельный атом, содержащийся в формуле (I), или в формулах, представленных ниже в настоящем изобретении, в действительности может содержаться в форме любого из его изотопов, встречающихся в природе, причем наиболее часто встречающийся изотоп (изотопы) является предпочтительным. Так, например, каждый отдельный атом водорода, содержащийся в формуле (I), или в формулах, представленных ниже в настоящем изобретении, может содержаться в виде атома 1Н, 2Н (дейтерий) или 3Н (тритий), предпочтительно в виде 1Н или 2Н. Аналогичным образом, например, каждый отдельный атом углерода, содержащийся в формуле (I), или в формулах, представленных ниже в настоящем изобретении, может содержаться в виде атома 12С, 13С или 14С, предпочтительно в виде 12С.
В объем настоящего изобретения входят сольваты соединений формулы (I), приведенной выше. Такие сольваты можно получить с обычными органическими растворителями или с водой.
В объем настоящего изобретения также входят совместные кристаллы соединений формулы (I), приведенной выше. Технический термин "совместный кристалл" используют для описания случая, когда нейтральные молекулярные компоненты содержатся в кристаллическом соединении при определенном стехиометрическом соотношении. Получение фармацевтических совместных кристаллов позволяет модифицировать кристаллическую форму активного фармацевтического ингредиента, что, в свою очередь, может изменить его физико-химические характеристики без ухудшения его необходимой биологической активности (см. публикацию Pharmaceutical Salts and Co-crystals, ed. J. Wouters & L. Quere, RSC Publishing, 2012).
Соединения, предлагаемые в настоящем изобретении, могут находиться в различных полиморфных формах. Хотя это явно не указано в приведенной выше формуле, такие формы входят в объем настоящего изобретения.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) выделяют в форме моногидрата, как это дополнительно описано в примерах.
В объем настоящего изобретения также входят пролекарственные формы соединений формулы (I) и различные их подклассы и подгруппы.
Активность при любом из указанных выше терапевтических показаний или нарушений, разумеется, можно оценить путем проведения соответствующих клинических исследований по методикам, известным специалисту в соответствующей области техники применительно к конкретному показанию и/или при проведении общих клинических исследований.
Для лечения заболеваний соединение формулы (I) или его фармацевтически приемлемые соли можно использовать в эффективной суточной дозе и вводить в форме фармацевтической композиции.
Поэтому настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы (I), представленной выше, или его фармацевтически приемлемую соль совместно с одним или большим количеством фармацевтически приемлемых носителей.
Для приготовления фармацевтической композиции, предлагаемой в настоящем изобретении, одно или большее количество соединений формулы (I) или их фармацевтически приемлемых солей тщательно смешивают с фармацевтическим разбавителем или носителем по обычным фармацевтическим методикам приготовления смесей, известным опытным специалистам-практикам.
Подходящие разбавители и носители могут находиться в самых различных формах в зависимости от необходимого пути введения, например, перорального, ректального, парентерального или назального.
Фармацевтические композиции, содержащие соединения, предлагаемые в настоящем изобретении, можно, вводить, например, перорально, парентерально, т.е. внутривенно, внутримышечно или подкожно, внутриоболочечно, путем ингаляции или назально.
Фармацевтические композиции, подходящие для перорального введения, могут быть твердыми или жидкими и могут, например, находиться в форме таблеток, пилюль, драже, желатиновых капсул, растворов, сиропов, жевательных резинок и т.п.
Для этого активный ингредиент можно смешать с инертным разбавителем или нетоксичным фармацевтически приемлемым носителем, таким как крахмал или лактоза. Эти фармацевтические композиции также необязательно могут содержать связующее, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин, разрыхлитель, такой как альгиновая кислота, смазывающее вещество, такое как стеарат магния, агент, придающий скользкость, такое как коллоидный диоксид кремния, подсластитель, такой как сахароза или сахарин, или окрашивающие агенты, или вкусовую добавку, такую как экстракт мяты перечной или метилсалицилат.
В объем настоящего изобретения также входят композиции, которые могут высвобождать активное вещество регулируемым образом. Фармацевтические композиции, которые можно использовать для парентерального введения, находятся в обычной форме, такой как водные или масляные растворы или суспензии, обычно содержащиеся в ампулах, одноразовых шприцах, стеклянных или пластмассовых флаконах или контейнерах для вливания.
В дополнение к активному ингредиенту эти растворы или суспензии также необязательно могут содержать стерильный разбавитель, такой как вода для инъекций, физиологический раствор, масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители, бактерицидные средства, такие как бензиловый спирт, антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия, хелатные реагенты, такие как этилендиаминтетрауксусная кислота, буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования осмоляльности, такие как хлорид натрия или декстроза.
Эти фармацевтические формы готовят по методикам, которые стандартным образом используются фармацевтами.
Количество соединения, предназначенного для применения в настоящем изобретении, необходимое для профилактики или лечения конкретного патологического состояния, будет меняться в зависимости от выбранного соединения и состояния подвергающегося лечению пациента. Однако обычно суточная доза может находиться в диапазоне от 0,05 до 3000 мг, обычно от 0,5 до 1000 мг для композиций, предназначенных для парентерального введения.
Соединение, предлагаемое в настоящем изобретении, или его фармацевтически приемлемую соль можно вводить по отдельности (монотерапия) или в комбинации с L-допа (комбинированная терапия). При введении по отдельности или в комбинации с долями доз L-допа, необходимыми для уменьшения недостатка двигательной способности у пациентов, соединения формулы (I), предлагаемые в настоящем изобретении, или их фармацевтически приемлемые соли, могут быть применимы для лечения дискинезии, связанной с введением L-допа. Так, например, полагают, что, если соединение формулы (I), предлагаемое в настоящем изобретении, применяют с долями доз L-допа, вводимыми пациенту, или его применяют отдельно для замены L-допа, то соединение формулы (I), предлагаемое в настоящем изобретении, будет являться эффективным для устранения недостатка двигательной способности и не будет вызывать опасную дискинезию. Поэтому полагают, что соединение, предлагаемое в настоящем изобретении, может быть применимым для лечения недостатков двигательной способности и вызванной леводопой дискинезии (ВЛД).
Поэтому одним предпочтительным объектом настоящего изобретения также является соединение формулы (I), которое применимо для лечения вызванной леводопой дискинезии (ВЛД).
Соединение формулы (I) можно получить по методике, включающей реакцию промежуточного продукта формулы (II) с промежуточным продуктом формулы (III).
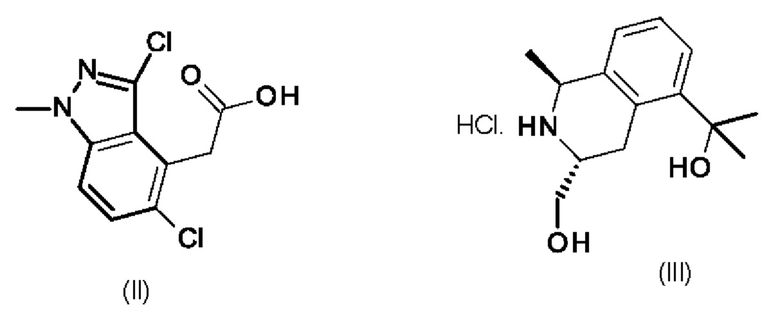
Промежуточный продукт (III) обычно можно ввести в реакцию с промежуточным продуктом формулы (II) в присутствии (2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфата (HBTU) или другого реагента реакции сочетания, известного специалисту в данной области техники, в подходящем растворителе, например, в диметилформамиде, с использованием избыточного количества основания, например, N,N-диизопропилэтиламина.
Промежуточный продукт формулы (III) можно получить по методике, включающей реакцию промежуточных продуктов формулы (IV),
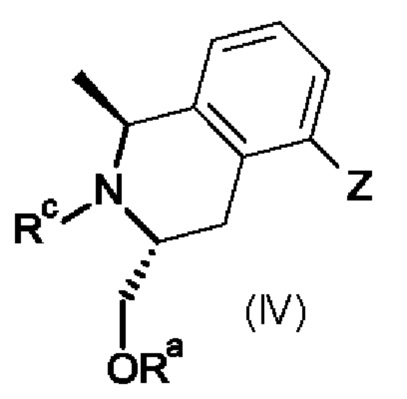
в которой
Z обозначает галоген или 1-гидрокси-1-метилэтил;
Ra обозначает трет-бутилдиметилсилил; и
Rc обозначает водород или трет-бутоксикарбонил.
На первой стадии в промежуточный продукт формулы (IV), в которой Z обозначает бром и Rc обозначает водород, ниже в настоящем изобретении называющийся промежуточным продуктом (IVa), можно ввести соответствующую защитную группу по методикам, известным специалистам в данной области техники, и получить соединение формулы (IV), в которой Z обозначает бром и Rc обозначает трет-бутоксикарбонил, ниже в настоящем изобретении называющееся промежуточным продуктом (IVb).
На второй стадии можно провести реакцию обмена металл-галоген, например, в присутствии n-BuLi, в подходящем растворителе, например, в тетрагидрофуране, при низкой температуре, в присутствии сухого ацетона в непрерывном потоке, по методике, описанной в прилагаемых примерах, и получить соответствующий промежуточный продукт (IV), описанный выше, в котором Z обозначает 1-гидрокси-1-метилэтил, ниже в настоящем изобретении называющийся промежуточным продуктом (IVc).
Затем сначала можно удалить защитную трет-бутоксикарбонильную (Boc) группу (Rc) по методикам, известным специалисту в данной области техники, или дополнительно описанным в прилагаемых примерах, и затем удалить защитную триметилсилильную группу, образовавшуюся во время удаления защитной группы Boc, и трет-бутилдиметилсилильную группу (Ra) и получить промежуточный продукт (III).
Промежуточный продукт формулы (IVa) можно получить по методике, включающей реакцию промежуточного продукта формулы (V), в которой Y обозначает галоген, например, бром, и Ra является таким, как определено выше для промежуточного продукта формулы (IV).
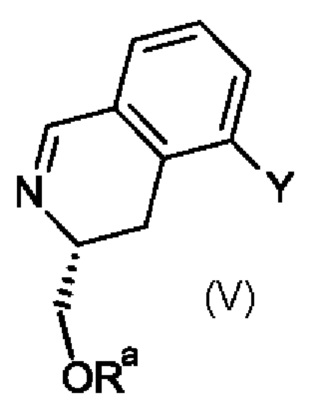
Реакцию обычно проводят в присутствии метилхлорида магния в подходящем растворителе например, в тетрагидрофуране, при низкой температуре.
Промежуточный продукт (V) можно получить по 2-стадийной методике, включающей реакцию промежуточного продукта формулы (VI),
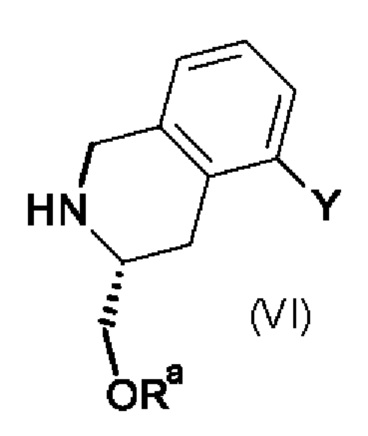
в которой Y является таким, как определено выше для промежуточного продукта формулы (V), и Ra обозначает водород или трет-бутилдиметилсилил.
На первой стадии промежуточный продукт (VI), в котором Ra обозначает водород, вводят в реакцию с трет-бутилдиметилсилилхлоридом в присутствии подходящего основания например, 4-диметиламинопиридина, при комнатной температуре и получают промежуточный продукт (VI), в котором Ra обозначает трет-бутилдиметилсилил.
На второй стадии промежуточный продукт (VI), в котором Ra обозначает трет-бутилдиметилсилил, вводят в реакцию с N-хлорсукцинимидом (NCS) в подходящем растворителе, например, в ТГФ (тетрагидрофуран), и получают промежуточный продукт (V).
Промежуточный продукт (VI), в котором Ra обозначает водород, можно получить по методике, включающей использование промежуточного продукта формулы (VII), в которой Y является таким, как определено выше для промежуточного продукта (V).
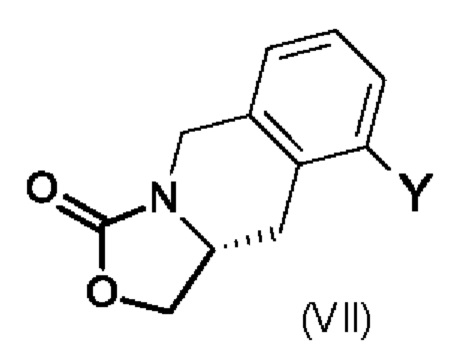
Реакцию обычно проводят в присутствии сильного основания, например, гидроксида натрия, в подходящем растворителе, например, в смеси этанола и воды, при высокой температуре.
Промежуточный продукт формулы (VII) можно получить по методике, включающей реакцию промежуточного продукта (VIII),
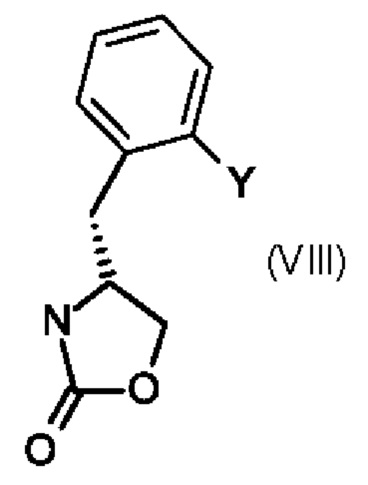
в котором Y является таким, как определено выше в настоящем изобретении для промежуточного продукта формулы (V).
Реакцию обычно проводят в присутствии триметилсилилтрифлата и параформальдегида в подходящем растворителе например, в дихлорметане.
Промежуточный продукт (VIII) можно получить по 2-стадийной методике, включающей использование имеющегося в продаже промежуточного продукта (IX),
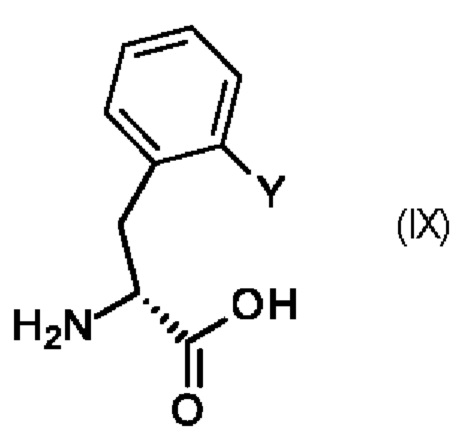
в котором Y является таким, как определено выше для промежуточного продукта (V).
Реакцию обычно проводят по методикам, описанным в прилагаемых примерах, или по методикам, известным специалисту в данной области техники.
Промежуточный продукт формулы (II) можно получить по многостадийной методике, включающей реакцию промежуточных продуктов формулы (X),
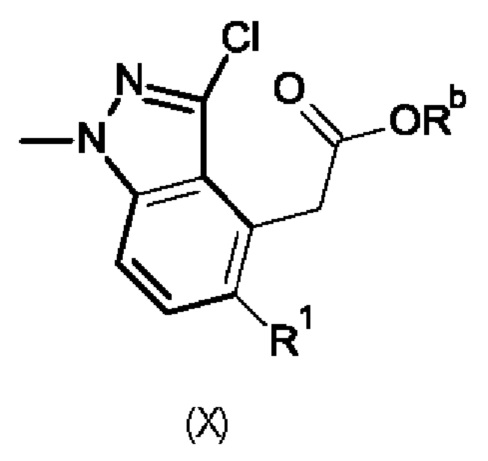
в которой
R1 обозначает хлор, аминогруппу или нитрогруппу; и
Rb обозначает водород или трет-бутил.
На первой стадии промежуточный продукт формулы (X), в которой R1 обозначает нитрогруппу и Rb обозначает трет-бутил, ниже в настоящем изобретении называющийся промежуточным продуктом (Ха), восстанавливают с получением соответствующего промежуточного продукта (X), в котором R1 обозначает аминогруппу и Rb обозначает трет-бутил, ниже в настоящем изобретении называющегося промежуточным продуктом (Xb). Реакцию обычно проводят путем гидрирования, катализируемого с помощью Pd/C, при высоком давлении в подходящем растворителе, например, в метаноле.
Промежуточный продукт (Xb) превращают в соответствующий промежуточный продукт (X), в котором R1 обозначает хлор и Rb обозначает водород, ниже в настоящем изобретении называющийся промежуточным продуктом (Хс), путем добавления концентрированной хлористоводородной кислоты и нитрита натрия и последующего добавления хлористоводородной кислоты и хлорида меди(II). Реакцию обычно проводят при низкой температуре.
Затем промежуточный продукт (II) можно получить непосредственно из промежуточного продукта (Хс) по реакции с N-хлорсукцинимидом, проводимой по методикам, описанным в прилагаемых примерах, или по методикам, известным специалисту в данной области техники.
Промежуточный продукт формулы (Ха) можно получить по методике, включающей использование промежуточного продукта формулы (XI),
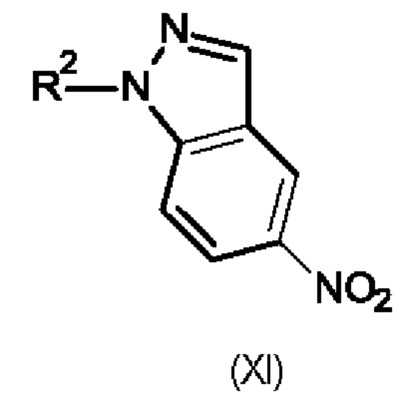
в которой R2 обозначает водород или метил.
На первой стадии имеющийся в продаже промежуточный продукт формулы (XI), в которой R2 обозначает водород, ниже в настоящем изобретении называющийся промежуточным продуктом (XIa), вводят в реакцию с метилйодидом в присутствии сильного основания, например, гидроксида калия, в подходящем растворителе, например, в диметилформамиде. Затем полученный промежуточный продукт (XI), в котором R2 обозначает метил, ниже в настоящем изобретении называющийся промежуточным продуктом (XIb), вводят в реакцию с трет-бутил-2-хлорацетатом в присутствии трет-бутоксида калия в подходящем растворителе, например, в тетрагидрофуране, при низкой температуре и получают промежуточный продукт (Ха).
Если при использовании любой из описанных выше методик получения соединений, предлагаемых в настоящем изобретении, образуется смесь продуктов, то искомый продукт можно из нее выделить на подходящей стадии с помощью обычных методик, таких как препаративная ВЭЖХ (высокоэффективная жидкостная хроматография) или колоночная хроматография с использованием, например, диоксида кремния и/или оксида алюминия вместе с подходящей системой растворителей.
Если при использовании описанных выше методик получения соединений, предлагаемых в настоящем изобретении, образуется смесь стереоизомеров, то эти изомеры можно разделить по обычным методикам. В частности, когда необходимо получить конкретный энантиомер соединения формулы (I), то его можно получить из соответствующей смеси энантиомеров по любой обычной методике разделения энантиомеров. Так, например, диастереоизомерные производные, например, соли можно получить по реакции смеси энантиомеров формулы (I), например, рацемата с соответствующим хиральным соединением, например, хиральным основанием. Затем диастереоизомеры можно разделить по любым обычным методикам, например, путем кристаллизации и выделить необходимый энантиомер, например, путем обработки кислотой, в случае, если диастереоизомер является солью. В другой методике разделения рацемат формулы (I) можно разделить с помощью хиральной ВЭЖХ. Кроме того, при необходимости конкретный энантиомер можно получить путем использования подходящего хирального промежуточного продукта в одной из методик, описанных выше. Альтернативно, конкретный энантиомер можно получить путем проведения энантиомерно специфического ферментативного биологического превращения, например, гидролиза сложного эфира с использованием эстеразы с последующей очисткой только энантиомерно чистой образовавшейся вследствие гидролиза кислоты от непрореагировавшего антипода - сложного эфира. Если необходимо получить конкретный геометрический изомер, предлагаемый в настоящем изобретении, то для промежуточных продуктов или конечных продуктов также можно использовать хроматографию, перекристаллизацию и другие обычные методики разделения. Альтернативно, нежелательный энантиомер можно рацемизировать в присутствии кислоты или основания по методикам, известным специалисту в данной области техники, или по методикам, описанным в прилагаемых примерах, и получить желательный энантиомер.
В ходе проведения любой из указанных выше последовательностей синтеза может оказаться необходимой и/или желательной защита чувствительных или реакционноспособных групп, содержащихся в любой из участвующих в реакциях молекул. Это можно выполнить с помощью обычных защитных групп, таких как описанные в публикациях Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973; и T.W. Greene & P.G.M. Wuts, Protective Croups in Organic Synthesis, John Wiley & Sons, 3rd edition, 1999. Защитные группы можно удалить на любой подходящей последующей стадии по методикам, известным в данной области техники
Соединения формулы (I), предлагаемые в настоящем изобретении, не активируют допаминовый рецептор D1 непосредственно, а усиливают воздействие агонистов D1 или эндогенного лиганда на допаминовые рецепторы D1 по аллостерическому механизму и поэтому они являются позитивными аллостерическими модуляторами D1 (ПАМ D1).
Допамин и другие агонисты D1 сами непосредственно активируют допаминовый рецептор D1.
Исследования предназначены для изучения воздействия соединений, предлагаемых в настоящем изобретении, при отсутствии допамина ("исследование активации") и в присутствии допамина ("исследование увеличения активности").
В исследовании активации определяют стимулирование продуцирования циклического аденозинмонофосфата (цАМФ) при исследовании с помощью однородной флуоресценции с разрешением по времени (ОФРВ), причем максимальное увеличение количества цАМФ обеспечивают путем увеличения концентраций эндогенного агониста, допамина, и его определяют как активацию, составляющую 100%.
По данным исследования соединения формулы (I), предлагаемые в настоящем изобретении, не оказывают существенного прямого подобного агонистическому воздействия, поскольку они обеспечивают активацию, составляющую менее 20% (от максимального ответа на допамин) при концентрации, равной 10 мкМ.
В исследовании увеличения активности определяют способность соединений увеличивать количество цАМФ, продуцируемого при низкой пороговой концентрации допамина. Такую концентрацию допамина ([ЕС20]) используют, чтобы обеспечить стимулирование, составляющее 20% от максимального ответа (100%), полученного при увеличивающейся концентрации допамина. Для определения этого увеличения соединения при увеличивающихся концентрациях инкубируют с допамином [ЕС20] и увеличение определяют, как увеличение продуцирования цАМФ, и определяют концентрацию соединения, которая обеспечивает увеличение концентрации цАМФ на 50%.
По данным исследования цАМФ с помощью ОФРВ соединение формулы (I), предлагаемое в настоящем изобретении обладает значением рЕС50, равным более примерно 6,5, это показывает, что оно является позитивным аллостерическим модулятором D1.
Известно, что ингибирование рецептора GABAA непосредственно связано с припадками и эпилепсией. Поэтому необходимо разработать соединения, которые являются позитивными аллостерическими модуляторами D1, и которые в то же самое время обеспечивают сведение к минимуму таких эффектов.
Поэтому предпочтительно, если по данным исследования ингибирования рецептора GABA-A, описанного в настоящем изобретении, соединение формулы (I) обеспечивает выраженное в процентах ингибирование рецептора GABAA, меньшее или равное примерно 20%, в идеальном случае меньшее примерно 10%, предпочтительно меньшее примерно 5%, при исследовании соединения формулы (I) при концентрации, равной 10 мкМ.
Затруднением, которое может возникнуть при разработке соединений, предназначенных для применения для лечения, является способность определенных соединений индуцировать ферменты CYP450. Индуцирование таких ферментов может оказывать влияние на воздействие таких соединений или других соединений, которые можно вводить пациенту совместно с ними, при этом, вероятно, изменяются их соответствующие безопасность и эффективность. Поэтому необходимо разработать соединения, которые обеспечивают сведение к минимуму вероятности индуцирования.
Способность соединения формулы (I), предлагаемого в настоящем изобретении, индуцировать CYP450 исследуют путем определения возможного увеличения активностей CYP450 в гепатоцитах человека, обработанных соединениями, предлагаемыми в настоящем изобретении, при увеличивающихся концентрациях.
Предпочтительно, если по данным исследования индуцирования CYP3A4, проводимого в соответствии с протоколами, описанными в настоящей заявке на патент, соединение формулы (I), предлагаемое в настоящем изобретении, обладает значением кратности индуцирования, по меньшей мере примерно в 2 раза меньшим, в идеальном случае по меньшей мере примерно в 3 раза меньшим, предпочтительно по меньшей мере примерно в 4 раза меньшим, чем значение, полученное для использующегося в качестве положительного контроля соединения (рифампицина).
При разработке соединения, предназначенного для применения для лечения, важно иметь сведения о его выведении после его введения в организм.
Клиренс является параметром, который обеспечивает такие сведения, поскольку он означает объем плазмы (или крови), полностью очищенный от исследуемого соединения за единицу времени. Обычно его выражают в мл/мин/кг или л/ч. Затем его можно сопоставить с любым физиологическим кровотоком (например, печеночным кровотоком) для определения того, является ли клиренс низким, средним или высоким.
Если клиренс является низким, то в зависимости от объема распределения можно ожидать необходимость низкой дозы и обеспечение сравнительно продолжительного воздействия. Если клиренс является высоким, то также в зависимости от объема распределения можно ожидать необходимость высокой дозы и обеспечение сравнительно непродолжительного воздействия.
Клиренс обычно оценивают в соответствии с протоколами, описанными в настоящем изобретении, с использованием инкубирования с гепатоцитами и расчетов путем масштабирования данных, полагая, что основным путем выведения является метаболизм. Собственный клиренс, рассчитанный с использованием гепатоцитов, выражают в мкл/мин/106 клеток.
Предпочтительно, если по данным исследования клиренса, описанного в настоящем изобретении, соединение формулы (I), предлагаемое в настоящем изобретении, обладает клиренсом, равным менее примерно 10 мкл/мин/106 клеток.
Исследование цАМФ с помощью ОФРВ
Конкретные условия, при которых исследовали соединения, приведены ниже в настоящем изобретении.
а. Методики с использованием культуры клеток, экспрессирующих D1
Клетки выращивали при 37°С в увлажненной атмосфере, содержащей 5% СО2. Клетки выращивали в среде DMEM-F12+GlutaMAX™-I (GIBCO®, Invitrogen, Merelbeke, Belgium), содержащей 10% фетальной бычьей сыворотки (Bio Whittaker®, Lonza, Verviers, Belgium), 400 мкг/мл генетицина (GIBCO®), 100 МЕ/мл пенициллина (ME = международные единицы) и 100 МЕ/мл стрептомицина (раствор Pen-Strep, Bio Whittaker®). Использовали фибробласты мышей LMtk (Ltk-), экспрессирующие допаминовый рецептор D1 (BioSignal Inc, Montreal, Canada, в настоящее время Perkin Elmer) и установлено, что они эффективно связываются и дают устойчивые функциональные реакции (Watts et al, 1995).
b. Исследование цАМФ
Исследование изменения концентрации внутриклеточного циклического аденозинмонофосфата (цАМФ) определяли с использованием набора для динамического исследования цАМФ с помощью ОФРВ, выпускающегося фирмой CisBio (Codolet, France). В исследовании использовали методику однородной флуоресценции с разрешением по времени и оно основано на определении конкурентности между нативным цАМФ, продуцируемым клетками, и цАМФ, меченым с помощью красителя d2. Связывание метки определяли с использованием антител к цАМФ, меченых криптатом. Воздействие только соединения (агонизм) определяли путем проведения исследования при отсутствии допамина, тогда как воздействие соединения, как позитивного аллостерического модулятора (ПАМ), определяли в присутствии допамина, при концентрации, равной ЕС20. Клетки (20000 клеток/лунка) инкубировали в 384-луночных планшетах при комнатной температуре в течение 1 ч при конечном объеме в ССРХ (сбалансированный солевой раствор Хенкса) (Lonza, содержащий кальций, магний и 20 мМ буфера HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), рН 7,4), равном 20 мкл, содержащих: изобутилметилксантин (Sigma, конечная концентрация: 0,1 мМ), исследуемое соединение при разных концентрациях (обычно от 10-9,5 до 10-4,5 М) в присутствии и при отсутствии допамина (конечная концентрация: 1,1 нМ). Затем реакцию останавливали и клетки лизировали путем добавления реагента для детектирования d2 в буфере для лизиса (10 мкл) и содержащего криптат реагента в буфере для лизиса (10 мкл) в соответствии с инструкциями изготовителя. Затем смеси инкубировали при комнатной температуре в течение еще 60 мин и определяли изменение сигналов испускания флуоресценции в ОФРВ в соответствии с инструкциями изготовителя с использованием устройства для считывания планшетов Envision (Perkin Elmer, Zaventem, Belgium) при возбуждении лазером. Все инкубирования проводили дважды и результаты сравнивали с полученной для допамина зависимостью концентрация-эффект (от 10-11 до 10-6 М).
с. Анализ результатов
Результаты анализировали с помощью Excel и PRISM (GraphPad Software) и получали значения рЕС50 и Еотн с использованием 4-параметрического логистического уравнения (DeLean et al, 1978), где Еотн обозначает аппроксимированный максимальный ответ на исследуемое соединение за вычетом фона, выраженный в процентах от значения, полученного при использованием допамина, который определен, как 100%.
Значение рЕС50 соединения равно -log10 концентрации соединения, которая обеспечивает увеличение концентрации цАМФ на 50%.
Еотн обозначает относительную эффективность, определенную, как выраженное в % максимальное значение увеличения активности, обеспечиваемое соединением, по сравнению с максимальным ответом, полученным при увеличивающихся концентрациях допамина (Еотн, равное 1, = максимальный ответ на допамин).
По данным приведенного выше исследования соединение формулы (I) обладает значением рЕС50, равным примерно 8,1, и значением Еотн, равным примерно 68%.
Автоматизированные исследования фиксации потенциала с использованием клеток, содержащих рецептор GABAA
Использовали клетки СНО-K1, стабильно экспрессирующие субъединицы α1, α2 и α2 рецептора GABAA человека. Клетки выращивали с использованием трипсина и выдерживали в бессывороточной среде при комнатной температуре. До проведения исследования клетки промывали и повторно суспендировали во внеклеточном растворе.
Исследования фиксации потенциала
Эксперименты с использованием каналов GABAA (α1β2γ2) человека проводили путем автоматизированного исследования фиксации потенциала (IonFlux™ НТ). Соединения исследовали при 3 концентрациях (0,1, 1 и 10 мкМ) с использованием от 3 до 4 клеток. Внешний раствор, предназначенный для регистрации токов GABAA, состоял из 137 мМ хлорида натрия, 4 мМ хлорида калия, 1,8 мМ хлорида кальция, 1 мМ хлорида магния, 10 мМ HEPES и 10 мМ глюкозы. Внешний и внутренний растворы титровали с помощью NaOH или KOH и обеспечивали значение рН, равное 7,35 или 7,3 соответственно. Внутренний подаваемый пипеткой раствор содержал 70 мМ фторида калия, 60 мМ хлорида калия, 70 мМ хлорида натрия, 5 мМ HEPES, 5 мМ ЭГТК (этиленгликольтетрауксусная кислота) и 4 мМ комплекса магний-АТФ (аденозинтрифосфат). Конечная концентрация растворителя, ДМСО, использующегося для разведения соединений составляла 0,33% в каждой лунке. В качестве ингибитора-положительного контроля использовали бикукуллин (от 0,032 до 100 мкМ). В качестве агониста использовали GABA (15 мкМ). Все измерения проводили при исходном потенциале, равном -60 мВ.
Последовательность добавления соединения являлась следующей: для определения базового ответа добавляли GABA при концентрации, соответствующей EC80. Обрабатывали соединением при каждой концентрации в течение 30 с, затем обрабатывали с помощью 15 мкМ GABA в присутствии соединения в течение 2 с. Процедуру повторяли с использованием следующей более высокой концентрации соединения. Определяли максимальные значения входящего тока, как ответ на добавления GABA в присутствии соединения при одной концентрации. Все полученные для соединения результаты нормировали на базовое максимальное значение тока, полученное при обработке с помощью 15 мкМ GABA в течение 2 с.
По данным описанного выше исследования соединение, описывающееся формулой (I), при концентрации, равной 10 мкМ, обеспечивает выраженное в процентах ингибирование рецептора GABAA, составляющее менее примерно 0,1%, определенное при концентрации соединения формулы (I), равной 10 мкМ.
Исследование индуцирования
Приведенные ниже исследования предназначены для определения способности соединения, предлагаемого в настоящем изобретении, индуцировать фермент CYP3A4.
А. Исследование способности индуцирования CYP3A4 in vitro с использованием замороженных гепатоцитов человека
Задачей исследования с использованием гепатоцитов человека являлось определение индуцирующей способности соединения формулы (I), проводимое путем определения активностей CYP3A4 после проводимой в течение 3 дней обработки гепатоцитов только культуральной средой или культуральной средой, содержащей НХВ (новое химическое вещество).
Для этой цели замороженные гепатоциты человека, взятые у одного донора, высевали в лунки 48-луночного планшета с покрытием из коллагена таким образом, что конечная плотность при высевании составляла 0,2×106 клеток/лунка (конечный объем в каждой лунке составлял 0,25 мл). Затем клетки инкубировали в среде для высевания при 37°С, влажности, равной 95%, 5% CO2, для обеспечения прилипания клеток. Через 4 ч среду для высевания заменяли на предварительно нагретую бессывороточную среду Вильямса Е объемом 0,25 мл, содержащую 100 МЕ/мл пенициллина, 100 мкг/мл стрептомицина, 10 мкг/мл инсулина, 2 мМ L-глутамина и 0,1 мкМ гидрокортизона.
На следующий день к клеткам добавляли исследуемое соединение в среде для анализа при концентрации, равной 1 мкМ (конечная концентрация ДМСО равна 0,1%). Использующийся в качестве положительного контроля индуктор, рифампицин (10 мкМ), инкубировали одновременно с исследуемым соединением. В исследование также включали лунки с отрицательным контролем, в которых исследуемое соединение заменяли на являющийся разбавителем растворитель (0,1% ДМСО в среде для анализа). Каждое исследуемое соединение исследовали трижды. Клетки обрабатывали культуральной средой в течение 72 ч, причем каждые 24 ч проводили замену среды.
Для определения каталитической активности готовили раствор маркерного субстрата для CYP3A4, мидазолама (конечная концентрация равна 2,5 мкМ), в предварительно нагретой среде для анализа. После завершения обработки, проводимой в течение 72 ч, среду заменяли на маркерный субстрат для CYP3A4. Гепатоциты инкубировали в течение 30 мин. После завершения инкубирования аликвоту отбирали и помещали в такой же объем метанола, содержащего внутренний стандарт. Образцы центрифугировали при скорости, равной 2500 об/мин, при 4°С в течение 20 мин. Аликвоту надосадочной жидкости разводили деионизированной водой и определяли концентрации 1-гидроксиимидазолама с помощью типичных методик ЖХ-МС/МС.
Гепатоциты солюбилизировали в 0,1М растворе гидроксида натрия при комнатной температуре и содержание белка в каждой лунке определяли с помощью набора для анализа белков Pierce™ ВСА (Thermo Scientific) с использованием бычьего сывороточного альбумина в качестве стандарта.
Анализ результатов
Кратность индуцирования (увеличение активности CYP) получали путем определения отношения ферментативной активности, наблюдающейся в гепатоцитах, обработанных соединением формулы (I), к наблюдающейся в контрольных гепатоцитах, обработанных разбавителем. Кратность индуцирования для положительного контроля (рифампицина) рассчитывали таким же образом. Затем индуцирующую способность соединения формулы (I) сопоставляли с индуцирующей способностью рифампицина путем расчета отношения кратности индуцирования, полученной для рифампицина, к кратности индуцирования, полученной для соединения формулы (I).
По данным исследования индуцирования CYP3A4, проводимого в соответствии с описанными выше в настоящем изобретении протоколами, соединение формулы (I), предлагаемое в настоящем изобретении, при концентрации, равной 1 мкМ, обладает значением кратности индуцирования, по меньшей мере примерно в 7 раз меньшим, чем значение, полученное для использующегося в качестве положительного контроля соединения (рифампицина) при концентрации, равной 10 мкМ.
С использованием такого же протокола, как описанный выше, к клеткам добавляли исследуемое соединение в среде для анализа при концентрациях, находящихся в диапазоне от 0,03 до 10 мкМ (конечная концентрация ДМСО равна 0,1%). Использующийся в качестве положительного контроля индуктор, рифампицин, при концентрациях, находящихся в диапазоне примерно от 0,1 до 30 мкМ, инкубировали одновременно с исследуемым соединением.
Результаты сопоставления наибольших значений кратности индуцирования, полученных в соответствии с описанным выше протоколом, т.е. полученных при максимально возможной для обеспечения растворения концентрации (3 мкМ) соединения формулы (I), предлагаемого в настоящем изобретении, и при концентрации положительного контроля (рифампицина), равной 10 мкМ, показывают, что соединение формулы (I) обладает индуцирующей способностью, примерно в 4 раза меньшей, чем индуцирующая способность использующегося в качестве положительного контроля соединения (рифампицина).
Исследование с использованием азамулина
Замороженные гепатоциты человека (смесь образцов, взятых у 20 доноров, партия BSU фирм Celsis/IVT/Bioreclamation) оттаивали в соответствии с рекомендациями поставщика. Жизнеспособность (отбор с помощью красителя трипановый синий) превышала 75%. Предварительное инкубирование (250 мкл суспензии гепатоцитов при концентрации, равной 2×106 гепатоцитов/мл) проводили в среде Вильямса, содержащей 2 мМ глутамина и 15 мМ HEPES, в 48-луночных планшетах при +37°С, в инкубаторе (5% СО2), при осторожном встряхивании (вибрирующее встряхивающее устройство, Titramax 100, примерно 300 об/мин) в течение 30 мин. После предварительного инкубирования начинали инкубирование путем добавления к гепатоцитам 250 мкм культуральной среды (состав описан выше), содержащей соединение фирмы UCB (1 мкМ) или мидазолам (положительный контроль) с добавлением или без добавления азамулина (6 мкМ, специфичный ингибитор CYP3A4/5). Конечные концентрации соединения фирмы UCB и азамулина в инкубируемых смесях составляли 0,5 мкМ и 3 мкМ соответственно. Суспензии клеток быстро повторно гомогенизировали путем проводимого 2 раза втягивания пипеткой и выпускания из нее. После инкубирования в течение 0, 30, 60, 120, 180 и 240 мин реакции останавливали путем переноса 50 мкл инкубированных смесей в соответствующие лунки 96-луночного планшета, содержащие 50 мкл охлажденного льдом ацетонитрила с добавлением 1 мкМ кетоконазола, использующегося в качестве внутреннего стандарта. Перед каждым отбором образцов инкубированные клетки повторно гомогенизировали путем проводимого 2 раза втягивания пипеткой и выпускания из нее.
При инкубировании с суспензией гепатоцитов человека собственный клиренс (Clint) соединений формулы (I), предлагаемых в настоящем изобретении, при разных концентрациях составляет 2,1±0,6 мкл/мин/106 клеток.
В приведенных ниже примерах проиллюстрировано получение соединений формулы (I), предлагаемых в настоящем изобретении.
ПРИМЕРЫ
Аббревиатуры/многократно использовавшиеся реагенты
АЦН: ацетонитрил
Рассол: насыщенный водный раствор хлорида натрия
nBu: н-бутил
tBu: трет-бутил
ДХМ: дихлорметан
ДМАП: 4-диметиламинопиридин
ДМФ: N,N-диметилформамид
ДМСО: диметилсульфоксид
ЕС20/50: концентрация, при которой обеспечивается 20%/50% от максимального значения ответа
Еотн: относительная эффективность
ЭР+: ионизация электрораспылением в режиме положительных ионов
Et: этил
EtOH: этанол
Et2O: диэтиловый эфир
EtOAc: этилацетат
ч: час(ы)
ВЭЖХ: высокоэффективная жидкостная хроматография
ОФРВ: однородная флуоресценция с разрешением по времени
ЖХМС: жидкостная хроматография - масс-спектрометрия
МеОН: метанол
мин: минута (минуты)
NCS: N-хлорсукцинимид
ЯМР: ядерный магнитный резонанс
iPrOH: изопропанол
КТ: комнатная температура
НЖХ: надкритическая жидкостная хроматография
ТЭА: триэтиламин
ТГФ: тетрагидрофуран
ТСХ: тонкослойная хроматография
цАМФ: циклический аденозинмонофосфат.
Названия, соответствующие нормам IUPAC (Международный союз теоретической и прикладной химии), получают с помощью программного обеспечения Biovia Draw 16.1.
Методики анализа
Все реакции, в которых используют реагенты, чувствительные к воздействию воздуха или влаги, проводят в атмосфере азота или аргона с использованием высушенных растворителей и стеклянной посуды. Имеющиеся в продаже растворители и реагенты обычно используют без дополнительной очистки, включая безводные растворители, когда это является целесообразным (обычно продукты Sure-Seal™, выпускающиеся фирмой Aldrich Chemical Company, или AcroSeal™, выпускающиеся фирмой ACROS Organics). За протеканием реакций обычно следят с помощью тонкослойной хроматографии, ВЭЖХ или масс-спектрометрии.
Неочищенные вещества можно очистить с помощью хроматографии с нормальной фазой, хроматографии с обращенной фазой (в кислой или щелочной среде), хирального разделения или перекристаллизации.
Перед проведением окончательных анализов и биологических исследований продуктов их обычно сушат в вакууме.
Все спектры ЯМР снимают при 250 МГц, 300 МГц, 400 МГц или 500 МГц.
Соединения исследуют в растворе в ДМСО-d6, CDCl3 или MeOH-d4 при температуре датчика, равной 300 K, и при концентрации, равной 10 мг/мл. Прибор фиксируют на сигнал дейтерия, содержащегося в ДМСО-d6 CDCl3 или MeOH-d4. Химические сдвиги приведены в част./млн в слабопольную сторону от ТМС (тетраметилсилан), использующегося в качестве внутреннего стандарта.
1. Получение промежуточного продукта формулы (II) - 2-(3,5-дихлор-1-метилиндазол-4-ил)уксусной кислоты
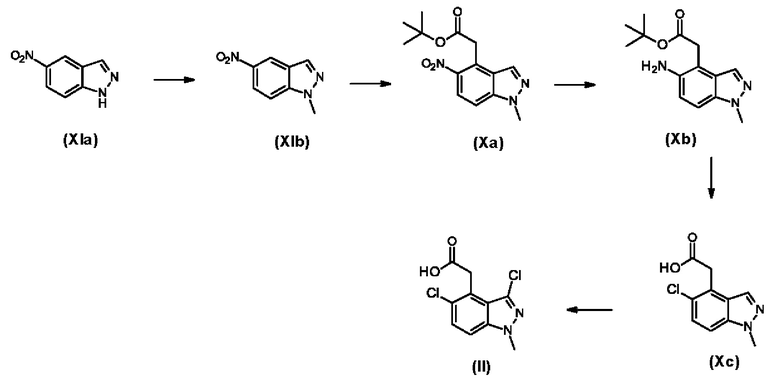
1.1. Получение промежуточного продукта (XIb) - 1-метил-5-нитроиндазола
В трехгорлую круглодонную колбу объемом 50 л при 15-30°С помещают 5-нитро-1Н-индазол (XIa) (3,00 кг, 18,4 моля) и ДМФ (30,0 л). В реактор при 0-5°С одной порцией добавляют КОН (2,06 кг, 36,7 моля). Смесь перемешивают при 0-50°С в течение 1 ч. Затем при 0-5°С добавляют метилйодид (2,87 кг, 20,2 моля) и смесь перемешивают при 15-30°С в течение 3 ч. Реакционную смесь при 0-10°С выливают в воду (30 л) и смесь перемешивают в течение 10 мин, затем фильтруют. Осадок на фильтре промывают водой (5 л) и сушат. Всю эту процедуру проводят одновременно с использованием 4 порций одинакового размера. Твердые вещества, полученные с использованием 4 порций, объединяют и получают 1-метил-5-нитроиндазол (XIb) в виде коричневого твердого вещества (10,0 кг, 42,3 моля, чистота 75% (ЖХ/МС), выход 57,5%), которое используют на следующей стадии без дополнительной очистки.
1Н ЯМР (400 МГц, CDCl3) δ 8,65 (s, 1Н), 8,21 (d, J=9,17 Гц, 1Н), 8,13 (s, 1Н), 7,39 (d, J=9,17 Гц, 1Н), 4,08 (s, 3Н).
1.2. Получение промежуточного продукта (Ха) - трет-бутил-2-(1-метил-5-нитроиндазол-4-ил)ацетата
В трехгорлую круглодонную колбу объемом 50 л помещают t-BuOK (4,43 кг, 39,5 моля) и ТГФ (30 л) и смесь в атмосфере азота охлаждают до -45/-35°С и перемешивают. Затем при -45/-35°С порциями добавляют 1-метил-5-нитроиндазол (XIb) (3,50 кг, 19,7 моля). При такой же температуре по каплям добавляют трет-бутил-2-хлорацетат (3,57 кг, 23,7 моля) и смесь перемешивают в течение 1 ч. Смесь нагревают до 15-30°С и перемешивают в течение 5 ч. Реакцию останавливают путем добавления насыщенного раствора хлорида аммония (9 л) и добавляют воду (2 л). Органический слой отделяют и водный слой экстрагируют этилацетатом (2×5 л). Органические фазы объединяют, промывают рассолом (2 л), сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают путем перекристаллизации из этилацетата (5 л). Всю эту процедуру проводят одновременно с использованием 2 порций одинакового размера. Твердые вещества, полученные с использованием 2 порций, объединяют и сушат вместе и получают трет-бутил-2-(1-метил-5-нитроиндазол-4-ил)ацетат в виде желтого твердого вещества (Ха) (5,30 кг, 17,7 моля, чистота 97,6% (ЖХ/МС), выход 44,9%).
1Н ЯМР (400 МГц, CDCl3) δ 8,18-8,20 (m, 2Н), 7,37 (d, J=9,21 Гц, 1Н), 4,27 (s, 2Н), 4,14 (s, 3Н), 1,44 (s, 9Н).
1.3. Получение промежуточного продукта (ХЬ) - трет-бутил-2-(5-амино-1-метилиндазол-4-ил)ацетата
В реактор помещают трет-бутил-2-(1-метил-5-нитроиндазол-4-ил)ацетат (Ха) (7,30 кг, 25,0 моля) и МеОН (76 л). Реактор продувают аргоном и добавляют Pd/C (50%, 760 г). Реактор трижды продувают водородом и смесь перемешивают при 50°С в атмосфере водорода (50 фунт-сила/дюйм2) в течение 3 ч. Реакционную смесь фильтруют и твердое вещество промывают с помощью МеОН (5 л). Смесь концентрируют и получают трет-бутил-2-(5-амино-1-метилиндазол-4-ил)ацетат (Xb) в виде коричневого масла (6,50 кг, 23,9 моля, чистота 96,2% (ЖХ/МС), выход 95,4%), которое используют на следующей стадии без дополнительной очистки.
1Н ЯМР (400 МГц, CDCl3) δ 7,72 (s, 1Н), 7,27 (d, J=8,80 Гц, 1Н), 6,91 (d, J=8,80 Гц, 1Н), 4,60 (s, 2Н), 3,93 (s, 3Н), 3,68 (s, 2Н), 1,38 (s, 9Н).
1.4. Получение промежуточного продукта (Хс) - 2-(5-хлор-1-метилиндазол-4-ил)уксусной кислоты
В трехгорлую круглодонную колбу объемом 50 л помещают трет-бутил-2-(5-амино-1-метилиндазол-4-ил)ацетат(Xb) (2,00 кг, 7,65 моля) и концентрированную HCl (10,0 л, 12М) и смесь охлаждают до -10/-5°С и перемешивают. При -10/-5°С по каплям добавляют водный раствор (5 л) нитрита натрия (686 г, 9,95 моля) и перемешивают в течение 30 мин. В трехгорлую круглодонную колбу объемом 20 л помещают CuCl (833 г, 8,42 моля) и концентрированную HCl (10,0 л, 12М) и смесь перемешивают при -10/-5°С в течение 30 мин, затем добавляют в другой реактор. Смесь перемешивают при -10/-5°С в течение 1 ч, затем при 10-30°С в течение 16 ч. Реакционную смесь фильтруют и твердые вещества промывают водой. Всю эту процедуру проводят одновременно с использованием 3 порций одинакового размера. Твердые вещества, полученные с использованием 3 порций, объединяют и сушат вместе и получают 2-(5-хлор-1-метилиндазол-4-ил)уксусную кислоту (Хс) в виде желтого твердого вещества (4,00 кг, 16,3 моля, чистота 92% (ЖХ/МС), выход 71,3%), которое используют на следующей стадии без дополнительной очистки.
1.5. Получение 2-(3,5-дихлор-1-метилиндазол-4-ил)уксусной кислоты (II)
В трехгорлую круглодонную колбу объемом 50 л при 20°С помещают 2-(5-хлор-1-метилиндазол-4-ил)уксусную кислоту (Хс) (1,30 кг, 5,79 моля) и ДМФ (6,5 л). При 20°С порциями добавляют N-хлорсукцинимид (772 г, 5,79 моля) и смесь перемешивают при 20°С в течение 2 ч. Реакционную смесь выливают в воду (25 л) и фильтруют. Неочищенный продукт растирают со смесью изопропиловый эфир:этилацетат (3:1) (7,0 л) при 20°С в течение 2 ч, затем фильтруют и сушат. Всю эту процедуру проводят одновременно с использованием 3 порций одинакового размера. Твердые вещества, полученные с использованием 3 порций, объединяют и получают 2-(3,5-дихлор-1-метилиндазол-4-ил)уксусную кислоту (II) (2,1 кг, 7,9 моля, чистота 97,5% (ЖХ/МС), выход 46%).
1Н ЯМР (400 МГц, CDCl3) δ 12,67 (s, 1Н), 7,68 (d, J=9,05 Гц, 1Н), 7,53 (d, J=9,05 Гц, 1Н), 4,20 (s, 2Н), 4,02 (s, 3Н).
2. Получение соединения формулы (I)
2-(3,5-Дихлор-1-метилиндазол-4-ил)-1-[(1S,3R)-3-(гидроксиметил)-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1Н-изохинолин-2-ил]этанон
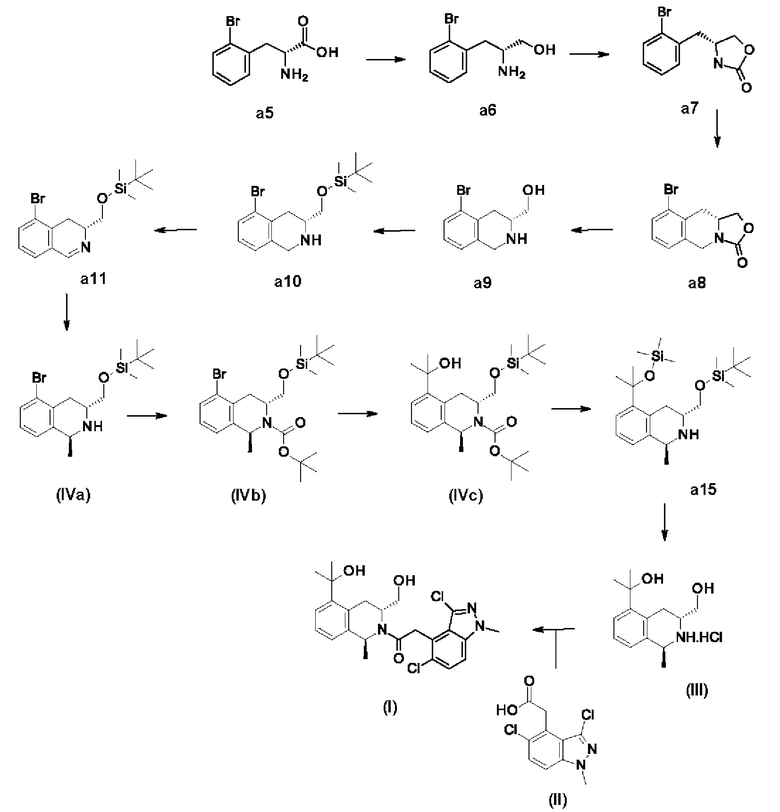
2.1. Получение промежуточного продукта (IX).
(2R)-2-Амино-3-(2-бромфенил)пропан-1-ол-аб
В реактор помещают (2R)-2-амино-3-(2-бромфенил)пропановую кислоту а5 (34,0 кг, 139 молей) и ТГФ (238 л). При 20-30°С медленно добавляют борогидрид натрия (15,6 кг, 413 молей). При 0-10°С медленно добавляют раствор йода (35,3 кг, 139 молей) в сухом ТГФ (20,0 л) и реакционную смесь перемешивают при 70°С в течение 12 ч. Реакцию при 0°С останавливают метанолом (70,0 л) и смесь нагревают при 80°С в течение 30 мин. Смесь охлаждают, концентрируют в вакууме и остаток суспендируют в NaOH (30,0 л, 2N), затем фильтруют. Осадок на фильтре сушат в вакууме и получают (2R)-2-амино-3-(2-бромфенил)пропан-1-ол а6 в виде белого твердого вещества (31,0 кг, 135 молей, выход 96,7%), которое используют на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,57 (d, J=7,7 Гц, 1Н), 7,21-7,29 (m, 2Н), 7,07-7,15 (m, 1H), 3,66 (dd, J=10,5, 3,6 Гц, 1H), 3,41 (dd, J=10,5, 7,2 Гц, 1Н), 3,18-3,29 (m, 1Н), 2,95 (dd, J=13,5, 5,5 Гц, 1Н), 2,70 (dd, J=13,5, 8,2 Гц, 1Н), 1,51-1,91 (m, 3Н).
2.2. Получение промежуточного продукта формулы (VIII).
(4R)-4-[(2-Бромфенил)метил]оксазолидин-2-он - а7
В реактор помещают (2R)-2-амино-3-(2-бромфенил)пропан-1-ол а6 (31,0 кг, 135 молей) и дихлорметан (220 л). При комнатной температуре добавляют трифосген (13,9 кг, 47,1 моля), затем при 0-10°С медленно добавляют N,N-диизопропилэтиламин (39,1 кг, 303 моля). Реакционную смесь перемешивают при 0-10°С в течение 1 ч, затем дважды промывают водой (50,0 л), сушат над безводным сульфатом натрия и фильтруют и получают (4R)-4-[(2-бромфенил)метил]оксазолидин-2-он а7 в виде раствора в дихлорметане, который используют на следующей стадии без обработки.
2.3. Получение промежуточного продукта (VII).
(10aR)-9-Бром-1,5,10,10а-тетрагидрооксазоло[3,4-b]изохинолин-3-он а8
В реактор помещают раствор (4R)-4-[(2-бромфенил)метил]оксазолидин-2-она а7 (135 молей) в дихлорметане (220 л) и охлаждают до 0-5°С. При 0-5°С добавляют триметилсилилтрифлат (35,9 кг, 162 моля) и параформальдегид (13,3 кг, 148 молей), затем перемешивают при 15-20°С в течение 2 ч. К смеси добавляют воду (170 л), затем ее дважды экстрагируют дихлорметаном (5 0,0 л). Органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Добавляют смесь петролейный эфир:этилацетат (1:1, 45,0 л) и смесь перемешивают при комнатной температуре в течение 6 ч и фильтруют. Твердое вещество сушат и получают (10aR)-9-бром-1,5,10,10а-тетрагидрооксазоло[3,4-b]изохинолин-3-он а8 в виде почти белого твердого вещества (29,0 кг, выход 80,2%).
1Н ЯМР (400 МГц, CDCl3) δ 7,45-7,52 (m, 1H), 7,08-7,14 (m, 2Н), 4,83 (d, J=17,0 Гц, 1H), 4,62 (t, J=8,4 Гц, 1H), 4,36 (d, J=17,0 Гц, 1Н), 4,21 (dd, J=8,6, 4,9 Гц, 1H), 3,91-3,99 (m, 1H), 3,25 (dd, J=16,3, 4,2 Гц, 1Н), 2,67 (dd, J=16,l, 11,0 Гц, 1Н).
2.4. Получение промежуточных продуктов (VI)
2.4.1. [(3R)-5-Бром-1,2,3,4-тетрагидроизохинолин-3-ил]метанол а9
В реактор помещают этанол (120 л) и воду (60,0 л). При 15-20°С добавляют (10aR)-9-бром-1,5,10,10а-тетрагидрооксазоло[3,4-b]изохинолин-3-он а8 (29,7 кг, 111 молей), затем медленно добавляют гидроксид натрия (13,3 кг, 332 моля). Реакционную смесь перемешивают при 90°С в течение 2 ч, затем охлаждают до комнатной температуры. К смеси добавляют воду (300 л), затем ее центрифугируют. Полученный после центрифугирования остаток сушат в сушильном шкафу с циркуляцией газов и получают [(3R)-5-бром-1,2,3,4-тетрагидроизохинолин-3-ил]метанол а9 в виде белого твердого вещества (23,7 кг, выход 88,3%), которое используют на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,37-7,47 (m, 1H), 6,95-7,08 (m, 2Н), 4,00-4,10 (m, 2Н), 3,85 (dd, J=10,9, 3,7 Гц, 1Н), 3,57 (dd, J=10,9, 7,9 Гц, 1H), 3,06 (ddt, J=11,3, 7,6, 4,1, 4,1 Гц, 1Н), 2,79 (dd, J=17,1, 4,4 Гц, 1H), 2,40 (dd, J=17,1, 10,9 Гц, 1H), 1,93 (br s, 2Н).
2.4.2. [(3R)-5-Бром-1,2,3,4-тетрагидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан а10
В реактор помещают [(3R)-5-бром-1,2,3,4-тетрагидроизохинолин-3-ил]метанол а9 (23,7 кг, 97,8 моля) и дихлорметан (240 л). Добавляют ДМАП (120 г, 0,98 моля) и имидазол (13,3 кг, 196 молей). При 15-20°С медленно добавляют трет-бутилдиметилсилилхлорид (TBSCl) (17,7 кг, 117 молей) и смесь перемешивают в течение 12 ч. К смеси добавляют хлорид аммония (100 л). Органическую фазу отделяют, промывают водой (50,0 л), сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме и получают [(3R)-5-бром-1,2,3,4-тетрагидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан а10 в виде желтого масла (37,6 кг, чистота 86%, выход 93%), которое используют на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,36-7,45 (m, 1H), 7,01 (d, J=4,6 Гц, 1H), 4,01-4,13 (m, 2Н), 3,84 (dd, J=9,9, 3,7 Гц, 1H), 3,64 (dd, J=9,8, 7,2 Гц, 1Н), 2,96-3,08 (m, 1Н), 2,75 (dd, J=17,0, 4,2 Гц, 1Н), 2,44 (dd, J=17,0, 10,8 Гц, 1Н), 1,76-2,20 (m, 2Н), 0,89-0,97 (m, 9Н), 0,08-0,14 (m, 6Н).
2.5. Получение промежуточного продукта (V).
[(3R)-5-Бром-3,4-дигидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан a11
В реактор помещают [(3R)-5-бром-1,2,3,4-тетрагидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан а10 (3,42 кг, 8,31 моля) и ТГФ (30,0 л). При комнатной температуре медленно добавляют N-хлорсукцинимид (NCS) (1,17 кг, 8,73 моля) и смесь перемешивают при 25°С в течение 30 мин. При комнатной температуре медленно добавляют раствор КОН (1,52 кг, 27,1 моля) в сухом метаноле (7,00 л) и реакционную смесь перемешивают при 25°С в течение 1 ч. Реакцию останавливают водой (10,0 л) и смесь экстрагируют смесью петролейный эфир:этилацетат (1:2, 5,00 л). Органический слой отделяют, промывают рассолом (10,0 л), сушат над безводным сульфатом натрия и фильтруют. Всю эту процедуру проводят одновременно с использованием 10 порций одинакового размера и полученные при проведении 10 реакций фильтраты объединяют и концентрируют в вакууме и получают [(3R)-5-бром-3,4-дигидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан all в виде коричневого масла (28,0 кг, неочищенное), которое используют на следующей стадии без дополнительной очистки.
1Н ЯМР (400 МГц, CDCl3) δ 8,24 (d, J=2,6 Гц, 1Н), 7,58 (dd, J=7,8, 1,2 Гц, 1Н), 7,12-7,25 (m, 2Н), 4,03 (dd, J=9,5, 4,0 Гц, 1Н), 3,67-3,77 (m, 2Н), 3,07 (dd, J=17,0, 6,2 Гц, 1H), 2,68 (dd, J=17,1, 10,9 Гц, 1Н), 0,88-0,91 (m, 9Н), 0,07 (d, J=1,5 Гц, 6Н).
2.6. Получение промежуточных продуктов формулы (IV)
2.6.1. [(1S,3R)-5-Бром-1-метил-1,2,3,4-тетрагидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан (IVa)
В реактор помещают [(3R)-5-бром-3,4-дигидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан all (3,10 кг, 8,75 моля) и ТГФ (20,0 л). Смесь охлаждают до 0°С и добавляют метилмагнийхлорид (ЗМ, 11,6 л). Смесь перемешивают при 20°С в течение 12 ч. Реакцию останавливают насыщенным раствором хлорида аммония. Фазы разделяют и водный слой дважды экстрагируют смесью петролейный эфир:этилацетат (3:1, 5,00 л). Объединенные органические фазы промывают рассолом (10,0 л), сушат над безводным сульфатом натрия и фильтруют. Всю эту процедуру проводят одновременно с использованием 9 порций одинакового размера и полученные при проведении 9 реакций фильтраты объединяют и концентрируют в вакууме. Неочищенную смесь очищают с помощью хроматографии на силикагеле с использованием смеси петролейный эфир:этилацетат (10:1) и получают [(1S,3R)-5-бром-1-метил-1,2,3,4-тетрагидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан (IVa) в виде коричневого масла (4,60 кг, чистота 99,7%, выход 15,7%).
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,41 (dd, J=7,7, 0,9 Гц, 1H), 7,12-7,18 (m, 1Н), 7,03-7,11 (m, 1H), 4,12 (q, J=6,8 Гц, 1H), 3,62 (d, J=5,7 Гц, 2Н), 3,07-3,17 (m, 1Н), 2,67-2,76 (m, 1H), 2,26 (dd, J=16,9, 10,0 Гц, 1Н), 2,12 (br s, 1Н), 1,32 (d, J=6,8 Гц, 3Н), 0,84-0,93 (m, 9Н), 0,07 (d, J=0,9 Гц, 6Н).
2.6.2. трет-Бутил-(1S,3R)-5-бром-3-[[трет-бутил(диметил)силил]оксиметил]-1-метил-3,4-дигидро-1H-изохинолин-2-карбоксилат (IVb)
В реактор помещают [(1S,3R)-5-бром-1-метил-1,2,3,4-тетрагидроизохинолин-3-ил]метокси-трет-бутилдиметилсилан (IVa) (1,85 кг, 4,99 моля) и дихлорметан (13,0 л). При комнатной температуре добавляют N,N-диизопропилэтиламин (1,94 кг, 14,9 моля) и ди-трет-бутилдикарбонат (1,14 кг, 5,24 моля) и смесь перемешивают в течение 12 ч. Реакционную смесь дважды промывают насыщенным раствором хлорида аммония (10,0 л), органический слой сушат над безводным сульфатом натрия и фильтруют. Всю эту процедуру проводят одновременно с использованием 2 порций одинакового размера и полученные при проведении 2 реакций фильтраты объединяют и концентрируют в вакууме. Неочищенную смесь очищают с помощью хроматографии на силикагеле с использованием смеси петролейный эфир:этилацетат (30:1) и получают трет-бутил-(1S,3R)-5-бром-3-[[трет-бутил(диметил)силил]оксиметил]-1-метил-3,4-дигидро-1Н-изохинолин-2-карбоксилат (IVb) в виде желтого масла (4,00 кг, чистота 99,5%, выход 85,2%).
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,50 (d, J=7,9 Гц, 1H), 7,22 (br d, J=6,7 Гц, 1H), 7,06-7,18 (m, 1H), 4,84 (br s, 1H), 4,12 (br s, 1H), 3,46 (br d, J=15,4 Гц, 2H), 2,94 (br dd, J=15,8, 5,2 Гц, 1H), 2,71 (br t, J=9,5 Гц, 1H), 1,45 (s, 9H), 1,28 (br s, 3H), 0,81 (s, 9H), -0,08 (s, 6H).
2.6.3. трет-Бутил-(1S,3R)-3-[[трет-бутил(диметил)силил]оксиметил]-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1Н-изохинолин-2-карбоксилат (IVc)
Раствор трет-бутил-(1S,3R)-5-бром-3-[[трет-бутил(диметил)силил]оксиметил]-1-метил-3,4-дигидро-1Н-изохинолин-2-карбоксилата (IVb) (42,5 г, 90,3 ммоля) в сухом ТГФ (0,5 М раствор) и имеющийся в продаже раствор н-бутиллития в гексанах (1,6 М раствор) подают с помощью насосов при скоростях, равных 6,0 мл/мин (1,0 экв.) и 2,46 мл/мин (1,3 экв.) соответственно, и смешивают в стеклянном микрореакторе, охлажденном до -40°С. Затем смешанный поток пропускают через зону реакции 1 микрореактора (0,3 мл) и затем объединяют с раствором сухого ацетона (13,5 М), подаваемым с помощью насоса при скорости, равной 6,0 мл/мин (27 экв.). Затем полученный поток при -40°С пропускают через зону реакции 2 микрореактора (0,7 мл). В заключение, объединенный поток, выходящий из реактора, собирают и реакцию останавливают при КТ насыщенным водным раствором хлорида аммония. После израсходования всех загруженных растворов получают двухслойную реакционную смесь. Водный слой отделяют от органического слоя и затем его дважды экстрагируют этилацетатом. Объединенные органические слои промывают рассолом, сушат над безводным сульфатом натрия и концентрируют в вакууме. Получают желтое масло (46,5 г) и его очищают с помощью НЖХ с использованием колонки GreenSep Nitro (10 мкм, 5×22,3, с использованием в качестве элюента смеси 98% CO2/2% EtOH). Растворитель удаляют в вакууме и получают белое твердое вещество, трет-бутил-(1S,3R)-3-[[трет-бутил(диметил)силил]оксиметил]-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1Н-изохинолин-2-карбоксилат(IVc) (25 г, 56 ммолей, выход 62%).
СЭЖХ (сверхэффективная жидкостная хроматография)-МС, щелочная среда, 1 пик при 3,83 мин (ЭР+): 350 (М-Вос+Н)+, 332 (М-Вос-H2O+Н)+, чистота 100%.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,44 (d, J=7,9 Гц, 1H), 7,19 (dt, J=8,l, 5,2 Гц, 1Н), 7,09 (t, J=9,0 Гц, 1Н), 4,99 (s, 1H), 4,87 (dq, J=13,4, 6,4 Гц, 1Н), 4,11 (s, 1H), 3,96 (t, J=14,9 Гц, 1H), 3,48 (dd, J=9,4, 4,1 Гц, 1H), 2,98 (dd, J=16,5, 5,0 Гц, 1H), 2,89 (t, J=9,6 Гц, 1H), 1,65 (s, 3Н), 1,58 (s, 3Н), 1,55 (d, J=2,5 Гц, 9Н), 1,34 (dd, J=20,5, 6,6 Гц, 3Н), 0,90 (s, 9Н), 0,08 (d, J=7,2 Гц, 3Н), -0,00 (s, 3Н).
2.7. Получение промежуточного продукта (III) - 2-[(1S,3R)-3-(гидроксиметил)-1-метил-1,2,3,4-тетрагидроизохинолин-2-ий-5-ил]пропан-2-олхлорида
2.7.1. трет-Бутилдиметил-[[(1S,3R)-1-метил-5-(1-метил-1-триметилсилилоксиэтил)-1,2,3,4-тетрагидроизохинолин-3-ил]метокси]силан -а15
трет-Бутил-(1S,3R)-3-[[трет-бутил(диметил)силил]оксиметил]-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1Н-изохинолин-2-карбоксилат (IVc) (148 г, чистота 87%, 287 ммолей) растворяют в 1000 мл дихлорметана и переносят в реактор с двойными стенками объемом 2 л. Добавляют 2,6-лутидин (100 мл, 860 ммолей) и температуру кожуха устанавливают равной -2°С. В течение 40 мин с помощью капельной воронки добавляют триметилсилилтрифторметансульфонат (154 г, 129 мл, 692 ммоля). Через 2 ч после начала добавления реакцию останавливают путем добавления 650 мл водного раствора лимонной кислоты (1М) и температуру смеси устанавливают равной 20°С. Через 1 ч после начала остановки реакции слои разделяют. Органический слой дважды промывают с помощью 350 мл водного раствора лимонной кислоты (1М). Органический слой перемешивают с 750 мл водного раствора карбоната натрия (10% мас./мас.) в течение 10 мин, затем слои разделяют. Органический слой сушат над безводным сульфатом натрия. Затем органический слой фильтруют и фильтрат концентрируют в вакууме при 40°С и получают желтое масло (128 г), трет-бутилдиметил-[[(1S,3R)-1-метил-5-(1-метил-1-триметилсилилоксиэтил)-1,2,3,4-тетрагидроизохинолин-3-ил]метокси]силан а15, который используют на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,19 (d, J=7,7 Гц, 1Н), 7,07 (t, J=7,7 Гц, 1Н), 7,00 (d, J=7,6 Гц, 1Н), 4,24 (q, J=6,8 Гц, 1Н), 3,75 (dd, J=9,7, 4,4 Гц, 1H), 3,60 (dd, J=9,7, 7,0 Гц, 1H), 3,54 (dd, J=16,3, 3,5 Гц, 1Н), 3,15 (ddt, J=10,9, 7,4, 4,0 Гц, 1Н), 2,52 (dd, J=16,3, 10,9 Гц, 1H), 1,66 (d, J=14,6 Гц, 6Н), 1,52-1,43 (m, 3Н), 0,92 (q, J=1,2 Гц, 9Н), 0,14 (q, J=1,2 Гц, 2Н), 0,09 (d, J=1,1 Гц, 6Н), 0,00 (q, J=1,2, 0,8 Гц, 9Н).
2.7.2. 2-[(1S,3R)-3-(Гидроксиметил)-1-метил-1,2,3,4-тетрагидроизохинолин-2-ий-5-ил]пропан-2-олхлорид - промежуточный продукт (III)
В трехгорлой круглодонной колбе, снабженной механической мешалкой, трет-бутилдиметил-[[(1S,3R)-1-метил-5-(1-метил-1-триметилсилилоксиэтил)-1,2,3,4-тетрагидроизохинолин-3-ил]метокси]силан а15 (20,0 г, 47,4 ммоля) растворяют в 220 мл изопропанола. К этому раствору добавляют 42,3 мл хлористоводородной кислоты в изопропаноле (5-6 М, примерно 5 экв.). Через 45 мин после добавления хлористоводородной кислоты добавляют 100 мг затравочных кристаллов искомого продукта. После выдерживания при комнатной температуре в течение 7 ч реакционную смесь фильтруют через фильтр из пористого стекла. Осадок на фильтре промывают с помощью 40 мл изопропанола и сушат в вакууме при комнатной температуре в течение ночи. Получают 11,1 г 2-[(1S,3R)-3-(гидроксиметил)-1-метил-1,2,3,4-тетрагидроизохинолин-2-ий-5-ил]пропан-2-олхлорида (III) в виде розоватого твердого вещества. Выход после проведения 2 стадий удаления защитных групп составляет 91%.
1H ЯМР (400 МГц, CD3OD) δ 7,46 (dd, J=7,8, 1,3 Гц, 1H), 7,28 (t, J=7,8 Гц, 1H), 7,21 (dd, J=7,8, 1,3 Гц, 1Н), 4,63 (q, J=6,9 Гц, 1H), 3,97 (dd, J=11,7, 3,8 Гц, 1H), 3,88 (dd, J=17,2, 4,3 Гц, 1Н), 3,78 (dd, J=11,8, 6,1 Гц, 1H), 3,66-3,56 (m, 1H), 3,14 (dd, J=17,2, 11,4 Гц, 1Н), 1,73 (d, J=6,8 Гц, 3Н), 1,64 (d, J=4,8 Гц, 6Н). Сигналы содержащихся в ОН и NH протонов отсутствуют.
2.8. Получение соединения формулы (I).
2-(3,5-Дихлор-1-метилиндазол-4-ил)-1-[(1S,3R)-3-(гидроксиметил)-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1Н-изохинолин-2-ил]этанон
В реактор Easymax объемом 100 мл, снабженный механической мешалкой, помещают 2-(3,5-дихлор-1-метилиндазол-4-ил)уксусную кислоту (II) (4,00 г, 15,4 ммоля), 2-[(1S,3R-3-(гидроксиметил)-1-метил-1,2,3,4-тетрагидроизохинолин-2-ий-5-ил]пропан-2-олхлорид (III) (4,46 г, 16,4 ммоля) и 48 мл ДМФ. Суспензию перемешивают при 20°С и затем охлаждают путем установления температуры кожуха, равной -2°С. После установления температуры смеси, равной ниже 3°С, добавляют N,N-диизопропилэтиламин (9,5 мл, 54 ммоля). В течение 1 ч 4 порциями добавляют (2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат (6,4 г, 17 ммолей). Смесь перемешивают в течение 1 ч и 45 мин, затем температуру кожуха устанавливают равной 15°С. Затем в течение нескольких минут добавляют 16 мл воды. Через 15 мин добавляют 30 мг твердого продукта, использующегося в качестве затравочных кристаллов для инициирования кристаллизации. Температуру кожуха устанавливают равной 20°С. Через 0,5 ч в течение 17 мин добавляют 16 мл воды. Суспензию перемешивают при 20°С в течение 2 ч и 15 мин и затем фильтруют через фильтр из пористого стекла. Осадок на фильтре промывают водой, двумя порциями по 20 мл, и затем сушат в вакууме при 50°С в течение ночи и получают 6,03 г 2-(3,5-дихлор-1-метилиндазол-4-ил)-1-[(1S,3R)-3-(гидроксиметил)-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1Н-изохинолин-2-ил]этанон (I) (неочищенное вещество).
Проводят перекристаллизацию 5,00 г неочищенного вещества, полученного путем проводимого сначала суспендирования в 50 мл ацетонитрила. Температуру кожуха устанавливают равной 70°С. После растворения твердого вещества и установления температуры смеси, равной 66°С, добавляют 720 мкл воды. Затем температуру смеси понижают до 59°С и в качестве затравочных кристаллов добавляют 125 мг твердого продукта. Затем температуру смеси в течение 25 мин понижают до 55°С, на этой стадии происходит кристаллизация. Затем температуру кожуха в течение 2 ч понижают от равной 58°С до равной 20°С. Через 50 мин суспензию фильтруют и осадок на фильтре промывают с помощью 7,5 мл ацетонитрила. Затем осадок на фильтре сушат в вакууме при 45°С в течение ночи и при 50°С в течение 2 ч и получают 4,04 г 2-(3,5-дихлор-1-метилиндазол-4-ил)-1-[(1S,3R)-3-(гидроксиметил)-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1H-изохинолин-2-ил]этанона (I) в виде почти белого порошкообразного вещества (в форме гидрата). Выход = 64%.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,65 (dd, J=9,0, 2,2 Гц, 1H), 7,52 (dd, J=9,0, 2,1 Гц, 1H), 7,37 (ddd, J=19,6, 7,6, 1,7 Гц, 1H), 7,25-7,03 (m, 2Н), 5,30 (q, J=6,5 Гц, 0,3Н), 5,16-4,99 (m, 1,7Н), 4,99-4,84 (m, 0,7Н), 4,63-4,30 (m, 3,3Н), 4,17-3,93 (m, 4Н), 3,28 (dt, J=10,5, 5,1 Гц, 1,3Н), 3,10-2,85 (m, 1,7Н), 1,56 (dd, J=13,2, 6,9 Гц, 6,7Н), 1,24 (d, J=6,5 Гц, 2,3Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ТЕТРАГИДРОИЗОХИНОЛИНА КАК ПОЗИТИВНЫЙ АЛЛОСТЕРИЧЕСКИЙ МОДУЛЯТОР D1 | 2020 |
|
RU2824527C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И БЕНЗОМОРФОЛИНА В КАЧЕСТВЕ МОДУЛЯТОРА МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2517181C2 |
| Новые пролекарства на основе катехоламина для применения в лечении болезни Паркинсона | 2018 |
|
RU2806075C2 |
| 1,3,5-ТРИЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ТРИАЗОЛА | 2008 |
|
RU2476433C2 |
| 3-АЗАБИЦИКЛО[3.1.0]ГЕКСИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2510396C2 |
| Гетероароматические соединения и их применение в качестве допаминовых D1 лигандов | 2013 |
|
RU2617842C2 |
| 1,2,4-ТРИАЗОЛО[4,3-a]ПИРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПОЛОЖИТЕЛЬНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ РЕЦЕПТОРОВ MGLUR2 | 2015 |
|
RU2695651C2 |
| МОДУЛЯТОРЫ МЕТАБОТРОПНОГО ГЛУТАМАТНОГО РЕЦЕПТОРА ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2008 |
|
RU2508107C2 |
| 5-АРОМАТИЧЕСКОЕ АЛКИНИЛЗАМЕЩЕННОЕ БЕНЗАМИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2695371C2 |
| СОЕДИНЕНИЯ, КОТОРЫЕ УСИЛИВАЮТ РЕЦЕПТОР ГЛУТАМАТА, И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2005 |
|
RU2403242C2 |
Изобретение относится к области химии и фармацевтики, а именно к 2-(3,5-дихлор-1-метилиндазол-4-ил)-1-[(1S,3R)-3-(гидроксиметил)-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1H-изохинолин-2-ил]этанону формулы (I) или его фармацевтически приемлемой соли, фармацевтической композиции, обладающей активностью позитивного аллостерического модулятора D1, на его основе, способу лечения и/или предупреждения связанных с познавательной способностью и негативных симптомов шизофрении, слабого нарушения познавательной способности (СНП), импульсивности, болезни Паркинсона и других нарушений движений, слабоумия с тельцами Леви, болезни Альцгеймера, для которых показано введение позитивного аллостерического модулятора D1, и способу лечения болезни Паркинсона и других нарушений движений, болезни Альцгеймера или связанных с познавательной способностью и негативных симптомов шизофрении, для которых показано введение позитивного аллостерического модулятора D1, которые включают введение соединения формулы (I) или его фармацевтически приемлемой соли в эффективном количестве. Использование изобретения позволяет эффективно лечить заболевания, для которых показано введение позитивного аллостерического модулятора D1. 4 н. и 4 з.п. ф-лы.
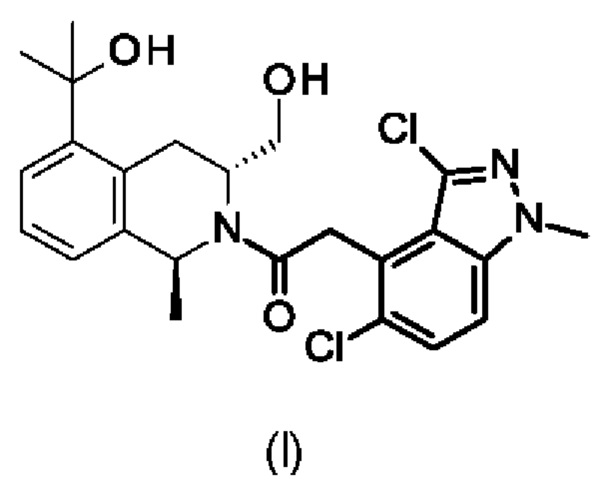
1. 2-(3,5-Дихлор-1-метилиндазол-4-ил)-1-[(1S,3R)-3-(гидроксиметил)-5-(1-гидрокси-1-метилэтил)-1-метил-3,4-дигидро-1H-изохинолин-2-ил]этанон формулы (I) или его фармацевтически приемлемая соль
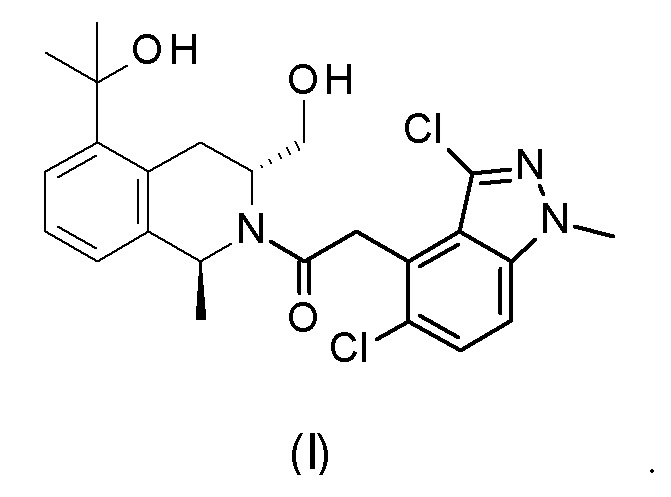
2. Соединение формулы (I) по п. 1 или его фармацевтически приемлемая соль для применения в терапии.
3. Соединение формулы (I) по п. 1 или его фармацевтически приемлемая соль для применения для лечения и/или предупреждения заболеваний и/или нарушений, в которых играют роль рецепторы D1.
4. Соединение формулы (I) по п. 1 или его фармацевтически приемлемая соль для применения для лечения и/или предупреждения связанных с познавательной способностью и негативных симптомов шизофрении, нарушения познавательной способности, связанного с лечением нейролептическим средством, слабого нарушения познавательной способности (СНП), импульсивности, синдрома дефицита внимания с гиперактивностью (СДВГ), болезни Паркинсона и других нарушений движений, дистонии, слабоумия при болезни Паркинсона, болезни Гентингтона, слабоумия с тельцами Леви, болезни Альцгеймера, привыкания к чрезмерному употреблению лекарственного средства или наркотика, нарушений сна, апатии, травматического повреждения спинного мозга или невропатической боли.
5. Соединение формулы (I) по п. 1 или его фармацевтически приемлемая соль для применения для лечения болезни Паркинсона и других нарушений движений, болезни Альцгеймера или связанных с познавательной способностью и негативных симптомов шизофрении.
6. Фармацевтическая композиция, обладающая активностью позитивного аллостерического модулятора D1, содержащая эффективное количество соединения формулы (I) по п. 1 или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем.
7. Способ лечения и/или предупреждения связанных с познавательной способностью и негативных симптомов шизофрении, слабого нарушения познавательной способности (СНП), импульсивности, болезни Паркинсона и других нарушений движений, слабоумия с тельцами Леви, болезни Альцгеймера, для которых показано введение позитивного аллостерического модулятора D1, который включает введение пациенту, нуждающемуся в таком лечении, соединения формулы (I) по п. 1 или его фармацевтически приемлемой соли в эффективном количестве.
8. Способ лечения болезни Паркинсона и других нарушений движений, болезни Альцгеймера или связанных с познавательной способностью и негативных симптомов шизофрении, для которых показано введение позитивного аллостерического модулятора D1, который включает введение пациенту, нуждающемуся в таком лечении, соединения формулы (I) по п. 1 или его фармацевтически приемлемой соли в эффективном количестве.
| WO 2014193781 A1, 04.12.2014 | |||
| WO 2017178377 A1, 19.10.2017 | |||
| WO 2016055479 A1, 14.04.2016 | |||
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |

