Область техники
Изобретение относится к области органической химии, фармакологии и медицины, а именно, к способу получения известного соединения - производного 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тиона формулы (I), обладающего свойством ингибирования репликации бета-коронавирусов, для лечения заболеваний, вызванных бета-коронавирусами, включая SARS-CoV-2. Изобретение может быть использовано для производства фармацевтической композиции для лечения указанных заболеваний.
Уровень техники
Вспышка COVID-19, вызванная SARS-CoV-2, по всему миру и связанные с ней последствия, являются угрозой для общественного здравоохранения и экономики многих стран. Отсутствие специальной терапии против нового вируса требует поиска лекарственных средств, противодействующих инфекции.
Коронавирусы (Coronaviridae) - это большое семейство РНК-содержащих вирусов, способных инфицировать человека и некоторых животных. Современная классификация включает четыре группы - альфа-, бета-, гамма- и дельта-коронавирусы [Helena Jane Maier, Erica Bickerton, Paul Britton / Coronaviruses: An Overview of Their Replication and Pathogenesis // Coronaviruses. – 2015; 1282; 1-23]. Род Betacoronavirus считается наиболее опасным для человека. У людей коронавирусы могут вызывать целый ряд заболеваний - от легких форм острой респираторной инфекции до тяжелого острого респираторного синдрома. В частности, три коронавируса из известных β-коронавирусов - SARS-CoV-1, MERS-CoV и SARS-CoV-2 (источник пандемии COVID-19), вызывают более тяжелые симптомы и приводят к более высокому уровню смертности. При этом отсутствуют специфические высокоэффективные терапевтические средства, проявляющие высокую биологическую активность в ингибировании репродукции бета-коронавируса.
Во всем мире ведется интенсивный поиск терапевтических средств для профилактики и лечения COVID-19. В этой связи Всемирная организация здравоохранения и национальные министерства здравоохранения выпускают постоянно обновляющиеся методические рекомендации по профилактике, диагностике и лечению коронавирусной инфекции с использованием существующих терапевтических средств, ранее предназначенных для других вирусных инфекций (грипп, СПИД, Эбола и др.).
Наиболее широкое распространение при лечении COVID-19 на сегодняшний день получил препарат Фавипиравир (ФВП) [https://static-0.rosminzdrav.ru/system/attachments/attaches/000/050/584/original/03062020_%D0%9CR_COVID-19_v7.pdf], где в качестве действующего вещества использован 6-фторо-3-гидрокси-2-пиразинкарбоксамид, а также Ремдесивир (РМД) [https://nypost.com/2020/05/01/fda-approves-remdesivir-as-emergeney-coronavirus-treatment/], и Апротинин (АПР) [A. Azimi.TMPRSS2 inhibitors, Bromhexine, Aprotinin, Camostat and Nafamostat as potential treatments for COVID-19, 10.31226/osf.io/a3rvm].
Известно применение даларгина (диацетата гексапептида) для лечения коронавирусной инфекции COVID-19 [RU2728939, 2020], который входит в состав коммерчески доступных средств, используемых ранее для лечения язвенной болезни желудка и двенадцатиперстной кишки, панкреатита и панкреонекроза. Препарат может быть использован в форме ингаляций или инъекций, при этом лекарственное средство содержит даларгин в количестве от 0,1 до 50 мг в одной дозе.
Известно применение статинов, используемых для снижения и контроля уровня холестерина, снижения смертности, вероятности развития и тяжести сердечно-сосудистых катастроф - таких, как инфаркт миокарда и инсульт, в терапии коронавирусной инфекции [Патент CN111603465, 2020; CN111632053, 2020; CN111588720, 2020; CN111617065, 2020; CN111588719, 2020]. Лекарственное средство находится в форме геля, мягкой капсулы, перорального препарата, инъекции, лиофилизированного порошка для инъекций или раствора для инфузии.
Однако известные до сих пор соединения и средства, предлагаемые для лечения COVID-19, имеют несколько недостатков, которые препятствуют их широкому применению. Так, Фавипиравир показал в экспериментах на культурах клеток Vero E6 почти полную неспособность подавлять репликацию коронавируса SARS-CoV-2, так как его эффективная концентрация 50% подавления репликации этого коронавируса равна всего лишь EC50 = 61.88 μM, а индекс селективности (отношение цитотоксичности к EC50) весьма невысок SI > 6.46 [M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong, G. Xiao / Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro // Cell Res. – 2020; 30; 269–271. https://doi.org/10.1038/s41422-020-0282-0]; кроме того, фавипиравир показал низкую эффективность лечения в клинических испытаниях, и для него нет пока надежных доказательств отсутствия побочных эффектов [Annoor Awadasseid, Yanling Wu, Yoshimasa Tanaka, Wen Zhang / Effective drugs used to combat SARS-CoV-2 infection and the current status of vaccines // Biomedicine & Pharmacotherapy. – 2021; 137; 111330; https://doi.org/10.1016/j.biopha.2021.111330]. Ремдесивир для повышения терапевтической эффективности необходимо вводить в виде внутривенных инфузий в диапазоне ежедневной дозы 100 - 200 мг в течение не менее 6 суток, что приводит к повышению аспартатаминотрансферазы и аланинтрансаминазы. Повышение уровня ферментов печени может быть признаком воспаления или повреждения клеток печени, низкий уровень альбумина, низкий уровень калия, низкое количество эритроцитов, низкое количество тромбоцитов, способствующих свертыванию, и пожелтение кожи. То есть Ремдесевир обладает достаточно высокой цитотоксичностью, недостаточно эффективно подавляет репликацию коронавируса SARS-CoV-2, обладает низким индексом селективности.
Известен ряд публикаций, в которых представлены данные о SARS – CoV, касающиеся пространственной атомистической модели его основной протеазы [Anand, K., Ziebuhr, J., Wadhwani, P., et al. / Coronavirus main proteinase (3CLpro) structure: basis for design of anti–SARS drugs // Science (New York, N.Y.). – 2003; 300 (5626); 1763–1767], экспериментальной кристаллической структуры [Yang, H., Yang, M., Ding, Y., et al. / The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor // Proceedings of the National Academy of Sciences of the United States of America. – 2003; 100 (23); 13190–13195], и ключевой роли Mpro в репликации SARS – CoV и MERS – CoV [Anand, K., Yang, H., Bartlam, M., et al. / Coronavirus main proteinase: target for antiviral drug therapy BT – Coronaviruses with Special Emphasis on First Insights Concerning SARS, Birkh¨auser Basel. – 2005. pp. 173–199]. В 2003–2015 гг. были найдены обратимые и необратимые ингибиторы основной протеазы SARS – CoV [Pillaiyar, T., Manickam, M., Namasivayam, V., et al. / An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS–CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy // Journal of Medicinal Chemistry. – 2016; 59 (14); 6595–6628], однако большинство из них были слабыми ингибиторами, и ни один из них не прошел клинических испытаний. Основная протеаза и несколько других белков SARS-CoV-2 были идентифицированы в качестве терапевтической мишени для препаратов против COVID – 19. Высококачественные 3D-структуры SARS – CoV – 2 Mpro в форме апо с различными ингибиторами депонированы в Protein Data Bank [Berman, H.M., Westbrook, J., Feng, Z., et al. / The Protein Data Bank // Nucleic Acids Research. – 2000; 28 (1); 235–242]. Выявлено, что SARS-CoV-2 Mpro имеет каталитическую диаду цистеин-гистидин: Cys145 и His41. Данные сведения были использованы при поиске соединений среди известных, в качестве основных ингибиторов протеазы SARS, которые можно было бы отнести к группе соединений-кандидатов, обладающих потенциальной противовирусной биологической активностью в отношении бета-коронавирусов для дальнейшей экспериментальной проверки.
Недостатком всех вышеперечисленных соединений является их низкая эффективность связывания с указанной мишенью, быстрое снижение концентрации с периодом полувыведения около 1 часа и, как следствие, необходимость использования доставляемых препаратов в более высоких концентрациях, что может вызывать нежелательные реакции у пациентов, перечисленные ранее.
Заявляемое изобретение основано на использовании известного соединения - производного 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тиона, для лечения заболеваний, вызванных бета-коронавирусами, в т.ч. SARS-CoV-2, которое продемонстрировало ингибирование репликации бета-коронавирусов, и может быть рекомендовано для использования в терапии COVID – 19.
Из уровня техники известны соединения, представляющие собой производные 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тиона, и способы их получения, основанные на участии различных реакционных центров, имеющихся в молекуле 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тиона, описанные ниже.
Впервые синтез производных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов был предложен при помощи нагревания 1-R-6-R’-2,2,4-триметил-1,2-дигидрохинолинов с 3-5 кратным избытком серы в N,N-диметилформамиде с образованием соответствующих 5-R-8-R’-4,5-дигидро-4,4-диметил-2,3-дитиоло[3,4-c]хинолин-1-тионов с выходом 15-50% [Brown, J. P. // J. Chem. Soc. C. – 1968; 1074. Патент FR1429915 (A), 1966]. Главным недостатком данного способа является достаточно низкий выход целевых продуктов. Далее было установлено, что увеличение выхода продуктов реакции до 45-55%, можно добиться заменой дигидрохинолинов, указанных ранее, на аналогичные 2,2,4-триметил-1,2,3,4-тетрагирохинолины и проведением взаимодействия с серой при нагревании в трихлорбензоле [Shikhaliev Kh. S., Kasaikina O.T., Shmyreva Zh. V. // Bulletin of the Academy of Sciences of the USSR Division of Chemical Science. – 1989; 38 (1.2); 176 - 178].
Получение производных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов было предложено реализовывать двухстадийно через образование йодида S-метилпроизводного с последующим взаимодействием с п-фенетидином при кипячении в этаноле около 6 часов [Brown, J. P. // J. Chem. Soc. C. – 1968; 1074-1075]. Далее данная реакция была распространена на ряд первичных аминов: анилин, этиламин, циклогексиламин [Патент GB1174830 (A), 1969]. В результате указанного взаимодействия были получены 1,2-дитиол-3-имины. Основной недостаток указанных способов заключается в использовании очень ограниченного числа аминов для синтеза производных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов. В работе [Medvedeva S.M., Zubkov F.I., Yankina K.Yu., Grudinin D.G. / Reaction of substituted 1-methylthio-4,5-dihydro[1,2]dithiolo[3,4-c]-quinolin iodides with arylamines. Synthesis of novel 1,2-dithiolo[3,4-c]-quinolin-1-ylidene(aryl)amines and 10-(arylimino)-7,10-dihydro[1,2]dithiolo[3,4-c]-pyrrolo[3,2,1-ij]quinoline-4,5-diones // Arkivoc. – 2017; III; 269-278] представлены возможности синтеза (8-R-7-R’-4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-c]хинолин-1-илиден)(4(2)-R”-фенил)аминов с выходом 69-81% в результате взаимодействия с различными ариламинами при кипячении в смеси изопропилового спирта и пиридина в течение 2-3 часов.
При N-ацилировании 4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тионов ангидридами или хлорангидридами акриловой, бензойной и уксусной кислот при кипячении в абсолютированном толуоле в течение 6 часов происходит образование 5-R-4,5-дигидро-4,4-диметил-2,3-дитиоло-[3,4-c]хинолин-1-тионов. Проведение N-ацилирования в ранее указанных условиях возможно и при использовании в качестве ацилирующего агента 4-хлор-3-нитробензоилхлорида или никотиноилхлорида [Шихалиев, Х.С., Шмырева Ж.В., Залукаев Л.П. / Ацилирование 4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тиона // Журн. орган. химии. – 1988; 24 (1); 232-233]. В данной работе показано, что аналогичные структуры можно получать не только за счет модификации 4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тиона, а на этапе формирования гетероциклической матрицы за счет реакции замещенных хинолинов, например, 1-(2,2,4-триметилхинолин-1(2Н)-ил)этан-1-она или 1-бензоил-2,2,4-триметил-1,2-дигидрохинолина при кипячении с серой в среде N,N-диметилформамида в течение 11 часов. Главный недостаток данного способа – использование ограниченного числа ацилирующих агентов и высокая температура проведения процесса. N-Ацилирование 8-R-4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тиона можно осуществить хлорацетилхлоридом в среде толуола с выходом продукта 65% [Х.С.Шихалиев, С.М.Медведева, В.В.Пигарев, А.С.Соловьев, Г.В.Шаталов / Новые гетероциклические соединения на основе 8-R-4,4-диметил-2,3-дитиоло[5,4-c]хинолин-1-тионов // Журн. общ. химии. – 2000; 70 (3); 484-486]. Для полученного 8-R-N-хлорацетил-4,4-диметил-2,3-дитиоло[5,4-c]-хинолин-1-тиона указана возможность проведения последующей реакции замещения при взаимодействии с гетероциклическими меркаптанами, в том числе 1,3-бензоксазол-2-тиолом, 1,3-бензотиазол-2-тиолом, 4,5-дигидротиазол-2-тиолом, 2-меркапто-4-фенилтиазол-5(4Н)-тионом в присутствии карбоната калия в среде 1,4-диоксана при 40-50 °С [Шихалиев Х.С., Пигарев В.В., Соловьев А.С., Шаталов Г.В. / Синтез N-(2-гетарилацетил)-8-R-4,5-дигидро-4,4-диметил-2,3-дитиоло[5,4-с]хинолин-1-тионов // Изв. вузов.Химия и хим. технология. – 2000; 43 (2); 93-94]. К недостаткам данного способа можно отнести ограниченный ряд получаемых производных.
В качестве еще одного реакционного центра в 4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тионах выступает бензольное кольцо, где можно реализовать реакцию нитрования смесью азотной (ρ= 1.4 г/см3) и уксусной кислот, что приводит к образованию 4,4-диметил-8-нитро-4,5-дигидро-[1,2]дитиоло[3,4-с]хинолин-1-тионов [Kasaikina O.T., Golovina N.A., Shikhaliev Kh.S., Smyreva Zh.V. / Kinetic description of the oxidation of hydrocarbons inhibited by sulfur-containing hydrogenated quinolines // Russian Chemical Bulletin. – 1994; 43 (5); 755 – 759. О.Т. Касаикина, Н.А. Головина, Ж.В. Шмырева, Х.С. Шихалиев / Феноменология ингибирования окисления углеводородов серосодержащими гидрированными хинолинами // Изв. АН СССР. Сер. хим. – 1994; 5; 814-818]. Там же указана возможность осуществления алкилирования по бензольному кольцу 4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тиона трифенилметанолом с образованием 4,4-диметил-8-тритил-4,5-дигидро-[1,2]дитиоло[3,4-с]хинолин-1-тиона. Главный недостаток данного способа – использование ограниченного числа ацилирующих и алкилирующих агентов и проведение процесса при высоких температурах в течение длительного времени. Нитрование в бензольное кольцо можно реализовать взаимодействием с ацетилнитратом в хлороформе с образованием 6-нитро-[1,2]дитиоло[3,4-с]хинолин-1-тиона. [Шихалиев Х.С., Пигарев В.В., Шмырева Ж.В., Медведева С.М., Ермолова Г.И. / Нитрование 4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тиона и его производных // Известия ВУЗов. Химия и химическая технология. – 1998; 41; 48-51].
Показано, что 8-алкил-4,4-диметил-4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионы, N-ацил-[1,2]дитиоло[3,4-c]хинолин-1-тионы, N-гетарил(тио)ацетил-[1,2]дитиоло[3,4-c]хинолин-1-тионы обладают антимикробной и антигрибковой активностью. Среди протестированных бактерий наиболее чувствительными оказались En.cloacae, за которыми следовали S. aureus, а наиболее устойчивыми оказались L.monocytogenes. Среди грибов наиболее чувствительным оказался T. viride, а наиболее устойчивым - Penicillium v.c. [Kartsev V., Shikhaliev Kh. S., Geronikaki A., Medvedeva S. M., Ledenyova I. V., Krysin M. Yu., Petrou A., Ciric A., Glamoclija J., Sokovic M. / Appendix A. dithioloquinolinethiones as new potential multitargeted antibacterial and antifungal agents: Synthesis, biological evaluation and molecular docking studies // European Journal of Medicinal Chemistry. – 2019; 175; 201-214].
Известны различные способы получения производных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов, основанные на замещении экзо-атома серы или N-ацилировании. Одним из способов получения искомых соединений является синтез N-ацильных производных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов на основе взаимодействия дитиолохинолинтионов с карбонилхлоридами, включая хлорацетилхлорид, в толуоле с возможностью последующего взаимодействия с сукцинимидом или некоторыми другими гетероциклическими меркаптанами с образованием производных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов [Kartsev V., Shikhaliev Kh. S., Geronikaki A., Medvedeva S. M., Ledenyova I. V., Krysin M. Yu., Petrou A., Ciric A., Glamoclija J., Sokovic M. / Appendix A. dithioloquinolinethiones as new potential multitargeted antibacterial and antifungal agents: Synthesis, biological evaluation and molecular docking studies // European Journal of Medicinal Chemistry. – 2019; 175; 201-214]. К недостаткам данного способа можно отнести использование абсолютированного толуола и диоксана в качестве среды для проведения указанных взаимодействий. Замена растворителя и добавление каталитических количеств йодида калия позволяет сократить время проведения реакции и увеличить выходы целевых продуктов, как это было достигнуто в данном изобретении.
Наиболее близким к заявляемому способу является способ получения, основанный на взаимодействии соли 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов (метилтиодитиолия) с первичными аминами, такими как анилин, этиламин, циклогексиламин, п-фенетидин при кипячении в этаноле около 6 часов с образованием 1,2-дитиол-3-иминов [Патент GB1174830 (A), 1969]. По предлагаемому изобретению время проведения этой стадии процесса сокращается до 2-3 ч при использовании бутанола в качестве растворителя и проведения реакции в присутствии триэтиламина. Более широкий спектр производных получают за счет дальнейшей модификации солей 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов (метилтиодитиолия).
Технической проблемой, решаемой заявляемым изобретением, является разработка способа получения соединений для лечения заболеваний, вызванных бета-коронавирусом, включая SARS-CoV-2, вызвавшего пандемию COVID-19, характеризующихся высокой эффективностью за счет подавления репликации коронавируса, и обладающих низкой токсичностью.
Раскрытие изобретения
Техническим результатом изобретения является сокращение времени проведения процесса до 2-3 ч. и значительное расширение спектра используемых для синтеза аминов, а также проведение дальнейшей модификации для получения новых производных с улучшенным биологическим действием.
Полученные заявляемым способом производные 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тиона для подавления репликации бета-коронавирусов, включая SARS-CoV-2, характеризуются высокой эффективностью подавления репликации коронавируса в культуре клеток Vero E6 при низких значениях эффективной концентрации EC50 (концентрация соединения, которая ингибирует репликацию вируса на 50% и более по сравнению с контролем)  0.51 μM, низкой цитотоксичностью и высоким значением индекса селективности SI > 400. Данное соединение обладает низкой токсичностью, о чем свидетельствует отсутствие активности на клеточной линии здоровых клеток легочного эпителия человека, подтвержденное примерами.
0.51 μM, низкой цитотоксичностью и высоким значением индекса селективности SI > 400. Данное соединение обладает низкой токсичностью, о чем свидетельствует отсутствие активности на клеточной линии здоровых клеток легочного эпителия человека, подтвержденное примерами.
Технический результат достигается способом получения трициклического серусодержащего производного 1,2-дигидрохинолина общей формулы I для подавления репликации бета-коронавируса, включая SARS-CoV-2 :
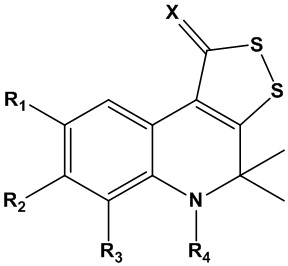 (I)
(I)
где R1, R2, R3 представляют собой заместитель, выбранный из водорода; алкила (С1-С10) неразветвленного или разветвленного строения; алкоксигруппы (С1-С10) неразветвленного или разветвленного строения; арила без заместителей или замещенного по положениям 2,3,4,5,6; гидроксила; аминогруппы; алкиламиногруппы С1-С10 неразветвленного или разветвленного строения; нитрогруппы; карбоксильной группы; алкилкарбоксильной группы С1-С10, неразветвленного или разветвленного строения; незамещенной или замещенной по 2,3,4,5,6 положениям арилкарбоксильной группы; гетарилкарбоксильной группы, где гетарил представляет собой азот- и/или серу и/или кислородсодержащий пяти- или шестичленный гетероцикл,
R4 - заместитель, выбранный из водорода, С1-С10 алкила неразветвленного или разветвленного строения; незамещенного или замещенного по положениям 2,3,4,5,6 арил-CH2-;
или R4 – заместитель COR5, где R5 представляет собой С1-С10 алкил неразветвленного, разветвленного или циклического строения без заместителей, или содержащий заместители; замещенный по положениям 2,3,4,5,6 или незамещенный арила; азот- и/или серу и/или кислородсодержащий пяти- или шестичленный гетероцикл; замещенный по положениям 2,3,4,5,6 или незамещенный ариламин;
или R4 – заместитель СOCH2R6, где R6 представляет собой замещенную по положениям 2,3,4,5,6 или незамещенную арилоксигруппу; алкиламиногруппу С1-С10 неразветвленного, разветвленного или циклического строения; замещенную по положениям 2,3,4,5,6 или незамещенную ариламиногруппу; гетариламиногруппу, где гетарил представляет собой азот- и/или серу и/или кислородсодержащий пяти- или шестичленный гетероцикл;
или R4 – заместитель COCH2SR7, где R7 представляет собой представляет собой С1-С10 алкил неразветвленного, разветвленного или циклического строения без заместителей, или содержащий заместители; арил без заместителей или замещенный по положениям 2,3,4,5,6; азот- и/или серу и/или кислородсодержащий пяти- или шестичленный гетероцикл;
Х представляет собой заместитель, выбранный из серы, незамещенного или замещенного по положениям 2,3,4,5,6 арилимина.
заключающийся в модификации трициклического серусодержащего производного 1,2-дигидрохинолина предварительным алкилированием йодистым метилом с последующим замещением серы под действием ариламинов и/или алкилированием или ацилированием по атому азота. При этом синтез соединений I при X=S осуществляют реакцией N-ацилирования хлорангидридами или ангидридами карбоновых кислот исходных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов. В качестве хлорангидрида используют фуран-2-карбонилхлорид, 2-(нафтален-2-илокси)ацетилхлорид, 3-фенилпропаноилхлорид, 2-(4-хлорфенокси)ацетилхлорид, а реакцию N-ацилирования хлорангидридами карбоновых кислот проводят в смеси толуола и пиридина с последующим удалением соли пиридиния и выделением продукта реакции кристаллизацией из фильтрата. Для приготовления смеси толуола и пиридина используют абсолютированные толуол и пиридин, взятые в объемном соотношении на 10 частей абсолютированного толуола берут от 0,8 до 1,2 частей пиридина. Реакцию N-ацилирования ангидридами карбоновых кислот проводят в абсолютированном диоксане с последующим выделением продукта реакции и перекристаллизацией его из толуола, при этом при использовании в реакции N-ацилирования ангидрида 2-хлоруксусной кислоты проводят дальнейшее алкилирование меркаптанов в результате их взаимодействия с полученным хлорацетамидом 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в абсолютированном ацетонитриле с добавлением от 1,8 до 2,2 эквимоль карбоната калия, и каталитических количеств йодида калия с последующим выделением продукта реакции и перекристаллизацией его из толуола. При использовании в реакции N-ацилирования ангидрида 2-хлоруксусной кислоты, проводят алкилирование гетероциклических аминов в результате их взаимодействия с полученным хлорацетамидом 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в абсолютированном ацетонитриле с добавлением каталитических количеств йодида калия с последующим выделением продукта реакции и перекристаллизацией его из толуола.
Предпочтительно когда R1 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этоксигруппы; R2 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этоксигруппы; R3 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этокси-группы; R4 представляет собой заместитель, выбранный из метил-, бензил-, 3-фенилакрилоил-, 3-(4-метоксифенил)акрилоил-, 3-фенилпропаноил-, фенилэтила, 2-(4-фторфенокси)ацетил-, 4-хлорфеноксиацетил-, 2-(2,4-дихлорфенокси)ацетил-, 4-нитробензоил-, 2-(1,3-диоксоизоиндолин-2-ил)-3-фенилпропаноил-) группы; 3,4-дихлорфенилкарбамоил-, фуран-2-карбонил-, этилкарбамоил-, 2-нафтилилоксиацетил-, 2-(1,3-диоксоизоиндолин-2-ил)ацетил-, 2-(1,3-диоксогексагидро-1H-изоиндол-2(3H)-ил)ацетил-, 2-морфолиноацетил-, пиперидин-1-илацетил-, 2-(4-карбамоилпиперидин-1-ил)ацетил-, 2-(2,5-диоксопирролидин-1-ил)ацетил-,2-феноксиацетил-,2-1,3-диоксо-1H-бензо[de]изохинолин-2(3H)-ил)ацетил-, 2-бензо[d]оксазол-2-илтио)ацетил-, 2-(бензо[d]тиазол-2-илокси)ацетил-, бензоксазол-2-илмеркаптоацетил-, фураноил-, 2-нафлинилиоксиметилкарбонил, 3,4-дихлорфенилкарбамоил;
Х представляет собой заместитель, выбранный из 4-(4-хлорбензилокси)фенилимино-, 4-изопропилфенилимино-, 4-(6-метилбензо[d]тиазол-2-ил)фенилимино-, 4-(N-бензоилсульфамоил)фенилимино-, 4-этоксифенилимино-, 4-этоксикарбонилфенилимино-, фенилимино-, 4-хлорфенилимино-, 3-хлорфенилимино-, 4-сульфамоилфенилимино-, 4-(N-ацетилсульфамоил)фенилимино-, 4-(N-(тиазол-2-ил)сульфамоил)фенилимино-, 2-никотиноилгидразоно-, 4-((4-хлорбензил)окси)фенилимино-, 2-метоксифенилимино-, 4-(N-(пиримидин-2-ил)сульфамоил)фенилимино-, 4-(N-(4,6-диметилпиримидин-2-ил)сульфамоил)фенилимино-.
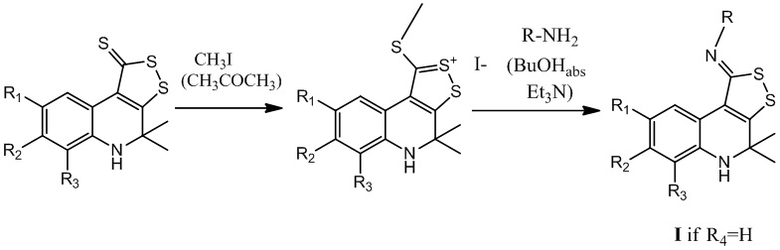
Получение соединения формулы I при R4=H осуществляют путем синтеза иминов 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов через образование соли метилтиодитиолия за счет взаимодействия 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона с йодистым метилом с последующим замещением серы в результате реакции с первичными аминами в присутствии избытка триэтиламина (1,8 – 2,2 эквимоль), преимущественно 2 эквимоль, при кипячении в абсолютированном бутаноле с последующим выделением искомого продукта реакции и перекристаллизацией его из изопропилового спирта.
Соединение формулы I при X=S получают путем синтеза производных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов через последовательное проведение замещения и N-ацилирование.
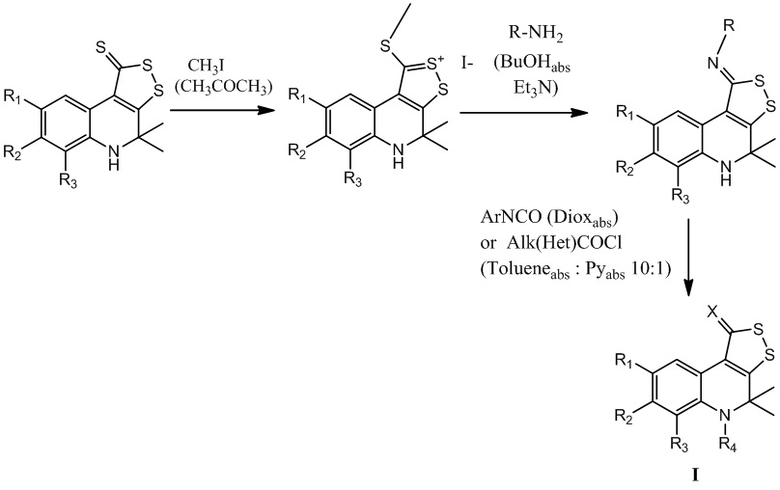
Заявляемый способ получения соединения формулы (I), заключающемуся во взаимодействии соли 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов (метилтиодитиолия) с первичными аминами в среде бутанола, в присутствии триэтиламина, позволяет сократить время проведения процесса до 2-3 ч. и значительно расширить спектр используемых аминов, а также в проведении дальнейшей модификации для получения новых производных с улучшенным биологическим действием.
Полученные заявляемым способом трициклическое серусодержащее производное 1,2-дигидрохинолина общей формулы I может быть использовано для лечения заболеваний, вызванных бета-коронавирусами, включая SARS-CoV-2, а также в комплексной терапии заболеваний, вызванных бета-коронавирусами.
Применение полученного заявляемым способом трициклического серусодержащего производного 1,2-дигидрохинолина общей формулы I для лечения заболеваний, вызванных бета-коронавирусами предпочтительно в виде фармацевтической композиция, включающей терапевтически эффективное количество соединения формулы (I) и фармацевтически приемлемых добавок. При этом фармацевтическая композиция может быть представлена в виде единичной дозированной формы или в виде двух или более отдельных готовых фармацевтических форм для последовательного или одновременного введения.
Соединение формулы I может быть использовано, как в чистом виде, так и в качестве активнодействующего компонента новых лекарственных форм против коронавирусов.
Полученные заявляемым способом соединения используют для лечения заболеваний, вызванных бета-коронавирусом, включая SARS-CoV-2, заключающийся во введении фармацевтической композиции в терапевтически эффективном количестве.
Изобретение расширяет арсенал средств для лечения и профилактики заболеваний, вызванных коронавирусами.
Согласно данному изобретению производное 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тиона может быть использовано для подавления репликации бета- коронавируса, включая SARS-CoV-2, следовательно, для лечения заболеваний, вызванных бета-коронавирусами.
Краткое описание чертежей
На фиг. 1 представлены график эффективности ингибирования репликации коронавируса SARS-CoV-2 в культуре клеток Vero E6 и цитотоксичности для соединения 2-(нафтален-2-илокси)-1-(4,4,7,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанон (Id) и препарата сравнения Ремдесивир. Значения EC50 составляют 0,51 μM и 3,76 μM для соединения 2-(нафтален-2-илокси)-1-(4,4,7,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанон (Id) и препарата сравнения Ремдесивир, соответственно.
Осуществление изобретения
Ниже приведены определения терминов, которые используются в описании настоящего изобретения.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции), в виде таблеток капсул инъекций, мазей и др. готовых форм предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных средств, средств доставки, консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, внутримышечного, внутривенного, подкожного, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные формы введения.
Термин «терапевтически эффективное количество» означает количество действующего вещества, которое (1) лечит или предупреждает конкретное заболевание, состояние или расстройство, (2) ослабляет, улучшает или устраняет один или более симптомов конкретного заболевания, состояния или расстройства, или (3) предупреждает или задерживает наступление одного или более симптомов конкретного заболевания, состояния или расстройства, изложенного в данном описании. Термин «терапевтически эффективное количество» означает такое количество соединения формулы (I), которое достаточно для того, чтобы обеспечить желаемый терапевтический эффект. При этом суточная доза у взрослых обычно составляет 1,0 ~ 500 мг, предпочтительно - 10 ~ 300 мг. Поэтому во время приготовления из фармацевтической композиции лекарственного средства по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку, при этом каждая единичная дозировка препарата должна содержать 10 ~ 500 мг соединения общей формулы I предпочтительно - 50 ~ 300 мг. В предпочтительном варианте, терапевтически эффективное количество составляет от 0,01 до 1 мг/кг веса тела субъекта. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно – от одного до шести раз). При этом, дозировка средства, содержащего соединение общей формулы (I), у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активного ингредиента в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента.
Лекарственные средства могут вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно).
EC50, или концентрация полумаксимального ингибирования - показатель эффективности лиганда при ингибирующем биохимическом или биологическом взаимодействии. EC50 является количественным индикатором, который показывает, сколько нужно лиганда-ингибитора для ингибирования биологического процесса на 50%, в том числе подавления репликации вируса в культуре клеток.
IC50 – концентрация полумаксимального ингибирования белка-мишени в экспериментах с флюорогенным субстратом в тестовой системе белок-субстрат-ингибитор.
ТC50 или CC50 – концентрация препарата, при которой наблюдается 50% гибель исследуемой клеточной культуры.
SI – индекс селективности, отношение токсичности соединения и ингибирующей активности против бета-коронавируса (TC50 / IC50).
Каталитические количества катализатора – минимальное количество вещества, обеспечивающее проведение реакции с учетом различных технологических факторов проведения реакции, прежде всего зависимости температуры/продолжительности реакции (чем больше катализатора, тем быстрее идет реакция, чем выше температура – ускорение протекания реакции, катализатора можно брать меньше).
Биологическая активность соединения формулы (I), заключающаяся в ингибировании репродукции бета-коронавируса, была предварительно выявлена при помощи метода молекулярного моделирования. На основании докинга были выявлены производные 4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тионов с лучшими параметрами, которые в последующем нашли экспериментальные подтверждения их ингибирующей активности. Молекулярный докинг был реализован с использованием разработанной в МГУ имени М.В.Ломоносова программы SOL [Романов А.Н., Кондакова О.А., Григорьев Ф.В., Сулимов А.В., Лущекина С.В., Мартынов Я.Б., Сулимов В.Б. / Компьютерная разработка лекарств: программа докинга SOL // Вычислительные методы и программирование. – 2008; 9 (2); 64-84; Sulimov V.B., Ilin I.S., Kutov D.C., Sulimov A.V. / Development of docking programs for Lomonosov supercomputer // Journal of the Turkish Chemical Society Section A: Chemistry. – 2020; 7 (1); 259-276], определяющей энергию связывания белок-лиганд, и получившей международное признание среди аналогичных программных продуктов [Alexey V. Sulimov, Danil C. Kutov, Igor V. Oferkin, Ekaterina V. Katkova, and Vladimir B. Sulimov / Application of the Docking Program SOL for CSAR Benchmark // J. Chem. Inf. Model. – 2013; 53; 1946−1956]. Выявленная группа соединений затем была дополнительно проверена с помощью квантово-химического полуэмпирического метода PM7 [Sulimov A.V., Kutov D.C., Taschilova A.S., Ilin I.S., Stolpovskaya N.V., Shikhaliev Kh S., Sulimov V.B. / In search of non-covalent inhibitors of SARS-CoV-2 main protease: Computer aided drug design using docking and quantum chemistry // Supercomputing Frontiers and Innovations. – 2020; 7 (3); 41-56] с континуальной моделью растворителя: дополнительная локальная оптимизация из найденного при докинге положения лиганда в белке и вычисление энтальпии связывания белок-лиганд. Для эксперимента были отобраны соединения, молекулы которых по данной мишени имели достаточно отрицательный скор докинга и достаточно отрицательную энтальпию связывания белок-лиганд. Именно среди этих соединений, отобранных на основании их высокой энергии связывания с главной протеазой коронавируса SARS-CoV-2, были обнаружены соединения эффективно подавляющие репликацию этого коронавируса в культуре клеток Vero E6. Очень близкий аналог соединения 2-(нафтален-2-илокси)-1-(4,4,7,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанон (Id), а именно соединение 2-(нафтален-2-илокси)-1-(4,4,7-триметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанон (Ic), не только подавляет репликацию коронавируса SARS-CoV-2 в культуре клеток Vero E6 с эффективной концентрацией EC50 = 1.16 μM, имеет высокий индекс селективности SI > 86, но и ингибирует главную протеазу коронавируса SARS-CoV-2 с эффективной ингибирующей концентрацией IC50 = 2.74 μM.
Отобранные соединения эффективно подавляют репликацию бета-коронавирусов в клеточных культурах.
Далее представлено более подробное описание примеров осуществления данного изобретения с достижением заявленного результата. Приведенные ниже примеры иллюстрируют, но не ограничивают данное изобретение.
Все используемые реагенты являются коммерчески доступными, контроль за ходом реакции осуществляли при помощи тонкослойной хроматографии (ТСХ), и время реакции указано только для иллюстрации; структуру и чистоту всех выделенных соединений подтверждали, по меньшей мере, одним из следующих методов: ТСХ (пластины для ТСХ с предварительно нанесенным силикагелем 60 F254 Merck), масс-спектрометрия или ядерный магнитный резонанс (NMR). Выход продукта приведен только для иллюстрации. 1H NMR спектры были зарегистрированы на спектрометре Agilent MR 400+ (на рабочих частотах 400 и 100 MHz, соответственно) при нормальных условиях в растворах DMSO-D6 если не указано иное, относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, миллионных долях (м.д.). Хроматографический анализ проводился на приборе Agilent Technologies 1260 infinity с масс-детектором Agilent 6230 TOF LC/MS (времяпролетный детектор масс высокого разрешения), метод ионизации – двойное электрораспыление (dual-ESI). Запись и регистрация сигналов проводилась в положительной полярности; небулайзер (N2) 20 psig, газ-осушитель (N2) 6 мл/мин, 325 °C; диапазон обнаружения масс составляет 50-2000 Дальтон. Напряжение на капилляре 4.0 кВ, фрагментаторе +191 В, скиммере +66 В, OctRF 750 В. Условия хроматографирования: колонка Poroshell 120 EC-C18 (4.6 x 50 мм; 2.7 мкм). Градиентное элюирование: ацетонитрил/вода (0,1 % муравьиной кислоты); скорость потока 0.4 мл/мин. Программное обеспечение для обработки результатов исследований – MassHunter Workstation / Data Acquisition V.06.00. Температуры плавления определены на аппарате Stuart SMP30.
Все процедуры, если не оговорено отдельно проводили при комнатной температуре или температуре окружающей среды, т.е. в диапазоне 20-25°С.
Высушивание продуктов до постоянного веса проводили при температуре 35-45°С при атмосферном давлении или с использованием вакуум-сушильного шкафа при остаточном давлении 0,35±0,005 кг/см2 (35±5 кПа).
Для промывания осадков/фильтрата использовали дистиллированную воду, если не оговорено особо.
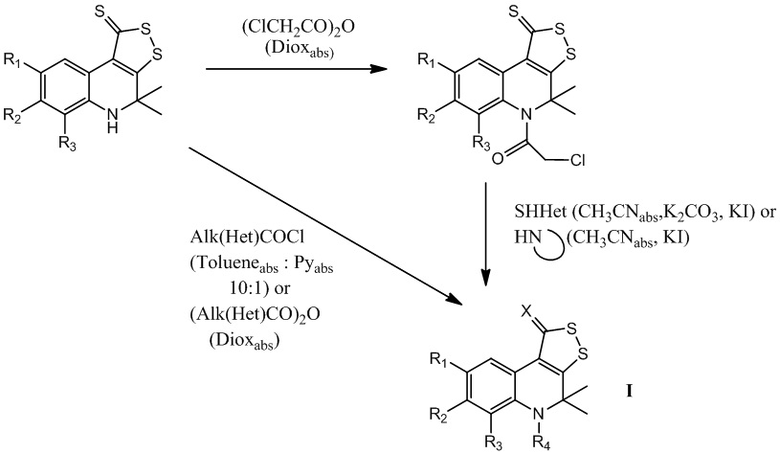
Целевые серусодержащие производные 1,2-дигидрохинолина I получены многостадийным синтезом, включающим получение исходных дигидрохинолинов с последующей их гетероциклизацией с молекулярной серой с образованием 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов и дальнейшим проведением реакций их структурной модификации по трем путям (А, В, С), представленным на схеме 1.
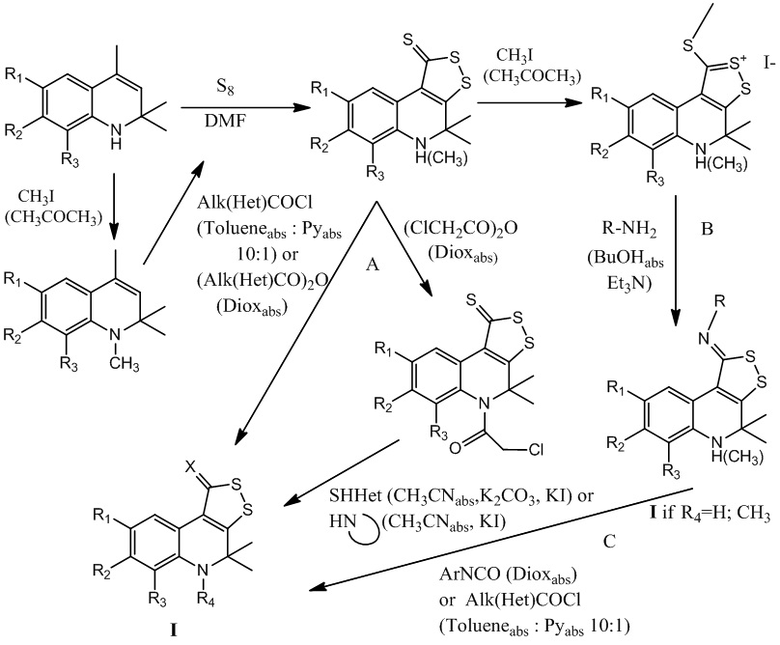
Схема 1
Синтез исходных 4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тионов осуществляют в две стадии из соответствующих анилинов по известным методикам [Brown, J. P. J. Chem. Soc. C, 1968, 1074-1075; Reddelin С., Thurm A. // Ber., 1932, 65, 1511-1521; Шихалиев, Х.С., Шмырева Ж.В., Залукаев Л.П. / Ацилирование 4,5-дигидро-4,4-диметил-5Н-2,3-дитиоло[5,4-c]хинолин-1-тиона // Журн. орган. химии. – 1988; 24 (1); 232-233; Шихалиев Х.С., Пигарев В.В., Соловьев А.С., Шаталов Г.В. / Синтез N-(2-гетарилацетил)-8-R-4,5-дигидро-4,4-диметил-2,3-дитиоло[5,4-с]хинолин-1-тионов // Изв. вузов.Химия и хим. технология. – 2000; 43 (2); .93-94; Medvedeva S.M.; Leshcheva E.V.; Shikhaliev Kh.S. Solov'ev A.S. / Novel heterocyclic systems based on 8-R-4,5-dihydro-4,4-dimethyl[1,2]dithiolo[3,4-c]quinoline-1-thiones // Chem Heterocycl Compd. – 2006; 42; 534-539; Kartsev V., Shikhaliev Kh. S., Geronikaki A., Medvedeva S. M., Ledenyova I. V., Krysin M. Yu., Petrou A., Ciric A., Glamoclija J., Sokovic M. / Appendix A. dithioloquinolinethiones as new potential multitargeted antibacterial and antifungal agents: Synthesis, biological evaluation and molecular docking studies // European Journal of Medicinal Chemistry. – 2019; 175; 201-214]. При выполнении данных стадий синтеза (получение 2,2,4-триметил-1,2-дигидрохинолинов и 4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тионов) могут быть использованы любые известные из уровня техники способы и средства.
Путь А : получение трициклического серусодержащего производного 1,2-дигидрохинолина на основе реакций N-ацилирования хлорангидридами или ангидридами карбоновых кислот с образованием конечных соединений I при X=S.
Стадия 1: готовят смесь 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона, содержащего заместители в бензольном кольце, в том числе алкильные группы (С1-С10) неразветвленного или разветвленного строения; алкоксигруппы (С1-С10) неразветвленного или разветвленного строения; арилы без заместителей или замещенного по положениям 2,3,4,5,6; гидроксильную группу; аминогруппу; алкиламиногруппу, где алкил С1-С10 неразветвленного или разветвленного строения; нитрогруппу; карбоксильную группу; алкилкарбоксильную группу, где алкил С1-С10 неразветвленного или разветвленного строения; арилкарбоксильную группу, где арил без заместителей или замещен по положениям 2,3,4,5,6; гетарилкарбоксильную группу, где гетарил азот- и/или серу и/или кислородсодержащий пяти- или шестичленный гетероцикл и соответствующего хлорангидрида карбоновой кислоты, взятых из расчета, что на 0,01 моль 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона берут 0,012 – 0,013 моль галогенангидрида карбоновой кислоты, в смеси абсолютированных толуола и пиридина, взятых в объемном соотношении 10 : 1 в количестве 70-100 мл. Кипятят в течение 3-5 часов, отфильтровывают полученную соль хлорида пиридиния, фильтрат упаривают, выпавший осадок отфильтровывают и перекристаллизовывают из толуола. Выход полученного продукта составляет 70-85%.
В качестве галогенангидрида карбоновой кислоты могут быть использованы хлорангидриды и бромангидриды алифатических кислот, содержащих углеводородные радикалы с разветвленной и неразветвленной цепью, а также циклические углеводородные радикалы, ненасыщенные углеводородные радикалы, в том числе хлорангидрид уксусной, трифторуксусной, пропановой, бутановой, изобутановой, пентановой, 2-метилбутановой, 3-метилбутановой, гексановой, пропеновой, бутеновой, коричной и т.д. кислот, хлорангидриды и бромангидриды ароматических карбоновых кислот, в том числе бензойной, 2- хлорбензойной, 3-хлорбензойной, 4-хлорбензойной, 2,4-дихлорбензойной, 2-бромбензойной, 3-бромбензойной, 4-бромбензойной, 4-йодбензойной, 2-йодбензойной, 3-йодбензойной, 2-фторбензойной, 3-фторбензойной, 4-фторбензойной, 2-метилбензойной, 3-метилбензойной, 4-метилбензойной, 2,4-диметилбензойной, 3,4-диметилбензойной, 2,5-диметилбензойной, 2-метоксибензойной, 3-метоксибензойной, 4-метоксибензойной, 4-нитробензойной, нафталин-2-карбоной, нафталин-3-карбоновой и т.д. кислот, хлорангидриды и бромангидриды гетероциклических карбоновых кислот, в том числе фуран-2-карбоновой, тиофен-2-карбоновой и т.д. кислот.
Или готовят смесь 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и соответствующего ангидрида карбоновой кислоты, в том числе ангидридов алифатических кислот, содержащих углеводородные радикалы с разветвленной и неразветвленной цепью, а также циклические углеводородные радикалы, включая ангидрид 2-хлоруксусной кислоты, взятых из расчета на 0,01 моль 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона берут 0,012 – 0,013 моль ангидрида карбоновой кислоты, кипятят в 70-100 мл абсолютированного диоксана в течение 3-6 часов, выпавший осадок отфильтровывают, промывают водой, сушат и перекристаллизовывают из толуола. Выход полученного продукта составляет 60-85%.
В случае, если в качестве ацилирующего агента выступает ангидрид 2-хлоруксусной кислоты, возможно проведение дальнейшей модификации (стадия 2): алкилирование различных меркаптанов и гетероциклических аминов полученным на первой стадии пути А хлорацетамидом 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона.
Стадия 2: Данная стадия заключается в алкилировании различных гетероциклических аминов или меркаптанов хлорацетамидом 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона, полученным на первой стадии.
В качестве меркаптанов может быть использован бензо[d]оксазол-2-илтиол, бензо[d]тиазол-2-илтиол, пиридин-2-илтиол, пиримидин-2-илтиол, имидазол-2-илтиол, 1,2,4-триазол-2-илтиол, 1,3,4-тиазол-2-илтиол и т.д.
В качестве гетероциклического амина могут быть использованы морфолин, пиперидин, пиперидин-4-карбоксамид, 4-метилпиперидин, пирролидин, 4-метилпиперазин, 3-метилпиперидин-, этил пиперидин-4-карбоксилат, 4-фенилпиперазин, 4-(2-хлорфенил)пиперазин, 4-(4-хлорфенил)пиперазин, 4-(4-метилфенил)пиперазин и т.д.
Готовят смесь хлорацетамида 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и соответствующего меркаптана, взятых из расчета, что на 0,01 моль хлорацетамида 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона берут не менее 0,01 моль меркаптана, полученную смесь растворяют в 70-100 мл абсолютированного ацетонитрила, добавляют не менее 0,02 моль карбоната калия и 0,001 – 0,0015 моль йодида калия, кипятят в течение 3-5 часов, охлаждают, отфильтровывают, выпавший осадок перекристаллизовывают из толуола. Выход полученного продукта составляет 70-80%.
Или готовят смесь хлорацетамида 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и соответствующего гетероциклического амина, взятых из расчета, что на 0,01 моль хлорацетамида 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона берут 0,022 – 0,023 моль гетероциклического амина, полученную смесь растворяют в 70-100 мл абсолютированного ацетонитрила, добавляют 0,001 – 0,0015 моль йодида калия, кипятят в течение 3-5 часов, охлаждают, отфильтровывают, выпавший осадок перекристаллизовывают из толуола. Выход полученного продукта составляет 50-70%.
Путь В: получение трициклического серусодержащего производного 1,2-дигидрохинолина в результате замещения серы при взаимодействии соли метилтиодитиолия с первичными аминами и образованием иминов 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона I при R4=H.
Готовят раствор 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в ацетоне (0,01 моль в 50-100 мл), добавляют йодистый метил, взятый из расчета что на 0,01 моль 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона берут не менее 0,015 моль йодистого метила, кипятят в течение 3-5 часов, охлаждают, отфильтровывают соль метилтиодитиолия. Готовят смесь полученной соли метилтиодитиолия, соответствующего первичного амина и триэтиламина, взятых из расчета что на 0,01 моль соли метилтиодитиолия берут 0,011 – 0,012 моль первичного амина и не менее 0,02 моль триэтиламина, полученную смесь кипятят в абсолютированном бутаноле в течение 2-3 часов, охлаждают, растворитель отгоняют при пониженном давлении, осадок отфильтровывают, перекристаллизовывают из изопропилового спирта. Выход продуктов реакции составляет 60-80%.
В качестве первичных аминов могут быть использованы анилин, 2-метилфениламин, 3-метилфениламин, 4-метилфениламин, 2,4-диметилфениламин, 3,4-диметилфениламин, 2-метоксифениламин, 3-метоксифениламин, 4-метоксифениламин, 2-этоксифениламин, 3-этоксифениламин, 4-этоксифениламин, 2-хлорфениламин, 3-хлорфениламин, 4-хлорфениламин, 2-бромфениламин, 3-бромфениламин, 4-бромфениламин, 2-нитрофениламин, 3-нитрофениламин, 4-нитрофениламин, 4-феноксиметилфениламин, 4-(4-хлорфеноксиметил)-фениламин, N-ацетил-4-аминобензолсульфамид, N-пиримидин-2-ил-4-аминобензолсульфамид и т.д.
Путь С (В+А): последовательное проведение замещения серы (1 стадия) и N-ацилирования или N-карбамоилирования (2 стадия) с образованием конечных соединений I.
Стадия 1: готовят раствор 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в ацетоне (0,01 моль в 50-100 мл), добавляют йодистый метил, взятый из расчета что на 0,01 моль 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона берут не менее 0,015 моль йодистого метила, и кипятят полученную смесь в течение 3-5 часов, охлаждают, отфильтровывают соль метилтиодитиолия. Готовят смесь полученной соли метилтиодитиолия, соответствующего первичного амина и триэтиламина, взятых из расчета что на 0,01 моль соли метилтиодитиолия берут 0,011 – 0,012 моль первичного амина и не менее 0,02 моль триэтиламина, полученную смесь кипятят в абсолютированном бутаноле в течение 2-3 часов, охлаждают, отфильтровывают, растворитель отгоняют при пониженном давлении, осадок отфильтровывают, перекристаллизовывают из изопропилового спирта. Выход продуктов реакции составляет 60-80%.
В качестве первичных аминов могут быть использованы анилин, 2-метилфениламин, 3-метилфениламин, 4-метилфениламин, 2,4-диметилфениламин, 3,4-диметилфениламин, 2-метоксифениламин, 3-метоксифениламин, 4-метоксифениламин, 2-этоксифениламин, 3-этоксифениламин, 4-этоксифениламин, 2-хлорфениламин, 3-хлорфениламин, 4-хлорфениламин, 2-бромфениламин, 3-бромфениламин, 4-бромфениламин, 2-нитрофениламин, 3-нитрофениламин, 4-нитрофениламин, 4-феноксиметилфениламин, 4-(4-хлорфеноксиметил)-фениламин, N-ацетил-4-аминобензолсульфамид, N-пиримидин-2-ил-4-аминобензолсульфамид и т.д.
Стадия 2. Данная стадия заключается в ацилировании или карбамоилировании имина 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона, полученного на первой стадии хлорангидридом карбоновой кислоты или изоцианатом, соответственно.
В качестве галогенангидрида карбоновой кислоты могут быть использованы хлорангидриды и бромангидриды алифатических кислот, содержащих углеводородные радикалы с разветвленной и неразветвленной цепью, а также циклические углеводородные радикалы, ненасыщенные углеводородные радикалы, в том числе хлорангидрид уксусной, трифторуксусной, пропановой, бутановой, изобутановой, пентановой, 2-метилбутановой, 3-метилбутановой, гексановой, пропеновой, бутеновой, коричной и т.д. кислот, хлорангидриды и бромангидриды ароматических карбоновых кислот, в том числе бензойной, 2- хлорбензойной, 3-хлорбензойной, 4-хлорбензойной, 2,4-дихлорбензойной, 2-бромбензойной, 3-бромбензойной, 4-бромбензойной, 4-йодбензойной, 2-йодбензойной, 3-йодбензойной, 2-фторбензойной, 3-фторбензойной, 4-фторбензойной, 2-метилбензойной, 3-метилбензойной, 4-метилбензойной, 2,4-диметилбензойной, 3,4-диметилбензойной, 2,5-диметилбензойной, 2-метоксибензойной, 3-метоксибензойной, 4-метоксибензойной, 4-нитробензойной, нафталин-2-карбоной, нафталин-3-карбоновой и т.д. кислот, хлорангидриды и бромангидриды гетероциклических карбоновых кислот, в том числе фуран-2-карбоновой, тиофен-2-карбоновой и т.д. ксилот.
В качестве изоцианата может быть использован фенилизоцианат, 2-метилфенилизоцианат, 3-метилфенилизоцианат, 4-метилфенилизоцианат, 2-этилфенилизоцианат, 3-этилфенилизоцианат, 4-этилфенилизоцианат, 2-метоксифенилизоцианат, 3-метоксифенилизоцианат, 4-метоксифенилизоцианат, 2-хлорфенилизоцианат, 3-хлорфенилизоцианат, 4-хлорфенилизоцианат, 3,4-дихлорфенилизоцианат, 2-бромфенилизоцианат, 3-бромфенилизоцианат, 4-бромфенилизоцианат, нафталин-2-илизоцианат, нафталин-1-илизоцианат и т.д.
Готовят смесь полученного на 1 стадии имина 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и соответствующего хлорангидрида карбоновой кислоты, взятых из расчета, что на 0,01 моль имина 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона берут 0,012 – 0,013 моль соответствующего хлорангидрида карбоновой кислоты, кипятят в смеси абсолютированных толуола и пиридина, взятых в объемном соотношении 10 : 1 в количестве 70-100 мл, в течение 3-5 часов, отфильтровывают соль хлорида пиридиния, фильтрат упаривают, выпавший осадок отфильтровывают и перекристаллизовывают из толуола. Выходы целевых продуктов составляют 70-85%.
Или готовят смесь полученного на 1 стадии имина 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и соответствующего изоцианата, взятых из расчета что на 0,01 моль имина 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона берут 0,011 – 0,012 моль изоцианата, полученную смесь кипятят в 70-100 мл абсолютированного диоксана в течение 5-6 часов, выпавший осадок отфильтровывают, промывают водой, после высыхания перекристаллизовывают из толуола. Выход 60-85%.
Методики синтеза конечных соединений I, способы их получения, а также спектральные данные, физико-химические характеристики представлены в примерах ниже.
Производные трициклического серусодержащего производного хинолин-2(1Н)-она I указанными выше способами получены впервые.
ПРИМЕР 1
Синтез 2-(нафтален-2-илокси)-1-(4,4,8-триметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанона (Ia) проводили по пути А.
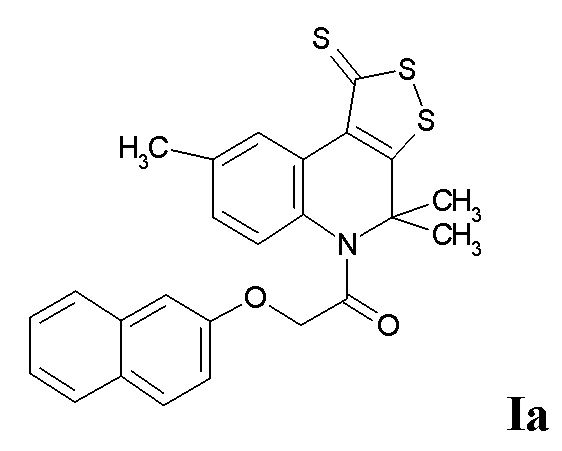
Смесь 0,01 моль 4,4,8-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и 0,012 моль 2-(нафтален-2-илокси)ацетилхлорида кипятили в 100 мл смеси абсолютированных толуола и пиридина (10 : 1) в течение 5 часов, отфильтровывали соль пиридиния, фильтрат упаривали, выпавший осадок отфильтровывали и перекристаллизовывали из толуола. Выход 80%.
Оранжевый порошок, температура плавления 153-154 °C; 1.65 (уш. с, 6H, (CH3)2), 2.35 (с, 3H, CH3), 4.79 (уш. с, 2H, CH2CO), 6.77 (с, 1H, H-Ar), 6.81 (д, J = 8.78 Гц, 1H, H-6(7)), 7.22 (д, J = 8.78 Гц, 1H, H-7(6)), 7.28 (т, J = 8.04 Гц, 1H, H-Ar), 7.38 (т, J = 8.04 Гц, 1H, H-Ar), 7.47 (д, J = 8.04 Гц, 1H, H-Ar), 7.58 (д, J = 8.04 Гц, 1H, H-Ar), 7.66 (д, J = 8.04 Гц, 1H, H-Ar), 7.75 (д, J = 8.04 Гц, 1H, H-Ar), 8.98 (с, 1H, H-9);HPLC-HRMS (ESI) вычислено для C25H21NO2S3+H+, 464.0808; найдено, 464.0811.
ПРИМЕР 2
Синтез 1-(8-метокси-4,4-диметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)-2-(нафтален-2-илокси)этанона (Ib).
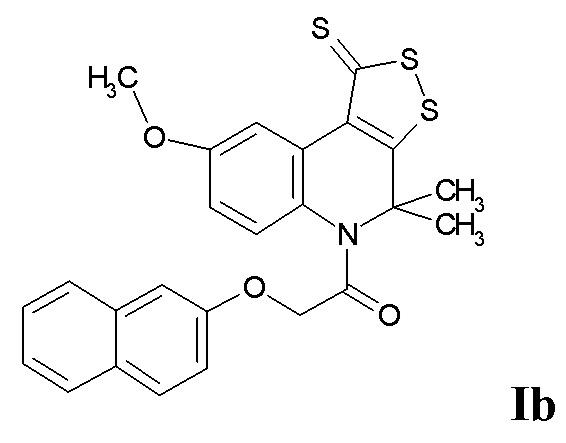
Синтез проводили аналогично примеру 1, при этом в качестве исходных реагентов использовали 8-метокси-4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и 2-(нафтален-2-илокси)ацетилхлорид. Выход 83%.
Оранжевый порошок, температура плавления 136-137 °C; 1.33 (уш. с, 3H, (CH3)2), 2.04 (уш. с, 3H, (CH3)2), 3.79 (с, 3H, CH3O), 4.78 (уш. с, 2H, CH2CO), 6.74 (с, 1H, H-Ar), 6.82 (д, J = 8.75 Гц, 1H, H-6(7)), 7.00 (д, J = 8.75 Гц, 1H, H-7(6)), 7.30 (т, J = 8.23 Гц, 1H, H-Ar), 7.38 (т, J = 8.23 Гц, 1H, H-Ar), 7.55 (д, J = 8.23 Гц, 1H, H-Ar), 7.58 (д, J = 8.23 Гц, 1H, H-Ar), 7.67 (д, J = 8.23 Гц, 1H, H-Ar), 7.75 (д, J = 8.23 Гц, 1H, H-Ar), 9.03 (с, 1H, H-9); HPLC-HRMS (ESI) вычислено для C25H21NO3S3 +H+, 480.0758; найдено, 480.0755.
ПРИМЕР 3
Синтез 2-(нафтален-2-илокси)-1-(4,4,7-триметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанона (Ic)
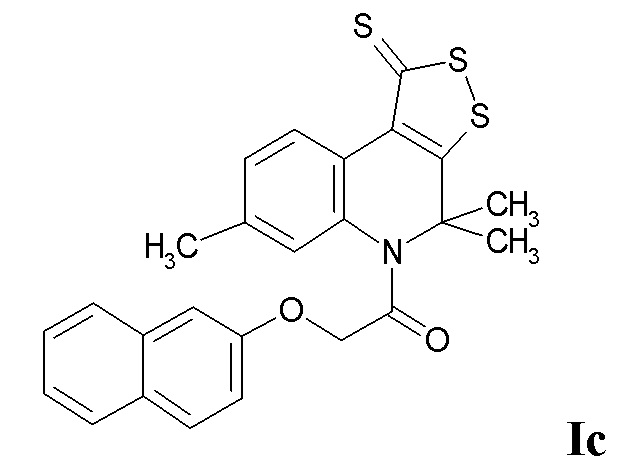
Синтез проводили аналогично примеру 1, при этом в качестве исходных реагентов использовали 4,4,7-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и 2-(нафтален-2-илокси)ацетилхлорид. Выход 77%.
Оранжевый порошок, температура плавления 144 - 145 °C; 1.65 (уш. с, 6H, (CH3)2), 2.32 (с, 3H, CH3), 4.84 (уш. с, 2H, CH2CO), 6.77 (с, 1H, H-6), 6.79 (д, J = 8.15 Гц, 1H, H-Ar), 7.21 (д, J = 8.35 Гц, 1H, H-8), 7.30 (т, J = 8.15 Гц, 1H, H-Ar), 7.36 (с, 1H, H-Ar), 7.39 (т, J = 8.15 Гц, 1H, H-Ar), 7.56 (д, J = 8.15 Гц, 1H, H-Ar), 7.62 (д, J = 8.15 Гц, 1H, H-Ar), 7.75 (д, J = 8.15 Гц, 1H, H-Ar), 9.03 (д, J = 8.35 Гц, 1H, H-9); HPLC-HRMS (ESI) вычислено для C25H21NO2S3 +H+, 464.0808; найдено, 464.0812.
ПРИМЕР 4
Синтез 2-(нафтален-2-илокси)-1-(4,4,7,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанона (Id)
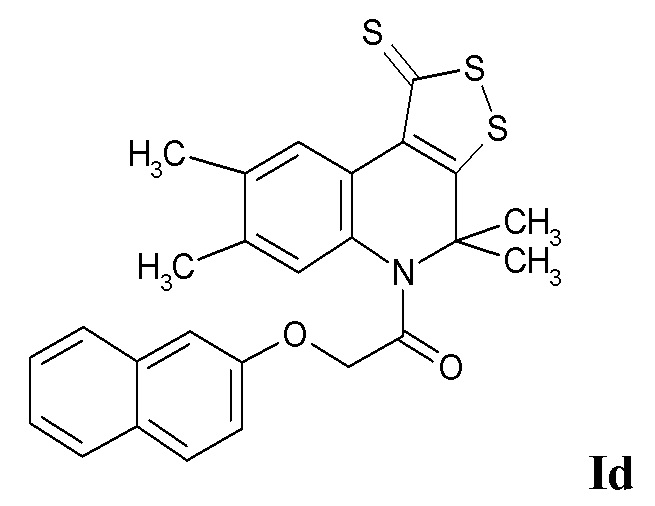
Синтез проводили аналогично примеру 1, при этом в качестве исходных реагентов использовали 4,4,7,8-тетраметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и 2-(нафтален-2-илокси)ацетилхлорид. Выход 80 %.
Оранжевый порошок, температура плавления 189 – 190 °C; 1.64 (уш. с, 6H, (CH3)2), 2.24 (с, 6H, 2CH3), 4.82 (уш. с, 2H, CH2CO), 6.76 (с, 1H, H-6), 6.82 (д, J = 8.71 Гц, 1H, H-Ar), 7.30 (д, J = 8.71 Гц, 1H, H-Ar), 7.32 (с, 1H, H-Ar), 7.38 (д, J = 8.71 Гц, 1H, H-Ar), 7.57 (д, J = 8.71 Гц, 1H, H-Ar), 7.66 (д, J = 8.71 Гц, 1H, H-Ar), 7.75 (д, J = 8.71 Гц, 1H, H-Ar), 8.93 (с, 1H, H-9); HPLC-HRMS (ESI) вычислено для C26H23NO2S3+ H+, 478.0965; найдено, 478.0964.
ПРИМЕР 5
Синтез 3-фенил-1-(4,4,6-триметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)пропан-1-она (Ie)
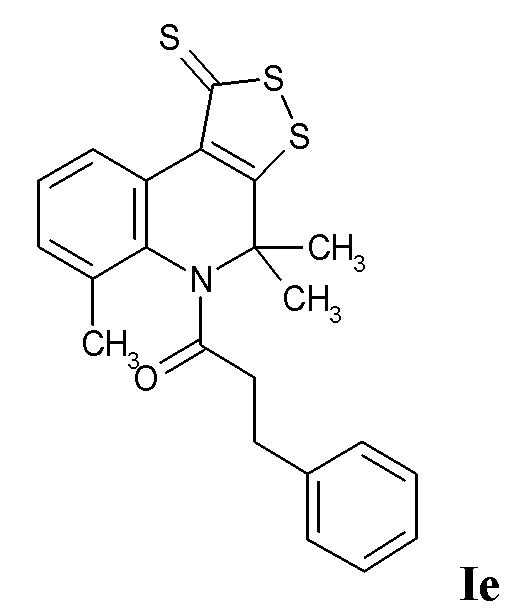
Синтез проводили аналогично примеру 1, при этом в качестве исходных реагентов использовали 4,4,6-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и 3-фенилпропаноилхлорид. Выход 74%.
Оранжевый порошок, температура плавления 117 – 118 °C; 1.04 (с, 3H, CH3), 2.07-2.15 (м, 1H, CH2), 2.22 (с, 3H, (CH3)2), 2.28 (с, 3H, (CH3)2), 2.32-2.39 (м, 1H, CH2), 2.52 (д, J = 7.66 Гц, 2H, CH2CO), 6.84 (д, J = 7.01 Гц, 2H, H-Ar), 7.04 (т, J = 7.01 Гц, 1H, H-Ar), 7.12 (т, J = 7.01 Гц, 2H, H-Ar), 7.32 (т, J = 6.18 Гц, 1H, H-8), 7.35 (д, J = 6.18 Гц, 1H, H-7), 8.91 (д, J = 6.18 Гц, 1H, H-9);HPLC-HRMS (ESI) вычислено для C22H21NOS3+ H+, 412.0859; найдено, 412.0862.
ПРИМЕР 6
Синтез 2-(4-хлорфенокси)-1-(4,4,7,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанона (If)
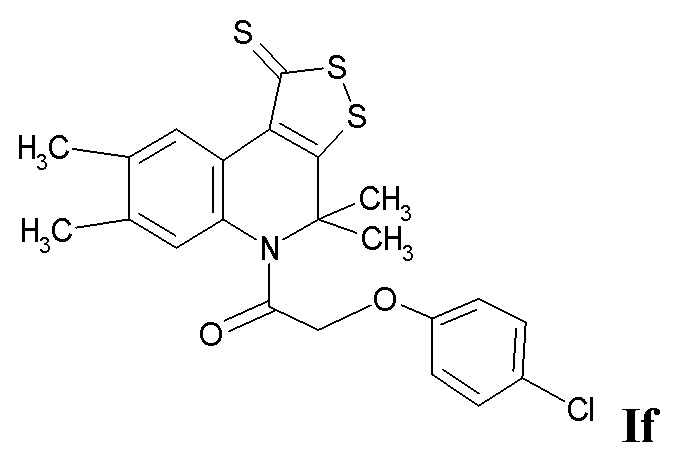
Синтез проводили аналогично примеру 1, при этом в качестве исходных реагентов использовали 4,4,7,8-тетраметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и 2-(4-хлорфенокси)ацетилхлорид. Выход 70%.
Оранжевый порошок, температура плавления 181 – 182 °C; 1H ЯМР (ДМСО-d6, 500 MГц) δ ppm: 1.63 (уш. с, 6H, (CH3)2), 2.22 (с, 6H, 2CH3), 4.66 (с, 2H, CH2CO), 6.54 (д, J = 9.01 Гц, 2H, H-Ar), 7.13 (д, J = 9.01 Гц, 2H, H-Ar), 7.32 (с, 1H, H-6), 8.82 (с, 1H, H-9); HPLC-HRMS (ESI) вычислено для C22H20ClNO2S3+ H+, 462.0419; найдено, 462.0420.
ПРИМЕР 7
Синтез 1-(8-метокси-4,4-диметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)-2-(пиперидин-1-ил)этанона (Ig)
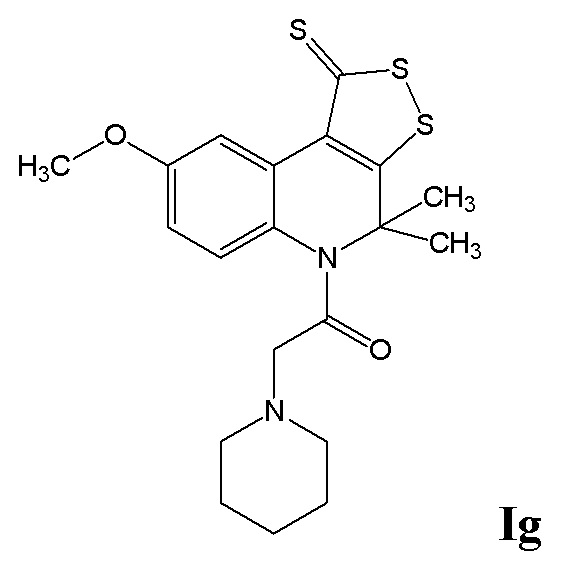
Смесь 0,01 моль 8-метокси-4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и 0,012 моль ангидрида 2-хлоруксусной кислоты кипятили в 100 мл абсолютированного диоксана в течение 6 часов, выпавший осадок отфильтровывали, промывали водой, сушили и перекристаллизовывали из толуола. К смеси 0,01 моль хлорацетамида 8-метокси-4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и 0,022 моль пиперидина, растворенной в 100 мл абсолютированного ацетонитрила, добавляли 0,001 моль йодида калия, полученную смесь кипятили в течение 5 часов, охлаждали, отфильтровывали выпавший осадок, сушили и перекристаллизовывали из толуола. Выход 70%.
Оранжевый порошок, температура плавления 124-125 °C; 1H ЯМР (ДМСО-d6, 500 MГц) δ ppm: 1.15-1.17 (м, 8H, 4 CH2), 1.99 (уш. с, 6H, (CH3)2), 2.51 (м, 2H, CH2), 2.95 (уш. с, 2H, COCH2), 3.77 (с, 3H, CH3O), 6.93 (д, J = 8.66 Гц, 1H, H-6(7)); 7.39 (д, J = 8.66 Гц, 1H, H-7(6)), 8.72 (с, 1H, H-9); HPLC-HRMS (ESI) вычислено для C20H24N2O2S3+ H+, 421.1074; найдено, 421.1074.
ПРИМЕР 8
Синтез 1-(2-оксо-2-(4,4,8-триметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этил)пиперидин-4-карбоксамида (Ih)
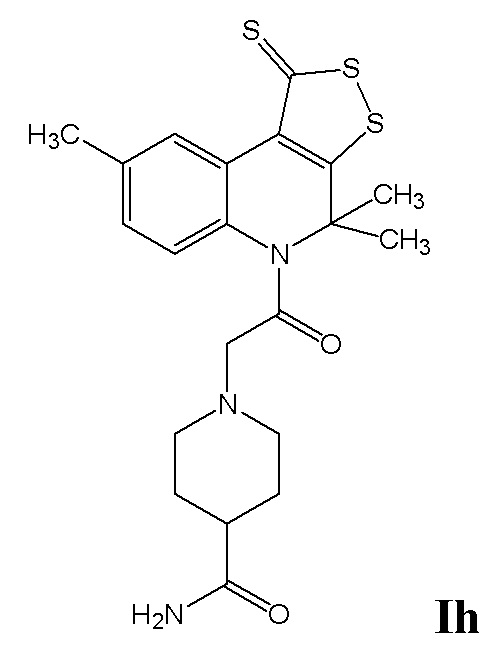
Синтез проводили аналогично примеру 7, при этом в качестве исходных реагентов использовали 4,4,8-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и пиперидин-4-карбоксамид. Выход 50%.
Оранжевый порошок, температура плавления 115-116 °C; 1H ЯМР (ДМСО-d6, 500 MГц) δ ppm: 1.26-1.34 (м, 2H, CH2), 1.46-1.49 (м, 2H, CH2), 1.70 (уш. с, 6H, (CH3)2), 1.85-1.87 (м, 3H, CH2), 2.34 (с, 3H, CH3), 2.51 (м, 1H, CH2), 3.00 (уш. с, 2H, COCH2), 6.63 (уш. с, 1H, NH2), 7.02 (уш. с, 1H, NH2), 7.20 (д, J = 8.06 Гц, 1H, H-6(7)); 7.36 (д, J = 8.06 Гц, 1H, H-7(6)), 8.90 (с, 1H, H-9); HPLC-HRMS (ESI) вычислено для C21H25N3O2S3+ H+, 448.1183; найдено, 448.1186.
ПРИМЕР 9
Синтез 2-(бензо[d]оксазол-2-илтио)-1-(4,4,6,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанона (Ii)
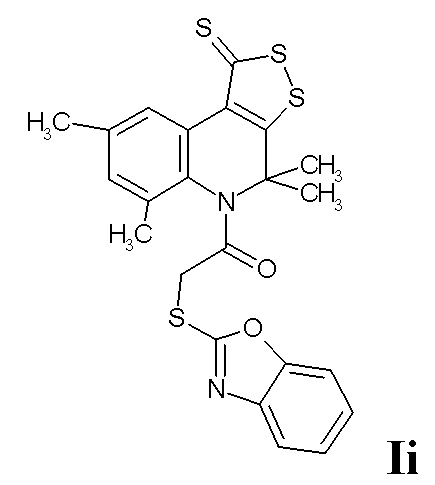
Смесь 0,01 моль 4,4,6,8-тетраметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и 0,012 моль ангидрида 2-хлоруксусной кислоты, кипятили в 100 мл абсолютированного диоксана в течение 6 часов, выпавший осадок отфильтровывали, промывали водой, после высыхания перекристаллизовывали из толуола. К смеси 0,01 моль хлорацетамида 4,4,6,8-тетраметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и 0,01 моль бензо[d]оксазола-2-тиола в 100 мл абсолютированного ацетонитрила, добавляли 0,02 моль карбоната калия и 0,001 моль йодида калия, кипятили в течение 4 часов, охлаждали, отфильтровывали выпавший осадок, сушили и перекристаллизовывали из толуола. Выход 78%.
Оранжевый порошок, температура плавления 156-157 °C; 1H ЯМР (ДМСО-d6, 500 MГц) δ ppm: 1.16 (с, 3H, (CH3)2), 2.23 (с, 3H, (CH3)2), 2.33 (с, 3H, CH3), 2.40 (с, 3H, CH3), 3.79 (д, J = 16.27 Гц, 1H, CH2), 4.11 (d, J = 16.27 Гц, 1H, CH2), 7.24 (т, J = 7.76 Гц, 1H, H-Ar), 7.26 (с, 1H, H-7); 7.28 (т, J = 7.76 Гц, 1H, H-Ar), 7.52 (d, J = 7.76 Гц, 1H, H-Ar), 7.54 (d, J = 7.76 Гц, 1H, H-Ar), 8.80 (с, 1H, H-9); HPLC-HRMS (ESI) вычислено для C23H20N2O2S4+ H+, 485.0481; найдено, 485.0479.
ПРИМЕР 10
Синтез 4-((4,4,7-триметил-4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-илиден)амино)бензолсульфамида (Ij) проводили по пути В.
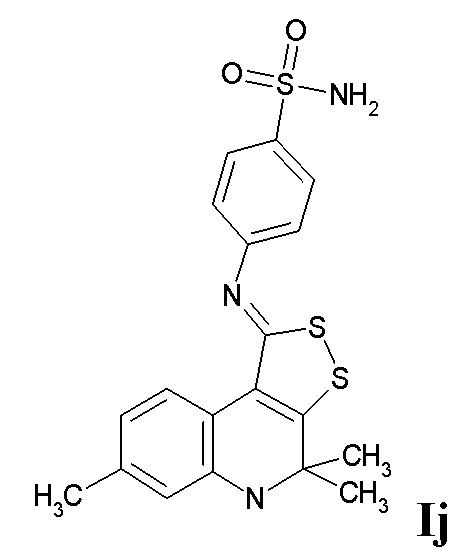
К раствору 0,01 моль 4,4,7-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в 100 мл ацетона добавляли 0,015 моль йодистого метила и кипятили в течение 3 часов, охлаждали, отфильтровывали соль метилтиодитиолия, 0,01 моль которой после высушивания кипятили с 0,011 моль 4-аминобензолсульфамида и 0,02 моль триэтиламина в 100 мл сухого бутанола в течение 3 часов, охлаждали, растворитель отгоняли при пониженном давлении, осадок отфильтровывали, сушили и перекристаллизовывали из диоксана. Выход 76%.
Желтый порошок, температура плавления 306-307 °C; 1H ЯМР (ДМСО-d6, 500 Гц) δ ppm: 1.42 (уш. с, 6H, (CH3)2), 2.19 (с, 3H, CH3), 6.20 (уш. с, 1H, NH), 6.47 (д, J = 8.10 Гц, 1H, H-8); 6.56 (с, 1H, H-6), 7.18 (д, J = 8.60 Гц, 2H, H-Ar), 7.32 (уш. с, 2H, NH2), 7.85 (д, J = 8.60 Гц, 2H, H-Ar), 8.40 (д, J = 8.10 Гц, 1H, H-9) ; HPLC-HRMS (ESI) вычислено для C19H19N3O2S3+ H+, 418.0713; найдено, 418.0709.
ПРИМЕР 11
Синтез N-((4-((4,4,8-триметил-4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-илиден)амино)фенил)сульфонил)ацетамида (Ik).
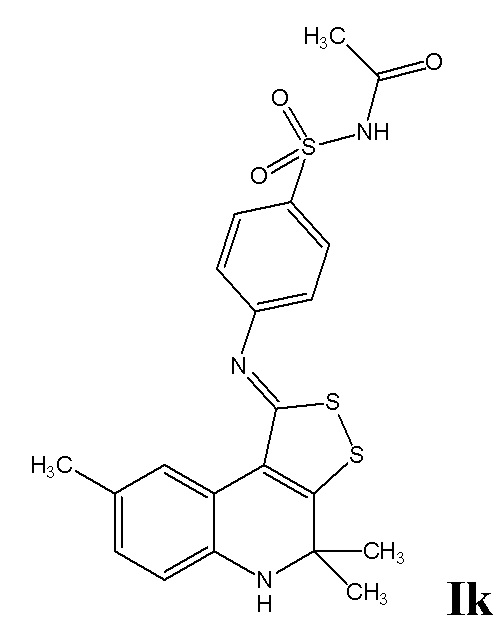
Синтез проводили аналогично примеру 10, но в качестве исходных реагентов использовали 4,4,8-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и N-((4-аминофенил)сульфонил)ацетамид. Выход 68%.
Желтый порошок, температура плавления 288-289 °C; 1H ЯМР (ДМСО-d6, 500 Гц) δ ppm: 1.42 (уш. с, 6H, (CH3)2), 1.93 (с, 3H, CH3CO), 2.19 (с, 3H, CH3), 6.12 (уш. с, 1H, NH), 6.67 (д, J = 8.06 Гц, 1H, H-6(7)); 6.88 (д, J = 8.06 Гц, 1H, H-7(6)), 7.24 (д, J = 8.67 Гц, 2H, H-Ar), 7.93 (д, J = 8.67 Гц, 2H, H-Ar), 8.33 (с, 1H, H-9 ), 12.05 (уш. с, 1H, CONH); HPLC-HRMS (ESI) вычислено для C21H21N3O3S3+ H+, 460.0819; найдено, 460.0816.
ПРИМЕР 12
Синтез 4-((8-этокси-4,4-диметил-4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-илиден)амино)-N-(пиримидин-2-ил)бензолсульфамида (Il)
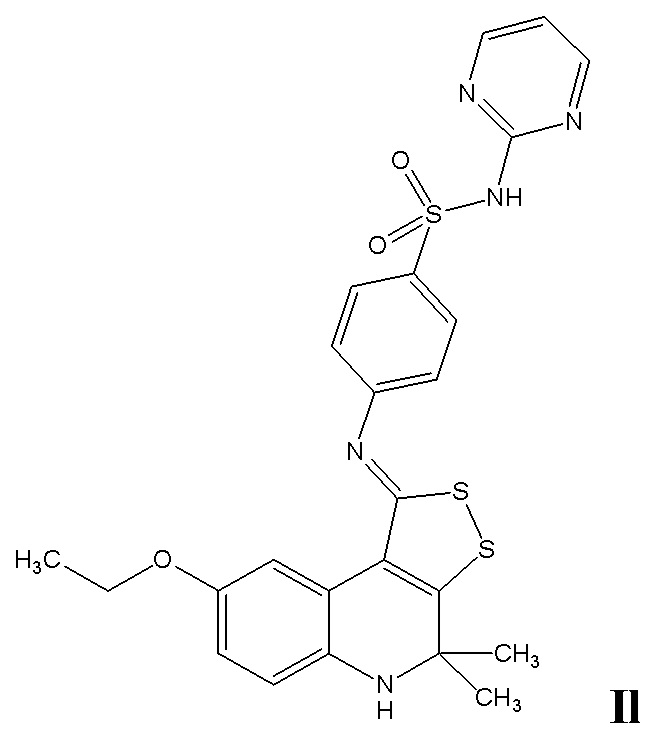
Синтез проводили аналогично примеру 10, но в качестве исходных реагентов использовали 8-этокси-4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и 4-амино-N-(пиримидин-2-ил)бензолсульфамид. Выход 80%.
Желтый порошок, температура плавления 272-273 °C; 1H ЯМР (ДМСО-d6, 500 Гц) δ ppm: 1.22 (т, 3H, J = 13.89 Гц, CH3CH2O), 1.39 (уш. с, 6H, (CH3)2), 3.84 (кв, 2H, J = 13.89 Гц, CH2O), 5.95 (уш. с, 1H, NH), 6.67 (д, J = 8.11 Гц, 1H, H-6(7)); 6.73 (д, J = 8.11 Гц, 1H, H-7(6)), 7.05 (т, J = 4.89 Гц, 1H, H-Het), 7.20 (д, J = 8.60 Гц, 2H, H-Ar), 8.01 (д, J = 8.60 Гц, 2H, H-Ar), 8.16 (с, 1H, H-9 ), 8.51 (д, J = 4.89 Гц, 2H, H-Het), 11.79 (уш. с, 1H, SNH); HPLC-HRMS (ESI) вычислено для C24H23N5O3S3+ H+, 526.1037; найдено, 526.1037.
ПРИМЕР 13
Синтез 4-((4-хлорбензил)окси)-N-(4,4,8-триметил-4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-илиден)анилина (Im)
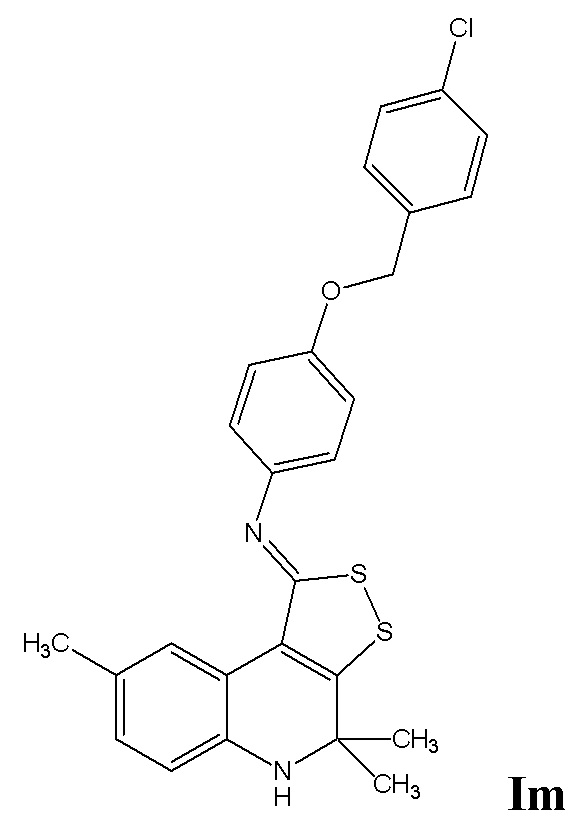
Синтез проводили аналогично примеру 10, но в качестве исходных реагентов использовали 4,4,8-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тион и 4-((4-хлорбензил)окси)анилин. Выход 73%.
Желтый порошок, температура плавления 142-143 °C; 1H ЯМР (ДМСО-d6, 500 Гц) δ ppm: 1.42 (уш. с, 6H, (CH3)2), 2.17 (с, 3H, CH3), 5.12 (уш. с, 2H, CH2O), 6.09 (уш. с, 1H, NH), 6.67 (д, J = 8.0 Гц, 1H, H-6(7)) 6.90 (д, J = 8.0 Гц, 1H, H-7(6)), 6.99 (д, J = 8.68 Гц, 2H, H-Ar), 7.08 (д, J = 8.68 Гц, 2H, H-Ar), 7.76 (д, J = 8.38 Гц, 2H, H-Ar), 7.51 (д, J = 8.38 Гц, 2H, H-Ar), 8.41 (с, 1H, H-9); HPLC-HRMS (ESI) вычислено для C26H23ClN2OS2+ H+, 479,1014; найдено, 479,1016.
ПРИМЕР 14
Синтез N-((4-((4,4,5,8-тетраметил-4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-илиден)амино)фенил)сульфонил)ацетамида (In). Для получения соединения In предварительно проводят: 1) стадию алкилирования атома азота дигидрохинолина йодистым метилом, 2) взаимодействием с серой получают 4,4,5-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона.
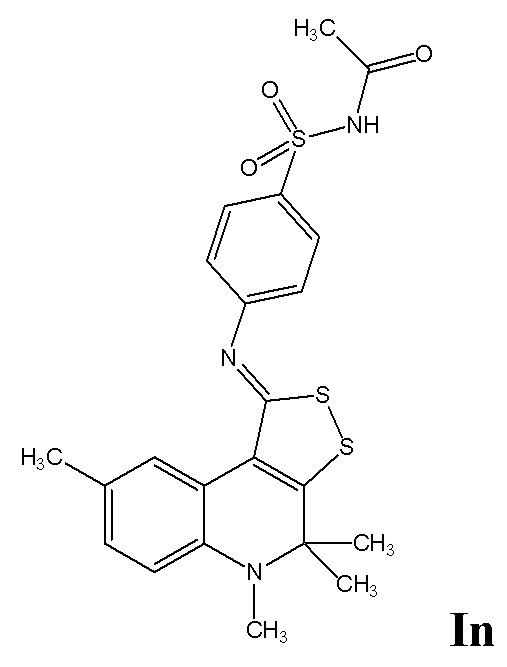
К раствору 0,01 моль 2,2,4-триметил-1,2-дигидрохинолина в 100 мл ацетона добавляли 0,015 моль йодистого метила и кипятили в течение 10 часов, охлаждали, отфильтровывали 1,2,2,4-тетраметил-1,2-дигидрохинолин (предварительная стадия 1), 0,01 моль которого после высушивания кипятили с 0,05 моль молекулярной серы в 30 мл N,N-диметилформамида в течение 12 часов, охлаждали, реакционную смесь выливали в воду, осадок отфильтровывали, сушили, полученный 4,4,5-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона перекристаллизовывали из смеси толуол-изопропиловый спирт (3:1) (предварительная стадия 2). К раствору 0,01 моль 4,4,5-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в 100 мл ацетона добавляли 0,015 моль йодистого метила и кипятили в течение 5 часов, охлаждали, отфильтровывали соль метилтиодитиолия, 0,01 моль которой после высушивания кипятили с 0,011 моль N-((4-аминофенил)сульфонил)ацетамида и 0,02 моль триэтиламина в 100 мл сухого бутанола в течение 3 часов, охлаждали, отфильтровывали, растворитель отгоняли при пониженном давлении, осадок отфильтровывали, перекристаллизовывали из диоксана. Выход 65%.
Желтый порошок, температура плавления 247-248 °C; 1H ЯМР (ДМСО-d6, 500 Гц) δ ppm: 1.40 (уш. с, 6H, (CH3)2), 1.93 (с, 3H, CH3CO), 2.20 (с, 3H, CH3), 2.78 (с, 3H, NCH3), 6.76 (д, J = 8.36 Гц, 1H, H-6(7)); 7.08 (д, J = 8.36 Гц, 1H, H-7(6)), 7.24 (д, J = 8.60 Гц, 2H, H-Ar), 7.94 (д, J = 8.60 Гц, 2H, H-Ar), 8.39 (с, 1H, H-9 ), 12.05 (уш. с, 1H, CONH); HPLC-HRMS (ESI) вычислено для C22H23N3O3S3+ H+, 474.0976; найдено, 474.0980.
ПРИМЕР 15
Синтез (1-((4-этоксифенил)имино)-4,4,8-триметил-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)(фуран-2-ил)метанона (Io) осуществляли по пути С.
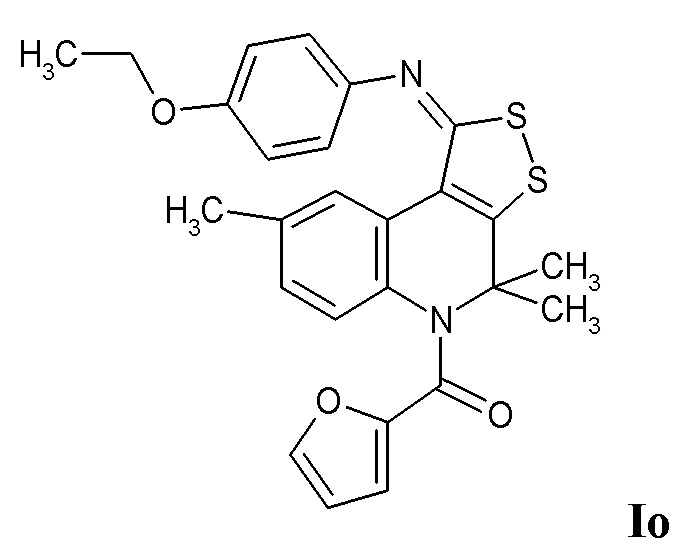
К раствору 0,01 моль 4,4,8-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в 100 мл ацетона добавляли 0,015 моль йодистого метила и кипятили в течение 3 часов, охлаждали, отфильтровывали соль метилтиодитиолия, 0,01 моль которой после высушивания кипятили с 0,011 моль 4-этоксианилина и 0,02 моль триэтиламина в 100 мл сухого бутанола в течение 3 часов, охлаждали, отфильтровывали, растворитель отгоняли при пониженном давлении, осадок отфильтровывали, перекристаллизовывали из изопропилового спирта. Смесь 0,01 моль полученного имина 4,4,8-триметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и 0,012 моль фуран-2-карбонилхлорида кипятили в 100 мл смеси абсолютированного толуола и пиридина (10:1) в течение 5 часов, отфильтровывали соль пиридиния, фильтрат упаривали, выпавший осадок отфильтровывали и перекристаллизовывали из толуола. Выход 82%.
Желтый порошок, температура плавления 203-204 °C; 1H ЯМР (ДМСО-d6, 500 Гц) δ ppm: 1.32 (т, 3H, J = 6.95 Гц, CH3CH2O), 1.69 (уш. с, 6H, (CH3)2), 2.23 (с, 3H, CH3), 4.02 (кв, 2H, J = 6.95 Гц, CH2O), 6.65 (д, J = 8.15 Гц, 1H, H-6(7)); 6.87 (д, J = 8.15 Гц, 1H, H-7(6)), 6.96 (т, J = 4.36 Гц, 1H, H-Het), 7.00 (д, J = 8.96 Гц, 2H, H-Ar), 7.06 (д, J = 8.96 Гц, 2H, H-Ar), 7.16 (д, J = 4.36 Гц, 1H, H-Het), 7.74 (д, J = 4.36 Гц, 1H, H-Het), 8.45 (с, 1H, H-9); HPLC-HRMS (ESI) вычислено для C26H24N2O3S2+ H+, 477.1302; найдено, 477.1300.
ПРИМЕР 16
Синтез N-(3,4-дихлорфенил)-8-этокси-1-((4-изопропилфенил)имино)-4,4-диметил-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-карбоксамида (Ip)
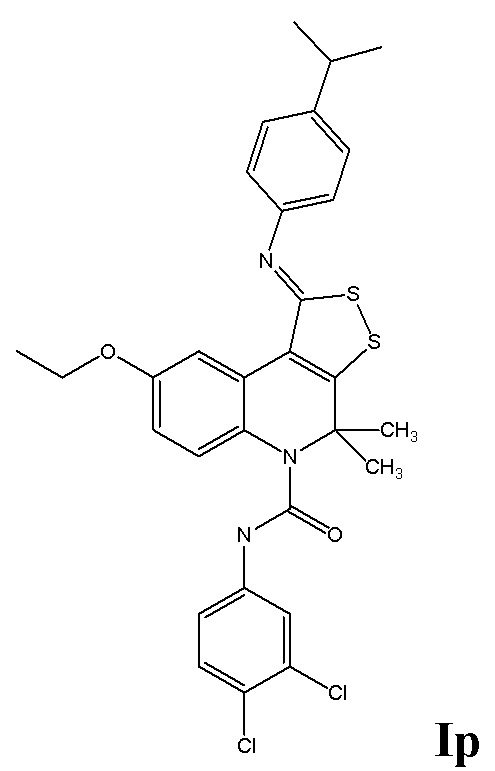
К раствору 0,01 моль 8-этокси-4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в 100 мл ацетона добавляли 0,015 моль йодистого метила и кипятили в течение 5 часов, охлаждали, отфильтровывали соль метилтиодитиолия, 0,01 моль которой после высушивания кипятили с 0,011 моль 4-изопропиланилина и 0,02 моль триэтиламина в 100 мл сухого бутанола в течение 3 часов, охлаждали, отфильтровывали, растворитель отгоняли при пониженном давлении, осадок отфильтровывали, перекристаллизовывали из изопропилового спирта. Смесь 0,01 моль полученного имина 8-этокси-4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона и 0,011 моль 3,4-дихлорфенилизоцианат кипятили в 100 мл абсолютированного диоксана в течение 6 часов, выпавший осадок отфильтровывали, промывали водой, сушили и перекристаллизовывали из толуола. Выход 80%.
Желтый порошок, температура плавления 189-190 °C; 1H ЯМР (ДМСО-d6, 500 Гц) δ ppm: 1.22 (д, 6H, J = 6.79 Гц, 2 CH3), 1.29 (т, 3H, J = 6.85 Гц, CH3CH2O), 1.65 (уш. с, 6H, (CH3)2), 2.92 (м, 1H, CH), 3.97 (кв, 2H, J = 6.85 Гц, CH2O), 6.85 (с, 2H, CH3, H-6 + H-7); 7.01 (д, J = 8.10 Гц, 2H, H-Ar), 7.34 (д, J = 8.10 Гц, 2H, H-Ar), 7.42 (д, J = 8.76 Гц, 1H, H-Ar), 7.55 (д, J = 8.76 Гц, 1H, H-Ar), 7.89 (с, 1H, H-Ar), 8.29 (с, 1H, H-9), 10.14 (уш. с, 1H, CONH); HPLC-HRMS (ESI) вычислено для C30H29Cl2N3O2S2+ H+, 598,1152; найдено, 598,1150.
Также были синтезированы соединения и с другими вариантами радикалов, приведенных в общей формуле I, которые продемонстрировали схожую активность в ингибировании репликации бета-коронавирусов, включая SARS-Cov-2.
ПРИМЕР 17
Исследование активности антивирусных химически синтезированных соединений против коронавируса SARS - CoV- 2 in vitro
Химически синтезированные соединения растворяли в диметилсульфоксиде (ДМСО) до концентрации 10 мг/мл (исходный раствор) и хранили при -200C.
В этом исследовании был использован штамм коронавируса SARS - CoV- 2 гомологичный вирусному изоляту, выделенному в начале пандемии в г. Ухань (КНР).
Исследование противовирусной активности проводили с использованием перевиваемой линии клеток Vero (эпителиальные из почки Африканской зеленой мартышки). Клетки выращивали в модифицированной среде Дульбеко (ДМЕМ) с 10% эмбриональной сыворотки телят, 1% смесью антибиотиков и антимикотиков и 1% глутамина (Gibco, Life Technologies, UK).
Клетки Vero высевали в 96-луночные планшеты (примерно 2 × 104 клеток на лунку) и инкубировали в течение 24 ч при 37°С до образования конфлюэнтного монослоя. Затем среду удаляли из лунок и заменяли 200 мкл свежей среды, содержащей 0-100 мкM соединения, и инкубировали в течение 2 ч. Затем среду удаляли из лунок и заменяли свежей средой, содержащей вирус множественностью заражения (MOI) 0,1. После 1-часовой инкубации среду заменяли свежей средой, содержащей соединение, и инкубировали в течение 72 - 96 часов при 37°С до появления цитопатического действие (ЦПД) в лунках с контролем вируса (без ингибитора).
Во всех экспериментах добавляли в инфицированные вирусом клетки в качестве отрицательного контроля в концентрации, соответствующей разведению исходного раствора соединение-ДМСО.
Количество жизнеспособных клеток (защищенных от ЦПД вируса соединением) определяли с помощью 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил-2Н-тетразолий бромида (МТТ). В основе теста лежит реакция превращения бледно-желтого 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил-2Н-тетразолий бромида (Sigma-Aldrich, USA) в формазан фиолетового цвета под действием фермента сукцинатдегидрогеназы.
Для этого к супернатанту клеток добавляем раствор МТТ (50 мкл, 5 мг/мл).
Планшет инкубировали в течение 90 мин при 37 °С.
Супернатант полностью удаляли, а клетки фиксировали 4% - ным формальдегидом в течение 30 мин.
Кристаллы МТТ растворяли в 1 мл 96 % этанола в течение 10 мин.
На многофункциональном спектрофотометре xMark (Bio-Rad, USA) измеряли значения оптической плотности (экстинкцию) в 96-луночных планшетах при длине волны 450 нм. В качестве контроля интактного монослоя клеток использовали лунки, не зараженные вирусом.
Эффективность ингибирования вирусной репродукции изучаемым соединением определяли по формуле:
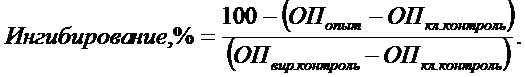
где ОП – оптическая плотность
На каждое разведение соединения использовали по 3 лунки планшета, по которым определяли среднее значение.
В таблице 1 приведены результаты определения активности синтезированных соединений против коронавируса SARS - CoV- 2, в качестве реверсного соединения протестирован Ремдесевир.
Таблица 1.
(TC50/ EC50)
Из приведенной таблицы видно, что по крайней мере два соединения, а именно 2-(нафтален-2-илокси)-1-(4,4,7,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанон Id (EC50 = 0.51 μM, SI = 411.5) и 2-(нафтален-2-илокси)-1-(4,4,7-триметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанон Ic (EC50 = 1.16 μM, SI = 86.21), существенно превосходят Ремдесевир как по эффективности подавления репликации коронавируса SARS-CoV-2, так и по индексу селективности, т.е. у этих соединений цитотоксичность существенно ниже, чем у Ремдесевира, при более высокой эффективности подавления репликации коронавируса в культуре клеток. Все указанные соединения общей формулы I сохраняют стабильность в течение длительного времени (1 года) на воздухе при комнатной температуре. Анализ стабильности осуществляли на основе данных хроматографических и спектральных методов анализа.
На фиг. 1 представлены график эффективности ингибирования репликации коронавируса SARS-CoV-2 в культуре клеток Vero E6 и цитотоксичности для соединения 2-(нафтален-2-илокси)-1-(4,4,7,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанон (Id) и препарата сравнения Ремдесивир. Из графика видно, что соединение 2-(нафтален-2-илокси)-1-(4,4,7,8-тетраметил-1-тиоксо-1H-[1,2]дитиоло[3,4-c]хинолин-5(4H)-ил)этанон (Id), существенно превосходит Ремдесевир как по эффективности подавления репликации коронавируса SARS-CoV-2, так и по индексу селективности, цитотоксичность Id существенно ниже, чем у Ремдесевира, при более высокой эффективности подавления репликации коронавируса в культуре клеток.
ПРИМЕР 18.
Для получения лекарственного средства в форме таблеток смешивают 1600 мг крахмала, 1600 мг измельченной лактозы, 400 мг талька и 1000 мг соединения Id, спрессовывают в брусок. Полученный брусок измельчают в гранулы и просеивают через сита, собирая гранулы размером 14-16 меш. Полученные гранулы таблетируют в подходящую форму таблетки весом 560 мг.
ПРИМЕР 19.
Для получения лекарственного средства в форме капсул тщательно смешивают соединение Id с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывают по 250 мг в желатиновые капсулы подходящего размера.
ПРИМЕР 20.
Получение лекарственного средства в форме инъекционных композиций для внутримышечного, внутрибрюшинного или подкожного введения.
Смешивают 500 мг соединения Id с 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл водя для инъекций. Полученный раствор фильтруют и помещают по 1 мл в ампулы и запаивают.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ТРИЦИКЛИЧЕСКОЕ СЕРУСОДЕРЖАЩЕЕ ПРОИЗВОДНОЕ 1,2-ДИГИДРОХИНОЛИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-COV-2, И СПОСОБ ЕЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2814434C1 |
| ПРИМЕНЕНИЕ ТРИЦИКЛИЧЕСКОГО СЕРУСОДЕРЖАЩЕГО ПРОИЗВОДНОГО 1,2-ДИГИДРОХИНОЛИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-CoV-2 | 2021 |
|
RU2780247C1 |
| СПОСОБ ПОЛУЧЕНИЯ КИСЛОРОДСОДЕРЖАЩЕГО ПРОИЗВОДНОГО 6-ГАЛОГЕНХИНОЛИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-COV-2 | 2021 |
|
RU2827892C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ КИСЛОРОДСОДЕРЖАЩЕЕ ПРОИЗВОДНОЕ 6-ГАЛОГЕНХИНОЛИНА, ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-COV-2, И СПОСОБ ЕЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2827893C1 |
| ПРИМЕНЕНИЕ КИСЛОРОДСОДЕРЖАЩЕГО ПРОИЗВОДНОГО 6-ГАЛОГЕНХИНОЛИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-COV-2, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2021 |
|
RU2780245C1 |
| Способ получения амидов 6-амино-7-фенил-3-(фенилимино)-4,7-дигидро-3H-[1,2]дитиоло[3,4-b]пиридин-5-карбоновой кислоты | 2022 |
|
RU2802515C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ПРОИЗВОДНОЕ 1,3,5-ТРИАЗИН-2,4-ДИАМИНА, ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-COV-2, И СПОСОБ ЕЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2827891C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТИОПРОИЗВОДНОГО ПИРРОЛИДИН-2,5-ДИОНА С ФРАГМЕНТОМ ХИНАЗОЛИНА | 2021 |
|
RU2807057C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНОГО 1,3,5-ТРИАЗИН-2,4-ДИАМИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-COV-2 | 2021 |
|
RU2780249C1 |
| 5,15-БИС(4'-БИС-L-ТИРОЗИНИЛАМИДОФЕНИЛ)-10,20-БИС(N-МЕТИЛПИРИДИНИЙ-3'-ИЛ)ПОРФИН ДИИОДИД, ПРОЯВЛЯЮЩИЙ СВОЙСТВО СВЯЗЫВАНИЯ S-БЕЛКА ВИРУСА SARS-CoV-2 | 2022 |
|
RU2784940C1 |
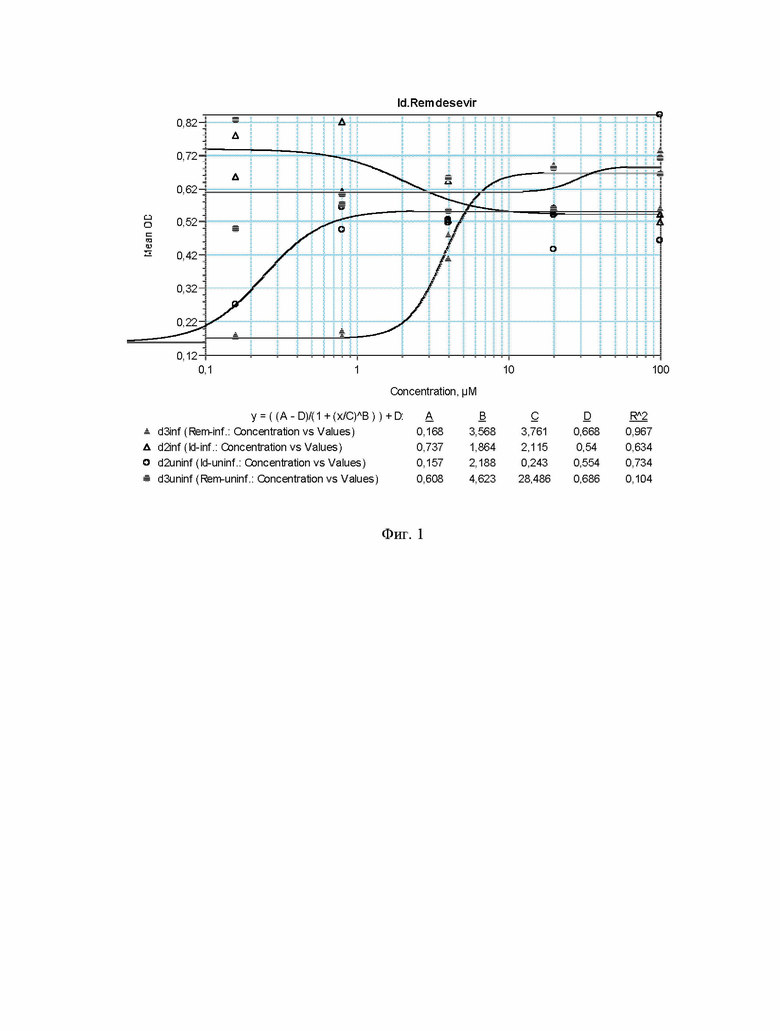
Изобретение относится к области органической химии, а именно к способу получения трициклического серусодержащего производного 1,2-дигидрохинолина общей формулы I, обладающего свойством ингибирования репликации бета-коронавирусов, включая SARS-CoV-2, заключающемуся в модификации трициклического серусодержащего производного 1,2-дигидрохинолина предварительным алкилированием йодистым метилом с последующим замещением серы под действием ариламинов и/или алкилированием или ацилированием по атому азота, следующим образом: синтез иминов 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов проводят через образование соли метилтиодитиолия за счет взаимодействия 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона с йодистым метилом с последующим замещением серы в результате реакции с первичными аминами в присутствии 1,8-2,2 эквимоль триэтиламина, при кипячении в абсолютированном бутаноле с последующим выделением искомого продукта реакции и перекристаллизацией его из изопропилового спирта; синтез соединений I при X=S осуществляют реакцией N-ацилирования хлорангидридами карбоновых кислот в смеси абсолютированного толуола и пиридина, взятых в объемном соотношении 10 частей абсолютированного толуола : 0,8-1,2 частей пиридина, с последующим удалением соли пиридиния и выделением продукта реакции кристаллизацией из фильтрата, или ангидридами карбоновых кислот исходных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов в абсолютированном диоксане с последующим выделением продукта реакции и перекристаллизацией его из толуола. Использование изобретения позволяет сократить время синтеза до 2-3 ч и значительно расширить спектр используемых аминов. В общей формуле (I) R1 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этоксигруппы; R2 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этоксигруппы; R3 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этокси-группы; R4 представляет собой заместитель, выбранный из метил-, бензил-, 3-фенилакрилоил-, 3-(4-метоксифенил)акрилоил-, 3-фенилпропаноил-, фенилэтила, 2-(4-фторфенокси)ацетил-, 4-хлорфеноксиацетил-, 2-(2,4-дихлорфенокси)ацетил-, 4-нитробензоил-, 2-(1,3-диоксоизоиндолин-2-ил)-3-фенилпропаноил-) группы; 3,4-дихлорфенилкарбамоил-, фуран-2-карбонил-, этилкарбамоил-, 2-нафтилилоксиацетил-, 2-(1,3-диоксоизоиндолин-2-ил)ацетил-, 2-(1,3-диоксогексагидро-1H-изоиндол-2(3H)-ил)ацетил-, 2-морфолиноацетил-, пиперидин-1-илацетил-, 2-(4-карбамоилпиперидин-1-ил)ацетил-, 2-(2,5-диоксопирролидин-1-ил)ацетил-,2-феноксиацетил-,2-1,3-диоксо-1H-бензо[de]изохинолин-2(3H)-ил)ацетил-, 2-бензо[d]оксазол-2-илтио)ацетил-, 2-(бензо[d]тиазол-2-илокси)ацетил-, бензоксазол-2-илмеркаптоацетил-, фураноил-, 2-нафлинилиоксиметилкарбонил, 3,4-дихлорфенилкарбамоил; Х представляет собой заместитель, выбранный из серы, незамещенного или замещенного по положениям 2,3,4,5,6 арилимина, выбранного из группы, включающей 4-(4-хлорбензилокси)фенилимино-, 4-изопропилфенилимино-, 4-(6-метилбензо[d]тиазол-2-ил)фенилимино-, 4-(N-бензоилсульфамоил)фенилимино-, 4-этоксифенилимино-, 4-этоксикарбонилфенилимино-, фенилимино-, 4-хлорфенилимино-, 3-хлорфенилимино-, 4-сульфамоилфенилимино-, 4-(N-ацетилсульфамоил)фенилимино-, 4-(N-(тиазол-2-ил)сульфамоил)фенилимино-, 2-никотиноилгидразоно-, 4-((4-хлорбензил)окси)фенилимино-, 2-метоксифенилимино-, 4-(N-(пиримидин-2-ил)сульфамоил)фенилимино-, 4-(N-(4,6-диметилпиримидин-2-ил)сульфамоил)фенилимино-. 3 з.п. ф-лы, 1 ил., 1 табл., 20 пр.
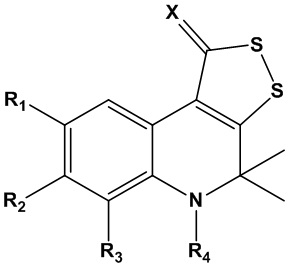 (I)
(I)
1. Способ получения трициклического серусодержащего производного 1,2-дигидрохинолина общей формулы I:
 (I),
(I),
где R1 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этоксигруппы; R2 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этоксигруппы; R3 представляет собой заместитель, выбранный из метил-, этил-, метокси-, этокси-группы; R4 представляет собой заместитель, выбранный из метил-, бензил-, 3-фенилакрилоил-, 3-(4-метоксифенил)акрилоил-, 3-фенилпропаноил-, фенилэтила, 2-(4-фторфенокси)ацетил-, 4-хлорфеноксиацетил-, 2-(2,4-дихлорфенокси)ацетил-, 4-нитробензоил-, 2-(1,3-диоксоизоиндолин-2-ил)-3-фенилпропаноил-) группы; 3,4-дихлорфенилкарбамоил-, фуран-2-карбонил-, этилкарбамоил-, 2-нафтилилоксиацетил-, 2-(1,3-диоксоизоиндолин-2-ил)ацетил-, 2-(1,3-диоксогексагидро-1H-изоиндол-2(3H)-ил)ацетил-, 2-морфолиноацетил-, пиперидин-1-илацетил-, 2-(4-карбамоилпиперидин-1-ил)ацетил-, 2-(2,5-диоксопирролидин-1-ил)ацетил-,2-феноксиацетил-,2-1,3-диоксо-1H-бензо[de]изохинолин-2(3H)-ил)ацетил-, 2-бензо[d]оксазол-2-илтио)ацетил-, 2-(бензо[d]тиазол-2-илокси)ацетил-, бензоксазол-2-илмеркаптоацетил-, фураноил-, 2-нафлинилиоксиметилкарбонил, 3,4-дихлорфенилкарбамоил;
Х представляет собой заместитель, выбранный из серы, незамещенного или замещенного по положениям 2,3,4,5,6 арилимина, выбранного из группы, включающей 4-(4-хлорбензилокси)фенилимино-, 4-изопропилфенилимино-, 4-(6-метилбензо[d]тиазол-2-ил)фенилимино-, 4-(N-бензоилсульфамоил)фенилимино-, 4-этоксифенилимино-, 4-этоксикарбонилфенилимино-, фенилимино-, 4-хлорфенилимино-, 3-хлорфенилимино-, 4-сульфамоилфенилимино-, 4-(N-ацетилсульфамоил)фенилимино-, 4-(N-(тиазол-2-ил)сульфамоил)фенилимино-, 2-никотиноилгидразоно-, 4-((4-хлорбензил)окси)фенилимино-, 2-метоксифенилимино-, 4-(N-(пиримидин-2-ил)сульфамоил)фенилимино-, 4-(N-(4,6-диметилпиримидин-2-ил)сульфамоил)фенилимино-,
заключающийся в модификации трициклического серусодержащего производного 1,2-дигидрохинолина предварительным алкилированием йодистым метилом с последующим замещением серы под действием ариламинов и/или алкилированием или ацилированием по атому азота, следующим образом:
- синтез иминов 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов проводят через образование соли метилтиодитиолия за счет взаимодействия 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона с йодистым метилом с последующим замещением серы в результате реакции с первичными аминами в присутствии 1,8-2,2 эквимоль триэтиламина, при кипячении в абсолютированном бутаноле с последующим выделением искомого продукта реакции и перекристаллизацией его из изопропилового спирта;
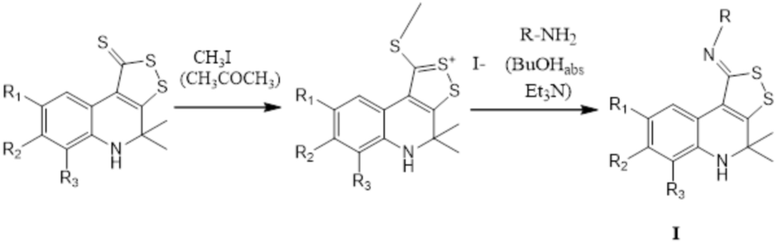
- синтез соединений I при X=S осуществляют реакцией N-ацилирования хлорангидридами карбоновых кислот в смеси абсолютированного толуола и пиридина, взятых в объемном соотношении 10 частей абсолютированного толуола 0,8-1,2 частей пиридина, с последующим удалением соли пиридиния и выделением продукта реакции кристаллизацией из фильтрата,
или ангидридами карбоновых кислот исходных 4,5-дигидро-1H-[1,2]дитиоло[3,4-c]хинолин-1-тионов в абсолютированном диоксане с последующим выделением продукта реакции и перекристаллизацией его из толуола.
2. Способ по п. 1, характеризующийся тем, что в качестве хлорангидрида используют фуран-2-карбонилхлорид, 2-(нафтален-2-илокси)ацетилхлорид, 3-фенилпропаноилхлорид, 2-(4-хлорфенокси)ацетилхлорид.
3. Способ по п. 1, характеризующийся тем, что при использовании в реакции N-ацилирования ангидрида 2-хлоруксусной кислоты проводят дальнейшее алкилирование меркаптанов в результате их взаимодействия с полученным хлорацетамидом 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в абсолютированном ацетонитриле с добавлением от 1,8 до 2,2 эквимоль карбоната калия и каталитических количеств йодида калия с последующим выделением продукта реакции и перекристаллизацией его из толуола.
4. Способ по п. 1, характеризующийся тем, что при использовании в реакции N-ацилирования ангидрида 2-хлоруксусной кислоты проводят алкилирование гетероциклических аминов в результате их взаимодействия с полученным хлорацетамидом 4,4-диметил-4,5-дигидро-1Н-[1,2]дитиоло[3,4-с]хинолин-1-тиона в абсолютированном ацетонитриле с добавлением каталитических количеств йодида калия с последующим выделением продукта реакции и перекристаллизацией его из толуола.
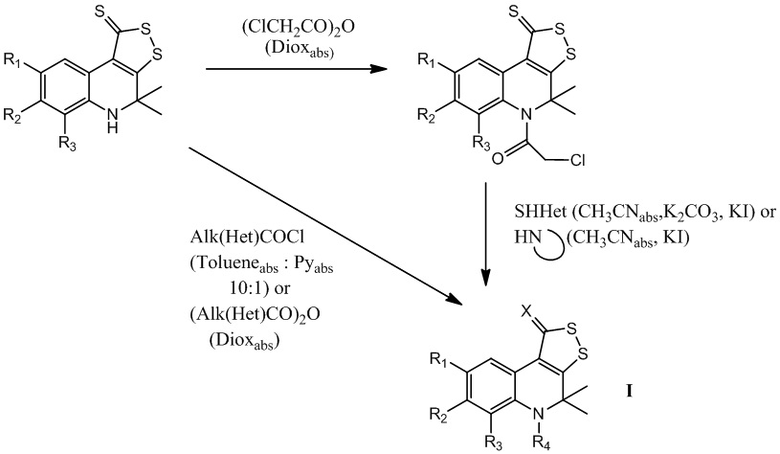
| Устройство для исследования изнашивания | 1984 |
|
SU1174830A1 |
| Kartsev V.; Shikhaliev, Khidmet S | |||
| et al | |||
| "Appendix A | |||
| dithioloquinolinethiones as new potential multitargeted antibacterial and antifungal agents: Synthesis, biological evaluation and molecular docking studies", European Journal of Medicinal Chemistry, 2019, N175, pp.201-214 | |||
| SHIKHALIEV K | |||
| S., SHMYREVA Z | |||
| V., & ZALUKAEV | |||

