Для настоящей заявки испрашивается приоритет в соответствии с патентной заявкой Норвегии №20171944, поданной 6 декабря 2017 г.; патентной заявкой Норвегии №20171945, поданной 6 декабря 2017 г.; и предварительной патентной заявкой США №62/734013, поданной 9 октября 2018 года. Все вышеуказанные заявки полностью включены в настоящий документ посредством ссылки.
Область изобретения
Настоящее раскрытие относится к способу лечения неалкогольного стеатогепатита-НАСГ (NASH), алкогольного стеатогепатита-АСГ (ASH) и других заболеваний печени, характеризующихся фиброзом и/или воспалением печени у субъекта, нуждающегося в лечении. Кроме того, настоящее раскрытие относится к соединению и композиции, содержащей соединение, для применения при лечении неалкогольного стеатогепатита (НАСГ), алкогольного стеатогепатита (АСГ) и других заболеваний печени, характеризующихся фиброзом и/или воспалением печени у субъекта, нуждающегося в лечении. Соединение для применения согласно изобретению представляет собой ненасыщенную жирную кислоту с кислородом, включенным в β-положение и дополнительно содержащую α-заместитель.
Предпосылки создания изобретения
Длинноцепочечные омега-3 жирные кислоты, например, (5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаеновая кислота (EPA) и (4Z, 7Z, 10Z, 13Z, 16Z, 19Z) -докоза-4,7, 10,13,16,19-гексаеновая кислота (DHA) обладают биологическим эффектом широкого спектра, влияя на уровень липидов в плазме, сердечно-сосудистые и иммунные функции, действие инсулина, развитие нейронов и зрительные функции. Высокие дозы EPA/DHA в настоящее время назначаются для лечения тяжелой гипертриглицеридемии (HTG). Эти эффекты опосредованы, по крайней мере частично, их воздействием на метаболизм жирных кислот в печени.
Использование соединений омега-3, таких как EPA и DHA, для лечения неалкогольного стеатогепатита (НАСГ) было предложено в предшествующем уровне техники. Например, WO 2014/057522 (Mochida) описывает композиции, содержащие этилкосапентат, для применения при лечении или облегчении симптомов НАСГ.
Dignity Science LTD (WO2014/118097) предлагает использовать модифицированные омега-3 соединения, такие как 15-гидрокси-эйкозапентаеновая кислота (15-OHEPA), для лечения жировых заболеваний печени, таких как неалкогольная жировая болезнь печени (НАЖБП) и НАСГ. Также Krisani Biosciences (WO2014/045293) предлагают использовать модифицированные соединения омега-3 для лечения различных заболеваний, включая НАСГ. Совсем недавно компания Pronova Biopharma AS (WO2016173923 A1) предложила использовать серосодержащие структурно модифицированные жирные кислоты для лечения НАСГ.
Термины «жировая болезнь печени (НАЖБП)» и «неалкогольный стеатогепатит (НАСГ)» часто используются взаимозаменяемо, несмотря на то, что НАЖБП охватывает гораздо более широкий спектр заболеваний печени, включая изолированный гепатостеатоз (>5% гепатоцитов гистологически). Гепатостеатоз, скорее всего, является относительно доброкачественным заболеванием, если он не сопровождается воспалительным ответом и повреждением клеток. Тем не менее подгруппа пациентов с НАЖБП демонстрирует повреждения клеток печени и воспаление таковой в дополнение к гепатостеозу, т.е. наблюдается состояние, известное как неалкогольный стеатогепатит (НАСГ). НАСГ гистологически практически неотличим от алкогольного стеатогепатита (АСГ). В то время как простой стеатоз, наблюдаемый при НАЖБП, не коррелирует ни с повышенной кратковременной заболеваемостью, ни со смертностью, НАСГ резко повышает риск развития цирроза, печеночной недостаточности и гепатоцеллюлярной карциномы (ГЦК). Цирроз печени, возникающий вследствие НАСГ, становится все более частой причиной трансплантации печени. В то время как заболеваемость и смертность от заболеваний печени значительно повышаются у пациентов с НАСГ, нарушения печени коррелируют еще сильнее с заболеваемостью и смертностью от сердечно-сосудистых заболеваний.
Единые критерии диагностики и постановки диагноза НАСГ все еще обсуждаются (подробности см. в последующих разделах). Ключевыми гистологическими компонентами НАСГ являются стеатоз, гепатоцеллюлярное баллонирование и лобулярное воспаление; фиброз не является частью гистологического диагноза НАСГ. Тем не менее, степень (стадия) фиброза, определяемая при биопсии печени, является определяющей для прогноза, тогда как степень воспаления и некроза (степень поражения) при биопсии печени таковой не является.
Что касается различных гистологических компонентов, то было показано, что лечение омега-3 жирными кислотами эффективно снижает гепатостеатоз у пациентов с НАЖБП (Scorletti E, et al., Effects of purified eicosapentaenoic and docosahexanoic acids in non-alcoholic fatty liver disease: Results from the *WELCOME stud, Hepatology, 2014 Oct;60(4):1211-21), и, если лечение начато на ранней стадии заболевания, то может предположительно замедлить прогрессирование, не его доводя до последних, более тяжелых стадий заболевания. Однако непонятно, окажутся ли омега-3 жирные кислоты достаточно эффективными для лечения и/или реверсии НАСГ, если уже развились выраженные гистологические/воспалительные изменения (Sanyal AJ, et al; EPE-A Study Group, Gastroenterology. 2014 Aug; 147 (2): 377-84.e1).
Умеренная эффективность омега-3 жирных кислот в лечении НАСГ может быть вторичной по сравнению с их мягким воздействием на другие каскады реакций, лежащие в основе патогенеза НАСГ. Исследования НАСГ как на людях, так и на животных, убедительно демонстрируют, что в развитии стеатогепатита и фиброза участвует множество факторов, а не только изолированный гепатостеатоз. К таким факторам относятся инсулинорезистентность, окислительный стресс, воспаление, эндотоксины кишечника и повышенный уровень холестерина и желчных кислот в печени. Было показано, что все эти факторы играют важную способствующую роль у генетически восприимчивых людей, и, соответственно, для лечения НАСГ разрабатываются препараты, нацеленные на эти механизмы (каскады реакций).
Как и в случае с НАСГ, алкогольное заболевание печени (АСГ) может быть разделено на гистологические стадии, представляющие собой переход от жировой болезни печени или простого стеатоза к алкогольному гепатиту (то есть, АСГ) и, наконец, к хроническому гепатиту с фиброзом или циррозом печени. Таким образом, хотя происхождение АСГ и НАСГ может различаться, ответ печени на соответствующее постоянное повреждающее воздействие имеет много общего, включая провоспалительные и профиброзные каскады, включающие активацию макрофагов и продукцию цитокинов, и возникающие в результате этого активированные звездчатые клетки, т.е. пролиферирующие миофибробласты. (см., например, Friedman, SL; Alcoholism: Clinical and Experimental Research. 1999 May; 23(5):904-910.)
Проблемой в разработке лекарств, предназначенных для НАСГ и АСГ, является отсутствие доклинических моделей, которые воспроизводят течение этих заболеваний подобно человеческим. Модели грызунов, которые более соответствуют характеру метаболических нарушений, обычно сопровождающих НАСГ, таких как дислипидемия и резистентность к инсулину, характеризуются очень легким воспалением печени и фиброзом. На другом конце спектра представлены модели химически индуцированного фиброза, например, фиброз, вызванный тетрахлоридом углерода (CCl4) или тиоацетамидом, в которых развивается более тяжелый фиброз, но может отсутствовать всякое соответствие с человеческими пациентами в отношении обоих метаболических компонентов, таких как резистентность к инсулину и ожирение. Таким образом, для выявления потенциальных лекарств для лечения НАСГ и АСГ необходимы многочисленные доклинические модели, захватывающие оба конца этиопатогенного спектра (метаболические нарушения - воспалительный ответ - фиброз).
Другим элементом, который необходимо учитывать при сравнении фиброза печени у грызунов и человека, является местоположение и функциональная значимость индуцированного фиброза. Например, модели тяжелого фиброза с более короткой продолжительностью могут демонтировать только коллагеновые плотные большие портальные тракты, а не паренхимные отложения коллагена, которые функционально более актуальны и часто представляют собой большую часть коллагена печени. Таким образом, в оценке фиброза на моделях грызунов использование биохимической составляющей (например, оценка гидроксипролина) в комбинации с гистологической составляющей (например, морфометрией Sirius Red) является оптимальным с точки зрения определения количества, местоположения и функциональной значимости отложений коллагена в печени.
Обнаружение лекарств, нацеленных на фиброзный компонент НАСГ и АСГ, имеет решающее значение, так как фиброз печени может прогрессировать до цирроза, что, в свою очередь, связано с очень высокой степенью морбидности заболевания и смертностью. Фиброз также представляет собой основную жесткую конечную точку (основной исход) в клинических исследованиях хронических заболеваний печени. Например, появляющиеся данные позволяют предполагать, что именно фиброз, а не НАСГ как таковая, является наиболее важным гистологическим предиктором смерти, как при заболеваниях печени, так и в заболеваниях, не связанных с печенью. Дополнительно, цирроз является главным кофактором первичного рака печени. Таким образом, крайне важно, чтобы новые лекарственные средства, разрабатываемые для лечения НАСГ и АСГ, были достаточно эффективными для предотвращения развития и/или регрессии установленного фиброза.
WO2016173923A1, Pronova Biopharma Norge AS раскрывает, что серосодержащие структурно модифицированные жирные кислоты, такие как 2-этил-2-((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаенилтио)бутановая кислота (соединение N), могут быть полезны при лечении НАСГ. Это основывается на обнаружении того факта, что соединение N дает более значимые эффекты, чем росиглитазон, агонист PPAR-гамма, в профилактике фиброза печени, вызванного диетой. Это также указывает на то, что соединение N предотвращает приток воспалительных клеток в печень. Было продемонстрировано, что соединение N обладает эффективностью в снижении фиброза (определяемого по гидроксипролин/пролин соотношению) у мышей APOE*3Leiden.CETP. Важно отметить, что у мышей APOE*3Leiden.CETP развивается только очень мягкий фиброзный ответ печени (увеличение внеклеточного матрикса на 20-30%), что подтверждается с помощью биохимического анализа, где определяется содержание гидроксипролина (HYP). Кроме того, в конкретной модели, описанной в WO2016173923A1 (содержание которой включено в настоящее описание посредством ссылки), степень (размер) фиброза печени, измеренная морфометрией Sirius Red (SR), была намного ниже, чем обнаруживалось в 5 предыдущих исследованиях с использованием аналогичных экспериментов. В этих исследованиях было обнаружено около 4-5% фиброза через 20 недель и 7-8% через 25-30 недель по результатам измерений посредством морфометрии SR, по сравнению с 1,5% в исследовании, представленном в WO2016173923A1. Для измерения фиброза SR морфометрия является более чувствительным методом, чем биохимический анализ (HYP), который способен занижать значения более функционально релевантного коллагена, являющегося в основном паренхиматозным, в отличие от функционально менее значимого, но количественно преобладающего портального коллагена.
Таким образом, исходя из потребности в сильнодействующих препаратах для лечения НАСГ и АСГ, способных обеспечить профилактическое лечение или регрессию фиброза печени, было проведено несколько новых исследований для определения соединений, которые могут быть использованы для лечения данных аспектов заболевания печени, а также для подтверждения эффекта на основе новых расчетов/оценок и улучшенных моделей, демонстрирующих более высокий уровень фиброза.
Исходя из потребности в сильнодействующих препаратах для лечения НАСГ и АСГ, например, для профилактического лечения или регрессии фиброза печени, было проведено несколько новых исследований с целью выявления соединений, которые могут использоваться при лечении этих аспектов заболевания печени, и для подтверждения эффекта на основе новых расчетов/оценок и улучшенных моделей.
Краткое описание изобретения
Настоящее раскрытие предоставляет способ лечения неалкогольного стеатогепатита и/или алкогольного стеатогепатита у субъекта, нуждающегося в лечении, включающий введение субъекту фармацевтически эффективного количества соединения Формулы (II):
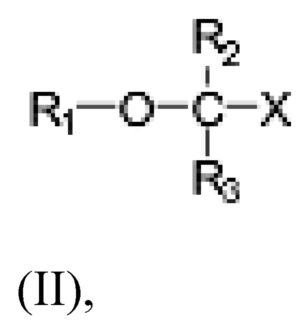
где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей;
R2 и R3 являются одинаковыми или разными и выбраны из группы заместителей, состоящей из атома водорода, гидроксигруппы, алкильной группы, атома галогена, алкоксигруппы, ацилоксигруппы, ацильной группы, алкенильной группы, алкинильной группа, арильной группы, алкилтиогруппы, алкоксикарбонильной группы, карбоксигруппы, алкилсульфинильной группы, алкилсульфонильной группы, аминогруппы и алкиламиногруппы, при условии, что R2 и R3 могут быть связаны для образования циклоалкана, такого как циклопропан, циклобутан, циклопентан или циклогексан;
Х представляет собой: карбоновую кислоту или производное таковой, где производное является карбоксилатом, таким как сложный эфир карбоновой кислоты; глицерид; ангидрид; карбоксамид; фосфолипид; или гидроксиметил; или пролекарство таковых;
или фармацевтически приемлемой соли такового, сольвата или сольвата такой соли. Раскрытие изобретения предусматривает, что соединение может быть введено в виде монотерапии или в комбинации с одним или несколькими дополнительными активными агентами.
Равноценный аспект раскрытия относится к соединению Формулы (II)

где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей;
R2 и R3 являются одинаковыми или различными и выбраны из группы заместителей, состоящей из атома водорода, гидроксигруппы, алкильной группы, атома галогена, алкоксигруппы, ацилоксигруппы, ацильной группы, алкенильной группы, алкинильной группа, арильной группы, алкилтиогруппы, алкоксикарбонильной группы, карбоксигруппы, алкилсульфинильной группы, алкилсульфонильной группы, аминогруппы и алкиламиногруппы, при условии, что R2 и R3 могут быть связаны с образованием циклоалкана, такого как циклопропан, циклобутан, циклопентан или циклогексан;
Х представляет собой: карбоновую кислоту или производное таковой, где производное является карбоксилатом, таким как сложный эфир карбоновой кислоты; глицерид; ангидрид; карбоксамид; фосфолипид; или гидроксиметил; или пролекарство таковых;
или фармацевтически приемлемой соли такового, сольвата или сольвата такой соли для использования в терапевтическом и/или профилактическом лечении неалкогольного стеатогепатита и/или алкогольного стеатогепатита. Раскрытие также предусматривает, что соединение для применения можно вводить как в виде монотерапии, так и в комбинации с одним или несколькими дополнительными активными агентами.
По меньшей мере, в одном варианте осуществления R2 и R3 являются одинаковыми или различными и могут быть выбраны из группы заместителей, состоящей из атома водорода, алкильной группы, алкоксигруппы, алкенильной группы; или R2 и R3 могут быть связаны с образованием циклоалкана, такого как циклопропан, циклобутан, циклопентан или циклогексан;
Х представляет собой карбоновую кислоту или производное таковой, где производным является эфир карбоновой кислоты, глицерид или фосфолипид;
или фармацевтически приемлемой соли такового, сольвата или сольвата такой соли.
Более конкретно, настоящее раскрытие относится к способу лечения неалкогольного стеатогепатита и/или алкогольного стеатогепатита у субъекта, в этом нуждающегося, и включающему введение субъекту фармацевтически эффективного количества соединения Формулы (I):
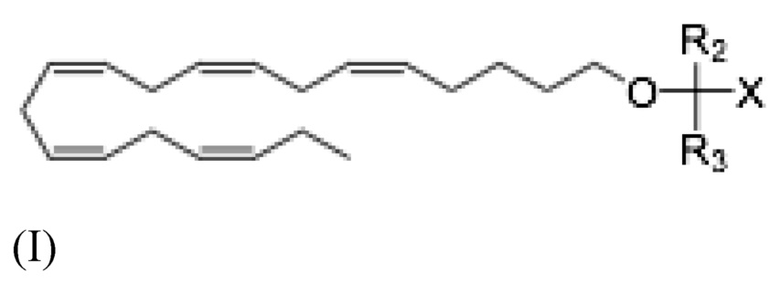
где R2 и R3 и X определены так же, как и для Формулы II. В частности,
R2 и R3 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп;
Х представляет собой карбоновую кислоту или производное таковой, где производным является эфир карбоновой кислоты, глицерид или фосфолипид;
или фармацевтически приемлемой соли такового, сольвата или сольвата такой соли.
Аналогично, настоящее раскрытие относится к соединению Формулы (I):
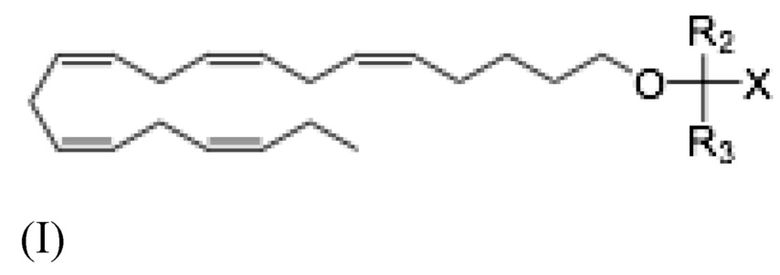
где R2 и R3 и X определены так же, как и для Формулы II. В частности,
R2 и R3 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп;
Х представляет собой карбоновую кислоту или ее производное, где производное представляет собой сложный эфир карбоновой кислоты, глицерид или фосфолипид;
или фармацевтически приемлемой соли такового, сольвата или сольвата такой соли,
для использования при лечении неалкогольного стеатогепатита.
Настоящее изобретение также относится к способу лечения неалкогольного стеатогепатита и/или алкогольного стеатогепатита у субъекта, нуждающегося в этом, причем способ включает введение субъекту фармацевтически эффективного количества 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (соединение А):
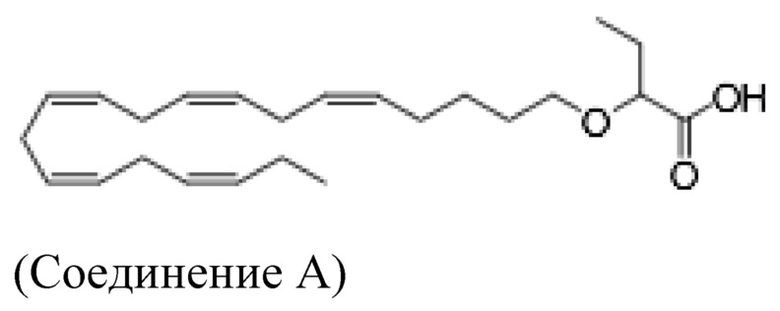
или фармацевтически приемлемой соли или сложного эфира такового.
Настоящее изобретение также предоставляет 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановую кислоту (Соединение A) или фармацевтически приемлемую соль или сложный эфир таковой для использования при лечении неалкогольного стеатогепатита и/или алкогольного стеатогепатита.
Краткое описание графиков
Фиг.1A-1C иллюстрируют влияние Соединения A на количество волокон коллагена (Фиг. 1A), их толщину (Фиг. 1B) и длину (Фиг. 1C) в мышиной модели CDAA на диете с высоким содержанием жира.
Фиг. 2A-2D иллюстрируют влияние Соединения A на содержание липидов в печени для триглицеридов (TG, Фиг. 2A), диглицеридох (DG, Фиг. 2B), свободных жирных кислот (FFA, Фиг.2C) и эфира холестерина (Фиг. 2D) на мышиной модели CDAA на диете с высоким содержанием жиров.
Фиг. 3A-3D показывают, что Соединение A не оказывает значительного влияния на ob/ob мышей, получавших AMLN диету, в отношении массы тела (Фиг. 3A) или веса печени (Фиг. 3B) после 4 недель лечения, а также на массу тела (Фиг. 3C) или вес печени (Фиг. 3D) после 8 недель лечения.
На Фиг.4А-4F показано влияние 4-недельного лечения Соединением А на экспрессию канонических генов, регулирующих фиброгенез печени, у ob/ob мышей, получавших AMLN диету.
На Фиг.5А-5D показано влияние 4-недельного лечения Соединением А на экспрессию генов, регулирующих стабильность внеклеточного матрикса (ЕСМ) или фибролиз у ob/ob мышей, получавших AMLN диету.
На Фиг. 6A-6F показано, что 8 недель лечения Соединением A снижает содержание печеночной α-SMA (Фиг. 6A-6B), col1a1 (Фиг. 6C-6D) и коллагена (Фиг. 6E-6F) у ob/ob мышей на AMLN-диете.
На Фиг.7А-7В показано, что после 8 недель лечения Соединением А у ob/ob мышей на AMLN-диете уменьшается количество активированных звездчатых клеток печени (миофибробластов) и начинается регрессия фиброза, что демонстрируется снижением содержания печеночных α-SMA (Фиг.7А) и col1a1 (Фиг. 7B) соответственно по сравнению с уровнями таковых до лечения.
Фиг. 8A-8F показывают, что 4 недели лечения 112 мг/кг Соединения A приводят к сниженной экспрессии печеночных генов, связанных с воспалением, у ob/ob мышей на AMLN-диете.
Фиг. 9А-9В показывают влияние 8-недельного лечения Соединением А на уровень галектина-3 (Gal-3) в печени у ob/ob мышей на AMLN-диете.
Фиг.10А-10В демонстрируют влияние 8-недельного лечения Соединением А на гепатоцеллюлярное повреждение у ob/ob мышей на AMLN-диете, определяемое по уровням печеночных ферментов аминотрансферазы (ALT) (Фиг. 10A) и аспартаттрансаминазы (AST) (Фиг. 10B).
Фиг. 11А-11Е показывают влияние 8 недельного лечения Соединением А на стеатоз печени, определяемый в процентах от общей площади (Фиг.11А), на основе общего содержания липидов в печени (Фиг.11В), содержания холестерина (Фиг.11С), и уровней триглицерида (Фиг. 11D) и холестерина в плазме (Фиг. 11E) у ob/ob мышей на AMLN-диете.
Фиг. 12A-12C изображают влияние 3-недельного лечения Соединением A на гликемический контроль у ob/ob мышей на AMLN-диете.
Фиг. 13А-13В показывают влияние Соединения А на звездчатые клетки печени человека (LX-2) in vitro.
Подробное описание
Следует отметить, что варианты осуществления и признаки, описанные в контексте одного аспекта настоящего раскрытия, также применимы к другим аспектам изобретения. В частности, варианты осуществления, применимые к способу лечения неалкогольного стеатогепатита или алкогольного стеатогепатита в соответствии с настоящим изобретением, также применимы к аспекту, относящемуся к соединению или композиции, содержащей соединение для применения в лечении неалкогольного стеатогепатита или алкогольного стеатогепатита, согласно настоящему раскрытию. В некоторых вариантах осуществления соединение или композицию, содержащую соединение, вводят в комбинации с одним или несколькими дополнительными активными агентами.
Конкретные аспекты раскрытия описаны более подробно ниже. Термины и определения, используемые в настоящей заявке и поясненные в данном документе, даны для определения понятий настоящего раскрытия.
Единственные формы «а», «an» и «the» также подразумевают использование множественного числа, если контекст не предписывает иное.
Термины «приблизительно», «примерно» и «около» означают, что величина почти совпадает с указанным числом или значением. Используемые здесь термины «приблизительно», «примерно» и «около», как правило, следует понимать как охватывающие интервал ±5% от определенного количества, частоты или значения.
Термины «лечить», «лечение» и «обработка» включают любое терапевтическое или профилактическое применение, которое может принести пользу млекопитающему, являющемуся или не являющемуся человеком. Как медицинское (для человека), так и ветеринарное лечение входят в объем настоящего изобретения. Лечение можно проводить в ответ на существующее состояние или профилактически, то есть для предотвращения возникновения заболевания.
Термины «вводить» и «введение», используемые в данном документе, относятся к (1) предоставлению, введению, дозировке и/или назначению врачом или лицензированным медицинским персоналом, находящимся в подчинении врача, соединения или композиции в соответствии с настоящим раскрытием и (2) введением, приемом или потреблением человеком или млекопитающим, не являющимся человеком, соединения или композиции в соответствии с настоящим раскрытием.
Термины «предотвращение и/или лечение» и «терапевтическое и/или профилактическое лечение» могут использоваться взаимозаменяемо. Как правило, соединения Формулы (I) или Формулы (II) используются для лечения, то есть терапевтического излечения НАСГ или АСГ. Однако также предусмотрено, что в некоторых случаях соединения Формулы (I) или Формулы (II) будут использоваться для профилактического лечения НАСГ или АСГ, например, в случаях, когда у пациента имеется один или несколько факторов риска, связанных с НАСГ или АСГ.
Термины «вводимый в комбинации» и «совместное введение» используются взаимозаменяемо и относятся к введению (а) соединения Формулы (I) или (II) или фармацевтически приемлемой соли таковых, сольвата или сольвата такой соли; и (b) по меньшей мере, одного дополнительного активного агента, совместно скоординированным образом. Например, совместное введение может быть одновременным введением, последовательным введением, совмещенным введением, интервальным введением, непрерывным введением или комбинацией таковых. Способ введения может быть различным для соединений и дополнительного агента(ов), и совместное введение означает любой способ введения, такой как пероральный, подкожный, подъязычный, трансмукозальный, парентеральный, внутривенный, внутриартериальный, внутрибрюшинный, буккальный, сублингвальный, местный, вагинальный, ректальный, офтальмологический, ушной, назальный, ингаляционный и трансдермальный, или является комбинацией таковых. Примеры парентерального введения включают, не ограничиваясь таковыми: внутривенное (IV) введение, внутриартериальное введение, внутримышечное введение, подкожное введение, внутрикостное введение, интратекальное введение или комбинацию таковых. Соединение Формулы (I) или (II) и дополнительный активный агент можно вводить независимо, например, орально или парентерально. В одном варианте осуществления соединение Формулы (I) или (II) вводят перорально, а дополнительный активный агент вводят парентерально. Парентеральное введение может проводиться путем инъекции или инфузии. В некоторых вариантах осуществления способ и/или применение по настоящему раскрытию направлено на терапевтическое и/или профилактическое лечение НАСГ или АСГ с использованием по меньшей мере двух разных активных агентов: соединения Формулы (I) или (II) и дополнительного активного агента соответственно. По меньшей мере, два активных агента можно рассматривать как «комбинированный продукт», где агенты являются, например, отдельно упакованными, и где оба агента необходимы для достижения оптимального предполагаемого эффекта.
Термин «фармацевтически эффективное количество» означает количество, достаточное для достижения желаемых фармакологических и/или терапевтических эффектов, то есть то количество раскрытого соединения, которое эффективно для достижения предполагаемого эффекта. Хотя потребности индивидуальных субъектов/пациентов могут варьироваться, определение оптимальных диапазонов эффективных количеств раскрытого соединения находится в пределах квалификации специалиста в данной области техники. Как правило, режим дозирования для лечения заболевания и/или состояния с помощью раскрытых в настоящее время соединений может быть определен в соответствии с различными параметрами, такими как тип, возраст, вес, пол, диета и/или состояние здоровья субъекта/пациента.
Термин «фармацевтическая композиция» означает соединение согласно настоящему раскрытию в любой форме, подходящей для медицинского применения.
Соединения Формулы (I) и (II) могут существовать в различных стереоизомерных формах, включая энантиомеры, диастереомеры или смеси таковых. Должно быть понятно, что изобретение охватывает все оптические изомеры соединений Формул (I) и (II), а также смеси таковых. Следовательно, соединения Формулы (I) и (II), существующие в виде диастереомеров, рацематов и/или энантиомеров, включаются в объем настоящего изобретения.
В одном аспекте соединение для применения согласно изобретению представляет собой соединение Формулы (II)
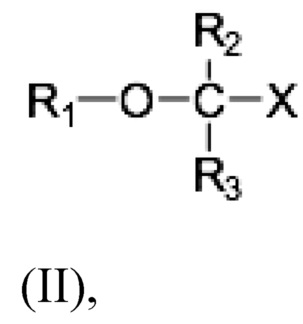
где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей;
R2 и R3 являются одинаковыми или различными и могут быть выбраны из группы заместителей, состоящей из атома водорода, гидроксигруппы, алкильной группы, атома галогена, алкоксигруппы, ацилоксигруппы, ацильной группы, алкенильной группы, алкинильной группы, арильной группы, алкилтиогруппы, алкоксикарбонильной группы, карбоксигруппы, алкилсульфинильной группы, алкилсульфонильной группы, аминогруппы и алкиламиногруппы, и где R2 и R3 могут быть связаны с образованием циклоалкана, такого как циклопропан, циклобутан, циклопентан или циклогексан;
Х представляет собой: карбоновую кислоту или ее производное, где производным является карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; ангидрид; карбоксамид; фосфолипид; или гидроксиметил; или пролекарство таковых;
или фармацевтически приемлемую соль таковых, сольват или сольват такой соли.
В одном варианте осуществления R1 представляет собой C18-C22 алкенил, имеющий 3-6 двойных связей, такой как с 5 или 6 двойными связями, где одна двойная связь, предпочтительно, находится в положении омега-3.
R2 и R3 более предпочтительно независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп. В одном варианте осуществления, по меньшей мере, один из R2 и R3 представляет собой атом водорода, метильную группу, этильную группу, n-пропильную группу или изопропильную группу, бутильную группу или пентильную группу.
Х предпочтительно представляет собой карбоновую кислоту или сложный эфир карбоновой кислоты; или фармацевтически приемлемую соль таковых, сольват или сольват такой соли.
Соединение Формулы (II) для применения в лечении можно вводить как в виде монотерапии, так и в комбинации с одним или несколькими дополнительными активными агентами.
Более конкретно, настоящее раскрытие относится к способу лечения неалкогольного стеатогепатита или алкогольного стеатогепатита у субъекта, нуждающегося в этом, включающему введение субъекту фармацевтически эффективного количества соединения Формулы (I):
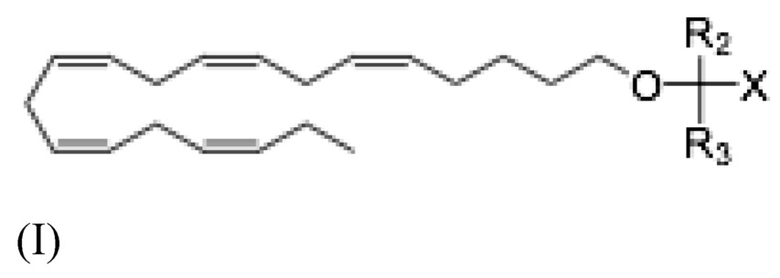
где R2, R3 и X определены как для Формулы (II).
Предпочтительно, для соединений Формулы (I), R2 и R3 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп; а Х представляет собой карбоновую кислоту или сложный эфир карбоновой кислоты; или фармацевтически приемлемую соль таковых, сольват или сольват такой соли.
Соединение Формулы (I) можно вводить в виде монотерапии или в комбинации с одним или несколькими дополнительными активными агентами.
В некоторых вариантах осуществления настоящее раскрытие относится к соединению Формулы (I):
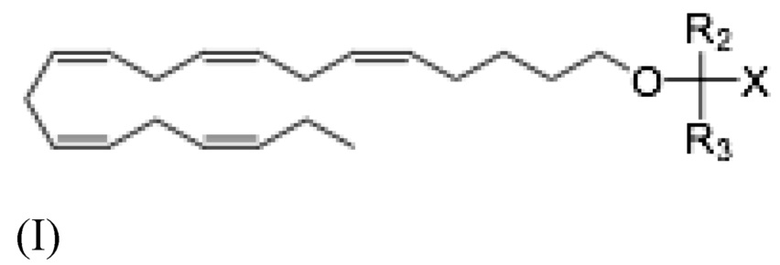
где R2, R3 и X определены как для Формулы (II),
для использования при лечении неалкогольного стеатогепатита или алкогольного стеатогепатита.
Предпочтительно, для соединений Формулы (I) R2 и R3 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп;
Х представляет собой карбоновую кислоту или сложный эфир карбоновой кислоты; или фармацевтически приемлемую соль таковых, сольват или сольват такой соли.
В тех случаях, когда R2 и R3 различны, соединения Формулы (I) и Формулы (II) могут существовать в стереоизомерных формах. Следует понимать, что изобретение охватывает все оптические изомеры соединений Формулы (I) и Формулы (II) и смесей таковых.
По меньшей мере, в одном варианте осуществления R2 и R3 независимо выбраны из группы, включающей атом водорода, метильную группу, этильную группу, n-пропильную группу, изопропильную группу, бутильную группу и пентильную группу.
По меньшей мере, в одном варианте осуществления R2 и R3 независимо выбраны из группы, включающей атом водорода, метильную группу и этильную группу.
По меньшей мере, в одном варианте один из R2 и R3 представляет собой атом водорода, а другой из R2 и R3 выбран из C1-C3 алкильной группы. В одном варианте осуществления один из R2 и R3 представляет собой атом водорода, а другой из R2 и R3 выбран из списка, включающего метильную группу и этильную группу, и, наиболее предпочтительно, один из R2 и R3 представляет собой атом водорода, и другой является этильной группой.
Для соединений как Формулы I, так и Формулы II, R2 и R3 в некоторых вариантах осуществления независимо представляют собой C1-C6 алкильные группы. В некоторых вариантах осуществления оба R2 и R3 представляют собой C1-C3 алкильные группы. В некоторых вариантах осуществления R2 и R3 являются одинаковыми или различными, и каждый независимо выбран из метильной группы, этильной группы, n-пропильной группы или изопропильной группы. В некоторых вариантах осуществления R2 и R3 являются одинаковыми и выбраны из пары метильных групп, пары этильных групп, пары n-пропильных групп или пары изопропильных групп. По меньшей мере, в одном предпочтительном варианте осуществления R2 и R3 представляют собой этильные группы. В некоторых вариантах осуществления один из R2 и R3 представляет собой метильную группу, а другой представляет собой этильную группу. В некоторых вариантах осуществления один из R2 и R3 представляет собой этильную группу, а другой представляет собой n-пропильную группу.
По меньшей мере, в одном варианте осуществления соединение присутствует в его различных стереоизомерных формах, таких как энантиомер (R или S), диастереомер или смесь таковых. По меньшей мере, в одном варианте осуществления соединение присутствует в рацемической форме.
В тех случаях, когда соединение Формулы (I) представляет собой соль противоиона с, по меньшей мере, одним стереогенным центром или сложный эфир спирта с, по меньшей мере, одним стереогенным центром, соединение может иметь несколько стереоцентров. В этих ситуациях соединения по настоящему изобретению могут существовать в виде диастереомеров. Таким образом, по меньшей мере, в одном варианте осуществления соединения по настоящему изобретению присутствуют в качестве соединения, где имеется, по меньшей мере, один диастереомер.
По меньшей мере, в одном варианте осуществления, соединение по настоящему изобретению представляет собой 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановую кислоту (соединение А):
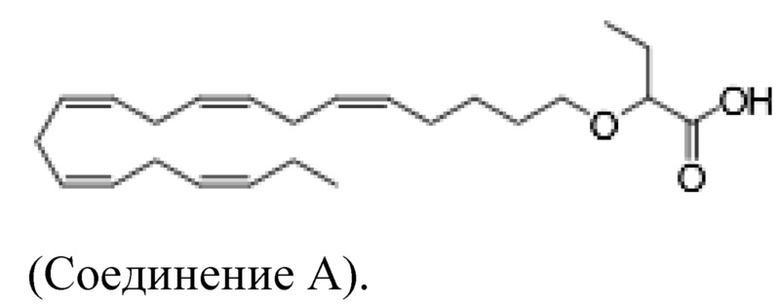
По меньшей мере, в одном варианте осуществления соединение по настоящему изобретению представляет собой 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановую кислоту (соединение А), присутствующую в ее S и/или R форме, представленной Формулами:
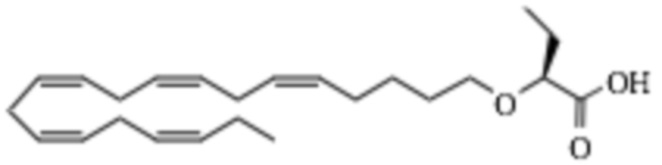 и
и 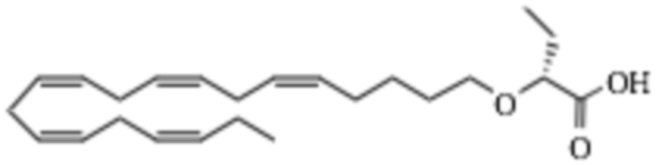 .
.
В некоторых вариантах осуществления 2 - ((((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановая кислота (соединение A) вводится в виде монотерапии. В некоторых вариантах осуществления 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановая кислота (соединение А) вводится в сочетании с одним или несколькими дополнительными активными агентами.
Как описано ранее, многочисленные независимые и взаимозависимые метаболические, воспалительные и в конечном итоге фиброзные компоненты действуют конвергентно в развитии НАСГ человека. Вполне вероятно, что любое успешное лечение должно быть направлено на несколько аспектов НАСГ, предпочтительно через метаболические/воспалительные мишени. Тем не менее, поскольку развитие фиброза связано с клиническими проявлениями, идеальная терапия НАСГ должна быть направлена на воспалительный компонент, а также в идеале уменьшать или профилактически излечивать как развитие фиброза, так и способствовать регрессии существующего фиброза. Прилагаемые Примеры показывают удивительные и неожиданно значимые противовоспалительные и антифиброзные эффекты кислородсодержащих структурно-модифицированных жирных кислот, таких как соединение А. Эти результаты, которые были показаны на нескольких доклинических моделях НАСГ, подтверждают использование кислородосодержащих соединений данного раскрытия в терапевтическом и профилактическом лечении НАСГ и АСГ у людей.
Неожиданно обнаружилось, что Соединение А является удивительно активным в замедлении фиброза, профилактическом лечении развития фиброза и реверсии фиброза печени, например, как подтверждено биохимическим анализом, оценивающим содержание гидроксипролина на модели мышей с фиброзом, индуцированным диетой CDAA (метионин-холин-дефицитная диета) (наблюдалось увеличение внеклеточного матрикса (ECM) на 2-400% по сравнению с увеличением ECM на 20-30% в более щадящей мышиной модели APOE*3Leiden.CETP).
Новое открытие в модели НАСГ, индуцированной CDAA, заключалось также в том, что соединение A также уменьшало фиброз печени, измеренный гистологически с использованием морфометрии Sirius Red (далее-морфометрии SR). В отличие от биохимической оценки содержания гидроксипролина (HYP), измеряющей коллаген в крупных сосудах, морфометрия SR количественно определяет более функционально значимые синусоидальные отложения коллагена.
Учитывая ключевую важность фиброза в морбидности (заболеваемости) и смертности, связанной с НАСГ, результаты, полученные на модели НАСГ, индуцированной CDAA, подтверждают вывод о том, что кислородосодержащие структурно-модифицированные жирные кислоты, такие как соединение А, эффективны для лечения осложнений, связанных с НАСГ. Это открытие также подтверждается изменениями экспрессии гена печени, ассоциированного с фиброзом (Col1a1), и гена, связанного с воспалительными реакциями (экспрессия гена TNF-a в печени). Значительное снижение более функционально значимого синусоидального коллагена, измеренное с помощью морфометрии SR, также свидетельствует в поддержку необходимости тестирования и использования Соединения A при лечении НАСГ человека.
Несмотря на критическую важность наличия фиброза в клинических проявлениях, нормативное одобрение новых эффективных лекарств для лечения НАСГ основывается на лечении НАСГ без ухудшения фиброза. Также важно, чтобы улучшения по параметрам оценки НАСГ достигались с помощью новых соединений. Таким образом, улучшения по параметрам стеатоза, лобулярного воспаления и гепатоцеллюлярного баллонирования в идеале должны происходить в дополнение к желаемому улучшению состояния фиброза.
Новая находка в другой модели НАСГ, модели мыши STAM, заключалась в том, что, в дополнение к улучшениям при стеатозе и воспалении, Соединение А улучшало гепатоцеллюлярное баллонирование. Гепатоцеллюлярное баллонирование обычно характеризуется как увеличение клеток в 1,5-2 раза от нормального диаметра гепатоцитов за счет разреженной цитоплазмы и, как было показано, коррелирует с фиброзом и связано с повреждением печени.
Клеточное раздувание (определяемое как гепатоцеллюлярная гипертрофия) также предотвращалось Соединением А у двойных трансгенных мышей APOE*3L.CETP. Определение гипертрофии по сравнению с баллонированием относится к появлению определенных гистологических различий, характерным для гепатоцитов грызунов и человека.
В сочетании с описанными противовоспалительными эффектами, эти новые результаты, связанные с гепатоцеллюлярным(ой) баллонированием/гипертрофией, подчеркивают потенциальную полезность Соединения A для улучшения всех парамертов оценки НАСГ, что, в свою очередь, должно способствовать положительному результату в клинической разработке и последующему одобрению регуляторным органом.
Неожиданно обнаружилась эффективность Соединения А в модулировании как воспалительных, так и фиброзных компонентов на множественных моделях грызунов НАСГ различной степени тяжести, как описано выше и как показано в Примерах.
Сложное взаимодействие адаптивного и неадаптивного рекрутирования, активации, дифференцировки и пролиферации иммунных клеток в НАСГ указывает на то, что измерение значений (мониторинг) какого-либо одного воспалительного параметра требует сопутствующего измерения фиброза для интерпретации функциональной значимости таких измерений. Таким образом, антифиброзные эффекты Соединения А в модели CDAA могут усиливать клиническую значимость противовоспалительных эффектов, наблюдаемых при использовании кислородозамещенных структурно-модифицированных жирных кислот.
Так как нормативная легализация новых эффективных препаратов для лечения НАСГ зависят от улучшения НАСГ без ухудшения фиброза, улучшения гепатоцеллюлярного баллонирования/гипертрофии, наблюдаемые при лечении Соединением А в двух разных моделях НАСГ грызунов, также являются важной и новой находкой (баллонирование/гипертрофия не замерялась в модели мыши CDAA).
В сочетании с описанными противовоспалительными эффектами, эти новые результаты, связанные с гепатоцеллюлярным(ой) баллонированием/гипертрофией, подчеркивают потенциальную полезность Соединения A для улучшения всех параметров оценки НАСГ, что, в свою очередь, должно способствовать положительному результату в клинической разработке и последующему одобрению регулирующими органами.
Важно отметить, что способность соединений, таких как соединение А, улучшать все параметры оценки НАСГ и фиброза, связанных как с портальными трактами, так и с паренхимой, в многочисленных доклинических моделях НАСГ с различным этиопатогенезом служит сильной поддержкой в пользу проведения тестирования на человеческих пациентах с НАСГ.
Кроме того, поскольку значительная часть пациентов с НАСГ также страдает диабетом 2 типа, важно изучить влияние отсроченного начала лечения препаратом, таким как соединение А, не только на стеатоз печени, воспаление и фиброз, а также его влияние на гликемический контроль в модели НАСГ, вызванной ожирением. Описанные выше противовоспалительные, антифиброзные и снижающие стеатоз эффекты Соединения A были дополнительно продемонстрированы на модели НАСГ, индуцированной диетой с компонентом ожирения (ob/ob мыши на диете AMLN с высоким содержанием жира). Соединение А также оказывало положительное влияние на гликемический контроль в этой модели, не влияя на массу тела, в отличие от лечения тиазолидиндионами (например, пиоглитазоном), которые отрицательно влияют на массу тела. Кроме того, Соединение А снижало плазменные уровни аланинаминотрансферазы (ALT) и аспартаттрансаминазы (AST), указывая на то, что гепатоцеллюлярное повреждение и/или урон уменьшаются.
На основании этих результатов, Соединения Формулы (II) или, предпочтительно, Формулы (I) можно вводить для лечения и/или регрессии неалкогольного стеатогепатита (НАСГ) или других заболеваний печени, характеризующихся фиброзом, воспалением и/или гепатоцеллюлярным баллонированием. В некоторых вариантах лечение НАСГ может быть профилактическим. Кроме того, соединения могут вводиться для лечения по меньшей мере одного заболевания, состояния или при наличии фактора риска, связанного с НАСГ. В некоторых вариантах осуществления лечение по меньшей мере одного заболевания, состояния или при наличии фактора риска, связанного с НАСГ, может быть профилактическим.
Ввиду сходства провоспалительных и профиброзных механизмов НАСГ и алкогольного стеатогепатита (АСГ), противовоспалительные и антифиброзные эффекты описанных здесь соединений, описанных в моделях НАСГ и в экспериментах in vitro, также наблюдаются при лечении и/или регрессии АСГ, в частности, при предотвращении прогрессирования и индукции регрессии прогрессирующей АСГ и ассоциированного с ним фиброза. Например, ингибирующее действие Соединения А на пролиферацию изолированных клеток LX-2 (звездчатых клеток печени человека) in vitro не зависит от передачи паракринных сигналов от паренхиматозных клеток и/или клеток Купфера, что свидетельствует о том, что антифиброзные эффекты Соединения А могут достигаться независимо от того, исходят ли предшествующие сигналы от НАСГ- или АСГ-ассоциированных поражений печени.
Таким образом, соединения Формулы (II) или предпочтительно Формулы (I) можно применять для лечения и/или реверсии АСГ. В некоторых вариантах лечение АСГ может быть профилактическим. Кроме того, соединения могут вводиться для лечения по меньшей мере одного заболевания, состояния, или при наличии фактора риска, связанного с АСГ. В некоторых вариантах осуществления лечение по меньшей мере одного заболевания, состояния или при наличии фактора риска, связанного с АСГ, может быть профилактическим.
Соответственно, настоящее раскрытие охватывает способ приостановки развития или профилактического лечения при риске развития фиброза печени и уменьшения уже существующего фиброза печени. В соответствии с этим методом фиброз излечивается путем уменьшения фиброзной области, фиброзного контента/параметров или степени тяжести фиброза. По меньшей мере, в одном варианте осуществления способ обеспечивает уменьшение, иногда значительное уменьшение процентной площади поверхности фиброза печени. По меньшей мере, в одном варианте осуществления способ обеспечивает снижение суммарной оценки по шкале НАСГ, например, значительное уменьшение показателей шкалы НАСГ (NAS score). Кроме того, способ включает уменьшение воспаления печени, такого как лобулярное воспаление; уменьшение гепатоцеллюлярного баллонирования; снижение стеатогепатита; все в дополнение к улучшению состояния фиброза.
В некоторых вариантах осуществления соединение по настоящему изобретению уменьшает фиброзную область печени на 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65% или 70% по результатам морфометрии Sirius Red. В некоторых вариантах осуществления соединение по настоящему изобретению уменьшает фиброзную область печени на 20-30%, 30-40%, 10-40%, 40-50%, 40-60%, 50-60%, 50-70%, или 60-70%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает фиброзный контент печени на 20%, 25%, 30%, 35% или 40% по результатам измерения содержания гидроксипролина в печени. В некоторых вариантах осуществления применение настоящего изобретения снижало фиброзный контент печени на 20-30%, 20-25%, 24-30% или 30-40%, 30-35% или 35-40%. В некоторых вариантах осуществления соединение по настоящему изобретению снижало содержание печеночного коллагена на 20%, 25%, 30%, 35% или 40%. В некоторых вариантах осуществления соединение по настоящему изобретению снижало содержание печеночного коллагена на 20-30%, 20-25%, 25-30%, 30-40%, 30-35% или 35-40%. В некоторых вариантах осуществления соединение по настоящему изобретению уменьшало область содержания α-SMA в печени по сравнению с уровнями до начала лечения на 3%.
В некоторых вариантах осуществления соединение по настоящему изобретению уменьшает стеатоз на 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85% или 90%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает стеатоз на 40-50%, на 50-60%, на 60-70%, на 50-70%, на 70-80%, на 60-80%, на 70 -90% или на 80-90%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает общее содержание липидов в печени на 20%, 25%, 30%, 35%, 40%, 45% или 50%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает общее содержание липидов в печени на 20-30%, на 30-40% или на 40-50%.
В некоторых вариантах осуществления соединение по настоящему изобретению уменьшает гепатоцеллюлярное баллонирование на 30%, 35%, 40%, 45% или 50%. В некоторых вариантах осуществления соединение по настоящему изобретению уменьшает гепатоцеллюлярное баллонирование на 30-40%, 30-35%, 35-40%, 40-50%, 40-45% или 45-50%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает гепатоцеллюлярную гипертрофию на 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или 80%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает гепатоцеллюлярную гипертрофию на 40-50%, 50-60%, 60-70% или 70-80%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает показатели шкалы НАСГ на 30%, на 35%, на 40%, на 45%, на 50%, на 55%, на 60%, на 65% или на 70%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает показатели шкалы НАСГ на 30-40%, на 40-50%, на 30-50%, на 50-60%, на 60-70% или на 50-70%.
В некоторых вариантах осуществления соединение по настоящему изобретению уменьшает воспаление печени на 20%, 30% или 40%, что определяется уровнями галектина (Gal-3). В некоторых вариантах осуществления соединение по настоящему изобретению уменьшает воспаление печени на 20-30%, 20-25%, 25-30%, 30-40%, 30-35% или 35-40%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает уровни аланинаминотрансферазы (ALT) в плазме на 20%, 35%, 30%, 35% или 40%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает уровни ALT на 20-30%, 20-25%, 25-30%, 30-40%, 30-35% или 35-40%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает уровни аспартаттрансаминазы (AST) в плазме на 10%, 15% или 20%. В некоторых вариантах осуществления соединение по настоящему изобретению снижает уровни AST на 10-15%, на 10-20% или на 15-20%.
В некоторых вариантах осуществления соединение по настоящему раскрытию и/или лечение соединением по настоящему раскрытию, например, соединением А, улучшает показатели шкалы НАСГ по биопсии печени, оценке МРТ LiverMultiScan PDFF и cT1, тесту(ам) функции печени, HOMA-IR и/или биомаркерам воспаления и фиброза (включая hsCRP, Pro-C3, панель ELF и другие подходящие биомаркеры) по сравнению с контролем.
В некоторых вариантах осуществления соединение по настоящему раскрытию и/или обработка соединением по настоящему раскрытию, например, соединением А, приводит к улучшению, например, уменьшению баллонирования (например, оценка=0), например, с сопутствующим показателем лобулярного воспаления 0 или 1 и отсутствием ухудшения фиброза по сравнению с контролем. В некоторых вариантах осуществления соединение по настоящему раскрытию и/или лечение соединением по настоящему раскрытию, например, соединением А, приводит к изменениям, например, улучшениям по сравнению с исходными показателями шкалы НАСГ по результатам биопсии печени, по отдельным гистологическим показателям стеатоза, баллонированию, воспалению и фиброзу, по ферментам печени, по параметрам визуализации и/или по биомаркерам (включая hsCRP, Pro-C3, панель ELF, цитокины и другие подходящие биомаркеры) по сравнению с контролем.
В некоторых вариантах осуществления безопасность и переносимость соединения по настоящему изобретению, например соединения А, у пациентов можно оценить с помощью мониторинга побочных эффектов у пациентов, мониторинга лабораторных показателей (гематология, биохимия и анализ мочи), показателей жизнедеятельности (артериальное давление, частота пульса и температура), hsCRP и/или ЭКГ с 12 отведениями в состоянии покоя по сравнению с контролем. Фармакокинетику Соединения А у пациентов можно изучать путем сравнения средних минимальных концентраций Соединения А в плазме в стабильном состоянии, например, с помощью проб, взятых через регулярные промежутки времени. Например, средние минимальные концентрации в плазме могут быть взяты с интервалами в 8 недель для определения фармакокинетики соединения А.
Пациенты, соответствующие одному или нескольким из следующих критериев, могут показать улучшенный ответ на лечение Соединением А по сравнению с пациентами, несоответствующими одному или нескольким критериям из списка: гистологический диагноз НАСГ, показатель фиброза 1-3 включительно (F1 ограничен на уровне 30%), PDFF> 10% по показанием МРТ, скомпенсированное заболевание печени со следующими гематологическими и биохимическими критериями при поступлении на лечение: ALT <5 х ULN, AST> 30, гемоглобин >11 г/дл для женщин и >12 г/дл для мужчин, лейкоциты (WBC) >2,5 тыс/мкл, количество нейтрофилов >1,5 тыс/мкл, тромбоцитов >100 тыс/мкл, общий билирубин <35 мкмоль/л (хотя пациенты с билирубином >35 мкмоль/л могут быть включены при условии, что это это неконъюгированный билирубин при синдроме Гилберта), альбумин >36 г/л, международное нормализованное отношение (МНО) <1,4, креатинин сыворотки <1,3 мг/дл (мужчины) или <1,1 (женщины) или предполагаемая скорость клубочковой фильтрации ≥60 мл/мин/1,73 м2, при отсутствии других причин хронического заболевания печени (таких как, например, аутоиммунный, первичный желчный холангит, HBV, HCV, болезнь Вильсона, дефицит α-1-антитрипсина, гемохроматоз) и/или, если применимо, компенсированный диабет 2 типа (с показателями HgbA1c <9,5% и гликемии натощак <10 ммоль/л, при отсутствии изменений в лечении в течение предыдущих 6 месяцев, и/или при отсутствии новых симптомов, связанных с декомпенсированным диабетом в течение предыдущих 3 месяцев).
И, наоборот, у пациентов, удовлетворяющих одному или нескольким из следующих критериев из следующего списка, может не проявляться ответ на лечение Соединением А по сравнению с пациентами, у которых такой же один или несколько критериев отсутствуют. Список критериев включает: в анамнезе - длительное употребление избыточного алкоголя, нестабильное метаболическое состояние (например, прирост или потеря веса более чем на 5 кг за последние три месяца, диабет с плохим гликемическим контролем (HgbA1c> 9,5%) или применение антидиабетического или лекарственного средства против ожирения, мальабсорбционный синдром или рестриктивное бариатрическое (потеря веса) хирургическое вмешательство в течение последних 6 месяцев до скрининга); наличие в анамнезе желудочно-кишечной мальабсорбционной бариатрической операции в течение менее 5 лет до скрининга или прием препаратов, вызывающих стеатоз печени, включая кортикостероиды, высокие дозы эстрогенов, метотрексат, тетрациклин или амиодарон в предыдущие 6 месяцев; антиген HB>0, PCR HCV>0 (пациенты с инфекцией HCV в анамнезе могут быть включены, если PCR HCV отрицательна в течение более 3 лет), или ВИЧ-инфекция, диабет тип 1 или диабет тип 2 при текущем лечении препаратами инсулина, диабетический кетоацидоз, триглицериды натощак> 300 мг/дл, нарушения гемостаза или текущее лечение антикоагулянтами, текущие сердечные дисритмии или наличие таковых и/или сердечно-сосудистых заболеваний в анамнезе, включая инфаркт миокарда, за исключением пациентов с хорошо контролируемой артериальной гипертензией; любая клинически значимая аномалия ЭКГ и/или прием противодиабетических средств, которые, как известно, обладают активностью против НАСГ, как, например, пиоглитазон и агонисты рецептора GLP-1.
Соединения Формулы (I) и Формулы (II) могут быть получены, как описано, например, в заявках PCT WO2009/061208, WO2010/128401, WO2011/089529, WO2016/156912 и в соответствии с приведенными ниже Примерами. Кроме того, Соединение A может быть получено, как описано, например, в PCT WO2010/128401 и WO2014/132135 и в соответствии с Примером 2 ниже.
Приведенные ниже Примеры являются иллюстративными, и специалист в данной области техники поймет, как применять эти общие методы для получения других соединений в рамках Формулы (I) и Формулы (II). Соединения по настоящему изобретению могут находится в форме фармацевтически приемлемой соли или эфира. Например, соединения Формулы (I) и Формулы (II) могут быть в форме сложных эфиров, таких как фосфолипид, глицерид или C1-C6-алкиловый эфир. По меньшей мере, в одном варианте осуществления сложный эфир выбирают из глицерида или C1-C6-алкилового эфира. По меньшей мере, в одном варианте осуществления сложный эфир выбирают из триглицерида, 1,2-диглицерида, 1,3-диглицерида, 1-моноглицерида, 2-моноглицерида, метилового эфира, этилового эфира, пропилового эфира, изопропилового эфира, n-бутилового эфира и трет-бутилового эфира. По меньшей мере, в одном варианте осуществления Соединение Формулы (I) присутствует в виде метилового эфира, этилового эфира, изопропилового эфира, n-бутилового эфира или трет-бутилового эфира, например, в виде метилового эфира или этилового эфира. Как правило, сложные эфиры, представленные Формулой (I) (например, этиловые эфиры), будут гидролизоваться в желудочно-кишечном тракте.
Соли, подходящие для настоящего раскрытия, включают, не ограничиваясь таковыми, соли NH4+; ионы металлов, такие как Li+, Na+, K+, Mg2+ или Ca2+; протонированные первичные амины, такие как трет-бутиламмоний, (3S, 5S, 7S)-адамантан-1-аммоний, 1,3-дигидрокси-2- (гидроксиметил) пропан-2-аммоний, протонированный аминопиридин (например, пиридин-2- аммоний); протонированные вторичные амины, такие как диэтиламмоний, 2,3,4,5,6-пентагидрокси-N-метилгексан-1-аммоний, N-этилнафталин-1-аммоний, протонированные третичные амины, такие как 4-метилморфолин-4-ий, протонированные четвертичные амины, такие как 2-гидрокси-N, N,N-триметилэтан-1-аминиум и протонированный гуанидин, такой как амино((4-амино-4-карбоксибутил)амино)метаниминий, или протонированные гетероцикличные соединения, такие как 1H-имидазол-3-иум. Дополнительные примеры подходящих солей включают соли дипротонированных диаминов, таких как этан-1,2-диаммоний или пиперазин-1,4-дииум. Другие соли, согласно настоящему раскрытию, могут включать протонированный Хитозан:
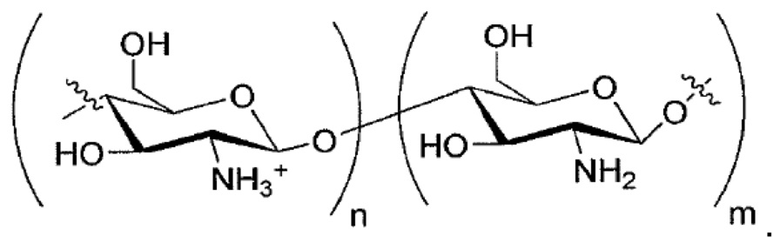
По меньшей мере, в варианте осуществления соли выбраны из соли натрия, соли кальция и соли холина. В одном варианте осуществления соль представляет собой соль натрия или кальция.
Настоящее изобретение относится к способу лечения НАСГ или АСГ у субъекта, нуждающегося в этом, и включающему введение субъекту фармацевтически эффективного количества соединения Формулы (I) или Формулы (II). Субъект может быть человеком или млекопитающим, не являющимся человеком. Соединения, раскрытые в настоящее время, могут быть введены в виде лекарственного средства, такого как фармацевтическая композиция. В одном аспекте изобретение относится к фармацевтической композиции, содержащей соединение Формулы (II), такое как соединение Формулы (I), такое как соединение A, для применения при лечении неалкогольного стеатогепатита. Композиция, раскрытая в настоящее время, может содержать по меньшей мере одно раскрытое здесь соединение и, необязательно, по меньшей мере, один неактивный фармацевтический ингредиент, то есть эксципиент. Неактивные ингредиенты могут растворять, суспендировать, сгущать, разбавлять, эмульгировать, стабилизировать, сохранять, защищать, окрашивать, ароматизировать и/или по-другому превращать активные ингредиенты в подходящий и эффективный препарат, таким образом, что препарат становится безопасным, удобным и/или, в противном случае, приемлемым для использования. Примеры эксципиентов включают, но не ограничиваются ими, растворители, носители, разбавители, связующие вещества, наполнители, подсластители, ароматы, модификаторы рН, модификаторы вязкости, антиоксиданты, наполнители, увлажнители, дезинтегрирующие агенты, агенты, замедляющие растворение, ускорители абсорбции, смачивающие агенты, абсорбенты, смазки, красители, диспергаторы и консерванты. Вспомогательные вещества могут иметь более одной роли или функции или могут быть отнесены к более чем одной группе; классификации носят только описательный характер и не предназначены для ограничения объема изобретения. В некоторых вариантах, например, по меньшей мере, один эксципиент может быть выбран из кукурузного крахмала, лактозы, глюкозы, микрокристаллической целлюлозы, стеарата магния, поливинилпирролидона, лимонной кислоты, винной кислоты, воды, этанола, глицерина, сорбита, полиэтиленгликоля, пропиленгликоля цетилстеарилового спирта, карбоксиметилцеллюлозы и жирных веществ, таких как твердые жиры, или подходящих смесей таковых. В некоторых вариантах осуществления раскрытые в настоящее время композиции содержат, по меньшей мере, одно соединение Формулы (II), такое как одно из соединений Формулы (I), и, по меньшей мере, один фармацевтически приемлемый антиоксидант, например, токоферол, такой как альфа-токоферол, бета-токоферол, гамма-токоферол и дельта-токоферол или смесь таковых, BHA, такие как 2-трет-бутил-4-гидроксианизол и 3-трет-бутил-4-гидроксианизол, или смеси таковых, и BHT (3,5-ди-трет-бутил)-4-гидрокситолуол) или смеси таковых.
Композиции, раскрытые в данном документе, могут быть составлены в форме для перорального введения, например, в виде таблеток или желатиновых мягких или твердых капсул. Лекарственная форма может иметь любую форму, подходящую для перорального введения, такую как сферическая, овальная, эллипсоидальная, кубическая, правильная и/или неправильная форма. Обычные методики приготовления, известные в данной области техники, могут быть использованы для приготовления соединений в соответствии с настоящим раскрытием. В некоторых вариантах осуществления композиция может быть в форме желатиновой капсулы или таблетки.
Подходящая суточная дозировка соединения, раскрытого в данном документе, такого как соединение Формулы (I) или соединение Формулы (II), может составлять от около 5 мг до около 4 г, например от около 5 мг до около 2 г. Например, в некоторых вариантах осуществления суточная доза составляет от около 10 мг до около 1,5 г, от около 50 мг до около 1 г, от около 100 мг до около 1 г, от около 150 мг до около 900 мг, от около 50 мг до около 800 мг, от около 100 мг до около 800 мг, от около 100 мг до около 600 мг, от около 150 до около 550 мг или от около 200 до около 500 мг. По меньшей мере, в одном варианте осуществления суточная доза составляет от около 200 до около 600 мг. По меньшей мере, в одном варианте осуществления суточная доза составляет около 50 мг, около 100 мг, около 150 мг, около 200 мг, около 250 мг, около 300 мг, около 350 мг, около 400 мг, около 450 мг, около 500 мг, около 550 мг, около 600 мг, около 650 мг, около 700 мг, около 750 мг, около 800 мг, около 850 мг или около 900 мг. Соединение(я) можно вводить, например, один, два или три раза в день.
По меньшей мере, в одном варианте осуществления соединение Формулы (I) вводят в количестве от около 200 до около 800 мг на дозу. По меньшей мере, в одном варианте осуществления соединение Формулы (I) вводят один раз в день. По меньшей мере, в одном варианте осуществления соединение Формулы (I) вводят один раз в день в дозе 750 мг. В некоторых вариантах осуществления соединение Формулы (I) вводят один раз в день в дозе 600 мг. В некоторых вариантах осуществления соединение Формулы (I) вводят один раз в день в дозе 500 мг. В некоторых вариантах осуществления соединение Формулы (I) вводят один раз в день в дозе 300 мг. В некоторых вариантах осуществления соединение Формулы (I) вводят один раз в день в дозе 250 мг. Предпочтительно, соединение Формулы (I) вводят один раз в день в дозе 300 или 600 мг.
По меньшей мере, в одном варианте осуществления соединение Формулы (II) вводят в количестве от около 200 до около 800 мг на дозу. По меньшей мере, в одном варианте соединение Формулы (II) вводят один раз в день. По меньшей мере, в одном варианте осуществления соединение Формулы (II) вводят один раз в день в дозе 750 мг. В некоторых вариантах осуществления соединение Формулы (II) вводят один раз в день в дозе 600 мг. В некоторых вариантах осуществления соединение Формулы (II) вводят один раз в день в дозе 500 мг. В некоторых вариантах осуществления соединение Формулы (II) вводят один раз в день в дозе 300 мг. В некоторых вариантах осуществления соединение Формулы (II) вводят один раз в день в дозе 250 мг. Предпочтительно, соединение Формулы (II) вводят один раз в день в дозе 300 или 600 мг.
Согласно настоящему раскрытию, соединение Формулы (I) или (II) можно вводить в виде монотерапии или в комбинации с одним или несколькими дополнительными активными агентами. Для совместного введения дополнительный активный агент предпочтительно представляет собой терапевтически активный агент, такой как лекарственное средство, которое, предпочтительно, оказывает терапевтическое воздействие при НАСГ или АСГ, например, влияет на один или несколько факторов, участвующих в развитии и/или ухудшении состояния при НАСГ или АСГ. Предпочтительно, использование комбинированного продукта, то есть соединения Формулы (I) или (II) в сочетании с одним или несколькими дополнительными активными агентами, оказывает синергетический эффект при профилактике и/или лечении НАСГ или АСГ. В одном варианте осуществления один или несколько дополнительных терапевтических агентов независимо выбраны из группы ингибиторов аллостерической ацетил-СоА-карбоксилазы (ACC), антагонистов рецептора ангиотензина II, ингибиторов ангиотензинпревращающего фермента (ACE), ингибиторов киназы-1 (ASK1), регулирующей сигнал апоптоза, ингибиторов каспазы, ингибиторов катепсина B, антагонистов хемокинов CCR2, антагонистов хемокинов CCR5, стимуляторов хлоридных каналов, солюбилизаторов холестерина, ингибиторов диацилглицерола O-ацилтрансферазы 1 (DGAT1), ингибиторов дипептидилпептидазы IV (ингибитор DPP IV), агонистов фактора роста фибробластов (FGF) -21, агонистов фарнезоидного рецептора X (FXR), анти-CD3 mAb, ингибиторов галектина-3, агонистов глюкагоноподобного пептида 1 (GLP1), предшественников глутатиона, ингибиторов протеазы NS3 вируса гепатита C, ингибиторов HMG, ингибиторов CoA-редуктазы, ингибиторов 1-β-гидроксистероиддегидрогеназы (I--HSD1), ингибиторов белка теплового шока (Hsp) 47, антагонистов IL-Iβ, антагонистов IL-6, агонистов IL-10, антагонистов IL-17, ингибиторов ко-транспортера желчных кислот, аналогов лептина, ингибиторов 5-липоксигеназы, стимуляторов гена LPL, ингибиторов гомолога (LOXL2) лизилоксидазы 2, антагонистов рецепторов лизофосфатидной кислоты 1 (LPA1), омега-3 жирных кислот, ингибиторов PDE3, ингибиторов PDE4, ингибиторов фосфолипазы C (PLC), агонистов PPARa, агонистов PPARy, агонистов PPAR5, рекомбинантного человеческого белка пентраксина-2 (PRF-1), ингибиторов Rho-ассоциированной протеинкиназы 2 (ROCK2), ингибиторов чувствительной к семикарбазиду аминоксидазы (SSAO), ингибиторов натрий-глюкозного транспортера типа 2 (SGLT2), ингибиторов стеароил-СоА десатуразы-1, агонистов рецепторов гормонов щитовидной железы, ингибиторов лигандов фактора некроза опухолей (TNFα), ингибиторов трансглутаминазы, предшественников ингибиторов трансглутаминазы и малых активирующих РНК (saRNA).
В частности, в некоторых вариантах осуществления один или несколько дополнительных активных агентов выбраны из группы, включающей: агонист глюкагоноподобного пептида 1 (GLP-1), ингибитор дипептидилпептидазы (антагонисты DPP-4) и омега-3 (n-3) жирные кислоты.
Агонисты рецептора глюкагоноподобного пептида-1, также известные как агонисты рецептора GLP-1 или миметики инкретина, являются агонистами рецептора GLP-1. Этот класс лекарств обычно используется для лечения диабета 2 типа. Неограничивающий список примеров GLP-агонистов включает: эксенатид, лираглутид, ликсисенатид, альбиглютид, дулаглутид, таспоглютид и семаглутид.
В некоторых вариантах осуществления дополнительный активный агент представляет собой ингибитор дипептидилпептидазы (антагонист DPP-4). Антагонисты DPP-4 представляют собой класс пероральных гипогликемических средств, блокирующих DPP-4 (DPP-IV). Их можно использовать для лечения сахарного диабета 2 типа. Глюкагон повышает уровень глюкозы в крови, а ингибиторы DPP-4 снижают уровень глюкагона и глюкозы в крови. Механизм ингибиторов DPP-4 заключается в повышении уровней инкретина (GLP-1 и GIP), ингибирующих высвобождение глюкагона, что, в свою очередь, увеличивает секрецию инсулина, уменьшает опорожнение желудка и снижает уровень глюкозы в крови. Неограничивающий Примерный список ингибиторов дипептидилпептидазы включает: Ситаглиптин, Вилдаглиптин, Саксаглиптин, Линаглиптин, Гемиглиптин, Анаглиптин, Тенелиглиптин, Алоглиптин, Трелаглиптин, Омариглиптин, Эвоглиптин, Эвтоглиптин, Дутоглиптин.
В некоторых вариантах осуществления дополнительный агент представляет собой омега-3 жирную кислоту. Когда дополнительный активный агент представляет собой омега-3 жирную кислоту, омега-3 жирная кислота обычно является длинноцепочечной полиненасыщенной омега-3 жирной кислотой (LC n-3 PUFA). Предпочтительно, это включает, по меньшей мере, одну из (все-Z омега-3) -5,8,11,14,17-эйкозапентаеновых кислот (EPA) и одну из (все-Z омега-3) -4,7,10,13, 16,19-докозагексаеновых кислот (DHA) или производные таковых. Кислоты n-3 PUFA, включая EPA и DHA, могут быть в разных формах и представлены, по меньшей мере, одной из свободных форм жирных кислот; этерифицированной формой, представленной С1-С4 алкиловым эфиром и, предпочтительно, этиловым эфиром; фосфолипидами; моно/ди/триглицеридами; и солями таковых. Жирная кислота омега-3 может быть предоставлена в форме композиции, такой, как композиция для перорального введения. Такая композиция может содержать, по меньшей мере, 40%, например, по меньшей мере, 50, 60, 70 или 80% активной омега-3 жирной кислоты. В некоторых вариантах осуществления дополнительный активный агент представляет собой композицию, содержащую, по меньшей мере, одну из EPA и DHA, предпочтительно в форме этилового эфира, в концентрации, по меньшей мере, 70%.
В некоторых вариантах осуществления один или несколько дополнительных активных агентов независимо выбраны из группы, включающей ацетилсалициловую кислоту, алипоген типарвовек, арамхол, аторвастатин, BI 1467335, BLX-1002, BMS-986036, BMS-986020, ценицевирок (cenicriviroc), кобипростон, колесевелам, эмриказан., эналаприл, форамулаб, GFT-505, GR-MD-02, GS-0976, GS-9674, гидрохлоротиазид, этиловый эфир эйкозапента (этиловый эфир эйкозапентаеновой кислоты, этиловый эфир EPA), IMM-124E, IVA337, K-877, KD- 025, линаглиптин, лираглутид, меркаптамин, MGL-3196, ND-L02-s0201, обетихоловую кислоту, олесоксим, пегилодекакин, пиоглитазон, PRM-151, PX-102, ремоглифлозин этабонат, SHE-S26TUSB 6 солитромицин, типелукаст, TRX-318, урсодезоксихолевую кислоту и VBY-376. Первый компонент комбинированного продукта, то есть соединение Формулы (I) или (II), можно вводить или формулировать любым способом, как описано выше. Второй компонент комбинированного продукта, дополнительный активный агент, может быть сформулирован так, чтобы способ подходил для того типа агента, каковым он является в зависимости от нескольких факторов, включающих способ введения агента. Доза дополнительного активного агента зависит от типа выбранного агента и должна соответствовать утвержденным количествам для конкретного агента. Как указано в Примерах, модель НАСГ, индуцированная диетой CDAA (метионин-холин-дефицитная), представляет собой модель как фиброза, так и воспаления печени. Как подтверждается Примерами, комбинация агониста GLP-1 с соединением Формулы (I) или (II), таким как соединение A, обладает превосходящим эффектом по сравнению с лечением одним только GLP-1 при лечении фиброза и воспаления, связанных с НАСГ. Поскольку фиброз печени является в значительной степени ответом на воспалительные реакции, данный эффект также подтверждается продемонстрированными изменениями в экспрессии гена печени, ассоциированной с воспалением (TNF-α), таким образом подтверждая превосходство кислород-содержащих структурно-модифицированных жирных кислот (Соединение А) в сочетании с агонистом GLP-1 по сравнению с применением только одного агониста GLP-1. В целом данные свидетельствуют о том, что комбинация агониста GLP-1 (или, альтернативно, антагонистов DPP-4) с соединением по настоящему изобретению может обладать синергетическим эффектом при лечении как воспаления печени, так и фиброза.
Хотя стеатоз печени сам по себе может быть доброкачественным состоянием, он способен повышать чувствительность печени ко «второму удару» при действии других факторов, вызывающих воспалительный ответ. Поэтому желательно снижение стеатоза как с точки зрения его вклада в оценку по шкале НАСГ, так и с точки зрения определения НАСГ как потенциального «первичного» фактора для других уже имеющихся или повторных поражений печени. Новые значительные улучшения в отношении липидов печени, достигнутые с помощью комбинации высоких доз омега-3 этиловых эфиров и низкой дозы кислород-содержащей структурно-модифицированной жирной кислоты (соединение А), дают основание предполагать, что достигается синергетический эффект, тем более, что ни одно соединение не вызывает значительного снижения уровня печеночных триглицеридов (TG) у трансгенных мышей ApoE*3L-CETP.
Примечательно, что Соединение А, в количестве доли дозы этиловых эфиров омега-3 значительно повышало или понижало >1094 зондов для определения экспрессии генов в печени трансгенных мышей APOE*3.CETP по сравнению с количеством <10 для омега-3 этиловых эфиров (данные не показаны). Не будучи связанными теорией, этот различающийся эффект на транскриптом печени может свидетельствовать о том, что наблюдаемый синергетический эффект является вторичным по отношению к различным каскадам реакций, а не эффектом накопления дозы. Его существенное влияние на содержание холестерина в печени может также распространятся на воспалительные реакции, связанные с патогенезом НАСГ и АСГ. В целом, данные, полученные на мышах APOE*3.CETP говорят в пользу комбинации омега-3 PUFA, такой, как комбинации этиловых эфиров EPA и кислород-содержащей структурно модифицированной жирной кислоты в соответствии с Формулой (I) или (II) в качестве сильнодействующей терапии для уменьшения стеатоза печени и, следовательно, лечения НАСГ и/или АСГ.
Соединения Формулы (II) или, предпочтительно, Формулы (I) можно вводить в виде монотерапии или в комбинации по меньшей мере с одним дополнительным активным агентом для лечения и/или реверсии неалкогольного стеатогепатита (НАСГ) или алкогольного стеатогепатита (АСГ). В некоторых вариантах, по меньшей мере, один дополнительный активный агент представляет собой агонист GLP-1. Кроме того, на основании результатов, полученных на трансгенных мышах APOE*3.CETP, соединения Формулы (II) или, предпочтительно, Формулы (I) можно вводить в комбинации с, по меньшей мере, одним дополнительным активным агентом для терапевтического и/или профилактического лечения и/или реверсии стеатоза печени (содержание триглицеридов и/или холестерина). В некоторых вариантах осуществления по меньшей мере один дополнительный активный агент представляет собой омега-3 жирную кислоту, такую как этиловый эфир омега-3 PUFA.
Вышеприведенные Примеры подчеркивают возможность сочетания кислород-содержащих структурно модифицированных жирных кислот с рядом других активных агентов для лечения НАСГ и/или АСГ. Эти комбинации могут не только улучшить результаты, связанные с эффективностью действия комбинации по сравнению с монотерапией, но также могут улучшить безопасность применения. В качестве примера улучшения безопасности продемонстрировано, что Соединение A значительно снижает экспрессию CETP у мышей APOE*3.CETP и улучшает липидные профили как у грызунов, так и у людей. Оно может быть выгодно для применения в комбинации с обетихоловой кислотой (агонистом FXR), которая, в отличие от Соединения А, увеличивает экспрессию CETP и ухудшает липидные профили как у мышей APOE * 3.CETP, так и у людей.
Авторы настоящего изобретения обнаружили, что соединения Формулы (I), такие как 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановая кислота, обладают удивительно хорошей фармацевтической активностью. Неожиданно, соединения Формулы (I), раскрытые в настоящем документе, проявляют улучшенную биологическую активность по сравнению с агонистом рецептора глюкагоноподобного пептида 1 (GLP-1R) в лечении связанного с НАСГ фиброза и воспаления печени. Соединения Формулы (I) также, возможно, проявляют синергетический эффект при введении в комбинации с дополнительными активными агентами.
Примеры
Настоящее раскрытие может быть дополнительно описано следующими неограничивающими Примерами, в которых стандартные методы, известные специалисту-химику, и методы, аналогичные описанным в этих Примерах, могут быть использованы там, где это применимо. Понятно, что специалисту в данной области техники будут понятны дополнительные варианты осуществления, согласующиеся с раскрытым здесь описанием.
Если не указано иное, реакции проводили при комнатной температуре, обычно в интервале 18-25°С с растворителями класса ВЭЖХ (HPLC) в безводных условиях. Выпаривание проводили роторным испарением в вакууме. Колоночную хроматографию выполняли с помощью процедуры флэш-хроматографии на силикагеле 40-63 мкм (Merck) или с помощью Armen Spotflash с использованием предварительно упакованных колонок с силикагелем «MiniVarioFlash», «SuperVarioFlash», «SuperVarioPrep» или «EasyVarioPrep» (Merck). Значения сдвига ядерного магнитного резонанса (ЯМР) регистрировали на приборе Bruker Avance DPX 200 или 300 с кратностями пиков, описанными следующим образом: s, синглет; d, дублет; dd, двойной дублет; t, триплет; q, квартет; p, пентет; m, мультиплет; br- уширенный спектр. Масс-спектры регистрировали с помощью LC/MS-спектрометра. Разделение проводили с использованием модуля серии Agilent 1100 на колонке Eclipse XDB-C18 2,1×150 мм с градиентным элюированием. В качестве элюента использовали градиент 5-95% ацетонитрила в буферах, содержащих 0,01% трифторуксусной кислоты или 0,005% формиата натрия. Масс-спектры регистрировали с помощью масс-спектрометра Gl956A (электрораспыление, 3000 В), переключающего режим положительной и отрицательной ионизации. Указанный выход продукции является иллюстративным и необязательно отражает максимально возможный выход.
Приготовление соединений
Пример 1. Получение трет-бутил-2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -икозо-5,8,11,14,17-пентаен-1-илокси) бутаноата:
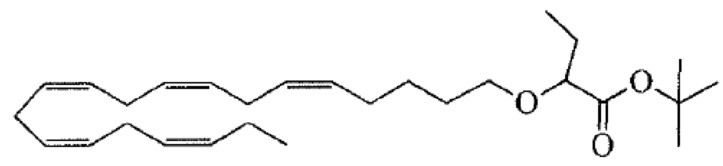
Хлорид тетрабутиламмония (0,55 г, 1,98 ммоль) добавляли к раствору (5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ола (3,50 г, 12,1) ммоль) в толуоле (35 мл) при комнатной температуре в атмосфере азота. Водный раствор гидроксида натрия (50% (вес/вес), 11,7 мл) добавляли при интенсивном перемешивании при комнатной температуре, а затем трет-бутил-2-бромбутират (5,41 г, 24,3 ммоль). Полученную смесь нагревали до 50°С, и через 1,5 часа (2,70 г, 12,1 ммоль), 3,5 часа (2,70 г, 12,1 ммоль) и 4,5 часа (2,70 г, 12,1 ммоль) добавляли дополнительный трет-бутил-2-бромбутират и перемешивали в течение 12 часов. После охлаждения до комнатной температуры добавляли ледяную воду (25 мл) и полученные две фазы разделяли. Органическую фазу промывали смесью NaOH (5%) и насыщенным солевым раствором, сушили (MgSO4), фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (100: 0 -> 95: 5). Концентрирование соответствующих фракций дало 1,87 г (36% выход) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,10 (м, 6H), 1,35-1,54 (м, 11H), 1,53-1,87 (м, 4H), 1,96-2,26 (м, 4H), 2,70-3,02 (м, 8Н), 3,31 (дт, 1Н), 3,51-3,67 (м, 2Н), 5,10-5,58 (м, 10Н).
Пример 2. Получение 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутановой кислоты (соединение A):
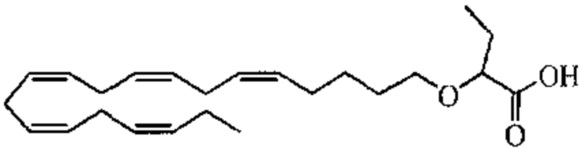
Трет-бутил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноат (19,6 г, 45,5 ммоль) растворяли в дихлорметане (200 мл) и помещали в азот. Затем добавляли трифторуксусную кислоту (50 мл) и реакционную смесь перемешивали при комнатной температуре в течение одного часа. Вода была добавлена и водную фазу дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана, этилацетата и муравьиной кислоты (90: 10: 1 -> 80: 20: 1). Концентрирование соответствующих фракций дало 12,1 г (выход 71%) указанного в заголовке соединения в виде масла. 1H-ЯМР (300 МГц, CDCl3): δ 0,90-1,00 (м, 6H), 1,50 (м, 2H), 1,70 (м, 2H), 1,80 (м, 2H), 2,10 (м, 4H), 2,80- 2,90 (м, 8H), 3,50 (м, 1H), 3,60 (м, 1H), 3,75 (т, 1H), 5,30-5,50 (м, 10H); МС (электрораспыление): 373,2 [М-Н] -.
Пример 3. Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата кальция
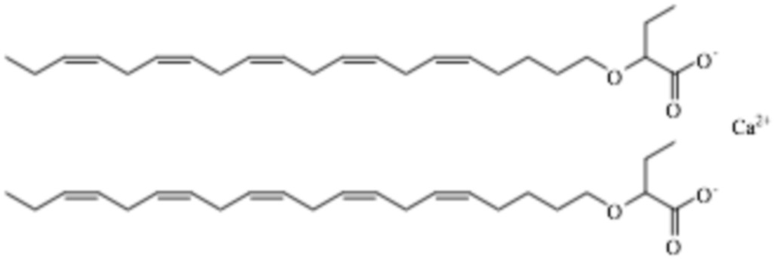
2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -Эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановая кислота (1,87 г, 4,99 ммоль, 93%) была смешана с CaCO3 (0,25 г, 2,50 ммоль). Добавляли воду (1 мл) и смесь перемешивали при механическом перемешивании при комнатной температуре в течение 1 часа. CO2 образовался. Плотная однородная паста была сформирована. При перемешивании добавляли ацетон (7 мл). Твердый материал начинал отделяться. Твердое вещество отфильтровывали и сушили над азотом, герметично закрывали и хранили в холодильнике при 4°С. Выход: 1,86 г (95%). Твердое вещество не было дополнительно охарактеризовано аналитическими или спектроскопическими методами, но было проведено несколько экспериментов, показывающих, что образовалась соль кальция.
• Твердый материал плавится на горячей плите при температуре ниже 100°C. Конкретная точка плавления не была определена
• Материал не выделяет CO2 при добавлении кислоты, но «растворяется» и осаждается в виде масла.
Пример 4. Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата натрия
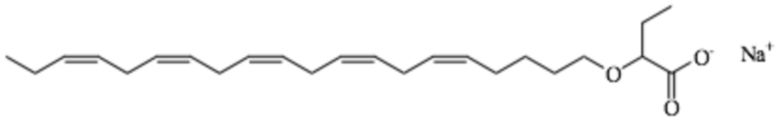
2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановая кислота (1,87 г, 4,99 ммоль, 93%) была смешана с NaHCO3 (0,420 г, 5,00 ммоль). Добавляли воду (1 мл) и смесь перемешивали при механическом перемешивании при комнатной температуре в течение 1 часа. Выделялся CO2 и сформировалась густая однородная паста. При перемешивании в реакционную колбу добавляли этанол (7 мл). Натриевая соль, образованная из 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -Эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты, переходит в раствор при добавлении этанола (7 мл). Небольшие количества непрореагировавшего NaHCO3 отфильтровывали и раствор упаривали досуха. Сырое, слегка вязкое масло упаривали два раза с 96% этанолом для удаления следов воды.
Пример 5. Получение 2-гидрокси-N, N, N-триметилэтан-1-аминия 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен- 1-ил) окси) бутановой кислоты
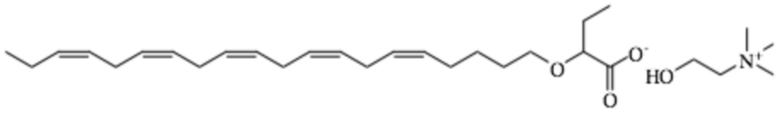
Гидроксид холина (327,7 мкл) в воде пипеткой переносили в сцинтилляционный флакон, содержащий Примерно 2,5 мл MTBE и 7,5 мл n-гептана. Внутри азотной камеры 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановая кислота (500 мг, 95,8%) была перенесена во флакон. В азотной камере медленно и при перемешивании добавляли приблизительно 1,0 мл воды во флакон. Флакон был затем запечатан. Реакционную смесь перемешивали в течение приблизительно 30 минут. Образовавшаяся 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) холиновая соль бутановой кислоты представляла собой жесткий гелеобразный материал, который был отфильтрован на воронке Бюхнера. Влажный материал на фильтре промывали 3 раза, используя 1 мл MTBE. Промытый материал представлял собой твердое гелеобразное твердое вещество.
Пример 6. Получение (4S, 5R) -3 - ((S) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутаноила) - 4-метил-5-фенилоксазолидин-2-он и (4S, 5R) -3 - ((R) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14, 17-pentaenyloxy) бутаноил) -4-метил-5-фенилоксазолидин-2-она:
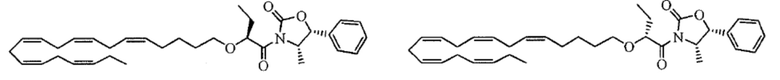
DMAP (1,10 г, 8,90 ммоль) и DCC (1,90 г, 9,30 ммоль) добавляли к смеси 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутановой кислоты (3,20 г, 8,50 ммоль) в сухом дихлорметане (100 мл), выдерживаемую при 0°C в атмосфере азота. Полученную смесь перемешивали при 0°С в течение 20 минут. Добавляли (4S, 5R) -4-метил-5-фенилоксазолидин-2-он (1,50 г, 8,50 ммоль) и полученную мутную смесь перемешивали при температуре окружающей среды в течение пяти дней. Смесь фильтровали и концентрировали при пониженном давлении, получая сырой продукт, содержащий желаемый продукт в виде смеси двух диастереомеров. Остаток очищали флэш-хроматографией на силикагеле, используя 15% этилацетат в гептане в качестве элюента. Два диастереомера разделяли и соответствующие фракции концентрировали. (4S, 5R) -3 - ((S) -2- ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутаноил) -4-метил-5 - фенилоксазолидин-2-он элюировался первым с выходом в количестве 1,1 г (выход 40%) в виде масла. (4S, 5R) -3 - ((R) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутаноил) -4-метил-5 -фенилоксазолидин-2-он был получен в виде масла с выходом 0,95 г (выход 34%).
(4S, 5R) -3 - ((S) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутаноил) -4-метил-5 -фенилоксазолидин-2-он (E1): 1H-ЯМР (300 МГц, CDCl3): δ 0,90 (д, 3H), 1,00 (т, 3H), 1,07 (т, 3H), 1,45-1,57 (м, 2H), 1,62-1,76 (м, 3H), 1,85-1,95 (м, 1H), 2,05-2,15 (м, 4H), 2,87 (м, 8H), 3,39 (м, 1H), 3,57 (м, 1H), 4,85-4,92 (м, 2H), 5,30- 5,45 (м, 10H), 5,75 (д, 1H), 7,32 (м, 2H), 7,43 (м, 3H).
(4S, 5R) -3 - ((R) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутаноил) -4-метил-5 -фенилоксазолидин-2-он (E2): 1H-ЯМР (300 МГц, CDCl3): δ 0,98 (д, 3H), 0,99 (т, 3H), 1,08 (т, 3H), 1,40-1,52 (м, 2H), 1,55-1,75 (м, 3H), 1,80-1,90 (м, 1H), 2,05-2,15 (м, 4H), 2,84 (м, 8H), 3,39 (м, 1H), 3,56 (м, 1H), 4,79 (пент, 1H), 4,97 (дд, 1H), 5,30-5,45 (м, 10H), 5,71 (д, 1H), 7,33 (м, 2H), 7,43 (м, 3H).
Пример 7: Получение (S) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутановой кислоты:
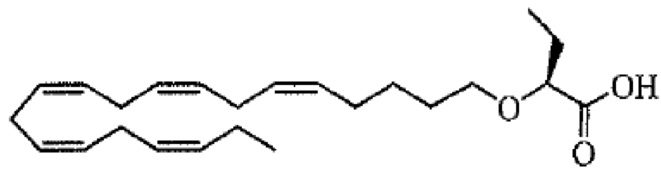
Перекись водорода (35% в воде, 0,75 мл, 8,54 ммоль) и моногидрат гидроксида лития (0,18 г, 4,27 ммоль) добавляли к раствору (4S, 5R) -3 - ((S) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутаноил) -4-метил-5-фенилоксазолидин-2-он (1,10 г, 2,13 ммоль) в тетрагидрофуране (12 мл) и воду (4 мл) выдерживали при 0°С в атмосфере азота. Реакционную смесь перемешивали при 0°С в течение 30 минут. Добавляли 10% Na2SO3 (водный) (30 мл), рН доводили до 2 с помощью 2 М HCl и смесь дважды экстрагировали гептаном (30 мл). Объединенный органический экстракт сушили (Na2SO4), фильтровали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (98: 8 -> 1: 1). Концентрирование соответствующих фракций дало 0,48 г (выход 60%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3): δ 0,90-1,00 (м, 6H), 1,48 (м, 2H), 1,65 (м, 2H), 1,85 (м, 2H), 2,10 (м, 4H), 2,80-2,90 (м, 8Н), 3,55 (м, 1Н), 3,60 (м, 1Н), 3,88 (т, 1Н), 5,35-5,45 (м, 10Н); МС (электрораспыление): 373,3 [М-Н]-; [α]D -37°; (с=0,104, этанол).
Пример 8. Получение (R) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутановой кислоты:
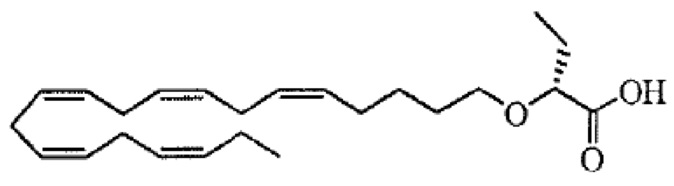
Перекись водорода (35% в воде, 0,65 мл, 7,37 ммоль) и моногидрат гидроксида лития (0,15 г, 3,69 ммоль) добавляли к раствору, содержащему (4S, 5R) -3 - ((R) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) бутаноил) -4-метил-5-фенилоксазолидин-2-он (0,95 г, 1,84 ммоль) в тетрагидрофуране (12 мл) и воду (4 мл), выдерживанному при 0°С в атмосфере азота. Реакционную смесь перемешивали при 0°С в течение 30 минут. Добавляли 10% Na2SO3 (водный) (30 мл), pH доводили до 2 с помощью 2 М HCl и смесь дважды экстрагировали гептаном (30 мл). Объединенный органический экстракт сушили (Na2SO4), фильтровали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (98: 8 -> 50:50). Концентрирование соответствующих фракций дала 0,19 г (выход 29%) указанного в заголовке соединения в виде масла. 1H-ЯМР (300 МГц, CDCl3): δ 0,90-1,00 (м, 6H), 1,48 (м, 2H), 1,65 (м, 2H), 1,85 (м, 2H), 2,10 (м, 4H), 2,80- 2,90 (м, 8H), 3,55 (м, 1H), 3,60 (м, 1H), 3,88 (т, 1H), 5,35-5,45 (м, 10H); МС (электрораспыление): 373,3 [М-Н]-; [α]D -31° (с=0,088, этанол).
Пример 9. Получение этил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата:
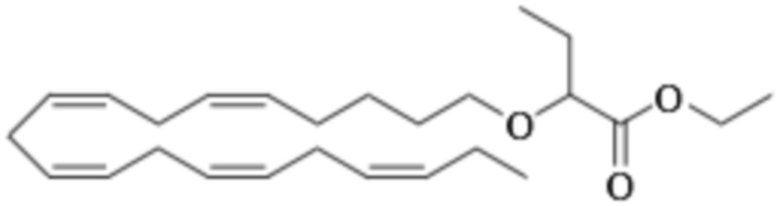
Дициклогексилметандимин (DCC) (304 мг, 1,47 ммоль) и N, N-диметилпиридин-4-амин (DMAP) (10 мг, 0,067 ммоль) добавляли к перемешиваемому раствору 2 - (((5Z, 8Z, 11Z, 14Z), 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (501,3 мг, 1,335 ммоль) в дихлорметане (DCM) (4 мл) при 0°C в атмосфере азота. Смесь перемешивали в течение 5 минут перед добавлением этанола (EtOH) (0,16 мл, 2,67 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь очищали флэш-хроматографией на силикагеле, используя прогрессивно полярные смеси гептана и этилацетата (100: 0 99: 1) в качестве элюента. Концентрирование соответствующих фракций дало 473 мг (выход 88%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, хлороформ-d) δ 0,95 (2х, 6H), 1,37-1,48 (м, 2H), 1,54-1,79 (м, 4H), 2,01-2,10 (м, 4H), 2,77-2,84 (м, 8H), 3,27-3,34 (м, 1H), 3,53-3,60 (м, 1H), 3,69-3,73 (дд, 1H), 4,13-4,24 (м, 2H), 5,25-5,33 (м, 10H), МС (электрораспыление); 425,3 [М+Na]+; HRMS (электрораспыление): найдено 425,3021 [M+Na]+, рассчитано для [C26H42O3+Na]+ 425.3031.
Пример 10. Получение изопропил-2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата
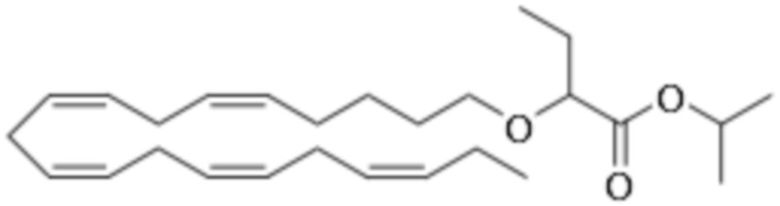
DCC (310 мг, 1,47 ммоль) и DMAP (9 мг, 0,067 ммоль) добавляли к перемешиваемому раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14, 17-пентаен-1-ил) окси) бутановой кислоты (501 мг, 1,335 ммоль) в DCM (4 мл) при 0°C в атмосфере азота. Смесь перемешивали в течение 5 минут перед добавлением изопропанола (iPrOH) (0,16 мл, 2,67 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 20 часов. Смесь фильтровали и концентрировали в вакууме. К остатку добавляли гептан (50 мл), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 1% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 496 мг (выход 89%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3): δ 0,97 (2х, 6Н), 1,25 (м, 6Н), 1,42-1,50 (м, 2Н), 1,61-1,70 (м, 2Н), 1,70-1,77 (м, 2Н), 2,05-2,12 (м, 4H), 2,79-2,86 (м, 8H), 3,29-3,34 (м, 1H), 3,54-3,59 (м, 1H), 3,67-3,71 (м, 1H), 5,06-5,10 (м, 1Н), 5,31-5,42 (м, 10Н); МС (электрораспыление): 439,3 [М+Na]+.
Пример 11. Получение метил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата:
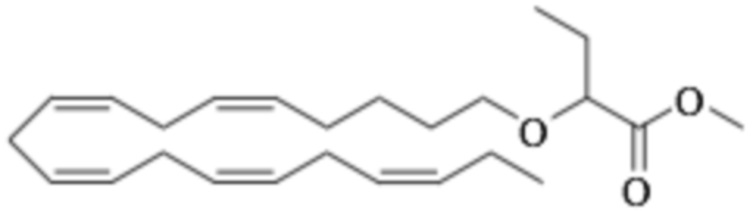
Серную кислоту (0,049 мл, 0,918 ммоль) добавляли к раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановуой кислоты ((385 мг, 1028 ммоль) в метаноле (20 мл) при комнатной температуре в атмосфере азота и полученную смесь перемешивали при комнатной температуре в течение ночи. MS (электрораспыление): 389,3 [М+1]+.
Пример 12. Получение бутил-2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата:
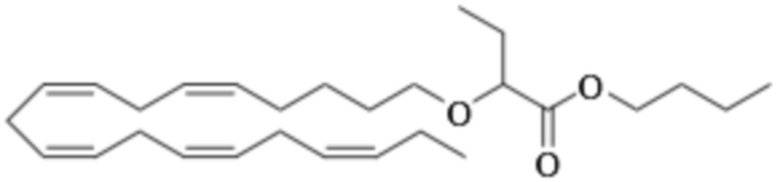
Серную кислоту (0,049 мл, 0,918 ммоль) добавляли к раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты ((33 г, 88 ммоль) в бутан-1-оле (400 мл, 4,37 моль) при комнатной температуре в атмосфере азота и реакционную смесь перемешивали в течение 120 ч. Добавляли гептан (400 мл) и этилацетат (400 мл) и раствор промывали насыщенным водным NaHCO3 (3х300 мл) и водой (2х300 мл). Объединенную водную фазу экстрагировали смесью гептан/эфир (1: 1) (2х300 мл). Фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (99: 1, 96: 4). Концентрация соответствующих фракций давала 26,3 г (67). % выхода) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 0,93-1,02 (м, 9H), 1,36-1,51 (м, 4H), 1,60-1,70 (м, 4H), 1,72-1,84 (м, 2H), 2,05-2,16 (м, 4H), 2,78-2,92 (м, 8H), 3,28-3,39 (м, 1H), 3,54-3,65 (м, 1H), 3,70-3,82 (м, 1H), 4,08-4,24 (м, 2H), 5,27 -5,48 (м, 10Н), МС (электрораспыление): 453,2 [М+Na]+.
Пример 13: Получение 2,3-дигидроксипропил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата:
Стадия а) Получение (2,2-диметил-1,3-диоксолан-4-ил) метил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17- пентаен-1-ил) окси) бутаноата
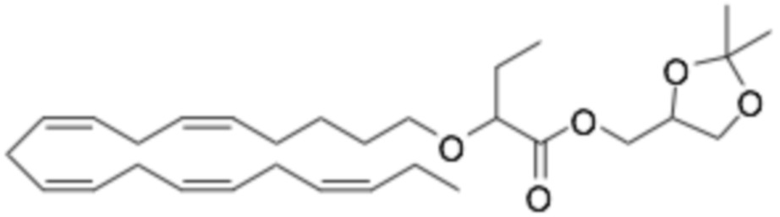
2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановая кислота (25 г, 66,7 ммоль) и DMAP (8,15 г, 66,7 ммоль) добавляли к раствору 2,2-диметил-1,3-диоксолан-4-метанола (7,54 мл, 60,7 ммоль) в хлороформе (150 мл) в атмосфере азота. Затем по каплям добавляли раствор DCC (13,77 г, 66,7 ммоль) в хлороформе (65 мл) при температуре окружающей среды. Смесь перемешивали в течение ночи и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси 10% этилацетата в гептане. Концентрирование соответствующих фракций дало 19,6 г (выход 66%) указанного в заголовке продукта в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,99 (т, 6Н), 1,37-1,40 (м, 3Н), 1,41-1,53 (м, 5Н), 1,59-1,71 (м, 2Н), 1,72-1,85 (м, 2Н), 2,05-2,14 (м, 4H), 2,74-2,95 (м, 8H), 3,31-3,38 (м, 1H), 3,57-3,65 (м, 1H), 3,72-3,86 (м, 2H), 4,07-4,12. (м, 1Н), 4,15-4,27 (м, 2Н), 4,29-4,40 (м, 1Н), 5,23-5,50 (м, 10Н). МС (электрораспыление): 511,3 [М+Na]+.
Стадия b) Получение 2,3-дигидроксипропил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата
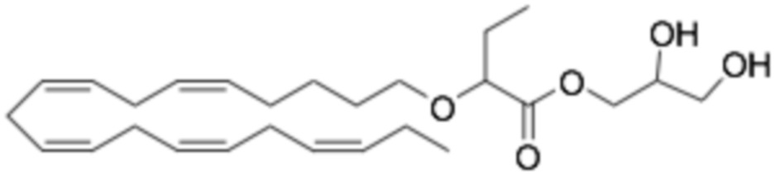
К раствору (2,2-диметил-1,3-диоксолан-4-ил) метил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17- пентаен-1-ил) окси) бутаноата (27,5 г, 56,3 ммоль) в диоксане (280 мл) при комнатной температуре в атмосфере азота добавляли водный раствор HCl (37% (вес/вес), 28 мл, 341 ммоль) и смесь перемешивали в течение 60 минут. Затем смесь осторожно выливали в сатурированный водный NaHCO3 (500 мл) и экстрагировали EtOAc (2х300 мл). Органическую фазу промывали 1 М HCl (200 мл), насыщенным солевым раствором (200 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя гептан и этилацетат (50:50) в качестве элюента. Концентрирование соответствующих фракций дало 19 г целевого продукта в виде масла, загрязненного ~ 10% изомера 1,3-дигидроксипропан-2-ил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза -5,8,11,14,17-пентаен-1-ил) окси) бутаноатом. Материал смешивали с 1,35 г другой партии перед дальнейшей очисткой препаративной ВЭЖХ. Изократическая смесь воды/ацетонитрила (9: 1) и ацетонитрила (100%) в соотношении 17:83 использовалась в качестве элюента. Концентрирование соответствующих фракций дало 11,3 г (выход 38%) указанного в заголовке продукта в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,97-1,03 (м, 6H), 1,41-1,51 (м, 2H), 1,59-1,69 (м, 2H), 1,72-1,87 (м, 2H), 2,05-2,14 (м., 5H), 2,56 (с, 1H), 2,73-2,94 (м, 8H), 3,33-3,40 (м, 1H), 3,55-3,68 (м, 2H), 3,69-3,77 (м, 1H), 3,79-3,85 (м, 1Н), 3,93-4,03 (м, 1Н), 4,15-4,37 (м, 2Н), 5,25-5,51 (м, 10Н). МС (электрораспыление): 471,3 [М+Na] +.
Пример 14. Получение 1,3-дигидроксипропан-2-ил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата
Стадия а) Получение оксиран-2-илметил-2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата
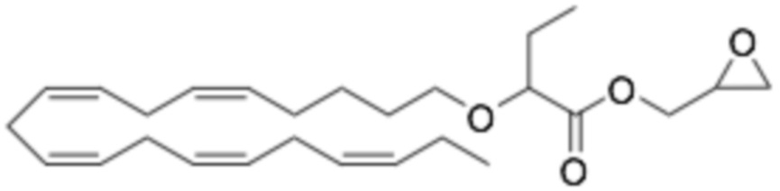
Смесь 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (800 мг, 2. 14 ммоль), глицидола (0,17 мл, 2,56 ммоль), 1- (3-диметиламинопропил) -3-этилкарбодиимид гидрохлорид (EDC * HCl) (491 мг, 2,56 ммоль) и 4-диметиламинопиридина (DMAP) (313 мг, 2,56 ммоль) в сухом DCM (7 мл) перемешивали при комнатной температуре в атмосфере азота. Реакционную смесь концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента все прогрессивно полярные смеси гептана и этилацетата (99: 1 ~ 95: 5). Концентрирование соответствующих фракций дало 527 мг (выход 57%) указанного в заголовке продукта в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 0,94-0,98 (м, 6H), 1,40-1,44 (м, 2H), 1,57-1,64 (м, 2H), 1,70-1,82 (м, 2H), 2,02-2,12 (м, 4H), 2,63 (ушир.с, 1H), 2,78-2,84 (м, 9H), 3,20 (ушир.с, 1H), 3,30-3,35 (м, 1H), 3,55-3,61 (м, 1H), 3,77-3,80 (м., 1H), 3,94-4,01 (м, 1H), 4,42-4,48 (м, 1H), 5,36-5,26 (м, 10H). МС (электрораспыление): 453,3 [М+Na] +.
Стадия b) Получение 2 - ((2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноил) окси) пропана -1,3-диил-бис (2,2,2-трифторацетата)

Трифторуксусный ангидрид (TFAA) (0,55 мл, 3,96 ммоль) в сухом DCM (3 мл) добавляли порциями к предварительно охлажденному раствору оксиран-2-илметил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза -5,8,11,14,17-пентаен-1-ил) окси) бутаноата (286 мг, 0,66 ммоль) в сухом DCM (3 мл) при -20°C в атмосфере азота. Охлаждающую баню убирали и смесь перемешивали в течение 19 часов при температуре окружающей среды, после чего реакционную смесь концентрировали в вакууме. Остаток растворяли в толуоле (6 мл) и пропускали через слой диоксида кремния (6,5 г), элюируя толуолом (150 мл). Концентрирование в вакууме дает 357 мг (выход 84%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 0,95 (2x т, 6H), 1,38-1,45 (м, 2H), 1,57-1,63 (м, 2H), 1,66-1,78 (м, 2H), 2,09-2,02 (м, 4H)), 2,78-2,84 (м, 8H), 3,27-3,33 (м, 1H), 3,51-3,56 (м, 1H), 3,77 (дд, 1H), 4,30-4,53 (м, 2H), 4,60-4,69 (м, 2H), 5,17-5,43 (м, 10H), 5,43-5,55 (м, 1H). МС (электрораспыление): 661,1 [М+Na]+.
Стадия c) Получение 1,3-дигидроксипропан-2-ил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата
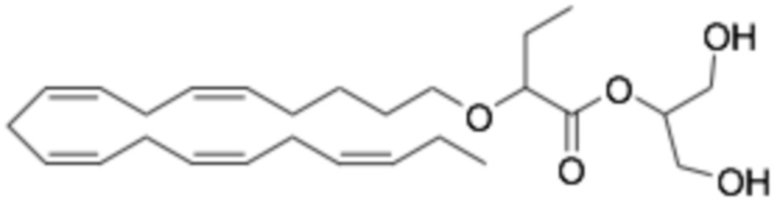
Раствор пиридина (0,4 мл, 4,95 ммоль) и метанола (0,3 мл, 7,41 ммоль) в пентане/DCM (2: 1) (4,5 мл) добавляли по каплям к раствору 2 - ((2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноил) окси) пропан-1,3-диил-бис (2,2,2- трифторацетат) (354 мг, 0,552 ммоль) в пентане/DCM (2: 1) (5 мл), охлажденный до -20°C в атмосфере азота. Охлаждающую баню убирали и смесь перемешивали в течение 3 часов при комнатной температуре, затем концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (95: 5 90:10 80:20 50:50). Концентрирование соответствующих фракций дало 223 мг указанного в заголовке продукта в виде сырой нефти. Очистка препаративной ВЭЖХ дает 58 мг (выход 22%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 0,95 (т, 3Н), 0,96 (т, 3Н), 1,38-1,45 (м, 2Н), 1,54-1,64 (м, 2Н), 1,67-1,84 (м, 2Н), 2,01-2,09 (м, 4H), 2,45 (ушир.с, 2H), 2,83-2,77 (м, 8H), 3,36-3,30 (м, 1H), 3,60-3,55 (м, 1H), 3,84-3,78 (м, 5H).), 4,98-4,93 (м, 1H), 5,65-5,09 (м, 10H). МС (электрораспыление): 471,1 [М+Na] +.
Пример 15. Получение 3-гидроксипропан-1,2-диил-бис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ила) окси) бутаноата)
Стадия а) Получение трет-бутил ((2,2-диметил-1,3-диоксолан-4-ил) метокси) диметилсилана
Трет-бутилхлордиметилсилан (14,41 г, 91 ммоль) добавляли к раствору (2,2-диметил-1,3-диоксолан-4-ил) метанола (10 г, 76 ммоль) и имидазола (7,73 г, 114). ммоль) в THF (100 мл) при температуре окружающей среды в атмосфере азота. Смесь перемешивали в течение 1,5 часов, выливали в воду (200 мл) и экстрагировали трет-бутилметиловым эфиром (2 × 150 мл). Фазы разделяли и органический слой промывали водой (100 мл), насыщенным солевым раствором (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 3% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 18 г (выход 97%) названного соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,02-0,05 (м, 6H), 0,85-0,89 (м, 9H), 1,31-1,35 (м, 3H), 1,35-1,40 (м, 3H), 3,50-3,60 (м, 1H), 3,63-3,72 (м, 1H), 3,75-3,85 (м, 1H), 3,96-4,05 (м, 1H), 4,07-4,18 (м, 1H). МС (электрораспыление): 229,2 [М+Na] +.
Стадия b). Получение 3 - ((трет-бутилдиметилсилил) окси) пропан-1,2-диола.
К раствору трет-бутил ((2,2-диметил-1,3-диоксолан-4-ил) метокси) диметилсилана в хлороформе (60 мл) добавляли FeCl3×6H2O, абсорбированный на силикагеле (30 г, 11,9 ммоль), и смесь перемешивали в течение ночи. Смесь фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (50:50 ~ 25:75). Концентрирование соответствующих фракций дало 760 мг (выход 9%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,09-0,12 (м, 6H), 0,91-0,95 (м, 9H), 2,11-2,17 (м, 1H), 2,60 (д, 1H), 3,57-3,85 (м, 5H). МС (электрораспыление): 229,2 [М+Na] +.
Стадия c) Получение 3 - ((трет-бутилдиметилсилил) окси) пропан-1,2-диил-бис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14, 17-пентаен-1-ил) окси) бутаноата)
К раствору 3 - ((трет-бутилдиметилсилил) окси) пропан-1,2-диола (0,91 г, 4,41 ммоль) в DMF (20 мл) в атмосфере азота при температуре окружающей среды добавляли 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановую кислоту (3,47 г, 9,3 ммоль), DMAP (1,13 г, 9,3 ммоль), 1- (3-диметиламинопропил) -3-этилкарбодиимид гидрохлорид (DCI) (1,776 г, 9,26 ммоль) и сухой DCM (60 мл). Смесь перемешивали в течение ночи, прежде чем реакционную смесь разбавляли диэтиловым эфиром (200 мл). Смесь промывали 1 М HCl (100 мл) и насыщенным солевым раствором (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 3% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 2,26 г (выход 56%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,08 (с, 6H), 0,90 (д, 9H), 0,95-1,03 (м, 12H), 1,40-1,52 (м, 4H), 1,58-1,69 (м, 4H), 1,70-1,83 (м, 4H), 2,04-2,15 (м, 8H), 2,77-2,92 (м, 16H), 3,27-3,37 (м, 2H), 3,57-3,67 (м, 2H), 3,72-3,80 (м, 4H), 4,14-4,32 (м, 1H), 4,41-4,56 (м, 1H), 5,09-5,22 (м, 1H), 5,23-5,49 (м, 20H). МС (электрораспыление): 941,6 [М+Na]+.
Стадия d) Получение 3-гидроксипропан-1,2-диил-бис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ила) окси) бутаноата)
К раствору 3 - ((трет-бутилдиметилсилил) окси) пропан-1,2-диил-бис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17 -пентаен-1-ил) окси) бутаноата) (2,26 г, 2,46 ммоль) в диоксане (100 мл). HCl (37% (вес/вес, 2 мл) и смесь перемешивали в течение 3 часов в атмосфере азота при температуре окружающей среды, затем концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 15% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 0,83 г (выход 42%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,96-1,03 (м, 12H), 1,40-1,53 (м, 4H), 1,58-1,68 (м, 4H), 1,70-1,85 (м, 4H), 1,87-2,01 (м, 1H), 2,05-2,15 (м, 8H), 2,75-2,95 (м, 16H), 3,28-3,41 (м., 2H), 3,56-3,65 (м, 2H), 3,73-3,85 (м, 4H), 4,24-4,37 (м, 1H), 4,42-4,53 (м, 1H), 5,14-5,23 (м, 1H), 5,26 -5,51 (м, 20Н). МС (электрораспыление): 827,5 [М+Na]+.
Пример 16. Получение 2-гидроксипропан-1,3-диил-бис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ила) окси) бутановой кислоты):
Стадия а) 2-оксопропан-1,3-диил-бис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноат)
2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановую кислоту (5,0 г, 13,4 ммоль) и DMAP (1,63 г) 13,4 ммоль) добавляли к раствору димера 1,3-дигидроксиацетона (1,145 г, 6,36 ммоль) в хлороформе (25 мл) в атмосфере азота. Затем по каплям добавляли раствор DCC (2,75 г, 13,35 ммоль) в хлороформе (10 мл) при температуре окружающей среды. Смесь перемешивали в течение ночи при комнатной температуре, затем концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя прогрессивно полярные смеси гептана и этилацетата (90:10-88:12) в качестве элюента. Концентрирование соответствующих фракций дало 2,4 г (выход 47%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,97-1,06 (м, 12H). 1,38-1,53 (м, 4H), 1,57-1,73 (м, 4H), 1,73-1,96 (м, 4H), 2,03-2,17 (м, 8H), 2,76-2,92 (м, 16H), 3,35-3,42 (м, 2H), 3,63-3,70 (м, 2H), 3,89 (дд, 2H), 4,75-4,93 (м, 4H), 5,27-5,49 (м, 20H). МС (электрораспыление): 827,5 [М+Na]+.
Стадия b) 2-гидроксипропан-1,3-диил-бис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановой)
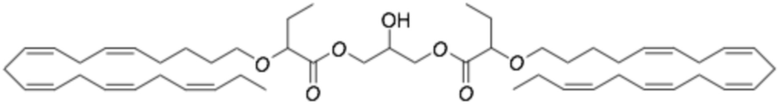 .
.
NaBH4 (0,336 г, 8,87 ммоль) осторожно добавляли к раствору 2-оксопропан-1,3-диил-бис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11, 14,17-пентаен-1-ил) окси) бутаноат) (3,24 г, 4,03 ммоль) в THF (55 мл) и воде (4 мл) при 0°С. Смесь перемешивали в течение 15 минут при 0°С. Затем осторожно добавляли уксусную кислоту (1 мл), а затем этилацетат (100 мл). Смесь промывали водой (100 мл), насыщенным водным NaHCO3 (100 мл) и насыщенным солевым раствором перед сушкой (Na2SO4) фильтровали и концентрировали в вакууме. Остаток объединяли с другой порцией материала перед очисткой флэш-хроматографией на силикагеле, используя 15% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 1,62 г (выход 50%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,97-1,03 (м, 12H), 1,41-1,52 (м, 4H), 1,58-1,69 (м, 6H), 1,71-1,87 (м, 4H), 2,05-2,14 (м, 8H), 2,38-2,42 (м, 1H), 2,78-2,92 (м, 16H), 3,32-3,39 (м, 2H), 3,57-3,64 (м, 2H), 3,80-3,84 (м, 2H), 4,05 -4,34 (м, 5Н), 5,26-5,49 (м, 20Н). МС (электрораспыление): 827,5 [М+Na]+.
Пример 17: Получение пропан-1,2,3-триил трис (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ила) окси) бутаноата)
2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17 -пентаен-1-ил) окси) бутановой кислоты (4,0 г, 10,7 ммоль), 4-диметиламинопиридина (1,305 г, 10,7 ммоль), гидрохлорида 1- (3-диметиламинопропил) -3-этилкарбодиимида (2,047 г, 10,7 ммоль) и сухой DCM (30 мл) добавляли к раствору глицерина (0,173 мл, 2,373 ммоль) в DMF (10 мл) в атмосфере азота при комнатной температуре. Смесь перемешивали в течение ночи, затем реакционную смесь разбавляли диэтиловым эфиром (250 мл). Смесь промывали водным 1 М HCl (100 мл) и насыщенным солевым раствором (100 мл) перед сушкой (Na2SO4) фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 5% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 2,1 г (выход 73%) названного соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 0,91-1,05 (м, 18H), 1,40-1,52 (м, 6H), 1,57-1,69 (м, 6H), 1,69-1,86 (м, 6H), 2,01-2,17 (м, 12H), 2,69-2,96 (м, 24H), 3,27-3,38 (м, 3H), 3,53-3,67 (м, 3H), 3,73-3,81 (м, 3H), 4,17-4,27 (м, 2H), 4,37-4,54 (м, 2Н), 5,28-5,47 (м, 30Н). МС (электрораспыление): 1183,8 [М+Na]+.
Пример 18: Получение трет-бутил 2 - ((2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноил) окси) бензоата:
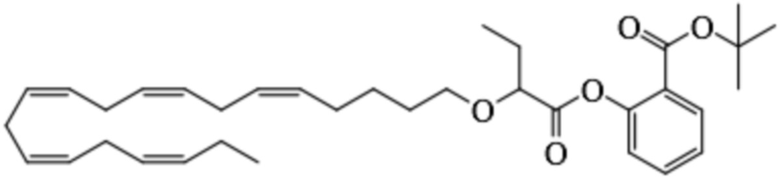
1-этил-3- (3-диметиламинопропил) карбодиимид (EDC) (316 мг, 1,65 ммоль) и 4-диметиламинопиридин (DMAP) (20 мг, 0,15 ммоль) добавляли к раствору 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутановой кислоты (561 мг, 1,5 ммоль) в DCM (10 мл) и реакционную смесь перемешивали в течение 10 минут. Добавляли трет-бутил-2-гидроксибензоат (291 мг, 1,5 ммоль) и смесь перемешивали в течение 3 часов. Добавляли насыщенный солевой раствор и полученные две фазы разделяли. Водную фазу экстрагировали DCM, и объединенные органические фазы сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя смесь 5% EtOAc в гептане в качестве элюента. Концентрация соответствующих фракций дало 500 мг (выход 61%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,98 (т, 3Н), 1,11 (т, 3Н), 1,13 (т, 3Н), 1,45-1,75 (м, 4Н), 1,56 (с, 9Н), 1,90-2,20 (м, 6H), 2,80-2,90 (м, 8H), 3,45-3,60 (м, 1H), 3,80-3,90 (м, 1H), 4,05-4,15 (м, 1H), 5,25-5,50 (м, 10H) 7,06 (м, 1Н), 7,28 (м, 1Н), 7,52 (м, 1Н), 7,89 (м, 1Н). МС (ESI): 573 [М+Na]+.
Пример 19: Получение 2 - ((2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси)бутаноил)окси) бензойной кислоты:
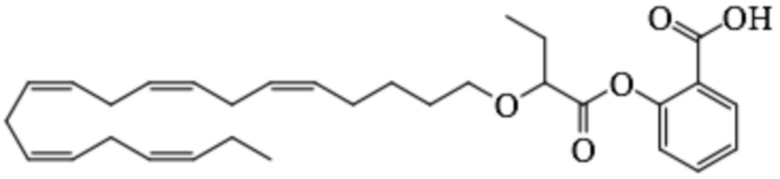 .
.
Раствор трет-бутил 2 - ((2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноил) окси) бензоата (500 мг (0,9 ммоль) в FA (10 мл) перемешивали в течение 2 дней. Растворитель удаляли при пониженном давлении и остаток подвергали флэш-хроматографии с использованием градиента 1-5% EtOAc (содержащего 5% FA) в гептане (также содержащем 5% FA) в качестве элюента. Соответствующие фракции объединяли и концентрировали, а остаток очищали дополнительно второй флэш-хроматографией, используя градиент 1-10% EtOAc в гептане в качестве элюента. Концентрация соответствующих фракций дало 75 мг (17% выход) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,99 (т, 3Н), 1,13 (т, 3Н), 1,45-1,60 (м, 2Н), 1,65-1,70 (м, 2Н), 1,90-2,15 (м, 6Н) 2,80-2,90 (м, 8H), 3,45-3,55 (м, 1H), 3,80-3,90 (м, 1H), 4,05-4,15 (м, 1H), 5,30-5,45 (м, 10H), 7,14 (д, 1Н), 7,35-7,45 (м, 1Н), 7,60-7,70 (м, 1Н), 8,10-8,15 (м, 1Н). МС (ESI): 517 [М+Na]+.
Пример 20. Получение 2 - ((трет-бутоксикарбонил) амино) этил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноата:
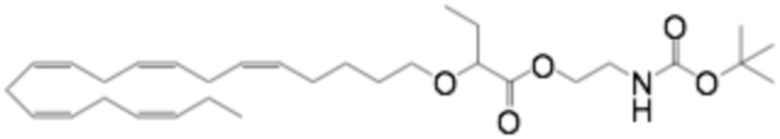 .
.
O- (7-Азабензотриазол-1-ил) -N, N, N ', N'-тетраметилуронийгексафторфосфат (HATU) (800 мг, 2,1 ммоль) и TEA (0,56 мл, 4 ммоль) добавляли к раствору 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутановой кислоты (748 мг, 2 ммоль) в DCM (10 мл) и реакционную смесь перемешивали в течение 20 минут. Добавляли раствор трет-бутил N- (2-гидроксиэтил) карбамата (340 мг, 2,1 ммоль) в DCM (1 мл) и полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли Et2O (100 мл) и смесь промывали водой, 1М HCl (водный), насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили ((Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 10-20% EtOAc в гептане в качестве элюента. Концентрация соответствующих фракций дала 780 мг (выход 76%) названного соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,98 (т, 6Н), 1,40-1,55 (м, 2Н), 1,45 (с, 9Н), 1,60-1,85 (м, 4Н), 2,05-2,20 (м, 4Н), 2,75-2,90 (м, 8H), 3,30-3,45 (м, 3H), 3,55-3,65 (м, 1H), 3,75-3,80 (м, 1H), 4,20-4,25 (м, 2H), 4,76 (ушир. С 1Н) 5,30-5,45 (м, 10Н). МС (ESI): 540 [М+Na]+.
Пример 21. Получение 2- (2-ацетоксибензамидо) этил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноата:
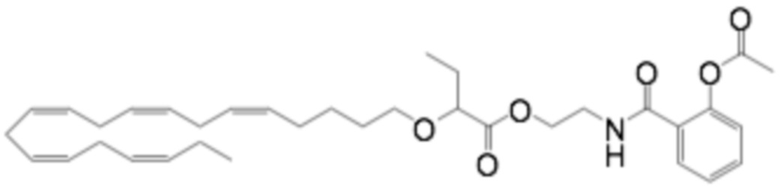 .
.
Ацетилхлорид (1 мл) добавляли к раствору 2 - ((трет-бутоксикарбонил) амино) этил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17- пентаен-1-илокси) бутаноата (300 мг, 0,58 ммоль) в МеОН (5 мл) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали в вакууме до получения HCl соли 2-аминоэтил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноата (286 мг) в виде неочищенного продукта. TEA (109 мкл, 0,78 ммоль) добавляли к раствору ацетилсалициловой кислоты (137 мг, 0,76 ммоль) в DCM (10 мл) и смесь охлаждали до 0°C. Этилхлорформиат (75 мкл, 0,78 ммоль) добавляли по каплям и реакционную смесь перемешивали в течение 2 часов. Этот раствор затем добавляли к раствору неочищенного продукта HCl-соли 2-аминоэтил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1 -илокси) бутаноата в смеси DCM (10 мл) и TEA (2 мл). Полученную смесь перемешивали при комнатной температуре в течение 3,5 часов и затем добавляли воду. Полученные две фазы разделяли и водную фазу дважды экстрагировали DCM. Объединенные органические фазы промывали 1 М HCl (водным), насыщенным NaHCO3 (водн.) и водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя 20% EtOAc в гептане в качестве элюента. Концентрирование соответствующих фракций дало 90 мг (выход 23%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,95-1,05 (м, 6H), 1,35-1,45 (м, 2H), 1,55-1,70 (м, 2H), 1,70-1,85 (м, 2H), 2,05-2,15 (м, 4Н), 2,35 (с, 3Н), 2,80-2,90 (м, 8Н), 3,30-3,40 (м, 1Н), 3,55-3,65 (м, 1Н), 3,70-3,85 (м, 3Н), 4,33 (т, 2Н), 5,30-5,45 (м, 10Н), 6,61 (ушир. с, 1Н), 7,10-7,15 (м, 1Н), 7,25-7,35 (м, 1Н), 7,45-7,50 (м, 1Н), 7,70. -7,75 (м, 1Н). MS (ESI); 602 [М+Na]+.
Пример 22. Получение 2- (2-гидроксибензамидо) этил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноата:
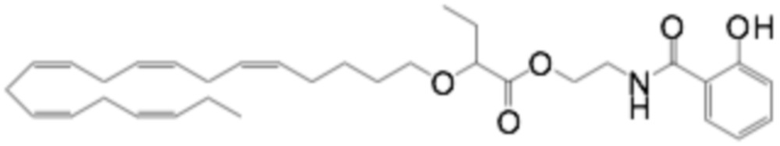 .
.
Аммиак (водный, 28%, 20 капель) добавляли по каплям к раствору 2- (2-ацетоксибензамидо) этил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14, 17-пентаен-1-илокси) бутаноата (99 мг, 0,17 ммоль) в 2-пропаноле (9 мл) и воде (3 мл) и реакционную смесь перемешивали в течение 10 минут. Добавляли воду и полученную смесь дважды экстрагировали Et2O. Объединенные органические фазы промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 0-40% EtOAc в гептане в качестве элюента. Концентрирование соответствующих фракций дало 33 мг (36% выход) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,95-1,05 (м, 6H), 1,35-1,45 (м, 2H), 1,60-1,70 (м, 2H), 1,70-1,85 (м, 2H), 2,05-2,20 (м, 4Н), 2,80-2,90 (м, 8Н), 3,35-3,45 (м, 1Н), 3,50-3,60 (м, 1Н), 3,75-3,85 (м, 3Н), 4,40-4,50 (м, 2Н), 5,30-5,45 (м, 10H), 6,80-6,90 (м, 2H), 6,95-7,05 (м, 1H), 7,35-7,45 (м, 1H), 12,22 (с, 1H). МС (ESI): 560 [М+Na]+.
Пример 23. Получение N-бензил-2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутанамида:
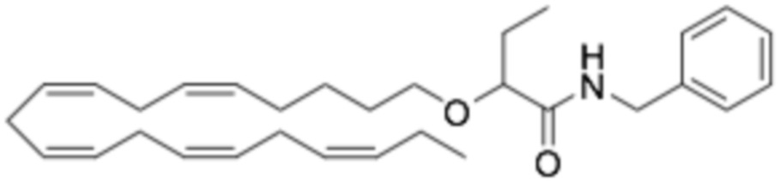 .
.
К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (302,1 мг, 0,807 ммоль) и о-бензотриазол-1-ил-N, N, N', N'-тетраметилурония тетрафторбората (TBTU) (285 мг, 0,888 ммоль) в DCM (10 мл) при комнатной температуре в атмосфере азота добавляли триэтиламин (123 мкл) 0,887 ммоль). После перемешивания в течение 15 минут к смеси добавляли бензиламин (97 мкл, 0,887 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NaHCO3 (25 мл) и смесь экстрагировали трет-бутилметиловым эфиром (2 × 50 мл). Органические фазы объединяли, сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя прогрессивно полярные смеси гептана и этилацетата (95: 5 × 50:50) в качестве элюента. Концентрация соответствующих фракций дала 0,253 г (выход 66%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) d 0,97-0,90 (м, 6H), 1,41-1,35 (м, 2H), 1,59-1,52 (м, 2Н), 1,75-1,68 (м, 1Н), 1,86-1,80 (м, 1Н), 2,09-2,03 (м, 4Н), 2,82-2,77 (м, 8Н), 3,44 (т, J=6,4, 2Н), 3,72-3,71 (м, 1Н), 4,46 (д, J=5,8, 2Н), 5,36-5,28 (м, 10Н), 6,83 (с, 1Н), 7,26-7,24 (м, 3Н), 7,33. - 7,30 (м, 2Н).
Пример 24. Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) -N, N-диметилбутанамида

К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (300,2 мг, 0,801 ммоль) и о-бензотриазол-1-ил-N, N, N', N'-тетраметилуроний тетрафторбората (TBTU) (286,4 мг, 0,892 ммоль) в DCM (10 мл) при комнатной температуре в атмосфере азота добавляли триэтиламин (123 мкл) 0,884 ммоль). После перемешивания в течение 15 минут к смеси добавляли диметиламин (2,0 М в THF) (0,80 мл, 1,60 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NaHCO3 (25 мл) и смесь экстрагировали трет-бутилметиловым эфиром (2 × 50 мл). Органические фазы объединяли, сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (95: 5 × 50:50). Концентрирование соответствующих фракций дало 0,268 г (выход 81%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) d 0,95 (т, 3Н), 0,96 (т, 3 H), 1,35-1,46 (м, 2H), 1,53-1,63 (м, 2H), 1,66-1,79 (м, 2H), 2,01-2,10 (м, 4H), 2,77-2,87 (м, 8H), 2,93. (с, 3Н), 3,10 (с, 3Н), 3,28 (дт, J=9,1, 6,5, 1Н), 3,46 (дт, J=9,1, 6,3, 1Н), 3,96 (дд, J=7,7, 6,1, 1Н), 4,97-5,70 (м, 10H).
Пример 25. Получение N-этил-2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутанамида:
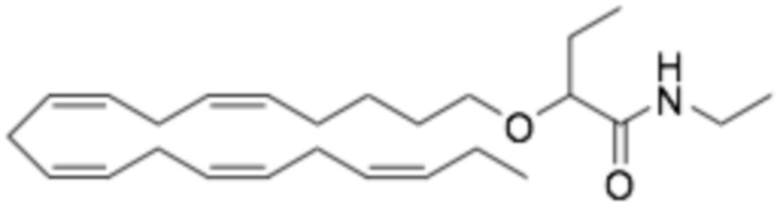
К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (304,6 мг, 0,813 ммоль) и о-бензотриазол-1-ил-N, N, N', N'-тетраметилурония тетрафторбората (TBTU) (286,9 мг, 0,894 ммоль) в DCM (10 мл) при комнатной температуре в атмосфере азота добавляли триэтиламин (123 мкл) 0,884 ммоль). После перемешивания в течение 15 минут к смеси добавляли этиламин (2,0 М в THF) (0,80 мл, 1,60 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NaHCO3 (25 мл) и смесь экстрагировали трет-бутилметиловым эфиром (2 × 50 мл). Органические фазы объединяли, сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (95: 5 × 50:50). Концентрирование соответствующих фракций дала 0,186 г (выход 56%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3) d 0,90 (т, 3Н), 0,95 (т, 3Н), 1,13 (т, 3Н), 1,55-1,62 (м, 2Н), 1,64-1,66 (м, 2Н), 1,69-1,69 (м, 1H), 1,74-1,83 (м, 1H), 2,01-2,12 (м, 4H), 2,78-2,84 (м, 8H), 3,25-3,34 (м, 2H), 3,44 (т, 2H), 3,64 (дд, 1H), 5,50-5,13 (м, 10H), 6,50 (с, 1H).
Пример 26: Получение трет-бутил (2- (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутанамидо) этил) карбаминовой кислоты:
Дициклогексилметандимин (DCC) (1,73 г, 8,4 ммоль) и 1H-бензо [d] [1,2,3] триазол-1-ол (HOBt) (1,14 г, 8,4 ммоль) добавляли к перемешиваемому раствору 2- (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (3,14 г, 8,4 ммоль) в THF (25 мл) при 0°С в атмосфере азота. Смесь перемешивали в течение 20 минут, после чего по каплям добавляли раствор N-Boc-этилендиамина (1,12 г) в THF (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли диэтиловый эфир (200 мл) и смесь промывали водой, 1 М HCl, насыщенным NaHCO3 и насыщенным солевым раствором. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 25% этилацетат в гептане (с добавлением 0,5% HCOOH) в качестве элюента. Концентрирование соответствующих фракций дало 3,0 г (выход 83%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): д 0,90 (т, 3Н), 0,96 (т, 3Н), 1,43 (с, 9Н), 1,40-1,80 (м, 6Н), 2,05-2,20 (м, 4Н), 2,75 2,90 (м, 8H), 3,20-3,60 (м, 6H), 3,65-3,75 (м, 1H), 5,02 (ушир. С, 1H), 5,30-5,45 (м, 10H), 7,01 (ушир. С, 1H), МС (электрораспыление); 539 [М+Na]+.
Пример 27: Получение N- (2-аминоэтил) -2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутанамида:
К раствору трет-бутил (2- (2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутанамидо) этил) карбамата(774 мг, 1,5 ммоль) в DCM (8 мл) в атмосфере азота при температуре окружающей среды, добавляли TFA (2 мл), реакционную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме. К остатку добавляли диэтиловый эфир (50 мл) и NaOH (водный 1М, 50 мл) и смесь энергично перемешивали в течение 30 минут. Фазы разделяли и органическую фазу промывали насыщенным солевым раствором, сушили (NaSO4), фильтровали и концентрировали в вакууме с получением 560 мг (90%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,05 (2xт, 6H), 1,35-1,50 (м, 2H), 1,55-1,85 (м, 4H), 2,05-2,20 (м, 4H), 2,75-2,95 (м, 8Н), 3,10-3,20 (м, 1Н), 3,30-3,80 (м, 6Н), 4,05-4,20 (ушир. м, 1Н), 4,25-4,75 (ушир. с, 2Н), 5,30-5,50 (м, 10Н). МС (электрораспыление); 417 [М+Н]+, 439 [М+Na].
Пример 28: Получение N- (2-гидроксиэтил) -2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутанамида:
 .
.
К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (4,5 г, 12 ммоль) в DCM (100 мл) в атмосфере азота при комнатной температуре добавляли оксалилхлорид (8,4 мл, 100 ммоль), а затем 2 капли DMF. Смесь перемешивали в течение 30 минут при комнатной температуре и концентрировали в вакууме. Остаток растворяли в DCM (100 мл) в атмосфере азота. Добавляли триэтиламин (3,34 мл, 24 ммоль), а затем этаноламин (1,08 мл, 18 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Добавляли воду (300 мл) и смесь дважды экстрагировали дихлорметаном. Органические фазы объединяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (50:50 × 20:80). Концентрирование соответствующих фракций дало 4,6 г (выход 92%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,90-1,05 (м, 6H), 1,40-1,55 (м, 2 H), 1,60-1,90 (м, 4H), 2,05-2,20 (м, 4H), 2,75-2,90 (м, 8H), 3,05 (ушир. с, 1H), 3,40-3,55 (м, 4H), 3,70-3,80. (м, 3Н), 5,30-5,45 (м, 10Н), 7,04 (ушир. с, 1Н). МС (электрораспыление); 440 [М+Na]+
Пример 29. Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) -N-изопропилбутанамида:
 .
.
К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (300 мг, 0,801 ммоль) и о-бензотриазол-1-ил-N, N, N ', N'-тетраметилуроний тетрафторбората (TBTU) (284 мг, 0,884 ммоль) в DCM (10 мл) при комнатной температуре в атмосфере азота добавляли триэтиламин (123 мкл) 0,884 ммоль). После перемешивания в течение 15 минут при комнатной температуре в атмосфере азота добавляли изопропиламин (76 мкл, 0,884 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NaHCO3 (25 мл) и смесь экстрагировали трет-бутилметиловым эфиром (2 × 50 мл). Органические фазы объединяли, сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (95: 5 × 50:50). Концентрирование соответствующих фракций дало 0,207 г (выход 59%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 0,83-0,93 (м, 3Н), 0,95 (т, J=7,5, 3Н), 1,14 (т, J=6,1, 6Н), 1,39-1,47 (м, 2Н), 1,56. 1,73 (м, 3Н), 1,73-1,83 (м, 1Н), 2,02-2,11 (м, 4Н), 2,80-2,84 (м, 8Н), 3,43 (т, J=6,4, 2Н), 3,60 (дд, J=6,6, 4,4, 1H), 4,02-4,11 (м, 1H), 5,66-5,01 (м, 10H), 6,33 (д, J=5,9, 1H).
Пример 30. Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) -N-метилбутанамида:
 .
.
К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (303,6 мг, 0,811 ммоль) и о-бензотриазол-1-ил-N, N, N', N'-тетраметилурония тетрафторбората (TBTU) (285,3 мг, 0,889 ммоль) в DCM (10 мл) при комнатной температуре в атмосфере азота добавляли триэтиламин (123 мкл) 0,884 ммоль). После перемешивания в течение 15 минут при комнатной температуре в атмосфере азота добавляли метиламин 2,0 М в THF (0,8 мл, 1600 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NaHCO3 (25 мл) и смесь экстрагировали трет-бутилметиловым эфиром (2 × 50 мл). Органические фазы объединяли, сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (95: 5 × 50:50). Концентрирование соответствующих фракций дала 0,235 г (выход 72%) указанного в заголовке соединения в виде масла.
1H ЯМР (300 МГц, CDCl3) d 0,89 (т, J=7,4, 3Н), 0,95 (т, J=7,5, 3Н), 1,37-1,47 (м, 2Н), 1,56-1,72 (м, 3Н), 1,74 1,84 (м, 1H), 2,01-2,15 (м, 4H), 2,78-2,84 (м, 11H), 3,44 (т, J=6,4, 2H), 3,66 (дд, J=6,4, 4,4, 1H), 5,18-5,52 (м, 10H), 6,51 (с, 1H).
Пример 31: Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) -1- (пиперидин-1-ила) бутан-1-она:

К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (302,2 мг, 0,807 ммоль) и о-бензотриазол-1-ил-N, N, N', N'-тетраметилурония тетрафторбората (TBTU) (284 мг, 0,884 ммоль) в DCM (8 мл) при комнатной температуре в атмосфере азота добавляли триэтиламин (123 мкл) 0,884 ммоль). После перемешивания в течение 15 минут при комнатной температуре в атмосфере азота добавляли пиперидин (87 мкл, 0,884 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NaHCO3 (25 мл) и смесь экстрагировали трет-бутилметиловым эфиром (2 × 50 мл). Органические фазы объединяли, сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (95: 5 × 50:50). Концентрирование соответствующих фракций дало 0,270 г (выход 73%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) d 0,93-0,98 (м, 6H) 1,37-1,46 (м, 2H), 1,51-1,62 (м, 8H), 1,66-1,77 (м, 2H), 2,02-2,09 (м, 4H), 2,77-2,83 (м, 8H), 3,30 (дт, J=9,1, 6,6, 1H), 3,47-3,52 (м, 2H), 3,54-3,62 (м, 3H), 3,95 (дд, J=7,9 6,1, 1Н), 5,02-5,65 (м, 10Н).
Пример 32. Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутанамида:
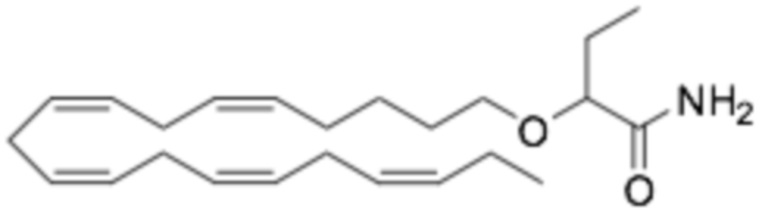 .
.
К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (320 г, 854 ммоль) в толуоле (2500 мл) добавляли оксалилхлорид (276 мл, 3,15 моль). Затем осторожно добавляли по каплям DMF (10 мл), затем смесь перемешивали при температуре окружающей среды в течение 1 часа и концентрировали в вакууме. Остаток растворяли в DCM (750 мл) и медленно добавляли к перемешиваемому, охлажденному (0°C) раствору аммония (28%, водн.) (528 мл, 6,84 моль). Смесь перемешивали в течение 30 минут при температуре окружающей среды. Фазы разделяли и водную фазу экстрагировали метил-трет-бутиловым эфиром (700 мл). Объединенную органическую фазу промывали с помощью 3М HCl (2х300 мл), насыщенным водным NaHCO3 (500 мл), насыщенным солевым раствором (2х300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (70:30 до 60:40). Концентрирование соответствующих фракций дало 249,8 г (выход 76%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) d 0,87-0,92 (м, 6H), 1,32-1,46 (м, 2H), 1,52-1,59 (м, 2H), 1,62-1,79 (м, 2H), 1,97-2,06 (м, 4H), 2,68-2,85 (м, 8H), 3,34-3,51 (м, 2H), 3,60 (дд, J=6,5, 4,5, 1H), 5,18-5,42 (м, 10H), 5,61 (с, 1H) 6,42 (с, 1Н).
Пример 33: Получение N- (трет-бутил) -2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутанамида:
 .
.
К раствору 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (301,3 мг, 0,804 ммоль) в DCM (10 мл) при комнатной температуре в атмосфере азота добавляли о-бензотриазол-1-ил-N, N, N ', N'-тетраметилуроний тетрафторборат (TBTU) (285,6 мг, 0,889 ммоль) с последующим добавлением триэтиламина (0,123). мл, 0,885 ммоль). Смесь перемешивали в течение 15 минут при комнатной температуре в атмосфере азота, после чего добавляли 2-амино-2-метилпропан (0,10 мл, 0,952 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NaHCO3 (25 мл) и смесь экстрагировали трет-бутилметиловым эфиром (2 × 50 мл). Объединенную органическую фазу сушили (Na2SO4), фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (95: 5 × 50:50). Концентрирование соответствующих фракций дало 244,1 мг (выход 70%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 0,89 (т, J=7,4, 3H), 0,96 (т, J=7,5, 3H), 1,34 (с, 9H), 1,41-1,46 (м, 2H), 1,57-1,70 (м, 3Н), 1,74-1,81 (м, 1Н), 2,02-2,11 (м, 4Н), 2,80-2,83 (м, 8Н), 3,27-3,48 (м, 2Н), 3,48-3,56 (м, 1Н) 4,93-5,55 (м, 10H), 6,38 (с, 1H).
Пример 34: Получение (R) -2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) -N - ((R) -1-фенилэтил) бутанамида:
К раствору (R) -2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (40 мг, 0,11 ммоль) и (R) -1-фенилэтан-1-амина (14 мкл, 0,11 ммоль) в сухом DMF (3 мл) при 0oC в атмосфере азота добавляли N-метилморфолин (14 мкл, 0,13 ммоль) с последующим добавлением o - (бензотриазол-1-ил) -N, N, N', N'-тетраметилурония гексафторфосфата (HBTU) (45 мг, 0,14 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 30 минут. Добавляли воду (10 мл) и смесь экстрагировали гептаном (10 мл). Фазы разделяли и водную фазу экстрагировали гептаном (10 мл), а затем диэтиловым эфиром (10 мл). Органические фазы объединяли и промывали насыщенным водным NH4Cl (10 мл) и насыщенным солевым раствором (10 мл). Органическую фазу сушили (Na2SO4), фильтровали и упаривали в вакууме, получая указанное в заголовке соединение (20 мг, 0,042 ммоль, 38% выход) в виде масла.
Пример 35: Получение (2S) -этил 2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) -4 - метилпентаноата:
 .
.
N, N'-дициклогексилкарбодиимид (DCC) (1,13 г, 5,5 ммоль), гидроксибензотриазол (HOBt) (0,74 г, 5,5 ммоль) и триэтиламин (TEA) (1,58 мл, 11,4 ммоль) добавляли к раствору 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутановой кислоты (1,87 г, 5,0 ммоль) в тетрагидрофуране (THF) (20 мл) и смесь перемешивали в течение 10 минут. Добавляли гидрохлорид этилового эфира L-лейцина (0,89 г, 4,6 ммоль) и полученную смесь перемешивали в течение 2 часов. Смесь концентрировали в вакууме, растворяли в Et2O (100 мл) и промывали 1М HCl (водный раствор), насыщенным NaHCO3 (водный) и насыщенным солевым раствором. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 10-15% EtOAc в гептане в качестве элюента. Концентрирование соответствующих фракций дало 1,88 г (выход 80%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,90-1,05 (м, 12H), 1,25-1,35 (м, 3H), 1,45-1,85 (м, 9H), 2,05-2,20 (м, 4H), 2,80-2,90 (м, 8Н), 3,40-3,70 (м, 3Н), 4,18 (кв, 2Н), 4,55-4,70 (м, 1Н), 5,30-5,45 (м, 10Н), 6,80-6,95 (м, 1Н). МС (ESI): 538 [М+Na]+.
Пример 36. Получение (2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) -4- метилпентановой кислоты:
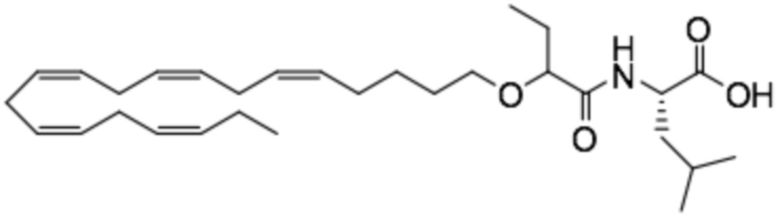 .
.
Раствор LiOH (700 мг, 29 ммоль) в воде (10 мл) добавляли к раствору (2S) -этил 2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z)-эйкоза-5, 8,11,14,17-пентаен-1-илокси) бутанамидо) -4-метилпентаноата (1,88 г, 3,65 ммоль) в EtOH (20 мл) и реакционную смесь перемешивали при 40°C в течение 1 часа. Смесь охлаждали до температуры окружающей среды и добавляли 3 М HCl (водный раствор) до рН ~ 2. Полученную смесь дважды экстрагировали Et2O и объединенные органические фазы сушили (NaSO4), фильтровали и концентрироали при пониженном давлении, получая 1,55 г (выход 87%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,90-1,05 (м, 12H), 1,45-1,55 (м, 2H), 1,65-1,85 (м, 7H), 2,05-2. 20 (м, 4H), 2,80-2,90 (м, 8H), 3,45-3,65 (м, 2H), 3,70-3,80 (м, 1H), 4,55-4,70 (м, 1H), 5,30-5,45 (м, 10H), 6,85-7,00 (м, 1Н), 10,30 (ушир. с, 1Н). MS (ESI); 510 [М+Na]+.
Пример 37. Получение трет-бутил 2 - (((2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1- илокси) бутанамидо) -4-метилпентаноил) окси) бензоата:
Трет-бутиловый эфир 2-гидроксибензойной кислоты (291 мг, 1,5 ммоль), TEA (0,46 мл, 3,3 ммоль) и O- (бензотриазол-1-ил) -N, N, N´, N´-тетраметилурония гексафторфосфат (TBTU) (500 мг, 1,65 ммоль) добавляли к раствору (2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаена -1-илокси) бутанамидо) -4-метилпентановой кислоты (730 мг, 1,5 ммоль) в диметилформамиде (DMF) (10 мл). Реакционную смесь нагревали в течение 2 часов при 65°C, охлаждали до комнатной температуры и добавляли Et20 (100 мл). Полученные две фазы разделяли и органическую фазу промывали водным 10% NH4Cl и насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя 10% EtOAc в гептане в качестве элюента. Концентрирование соответствующих фракций дало 404 мг (выход 41%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,90-1,05 (м, 12H), 1,40-1,55 (м, 2H), 1,57 (с, 9H), 1,60-1,85 (м, 6H), 2,00-2,20 (м, 5H), 2,80-2,90 (м, 8H), 3,40-3,60 (м, 2H), 3,70-3,80 (м, 1H), 4,90-5,05 (м, 1H), 5,30-5,45 (м, 10H), 7,10 (д, 2Н), 7,25-7,35 (м, 1Н), 7,50-7,55 (м, 1Н), 7,85-7,95 (м, 1Н). МС (ESI): 686 [М+Na]+.
Пример 38. Получение 2 - (((2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) -4-метилпентаноил) окси) бензойной кислоты:
 .
.
TFA (1 мл) добавляли к раствору трет-бутил 2 - (((2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14, 17-пентаен-1-илокси) бутанамидо) -4-метилпентаноил) окси) бензоата (150 мг, 0,23 ммоль) в DCM (4 мл) при 0°C и реакционную смесь перемешивали в течение 40 минут. Добавляли толуол (10 мл) и смесь концентрировали в вакууме. Остаток очищали флэш-хроматографией с использованием 13% EtOAc (содержащего 0,1% FA) в гептане (также содержащем 0,1% FA) в качестве элюента. Соответствующие фракции концентрировали и остаток (100 мг) растворяли в Et2O (50 мл). Раствор промывали насыщенным водным NaHCO3, сушили (Na2SO4), фильтровали и концентрировали в вакууме, получили 60 мг желаемого продукта. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,05 (м, 12H), 1,35-2,15 (м, 13H), 2,75-2,90 (м, 8H), 3,40-3,60 (м, 2H), 3,70-3,80 (м, 1Н), 4,85-5,05 (м, 1Н), 5,30-5,45 (м, 10Н), 7,0-7,20 (м, 2Н), 7,25-7,40 (м, 1Н), 7,50-7,65 (м, 1Н), 8,00-8,15 (м, 1Н). MS (ESI); 630 [М+Na]+. HR-MS (ESI): рассчитано для C37H53NO6+Na: 630,3770, найдено: 630,3786.
Пример 39. Получение трет-бутил (2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) этил) карбамата:
DCC (1,73 г, 8,4 ммоль) и HOBt (1,14 г, 8,4 ммоль) добавляли к раствору 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17- пентаен-1-илокси) бутановой кислоты (3,14 г, 8,4 ммоль) в THF (25 мл) при 0°С и смесь перемешивали в течение 20 минут. Раствор N-Boc-этилендиамина в THF (1 мл) добавляли по каплям и полученную смесь перемешивали при комнатной температуре в течение 1,5 часов. Добавляли Et2O (200 мл) и смесь промывали водой, 1М HCl (водной), насыщенным NaHCO3 (водная) и насыщенным солевым раствором. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией с использованием 25% EtOAc (содержащего 0,5% FA) в гептане (также содержащем 0,5% FA) в качестве элюента. Концентрирование соответствующих фракций дало 3,0 г (выход 83%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,90 (т, 3Н), 0,96 (т, 3Н), 1,43 (с, 9Н), 1,40-1,80 (м, 6Н), 2,05-2,20 (м, 4Н), 2,75 -2,90 (м, 8H), 3,20-3,60 (м, 6H), 3,65-3,75 (м, 1H), 5,02 (ушир. с, 1H), 5,30-5,45 (м, 10H), 7,01 (ушир. с, 1H), МС (ESI): 539 [М+Na]+.
Пример 40: Получение 2-гидрокси-N- (2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) этил) бензамида:
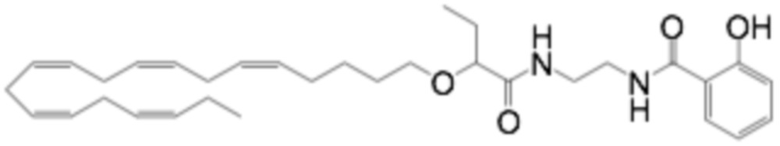 .
.
TFA (2 мл) добавляли к раствору трет-бутил (2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси)) бутанамидо) этил) карбамата (668 мг, 1,3 ммоль) в DCM (8 мл) и полученную смесь перемешивали в течение 1,5 часов. Смесь концентрировали в вакууме с получением TFA-соли N- (2-аминоэтил) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1 -илокси) бутанамида в виде сырого продукта (800 мг, МС (ESI): 417 [М+Н]+, 439 [М+Na]+). Остаток растворяли в DCM (10 мл) и добавляли DCC (320 мг, 1,55 ммоль), TEA (0,36 мл, 2,6 ммоль) и HOBt (210 мг, 1,55 ммоль). Через 15 минут по каплям добавляли раствор салициловой кислоты (214 мг, 1,55 ммоль) в DCM (1 мл) и полученную смесь перемешивали в течение ночи. Смесь концентрировали и остаток растворяли в Et2O (100 мл), промывали водой, 1М HCl (водной), насыщенным водным NaHCO3 и насыщенным солевым насыщенным солевым раствором. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное масло очищали флэш-хроматографией, используя градиент 4-15% EtOAc (содержащий 5% FA) в гептане (также содержащем 5% FA) в качестве элюента. Концентрация соответствующих фракций дала 110 мг (16% выход за две стадии) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,89 (т, 3Н), 0,99 (т, 3Н), 1,35-1,50 (м, 2Н), 1,60-1,90 (м, 4Н), 2,05-2,20 (м, 4Н) 2,8-2,9 (м, 8Н), 3,47 (т, 2Н), 3,50-3,70 (м, 4Н), 3,70-3,80 (м, 1Н), 5,30-5,45 (м, 10Н), 6,85-6,95 (м, 1H), 6,97 (дд, 1H), 7,14 (ушир. с, 1H), 7,35-7,45 (м, 1H), 7,48 (дд, 1H), 7,95 (ушир. с, 1H), 12,51 (ушир. С, 1H). МС (ESI): 537 [М+Н]+, 559 [М+Na]+.
Пример 41: Получение 2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) этил 2-ацетоксибензоата:
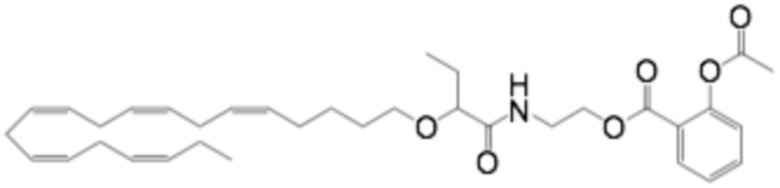 .
.
TBTU (0,85 г, 2,64 ммоль) и TEA (0,8 мл, 5,3 ммоль) добавляли к раствору ацетилсалициловой кислоты (0,4 г, 2,4 ммоль) в DCM (20 мл) и смесь перемешивали в течение 10 минут. Раствор N- (2-гидроксиэтил) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамида (1,0 г, 1,4 ммоль) в DCM (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем кипятили с обратным холодильником в течение ночи. Добавляли воду и полученную смесь дважды экстрагировали DCM. Объединенные органические фазы сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 0-20% EtOAc в гептане в качестве элюента. Концентрирование соответствующих фракций дало 900 мг (выход 65%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,00 (м, 6H), 1,35-1,50 (м, 2H), 1,50-1,85 (м, 4H), 2,00-2,20 (м, 4H), 2,37 (с, 3H), 2,75-2,90 (м, 8H), 3,40-3,55 (м, 2H), 3,55-3,75 (м, 3H), 4,15-4,45 (м, 2H), 5,30-5,50 (м, 10H), 6,80- 95 (м, 1Н), 7,10-7,15 (м, 1Н), 7,30-7,40 (м, 1Н), 7,55-7,65 (м, 1Н), 8,00-8,05 (м, 1Н). МС (ESI): 602 [М+Na]+.
Пример 42. Получение 2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) этил 2-гидроксибензоата:
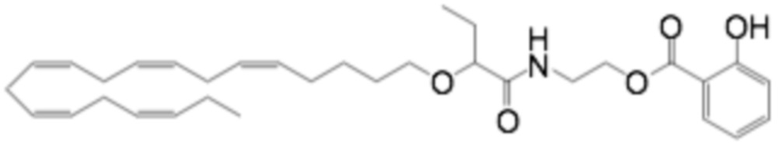 .
.
Аммиак (водный, 28%, 3 мл) добавляли по каплям к раствору 2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1 -илокси) бутанамидо) этил 2-ацетоксибензоата (390 мг, 0,67 ммоль) в 2-пропаноле (27 мл) и воде (9 мл), и смесь перемешивали в течение 10 минут. Добавляли воду и полученную смесь дважды экстрагировали Et2O. Объединенные органические фазы промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 0-20% EtOAc в гептане в качестве элюента. Концентрирование соответствующих фракций дало 63 мг (выход 18%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,91 (т, 3Н), 0,97 (т, 3Н), 1,35-1,45 (м, 2Н), 1,55-1,90 (м, 4Н), 2,00-2,15 (м, 4Н) 2,80-2,90 (м, 8H), 3,44 (т, 2H), 3,65-3,80 (м, 3H), 4,40-4,50 (м, 2H), 5,30-5,45 (м, 10H), 6,85-6,95 (м, 2H), 6,95-7,05 (м, 1H), 7,40-7,50 (м, 1H), 7,80-7,85 (м, 1H), 10,63 (с, 1H). МС (ESI): 560 [М+Na]+.
Пример 43. Получение метил 2-гидрокси-5- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) бензоата:
 .
.
2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -Эйкоза-5,8,11,14,17-пентаен-1-илокси) бутановую кислоту (300 мг, 0,80 ммоль), метил 5-аминосалицилат (140 мг 0,83 ммоль) и TEA (0,22 мл, 1,60 ммоль) растворяли в MeCN (3 мл). Добавляли HATU (320 мг, 0,83 ммоль) и реакционную смесь перемешивали в течение ночи. Затем смесь концентрировали при пониженном давлении и остаток распределяли между Et2O (30 мл) и насыщенным солевым раствором (20 мл). Водную фазу экстрагировали Et2O (20 мл) и объединенные органические фазы промывали 2М HCl (водный, 15 мл), насыщенным NaHCO3 (водный, 15 мл) и насыщенным солевым раствором (15 мл), сушили (Na2SO4), фильтровали и концентроровали в вакууме. Остаток очищали флэш-хроматографией, используя 5% EtOAc в гептане в качестве элюента. Концентрация соответствующих фракций дала 320 мг (77% выход) указанного в заголовке соединения. МС (ESI): 546 [М+Na]+.
Пример 44. Получение 2-гидрокси-5- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) бензойной кислоты:
 .
.
2M NaOH (водный, 6 мл) добавляли к раствору метил-2-гидрокси-5- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен -1-илокси) бутанамидо) бензоата (320 мг, 0,61 ммоль) в МеОН (3 мл) и реакционную смесь нагревали при 50°С в течение ночи. Смесь охлаждали до температуры окружающей среды и подкисляли до pH ~ 2 с помощью 5 М HCl (водный раствор). Полученную смесь экстрагировали EtOAc и органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 1-2% МеОН в EtOAc в качестве элюента. Концентрация соответствующих фракций дала 85 мг (выход 27%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,90-1,05 (т, 3Н), 1,20-1,30 (м, 1Н), 1,45-1,60 (м, 2Н), 1,65-1,75 (м, 2Н), 1,80-1,95 (м, 2Н), 2,05-2,20 (м, 4Н), 2,80-2,90 (м, 8Н), 3,50-3,65 (м, 2Н), 3,85-3,95 (м, 1Н), 5,30-5,45 (м, 10Н), 6,90-7,00 (м, 1Н), 7,58 (ушир. с, 1Н), 8,11 (ушир. с, 1Н), 8,40 (с, 1Н). МС (ESI): 508 [М-Н] -.
Пример 45. Получение (2S) -этил-2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) -4 -метилпентаноата:
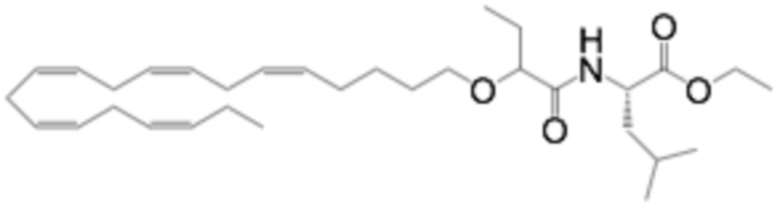 .
.
DCC (1,13 г, 5,5 ммоль) и HOBt (0,74 г, 5,5 ммоль), а затем TEA (1,58 мл, 11,4 ммоль) добавляли к раствору 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутановой кислоты (1,87 г, 5,0 ммоль) в THF (20 мл) и смесь перемешивали в течение 10 минут. Добавляли гидрохлорид этилового эфира L-лейцина (0,89 г, 4,6 ммоль) и полученную смесь перемешивали в течение 2 часов. Добавляли EtOAc (100 мл) и смесь промывали водой, 1 М HCl (водн.), насыщенным NaHCO3 (водн.) и насыщенным солевым раствором. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 10-15% EtOAc в гептане в качестве элюента. Концентрирование соответствующих фракций дало 1,84 г (выход 79%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,05 (м, 12H), 1,25-1,35 (м, 3H), 1,40-1,85 (м, 9H), 2,05-2,20 (м, 4H), 2,80-2,90 (м, 8Н), 3,45-3,75 (м, 3Н), 4,15-4,25 (м, 2Н), 4,55-4,75 (м, 1Н), 5,30-5,45 (м, 10Н), 6,80-6,95 (м, 1Н). МС (ESI): 538 [М+Na]+.
Пример 46. Получение (2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) -4- метилпентановой кислоты:
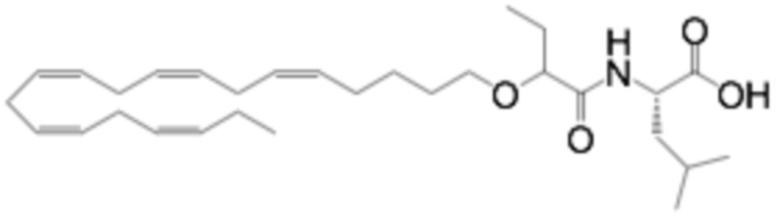 .
.
1M LiOH (водн., 28 мл) добавляли к раствору (2S) -этил 2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен -1-илокси) бутанамидо) -4-метилпентаноата (1,79 г, 3,5 ммоль) в EtOH (50 мл) и реакционную смесь нагревали до 50°C в течение 2 часов. Смесь охлаждали до температуры окружающей среды и добавляли 6М HCl (водн.) до рН ~ 2. Полученную смесь дважды экстрагировали Et2O и объединенные органические фазы сушили (NaSO4), фильтровали и концентрировали при пониженном давлении, получая 1,61 г (выход 95%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,05 (м, 12H), 1,45-1,55 (м, 2H), 1,60-1,85 (м, 7H), 2,05-2,20 (м, 4H), 2,80-2,90 (м, 8H), 3,45-3,65 (м, 2H), 3,70-3,80 (м, 1H), 4,60-4,75 (м, 1H), 5,30-5,45 (м, 10H), 6,90-7,05 (м, 1H), 10,15 (ушир. С, 1H). МС (ESI): 486 [М-Н] -.
Пример 47. Получение метил 2-гидрокси-5 - ((2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1 илокси) бутанамидо) -4-метилпентанамидо) бензоата:
 .
.
DCC (248 мг, 1,2 ммоль) и HOBt (163 мг, 1,2 ммоль) добавляли к раствору (2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8 11,14,17-пентаен-1-илокси) бутанамидо) -4-метилпентановой кислоты (487 мг, 1,0 ммоль) в THF (8 мл) при 0°C. Раствор метил 5-аминосалицилата (201 мг, 1,2 ммоль) в THF (1 мл) добавляли по каплям и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли Et2O (100 мл) и смесь промывали водой, 1М HCl (водн.), насыщенным NaHCO3 (водн.) и насыщенным солевым раствором. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 3-15% EtOAc (содержащий 5% FA) в гептане (также содержащий 5% FA) в качестве элюента. Концентрация соответствующих фракций дало 197 мг (31% выход) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,05 (м, 12H), 1,40-1,50 (м, 2H), 1,60-1,90 (м, 7H), 2,05-2,20 (м, 4H), 2,80-2,90 (м, 8Н), 3,45-3,55 (м, 2Н), 3,75-3,80 (м, 1Н), 3,94 (с, 1Н), 4,55-4,65 (м, 1Н), 5,30-5,45 (м, 10Н), 6,90- 7,00 (м, 2Н), 7,45-7,50 (м, 1Н), 8,00-8,10 (м, 1Н), 8,66 (с, 1Н), 10,59 (с, 1Н). МС (ESI): 659 [М+Na]+.
Пример 48: Получение 2-гидрокси-5 - ((2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1- илокси) бутанамидо) -4-метилпентанамидо) бензойнай кислоты:
 .
.
1M LiOH (водный 2,5 мл) добавляли к раствору метил 2-гидрокси-5 - ((2S) -2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8, 11,14,17-пентаен-1-илокси) бутанамидо) -4-метилпентанамидо) бензоата (190 мг, 0,3 ммоль) в МеОН (5 мл). Реакционную смесь нагревали при 50°С в течение 5 часов и затем перемешивали при комнатной температуре в течение 3 дней. Смесь подкисляли 6М HCl (водн.) до рН ~ 2 и большую часть растворителя удаляли в вакууме. Остаток дважды экстрагировали Et2O и объединенные органические фазы сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией с использованием градиента 5-25% EtOAc (содержащего 5% FA) в гептане (также содержащем 5% FA) в качестве элюента. Концентрирование соответствующих фракций дало 100 мг (выход 54%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,90-1,05 (м, 12H), 1,40-1,50 (м, 2H), 1,60-1,90 (м, 7H), 2,05-2,20 (м, 4H), 2,80-2,90 (м, 8H), 3,45-3,60 (м, 2H), 3,80-3,90 (м, 1H), 4,60-4,75 (м, 1H), 5,30-5,45 (м, 10H), 6,89 (д, 1H), 7,24 (д, 1Н), 7,70-7,90 (м, 2Н), 9,02 (д, 1Н), 10,37 (д, 1Н). МС (ESI): 623 [М+Н]+.
Пример 49. Получение метил 2-гидрокси-5 - (((2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z)) -эйкоза-5,8,11,14,17-пентаен-1-илокси) butanamido) этокси) карбонил) амино) бензоата:
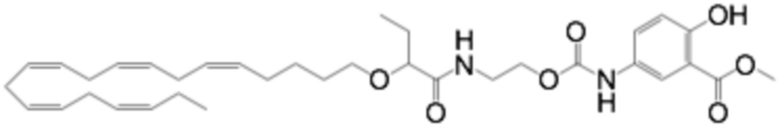 .
.
Дифосген (0,16 мл, 1,3 ммоль) и диизопропиламин (0,16 мл, 0,96 ммоль) добавляли к раствору N- (2-гидроксиэтил) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5, 8,11,14,17-пентаен-1-илокси) бутанамида (0,4 г, 0,96 ммоль) в DCM (15 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа и затем при комнатной температуре в течение 2 часов. Смесь концентрировали при пониженном давлении и остаток суспендировали в THF (20 мл). Суспензию фильтровали и фильтрат концентрировали при пониженном давлении. Затем остаток суспендировали в Et2O, фильтровали сквозь слой диоксида кремния и концентрировали в вакууме, получая 0,66 г 2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14 17-пентаен-1-илокси) бутанамидо) этилкарбонохлоридата в виде неочищенного продукта. Остаток растворяли в DCM (15 мл) и добавляли метил 5-аминосалицилат (0,13 г, 0,77 ммоль) и TEA (0,2 мл, 1,54 ммоль). Реакционную смесь перемешивали в течение 2 часов и затем добавляли воду. Полученную смесь дважды экстрагировали DCM и объединенные органические фазы сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 30-50% EtOAc в гептане в качестве элюента. Концентрирование соответствующих фракций дало 145 мг (30% выход) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,00 (м, 6H), 1,35-1,50 (м, 2H), 1,50-1,85 (м, 4H), 2,00-2,20 (м, 4H), 2,75-2,90 (м, 8Н), 3,40-3,55 (м, 2Н), 3,55-3,75 (м, 3Н), 3,95 (с, 3Н), 4,25-4,35 (м, 2Н), 5,25-5,45 (м, 10Н), 6,68 (ушир. с, 1Н), 6,85-7,00 (м, 2Н), 7,35-7,45 (м, 1Н), 7,91 (ушир. с, 1Н), 10,57 (с, 1Н). МС (ESI): 633 [М+Na]+.
Пример 50. Получение 2-гидрокси-5 - (((2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) этокси) карбонил) амино) бензойная кислоты:
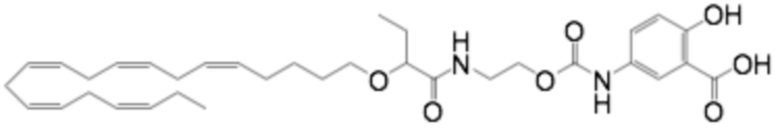 .
.
1M LiOH (водный раствор, 1,9 мл) добавляли к раствору метил 2-гидрокси-5 - (((2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11) 14,17-пентаен-1-илокси) бутанамидо) этокси) карбонил) амино) бензоата (145 мг, 0,24 ммоль) в МеОН (20 мл) и смесь нагревали при 50°С в течение ночи. Реакционную смесь подкисляли 1 М HCl (водн.) до рН ~ 2, а затем дважды экстрагировали Et2O. Объединенные органические фазы сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя 20% EtOAc (содержащий 5% FA) в гептане (также содержащий 5% FA) в качестве элюента. Соответствующие фракции объединяли и концентрировали. Остаток (46 мг) дополнительно очищали препаративной ВЭЖХ, используя градиент 30-95% MeCN в воде (содержащей 5% MeCN и 0,01% TFA) в качестве элюента. Соответствующие фракции объединяли и концентрировали, а остаток растворяли в толуоле. Концентрирование раствора дает 24 мг (17% выход) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,91-1,01 (м, 6H), 1,35-1,50 (м, 2H), 1,55-1,90 (м, 4H), 2,05-2,15 (м, 4H), 2,80-2,90 (м, 8H), 3,45-3,55 (м, 2H), 3,65-3,75 (м, 2H), 3,75-3,85 (м, 1H), 4,25-4,35 (м, 2H), 5,30-5,45 (м, 10H), 6,90-7,00 (м, 2Н), 7,00-7,10 (м, 1Н), 7,63 (ушир. С, 1Н), 7,83 (с, 1Н), 10,43 (ушир. С, 1Н). МС (ESI): 597 [М+Н]+.
Пример 51: Получение 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) -N- (2-изоцианатоэтил) бутанамида:
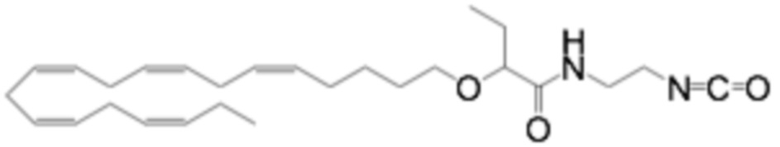 .
.
1,8-бис (диметиламино) нафталин (577 мг, 2,7 ммоль) добавляли к раствору N- (2-аминоэтил) -2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8 11,14,17-пентаен-1-илокси) бутанамида (560 мг, 1,35 ммоль) в DCM (10 мл) и смесь охлаждали до 0°С. Трихлорметилхлорформиат (98 мкл, 0,81 ммоль) добавляли по каплям, прежде чем убрать охлаждающую баню и смесь перемешивали в течение 15 минут. Добавляли 1 М HCl (водный, 30 мл) и DCM (30 мл). Полученные две фазы разделяли и органическую фазу промывали 4 раза 1М HCl (водн.) и один раз 1М NaOH (водн.), сушили (NaSO4), фильтровали и концентрировали при пониженном давлении, получая 500 мг (выход 84%) указанное в заголовке соединение в виде сырого продукта. MS (ESI): 465 [М+Na]+.
Пример 52: Получение метил-2-гидрокси-5- (3- (2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1- илокси) бутанамидо) этил) уреидо) бензоата:
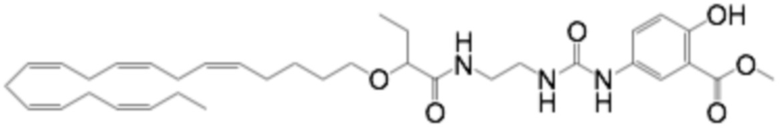 .
.
Метил 5-аминосалициат (189 мг, 1,13 ммоль) добавляли к раствору 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) -N- (2-изоцианатоэтил) бутанамида (500 мг, 1,13 ммоль) в DCM (5 мл). Реакционную смесь перемешивали в течение 2 часов и затем концентрировали в вакууме. Остаток очищали флэш-хроматографией, используя градиент 40-0% гептана в EtOAc в качестве элюента. Концентрирование соответствующих фракций дало 230 мг (выход 33%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,85-1,05 (2х, 6H), 1,35-1,50 (м, 2H), 1,55-1,85 (м, 4H), 2,05-2,20 (м, 4H), 2,75-2,95 (м, 8Н), 3,35-3,50 (м, 6Н), 3,65-3,75 (м, 1Н), 3,94 (с, 3Н), 5,30-5,50 (м, 10Н), 6,95 (д, 1Н), 7,10 (ушир. с., 1Н), 7,40 (дд, 1Н), 7,88 (д, 1Н), 10,62 (с, 1Н). MS (ESI); 632 [М+Na]+.
Пример 53. Получение 2-гидрокси-5- (3- (2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z)) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутанамидо) этил) уреидо) бензойной кислоты:
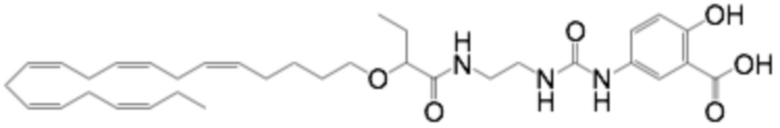 .
.
1M LiOH (водный раствор, 2,9 мл) добавляли по каплям к раствору метил 2-гидрокси-5- (3- (2- (2 - ((5Z, 8Z, 11Z, 14Z, 17Z)) эйкоза-5,8, 11,14,17-пентаен-1-илокси) бутанамидо) этил) уреидо) бензоата (220 мг, 0,36 ммоль) в МеОН (10 мл) и смесь нагревали при 50°С в течение ночи. Смесь охлаждали до температуры окружающей среды и затем подкисляли до рН ~ 2 с помощью 6 М HCl (водн.). Полученную смесь дважды экстрагировали EtOAc и объединенные органические фазы промывали насыщенным солевым раствором, сушили (NaSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией при использовании градиента 5-40% EtOAc (содержащего 5% FA) в гептане (также содержащего 5% FA) в качестве элюента. Концентрирование соответствующих фракций дало 92 мг (выход 43%) указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3): δ 0,91 (т, 3Н), 0,98 (т, 3Н), 1,40-1,50 (м, 2Н), 1,60-1,90 (м, 4Н), 2,05-2,20 (м, 4Н) 2,80-2,90 (м, 8H), 3,40-3,60 (м, 6H), 3,75-3,80 (м, 1H), 5,30-5,45 (м, 10H), 6,90 (д, 1H), 7,25-7,35 (м, 1H), 7,45-7,55 (м, 1H), 7,80-7,95 (м, 2H), 10,56 (ушир. с, 1H). МС (ESI): 596 [М+Н]+, 618 [М+Na]+.
Пример 54. Получение трет-бутил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) пропаноата:
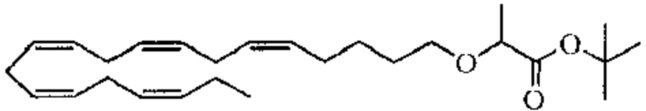 .
.
Смесь (5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ола, (1,00 г, 3,47 ммоль), хлорида тетрабутиламмония (0,24 г, 0,87 ммоль) и трет-бутил-2-бромпропионата (3,62 г, 17,3 ммоль) растворяли в толуоле (36 мл) и помещали в атмосферу азота. При интенсивном перемешивании медленно добавляли водный раствор гидроксида натрия (50%, 8 мл) и полученную смесь перемешивали при температуре окружающей среды в течение двадцати часов. Добавляли воду и смесь трижды экстрагировали эфиром. Объединенный органический экстракт промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, используя 2% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 1,40 г (выход 90%) указанного в заголовке соединения в виде масла. 1H-ЯМР (300 МГц, CDCl3): δ 0,95 (т, 3Н), 1,41 (д, 3Н), 1,48 (с, 9Н), 1,48-1,66 (м, 4Н), 2,05 (м, 4Н), 2,83 (м, 8Н), 3,35 (м, 1Н), 3,55 (м, 1Н), 3,79 (кв, 1Н), 5,32-5,44 (м, 10Н).
Пример 55. Получение 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) пропановой кислоты:
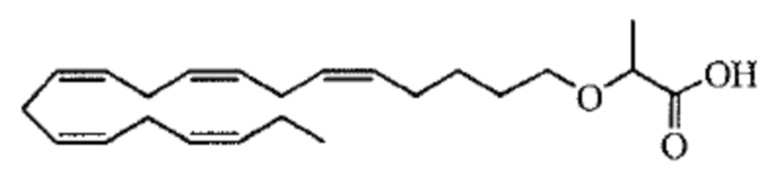 .
.
Трифторуксусную кислоту (2 мл) добавляли к раствору 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) пропаноата (1,40 г, 3,36 ммоль) в дихлорметане (10 мл), выдерживали в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение трех часов. Добавляли диэтиловый эфир (50 мл) и органическую фазу промывали водой (30 мл), сушили (Na2SO4) и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана, этилацетата и муравьиной кислоты (95: 5: 0,25 -> 80: 20: 1). Концентрирование соответствующих фракций дало 0,67 г слегка загрязненного продукта. Это вещество растворяли в гептане (15 мл), трижды промывали водой (5 мл), сушили (Na2SO4), фильтровали и концентрировали, получая 0,50 г (выход 41%) указанного в заголовке соединения в виде масла. 1H-ЯМР (300 МГц, CDCl3): δ 0,99 (т, 3H), 1,40-1,48 (м, 5H), 1,67 (м, 2H), 2,09 (м, 4H), 2,80-2,60 (м, 8H), 3,53 (м, 2Н), 4,01 (кв, 1Н), 5,31-5,47 (м, 10Н); МС (электрораспыление): 359,2 [М-Н]-.
Пример 56. Получение трет-бутил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) -2-метилпропаноата:
 .
.
Смесь (5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ола (0,83 г, 3,14 ммоль), хлорида тетрабутиламмония (0,24 г, 0,85 ммоль) и трет-бутил-2-бромизобутирата (3,50 г, 15,7 ммоль) растворяли в толуоле (15 мл) и помещали в атмосферу азота. Водный раствор гидроксида натрия (50%, 5 мл) медленно добавляли при интенсивном перемешивании при комнатной температуре. Полученную смесь нагревали до 60°С и перемешивали в течение шести часов. Смесь охлаждали, добавляли воду и трижды экстрагировали эфиром. Объединенный органический экстракт промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, используя градиент 5-10% этилацетата в гептане в качестве элюента. Концентрирование соответствующих фракций дало 0,60 г (выход 44%) указанного в заголовке соединения в виде масла. МС (электрораспыление): 453,3 [М+Na]+.
Пример 57. Получение 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) -2-метилпропановой кислоты:
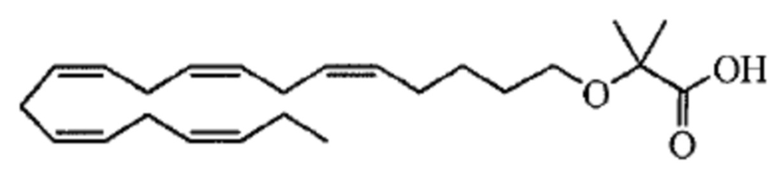 .
.
Трифторуксусную кислоту (5 мл) добавляли к раствору трет-бутил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаенилокси) -2-метилпропаноата (600 мг (1,39 ммоль) в дихлорметане (20 мл) в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение двух часов. Добавляли воду и водную фазу дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, используя смесь гептана, этилацетата и муравьиной кислоты (80: 20: 1) в качестве элюента. Соответствующие фракции концентрировали и остаток (135 мг) дополнительно очищали флэш-хроматографией на силикагеле, используя градиент 5-10% смеси этилацетата и муравьиной кислоты (95: 5) в гептане в качестве элюента. Концентрация соответствующих фракций давала 80 мг слегка загрязненного продукта. Это вещество растворяли в гептане (5 мл), дважды промывали водой (5 мл), сушили (Na2SO4), фильтровали и концентрировали, получая 40 мг (выход 8%) указанного в заголовке соединения в виде масла. 1H-ЯМР (300 МГц, CDCl3): δ 0,99 (т, 3Н), 1,47 (с, 6Н), 1,64 (м, 2Н), 2,07 (м, 4Н), 2,81-2,88 (м, 8Н), 3,46 (т, 2Н), 5. 29-5,44 (м, 10Н); МС (электрораспыление): 373,3 [М-Н] -.
Пример 58. Получение трет-бутил-2-этил-2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноата:
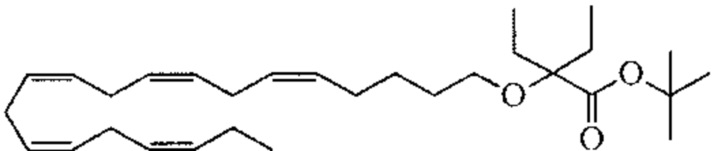 .
.
Трет-бутил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноат (480 мг, 1,11 ммоль) добавляли по каплям в течение 30 минут к раствору диизопропиламина лития (LDA) (2,0 М, 750 мкл, 1,50 ммоль) в сухом тетрагидрофуране (10 мл), выдерживаемом при -70°C в атмосфере азота. Реакционную смесь перемешивали в течение 30 минут. Этилйодид (312 мг, 2,00 ммоль) добавляли одной порцией и полученную смесь нагревали до температуры окружающей среды в течение 1 часа. Реакционную смесь перемешивали при температуре окружающей среды в течение 17 часов. Смесь выливали в насыщенный NH4Cl (водный) (50 мл) и экстрагировали гептаном (2 × 50 мл). Объединенные органические фазы последовательно промывали насыщенным солевым раствором (50 мл), 0,25 М HCl (50 мл) и снова насыщенным солевым раствором (50 мл), сушили (MgSO4), фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (100: 0 -> 95: 5). Концентрирование соответствующих фракций дало 343 мг (выход 67%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3): δ 0,84 (т, 6Н), 0,99 (тд, 3Н), 1,35-1,55 (м, 11Н), 1,54-1,69 (м, 2Н), 1,68-1,87 (м, 4Н), 1,99-2,24 (м, 4H), 2,74-2,99 (м, 8H), 3,31 (т, 2H), 5,23-5,52 (м, 10H); МС (электрораспыление): 401,3 [М-1] -.
Пример 59. Получение 2-этил-2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -икозо-5,8,11,14,17-пентаен-1-илокси) бутановой кислоты:
 .
.
Смесь муравьиной кислоты (5 мл) и трет-бутил-2-этил-2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноата (250 мг, 0,55 ммоль) энергично перемешивали в атмосфере азота при комнатной температуре в течение 4,5 часов. Муравьиную кислоту удаляли в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата (100: 0 -> 80:20). Концентрирование соответствующих фракций дало 163 мг (выход 74%) указанного в заголовке соединения в виде масла. 1H ЯМР (300 МГц, CDCl3): δ 0,86 (т, 6Н), 0,99 (т, 3Н), 1,36-1,57 (м, 2Н), 1,68 (дд, 2Н), 1,73-1,98 (м, 4Н), 2,11 (тт, 4H), 2,70-3,01 (м, 8H), 3,39 (т, 2H), 5,20-5,56 (м, 10H). МС (электрораспыление): 481,4 [М+Na]+.
Пример 60: Получение трет-бутил 2 - (((3Z, 6Z, 9Z, 12Z) -пентадека-3,6,9,12-тетраен-1-ил) окси) бутаноата
Стадия а) (8Z, 11Z, 14Z, 17Z) -5,6-дигидроксиэйкоза-8,11,14,17-тетраеновая кислота
 .
.
Раствор, содержащий 6 - ((3Z, 6Z, 9Z, 12Z) -1-йодопентадека-3,6,9,12-тетраен-1-ил) тетрагидро-2Н-пиран-2-он (12,5 г, 29,2 ммоль)) и КОН (5,73 г, 102 ммоль) в смеси МеОН (150 мл) и воды (7,5 мл) перемешивали при 60°С в атмосфере азота в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, подкисляли 2 М HCl (водн.) и экстрагировали EtOAc (300 мл). Органическую фазу промывали водой (300 мл с небольшим количеством солевого раствора), сушили (Na2SO4), фильтровали и концентрировали в вакууме, получая 9,8 г (выход 100%) в виде масла.
Стадия b) (3Z, 6Z, 9Z, 12Z) -пентадека-3,6,9,12-тетраен-1-ол
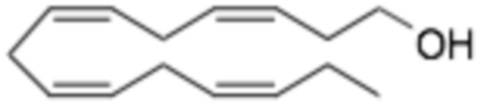 .
.
Периодат натрия (9,34 г, 43,7 ммоль) добавляли к раствору (8Z, 11Z, 14Z, 17Z) -5,6-дигидроксиэйкоза-8,11,14,17-тетраеновой кислоты (9,8 г, 29,1 ммоль) в THF: вода (150 мл) при 0°С в атмосфере азота и перемешивали в течение 1 часа. Реакционную смесь экстрагировали EtOAc (300 мл). Органический слой отделяли и промывали разбавленным солевым раствором (200 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток растворяли в МеОН (100 мл) и охлаждали до 0°С. NaBH4 (3,31 г, 87 ммоль) осторожно добавляли порциями и смесь перемешивали в течение 30 минут при 0°C. Осторожно добавляли воду (300 мл) и смесь экстрагировали EtOAc (2 × 200 мл). Органические фазы объединяли и промывали водой (200 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 20% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 4,06 г (выход 63%) названного соединения в виде масла.
1H ЯМР (400 МГц, CDCl3) δ 0,91 (т, 3Н), 1,97-2,05 (м, 2Н), 2,27-2,32 (м, 2Н), 2,73-2,81 (м, 6Н), 3,57-3,62 (м, 2Н), 5,17-5,41 (м, 7H), 5,42-5,56 (м, 1H).
Стадия c) трет-бутил 2 - (((3Z, 6Z, 9Z, 12Z) -пентадека-3,6,9,12-тетраен-1-ил) окси) бутаноат
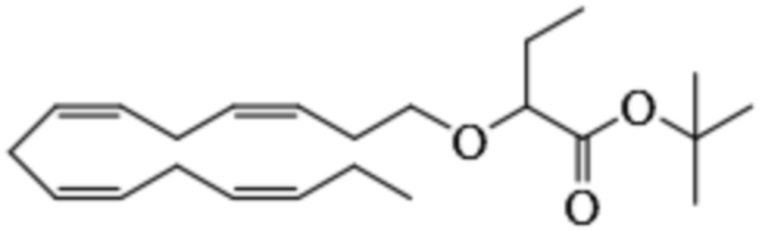 .
.
Хлорид тетрабутиламмония (0,10 г, 0,33 ммоль) добавляли к раствору ((3Z, 6Z, 9Z, 12Z) -пентадека-3,6,9,12-тетраен-1-ола (0,22 г, 1,00 ммоль) в толуоле (10 мл) при температуре окружающей среды в атмосфере азота, с последующим добавлением трет-бутил-2-бромбутирата (1,11 г, 5,00 ммоль). Водный раствор гидроксида натрия (50% (вес/вес), 4 мл) добавляли при энергичном перемешивание при комнатной температуре. Полученную смесь нагревали до 50°C и перемешивали в течение двух часов с последующим перемешиванием при температуре окружающей среды в течение ночи. После охлаждения до комнатной температуры добавляли воду и водную фазу экстрагировали диэтиловым эфиром. Фазы разделяли и водную фазу экстрагировали еще два раза диэтиловым эфиром. Органические фазы объединяли и промывали водой, а затем насыщенным солевым раствором, затем сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 5% этилацетат в гептане в качестве элюента. Концентрирование соответствующих фракций дало 0,30 г (83%) названного соединения в виде масла.
Пример 61: Получение 2 - (((3Z, 6Z, 9Z, 12Z) -пентадека-3,6,9,12-тетраен-1-ил) окси) бутановой кислоты:
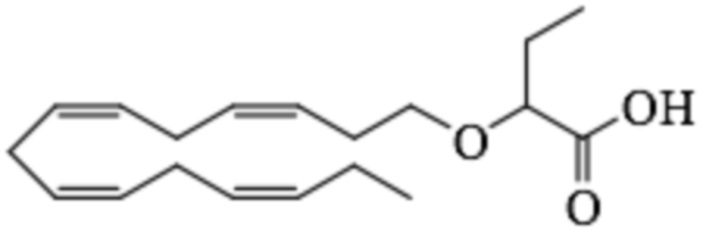 .
.
Трифторуксусную кислоту (2 мл) добавляли к раствору трет-бутил 2 - (((3Z, 6Z, 9Z, 12Z) -пентадека-3,6,9,12-тетраен-1-ил) окси) бутаноата в дихлорметане (10 мл) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение одного часа, затем добавляли воду и водную фазу дважды экстрагировали дихлорметаном. Органические фазы объединяли и промывали насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя 5% этилацетат в гептане (добавляли 1% HCOOH) в качестве элюента. Концентрирование соответствующих фракций дало 0,18 г (выход 71%) указанного в заголовке соединения в виде масла. 1H-ЯМР (300 МГц, CDCl3): δ 0,90-1,05 (2xт, 6H), 1,75-1,90 (м, 2H), 2,05-2,15 (м, 2H), 2,30-2,50 (м, 2H), 2,85 (м, 6H), 3,60 (м, 2H), 3,85 (т, 1H), 5,25-5,60 (м, 8H).
Пример 62: Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -нонадека-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты:
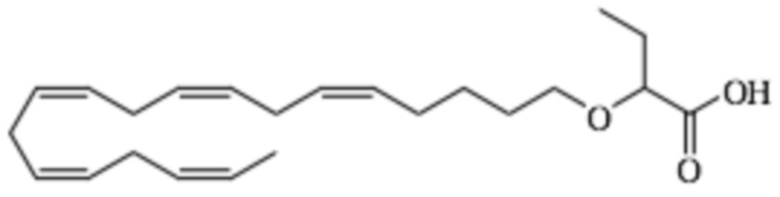 .
.
Шаг 1:
Раствор дека-2,5,8-триин-1-ола (0,94 г, 6,43 ммоль) в THF (100 мл), охлажденный до 0°C в атмосфере азота, добавляли к метансульфонилхлориду (720 мкл, 9,24 ммоль) с последующим добавлением по каплям TEA (1,97 мл, 14,13 ммоль) в течение 15 минут. Смесь перемешивали при 0°С в течение 2 часов. Затем смесь выливали в воду: лед 1: 1 (200 грамм) и экстрагировали диэтиловым эфиром (2х100). Объединенные органические фазы промывали насыщенным водным NaHCO3 (200 мл), воду (200 мл) и насыщенным солевым раствором (200 мл), сушили (Na2SO4), фильтровали и упаривали в вакууме, получая сырой продукт, который затем очищали сухим испарением на силикагеле. Фракции 1-3 собирали, используя гептан, фракции 4-6, используя EtOAc: гептан 2,5: 100, фракции 7-10, используя EtOAc: гептан (5: 100), и фракцию 11, используя EtOAc. Соответствующие фракции объединяли и концентрировали. Получено 1,07 г (4,77 ммоль, выход 74,1%) нона-2,5,7-триин-1-ил метансульфоната.
Шаг 2:
К раствору этилмагнийбромида (4,00 мл, 12,00 ммоль) комнатной температуры, разбавленному сухим THF (25 мл), добавляли порциями раствор пропаргилового спирта (0,35 мл, 5,99 ммоль) в течение 20 минут. Реакционную смесь кипятили с обратным холодильником в течение 90 минут, затем охлаждали до 0°С в течение 30 минут. Добавляли каталитическое количество бром(диметилсульфид) меди (I) (330 мг, 1,605 ммоль) и охлаждающую баню убирали. После 15-минутного перемешивания по каплям в течение 5 минут добавляли нона-2,5,7-триин-1-илметансульфонат (1,05 г, 4,68 ммоль), растворенный в THF (5 мл) при нагревании. Реакционную смесь нагревали до температуры кипения с обратным холодильником и перемешивали при температуре кипения с обратным холодильником в течение ночи. Реакционную смесь охлаждали до температуры окружающей среды и реакцию останавливали осторожным добавлением 50 мл насыщенного раствора водного NH4Cl. Полученную смесь переносили в делительную воронку, содержащую диэтиловый эфир (100 мл). Органическую фазу отделяли, а водную фазу дважды экстрагировали диэтиловым эфиром (2 × 100 мл). Объединенные органические фазы промывали насыщенным солевым раствором (50 мл) и сушили (Na2SO4). Остаток концентрировали в вакууме перед очисткой флэш-хроматографией на силикагеле. Соответствующие фракции объединяли и концентрировали, получая 0,49 г (2,65 ммоль, выход 56,6%) додека-2,5,8,10-тетраин-1-ола.
Шаг 3:
Раствор додека-2,5,8,10-тетраин-1-ола (465 мг, 2,52 ммоль) в THF(30 мл), охлажденный до 0°С в атмосфере азота, добавляли метансульфонилхлорид (0,256 мл, 3,28 ммоль) с последующим добавлением по каплям TEA (0,704 мл, 5,05 ммоль) в течение 15 минут. Смесь перемешивали при 0°С в течение 1 часа. Затем смесь выливали в смесь вода: лед 1: 1 (100 грамм) и экстрагировали диэтиловым эфиром (2х100 мл). Объединенные органические фазы промывали насыщенным водным NaHCO3 (200 мл), водой (200 мл) и насыщенным солевым раствором (200 мл), сушили (Na2SO4), фильтровали и упаривали в вакууме, получая 622 мг неочищенного продукта, который затем очищали сухим испарением. Фракции 1-3 собирали с использованием гептана, фракции 4-6 с использованием EtOAc: гептан (2,5: 100), фракции 7-10 с использованием EtOAc: гептан (5: 100) и фракции 11 с использованием EtOAc. Соответствующие фракции объединяли и концентрировали. Получено 466 мг (1,78 ммоль, выход 70,4%) тридека-2,5,8,11-тетраин-1-ил метансульфоната.
Шаг 4:
К раствору/суспензии карбоната цезия (553 мг, 1,696 ммоль), йодида натрия (280 мг, 1,866 ммоль) и иодида меди (I) (323 мг, 1,696 ммоль) в DMF (сухой) (15 мл) добавляли этил-гекс-5-иноат (262 мг, 1,866 ммоль) в DMF (сухой) (5 мл). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 40 минут. Добавляли тридека-2,5,8,11-тетраин-1-ил метансульфонат (445 мг, 1,696 ммоль) в DMF (сухой) (5 мл) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NH4Cl (200 мл) и смесь экстрагировали смесью EtOAc: гептан (1: 1, 3 × 150 мл). Объединенные органические фазы промывают насыщенным солевым раствором (200 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и этилацетата, начиная с раствора 2% EtOAc в гептане. Соответствующие фракции объединяли и концентрировали, получая 51 мг (0,166 ммоль, выход 9,8%) этилнонадека-5,8,11,14,17-пентайноата.
Шаг 5:
Раствор этилнонадека-5,8,11,14,17-пентайноата (60 мг, 0,196 ммоль) в гептане (10 мл) и толуоле (15 мл) барботировали с помощью N2-газа перед добавлением хинолина (10 мкл, 0,084 ммоль) и катализатора Линдлара (50 мг, 0,023 ммоль), которые добавляли в атмосфере азота. Суспензию перемешивали в атмосфере H2 (1 атм) в течение 15 часов. Реакционную смесь фильтровали через целит и упаривали в вакууме. Неочищенный материал очищали флэш-хроматографией на силикагеле, используя в качестве элюента смесь 2% EtOAc в гептане. Соответствующие фракции объединяли и концентрировали, получая (5Z, 8Z, 11Z, 14Z, 17Z) -этилнонадека-5,8,11,14,17-пентаеноат (24,6 мг, 0,078 ммоль, выход 39,7%) в виде масла. 1H ЯМР (400 МГц, хлороформ-d) δ 1,27 (т, 2Н), 1,59-1,70 (м, 2Н), 1,72 (с, 0Н), 2,31 (д, 1Н), 2,72-2,95 (м, 3Н), 4,15 (д, 1H), 5,40 (дкв, 4H).
Шаг 6:
К раствору (5Z, 8Z, 11Z, 14Z, 17Z) -этилнонадека-5,8,11,14,17-пентаеноата (22 мг, 0,070 ммоль) в тетрагидрофуране (5 мл), охлажденному до 0°С, добавили ЛАГ (LAH, алюмогидрид лития) (8 мг, 0,211 ммоль) одной порцией. Реакционную смесь перемешивали 30 минут при 0°С, а затем 90 минут при комнатной температуре. Реакционную смесь выливали в насыщенный NH4Cl (водн.) (10 мл) со льдом (10 г) и смесь подкисляли, используя 10% HCl (водн.), до рН ~ 1-2. Смесь дважды экстрагировали Et2O (25 мл) и объединенные органические фазы промывали водой (25 мл) и насыщенным солевым раствором (25 мл), затем сушили (MgSO4), фильтровали и концентрировали в вакууме, получая (5Z, 8Z, 11Z, 14Z, 17Z) -нонадека-5,8,11,14,17-пентаен-1-ол (15,5 мг, 0,056 ммоль, выход 81%) в виде масла.
Шаг 7:
2-Бромомасляную кислоту (100 мг, 0,599 ммоль) добавляли к раствору (5Z, 8Z, 11Z, 14 Z, 17Z) -нонадека-5,8,11,14,17-пентаен-1-ола (8 мг, 0,029 ммоль) в тетрагидрофуране (10 мл). Смесь охлаждали до 5°C, после чего по каплям в течение 5 минут добавляли 17% (вес/вес) раствор трет-бутоксида натрия (500 мкл, 0,678 ммоль) в трет-бутилметиловом эфире (30 мл). Через 3 часа добавляли дополнительное количество трет-бутоксида натрия (100 мкл 0,136 ммоль) и реакционную смесь перемешивали в течение еще 3 часов. Реакцию останавливали добавлением муравьиной кислоты (0,100 мл, 2,61 ммоль) и воды (10 мл), и полученную смесь экстрагировали трет-бутилметиловым эфиром (30 мл). Органическую фазу промывали четыре раза водой (4 × 10 мл), сушили (MgSO4), фильтровали и упаривали в вакууме. Концентрат растворяли в Me-THF (50 мл) и промывали насыщенным водным NH4Cl (20 мл) четыре раза. Органическую фазу сушили (MgSO4), фильтровали и упаривали в вакууме, получая 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -нонадека-5,8,11,14,17-пентаен-1-ил) окси) бутановую кислоту (6 мг, 0,017 ммоль, 57,1%) в виде масла. HRMS (электрораспыление), [M-H] -: Рассчитано: 359,2586, найдено: 359,2578.
Пример 63: Получение 2 - (((6Z, 9Z, 12Z, 15Z) -октадека-6,9,12,15-тетраен-1-ил) окси) бутановой кислоты:
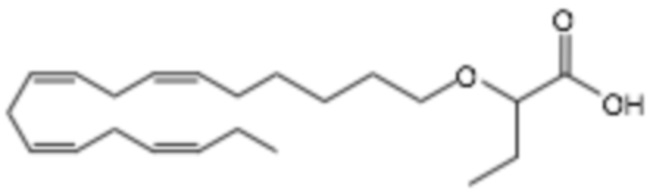 .
.
К раствору (6Z, 9Z, 12Z, 15Z) -октадека-6,9,12,15-тетраен-1-ола (90 мг, 0,343 ммоль) в толуоле (3 мл) добавляли гидроксид тетрабутиламмония (TBAOH) (2 мг, 0,008 ммоль), трет-бутил-2-бромбутаноат (190 мг, 0,819 ммоль) и NaOH (50 мас.%, 1 мл) и смесь перемешивали в течение 3 часов при 45°С. К реакционной смеси добавляли еще трет-бутил-2-бромбутаноат (190 мг, 0,819 ммоль) и реакционную смесь перемешивали в течение 24 часов при 45°C. Смесь охлаждали до комнатной температуры и дважды экстрагировали Et2O (2 × 30 мл). Объединенные органические экстракты промывали водой (3 × 25 мл) и насыщенным солевым раствором (25 мл), фильтровали и упаривали в вакууме, получая сырой продукт трет-бутил 2 - (((6Z, 9Z, 12Z, 15Z) -октадека-6)., 9,12,15-тетраен-1-ил) окси) бутаноата. Неочищенный продукт трет-бутил 2 - (((6Z, 9Z, 12Z, 15Z) -октадека-6,9,12,15-тетраен-1-ил) окси) бутаноата растворяли в муравьиной кислоте (FA, 5 мл) и полученный раствор перемешивали при комнатной температуре в течение 3 часов. Смесь концентрировали в вакууме при 25°C и остаток растворяли в EtOAc (30 мл). Раствор промывали водой (4х25 мл), разбавляли EtOAc (20 мл) и сушили (MgSO4). Растворитель удаляли в вакууме и неочищенный продукт очищали флэш-хроматографией на силикагеле, используя в качестве элюента прогрессивно полярные смеси гептана и ацетона (1: 0 до 5: 1). Фракции объединяли и упаривали в вакууме. Остаток растворяли в FA (5 мл) и перемешивали при комнатной температуре в течение 3 часов, после чего растворитель выпаривали в вакууме. К остатку добавляли EtOAc (30 мл), промывали водой (4 × 25 мл), насыщенным солевым раствором (25 мл), сушили (MgSO4) и упаривали в вакууме. Остаток очищали препаративной ВЭЖХ, используя 85% MeCN в воде в качестве элюента. Соответствующие фракции объединяли и концентрировали. Остаток растворяли в EtOAc, сушили (MgSO4), фильтровали и концентрировали в вакууме. Продукт растворяли в Et2O и концентрировали в вакууме, получая 10 мг (выход 8%) указанного в заголовке соединения. 1H ЯМР (400 МГц, CDCl3) δ 1,09-0,81 (м, 6H), 1,41 (дт, 4H), 1,67 (д, 2H), 1,97-1,74 (м, 2H), 2,10 (т, 4H), 2,84 (кв, 6H), 3,55-3,42 (м, 1H), 3,65-3,54 (м, 1H), 3,88 (дд, 4,8 Гц, 1H), 5,53-5,25 (м, 8H). HRMS (электрораспыление), [M-H] -: Рассчитано: 347,2592, обнаружено: 347,2576.
Пример 64: Получение 2 - (((4Z, 7Z, 10Z, 13Z, 16Z) -нонадека-4,7,10,13,16-пентаен-1-ил) окси) бутановой кислоты
Шаг 1:
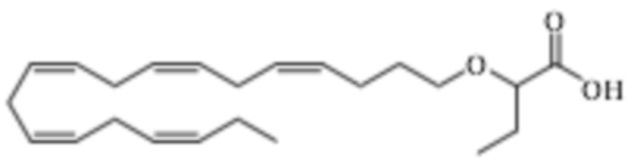 .
.
К раствору (4Z, 7Z, 10Z, 13Z, 16Z) -нонадека-4,7,10,13,16-пентаен-1-ола (446 мг, 1,625 ммоль) и трет-бутил-2-бромбутирата (501 мг)., 2,246 ммоль) в толуоле (5 мл) и добавляли гидросульфат тетрабутиламмония (102 мг, 0,300 ммоль). При перемешивании добавляли гидроксид натрия (2,5 мл, 46,9 ммоль) и реакционную смесь нагревали непосредственно до 50°С. Через 1 час добавляли дополнительный трет-бутил-2-бромбутират (533 мг, 2,389 ммоль), и реакцию оставляли еще на 3 часа, затем охлаждали при перемешивании в течение ночи. Добавляли Et2O (50 мл) и эмулировали. Добавляли насыщенный NH4Cl (водн.) (50 мл) и смесь встряхивали. Фазы разделяли и водную фазу экстрагировали Et2O (50 мл). Объединенную органическую фазу промывали 5% (мас %) NaOH (водн., 2 × 50 мл), сушили (MgSO4), фильтровали и упаривали. Остаток сушили под вакуумом в течение 5 дней, затем разбавляли гептаном (1 мл) и очищали методом сухого испарения с использованием силикагеля (5 грамм), элюируя сначала 2х50 мл гептана, а затем 6х50 мл смеси гептан: EtOAc (95: 5) всего 8 фракций. Фракцию 3-4 собирали с получением трет-бутил 2 - ((4Z, 7Z, 10Z, 13Z, 16Z) -нонадека-4,7,10,13,16-пентаенилокси) бутаноата (455 мг, 1,092 ммоль, выход 67,2%) после выпаривания. 1H ЯМР (300 МГц, хлороформ-d) δ 0,98 (тд, 6H), 1,49 (с, 9H), 1,57-1,84 (м, 4H), 2,00-2,28 (м, 4H), 2,74-2,94 (м, 8H).), 3,33 (дт, 1H), 3,50-3,70 (м, 2H), 5,24-5,50 (м, 10H).
Шаг 2:
Смесь трет-бутил 2 - ((4Z, 7Z, 10Z, 13Z, 16Z) -нонадека-4,7,10,13,16-пентаенилокси) бутаноата (373 мг, 0,895 ммоль) в HCOOH (10 мл) энергично перемешивали в течение 2,5 часов. Смесь концентрировали через 3 часа общего времени реакции, разбавляли этилацетатом (50 мл) и промывали водой (3 × 50 мл), до получения нейтрального значения рН в водной фазе. Органическую фазу сушили (MgSO4), фильтровали и концентрировали в вакууме. Очистку проводили сухим способом флэш-хроматографии с использованием силикагеля (8 г) и элюирование гептаном: EtoAc 90:10 (3 × 30 мл), затем 80:20 (7 × 50 мл). Фракции 4-6 собирали и концентрировали в вакууме, получая 2 - ((4Z, 7Z, 10Z, 13Z, 16Z) -нонадека-4,7,10,13,16-пентаенилокси) бутановую кислоту (122 мг, 0,338 ммоль, выход 37,8%). 1H ЯМР (300 МГц, хлороформ-d) δ 1,00 (тд, 2,6 Гц, 6H), 1,62-1,79 (м, 2H), 1,79-1,92 (м, 2H), 1,99-2,13 (м, 2H), 2,19 (дд, 2H), 2,85 (дтд, 8H), 3,47-3,67 (м, 2H), 3,88 (дд, 1H), 5,31-5,47 (м, 10H). HRMS (электрораспыление), [M-H] -: Рассчитано: 359,2586, найдено: 359,2590.
Пример 65: Получение 2 - (((8Z, 11Z, 14Z) -октадека-8,11,14,17-тетраен-1-ил) окси) бутановой кислоты:
 .
.
Шаг 1:
К раствору карбоната цезия (2,67 г, 8,20 ммоль), NaI (1,23 г, 8,20 ммоль) и иодида меди (I) (1,56 г, 8,20 ммоль) в DMF (20 мл) добавляли метил нон-8- иноат (1,38 г, 8,20 ммоль) в DMF (5 мл). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 40 минут. Добавляли нона-8-ен-2,5-диин-1-ил метансульфонат (1,74 г, 8,20 ммоль) в DMF (5 мл) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный NH4Cl (200 мл) и смесь экстрагировали EtOAc: гептан (1: 1, 3 × 150 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. На выходе флэш-хроматография (гептан/EtOAc 92/8) давала метилоктадека-17-ен-8,11,14-трийноат (1,43 г, 5,03 ммоль, выход 61,3%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,80-5,67 (м, 1Н), 5,29-5,18 (м, 1Н), 5,08-4,99 (м, 1Н), 3,60 (с, 3Н), 3,15-3,09 (м, 2Н)), 3,09-3,05 (м, 2H), 2,93-2,85 (м, 2H), 2,24 (т, 2H), 2,11-2,05 (м, 2H), 1,60-1,52 (м, 2H), 1,46-1,38 (м., 2H), 1,35-1,23 (м, 4H). МС (электрораспыление): 307,1 [М+Na]+.
Шаг 2:
Суспензию Lindlar Catalyst (1,063 г, 0,499 ммоль) в гептане (15 мл) в атмосфере азота добавляли к хинолину (0,177 мл, 1,498 ммоль) и раствору метилоктадека-17-ен-8,11,14- трийноата (1,42 г, 4,99 ммоль) в гептане (5 мл). Смесь перемешивали в атмосфере H2 (1 атм) в течение ночи. Смесь фильтровали и концентрировали в вакууме. Флэш-хроматография (гептан/EtOAc 98,5/1,5) давала (8Z, 11Z, 14Z) -метооктадека-8,11,14,17-тетраеноат (0,830 г, 2,86 ммоль, выход 57,2%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,84-5,65 (м, 1H), 5,43-5,19 (м, 6H), 5,05-4,85 (м, 2H), 3,60 (с, 3H), 2,81-2,68 (м, 6H)), 2,23 (т, 2Н), 2,02-1,92 (м, 2Н), 1,59-1,52 (м, 2Н), 1,34-1,18 (м, 6Н). МС (электрораспыление): 313,2 [М+Na]+.
Шаг 3:
Раствор (8Z, 11Z, 14Z) -метилоктадека-8,11,14,17-тетраеноата (830 мг, 2,86 ммоль) в THF (2 мл) по каплям добавляли к охлажденной (0°С) суспензии LAH (114 мг, 3,00 ммоль) в THF (10 мл) в атмосфере азота. Смесь перемешивали в течение 40 минут при 0°C и осторожно вливали в холодный насыщенный NH4Cl (20 мл). Водный слой подкисляли HCl (2М) до рН ~ 2. Фазы разделяли и водную фазу экстрагировали гептаном (3 × 20 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме, получая (8Z, 11Z, 14Z) -октадека-8,11,14,17-тетраен-1-ол (730 мг, 2,78 ммоль, выход 97%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,84-5,66 (м, 1H), 5,43-5,20 (м, 6H), 5,03-4,82 (м, 2H), 3,57 (т, 2H), 2,84-2,62 (м, 6H).), 2,06-1,90 (м, 4H), 1,58-1,42 (м, 3H), 1,33-1,20 (м, 6H). МС (электрораспыление): 285,1 [М+Na]+.
Шаг 4:
Раствор NaOH (2 мл, 37,9 ммоль) медленно добавляли к энергично перемешиваемому раствору (8Z, 11Z, 14Z) -октадека-8,11,14,17-тетраен-1-ола (720 мг, 2,74 ммоль) трет-бутил-2-бромбутаноата (918 мг, 4,12 ммоль) и гидросульфата тетрабутиламмония (279 мг, 0,823 ммоль) в толуоле (8 мл) в атмосфере азота. Смесь нагревали при 40°С в течение ночи. Дополнительный трет-бутил-2-бромбутаноат (306 мг, 1,372 ммоль) добавляли дважды через 5 часов и 24 часа. Через 48 часов смесь охлаждали до 5-10°C и добавляли насыщенный NH4Cl (20 мл). Фазы разделяли и водную фазу экстрагировали EtOAc (50 мл). Объединенную органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Флэш-хроматография (гептан/EtOAc 99/1) давала трет-бутил 2 - (((8Z, 11Z, 14Z) октадека-8,11,14,17-тетраен-1-ил) окси) бутаноат (675 мг. 1,666 ммоль, выход 60,8%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,86-5,65 (м, 1H), 5,41-5,21 (м, 6H), 5,04-4,83 (м, 2H), 3,57-3,46 (м, 2H), 3,30-3,19 (м, 1Н), 2,81-2,68 (м, 6Н), 2,02-1,89 (м, 4Н), 1,71-1,60 (м, 2Н), 1,54-1,50 (м, 2Н), 1,41 (с, 12Н), 1,32-1,20. (м, 8Н), 0,89 (т, 3Н).
Шаг 5:
Раствор трет-бутил 2 - (((8Z, 11Z, 14Z) -октадека-8,11,14,17-тетраен-1-ил) окси) бутаноата (675 мг, 1,666 ммоль) в HCOOH (6,75 мл, 179 ммоль) перемешивали в течение 21 часа при 40°C в атмосфере азота. Реакционную смесь упаривали при пониженном давлении и очищали препаративной ВЭЖХ, элюируя смесью CH3CN: H2O (90: 10) с 0,02% HCOOH. Фракции, содержащие чистый продукт, выпаривали при пониженном давлении для удаления CH3CN. К остатку добавляли трет-бутилметиловый эфир и насыщенный солевой раствор и фазы разделяли. Органическую фазу сушили (Na2SO4), фильтровали и упаривали при пониженном давлении, получая 2 - ((8Z, 11Z, 14Z) -октадека-8,11,14,17-тетраен-1-илокси) бутановую кислоту (163 мг, 0,464 ммоль, выход 27,8%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,89-5,70 (м, 1H), 5,51-5,22 (м, 6H), 5,00 (дд, 2H), 3,84 (т, 1H), 3,62-3,38 (м, 2H), 2,79 (м, 6H), 2,12-1,95 (м, 2H), 1,88-1,75 (м, 2H), 1,64-1,58 (м, 2H), 1,31 (с, 8H), 0,96 (т, 3H). HRMS (электрораспыление) [M-H] -: Рассчитано: 347,2586, обнаружено: 347,2589.
Пример 66: Получение 2 - (((5Z, 8Z, 11Z, 14Z) -нонадека-5,8,11,14-тетраен-1-ил) окси) бутановой кислоты:
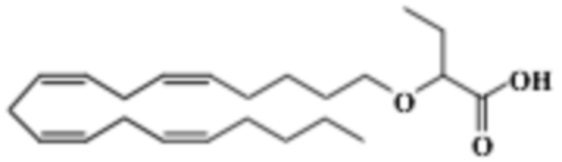 .
.
Шаг 1:
Раствор (5Z, 8Z, 11Z, 14Z) -этилнонадека-5,8,11,14-тетраеноата (21 г, 65,9 ммоль) в THF (50 мл) добавляли по каплям в течение 15 минут к охлажденной (0°С) суспензия LAH (2,503 г, 65,9 ммоль) в сухом THF (300 мл) в атмосфере азота. Смесь перемешивали в течение 1 часа при 0°C и осторожно вливали в холодный насыщенный NH4Cl (300 мл). Водный слой подкисляли HCl (2 М) до pH ~ 2. Фазы разделяли и водную фазу экстрагировали гептаном (2 × 250 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме, получая (5Z, 8Z, 11Z, 14Z) -нонадека-5,8,11,14-тетраен-1-ол. (17,9 г, 64,7 ммоль, выход 98%) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 5,46-5,11 (м, 8H), 3,68-3,57 (м, 2H), 2,89-2,69 (м, 6H), 2,12-1,98 (м, 4H), 1,62-1,51 (м., 2H), 1,47-1,23 (м, 6H), 0,91-0,85 (м, 3H).
Шаг 2:
К раствору (5Z, 8Z, 11Z, 14Z) -нонадека-5,8,11,14-тетраен-1-ола (17,9 г, 64,7 ммоль) в THF (350 мл) добавляли 2-бромбутановую кислоту (10,35) мл, 97 ммоль). Реакционную смесь охлаждали до 8-10°С. Раствор трет-бутоксида натрия (17,42 г, 181 ммоль) (17% в МТВЕ (120 мл)) добавляли в течение 25 минут, поддерживая температуру в пределах 8-10°С. Реакционную смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (2,489 г, 25,9 ммоль) (17% в МТВЕ (25 мл)). Смесь перемешивали в течение 30 минут при 8-10°C с последующим добавлением 2-бромомасляной кислоты (6,90 мл, 64,7 ммоль) в течение 5 минут. Затем смесь перемешивали при 8-10°С в течение 30 минут. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (4,98 г, 51,8 ммоль) (17% в МТВЕ (40 мл)). Смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (2,489 г, 25,9 ммоль) (17% в МТВЕ (25 мл)). Смесь перемешивали в течение 30 минут при 8-10°С. HCOOH (3 мл), а затем 2 М HCl (водн.) до получения рН ~ 2. Фазы разделяли и водную фазу экстрагировали МТВЕ (2 × 300 мл). Объединенную органическую фазу промывали водой (300 мл), насыщенным NaHCO3 (4х200 мл) и насыщенным солевым раствором (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (гептан/EtOAc/HCOOH 90/10/0,5), соответствующие фракции объединяли и концентрировали в вакууме. Затем остаток очищали дополнительно препаративной ВЭЖХ, используя 88% MeCN (содержащий 0,02% HCOOH) в воде (содержащей 10% MeCN и 0,02% HCOOH) в качестве элюента. После удаления растворителей было получено 10,16 г 2 - (((5Z, 8Z, 11Z, 14Z) -нонадека-5,8,11,14-тетраен-1-ил) окси) бутановой кислоты (26,8 ммоль, выход 41,5%) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 9,93 (с, 1Н), 5,51-5,21 (м, 8Н), 3,84-3,80 (м, 1Н), 3,62-3,54 (м, 1Н), 3,50-3,39 (м, 1Н), 2,96-2,61 (м, 6H), 2,12-2,01 (м, 4H), 1,90-1,71 (м, 2H), 1,69-1,57 (м, 2H), 1,49-1,37 (м, 2H), 1,36-1,23. (м, 4Н), 0,97 (т, 3Н), 0,91-0,84 (м, 3Н). МС (электрораспыление): 361,2 [М-Н] -.
Пример 67: Получение 2 - (((8Z, 11Z, 14Z) октадека-8,11,14-триен-1-ил) окси) бутановой кислоты
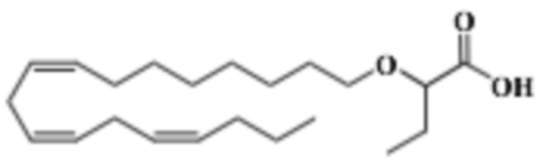 .
.
Шаг 1
К перемешиваемому раствору Cs2CO3 (106 г, 326 ммоль), йодида натрия (44,8 г, 299 ммоль) и йодида меди (I) (56,9 г, 299 ммоль) в DMF (750 мл) в атмосфере азота при температуре окружающей среды добавляли этил-нон-8-иноат (49,5 г, 272 ммоль), а затем 1-бромонона-2,5-диин (54,1 г, 272 ммоль). Смесь перемешивали в течение ночи при температуре окружающей среды и вливали в насыщенный NH4Cl (600 мл). Смесь экстрагировали МТВЕ (3х300 мл) и объединенную органическую фазу промывали насыщенным солевым раствором (400 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Флэш-хроматография (гептан/EtOAc 95/5-92/8-90/10) позволила получить этилоктадека-8,11,14-трийноат (52,8 г, 176 ммоль, выход 64,7%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 4,09 (кв, 2Н), 3,15-3,06 (м, 4Н), 2,26 (т, 2Н), 2,15-2,06 (м, 4Н), 1,63-1,56 (м, 2Н), 1,53-1,42 (м, 4H), 1,40-1,26 (м, 4H), 1,22 (т, 3H), 0,93 (т, 3H). МС (электрораспыление): 323,2 [М+Na]+.
Шаг 2:
К перемешиваемому раствору никеля ацетат тетрагидрата (7,67 мл, 55,4 ммоль) в абсолютном EtOH (150 мл) при комнатной температуре добавляли твердый NaBH4 (2,097 г, 55,4 ммоль) в атмосфере H2 (1 атм). Полученную суспензию перемешивали в течение 30 минут перед добавлением этилендиамина (12,38 мл, 185 ммоль). После завершения добавления суспензию перемешивали в течение еще 15 минут, после чего добавляли этилоктадека-8,11,14-трииноат (11,1 г, 36,9 ммоль) в абсолютном EtOH (50 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере H2 (1 атм) в течение 3 ночей. Диэтиловый эфир (500 мл) добавляли для разбавления реакционной смеси, и полученную смесь затем пропускали через короткую колонку с силикагелем. Фильтрат концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (гептан/EtOAc 98/2), получая этил (8Z, 11Z, 14Z) -октадека-8,11,14-триеноат (10 г, 32,6 ммоль, выход 88%) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 5,46-2,23 (м, 6H), 4,10 (кв, 2H), 2,87-2,67 (м, 3H), 2,26 (т, 2H), 2,07-1,97 (м, 4H), 1,66-1,55 (м, 2H), 1,41-1,18 (м, 11H), 0,91-0,84 (м, 3H).
Шаг 3:
Раствор этил (8Z, 11Z, 14Z) -октадека-8,11,14-триеноата (24,6 г, 80 ммоль) в THF (50 мл) добавляли по каплям в течение 15 минут к охлажденной (0°C) суспензии LAH (3,05 г, 80 ммоль) в сухом THF (300 мл) в атмосфере азота. Смесь перемешивали в течение 1 часа при 0°С. Смесь осторожно вливали в холодный насыщенный NH4Cl (300 мл). Водный слой подкисляли HCl (2 М) до pH ~ 2. Фазы разделяли и водную фазу экстрагировали гептаном (2 × 250 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме, получая (8Z, 11Z, 14Z) -октадека-8,11,14-триен-1-ол (20,7 г, 78 ммоль, выход 98%) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 5,49-2,23 (м, 6H), 3,61 (т, 2H), 2,86-2,67 (м, 4H), 2,11-1,92 (м, 4H), 1,59-1,49 (м, 2H).), 1,43-1,21 (м, 10H), 0,94-0,83 (м, 3H).
Шаг 4:
К раствору (8Z, 11Z, 14Z) -октадека-8,11,14-триен-1-ола (20,7 г, 78 ммоль) в THF (350 мл) добавляли 2-бромбутановую кислоту (12,51 мл, 117 ммоль). Реакционную смесь охлаждали до 8-10°С. Раствор трет-бутоксида натрия (21,06 г, 219 ммоль) (17% в МТВЕ (130 мл)) добавляли в течение 15 минут, поддерживая температуру в пределах 8-10°С. Реакционную смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (3,01 г, 31,3 ммоль) (17% в МТВЕ (25 мл)). Смесь перемешивали в течение 30 минут при 8-10°C с последующим добавлением 2-броммасляной кислоты (8,34 мл, 78 ммоль) в течение 5 минут. Затем смесь перемешивали при 8-10°С в течение 30 минут. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (6,02 г, 62,6 ммоль) (17% в МТВЕ (40 мл)). Смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (3,01 г, 31,3 ммоль) (17% в МТВЕ (25 мл)). Смесь перемешивали в течение 30 минут при 8-10°С.
Добавляли НСООН (3 мл), а затем 2 М HCl (водный раствор) до рН ~ 2. Фазы разделяли и водную фазу экстрагировали МТВЕ (2 × 300 мл). Объединенную органическую фазу промывали водой (300 мл), насыщенным NaHCO3 (4 × 200 мл) и насыщенным солевым раствором (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (гептан/EtOAc/HCOOH в соотношении 90/10/0,5) и соответствующие фракции объединяли и концентрировали в вакууме. Затем остаток очищали дополнительно препаративной ВЭЖХ, используя 88% MeCN (содержащий 0,02% HCOOH) в воде (содержащей 10% MeCN и 0,02% HCOOH) в качестве элюента. После удаления растворителей было получено 10,55 г 2 - (((8Z, 11Z, 14Z) -октадека-8,11,14-триен-1-ил) окси) бутановой кислоты (28,8 ммоль, выход 36,8%) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 9,70 (с, 1Н), 5,47-5,25 (м, 6Н), 3,85-3,81 (м, 1Н), 3,60-3,52 (м, 1Н), 3,49-3,42 (м, 1Н)), 2,87-2,68 (м, 4H), 2,10-1,94 (м, 4H), 1,89-1,69 (м, 2H), 1,66-1,54 (м, 2H), 1,43-1,24 (м, 10H), 0,96 (т, 3Н), 0,89 (т, 3Н). МС (электрораспыление): 349,2 [М-Н] -.
Пример 68. Получение 2 - (((9Z, 12Z, 15Z) -октадека-9,12,15-триен-1-ил) окси) бутановой кислоты:
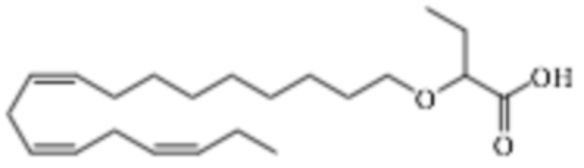 .
.
К раствору (9Z, 12Z, 15Z) -октадека-9,12,15-триен-1-ола (29,6 г, 112 ммоль) в THF (400 мл) добавляли 2-бромбутановую кислоту (17,89 мл, 168 ммоль). Реакционную смесь охлаждали до 8-10°С. Раствор трет-бутоксида натрия (30,1 г, 313 ммоль) (17% в МТВЕ (180 мл)) добавляли в течение 5 минут, поддерживая температуру в пределах 8-10°С. Реакционную смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (4,30 г, 44,8 ммоль) (17% в МТВЕ (30 мл)). Смесь перемешивали в течение 30 минут при 8-10°С с последующим добавлением 2-броммасляной кислоты (11,93 мл, 112 ммоль) в течение 5 минут. Затем смесь перемешивали при 8-10°С в течение 30 минут. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (8,61 г, 90 ммоль) (17% в МТВЕ (55 мл)). Смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (4,30 г, 44,8 ммоль) (17% в МТВЕ (30 мл)). Смесь перемешивали в течение 30 минут при 8-10°С.
Добавляли НСООН (3 мл), а затем 2 М HCl (водный раствор) до рН ~ 2. Фазы разделяли и водную фазу экстрагировали МТВЕ (2 × 300 мл). Объединенную органическую фазу промывали водой (300 мл), насыщенным NaHCO3 (4 × 200 мл) и насыщенным солевым раствором (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (гептан/EtOAc/HCOOH в соотношении 90/10/0,5) на силикагеле. Соответствующие фракции объединяли и концентрировали в вакууме с получением 2 - (((9Z, 12Z, 15Z) -октадека-9,12,15-триен-1-ил) окси) бутановой кислоты (28 г, 78 ммоль, 69,9% выход) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 10,07 (с, 1Н), 5,48-5,16 (м, 6Н), 3,84-3,80 (м, 1Н), 3,60-3,53 (м, 1Н), 3,48-3,40 (м, 1Н)), 2,87-2,70 (м, 4H), 2,12-1,97 (м, 4H), 1,91-1,70 (м, 2H), 1,66-1,53 (м, 2H), 1,38-1,27 (м, 10H), 0,99 0,93. (м, 6Н). МС (электрораспыление): 373,3 [М+Na]+.
Пример 69: Получение 2 - (((6Z, 9Z, 12Z) октадека-6,9,12-триен-1-ил) окси) бутановой кислоты:
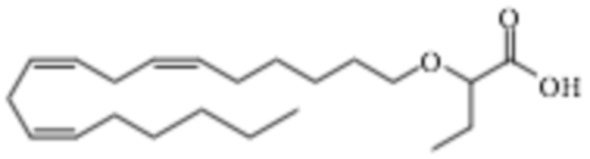 .
.
К раствору (6Z, 9Z, 12Z) -октадека-6,9,12-триен-1-ола (29,6 г, 112 ммоль) в THF (400 мл) добавляли 2-бромбутановую кислоту (17,89 мл, 168 ммоль). Реакционную смесь охлаждали до 8-10°С. Раствор трет-бутоксида натрия (30,1 г, 313 ммоль) (17% в МТВЕ (180 мл)) добавляли в течение 25 минут, поддерживая температуру в пределах 8-10°С. Реакционную смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (4,30 г, 44,8 ммоль) (17% в МТВЕ (30 мл)). Смесь перемешивали в течение 30 минут при 8-10°С с последующим добавлением 2-броммасляной кислоты (11,93 мл, 112 ммоль) в течение 5 минут. Затем смесь перемешивали при 8-10°С в течение 30 минут. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (8,61 г, 90 ммоль) (17% в МТВЕ (55 мл)). Смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (4,30 г, 44,8 ммоль) (17% в МТВЕ (30 мл)). Смесь перемешивали в течение 30 минут при 8-10°С.
Добавляли НСООН (3 мл), а затем 2 М HCl (водный раствор) до рН ~ 2. Фазы разделяли и водную фазу экстрагировали МТВЕ (2 × 300 мл). Объединенную органическую фазу промывали водой (300 мл), насыщенным NaHCO3 (4х200 мл) и насыщенным солевым раствором (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (гептан/EtOAc/HCOOH 90/10/0,5) на силикагеле. Соответствующие фракции объединяли и концентрировали в вакууме с получением 2 - ((6Z, 9Z, 12Z) -октадека-6,9,12-триен-1-илокси) бутановой кислоты (30,2 г, 83 ммоль, 74, 1% выход) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 9,56 (с, 1H), 5,46-5,22 (м, 6H), 3,84-3,80 (м, 1H), 3,61-3,54 (м, 1H), 3,47-3,40 (м, 1H).), 2,86-2,71 (м, 4H), 2,10-1,99 (м, 4H), 1,91-1,70 (м, 2H), 1,65-1,56 (м, 2H), 1,45-1,18 (м, 10H), 0,97 (т, 3Н), 0,87 (т, 3Н). МС (электрораспыление): 373,2 [М+Na]+.
Пример 70. Получение 2 - (((6Z, 9Z, 12Z, 15Z, 18Z) -хенэйкоза- 6,9,12,15,18-пентаен-1-ил) окси) бутановой кислоты:
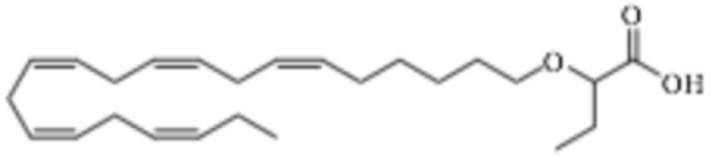 .
.
К раствору (6Z, 9Z, 12Z, 15Z, 18Z) -хенэйкоза-6,9,12,15,18-пентаен-1-ола (23 г, 76 ммоль) в THF (350 мл) добавляли 2- бромбутановую кислоту (12,15 мл, 114 ммоль). Реакционную смесь охлаждали до 8-10°С. Раствор трет-бутоксида натрия (20,46 г, 213 ммоль) (17% в МТВЕ (140 мл)) добавляли в течение 25 минут, поддерживая температуру в пределах 8-10°С. Реакционную смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (2,92 г, 30,4 ммоль) (17% в МТВЕ (25 мл)). Смесь перемешивали в течение 30 минут при 8-10°C с последующим добавлением 2-бромомасляной кислоты (8,10 мл, 76 ммоль) в течение 5 минут. Затем смесь перемешивали при 8-10°С в течение 30 минут. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (5,85 г, 60,8 ммоль) (17% в МТВЕ (45 мл)). Смесь перемешивали в течение 30 минут при 8-10°С. В течение 5 минут добавляли дополнительное количество трет-бутоксида натрия (2,92 г, 30,4 ммоль) (17% в МТВЕ (25 мл)). Смесь перемешивали в течение 30 минут при 8-10°С. Добавляли НСООН (3 мл), а затем 2 М HCl (водный) до рН ~ 2. Фазы разделяли и водную фазу экстрагировали МТВЕ (2 × 300 мл). Объединенную органическую фазу промывали водой (300 мл), насыщенным NaHCO3 (4х200 мл) и насыщенным солевым раствором (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (гептан/EtOAc/HCOOH 90/10/0,5) на силикагеле. Соответствующие фракции объединяли и концентрировали в вакууме с получением 2 - (((6Z, 9Z, 12Z, 15Z, 18Z) -хенэйкоза-6,9,12,15,18-пентаен-1-ил) окси) бутановой кислоты (18,9 г, 47,3 ммоль, 62,2% выход) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 9,85 (с, 1Н), 5,48-5,21 (м, 10Н), 3,84-3,81 (м, 1Н), 3,61-3,53 (м, 1Н), 3,48-3,41 (м, 1Н), 2,93-2,71 (м, 8H), 2,18-1,96 (м, 4H), 1,91-1,70 (м, 2H), 1,66-1,57 (м, 2H), 1,45-1,30 (м, 4H), 0,99 0,93. (м, 6Н). МС (электрораспыление): 411,4 [М+Na]+.
Пример 71: Получение 2 - (((4Z, 7Z, 10Z, 13Z, 16Z, 19Z) -докоса-4,7,10,13,16,19-гексаен-1-ил) окси) бутановой кислоты:
 .
.
К раствору (4Z, 7Z, 10Z, 13Z, 16Z, 19Z) -докоса-4,7,10,13,16,19-гексаен-1-ола (30 г, 95 ммоль) в толуоле (11 мл) и THF (400 мл) добавляли 2-бромасляную кислоту (17,28 мл, 162 ммоль) с последующим добавлением раствора натрий-трет-бутоксида (25,7 г, 267 ммоль) в THF (160 мл) в течение 35 минут, поддерживая температуру в пределах 8-10°C. Реакционную смесь перемешивали в течение 40 минут при 8-10°С. В течение 10 минут добавляли натрий-трет-бутоксид (4,17 г, 43,4 ммоль) в THF (60 мл). Полученную смесь перемешивали при 8-10°С в течение 40 минут, после чего добавляли еще 2-бромомасляную кислоту (10,17 мл, 95 ммоль) одной порцией. Смесь перемешивали при 8-10°С в течение 1 часа, после чего в течение 10 минут добавляли еще трет-бутоксид натрия (7,33 г, 76 ммоль) в THF (100 мл), поддерживая температуру ниже 12°С. Реакционную смесь перемешивали при 10°С в течение 60 минут. Натрий-трет-бутоксид (4,17 г, 43,4 ммоль) добавляли одной порцией и затем перемешивали в течение дополнительного 1 часа. Добавляли 2-бромбутириновую кислоту (8,5 мл, 50 ммоль) и трет-бутоксид натрия (4,8 г, 50 ммоль) и смесь перемешивали в течение 1 часа при 8-10°С. Добавляли 2-бромбутириновую кислоту (8,5 мл, 50 ммоль) и трет-бутоксид натрия (4,8 г, 50 ммоль) и смесь перемешивали в течение 1 часа при 8-10°С. Добавляли THF (200 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли 2-бромбутириновую кислоту (17 мл, 100 ммоль) и трет-бутоксид натрия (9,6 г, 100 ммоль) и реакционную смесь перемешивали в течение 1 часа при 8-10°С. Затем к реакционной смеси добавляли (1 мл) муравьиную кислоту и полученную смесь перемешивали при 8-10°С в течение 10 минут, затем добавляли 6 М HCl, чтобы получить рН ~ 2. Добавляли воду (100 мл), затем разделяли водную и органическая фазы. Водную фазу дважды экстрагировали диэтиловым эфиром (2х500 мл). Объединенную органическую фазу промывали водой (3х1000 мл), насыщенным NaHCO3 (3х500 мл) и насыщенным солевым раствором 1х100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (EtOAc/гептан/5% HCOOH) (90/10) на силикагеле с получением 2 - (((4Z, 7Z, 10Z, 13Z, 16Z, 19Z) -докоса-4,7, 10,13,16,19-гексаен-1-ил) окси) бутановой кислоты (10,3 г, 25,07 ммоль, 26,3% выход) в виде масла. 1H ЯМР (300 МГц, CDCl3) δ 5,49-5,20 (м, 12H), 3,90-3,75 (м, 1H), 3,65-3,52 (м, 1H), 3,51-3,35 (м, 1H), 2,91-2,69 (м 10Н), 2,21-1,98 (м, 4Н), 1,91-1,59 (м, 4Н), 1,05-0,88 (м, 6Н). МС (электрораспыление): 423,3 [М+Na]+.
Пример 72: Получение 2 - (((8Z, 11Z, 14Z, 17Z) -эйкоза-8,11,14,17-тетраен-1-ил) окси) бутановой кислоты:
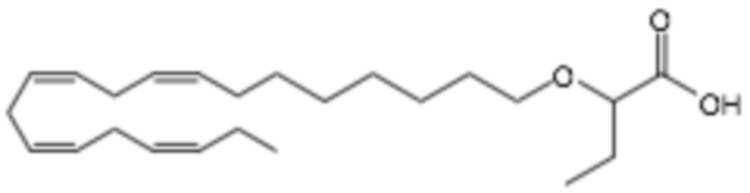 .
.
Шаг 1:
К нагретой суспензии магния (0,217 г, 8,93 ммоль) в THF (3 мл) добавляли 1,2-дибромэтан (2 капли) с последующим добавлением по каплям раствора 2 - ((5-бромпентил) окси) тетрагидро-2Н-пирана (2,07 г, 8,24 ммоль) в THF (15 мл) в атмосфере азота. Смесь кипятили с обратным холодильником в течение 1 часа и охлаждали до температуры окружающей среды. Добавляли раствор (3Z, 6Z, 9Z, 12Z) -пентадека-3,6,9,12-тетраенала (1,5 г, 6,87 ммоль) в THF (10 мл). Затем смесь перемешивали в течение 1 часа при температуре окружающей среды. Добавляли 1 М HCl и смесь экстрагировали EtOAc (2 × 50 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Флэш-хроматография на силикагеле (гептан/EtOAc 80/20-75/25) давала (8Z, 11Z, 14Z, 17Z) -1 - ((тетрагидро-2H-пиран-2-ил) окси) эйкоза-8, 11,14,17-тетраен-6-ол (0,74 г, 1,895 ммоль, выход 27,6%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,59-5,49 (м, 1H), 5,49-5,25 (м, 7H), 4,56-4,53 (м, 1H), 3,88-3,82 (м, 1H), 3,77-3,67 (м, 1Н), 3,65-3,59 (м, 1Н), 3,52-3,43 (м, 1Н), 3,40-3,34 (м, 1Н), 2,85-2,77 (м, 6Н), 2,23 (т, 2Н), 2,13-1,97. (м, 2Н), 1,84-1,77 (м, 1Н), 1,73-1,67 (м, 1Н), 1,63-1,34 (м, 12Н), 0,96 (т, 3Н). МС (электрораспыление): 413,3 [М+Na]+.
Шаг 2:
К раствору (8Z, 11Z, 14Z, 17Z) -1 - ((тетрагидро-2H-пиран-2-ил) окси) эйкоза-8,11,14,17-тетраен-6-ола (0,74 г, 1,895 ммоль) в THF (20 мл) добавляли TEA (0,528 мл, 3,79 ммоль), а затем метансульфонилхлорид (0,192 мл, 2,446 ммоль). Смесь перемешивали в атмосфере азота в течение 3 часов при температуре окружающей среды. Добавляли лимонную кислоту (5%, водн., 50 мл) и смесь экстрагировали EtOAc (2×50 мл). Объединенную органическую фазу промывали насыщенным Na-HCO3 (50 мл) и насыщенным солевым раствором (50 мл), суштили (Na2SO4), фильтровали и концентрировали в вакууме, получая 0,82 г промежуточного мезилата. Промежуточный мезилат растворяли в Et2O (10 мл) и добавляли по каплям к перемешиваемой суспензии LAH (0,288 г, 7,58 ммоль) в Et2O (25 мл). Смесь перемешивали в течение 1 часа при температуре окружающей среды. Осторожно добавляли 1 М HCl (50 мл). Смесь экстрагировали МТВЕ (2 × 50 мл). Объединенную органическую фазу промывали насыщенным NaHCO3 (50 мл) и насыщенным солевым раствором (50 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Флэш-хроматография на силикагеле (гептан/EtOAc 97/3) давала 2 - (((8Z, 11Z, 14Z, 17Z) -эйкоза-8,11,14,17-тетраен-1-ил) окси) тетрагидро-2H- пиран (0,46 г, 1,228 ммоль, выход 64,8%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,46-5,20 (м, 8H), 4,57-4,54 (м, 1H), 3,88-3,82 (м, 1H), 3,74-3,68 (м, 1H), 3,51-3,44 (м, 1Н), 3,39-3,33 (м, 1Н), 2,84-2,77 (м, 6Н), 2,14-1,96 (м, 4Н), 1,85-1,77 (м, 1Н), 1,73-1,65 (м, 1Н), 1,62. 1,46 (м, 6H), 1,37-1,24 (м, 8H), 0,96 (т, 3H). МС (электрораспыление): 397,3 [М+Na]+.
Шаг 3:
К раствору 2 - (((8Z, 11Z, 14Z, 17Z) -эйкоза-8,11,14,17-тетраен-1-ил) окси) тетрагидро-2Н-пирана (0,46 г, 1,228 ммоль) в EtOH (5 мл) добавляли пиридиний-п-толуолсуфонат (PPTS, 0,309 г, 1,228 ммоль) и смесь нагревали при 50°C в течение 2 часов. Смесь охлаждали до температуры окружающей среды. Добавляли насыщенный NaHCO3 (50 мл) и смесь экстрагировали EtOAc (2 × 50 мл). Объединенную органическую фазу промывают насыщенным солевым раствором (50 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Флэш-хроматография на силикагеле (гептан/EtOAc 80/20) давала (8Z, 11Z, 14Z, 17Z) -эйкоза-8,11,14,17-тетраен-1-ол (0,26 г, 0,895 ммоль, выход 72,9%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,41-5,17 (м, 8H), 3,57 (т, 2H), 2,84-2,68 (м, 6H), 2,06-1,94 (м, 4H), 1,54-1,46 (м, 2H), 1,34-1,21 (м, 8H), 1,20-1,13 (м, 1H), 0,91 (т, 3H).
Шаг 4:
К раствору (8Z, 11Z, 14Z, 17Z) -эйкоза-8,11,14,17-тетраен-1-ола (93 мг, 0,32 ммоль) в толуоле (3 мл) добавляли трет-бутил 2-бромбутаноат (143 мг, 0,64 ммоль), гидроксид тетрабутиламмония (TBAOH) (5 мг, 0,02 ммоль) и NaOH (50 мас.%, 1 мл), и смесь нагревали до 45°C. 71,4 мг (0,32 ммоль) трет-бутил-2-бромбутаноата добавляли к реакционной смеси после 1,5 часов перемешивания, 71,4 мг (0,32 ммоль) трет-бутил-2-бромбутаноата добавляли к реакционной смеси через 3 часа и 143 мг (0,64 ммоль) трет-бутил-2-бромбутаноата добавляли к реакционной смеси через 8 часов при 45°C. Через 24 часа смесь охлаждали до комнатной температуры и добавляли воду (40 мл). Полученную смесь дважды экстрагировали Et2O (25+40 мл) и объединенную органическую фазу промывали водой (4×25 мл) и насыщенным солевым раствором (25 мл), сушили (MgSO4), фильтровали и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (гептан-гептат: ацетон-5: 1). Соответствующие фракции объединяли и концентрировали, получая 15 мг трет-бутил 2 - (((8Z, 11Z, 14Z, 17Z) -эйкоза-8,11,14,17-тетраен-1-ил) окси) бутаноата. Это промежуточное соединение растворяли в муравьиной кислоте (5 мл) и полученную смесь перемешивали при комнатной температуре в течение 1 часа и 45 минут. Растворитель удаляли в вакууме и к остатку добавляли EtOAc (30 мл). Раствор промывали водой (4х25 мл) и насыщенным солевым раствором (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали препаративной ВЭЖХ, используя 85% MeCN в воде в качестве элюента. Соответствующие фракции объединяли и концентрировали, получая 15 мг (0,04 ммоль, 12% выход) 2 - (((8Z, 11Z, 14Z, 17Z) -эйкоза-8,11,14,17-тетраен-1-ила) окси) бутановой кислоты. 1H ЯМР (400 МГц, CDCl3) δ 1,00 (тд, 6H), 1,49-1,20 (м, 8H), 1,64 (т, 2H), 1,96-1,73 (м, 2H), 2,08 (дт, 4H), 2 0,84 (кв, 6H), 3,55-3,43 (м, 1H), 3,65-3,54 (м, 1H), 3,87 (дд, 1H), 5,49-5,24 (м, 8H). HRMS (электрораспыление), [M-H] -: Рассчитано: 375,2899, обнаружено: 375,2892.
Пример 73. Получение 2 - (((5Z, 8Z, 11Z, 14Z) -эйкоза-5,8,11,14-тетраен-1-ил) окси) бутановой кислоты:
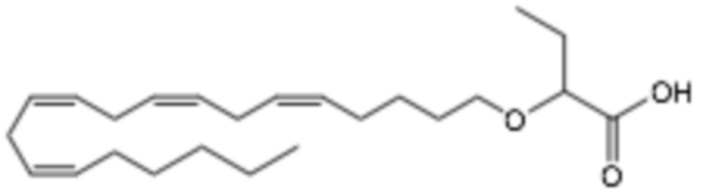 .
.
Шаг 1:
Раствор (5Z, 8Z, 11Z, 14Z) -этэйкоза-5,8,11,14-тетраеноата (501,9 мг, 1,509 ммоль) в сухом THF (1 мл) добавляли по каплям к охлажденной (0°C) суспензии LiAlH4 (64,8 мг, 1,707 ммоль) в сухом THF (3 мл) в атмосфере азота. Смесь перемешивали в течение 55 минут при 0°С. По каплям добавляли воду (5 мл) и затем добавляли 1 М HCl (10 мл). Охлаждающую баню убирали и реакционную смесь перемешивали в течение 5 минут. Реакционную смесь экстрагировали трет-бутилметиловым эфиром (2×50 мл), органическую фазу промывали 1 М HCl (20 мл), сушили (Na2SO4), фильтровали и упаривали при пониженном давлении, получая (5Z, 8Z, 11Z). 14Z) -эйкоза-5,8,11,14-тетраен-1-ол (397,7 мг, 1,369 ммоль, выход 91%) в виде жидкости. 1H ЯМР (400 МГц, CDCl3) δ 5,41-5,29 (м, 8H), 3,63 (т, 2H), 2,83-2,77 (м, 6H), 2,20-1,91 (м, 4H), 1,61-1,54 (м, 2H), 1,46-1,41 (м, 2H), 1,35-1,28 (м, 8H), 0,87 (т, 3H).
Шаг 2:
К раствору (5Z, 8Z, 11Z, 14Z) -эйкоза-5,8,11,14-тетраен-1-ола (349 мг, 1,20 ммоль) в толуоле (5 мл) добавляли трет-бутил 2-бромбутаноат (535 мг, 2,40 ммоль), гидроксид тетрабутиламмония (TBAOH) (156 мг, 0,24 ммоль, 40% мас./мас.) и NaOH (50% мас., 2 мл), и смесь нагревали до 45°C. Смесь охлаждали до комнатной температуры через 12 часов и добавляли ледяную воду (25 мл) и Et2O (20 мл). Фазы разделяли и водную фазу экстрагировали Et2O (20 мл). Объединенную органическую фазу промывали водой (25 мл) и насыщенным солевым раствором (25 мл), сушили (MgSO4), фильтровали и упаривали в вакууме. Остаток растворяли в EtOH (20 мл) и упаривали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (гептан: ацетон-1:20, гептан-ацетон-1: 10). Соответствующие фракции объединяли и концентрировали, получая 360 мг (0,83 ммоль, выход 69%) промежуточной трет-бутил 2 - (((5Z, 8Z, 11Z, 14Z) -эйкоза-5,8,11,14 -тетраен-1-ил) окси) бутановой кислоты. 342 мг (0,79 ммоль) этого промежуточного вещества растворяли в муравьиной кислоте (7 мл) и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли в вакууме и остаток добавляли к EtOAc (75 мл). Раствор промывали водой (3×50 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле (ацетон: гептан: HOAc-10: 10: 1, ацетон: гептан: HOAc-30: 70: 1). Соответствующие фракции объединяли и концентрировали, получая при этом 181 мг (0,48 ммоль, выход 58%) 2 - (((5Z, 8Z, 11Z, 14Z) -эйкоза-5,8,11,14-тетраен-1-ила) окси) бутановой кислоты.
Пример 74: Получение 2 - (((7Z, 10Z, 13Z, 16Z, 19Z) -докоса-7,10,13,16,19-пентаен-1-ил) окси) бутановой кислоты:
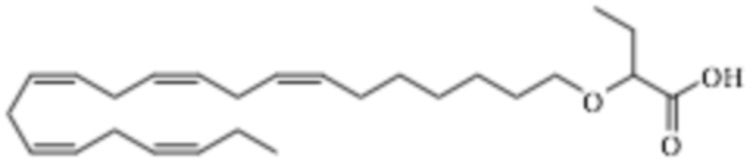 .
.
Шаг 1:
Раствор (7Z, 10Z, 13Z, 16Z, 19Z) -этилдокоса-7,10,13,16,19-пентаеноата (1,5 г, 4,18 ммоль) в THF (4 мл) добавляли по каплям к охлажденной (0°C) суспензии LAH (0,159 г, 4,18 ммоль) в сухом THF (20 мл) в атмосфере азота. Смесь перемешивали в течение 1 часа при 0°С. Смесь осторожно вливали в 1 М HCl (100 мл). Фазы разделяли и водную фазу экстрагировали гептаном (100 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (30 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме, получая (7Z, 10Z, 13Z, 16Z, 19Z) -докоса-7,10,13,16,19-пентаен -1-ол (1,16 г, 3,66 ммоль, выход 88%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,44-2,23 (м, 10H), 3,62 (т, 2H), 2,87-2,73 (м, 8H), 2,10-1,99 (м, 4H), 1,60-1,51 (м, 2H)), 1,40-1,29 (м, 6H), 0,95 (т, 3H).
Шаг 2:
К раствору (7Z, 10Z, 13Z, 16Z, 19Z) -докоса-7,10,13,16,19-пентаен-1-ола (1,16 г, 3,66 ммоль) в THF (50 мл) добавляли 2- бромбутановую кислоту (0,918 г, 5,50 ммоль). Реакционную смесь охлаждали до 8-10°С. Раствор трет-бутоксида натрия (0,986 г, 10,26 ммоль) в МТВЕ (15 мл) добавляли в течение 5 минут. Реакционную смесь перемешивали в течение 30 минут при 8-10°С. Добавляли дополнительное количество трет-бутоксида натрия (0,141 г, 1,466 ммоль) в МТВЕ (4 мл). Смесь перемешивали в течение 30 минут при 8-10°C с последующим добавлением 2-бромбутановой кислоты (0,612 г, 3,66 ммоль) в течение 5 минут. Затем смесь перемешивали при 8-10°С в течение 30 минут. Добавляли дополнительное количество трет-бутоксида натрия (0,282 г, 2,93 ммоль) в МТВЕ (8 мл). Смесь перемешивали в течение 30 минут при 8-10°С. Добавляли дополнительное количество трет-бутоксида натрия (0,141 г, 1,466 ммоль) в МТВЕ (4 мл). Смесь перемешивали в течение 30 минут при 8-10°С. Добавляли HCOOH (0,5 мл), а затем 1 М HCl (водный раствор), чтобы получить pH ~ 2. Фазы разделяли и водную фазу экстрагировали МТВЕ (2 × 50 мл). Объединенную органическую фазу промывали водой (50 мл), насыщенным NaHCO3 (4 × 30 мл) и насыщенным солевым раствором (30 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. При препаративной ВЭЖХ с использованием 90% MeCN (содержащего 0,02% HCOOH) в воде (содержащей 10% MeCN и 0,02% HCOOH) в качестве элюента получали 2 - (((7Z, 10Z, 13Z, 16Z, 19Z) -докоса-7,10 13,16,19-пентаен-1-ил) окси) бутановую кислоту (1 г, 2,439 ммоль, выход 66,6%) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,44-5,24 (м, 10H), 3,84-3,81 (м, 1H), 3,59-3,64 (м, 1H), 3,47-3,41 (м, 1H), 2,90-2,73 (м, 8H), 2,11-1,98 (м, 4H), 1,90-1,71 (м, 2H), 1,64-1,57 (м, 2H), 1,41-1,26 (м, 6H), 0,98-0,93 (м, 6H). HRMS (электрораспыление), [M]+: Рассчитано: 402,3134, обнаружено: 402,3141.
Пример 75. Получение (5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) - Эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноата:
 .
.
2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутановую кислоту (14,28 г, 38,1 ммоль) и (5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ол (10,00 г, 34,7 ммоль) добавляли к смеси D (+) - 10-камфорсульфоновой кислоты (0,530 г, 2,282 ммоль) в циклогексане (50 мл) и нагревали до температуры кипения с обратным холодильником, удаляя воду с помощью аппарата Дина-Старка. Реакционную смесь охлаждали до комнатной температуры и очищали сухой флэш-хроматографией, используя силикагель (100 г) и элюируя циклогексаном (50 мл), затем гептаном (50 мл), 2% EtOAc в гептане (2 × 200 мл) и 4%. EtOAc в гептане (2х200 мл). Соответствующие фракции объединяли и концентрировали в вакууме с получением (5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутаноата (20,3 г, 30,5 ммоль, выход 88%) в виде масла. 1H ЯМР (300 МГц, хлороформ-d) δ 0,99 (т, 9Н), 1,45 (ддд, 4Н), 1,56-1,93 (м, 6Н), 2,09 (р, 8Н), 2,85 (дт, 16Н), 3,34 (дт, 1H), 3,60 (дт, 1H), 3,76 (дд, 1H), 4,16 (тд, 2H), 5,05-5,76 (м, 20H). МС (электро-спрей): 645,5 [М+Н]+.
Пример 76. Получение (2R) -2 - ((2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноила) окси) бутановой кислоты:
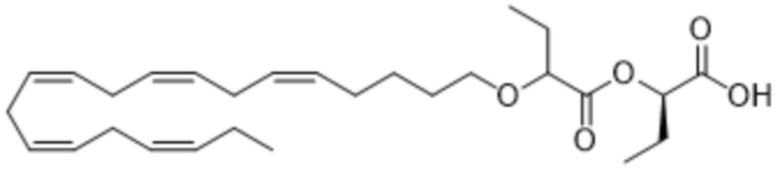 .
.
Шаг 1:
К раствору 2 - ((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-илокси) бутановой кислоты (400 мг, 1,07 ммоль) в CDM (10) мл) добавляли N-метилморфолин (235 мкл, 2,15 ммоль) и TBTU (тетрафторборат 2- (1H-бензотриазол-1-ил) -1,1,3,3-тетраметиламиния) (344 мг, 1,07 ммоль). Реакционную смесь перемешивали в течение 30 минут перед добавлением трет-бутил (R) -2-гидроксибутаноата (250 мг, 2,14 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 22 часов и затем промывали водой (10 мл), 1М HCl (10 мл), насыщенным NaHCO3 (10 мл) и солевым раствором (10 мл). Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Было получено 161 мг (0,312 ммоль, выход 29,2%) промежуточного соединения (R) -1- (трет-бутокси) -1-оксобутан-2-ил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза- 5,8,11,14,17-пентаен-1-ил) окси) бутаноат выделяли в виде масла. МС (электрораспыление): 539,4 [М+Na]+.
Шаг 2:
(R) -1- (трет-бутокси) -1-оксобутан-2-ил 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1 ил) окси) бутаноат (161 мг, 0,312 ммоль) растворяли в муравьиной кислоте (4 мл) и реакционную смесь перемешивали при комнатной температуре в течение 17 часов. К смеси добавляли EtOAc (10 мл) и полученную смесь промывали водой (10 мл) и солевым раствором (10 мл). Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Было получено 60 мг (0,130 ммоль, выход 41,8%) (2R) -2 - ((2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1) ил) окси) бутаноил) окси) бутановой кислоты в виде масла. МС (электрораспыление): 459,3 [М-Н] -.
Пример 77: Получение (2S, 3S, 4S, 5R, 6S) -3,4,5-тригидрокси-6 - ((2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8, 11,14,17-пентаен-1-ил) окси) бутаноил) окси) тетрагидро-2Н-пиран-2-карбоновой кислоты:
 .
.
Шаг 1:
К смеси 4-метоксибензил (2S, 3S, 4S, 5R, 6R) -3,4,5,6-тетрагидрокситетрагидро-2H-пиран-2-карбоксилата (2,47 г, 7,86 ммоль), 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутановой кислоты (2,94 г, 7,85 ммоль) и HATU (1- [бис (диметиламино) метилен ] -1Н-1,2,3-триазоло [4,5-b] пиридиния 3-оксид гексафторфосфат) (3,00 г, 7,90 ммоль) в ацетонитриле (85 мл) добавляли 4-метилморфолин (1,75 мл, 15,92 ммоль). Реакционную смесь перемешивали в течение 23,5 часов в атмосфере азота при комнатной температуре. Amberlyst-15 (4,7 мг-экв/г) (3,35 г) добавляли и смесь перемешивали в течение приблизительно 5 минут. Реакционную смесь фильтровали и концентрировали при пониженном давлении и остаток очищали флэш-хроматографией на силикагеле, элюируя с помощью CH2Cl2- CH2Cl2: EtOH (95: 5). Соответствующие фракции объединяли и концентрировали в вакууме, получая 2,64 г загрязненной смеси. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью CH2Cl2- CH2Cl2: EtOH (95: 5) и затем CH2Cl2- CH2Cl2: EtOH (97: 3). Соответствующие фракции объединяли и концентрировали в вакууме, получая промежуточный (4-метоксициклогексил)метил (2S, 3S, 4S, 5R, 6S) -3,4,5-тригидрокси-6 - ((2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутаноил) окси) тетрагидро-2Н-пиран-2-карбоксилат (1,27 г, 1,78 ммоль, 22,4% выхода) в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 7,26-7,22 (м, 2H), 6,83-6,81 (м, 2H), 5,60-5,57 (м, 1H), 5,40-5,26 (м, 10H), 5,12-5,10 (м., 2Н), 4,60-4,56 (м, 1Н), 4,14-4,13 (м, 1Н), 3,98 (т, 1Н), 3,93-3,67 (м, 3Н), 3,74 (с, 3Н), 3,61-3,56 (м, 3Н), 3,31-3,25 (м, 1Н), 2,82-2,77 (м, 8Н), 2,07-2,02 (м, 4Н), 1,80-1,67 (м, 2Н), 1,57-1,54 (с, 2Н), 1,43-1,36 (м, 2Н), 1,05-0,77 (м, 6Н). МС (электрораспыление): 693,4 [М+Na]+.
Шаг 2:
(4-Метоксициклогексил) метил (2S, 3S, 4S, 5R, 6S) -3,4,5-тригидрокси-6 - ((2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5, 8,11,14,17-пентаен-1-ил) окси) бутаноил) окси) тетрагидро-2Н-пиран-2-карбоксилат (1,20 г, 1,79 ммоль) обрабатывали раствором 10% TFA в DCM (8 мл)) при 0°С в атмосфере азота. После перемешивания в течение 3 ч при температуре окружающей среды растворитель удаляли в вакууме. Флэш-хроматография (DCM/MeOH/HCOOH - 95/5/0,2) дала 500 мг загрязненного продукта. Продукт дополнительно очищали препаративной ВЭЖХ с использованием 85% MeCN (содержащего 0,02% HCOOH) в воде (содержащей 10% MeCN и 0,02% HCOOH) в качестве элюента. Соответствующие фракции объединяли и концентрировали с получением (2S, 3S, 4S, 5R, 6S) -3,4,5-тригидрокси-6 - ((2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза -5,8,11,14,17-пентаен-1-ил) окси) бутаноил) окси) тетрагидро-2Н-пиран-2-карбоновой кислоты (80 мг, 0,139 ммоль, выход 7,8%) в виде твердого вещества. 1H ЯМР (400 МГц, MeOD) δ 5,60-5,56 (м, 1H), 5,51-5,27 (м, 10H), 3,99-3,88 (м, 2H), 3,71-3,63 (м, 1H), 3,62-3,54 (м, 1Н), 3,53-3,38 (м, 3Н), 2,93-2,83 (м, 8Н), 2,19-2,07 (м, 4Н), 1,93-1,83 (м, 1Н), 1,81-1,72 (м, 1Н), 1,69. 1,60 (м, 2Н), 1,53-1,45 (м, 2Н), 1,04-0,98 (м, 6Н). МС (электрораспыление): 549,3 [М-Н]-.
Пример 78. Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутан-1-ола:
 .
.
Раствор бутил 2-(((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил)окси) бутаноата (1,008 г, 2,34 ммоль) в сухом THF (3 мл) добавляли по каплям к суспензии LiAlH4 (94,9 мг, 2,50 ммоль) в сухом THF (10 мл) при 0°С в атмосфере азота. Реакционную смесь перемешивали при 0°С в течение 1,5 часов, после чего охлаждающую баню убирали и реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь охлаждали на ледяной бане перед добавлением по каплям воды (2,5 мл) и 1 М HCl (10 мл). Охлаждающую баню убирали и смесь перемешивали в течение 10 минут при комнатной температуре. Полученную смесь экстрагировали гептаном (2×50 мл), промывали 1 М HCl (10 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Флэш-хроматография на силикагеле с элюированием смесью гептан-EtOAc (90:10) давала 0,705 г (выход 83%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,71-4,93 (м, 10Н), 3,63 (дд, 1Н), 3,58-3,34 (м, 3Н), 3,29-3,23 (м, 1Н), 2,95-2,65 (м, 8Н), 2,18-1,98 (м, 4H), 1,88 (ушир.с, 1H), 1,66-1,51 (м, 3H), 1,45 (дт, 3H), 0,95 (т, 3H), 0,89 (т, 3H). МС (ESI, pos) 383 [М+Na]+.
Пример 79. Получение 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутилацетата:
 .
.
Смесь 2 - (((5Z, 8Z, 11Z, 14Z, 17Z) -эйкоза-5,8,11,14,17-пентаен-1-ил) окси) бутан-1-ола (300,9 мг, 0,834 ммоль), триэтиламина (140 мл, 1,00 ммоль) и DMAP (4,7 мг, 0,038 ммоль) в сухом CH2Cl2 (5 мл) добавляли к уксусному ангидриду (95 мл, 1,00 ммоль). Реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 80 минут. Добавляли воду (10 мл) и смесь экстрагировали гептаном (2 × 50 мл), промывали насыщенным солевым раствором (20 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Флэш-хроматография на силикагеле с элюированием смесью гептан-EtOAc (100: 1) дала 0,319 г (95%) указанного в заголовке соединения в виде масла. 1H ЯМР (400 МГц, CDCl3) δ 5,59-5,14 (м, 10H), 4,10 (дд, 1H), 4,02 (дд, 1H), 3,54 (дт, 1H), 3,43 (дт, 1H), 3,38-3,29 (м, 1Н), 2,92-2,65 (м, 8Н), 2,14-1,95 (м, 4Н), 2,05 (с, 3Н), 1,63-1,47 (м, 4Н), 1,45-1,38 (м, 2Н), 0,96 (т, 3Н), 0,92 (т, 3Н). МС (ESI, pos) 425 [М+Na]+.
Биологические Примеры
Оценка соединения A на НАСГ мышиной модели, индуцированной диетой (CDAA/диета с высоким содержанием жира)
Мышиные модели с дефицитом метионина и/холина (MCD и CDAA соответственно) хорошо известны для изучения развития и лечения НАСГ при доклинической разработке лекарств. Дефицит метионина/холина, являющихся предшественниками для синтеза фосфатидилхолина, в рационе питания приводит к неспособности синтезировать печеночные липопротеины для экспорта триглицеридов, что приводит к тяжелому стеатозу печени, воспалению и фиброзу.
Хотя широко используемая диета с дефицитом метионин-холина (MCD) способна постоянно индуцировать тяжелое НАСГ-подобное воспаление печени и фиброз у мышей, она также связана с серьезной потерей веса (потерей как скелетных мышц, так и массы жира). Это приводит к повышеннию риска смертности, что создает серьезные проблемы для длительных экспериментов с фиброгенезом. При добавлении субоптимальной дозы метионина (0,17%), диетическая модель CDAA способна преодолеть эти проблемы и, как было продемонстрировано, имитирует НАСГ человека как у мышей, так и у крыс, последовательно вызывая стеатогепатит, фиброз печени и рак печени с менее серьезной потерей веса тела.
В биологических Примерах 1-7 исследования были выполнены на самцах мышей C56BL/6J (9 недель). Мышей кормили либо диетами с высоким содержанием холина (CS), либо с дефицитом холина при высоком содержании жира (31% от общего количества калорий); «CDAA/с высоким содержанием жира»). Диета, индуцирующая НАСГ, была инициирована за 6 недель до начала лечения, чтобы спланировать исследование как схему лечения/возвращения к норме, а не как профилактику заболевания.
В биологических Примерах №1-5 мышей разделили на 6 экспериментальных групп (n=9 на группу) и кормили либо CS, либо CDAA/рационом с высоким содержанием жира. После 6 недель диеты CS- или CDAA мышам перорально вводили соединение A или соединение N, оба соединения в 2 дозах, 0,15 ммоль/кг массы тела/день (низкие дозы, LD) и 0,3 ммоль/кг массы тела в день (высокая доза, HD). Испытуемые вещества вводили перорально в качестве добавки к диете с высоким содержанием жиров. Фиброз печени оценивали как путем измерения содержания гидроксипролина (HYP), так и морфометрии Sirius Red (SR). Уровни транскриптов, связанных с фиброзом, воспалением и метаболизмом, измеряли с помощью количественной полимеразной цепной реакции в реальном времени (кПЦР).
В биологических Примерах №6 и 7 мышей кормили либо CS, либо CDAA/диетой с высоким содержанием жира, как описано выше. В Примере №6 после 6 недель диеты CS или CDAA/с высоким содержанием жира мышам перорально вводили 56 мг/кг массы тела в день Соединения А или Bydureon® (эксенатид) который является агонистом GLP-1R пролонгированного действия, и который одобрен для лечения диабета 2 типа и вводится подкожно в дозе 0,4 мг/кг в неделю в течение шести недель. Влияние на количество, толщину и длину волокон коллагена в печени определяли с помощью нелинейной многофотонной оптической системы визуализации в изолированных образцах печени из групп контроля носителя (n=8) и групп, которым вводили соединения A (n=9) и GLP-1R (n=8).
В биологическом Примере 7 после 6 недель диеты CS или CDAA/с высоким содержанием жира мышам перорально вводили 112 мг/кг массы тела в день соединения A или эквимолярную дозу эйкозапентаеновой кислоты (EPA). Липидомный анализ печени проводили в изолированных образцах печени из групп CS (n=9), соединения A (n=9) и EPA (n=8).
В биологических Примерах №21-24 изучали эффекты Соединения А отдельно и в комбинации с агонистом эксенатида GLP-1. Исследования проводились на самцах мышей C56BL/6J (9 недель). Мышей делили на 6 экспериментальных групп (n=9 на группу), находящихся на либо CS, либо CDAA/диете с высоким содержанием жира, как описано выше. НАСГ-индуцирующая CDAA/диета с высоким содержанием жиров была начата за 6 недель до начала лечения, чтобы оценить лечение/реверс синдрома, а не профилактику. Таким образом, после 6 недель диеты CS или CDAA/с высоким содержанием жира мышей обрабатывали (A) пероральным соединением A в дозе 0,3 ммоль/кг массы тела/день отдельно, (B) одним агонистом GLP-1, вводимым внутрибрюшинно, в дозе 0,4 мг/кг один раз в неделю или (C) комбинация соединения A в дозе 0,3 ммоль/кг массы тела/сутки и агониста GLP-1, вводимого внутрибрюшинно. в дозе 0,4 мг/кг один раз в неделю (снижен до 0,1 мг/кг в течение последних 3 недель из-за чрезмерной потери веса). Соединение А вводили перорально в качестве добавки к рациону с высоким содержанием жира, в то время как агонист GLP-1 доставляли посредством внутрибрюшинной инъекции (т.е. внутрибрюшинно). Уровни транскриптов, связанных с фиброзом, воспалением и метаболизмом, измеряли с помощью количественной полимеразной цепной реакции в реальном времени (кПЦР) в изолированной ткани печени.
Оценка соединения A в модели НАСГ, инъецированной инъекцией стрептозотоцина и диетой с высоким содержанием жиров (модель STAM)
В биологическом Примере №8 15-дневных беременных мышей C57BL/6 без патогенов получали от Charles River Laboratories в Japan Inc. (Канагава, Япония). НАСГ был индуцирован у самцов мышей путем однократной подкожной инъекции стрептозотоцина (STZ) (Sigma, США) после рождения и кормления диетой с высоким содержанием жира (HFD; CLEA) Япония, Япония) ad libitum после 4-недельного возраста (день 28 ±). 2). Мышей разбивали на 3 группы случайным образом по 8 мышей в возрасте 6 недель за день до начала лечения, на группы контроля носителя (физиологического раствора), Соединения А и телмисартана (положительный контроль). Лечение Соединением А в дозе 37 мг/кг массы тела в день через оральный зонд в сочетании с диетой с высоким содержанием жиров проводилось в течение 6 недель до умерщвления. Показатель активности по шкале оценки НАСГ (NAFLD (NAS)) рассчитывали в соответствии со стандартизированными критериями, а площадь фиброза оценивали с помощью окрашивания Sirius Red.
Оценка действия Соединения А на мышиной модели НАСГ, индуцированной диетой (APOE * 3Leiden.CETP, двойные трансгенные мыши)
Двойная трансгенная мышь APOE * 3Leiden.CETP экспрессирует вариант человеческого аполипопротеина E3 (APOE3), APOE * 3Leiden в дополнение к человеческому аполипопротеину C1 (APOC1) и CETP. APOE * 3Leiden.CETP двойные трансгенные мыши демонстрируют повышенные уровни холестерина и триглицеридов в плазме, в основном ограниченные фракцией липопротеинов размера VLDL/LDL. При увеличении содержания холестерина в рационе в этой модели, развиваются все характеристики человеческого НАСГ.
В биологическом Примере 9 исследования были выполнены на мышах APOE * 3Leiden.CETP, помещенных на диету с высоким содержанием жира (24% жира по массе) с различным содержанием холестерина (0,25-1% холестерина по массе). В одном исследовании (с использованием 1% холестерина по массе) после 3-недельного периода на диете мышей с низким уровнем ответа (20% от общего числа) исключали из исследования, а оставшихся мышей разделяли на 4 группы по 12 мышей каждая (+5 в контрольной группе), сопоставимые по холестерину в плазме, триглицеридам, глюкозе в крови, массе тела и возрасту (t=0), после чего начинали лечение. Мышам вводился каждые сутки желудочный зонд между 07 ч. 00 м. и 10ч. 00м. с Соединением А в дозе 0,3 ммоль/кг массы тела в день, соединением N (0,3 ммоль/кг массы тела в день), росиглитазоном (13 мг/кг массы тела в день) или контролем (кукурузное масло). После 20 недель лечения мышей умерщвляли СО2 -асфиксией, брали печень и оценивали уровень гепатоцеллюлярной гипертрофии.
В биологическом Примере 25 были изучены эффекты соединения А отдельно и в сочетании с омега-3 жирной кислотой. Мышей APOE * 3Leiden.CETP помещали на полусинтетическую диету с высоким содержанием жира (24% жира по массе) с 0,25 холестерина по массе. После 4-недельного периода на диете мышей с низким уровнем ответа исключили из исследования, а оставшихся мышей разбили на группы по 8 мышей в каждой, сопоставимые по холестерину в плазме, триглицеридам, глюкозе в крови, массе тела и возрасту (t=0) и начинали лечение. Для облегчения смешивания соединений подсолнечное масло было добавлено до объема 10 мл/кг рациона. Соединение А вводили в дозе 0,3 ммоль/кг массы тела/день, тогда как этиловые эфиры омега-3 жирных кислот (85% мас./мас. от EPA/DHA этиловых эфиров) вводили в количестве 3,0 ммоль/кг массы тела/день. Соединение А и этиловые эфиры омега-3 вводили перорально в качестве добавки к рациону с высоким содержанием жиров. После 4 недель лечения мышей умерщвляли CO2-асфиксией, брали печень и измеряли содержание свободного холестерина, сложных эфиров холестерина и триглицеридов в печени после их экстракции и разделения с помощью высокоэффективной тонкослойной хроматографии.
Оценка эффектов соединения A на модели мышей, страдающих ожирением и НАСГ, индуцированным диетой (модель ob/ob AMLN)
Мыши B6.V-Lepob/Jrj, страдающие ожирением, склонны к фиброзу (печени), при добавлении холестерина (2%), 40% жира (содержащего 18% транс-жирных кислот) и 20% фруктозы к высококалорийной диета (т.е. к AMLN -диете с высоким содержанием жиров). У мышей ob/ob на диете AMLN (называемых «мыши ob/ob AMLN») развивается стеатогепатит и фиброз в более короткие сроки (≤12 недель) по сравнению с мышами C57BL/6 дикого типа (мыши AMLN), получавшими ту же диету. Мыши AMLN ob/ob предоставляют модель НАСГ, индуцированную ожирением, которая также позволяет изучать влияние на гликемический контроль. Развитие НАСГ в модели ob/ob AMLN подтверждается биопсией.
В биологических Примерах 10-12, 15 и 19 исследования проводились на самцах мышей ob/ob (5 недель), которых кормили рационом с высоким содержанием жира (диета AMLN) в течение 15 недель, затем разбили на 3 группы случайным образом (n=10 на группу) для введения либо 112 мг/кг массы тела в день Соединения А с помощью диеты, либо пиоглитазона с помощью диеты в дозе 30 мг/кг массы тела в день, либо (наполнителя) в течение следующих 4 недель. Пероральный тест на толерантность к глюкозе (OGTT) был проведен на 3-ей неделе. Фиброз печени оценивали путем измерения экспрессии фиброгенного/фибролитического гена в печени путем секвенирования РНК через 4 недели.
В биологических Примерах 10, 13-14 и 16-18 исследования проводились на самцах мышей ob/ob (5 недель), которых кормили рационом с высоким содержанием жира AMLN в течение 18 недель, затем разбивали на 6 групп случайным образом (n=10 на группу) для введения либо Соединения А (в 3 различных суточных дозах, 45 мг/кг, 90 мг/кг или 135 мг/кг) с помощью диеты, либо обетихоловой кислоты (ОСА) в дозе 30 мг/кг массы тела в день с помощью диеты, либо ведение наполнителя (лечение отсутствуе) еще в течение 8 недель. Оценка связанных с НАСГ параметров и фиброза производилась в конце исследования с помощью количественных измерений иммуногистохимии (IHC).
В соответствии с медицинскими параметрами, используемые здесь дозы Соединения А (45 мг/кг, 90 мг/кг и 135 мг/кг в 8-недельном исследовании и 112 мг/кг в 4-недельном исследовании) соответствуют человеческим дозам приблизительно 250 мг/день, 500 мг/день и 750 мг/день (и 600 мг/день в 4-недельном исследовании) для человека весом 70 кг, с поправкой на межвидовую разницу. См. Nair, A.B., и Jacob, S., J Basic Clin Pharm, 2016; 7 (2): 27-31, для руководства по межвидовой конверсии дозы.
Оценка влияния Соединения А на звездчатые клетки печени человека in vitro.
Звездчатые клетки являются резидентными клетками печени и основным типом клеток, участвующих в инициации и прогрессировании фиброза печени. В биологическом Примере 20 исследования in vitro на культивируемых звездчатых клетках человека (клетки LX-2) проводились для оценки прямого воздействия Соединения А на эти клетки. Клетки обрабатывали в течение 24 часов Соединением А (10 мкМ, 25 мкМ, 50 мкМ или 75 мкМ) или олеиновой кислотой (ОА; 75 мкМ), и оценивалось включение бромодезоксиуридина (BrdU), с целью определения влияние Соединения А на пролиферацию. Анализ МТС (3- (4,5-диметилтиазол-2-ил) -5- (3-карбоксиметоксифенил) -2- (4-сульфофенил) -2Н-тетразолия) использовали для определения жизнеспособности клеток.
Биологический Пример 1. Влияние соединения A и соединения N на состояние фиброза печени, по результатам содержания общего гидроксипролина (HYP) в печени
Соединение А в дозах как низкой (LD), так и высокой (HD), вводимых мышам, получавшим CDAA/жирную пищу, вызывало значительное снижение фиброза печени (содержание HYP в печени) по сравнению с контролем (р <0,01 * и 0,001 ** соответственно).). Никаких значительных эффектов не наблюдалось для соединения N, взятому для сравнения. Влияние Соединения А на содержание печеночного HYP показано в таблице 1.
Биологический Пример 2. Влияние соединения A и соединения N на состояние фиброза печени, по результатам относительного содержания гидроксипролина (HYP) (мкг HYP на 100 мг печени).
Соединение A как в низких (LD), так и в высоких (HD) дозах, вводимых мышам, получавшим CDAA/жирную пищу, уменьшало фиброз печени (содержание HYP на печень), тогда как для соединения N не наблюдалось никаких эффектов. Влияние соединения A на относительное содержание HYP показано в Таблице 2.
Содержание соединения гидроксипролина (мкг) на 100 мг печени
Биологический Пример 3. Влияние соединения A и соединения N на экспрессию гена воспалительного процесса в печени (экспрессия мРНК TNF-a).
Соединение А в дозах как низкой (LD), так и высокой (HD), вводимых мышам, получавшим CDAA/жирную пищу, вызывало значительное уменьшение воспаления печени (мРНК TNF-α) по сравнению с контролем (обе p<0,001 ***). Значительное снижение (р<0001) наблюдалось также для соединения N, но не для высокой дозы (HD). Влияние соединения A на печеночный TNF-α показано в Таблице 3.
Биологический Пример 4. Влияние соединения A и соединения N на экспрессию гена, связанного с фиброзом (экспрессия мРНК Col1a1)
Соединение А в дозах, как низкой (LD), так и высокой (HD), вводимых мышам, получавшим CDAA/жирную пищу, вызывало значительное снижение экспрессии гена, связанного с фиброзом печени (мРНК Col1a1), по сравнению с контролем (обе р<0,001 *** соответственно)). Никаких существенных эффектов не наблюдалось для Соединения N. Эффекты Соединения А на печеночный Col1a1 показаны в Таблице 4.
Биологический Пример 5. Влияние Соединения А на содержание фиброза печени, измеренное с помощью морфометрии Sirius Red.
Чтобы более точно определить количество локализации и функциональную значимость фиброза печени, была выполнена морфометрия Sirius Red (SR). В отличие от Примеров 1 и 2, где была использована биохимическая оценка содержания гидроксипролина (HYP) для измерения коллагена в крупных сосудах, морфометрия SR количественно определяет более функционально значимые синусоидальные отложения коллагена. 10 областей на слайде были измерены с использованием 100-кратного увеличения. Соединение А в дозах как низкой (LD), так и высокой (HD), вводимых мышам, получавшим CDAA/жирную пищу, вызывало значительное уменьшение площади поверхности в процентах фиброза печени по сравнению с контролем (р <0,001 *** и 0,05 * соответственно).). Влияние Соединения А на фиброз печени, измеренное с помощью морфометрии SR, показано в таблице 5.
Биологический Пример 6. Воздействие Соединения A по сравнению с Bydureon® на коллагеновые волокна
При введении в низких дозах (56 мг/кг) CDAA/мышам на диете с высоким содержанием жира Соединение А значительно снижало количество коллагеновых волокон (Фиг. 1А) на 51% (р=0,01). Этот эффект был отражен в почти полном исчезновении коротких (<8,5 мкм) и тонких (<3,5 мкм) коллагеновых волокон, и выражался в незначительном увеличении толщины и длины волокон соединением A на Фиг. 1B и Фиг. 1C, соответственно. Bydureon® (0,4 мг/кг еженедельно вводимый подкожно) не оказал значительного эффекта.
Биологический Пример 7. Влияние соединения A и EPA на липидный обмен в печени.
Чтобы дополнительно понять влияние Соединения А на липидный обмен в печени, был проведен липидомный анализ мышей, получавших CS- и CDAA/с высоким содержанием жира («CD»), в изолированной ткани печени. При введении в высокой дозе (112 мг/кг) CDAA/мышам с высоким содержанием жира, Соединение А значительно уменьшало эффекты дефицита холина, по результатам определения печеночных триглицеридов (TG) (Фиг. 2A), печеночных диглицеридов (DG) (Фиг. 2B), свободных жирных кислот печени (FFA) (Фиг. 2C) или сложных эфиров холестерина (Фиг. 2D). Напротив, лечение эквимолярным количеством EPA не уменьшало увеличение печеночной TG, DG, FFA или холестериновых эфиров, связанных с холин-дефицитной диетой.
Биологический Пример 8. Воздействие соединения A на гепатоцеллюлярное баллонирование, лобулярное воспаление, стеатоз, композитный балл по шкале оценуи НАСТ (NAS) и площади фиброза.
Соединение А, вводимое STAM (модель) мышам уменьшало гепатоцеллюлярное баллонирование, а также стеатоз и лобулярное воспаление. Как составной балл по шкале NAS (p <0,001 *), так и площадь фиброза (положительная область, окрашенная Sirius Red) были снижены при введении соединениея A.
Биологический Пример 9. Воздействие соединения A и соединения N на гепатоцеллюлярную гипертрофию.
Соединение А, вводимое мышам ApoE * 3L-CETP (0,25% мас. холестериновой диеты), вызывало значительное снижение гепатоцеллюлярной гипертрофии (р<0,01 *), которое составляло 83% по сравнению с контролем (р <0,01). Никакого эффекта не наблюдалось при введении Соединения N.
Биологический Пример 10. Влияние соединения А, пиоглитазона и ОСА на массу печени и массу тела.
Как показано на Фиг.3А, через 4 недели Соединение А (112 мг/кг) не оказывало влияния на массу тела ob/ob мышей AMLN, тогда как пиоглитазон вызывал значительное увеличение массы тела на 15% (р <0,01). Вес печени был неизменным во всех группах на 4 неделе, как показано на Фиг. 3B. Через 8 недель ни Соединение А (45 мг/кг, 90 мг/кг и 135 мг/кг), ни ОСА существенно не влияли на массу тела, как показано на Фиг. 3C, и потребление пищи не изменилось ни в одной группе. ОСА значительно (р <0,05) снижала массу печени через 8 недель, тогда как Соединение А не оказывало значительного эффекта ни при какой дозе по сравнению с носителем, как показано на Фиг. 3D.
Биологический Пример 11. Влияние Соединения А и пиоглитазона на печеночную фиброгенную и фибролитическую экспрессию генов.
Влияние 4-недельного лечения Соединением А (112 мг/кг) или пиоглитазоном (30 мг/кг) на экспрессию фиброгенных и фибролитических генов в изолированных пробах печени мышей ob/ob AMLN- показано на Фиг. 4A-4F. Соединение А значительно снижало экспрессию канонических генов, регулирующих фиброгенез печени, включая бета-фактор роста тромбоцитов (PDGF-β) (Фиг. 4А), β-рецептор фактора роста тромбоцитов- (PDGFR-β) (Фиг. 4В), коллаген -1 α1 (col1A1) (Фиг. 4C), col3A1 (Фиг. 4D), col4A1 (Фиг. 4E) и белок теплового шока-47 (HSP47) (Фиг. 4F). Пиоглитазон значительно снижал экспрессию исследованных изоформ коллагена (col1A1, col3A1, col4A1) и HSP47, но не оказывал влияния на экспрессию PDGF-β или PDGFR-β.
Биологический Пример 12. Влияние Соединения А и пиоглитазона на стабильность внеклеточного матрикса печени и экспрессию генов фибролиза.
Как показано на Фиг. 5A-5D, в отношении генов, регулирующих стабильность внеклеточного матрикса (ECM) или фибролиз, 4 недели лечения соединением A значительно снижало печеночную экспрессию лизилоксидазы (LOX) (Фиг. 5A), LOX-like 2 (LOXL2) (Фиг. 5B) и LOXL1 (Фиг. 5C), а также тканевой ингибитор металлопротеиназы (TIMP1) (Фиг. 5D) у ob/ob мышей на AMLN-диете . Пиоглитазон также значительно снижал уровни этих транскриптов, хотя эффект был слабее, чем эффект, полученный при введении соединениея А.
Биологический Пример 13. Влияние Соединения А и ОСА на прогрессирование фиброза печени.
Чтобы оценить влияние 8 недель введения Соединения А (45 мг/кг, 90 мг/кг или 135 мг/кг) и ОСА (30 мг/кг) на прогрессирование фиброза печени у ob/ob мышей на AMLN-диете, количественную иммуногистохимию (IHC) проводили, измеряя содержание col1A1 и α-актина гладких мышц (α-smooth muscle actin, SMA). Как показано на Фиг.6А и 6В, при дозах 90 мг/кг и 135 мг/кг Соединение А заметно снижало маркер миофибробласта α-SMA (выраженный как в виде общего, так и в процентном отношении фракции), тогда как введение ОСА не показало какой-либо значительный эффект.
Аналогично, соединение А, вводимое в дозе 90 и 135 мг/кг, уменьшало общее содержание col1A1 (Фиг. 6С), тогда как ОСА не оказывало значительного эффекта. Если рассматривать в процентах от общей площади поверхности (Фиг. 6D) то доза 90 мг/кг вызывала значительное снижение уровней col1A1, тогда как снижение при дозах 45 и 135 мг/кг не было статистически значимым. Для подтверждения антифиброзных эффекты, наблюдаемых при количественном IHC, измеряли концентрации гидроксипролина (HYP) в печени, как показано на Фиг. 6E и Фиг. 6F. Только Соединение А снижало содержание HYP, выраженное либо как общее (р <0,01 для доз 90 и 135 мг/кг), так и относительное (р <0,05 для доз 90 и 135 мг/кг) содержание.
Биологический Пример 14. Влияние Соединений А и ОСА на регресс печеночного фиброза.
Поскольку заранее были сделаны исходные (до лечения) биопсии мышей ob/ob AMLN, включающие измерения IHC как печеночного col1A1, так и SM-SMA, конечные значения col1A1 и α-SMA сравнивались с исходными значениями (биопсия до лечения) во всех группах, чтобы установить, были ли значимые снижение, которые наблюдались после лечения Соединением А, по сравнению с носителем в конце исследования, связаны с регрессом фиброза. Как показано на Фиг.7А, обработка Соединением А в дозе 135 мг/кг снижала процентную долю содержания α-SMA, оцененную по иммуногистохимическому окрашиванию, по сравнению с содержанием α-SMA в биопсии перед введением препарата. Дозы Соединения А в дозе 45 мг/кг и 90 мг/кг приводили к незначительной разнице тенденций. ОСА не оказала существенного влияния. Таким образом, все дозы соединения A показали тенденцию к снижению содержания col1A1 по сравнению с содержанием до биопсии, хотя только доза 90 мг/кг была статистически значимой (p <0,005) (Фиг. 7B). Обе терапии носителем и ОСА показали умеренное увеличение содержания col1A1 в печени после 8-недельного периода введения, хотя увеличение было значимым только для ОСА (р <0,05).
Биологический Пример 15. Влияние Соединения А и пиоглитазона на экспрессию генов воспаления.
Особенно важным фактом для прогрессирования фиброза у ob/ob мышей на AMLN-диете, было то, что 4 недели лечения 112 мг/кг соединением A значительно снижали экспрессию воспалительных генов по сравнению с контрольными мышами, которым вводили носитель. В частности, Соединение А снижало уровни транскриптов как трансформирующего ростового фактора бета 1 (TGF-β1) (р <0,05), так и рецептора TGF-β1 (TGFRβ (р <0,01), как показано на Фиг. 8A и Фиг. 8В, соответственно. Пиоглитазон уменьшал экспрессию TGFRβ (p <0,05), но не TGF-β1. Значительное снижение CCR2, MAC-2 (данные не показаны), CD68 и CD14 (все p <0,001) было обнаружены после лечения Соединением А по сравнению с контролем, как показано на Фиг. 8C, 8D и 8F. Обработка соединением A в группах также значительно снижала уровни галектина-3 (Gal-3) по сравнению с контрольными группами, где вводился носитель (Фиг. 8E). Пиоглитазон значительно снижал CCR2 (р <0,01) (Фиг. 8C), CD68 (р <0,01) (Фиг. 8D) и CD14 (р <0,001) (Фиг. 8F). Ни одно из соединений значительно не снижало уровни транскриптов интерлейкина (IL)-1β или МСР-1 (данные не показаны).
Биологический Пример 16. Влияние Соединения А и ОСА на воспаление печени по результатам измеренний печеночного Gal-3.
Для оценки влияния 8-недельного лечения Соединением А (45 мг/кг, 90 мг/кг или 135 мг/кг) или ОСА (30 мг/кг) на воспаление печени у ob/ob мышей на AMLN-диете проводили IHC для оценки уровней галектина-3 (Gal-3). Как показано на Фиг. 9A и 9B, обработка соединением A приводила к дозозависимому снижению уровня экспрессии Gal-3 при измерении либо в виде общего содержания, либо в процентах от площади, тогда как OCA только снижала уровни экспрессии Gal-3 только тогда, когда измерялась общее содержание.
Биологический Пример 17. Влияние Соединения А и ОСА на гепатоцеллюлярное повреждение.
Для оценки влияния Соединения А и пиоглитазона на гепатоцеллюлярное повреждение или повреждение у ob/ob мышей на AMLN-диете, плазменную аланинаминотрансферазу (ALT) и аспартаттрансаминазу (AST) измеряли после 8 недель введения. Как показано на Фиг.10А, заметное и значимое снижение ALT было достигнуто при дозах Соединения А 90 мг/кг и 135 мг/кг (р <0,01 и р <0,001 соответственно), тогда как ОСА не оказываал эффекта. В дозах 90 мг/кг и 135 мг/кг Соединение A также значительно снижало AST (p <0,05 и p <0,01 соответственно), тогда как OCA не оказывала влияния на уровни AST (Фиг. 10B).
Биологический Пример 18. Влияние Соединения А и ОСА на стеатоз печени, уровень липидов в печени и уровень липидов в плазме.
Как показано на Фиг. 11A и Фиг. 11B, и Соединение A, и OCA значительно снижали стеатоз у мышей ob/ob AMLN после 8 недель введения (как выраженный в процентах размер площади или общее содержание липидов), причем OCA демонстрировала сопоставимую эффективность с наибольшей дозой (135 мг/кг) Соединения А (оба р <0,001). Аналогично, как и ОСА, так и Соединение А (в дозах 90 мг/кг и 135 мг/кг) продемонстрировали сопоставимое снижение уровней триглицеридов и общего уровня холестерина в плазме, как показано на Фиг.11D и Фиг.11E. Лечение ОСА, но не соединением А, показало снижение общего содержания холестерина в печени (р <0,01) (Фиг. 11С).
Биологический Пример 19. Влияние Соединения А и пиоглитазона на гликемический контроль.
Через 3 недели после введения Соединения A (112 мг/кг), пиоглитазона или контроля носителя был проведен оральный тест на толерантность к глюкозе (OGTT) у мышей ob/ob AMLN. Как показано на Фиг. 12A, и соединение A, и пиоглитазон значительно уменьшали отклонение глюкозы в течение первых 240 минут после нагрузки глюкозой (AUC 0-240 минут), хотя эффект был более выраженным при использовании пиоглитазона. Подобным образом, значительное снижение уровня глюкозы в плазме натощак было достигнуто с обоими соединениями, хотя эффект был более выраженным у пиоглитазона по сравнению с Соединением A (Фиг. 12B). Пиоглитазон, но не Соединение А, снижает уровень инсулина натощак (р <0,001) (Фиг. 12С).
Биологический Пример 20. Влияние соединения А и олеиновой кислоты на пролиферацию и жизнеспособность звездчатых клеток in vitro.
Клеточную пролиферацию звездчатых клеток печени человека LX-2, обработанных в течение 24 часов 10-75 мкм Соединениея А или олеиновой кислотой (ОА) (75 мкМ), оценивали путем включения BrdU. Результаты являются нормированными средними значениями ± S.E.M. пяти независимых экспериментов для проведенных для эйкозабутата и двух независимых экспериментов для ОА. Как показано на Фиг. 13A, клетки LX-2, обработанные 25 мкМ (р <0,005), 50 мкМ (р <0,0001) и 75 мкМ (р <0,005) соединения А были менее пролиферативными по сравнению с клетками, обработанными ОА или носителем. Снижение пролиферации Соединением А составило 25-34% через 24 часа, тогда как ОА не оказывала эффекта. Сравнения были сделаны односторонним ANOVA с поправкой Даннетта для множественных сравнений. Как показано на Фиг. 13B, ни Соединение A, ни OA не оказывали значительного влияния на жизнеспособность клеток, как измерено анализом MTS в двух независимых экспериментах.
Биологический Пример 21. Влияние соединения А и агониста GLP-1 (GLP-1a, экзенатид), отдельно и в комбинации, на экспрессию генов, связанных с фиброзом печени (экспрессия мРНК Col1a1).
Соединение A, GLP-1a и Соединение A+GLP-1a в комбинации, вводимой мышам, получавшим CDAA/жирную пищу, вызывало значительное снижение экспрессии мРНК Col1a1 по сравнению с мышами, получавшими CDAA (все p<0,001 ***). Влияние Соединения А отдельно и в комбинации с GLP-1a на печеночную мРНК Col1a1 показано в Таблице 8.
Биологический Пример 22. Влияние соединения А и агониста GLP-1 (GLP-1a, экзенатид), отдельно и в комбинации, на экспрессию гена, связанного с фиброзом печени (на экспрессию мРНК TIMP (тканевой ингибитор матричной металлопротеиназы) -1a)
Соединение A, GLP-1a и соединение A+GLP-1a в комбинации, вводимые мышам, получавшим CDAA с высоким содержанием жира, вызывали значительное снижение экспрессии мРНК печеночного TIMP-1a по сравнению с мышами, получавшими CDAA (все p <0,001 ***). Влияние Соединения А отдельно и в комбинации с GLP-1a на экспрессию мРНК TIMP-1a в печени показано в Таблице 9.
Биологический Пример 23. Влияние Соединения А и агониста GLP-1 (GLP-1a, экзенатид), отдельно и в комбинации, на экспрессию генов, связанных с фиброзом печени (экспрессия мРНК ММР-13 (матриксной металлопротеиназы))
Соединение A, GLP-1a и Соединение A+GLP-1a в комбинации, вводимой мышам, получавшим CDAA диету с высоким содержанием жира, вызывали значительное снижение экспрессии мРНК печеночной ММР-13 по сравнению с мышами, получавшими CDAA (все p <0,001 ***). Влияние Соединения А отдельно и в комбинации с GLP-1a на печеночную мРНК ММР-13 в печени показано в Таблице 10.
Биологический Пример 24. Влияние соединения А и агониста GLP-1 (GLP-1a, экзенатид), отдельно и в комбинации, на экспрессию гена, связанного с воспалением печени (экспрессия мРНК TNF-α)
Соединение A, GLP-1a и Соединение A+GLP-1a в комбинации, вводимой мышам, получавшим CDAAc высоким содержанием жира, вызывали значительное снижение экспрессии мРНК печеночного TNF-α по сравнению с мышами, получавшими CDAA (p <0,001 для соединения A отдельно или в комбинации с GLP-1a, р <0,05 только для GLP-1a). Влияние Соединения А отдельно и в комбинации с GLP-1a на печеночную мРНК печеночного TNF-α показано в Таблице 11.
Биологический Пример 25. Воздействие соединения А и концентрированных омега-3 этиловых эфиров, отдельно и в комбинации, на липиды печени (сложный эфир холестерина-CE, свободный холестерин- FC, триглицериды-TG).
Низкая доза (0,3 ммоль/кг массы тела/день) Соединения A (LD), вводимая трансгенным мышам ApoE*3L-CETP, получавшим Западную Диету с 0,25% холестерина в сочетании с высокой дозой (3 ммоль/кг массы тела в день) 85% EPA/DHA омега-3 этиловых эфиров (HD) вызывала значительное снижение FC (р<0,01), СЕ (р<0,001) и TG (р<0,01). Ни одно из соединений значительно не снижало содержание TG в печени в качестве монотерапии в этой модели.
EPA/DHA HD
Биологический Пример 26: Многоцентровое рандомизированное двойное слепое плацебо-контролируемое исследование в параллельных группах эффективности Соединения А у пациентов с неалкогольным стеатогепатитом (НАСГ)
Целью этого клинического испытания являлась оценка эффективности различных доз Соединения А в лечении НАСГ у пациентов без ухудшения фиброза и оценка безопасности и переносимости Соединения А у пациентов, страдающих НАСГ.
Количество пациентов
600 пациентов, страдающих НАСГ, обследованы в примерно 30 центрах для определения их пригодности для исследования; ожидается, что будет найдено около 264 пациента, отвечающим критериям отбора, описанным ниже, из которых приблизительно 198 по прогнозу завершат полное испытание (в результате получая 66 субъектов на группу лечения, и допуская, что 25% пациентов не закончат лечение).
Назначение лечения
Пациенты, признанные подходящими для исследования, случайным образом делятся на 3 параллельные группы, состоящие из 88 пациентов в каждой. Группе 1 вводят плацебо Соединения А, группе 2 вводят 300 мг Соединения А, а группе 3 вводят 600 мг Соединения А. Соединение А или плацебо вводят один раз в день в одной (300 мг) или двух (600 мг) капсулах в течение 52 недель. Распределение для F1 против F2/F3.
Эффективность лечения соединением А оценивают, например, по шкале NAS по результатам биопсии печени, по оценке МРТ LiverMultiScan PDFF и cT1, тестам функции печени, HOMA-IR и биомаркерам воспаления и фиброза (включая hsCRP, Pro-C3, панель ELF, и другие подходящие биомаркеры).
Безопасность и переносимость оцениваются по сообщениям о побочных эффектах, мониторингу жизненно важных функций, физичесому обследованию, гематологии и биохимии (почки, печень), анализу мочи и ЭКГ по 12 отведениям в состоянии покоя.
Артериальное давление контролируется тремя показаниями артериального давления, которые снимаются при каждом посещении.
Дизайн исследования
Исследование проводится в течение 58 недель и включает 10 клинических посещений на пациента, включая скрининг, двойной слепой период лечения и последующее наблюдение. Процедуры, выполняемые при каждом посещении, описаны ниже и суммированы в Тблице 13.
Визит 1 (от 1 до 42 дней до начала введения лечения): пациенты проверяются на пригодность для исследования в соответствии с критериями, описанными ниже, от пациента получают согласие на исследование. Оценка пригодности может проводиться более чем за одно посещение.
Визит 2: пациенты разбиваются случайным образом (рандомизируются) на 3 группы лечения. Каждой из групп лечения вводится 300 или 600 мг соединения А или плацебо ежедневно в течение 52 недель. Проводятся базовые оценки, в том числе печеночная томография (МРТ - PDFF и cT1), берутся показатели жизненно важных функций, берутся анализы на безопасность лечения (базовая гематология, биохимия и анализ мочи), проводится ЭКГ в 12 отведениях в состоянии покоя и анализ биомаркеров (HOMA-IR, hsCRP, Pro-C3 и панель ELF).).
Визит 3 (после 4 недель лечения): пациенты обследуются на наличие побочных эффектов, показателей жизненно важных функций, берутся анализы на безопасность лечения (базовая гематология, биохимия и анализ мочи), проводится ЭКГ в 12 отведениях в состоянии покоя и детальное изучение фармакинетики (PK samples).
Визит 4 (после 10 недель лечения): пациенты обследуются на наличие побочных эффектов, показателей жизненно важных функций, берутся анализы на безопасность лечения (базовая гематология, биохимия и анализ мочи), проводится ЭКГ в 12 отведениях в состоянии покоя и детальное изучение фармакинетики (PK samples).
Визит 5 (после 16 недель лечения): пациенты обследуются на наличие побочных эффектов, проводится томография печени (МРТ - PDFF и cT1), берутся показатели жизненно важных функций, берутся анализы на безопасность лечения (базовая гематология, биохимия и анализ мочи), проводится ЭКГ в 12 отведениях в состоянии покоя, проводится анализ биомаркеров (HOMA-IR, hsCRP, Pro-C3 и панель ELF), и детальное изучение фармакинетики (PK samples).
Посещения 6, 7 и 8 (после 24, 32 и 40 недель лечения): пациенты обследуются на наличие побочных эффектов, берутся показатели жизненно важных функций, берутся анализы на безопасность лечения (базовая гематология, биохимия и анализ мочи), проводится ЭКГ в 12 отведениях в состоянии покоя, проводится анализ биомаркеров (hsCRP) и детальное изучение фармакинетики (PK samples).
Визит 9 (после 52 недель лечения): пациентам проводится биопсии печени, визуализация печени (МРТ - PDFF и cT1), берутся показатели жизненно важных функций, берутся анализы на безопасность лечения (базовая гематология, биохимия и анализ мочи), проводится ЭКГ в 12 отведениях в состоянии покоя, проводится анализ биомаркеров (HOMA-IR, hsCRP, Pro-C3 и панель ELF), и детальное изучение фармакинетики (PK samples).
Визит 10 (через 14-21 день после прекращения приема пробного лекарства): пациенты обследуются на предмет безопасности лечения и оцениваются на наличие нежелательных явлений, берутся показатели жизненно важных функций, берутся анализы на безопасность лечения (базовая гематология, биохимия и анализ мочи), проводится ЭКГ в 12 отведениях в состоянии покоя и детальное изучение фармакинетики (PK samples).
Пациенты, которые не проходят исследования до завершения, оцениваются на наличие побочных эффектов, у них берутся показатели жизненно важных функций, берутся анализы на безопасность лечения (базовая гематология, биохимия и анализ мочи), проводится ЭКГ в 12 отведениях в состоянии покоя, проводится анализ биомаркеров (HOMA-IR, hsCRP, Pro-C3 и панель ELF) и детальное изучение фармакинетики (PK samples).
Критерии включения пациента:
Пациенты соответствуют приведенным ниже критериям для участия в исследовании.
Возраст: от 18 до 75 лет.
Женщины не имеют детородного потенциала или используют приемлемые средства контрацепции.
Если женщины-партнеры пациентов мужского пола имеют детородный потенциал, пациенты мужского пола готовы использовать контрацепцию (например, презервативы). Кроме того, их партнерша должна использовать противозачаточные средства (например, гормональные, внутриматочные и т. д.). Двойная контрацепция используется от первой дозы препарата соединения и в течение 90 дней после введения последней дозы исследуемого препарата.
Письменное информированное согласие.
Необязательные критерии сканирования фиброза перед биопсией.
Гистологический диагноз НАСГ при скрининге или в течение 6 месяцев после скрининга.
Оценка по шкале NAS ≥ 4.
Показатель фиброза: 1-3 включительно (F1 ограничен 30%).
Дополнительный PDFF> 10% на МРТ
Согласен на биопсию печени после 52 недель лечения.
Компенсированное заболевание печени по следующим гематологическим и биохимическим критериям при вступлении в протокол:
ALT <5 х ULN
AST> 30
Гемоглобин> 11 г/дл для женщин и> 12 г/дл для мужчин
лейкоциты (WBC)> 2,5 К/мкл
Количество нейтрофилов> 1,5 К/мкл
Тромбоциты> 100 К/мкл
Общий билирубин <35 мкмоль/л. Пациенты остроумие h билирубин> 35 мкмоль/л может быть включен, если неконъюгированный билирубин при синдроме Гилберта.
Альбумин> 36 г/л
Международный нормализованный коэффициент (МНО) <1,4
креатинин сыворотки <1,3 мг/дл (мужчины) или <1,1 (женщины) или предполагаемая скорость клубочковой фильтрации ≥ 60 мл/мин/1,73 м2
Отсутствие других причин хронического заболевания печени (аутоиммунные заболевания, первичный желчный холангит, ВГВ, ВГС, болезнь Вильсона, α-1-антитрипсиновый дефицит, гемохроматоз и т. д.)
Если применимо, стабильный диабет 2 типа (определяется как HgbA1c <9,5% и гликемия натощак <10 ммоль/л, при отсутствии изменений в приеме лекарств в предыдущие 6 месяцев и при отсутствии новых симптомов, связанных с декомпенсированным диабетом в предыдущие 3 месяца).
С момента биопсии печени стабильность веса, причем стабильным считается потеря не более 5% начальной массы тела.
Критерии исключения пациентов
Пациенты не соответствуют какому-либо из следующих критериев исключения:
История постоянного чрезмерного употребления алкоголя, оценивается с помощью анкеты потребления алкоголя.
Нестабильное метаболическое состояние, определяемое по: увеличению или снижению веса более чем на 5 кг за последние три месяца, диабету с плохим гликемическим контролем (HgbA1c>9,5%) или введению противодиабетического средства или препарата против ожирения/мальабсорбционная или рестриктивная бариатрическая (потеря веса) операция в течение 6 месяцев, предшествующие скринингу.
История желудочно-кишечной мальабсорбционной бариатрической хирургии в течение менее 5 лет или прием препаратов, вызывающих стеатоз печени, включая кортикостероиды, высокие дозы эстрогенов, метотрексат, тетрациклин или амиодарон в предыдущие 6 месяцев.
Значительные системные или другие серьезные заболевания, отличные от заболеваний печени, включая застойную сердечную недостаточность (класс C и D AHA), нестабильное заболевание коронарной артерии, цереброваскулярное заболевание, легочную недостаточность, почечную недостаточность, трансплантацию органов, серьезные психические заболевания или злокачественные новообразования, которые, по мнению исследователя, исключают лечение соединением А.
Антиген HB>0, PCR HCV>0 (пациенты с историей HCV-инфекции могут быть включены, если ПЦР HCV более 3 лет имеет отрицательный результат), или ВИЧ-инфекция.
Любое другое условие, которое, по мнению исследователя, может помешать проведению или соблюдению условий исследования или может помешать завершению исследования.
Индекс массы тела (ИМТ)> 45 кг/м2.
Диабет 1 типа или диабет 2 типа, которые лечат инсулином.
Диабетический кетоацидоз.
Триглицериды натощак >300 мг/дл.
Нарушения гемостаза или текущее лечение антикоагулянтами.
Противопоказания к биопсии печени.
наличие в анамнезе или текущие сердечные дисритмии и/или наличие в анамнезе сердечно-сосудистых заболеваний, включая инфаркт миокарда, за исключением пациентов с хорошо контролируемой гипертензией и любой клинически значимой аномалией ЭКГ
Участие в любом другом исследовании фармакологических препаратов в течение предыдущих 3 месяцев или 5 периодов полураспада указанного препарата.
Наличие гиперчувствительности к любому из ингредиентов или наполнителей IMP.
Прием противодиабетических средств, обладающих активностью против НАСГ, например, пиоглитазона и агонистов рецептора GLP-1.
Критерии конечной точки
Первичной конечной точкой эффективности соединения А является процент пациентов с излечением НАСГ, определяемым по исчезновению баллонирования (балл=0) с оценкой лобулярного воспаления «0 или 1» без ухудшения фиброза.
Вторичной конечной точкой эффективности соединения А являются изменение по сравнению с исходным значением в баллах по шкале NAS, изменения в отдельных гистологических баллах по стеатозу, баллонированию, воспалению и фиброзу, изменения в ферментах печени, изменения в параметрах визуализации и изменения в биомаркерах (включая hsCRP, Pro-C3, ELF панель и цитокины.
Вторичные конечные точки для оценки безопасности/переносимости включают в себя сообщаемые побочные эффекты, лабораторные показатели (гематология, биохимия и анализ мочи), показатели жизненно важных функций (артериальное давление, частота пульса и температура) и hsCRP.
Вторичные конечные точки для фармакокинетики включают сравнение средних минимальных концентраций в плазме в устойчивом состоянии для трех доз Соединения А, взятых при посещениях 4, 5, 6, 7 и 8.
Статистические методы
Размер выборки из 88 пациентов на группу обеспечивает 80% -ную вероятность для выявления 40%-ой доли респондеров на активное соединение vs. плацебо, при условии, что коэффициент ответа на плацебо составляет 18%, а коэффициент отсева составляет 25%. После 16-недельного промежуточного анализа может быть сделана переоценка размера выборки. Анализ эффективности проводится по измененному (modified ITT-concept) ITT-концепту (intention to treat population). Популяция состоит из пациентов с измеренным базовым уровнем и, по крайней мере, с одним измерением эффективности после базового уровня. Сравнения делаются между каждой отдельной дозой и плацебо.
2Гематология, биохимия, коагуляция, анализ мочи, липиды;
3hsCRP, Pro-c3, ELF панель, HOMA-IR, цитокины;
* период обследования до 6 недель может включать более одного фактического визита, если планируется биопсия.
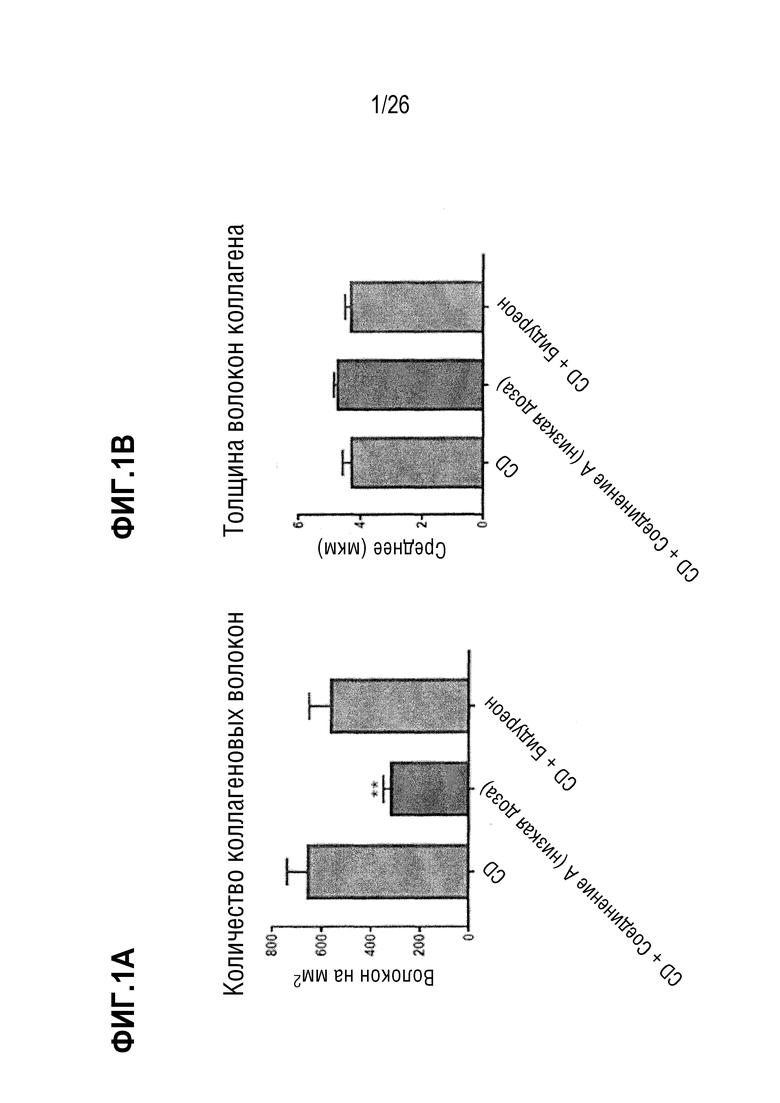


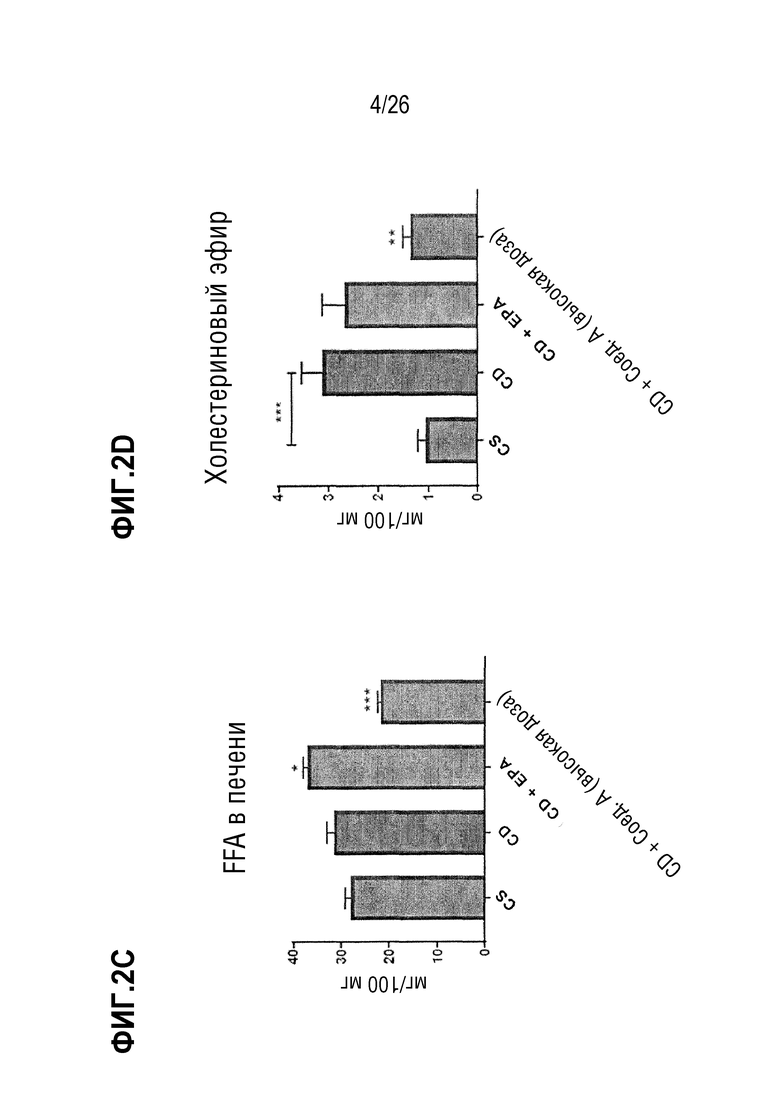

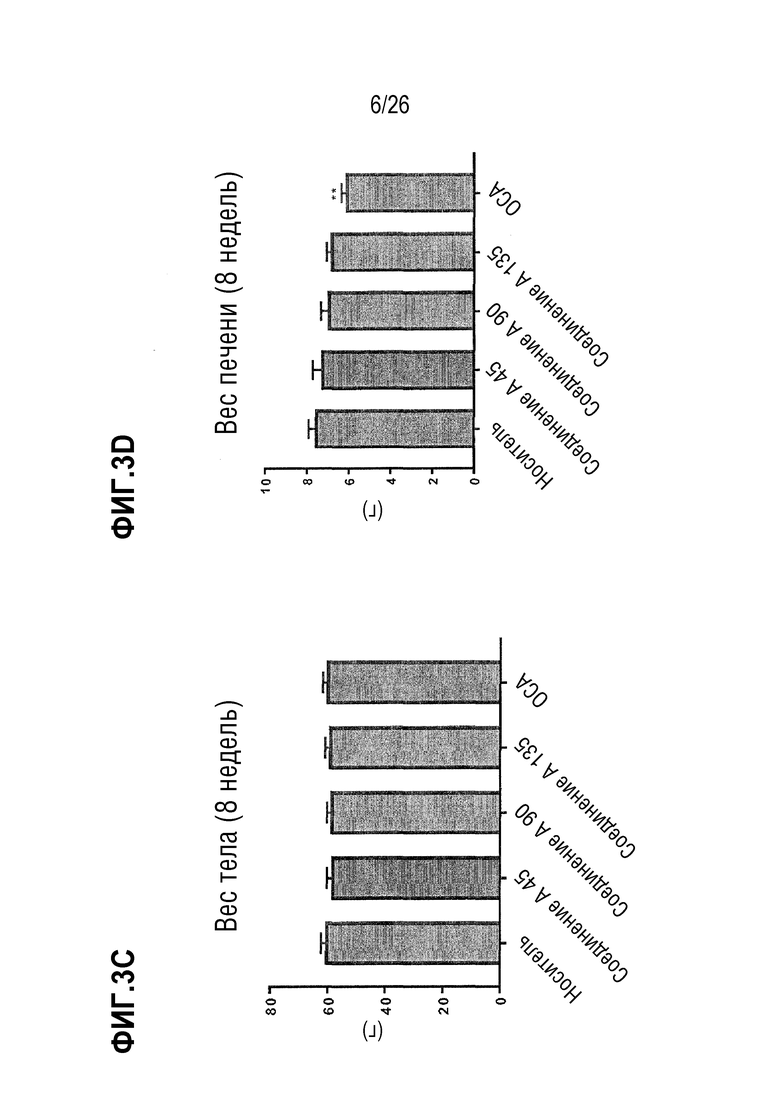
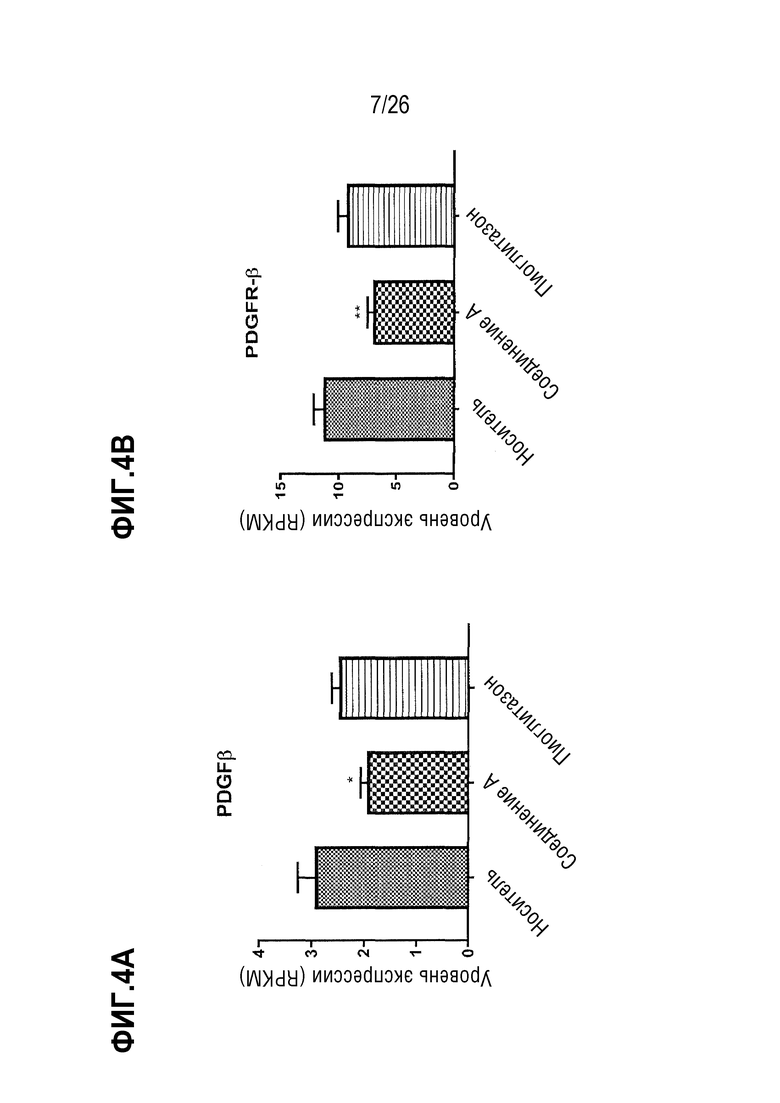

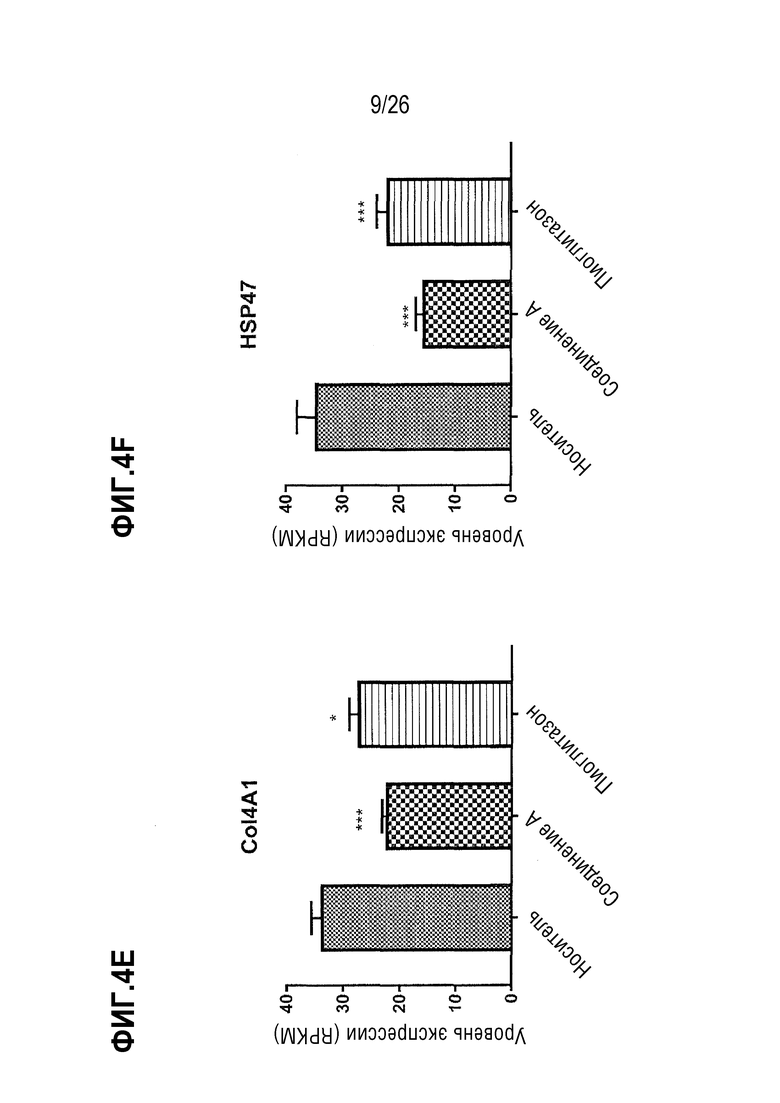

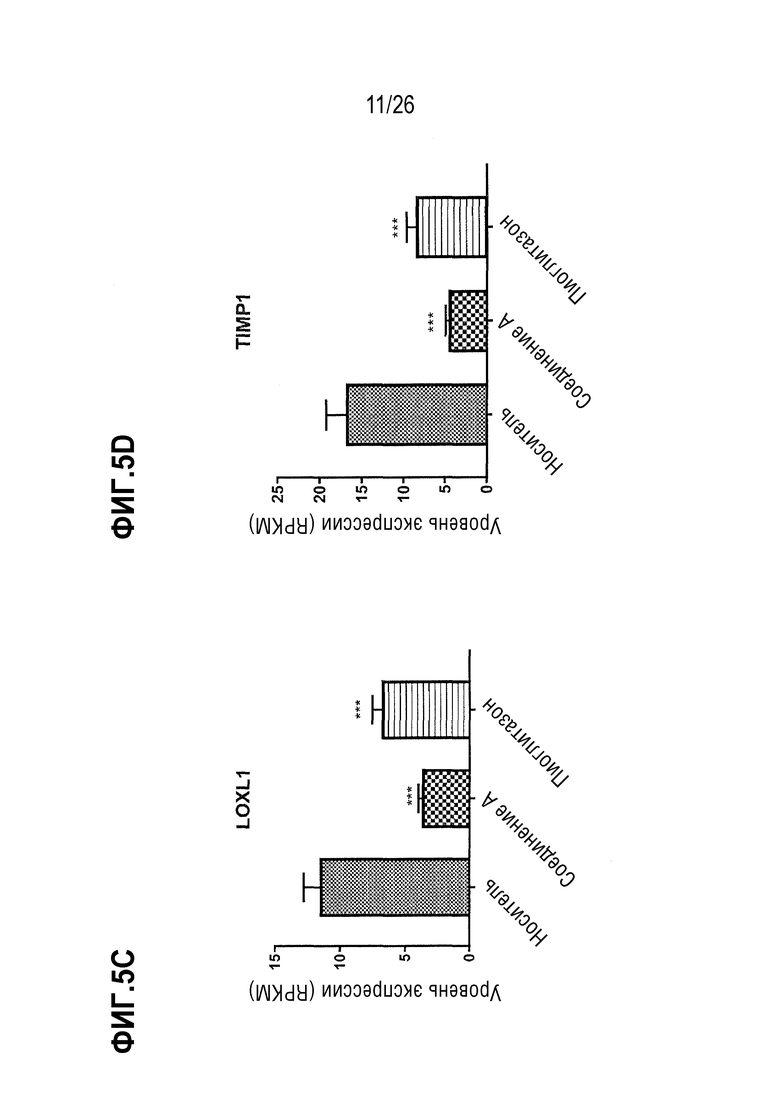
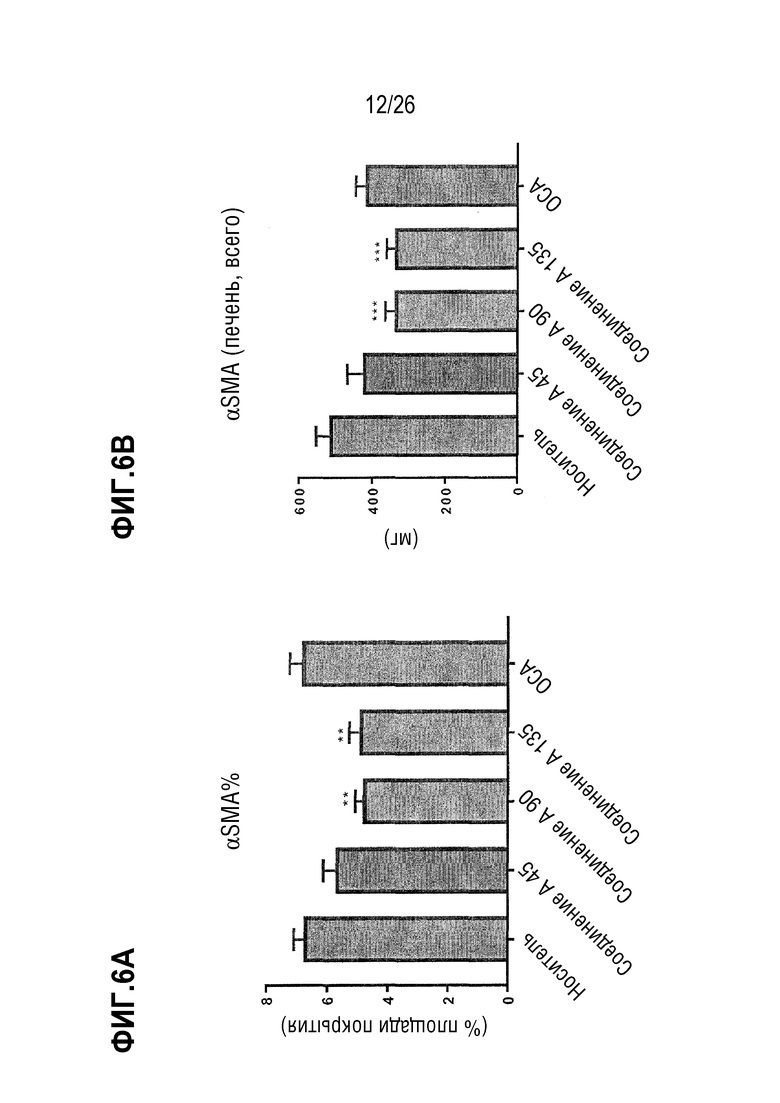
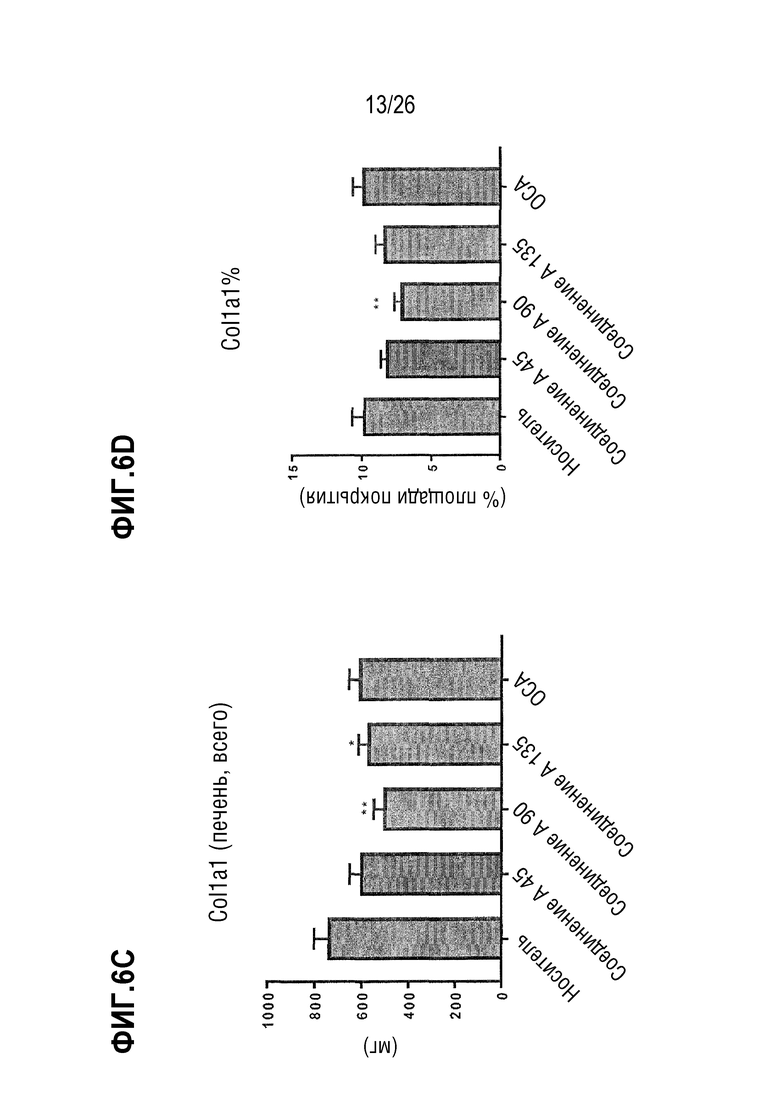
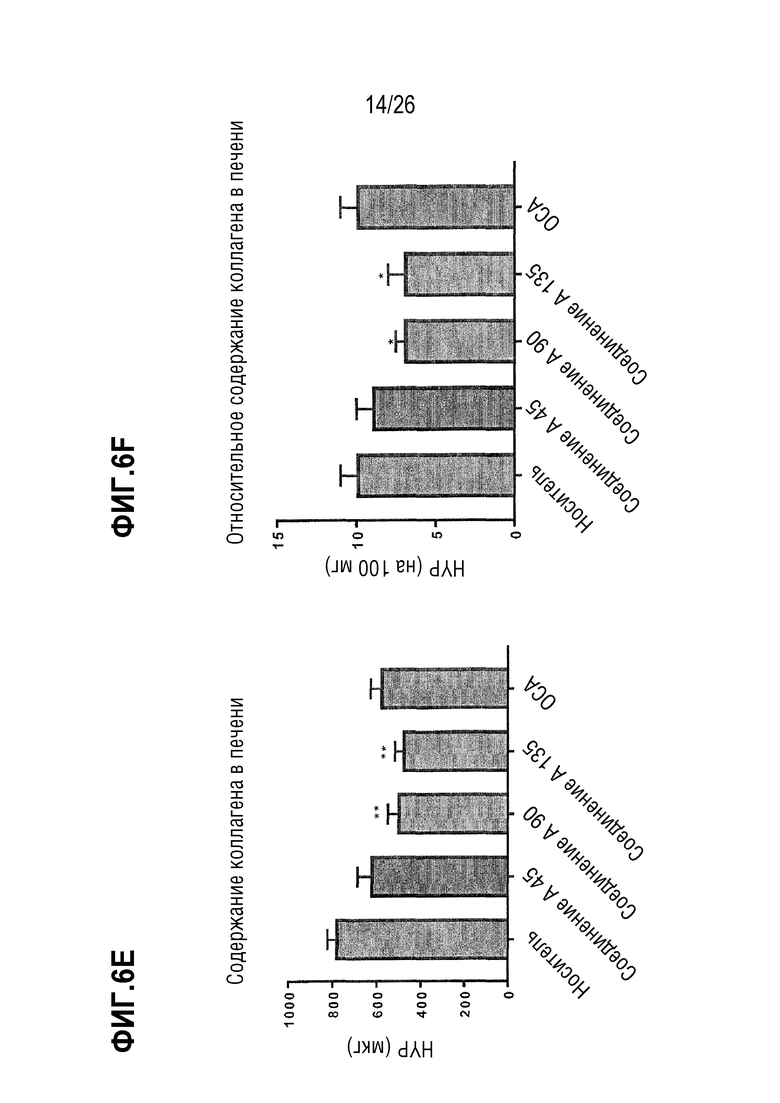
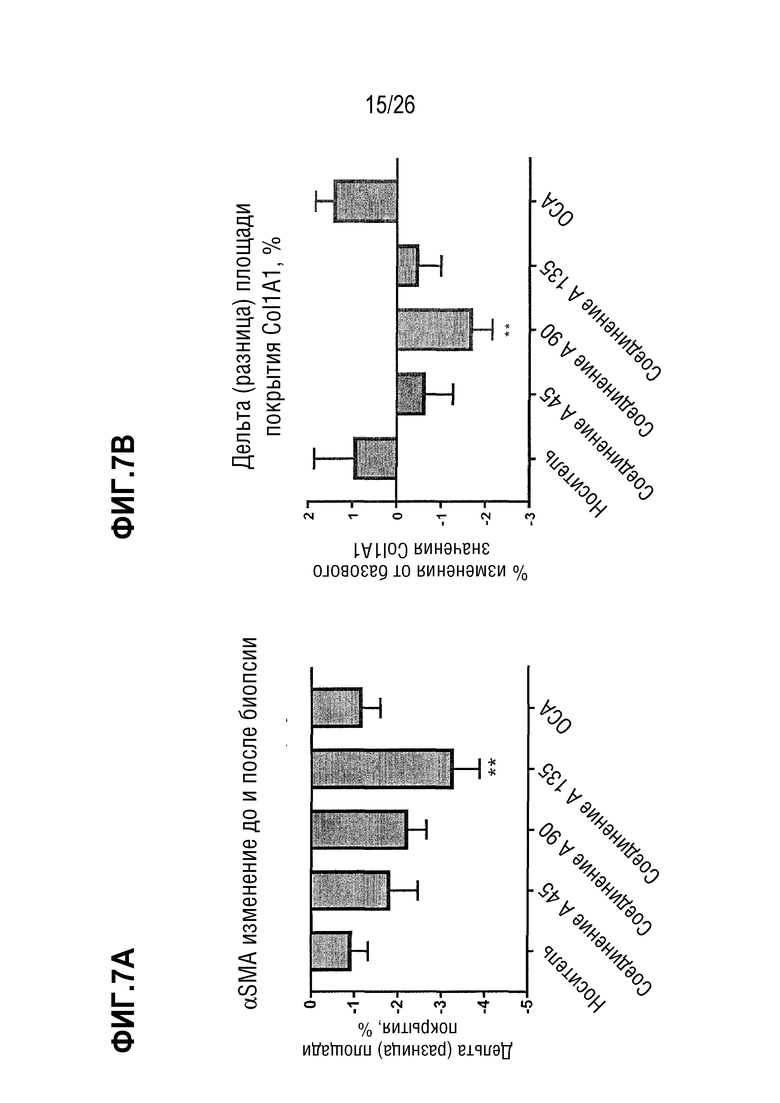
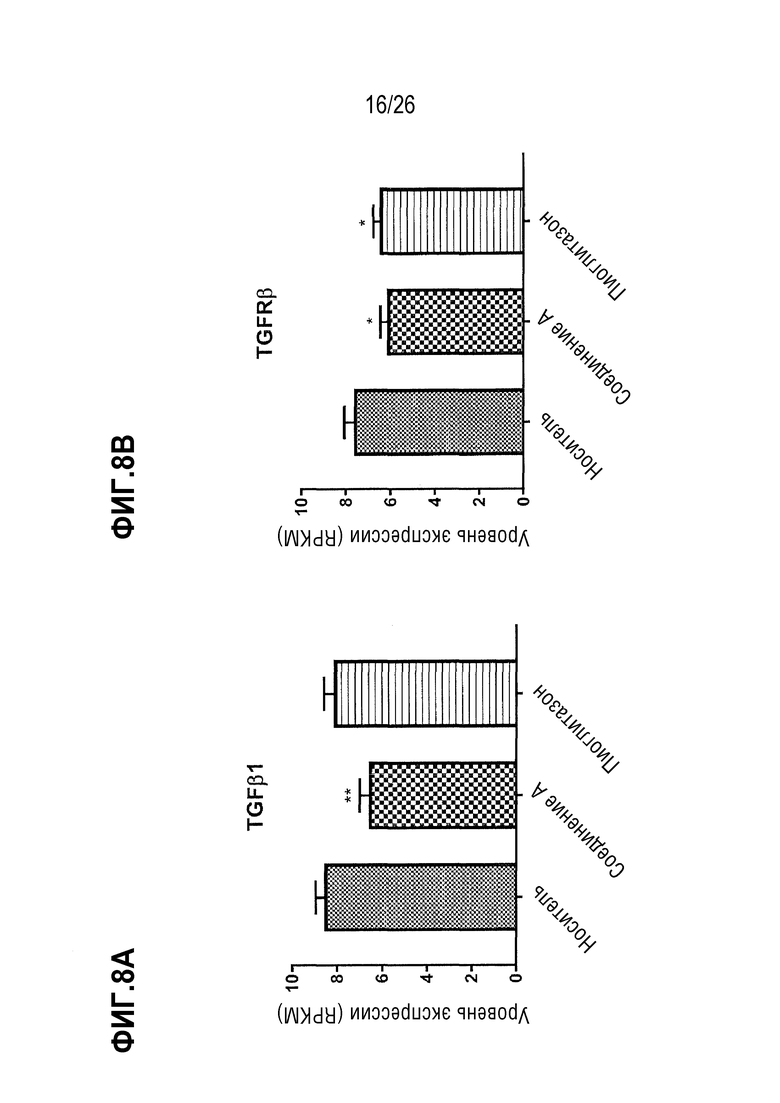
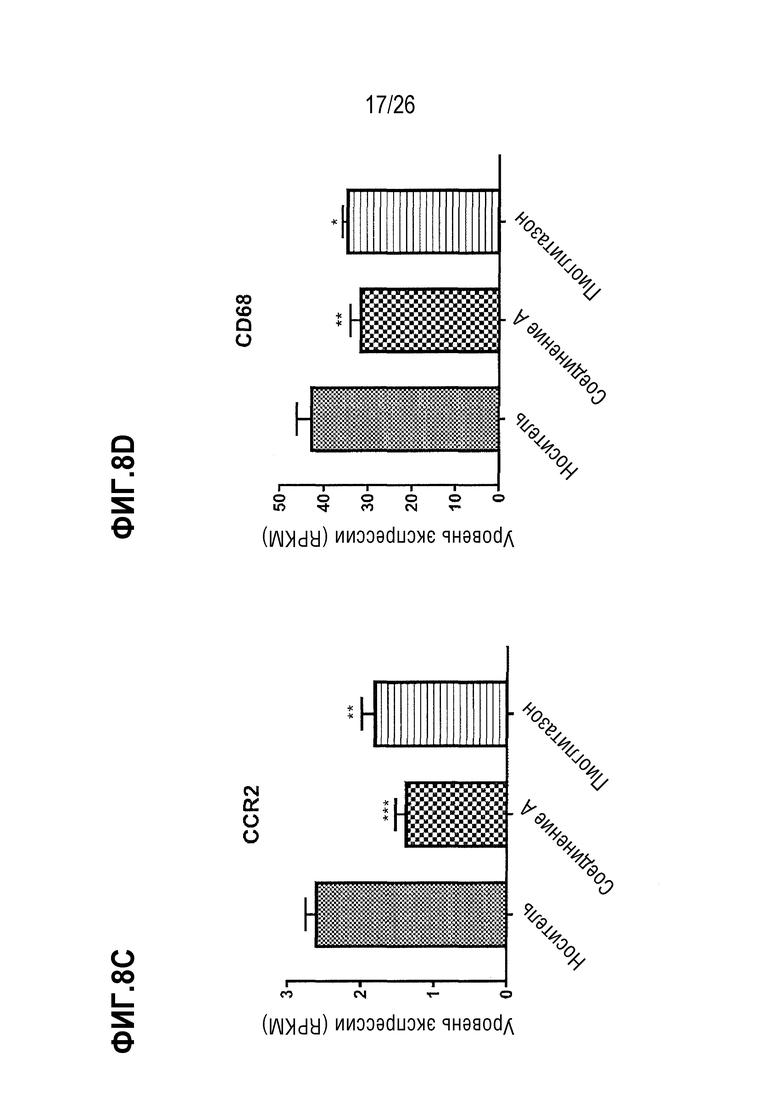
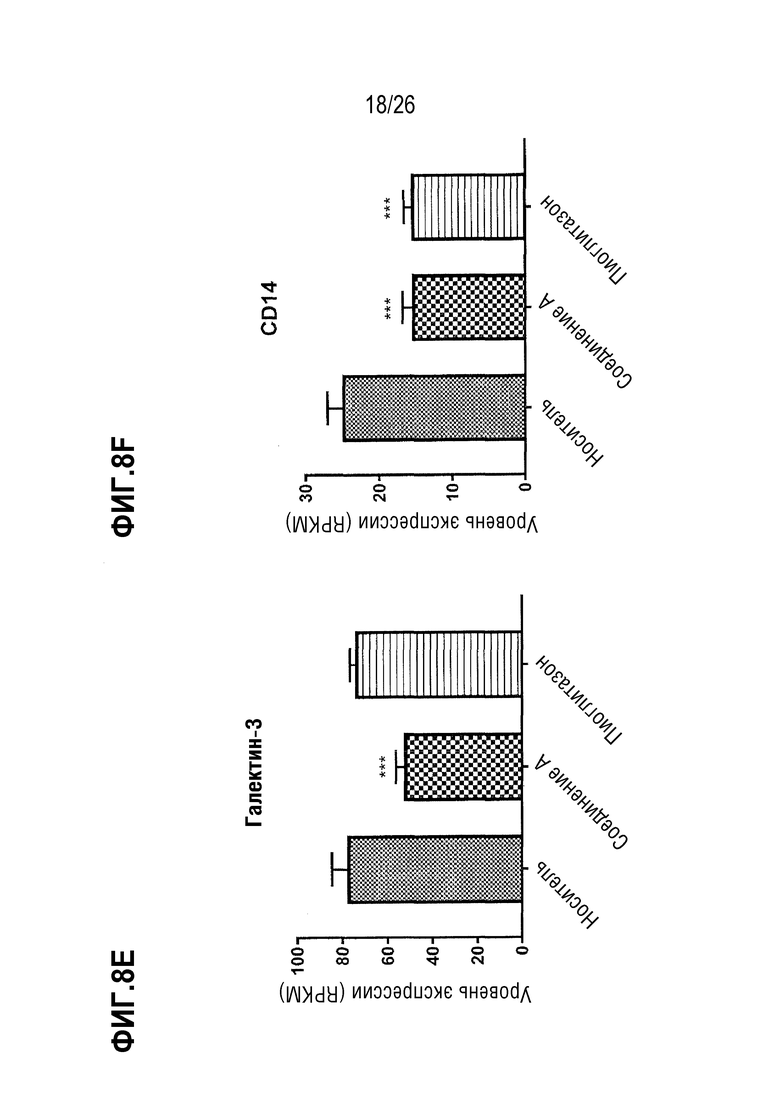
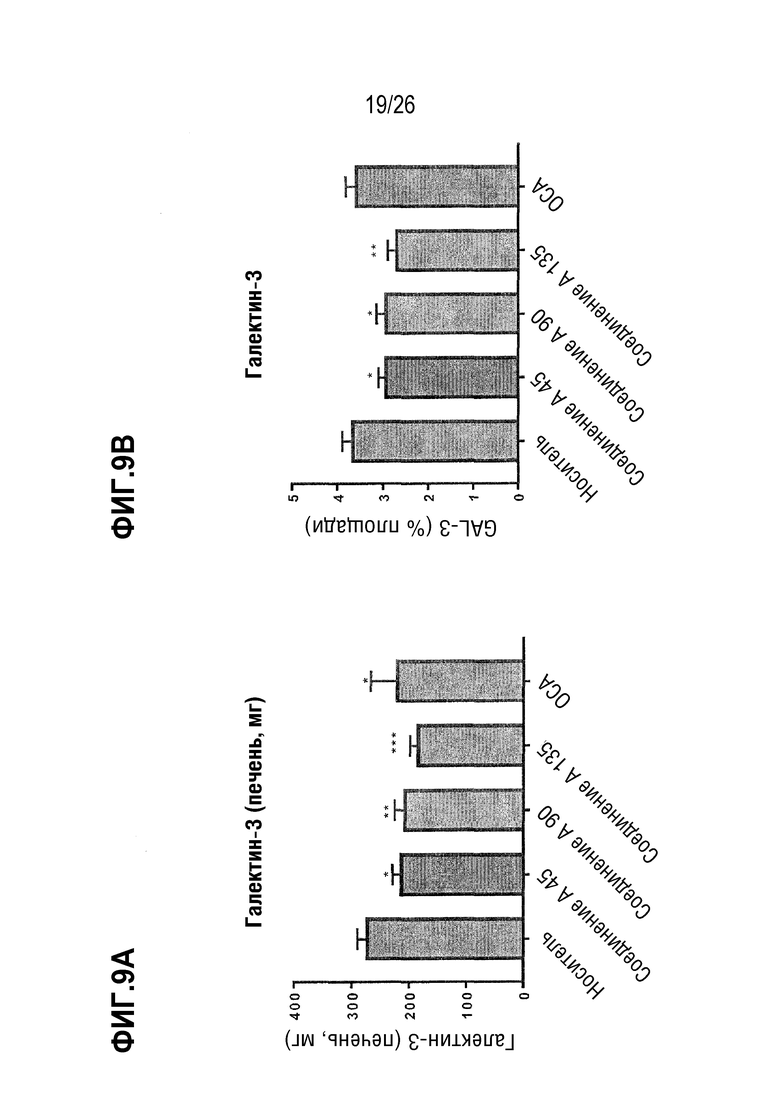
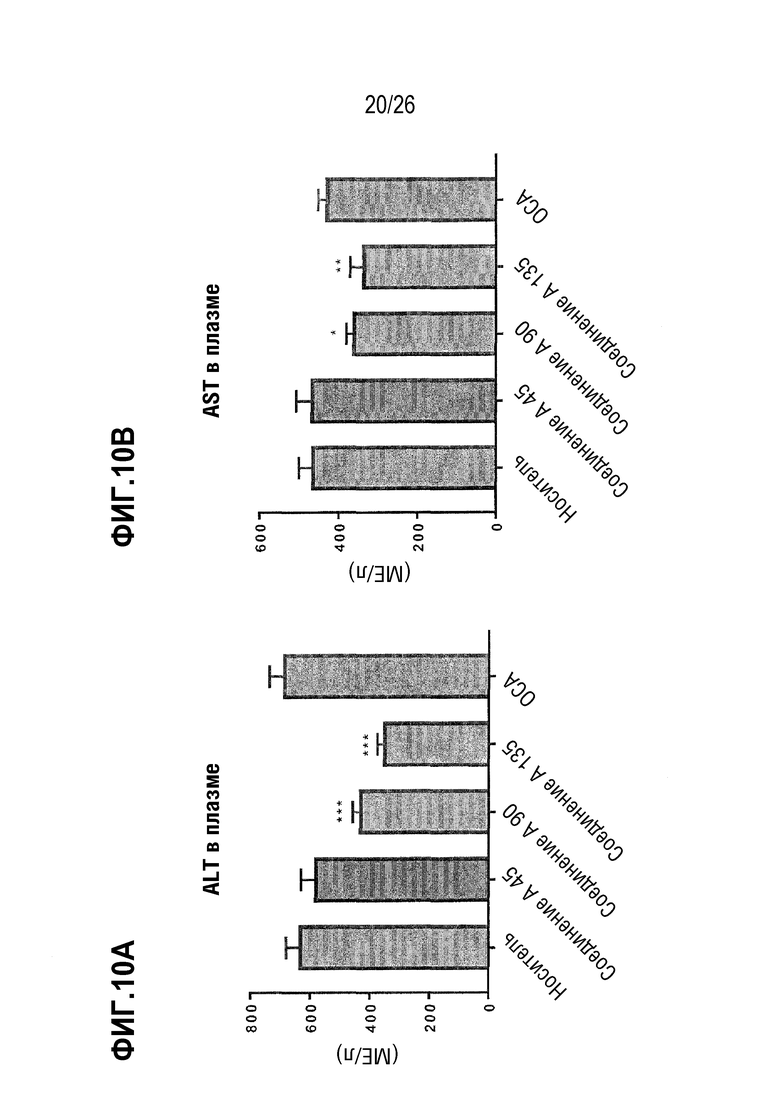
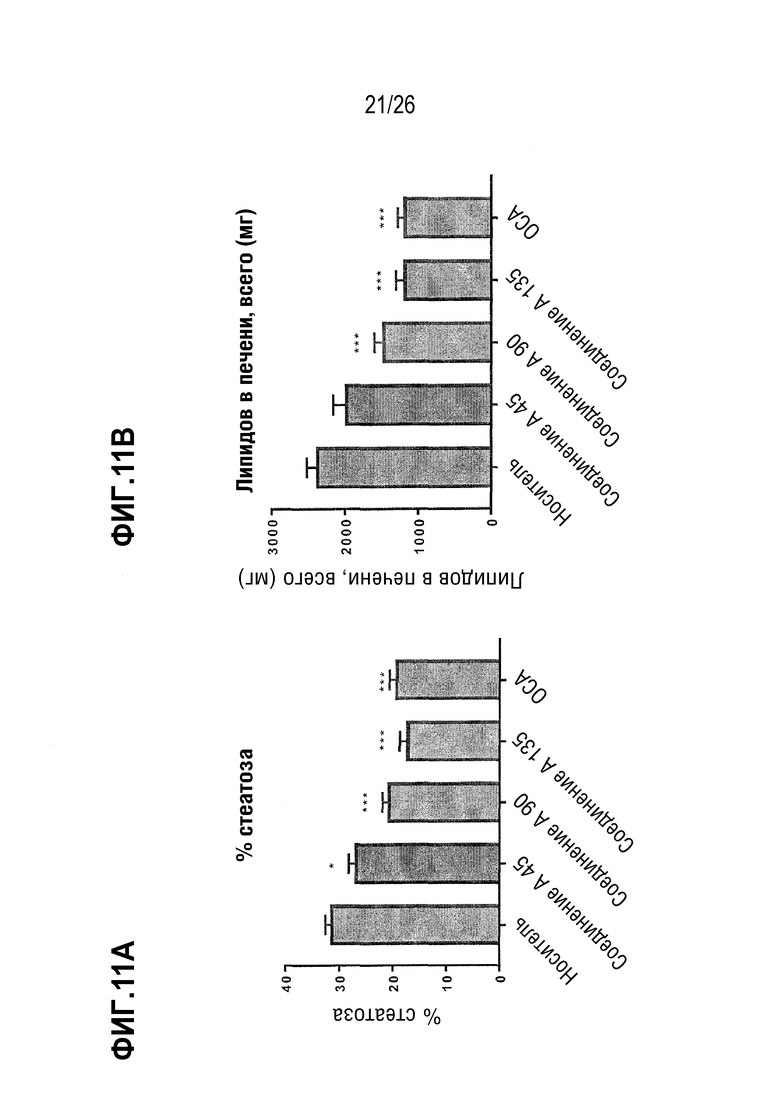
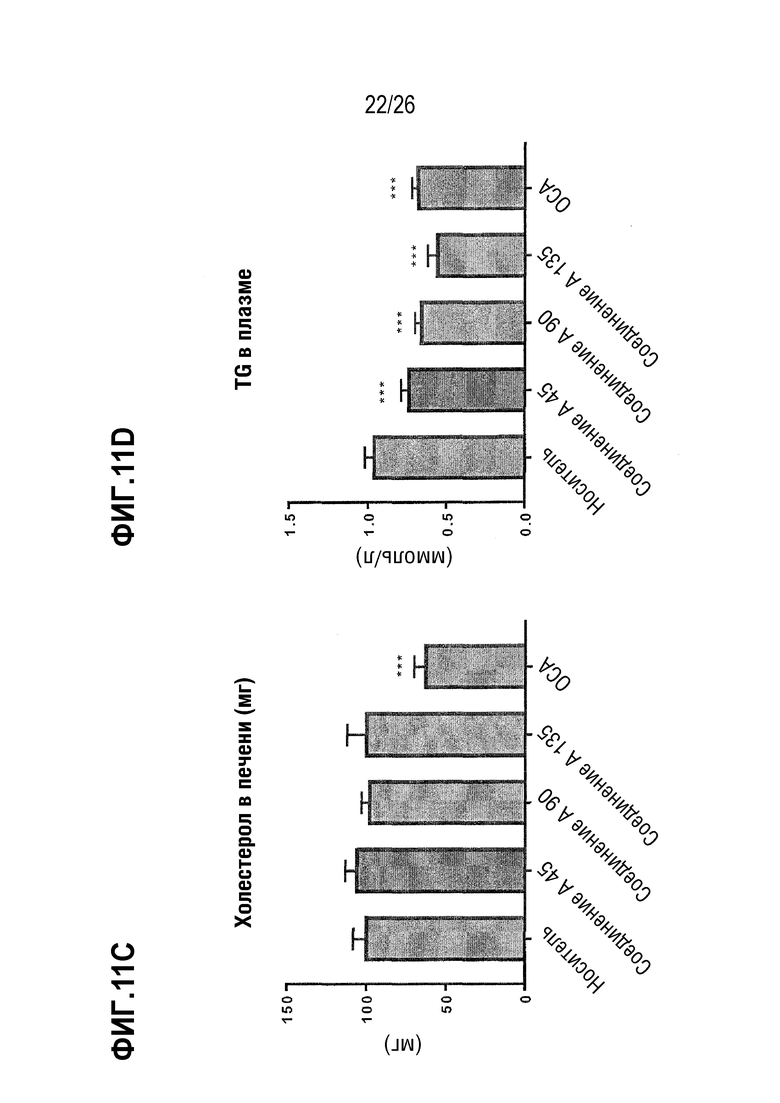
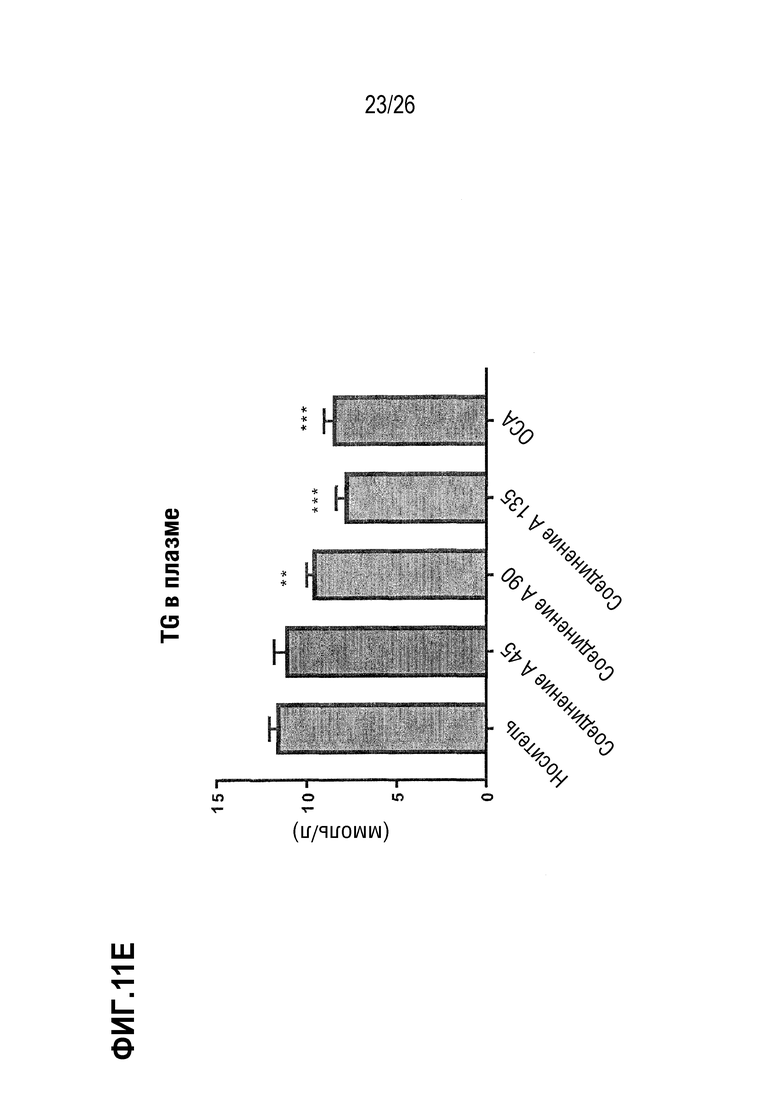
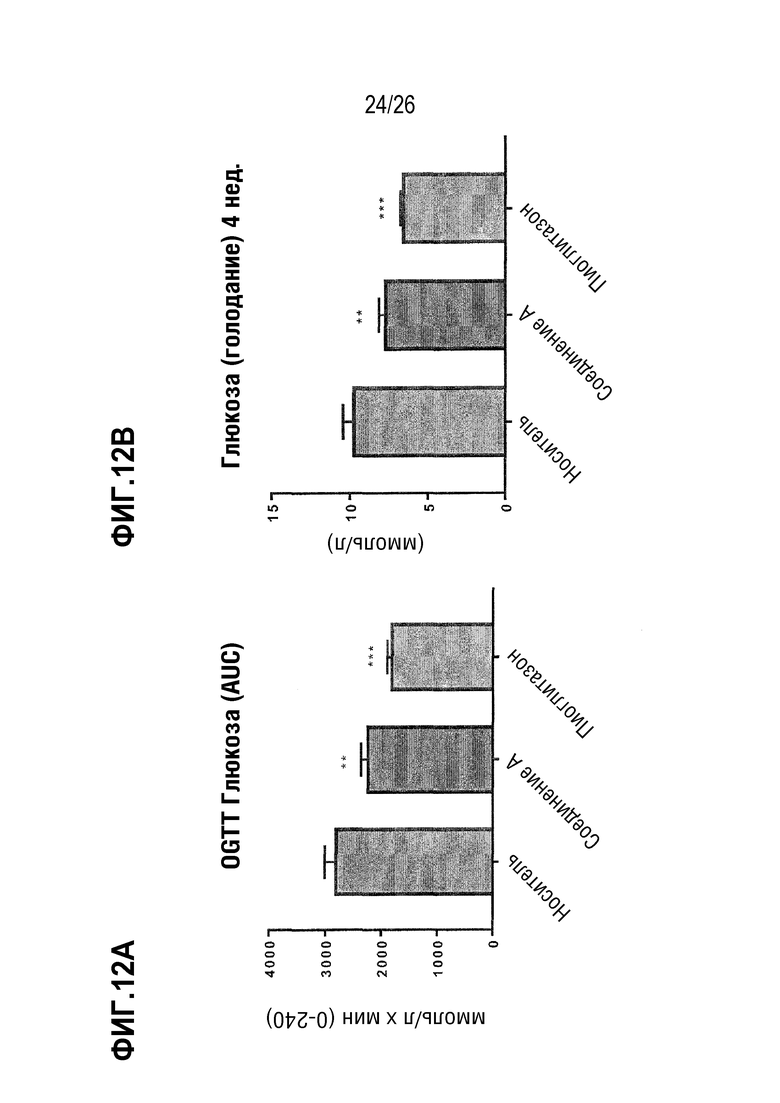
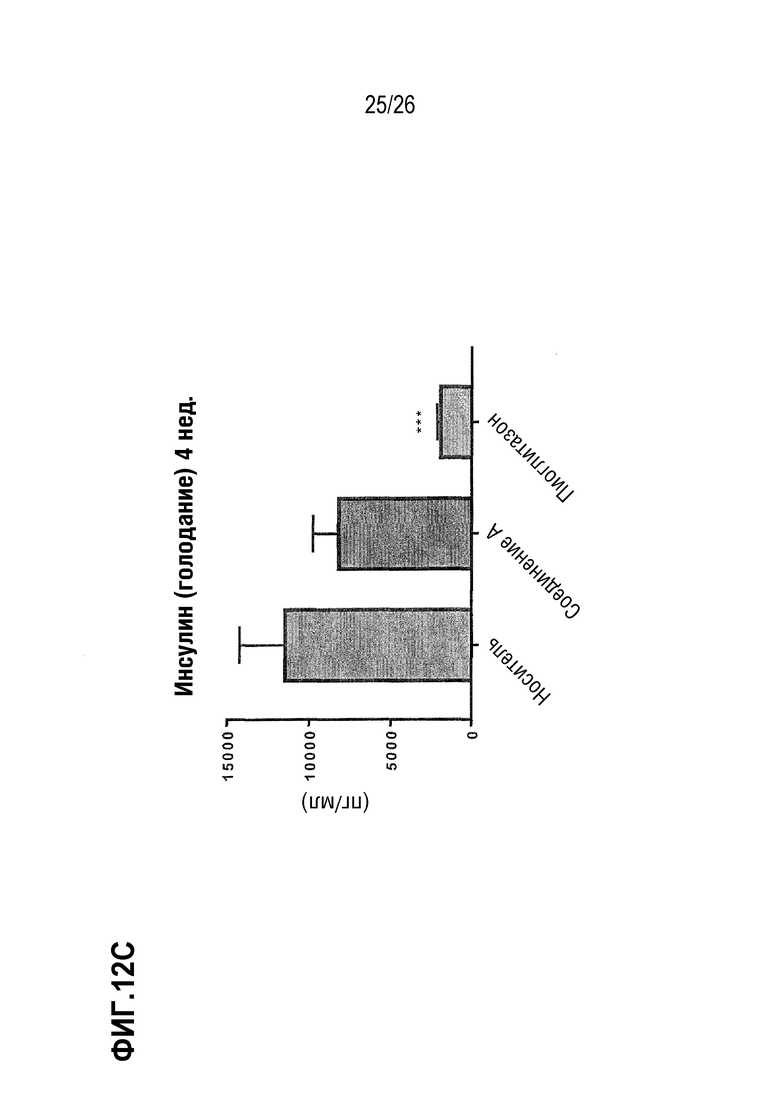
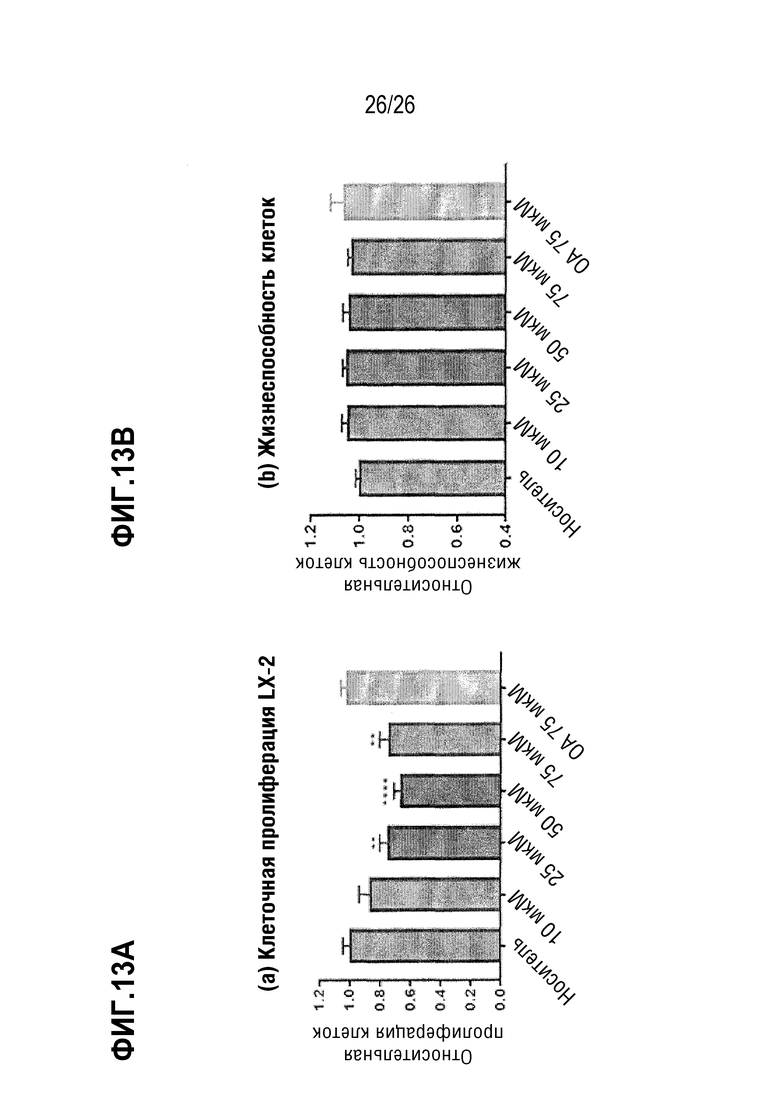
Настоящая группа изобретений относится к области фармакологии, конкретно к применению соединения формулы (II) и его композиции для получения лекарственного средства и для лечения неалкогольного стеатогепатита (НАСГ) или алкогольного стеатогепатита (АСГ), а также к способу замедления фиброза печени. Применение ненасыщенной жирной кислоты с кислородом, включенным в β-положение, и дополнительно содержащую α-заместитель со структурой соединения формулы (II), где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей; R2 и R3 являются одинаковыми или различными и выбраны из группы, включающей атом водорода и алкильную группу; Х представляет собой карбоновую кислоту или производное таковой, где производное представляет собой карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид; или фармацевтически приемлемая соль, осуществляют в терапевтическом и/или профилактическом лечении НАСГ или АСГ. Применение соединения формулы (II) замедляет или профилактически предотвращает развитие фиброза печени или уменьшает уже имеющийся фиброз печени. Техническим результатом изобретения является обеспечение способом лечения НАСГ или АСГ субъекта, который в этом нуждается. 5 н. и 83 з.п. ф-лы, 26 ил., 13 табл., 105 пр.
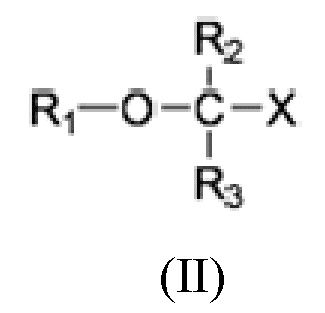
1. Применение соединения Формулы (II)
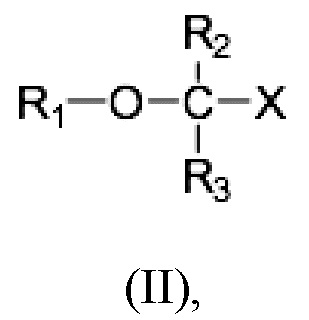
где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей;
R2 и R3 являются одинаковыми или различными и выбраны из группы, включающей атом водорода и алкильную группу;
Х представляет собой карбоновую кислоту или производное таковой, где производное представляет собой карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид;
или его фармацевтически приемлемой соли,
в терапевтическом и/или профилактическом лечении неалкогольного стеатогепатита или
алкогольного стеатогепатита,
где применение соединения замедляет или профилактически предотвращает развитие фиброза печени или уменьшает уже имеющийся фиброз печени.
2. Применение по п. 1, где соединение имеет Формулу (I), и R2, R3 и X имеют значения, определенные для Формулы (II)
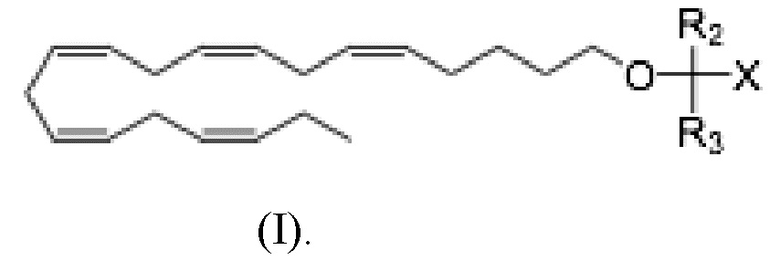
3. Применение по п. 1 или 2,
где R2 и R3 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп;
Х представляет собой карбоновую кислоту или производное таковой, где производное представляет собой карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид;
или фармацевтически приемлемой соли такового,
для использования при лечении неалкогольного стеатогепатита.
4. Применение по любому из пп. 1-3, где R2 и R3 независимо выбраны из атома водорода, метильной группы, этильной группы, n-пропильной группы и изопропильной группы.
5. Применение по любому из пп.1-3, где оба R2 и R3 независимо представляют собой C1-C6 алкильные группы.
6. Применение по любому из пп.1-4, где один из R2 и R3 представляет собой атом водорода, а другой представляет собой этильную группу.
7. Применение по любому из пп. 1-6, где Х представляет собой карбоновую кислоту.
8. Применение по любому из пп. 1-6, где Х представляет собой C1-C6 алкиловый эфир.
9. Применение по п. 8, где Х выбран из метилового эфира, этилового эфира, изопропилового эфира, n-бутилового эфира и трет-бутилового эфира.
10. Применение по любому из пп. 1, 8 и 9, где Х выбран из группы, включающей метиловый эфир и этиловый эфир.
11. Применение по любому из пп. 1-6, где Х представляет собой глицерид, выбранный из триглицерида, 1,2-диглицерида, 1,3-диглицерида, 1-моноглицерида и 2-моноглицерида.
12. Применение по любому из пп. 1-11, где соединение присутствует в форме энантиомера, диастереомера или смеси таковых.
13. Применение по п. 12, где соединение присутствует в форме R, в форме S или в рацемической форме.
14. Применение по п. 1 или 2, где соединение представляет собой 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (Соединение А) или фармацевтически приемлемую соль или сложный эфир таковой, и формула представляет собой
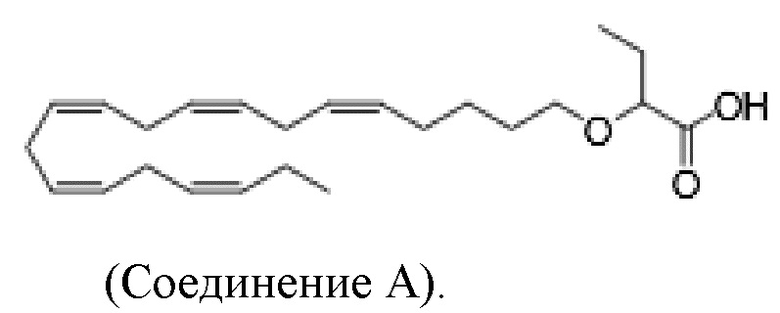
15. Применение по любому из пп. 1-14, где указанное соединение вводят в дозе в диапазоне от примерно 5 мг до примерно 4 г на дозу.
16. Применение по любому из пп. 1-15, где указанное соединение вводят один раз в день.
17. Применение по любому из пп. 1-16, где применение приводит к излечению или вызывает уменьшение неалкогольного стеатогепатита (НАСГ).
18. Применение по любому из пп. 1-17, где применение приводит к: уменьшению воспаления печени, такому как лобулярное воспаление; уменьшение гепатоцеллюлярного баллонирования; и/или снижение стеатогепатита.
19. Применение фармацевтической композиции, содержащей соединение Формулы (II) или его фармацевтически приемлемой соли
где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей;
R2 и R3 являются одинаковыми или различными и выбраны из группы заместителей, состоящей из атома водорода и алкильной группы;
Х представляет собой карбоновую кислоту или производное таковой, где производное представляет собой карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид,
в терапевтическом и/или профилактическом лечении неалкогольного стеатогепатита и/или алкогольного стеатогепатита,
где применение замедляет или профилактически предотвращает развитие фиброза печени или уменьшает уже имеющийся фиброз печени.
20. Применение по п. 19, где соединение имеет Формулу (I)
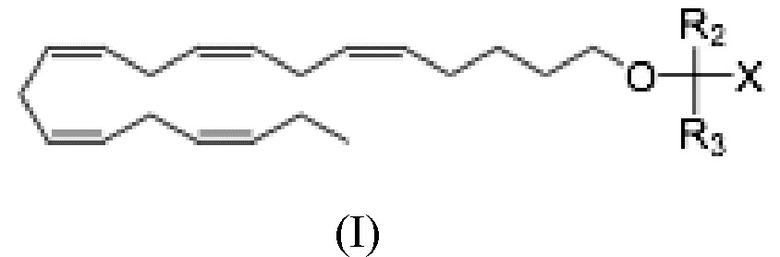
и где R2 и R3 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп;
Х представляет собой карбоновую кислоту или производное таковой, где производным является карбоксилат, такой как сложный эфир карбоновой кислоты, глицерид или карбоксамид;
или фармацевтически приемлемая соль такового.
21. Применение по п. 19 или 20, где указанная фармацевтическая композиция составлена для перорального введения.
22. Применение по любому из пп. 19-21, где фармацевтическая композиция дополнительно содержит по меньшей мере одно связующее вещество, эксципиент, разбавитель или антиоксидант или любые комбинации таковых.
23. Применение по любому из пп. 19-22, где соединение представляет собой 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (соединение А), или ее фармацевтически приемлемую соль или эфир, и представлено формулой
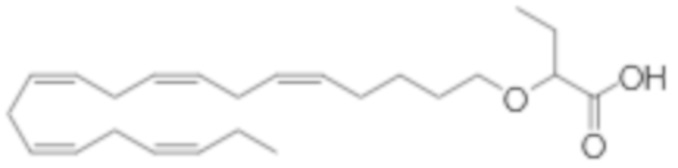 .
.
24. Применение по любому из пп. 1-16 и 18, где применение имеет целью профилактическое лечение неалкогольного стеатогепатита (НАСГ) и/или алкогольного стеатогепатита.
25. Способ замедления фиброза печени у субъекта, нуждающегося в лечении, включающий введение субъекту фармацевтически эффективного количества соединения Формулы (II):
где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей;
R2 и R3 являются одинаковыми или различными и выбраны из группы, включающей атом водорода и алкильную группу;
Х представляет собой карбоновую кислоту или производное таковой, где производное представляет собой карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид;
или фармацевтически приемлемой соли такового,
где субъект имеет неалкогольный стеатогепатит или алкогольный стеатогепатит.
26. Способ по п. 25, в котором соединение имеет Формулу (I):
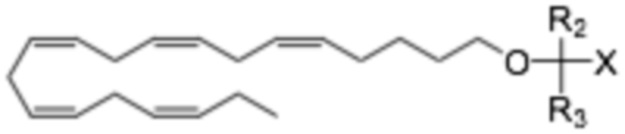 .
.
27. Способ по п. 25 или 26,
где R2 и R3 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп; а также
Х представляет собой карбоновую кислоту или производное таковой; где производное представляет собой карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид;
или фармацевтически приемлемой соли такового.
28. Способ по любому из пп. 25-27, в котором R2 и R3 независимо выбраны из атома водорода, метильной группы, этильной группы, n-пропильной группы и изопропильной группы.
29. Способ по любому из пп. 25-27, в котором R2 и R3 оба независимо представляют собой C1-C6 алкильные группы.
30. Способ по любому из пп. 25-28, в котором один из R2 и R3 представляет собой атом водорода, а другой представляет собой этильную группу.
31. Способ по любому из пп. 25-30, в котором Х представляет собой карбоновую кислоту.
32. Способ по любому из пп. 25-30, в котором Х представляет собой C1-C6-алкиловый эфир.
33. Способ по п. 32, в котором Х выбран из метилового эфира, этилового эфира, изопропилового эфира, n-бутилового эфира и трет-бутилового эфира.
34. Способ по любому из пп. 25, 32 или 33, в котором Х выбран из метилового эфира и этилового эфира.
35. Способ по любому из пп. 25-30, в котором Х представляет собой глицерид, выбранный из триглицерида, 1,2-диглицерида, 1,3-диглицерида, 1-моноглицерида и 2-моноглицерида.
36. Способ по любому из пп. 25-35, в котором соединение присутствует в форме энантиомера, диастереомера или смеси таковых.
37. Способ по п. 36, в котором соединение присутствует в форме R, в форме S или в рацемической форме.
38. Способ по п. 25, в котором соединение представляет собой 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (Соединение А) или фармацевтически приемлемую соль или сложный эфир такового, а формула представляет собой
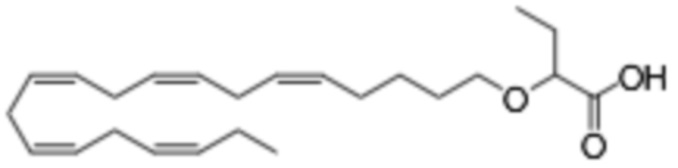 .
.
39. Способ по любому из пп. 25-38, где указанное соединение вводят в дозе в диапазоне от примерно 5 мг до примерно 4 г на дозу.
40. Способ по любому из пп. 25-39, в котором соединение вводят один раз в день.
41. Способ по любому из пп. 25-40, в котором способ излечивает или вызывает регрессию неалкогольного стеатогепатита и/или алкогольного стеатогепатита.
42. Способ по любому из пп. 25-41, где способ включает профилактическое лечение неалкогольного стеатогепатита и/или алкогольного стеатогепатита.
43. Способ по любому из пп. 25-42, в котором способ замедляет или профилактически предотвращает развитие фиброза печени или уменьшает уже существующий фиброз печени.
44. Способ по любому из пп. 25-43, в котором способ обеспечивает уменьшение воспаления печени, гепатоцеллюлярного баллонирования, стеатогепатита, фиброза и/или биомаркеров воспаления и/или фиброза, например hsCRP, Pro-C3, панель ELF; и/или улучшение показателей по шкале оценки NAS на основе биопсии печени, теста функции печени, МРТ-анализа LiverMultiScan PDFF и cT1 и/или HOMA-IR.
45. Способ по п. 44, в котором уменьшение воспаления печени представляет собой уменьшение лобулярного воспаления.
46. Способ по любому из пп. 25-45, в котором соединение сформулировано в виде фармацевтической композиции.
47. Способ по п. 46, где фармацевтическая композиция составлена для перорального введения.
48. Способ по п. 46 или 47, в котором фармацевтическая композиция дополнительно содержит, по меньшей мере, одно связующее вещество, эксципиент, разбавитель или антиоксидант или любую комбинацию таковых.
49. Способ по любому из пп. 46-48, где соединение представляет собой 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (соединение А), или его фармацевтически приемлемую соль или эфир, и представлено формулой
.
50. Способ по любому из пп. 25-49, в котором способ лечения является профилактическим.
51. Применение соединения Формулы (II)
где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей;
R2 и R3 являются одинаковыми или различными и выбраны из группы, включающей атом водорода и алкильную группу;
Х представляет собой карбоновую кислоту или производное таковой; где производное представляет собой карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид;
или фармацевтически приемлемой соли такового,
в получении лекарственного средства для терапевтического и/или профилактического лечения алкогольного стеатогепатита или неалкогольного стеатогепатита,
где лекарственное средство замедляет или профилактически предотвращает развитие фиброза печени или уменьшает уже имеющийся фиброз печени.
52. Применение по п. 51, где соединение имеет Формулу (I), а R2, R3 и X имеют значения, определенные для Формулы (II)
53. Применение по п. 51 или 52,
где R2 и R3 независимо выбраны из атома водорода или линейных, разветвленных и/или циклических C1-C6 алкильных групп;
Х представляет собой карбоновую кислоту или производное таковой; где производное представляет собой карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид;
или фармацевтически приемлемой соли такового,
для использования в профилактике и/или лечении алкогольного стеатогепатита.
54. Применение по любому из пп. 51-53, где R2 и R3 независимо выбраны из атома водорода, метильной группы, этильной группы, n-пропильной группы и изопропильной группы.
55. Применение по любому из пп. 51-53, где R2 и R3 оба независимо представляют собой C1-C6 алкильные группы.
56. Применение по любому из пп. 51-54, где один из R2 и R3 представляет собой атом водорода, а другой представляет собой этильную группу.
57. Применение по любому из пп. 51-56, где Х представляет собой карбоновую кислоту.
58. Применение по любому из пп. 51-56, где Х представляет собой C1-C6 алкиловый эфир.
59. Применение по п. 58, где Х выбран из метилового эфира, этилового эфира, изопропилового эфира, n-бутилового эфира и трет-бутилового эфира.
60. Применение по любому из пп. 51-56, 58 и 59, где Х выбран из группы, включающей метиловый эфир и этиловый эфир.
61. Применение по любому из пп. 51-56, где Х представляет собой глицерид, выбранный из триглицерида, 1,2-диглицерида, 1,3-диглицерида, 1-моноглицерида и 2-моноглицерида.
62. Применение по любому из пп. 51-61, где соединение присутствует в форме энантиомера, диастереомера или смеси таковых.
63. Применение по п. 62, где соединение присутствует в форме R, в форме S или в рацемической форме.
64. Применение по п. 51 или 52, где соединение представляет собой 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (Соединение А) или фармацевтически приемлемую соль или сложный эфир такового, с формулой
65. Применение по любому из пп. 51-64, где указанное соединение вводят в дозе в диапазоне от примерно 5 мг до примерно 4 г на дозу.
66. Применение по любому из пп. 51-65, где указанное соединение вводят один раз в день.
67. Применение по любому из пп. 51-66, применение которого лечит или вызывает регрессию неалкогольного стеатогепатита.
68. Применение по любому из пп. 51-67, которое обеспечивает: уменьшение воспаления печени, такого как лобулярное воспаление; уменьшение гепатоцеллюлярного баллонирования; и/или уменьшение стеатогепатита.
69. Применение и способ по любому из пп. 1-50, где соединение вводят в дозе 600 мг в день.
70. Применение и способ по любому из пп. 1-50, где соединение вводят в дозе 300 мг в день.
71. Применение и способ по любому из пп. 1-50, 69 и 70, в которых уровни аланинаминотрансферазы в плазме снижаются на 30-40%.
72. Применение и способ по любому из пп. 1-50 и 69-71, в которых уровни аспартаттрансаминазы в плазме снижаются на 10-20%.
73. Применение и способ по любому из пп. 1-50 и 69-72, при которых стеатоз печени уменьшается на 70-90%.
74. Применение и способ по любому из пп. 1-50 и 69-73, в которых показатели шкалы NAS снижаются на 50-70%.
75. Применение и способ по любому из пп. 1-50 и 69-74, в которых фиброзная область в печени уменьшается на 40-60%.
76. Способ по любому из пп. 25-50, в котором соединение вводят в виде монотерапии.
77. Способ по любому из пп. 25-50, дополнительно включающий совместное введение по меньшей мере одного дополнительного активного агента, выбранного из группы, включающей агониста глюкагоноподобного пептида 1 (GLP-1); ингибитора дипептидилпептидазы (DPP IV) и омега-3 жирных кислот (n-3 PUFA).
78. Способ по п. 77, в котором совместное введение осуществляется одновременным введением, последовательным введением, совместным введением, интервальным введением, непрерывным введением или комбинацией таковых.
79. Применение по любому из пп. 1-18, где соединение вводят в виде монотерапии.
80. Применение по любому из пп. 1-18, где соединение вводят по меньшей мере с одним дополнительным активным агентом, выбранного из группы, включающей агониста глюкагоноподобного пептида 1 (GLP-1); ингибитора дипептидилпептидазы (DPP IV) и омега-3 жирных кислот (n-3 PUFA).
81. Применение по п. 80, где совместное введение осуществляется путем одновременного введения, последовательного введения, совмещенного введения, интервального введения, непрерывного введения или комбинацией таковых.
82. Применение соединения Формулы (II)
где R1 выбран из C10-C22 алкенила, имеющего 3-6 двойных связей;
R2 и R3 являются одинаковыми или различными и выбраны из группы заместителей, состоящей из атома водорода, или алкильной группы;
Х представляет собой карбоновую кислоту или производное таковой, где производным является карбоксилат, такой как сложный эфир карбоновой кислоты; глицерид; или карбоксамид;
или фармацевтически приемлемой соли такового,
в терапевтическом и/или профилактическом лечении неалкогольного стеатогепатита или алкогольного стеатогепатита,
где указанное соединение вводят совместно с одним или несколькими дополнительными активными агентами, выбранными из группы, включающей агониста глюкагоноподобного пептида 1 (GLP-1); ингибитора дипептидилпептидазы (DPP IV) и омега-3 жирных кислот (n-3 PUFA).
83. Применение по п. 82, где соединение имеет Формулу (I), и R2, R3 и X имеют значения, определенные для Формулы (II)
84. Применение по п. 82 или 83, где R2 и R3 независимо выбраны из атома водорода, метильной группы, этильной группы, n-пропильной группы и изопропильной группы.
85. Применение по любому из пп. 82-84, где соединение представляет собой 2-(((5Z,8Z,11Z,14Z,17Z)-эйкоза-5,8,11,14,17-пентаен-1-ил)окси)бутановую кислоту (Соединение А) или ее фармацевтически приемлемую соль или сложный эфир таковой, а формула представляет собой
86. Применение по любому из пп. 82-85, в котором указанное совместное введение осуществляется путем одновременного введения, последовательного введения, совмещенного введения, интервального введения, непрерывного введения или комбинацией таковых.
87. Применение по любому из пп. 82-85, где применение предотвращает, лечит или вызывает реверсию неалкогольного стеатогепатита (НАСГ).
88. Применение по любому из пп. 82-86, где применение предотвращает развитие фиброза печени или уменьшает уже существующий фиброз печени.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| EA 23207 B1, 31.05.2016. | |||

