ОБЛАСТЬ ТЕХНИКИ
Настоящая заявка относится к области техники химического получения и кристаллизации лекарственного препарата. В частности, она относится к солевой форме лекарственного препарата от заболевания или состояния, опосредованных рецептором S1P1, и его кристаллической форме, и дополнительно относится к способу получения солевой формы или кристаллической формы и фармацевтической композиции и применению солевой формы или кристаллической формы.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Химическая формула 1-{2-фтор-4-[5-(4-изобутилфенил)-1,2,4-оксадиазол-3-ил] бензил}-3-азетидинкарбоновой кислоты является следующей C23H24FN3O3, при этом она имеет молекулярную массу 409,45 и химическую структуру, представленную следующей формулой A.
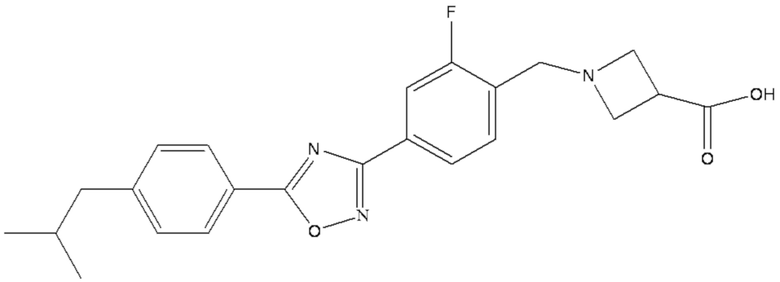
A
В данном документе термин «1-{2-фтор-4-[5-(4-изобутилфенил)-1,2,4- оксадиазол-3-ил]бензил}-3-азетидинкарбоновая кислота» и термин «соединение, представленное формулой A», являются взаимозаменяемыми.
Соединение, представленное формулой A, обладает агонистической активностью и избирательной специфичностью в отношении рецептора S1P1 и характеризуется значительно укороченным периодом полувыведения in-vivo, поэтому оно является превосходным агонистом рецептора S1P1 второго поколения. Большое число исследований показало, что существует множество видов агонистов рецептора S1P1, которые могут связываться с гомологичными рецепторами, экспрессирующимися на лимфоцитах, приводя к интернализации рецептора S1P1 и, в свою очередь, предотвращая экспорт лимфоцитов. Таким образом, агонисты рецептора S1P1 могут снижать способность организма человека к инициации иммунного ответа путем блокирования транспорта лимфоцитов, следовательно, их можно применять в качестве иммунодепрессантов при лечении различных аутоиммунных заболеваний.
Теоретически, соль может образовываться соединением, представленным формулой A, и одним или более кислотными соединениями, представленными формулой XmHn, в которой H представляет собой диссоциирующий ион водорода, X представляет собой фармацевтически приемлемый анион, а m и n представляют собой натуральные числа. Соль может также образовываться соединением, представленным формулой A, и одним или более фармацевтически приемлемыми катионами, такими как ионы щелочных металлов или другие фармацевтически приемлемые органические катионы.
Идентификация, получение, составление и применение соединения, представленного формулой A, раскрыты в патентном документе CN103450171A (который включен в настоящий документ посредством ссылки во всей своей полноте). В частности, способ получения соединения раскрыт в примере 2. 12 кристаллических форм соединения, представленного формулой A, раскрыты в патентном документе CN105315266A (который включен в настоящий документ посредством ссылки во всей своей полноте). Однако авторы настоящего изобретения в своих исследованиях обнаружили, что эти свободные основания обладают чрезвычайно низкой растворимостью в воде, при этом их растворимость в воде при 25ºC составляет 1,1 мкг/мл, и предоставили различные формы, являющиеся стабильными в средах различных растворителей. Например, наиболее стабильной кристаллической формой в воде являлась кристаллическая форма I, тогда как наиболее стабильной кристаллической формой в органическом растворителе являлась кристаллическая форма IV. Таким образом, ограничения соединения включают то, что свободные основания соединения являются нерастворимыми в воде и характеризуются выраженным кристаллическим полиморфизмом. Таким образом, большое практическое значение имеет изучение солевых форм соединения, представленного формулой A, для обеспечения улучшения в отношении некоторых нежелательных физико-химических или биофармацевтических свойств лекарственного препарата, таких как солюбилизация или растворимость лекарственного препарата и явление полиморфизма и т. п., путем образования соли соединения, представленного формулой A.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Ввиду недостатков, существующих в уровне техники, первой целью настоящей заявки является обеспечение солевой формы соединения, представленного формулой A, и ее кристаллической формы. Солевая форма соединения, представленного формулой A, и ее кристаллическая форма обладают одним или более улучшенными свойствами, в частности в отношении полиморфизма, растворимости, стабильности кристаллической формы и химической стабильности и т. п. Например, по сравнению с другими общепринятыми солевыми формами, такими как калиевая соль, кальциевая соль, гидрохлорид, цитрат и фосфат, солевая форма соединения, представленного формулой A, в соответствии с настоящей заявкой обладает одним или более улучшенными свойствами в отношении гигроскопичности, растворимости и термической стабильности (температура плавления и температура разложения).
Второй целью настоящей заявки является обеспечение способа получения солевой формы соединения, представленного формулой A. Поскольку соединение, представленное формулой A, характеризуется низкой растворимостью в большинстве растворителей и температура не оказывает существенного эффекта в отношении улучшения растворимости, сложно получать соль с помощью общепринятой реакции смешивания раствора с раствором. В данном способе получения солевой формы в соответствии с настоящей заявкой предусмотрен ряд путей, включая реакции смешивания суспензии с раствором, твердого вещества с раствором, твердого вещества с твердым веществом и растворителем, суспензии с суспензией и твердого вещества с суспензией с образованием соли, используется способ обнаружения кристаллической формы для мониторинга завершения образования соли и предусмотрена ионообменная хроматография для подтверждения соотношения соединения, представленного формулой A, и противоиона. По сравнению с общепринятыми способами образования соли, данный способ получения солевой формы соединения, представленного формулой A, характеризуется хорошей управляемостью при образовании соли слаборастворимого соединения.
Третьей целью настоящей заявки является обеспечение фармацевтической композиции на основе солевой формы соединения, представленного формулой A, и ее кристаллической формы и их применение.
В соответствии с целями настоящей заявки, настоящая заявка предусматривает натриевую соль 1-{2-фтор-4-[5-(4-изобутилфенил)-1,2,4-оксадиазол-3-ил]бензил}-3-азетидинкарбоновой кислоты, которая представляет собой соединение, образованное соединением, представленным формулой A, и ионом натрия в молярном соотношении 1:1, имеющее структуру, представленную следующей формулой:
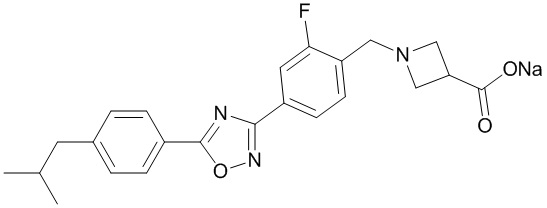
В данном документе термин «натриевая соль 1-{2-фтор-4-[5-(4-изобутилфенил)- 1,2,4-оксадиазол-3-ил]бензил}-3-азетидинкарбоновой кислоты» и термин «натриевая соль соединения, представленного формулой A», являются взаимозаменяемыми.
Натриевая соль соединения, представленного формулой A, в соответствии с настоящей заявкой по существу находится в кристаллическом состоянии и предпочтительно является негидратированной, гидратированной или несольватированной. Более предпочтительно, в соответствии с целями настоящей заявки, настоящая заявка предусматривает кристаллическую форму натриевой соли соединения, представленного формулой A, при этом кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ: 4,4±0,2°, 6,6±0,2°, 14,7±0,2° и 17,2±0,2°.
Еще более предпочтительно настоящая заявка предусматривает кристаллическую форму натриевой соли соединения, представленного формулой A, при этом кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ со следующими относительными значениями интенсивности:

Без ограничения типичный пример кристаллической формы натриевой соли соединения, представленного формулой A, характеризуется порошковой рентгенограммой (XRPD), показанной на фиг. 2. Более предпочтительно кристаллическая форма натриевой соли соединения, представленного формулой A, характеризуется инфракрасным спектром, полученным с использованием преобразования Фурье, имеющим характеристические пики при длинах волн 1560 см-1, 1505 см-1, 1476 см-1, 1417 см-1, 1365 см-1, 1276 см-1, 885 см-1, 849 см-1 и 756 см-1.
В соответствии с целями настоящей заявки, настоящая заявка предусматривает способ получения натриевой соли соединения, представленного формулой A, или ее кристаллической формы. Способ включает следующие стадии: смешивание соединения, представленного формулой A, и гидроксида натрия в молярном соотношении 1:1-1:5 в растворителе, выбранном из группы, состоящей из спирта, кетона, эфира, воды, нитрила или их смеси, для осуществления реакции; удаление растворителя после завершения реакции; и выполнение высушивания.
В соответствии с конкретными вариантами осуществления настоящей заявки, для получения солевой формы при операции удаления растворителя после завершения реакции сначала можно удалить часть растворителя, затем после предварительного охлаждения провести центрифугирование и высушить полученное твердое вещество; или после завершения реакции удаляют весь растворитель , к полученному твердому веществу снова добавляют растворитель с получением суспензии, затем проводят центрифугирование и высушивают полученное твердое вещество.
В соответствии с конкретными вариантами осуществления настоящей заявки, для получения кристаллической формы при операции удаления растворителя после завершения реакции сначала можно удалить часть растворителя, затем провести охлаждение (например, до комнатной температуры) для осаждения твердого вещества и высушить полученное твердое вещество.
Предпочтительно растворитель выбран из группы, состоящей из метанола, этанола, ацетона, диэтилового эфира, воды, ацетонитрила или их смеси.
Предпочтительно молярное соотношение соединения, представленного формулой A, и гидроксида натрия составляет 1:1,0-1:1,3.
Предпочтительно реакцию проводят при 10-60ºC, более предпочтительно при комнатной температуре. Предпочтительно реакцию проводят при перемешивании, и время перемешивания составляет 1-48 ч., более предпочтительно 3-24 ч.
Предпочтительно высушивание выполняют под вакуумом, и температура высушивания составляет 10-60ºC, более предпочтительно 10-40ºC.
Предпочтительно время высушивания составляет 1-48 ч., более предпочтительно 1-24 ч.
Предпочтительно отношение массы соединения, представленного формулой A, к объему растворителя в данном способе составляет 1 мг:1 мл- 50 мг:1 мл, более предпочтительно 2,5 мг:1 мл- 41 мг:1 мл.
«Удаление растворителя» можно осуществлять с применением общепринятых технических средств из области техники, например фильтрации, испарения, центрифугирования, продувки азотом или центробежной сушки. Предпочтительно растворитель удаляют посредством продувки азотом, испарения или фильтрации. Предпочтительно «удаление растворителя» проводят при экспериментальной температуре 10-60ºC.
Натриевая соль соединения, представленного формулой A, и ее кристаллическая форма обладают следующими благоприятными эффектами.
1) Кристаллический полиморфизм натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой является невыраженным.
2) Натриевая соль соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется растворимостью в воде при 25ºC, составляющей 10 мг/мл . По сравнению с известным соединением в свободном состоянии, представленным формулой A, его натриевая соль характеризуется существенно улучшенной растворимостью в воде и лучшей биодоступностью.
3) Натриевая соль соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется растворимостью в воде при 25ºC, составляющей 10 мг/мл . По сравнению с общепринятыми солевыми формами, такими как кальциевая соль соединения, представленного формулой A, гидрохлорид соединения, представленного формулой A, цитрат соединения, представленного формулой A, и фосфат соединения, представленного формулой A, и т. д., его натриевая соль характеризуется существенно улучшенной растворимостью в воде и лучшей биодоступностью.
4) По сравнению с соединением в свободном состоянии, представленным формулой A, кристаллическая форма натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой является стабильной в водных системах, поэтому она обладает большей практической ценностью при влажной грануляции или составлении суспензии.
5) Кристаллическая форма натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой остается неизменной по внешнему виду, рентгенограмме XRPD и температуре плавления после хранения в течение 4 месяцев при условиях комнатной температуры и относительной влажности 10-90%. Показано, что натриевая соль соединения, представленного формулой A, и ее кристаллическая форма в соответствии с настоящей заявкой характеризуются хорошей стабильностью при хранении и могут обеспечивать более эффективное предотвращение проблем с качеством, безопасностью и стабильностью отдельно активного ингредиента и препаратов, содержащих натриевую соль соединения, представленного формулой A, или ее кристаллическую форму, при изготовлении и/или хранении лекарственного средства и т. д., например, примесных кристаллических форм и разницы в растворимости и т. д.
Настоящая заявка дополнительно предусматривает фармацевтическую композицию, содержащую натриевую соль соединения, представленного формулой A, и/или ее кристаллическую форму и необязательно по меньшей мере один фармацевтически приемлемый носитель или вспомогательное вещество.
Настоящая заявка дополнительно предусматривает применение натриевой соли соединения, представленного формулой A, и/или ее кристаллической формы при изготовлении лекарственного препарата для лечения и/или предупреждения заболевания или состояния, опосредованных рецептором S1P1.
Настоящая заявка дополнительно предусматривает способ лечения и/или предупреждения заболевания или состояния, опосредованных рецептором S1P1, включающий введение субъекту, нуждающемуся в этом, натриевой соли соединения, представленного формулой A, и/или ее кристаллической формы, предусмотренных настоящей заявкой. Предпочтительно субъект представляет собой млекопитающее; более предпочтительно субъект представляет собой человека.
В соответствии с целями настоящей заявки, настоящая заявка предусматривает сульфат 1-{2-фтор-4-[5-(4-изобутилфенил)-1,2,4-оксадиазол-3-ил]бензил}- 3-азетидинкарбоновой кислоты, который представляет собой соединение, образованное соединением, представленным формулой A, и серной кислотой в молярном соотношении 2:1, и имеет структуру, представленную следующей формулой:
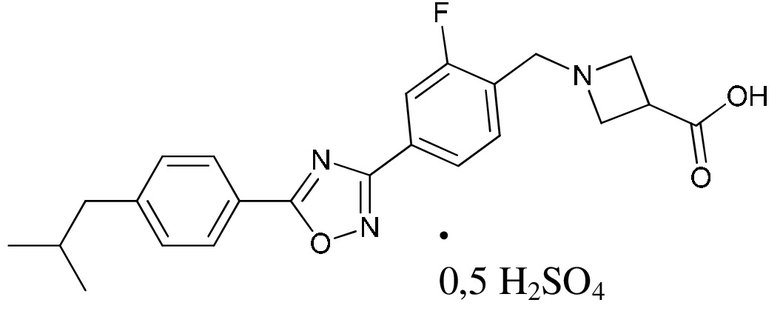
В данном документе термин «сульфат 1-{2-фтор-4-[5-(4-изобутилфенил)- 1,2,4-оксадиазол-3-ил]бензил}-3-азетидинкарбоновой кислоты» и термин «сульфат соединения, представленного формулой A», являются взаимозаменяемыми.
Сульфат соединения, представленного формулой A, в соответствии с настоящей заявкой по существу находится в кристаллическом состоянии и предпочтительно является негидратированным, гидратированным или несольватированным. Более предпочтительно, в соответствии с целями настоящей заявки, настоящая заявка предусматривает кристаллическую форму сульфата соединения, представленного формулой A. При облучении Cu-Kα-излучением кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ: 5,4±0,2°, 8,1±0,2°, 14,8±0,2°, 16,7±0,2° и 18,3±0,2°.
Более предпочтительно кристаллическая форма сульфата соединения, представленного формулой A, характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ: 5,4±0,2°, 8,1±0,2°, 14,8±0,2°, 15,6±0,2°, 16,7±0,2°, 18,3±0,2°, 21,0±0,2°, 22,0±0,2°, 22,9±0,2°, 25,2±0,2° и 26,3±0,2°.
Еще более предпочтительно настоящая заявка предусматривает кристаллическую форму сульфата соединения, представленного формулой A, при этом кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ со следующими относительными значениями интенсивности:


Без ограничения типичный пример кристаллической формы сульфата соединения, представленного формулой A, характеризуется порошковой рентгенограммой, показанной на фиг. 6.
Кристаллическая форма сульфата соединения, представленного формулой A, характеризуется инфракрасным спектром, полученным с использованием преобразования Фурье, имеющим характеристические пики при длинах волн 1733 см-1, 1438 см-1, 1346 см-1, 1230 см-1, 1184 см-1, 1109 см-1, 1063 см-1, 1009 см-1, 885 см-1, 854 см-1 и 758 см-1.
В соответствии с целями настоящей заявки, настоящая заявка предусматривает способ получения сульфата соединения, представленного формулой A, или его кристаллической формы. Способ включает следующие стадии: получение суспензии или раствора соединения, представленного формулой A, и суспензии или раствора серной кислоты в растворителе, выбранном из группы, состоящей из спирта, кетона, циклического эфира, нитрила, воды или их смеси соответственно; смешивание суспензии или раствора соединения, представленного формулой A, и серной кислоты в молярном соотношении 1:0,4-1:10 для осуществления реакции; удаление растворителя после завершения реакции и выполнение высушивания.
В соответствии с конкретными вариантами осуществления настоящей заявки для получения солевой формы при операции удаления растворителя после завершения реакции сначала можно удалить часть растворителя, затем провести охлаждение или центрифугирование и высушить полученное твердое вещество; или после завершения реакции удаляют весь растворитель, необязательно к полученному твердому веществу снова добавляют растворитель с получением суспензии, затем проводят центрифугирование и высушивают полученное твердое вещество.
В соответствии с конкретными вариантами осуществления настоящей заявки для получения кристаллической формы при операции удаления растворителя после завершения реакции сначала можно удалить весь растворитель, затем добавить воду для обработки ультразвуком, провести центрифугирование и высушить полученное твердое вещество.
Предпочтительно растворитель выбран из группы, состоящей из метанола, этанола, н-пропанола, ацетона, тетрагидрофурана, воды, ацетонитрила или их смеси.
Предпочтительно молярное соотношение соединения, представленного формулой A, и серной кислоты составляет 1:0,4-1:7,9.
Предпочтительно реакцию проводят при -10-60ºC, более предпочтительно при 10-40ºC. Предпочтительно реакцию проводят при перемешивании, и время перемешивания составляет 1-72 ч., более предпочтительно 1-24 ч.
Предпочтительно температура высушивания составляет 10-60ºC, более предпочтительно 10-40ºC.
Предпочтительно время высушивания составляет 1-48 ч., более предпочтительно 1-24 ч.
Предпочтительно отношение массы соединения, представленного формулой A, к объему растворителя в данном способе составляет 1 мг:1 мл- 50 мг:1 мл, более предпочтительно 4 мг:1 мл- 35 мг:1 мл.
«Удаление растворителя» можно осуществлять с применением общепринятых технических средств из области техники, например фильтрации, испарения, центрифугирования, продувки азотом или центробежной сушки. Предпочтительно растворитель удаляют посредством продувки азотом, испарения или фильтрации. Предпочтительно удаление растворителя проводят при экспериментальной температуре 10-60ºC.
«Серная кислота» относится к концентрированной серной кислоте, которая имеет концентрацию 98% (вес. %) и доступна коммерчески.
Сульфат соединения, представленного формулой A, и его кристаллическая форма обладают следующими благоприятными эффектами.
1) Кристаллический полиморфизм сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой является невыраженным.
2) Сульфат соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется растворимостью в воде при 25ºC, составляющей 19 мкг/мл. По сравнению с известным соединением в свободном состоянии, представленным формулой A, его сульфат характеризуется существенно улучшенной растворимостью в воде и лучшей биодоступностью.
3) Сульфат соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется растворимостью в воде при 25ºC, составляющей 19 мкг/мл. По сравнению с общепринятыми солевыми формами, такими как кальциевая соль соединения, представленного формулой A, гидрохлорид соединения, представленного формулой A, цитрат соединения, представленного формулой A, и фосфат соединения, представленного формулой A, и т. д., его сульфат характеризуется значительно улучшенной растворимостью в воде и лучшей биодоступностью.
4) Сульфат соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется увеличением веса на 0,7% при относительной влажности 20-80%. По сравнению с общепринятыми солевыми формами, такими как калиевая соль соединения, представленного формулой A, кальциевая соль соединения, представленного формулой A, гидрохлорид соединения, представленного формулой A, цитрат соединения, представленного формулой A, и фосфат соединения, представленного формулой A, и т. д., его сульфат характеризуется более низким значением увеличения веса вследствие гигроскопичности и, таким образом, лучшей стабильностью при хранении.
5) Кристаллическая форма сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой является стабильной в водных системах, поэтому она обладает большей практической ценностью при влажной грануляции или составлении суспензии.
6) Кристаллическая форма сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой является неизменной по внешнему виду, рентгенограмме XRPD и температуре плавления после хранения в течение 1 месяца при общепринятых условиях высокой температуры (60ºC) и ускоренного старения (40ºC, относительная влажность 75%). Показано, что сульфат соединения, представленного формулой A, и его кристаллическая форма в соответствии с настоящей заявкой характеризуются хорошей стабильностью при хранении и могут обеспечивать более эффективное предотвращение проблем с качеством, безопасностью и стабильностью отдельно активного ингредиента и препаратов, содержащих сульфат соединения, представленного формулой A, или его кристаллическую форму, при изготовлении и/или хранении лекарственного средства и т. д. например примесных кристаллических форм и разницы в растворимости и т. д.
Настоящая заявка дополнительно предусматривает фармацевтическую композицию, содержащую сульфат соединения, представленного формулой A, и/или его кристаллическую форму и необязательно по меньшей мере один фармацевтически приемлемый носитель или вспомогательное вещество.
Настоящая заявка дополнительно предусматривает применение сульфата соединения, представленного формулой A, и/или его кристаллической формы при изготовлении лекарственного препарата для лечения и/или предупреждения заболевания или состояния, опосредованных рецептором S1P1.
Настоящая заявка дополнительно предусматривает способ лечения и/или предупреждения заболевания или состояния, опосредованных рецептором S1P1, включающий введение субъекту, нуждающемуся в этом, сульфата соединения, представленного формулой A, и/или его кристаллической формы, предусмотренной настоящей заявкой. Предпочтительно субъект представляет собой млекопитающее; более предпочтительно субъект представляет собой человека.
В соответствии с целями настоящей заявки, настоящая заявка предусматривает малеат 1-{2-фтор-4-[5-(4-изобутилфенил)-1,2,4-оксадиазол-3-ил]бензил}-3-азетидинкарбоновой кислоты, который является соединением, образованным соединением, представленным формулой A, и малеиновой кислотой в молярном соотношении 1:1, и имеет структуру, представленную следующей формулой:
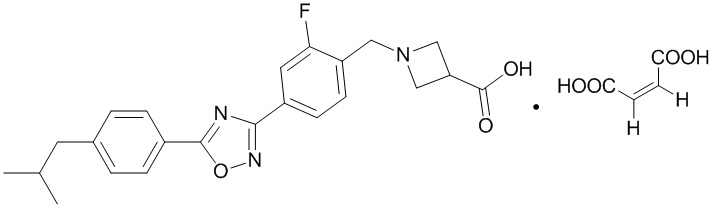
В данном документе термин «малеат 1-{2-фтор-4-[5-(4-изобутилфенил)- 1,2,4-оксадиазол-3-ил]бензил}-3-азетидинкарбоновой кислоты» и термин «малеат соединения, представленного формулой A», являются взаимозаменяемыми.
Малеат соединения, представленного формулой A, в соответствии с настоящей заявкой по существу находится в кристаллическом состоянии и предпочтительно является негидратированным, гидратированным или несольватированным. Более предпочтительно, в соответствии с целями настоящей заявки, настоящая заявка предусматривает кристаллическую форму малеата соединения, представленного формулой A, при этом при облучении Cu-Kα-излучением кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ: 10,6±0,2°, 16,3±0,2°, 19,5±0,2°, 21,5±0,2° и 26,9±0,2°.
Более предпочтительно кристаллическая форма малеата соединения, представленного формулой A, характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ: 7,0±0,2°, 10,6±0,2°, 13,6±0,2°, 16,3±0,2°, 19,5±0,2°, 20,1±0,2°, 21,5±0,2°, 24,5±0,2° и 26,9±0,2°.
Еще более предпочтительно настоящая заявка предусматривает кристаллическую форму малеата соединения, представленного формулой A, при этом кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ со следующими относительными значениями интенсивности:

Без ограничения типичный пример кристаллической формы малеата соединения, представленного формулой A, характеризуется порошковой рентгенограммой, показанной на фиг. 10.
Кристаллическая форма малеата соединения, представленного формулой A, характеризуется инфракрасным спектром, полученным с использованием преобразования Фурье, имеющим характеристические пики при длинах волн 1734 см-1, 1574 см-1, 1485 см-1, 1439 см-1, 1364 см-1, 1346 см-1, 1080 см-1, 1003 см-1, 893 см-1, 871 см-1, 757 см-1 и 729 см-1.
В соответствии с целями настоящей заявки, настоящая заявка предусматривает способ получения малеата соединения, представленного формулой A, или его кристаллической формы. Способ включает следующие стадии: получение суспензии или раствора соединения, представленного формулой A, и суспензии или раствора малеиновой кислоты в растворителе, выбранном из группы, состоящей из спирта, кетонов, эфира (включая циклический эфир), сложного эфира, воды или их смеси соответственно; смешивание суспензии или раствора соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1-1:5 для осуществления реакции; удаление растворителя после завершения реакции; и выполнение высушивания.
Предпочтительно растворитель выбран из группы, состоящей из этанола, ацетона, диэтилового эфира, воды, этилацетата, 1,4-диоксана или их смеси.
Предпочтительно молярное соотношение соединения, представленного формулой A, и малеиновой кислоты составляет 1:1,0-1:2,6.
Предпочтительно реакцию проводят при -10-60ºC, более предпочтительно при 10-40ºC. Предпочтительно реакцию проводят при перемешивании, и время перемешивания составляет 10-72 ч., более предпочтительно 10-24 ч.
Предпочтительно температура высушивания составляет 10-60ºC, более предпочтительно 10-40ºC.
Предпочтительно время высушивания составляет 1-48 ч., более предпочтительно 1-24 ч.
Предпочтительно отношение массы соединения, представленного формулой A, к объему растворителя в данном способе составляет 1 мг:1 мл-50 мг:1 мл, более предпочтительно 4 мг:1 мл-26 мг:1 мл.
Малеат соединения, представленного формулой A, и его кристаллическая форма обладают следующими благоприятными эффектами.
1) Кристаллический полиморфизм малеата соединения, представленного формулой A, в соответствии с настоящей заявкой является невыраженным.
2) Малеат соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется растворимостью в воде при 25ºC, составляющей 16 мкг/мл. По сравнению с известным соединением в свободном состоянии, представленным формулой A, его малеат характеризуется существенно улучшенной растворимостью в воде и лучшей биодоступностью.
3) Малеат соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется растворимостью в воде при 25ºC, составляющей 16 мкг/мл. По сравнению с общепринятыми солевыми формами, такими как кальциевая соль соединения, представленного формулой A, гидрохлорид соединения, представленного формулой A, цитрат соединения, представленного формулой A, и фосфат соединения, представленного формулой A, и т. д., его малеат характеризуется значительно улучшенной растворимостью в воде и лучшей биодоступностью.
4) Малеат соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется увеличением веса на 0,4% при относительной влажности 20-80%. По сравнению с общепринятыми солевыми формами, такими как калиевая соль соединения, представленного формулой A, кальциевая соль соединения, представленного формулой A, гидрохлорид соединения, представленного формулой A, цитрат соединения, представленного формулой A, и фосфат соединения, представленного формулой A, и т. д., его малеат характеризуется более низким значением увеличения веса вследствие гигроскопичности и, таким образом, лучшей стабильностью при хранении.
5) Кристаллическая форма малеата соединения, представленного формулой A, в соответствии с настоящей заявкой является стабильной в водных системах, поэтому она обладает большей практической ценностью при влажной грануляции или составлении суспензии.
6) Кристаллическая форма малеата соединения, представленного формулой A, в соответствии с настоящей заявкой является неизменной по внешнему виду, рентгенограмме XRPD и температуре плавления после хранения в течение 1 месяца при общепринятых условиях высокой температуры (60ºC) и ускоренного старения (40ºC, относительная влажность 75%). Показано, что малеат соединения, представленного формулой A, и его кристаллическая форма в соответствии с настоящей заявкой характеризуются хорошей стабильностью при хранении и могут обеспечивать более эффективное предотвращение проблем с качеством, безопасностью и стабильностью отдельно активного ингредиента и препаратов, содержащих малеат соединения, представленного формулой A, или его кристаллическую форму, при изготовлении и/или хранении лекарственного средства и т. д., например примесных кристаллических форм и разницы в растворимости и т. д.
Настоящая заявка дополнительно предусматривает фармацевтическую композицию, содержащую малеат соединения, представленного формулой A, и/или его кристаллическую форму и необязательно по меньшей мере один фармацевтически приемлемый носитель или вспомогательное вещество.
Настоящая заявка дополнительно предусматривает применение малеата соединения, представленного формулой A, и/или его кристаллической формы при изготовлении лекарственного препарата для лечения и/или предупреждения заболевания или состояния, опосредованных рецептором S1P1.
Настоящая заявка дополнительно предусматривает способ лечения и/или предупреждения заболевания или состояния, опосредованных рецептором S1P1, включающий введение субъекту, нуждающемуся в этом, малеата соединения, представленного формулой A, и/или его кристаллической формы, предусмотренной настоящей заявкой. Предпочтительно субъект представляет собой млекопитающее; более предпочтительно субъект представляет собой человека.
В любом способе получения натриевой соли соединения, представленного формулой A, и кристаллической формы натриевой соли соединения, представленного формулой A, сульфата соединения, представленного формулой A, и кристаллической формы сульфата соединения, представленного формулой A, малеата соединения, представленного формулой A, и кристаллической формы малеата соединения, представленного формулой A, в соответствии с настоящей заявкой справедливо следующее.
Если не указано иное, «комнатная температура» относится к температуре приблизительно 10-30ºC.
«Циклический эфир» может представлять собой тетрагидрофуран, 1,4-диоксан и т. д.
«Перемешивание» можно осуществлять с применением общепринятого в данной области техники способа. Например, перемешивание включает перемешивание с помощью магнитной мешалки и механическое перемешивание; и скорость перемешивания составляет 50-1800 об./мин., предпочтительно 300-900 об./мин.
«Удаление растворителя» можно проводить с применением общепринятого в данной области техники способа, такого как фильтрация, испарение, центрифугирование, продувка азотом или центробежная сушка. «Фильтрация» обычно относится к фильтрации с отсасыванием, проводимой при комнатной температуре под давлением меньше атмосферного, предпочтительно под давлением менее 0,09 МПа. «Центробежная сушка» обычно относится к роторному испарению под давлением меньше атмосферного, предпочтительно под давлением менее 0,09 МПа. «Продувка азотом» обычно относится к подаче азота с помощью прибора для продувки азотом, при этом жидкость испаряется с обеспечением высушивания с помощью быстрого потока подаваемого азота. Конкретная операция «центрифугирование» является следующей: образец, который необходимо разделить, помещают в центрифужную пробирку и центрифугируют, например со скоростью 6000 об./мин., пока твердое вещество полностью не осядет на дне центрифужной пробирки. Конкретная операция «испарение» является следующей: раствор образца, помещенный в открытый контейнер, испаряют при различных температурах до высыхания растворителя. «Удаление растворителя» проводят при экспериментальной температуре, составляющей предпочтительно 10-60ºC.
«Высушивание» можно осуществлять с применением общепринятых технических средств, известных в данной области техники, таких как высушивание при комнатной температуре, высушивание в потоке воздуха или высушивание под пониженным давлением. Его можно проводить под пониженным давлением или атмосферным давлением, предпочтительно под давлением менее 0,09 МПа. Устройство и способ для высушивания не ограничены. Устройство для высушивания может представлять собой вентилятор, сушильную печь с продувкой воздухом, распылительную сушилку, сушилку с кипящим слоем или вакуумную печь; и высушивание можно проводить под пониженным давлением или давлением, не являющимся пониженным, предпочтительно под давлением менее 0,09 МПа.
«Кристаллическая форма» в настоящей заявке означает, что соединение характеризуется уникальным и упорядоченным расположением молекул или конфигурацией в виде решетки, что подтверждается проиллюстрированной порошковой рентгеновской дифрактограммой. Как хорошо известно специалистам в данной области техники, может присутствовать погрешность эксперимента, величина которой зависит от условий эксплуатации прибора, пробоподготовки и чистоты образца. Значение угла 2θ пика на дифрактограмме XRD обычно несколько изменяется в зависимости от прибора и образца. При использовании различных приборов и различных образцов и т. п. разница в значении угла для пика может составлять 1°, 0,8°, 0,5°, 0,3°, 0,1° и т. д., и обычно допустимой является погрешность, составляющая ±0,2°; таким образом, разницу в значениях углов для пиков нельзя использовать в качестве единственного стандарта. Относительное значение интенсивности пика может изменяться в зависимости от образца, пробоподготовки и других условий эксперимента, поэтому порядок значений интенсивности пиков нельзя использовать в качестве единственного или решающего фактора. Влияние высоты пика образца и других факторов эксперимента может приводить к общему сдвигу углов для пиков, и обычно некоторая величина сдвига является допустимой. Таким образом, специалисты в данной области техники смогут понять, что любая кристаллическая форма с такими же характеристическими пиками, как и на порошковой рентгеновской дифрактограмме, представленной в настоящей заявке, или подобными им, относится к объему настоящей заявки. «Одна кристаллическая форма» относится к одной кристаллической форме, обнаруженной с помощью рентгеновской порошковой дифрактометрии.
Новая солевая форма соединения, представленного формулой A, в соответствии с настоящей заявкой является по существу чистой и отдельной и является по существу не смешанной с какой-либо другой кристаллической формой или аморфным состоянием. «По существу чистый» в настоящей заявке, при использовании для ссылки на новую кристаллическую форму, означает, что новая кристаллическая форма составляет по меньшей мере 80% (по весу) соединения, кроме того, по меньшей мере 90% (по весу), в частности, по меньшей мере 95% (по весу) и, в частности, по меньшей мере 99% (по весу).
Исходный материал в настоящей заявке, т. е. соединение, представленное формулой A, можно получить в соответствии со способом получения, раскрытым в патентном документе CN103450171A.
Кроме того, настоящая заявка предусматривает фармацевтическую композицию, содержащую терапевтически или профилактически эффективное количество одного или более компонентов, выбранных из группы, состоящей из солевых форм или их кристаллических форм и аморфных форм в соответствии с настоящей заявкой, или солевых форм и/или их кристаллических форм и аморфных форм, полученных с помощью способа в соответствии с настоящей заявкой, и необязательно по меньшей мере один фармацевтически приемлемый носитель или вспомогательное вещество. Солевые формы соединения, представленного формулой A, и их кристаллические формы включают натриевую соль соединения, представленного формулой A, кристаллическую форму натриевой соли соединения, представленного формулой A, сульфат соединения, представленного формулой A, кристаллическую форму сульфата соединения, представленного формулой A, малеат соединения, представленного формулой A, и кристаллическую форму малеата соединения, представленного формулой A. Кроме того, фармацевтическая композиция может также содержать другие фармацевтически приемлемые соли соединения, представленного формулой A, кристаллические формы солей или аморфные формы солей.
Вышеуказанную фармацевтическую композицию можно получать в виде определенных лекарственных форм, предпочтительно в виде лекарственных форм для перорального введения, парентерального введения (включая подкожное, внутримышечное и внутривенное), ректального введения, трансдермального введения, буккального введения и назального введения, включая помимо прочего твердую лекарственную форму, жидкую лекарственную форму, полужидкую лекарственную форму, аэрозоль или суппозиторий и т. д. Например, лекарственные формы, подходящие для перорального введения, включают таблетки, капсулы, гранулы, порошкообразные препараты, пилюли, порошки, пастилки, сиропы или суспензии; лекарственные формы, подходящие для парентерального введения, включают водные или безводные растворы или эмульсии; лекарственные формы, подходящие для ректального введения, включают суппозитории, в которых используются гидрофильные или гидрофобные носители; лекарственные формы, подходящие для трансдермального введения, включают мази и кремы; лекарственные формы, подходящие для назального введения, включают аэрозоли и спреи. При необходимости, вышеуказанные лекарственные формы можно видоизменить для обеспечения быстрого высвобождения, отсроченного высвобождения или регулируемого высвобождения активных ингредиентов.
Фармацевтически приемлемые носители в настоящей заявке включают твердые носители, в частности включающие помимо прочего разбавители, такие как крахмал, желатинированный крахмал, лактоза, порошкообразная целлюлоза, микрокристаллическая целлюлоза, гидрофосфат кальция, трикальцийфосфат, маннит, сорбит, сахар и т. п.; связующие, такие как гуммиарабик, гуаровая камедь, желатин, поливинилпирролидон, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, полиэтиленгликоль и т. п.; разрыхлители, такие как крахмал, натрия крахмалгликолят, прежелатинизированный крахмал, поперечно сшитый повидон, поперечно сшитая натрийкарбоксиметилцеллюлоза, коллоидный диоксид кремния и т. п.; смазывающие вещества, такие как стеариновая кислота, стеарат магния, стеарат цинка, бензоат натрия, ацетат натрия и т. п.; средства для повышения текучести, такие как коллоидный диоксид кремния и т. п.; комплексообразующие средства, такие как различные сорта циклодекстринов и смол; средства контроля скорости высвобождения, такие как гидроксипропилцеллюлоза, гидроксиметилцеллюлоза, гидроксипропилметилцеллюлоза, этилцеллюлоза, метилцеллюлоза, метилметакрилат и воск и т. п. Фармацевтически приемлемые носители в настоящей заявке также включают жидкие носители, в частности включающие помимо прочего растворители на основе водного, масляного или спиртового раствора, такие как стерильная вода, физиологический солевой раствор, раствор глюкозы, раствор маннита, растительное масло, жир печени трески, этанол, пропанол и глицерин и т. п. Кроме того, можно использовать другие носители, такие как полиэтиленгликоль и полипропиленгликоль и т. п. В зависимости от различных лекарственных форм можно также выбирать другие фармацевтически приемлемые носители, включающие, например, помимо прочего пленкообразующие средства, пластификаторы, красители, вкусоароматические вещества, регуляторы вязкости, консерванты, антиоксиданты, пенетранты, буферы и т. д. Каждый носитель должен являться приемлемым, совместимым с другими ингредиентами в составе и безвредным для пациентов.
Фармацевтическую композицию можно получать с применением способа, хорошо известного специалистам в данной области техники. При получении фармацевтической композиции натриевую соль соединения, представленного формулой A, кристаллическую форму натриевой соли соединения, представленного формулой A, сульфат соединения, представленного формулой A, кристаллическую форму сульфата соединения, представленного формулой A, малеат соединения, представленного формулой A, кристаллическую форму малеата соединения, представленного формулой A, или их комбинацию смешивают с одним или более фармацевтически приемлемыми носителями и необязательно смешивают с одним или более другими активными фармацевтическими ингредиентами. Твердые препараты можно получать с помощью процессов, таких как смешивание и гранулирование и т. д. тогда как жидкие или полужидкие лекарственные формы можно получать с помощью процессов, таких как смешивание, растворение, диспергирование и эмульгирование и т. д.
Кроме того, настоящая заявка предусматривает применение солевой формы и/или ее кристаллической формы и аморфной формы в соответствии с настоящей заявкой, или солевой формы и/или ее кристаллической формы и аморфной формы, полученных с применением способа получения в соответствии с настоящей заявкой, при изготовлении лекарственного препарата для лечения и/или предупреждения заболевания или состояния, опосредованных рецептором S1P1. Солевая форма и ее кристаллическая форма и аморфная форма включают натриевую соль соединения, представленного формулой A, кристаллическую форму натриевой соли соединения, представленного формулой A, сульфат соединения, представленного формулой A, кристаллическую форму сульфата соединения, представленного формулой A, малеат соединения, представленного формулой A, кристаллическую форму малеата соединения, представленного формулой A, или их комбинацию. Заболевание или состояние, опосредованные рецептором S1P1, выбрано из группы, состоящей из ревматоидного артрита, рассеянного склероза, воспалительного энтерита, аутоиммунных заболеваний, хронических воспалительных заболеваний, астмы, воспалительной невропатии, артрита, трансплантации, сегментарного илеита, язвенного колита, красной волчанки, псориаза, ишемически-реперфузионного повреждения, солидных опухолей, связанных с ангиогенезом заболеваний, сосудистых заболеваний, болевых симптомов, острых вирусных заболеваний, воспалительных заболеваний кишечника, инсулинозависимого и инсулинонезависимого сахарного диабета и других родственных иммунопатологических заболеваний. Предпочтительно заболевание или состояние выбрано из группы, состоящей из рассеянного склероза, ревматоидного артрита, воспалительного энтерита и псориаза.
Кроме того, настоящая заявка предусматривает способ лечения и/или предупреждения заболевания или состояния, опосредованных рецептором S1P1, включающий введение субъекту, нуждающемуся в этом, терапевтически или профилактически эффективного количества соли и/или ее кристаллической формы, или их комбинации, или фармацевтической композиции в соответствии с настоящей заявкой. Соль и ее кристаллическая форма и аморфная форма включают натриевую соль соединения, представленного формулой A, кристаллическую форму натриевой соли соединения, представленного формулой A, сульфат соединения, представленного формулой A, кристаллическую форму сульфата соединения, представленного формулой A, малеат соединения, представленного формулой A, кристаллическую форму малеата соединения, представленного формулой A, или их комбинацию. Заболевание или состояние, опосредованные рецептором S1P1, выбрано из группы, состоящей из ревматоидного артрита, рассеянного склероза, воспалительного энтерита, аутоиммунных заболеваний, хронических воспалительных заболеваний, астмы, воспалительной невропатии, артрита, трансплантации, сегментарного илеита, язвенного колита, красной волчанки, псориаза, ишемически-реперфузионного повреждения, солидных опухолей, связанных с ангиогенезом заболеваний, сосудистых заболеваний, болевых симптомов, острых вирусных заболеваний, воспалительных заболеваний кишечника, инсулинозависимого и инсулинонезависимого сахарного диабета и других родственных иммунопатологических заболеваний. Предпочтительно заболевание или состояние выбрано из группы, состоящей из рассеянного склероза, ревматоидного артрита, воспалительного энтерита и псориаза. Субъект включает без ограничения млекопитающих. Кристаллическую форму и аморфную форму или их комбинацию или фармацевтическую композицию, предусмотренные в настоящей заявке, можно применять вместе с другими видами терапии или терапевтическими средствами. Кроме того, дозировка соединения или фармацевтической композиции, необходимая для лечения, предупреждения или облегчения состояния и т. д., обычно зависит от конкретного вводимого соединения, пациента, конкретного заболевания или состояния и его тяжести, пути и частоты введения и должна определяться лечащим врачом в соответствии с конкретной ситуацией.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фиг. 1 представлен ИК-спектр натриевой соли соединения, представленного формулой A, в соответствии с примером 3 настоящей заявки.
На фиг. 2 представлена дифрактограмма XRPD натриевой соли соединения, представленного формулой A, в соответствии с примером 3 настоящей заявки.
На фиг. 3 представлена термограмма TGA натриевой соли соединения, представленного формулой A, в соответствии с примером 3 настоящей заявки.
На фиг. 4 представлена термограмма DSC натриевой соли соединения, представленного формулой A, в соответствии с примером 3 настоящей заявки.
На фиг. 5 представлен ИК-спектр сульфата соединения, представленного формулой A, в соответствии с примером 13 настоящей заявки.
На фиг. 6 представлена дифрактограмма XRPD сульфата соединения, представленного формулой A, в соответствии с примером 13 настоящей заявки.
На фиг. 7 представлена термограмма TGA сульфата соединения, представленного формулой A, в соответствии с примером 13 настоящей заявки.
На фиг. 8 представлена термограмма DSC сульфата соединения, представленного формулой A, в соответствии с примером 13 настоящей заявки.
На фиг. 9 представлен ИК-спектр малеата соединения, представленного формулой A, в соответствии с примером 21 настоящей заявки.
На фиг. 10 представлена дифрактограмма XRPD малеата соединения, представленного формулой A, в соответствии с примером 21 настоящей заявки.
На фиг. 11 представлена термограмма TGA малеата соединения, представленного формулой A, в соответствии с примером 21 настоящей заявки.
На фиг. 12 представлена термограмма DSC малеата соединения, представленного формулой A, в соответствии с примером 21 настоящей заявки.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Следующие примеры облегчат дальнейшее понимание настоящей заявки и не используются для ограничения настоящей заявки.
Приборы для обнаружения и способы обнаружения
Рентгеновская порошковая дифрактометрия (XPRD). Прибор представлял собой дифрактометр D8 Advance от Bruker. Образцы тестировали при комнатной температуре. Условия обнаружения являлись следующими: диапазон значений угла: 3-40° 2θ; размер шага: 0,02°2θ; скорость: 0,2 с/шаг.
Данные дифференциальной сканирующей калориметрии (DSC) собирали с помощью прибора Q200 MDSC от TA Instruments. Применяемый способ обнаружения являлся следующим: 1-10 мг образца помещали в герметичный алюминиевый тигель с небольшим отверстием и температуру образца повышали от комнатной температуры до 250ºC при скорости нагревания 10ºC/мин. под защитой сухого N2, 40 мл/мин.
Данные термогравиметрического анализа (TGA) собирали с помощью прибора Q500 TGA от TA Instruments. Применяемый способ обнаружения являлся следующим: 5-15 мг образцов помещали в платиновый тигель, и использовали способ на основе сегментирования для обнаружения с высоким разрешением, в котором температуру образца повышали от комнатной температуры до 300ºC при скорости нагревания 10ºC/мин. под защитой сухого N2, 40 мл/мин.
Данные 1H-ядерного магнитного резонанса (1H-ЯМР) получали с помощью спектрометра ядерного магнитного резонанса Avance II DMX 400MHZ от Bruker. Взвешивали 1-5 мг образца и растворяли в приблизительно 0,5 мл дейтерированного реагента в пробирке для образцов для ядерного магнитного резонанса и проводили обнаружение.
Данные ИК-спектроскопии (IR) собирали с помощью прибора Tensor 27 от Bruker. Как программное обеспечение для управления приборами, так и программное обеспечение для анализа данных являлось продуктами от OPUS. Данные по поглощению в инфракрасной области спектра в диапазоне 600-4000 см-1 обычно собирали с помощью оборудования от ATR.
Данные динамической сорбции паров (DVS) и данные анализа изотермической сорбции собирали с помощью прибора Q5000 TGA от TA Instruments. Применяемый способ обнаружения являлся следующим: В платиновый тигель помещали 1-10 мг образца и проводили обнаружение изменения веса при изменении относительной влажности от 20% до 80%.
Данные растворимости согласно HPLC собирали с помощью высокоэффективного жидкостного хроматографа Agilent 1260. Применяемая хроматографическая колонка представляла собой Poroshell 120 EC-C18 (2,7*50 мм, 4,6 мкм), длина волны обнаружения составляла 254 нм, температура колонки для обнаружения составляла 40ºC, расход составлял 1,5 мл/мин., а объем образца составлял 5 мкл. Образец растворяли в подвижной фазе B для получения раствора образца с концентрацией приблизительно 0,45 мг/мл, и обнаружение с помощью HPLC проводили в соответствии со следующим режимом градиентного элюирования с получением значения концентрации в образце.
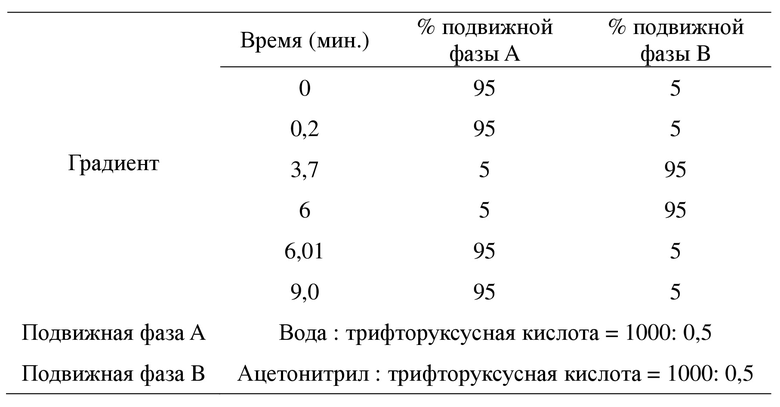
Данные ионообменной хроматографии (IC) собирали с помощью системы Dionex ICS-900. Как рабочая станция, так и программное обеспечение для анализа представляли собой продукцию Chromeleon Console. Определение содержания ионов проводили с применением способа с внешним стандартом.
Операции ультразвуковой обработки, описанные в примерах, могут облегчать растворение образцов. Оборудование представляло собой ультразвуковой очиститель, и обработку ультразвуком проводили в течение 15 минут при мощности 40 кГц.
Пример получения 1: получение соединения, представленного формулой A
Соединение, представленное формулой A, можно получать в соответствии со способом получения, описанным в примере 2 патентного документа CN103450171A.
В частности, при комнатной температуре раствор 2-фтор-4-[5-(4-изобутилфенил)-1,2,4-оксадиазол-3-ил]-бензальдегида (9,0 г, 27,8 ммоль), азетидин-3-карбоновой кислоты (2,8 г, 27,8 ммоль) и уксусной кислоты (10 мл) в смеси метанола и тетрагидрофурана (200 мл/200 мл) перемешивали в течение 2 ч. Затем раствор (600 мл) цианоборгидрата натрия (10,3 г, 163,5 ммоль) в метаноле (600 мл) добавляли в реакционную смесь, а затем полученную смесь перемешивали в течение еще 16 ч. при комнатной температуре. Проводили фильтрацию с получением осадка на фильтре, и осадок на фильтре промывали метанолом (100 мл) и сушили с получением 2,0 г продукта в виде белого твердого вещества.
1H-ЯМР (400 МГц, CD3OD) δ : 8,13 (d, J=8,4 Гц, 2H), 8,05 (m, 1H), 7,97 (m, 1H), 7,68 (t, J=8,0 Гц, 7,6 Гц, 1H), 7,42 (d, J=8,4 Гц, 2H), 4,40 (s, 2H), 4,15 (m, 4H), 3,41 (m, 1H), 2,61 (d, J=7,2 Гц, 2H), 1,95 (m, 1H), 0,94 (d, J=7,2 Гц, 6H) показал, что продукт представлял собой соединение, представленное формулой A, т. е. 1-{2-фтор-4-[5-(4-изобутилфенил)-1,2,4-оксадиазол-3-ил]бензил}-3-азетидинкарбоновую кислоту.
Пример получения 2: выбор и получение солевых форм соединения, представленного формулой A
2.1 Выбор соли
В соответствии со структурой соединения, представленного формулой A, для эксперимента выбора соли были выбраны 12 кислот типа I и 3 щелочи типа I. Установки и результаты эксперимента показаны в таблице 1.
Таблица 1. Установки и результаты эксперимента выбора соли
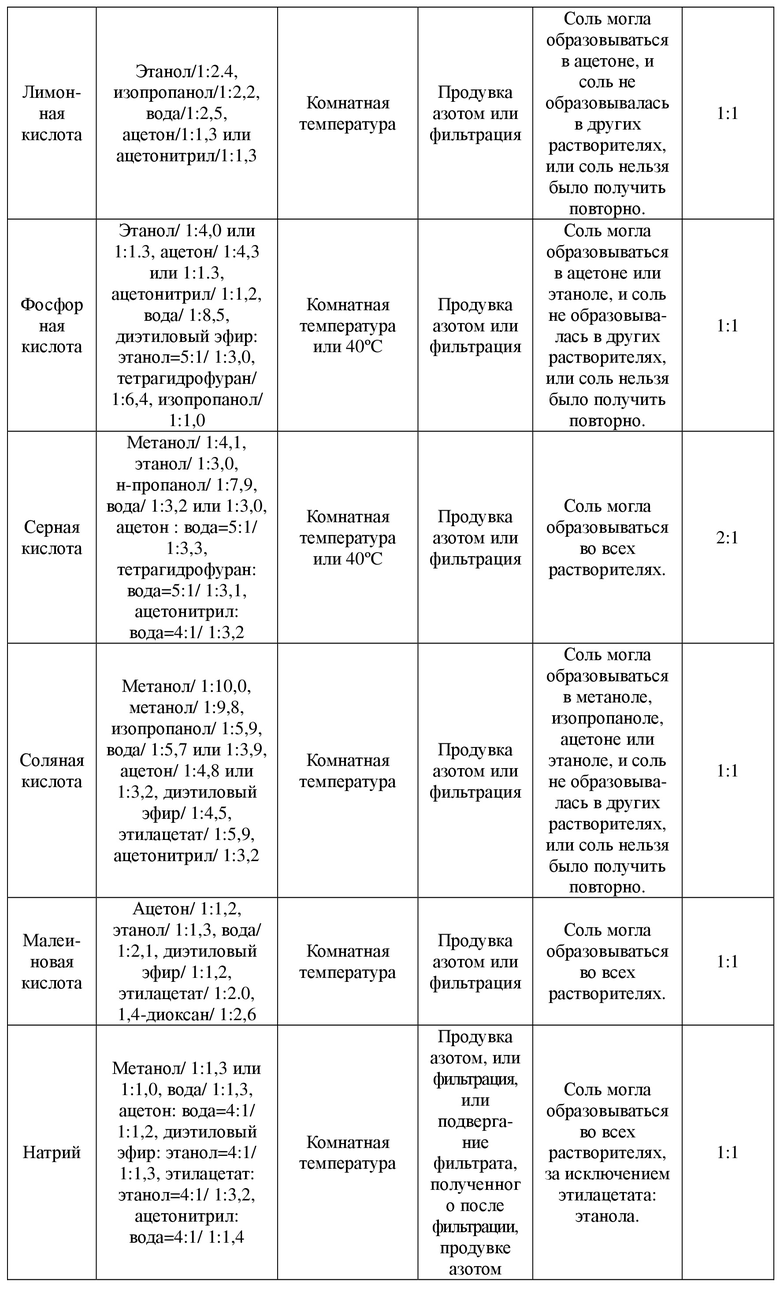
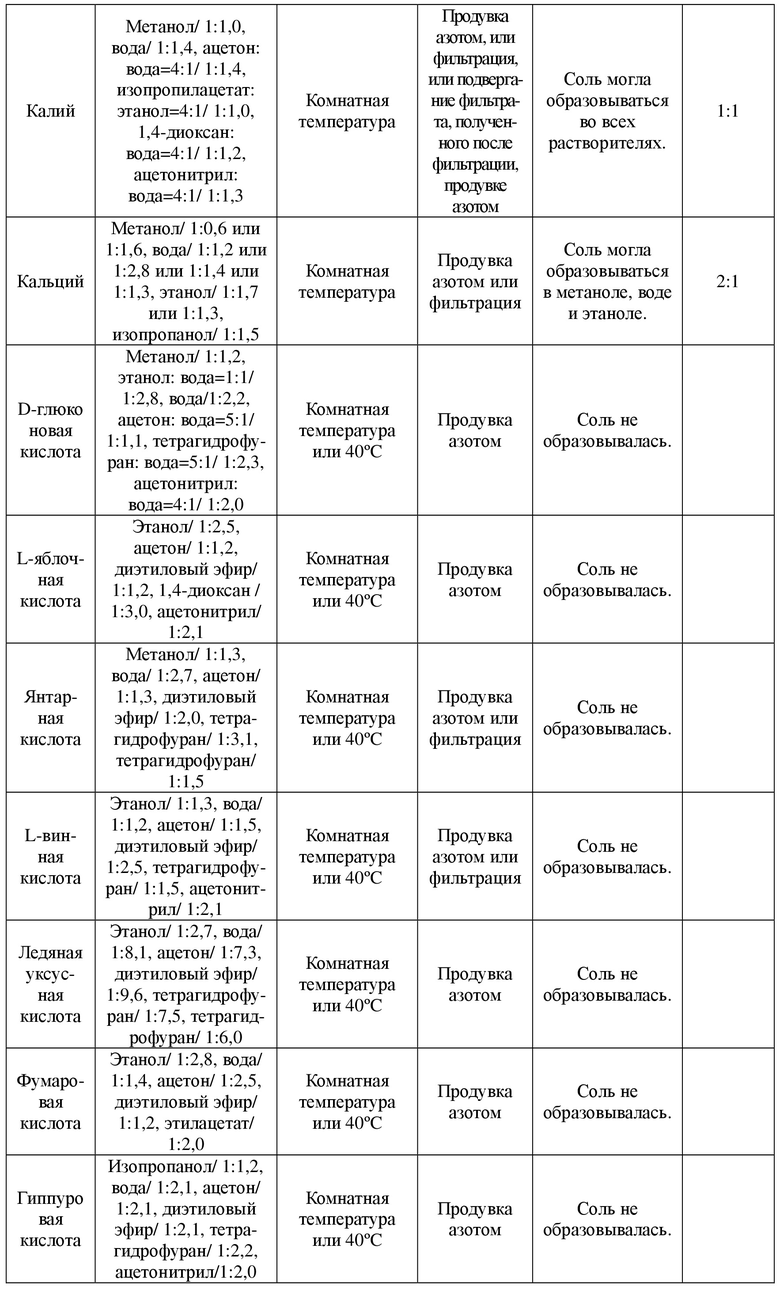
2.2 Получение некоторых солей
Ацетон и вода были выбраны в качестве растворителей для осуществления реакции, для образования соли использовали соединение в свободном состоянии, представленное формулой A, и противоионы в молярном соотношении 1:1,2 , и их соотношение в образованной соли определяли с применением IC. Получали цитрат соединения, представленного формулой A, фосфат соединения, представленного формулой A, гидрохлорид соединения, представленного формулой A, калиевую соль соединения, представленного формулой A, и кальциевую соль соединения, представленного формулой A.
Пример 1: получение натриевой соли соединения, представленного формулой A
Взвешивали 14,50 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,5 мл метанола, и полученную смесь перемешивали с образованием суспензии. Раствор гидроксида натрия (1,75 мг гидроксида натрия добавляли в 0,45 мл метанола) по каплям добавляли в суспензию соединения, представленного формулой A, в метаноле, и полученную смесь перемешивали в течение приблизительно 10 мин. при комнатной температуре с образованием прозрачного раствора, который перемешивали в течение еще 3 ч. Затем растворитель удаляли из раствора путем продувки азотом при комнатной температуре с получением 0,2 мл бесцветного прозрачного раствора, который охлаждали до 5ºC с получением суспензии. Проводили центрифугирование и полученное твердое вещество сушили в течение 16 ч. при комнатной температуре под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 2: получение натриевой соли соединения, представленного формулой A
Взвешивали 40,71 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,4 мл метанола, и полученную смесь перемешивали с образованием суспензии. Раствор гидроксида натрия (4,0 мг гидроксида натрия добавляли в 2,8 мл метанола) по каплям добавляли в суспензию соединения, представленного формулой A, в метаноле, и полученную смесь перемешивали в течение приблизительно 1 ч. при комнатной температуре с получением прозрачного раствора. Раствор перемешивали в течение еще 2 ч., затем проводили фильтрацию, и растворитель удаляли из фильтрата с помощью испарения при комнатной температуре с получением 0,2 мл суспензии. Проводили центрифугирование и полученное твердое вещество сушили в течение 24 ч. при комнатной температуре под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 3: получение натриевой соли соединения, представленного формулой A
Взвешивали 4,9 мг гидроксида натрия и добавляли в 1,0 мл воды, и ультразвуковую обработку проводили с получением прозрачного раствора. Прозрачный раствор по каплям добавляли в 40,7 мг соединения, представленного формулой A, полученного в примере получения 1, и полученную смесь перемешивали в течение 24 ч. при комнатной температуре. Проводили фильтрацию и растворитель удаляли из фильтрата путем продувки азотом при 60ºC с получением 0,2 мл светло-желтого прозрачного раствора. Раствор охлаждали до комнатной температуры для осаждения твердого вещества, затем проводили центрифугирование, и полученное твердое вещество сушили в течение 1 ч. при 40ºC под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
ИК-спектр натриевой соли показан на фиг. 1.
Дифрактограмма XRD натриевой соли показана на фиг. 2.
Термограмма TGA натриевой соли показана на фиг. 3.
Термограмма DSC натриевой соли показана на фиг. 4.
Пример 4: получение натриевой соли соединения, представленного формулой A
Взвешивали 3,5 мг гидроксида натрия и добавляли в 1,0 мл смеси ацетона: воды (4:1), и ультразвуковую обработку проводили с получением прозрачного раствора. Прозрачный раствор по каплям добавляли в 29,2 мг соединения, представленного формулой A, полученного в примере получения 1, и полученную смесь перемешивали в течение 16 ч. при комнатной температуре. Проводили фильтрацию, и полученный осадок на фильтре сушили в течение 1 ч. при 40ºC под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 5: получение натриевой соли соединения, представленного формулой A
Взвешивали 5,05 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,2 мл смеси диэтилового эфира: этанола (4:1), и полученную смесь перемешивали с получением суспензии. Раствор гидроксида натрия (0,65 мг гидроксида натрия добавляли в 0,3 мл смеси диэтилового эфира: этанола (4:1 по объему)) по каплям добавляли в суспензию соединения, представленного формулой A, в смеси диэтилового эфира: этанола, и полученную смесь перемешивали в течение 24 ч. при комнатной температуре. Проводили фильтрацию и растворители удаляли из фильтрата путем испарения при 60ºC. Полученное твердое вещество суспендировали с 0,2 мл диэтилового эфира в течение 1 ч., затем проводили центрифугирование, и твердое вещество, полученное после центрифугирования, сушили в течение 19 ч. при комнатной температуре под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 6: получение натриевой соли соединения, представленного формулой A
Взвешивали 8,02 мг соединения, представленного формулой A, полученного в примере получения 1. В соединение добавляли 8,0 мл смеси н-бутанола: метил-трет-бутилового эфира (1:1) и 2,5 мг гидроксида натрия и полученную смесь перемешивали в течение 1 ч. при 60ºC. Проводили фильтрацию и растворители удаляли из фильтрата путем роторного испарения при 60ºC. Полученное твердое вещество суспендировали с 0,2 мл смеси н-бутанола:метил-трет-бутилового эфира (1:1) в течение 1 ч., затем проводили центрифугирование, и твердое вещество, полученное после центрифугирования, сушили в течение 48 ч. при 40ºC под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 7: получение натриевой соли соединения, представленного формулой A
Взвешивали 45,01 мг соединения, представленного формулой A, полученного в примере получения 1. В соединение добавляли 0,9 мл смеси бутанона: н-пропанола (2:1) и 19,5 мг гидроксида натрия и полученную смесь перемешивали в течение 48 ч. при 60ºC. Проводили фильтрацию и растворители удаляли из фильтрата путем роторного испарения при комнатной температуре. Полученное твердое вещество суспендировали с 0,2 мл смеси н-бутанона:н-пропанола (2:1) в течение 1 ч., затем проводили центрифугирование, и твердое вещество, полученное после центрифугирования, сушили в течение 40 ч. при 60ºC под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 8: получение натриевой соли соединения, представленного формулой A
Взвешивали 4,69 мг гидроксида натрия и добавляли в 1,0 мл воды и ультразвуковую обработку проводили с получением прозрачного раствора. Прозрачный раствор по каплям добавляли в 38,77 мг соединения, представленного формулой A, полученного в примере получения 1, затем добавляли 14,0 мл воды, и полученную смесь перемешивали в течение 16 ч. при комнатной температуре. Проводили фильтрацию и растворитель удаляли из фильтрата путем продувки азотом при 50ºC с получением 0,2 мл светло-желтого прозрачного раствора. Раствор охлаждали до 5ºC для осаждения твердого вещества, затем проводили центрифугирование и полученное твердое вещество сушили в течение 24 ч. при 40ºC под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 9: получение натриевой соли соединения, представленного формулой A
Взвешивали 6,15 мг соединения, представленного формулой A, полученного в примере получения 1. В соединение добавляли 3,0 мл смеси метанола: изопропилового эфира (1:1) и 1,3 мг твердого гидроксида натрия и полученную смесь перемешивали в течение 1 ч. при 40ºC. Проводили фильтрацию и растворители удаляли из фильтрата путем роторного испарения при 50ºC. Полученное твердое вещество суспендировали с 0,1 мл смеси метанола:изопропилового эфира (1:1) в течение 1 ч., затем проводили центрифугирование и твердое вещество, полученное после центрифугирования, сушили в течение 24 ч. при 25ºC под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 10: получение натриевой соли соединения, представленного формулой A
Взвешивали 35,62 мг соединения, представленного формулой A, полученного в примере получения 1. В соединение добавляли 1,2 мл ацетонитрила и 8,7 мг твердого гидроксида натрия и полученную смесь перемешивали в течение 3 ч. при 35ºC. Проводили фильтрацию и растворитель удаляли из фильтрата путем роторного испарения при комнатной температуре с получением 0,2 мл бесцветного прозрачного раствора. Раствор охлаждали до 5ºC для осаждения твердого вещества, затем проводили центрифугирование и полученное твердое вещество сушили в течение 30 ч. при 40ºC под вакуумом с получением натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что натриевая соль соединения, представленного формулой A, образовывалась при реакции соединения, представленного формулой A, и иона натрия в молярном соотношении 1:1.
Пример 11: получение сульфата соединения, представленного формулой A
Взвешивали 76,02 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 5,2 мл метанола и полученную смесь перемешивали с образованием суспензии. Раствор серной кислоты (7,3 мг 98% серной кислоты добавляли в 7,6 мл метанола) по каплям добавляли в суспензию соединения, представленного формулой A, в метаноле и полученную смесь перемешивали в течение 5 ч. при комнатной температуре с получением суспензии. Суспензию перемешивали в течение еще 1 ч. после добавления 5,0 мл метанола, затем проводили фильтрацию, и растворитель удаляли из фильтрата путем продувки азотом при комнатной температуре с получением 1,0 мл суспензии. Проводили фильтрацию и полученное твердое вещество сушили в течение 20 ч. при комнатной температуре под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 12: получение сульфата соединения, представленного формулой A
Взвешивали 34,41 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 1,0 мл этанола и полученную смесь перемешивали с образованием суспензии. В суспензию соединения, представленного формулой A, в этаноле добавляли 24,82 мг 98% серной кислоты и полученную смесь перемешивали в течение 24 ч. при комнатной температуре. Проводили фильтрацию, и полученный осадок на фильтре сушили в течение 10 ч. при 40ºC под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 13: получение сульфата соединения, представленного формулой A
Взвешивали 4,63 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,2 мл н-пропанола и полученную смесь перемешивали с образованием суспензии. Раствор серной кислоты (8,79 мг 98% серной кислоты добавляли в 0,3 мл н-пропанола) по каплям добавляли в суспензию соединения, представленного формулой A, в н-пропаноле и полученную смесь перемешивали в течение 16 ч. при комнатной температуре. Проводили фильтрацию и растворитель удаляли из фильтрата путем продувки азотом при комнатной температуре с получением маслянистого вещества. В маслянистое вещество добавляли воду и проводили обработку ультразвуком с получением суспензии. Проводили центрифугирование и полученное твердое вещество сушили в течение 24 ч. при комнатной температуре под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
ИК-спектр сульфата показан на фиг. 5.
Дифрактограмма XRD сульфата показана на фиг. 6.
Термограмма TGA сульфата показана на фиг. 7.
Термограмма DSC сульфата показана на фиг. 8.
Пример 14: получение сульфата соединения, представленного формулой A
Взвешивали 10,02 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 1,0 мл воды, и полученную смесь перемешивали с образованием суспензии. В суспензию соединения, представленного формулой A, в воде добавляли 7,88 мг 98% серной кислоты и полученную смесь перемешивали в течение 24 ч. при 40ºC. Проводили фильтрацию и полученный осадок на фильтре сушили в течение 1 ч. при 60ºC под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 15: получение сульфата соединения, представленного формулой A
Взвешивали 34,4 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 1,0 мл воды, и полученную смесь перемешивали с образованием суспензии. Раствор серной кислоты (25,0 мг 98% серной кислоты добавляли в 0,5 мл воды) по каплям добавляли в суспензию соединения, представленного формулой A, в воде, и полученную смесь перемешивали в течение 24 ч. при комнатной температуре. Проводили фильтрацию, и полученный осадок на фильтре сушили в течение 1 ч. при 40ºC под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 16: получение сульфата соединения, представленного формулой A
Взвешивали 10,25 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,2 мл воды, и полученную смесь перемешивали с образованием суспензии. В суспензию соединения, представленного формулой A, в воде последовательно добавляли 8,25 мг 98% серной кислоты и 1,0 мл ацетона и полученную смесь перемешивали в течение 1 ч. при комнатной температуре с получением прозрачного раствора. Проводили фильтрацию, затем растворители удаляли из фильтрата путем продувки азотом при комнатной температуре и полученное твердое вещество сушили в течение 24 ч. при комнатной температуре под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 17: получение сульфата соединения, представленного формулой A
Взвешивали 10,40 мг соединения, представленного формулой A, полученного в примере получения 1. В соединение, представленное формулой A, последовательно добавляли 0,2 мл воды, 7,92 мг 98% серной кислоты и 1,0 мл тетрагидрофурана и полученную смесь перемешивали в течение 3 ч. при комнатной температуре с получением прозрачного раствора. Проводили фильтрацию и растворители удаляли из фильтрата путем продувки азотом при 60ºC с получением 0,3 мл суспензии. Проводили центрифугирование, и полученное твердое вещество сушили в течение 20 ч. при 40ºC под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 18: получение сульфата соединения, представленного формулой A
Взвешивали 4,15 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,2 мл смеси воды:ацетонитрила (1:4), и полученную смесь перемешивали с образованием суспензии. Раствор серной кислоты (3,2 мг 98% серной кислоты добавляли в 0,3 мл смеси воды:ацетонитрила (1:4)) по каплям добавляли в суспензию соединения, представленного формулой A, в смеси воды:ацетонитрила (1:4) и полученную смесь перемешивали в течение 24 ч. при комнатной температуре. Проводили фильтрацию и растворители удаляли из фильтрата путем продувки азотом при комнатной температуре с получением 0,1 мл суспензии. Проводили центрифугирование и полученное твердое вещество сушили в течение 1 ч. при 50ºC под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 19: получение сульфата соединения, представленного формулой A
Взвешивали 5,0 мг соединения, представленного формулой A, полученного в примере получения 1. В соединение добавляли 5,0 мл смеси втор-бутанола: бутанона (1:4) и 10,3 мг 98% серной кислоты и полученную смесь перемешивали в течение 30 ч. при -10ºC. Проводили фильтрацию и растворители удаляли из фильтрата путем продувки азотом при 40ºC с получением 0,1 мл суспензии. Проводили центрифугирование и полученное твердое вещество сушили в течение 10 ч. при 60ºC под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 20: получение сульфата соединения, представленного формулой A
Взвешивали 40,0 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,4 мл смеси 1,4-диоксана:воды (1:1), и полученную смесь перемешивали с образованием суспензии. Раствор серной кислоты (96,7 мг 98% серной кислоты добавляли в 0,4 мл 1,4-диоксана: воды (1:1)) по каплям добавляли в суспензию соединения, представленного формулой A, в 1,4-диоксане: воде (1:1), и полученную смесь перемешивали в течение 72 ч. при 60ºC. Проводили фильтрацию и растворители удаляли из фильтрата путем продувки азотом при 60ºC. Полученное твердое вещество суспендировали с 0,2 мл смеси 1,4-диоксана: воды (1:1) в течение 1 ч., затем проводили центрифугирование и твердое вещество, полученное после центрифугирования, сушили в течение 48 ч. при 40ºC под вакуумом с получением сульфата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что сульфат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и серной кислоты в молярном соотношении 2:1.
Пример 21: получение малеата соединения, представленного формулой A
Взвешивали 51,7 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 1,0 мл ацетона. Раствор малеиновой кислоты (17,7 мг малеиновой кислоты добавляли в 1,0 мл ацетона) по каплям добавляли в систему из соединения, представленного формулой A, в ацетоне при перемешивании и полученную смесь перемешивали в течение 24 ч. при комнатной температуре. Проводили фильтрацию и проводили высушивание в течение 16 ч. при 40ºC под вакуумом с получением малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
ИК-спектр малеата показан на фиг. 9.
Дифрактограмма XRD малеата показана на фиг. 10.
Термограмма TGA малеата показана на фиг. 11.
Термограмма DSC малеата показана на фиг. 12.
Пример 22: получение малеата соединения, представленного формулой A
Взвешивали 10,37 мг соединения, представленного формулой A, полученного в примере получения 1. Раствор малеиновой кислоты (3,91 мг малеиновой кислоты добавляли в 1,0 мл этанола) по каплям добавляли в соединение и полученную смесь перемешивали в течение 10 ч. при комнатной температуре. Проводили фильтрацию и полученный осадок на фильтре сушили в течение 20 ч. при 25ºC под вакуумом с получением малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
Пример 23: получение малеата соединения, представленного формулой A
Взвешивали 7,63 мг соединения, представленного формулой A, полученного в примере получения 1. Раствор малеиновой кислоты (4,47 мг малеиновой кислоты добавляли в 1,0 мл воды) по каплям добавляли в соединение и полученную смесь перемешивали в течение 24 ч. при 40ºC. Проводили фильтрацию и полученный осадок на фильтре сушили в течение 1 ч. при 40ºC под вакуумом с получением малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
Пример 24: получение малеата соединения, представленного формулой A
Взвешивали 10,70 мг соединения, представленного формулой A, полученного в примере получения 1. В соединение добавляли 3,52 мг малеиновой кислоты и 1,0 мл диэтилового эфира и полученную смесь перемешивали в течение 24 ч. при комнатной температуре. Проводили фильтрацию и полученный осадок на фильтре сушили в течение 24 ч. при 10ºC под вакуумом с получением малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
Пример 25: получение малеата соединения, представленного формулой A
Взвешивали 13,33 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 1,5 мл этилацетата. Раствор малеиновой кислоты (5,14 мг малеиновой кислоты добавляли в 1,0 мл этилацетата) по каплям добавляли в систему из соединения, представленного формулой A, в этилацетате при перемешивании и полученную смесь перемешивали в течение 18 ч. при комнатной температуре. Проводили фильтрацию и полученный осадок на фильтре сушили в течение 1 ч. при 40ºC под вакуумом с получением малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
Пример 26: получение малеата соединения, представленного формулой A
Взвешивали 6,04 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 1,0 мл 1,4-диоксана. Раствор малеиновой кислоты (4,4 мг малеиновой кислоты добавляли в 0,4 мл 1,4-диоксана) по каплям добавляли в систему из соединения, представленного формулой A, в 1,4-диоксане при перемешивании и полученную смесь перемешивали в течение 20 ч. при комнатной температуре. Проводили фильтрацию и полученный осадок на фильтре сушили в течение 24 ч. при 50ºC под вакуумом с получением 34,3 мг малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
Пример 27: получение малеата соединения, представленного формулой A
Взвешивали 5,0 мг соединения, представленного формулой A, полученного в примере получения 1. В соединение добавляли 4,7 мг малеиновой кислоты и 5,0 мл смеси бутанона: метилформиата (2:1) и полученную смесь перемешивали в течение 30 ч. при 60ºC. Проводили фильтрацию и проводили высушивание в течение 37 ч. при 56ºC под вакуумом с получением малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
Пример 28: получение малеата соединения, представленного формулой A
Взвешивали 40,5 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,6 мл смеси метанола: метил-трет-бутилового эфира (1:1). Раствор малеиновой кислоты (11,5 мг малеиновой кислоты добавляли в 0,4 мл смеси метанола: метил-трет-бутилового эфира (1:1)) по каплям добавляли в систему из соединения, представленного формулой A, в смеси метанола: метил-трет-бутилового эфира (1:1) при перемешивании и полученную смесь перемешивали в течение 48 ч. при 45ºC. Проводили фильтрацию и проводили высушивание в течение 48 ч. при 40ºC под вакуумом с получением малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
Пример 29: получение малеата соединения, представленного формулой A
Взвешивали 50,0 мг соединения, представленного формулой A, полученного в примере получения 1, и добавляли в 0,5 мл смеси н-бутанола: изопропилацетата (3:1). Раствор малеиновой кислоты (70,9 мг малеиновой кислоты добавляли в 0,5 мл смеси н-бутанола: изопропилацетата (3:1)) по каплям добавляли в систему из соединения, представленного формулой A, в смеси н-бутанола: изопропилацетата (3:1) при перемешивании и полученную смесь перемешивали в течение 72 ч. при -10ºC. Проводили фильтрацию и проводили высушивание в течение 30 ч. при 60ºC под вакуумом с получением малеата соединения, представленного формулой A, в соответствии с настоящей заявкой.
Определение характеристик с помощью IC показало, что малеат соединения, представленного формулой A, образовывался при реакции соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1.
Сравнительный пример 1: растворимость натриевой соли соединения, представленного формулой A
Для выполнения эксперимента на растворимость в воде брали натриевую соль соединения, представленного формулой A, в соответствии с настоящей заявкой. Конкретная операция являлась следующей: отбирали 5 мг натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой, и помещали в стеклянную бутылочку объемом 20 мл, и деионизированную воду постепенно добавляли при 25ºC в бутылочку, и проводили обработку ультразвуком с получением прозрачного раствора. Рассчитывали значение растворимости образца в воде.
Таблица 2. Растворимость в воде натриевой соли соединения, представленного формулой A, в соответствии с настоящей заявкой

Из таблицы 2 можно увидеть, что натриевая соль соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется высокой растворимостью, а следовательно она характеризуется лучшей биодоступностью.
Сравнительный пример 2: сравнение термической стабильности солевых форм соединения, представленного формулой A
Для выполнения анализов DSC и TGA брали натриевую соль соединения, представленного формулой A, в соответствии с настоящей заявкой и общепринятые соли (цитрат соединения, представленного формулой A, фосфат соединения, представленного формулой A, и гидрохлорид соединения, представленного формулой A) и получали данные о температуре плавления и температуре разложения каждой солевой формы.
Таблица 3. Данные о температуре плавления натриевой соли и других общепринятых солей соединения, представленного формулой A, в соответствии с настоящей заявкой
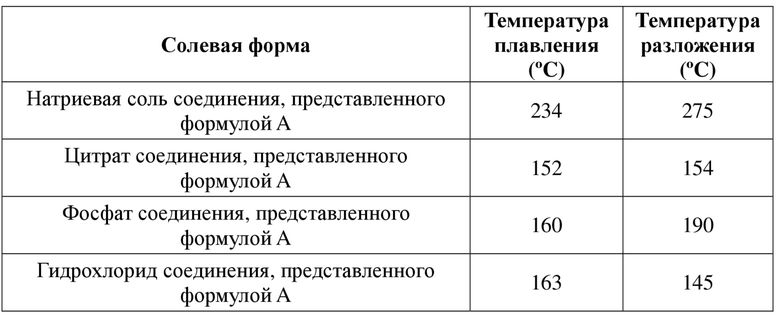
Из таблицы 3 можно увидеть, что по сравнению с общепринятыми солями (цитрат соединения, представленного формулой A, фосфат соединения, представленного формулой A, и гидрохлорид соединения, представленного формулой A) натриевая соль соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуется очень высокой температурой плавления и температурой разложения, а следовательно она характеризуется лучшей термической стабильностью.
Сравнительный пример 3: сравнение растворимости солевых форм соединения, представленного формулой A
Для выполнения эксперимента на растворимость в воде брали известное соединение в свободном состоянии, представленное формулой A, общепринятые соли (кальциевая соль соединения, представленного формулой A, цитрат соединения, представленного формулой A, фосфат соединения, представленного формулой A, и гидрохлорид соединения, представленного формулой A) и сульфат соединения, представленного формулой A, и малеат соединения, представленного формулой A, в соответствии с настоящей заявкой. Конкретная операция являлась следующей: брали 5 мг известного соединения в свободном состоянии, представленного формулой A, общепринятых солей (кальциевой соли соединения, представленного формулой A, цитрата соединения, представленного формулой A, фосфата соединения, представленного формулой A, и гидрохлорида соединения, представленного формулой A) и сульфата соединения, представленного формулой A, и малеата соединения, представленного формулой A, полученных в соответствии с настоящей заявкой, и помещали в стеклянные бутылочки объемом 20 мл, соответственно, и в каждую из бутылочек добавляли 15 мл деионизированной воды, и перемешивали в течение 2 ч. при 25ºC. Затем отбирали образцы и фильтровали их и определяли концентрации с помощью HPLC. В каждом из образцов рассчитывали растворимость активного ингредиента в воде.
Таблица 4. Растворимость в воде соединения в свободном состоянии, представленного формулой A, и его солевых форм
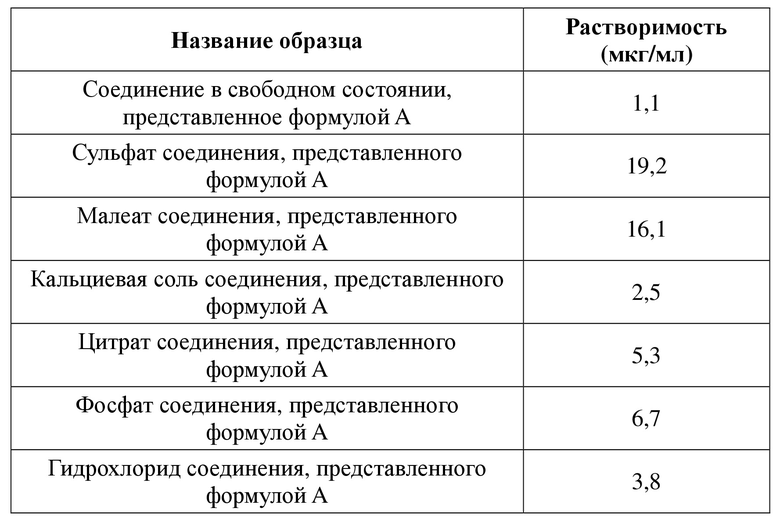
Из таблицы 4 можно увидеть, что растворимость в воде при 25ºC сульфата соединения, представленного формулой A, и малеата соединения, представленного формулой A, в соответствии с настоящей заявкой является приблизительно в 10-20 раз большей, чем у известного свободного состояния соединения, представленного формулой A; и является приблизительно в 3-8 раз большей, чем у других общепринятых солей (кальциевой соли соединения, представленного формулой A, цитрата соединения, представленного формулой A, фосфата соединения, представленного формулой A, и гидрохлорида соединения, представленного формулой A), поэтому сульфат и малеат характеризуются лучшей растворимостью и лучшей биодоступностью.
Сравнительный пример 4. Сравнение гигроскопичности солевых форм соединения, представленного формулой A
Для выполнения анализа DVS брали сульфат соединения, представленного формулой A, и малеат соединения, представленного формулой A, в соответствии с настоящей заявкой и общепринятые соли (калиевая соль соединения, представленного формулой A, кальциевая соль соединения, представленного формулой A, цитрат соединения, представленного формулой A, фосфат соединения, представленного формулой A, и гидрохлорид соединения, представленного формулой A) и для каждой солевой формы получали данные по гигроскопичности.
Таблица 5. Данные по гигроскопичности сульфата соединения, представленного формулой A, малеата соединения, представленного формулой A, в соответствии с настоящей заявкой и других общепринятых солей
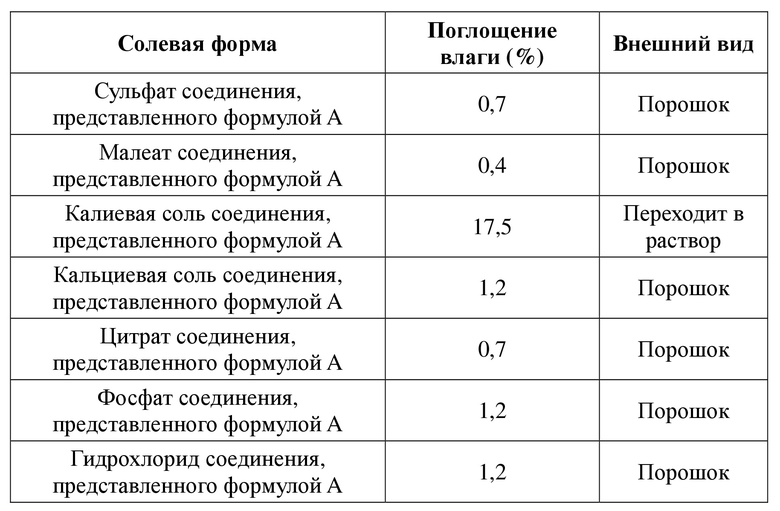
Из таблицы 5 можно увидеть, что при сравнении с общепринятыми солями (калиевой солью соединения, представленного формулой A, кальциевой солью соединения, представленного формулой A, цитратом соединения, представленного формулой A, фосфатом соединения, представленного формулой A, и гидрохлоридом соединения, представленного формулой A), сульфат соединения, представленного формулой A, и малеат соединения, представленного формулой A, в соответствии с настоящей заявкой имеют меньший прирост массы из-за гигроскопичности, таким образом, имеют лучшую стабильность при хранении и лучше могут избегать проблем с качеством, безопасностью и стабильностью при изготовлении и/или хранении лекарственного средства и т. д.
Сравнительный пример 5: сравнение стабильности кристаллических форм солей соединения, представленного формулой A
Для выполнения эксперимента на стабильность брали кристаллическую форму сульфата соединения, представленного формулой A, и кристаллическую форму малеата соединения, представленного формулой A, в соответствии с настоящей заявкой. Конкретная операция являлась следующей: брали по 60 мг образцов кристаллической формы сульфата соединения, представленного формулой A, и кристаллической формы малеата соединения, представленного формулой A, в соответствии с настоящей заявкой и выдерживали в течение 30 дней соответственно при общепринятых условиях (герметизация и помещение в темное место при 25ºC), высокотемпературных условиях (герметизация и помещение в темное место при 60ºC) и условиях ускоренного старения (открытый контейнер и помещение в темное место при 40ºC, относительной влажности 75%) для изучения стабильности кристаллической формы.
Таблица 6. Результаты испытания на стабильность кристаллической формы сульфата соединения, представленного формулой A, и кристаллической формы малеата соединения, представленного формулой A, в соответствии с настоящей заявкой
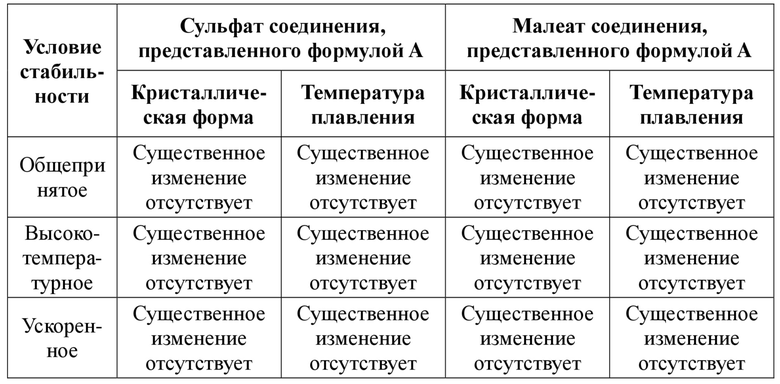
Из таблицы 6 можно увидеть, что кристаллическая форма сульфата соединения, представленного формулой A, и кристаллическая форма малеата соединения, представленного формулой A, в соответствии с настоящей заявкой характеризуются хорошей стабильностью, что является благоприятным для адаптации к различным условиям окружающей среды при изготовлении, хранении и перевозке.
Сравнительный пример 6: сравнение стабильности кристаллических форм солей соединения, представленного формулой A
Для получения суспензий в растворителях брали кристаллическую форму натриевой соли соединения, представленного формулой A, кристаллическую форму сульфата соединения, представленного формулой A, и кристаллическую форму малеата соединения, представленного формулой A, соответственно , как показано в таблице 7, и суспензии перемешивали в течение 3 дней при комнатной температуре. Изучали стабильность кристаллической формы и полученные результаты сравнивали с результатами в сравнительном примере 1 в патентном документе CN105315266A.
Таблица 7. Результаты испытания на стабильность кристаллической формы солевых форм соединения, представленного формулой A, в соответствии с настоящей заявкой и соединения в свободном состоянии, представленного формулой A, в растворителях
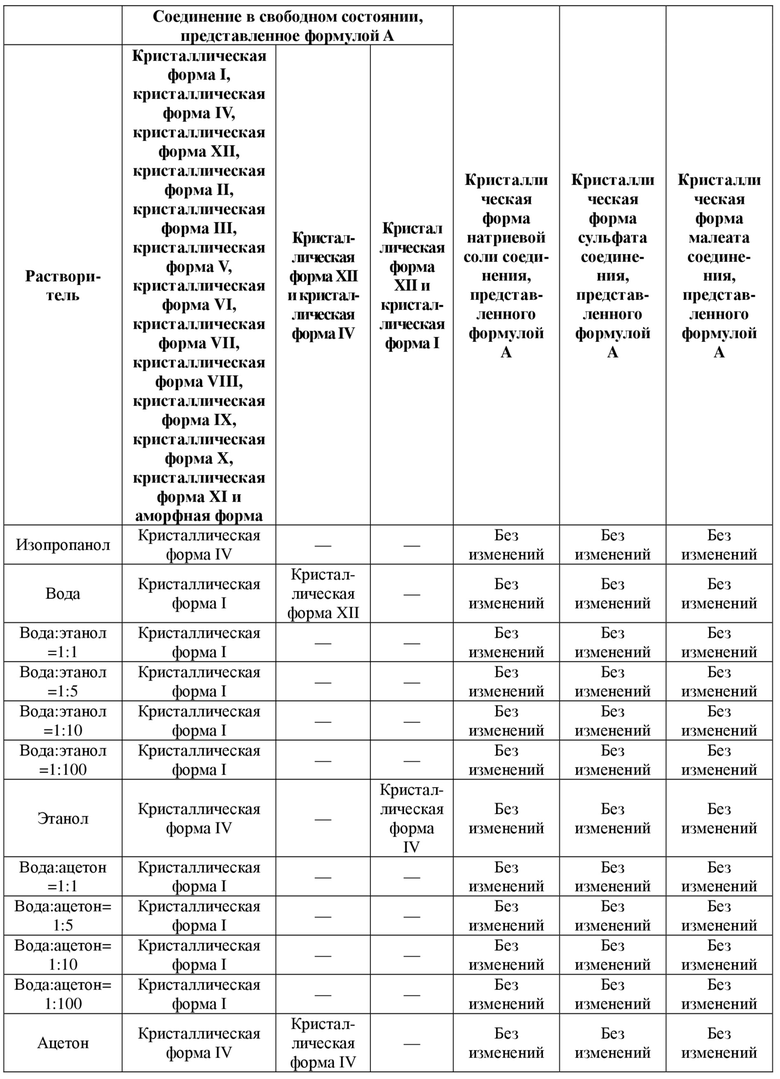
Из таблицы 7 можно увидеть, что соединение в свободном состоянии, представленное формулой A, в различных растворителях присутствует в виде различных конечных кристаллических форм, что показывает, что для соединения в свободном состоянии, представленного формулой A, характерными являются проблемы, связанные с образованием смешанных кристаллов, и кристаллическую форму сложно регулировать при получении лекарственного средства. Напротив, кристаллическая форма каждой солевой формы соединения, представленного формулой A, в соответствии с настоящей заявкой является относительно единственной, выбор растворителя, который следует использовать при получении, является более гибким, и кристаллическая форма является более стабильной.
Все патентные документы и непатентные публикации, цитируемые в описании, включены в данный документ посредством ссылки во всей их полноте.
Вышеописанные варианты осуществления являются только конкретными вариантами осуществления настоящей заявки, и объем защиты настоящей заявки не ограничен ими. Любое изменение или замена, которые могут без труда представить специалисты в данной области в пределах технического объема, раскрытого в настоящей заявке, охвачены объемом защиты настоящей заявки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 7Н-ПИРРОЛО[2,3-D] ПИРИМИДИНА И ЕГО ИНТЕРМЕДИАТА | 2016 |
|
RU2755618C2 |
| ПОЛИМОРФНЫЕ ФОРМЫ МАКРОЦИКЛИЧЕСКОГО ИНГИБИТОРА HCV | 2008 |
|
RU2533830C2 |
| СОЛЬ КОНДЕНСИРОВАННОГО ГЕТЕРОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО И ЕГО КРИСТАЛЛЫ | 2011 |
|
RU2557237C2 |
| СОЛИ N-(4-ФТОРБЕНЗИЛ)-N-(1-МЕТИЛПИПЕРИДИН-4-ИЛ)-N`-(2-МЕТИЛПРОПИЛОКСИ)ФЕНИЛМЕТИЛ)КАРБАМИДА И ИХ ПРИГОТОВЛЕНИЕ | 2005 |
|
RU2387643C2 |
| СОЛИ ПРОИЗВОДНОГО ХИНАЗОЛИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2720810C2 |
| СОЛИ ЭФИРОВ БЕНЗИМИДАЗОЛИЛПИРИДИЛА И ИХ СОДЕРЖАЩИЕ СОСТАВЫ | 2007 |
|
RU2457206C2 |
| СОЛИ ЭТИЛОВОГО ЭФИРА 3-(2-(4-(4-(АМИНОИМИНОМЕТИЛ)ФЕНИЛ)-4-МЕТИЛ-2,5-ДИОКСОИМИДАЗОЛИДИН-1-ИЛ)АЦЕТИЛАМИНО)-3-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2174119C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ НАТРИЕВОЙ СОЛИ(4-{ 4-[5-(6-ТРИФТОРМЕТИЛ-ПИРИДИН-3-ИЛАМИНО) ПИРИДИН-2-ИЛ] ФЕНИЛ} ЦИКЛОГЕКСИЛ) УКСУСНОЙ КИСЛОТЫ | 2011 |
|
RU2612556C2 |
| СОЛЬ АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ ИЛИ ЕЕ КРИСТАЛЛИЧЕСКАЯ ФОРМА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИНГИБИТОР FLT3 | 2014 |
|
RU2641106C2 |
| Твёрдые формы цефтолозана | 2014 |
|
RU2703457C2 |
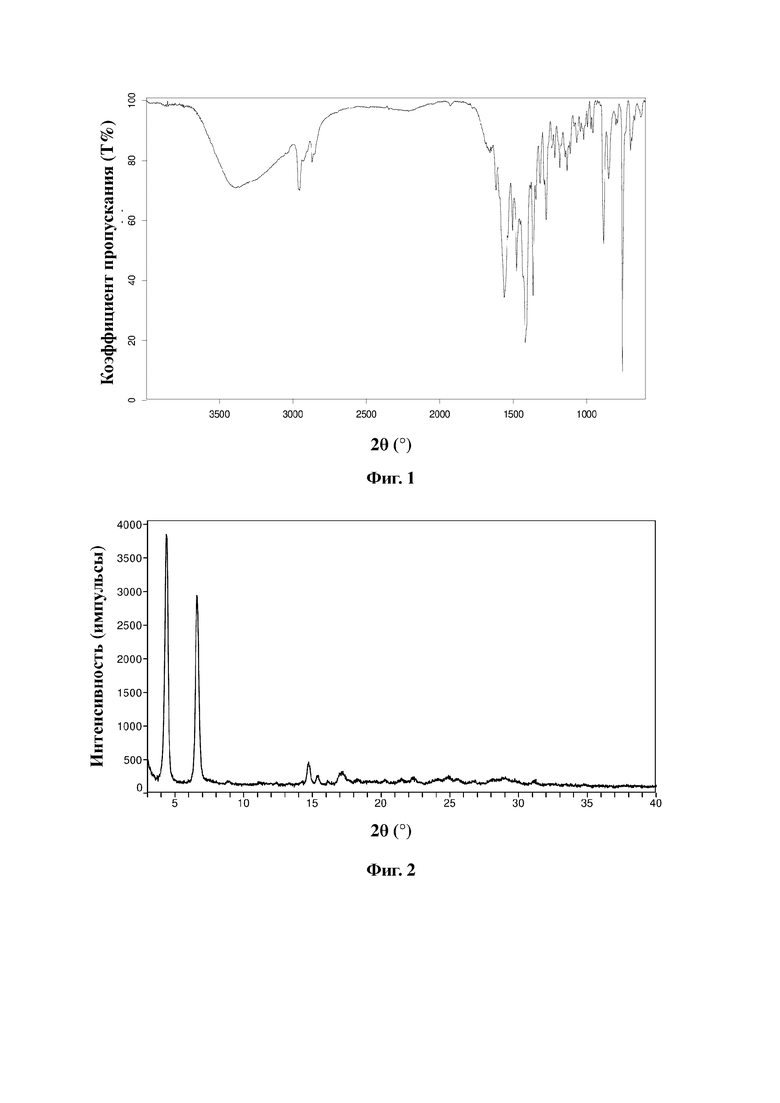
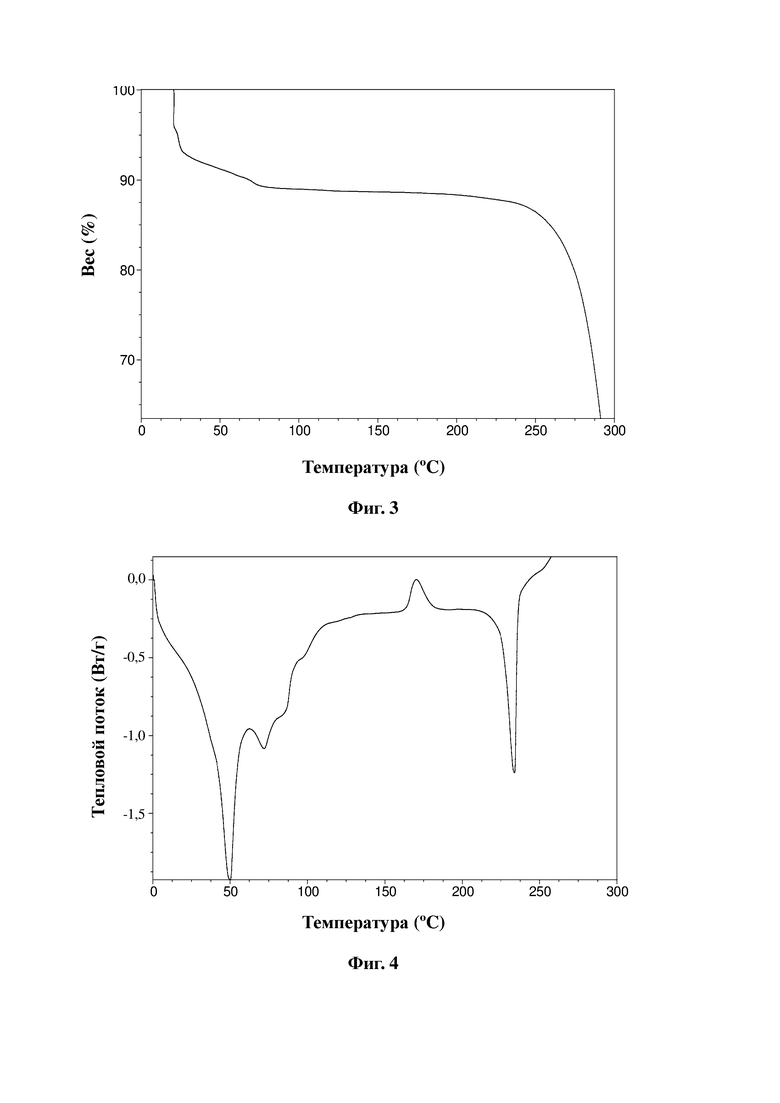
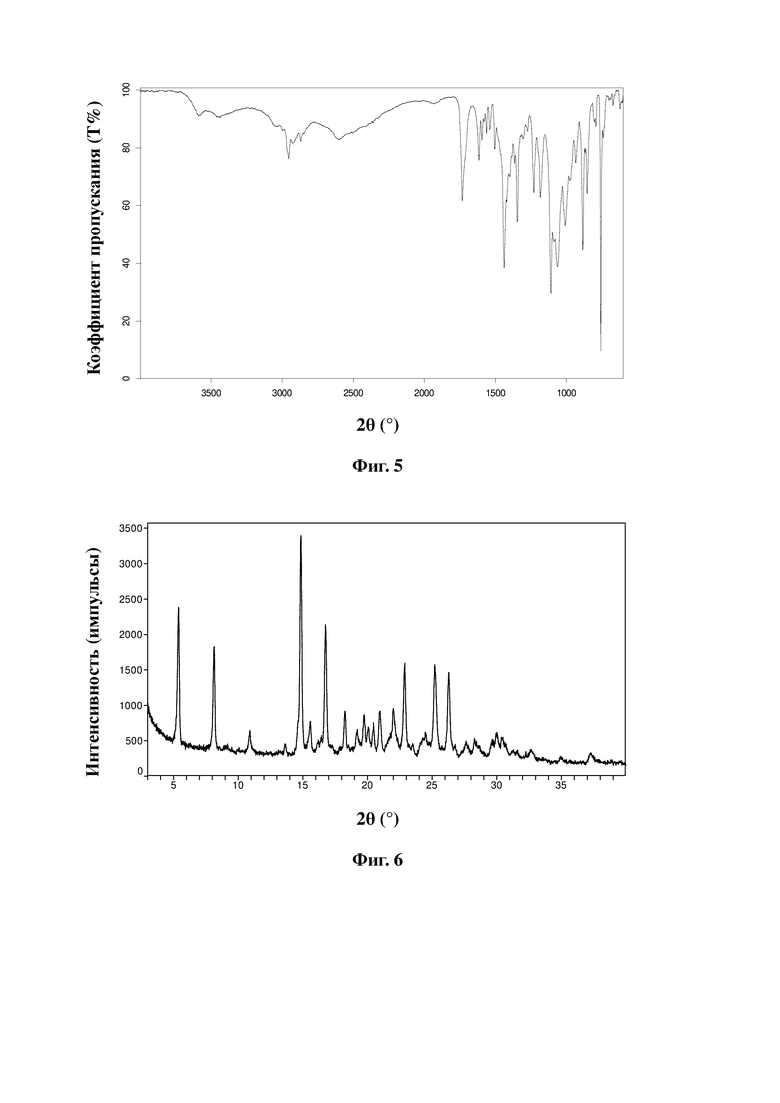
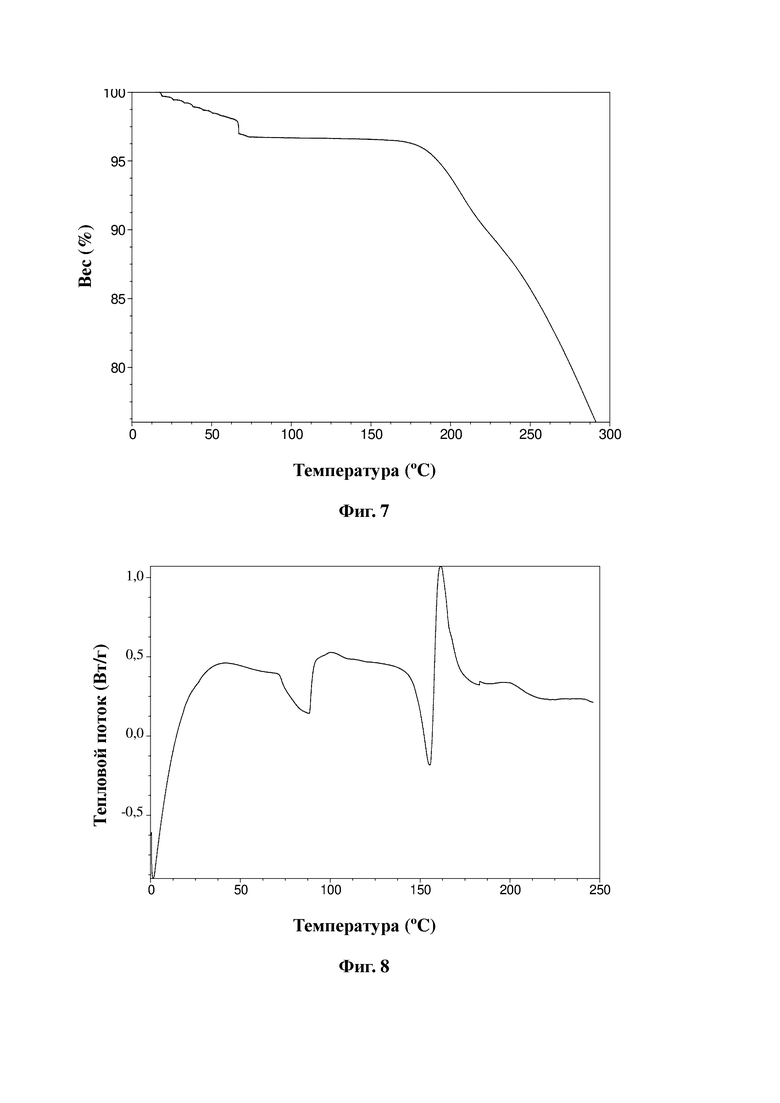
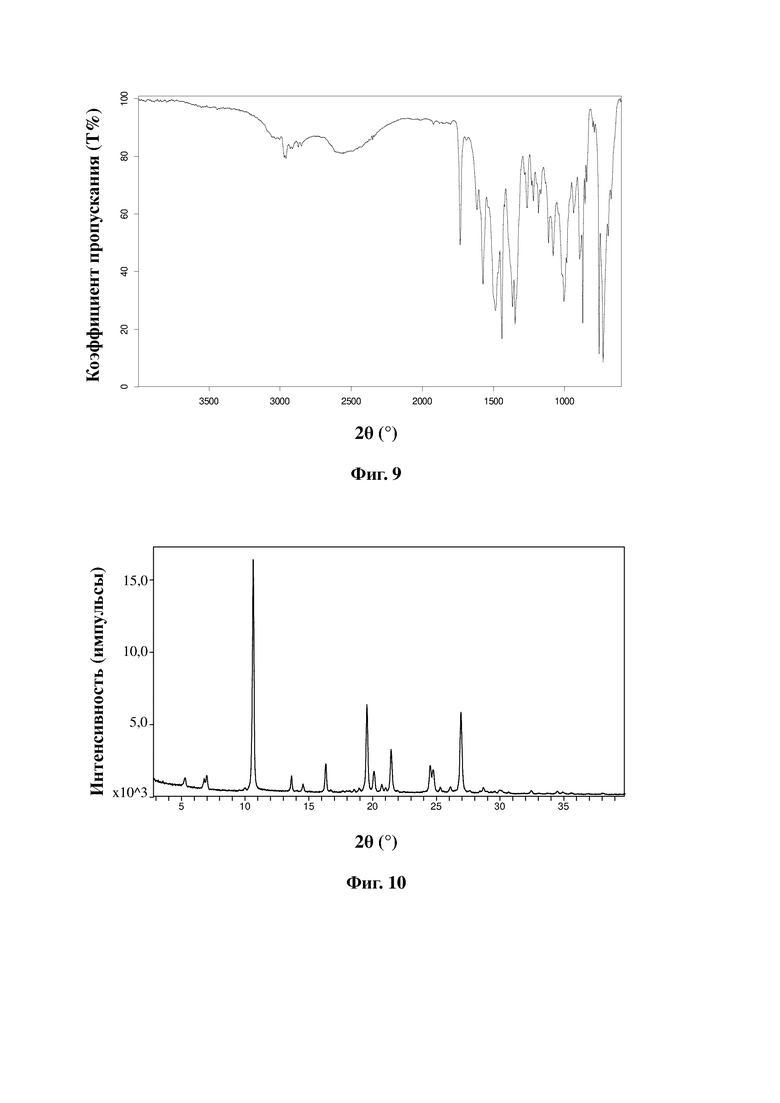
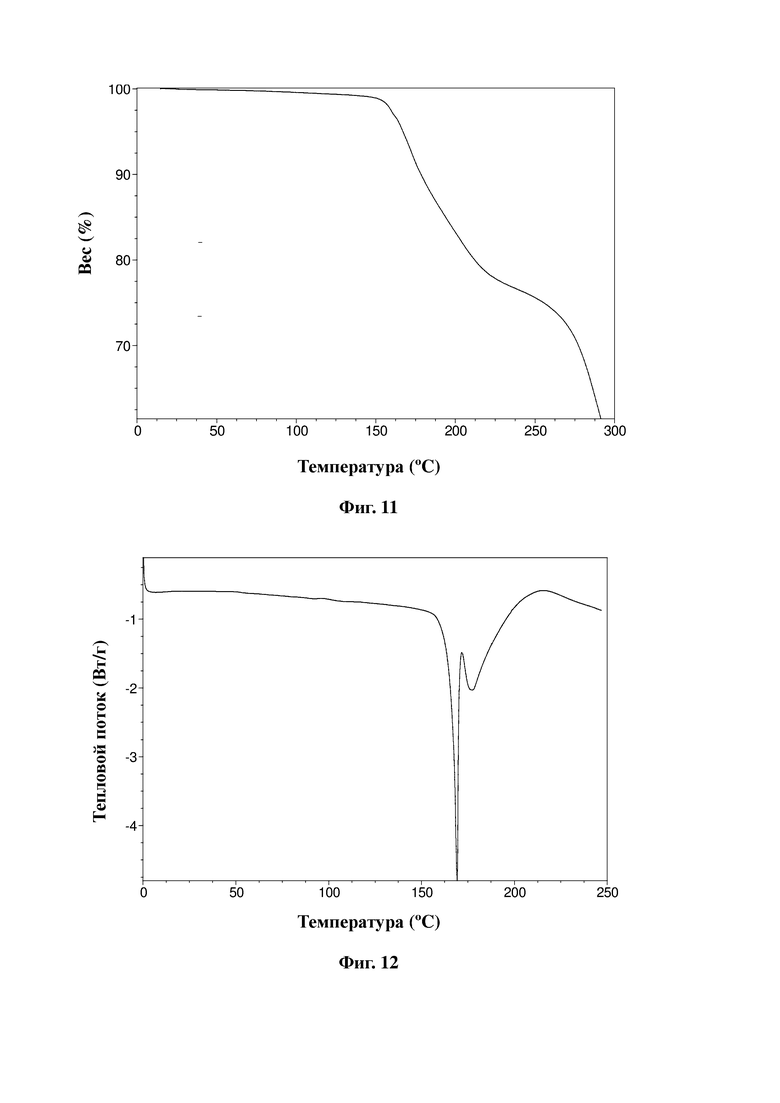
Изобретение относится к способу получения малеата соединения, имеющего структуру, представленную формулой I. Способ осуществляют путем получения суспензии или раствора соединения, представленного формулой A, и суспензии или раствора малеиновой кислоты в растворителе, выбранном из группы, состоящей из этанола, ацетона, диэтилового эфира, воды, этилацетата, 1,4-диоксана или их смеси, соответственно, смешивания суспензии или раствора соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1,0-1:2,6 для осуществления реакции; при этом реакцию проводят при 10-40°C при перемешивании, в течение 10-24 ч.. Затем удаляют растворитель после завершения реакции; и выполняют высушивания при 10-40°C, в течение 1-24 ч., и при этом отношение массы соединения, представленного формулой A, к объему растворителя в данном способе составляет 4 мг:1 мл-26 мг:1 мл. Технический результат – получение малеата 1-{2-фтор-4-[5-(4-изобутилфенил)-1,2,4-оксадиазол-3-ил]бензил}-3-азетидинкарбоновой кислоты, характеризующейся существенно улучшенной растворимостью в воде и лучшей биодоступностью. 3 з.п. ф-лы, 12 ил., 7 табл., 36 пр.
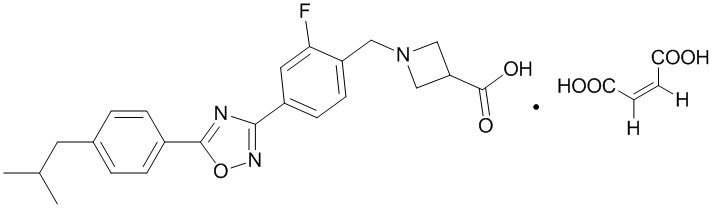
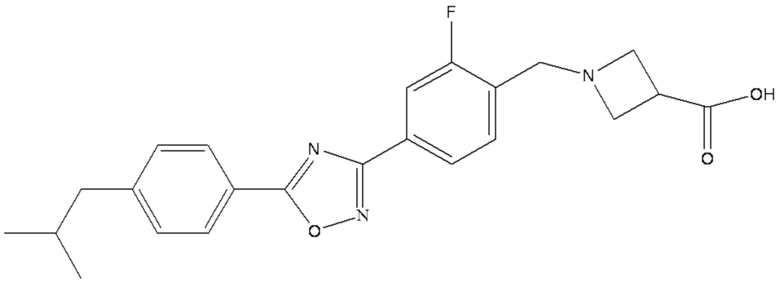 А
А
1. Способ получения малеата соединения, имеющего структуру, представленную формулой I:
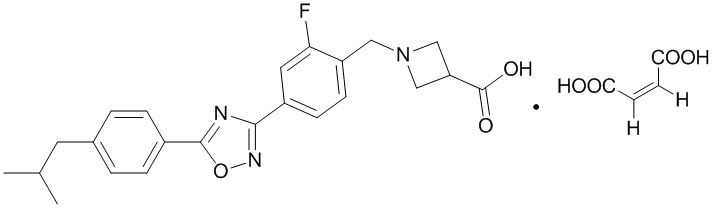 ,
,
включающий следующие стадии:
получение суспензии или раствора соединения, представленного формулой A, и суспензии или раствора малеиновой кислоты в растворителе, выбранном из группы, состоящей из этанола, ацетона, диэтилового эфира, воды, этилацетата, 1,4-диоксана или их смеси, соответственно;
смешивание суспензии или раствора соединения, представленного формулой A, и малеиновой кислоты в молярном соотношении 1:1,0-1:2,6 для осуществления реакции; при этом реакцию проводят при 10-40°C при перемешивании, в течение 10-24 ч.;
удаление растворителя после завершения реакции; и выполнение высушивания при 10-40°C, в течение 1-24 ч.;
и при этом отношение массы соединения, представленного формулой A, к объему растворителя в данном способе составляет 4 мг:1 мл-26 мг:1 мл.
 А.
А.
2. Способ по п.1, где малеат представляет собой кристаллическую форму, и при облучении Сu-Kα-излучением кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой с характеристическими пиками при следующих значениях угла 2θ со следующими относительными значениями интенсивности:
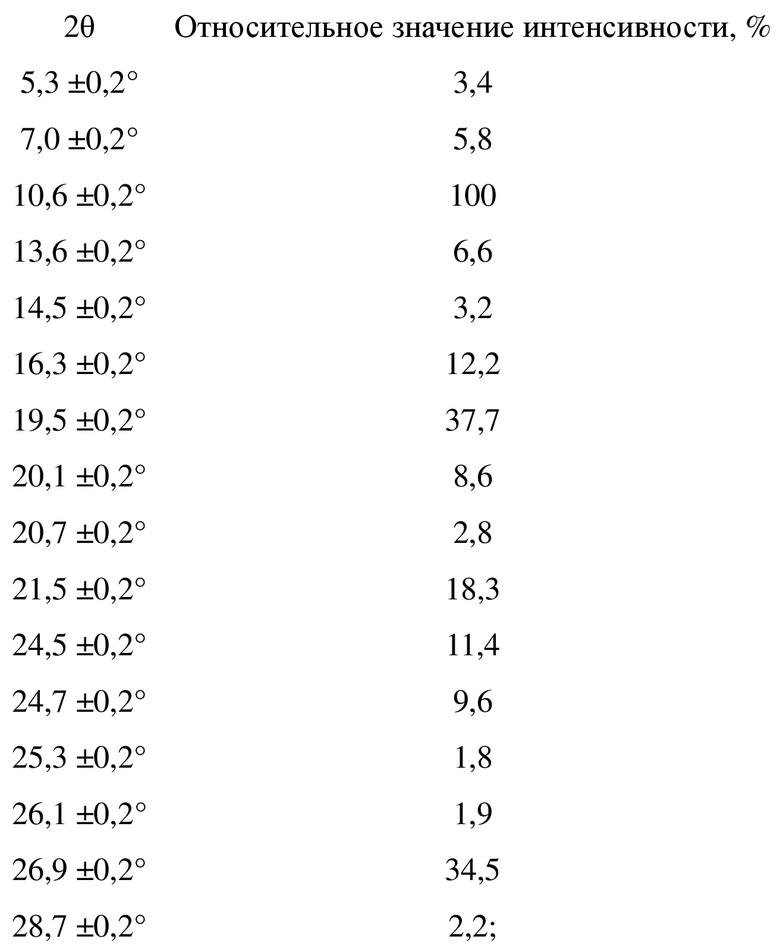
3. Способ по п.2, где кристаллическая форма характеризуется порошковой рентгеновской дифрактограммой, показанной на фиг. 10.
4. Способ по п.2 или п.3, где кристаллическая форма характеризуется инфракрасным спектром, полученным с использованием преобразования Фурье, имеющим характеристические пики при длинах волн 1734 см-1, 1574 см-1, 1485 см-1, 1439 см-1, 1364 см-1, 1346 см-1, 1080 см-1, 1003 см-1, 893 см-1, 871 см-1, 757 см-1 и 729 см-1.
| EP 3048103 A1, 27.07.2016 | |||
| CN 105315266 A, 10.02.2016 | |||
| Richard J.Bastin et al.: "Salt selection and Optimisation Procedures for Pharmaceutical New Chemical Entities", ORGANIC PROCESS RESEARCH & DEVELOPMENT, 2000, vol.4, p.427-435 | |||
| MINOR | |||
| CAIRA: "Crystalline Polymorphism of Organic Compounds", TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, |

