Область техники
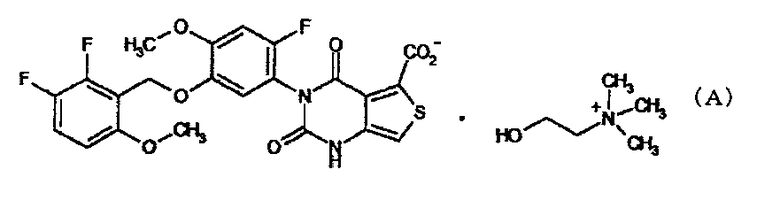
Настоящее изобретение относится к соединению (химическое название холиновая соль 3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-1,2,3,4-тетрагидротиено[3,4-d]пиримидин-5-карбоновой кислоты; далее именуемому как «соединение А»), представленному формулой:

которое имеет антагонистическую активность в отношении гонадотропинового-релизинг гормона и полезно в качестве превентивного или терапевтического средства для лечения половых гормонально-зависимых заболеваний, таких как доброкачественная гипертрофия предстательной железы, миома матки, эндометриоз, фибромиома матки, раннее половое созревание, аменоррея, предменструальный синдром, дисменоррея или подобные.
Предшествующий уровень техники
Соединение (далее именуемое как "соединение (B)"), представленное формулой:
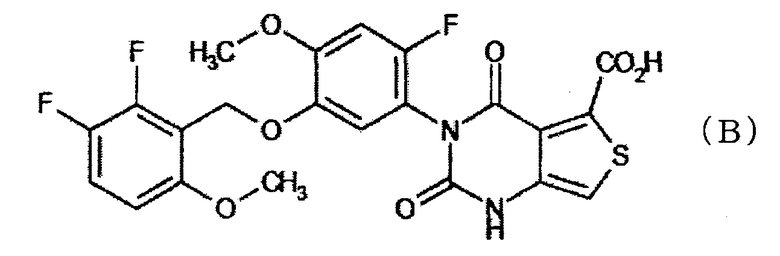
которое имеет антагонистическую активность в отношении гонадотропинового-релизинг гормона и полезно в качестве превентивного или терапевтического средства для лечения половых гормонально-зависимых заболеваний, таких как доброкачественная гипертрофия предстательной железы, миома матки, эндометриоз, фибромиома матки, раннее половое созревание, аменоррея, предменструальный синдром, дисменоррея или подобные, описано в патентной ссылке 1.
Публикация содержит только обычные описания солей, таких как фармакологически приемлемые соли, и не сообщает о конкретных солях соединения (В).
Патентная ссылка 1: International Publication pamphlet 2007/046392
Описание изобретения
Проблемы, решаемые настоящим изобретением
Авторы настоящего изобретения проведенной кропотливой работой подтвердили, что соединение (B), описанное в патентной ссылке 1, является аморфным или кристаллическим. Одним из аспектов аморфного состояния является то, что его трудно, например, выделить и очистить в промышленных масштабах в соответствии с определенными требованиями, поэтому кристаллы являются более предпочтительными в качестве вещества лекарственного препарата. Однако, как будет описано ниже в примере испытаний (тест на предел растворимости), у кристаллов соединения (B) существует проблема растворимости. Плохая растворимость часто вызывает проблемы всасываемости лекарственного препарата и может потребоваться изобретательность в разработке рецептуры с соединением (B), используемым как лекарственный препарат. Поэтому для применения соединения (B) в качестве вещества лекарственного препарата требуется улучшение его растворимости.
Средства для решения проблем.
Авторы настоящего изобретения провели интенсивные исследования для решения вышеупомянутых проблем и нашли, что холиновая соль 3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-l,2,3,4-тетрагидротиено-[3,4-d]пиримидин-5-карбоновой кислоты имеет значительно более высокую растворимость и желательную стабильность при хранении и, следовательно, представляет предпочтительное соединение в качестве вещества лекарственного препарата. Настоящее изобретение полностью основывается на этих полученных данных.
То есть, настоящее изобретение относится к:
(1) соединению, представленному вышеупомянутой формулой (A);
(2) соединению по п.(1), которое является кристаллическим;
(3) соединению по п.(2), которое имеет характерные пики при углах дифракции (2θ(°)) 7,1, 11,5, 19,4, 20,3, 21,5, 22,0, 22,6, 23,5 и 26,2 на диаграмме порошковой рентгеновской дифракции;
(4) соединению по п.(2), которое имеет характерные пики при значениях химических сдвигов (δ(ppm)) 1558, 149,8, 145,3, 118,0, 113,7, 111,6, 110,3, 98,1, 69,8, 58,7, 57,1 и 55,5 на диаграмме твердотельных спектров ЯМР 13C;
(5) соединению по п.(2), которое имеет характерные пики при значениях химических сдвигов (δ(ppm)) 131,6, -145,2 и -151,8 на диаграмме твердотельных спектров ЯМР 19F;
(6) соединению, описанному в любом одном из вышеупомянутых пп.(2)-(5), которое имеет эндотермический пик на уровне 213ºC на термограмме дифференциально-термического анализа;
(7) фармацевтической композиции, содержащей в качестве активного компонента соединение, как описано в любом одном из вышеупомянутых пп.(1)-(6);
(8) фармацевтической композиции по п.(7), которая является антагонистом гонадотропин-релизинг гормона;
(9) фармацевтической композиции по п.(7), которая является средством для предотвращения или лечения полового гормонально-зависимого заболевания, регулятором воспроизводства, контрацептивом, средством, вызывающим овуляцию, или средством для предотвращения послеоперационных рецидивов рака половых гормонально-зависимых органов;
(10) применению соединения, которое описано в любом одном из вышеупомянутых пп. (1)-(6), в производстве средства для предотвращения или лечения полового гормонально-зависимого заболевания, регулятора воспроизводства, контрацептива, средства, вызывающего овуляцию, или средства для предотвращения послеоперационных рецидивов рака половых гормонально-зависимых органов;
(11) способу предотвращения или лечения полового гормонально-зависимого заболевания, способу репродуктивного воспроизводства, способу контрацепции, способу, вызывающему овуляцию, или способу предотвращения послеоперационных рецидивов рака половых гормонально-зависимых органов, включающего введение эффективного количества соединения, которое описано в одном из вышеупомянутых пп. (1)-(6) или подобных.
Результат изобретения
Соединение (A) настоящего изобретения прекрасно растворяется и имеет пероральную всасываемость. Кроме того, соединение (A) имеет прекрасную кристалличность, отличную стабильность при хранении и текучесть. Соединение (A) также просто в обращении, например в технологии приготовления лекарственного средства.
Лучший вариант осуществления изобретения
Соединение (A) настоящего изобретения можно получать, например, используя следующий способ. В частности, например, в чистом виде соединение (B), которое может быть получено с помощью способа, описанного в патентной ссылке 1, или с помощью способов, соответствующих данному способу, смешивают с равным количеством (1,0 эквивалента) или малым избыточным количеством холина гидроксида в подходящем растворителе. Затем смесь растворяют при нагревании, а растворитель выпаривают или добавляют соответствующим образом по мере необходимости. Соединение (A) осаждается при охлаждении и затем может быть выделено. Кроме того, соединение (A) может быть очищено перекристаллизацией с использованием того же или аналогичного растворителя.
Растворитель может быть любым, при условии, что он не мешает образованию соли. Примеры пригодных для использования растворителей включают органические растворители, в том числе: спирты, такие как метанол, этанол, 1-пропанол, 2-пропанол, 1-бутанол и 2-бутанол; эфиры, такие как тетрагидрофуран и диизопропиловый эфир, и воду. Также может быть использована смесь этих растворителей.
Соединение (A) настоящего изобретения чрезвычайно полезно в качестве средства для предотвращения или лечения половых гармонально-зависимых заболеваний, таких как доброкачественная гипертрофия предстательной железы, миома матки, эндометриоз, фибромиома, раннее половое созревание, аменоррея, предменструальный синдром, дисменоррея, синдром поликистозных яичников, красная волчанка, гирсутизм, недостаточный рост, расстройство сна, акне, облысение, болезнь Альцгеймера, бесплодие, синдром раздраженной толстой кишки, рак простаты, рак матки, рак яичников, рак молочной железы и опухоль гипофиза, регулятор воспроизводства как средство контрацепции, средство, вызывающее овуляцию, средство для предотвращения послеоперационных рецидивов рака половых гормонально-зависимых органов или тому подобное.
Соединение (A), согласно настоящему изобретению, может соответствующим образом смешиваться с фармацевтическим носителем, традиционно применяемым для приготовления фармацевтической композиции.
Фармацевтический носитель может быть соответственно в комбинации с соответствующей лекарственной формой, как описано ниже. В качестве фармацевтического носителя могут, например, применяться инертные наполнители, такие как лактоза или подобные; смазывающие вещества, такие как стеарат магния или подобные; дезинтеграторы, такие как карбоксиметилцеллюлоза или подобные; связующие, такие как гидроксипропилметилцеллюлоза или подобные; поверхностно-активные вещества, такие как макрогол или подобные; пенообразователи, такие как бикарбонат натрия или подобные; вспомогательные средства, такие как циклодекстрин или подобные; кислоты, такие как лимонная кислота или подобные; стабилизаторы, такие эдентат натрия или подобные; pH регуляторы, такие как соль фосфорной кислоты или подобные.
Примеры лекарственной формы фармацевтической композиции согласно настоящему изобретению включают перорально-применяемые средства, такие как порошки, гранулы, мелкие гранулы, безводные сиропы, таблетки, капсулы и подобные; парентерально-применяемые средства, такие как инъекции, компрессы, свечи и подобные; причем перорально-применяемые средства являются предпочтительными.
Желательно, вышеупомянутые лекарственные средства приготавливать таким образом, чтобы соединение (A) согласно настоящему изобретению применялось в количестве от 0,1 до 1000 мг в день для взрослого человека в случае перорально-вводимых средств и от 0,01 до 100 мг в день для взрослых в случае инъекций.
Примеры
Настоящее изобретение далее иллюстрируется более подробно при помощи следующих примеров и примеров испытаний. Однако настоящее изобретение этим не ограничивается.
Пример 1
Соединение (A)
3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-1,2,3,4-тетрагидротиено[3,4-d]пиримидин-5-карбоновую кислоту (3,07 г) и 46% водный раствор гидроксида холина (1,64 г) суспедировали в смешанный раствор 1-пропанола и воды (объемное отношение приблизительно 1:1; 30 мл), смесь нагревали и перемешивали в течение 15 мин при 60°C. 1-пропанол (30 мл) добавляли к смеси при 60ºC и смесь перемешивали при комнатной температуре в течение 1 часа и в течение следующего одного часа охлаждали при помощи льда. После осаждения осадок был собран фильтрованием и промыт дважды 1-пропанолом (1 мл). Полученное твердое вещество сушили при пониженном давлении при 40ºC с получением соединения (A) (2,93 г). Далее это соединение нагревали и перемешивали при 60ºC в смешанном растворителе 1-пропанола и воды (объемное отношение приблизительно 1:1; 30 мл) и затем 1-пропанол (30 мл) добавляли к раствору, полученному после горячего фильтрования. Полученную смесь охлаждали до комнатной температуры и перемешивали в течение 1 часа и в течение следующего одного часа охлаждали при помощи льда. Выпавшие кристаллы были выделены фильтрованием и промыты дважды смешанным растворителем 1-пропанола и воды (объемное отношение приблизительно 3:1; 1 мл). Полученные кристаллы сушили на воздухе в течение 4 дней с получением соединения (A) (2,16 г).
1H-ЯМР (ДМСО-д6)(δ(ppm)): 3,10 (9H, с), 3,35-3,45 (2H, м), 3,70-3,90 (8H, м), 4,95 (2H, с), 5,47 (1H, шир.с), 6,44 (1H, с), 6,85-6,95 (1H, м), 7,05 (1H, д, J=11,5 Гц), 7,11 (1H, д, J=7,4 Гц), 7,48 (1H, дд, J=9,7 Гц, 19,5 Гц), 11,14 (1H, шир.с).
Полученное соединение (A) было исследовано с помощью порошковой рентгеновской дифракции, термического анализа, твердотельных спектров ЯМР 13C и твердотельных спектров ЯМР 19F при приведенных ниже условиях с получением соответствующих данных.
Для порошковой рентгеновской дифракции кристаллы измельчали в ступке и измеряли с помощью прибора порошковой рентгеновской дифракции X'Pert Pro MPD (Spectris pic, PANalytical Department) (метод отражения; CuKα лучи, напряжение трубки 45 кВ, ток трубки 40 мА).
Полученная дифракционная диаграмма показана на фигуре 1, углы дифракции (2θ(°)) и относительные интенсивности пиков (%), пики, у которых относительные интенсивности составляют приблизительно 20% или выше, показаны в таблице 1.
По причинам, связанным с природой данных порошковой ренгеновской дифракции, 2θ значения и полная картина дифракции являются важными результатами для подтверждения кристаллической идентичности. Общеизвестно, что характер относительной интенсивности рентгеновских дифракционных полос колеблется в зависимости от условий подготовки образца и условий измерений. Необходимо отметить, что 2θ значения дифракционных картин в порошковой дифракции рентгеновских лучей могут незначительно колебаться в зависимости от условий подготовки образца и условий измерений.
Для термического анализа измерения проводились с использованием термогравиметрического/дифференциального термического анализатора TG-DTA TG8120 (Rigaku Corporation) (Скорость нагрева: 10°C/мин; эталон: оксид алюминия). Полученная диаграмма показана на фигуре 2.
Эндотермический пик: около 213ºC.
Замечено, что эндотермический пик в термическом анализе может незначительно колебаться в зависимости от условий подготовки образца и условий измерений.
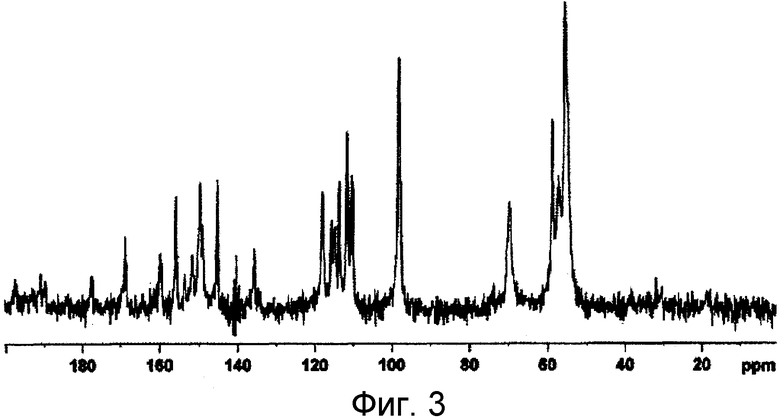
Для измерений твердотельных спектров ЯМР 13C образец загружали в 4-мм диоксидциркониевый ротор и проводили измерение с помощью спектрометра Bruker Avance DRX500 (скорость вращения 10 кГц), с применением CP/MAS техники. Полученная диаграмма спектров показана на фигуре 3.
Следует отметить, что значение химического сдвига твердотельного спектра ЯМР 13С может незначительно колебаться в зависимости от условий подготовки образца и условий измерений.
Для измерений твердотельных спектров ЯМР 19F образец загружали в 2,5-мм диоксидциркониевый ротор и проводили измерение с помощью спектрометра Bruker Avance III 400 WideBore (скорость вращения 30 кГц), с применением техники MAS. Используя образец поливинилиденфторида(PVDF) в качестве образца внешнего стандарта, наблюдали спектр, который давал резонанс на -91,2 ppm. Полученная диаграмма спектров показана на фигуре 5.
Следует отметить, что значение химического сдвига твердотельного спектра ЯМР 19F может незначительно колебаться в зависимости от условий подготовки образца и условий измерений.
Пример 2
Соединение (A)
3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-l,2,3,4-тетрагидротиено[3,4-d]пиримидин-5-карбоновую кислоту (4,07 г), 46% водный раствор гидроксида холина (2,18 г), 2-пропанол (200 мл) и воду (100 мл) смешивали и нагревали в течение 15 мин при 40ºC. После удаления нерастворимого вещества путем фильтрации полученный раствор концентрировали и добавляли 2-пропанол (80 мл) и диизопропиловый эфир (80 мл). Затем смесь перемешивали при комнатной температуре приблизительно в течение 1 часа, а затем в течение 4 часов охлаждали с использованием льда. Осажденное твердое вещество собирали и сушили при 90ºC в течение ночи при пониженном давлении (выход 3,59 г). К полученному твердому веществу (3,56 г) затем добавляли смешанный раствор этанола и воды (объемное отношение приблизительно 1:1; 25 мл). Смесь нагревали до 65ºC и после добавления при той же температуре смешанного раствора этанола и воды (объемное отношение 1:1; 10 мл) горячую смесь фильтровали. Полученный раствор перемешивали, давая в то же время остыть до комнатной температуры, в течение 2 часов, а затем после добавления этанола (20 мл) дополнительно перемешивали при комнатной температуре приблизительно 1 час. Далее смесь перемешивали при комнатной температуре в течение приблизительно 1 часа после добавления этанола (20 мл) и затем перемешивали в течение ночи при охлаждении с использованием льда. Твердое вещество выделяли из смеси, промывали этанолом (5 мл) и сушили с помощью продувки азотом. Твердое вещество далее высушивали при 40ºC в течение ночи при пониженном давлении с получением соединения (A) (2,43 г). У полученного соединения (A) измеряли порошковую дифракцию рентгеновских лучей таким же способом, как в примере 1. Результат подтвердил, что у соединения (A) была такая же кристаллическая форма, как наблюдалась в примере 1.
1H-ЯМР (ДМСО-д6)(δ(ppm)): 3,10 (9H, с), 3,35-3,45 (2H, м), 3,70-3,90 (8H, м), 4,95 (2H, с), 5,47 (1H, шир.с), 6,44 (1H, с), 6,85-6,95 (1H, м), 7,05 (1H, д, J=11,5 Гц), 7,10 (1H, д, J=7,4 Гц), 7,47 (1H, дд, J=9,7 Гц, 19,5 Гц), 11,11 (1H, шир.с).
Сравнительный пример 1
Кристаллы соединения (B)
Этилацетат (0,1 мл) добавляли к аморфной твердой 3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-l,2,3,4-тетрагидротиено[3,4-d]пиримидин-5-карбоновой кислоте (10 мг) и суспензию нагревали до 50ºC. Смесь далее сушили с помощью продувки азотом. Полученный сухой остаток затем нагревали до 70ºC и сушили в течение ночи при пониженном давлении с получением кристаллов соединения (B) (10 мг). Полученные кристаллы соединения (B) измеряли на аппарате порошковой дифракции рентгеновских лучей таким же способом, как в примере 1. Полученная дифракционная диаграмма показана на фигуре 4.
Сравнительный пример 2
Кристаллы соединения (B)
3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-l,2,3,4-тетрагидротиено[3,4-d]пиримидин-5-карбоновая кислота (0,69 г) была суспендирована в диизопропиловый эфир (10 мл) и смесь перемешивали при 60ºC в течение 3 часов при нагревании. Затем смесь перемешивали при комнатной температуре в течение ночи, далее в течение 1 часа при охлаждении льдом. Твердое вещество собирали с помощью фильтрования и сушили при пониженном давлении с получением кристаллов соединения (B) (0,65 г). Полученные кристаллы соединения (B) измеряли на аппарате порошковой дифракции рентгеновских лучей таким же способом, как в примере 1. Результат подтвердил, что у соединения (В) была такая же кристаллическая форма, как наблюдалась в сравнительном примере 1.
Сравнительный пример 3
3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-l,2,3,4-тетрагидротиено[3,4-d]пиримидин-5-карбоновой кислоты 1/2 N,N'-дибензилэтилендиаминовая соль (далее именуемое, как "соединение (C)").
Смешивали ацетонитрил (10 мл), N,N'-дибензилэтилендиамин (94,5 мг) и 3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-1,2,3,4-тетрагидротиено[3,4-d]пиримидин-5-карбоновая кислота (200 мг). Смесь суспендировали при температуре приблизительно 60ºC и перемешивали, давая возможность остыть до комнатной температуры. Твердое вещество (247 мг) выделили из смеси фильтрованием и сушили при температуре приблизительно 60ºC в течение ночи при пониженном давлении.
1H-ЯМР (ДМСО-д6)(δ(ppm)): 2,83 (2H, с), 3,79 (3H, с), 3,80 (3H, с), 3,87 (2H, с), 4,95 (2H, с), 6,85-6,95 (1H, м), 6,98 (1H, с), 7,09 (1H, д, J=11,5 Гц), 7,19 (1H, д, J=7,5 Гц), 7,25-7,55 (6H, м).
Тестовый пример 1
Испытание предела растворимости
Соединение (A) полученное в примере 2, кристаллы соединения (B), полученные в сравнительном примере 2, и соединение (C), полученное в сравнительном примере 3, суспендировали в воду или в 1-ю жидкость для испытания на растворимость (далее именуемая как "1-я жидкость") или во 2-ю жидкость (далее именуемая как "2-я жидкость"), как описано в документе Reagents-Test Solutions for General Tests, the Japanese Pharmacopoeia, 15th Edition. Суспензии выдерживали в термостате при 37ºC. После фильтрования части суспензий концентрации полученных фильтратов были исследованы методом HPLC (высокоэффективная жидкостная хроматография) и предел растворимости рассчитывали и сравнивали.
Условия проведения HPLC следующие:
Детектор: спектрофотометр ультрафиолетовой-видимой области/длина волны: 225 нм
Колонка: GL Science Inertsil ODS-3,5 мкм, 4,6×250 мм
Температура колонки: постоянная приблизительно 35ºC
Расход: 1,0 мл/мин
Подвижная фаза A: 10 мМ водный раствор дигидрофосфата калия, доведенный до pH 5,5 водным раствором гидроксида калия
Подвижная фаза B: Ацетонитрил
Подвижное фазовое соотношение от 0 до 30 мин: Подвижная фаза A/Подвижная фаза B=70/30.
Значения предела растворимости соединения (A), кристаллов соединения (B) и соединения (C) в воде, 1-й жидкости и 2-й жидкости показаны в таблице 2. В воде соединение (A) имеет предел растворимости приблизительно в 600 раз и приблизительно в 60 раз выше, чем у кристаллов соединения (B) и у соединения (C), соответственно. Предел растворимости соединения (A) в 1-й жидкости был приблизительно в 30 раз и приблизительно в 2 раза выше, чем у кристаллов соединения (B) и у соединения (C), соответственно. Во 2-й жидкости предел растворимости соединения (A) был приблизительно в 10 раз и приблизительно в 70 раз выше, чем у кристаллов соединения (B) и у соединения (C), соответственно. Эти результаты, таким образом, подтверждают значительные улучшения растворимости соединения (A) по сравнению с кристаллами соединения (B) и соединения (C).
Единица измерения: мкг/мл
Тестовый пример 2
Испытание на пероральную всасывающую способность
1) Приготовление образца для измерения концентрации лекарственного препарата путем введения его через хвостовую вену
Крысы с SD (Charles River, самец, возраст-7 недель, от 170 до 210 г) были использованы в качестве подопытных животных сразу после ночного голодания. N,N-диметилацетамид (0,2 мл), солевой раствор (0,798 мл) и 2N-NaOH (0,002 мл) были добавлены соответственно в этих количествах к 1 мг соединения (B) для приготовления 1,0 мг/мл раствора. Раствор затем вводили в дозе 1 мл/кг (1 мг/кг) через хвостовую вену без анастезии (3 пробы). Внутривенную инъекцию через хвост выполняли, используя инъекционную иглу 26G и 1 мл шприц. Кровь собирали через подключичные вены через 2, 15, 60, 120, 240 и 360 минут после внутривенного введения через хвост. Кровь центрифугировали и плазму использовали в качестве образца для измерения концентрации лекарственного препарата в крови.
2) Подготовка образца для измерения концентрации препарата при пероральном приеме
Крысы с SD (Charles River, самец, возраст-7 недель, от 220 до 290 г) были использованы в качестве подопытных животных сразу же после ночного голодания. 0,5% водный раствор метилцеллюлозы (5 мл) соответственно добавляли к 3 мг соединения (A) или соединения (B) (в пересчете на чистое вещество) для приготовления 0,6 мг/мл раствора лекарственного препарата. Каждый раствор перорально вводили крысам в дозе 5 мл/кг (3 мг/кг) (3 пробы каждого). Пероральное введение выполняли, применяя зонд для крысы и 2,5-мл шприц. Кровь собирали через подключичные вены без анестезии через 15, 30, 60, 120, 240, 360 и 480 минут после перорального введения. Кровь центрифугировали и плазму использовали в качестве образца для измерения концентрации лечебного препарата в крови.
3) Измерение концентрации лекарственного средства
Соответствующий раствор вещества внутреннего стандарта (0,1 мл) добавляли к плазме крови (0,025 мл), полученной в пп.1 и 2 с использованием обычного способа. Затем был добавлен ацетонитрил (0,875 мл) для удаления белка. После центрифугирования супернатант (надосадочная жидкость) (0,005 мл) ввели в LC-MS/MS. Концентрацию препарата в плазме крови измеряли с помощью оборудования LC-MS/MS при условиях, приведенных ниже. Отметим, что стандартная кривая была создана путем соответствующего добавления вещества внутреннего стандарта и вещества исследования к чистой плазме крови (0,05 мл) с использованием обычного способа в соответствии с предыдущими методиками проведения измерений.
LC
Устройство: Agilent 1100
Колонка: Capcellpak MGIII 5 мкм 4,6×50 мм
Подвижная фаза A: 10 мМ водный раствор ацетата аммония
Подвижная фаза B: Ацетонитрил
(Соотношения подвижных фаз приведены в таблице 3)
Температура колонки: 40ºC
Расход: 0,5 мл/мин MS/MS
Устройство: API-4000
Способ ионизации: ESI (ионизация) (Turbo Ion Spray)-(турбоэлектрический ионизатор)
Биологическая доступность соединения (A) составляла приблизительно 59% и желаемая всасываемость при пероральном приеме была подтверждена. Кроме того, время достижения максимальной концентрации лекарственного средства (T макс.) составляло 35 мин для соединения (A) по сравнению с 200 мин для соединения (B), ввиду того, что соединение (A) быстро всасывается после приема и ожидается проявление быстрого начала действия.
Биодоступность (%) рассчитывалась из величины области под кривой значений концентрации лекарственного препарата в крови от времени, определенной с использованием программ WinNonlin Professional фирмы (Pharsight Corporation), исходя из концентрации лекарственного препарата в крови в каждый момент времени, полученной, как указывалось выше, после внутривенного введения соединения (B) через хвост и перорального введения соединения (A) или соединения (B).
Тестовый пример 3
Испытание на устойчивость
Соединение (A), полученное в примере 2, хранилось в открытом виде при 90ºC для изучения стабильности. При измерении стабильности чистота образца измерялась с помощью HPLC в начале и после 8 дней, полученные результаты сопоставлялись.
Условия проведения измерений с помощью HPLC были следующие:
Детектор: спектрофотометр ультрафиолетовой и видимой области, длина волны: 225 нм
Колонка: GL Science Inertsil ODS-3, 5 мкм, 4,6×250 мм
Температура колонки: постоянная температура около 35ºC
Расход: 1,0 мл/мин
Подвижная фаза A: 10 мМ водный раствор дигидрофосфата калия доводят водным раствором гидроксида калия до pH 5,5
Подвижная фаза B: Ацетонитрил (отношения подвижных фаз приведены в таблице 4)
Диапазон измерений: 54 минуты от начала анализа. Площади пустых пиков из расчетов исключались.
Результаты измерений представлены в таблице 5.
Как описано выше, результаты тестовых примеров 1-3 показывают, что у соединения (A) настоящего изобретения прекрасная растворимость, пероральная всасываемость и стабильность при хранении и, следовательно, оно представляет собой прекрасное соединение, с помощью которого возможно решать проблемы, связанные с физическими свойствами чистого соединения (B).
Применение в промышленности
Соединение (A) согласно настоящему изобретению имеет прекрасную растворимость и другие желаемые физические свойства, поэтому является пригодным в качестве материала лекарственного препарата и подходит для промышленного производства лекарственных средств.
Краткое описание диаграмм
На фиг.1 показана диаграмма порошковой дифракции рентгеновских лучей соединения (A), полученного в примере 1. На вертикальной оси показана интенсивность дифракции рентгеновских лучей (число интервалов дискредитации измеряемого сигнала); на горизонтальной оси показан угол дифракции (2θ(°)).
На фиг. 2 показана диаграмма, представляющая TG-DTA измерения соединения (А), полученного в примере 1. На вертикальной оси (слева) показан масс. % на термогравиметрической кривой (TG); на вертикальной оси (правая) показан тепловой поток (УФ) на кривой дифференциально-термического анализа (DTA) и на горизонтальной оси показана температура (°С).
На фиг. 3 показаны спектры твердотельного ЯМР 13С соединения (А), полученного в примере 1. На вертикальной оси показана интенсивность; на горизонтальной оси показано значение химического сдвига (δ(ppm)). Характерные пики значений химических сдвигов (δ(ppm)) в спектре ЯМР 19F в твердой фазе составляют -131,6, -145 и -151,8.
На фиг. 4 показана диаграмма порошковой дифракции рентгеновских лучей соединения (А), полученного в сравнительном примере 1. На вертикальной оси показана интенсивность дифракции рентгеновских лучей (число интервалов дискредитации измеряемого сигнала); на горизонтальной оси показан угол дифракции (2θ(°)).
На фиг. 5 показаны спектры твердотельного ЯМР 19F соединения (А), полученного в примере 1. На вертикальной оси показана интенсивность; на горизонтальной оси показано значение химического сдвига (δ(ppm)). Соединение характеризуется эндотермическим пиком около 213°С в диаграмме дифференциально-термического анализа.




Изобретение относится к новой холиновой соли 3-[2-фтор-5-(2,3-дифтор-6-метоксибензилокси)-4-метоксифенил]-2,4-диоксо-1,2,3,4-тетрагидротиено[3,4-d]пиримидин-5-карбоновой кислоты, соответствующей формуле (А) и к ее кристаллической форме.
 .
.
Кристаллическая форма соли (А) имеет характерные пики при углах дифракции (2θ(Å)) 7,1, 11,5, 19,4, 20,3, 21,5, 22,0, 22,6, 23,5 и 26,2 в диаграмме порошковой дифракции рентгеновских лучей; характерные пики значений химических сдвигов (δ(ppm)) 155,8, 149,8, 145,3, 118,0, 113,7, 111,6, 110,3, 98,1, 69,8, 58,7, 57,1 и 55,5 в твердотельном спектре ЯМР 13С и характерные пики значений химических сдвигов (δ(ppm)) -131,6, -145, и -151,8 в спектре ЯМР 19F в твердой фазе, а также эндотермический пик около 213°С в диаграмме дифференциально-термического анализа. Соединение имеет прекрасную растворимость и стабильность при хранении. 4 з.п. ф-лы, 5 ил., 3 табл., 8 пр.
1. Соединение, представленное формулой
2. Соединение по п. 1, в кристаллической форме, имеющее характерные пики при углах дифракции (2θ(Å)) 7,1, 11,5, 19,4, 20,3, 21,5, 22,0, 22,6, 23,5 и 26,2 в диаграмме порошковой дифракции рентгеновских лучей.
3. Соединение по п. 2, имеющее характерные пики значений химических сдвигов (δ(ppm)) 155,8, 149,8, 145,3, 118,0, 113,7, 111,6, 110,3, 98,1, 69,8, 58,7, 57,1 и 55,5 в твердотельном спектре ЯМР 13С.
4. Соединение по п. 2, имеющее характерные пики значений химических сдвигов (δ(ppm)) -131,6, -145, и -151,8 в спектре ЯМР 19F в твердой фазе.
5. Соединение по любому одному из пп. 2-4, имеющее эндотермический пик около 213°С в диаграмме дифференциально-термического анализа.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| WO 9002748 A1, 22.03.1990 & EP0359058 A1 | |||
| US 4663348 A, 05.05.1987 & JP 62-5975A | |||
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |
| АННЕЛИРОВАННЫЕ АЗАГЕТЕРОЦИКЛИЧЕСКИЕ АМИДЫ, ВКЛЮЧАЮЩИЕ ПИРИМИДИНОВЫЙ ФРАГМЕНТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2007 |
|
RU2345996C1 |

