Область техники
Данное изобретение относится к соединениям, подходящим для хроматографического выделения редкоземельных элементов и/или металлов s-, p-, d- блоков, способу хроматографического выделения редкоземельных элементов и/или металлов s-, p-, d- блоков из смеси ионов металлов, где по меньшей мере один из них представляет собой редкоземельный металл, выбранный из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb и Y, щелочноземельный металл, Al, Ga, In, Tl, Sn, Pb или переходной металл, и к их применению для извлечения и выделения редкоземельных металлов и/или металлов s-, p-, d- блоков из смесей.
Предпосылки изобретения
Радионуклиды элементов металлов все чаще используются в ядерной медицине, в основном для диагностики и терапии онкологических заболеваний. Растет интерес к таргетной лучевой терапии, в которой используется таргетирующий вектор (пептид, антитело и т. д.) для доставки радиоактивной нагрузки конкретно к раковой ткани. Радионуклиды элементов металлов являются удобными, поскольку связь с таргетирующим вектором может быть легко достигнута посредством координирования с бифункциональным хелатором.
Чтобы снизить вероятность нежелательной токсичности и максимизировать эффективность лечения радионуклиды для медицинских применений предпочтительны в так называемой форме «без добавления носителя» (no-carrier-added, NCA), то есть не содержащей ненужных веществ. Однако достижение этой чрезвычайно высокой чистоты радионуклидов металлов является серьезной проблемой. Чаще всего медицинские радионуклиды получают из стабильного нуклида с помощью ядерной реакции, вызванной частицами. Приготовление радионуклида NCA требует полного удаления исходного нуклида и побочных продуктов, которые обычно присутствуют в количествах на несколько порядков больше. Следует строго избегать загрязнения следами металла из растворителей, химических реагентов и оборудования. Кроме того, обращение с радиоактивностью сопряжено со многими техническими трудностями. Обычные методы выделения либо непрактичны для работы с радиоактивностью, либо недостаточно эффективны для выделения радионуклидов NCA. Необходимы новые методы выделения, специально разработанные для радионуклидов металлов.
Редкоземельные элементы (скандий - Sc, иттрий - Y, лантан - La, церий - Ce, празеодим - Pr, неодим - Nd, прометий - Pm, самарий - Sm, европий - Eu, гадолиний - Gd, тербий - Tb, диспрозий - Dy, гольмий - Ho, эрбий - Er, тулий - Tm, иттербий - Yb и лютеций - Lu) представляют собой группу металлов, которые предлагают широкий выбор радионуклидов для медицинского применения. Радионуклиды 90Y и 153Sm одобрены FDA, продолжаются клинические испытания 166Ho и 177Lu, а другие демонстрируют полезные свойства (44Sc, 47Sc, 86Y, 149Pm, 159Gd, 149Tb, 161Tb, 165Dy, 161Ho, 169Er и 175Yb). Эти металлы близки по химическому составу, что дает то преимущество, что один и тот же вектор нацеливания, биоконъюгация и химия мечения могут использоваться с любым членом группы. Однако получение этих радионуклидов в виде NCA является общеизвестно трудным, поскольку обычно требует выделения двух соседних редкоземельных элементов с чрезвычайно похожими свойствами.
Методы, применяемые до сих пор для выделения радионуклидов редкоземельных элементов, включают ионообменную хроматографию, экстракционную хроматографию и жидкостно-жидкостную экстракцию (Nayak D., Lahiri S. (1999), Solvent Extr. Ion Exch. 17(5), 1133-1154). Эти методы используют небольшие различия в ионных радиусах, которые почти линейно уменьшаются от La3+ до Lu3+. Ионный радиус влияет на основность ионов и стерические требования к ним, свойства, которые используются в процессе выделения. Общей чертой этих методов выделения является то, что ион редкоземельного элемента участвует в относительно слабых взаимодействиях, которые позволяют быстро обмениваться с его непосредственным окружением. Эти взаимодействия включают ионные взаимодействия, сольватацию и координирование. Поскольку молекулярные взаимодействия повторяются много раз в процессе обмена, даже небольшие различия в свойствах между ионами металлов усиливаются, что в конечном итоге приводит к выделению. Важно отметить, что координирующие лиганды, используемые в этих методах, обеспечивают кинетически лабильные комплексы с ионами редкоземельных элементов, что позволяет осуществлять обмен. Типичными примерами таких лигандов являются ди-(2-этилгексил)фосфорная кислота (HDEHP) и α-гидроксиизомасляная кислота (α-HIBA) (Xie, F. et al. (2014), Miner. Eng. 56, 10-28). Сильно хелатирующие лиганды, такие как лиганды, полученные из 1,4,7,10-тетраазациклододекана (циклен), не используются, поскольку они образуют кинетически инертные комплексы, не допускающие обмена (типичным примером таких сильных хелаторов является 1,4,7,10-тетраазациклододекан-1,4,7,10-тетраксусная кислота (DOTA)).
Существуют также альтернативные методы выделения, которые используют преимущества более экзотических степеней окисления (кроме 3+) редкоземельных элементов, но они ограничены очень немногими случаями, когда такие состояния окисления возможны (Nayak D., Lahiri S. (1999), Solvent Extr. Ion Exch. 17(5), 1133-1154).
Методы выделения радионуклидов металлов s-, p- и d- блоков аналогичны упомянутым выше для редкоземельных элементов. Чаще всего используются ионообменная хроматография, экстракционная хроматография и жидкостно-жидкостная экстракция (Dietz M. L., Horwitz E. P. (2000), Ind. Eng. Chem. Res. 39(9), 3181-3188). Реже также осаждение, перегонка и электрохимическое осаждение. Как правило, ни один метод не может обеспечить удовлетворительный результат, и в качестве последней стадии необходимо использовать комбинацию методов с ионообменной хроматографией или экстракционной хроматографией (Medvedev D. G. et al. (2012), Appl. Radiat. Isot. 70(3), 423-429). Использование единого метода выделения значительно упрощает весь процесс и является весьма желательным. Также для этих металлов не используются сильно хелатирующие лиганды, такие как производные 1,4,7,10-тетраазациклододекана (циклена).
Таким образом, сохраняется потребность в эффективном и быстром выделении редкоземельных элементов и металлов s-, p- и d- блоков.
Описание изобретения
Несмотря на то, что согласно уровню техники не требуется использование сильных хелаторов для выделения редкоземельных элементов, авторы изобретения неожиданно обнаружили, что некоторые сильные хелаторы чрезвычайно эффективны при таком выделении, и, более того, они также могут быть использованы для выделения металлов s-, p-, d- блоков. s-, p-, d- металлы определяются как металлы, принадлежащие к группам II.A (щелочноземельные металлы), III.A (Al, Ga, In, Tl) и IV.A (Sn, Pb) и переходным металлам (группы от I.B до VIII.B). Настоящее изобретение относится к новым типам хелаторов и к способу их использования для выделения редкоземельных элементов и/или металлов s-, p-, d- блоков. Принцип выделения заметно отличается от вышеупомянутых существующих способов выделения и обеспечивает упрощенное (и, следовательно, более быстрое) обращение с радионуклидами редкоземельных металлов и/или металлов s-, p-, d-блоков в растворе, их переработку и очистку. Скорость и простота метода имеют решающее значение при работе с радионуклидами, которые подвергаются радиоактивному распаду. При связывании с ионами редкоземельных элементов и/или металлов s-, p-, d- блоков хелаторы по настоящему изобретению реагируют даже на очень небольшие различия ионных радиусов металлов выраженными различиями в полярности соответствующих образующихся хелатов. Из-за различной полярности хелаты можно выделить обычной хроматографией на нормальной или обращенной фазе. Таким образом, металлы выделяются в виде хелатов. Важно отметить, что хелаторы, описанные в этом изобретении, образуют хелаты, которые кинетически инертны в масштабе времени процесса выделения. Кинетическая инертность эффективно защищает радионуклид от дополнительного загрязнения другими металлами, поскольку радионуклид не может выйти из хелата и не может быть заменен другим ионом металла в процессе хроматографии. Важно отметить, что это свойство позволяет использовать обычные хроматографические колонки и приборы, состоящие из металлических частей. Способ выделения по настоящему изобретению может использоваться для выделения редкоземельных элементов независимо от конкретных изотопов задействованных элементов.
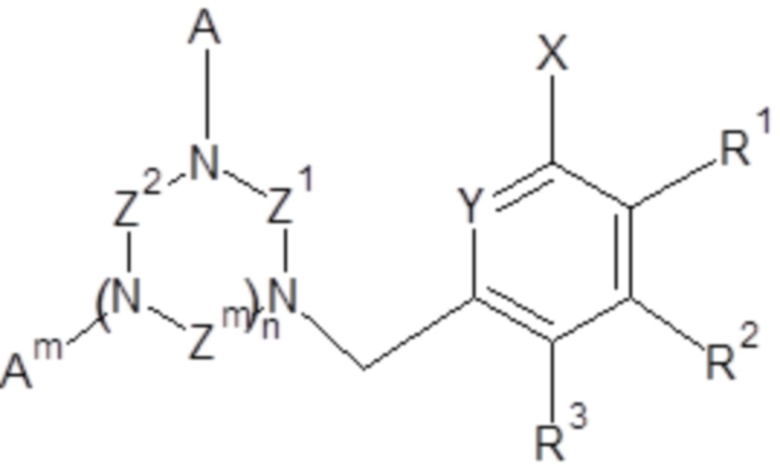
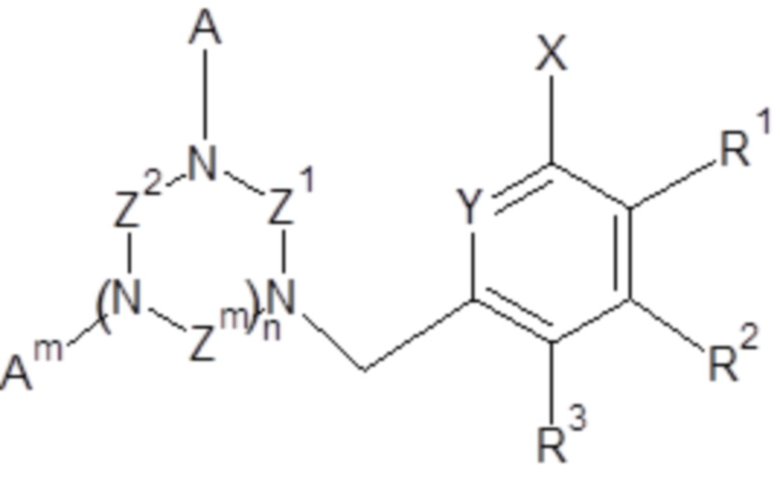
Объектом настоящего изобретения является применение соединений общей формулы (I)
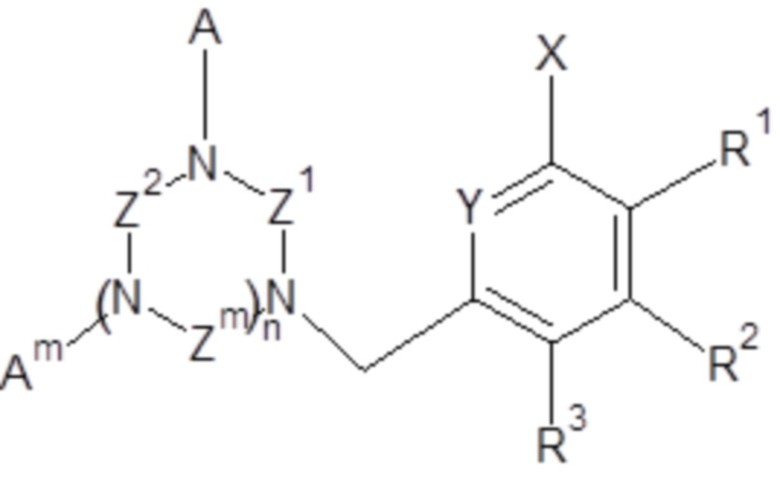
(I),
для хроматографического выделения редкоземельных элементов и/или металлов s-, p- и d- блоков,
где
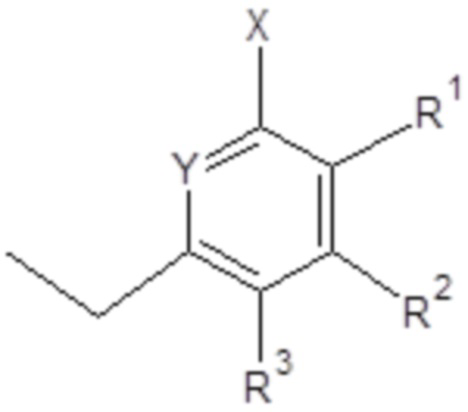
- X выбран из группы, включающей H; C1-C6 алкил; галоген (F, Cl, Br или I);
- Y выбран из группы, включающей азот; N-оксид;
- Z1, Z2, Zm, где m обозначает 1 или 2, независимо, выбраны из группы, включающей -CH2-CH2- и -CH2-CH2-CH2-;
- A, Am, где m обозначает 1 или 2, независимо, выбраны из H, -CH2COOH, -CH2C(O)NH2, -CH2P(O)(OH)2 и  ;
;
- n обозначает 1 или 2;
R1, R2, R3, независимо, представляют собой H, C1-C6 алкил, C1-C6 алкилокси, C6-C10 арилокси, бензилокси, C1-C6 алкилтио, C6-C10 арилтио, F, Cl, Br, I, OH, SH, NH2, C1-C6 алкиламино, ди(C1-C6 алкил)амино, C1-C6 ациламино, ди(C1-C6 ацил)амино, C6-C10 ариламино, ди(C6-C10 арил)амино, CN, OH, нитро, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn, независимо, представляет собой H, или C1-C10 алкил, или C6-C10 арил;
и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо, необязательно замещенное одним или несколькими заместителями, независимо выбранными из группы, включающей OH, SH, CF3, F, Cl, Br, I, C1-C6 алкил, C1-C6 алкилокси, C1-C6 алкилтио, NH2, C1-C6 алкиламино, ди(C1-C6 алкил)амино, NO2, COOH, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn, независимо, представляет собой H, или C1-C10 алкил, или C6-C10 арил;
при условии, что, когда n обозначает 2 и все Z1, Z2, Zm представляют собой -CH2-CH2-, тогда A не представляет собой -CH2COOH.
Редкоземельные элементы определяются как церий (Ce), диспрозий (Dy), эрбий (Er), европий (Eu), гадолиний (Gd), гольмий (Ho), лантан (La), лютеций (Lu), неодим (Nd), празеодим (Pr), прометий (Pm), самарий (Sm), скандий (Sc), тербий (Tb), тулий (Tm), иттербий (Yb) и иттрий (Y). Металлы s-, p- и d- блоков, предпочтительно, представляют собой металлы II.A, III.A, IV.A, V.A и переходные металлы, более предпочтительно, II.A, III.A (Al, Ga, In, Tl), IV.A (Sn, Pb), V.A (Bi), I.B, II.B и VIII. Металлы группы B, наиболее предпочтительно, выбраны из Ca2+, Fe2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Al3+, Pb2+, Bi3+.
Хроматографическое выделение редкоземельных элементов и/или металлов s-, p- и d- блоков определяется как выделение любого из упомянутых выше редкоземельных элементов и/или металлов s-, p- и d- блоков из смеси двух или более ионов металлов, по крайней мере, один из них является редкоземельным элементом и/или металлом s-, p- и d- блока.
Общая формула (I) по настоящему изобретению включает все изомеры, энантиомеры и диастереоизомеры.
В одном предпочтительном варианте изобретения применение по настоящему изобретению относится к хроматографическому выделению редкоземельных элементов, выбранных, таким образом, из церия (Ce), диспрозия (Dy), эрбия (Er), европия (Eu), гадолиния (Gd), гольмия (Ho), лантана (La), лютеция (Lu), неодима (Nd), празеодима (Pr), прометия (Pm), самария (Sm), скандия (Sc), тербия (Tb), тулия (Tm), иттербия (Yb) и иттрия (Y).
В одном предпочтительном варианте изобретения применение по настоящему изобретению относится к хроматографическому выделению металлов s-, p- и d- блоков, выбранных из групп металлов II.A, III.A, IV.A, V.A, переходных металлов (таких как I.B, II.B и VIII. B), предпочтительно, выбранных из Ca2+, Fe2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Al3+, Pb2+, Bi3+.
В одном предпочтительном варианте применения по настоящему изобретению X выбран из группы, включающей H, -CH3 и Cl.
В одном предпочтительном варианте применения по настоящему изобретению R1, R2, R3, независимо, представляют собой H, C1-C6 алкил, и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо, необязательно замещенное одним или несколькими заместителями, независимо выбранными из группы, включающей OH, SH, CF3, F, Cl, Br, I, C1-C6 алкил, C1-C6 алкилокси, C1-C6 алкилтио, NH2, C1-C6 алкиламино, ди(C1-C6 алкил)амино, NO2, COOH, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn, независимо, представляет собой H, или C1-C10 алкил, или C6-C10 арил.
В одном предпочтительном варианте применения по настоящему изобретению по меньшей мере два из A, Am представляют собой -CH2COOH и/или -CH2P(O)(OH)2.
В одном варианте применения по настоящему изобретению по меньшей мере один из A, Am представляет собой H.
В одном предпочтительном варианте применения по настоящему изобретению X представляет собой -CH3 или Cl, Y представляет собой азот и все R1, R2, R3 представляют собой H.
В другом предпочтительном варианте применения по настоящему изобретению X представляет собой H, Y представляет собой N-оксид и два из R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо, необязательно замещенное одним или несколькими заместителями, независимо выбранными из группы, включающей OH, SH, CF3, F, Cl, Br, I, C1-C6 алкил, C1-C6 алкилокси, C1-C6 алкилтио, NH2, C1-C6 алкиламино, ди(C1-C6 алкил)амино, NO2, COOH, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn, независимо, представляет собой H, или C1-C10 алкил, или C6-C10 арил.
В одном варианте применения по настоящему изобретению R1, R2, R3 независимо, выбраны из H, OH, OCH3, NO2, F, Cl, Br, I, CH3, COOH, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn определяется, как указано выше.
Предпочтительно, кольцо, содержащее атом Y, выбрано из изохинолин N-оксида, хинолин N-оксида, изохинолина, хинолина, пиридина, пиридин N-оксида; более предпочтительно, кольцо, содержащее атом Y, представляет собой пиридиновое кольцо, хинолин N-оксид или изохинолин N-оксид.
В одном предпочтительном варианте применения по настоящему изобретению соединения общей формулы (I) выбраны из группы, включающей:
1-((4,7-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид (1),
1-((4-(2-амино-2-oксoэтил)-7,10-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид (2),
1-((4,10-бис(2-амино-2-oксoэтил)-7-(фосфонометил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид (3),
1,1ʼ-((7,10-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1,4-диил)бис(метилен))бис(изохинолин 2-оксид) (4),
1-((1,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-4-ил)метил)изохинолин 2-оксид (5),
1-((4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-1-ил)метил)изохинолин 2-оксид (6),
1-((4,8,11-трис(карбоксиметил)-1,4,8,11-тетраазациклотетрадекан-1-ил)метил)изохинолин 2-оксид (7),
2,2ʼ,2ʺ-(11-((6-хлорпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота (8),
2,2ʼ,2ʺ-(11-((6-метилпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота (9),
1-((4,7-бис(карбоксиметил)-1,4,7-триазонан-1-ил)метил)изохинолин 2-оксид (10),
1-((5,9-бис(карбоксиметил)-1,5,9-триазациклододекан-1-ил)метил)изохинолин 2-оксид (11),
1-((4,8,12-трис(карбоксиметил)-1,4,8,12-тетраазациклопентадекан-1-ил)метил)изохинолин 2-оксид (12).
Объектом настоящего изобретения, кроме того, является способ хроматографического выделения редкоземельных элементов и/или металлов s-, p- и d- блоков, выбранных из групп металлов II.A, III.A, IV.A, V.A, переходных металлов (предпочтительно, группы I.B, II.B и VIII.B), из смеси по меньшей мере двух ионов металлов, где по меньшей мере один представляет собой металл, выбранный из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb, Y, щелочноземельных металлов, Al, Ga, In, Tl, Sn, Pb, Bi, переходных металлов (предпочтительно, где по меньшей мере один представляет собой металл, выбранный из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb, Y, Ca, Fe, Co, Ni, Cu, Zn, Al, Pb, Bi), и способ включает следующие стадии:
(a) предоставление смеси по меньшей мере одного иона металла, выбранного из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb, Y, щелочноземельных металлов, Al, Ga, In, Tl, Sn, Pb, Bi, переходных металлов, и по меньшей мере одного дополнительного иона металла, где указанный дополнительный ион металла выбран из ионов редкоземельных металлов, ионов переходных металлов, ионов непереходных металлов и ионов актинидов,
(b) ионы металлов, содержащиеся в указанной смеси, подвергают взаимодействию по меньшей мере с одним соединением общей формулы (I), как определено в любом из предшествующих пунктов, с образованием хелатов;
(c) хелаты со стадии (b) подвергают хроматографическому выделению,
предпочтительно, стационарная фаза выбрана из диоксида кремния (SiO2), оксида алюминия (Al2O3), диоксида титана (TiO2), диоксида циркония (ZrO2) или (C1-C18)дериватизированной обращенной фазы (такой как C1-C18, фенил, пентафторфенил, C1-C18 алкилфенил или обращенная фаза на основе полимера или углерод),
и, предпочтительно, подвижная фаза включает один или несколько растворителей, выбранных из воды, C1-C4 спирта, ацетонитрила, ацетона, N,N-диметилформамида, диметилсульфоксида, тетрагидрофурана, водного аммиака, подвижная фаза может в конечном итоге содержать одну или несколько добавок для регулирования pH, таких как кислоты, основания или буферы; где добавки для регулирования pH известны специалисту в данной области;
при этом, необязательно, стадия (c) может быть проведена по меньшей мере дважды, чтобы повысить чистоту по меньшей мере одного выделенного хелата металла;
и,
необязательно, (d) по меньшей мере один хелат металла, полученный в результате хроматографического выделения, подвергается кислотному разложению с получением иона металла, не входящего в комплекс.
Предпочтительно, фракции/пятна, содержащие отделенный хелат металла со стадии (с), объединяются вместе; предпочтительно, объединенные фракции, содержащие выделяемый хелат металла, концентрируют, например, упариванием перед повторением стадии (с).
В одном предпочтительном варианте изобретения способ хроматографического выделения по настоящему изобретению представляет собой способ хроматографического выделения редкоземельных элементов из смеси по меньшей мере двух ионов металлов, где по меньшей мере один из них представляет собой редкоземельный металл, выбранный из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb и Y, используя соединения общей формулы (I), определенные выше, и включающий следующие стадии:
(a) предоставление смеси по меньшей мере одного иона редкоземельного металла, выбранного из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb и Y, и по меньшей мере одного дополнительного иона металла, где указанный дополнительный ион металла выбран из ионов редкоземельных металлов, ионов переходных металлов, ионов непереходных металлов и ионов актинидов,
(b) ионы металлов, содержащиеся в указанной смеси, подвергают взаимодействию по меньшей мере с одним соединением общей формулы (I), определенным выше, с образованием хелатов;
(c) хелаты со стадии (b) подвергают хроматографическому выделению, такому как колоночная хроматография, тонкослойная хроматография или высокоэффективная жидкостная хроматография (ВЭЖХ); предпочтительно, стационарная фаза выбрана из диоксида кремния (SiO2), оксида алюминия (Al2O3), диоксида титана (TiO2), диоксида циркония (ZrO2) или (C1-C18)дериватизированной обращенной фазы (такой как C1-C18, фенил, пентафторфенил, C1-C18 алкилфенил или обращенная фаза на основе полимера или углерод)
и, предпочтительно, подвижная фаза включает один или несколько растворителей, выбранных из воды, C1-C4 спирта, ацетонитрила, ацетона, N,N-диметилформамида, диметилсульфоксида, тетрагидрофурана, водного аммиака, подвижная фаза может в конечном итоге содержать одну или несколько добавок для регулирования pH, такие как кислоты, основания или буферы; где добавки для регулирования pH известны специалисту в данной области;
при этом, необязательно, стадия (c) может быть проведена по меньшей мере дважды, чтобы повысить чистоту по меньшей мере одного выделенного хелата металла;
и,
необязательно, (d) по меньшей мере один хелат металла, полученный в результате хроматографического выделения, подвергается кислотному разложению с получением не входящего в комплекс иона редкоземельного металла.
Предпочтительно, фракции/пятна, содержащие отделенный хелат металла со стадии (с), объединяются вместе; предпочтительно объединенные фракции, содержащие выделяемый хелат металла, концентрируют, например, упариванием перед повторением стадии (c).
Дополнительный ион металла, упомянутый на стадии (а), выбран из ионов редкоземельных металлов, ионов переходных металлов, ионов непереходных металлов и ионов актинидов. Редкоземельные металлы представляют собой Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb и Y, переходные металлы представляют собой металлы d-блока периодической таблицы (группы от I.B до VIII.B), непереходные металлы представляют собой металлы из основных элементов группы (группы A) периодической таблицы и актиниды представляют собой от актиния до лоуренсия, химические элементы с атомными номерами от 89 до 103.
Кислота, используемая для разложения комплекса на стадии (d), предпочтительно выбирается из фтористоводородной, хлористоводородной, бромистоводородной, йодистоводородной, серной, азотной, пероксосерной, хлорной, метансульфоновой, трифторметансульфоновой, муравьиной, уксусной, трифторуксусной кислот или их смесей.
За стадией (d) может следовать хроматография полученной смеси для очистки свободных ионов редкоземельных металлов от молекул соединения общей формулы (I) или его фрагментов, образующихся в результате кислотного разложения. Метод хроматографического выделения осуществляется в растворе, и поиск подходящих условий для такой хроматографической очистки является рутинной работой специалиста в данной области.
В одном предпочтительном варианте изобретения хроматография на стадии a) представляет собой высокоэффективную жидкостную хроматографию (ВЭЖХ), осуществляемую с использованием стационарной обращенной фазы, предпочтительно, выбранной из C1-C18, фенила, пентафторфенила, C1-C18 алкил-фенила или обращенных фаз на основе полимера, и подвижной фазы, состоящей из воды и 0-40% (об.) смешивающегося с водой органического растворителя, выбранного из группы, включающей метанол, этанол, пропанол, изопропанол, ацетонитрил, ацетон, N,N-диметилформамид, диметилсульфоксид, тетрагидрофуран,
и, необязательно, подвижная фаза дополнительно содержит до 10% (мас./мас.) ионно-парной добавки, состоящей из катионной части и анионной части,
где катионная часть выбрана из группы, включающей H+, Li+, Na+, K+, Rb+, Cs+, NH4+, C1-C8 тетраалкиламмоний,
и где анионная часть выбрана из группы, включающей F-, Cl-, Br-, I-, сульфат, гидросульфат, нитрат, перхлорат, метансульфонат, трифторметансульфонат, (C2-C18 алкил)сульфонат, формиат, ацетат, (C2-C18 алкил)карбоксилат, лактат, малат, цитрат, 2-гидроксиизобутират, манделат, дигликолят, тартрат.
В предпочтительном варианте осуществления раствор, содержащий смесь, представленную на стадии (а), в виде солей (например, хлорид, бромид, сульфат, нитрат, метансульфонат, трифторметансульфонат, формиат, ацетат, лактат, малат, цитрат, 2-гидроксиизобутират, манделат, дигликолят, тартрат) или твердую фазу, содержащую смесь, представленную на стадии (а) (например, в виде оксида, гидроксида, карбоната),
смешивают с раствором соединения общей формулы (I) при молярном отношении ионов металла к соединению общей формулы (I) от 1:0,5 до 1:100, предпочтительно, от 1:0,7 до 1:50, более предпочтительно, от 1:0,9 до 1:10. Концентрации растворимых компонентов могут быть выбраны из диапазона концентраций, допускаемого растворимостью таких соединений в данном растворителе при данной температуре, предпочтительно, в диапазоне концентраций 0,000001-0,5 моль/л. Растворителем может быть вода, смешивающийся с водой органический растворитель, такой как метанол, этанол, пропанол, изопропанол, ацетон, ацетонитрил, N,N-диметилформамид, диметилсульфоксид, тетрагидрофуран или их смесь. Органическое или неорганическое основание, такое как LiOH, NaOH, KOH, водный NH3, триэтиламин, N,N-диизопропилэтиламин или пиридин, добавляют к реакционной смеси, чтобы компенсировать протоны, высвобождаемые во время комплексообразования, и в процессе растворения происходит комплексообразование. Предпочтительно, на молекулу соединения общей формулы (I) добавляют 1-10 молярных эквивалентов основания. В конце концов, реакция может происходить в буфере. В этом случае нет необходимости добавлять в реакционную смесь органическое или неорганическое основание. Смесь перемешивают или встряхивают при комнатной или повышенной температуре в течение до 24 часов до полного комплексообразования. Предпочтительно, смесь перемешивают или встряхивают при температуре 40ºC в течение 15 минут. Разумный избыток соединения общей формулы (I) можно использовать для ускорения комплексообразования и смещения равновесия в сторону образования хелатов. Результат стадии (b) представляет собой смесь различных хелатов металлов в растворе.
В предпочтительном варианте осуществления хроматографическое выделение хелатов на стадии (b) происходит на нормальной или обращенной неподвижной фазе. Нормальная фаза может быть диоксидом кремния (SiO2) или оксидом алюминия (Al2O3). Можно использовать ряд обращенных фаз, включая обращенные фазы на основе C1-C18, фенила, пентафторфенила, (C1-C18 алкил)-фенила и обращенные фазы на основе полимера. Раствор хелатов металлов необязательно можно центрифугировать или фильтровать перед хроматографией на стадии (b), чтобы удалить частицы, такие как нерастворимые примеси или пыль. Выделение может быть достигнуто с помощью различных хроматографических схем, включая колоночную хроматографию, тонкослойную хроматографию (ТСХ) и высокоэффективную жидкостную хроматографию (ВЭЖХ). В процессе хроматографии также выделяется избыток соединения общей формулы (I). Предпочтительно, хроматографическое выделение достигается с помощью ВЭЖХ на C8, C18 или фенилгексильной обращенной фазе. В предпочтительном варианте используется подвижная фаза, состоящая из воды и 3-40% метанола, этанола или ацетонитрила. Необязательно, в подвижной фазе используется 0,01-0,1 моль/л буфера, где буфер содержит ацетат натрия, pH=4,5, формиат аммония, pH=7,0, или ацетат аммония, pH=7,0. Фракции, содержащие желаемый хелат металла, собирают и объединяют, в результате чего получают раствор, значительно обогащенный содержанием желаемого хелата редкоземельного металла по сравнению с исходной смесью хелатов металлов до хроматографии. Процесс можно повторить для дальнейшего повышения чистоты продукта.
В предпочтительном варианте осуществления разложение очищенного хелата на стадии (d) выполняется обработкой раствора хроматографически очищенного хелата органической или неорганической кислотой для достижения декомплексации иона металла из хелата. Органическая или неорганическая кислота выбрана из группы, включающей фтористоводородную, хлористоводородную, бромистоводородную, йодистоводородную, серную, азотную, пероксосерную, хлорную, метансульфоновую, трифторметансульфоновую, муравьиную, уксусную, трифторуксусную кислоты или их смеси. Выбор кислоты и условий реакции для достижения полноты распада комплексов очевиден для специалиста в данной области. Предпочтительно распад комплексов достигается с помощью соляной кислоты (0,01-12 моль/л) при температуре 25-95ºC в течение периода времени от 5 минут до 24 часов. Затем проводят вторичную хроматографическую очистку для удаления молекулы свободного хелатора (соединения общей формулы (I)) от ионов редкоземельных металлов. Это может быть достигнуто с помощью колоночной хроматографии или твердофазной экстракции с использованием стационарной обращенной фазы. Предпочтительно, обращенная фаза представляет собой обращенную фазу на основе C18 или полимера. Предпочтительно использовать подвижную фазу, которая состоит из чистой воды или воды, содержащей 0,01-1% (об.) кислоты, используемой на стадии (d) для разложения хелата. Хелатор остается на обращенной фазе, в то время как свободные ионы металлов элюируются в виде соли с кислотой, используемой на стадии (d) для разложения хелата. В качестве альтернативы используется хроматографическое выделение, описанное на стадии (c). Еще одной альтернативой является минерализация очищенного хелата металла посредством окисления в азотной кислоте или пероксосерной кислоте. Предпочтительно, минерализация достигается путем смешивания 1 части раствора хелата металла с 4 или более частями 70%-ной азотной кислоты и инкубирования при температуре 25-95ºC в течение периода времени от 5 минут до 24 часов. В таком случае молекула хелатора озоляется, и выделения не требуется.
Увеличение концентрации объединенных фракций, содержащих хелат металла, выделяемый перед повторением стадии (с), может быть достигнуто путем частичного упаривания растворителя или адсорбции хелата на липофильных материалах, таких как обращенная фаза. Предпочтительно использовать ту же обращенную фазу, что и для хроматографического выделения на стадии (с). Когда водный раствор хелата приводят в физический контакт с обращенной фазой, это приводит к адсорбции хелата. Затем хелат может быть десорбирован из обращенной фазы с помощью более сильного элюента, где более сильный элюент содержит более высокий процент смешивающегося с водой органического растворителя, чем исходный раствор хелата, в котором смешивающийся с водой органический растворитель представляет собой метанол, этанол, пропанол, изопропанол, ацетон, ацетонитрил, N,N-диметилформамид, диметилсульфоксид, тетрагидрофуран или их смесь. Сила элюента регулируется процентным содержанием смешивающегося с водой органического растворителя в подвижной фазе.
В предпочтительном варианте осуществления раствор хелатов металлов соединений общей формулы (I) концентрируют путем адсорбции в обращенную фазу в два этапа: (i) разбавленный водный раствор хелата пропускают через обращенную фазу, что приводит к адсорбции хелата. Если раствор представляет собой хроматографическую фракцию, собранную после предыдущего хроматографического выделения, и, как таковой, содержит смешивающийся с водой органический растворитель, его сначала разбавляют дистиллированной водой перед адсорбцией для уменьшения силы элюента. Предпочтительно, раствор разбавляют равным или большим объемом воды, таким образом снижая процентное содержание смешиваемого с водой органического растворителя до половины или менее от первоначального значения. (ii) На второй стадии хелат десорбируется из обращенной фазы более сильным элюентом, содержащим более высокий процент смешивающегося с водой органического растворителя. Предпочтительно, в качестве элюента используется подвижная фаза, используемая для хроматографического выделения на стадии (c). В этом случае может быть непосредственно выполнено вторичное хроматографическое выделение. В качестве альтернативы используют более сильный элюент с объемом, меньшим, чем исходный объем адсорбированного раствора, и непосредственно собирают десорбированный хелат металла. В этом случае концентрация хелата металла увеличивается по сравнению с исходным раствором. Преимущество этого метода состоит в том, что он позволяет концентрировать растворы хелатов металлов без необходимости длительного упаривания, что не является предпочтительным, особенно при работе с радионуклидами. Важно отметить, что на хроматографической колонке с обращенной фазой этот метод приводит к сорбции хелатов металлов в узкой полосе в начале колонки и последовательно приводит к резким пикам и более эффективному хроматографическому выделению. Это контрастирует с широкими пиками и плохим выделением, которое могло бы быть результатом присутствия сильного элюента в ранее собранных фракциях, если бы такие фракции использовались без изменений для другого хроматографического выделения. Кроме того, этот способ позволяет быстро повторять хроматографические выделения ранее собранных хроматографических фракций. Быстрое повторение хроматографической очистки обеспечивает получение желаемого хелата металла высокой чистоты за более короткое время.
Объектом настоящего изобретения являются также соединения общей формулы (Ia),
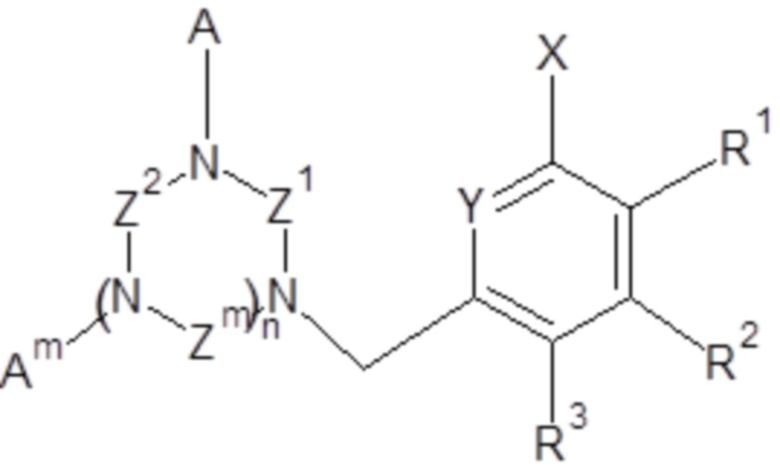 ,
,
(Ia),
где
- X выбран из группы, включающей H, C1-C6 алкил, галоген (F, Cl, Br или I); предпочтительно, X выбран из группы, включающей H, -CH3, галоген;
- Y выбран из группы, включающей азот, N-оксид;
- Z1, Z2, Zm, где m обозначает 1 или 2, независимо, выбраны из группы, включающей -CH2-CH2- и -CH2-CH2-CH2-;
- A, Am, где m обозначает 1 или 2, независимо, выбраны из H, -CH2COOH, -CH2C(O)NH2, -CH2P(O)(OH)2, и где по меньшей мере один из A, Am представляет собой H;
- n обозначает 1 или 2;
R1, R2, R3, независимо, представляют собой H, C1-C6 алкил, C1-C6 алкилокси, C6-C10 арилокси, бензилокси, C1-C6 алкилтио, C6-C10 арилтио, F, Cl, Br, I, OH, SH, NH2, C1-C6 алкиламино, ди(C1-C6 алкил)амино, C1-C6 ациламино, ди(C1-C6 ацил)амино, C6-C10 ариламино, ди(C6-C10 арил)амино, CN, OH, нитро, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn, независимо, представляет собой H, или C1-C10 алкил, или C6-C10 арил,
и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо, необязательно замещенное одним или несколькими заместителями, независимо выбранными из группы, включающей OH, SH, CF3, F, Cl, Br, I, C1-C6 алкил, C1-C6 алкилокси, C1-C6 алкилтио, NH2, C1-C6 алкиламино, ди(C1-C6 алкил)амино, NO2, COOH, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn, независимо, представляет собой H, или C1-C10 алкил, или C6-C10 арил;
при условии, что:
- когда n обозначает 2, все Z1, Z2, Zm представляют собой -CH2-CH2-, тогда A не представляет собой -CH2COOH;
- когда Y представляет собой азот, тогда X не представляет собой H;
- когда n обозначает 1, все Z1, Z2, Zm представляют собой -CH2-CH2- и Y представляет собой азот, тогда X не представляет собой галоген.
Подразумевается, что общая формула (Ia) по настоящему изобретению включает все изомеры, энантиомеры и диастереоизомеры.
В одном предпочтительном варианте соединений общей формулы (Ia) по настоящему изобретению X выбран из группы, включающей H; -CH3 и Cl.
В одном предпочтительном варианте соединений общей формулы (Ia) по настоящему изобретению R1, R2, R3, независимо, представляют собой H; C1-C6 алкил; и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо, необязательно замещенное одним или несколькими заместителями, независимо выбранными из группы, включающей OH, SH, CF3, F, Cl, Br, I, C1-C6 алкил, C1-C6 алкилокси, C1-C6 алкилтио, NH2, C1-C6 алкиламино, ди(C1-C6 алкил)амино, NO2, COOH, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn, независимо, представляет собой H, или C1-C10 алкил, или C6-C10 арил.
В одном предпочтительном варианте соединений общей формулы (Ia) по настоящему изобретению по меньшей мере два из A, Am представляют собой -CH2COOH и/или -CH2P(O)(OH)2.
В одном предпочтительном варианте соединений общей формулы (Ia) по настоящему изобретению по меньшей мере один из A, Am представляет собой H.
В одном предпочтительном варианте соединений общей формулы (Ia) по настоящему изобретению X представляет собой -CH3 или Cl, Y представляет собой азот и все R1, R2, R3 представляют собой H.
В другом предпочтительном варианте соединений общей формулы (Ia) по настоящему изобретению X представляет собой H, Y представляет собой N-оксид и два из R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо, необязательно замещенное одним или несколькими заместителями, независимо выбранными из группы, включающей OH, SH, CF3, F, Cl, Br, I, C1-C6 алкил, C1-C6 алкилокси, C1-C6 алкилтио, NH2, C1-C6 алкиламино, ди(C1-C6 алкил)амино, NO2, COOH, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn, независимо, представляет собой H, или C1-C10 алкил, или C6-C10 арил.
В одном предпочтительном варианте соединений общей формулы (Ia) по настоящему изобретению R1, R2, R3 независимо, выбраны из H, OH, OCH3, NO2, F, Cl, Br, I, CH3, COOH, COORn, C(O)NHRn, C(O)N(Rn)2, где Rn определяется, как указано выше.
Предпочтительно, кольцо, содержащее атом Y, выбрано из изохинолин N-оксида, хинолин N-оксида, изохинолина, хинолина, пиридина, пиридин N-оксида; более предпочтительно, кольцо, содержащее атом Y, представляет собой пиридиновое кольцо, хинолин N-оксид или изохинолин N-оксид.
В одном предпочтительном варианте соединений общей формулы (Ia) по настоящему изобретению соединения общей формулы (I) выбраны из группы, включающей:
1-((4,7-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид (1),
1-((4-(2-амино-2-oксoэтил)-7,10-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид (2),
1-((4,10-бис(2-амино-2-oксoэтил)-7-(фосфонометил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид (3),
1-((1,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-4-ил)метил)изохинолин 2-оксид (5),
1-((4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-1-ил)метил)изохинолин 2-оксид (6),
1-((4,8,11-трис(карбоксиметил)-1,4,8,11-тетраазациклотетрадекан-1-ил)метил)изохинолин 2-оксид (7),
2,2ʼ,2ʺ-(11-((6-хлорпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота (8),
2,2ʼ,2ʺ-(11-((6-метилпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота (9),
1-((4,7-бис(карбоксиметил)-1,4,7-триазонан-1-ил)метил)изохинолин 2-оксид (10),
1-((5,9-бис(карбоксиметил)-1,5,9-триазациклододекан-1-ил)метил)изохинолин 2-оксид (11),
1-((4,8,12-трис(карбоксиметил)-1,4,8,12-тетраазациклопентадекан-1-ил)метил)изохинолин 2-оксид (12).
Описанное изобретение представляет собой комплексный подход к манипуляции с ионами металлов в растворе, который значительно упрощает их перенос, очистку и уменьшение объема, одновременно предотвращая загрязнение другими металлами. Это особенно полезно при обращении с металлическими радионуклидами, когда эти операции проблематичны. Настоящее изобретение позволяет выполнять эти операции в быстрой последовательности, многократно и в изменяющемся порядке.
Способ хроматографического выделения ионов металлов согласно настоящему изобретению заметно отличается от существующих хроматографических методов. В существующих методах селективность по отношению к различным элементам, таким как редкоземельные элементы, обеспечивается неподвижной фазой или добавкой, которая добавляется к подвижной фазе в избытке по сравнению с выделенными металлами, или обоими одновременно (Kifle, D., Wibetoe, G. (2013), J. Chromatogr. A 1307, 86-90; Schwantes, J. M. et al. (2008) J. Radioanal. Nucl. Chem. 276(2), 533-542). Напротив, в способе по настоящему изобретению селективность происходит от молекулы хелатора, которая остается тесно связанной с ионом металла на протяжении всего процесса выделения. Таким образом, настоящее изобретение позволяет использовать обычные стационарные фазы (например, нормальная фаза: SiO2; обращенная фаза: C18, C8, фенилгексил, фенил, обращенная фаза на основе полимера) и подвижные фазы (такие как: вода/ацетонитрил, вода/метанол, вода/этанол, вода/изопропанол), не обладающие особой селективностью по отношению к конкретным элементам для их эффективного выделения.
У хелаторов, описанных в данном изобретении, есть несколько отличительных признаков, которые представляют собой важное отличие от хелаторов и лигандов, используемых в существующих технологиях выделения элементов, таких как редкоземельные элементы. Описанные хелаторы содержат ароматический фрагмент, который играет главную роль в полярности хелатов металлов. По этой причине ароматический фрагмент имеет решающее значение для способности хелаторов различать металлы на основе полярности хелатов. Кроме того, ароматическая составляющая служит хромофором, который облегчает обнаружение хелатора и хелатов металлов на основе УФ-поглощения или гашения флуоресценции на пластине для ТСХ. Другой важной особенностью описанных хелаторов является то, что они образуют хелаты с металлами, которые кинетически инертны в течение всего процесса выделения. Примечательно, что это свойство снижает риск загрязнения другими металлами, поскольку очищаемый металл не может легко выйти из хелата и не может быть заменен другим ионом металла.
Настоящее изобретение обеспечивает быстрый и удобный способ эффективного отделения даже соседних лантаноидов друг от друга, то есть отделение, которое, как известно, является сложной проблемой.
Все эти операции можно легко автоматизировать, чтобы ограничить облучение оператора в случае использования металлических радионуклидов. Присутствие ароматической части хромофора в структуре хелаторов облегчает обнаружение по УФ-поглощению или по гашению флуоресценции на пластине для ТСХ. Таким образом, настоящее изобретение представляет собой комплексный подход, позволяющий осуществлять быстрый перенос, очистку и уменьшение объема растворов радионуклидов металлов.
Краткое описание фигур
Фиг. 1: Набор хроматограмм ВЭЖХ хелатов Gd, Tb и Dy одного хелатора, подвергнутых анализу ВЭЖХ отдельно (три нижних) и в смеси (вверху), как описано в примере 15. Было достигнуто базовое выделение всех трех хелатов металлов.
Фиг. 2: Набор хроматограмм ВЭЖХ, показывающий распад комплекса хелата Tb (внизу) в кислых условиях, как описано в примере 16, и его сравнение с таким же свободным хелатирующим агентом (вверху) в тех же условиях. Присутствие пика свободного хелатора на хроматограмме хелата Tb подтверждает значительный распад комплекса хелата на свободный хелатор и свободные ионы металлов.
Примеры
Численные значения химического сдвига в спектрах ЯМР приведены в м. д. Обозначения, используемые в спектрах ЯМР: с (синглет), д (дублет), т (триплет), м (мультиплет), шир. с (широкий синглет). В качестве эталона были заданы следующие значения:
1H (25ºC): 7,26 м. д. (CDCl3); 3,75 м. д. (диоксан); 4,70 м. д. (HOD).
1H (90ºC): 3,75 м. д. (диоксан); 4,23 м. д. (HOD).
13C (90ºC): 67,2 м. д. (диоксан).
13C (25ºC): 77,16 м. д. (CDCl3); 66,6 м. д. (диоксан).
31P (25ºC): 0,0 м. д. (H3PO4).
Список сокращений
ESI (ионизация электрораспылением); ВЭЖХ (высокоэффективная жидкостная хроматография); МСРВ (масс-спектрометрия высокого разрешения); ЖХ-МС (жидкостная хроматография-масс-спектрометрия)); MOPS (3-морфолинопропан-1-сульфоновая кислота); NCA (без добавления носителя); ТФУ (трифторуксусная кислота); ТСХ (тонкослойная хроматография); УФ (ультрафиолетовое излучение).
I. Синтез соединений
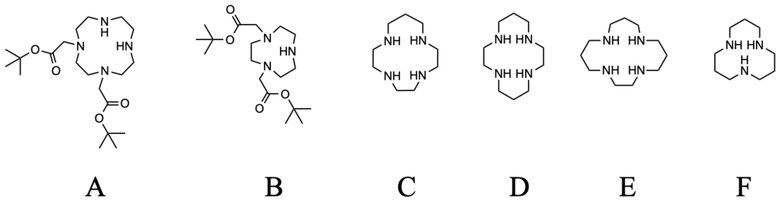
Структуры исходных макроциклических производных A, B, C, D, E и F
Пример 1: Получение 1-(бромметил)изохинолин 2-оксида (1a)
1-(Бромметил)изохинолин (150 мг, 0,675 ммоль) растворяли в хлороформе (15 мл) и охлаждали на ледяной бане. При перемешивании добавляли м-хлорпероксибензойную кислоту (77%, 0,230 г, 1,03 ммоль). Реакционной смеси давали постепенно нагреться до комнатной температуры и перемешивали в течение 24 часов. Растворитель упаривали и остаток очищали колоночной хроматографией на силикагеле в смеси метанол/этилацетат. Фракции, содержащие продукт, упаривали с получением 102 мг продукта в виде твердого вещества бледно-желтого цвета (0,430 ммоль, 64%-ный выход относительно 1-(бромметил)изохинолина).
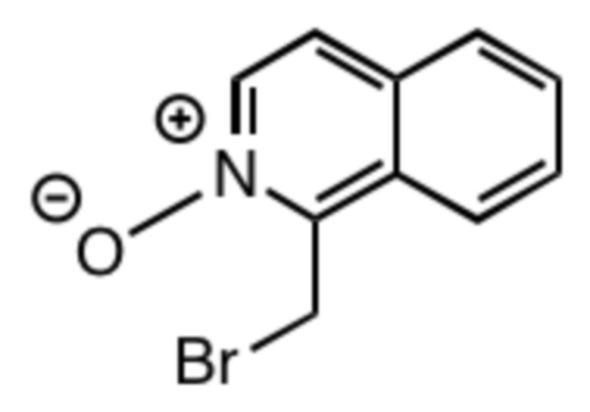
1H ЯМР (CDCl3, 25ºC, 500 МГц): δH 5,17 (CH2-аром., с, 2H); 7,61 (аром., ддд, 1H, 3JHH=8 Гц, 3JHH=7 Гц, 4JHH=1 Гц); 7,64 (аром., д, 1H, 3JHH=7 Гц); 7,73 (аром., ддд, 1H, 3JHH=9 Гц, 3JHH=7 Гц, 4JHH=1 Гц); 7,80-7,83 (аром., м, 1H); 7,95 (аром., ддд, 1H, 3JHH=9 Гц, 4JHH=2 Гц, 4JHH=1 Гц); 8,19 (аром., д, 1H, 3JHH=7 Гц); 13C{1H} ЯМР (CDCl3, 25ºC, 125 МГц): δC 20,9 (CH2-аром., с); 122,9 (аром., с); 124,0 (аром., с); 127,6 (аром., с); 127,8 (аром., с); 128,6 (аром., с); 128,8 (аром., с); 129,9 (аром., с); 136,9 (аром., с); 143,1 (аром., с).
МСРВ (ESI) m/z: [(M+H)+] (C10H9BrNO) вычислено: 237,9862, найдено: 237,9863.
Получение 1-((4,7-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксида (1)
Исходное соединение A (145 мг, 0,362 ммоль), 1-(бромметил)изохинолин 2-оксид (86 мг, 0,362 ммоль) и карбонат цезия (354 мг, 1,09 ммоль) растворяли в ацетонитриле (10 мл) и перемешивали в течение 24 часов при комнатной температуре. Твердые продукты отфильтровывали и фильтрат концентрировали на роторном испарителе. Образовавшееся масло очищали с помощью препаративной ВЭЖХ (колонка C18, градиент ацетонитрил/вода с 0,1% ТФУ в подвижной фазе). На этой стадии дважды алкилированный побочный продукт также собрали и обрабатывали отдельно. Фракции, содержащие чистый продукт в виде трет-бутилового эфира, объединяли, упаривали и сушили в высоком вакууме. Образовавшееся масло делили на две равные части, которые обрабатывали отдельно. Одну часть использовали для дальнейшего синтеза, как описано в примере 2. Вторую часть растворяли в чистой ТФУ (4 мл) и перемешивали в течение 24 ч при комнатной температуре. ТФУ упаривали на роторном испарителе, остаток растворяли в дистиллированной воде (2 мл) и очищали с помощью препаративной ВЭЖХ (условия, указанные выше). Фракции, содержащие продукт, объединяли и упаривали. Остаток растворяли в дистиллированной воде (2 мл) и лиофилизовали, получая 41,0 мг продукта в виде белого пушистого твердого вещества (0,055 ммоль, 30%-ный выход относительно A).
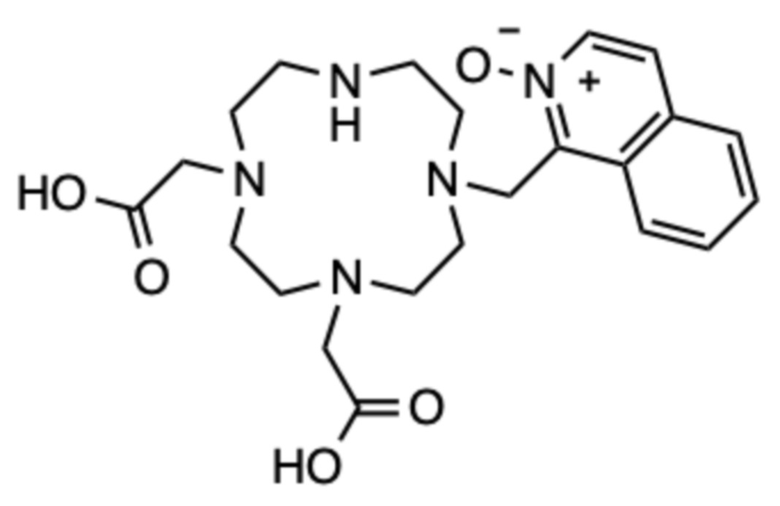
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 2,64-3,95 (8˟цикл CH2+2˟CH2-COOH, 20H); 4,13-4,39 (CH2-аром., м, 2H); 7,64-7,78 (2˟аром., м, 2H); 7,82-7,93 (2˟аром., м, 2H); 8,09 (аром., д, 1H, 3JHH=7 Гц); 8,14 (аром., д, 1H, 3JHH=8 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 42,5 (CH2‒COOH, с); 42,6 (CH2‒COOH, с); 45,3 (цикл, с); 47,1 (CH2‒аром., с); 48,1 (цикл, с); 49,1 (цикл, с); 50,1 (цикл, с); 50,9 (цикл, с); 52,8 (цикл, с); 53,1 (цикл, с); 53,2 (цикл, с); 123,5 (аром., с); 125,4 (аром., с); 127,7 (аром., с); 128,1 (аром., с); 130,6 (аром., с); 130,9 (аром., с); 131,0 (аром., с); 135,8 (аром., с); 144,0 (аром., с); 166,7 (CO, с); 173,5 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C22H32N5O5) вычислено: 446,2398, найдено: 446,2397.
Элем. анализ: M·2,3ТФУ·2,2H2O, вычислено: C (42,7), H (5,1), N (9,4), F (17,5), найдено: C (42,8), H (4,5), N (8,9), F (17,4).
Пример 2: 1-((4-(2-амино-2-оксоэтил)-7,10-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид (2)
Синтез начинали из соединений A и 1-(бромметил)изохинолин 2-оксида, как описано в примере 1, до стадии, на которой промежуточный бис(трет.бутиловый) эфир соединения 1, (t-Bu21), получали препаративной ВЭЖХ. Раствор t-Bu21 (приблизительно 57 мг, 0,102 ммоль, ½ от общего количества, полученного в примере 1), полученный с помощью препаративной ВЭЖХ, нейтрализовали карбонатом цезия до pH=7, упаривали и сушили в высоком вакууме. Полученный твердый продукт растворяли в ацетонитриле (10 мл), добавляли карбонат цезия (110 мг, 0,338 ммоль), затем добавляли йодацетамид (48,4 мг, 0,262 ммоль). Суспензию перемешивали в течение 48 ч при температуре 80ºC. Твердые продукты отфильтровывали и фильтрат концентрировали на роторном испарителе. Образовавшееся масло очищали с помощью препаративной ВЭЖХ (колонка C18, градиент ацетонитрил/вода с 0,1% ТФУ в подвижной фазе). Фракции, содержащие чистый продукт в виде трет-бутилового эфира, объединяли, упаривали и сушили в высоком вакууме. Остаток растворяли в чистой ТФУ (4 мл) и перемешивали в течение 24 ч при комнатной температуре. ТФУ упаривали на роторном испарителе, остаток растворяли в дистиллированной воде (2 мл) и очищали с помощью препаративной ВЭЖХ (условия, указанные выше). Фракции, содержащие продукт, объединяли и упаривали. Остаток растворяли в дистиллированной воде (2 мл) и лиофилизовали, получая 33,4 мг продукта в виде белого пушистого твердого вещества (0,041 ммоль, 33%-ный выход относительно A).
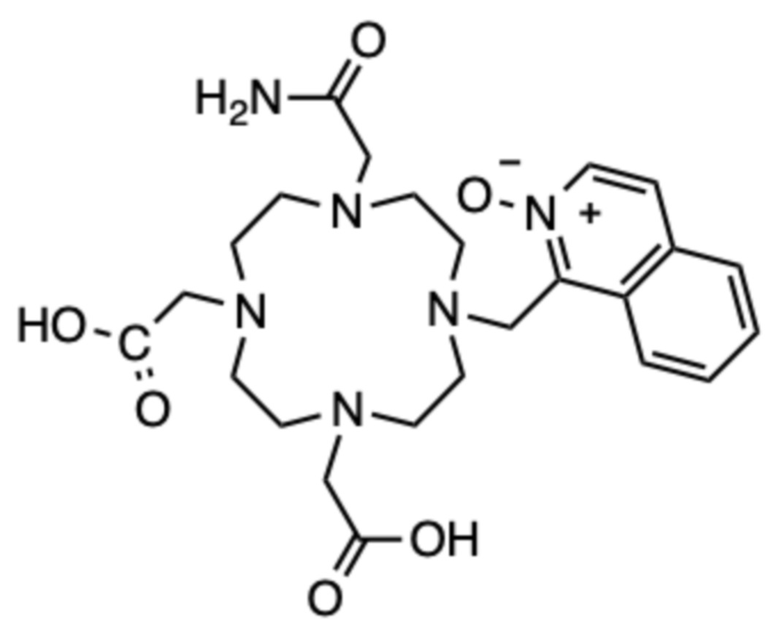
1H ЯМР (D2O с внутренним эталоном диоксаном, 90ºC, 500 МГц): δH 3,40-4,05 (8˟цикл CH2+CH2-COOH+CH2-CONH2, 20H); 4,40 (CH2-COOH, с, 2H); 5,42 (CH2-аром., с, 2H); 8,23-8,34 (2˟аром., м, 2H); 8,45-8,50 (аром., м, 1H); 8,52 (аром., д, 1H, 3JHH=7 Гц); 8,58-8,64 (аром., м, 1H); 8,64 (аром., д, 1H, 3JHH=7 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 90 °C, 125 МГц): 49,1 (цикл, с); 49,8 (цикл, с); 49,9 (цикл, с); 50,1 (цикл, с); 50,5 (CH2‒аром., с); 51,6 (цикл, с); 51,6 (цикл, с); 51,8 (цикл, с); 51,8 (цикл, с); 53,3 (CH2‒COOH, с); 54,4 (CH2‒CONH2, с); 55,2 (CH2‒COOH, с); 123,5 (аром., с); 127,4 (аром., с); 128,9 (аром., с); 129,0 (аром., с); 131,4 (аром., с); 131,6 (аром., с); 132,1 (аром., с); 135,8 (аром., с); 139,2 (аром., с); 170,2 (CO, с); 172,3 (CO, с); 172,4 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C24H35N6O6) вычислено: 503,2613, найдено: 503,2611.
Элем. анализ: M·2,5ТФУ·1,6H2O, вычислено: C (42,7), H (4,9), N (10,3), F (17,5), найдено: C (42,9), H (4,5), N (9,6), F (17,1).
Пример 3: 1-((4,10-бис(2-амино-2-оксоэтил)-7-(фосфонометил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид (3)
Циклен (300 мг, 1,74 ммоль) и параформальдегид (78,4 мг, 2,61 ммоль) смешивали в атмосфере аргона и растворяли в сухом тетрагидрофуране (12 мл). Добавляли три(трет.бутил)фосфит (654 мг, 2,61 ммоль) и смесь перемешивали при комнатной температуре в атмосфере аргона в течение 24 ч. Растворитель упаривали на роторном испарителе, остаток очищали с помощью препаративной ВЭЖХ (колонка C18, градиент ацетонитрил/вода с 0,1% ТФУ в подвижной фазе). Для обнаружения использовали масс-спектрометр. Промежуточный продукт с одной фосфонатной боковой цепью, то есть ди-трет-бутил ((1,4,7,10-тетраазациклододекан-1-ил)метил) фосфонат, собирали на основе массы (m/z для [M+H]+=379). Фракции, содержащие это промежуточное соединение, объединяли, нейтрализовали карбонатом цезия до pH=7, упаривали на роторном испарителе и сушили в высоком вакууме. Полученный твердый продукт смешивали с карбонатом цезия (400 мг, 1,84 ммоль), ацетонитрилом (10 мл) и перемешивали в течение 30 минут при комнатной температуре. Затем добавляли 1-(бромметил)изохинолин 2-оксид (55,3 мг, 0,232 ммоль) и смесь перемешивали 6 ч при температуре 80ºC. Твердые продукты отфильтровывали, растворитель упаривали на роторном испарителе и остаток очищали с помощью препаративной ВЭЖХ (так же, как описано выше). Собирали основной пик, соответствующий моноалкилированному промежуточному соединению (m/z для [M+H]+=536). Фракции, содержащие это промежуточное соединение, объединяли, нейтрализовали карбонатом цезия до pH=7, упаривали на роторном испарителе и сушили в высоком вакууме. Полученный твердый продукт смешивали с карбонатом цезия (73 мг, 0,224 ммоль), ацетонитрилом (4 мл) и йодацетамидом (10,4 мг, 0,056 ммоль) и перемешивали при комнатной температуре в течение 3 ч. Твердые продукты отфильтровывали, растворитель упаривали на роторном испарителе и остаток очищали с помощью препаративной ВЭЖХ (так же, как описано выше). Фракции, содержащие продукт в виде трет-бутилового эфира, объединяли и растворитель упаривали на роторном испарителе. Остаток растворяли в 2 мл ТФУ и перемешивали в течение 24 ч. ТФУ упаривали на роторном испарителе, остаток растворяли в дистиллированной воде (2 мл) и очищали с помощью препаративной ВЭЖХ (условия, указанные выше). Фракции, содержащие продукт, объединяли и упаривали. Остаток растворяли в дистиллированной воде (2 мл) и лиофилизовали, получая 10,3 мг продукта в виде белого пушистого твердого вещества (выход не определяли).
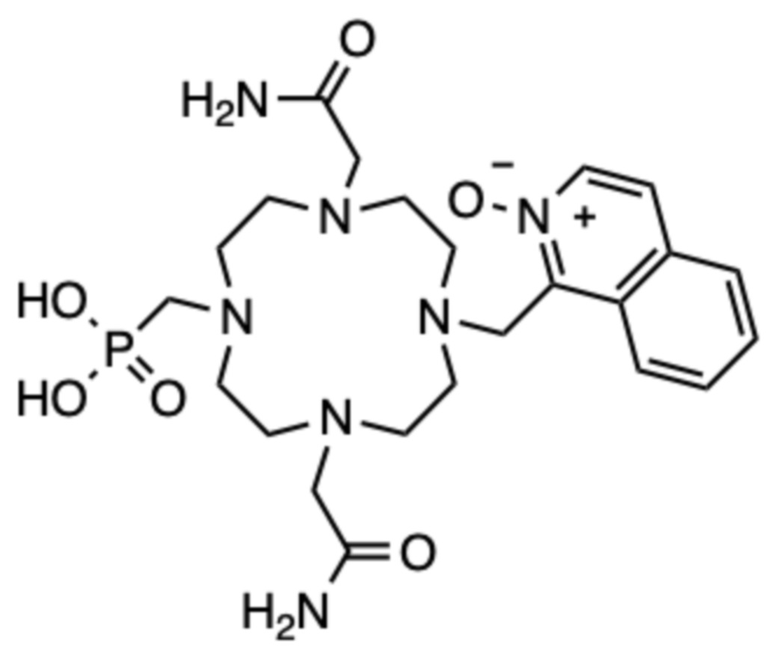
1H ЯМР (D2O с внутренним эталоном диоксаном, 90ºC, 500 МГц): δH 3,03-4,08 (8˟цикл CH2+2˟CH2-CONH2, 20H); 4,26 (CH2-PO3H2, шир. с, 2H); 5,03 (CH2-аром., шир. с, 2H); 8,23-8,36 (2˟аром., м, 2H); 8,45-8,52 (2˟аром., м, 2H); 8,60-8,65 (аром., м, 1H); 8,70 (аром., д, 1H, 3JHH=8 Гц). 31P{1H} ЯМР (D2O с внешним эталоном H3PO4, 25ºC, 202 МГц): δP 9,6 м. д. (шир. с).
МСРВ (ESI) m/z: [(M+H)+] (C23H37N7O6P) вычислено: 538,2537, найдено: 538,2531.
Пример 4: 1,1ʼ-((7,10-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1,4-диил)бис(метилен))бис(изохинолин 2-оксид) (4)
Соединение синтезировали в соответствии со способом, описанным в примере 1, в виде дважды алкилированного побочного продукта, аналогично получая 56,7 мг продукта в виде белого пушистого твердого вещества (0,061 ммоль, 17%-ный выход относительно A).
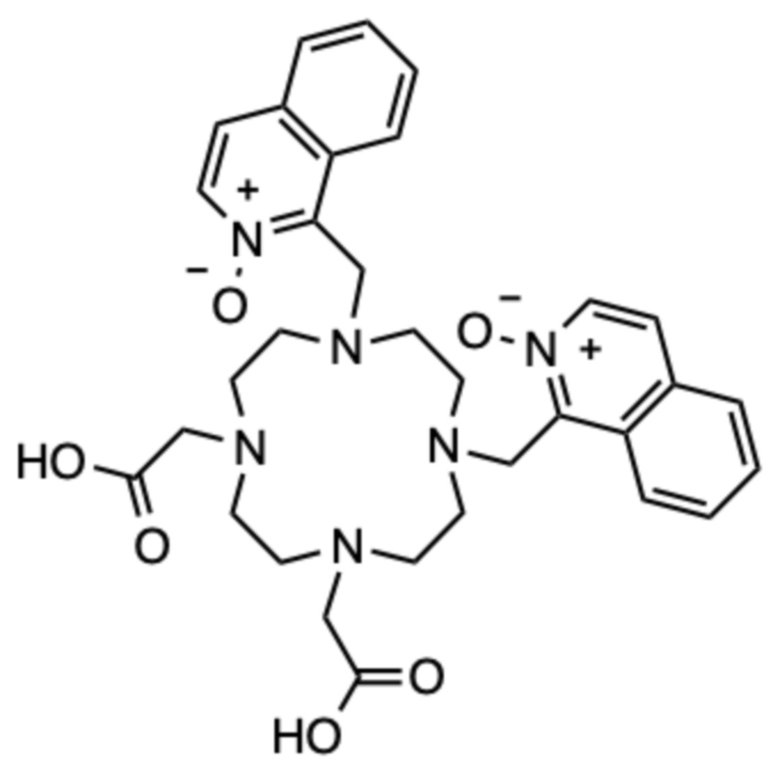
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 2,80-3,93 (8˟цикл CH2+2˟CH2-COOH, 20H); 4,71 (CH2-аром., с, 4H); 7,56-7,72 (2˟аром., м, 4H); 7,78-7,84 (аром., м, 2H); 7,84-7,90 (аром., м, 2H); 7,98-8,04 (аром., м, 2H); 8,04-8,10 (аром., м, 2H). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 48,2 (CH2‒аром., с); 49,8 (цикл, шир. с); 50,0 (цикл, шир. с); 50,2 (цикл, шир. с); 50,3 (цикл, шир. с); 53,2 (CH2‒COOH, с); 123,1 (аром., с); 126,3 (аром., с); 128,0 (аром., с); 128,5 (аром., с); 130,6 (аром., с); 130,6 (аром., с); 130,9 (аром., с); 135,1 (аром., с); 141,1 (аром., с); 172,4 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C32H39N6O6) вычислено: 603,2926, найдено: 603,2924.
Элем. анализ: M·2,6ТФУ·1,4H2O, вычислено: C (48,3), H (4,7), N (9,1), F (16,0), найдено: C (48,4), H (4,6), N (9,0), F (15,9).
Пример 5: 1-((1,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-4-ил)метил)изохинолин 2-оксид (5)
Исходное соединение C (42,7 мг, 0,229 ммоль) растворяли в смеси ацетонитрила (3 мл) и диметилформамида (0,5 мл). Затем добавляли 1-(бромметил)изохинолин 2-оксид (40 мг, 0,168 ммоль) и смесь перемешивали при комнатной температуре в течение 24 ч. Смесь концентрировали на роторном испарителе и остаток очищали с помощью препаративной ВЭЖХ (колонка C18, градиент ацетонитрил/вода с 0,1% ТФУ в подвижной фазе). Два изомера моноалкилированного промежуточного соединения (m/z для [M+H]+=344) успешно разделяли и обрабатывали отдельно. Фракции, содержащие эти две фракции, которые ранее были элюированы, объединяли, упаривали на роторном испарителе и сушили в высоком вакууме. Остаток растворяли в ацетонитриле (1 мл), добавляли карбонат цезия (84,7 мг, 0,260 ммоль) и смесь перемешивали в течение 10 мин при комнатной температуре. Затем добавляли трет.бутил бромацетат (29,6 мг, 0,152 ммоль) и смесь перемешивали при комнатной температуре в течение 24 ч. Твердые продукты отфильтровывали и фильтрат концентрировали на роторном испарителе. Образовавшееся масло очищали с помощью препаративной ВЭЖХ (колонка C18, градиент ацетонитрил/вода с 0,1% ТФУ в подвижной фазе). Фракции, содержащие продукт в виде трет-бутилового эфира, объединяли, упаривали на роторном испарителе и сушили в высоком вакууме. Остаток растворяли в чистой ТФУ (2 мл) и смесь перемешивали в течение 24 ч при комнатной температуре. ТФУ упаривали на роторном испарителе, остаток растворяли в дистиллированной воде (2 мл) и очищали с помощью препаративной ВЭЖХ (условия, указанные выше). Фракции, содержащие продукт, объединяли и упаривали. Остаток растворяли в дистиллированной воде (2 мл) и лиофилизовали, получая 8 мг продукта в виде белого пушистого твердого вещества (0,011 ммоль, 7%-ный выход относительно 1-(бромметил)изохинолин 2-оксида).
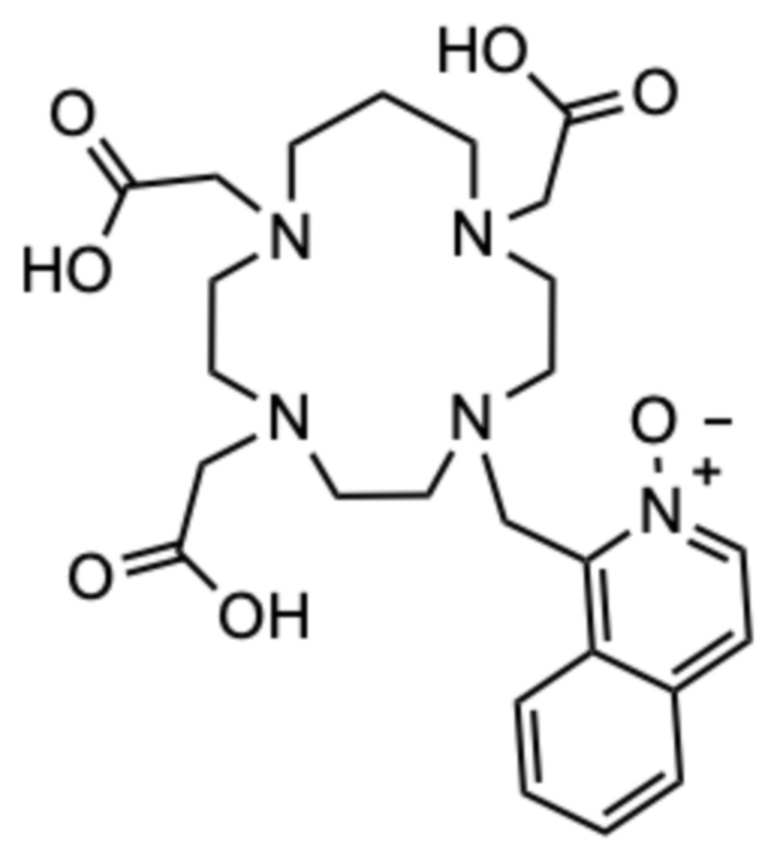
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 2,01 (CH2‒CH2‒CH2, м, 2H); 2,79-3,70 (8˟цикл CH2+2˟CH2-COOH, 20H); 3,97 (CH2-COOH, с, 2H); 5,03 (CH2-аром., с, 2H); 7,80-7,92 (2˟аром., м, 2H); 8,03-8,08 (аром., м, 1H); 8,14 (аром., д, 1H, 3JHH=7 Гц); 8,20-8,26 (аром., м, 1H); 8,35 (аром., д, 1H, 3JHH=7 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 21,9 (CH2‒CH2‒CH2, с); 49,6 (цикл, с); 49,9 (цикл, шир. с); 50,3 (цикл, с); 50,4 (CH2‒аром., с); 50,6 (цикл, с); 50,9 (цикл, с); 51,6 (цикл, с); 52,8 (цикл, с); 53,7 (CH2‒COOH, с); 53,9 (цикл, с); 54,8 (CH2‒COOH, шир. с); 55,0 (CH2‒COOH, с); 123,3 (аром., с); 127,2 (аром., с); 128,3 (аром., с); 128,5 (аром., с); 131,4 (аром., с); 131,5 (аром., с); 131,5 (аром., с); 135,1 (аром., с); 139,2 (аром., с); 169,8 (CO, с); 172,8 (CO, с); 172,9 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C25H36N5O7) вычислено: 518,2609, найдено: 518,2604.
Элем. анализ: M·1,5ТФУ·2,8H2O, вычислено: C (45,5), H (5,7), N (9,5), F (11,6), найдено: C (45,7), H (5,1), N (9,2), F (11,1).
Пример 6: 1-((4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-1-ил)метил)изохинолин 2-оксид (6)
Соединение получали в соответствии со способом, описанным в примере 5, в качестве второго изомера. Аналогичным образом, используя карбонат цезия (80,1 мг, 0,246 ммоль) и трет.бутил бромацетат (28 мг, 0,143 ммоль) на второй стадии и следуя тому же способу получали 7 мг конечного продукта в виде белого пушистого твердого вещества (0,0094 ммоль, 6%-ный выход относительно 1-(бромметил)изохинолин 2-оксида).
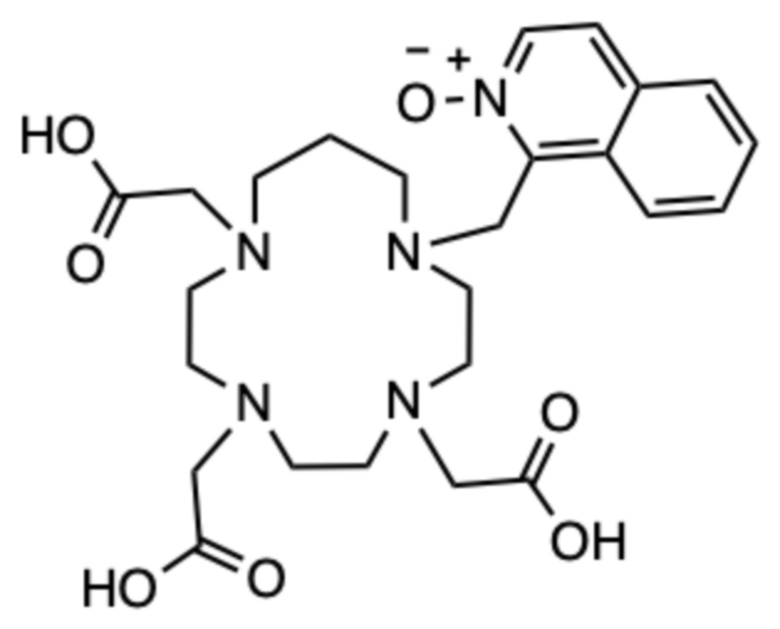
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 2,06-2,18 (CH2‒CH2‒CH2, м, 2H); 2,87-3,35 (6˟цикл CH2, 12H); 3,39-3,64 (2˟цикл CH2+2˟CH2-COOH, 8H); 3,93 (CH2-COOH, с, 2H); 5,02 (CH2-аром., с, 2H); 7,79-7,91 (2˟аром., м, 2H); 8,02-8,07 (аром., м, 1H); 8,10 (аром., д, 1H, 3JHH=7 Гц); 8,15-8,20 (аром., м, 1H); 8,23 (аром., д, 1H, 3JHH=7 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 21,3 (CH2‒CH2‒CH2, с); 49,8 (CH2‒аром., с); 49,8 (цикл, шир. с); 51,2 (цикл, с); 51,5 (цикл, с); 51,5 (цикл, с); 52,4 (цикл, с); 52,4 (CH2‒COOH, шир. с); 52,5 (цикл, с); 53,6 (CH2‒COOH, с); 53,6 (цикл, с); 54,1 (цикл, с); 55,1 (CH2‒COOH, с); 123,3 (аром., с); 126,9 (аром., с); 128,2 (аром., с); 128,4 (аром., с); 131,2 (аром., с); 131,4 (аром., с); 131,4 (аром., с); 135,1 (аром., с); 138,9 (аром., с); 170,7 (CO, шир. с); 172,8 (CO, с); 173,3 (CO, с).
МСРВ (ESI) m/z: [(M+Na)+] (C25H35N5O7Na) вычислено: 540,2429, найдено: 540,2429.
Элем. анализ: M·1,5ТФУ·2,9H2O, вычислено: C (45,4), H (5,8), N (9,5), F (11,5), найдено: C (45,5), H (5,1), N (9,1), F (11,3).
Пример 7: 1-((4,8,11-трис(карбоксиметил)-1,4,8,11-тетраазациклотетрадекан-1-ил)метил)изохинолин 2-оксид (7)
Исходное соединение D (67,3 мг, 0,336 ммоль) суспендировали в ацетонитриле (10 мл) и добавляли 1-(бромметил)изохинолин 2-оксид (40 мг, 0,168 ммоль), и смесь перемешивали при комнатной температуре в течение 24 ч. Смесь концентрировали на роторном испарителе и остаток очищали с помощью препаративной ВЭЖХ (колонка C18, градиент ацетонитрил/вода с 0,1% ТФУ в подвижной фазе). Фракции, содержащие моноалкилированное промежуточное соединение, (m/z для [M+H]+=358) объединяли, упаривали на роторном испарителе и сушили в высоком вакууме. Остаток растворяли в ацетонитриле (4 мл), добавляли карбонат цезия (204 мг, 0,625 ммоль) и смесь перемешивали в течение 10 мин при комнатной температуре. Затем добавляли трет.бутил бромацетат (71,1 мг, 0,365 ммоль) и смесь перемешивали при комнатной температуре в течение 72 ч. Твердые продукты отфильтровывали и фильтрат концентрировали на роторном испарителе. Образовавшееся масло очищали с помощью препаративной ВЭЖХ (колонка C18, градиент ацетонитрил/вода с 0,1% ТФУ в подвижной фазе). Фракции, содержащие продукт в виде трет-бутилового эфира, объединяли, упаривали на роторном испарителе и сушили в высоком вакууме. Остаток растворяли в чистой ТФУ (2 мл) и смесь перемешивали в течение 24 ч при комнатной температуре. ТФУ упаривали на роторном испарителе, остаток растворяли в дистиллированной воде (2 мл) и очищали с помощью препаративной ВЭЖХ (условия, указанные выше). Фракции, содержащие продукт, объединяли и упаривали. Остаток растворяли в дистиллированной воде (2 мл) и лиофилизовали, получая 30,2 мг продукта в виде белого пушистого твердого вещества (0,038 ммоль, 23%-ный выход относительно 1-(бромметил)изохинолин 2-оксида).
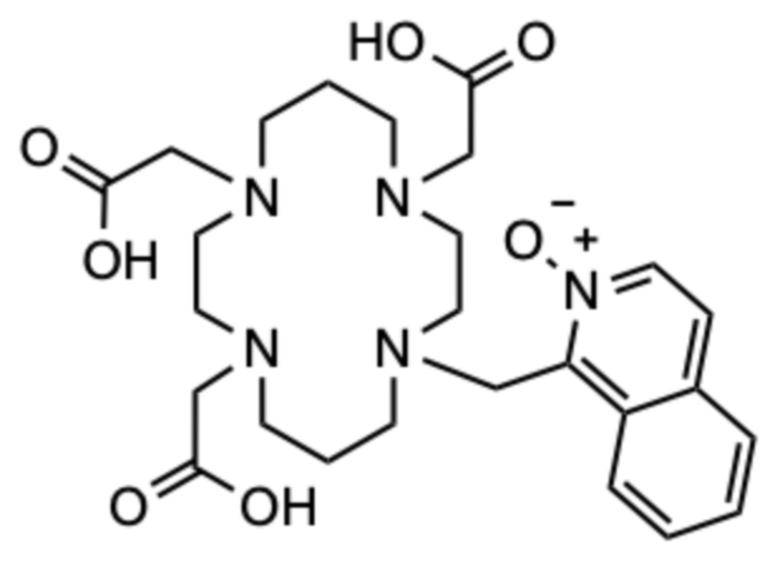
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 1,81-1,91 (CH2‒CH2‒CH2, м, 2H); 1,91-2,01 (CH2‒CH2‒CH2, м, 2H); 2,55-2,77 (цикл CH2, м, 2H); 2,97-3,08 (цикл CH2, м, 2H); 3,08-3,29 (5˟цикл CH2, м, 10H); 3,29-3,42 (цикл CH2, м, 2H); 3,54 (CH2-COOH, с, 2H); 3,64 (CH2-COOH, с, 2H); 3,72 (CH2-COOH, с, 2H); 4,72 (CH2-аром., с, 2H); 7,75-7,85 (2˟аром., м, 2H); 7,96-8,01 (аром., м, 1H); 8,03 (аром., д, 1H, 3JHH=7 Гц); 8,15-8,20 (аром., м, 1H); 8,21 (аром., д, 1H, 3JHH=7 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 21,0 (CH2‒CH2‒CH2, с); 21,1 (CH2‒CH2‒CH2, с); 48,3 (CH2‒аром., с); 48,3 (цикл, с); 49,9 (цикл, с); 50,6 (цикл, с); 50,7 (цикл, с); 51,0 (цикл, с); 51,1 (цикл, с); 52,7 (CH2‒COOH, шир. с); 53,1 (цикл, с); 53,2 (цикл, с); 54,0 (CH2‒COOH, с); 54,2 (CH2‒COOH, с); 123,6 (аром., с); 126,5 (аром., с); 128,1 (аром., с); 128,6 (аром., с); 131,1 (аром., с); 131,1 (аром., с); 131,2 (аром., с); 134,6 (аром., с); 141,2 (аром., с); 171,5 (CO, с); 171,9 (CO, с); 172,1 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C26H38N5O7) вычислено: 532,2766, найдено: 532,2764.
Элем. анализ: M·2,1ТФУ·1,6H2O, вычислено: C (45,3), H (5,3), N (8,8), F (15,0), найдено: C (45,5), H (4,6), N (8,3), F (14,6).
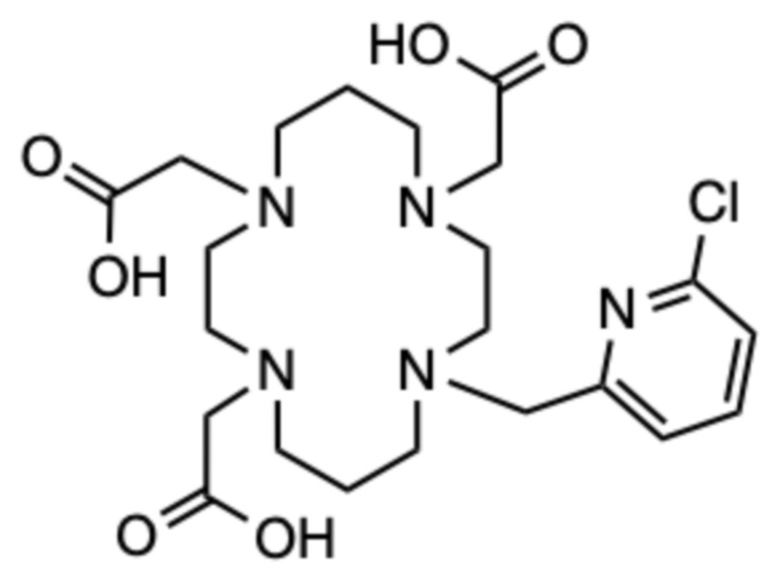
Пример 8: 2,2ʼ,2ʺ-(11-((6-хлорпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота (8)
Соединение получали способом, аналогичному способу примера 7, используя следующие количества: исходное соединение D (80 мг, 0,399 ммоль), 2-(бромметил)-6-хлорпиридин (41,2 мг, 0,199 ммоль); на второй стадии: карбонат цезия (294 мг, 0,901 ммоль) и трет.бутил бромацетат (103 мг, 0,526 ммоль). Следуя способу примера 7, аналогичным образом получали 27,3 мг продукта в виде белого пушистого твердого вещества (0,036 ммоль, 18%-ный выход относительно 2-(бромметил)-6-хлорпиридина).
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 1,87-2,04 (2˟CH2‒CH2‒CH2, м, 4H); 2,80-3,02 (2˟цикл CH2, м, 4H); 3,05-3,16 (2˟цикл CH2, м, 4H); 3,22-3,41 (4˟цикл CH2, м, 8H); 3,54 (2˟CH2-COOH, с, 4H); 3,78 (CH2-COOH, с, 2H); 4,30 (CH2-аром., с, 2H); 7,37 (аром., д, 1H, 3JHH=8 Гц); 7,43 (аром., д, 1H, 3JHH=8 Гц); 7,80 (аром., т, 1H, 3JHH=8 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 21,9 (CH2‒CH2‒CH2, с); 22,0 (CH2‒CH2‒CH2, с); 50,4 (цикл, с); 50,7 (цикл, с); 51,4 (цикл, с); 51,8 (цикл, с); 51,9 (цикл, с); 52,3 (цикл, с); 53,0 (цикл, с); 53,2 (цикл, с); 54,2 (CH2‒COOH, с); 54,4 (CH2‒COOH, с); 54,7 (CH2‒COOH, с); 56,9 (CH2‒аром., с); 123,3 (аром., с); 124,9 (аром., с); 141,2 (аром., с); 150,6 (аром., с); 152,0 (аром., с); 170,9 (CO, с); 173,4 (CO, с); 173,5 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C22H35N5O6Cl) вычислено: 500,2270, найдено: 500,2264.
Элем. анализ: M·2,0ТФУ·1,9H2O, вычислено: C (41,0), H (5,3), N (9,2), F (15,0), Cl (4,7), найдено: C (41,1), H (4,6), N (8,9), F (14,6), Cl (4,8).
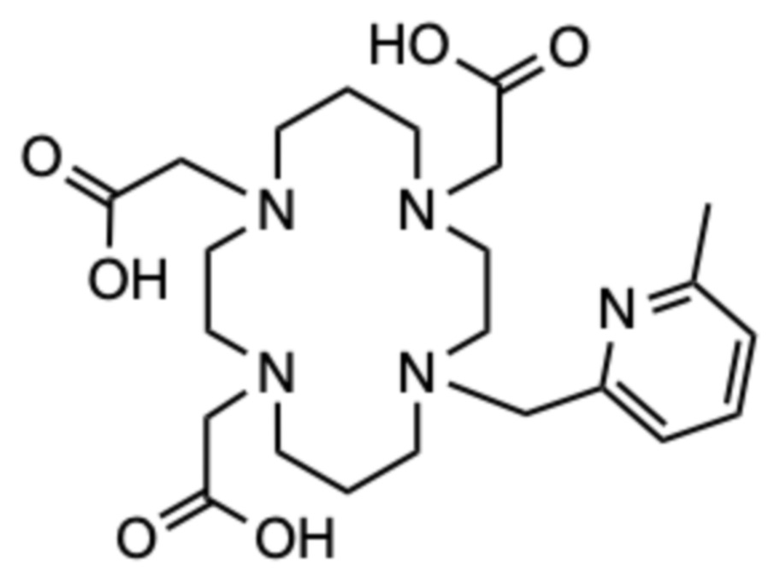
Пример 9: 2,2ʼ,2ʺ-(11-((6-метилпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота (9)
Соединение получали способом, аналогичному способу примера 7, используя следующие количества: исходное соединение D (80 мг, 0,399 ммоль), гидрохлорид 2-(хлорметил)-6-метилпиридина (35,6 мг, 0,199 ммоль); на второй стадии: карбонат цезия (232 мг, 0,711 ммоль) и трет.бутил бромацетат (80,9 мг, 0,415 ммоль). Следуя способу примера 7, аналогичным образом получали 21,4 мг продукта в виде белого пушистого твердого вещества (0,027 ммоль, 14%-ный выход относительно гидрохлорида 2-(хлорметил)-6-метилпиридина).
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 1,87-2,07 (2˟CH2‒CH2‒CH2, м, 4H); 2,53-2,62 (цикл CH2, м, 2H); 2,70 (CH3, с, 3H); 2,73-2,84 (цикл CH2, м, 2H); 2,89-3,08 (2˟цикл CH2, м, 4H); 3,15-3,29 (цикл CH2, м, 2H); 3,31-3,59 (3˟цикл CH2+CH2-COOH, 8H); 3,86 (2˟CH2-COOH, с, 4H); 3,96 (CH2-аром., шир. с, 2H); 7,71 (аром., д, 1H, 3JHH=8 Гц); 7,76 (аром., д, 1H, 3JHH=8 Гц); 8,32 (аром., т, 1H, 3JHH=8 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 19,0 (CH3, с); 21,7 (CH2‒CH2‒CH2, с); 22,3 (CH2‒CH2‒CH2, с); 47,3 (цикл, с); 48,6 (цикл, с); 50,9 (цикл, с); 51,8 (цикл, с); 51,8 (цикл, с); 52,1 (цикл, с); 52,6 (цикл, с); 52,8 (цикл, с); 54,7 (CH2‒COOH, с); 54,9 (CH2‒аром., с); 55,2 (CH2‒COOH, с); 55,4 (CH2‒COOH, с); 124,9 (аром., с); 127,1 (аром., с); 146,5 (аром., с); 151,0 (аром., с); 154,8 (аром., с); 169,4 (CO, с); 169,5 (CO, с); 174,7 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C23H38N5O6) вычислено: 480,2817, найдено: 480,2810.
Элем. анализ: M·2,7ТФУ·0,8H2O, вычислено: C (42,5), H (5,2), N (8,7), F (19,2), найдено: C (42,5), H (4,9), N (8,6), F (19,3).
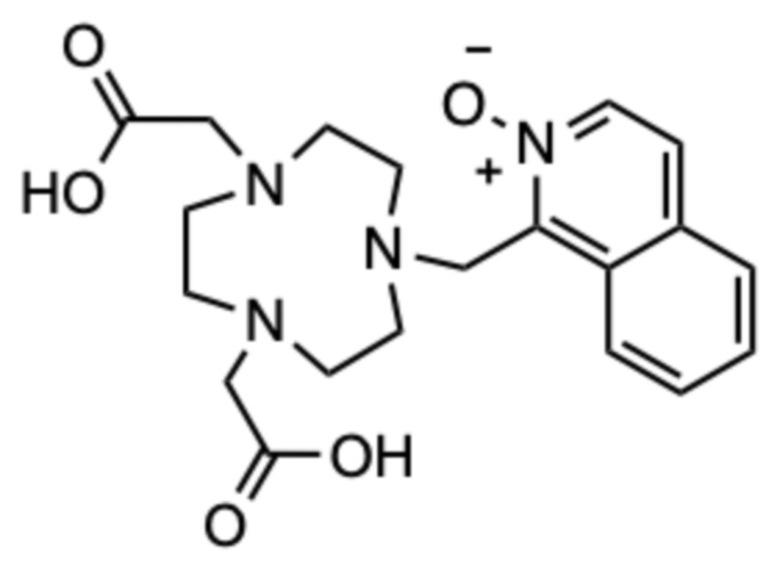
Пример 10: 1-((4,7-бис(карбоксиметил)-1,4,7-триазонан-1-ил)метил)изохинолин 2-оксид (10)
Исходное соединение B (54,6 мг, 0,153 ммоль) и 1-(бромметил)изохинолин 2-оксид (40 мг, 0,168 ммоль) растворяли вместе в ацетонитриле (4 мл) и перемешивали в течение 24 часов при комнатной температуре. Растворитель упаривали на роторном испарителе и образовавшееся масло очищали с помощью препаративной ВЭЖХ (колонка C18, градиент ацетонитрил/вода с 0,1% ТФУ в подвижной фазе). Фракции, содержащие продукт в виде трет-бутилового эфира, объединяли, упаривали на роторном испарителе и сушили в высоком вакууме. Остаток растворяли в чистой ТФУ (2 мл) и смесь перемешивали в течение 24 ч при комнатной температуре. ТФУ упаривали на роторном испарителе, остаток растворяли в дистиллированной воде (2 мл) и очищали с помощью препаративной ВЭЖХ (условия, указанные выше). Фракции, содержащие продукт, объединяли и упаривали. Остаток растворяли в дистиллированной воде (2 мл) и лиофилизовали, получая 54,6 мг продукта в виде пушистого твердого вещества не совсем белого цвета (0,092 ммоль, 60%-ный выход относительно B).
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 3,15-3,37 (6˟цикл CH2, м, 12H); 3,67 (CH2-COOH, с, 4H); 4,89 (CH2-аром., с, 2H); 7,73-7,84 (2˟аром., м, 2H); 7,94-7,99 (аром., м, 1H); 8,01 (аром., д, 1H, 3JHH=7 Гц); 8,12-8,16 (аром., м, 1H); 8,17 (аром., д, 1H, 3JHH=7 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 49,7 (цикл, с); 49,9 (цикл, с); 50,3 (цикл, с); 50,5 (CH2‒аром., с); 56,5 (CH2‒COOH, с); 123,5 (аром., с); 126,5 (аром., с); 128,0 (аром., с); 128,3 (аром., с); 131,1 (аром., с); 131,1 (аром., с); 131,1 (аром., с); 134,8 (аром., с); 141,0 (аром., с); 172,1 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C20H27N4O5) вычислено: 403,1976, найдено: 403,1973.
Элем. анализ: M·1,7ТФУ, вычислено: C (47,1), H (4,7), N (9,4), F (16,3), найдено: C (47,6), H (4,6), N (9,4), F (16,1).
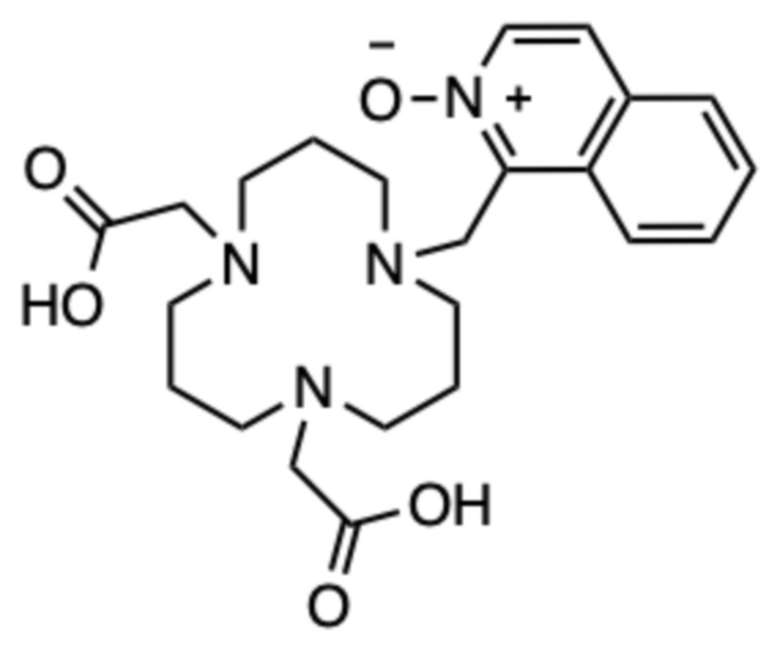
Пример 11: 1-((5,9-бис(карбоксиметил)-1,5,9-триазациклододекан-1-ил)метил)изохинолин 2-оксид (11)
Соединение получали способом, аналогичному способу примера 7, используя следующие количества: исходное соединение F (19,6 мг, 0,115 ммоль), 1-(бромметил)изохинолин 2-оксид (20 мг, 0,084 ммоль) в ацетонитриле (2 мл); на второй стадии: карбонат цезия (79 мг, 0,243 ммоль) и трет.бутил бромацетат (23,7 мг, 0,122 ммоль). Следуя способу примера 7, аналогичным образом получали 8,3 мг продукта в виде пушистого твердого вещества белого цвета (0,013 ммоль, 15%-ный выход относительно 1-(бромметил)изохинолин 2-оксида).
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 1,86-2,09 (CH2‒CH2‒CH2, м, 2H); 2,09-2,31 (CH2‒CH2‒CH2, м, 4H); 2,89-3,51 (6˟цикл CH2, м, 12H); 3,63 (CH2-COOH, с, 4H); 5,11 (CH2-аром., с, 2H); 7,79-7,91 (2˟аром., м, 2H); 8,00-8,07 (аром., м, 1H); 8,12 (аром., д, 1H, 3JHH=7 Гц); 8,14-8,18 (аром., м, 1H); 8,26 (аром., д, 1H, 3JHH=7 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 19,7 (CH2‒CH2‒CH2, с); 20,1 (CH2‒CH2‒CH2, с); 48,8 (цикл, с); 50,8 (CH2‒аром., с); 50,8 (цикл, шир. с); 53,2 (цикл, шир. с); 55,1 (CH2‒COOH, с); 123,2 (аром., с); 127,1 (аром., с); 128,2 (аром., с); 128,3 (аром., с); 131,4 (аром., с); 131,6 (аром., с); 131,7 (аром., с); 134,6 (аром., с); 138,3 (аром., с); 172,0 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C23H33N4O5) вычислено: 445,2446, найдено: 445,2445.
Элем. анализ: M·1,6ТФУ·1,4H2O, вычислено: C (48,2), H (5,6), N (8,6), F (14,0), найдено: C (48,5), H (5,3), N (8,5), F (13,7).
Пример 12: 1-((4,8,12-трис(карбоксиметил)-1,4,8,12-тетраазациклопентадекан-1-ил)метил)изохинолин 2-оксид (12)
Соединение получали способом, аналогичному способу примера 5, используя исходное соединение E (108 мг, 0,504 ммоль) и 1-(бромметил)изохинолин 2-оксид (60 мг, 0,252 ммоль) в ацетонитриле (10 мл). После выделения с помощью препаративной ВЭЖХ, как в примере 5, собирали только основной изомер моноалкилированного промежуточного соединения (m/z для [M+H]+=372). Аналогично примеру 5 вторую стадию реакции проводили со следующими количествами: карбонат цезия (309 мг, 0,948 ммоль) и трет.бутил бромацетат (108 мг, 0,553 ммоль). Затем, следуя способу примера 5, аналогичным образом получали 20,6 мг продукта в виде пушистого твердого вещества белого цвета (0,023 ммоль, 9%-ный выход относительно 1-(бромметил)изохинолин 2-оксида).
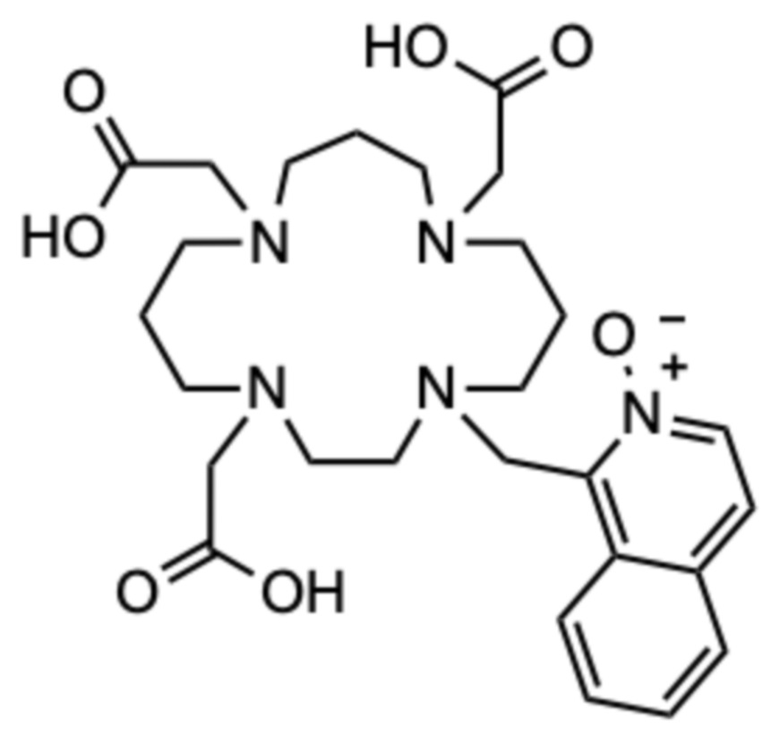
1H ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 500 МГц): δH 1,95-2,09 (CH2‒CH2‒CH2, м, 2H); 2,09-2,24 (2˟CH2‒CH2‒CH2, м, 4H); 2,87-2,97 (цикл CH2, м, 2H); 3,11-3,28 (3˟цикл CH2, м, 6H); 3,29-3,46 (4˟цикл CH2+CH2-COOH, 10H); 3,88 (CH2-COOH, с, 2H); 3,99 (CH2-COOH, с, 2H); 4,96 (CH2-аром., с, 2H); 7,78-7,89 (2˟аром., м, 2H); 8,01-8,06 (аром., м, 1H); 8,10 (аром., д, 1H, 3JHH=7 Гц); 8,16-8,21 (аром., м, 1H); 8,27 (аром., д, 1H, 3JHH=7 Гц). 13C{1H} ЯМР (D2O с внутренним эталоном диоксаном, 25ºC, 125 МГц): 18,6 (CH2‒CH2‒CH2, с); 21,2 (CH2‒CH2‒CH2, с); 22,0 (CH2‒CH2‒CH2, с); 50,1 (цикл, с); 50,2 (цикл, с); 50,4 (CH2‒аром., с); 50,4 (цикл, с); 50,4 (цикл, с); 51,1 (цикл, с); 51,4 (цикл, с); 51,6 (цикл, с); 52,1 (цикл, с); 54,2 (CH2‒COOH, с); 56,2 (CH2‒COOH, с); 56,3 (CH2‒COOH, с); 123,4 (аром., с); 127,0 (аром., с); 128,2 (аром., с); 128,4 (аром., с); 131,2 (аром., с); 131,4 (аром., с); 131,4 (аром., с); 134,9 (аром., с); 139,7 (аром., с); 168,4 (CO, с); 168,5 (CO, с); 172,3 (CO, с).
МСРВ (ESI) m/z: [(M+H)+] (C27H40N5O7) вычислено: 546,2922, найдено: 546,2920.
Элем. анализ: M·2,8ТФУ·2,2H2O, вычислено: C (43,3), H (5,1), N (7,7), F (17,6), найдено: C (43,5), H (4,4), N (7,3), F (17,1).
II Выделение металлов s-, p- и d- блоков
Молекулы хелатообразователя, описанные в данном изобретении, были исследованы на их способность разделять металлы s-, p- и d- блоков, сначала образуя хелаты с хелатором, который обеспечивает хроматографическую селективность по отношению к металлам, а затем подвергая хелаты традиционному хроматографическому выделению.
Пример 13: Вариабельность удерживания хелатов металлов при обращенно-фазовой ВЭЖХ, используемой для выделения
Комплексообразование выбранных металлов s-, p- и d- блоков (Ca2+, Fe2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Al3+, Pb2+) проводили параллельно в 96-луночных планшетах следующим образом. Буфер MOPS pH=7,0 (85 мкл), приблизительно 0,01 M водного раствора хелатора (5 мкл) и приблизительно 0,005 М водного раствора соли металла состава, приведенного в таблице 1 (10 мкл), пипеткой вносили в лунку. Планшет с лунками накрывали проницаемым герметизирующим матом, хорошо встряхивали для полного смешивания компонентов и оставляли на 16 часов при комнатной температуре. После этого его снова встряхивали и недолго центрифугировали, чтобы вся жидкость опустилась на дно лунки. Образцы для анализа ВЭЖХ отбирали непосредственно из лунок с помощью автоматического автосамплера. Условия для анализа ВЭЖХ были следующими: колонка Phenomenex Kinetex C18 (100˟3 мм, 2,6 мкм), объем инъекции 1 мкл, изократическое элюирование подвижной фазой, как указано в таблице 1, скорость потока 0,6 мл/мин и детектирование с помощью УФ-поглощения при 280 нм. Время удерживания для соответствующих хелатов металлов суммировано в таблице 1. Для данного хелатирующего агента различное время удерживания различных металлов означает, что такие металлы могут быть хроматографически выделены в виде хелатов с этим хелатирующим агентом. Результаты в таблице 1 демонстрируют, что согласно настоящему изобретению могут быть выделены различные комбинации металлов s-, p- и d- блоков.
Таблица 1
* Хелат металла нестабилен в данных условиях, обнаружен только свободный хелатор (значение в скобках).
a Элюент: 8% ацетонитрил в 10 мМ α-HIBA (pH=5,5).
b Элюент: 4,6% ацетонитрил в воде.
c Элюент: 11% ацетонитрил в воде.
III Выделение редкоземельных элементов
Молекулы хелаторов, описанные в настоящем изобретении, исследовали на их способность разделять редкоземельные элементы, сначала образуя хелаты с хелатором, который обеспечивает хроматографическую селективность по отношению к редкоземельным элементам, и затем подвергая хелаты традиционному хроматографическому выделению.
Пример 14: Изменчивость удерживания хелатов металлов на обращенно-фазовой ВЭЖХ, используемой для выделения
Растворы хелатов металлов готовили в соответствии со способом, приведенным в примере 13, в следующих количествах, помещаемых пипеткой: буфер MOPS pH=7,0 (90 мкл), 0,01 М раствор хелатора (5 мкл) и 0,01 М раствор металла. хлорид или нитрат (5 мкл). Условия для анализа ВЭЖХ были идентичны условиям в примере 13, за исключением состава подвижной фазы, указанного в таблице 2. Время удерживания для соответствующих хелатов металлов суммировано в таблице 2. Для данного хелатора разница во времени удерживания различных металлов означает, что такие металлы могут быть хроматографически выделены в виде хелатов с этим хелатором. Результаты в таблице 2 демонстрируют, что согласно настоящему изобретению могут быть выделены различные комбинации металлов из группы редкоземельных элементов.
Таблица 2
a Элюент: 6,2% ацетонитрил в воде.
b Элюент: 7,4% ацетонитрил в воде.
c Элюент: 4,6% ацетонитрил в 10 мМ α-HIBA (pH=5,5).
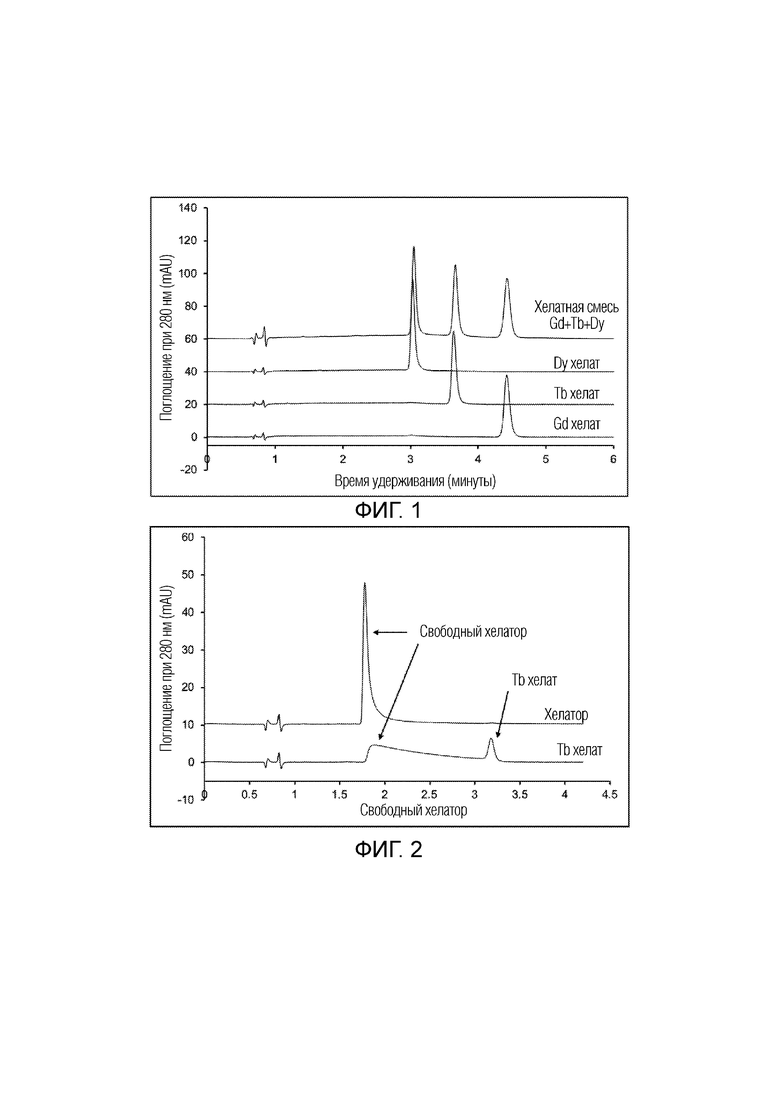
Пример 15: ВЭЖХ выделение Gd, Tb и Dy из смеси.
Растворы хелатов Gd, Tb и Dy были приготовлены в соответствии со способом по примеру 14 с хелатором 1-((4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-1-ил)метил)изохинолин 2-оксидом (получен в примере 6). Хелаты подвергали анализу ВЭЖХ отдельно и в виде смеси. Условия для анализа ВЭЖХ были идентичны условиям в примере 13, за исключением состава подвижной фазы, которая состояла из 7,4% ацетонитрила в воде без добавок. На фиг. 1 показано, что было успешно достигнуто выделение всех трех хелатов из смеси на исходном уровне.
Пример 16: Кислотное разложение и удаление свободного хелатора
Раствор хелата Tb хелатора 1-((4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-1-ил)метил)изохинолин 2-оксида (получен в примере 6) готовили в соответствии со способом по примеру 14 и подвергали анализу ВЭЖХ с использованием кислой подвижной фазы. Условия для анализа ВЭЖХ были идентичны условиям в примере 13, за исключением состава подвижной фазы, которая состояла из 7,4% ацетонитрила и 0,02% ТФУ в воде. На фиг. 2 показана хроматограмма этого анализа, наложенная на хроматограмму свободного хелатора в тех же условиях. Наблюдается очевидное (частичное) разложение хелата металла на свободный хелатор (видимый по УФ-поглощению) и свободные ионы Tb (невидимые по УФ-поглощению) из-за кислой подвижной фазы. Эти результаты подтверждают, что хелат металла может быть разложен на свободные ионы металла и свободный хелатор в кислых условиях, и что хелатор (имеющий удерживание на колонке) может быть отделен хроматографически от ионов металла (которые не удерживаются на колонке).
Промышленная применимость
Настоящее изобретение может быть использовано в промышленности при выделении и очистке металлов, выделении и очистке металлических радионуклидов, концентрировании разбавленных растворов радионуклидов металлов посредством твердофазной экстракции, извлечении изотопно-обогащенного металлического материала, используемого для производства металлических радионуклидов, очистке исходного металлического материала перед его использованием для производства металлических радионуклидов, дезактивации поверхностей, загрязненных металлическими радионуклидами, селективном извлечении металлов из ядерных отходов, селективном извлечении металлов из продуктов ядерного деления, гидрометаллургической переработке отработанного ядерного топлива и других радиоактивных отходов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ДЛЯ РАЗДЕЛЕНИЯ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ И S-, P-, D-МЕТАЛЛОВ, СПОСОБЫ РАЗДЕЛЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2783526C2 |
| НОВЫЕ АНТИМИКРОБНЫЕ СОЕДИНЕНИЯ, ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МЛЕКОПИТАЮЩИХ И НОВЫЙ МЕТАБОЛИЧЕСКИЙ МЕХАНИЗМ | 2017 |
|
RU2752568C2 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, РАДИОФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ИМЕЮЩИЕ РАДИОАКТИВНУЮ МЕТКУ | 1994 |
|
RU2145608C1 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2006 |
|
RU2443688C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОАЛКИЛАМИНОМ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА | 2007 |
|
RU2457203C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСИЛАМИНИЗОХИНОЛОНА В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2006 |
|
RU2440988C2 |
| ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНОВЫЕ И ИЗОХИНОЛИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2007 |
|
RU2455302C2 |
| ДИАГНОСТИКА, ЛЕЧЕНИЕ И ПРОФИЛАКТИКА СОСТОЯНИЙ, СВЯЗАННЫХ С РЕЦЕПТОРОМ НЕЙРОТЕНЗИНОМ | 2017 |
|
RU2796538C2 |
| ПИПЕРИДИНИЛ-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА КАК ИНГИБИТОРЫ Rho-КИНАЗЫ | 2006 |
|
RU2414467C2 |
| 2,5- ИЛИ 2,6-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ГИДРОХИНОНА, СОДЕРЖАЩИЕ ПО КРАЙНЕЙ МЕРЕ ОДНУ КАРБОКСИ-, СУЛЬФО- ИЛИ АМИДОГРУППУ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2021 |
|
RU2827702C1 |
Настоящее изобретение относится к применению соединений общей формулы (I), где X выбран из группы, включающей H, C1-C6 алкил, галоген (F, Cl, Br или I); Y выбран из группы, включающей азот, N-оксид; Z1, Z2, Zm, где m обозначает 1 или 2, независимо, выбраны из группы, включающей -CH2-CH2- и -CH2-CH2-CH2-; A, Am, где m обозначает 1 или 2, независимо, выбраны из H, -CH2COOH, -CH2C(O)NH2,
-CH2P(O)(OH)2 и ; n обозначает 1 или 2; R1, R2, R3, независимо, представляют собой H; и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо; при условии, что, когда n обозначает 2 и все Z1, Z2, Zm представляют собой -CH2-CH2-, тогда A не представляет собой -CH2COOH, для хроматографического выделения редкоземельных элементов и/или металлов s-, p- и d- блоков. Кроме того, изобретение относится к способу хроматографического выделения редкоземельных элементов и/или металлов s-, p- и d- блоков, к соединениям общей формулы (Ia). Технический результат изобретения заключается в эффективном и быстром выделении редкоземельных элементов и металлов s-, p- и d- блоков. 3 н. и 12 з.п. ф-лы, 2 ил., 2 табл., 16 пр.
(I) и (Ia)
1. Применение соединений общей формулы (I),
(I),
для хроматографического выделения редкоземельных элементов и/или металлов s-, p- и d- блоков,
где
- X выбран из группы, включающей H, C1-C6 алкил, галоген (F, Cl, Br или I);
- Y выбран из группы, включающей азот, N-оксид;
- Z1, Z2, Zm, где m обозначает 1 или 2, независимо, выбраны из группы, включающей -CH2-CH2- и -CH2-CH2-CH2-;
- A, Am, где m обозначает 1 или 2, независимо, выбраны из H, -CH2COOH, -CH2C(O)NH2, -CH2P(O)(OH)2 и  ;
;
- n обозначает 1 или 2;
R1, R2, R3, независимо, представляют собой H;
и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо;
при условии, что, когда n обозначает 2 и все Z1, Z2, Zm представляют собой -CH2-CH2-, тогда A не представляет собой -CH2COOH.
2. Применение по п.1 для хроматографического выделения редкоземельных элементов.
3. Применение по п.1 для хроматографического выделения металлов s-, p- и d- блоков, выбранных из групп металлов II.A, III.A, IV.A, V.A, I.B, II.B и VIII.B, предпочтительно, выбранных из Fe2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Pb2+.
4. Применение по любому из предшествующих пунктов, где X выбран из группы, включающей H, -CH3 и Cl.
5. Применение по любому из предшествующих пунктов, где R1, R2, R3, независимо, представляют собой H; и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо.
6. Применение по любому из предшествующих пунктов, где по меньшей мере один из A, Am представляет собой H.
7. Применение по любому из предшествующих пунктов, где X представляет собой -CH3 или Cl, Y представляет собой азот и все R1, R2, R3 представляют собой H.
8. Применение по любому из пп.1-6, где X представляет собой H, Y представляет собой N-оксид и два из R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо.
9. Применение по любому из предшествующих пунктов, где соединения общей формулы (I) выбраны из группы, включающей:
1-((4,7-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид,
1-((4-(2-амино-2-оксоэтил)-7,10-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид,
1-((4,10-бис(2-амино-2-оксоэтил)-7-(фосфонометил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид,
1,1ʼ-((7,10-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1,4-диил)бис(метилeн))бис(изохинолин 2-оксид),
1-((1,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-4-ил)метил)изохинолин 2-оксид,
1-((4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-1-ил)метил)изохинолин 2-оксид,
1-((4,8,11-трис(карбоксиметил)-1,4,8,11-тетраазациклотетрадекан-1-ил)метил)изохинолин 2-оксид,
2,2ʼ,2ʺ-(11-((6-хлорпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота,
2,2ʼ,2ʺ-(11-((6-метилпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота,
1-((4,7-бис(карбоксиметил)-1,4,7-триазонан-1-ил)метил)изохинолин 2-оксид,
1-((5,9-бис(карбоксиметил)-1,5,9-триазациклододекан-1-ил)метил)изохинолин 2-оксид,
1-((4,8,12-трис(карбоксиметил)-1,4,8,12-тетраазациклопентадекан-1-ил)метил)изохинолин 2-оксид.
10. Способ хроматографического выделения редкоземельных элементов и/или металлов s-, p- и d- блоков, выбранных из групп металлов II.A, III.A, IV.A, V.A, I.B, II.B и VIII.B, из смеси по меньшей мере двух ионов металлов, где по меньшей мере один из них представляет собой металл, выбранный из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb, Y, и переходных металлов, отличающийся тем, что он включает следующие стадии:
(a) предоставление смеси по меньшей мере одного иона металла, выбранного из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb, Y, и переходных металлов, и по меньшей мере одного дополнительного иона металла, где указанный дополнительный ион металла выбран из ионов редкоземельных металлов, ионов переходных металлов, ионов непереходных металлов и ионов актинидов,
(b) ионы металлов, содержащиеся в указанной смеси, подвергают взаимодействию по меньшей мере с одним соединением общей формулы (I), как определено в любом из предшествующих пунктов, с образованием хелатов;
(c) хелаты со стадии (b) подвергают хроматографическому выделению,
при этом, необязательно, стадия (c) может быть проведена по меньшей мере дважды, чтобы повысить чистоту по меньшей мере одного выделенного хелата металла;
и,
необязательно, (d) по меньшей мере один хелат металла, полученный в результате хроматографического выделения, подвергается кислотному разложению с получением иона металла, не входящего в комплекс.
11. Способ хроматографического выделения по п.10, где смесь по меньшей мере двух ионов металлов, подлежащих выделению, включает по меньшей мере один редкоземельный металл, выбранный из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb и Y, отличающийся тем, что он включает следующие стадии:
(a) предоставление смеси по меньшей мере одного иона редкоземельного металла, выбранного из Ce, Dy, Er, Eu, Gd, Ho, La, Lu, Nd, Pr, Pm, Sm, Sc, Tb, Tm, Yb и Y, и по меньшей мере одного дополнительного иона металла, где указанный дополнительный ион металла выбран из ионов редкоземельных металлов, ионов переходных металлов, ионов непереходных металлов и ионов актинидов,
(b) ионы металлов, содержащиеся в указанной смеси, подвергают взаимодействию по меньшей мере с одним соединением общей формулы (I), как определено в любом из предшествующих пунктов, с образованием хелатов;
(c) хелаты со стадии (b) подвергают хроматографическому выделению,
при этом, необязательно, стадия (c) может быть проведена по меньшей мере дважды, чтобы повысить чистоту по меньшей мере одного выделенного хелата металла;
и,
необязательно, (d) по меньшей мере один хелат металла, полученный в результате хроматографического выделения, подвергается кислотному разложению с получением не входящего в комплекс иона редкоземельного металла.
12. Соединения общей формулы (Ia),
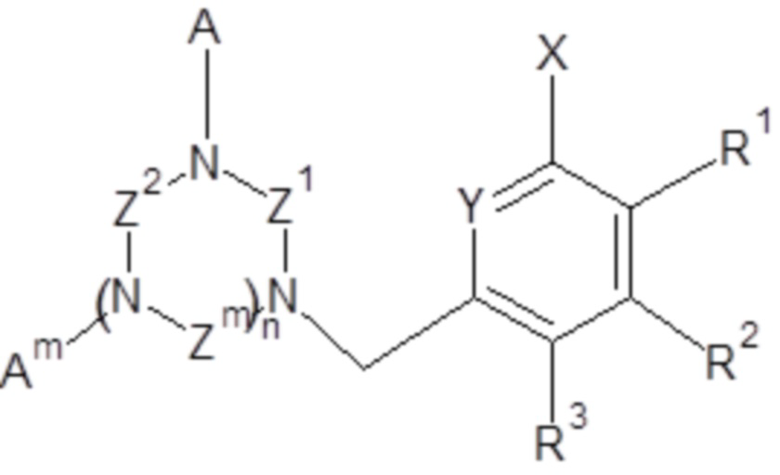
(Ia),
где
- X выбран из группы, включающей H, C1-C6 алкил, галоген;
- Y выбран из группы, включающей азот, N-оксид;
- Z1, Z2, Zm, где m обозначает 1 или 2, независимо, выбраны из группы, включающей -CH2-CH2- и -CH2-CH2-CH2-;
- A, Am, где m обозначает 1 или 2, независимо, выбраны из H, -CH2COOH, -CH2C(O)NH2, -CH2P(O)(OH)2, и где не более чем один из A, Am представляет собой H;
- n обозначает 1 или 2;
R1, R2, R3, независимо, представляют собой H;
и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо;
при условии, что:
- когда n обозначает 2 все Z1, Z2, Zm представляют собой -CH2-CH2-, тогда A не представляет собой -CH2COOH;
- когда Y представляет собой азот, тогда X не представляет собой H;
- когда n обозначает 1, все Z1, Z2, Zm представляют собой -CH2-CH2- и Y представляет собой азот, тогда X не представляет собой галоген.
13. Соединения общей формулы (Ia) по п.12, где X выбран из группы, включающей H, -CH3 и Cl.
14. Соединения общей формулы (Ia) по любому из пп.12 или 13, где R1, R2, R3, независимо, представляют собой H; и/или два соседних R1, R2, R3 вместе с двумя соседними атомами углерода ароматического цикла образуют шестичленное кольцо.
15. Соединения общей формулы (Ia) по любому из пп.12-14, где соединения общей формулы (Ia) представляют собой:
1-((4,7-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид,
1-((4-(2-амино-2-оксоэтил)-7,10-бис(карбоксиметил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид,
1-((4,10-бис(2-амино-2-оксоэтил)-7-(фосфонометил)-1,4,7,10-тетраазациклододекан-1-ил)метил)изохинолин 2-оксид,
1-((1,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-4-ил)метил)изохинолин 2-оксид,
1-((4,7,10-трис(карбоксиметил)-1,4,7,10-тетраазациклотридекан-1-ил)метил)изохинолин 2-оксид,
1-((4,8,11-трис(карбоксиметил)-1,4,8,11-тетраазациклотетрадекан-1-ил)метил)изохинолин 2-оксид,
2,2ʼ,2ʺ-(11-((6-хлорпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота,
2,2ʼ,2ʺ-(11-((6-метилпиридин-2-ил)метил)-1,4,8,11-тетраазациклотетрадекан-1,4,8-триил)триуксусная кислота,
1-((4,7-бис(карбоксиметил)-1,4,7-триазонан-1-ил)метил)изохинолин 2-оксид,
1-((5,9-бис(карбоксиметил)-1,5,9-триазациклододекан-1-ил)метил)изохинолин 2-оксид,
1-((4,8,12-трис(карбоксиметил)-1,4,8,12-тетраазациклопентадекан-1-ил)метил)изохинолин 2-оксид.
| XIE F., A critical review on solvent extraction of rare earths from aqueous solutions, Minerals Engineering, 2014, vol.56, p.10-28 | |||
| KIFLE D | |||
| et al, Selective liquid chromatographic separation of yttrium from heavier rare earth elements using acetic acid as a novel eluent, Journal of Chromatography A, 2013, no.1307, p.86-90 | |||
| WO 1994026275 A1, |

