Настоящая заявка претендует на приоритет китайской патентной заявки No. 201710900197.8, озаглавленной "PDE9 INHIBITOR AND USE THEREFOF (Ингибитор PDE9 и его применение)", поданной Национальным Управлением Китая по интеллектуальной собственности (China National Intellectual Property Administration) 28 сентября 2017 г., китайской патентной заявки No. 201810203538.0, озаглавленной "PDE9 INHIBITOR AND USE THEREFOF (Ингибитор PDE9 и его применение)", поданной Национальным Управлением Китая по интеллектуальной собственности 13 марта 2018 г., и китайской патентной заявки No. 201810871998.0, озаглавленной "PDE9 INHIBITOR AND USE THEREFOF (Ингибитор PDE9 и его применение)", поданной Национальным Управлением Китая по интеллектуальной собственности 2 августа 2018 г., содержания которых полностью включены в настоящее описание посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области техники, связанной с медициной, и, в частности, изобретение относится к ингибитору фосфодиэстеразы 9, представленному формулой (I), или к его фармацевтически приемлемой соли, стереоизомеру, а также к их применению.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Фосфодиэстеразы (англ. phosphodiesterase, сокращенно PDE) относятся к классу протеаз, которые в организме селективно разлагают важные вторичные мессенджеры cGMP (сокр. от англ. "cyclic guanosine monophosphate", что означает "циклический гуанозинмонофосфат", сокращенно цГМФ) и сАМР (сокр. от англ. "cyclic adenosine monophosphate", что означает "циклический аденозинмонофосфат", сокращенно цАМФ), то есть участвуют в важных физиологических процессах, протекающих в организме. На основании гомологии последовательностей генов и селективности в отношении цГМФ или цАМФ фосфодиэстеразы могут быть разделены на 11 видов (PDE1-PDE11). Важным видом семейства PDE является фосфодиэстераза PDE9A, которая в значительной степени экспрессируется в семенниках, мозге, тонком кишечнике, скелетной мускулатуре, сердце, легких, вилочковой железе и поджелудочной железе. Серьезные исследования последних лет, множество сообщений в имеющейся литературе и клинические данные показали, что ингибиторы PDE9A подходят для лечения заболеваний, связанных с когнитивными расстройствами, вызываемыми расстройствами центральной нервной системы, такими как болезнь Альцгеймера и шизофрения, и нейродегенеративных заболеваний мозга.
Оба нуклеотида, цАМФ и цГМФ, представляют собой важные вторичные мессенджеры, которые играют главную роль в процессах клеточной сигнализации. Они производят первоначальную активацию протеинкиназ: протеинкиназа, активируемая цАМФ, называется протеинкиназой А (англ. protein kinase А, сокращенно РКА), и протеинкиназа, активируемая цГМФ, называется протеинкиназой G (англ. protein kinase G, сокращенно PKG). Активированные РКА и PKG могут фосфорилировать различные клеточные эффекторные белки, такие как ионные каналы, рецепторы, связанные с G-белком, структурные белки и факторы трансдукции. Таким образом, посредством указанного механизма цАМФ и цГМФ могут регулировать большинство физиологических процессов во многих органах. В то же время цАМФ и цГМФ также могут непосредственно воздействовать на эффекторные белки, опосредуя процессы, описанные выше. Хорошо известно, что цГМФ могут непосредственно воздействовать на акцепторы ионов, влияя, таким образом, на концентрацию ионов в клетках. Фосфодиэстеразы (PDE) гидролизуют циклические монофосфаты цАМФ и цГМФ и превращают их в неактивируемые монофосфаты AMP и GMP.
Фосфодиэстераза PDE9 человека впервые была клонирована в 1998 году, и тогда же было произведено ее секвенирование; она представляет собой PDE, имеющую самую высокую из известных на настоящий момент селективность по отношению к цГМФ. Константа связывания (Km) цГМФ фосфодиэстеразой PDE9 составляет 170 нМ, величина константы связывания цАМФ фосфодиэстеразой PDE9 достигает 230000 нМ, то есть имеет селективность в 1000 раз больше. По сравнению с PDE2A и PDE5A, каталитическая активность PDE9 не может быть повышена при воздействии цГМФ, поскольку PDE9 не имеет участка связывания с цГМФ. Таким образом, ингибиторы PDE9 могут повышать базовую концентрацию цГМФ.
Традиционные ингибиторы PDE не могут ингибировать PDE9 человека, и поэтому такие лекарственные средства, как IBMX (ИБМК, то есть изобутилметилксантин), дипиридамол, SKF94120, ролипрам и винпоцетин не ингибируют или лишь очень слабо ингибируют активность PDE9.
В настоящее время в розничной продаже не имеется лекарственного средства, ингибирующего PDE9; лишь некоторые ингибиторы находятся в стадии клинических испытаний, и среди них можно отметить два ингибитора: PDE9, PF-04447943, созданный Pfizer, и BI-409306, созданный BI. В настоящее время эти два соединения находятся в стадиях I и II клинических испытаний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Одна из задач изобретения состоит в предоставлении класса соединений, подходящих для применения в качестве ингибиторов протеазы PDE9, или их фармацевтически приемлемых солей и стереоизомеров, которые обладают высокой ингибиторной активностью и селективностью в отношении протеазы PDE9 и пригодностью в качестве лекарственного средства (например, обладают хорошими фармакокинетическими свойствами и более высокой стабильностью в микросомах печени), которые могут применяться для лечения или профилактики соответствующих заболеваний, опосредованных PDE9, и играть важную роль в лечении заболеваний, связанных с когнитивными нарушениями, вызываемыми расстройствами центральной нервной системы.
Ниже приведены технические решения поставленной задачи, предлагаемые настоящим изобретением.
Настоящее изобретение относится к соединению, представленному формулой (I), или к его фармацевтически приемлемым солям или стереоизомерам:
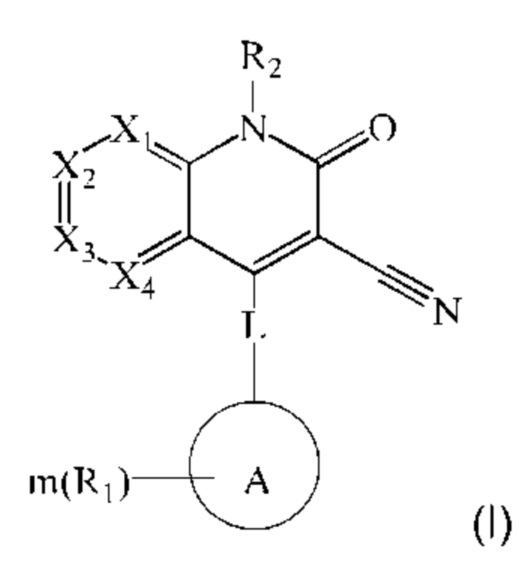
где каждый из X1, Х2, Х3 и Х4 независимо представляет собой CR3 или N, и Х1, Х2, Х3 и Х4 одновременно не представляют собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкила, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированного С1-6 алкила, галогенированного С1-6 алкокси, С2-8 алкенила, С2-8 алкинила, С1-6 алкилсульфонила, С1-6 алкилтио, С3-6 циклоалкила, 4-6-членного гетероциклила, С1-6 алкилкарбонила, аминокарбонила, С1-6 алкиламинокарбонила, (С1-6 алкил)2 аминокарбонила, 4-6-членного гетероциклилкарбонила и 5-6-членного гетероарилокси, где С1-6 алкил, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированный С1-6 алкокси, С2-8 алкенил, С2-8 алкинил, С1-6 алкилсульфонил, С1-6 алкилтио, С3-6 циклоалкил, 4-6-членный гетероциклил, С1-6 алкилкарбонил, аминокарбонил, С1-6 алкиламинокарбонил, (С1-6 алкил)2 аминокарбонил, 4-6-членный гетероциклилкарбонил и 5-6-членный гетероарилокси являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкилкарбониламино, C1-6 алкилсульфониламино, C1-6 алкилкарбонилокси, С3-6 циклоалкила, С2-8 алкинила, галогенированного C1-6 алкила, С2-8 алкенила, галогенированного C1-6 алкокси, 4-6-членного гетероциклила, который является незамещенным или необязательно замещен заместителем, и гетероарила, который является незамещенным или необязательно замещен заместителем;
заместитель названного выше 4-6-членного гетероциклила необязательно имеет заместитель, и гетероарил необязательно имеет заместитель, выбранный из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила и C1-6 алкокси;
L представляет собой связь, -NH-(CH2)t-, и t равен 0, 1, 2 или 3;
кольцо А выбрано из группы, состоящей из 3-12-членного гетероциклил, арил, 5-10-членного гетероарила, 3-12-членного циклоалкила и 3-12-членного циклоалкенила, где 3-12-членный гетероциклил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации, и атом S может быть необязательно окислен до S(O) или S(O)2 , и 5-10-членный гетероарил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкил, С1-6 алкокси, С1-6 алкиламино, (C1-6 алкил)2 амино, галогенированного C1-6 алкила, галогенированного C1-6 алкокси, С2-8 алкенила, С2-8 алкинила, C1-6 алкилсульфонила, C1-6 алкилтио, 3-12-членного циклоалкила, 3-12-членного циклоалкенила, 3-12-членного гетероциклила, арила и 5-10-членного гетероарила, где С1-6 алкил, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированный С1-6 алкил, галогенированный С1-6 алкокси, С2-8 алкенил, С2-8 алкинил, С1-6 алкилсульфонил, С1-6 алкилтио, 3-12-членный циклоалкил, 3-12-членный циклоалкенил, 3-12-членный гетероциклил, арил и 5-10-членный гетероарил являются незамещенными или необязательно замещены группой, выбранной из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкила, С1-6 алкокси, С1-6 алкокси С1-6 алкокси, С1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкилкарбониламино и C1-6 алкилсульфониламино;
m равен 0, 1, 2 или 3; и
R2 выбран из группы, состоящей из водорода, C1-6 алкила, С2-8 алкенила, С2-8 алкинила и галогенированного С1-6 алкила.
В одном из предпочтительных примеров осуществления Х2 представляет собой N, и каждый из Х1, Х3 и Х4 независимо представляет собой CR3.
В другом предпочтительном примере осуществления Х4 представляет собой N, и каждый из Х1, Х2 и Х3 независимо представляет собой CR3.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I), или его фармацевтически приемлемым солям или стереоизомерам,
где
каждый из X1, Х2, Х3 и Х4 независимо представляет собой CR3 или N, и Х1, Х2, Х3 и Х4 одновременно не представляют собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, галогенированного C1-6 алкила, галогенированного C1-6 алкокси, С2-8 алкенила, С2-8 алкинила, C1-6 алкилсульфонила, C1-6 алкилтио, С3-6 циклоалкила, 4-6-членного гетероциклила, C1-6 алкилкарбонила, аминокарбонила, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, 4-6-членного гетероциклилкарбонила и 5-6-членного гетероарилокси, где C1-6 алкил, C1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированный С1-6 алкокси, С2-8 алкенил, С2-8 алкинил, С1-6 алкилсульфонил, С1-6 алкилтио, С3-6 циклоалкил, 4-6-членный гетероциклил, С1-6 алкилкарбонил, аминокарбонил, С1-6 алкиламинокарбонил, (С1-6 алкил)2 аминокарбонил, 4-6-членный гетероциклилкарбонил и 5-6-членный гетероарилокси являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкила, С1-6 алкокси, С1-6 алкокси С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, С1-6 алкилкарбониламино, С1-6 алкилсульфониламино, С1-6 алкилкарбонилокси, С3-6 циклоалкила, С2-8 алкинила, галогенированного С1-6 алкила, С2-8 алкенила и галогенированного С1-6 алкокси;
заместитель названного выше 4-6-членного гетероциклила необязательно имеет заместитель, и гетероарил необязательно имеет заместитель, выбранный из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкила и С1-6 алкокси;
L представляет собой связь, -NH-(CH2)t-, и t равен 0, 1, 2 или 3;
кольцо А выбрано из группы, состоящей из 3-12-членного гетероциклила, арила и 5-10-членного гетероарила, где 3-12-членный гетероциклил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации, и атом S может быть необязательно окислен до S(O) или S(O)2 , и 5-10-членный гетероарил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, амино, циано, галогена, C1-6 алкила, C1-6 алкокси, 3-12-членного циклоалкила, 3-12-членного гетероциклила, арила, 5-10-членного гетероарила, где C1-6 алкил, C1-6 алкокси, 3-12-членный циклоалкил, 3-12-членный гетероциклил, арил и 5-10-членный гетероарил являются незамещенными или необязательно замещены группой, выбранной из группы, состоящей из С1-6 алкила, С1-6 алкокси, 3-12-членного циклоалкила, 3-12-членного гетероциклила, арила и 5-10-членного гетероарила;
m равен 0, 1, 2 или 3; и
R2 представляет собой водород или C1-6 алкил.
В одном из предпочтительных примеров осуществления Х2 представляет собой N, и каждый из Х1, Х3 и Х4 независимо представляет собой CR3.
В другом предпочтительном примере осуществления Х4 представляет собой N, и каждый из Х1, Х2 и Х3 независимо представляет собой CR3.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I), или его фармацевтически приемлемым солям или стереоизомерам:
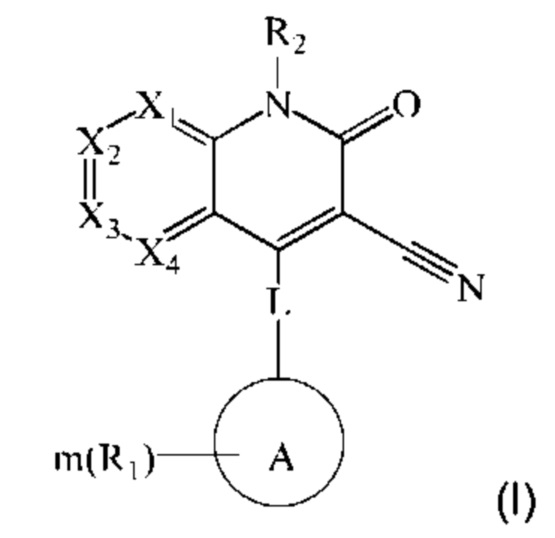
где
каждый из X1, Х2, Х3 и Х4 независимо представляет собой CR3 или N, и Х1, Х2, Х3 и Х4 одновременно не представляют собой CR3;
L представляет собой связь, -NH-(CH2)t-, и t равен 0, 1, 2 или 3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкила, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированного С1-6 алкила, галогенированного С1-6 алкокси, С2-8 алкенила, С2-8 алкинила, С1-6 алкилсульфонила, С1-6 алкилтио, С3-6 циклоалкила, 4-6-членного гетероциклила, С1-6 алкилкарбонила, С1-6 алкиламинокарбонила, (С1-6 алкил)2 аминокарбонила и аминокарбонила, где С1-6 алкил, 1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированный С1-6 алкокси, С2-8 алкенил, С2-8 алкинил, С1-6 алкилсульфонил, С1-6 алкилтио, С3-6 циклоалкил, 4-6-членный гетероциклил, C1-6 алкилкарбонил, C1-6 алкиламинокарбонил, (C1-6 алкил)2 аминокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, С3-6 циклоалкила, C1-6 алкилкарбониламино, C1-6 алкилсульфониламино, C1-6 алкилкарбонилокси и незамещенного или C1-6-алкилзамещенного 4-6-членного гетероциклила;
кольцо А выбрано из группы, состоящей из 3-12-членного гетероциклила, арила, 5-10-членного гетероарила, 3-12-членного циклоалкила и 3-12-членного циклоалкенила, где 3-12-членный гетероциклил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации, и атом S может быть необязательно окислен до S(O) или S(O)2 , и 5-10-членный гетероарил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкила, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированного С1-6 алкила, галогенированного С1-6 алкокси, С2-8 алкенила, С2-8 алкинила, С1-6 алкилсульфонила, С1-6 алкилтио, 3-12-членного циклоалкила, 3-12-членного циклоалкенила, 3-12-членного гетероциклила, арила и 5-10-членного гетероарила, где С1-6 алкил, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированный С1-6 алкил, галогенированный С1-6 алкокси, С2-8 алкенил, С2-8 алкинил, С1-6 алкилсульфонил, С1-6 алкилтио, 3-12-членный циклоалкил, 3-12-членный циклоалкенил, 3-12-членный гетероциклил, арил и 5-10-членный гетероарил являются незамещенными или необязательно замещены группой, выбранной из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкилкарбониламино и C1-6 алкилсульфониламино;
m равен 0, 1, 2 или 3; и
R2 выбран из группы, состоящей из водорода, С1-6 алкила, С2-8 алкенила, С2-8 алкинила и галогенированного С1-6 алкила.
В одном из предпочтительных примеров осуществления Х2 представляет собой N, и каждый из Х1, Х3 и Х4 независимо представляет собой CR3.
В другом предпочтительном примере осуществления Х4 представляет собой N, и каждый из Х1, Х2 и Х3 независимо представляет собой CR3.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (II), или к его фармацевтически приемлемым солям или стереоизомерам:
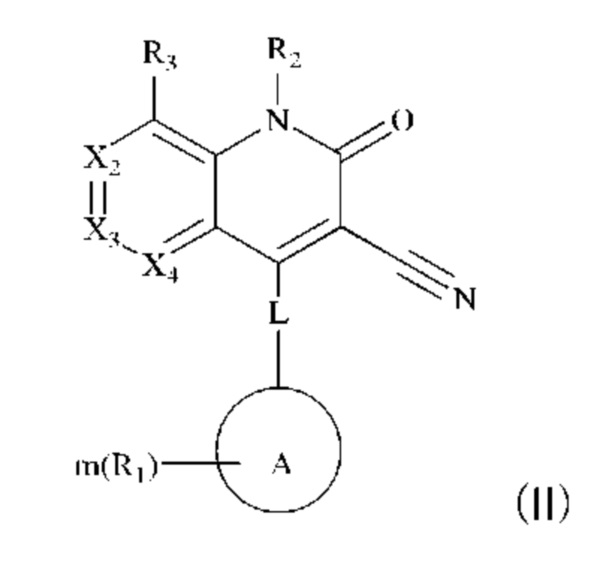
где
каждый из Х2, Х3 и Х4 независимо представляет собой CR3 или N, и Х2, Х3 и Х4 одновременно не представляют собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкила, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированного С1-6 алкила, галогенированного С1-6 алкокси, С2-8 алкенила, С2-8 алкинила, С1-6 алкилсульфонила, С1-6 алкилтио, С3-6 циклоалкила, 4-6-членного гетероциклила, С1-6 алкилкарбонила, С1-6 алкиламинокарбонила, (С1-6 алкил)2 аминокарбонила и аминокарбонила, где С1-6 алкил, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированный С1-6 алкокси, С2-8 алкенил, С2-8 алкинил, C1-6 алкилсульфонил, C1-6 алкилтио, С3-6 циклоалкил, 4-6-членный гетероциклил, C1-6 алкилкарбонил, C1-6 алкиламинокарбонил, (C1-6 алкил)2 аминокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, С3-6 циклоалкил, C1-6 алкилкарбониламино, C1-6 алкилсульфониламино, C1-6 алкилкарбонилокси и незамещенный или C1-6 алкилзамещенный 4-6-членный гетероциклил;
L представляет собой связь, -NH-(CH2)t-, и t равен 0, 1, 2 или 3;
кольцо А выбрано из группы, состоящей из 3-12-членного гетероциклила, арила, 5-10-членного гетероарила, 3-12-членного циклоалкила и 3-12-членного циклоалкенила, где 3-12-членный гетероциклил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации, и атом S может быть необязательно окислен до S(O) или S(O)2 , и 5-10-членный гетероарил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, С1-6 алкила, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированного С1-6 алкила, галогенированного С1-6 алкокси, С2-8 алкенила, С2-8алкинила, С1-6 алкилсульфонила, С1-6 алкилтио, 3-12-членного циклоалкила, 3-12-членного циклоалкенила, 3-12-членного гетероциклила, арила и 5-10-членного гетероарила, где С1-6 алкил, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галогенированный С1-6 алкил, галогенированный С1-6 алкокси, С2-8 алкенил, С2-8 алкинил, C1-6 алкилсульфонил, C1-6 алкилтио, 3-12-членный циклоалкил, 3-12-членный циклоалкенил, 3-12-членный гетероциклил, арил и 5-10-членный гетероарил являются незамещенными или необязательно замещены группой, выбранной из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкокси C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкилкарбониламино и C1-6 алкилсульфониламино;
m равен 0, 1, 2 или 3; и
R2 выбран из группы, состоящей из водорода, С1-6 алкила, С2-8 алкенила, С2-8 алкинила и галогенированного С1-6 алкила.
В одном из предпочтительных примеров осуществления Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3.
В другом предпочтительном примере осуществления Х4 представляет собой N, и каждый из Х2 и Х3 независимо представляет собой CR3.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
каждый из Х2, Х3 и Х4 независимо представляет собой CR3 или N, и Х2, Х3 и Х4 одновременно не представляют собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, карбоксила, циано, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, С2-8 алкенила, С2-8 алкинила, С1-6 алкилсульфонила, С1-6 алкилтио, С3-6 циклоалкила, 4-6-членного азотсодержащего гетероциклила, С1-6 алкилкарбонила, С1-6 алкиламинокарбонила, (С1-6 алкил)2 аминокарбонила и аминокарбонила, где С1-6 алкил, C1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, С2-8 алкенил, С2-8 алкинил, С1-6 алкилсульфонил, С1-6 алкилтио, С3-6 циклоалкил, 4-6-членный азотсодержащий гетероциклил, С1-6 алкилкарбонил, С1-6 алкиламинокарбонил, (С1-6 алкил)2 аминокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, галогена, С1-6 алкила, С1-6 алкокси, C1-6 алкиламино, (С1-6 алкил)2 амино, С1-6 алкилкарбонилокси, С3-6 циклоалкила и 4-6-членного гетероциклила, который является незамещенным или необязательно замещен C1-6 алкилом;
L представляет собой связь;
кольцо А представляет собой 3-12-членный гетероциклил, где 3-12-членный гетероциклил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации, и атом S может быть необязательно окислен до S(O) или S(O)2 ;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, C1-6 алкила, C1-6 алкокси и 5-6-членного гетероарила, где C1-6 алкил, C1-6 алкокси и 5-6-членный гетероарил не имеют заместителей или замещены гидроксигруппой;
m равен 0, 1, или 2; и
R2 представляет собой водород или С1-6 алкил.
В одном из предпочтительных примеров осуществления Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3.
В другом предпочтительном примере осуществления Х4 представляет собой N, и каждый из Х2 и Х3 независимо представляет собой CR3.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3 или N, предпочтительно CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, циано, галогена, карбоксила, алкила, алкокси, алкиламино, (С1-4 алкил)2 амино, С2-6 алкенила, С2-6 алкинила, С1-4 алкилкарбонила, алкиламинокарбонила, (С1-6 алкил)2 аминокарбонила, С1-4 алкилсульфонила, С1-4 алкилтио, аминокарбонила, циклопропила, азетидинила, морфолинила и пиперазинила, где алкил, алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С2-6 алкенил, С2-6 алкинил, С1-4 алкилкарбонил, С1-4 алкиламинокарбонил, (С1-6 алкил)2 аминокарбонил, С1-4 алкилсульфонил, С1-4 алкилтио, аминокарбонил, циклопропил, азетидинил, морфолинил и пиперазинил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, галогена, С1-4 алкила, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, циклопропила, С1-4 алкилкарбонилокси и 4-6-членного гетероциклила, который является незамещенным или необязательно замещен C1-6 алкилом;
L представляет собой связь;
кольцо А представляет собой 4-12-членный гетероциклил, где 4-12-членный гетероциклил включает гетероатом, выбранный из одного или комбинации двух из следующих атомов: О, S и N, и содержит по меньшей мере один N, и при этом кольцо А соединено с L через атом N, и атом S может быть необязательно окислен до S(O)2 ;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, С1-4 алкила, С1-4 алкокси, пиразолила, тиазолила и триазолила, где С1-4 алкил, C1-4 алкокси, пиразолил, тиазолил и триазолил не имеют заместителей или замещены гидроксигруппой; и
m равен 0, 1 или 2.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
Х2 представляет собой N, Х3 представляет собой CR3, и Х4 представляет собой CR3 или N, предпочтительно CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, циано, галогена, карбоксила, алкила, алкокси, алкиламино, (С1-4 алкил)2 амино, С2-6 алкенила, С2-6 алкинила, С1-4 алкилкарбонила, алкиламинокарбонила, (С1-6 алкил)2 аминокарбонила, С1-4 алкилсульфонила, С1-4 алкилтио, аминокарбонила, циклопропила, азетидинила, морфолинила и пиперазинила, где алкил, алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С2-6 алкенил, С2-6 алкинил, С1-4 алкилкарбонил, С1-4 алкиламинокарбонил, (С1-6 алкил)2 аминокарбонил, С1-4 алкилсульфонил, С1-4 алкилтио, аминокарбонил, циклопропил, азетидинил, морфолинил и пиперазинил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, галогена, С1-4 алкила, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, циклопропила, С1-4 алкилкарбонилокси и 4-6-членного гетероциклила, который является незамещенным или необязательно замещен C1-6 алкилом;
L представляет собой связь;
кольцо А представляет собой 4-12-членный гетероциклил, где 4-12-членный гетероциклил включает гетероатом, выбранный из одного или комбинации двух из следующих атомов; О, S и N, и содержит по меньшей мере один N, и при этом кольцо А соединено с L через атом N, и атом S может быть необязательно окислен до S(O)2 ;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, С1-4 алкила, С1-4 алкокси, пиразолила, тиазолила и триазолила, где С1-4 алкил, С1-4 алкокси, пиразолил, тиазолил и триазолил не имеют заместителей или замещены гидроксигруппой; и
m равен 0, 1 или 2.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
Х2 представляет собой N, Х3 представляет собой CR3, и Х4 представляет собой CR3 или N, предпочтительно CR3;
L представляет собой связь;
кольцо А представляет собой 4-7-членный моногетероциклил, где 4-7-членный моногетероциклил включает гетероатом, выбранный из одного или комбинации двух из следующих атомов: О, S и N, и содержит по меньшей мере один N, и при этом кольцо А соединено с L через атом N, и атом S может быть необязательно окислен до S(O)2;
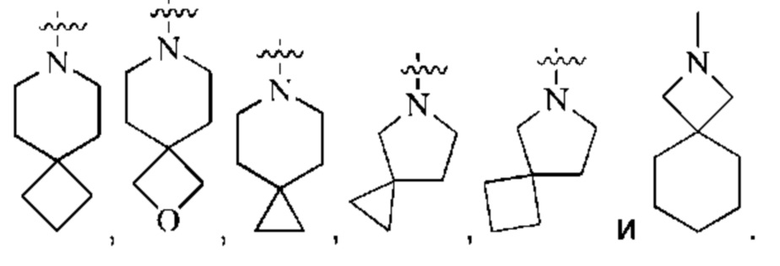
предпочтительно, кольцо А представляет собой насыщенный 4-7-членный азотсодержащий моногетероциклил, предпочтительнее:
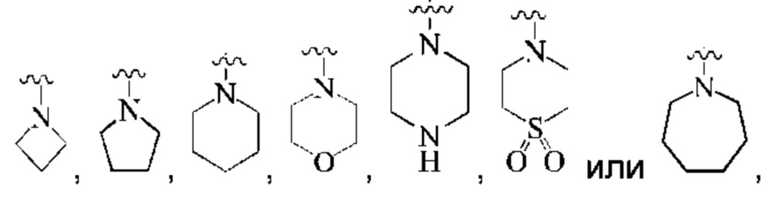
более предпочтительно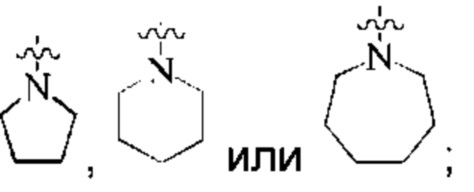
R3 в каждом случае независимо выбран из группы, состоящей из водорода, галогена, С1-4 алкила, С1-4 алкокси, морфолинила, С2-6 алкенила, С1-4 алкилкарбонила, С1-4 алкиламинокарбонила, (С1-4 алкил)2 аминокарбонила и аминокарбонила, где С1-4 алкил, С1-4 алкокси, морфолинил, С2-6 алкенил, С1-4 алкилкарбонил, С1-4 алкиламинокарбонил, (С1-4 алкил)2 аминокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, C1-4 алкокси, циклопропила, амино, алкиламино, (С1-4 алкил)2 амино, и 4-6-членного гетероциклила, который является незамещенным или необязательно замещен C1-4 алкилом;
каждый R1 независимо выбран из группы, состоящей из водорода, галогена, C1-4 алкила, C1-4 алкокси, пиразолила, тиазолила и триазолила, где C1-4 алкил, C1-4 алкокси, пиразолил, тиазолил и триазолил являются незамещенными или замещены гидроксигруппой; и
m равен 0, 1 или 2.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, галогена, С1-4 алкила, С1-4 алкокси, С2-6 алкенила, С1-4 алкилкарбонила, С1-4 алкиламинокарбонила и аминокарбонила, где С1-4 алкил, С1-4 алкокси, С2-6 алкенил, C1-4 алкилкарбонил, алкиламинокарбонил и аминокарбонил являются
незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, С1-4 алкокси, циклопропила и 4-6-членного гетероциклила, который является незамещенным или замещен С1-6 алкилом;
L представляет собой связь;
кольцо А представляет собой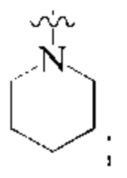
каждый R1 независимо выбран из группы, состоящей из водорода, С1-4 алкила и С1-4 алкокси; и
m равен 0, 1 или 2.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, галогена, С1-4 алкила и морфолинила, где С1-4 алкил является незамещенным или замещен одной или более гидроксигруппами;
L представляет собой связь;
кольцо А представляет собой 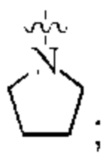 и
и
каждый R1 независимо выбран из группы, состоящей из пиразолила, тиазолила и триазолила.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
L представляет собой связь;
Х2 представляет собой N, и Х3 и Х4 независимо представляют собой CR3 или N, предпочтительно CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, циано, галогена, карбоксила, C1-4 алкила, C1-4 алкокси, С1-4 алкилкарбонила, С2-6 алкинила, C1-4 алкиламинокарбонила, (С1-4 алкил)2 аминокарбонила, C1-4 алкилтио, С1-4 алкилсульфонила, C1-4 алкиламино, (C1-4 алкил)2 амино, азетидинила, морфолинила, пиперазинила, С2-6 алкенила и циклопропила, где C1-4 алкил, алкокси, C1-4 алкилкарбонил, С2-6 алкинил, C1-4 алкиламинокарбонил, (C1-6 алкил)2 аминокарбонил, С1-4 алкилтио, C1-4 алкилсульфонил, алкиламино, (С1-4 алкил)2 амино, азетидинил, морфолинил, пиперазинил, С2-6 алкенил и циклопропил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, галогена, С1-4 алкила, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, циклопропила и С1-4 алкилкарбонилокси;
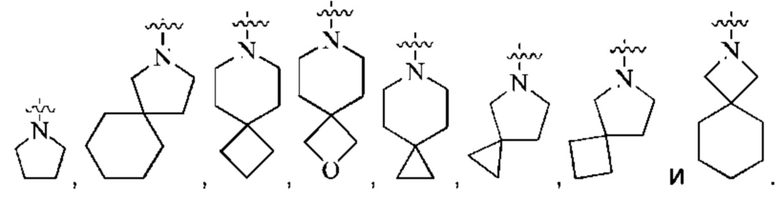
кольцо А представляет собой 7-12-членный спирогетероциклил, где спирогетероциклил включает один или более гетероатомов, выбранных из группы, состоящей из О, S и N, и содержит по меньшей мере один N, и кольцо А соединено с L через атом N, и атом S может быть необязательно окислен до S(O)2 ; предпочтительно, 7-12-членный спирогетероциклил представляет собой насыщенный 7-12-членный азотсодержащий спирогетероциклил; более предпочтительно, насыщенный 7-12-членный азотсодержащий спирогетероциклил выбран из группы, состоящей из:
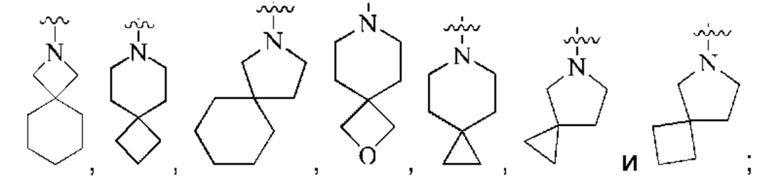
предпочтительно, кольцо А выбрано из группы, состоящей из 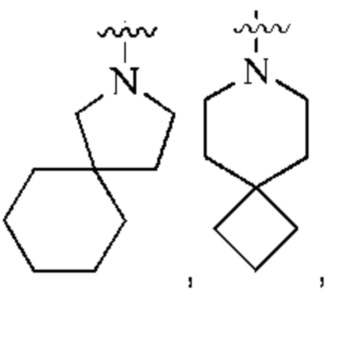
 и
и
более предпочтительно, кольцо А выбрано из группы, состоящей из 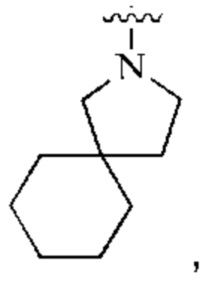
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
каждый из Х2, Х3 и Х4 независимо представляет собой CR3 или N;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, циано, амино, галогена, карбоксила, С1-4 алкила, С1-4 алкокси, С2-6 алкенила, С1-4 алкилкарбонила, С2-6 алкинила, С1-4 алкиламино, (С1-4 алкил)2 амино, С1-4 алкиламинокарбонила, С1-4 алкилтио, С1-4 алкилсульфонила, циклопропила, азетидинила, морфолинила и пиперазинила, где С1-4 алкил, С1-4 алкокси, С2-6 алкенил, С1-4 алкилкарбонил, С2-6 алкинил, С1-4 алкиламино, (С1-4 алкил)2 амино, С1-4 алкиламинокарбонил, С1-4 алкилтио, С1-4 алкилсульфонил, циклопропил, азетидинил, морфолинил и пиперазинил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, галогена, C1-4 алкила, C1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, циклопропила и C1-4 алкилкарбонилокси;
L представляет собой связь;
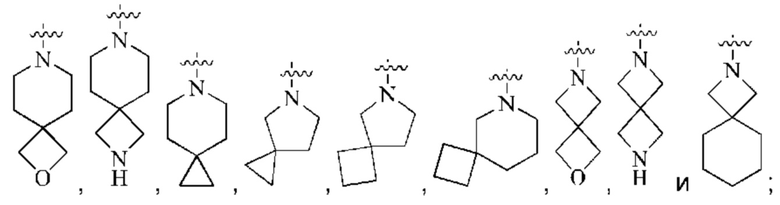
кольцо А выбрано из группы, состоящей из 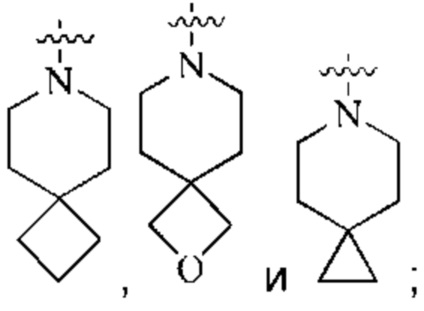 и
и
m равен 0.
В одном из предпочтительных примеров осуществления Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3.
В другом предпочтительном примере осуществления Х4 представляет собой N, и каждый из Х2 и Х3 независимо представляет собой CR3.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
каждый из Х2, Х3 и Х4 независимо представляет собой CR3 или N;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, циано, галогена, карбоксила, алкила, алкокси, алкиламино, (С1-4 алкил)2 амино, С2-6 алкенила, С2-6 алкинила, алкилкарбонила, алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила, С1-4 алкилсульфонила, С1-4 алкилтио, аминокарбонила, циклопропила, азетидинила, морфолинила и пиперазинила, где С1-4 алкил, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С2-6 алкенил, С2-6 алкинил, С1-4 алкилкарбонил, С1-4 алкиламинокарбонил, (C1-6 алкил)2 аминокарбонил, С1-4 алкилсульфонил, С1-4 алкилтио, аминокарбонил, циклопропил, азетидинил, морфолинил и пиперазинил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, галогена, С1-4 алкила, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, циклопропила, С1-4 алкилкарбонилокси, 4-6-членного гетероциклила, который является незамещенным или необязательно замещен С1-6 алкилом;
L представляет собой связь;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, C1-6 алкила, C1-6 алкокси, пиразолила, тиазолила и триазолила, где C1-6 алкил, C1-6 алкокси, пиразолил, тиазолил и триазолил не имеют заместителей или замещены гидроксигруппой;
m равен 0, 1 или 2;
кольцо А представляет собой группу, выбранную из группы, состоящей из:
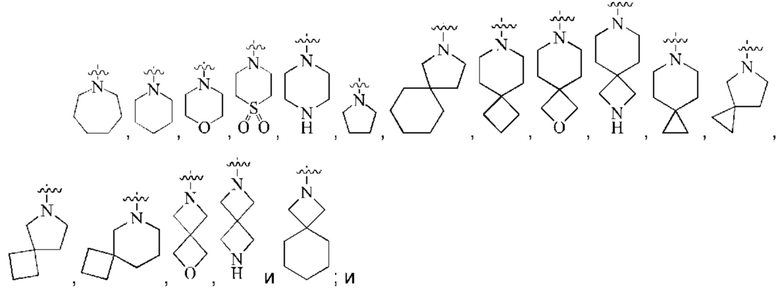
предпочтительно, кольцо А выбрано из группы, состоящей из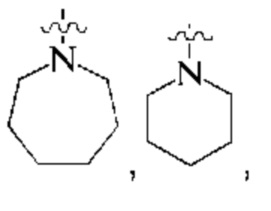
В одном из предпочтительных примеров осуществления Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3.
В другом предпочтительном примере осуществления Х4 представляет собой N, и каждый из Х2 и Х3 независимо представляет собой CR3.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3 или N;
L представляет собой -NH-(СН2)t- или связь, и t равен 0, 1 или 2;
кольцо А представляет собой фенил;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, карбоксила, циано, галогена, С1-6 алкила, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, С2-8 алкенила, С2-6 алкинила, С1-6 алкилсульфонила, С1-6 алкилтио, С3-6 циклоалкила, 4-6-членного азотсодержащего гетероциклила, С1-6 алкилкарбонила, С1-6 алкиламинокарбонила, (С1-6 алкил)2 аминокарбонила и аминокарбонила, где С1-6 алкил, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, С2-8 алкенил, С2-6 алкинил, С1-6 алкилсульфонил, С1-6 алкилтио, С3-6 циклоалкил, 4-6-членный азотсодержащий гетероциклил, С1-6 алкилкарбонил, С1-6 алкиламинокарбонил, (С1-6 алкил)2 аминокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, C1-6 алкилкарбонилокси, С3-6 циклоалкила и 4-6-членного гетероциклила, который является незамещенным или замещен C1-6 алкилом;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила и C1-6 алкокси, где C1-6 алкил и C1-6 алкокси являются незамещенными или необязательно замещены группой, выбранной из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила и C1-6 алкокси;
R2 представляет собой водород или С1-6 алкил; и
m равен 0, 1 или 2.
Некоторые примеры осуществления настоящего изобретения относятся к соединению, представленному формулой (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам,
где
Х2 представляет собой N, и каждый из Х3 и Х4 независимо представляет собой CR3 или N;
L представляет собой связь;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, карбоксила, циано, галогена, C1-6 алкила, C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, С2-8 алкенила, С2-6 алкинила, C1-6 алкилсульфонила, C1-6 алкилтио, С3-6 циклоалкила, 4-6-членного азотсодержащего гетероциклила, C1-6 алкилкарбонила, C1-6 алкиламинокарбонила, (C1-6 алкил)2 аминокарбонила и аминокарбонила, где C1-6 алкил, C1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2 амино, С2-8 алкенил, С2-6 алкинил, C1-6 алкилсульфонил, C1-6 алкилтио, С3-6 циклоалкил, 4-6-членный азотсодержащий гетероциклил, С1-6 алкилкарбонил, С1-6 алкиламинокарбонил, (С1-6 алкил)2 аминокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или более группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, галогена, С1-6 алкила, С1-6 алкокси, C1-6 алкиламино, (С1-6 алкил)2 амино, С1-6 алкилкарбонилокси, С3-6 циклоалкила и 4-6-членного гетероциклила, который является незамещенным или замещен С1-6 алкилом;
L представляет собой связь;
кольцо А представляет собой 5-10-членный гетероарил, и 5-10-членный гетероарил включает гетероатом, выбранный из одного из следующих элементов: О, S, N или любой их комбинации;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила, C1-6 алкокси, фенила и 5-6-членного гетероарила, где C1-6 алкил, C1-6 алкокси, фенил и 5-6-членный гетероарил являются незамещенными или необязательно замещены группой, выбранной из группы, состоящей из гидрокси, амино, карбоксила, циано, нитро, галогена, C1-6 алкила и C1-6 алкокси;
m равен 0, 1 или 2; и
R2 представляет собой водород или С1-6 алкил.
Предпочтительно, кольцо А представляет собой 9-10-членный гетероарил.
Более предпочтительно, кольцо А представляет собой 9-10-членный азотсодержащий гетероарил.
Наиболее предпочтительно, кольцо А представляет собой 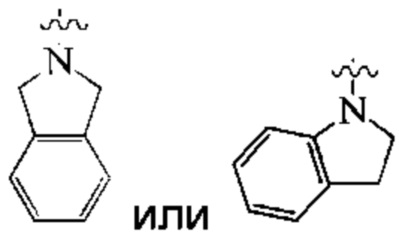
Один из примеров осуществления настоящего изобретения относится к приведенным выше соединениям, представленным формулами (I) или (II), или их фармацевтически приемлемым солям или стереоизомерам, которые приведены в Таблице 1:
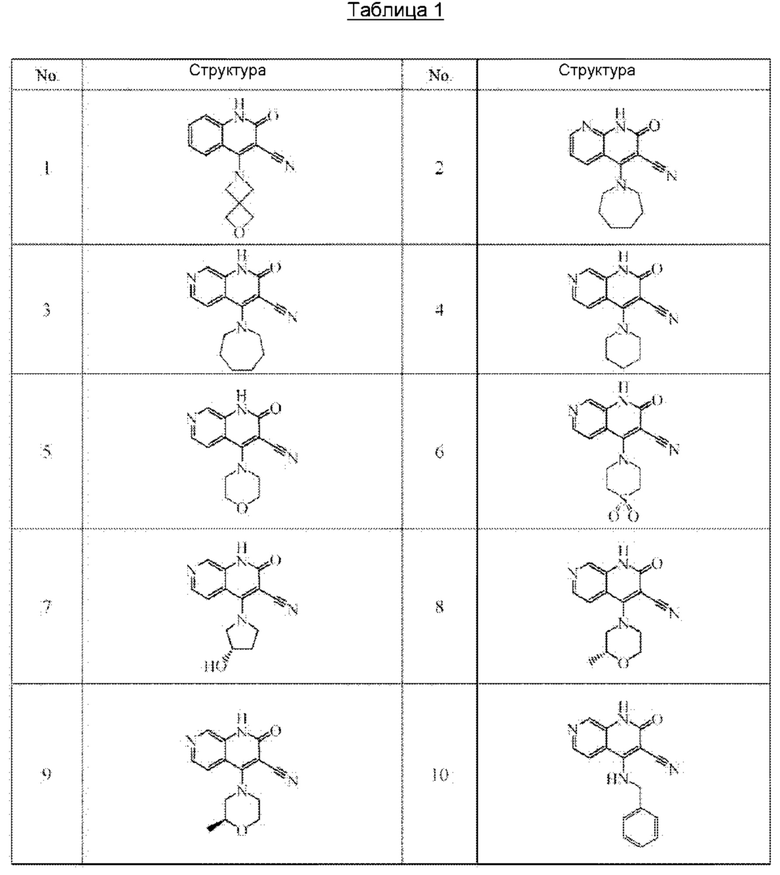
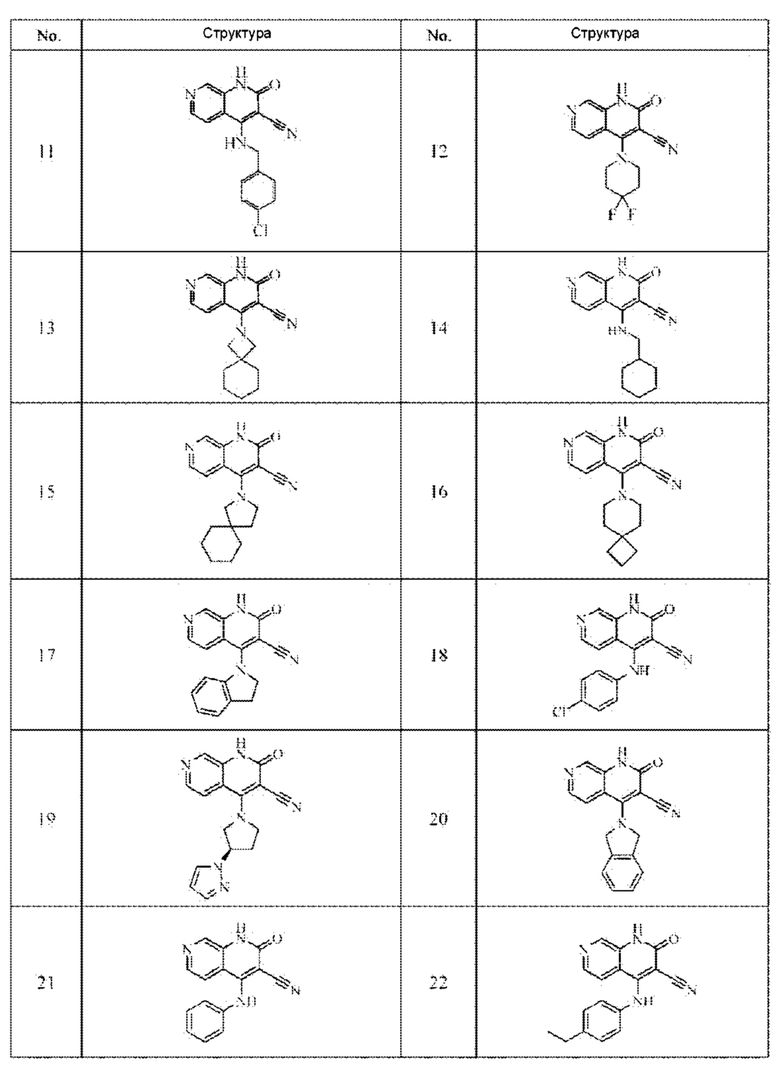
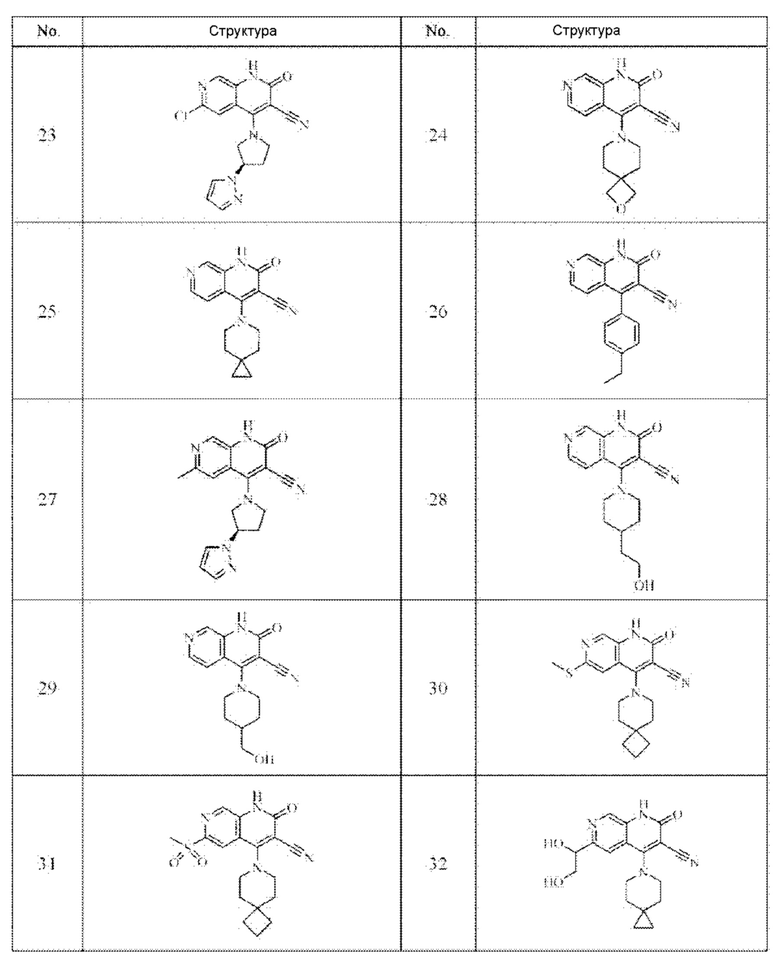
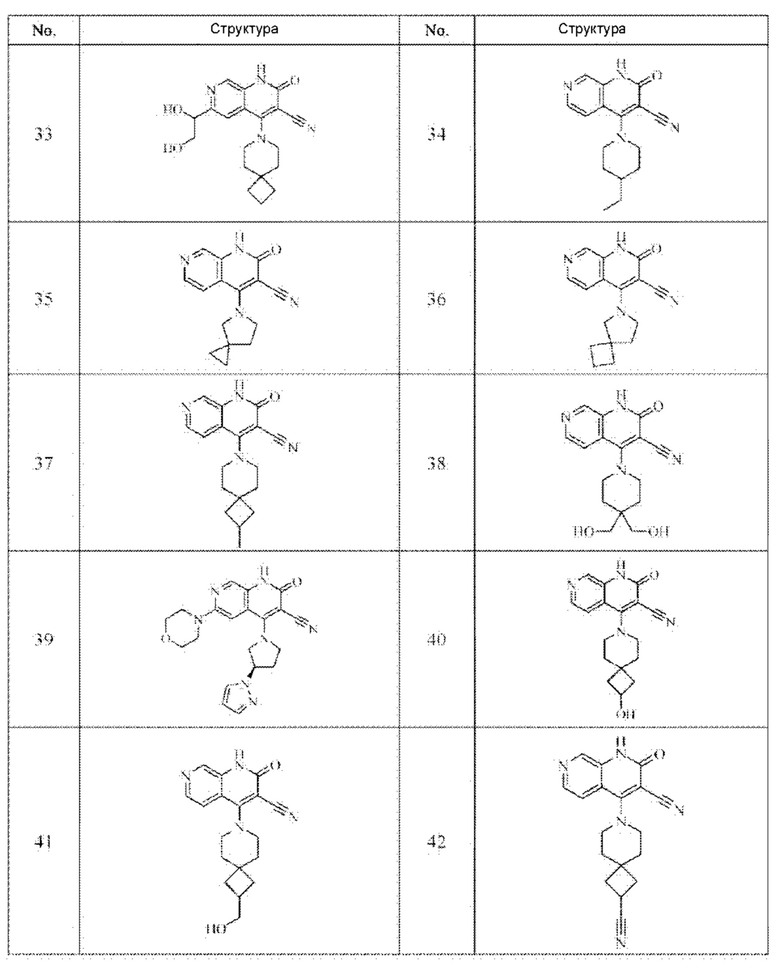
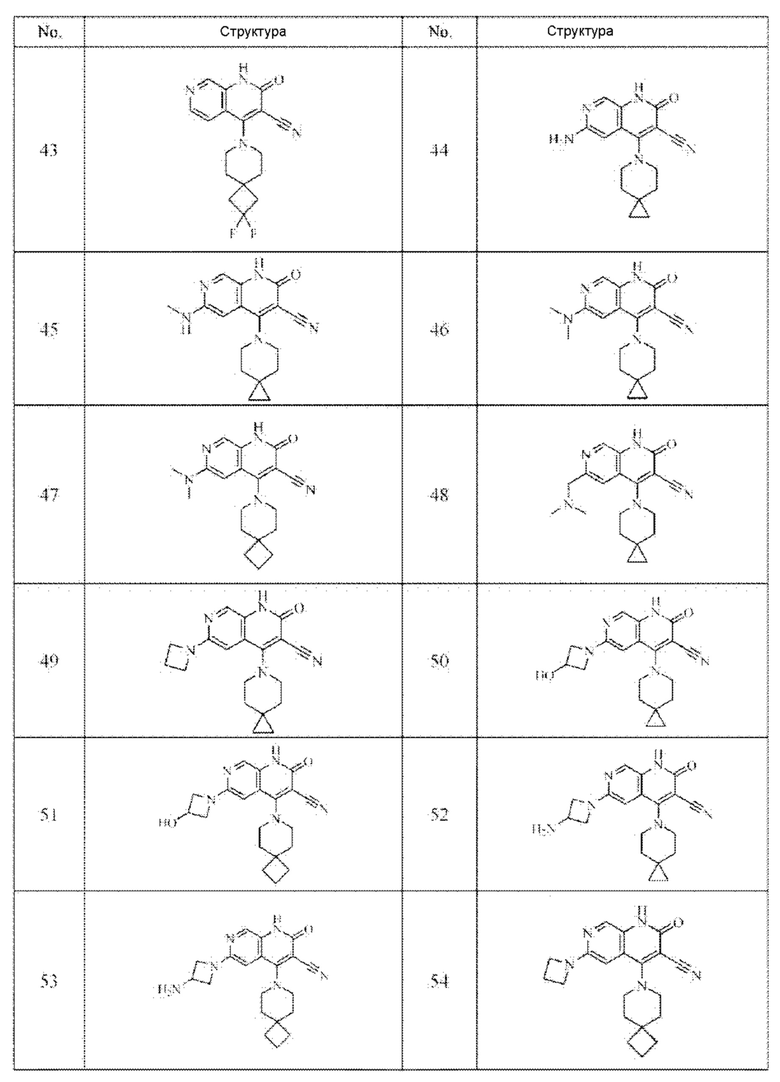
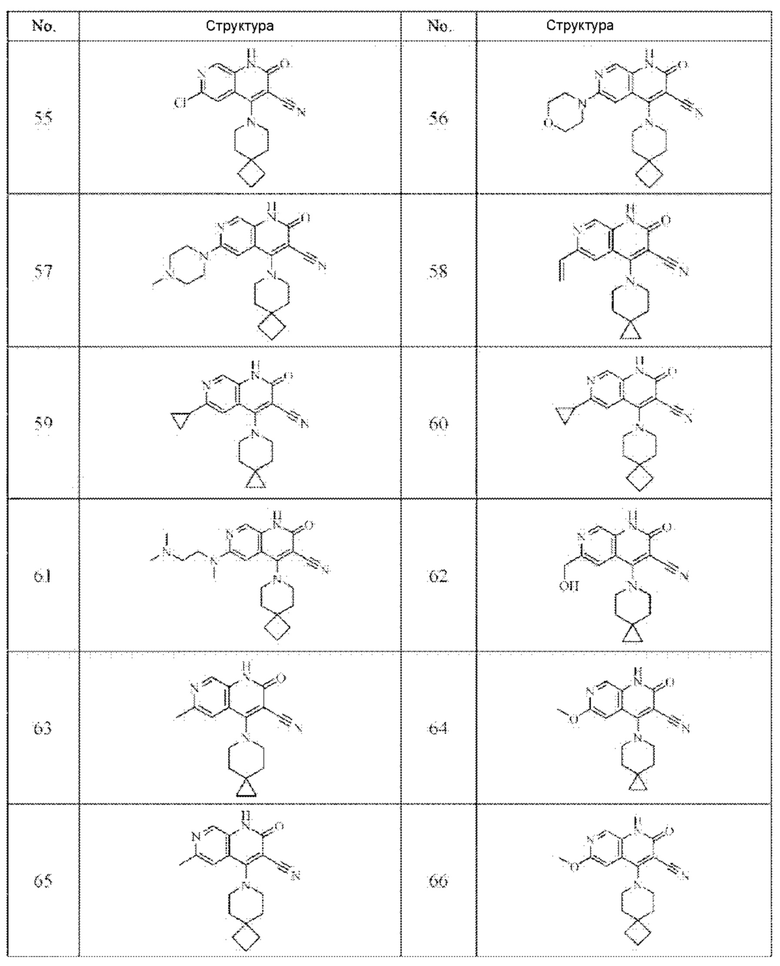
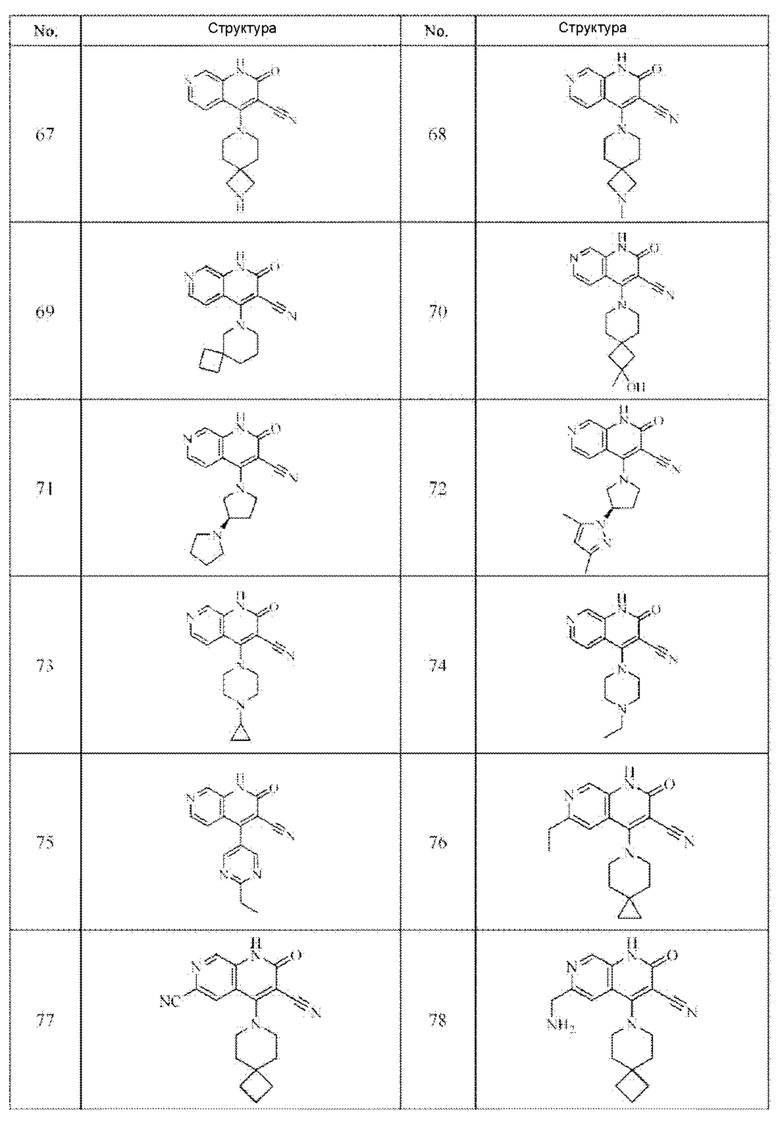
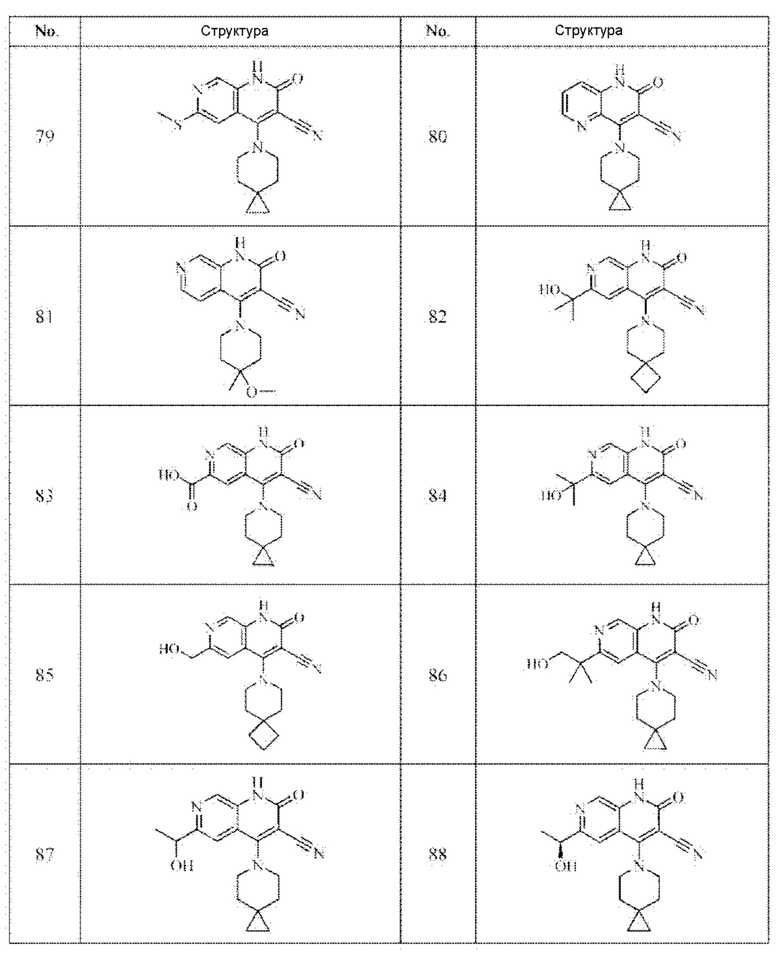
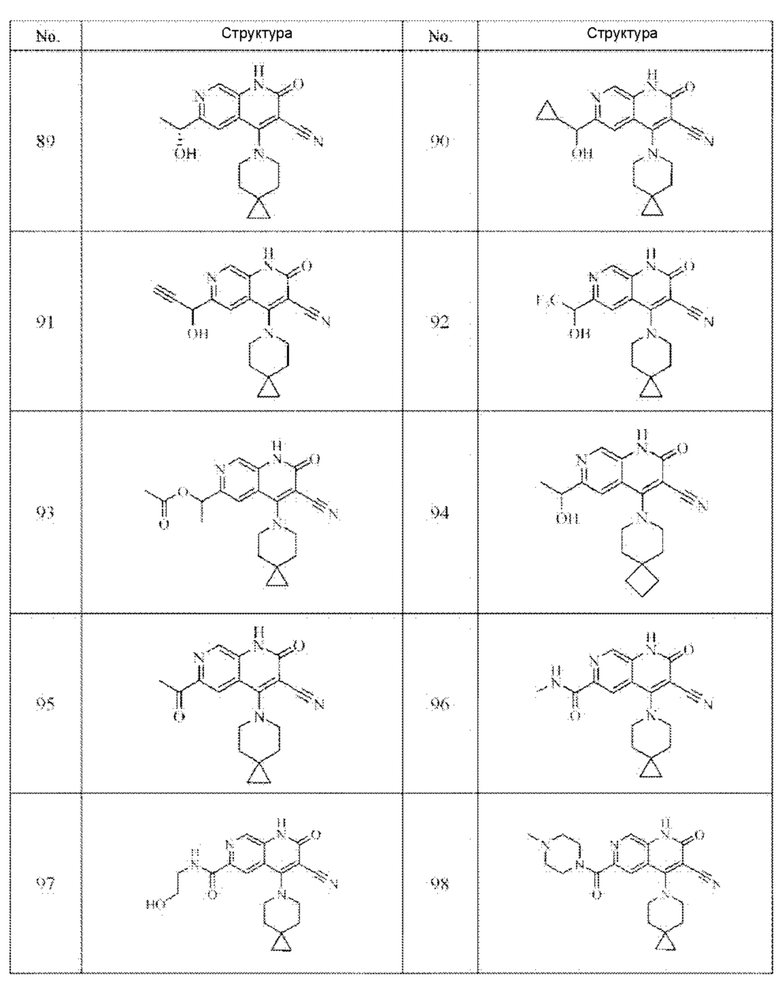
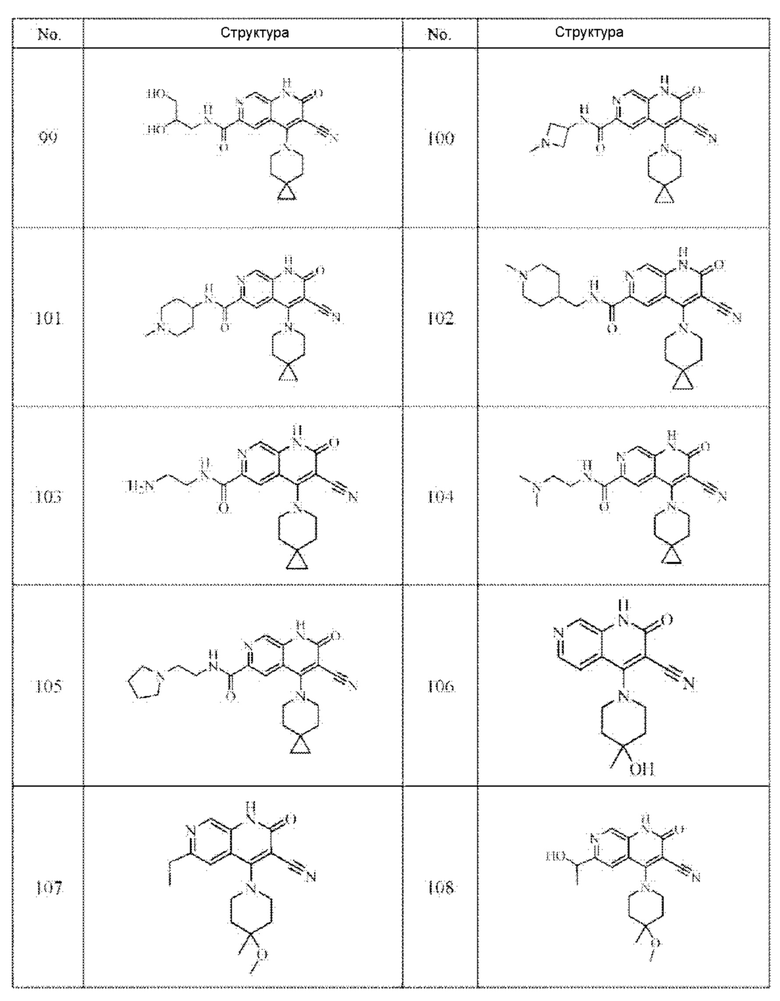
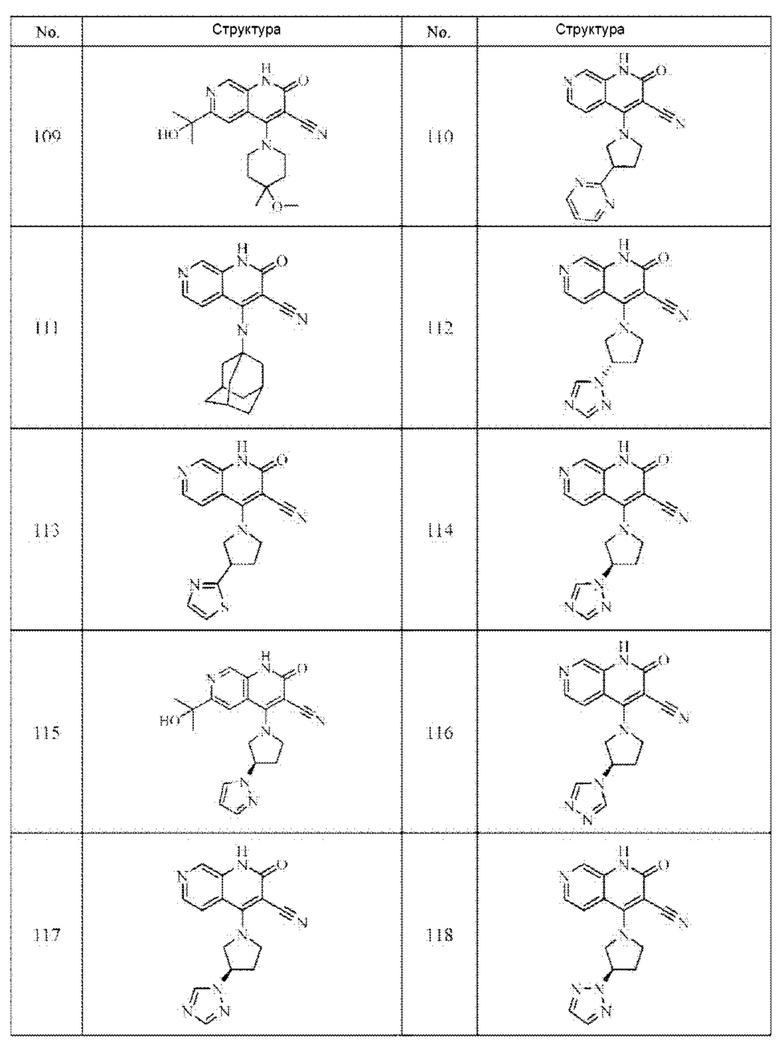
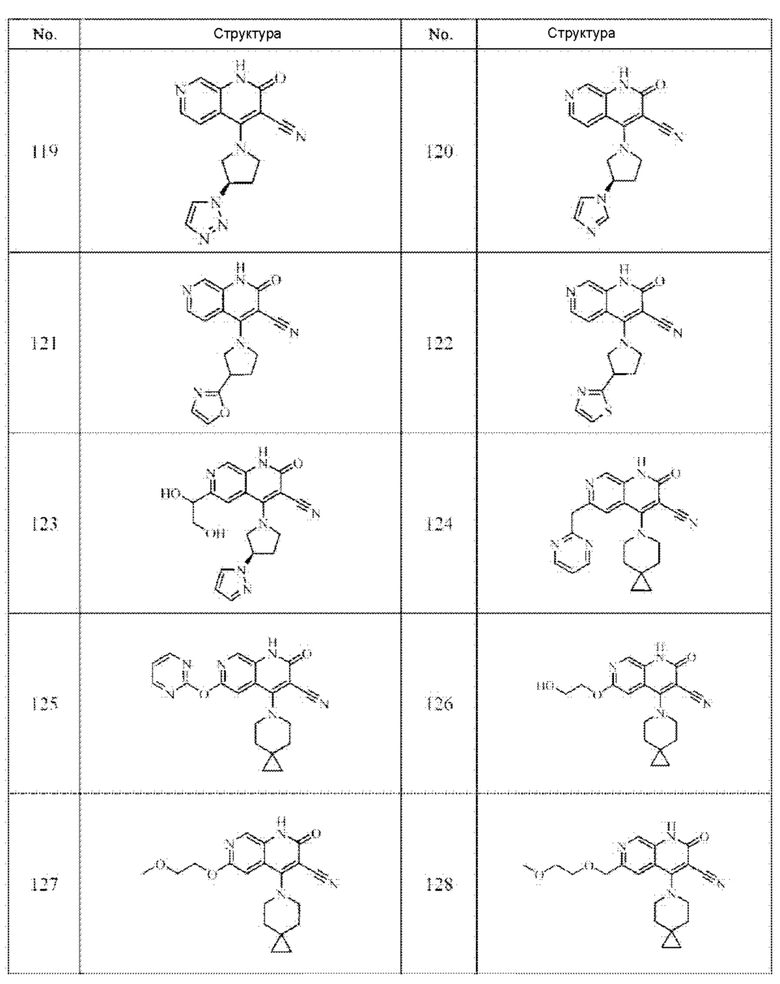
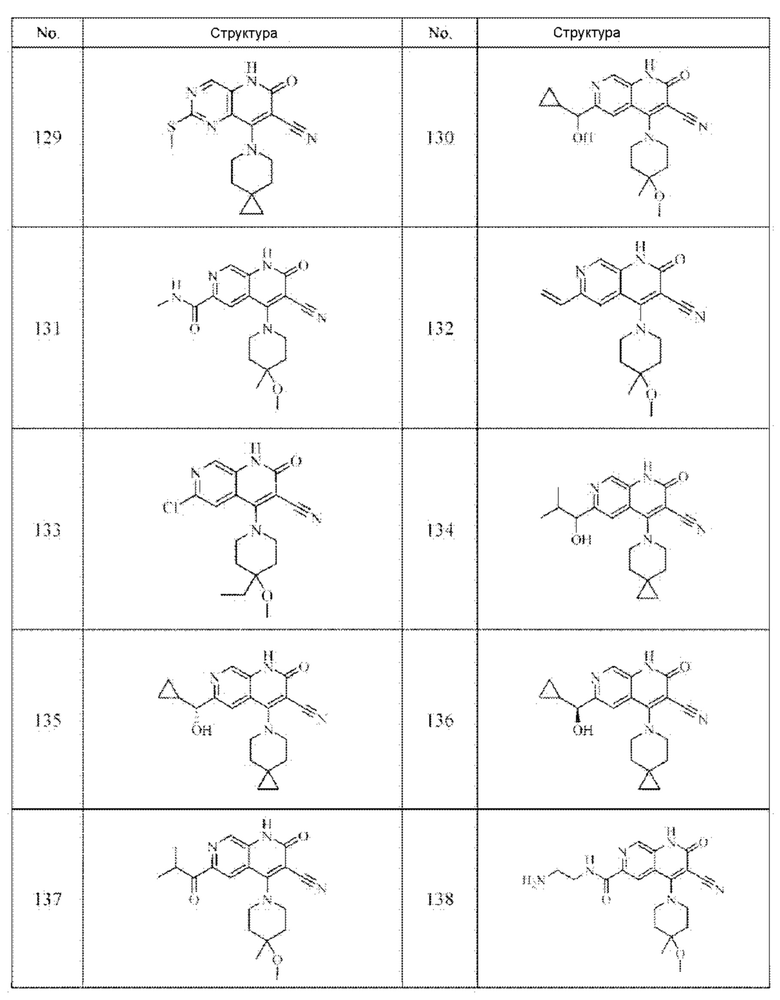
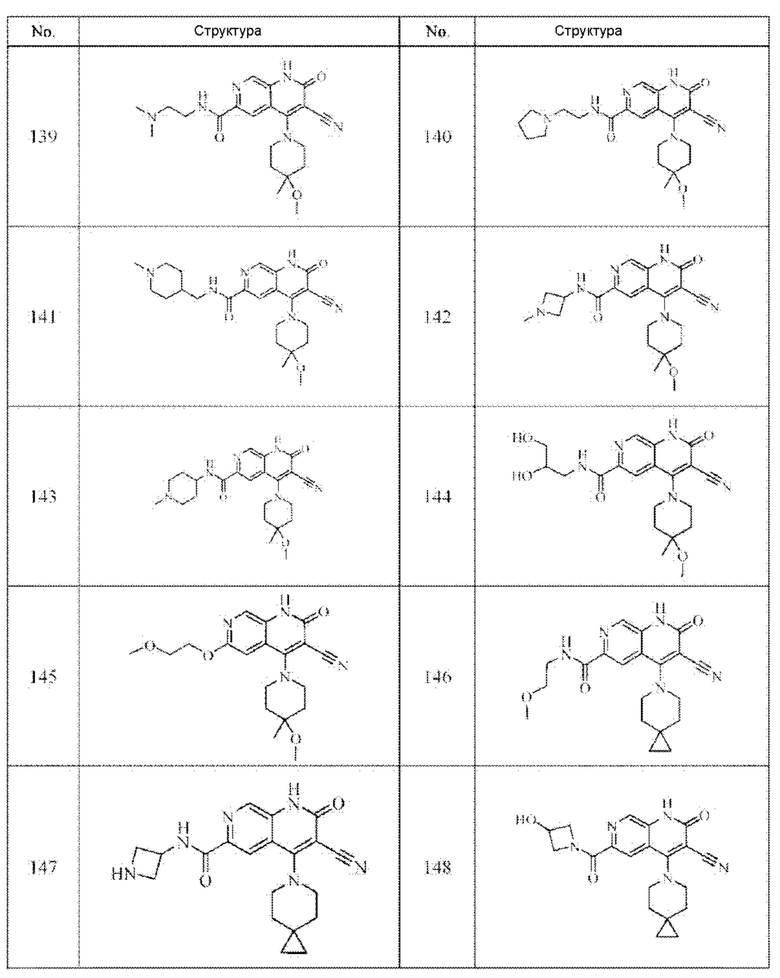
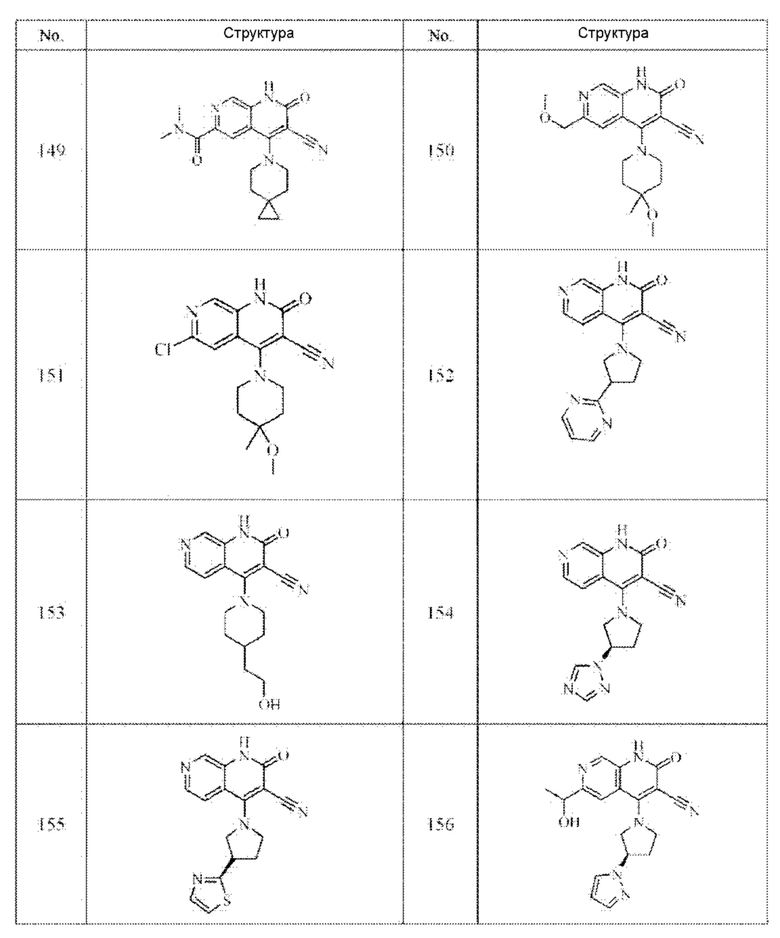
Настоящее изобретение также относится к фармацевтической композиции, включающей соединение, представленной формулой (I) или (II), или его фармацевтически приемлемые соли или стереоизомеры, и один или более дополнительных терапевтически активных агентов.
В одном из конкретных примеров осуществления настоящего изобретения композиция может быть применена для комбинированного введения "терапевтически эффективного количества" соединения, представленного формулами (I) или (II), или его фармацевтически приемлемых солей или стереоизомеров вместе с одном или более дополнительными терапевтически активными агентами, такого как последовательное введение, одновременное введение или введение состава соединения, содержащего соединение согласно изобретению или его фармацевтически приемлемых солей или стереоизомеров и дополнительного терапевтически активного агента.
Настоящее изобретение также относится к фармацевтическому составу, включающему соединение, представленное формулами (I) или (II), или его фармацевтически приемлемые соли или стереоизомеры.
В некоторых примерах осуществления изобретения фармацевтический состав может включать один или более фармацевтически приемлемых носителей.
Фармацевтически приемлемый носитель согласно настоящему изобретению может представлять собой один или более твердых или жидких наполнителей или гелеобразных материалов, подходящих для употребления человеком. Фармацевтически приемлемый носитель предпочтительно имеет достаточную чистоту и достаточно низкую токсичность, совместим с соединением или его фармацевтически приемлемыми солями или стереоизомерами согласно настоящему изобретению и не оказывает существенного понижающего влияния на их фармакологическое действие. Например, фармацевтически приемлемый носитель может представлять собой наполнитель, связующее вещество, дезинтегрирующий агент, скользящее вещество, водный растворитель или неводный растворитель и подобное вещество.
Фармацевтический состав согласно настоящему изобретению может быть изготовлен в виде любой фармацевтически приемлемой стандартной лекарственной формы и введен пациенту или индивидууму, нуждающемуся в такой терапии, с помощью любых подходящих средств введения, например, посредством перорального, парентерального, ректального или ингаляционного введения. Для перорального введения состав может быть приготовлен в виде таблеток, капсул, пилюль, гранул и подобных форм. Для парентерального введения состав может быть приготовлен в виде раствора для инъекций, стерильного порошка для инъекций и подобных композиций.
Изобретение также относится к применению соединения, представленного формулами (I) или (II), или его фармацевтически приемлемых солей или стереоизомеров, фармацевтической композиции или фармацевтического состава для получения лекарственного средства, предназначенного для лечения или профилактики заболевания, опосредованного PDE9. В частности, заболевание, опосредованное PDE9, представляет собой когнитивное расстройство, вызываемое расстройством центральной нервной системы. В частности, когнитивное расстройство включает: нарушение восприятия, внимания, памяти и способности к обучению, примеры которого включают, без ограничений, болезнь Альцгеймера, шизофрению, старческую потерю памяти, сосудистую деменцию, черепно-мозговую травму, инсульт, деменцию после инсульта, посттравматическую деменцию, общее нарушение внимания, нарушение внимания у детей, отягощенное проблемами, связанными со способностью к обучению и памятью, болезнь Альцгеймера, деменцию с тельцами Леви, лобно-долевую деменцию, кортикобазальную дегенерацию, боковой амиотрофический склероз, хорею Хантингтона, рассеянный склероз, дегенерацию таламуса, деменцию Крейтцфельда-Якоба, деменцию, вызванную ВИЧ, шизофрению, корсаковский психоз или депрессию или биполярное расстройство.
Изобретение также относится к соединению, представленному формулами (I) или (II), или к его фармацевтически приемлемым солям или стереоизомерам, фармацевтической композиции или фармацевтическому составу, предназначенному для применения в лечении или профилактике заболевания.
Изобретение также относится к применению соединения, представленного формулами (I) или (II), или его фармацевтически приемлемых солей или стереоизомеров, фармацевтической композиции или фармацевтического состава для применения в лечении или профилактике заболевания, опосредованного PDE9. В частности, заболевание, опосредованное PDE9, представляет собой когнитивное расстройство, вызываемое расстройством центральной нервной системы. В частности, когнитивное расстройство включает: нарушение восприятия, внимания, памяти и способности к обучению, примеры которого включают, без ограничений, болезнь Альцгеймера, шизофрению, старческую потерю памяти, сосудистую деменцию, черепно-мозговую травму, инсульт, деменцию после инсульта, посттравматическую деменцию, общее нарушение внимания, нарушение внимания у детей, отягощенное проблемами, связанными со способностью к обучению и памятью, болезнь Альцгеймера, деменцию с тельцами Леви, лобно-долевую деменцию, кортикобазальную дегенерацию, боковой амиотрофический склероз, хорею Хантингтона, рассеянный склероз, дегенерацию таламуса, деменцию Крейтцфельда-Якоба, деменцию, вызванную ВИЧ, шизофрению, корсаковский психоз или депрессию или биполярное расстройство.
Изобретение также относится к способу лечения или профилактики заболеваний. Способ включает введение пациенту, нуждающемуся в соответствующей терапии, терапевтически эффективного количества соединения, представленного формулами (I) или (II), или его фармакологически приемлемых солей или стереоизомеров, фармацевтической композиции или фармацевтического состава. Заболевание представляет собой заболевание, опосредованное PDE9. В частности, заболевание, опосредованное PDE9, представляет собой когнитивное расстройство, вызываемое расстройством центральной нервной системы. В частности, когнитивное расстройство включает: нарушение восприятия, внимания, памяти и способности к обучению, примеры которого включают, без ограничений, болезнь Альцгеймера, шизофрению, старческую потерю памяти, сосудистую деменцию, черепно-мозговую травму, инсульт, деменцию после инсульта, посттравматическую деменцию, общее нарушение внимания, нарушение внимания у детей, отягощенное проблемами, связанными со способностью к обучению и памятью, болезнь Альцгеймера, деменцию с тельцами Леви, лобно-долевую деменцию, кортикобазальную дегенерацию, боковой амиотрофический склероз, хорею Хантингтона, рассеянный склероз, дегенерацию таламуса, деменцию Крейтцфельда-Якоба, деменцию, вызванную ВИЧ, шизофрению, корсаковский психоз или депрессию или биполярное расстройство.
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Употребляемый в настоящей работе термин "галоген" означает фтор, хлор, бром, йод и подобные элементы, предпочтительно фтор и хлор.
Употребляемый в настоящей работе термин "галогенированный" означает, что любой атом водорода, имеющийся в заместителе, может быть замещен одним или более атомами галогена, которые могут быть как одинаковыми, так и различными. Определение термина "галоген" приведено выше.
Употребляемый в настоящей работе термин "C1-6 алкил" означает неразветвленную или разветвленную алкильную группу, полученную удалением одного атома водорода из углеводородной частицы, содержащей от 1 до 6 атомов углерода, такую как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, 1-метил-2-метилпропил и т.д. Термин "С1-4 алкил" относится к приведенным выше примерам, содержащим от 1 до 4 атомов углерода.
Употребляемый в настоящей работе термин "С2-8 алкенил" означает неразветвленную или разветвленную или циклическую алкенильную группу, полученную удалением одного атома водорода из олефина, содержащего от 2 до 8 атомов углерода и включающего углерод-углеродную двойную связь, такую как винил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 1,3-бутадиенил, 1-пентенил, 2-пентенил, 3-пентенил, 1,3-пентадиенил, 1,4-пентадиенил, 1-гексенил, 1,4-гексадиенил и т.д.
Употребляемый в настоящей работе термин "С2-8 алкинил" означает неразветвленную или разветвленную алкинильную группу, полученную удалением одного атома водорода из алкина, содержащего от 2 до 8 атомов углерода и включающего углерод-углеродную тройную связь, такую как этинил, пропинил, 2-бутинил, 2-пентинил, 3-пентинил, 4-метил-2-пентинил, 2-гексинил, 3-гексинил и т.д.
Употребляемый в настоящей работе термин "C1-6 алкокси" означает группу, в которой "С1-6 алкил", определение которого приведено выше, соединен с исходной частицей через атом кислорода, то есть группу "С1-6 алкил-О-", такую как метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси, н-пентилокси, неопентилокси, н-гексилокси и т.д. Термин "С1-6 алкокси" относится к приведенным выше примерам, содержащим от 1 до 4 атомов углерода, то есть к группе "С1-6 алкил-О-".
Употребляемые в настоящей работе термины "С1-6 алкиламино", "(С1-6 алкил)2 амино", "С1-6 алкилкарбониламино", "С1-6 алкилсульфониламино", "С1-6 алкиламинокарбонил", "(С1-6 алкил)2 аминокарбонил", "С1-6 алкоксикарбонил", "С1-6 алкилсульфонил", "С1-6 алкилтио", "С1-6 алкилкарбонил" означают, соответственно, группы С1-6 алкил-NH-, (С1-6 алкил)(С1-6 алкил)N-, С1-6 алкил-С(O)-NH-, С1-6 алкил-S(O)2 -NH2-, C1-6 алкил-NH-С(О)-, (C1-6 алкил)(С1-6 алкил)N-С(О)-, C1-6 алкил-О-С(О)-, С1-6 алкил-S(O)2 -, С1-6 алкил-S-, С1-6 алкил-С(О)-. Определение термина "С1-6 алкил" приведено выше, и предпочтительно он представляет собой "С1-4алкил".
Употребляемый в настоящей работе термин "конденсированная кольцевая структура" означает полициклическую структуру, образованную двумя или более циклическими структурами, соединенными в виде орто-конденсированной, спироциклической или мостиковой системы циклов. Орто-конденсированный цикл означает конденсированную циклическую структуру, образованную двумя или более циклическими структурами, содержащими два общих соседних атома, принадлежащих к разным циклам (т.е. содержащим одну общую связь). Мостиковый цикл означает конденсированную циклическую структуру, образованную двумя или более циклическими структурами, содержащими два общих несоседних атома, принадлежащих к разным циклам. Спироциклической структурой называется конденсированная циклическая структура, образованная двумя или более циклическими структурами, содержащими один атом, общий для разных циклов.
Если не указано иное, употребляемый в настоящей работе термин "3-12-членный циклоалкенил" включает все моноциклические и конденсированные кольца (включая конденсированные кольца в виде орто-конденсированных колец, спироциклических соединений и мостиковых циклических соединений), которые могут быть образованы, такие как 3-8-членный моноциклоалкенил, 7-11-членный спироциклоалкенил, 7-11-членный орто-конденсированный циклоалкенил, мостиковый 6-11-членный циклоалкенил и тому подобное.
Употребляемый в настоящей работе термин "циклоалкил" включает все моноциклические системы и конденсированные циклические системы (включая конденсированные циклы в виде орто-конденсированных циклов, спироциклических соединений и мостиковых циклических соединений), которые могут быть образованы. Например, "3-12-членный циклоалкил" может представлять собой моноциклическую, бициклическую или полициклическую циклоалкильную систему (также называемую системой конденсированных циклов). Если не указано иное, моноциклическая система представляет собой циклическую углеводородную группу, содержащую от 3 до 8 атомов углерода. Примеры 3-8-членных циклоалкилов включают, без ограничений, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и подобные группы. Конденсированный циклоалкил включает орто-конденсированный циклоалкил, мостиковый циклоалкил и спироциклоалкил. Орто-конденсированный циклоалкил может представлять собой 6-11-членный орто-конденсированный циклоалкил и 7-10-членный орто-конденсированный циклоалкил, и репрезентативные примеры таких циклов включают, без ограничений, бицикло[3,1,1]гептан, бицикло[2,2,1]гептан, бицикло[2,2,2]октан, бицикло[3,2,2]нонан, бицикло[3,3,1]нонан и бицикло[4,2,1]нонан. Спироциклоалкил может представлять собой 7-12-членный спироциклоалкил и 7-11-членный спироциклоалкил, и примеры таких систем включают, без ограничений:

Мостиковый циклоалкил может представлять собой 6-11-членный мостиковый циклоалкил и 7-10-членный мостиковый циклоалкил, и примеры таких систем включают, без ограничений: 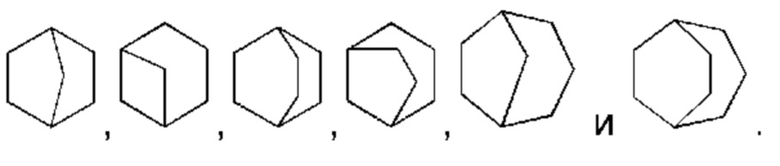
Употребляемый в настоящей работе термин "гетероциклил" означает 3-12-членную неароматическую циклическую группу, в которой по меньшей мере один атом углерода, находящийся в цикле, замещен гетероатом, выбранным из О, S и N, предпочтительно замещен 1-3 гетероатомами, причем атом углерода, атом азота и атом серы могут быть окислены.
Термин "3-12-членный гетероциклил" означает моноциклическую гетероциклильную, бициклическую гетероциклильную или полициклическую гетероциклильную систему (также называемую системой конденсированных циклов), включающую насыщенный и частично насыщенный гетероциклил, за исключением ароматических циклов. Если не указано иное, это определение включает все моноциклические кольца, конденсированные кольца (включая конденсированные кольца в виде орто-конденсированных колец, спироциклических систем мостиковых колец), насыщенные кольца и частично насыщенные кольца, которые могут быть образованы.
Моногетероциклил может представлять собой 3-8-членный гетероциклил, 3-8-членный насыщенный гетероциклил, 3-6-членный гетероциклил, 4-7-членный гетероциклил, 5-7-членный гетероциклил, 5-6-членный гетероциклил, 5-6-членный кислородсодержащий гетероциклил, 3-8-членный азотсодержащий гетероциклил, 5-6-членный азотсодержащий гетероциклил, 5-6-членный насыщенный гетероциклил и подобный радикал. Примеры "3-8-членного насыщенного гетероциклила" включают, без ограничений, азиридинил, оксиранил, тииранил, азетидинил, оксетанил, тиетанил, тетрагидрофуранил, пирролидинил, тетрагидротиофенил, имидазолидинил, пиразолидинил, 1,2-оксазолидинил, 1,3-оксазолидинил, 1,2-тиазолидинил, 1,3-тиазолидинил, тетрагидро-2Н-пиранил, тетрагидро-2Н-тиопиранил, пиперидинил, пиперазинил, морфолинил, 1,4-диоксанил, 1,4-оксатианил. Примеры "3-8-членного частично насыщенного гетероциклила" включают, без ограничений, 4,5-дигидроизооксазолил, 4,5-дигидрооксазолил, 2,5-дигидрооксазолил, 2,3-дигидрооксазолил, 3,4-дигидро-2Н-пирролил, 2,3-дигидро-1Н-пирролил, 2,5-дигидро-1Н-имидазолил, 4,5-дигидро-1Н-имидазолил, 4,5-дигидро-1Н-пиразолил, 4,5-дигидро-3Н-пиразолил, 4,5-дигидротиазолил, 2,5-дигидротиазолил, 2Н-пиранил, 4Н-пиранил, 2Н-тиопиранил, 4Н-тиопиранил, 2,3,4,5-тетрагидропиридил, 1,2-изооксазинил, 1,4-изооксазинил, или 6Н-1,3-оксазинил и подобные группы. Конденсированное гетероциклическое кольцо включает орто-конденсированный гетероциклил, спирогетероциклил, мостиковый гетероциклил и может быть насыщенным, частично насыщенным или ненасыщенным, но не ароматическим. Конденсированный гетероциклил представляет собой 5-6-членный моноциклический гетероциклический цикл, сконденсированный с бензольным кольцом, 5-6-членный моноциклический циклоалкил, 5-6-членный моноциклический циклоалкенил, 5-6-членный моноциклический гетероциклил или 5-6-членный моноциклический гетероарил. Орто-конденсированный гетероциклил может представлять собой 6-12-членный орто-конденсированный гетероциклил, 7-10-членный орто-конденсированный гетероциклил, 6-10-членный орто-конденсированный гетероциклил и 6-12-членный насыщенный орто-конденсированный гетероциклил, репрезентативные примеры которых включают, без ограничений, 3-азабицикло[3,1,0]гексил, 3,6-диазабицикло[3,2,0]гептил, 3,8-диазабицикло[4,2,0]октил, 3,7-диазабицикло[4,2,0]октил, октагидропирроло[3,4-с]пирролил, октагидропирроло[3,4-b]пирролил, октагидропирроло[3,4-b][1,4]оксазинил, октагидро-1Н-пирроло[3,4-с]пиридинил, 2,3-дигидробензофуран-2-ил, 2,3-дигидробензофуранил-3-ил, индолин-1-ил, индолин-2-ил, индолин-3-ил, 2,3-дигидробензотиофен-2-ил, октагидро-1Н-индолил, октагидробензофуранил. Спирогетероциклил может представлять собой 6-12-членный спирогетероциклил, 7-11-членный спирогетероциклил, 7-11-членный насыщенный спирогетероциклил, 6-12-членный насыщенный спироциклил, и примеры таких групп включают, без ограничений: 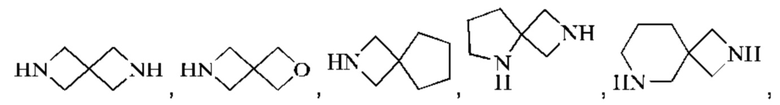
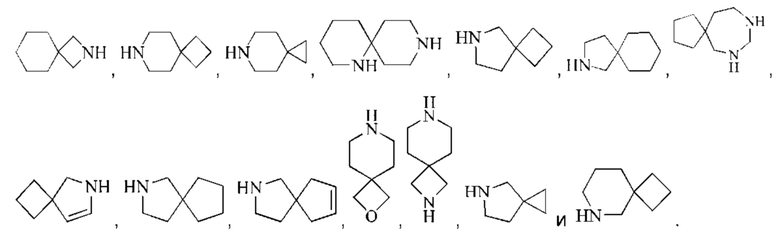
Мостиковый гетероциклил может представлять собой 6-12-членный мостиковый гетероциклил, 7-11-членный мостиковый гетероциклил и 6-12-членный насыщенный мостиковый гетероциклил, и примеры таких групп включают, без ограничений: 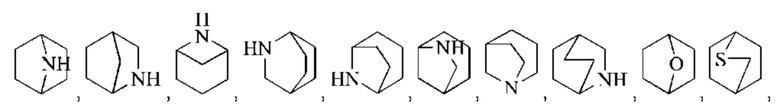
Употребляемый в настоящей работе термин "арил" означает циклическую ароматическую группу, содержащую от 6 до 14 атомов углерода, включающую фенил, нафталин, фенантрен и подобные группы.
Употребляемый в настоящей работе термин "гетероарил" включает все моноциклические циклы, конденсированные циклы и все ароматические и частично ароматические системы, которые могут быть образованы. Например, "5-10-членный гетероарил" означает ароматическую циклическую группу, в которой по меньшей мере один атом углерода, находящийся в цикле, замещен гетероатомом, выбранным из О, S и N, предпочтительно цикл содержит от 1 до 3 гетероатомов, и при этом атом углерода или атом серы может быть окислен, например, атом углерода замещен группой С(О), и атом серы замещен группой S(O) или S(O)2. Гетероарил включает моноциклический гетероарил и конденсированный гетероарил. Если не указано иное, моноциклический гетероарил может представлять собой 5-7-членный гетероарил или 5-6-членный гетероарил, и примеры моноциклических гетероарилов включают, без ограничений, фуранил, имидазолил, изоксазолил, тиазолил, изотиазолил, оксадиазолил, оксазолил, пиридил, пиридазинил, пиримидинил, пиразинил, пиразолил, пирролил, тетразолил, тиадиазолил, тиенил, триазолил и триазинил. В некоторых примерах осуществления, конденсированный гетероарил означает группу, в которой моноциклический гетероарил сконденсирован с фенилом, циклоалкенилом, гетероарилом, циклоалкилом или гетероциклилом. Конденсированный гетероарил может представлять собой 8-12-членный орто-конденсированный гетероарил или 9-10-членный орто-конденсированный гетероарил. Примеры конденсированных гетероарилов включают, без ограничений, бензимидазолил, бензофуранил, бензотиенил, бензооксадиазолил, бензотиадиазолил, бензотиазолил, циннолинил, 5,6-дигидрохинолин-2-ил, 5,6-дигидроизохинолин-1-ил, фуропиридинил, индазолил, индолил, изоиндолил, изохинолил, нафтиридинил, пуринил, хинолил, 5,6,7,8-тетрагидрохинол-2-ил, 5,6,7,8-тетрагидрохинолил, 5,6,7,8-тетрагидрохинол-4-ил, 5,6,7,8-тетрагидроизохинол-1-ил, тиенопиридинил,4,5,6,7-тетрагидро[с][1,2,5]оксадиазолил и 6,7-дигидро[с][1,2,5]оксадиазол-4(5Н)кето.
Употребляемый в настоящей работе термин "фармацевтически приемлемые соли" относится к фармацевтически приемлемым солям присоединения (аддитивным солям) и сольватам кислот и оснований. Фармацевтически приемлемые соли включают соли таких кислот, как соляная кислота, фосфорная кислота, бромоводородная кислота, серная кислота, сернистая кислота, муравьиная кислота, толуолсульфоновая кислота, метансульфоновая кислота, азотная кислота, бензойная кислота, лимонная кислота, винная кислота, малеиновая кислота, йодоводородная кислота, алкановая кислота (такая как уксусная кислота, НООС-(СН2)n-СООН (где n составляет от 0 до 4)), и подобных кислот. Соли оснований включают соли натрия, калия, кальция, аммония и подобных оснований. Специалистам в данной области техники известно множество различных нетоксичных фармацевтически приемлемых солей присоединения.
"Стереоизомер" соединения согласно настоящему изобретению, имеющего формулу (I), означает энантиомер в том случае, если соединение, имеющее формулу (I), содержит асимметрический атом углерода, цис-транс изомер в том случае, если соединение содержит углерод-углеродную двойную связь или циклическую структуру, таутомер в том случае, если в соединении присутствует группа кетона или оксима. В объем изобретения включены энантиомеры, диастереомеры, рацемические изомеры, цис-транс изомеры, таутомеры, геометрические изомеры, эпимеры соединения, имеющего формулу (I), и их смеси.
Употребляемый в настоящей работе термин "терапевтически эффективное количество" означает такое количество указанного выше соединения или его фармацевтически приемлемой соли, стереоизомера, композиции или фармацевтического состава, которое по меньшей мере может облегчать симптомы состояния пациента после введения его пациенту. Действительное количество, включающее "терапевтически эффективное количество", может быть различным в зависимости от ряда обстоятельств, которые включают, без ограничений, конкретное состояние, подвергаемое лечению, тяжесть состояния, физическое состояние и здоровье пациента, а также способ введения. Опытные медицинские работники могут с успехом определить подходящее количество, применяя способы известные в медицине.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Используемые в настоящей работе сокращения и обозначения означают следующее: "ДМФА" означает диметилформамид; "CDI" означает N,N'-карбонилдиимидазол (от англ. "N,N'-carbonyldiimidazole"); "DIPEA" означает N,N-диизопропилэтиламин (от англ. "N,N-diisopropylethylamine"); "ЭА" означает этилацетат; "ПЭ" означает петролейный эфир; "DIBAL-H" означает гидрид диизобутилалюминия (от англ. "diisobutylaluminum hydride"); "ТГФ" означает тетрагидрофуран; "ДХМ" означает дихлорметан; "TBAF" означает фторид тетрабутиламмония (от англ. "tetrabutylammonium fluoride"); "ДМАП" означает 4-диметиламинопиридин; "HATU" означает гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилмочевины (от англ. "2-(7-oxobenzotriazole)-N,N,N',N'-tetramethylurea hexafluorophosphate"); "AD-mix-β" означает смесь, содержащую 0,0016 моль (DHQD) 2PHAL (гидрированного простого хинидин-1,4-(2,3-нафтиридин)диэфира (англ. "hydrogenated quinidine 1,4-(2,3-naphthyridine)diether")), 0,4988 моль порошкообразного карбоната калия, 0,4988 моль феррицианида калия и 0,0007 моль дигидрата осмата калия; "ДМАА" означает диметилацетамид; "МТБЭ" означает простой метил-трет-бутиловый эфир; "Воc" означает трет-бутилоксикарбонил (от англ. "tert-butyloxycarbonyl"); "ТФУК" означает трифторуксусную кислоту; "Xphos" означает 2-дициклогексилфосфино-2,4,6-триизопропилбифенил (от англ. "2-dicyclohexylphosphino-2,4,6-triisopropylbiphenyl"); "DAST" означает трифторид диэтиламиносеры (от англ. "diethylaminosulfur trifluoride"); "LiHMDS" означает бис-триметилсилиламинлитий (от англ. "bistrimethylsilylamine lithium"); "TMSCF3" означает трифторметилтриметилсилан (от англ. "trifluoromethyl trimethylsilane").
Пример получения 1
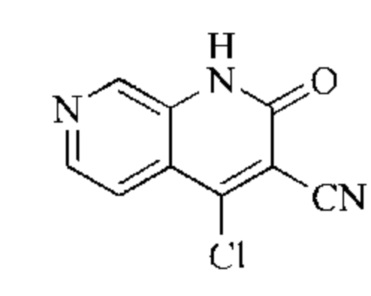
Синтез промежуточного 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
Этап 1: Синтез 2Н-пиридо[3,4-d][1,3]оксазин-2,4(1H)-диона
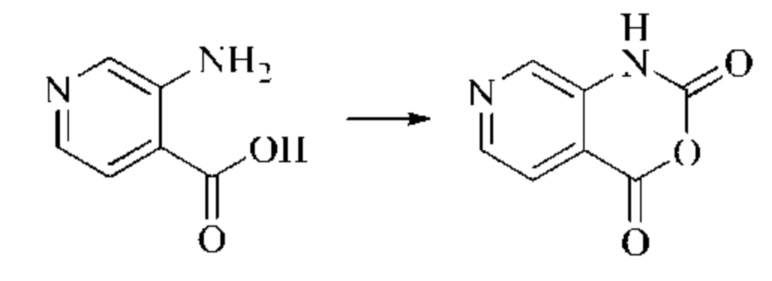
Исходное вещество, 3-аминоизоникотиновую кислоту (1000 мг, 7,240 ммоль, 1,0 экв.) растворяли в ДМФА (20 мл). Раствор охлаждали до 0°С, после чего порциями добавляли CDI (2000 мг, 12,334 ммоль, 1,7 экв.). Систему медленно нагревали до комнатной температуры, реакцию оставляли протекать в течение ночи, и завершение протекания реакции определяли способом ЖХ-МС (жидкостной хроматографии с масс-спектрометрией). Реакционный раствор охлаждали до комнатной температуры, и полученный реакционный раствор непосредственно использовали в следующем этапе без обработки.
Этап 2: Синтез 4-гидрокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
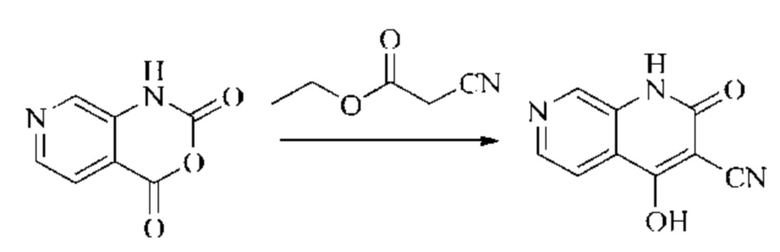
К реакционному раствору, содержащему промежуточный 2Н-пиридо[3,4-d][1,3]оксазин-2,4(1H)-дион (теоретически рассчитанное количество 1188 мг, 7,240 ммоль, 1 экв.), полученному в описанном выше этапе, добавляли этилцианоацетат (819 мг, 7,240 ммоль) и триэтиламин (1581 мг, 14,480 ммоль, 2 экв.). Смесь нагревали до 150°С, и оставляли реакцию протекать в течение приблизительно 4 часов, завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали до 50°С и концентрировали досуха при пониженном давлении, получая красное полутвердое вещество. Полутвердое вещество охлаждали до 10°С, после чего добавляли воду (5 мл) и перемешивали. Величину рН системы доводили до 1 добавлением соляной кислоты (1 моль/л). Смесь перемешивали в течение 15 минут и фильтровали с отсасыванием. Осадок на фильтре промывали водой, отсасывали и сушили при 40°С при пониженном давлении, получая 4-гидрокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (889 мг, выход 66%).
Этап 3: Синтез 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
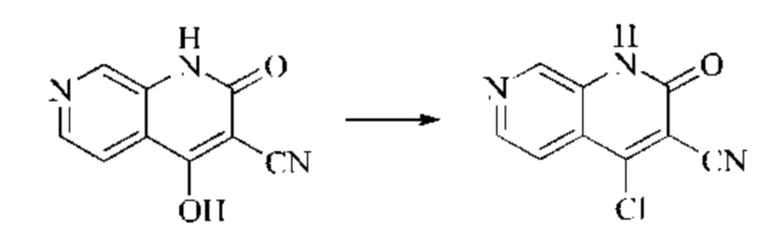
В реакционную колбу помещали 4-гидрокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (789 мг, 4,216 ммоль, 1 экв.), оксихлорид фосфора (2908 мг, 18,970 ммоль, 4,5 экв.) и пентахлорид фосфора (1756 мг, 8,431 ммоль, 2 экв.). Систему нагревали до 100°С, и реакцию проводили в течение 1 часа; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали до комнатной температуры, осторожно добавляли по каплям в лед до осаждения большого количества твердого вещества, и затем фильтровали с отсасыванием. Осадок на фильтре промывали водой и сушили при пониженном давлении при 40°С, получая 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (515 мг, выход: 60,0%).
Пример получения 2
Синтез промежуточного 4,6-дихлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
Этап 1: Синтез 6-хлор-2H-пиридо[3,4-d][1,3]оксазин-2,4(1H)-диона
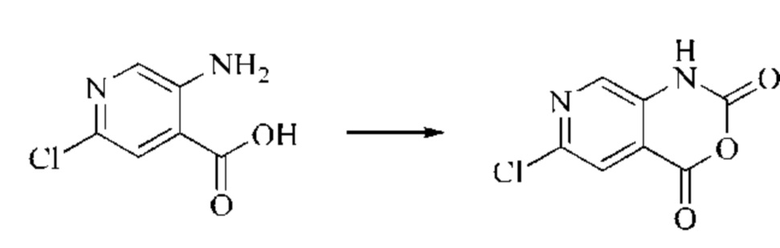
5-амино-2-хлоризоникотиновую кислоту (30 г, 0,1738 моль, 1,0 экв.) растворяли в N,N-диметилформамиде (300 мл) и затем порциями при 0°С добавляли N,N'-карбонилдиимидазол (48 г, 0,2955 моль, 1,7 экв.). Систему медленно нагревали до комнатной температуры, и реакцию оставляли протекать в течение ночи; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали до комнатной температуры и вводили непосредственно в следующий этап без обработки.
Этап 2: Синтез 6-хлор-4-гидрокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
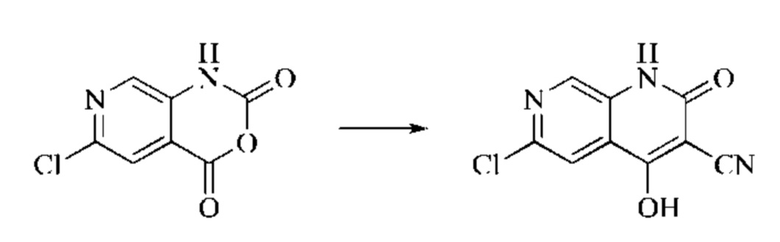
Триэтиламин (35,182 г, 0,3478 моль, 2 экв.) и этилцианоацетат (19,665 г, 0,1738 моль) добавляли в полученный как указано выше реакционный раствор. Реакцию в системе оставляли протекать при 150°С в течение 3 часов; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Добавляли воду (200 мл), и рН системы доводили до 1, добавляя соляную кислоту (1 моль/л). Смесь перемешивали в течение 15 минут и фильтровали с отсасыванием. Осадок на фильтре дважды промывали ЭА и сушили при 40°С, получая светлое, кирпично-красное твердое вещество (25,655 г, выход: 66%).
Этап 3: Синтез 4,6-дихлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
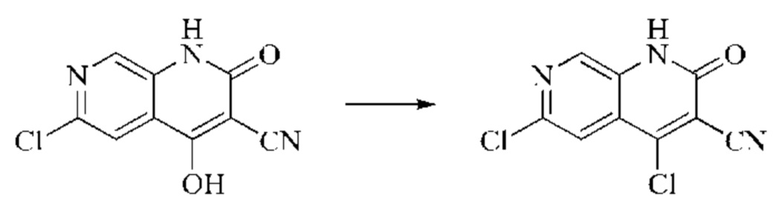
В реакционную колбу помещали 6-хлор-4-гидрокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (5,0 г, 0,0226 моль, 1 экв.) и оксихлорид фосфора (15 мл). Реакционную колбу помещали в масляную баню, которую нагревали до 100°С, и оставляли реагировать в течение 6 минут. Твердое вещество начинало медленно растворяться, и бледно-желтый цвет начал постепенно темнеть. Завершение протекания реакции определяли способом ЖХ-МС. Систему охлаждали до комнатной температуры, и в колбу добавляли подходящее количество ДХМ. Затем систему выливали в ледяную воду (100 мл) и перемешивали в течение 10 минут. Смесь фильтровали с отсасыванием, осадок на фильтре промывали простым метил-трет-бутиловым эфиром, отсасывали и сушили в вакууме при 40°С, получая бледно-желтое твердое вещество. Всего в реакцию пятью порциями вводили 25,655 г (0,1157 моль) 6-хлор-4-гидрокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила, получая 19,486 г продукта (выход: 70,1%).
Пример 1
Синтез 4-(азепан-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 3)
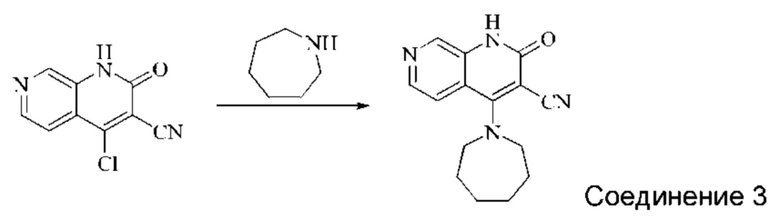
Исходное вещество, 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (19 мг, 0,092 ммоль, 1,0 экв.) растворяли в ДМФА (0,7 мл), после чего добавляли азепан (17,4 мг, 0,204 ммоль, 1,4 экв.) и DIPEA (48 мг, 0,370 ммоль, 4 экв.). Смесь нагревали до 100°С, и реакцию оставляли протекать в течение 4 часов; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали до комнатной температуры, в результате чего выпадал твердый осадок, который фильтровали с отсасыванием. Осадок на фильтре промывали водой (20 мл), отсасывали и сушили в вакууме, получая 4-(азепан-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (10,02 мг, 27,0%).
1Н ЯМР (400 МГц, ДМСО-d6 (ДМСО - сокр. от "диметилсульфоксид")) δ (м.д.): 11,96 (s, 1Н), 8,64 (s, 1Н), 8,31-8,32 (d, 1Н), 7,75-7,77 (d, 1Н), 3,80-3,83 (t, 4Н), 1,84 (s, 4Н), 1,71 (s, 4Н).
Молекулярная формула: C15H16N4O, Молекулярная масса: 268,32, ЖХ-МС (Pos, m/z)=269,2 [М+Н+].
Пример 2
Синтез 2-оксо-4-(пиперидин-1-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 4)
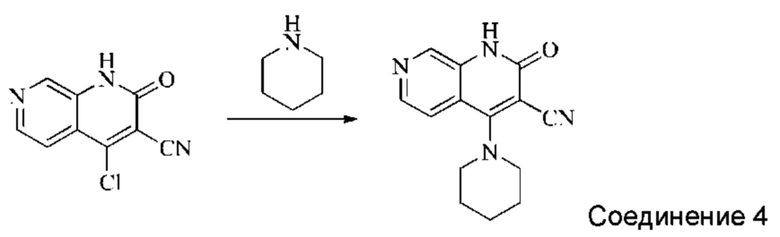
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (30 мг, 0,146 ммоль, 1,0 экв.) растворяли в ДМФА (1 мл), после чего добавляли пиперидин (17,4 мг, 0,204 ммоль, 1,4 экв.) и DIPEA (75,4 мг, 0,584 ммоль, 4 экв.). Систему нагревали до 100°С и оставляли реагировать в течение 1,5 часов; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали и фильтровали с отсасыванием. Осадок на фильтре промывали водой и сушили в вакууме, получая желтый твердый продукт (10,02 мг, выход: 27,0%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,99 (s, 1Н), 8,64 (s, 1Н), 8,33-8,35 (d, 1Н), 7,58-7,59 (d, 1Н), 3,60 (s, 4Н), 1,72-1,76 (m, 6Н).
Молекулярная формула: C14H14N4O, Молекулярная масса: 254,29, ЖХ-МС (Pos, m/z)=255,1 [М+Н+].
Пример 3
Синтез 4-(бензиламино)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 10)
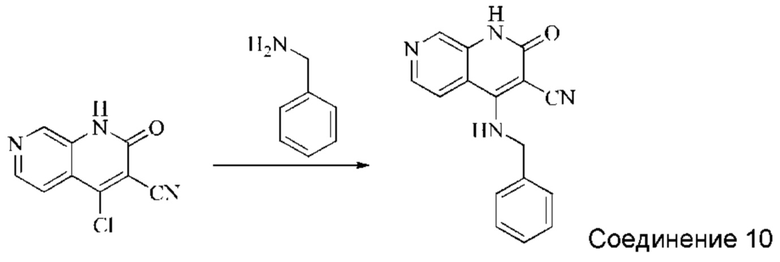
Исходное вещество, 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (30 мг, 0,146 ммоль, 1,0 экв.) растворяли в ДМФА (0,7 мл), после чего добавляли бензиламин (22 мг, 0,204 ммоль, 1,4 экв.) и DIPEA (75,4 мг, 0,584 ммоль, 4 экв.). Систему нагревали до 100°С и оставляли реагировать в течение 1,5 часов; завершение протекания реакции определяли способом ЖХ-МС. К реакционному раствору добавляли простой метил-трет-бутиловый эфир (2 мл) и затем добавляли воду (2 мл). Реакционный раствор перемешивали до тех пор, пока не выпадал твердый осадок, который фильтровали с отсасыванием. Осадок на фильтре сушили в вакууме, получая желтое твердое вещество, 4-(бензиламино)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (9 мг, выход: 22,3%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,62 (s, 1Н), 8,79-8,82 (t, 1Н), 8,60 (s, 1Н), 8,38-8,40 (d, 1Н), 8,10-8,11 (d, 1Н), 7,26-7,38 (m, 5Н), 5,05-5,06 (d, 2Н).
Молекулярная формула: C16H12N4O, Молекулярная масса: 276,30, ЖХ-МС (Pos, m/z)=277,02 [М+Н+].
Пример 4
Синтез 4-((4-хлорбензил)амино)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 11)
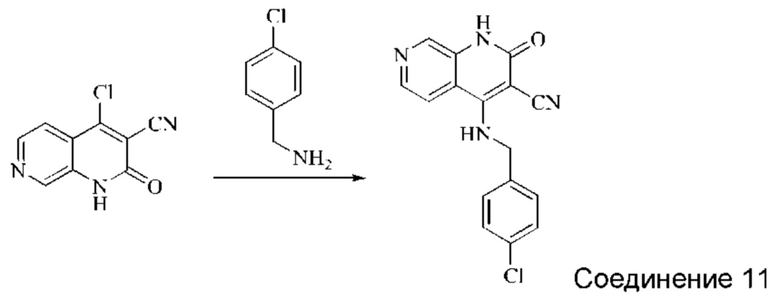
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (30 мг, 0,146 ммоль, 1,0 экв.) растворяли в ДМФА (0,7 мл), после чего добавляли (4-хлорфенил)метиламин (29 мг, 0,204 ммоль, 1,4 экв.) и DIPEA (75,4 мг, 0,584 ммоль, 4 экв.). Систему нагревали до 100°С и оставляли реагировать в течение 1,5 часов; завершение протекания реакции определяли способом ЖХ-МС. После добавления МТБЭ (2 мл), смесь перемешивали и затем добавляли воду (2 мл), перемешивали в течение 15 минут, в результате чего выпадал твердый осадок, который фильтровали с отсасыванием. Осадок на фильтре промывали водой и сушили в вакууме, получая желтый твердый продукт (19 мг, выход: 41,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,56 (s, 1Н), 8,80 (s, 1Н), 8,60 (s, 1Н), 8,38-8,40 (d, 1Н), 8,08-8,10 (d, 1Н), 7,34-7,44 (t, 4Н), 5,02 (s, 2Н).
Молекулярная формула: C16H11ClN4O, Молекулярная масса: 310,74 ЖХ-МС (Pos, m/z)=309,1 [М+Н+].
Пример 5
Синтез 2-оксо-4-(2-аза-спиро[3,5]нонан-2-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 13)
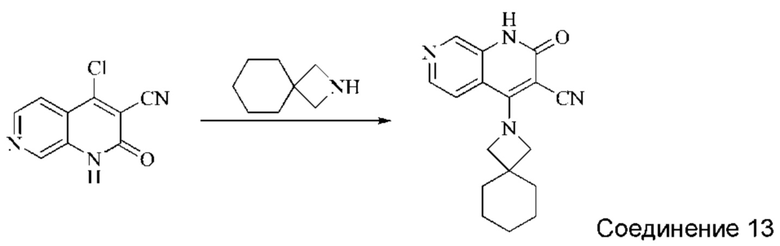
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,234 ммоль, 1,0 экв.) растворяли в ДМФА (1 мл), после чего добавляли 2-аза-спиро[3,5]нонан (43 мг, 0,340 ммоль, 1,4 экв.) и DIPEA (188 мг, 1,458 ммоль, 4 экв.). Систему нагревали до 80°С и оставляли реагировать в течение 2 часов; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали и фильтровали с отсасыванием. Осадок на фильтре промывали водой и сушили в вакууме, получая беловатый твердый продукт (27,42 мг, выход: 39,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,49 (s, 1Н), 8,58 (s, 1Н), 8,24-8,25 (d, 1Н), 7,74-7,76 (d, 1Н), 4,46 (s, 1Н), 1,69 (s, 4Н), 1,36-1,43 (d, 6Н).
Молекулярная формула: C17H18N4O, Молекулярная масса: 294,36 ЖХ-МС (Pos, m/z)=295,1 [М+Н+].
Пример 6
Синтез 2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 16)
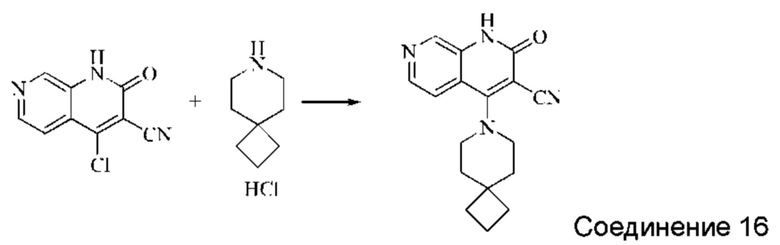
Исходное вещество, 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,234 ммоль, 1,0 экв.) растворяли в ДМФА (1 мл), после чего добавляли гидрохлорид 7-аза-спиро[3,5]нонана (55 мг, 0,340 ммоль, 1,4 экв.) и DIPEA (188 мг, 1,458 ммоль, 6 экв.). Систему нагревали до 80°С и оставляли реагировать в течение 2 часов; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали и фильтровали с отсасыванием. Осадок на фильтре промывали водой, отсасывали, и сушили при пониженном давлении, получая бледно-желтое твердое вещество, 2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (23,24 мг, 33,7%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,98 (s, 1Н), 8,64 (s, 1Н), 8,32-8,34 (d, 1Н), 7,59-7,60 (d, 1Н), 3,53 (s, 4Н), 3,41-3,47 (m, 4Н), 1,79-1,92 (m, 6Н).
Молекулярная формула: C17H18N4O, Молекулярная масса: 294,36 ЖХ-МС (Pos, m/z)=295,1 [М+Н+].
Пример 7
Синтез 4-((4-хлорфенил)амино)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 18)
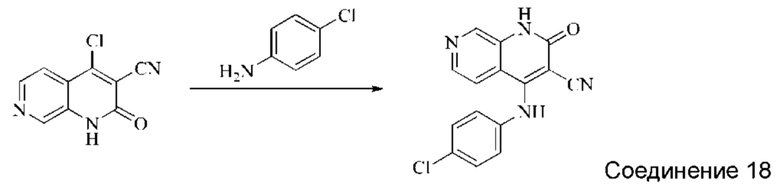
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (200 мг, 0,973 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), после чего добавляли пара-хлоранилин (174 мг, 1,362 ммоль, 1,4 экв.) и DIPEA (752 мг, 3,893 ммоль, 4 экв.). Систему нагревали до 80°С и оставляли реагировать в течение 1,5 часов. Реакционный раствор охлаждали, после чего добавляли воду (10 мл) и затем экстрагировали ЭА. Органическую фазу сушили безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт растворяли, добавляя небольшое количество ДХМ и метанольного раствора, и затем добавляли небольшое количество ЭА до образования твердого осадка, который фильтровали с отсасыванием. Осадок на фильтре промывали небольшим количеством ДХМ и сушили, получая желтый твердый продукт (44 мг, выход: 50,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,84 (s, 1Н), 9,94 (s, 1Н), 8,66 (s, 1Н), 8,41-8,42 (d, 1Н), 8,10-8,12 (d, 1Н), 7,47-7,49 (d, 2Н), 7,35-7,37 (d, 2Н).
Молекулярная формула: C15H9ClN4O, Молекулярная масса: 296,71, ЖХ-МС (Pos, m/z)=296,96 [М+Н+].
Пример 8
Синтез (R)-4-(3-(1Н-пиразол-1-ил)пирролидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 19)
Этап 1: Синтез трет-бутил-(S)-3-гидроксипирролидин-1-карбоксилата
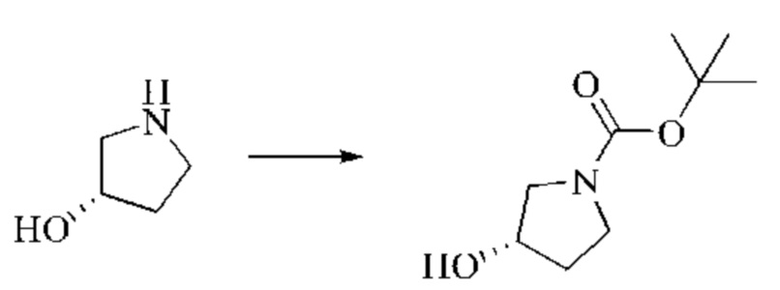
(S)-пирролидин-3-ол (3,0 г, 24,28 ммоль, 1,0 экв.) растворяли в ТГФ (54 мл), охлаждали до 0°С и затем добавляли по каплям триэтиламин (12,28 г, 121,38 ммоль, 5 экв.). В смесь медленно, при перемешивании добавляли по каплям (Вос)2O (5,83 г, 26,70 ммоль, 1,1 экв.). Систему медленно нагревали до комнатной температуры и оставляли реагировать в течение 2 суток. Реакционный раствор концентрировали при пониженном давлении, после чего добавляли воду, перемешивали и экстрагировали ДХМ три раза. Органические фазы объединяли, сушили безводным сульфатом натрия и концентрировали при пониженном давлении, получая желтое масло, трет-бутил-(S)-3-гидроксипирролидин-1-карбоксилат (4,42 г, выход: 97,2%).
Этап 2: Синтез трет-бутил-(S)-3-((метилсульфонил)окси)пирролидин-1-карбоксилата
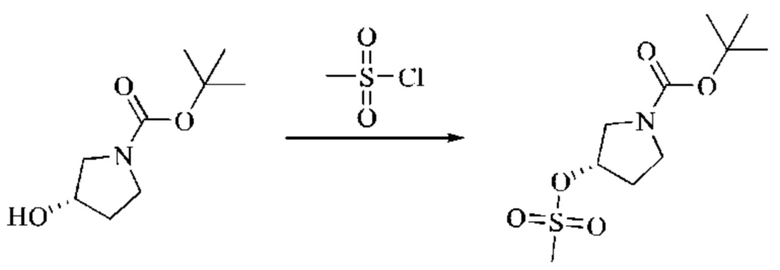
Трет-бутил-(S)-3-гидроксипирролидин-1-карбоксилат (3,0 г, 16,02 ммоль, 1 экв.) помещали в реакционную колбу и растворяли, добавляя ТГФ (30 мл). Систему охлаждали до 0°С и затем добавляли триэтиламин (3,24 г, 32,04 ммоль, 2 экв.). В смесь медленно добавляли по каплям метансульфонилхлорид (2,2 г, 19,23 ммоль, 1,2 экв.). Смесь медленно нагревали до комнатной температуры и оставляли реагировать в течение 3 часов; завершение протекания реакции определяли способом ТСХ (тонкослойной хроматографии). Реакционный раствор фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении, получая бледно-желтое масло, трет-бутил-(S)-3-((метилсульфонил)окси)пирролидин-1-карбоксилат, который вводили непосредственно в следующий реакционный этап без очистки.
Этап 3: Синтез трет-бутил-(R)-3-(1H-пиразол-1-ил)пирролидин-1-карбоксилата
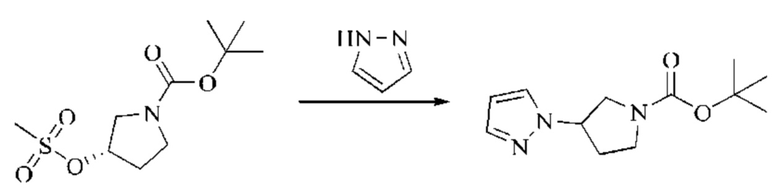
Пиразол (1,2 г, 17,62 ммоль, 1,1 экв.) растворяли в ДМАА (72 мл), охлаждали до 0°С, после чего добавляли гидрид натрия (2051 мг, 51,27 ммоль, 3,2 экв., содержание 60%), и затем реакцию оставляли протекать в течение 1 часа в атмосфере азота. Трет-бутил-(S)-3-((метилсульфонил)окси)пирролидин-1-карбоксилат, полученный в описанном выше этапе, растворяли в небольшом количестве ДМАА, и полученный раствор медленно добавляли к реакционному раствору по каплям, нагревали до 100°С и оставляли реагировать в течение ночи в атмосфере азота. Реакционный раствор охлаждали до комнатной температуры, разбавляли добавлением воды, перемешивали и экстрагировали ЭА. После разделения жидкостей органические фазы объединяли, промывали водой, сушили безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА=20:1), получая бесцветное масло, трет-бутил-(R)-3-(1H-пиразол-1-ил)пирролидин-1-карбоксилат (1,83 г, выход в результате двух этапов синтеза: 48,2%).
Этап 4: Синтез (R)-1-(пирролидин-3-ил)-1H-пиразола
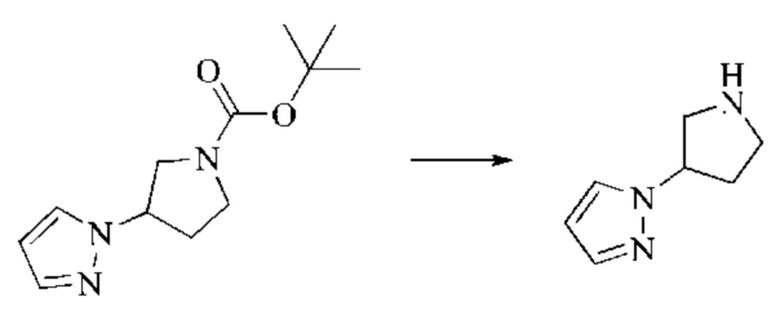
Трет-бутил-(R)-3-(1H-пиразол-1-ил)пирролидин-1-карбоксилат (200 мг, 0,8428 ммоль, 1,0 экв.) растворяли в ДХМ (4 мл), охлаждали до 0°С, затем добавляли по каплям трифторуксусную кислоту (2 мл), после чего реакцию оставляли протекать в течение 2-3 часов; завершение протекания реакции определяли способом ТСХ. Затем реакционную систему концентрировали при пониженном давлении, и подщелачивали добавлением подходящего количества DIPEA, получая неочищенный продукт, (R)-1-(пирролидин-3-ил)-1H-пиразол, который непосредственно использовали в следующем этапе.
Этап 5: Синтез (R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
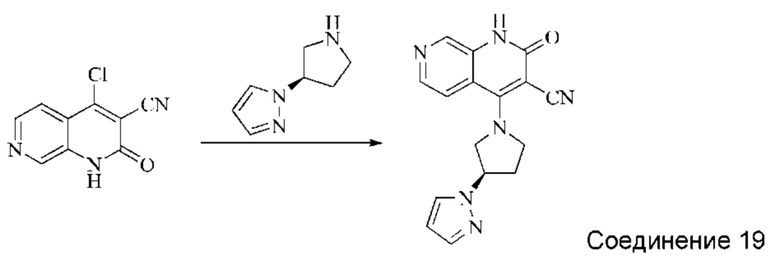
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (124 мг, 0,602 ммоль, 1,0 экв.) растворяли в ДМАА (2 мл), после чего добавляли неочищенный продукт, (R)-1-(пирролидин-3-ил)-1H-пиразол, полученный в описанном выше этапе, и DIPEA (467 мг, 3,612 ммоль, 6 экв.). Систему нагревали до 80°С и оставляли реагировать в течение 1,5 часов; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор разбавляли добавлением воды (10 мл) и экстрагировали ЭА (50 мл×3). Органические фазы объединяли, промывали водой (30 мл), сушили безводным Na2SO4 и концентрировали при пониженном давлении, получая коричневато-желтое твердое вещество. Твердое вещество суспендировали, добавляя небольшое количество ДХМ и ЭА, и фильтровали с отсасыванием. Осадок на фильтре промывали небольшим количеством ДХМ и сушили в вакууме, получая коричневато-желтое твердое вещество, (R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (35,96 мг, выход в результате двух этапов синтеза: 13,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,64 (s, 1Н), 8,61 (s, 1Н), 8,26-8,28 (d, 1Н), 7,97-7,98 (d, 1Н), 7,89-7,90 (d, 1Н), 7,50-7,51 (d, 1Н), 6,30 (s, 1Н), 5,14-5,20 (m, 1Н), 4,51-4,56 (q, 1Н), 4,32-4,36 (q, 1Н), 4,17-4,30 (m, 2Н), 2,43-2,49 (m, 2Н).
Молекулярная формула: C16H14N6O, Молекулярная масса: 306,33, ЖХ-МС (Pos, m/z)=307,0 [М+Н+].
Пример 9
Синтез 2-оксо-4-(фениламино)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 21)
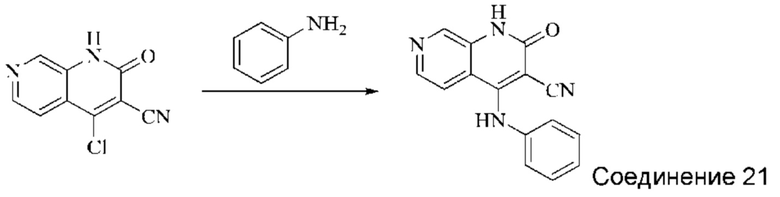
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50,0 мг, 0,243 ммоль, 1,0 экв.) и анилин (31,7 мг, 0,340 ммоль, 1,4 экв.) растворяли в N,N-диметилформамиде (1,0 мл), добавляли N,N-диизопропилэтиламин (125,8 мг, 0,973 ммоль, 4,0 экв.) и нагревали с обратным холодильником до 80°С в течение 2 часов. Завершение протекания реакции определяли способом ТСХ. Систему охлаждали до комнатной температуры и фильтровали с отсасыванием. Осадок на фильтре промывали водой (4,0 мл) в течение 30 минут, фильтровали с отсасыванием и сушили при 60°С, получая желтый твердый продукт (10,0 мг, выход: 20%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,80 (s, 1Н), 9,93 (s, 1Н), 8,67 (s, 1Н), 8,42-8,41 (m, 1Н), 8,15-8,14 (m, 1Н), 7,46-7,42 (m, 2Н), 7,35-7,29 (m, 3Н).
Молекулярная формула: C15H10N4O, Молекулярная масса: 262,27, ЖХ-МС (Pos, m/z)=263,59 [М+Н+].
Пример 10
Синтез (R)-4-(3-(1Н-пиразол-1-ил)пирролидин-1-ил)-6-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединения 23)
Этап 1: Синтез гидрохлорида (R)-1-(пирролидин-3-ил)-1H-пиразола
Трет-бутил-(R)-3-(1Н-пиразол-1-ил)пирролидин-1-карбоксилат (13,8 мг, 0,0583 ммоль, 1,0 экв.) растворяли в этаноле (1,0 мл) и затем при 0°С добавляли раствор хлорида водорода в этаноле (25%, 1,0 мл). Систему медленно нагревали до комнатной температуры и перемешивали в течение 1 часа; завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали, и неочищенный продукт вводили в следующий этап.
Этап 2: Синтез (R)-4-(3-(1Н-пиразол-1-ил)пирролидин-1-ил)-6-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
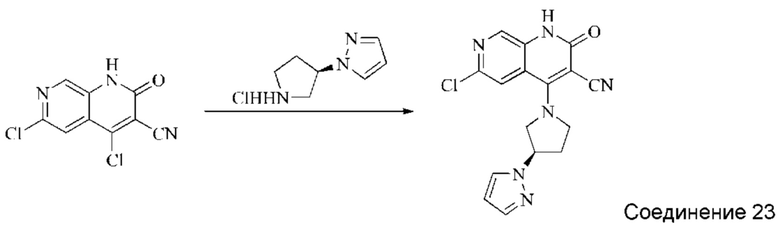
Полученный как указано выше неочищенный продукт растворяли в N,N-диметилформамиде (1,0 мл), после чего добавляли N,N-диизопропилэтиламин (32,3 мг, 0,250 ммоль, 6,0 экв.) и 4,6-дихлор-2 -оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (10,0 мг, 0,0416 ммоль, 1,0 экв.). Смесь нагревали с обратным холодильником до 80°С в течение 2 часов; завершение протекания реакции определяли способом ТСХ. Реакционную смесь концентрировали при пониженном давлении. Неочищенный продукт последовательно очищали тонкослойной хроматографией (ДХМ : МеОН = 20:1) и колоночной хроматографией на силикагеле (ДХМ : МеОН = 100:1-40:1), получая желтый твердый продукт (6,49 мг, выход: 64,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,77 (s, 1Н), 8,41 (s, 1Н), 8,00 (m, 1Н), 7,90-7,89 (m, 1Н), 7,52-7,51 (m, 1Н), 6,31-6,30 (m, 1Н), 5,18-5,15 (m, 1Н), 4,54-4,50 (m, 1Н), 4,36-4,16 (m, 3Н), 2,01-1,99 (m, 1Н), 0,88 (m, 1Н).
Молекулярная формула: C16H13N6O, Молекулярная масса: 340,77, ЖХ-МС (Pos, m/z)=341,0 [М+Н+].
Пример 11
Синтез 2-оксо-4-(2-оха-7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 24)
Этап 1: Синтез 2-оксо-4-(2-оха-7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
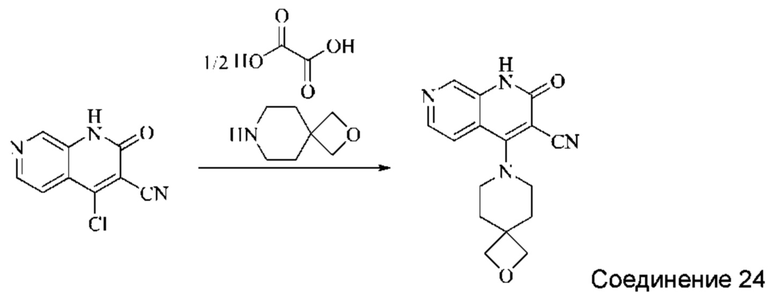
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50,0 мг, 0,243 ммоль, 1,0 экв.) и полуоксалат 2-окса-7-аза-спиро[3,5]нонана (58,6 мг, 0,170 ммоль, 0,7 экв.) растворяли в N,N-диметилформамиде (1,0 мл) и затем добавляли N,N-диизопропилэтиламин (188,4 мг, 1,458 ммоль, 6,0 экв.). Систему нагревали до 80°С с обратным холодильником в течение 2 часов, и завершение протекания реакции определяли способом ТСХ. Систему охлаждали до комнатной температуры, и фильтровали с отсасыванием. Осадок на фильтре промывали водой (4,0 мл) в течение 30 минут, фильтровали с отсасыванием и сушили при 60°С, получая желтый твердый продукт (10,0 мг, выход: 20%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,02 (s, 1Н), 8,66 (s, 1Н), 8,35-8,34 (m, 1Н), 7,59-7,58 (m, 1Н), 4,42 (s, 4Н), 3,56-3,54 (m, 4Н), 2,07-2,05 (m, 4Н).
Молекулярная формула: C16H16N4O2, Молекулярная масса: 296,33, ЖХ-МС (Pos, m/z)=297,61 [М+Н+].
Пример 12
Синтез 2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединения 25)
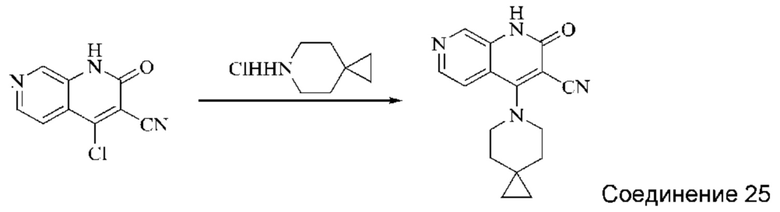
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50,0 мг, 0,243 ммоль, 1,0 экв.) и 6-аза-спиро[2,5]октан гидрохлорид (50,2 мг, 0,340 ммоль, 1,4 экв.) растворяли в N,N-диметилформамиде (1,0 мл) и добавляли N,N-диизопропилэтиламин (188,4 мг, 1,458 ммоль, 6,0 экв.). Систему нагревали до 80°С с обратным холодильником в течение 2 часов; завершение протекания реакции определяли способом ТСХ. Реакционную смесь охлаждали до комнатной температуры и фильтровали с отсасыванием. Осадок на фильтре промывали водой (4,0 мл) в течение 30 минут, фильтровали с отсасыванием и сушили при 60°С, получая желтый твердый продукт (18,0 мг, выход: 36%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,02 (s, 1Н), 8,66 (s, 1Н), 8,36-8,34 (m, 1Н), 7,65-7,64 (m, 1Н), 3,66-3,63 (m, 4Н), 1,62 (m, 4Н), 0,44 (s, 4Н).
Молекулярная формула: C16H16N4O, Молекулярная масса: 280,33, ЖХ-МС (Pos, m/z)=281,1 [М+Н+].
Пример 13
Синтез 4-(4-этилфенил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединения 26)
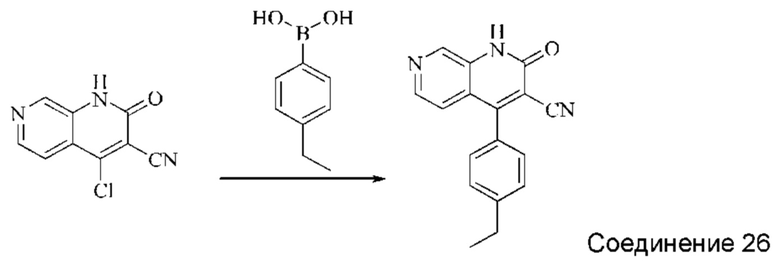
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (300,0 мг, 1,459 ммоль, 1,0 экв.), (4-этилфенил)борную кислоту (262,6 мг, 1,751 ммоль, 1,2 экв.), фосфат калия (619,4 мг, 2,918 ммоль, 2,0 экв.) и 1,1'-бис-дифенилфосфиноферроценпалладия дихлорид (47,5 мг, 0,0729 ммоль, 0,05 экв.) растворяли в диоксане (9,0 мл) и воде (4,5 мл). Систему нагревали до 115°С с обратным холодильником в течение ночи в атмосфере азота, и завершение протекания реакции определяли способом ТСХ. Реакционную смесь фильтровали с отсасыванием, и фильтрат экстрагировали этилацетатом (10,0 мл×3), сушили безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 200:1-40:1), получая желтый твердый продукт (100,0 мг, выход: 33,3%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,83 (s, 1Н), 8,80 (s, 1Н), 8,38-8,36 (m, 1Н), 7,49 (m, 1Н), 7,16-7,15 (m, 1Н), 2,77-2,75 (m, 2Н), 1,30-1,26 (m, 3Н).
Молекулярная формула: C17H13N3O, Молекулярная масса: 275,31, ЖХ-МС (Pos, m/z)=276,1 [М+Н+].
Пример 14
Синтез 4-(3-(1Н-пиразол-1-ил)пирролидин-1-ил)-6-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединения 27)
Этап 1: Синтез 4-(3-(1Н-пиразол-1-ил)пирролидин-1-ил)-6-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
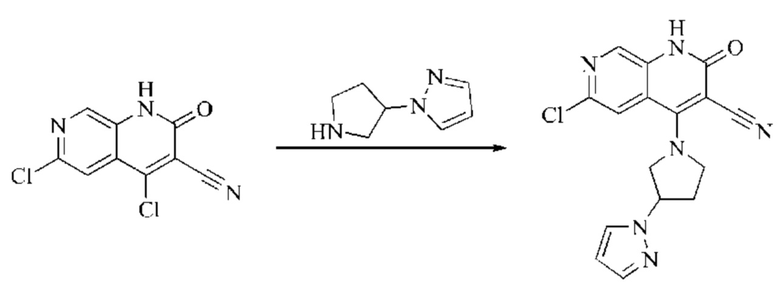
4,6-дихлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (180 мг, 0,75 ммоль, 1,0 экв.) и 1-(пирролидин-3-ил)-1Н-пиразол (303 мг, 1,28 ммоль, 1,7 экв., 57,6%) растворяли в ДМФА (5 мл) и затем добавляли DIPEA (387 мг, 3,0 ммоль, 4,0 экв.). Систему перемешивали при 80°С в течение 2 часов и охлаждали. Реакционный раствор очищали колоночной хроматографией с обращенной фазой (ацетонитрил : вода : раствор аммиака = 30:100:0,05%), получая желтый твердый продукт (200 мг, выход: 78,4%).
Этап 2: Синтез 4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-6-метил-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
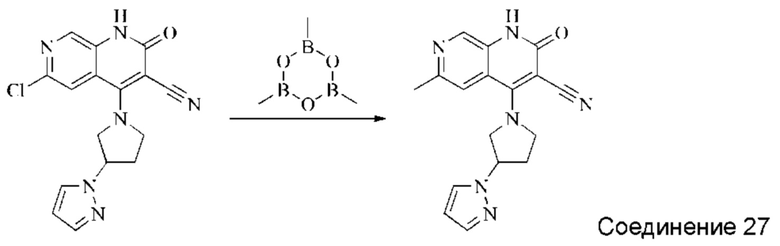
4-(3-(1Н-пиразол-1-ил)пирролидин-1-ил)-6-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (112,0 мг, 0,33 ммоль, 1,0 экв.), триметилциклотрибороксан (330,0 мг, 1,30 ммоль, 4,0 экв., 50%), карбонат цезия (322,0 мг, 0,99 ммоль, 3,0 экв.), фосфат калия (70,0 мг, 0,33 ммоль, 1,0 экв.) и Pd(dppf)2Cl2 (120 мг, 0,16 ммоль, 0,5 экв.) растворяли в 1,4-диоксане (4 мл). Систему нагревали до 105°С и перемешивали в течение ночи в атмосфере азота. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор тушили (т.е. быстро останавливали реакцию) добавлением H2O (10 мл), перемешивали в течение 30 минут, охлаждали до комнатной температуры и фильтровали с отсасыванием. Осадок на фильтре промывали небольшим количеством этилацетата. Фильтрат экстрагировали этилацетатом (10,0 мл×3). После разделения жидкостей органическую фазу промывали насыщенным солевым раствором (10,0 мл×2), сушили безводным Na2SO4, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 80:1-60:1), получая желтый твердый продукт (55,0 мг, выход: 52,1%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,04 (s, 1Н), 8,69 (s, 1Н), 7,57 (m, 3Н), 6,34 (s, 1Н), 5,11 (s, 1Н), 4,49-4,64 (m, 3Н), 4,49 (s, 1Н), 2,60-2,67 (m, 5Н).
Молекулярная формула: C17H16N6O, Молекулярная масса: 320,14, ЖХ-МС (Neg, m/z)=319,17 [М-Н]-.
Пример 15
Синтез 4-(4-(2-гидроксиэтил)пиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 28)
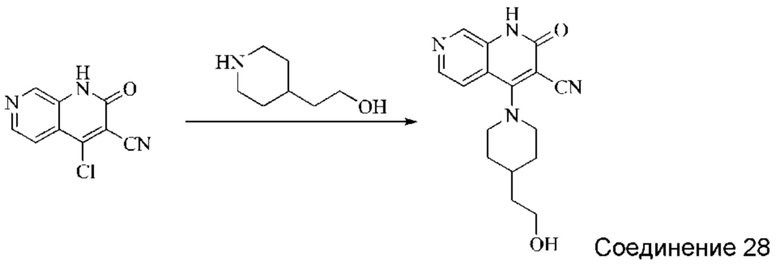
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (60 мг, 0,29 ммоль, 1,0 экв.) растворяли в N,N-диметилформамиде (1 мл). Затем добавляли N,N-диизопропилэтиламин (112 мг, 0,87 ммоль, 3,0 экв.) и 2-(пиперидин-4-ил)этан-1-ол (38 мг, 0,29 ммоль, 1,0 экв.). Систему нагревали до 80°С и оставляли реагировать в течение 2 часов, охлаждали до комнатной температуры, в результате чего выпадал твердый осадок, и затем фильтровали. Осадок на фильтре последовательно промывали тетрагидрофураном (1 мл) и петролейным эфиром (1 мл) и сушили при 45°С, получая желтый твердый продукт (16 мг, выход: 18%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,98 (s, 1Н), 8,65 (s, 1Н), 8,33-8,35 (d, 1Н), 7,58-7,80 (d, 1Н), 4,40-4,42 (m, 1Н), 3,83-3,86 (m, 2Н), 3,48-3,53 (m, 2Н), 3,37-3,43 (m, 2Н), 1,84-1,87 (m, 2Н), 1,74-1,80 (m, 1Н), 1,45-1,50 (m, 4Н).
Молекулярная формула: C16H18N4O2, Молекулярная масса: 298,35, ЖХ-МС (Neg, m/z)=297,15 [М-Н]-.
Пример 16
Синтез 6-(метилтио)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 30)
Этап 1: Синтез 2-бром-5-нитроизоникотиновой кислоты
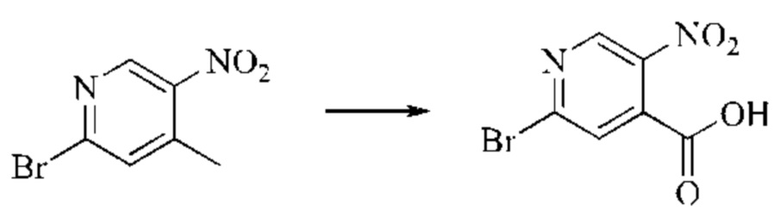
2-бром-4-метил-5-нитропиридин (2,5 г, 11,58 ммоль) растворяли в концентрированной серной кислоте (25 мл). Систему охлаждали до 0°С на ледяной бане, после чего добавляли триоксид хрома (3,88 г, 38,8 ммоль), затем медленно нагревали до комнатной температуры и перемешивали в течение ночи. Реакционный раствор выливали в ледяную воду (75 мл), перемешивали в течение 10 минут и фильтровали с отсасыванием, получая белый твердый продукт (2,5 г, выход: 87,8%).
Этап 2: Синтез 2-(метилтио)-5-нитроизоникотиновой кислоты
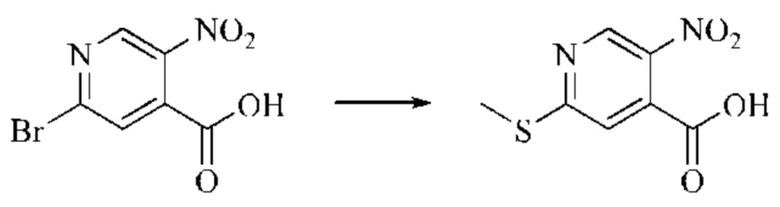
2-бром-5-нитроизоникотиновую кислоту (1,5 г, 6,1 ммоль) растворяли в ДМФА (30 мл), и систему охлаждали до 0°С на ледяной бане, после чего добавляли тиометилат натрия (1,07 г, 15,25 ммоль), затем медленно нагревали до комнатной температуры и перемешивали в течение ночи; завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор концентрировали при пониженном давлении, выливали в воду (8 мл), доводили до рН 2 добавлением соляной кислоты концентрацией 1 моль/л до образования твердого осадка и затем фильтровали с отсасыванием, получая красный твердый продукт (1,0 г неочищенного продукта).
Этап 3: Синтез 5-амино-2-(метилтио)изоникотиновой кислоты
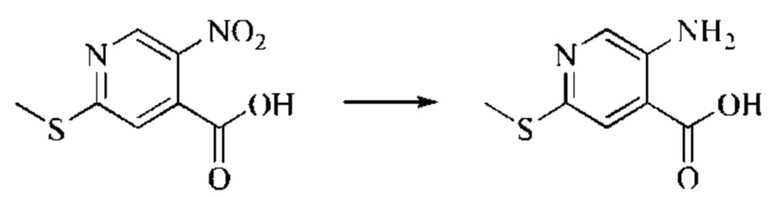
2-(метилтио)-5-нитроизоникотиновую кислоту (1,0 г неочищенного продукта) растворяли в этаноле (10 мл) и добавляли насыщенный водный раствор хлорида аммония (5 мл) и порошок железа (5,23 г, 93,5 ммоль). Систему нагревали до 70°С и оставляли реагировать в течение ночи; завершение протекания реакции определяли способом ЖХ-МС. Смесь фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией с обращенной фазой, получая продукт (400 мг, выход в результате двух этапов синтеза: 35,6%).
Этап 4: Синтез 6-(метилтио)-2H-пиридо[3,4-d][1,3]оксазин-2,4(1H)-диона
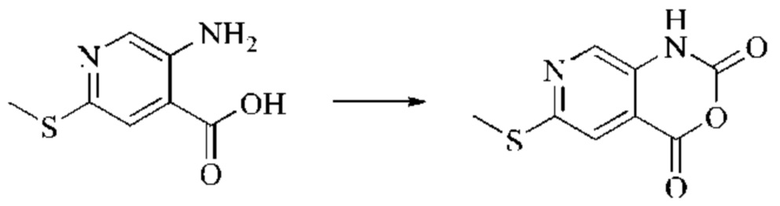
5-амино-2-(метилтио)изоникотиновую кислоту (300,0 мг, 1,63 ммоль) растворяли в ДМФА (3 мл) и охлаждали до 0°С на ледяной бане. Добавляли CDI (449,4 мг, 2,77 ммоль). Систему медленно нагревали до комнатной температуры и перемешивали в течение ночи; завершение протекания реакции определяли способом ТСХ. Продукт получали концентрированием при пониженном давлении и непосредственно вводили в следующий этап без очистки.
Этап 5: Синтез 4-гидрокси-6-метилтио-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
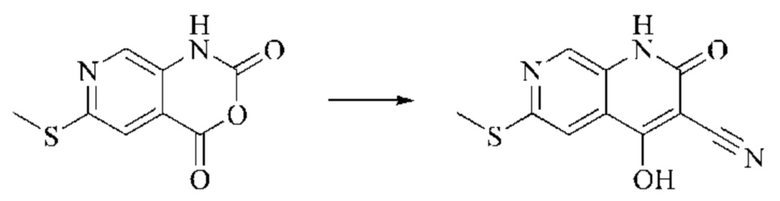
6-(метилтио)-2H-пиридо[3,4-d][1,3]оксазин-2,4(1H)-дион (300,0 мг неочищенного продукта) растворяли в ДМФА (3 мл). Добавляли этилцианоацетат (193,6 мг, 1,71 ммоль) и триэтиламин (329,9 мг, 3,26 ммоль), и систему перемешивали при 150°С в течение ночи; завершение протекания реакции определяли способом ТСХ. Смесь концентрировали при пониженном давлении, после чего добавляли воду (10 мл) и затем доводили до рН 1 добавлением соляной кислоты до образования твердого осадка, который фильтровали с отсасыванием, получая продукт (140,0 мг неочищенного продукта).
Этап 6: Синтез 2,4-дихлор-6-метилтио-1,7-нафтиридин-3-карбонитрила
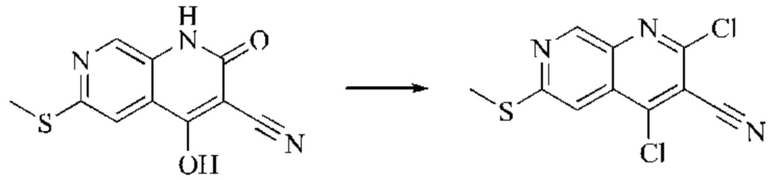
4-гидрокси-6-метилтио-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (130,0 мг неочищенного продукта) растворяли в оксихлориде фосфора (2 мл), после чего добавляли пентахлорид фосфора (168,7 мг, 1,1 ммоль) и затем нагревали до 100°С с обратным холодильником в течение ночи. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор выливали в ледяную воду (15 мл), доводили до рН 1, добавляя насыщенный водный раствор бикарбоната натрия, и фильтровали с отсасыванием. Фильтрат экстрагировали этилацетатом и концентрировали. Неочищенный продукт комбинировали с осадком на фильтре и очищали колоночной хроматографией на силикагеле (ПЭ:ЭА=20:1), получая желтый твердый продукт (60,0 мг, выход по завершении трех этапов: 13,7%).
Этап 7: Синтез 4-хлор-6-метилтио-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
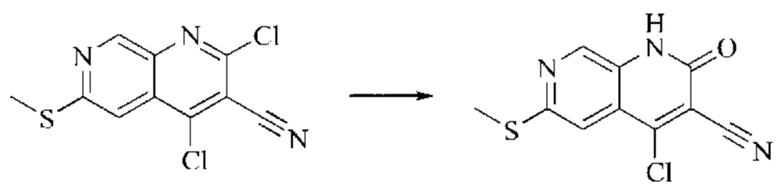
2,4-дихлор-6-метилтио-1,7-нафтиридин-3-карбонитрил (50,0 мг, 0,186 ммоль) растворяли в ТФУК (4 мл) и воде (1 мл). Систему нагревали при 100°С в течение 2 часов, и завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, получая продукт (68,0 мг неочищенного продукта), который непосредственно использовали в следующем этапе без очистки.
Этап 8: Синтез 6-метилтио-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
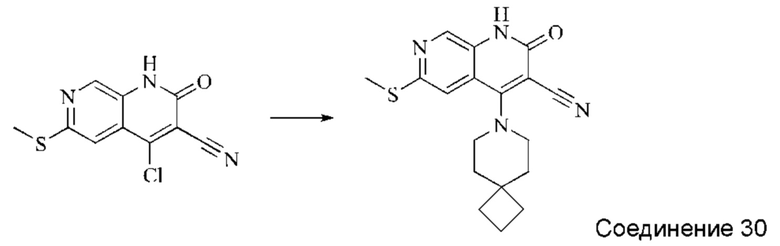
4-хлор-6-метилтио-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (60,0 мг неочищенного продукта) растворяли в ДМФА (2 мл). Добавляли гидрохлорид 7-аза-спиро[3,5]нонана (54,12 мг, 0,336 ммоль) и DIPEA (93,53 мг, 0,72 ммоль). Смесь нагревали при 70°С в течение 2 часов, и завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, после чего добавляли воду и этилацетат, и жидкость разделялась. Водную фазу экстрагировали этилацетатом. Органическую фазу объединяли, промывали водой и солевым раствором, сушили безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 80:1, 60:1, 40:1), получая продукт (50,0 мг, выход в результате двух этапов синтеза: 79%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,94 (s, 1Н), 8,54 (s, 1Н), 7,33 (s, 1Н), 3,50 (s, 4Н), 2,55 (s, 2Н), 1,92-1,80 (m, 10Н).
Молекулярная формула: C18H20N4OS, Молекулярная масса: 340,45, ЖХ-МС (Pos, m/z)=341,0 [М+Н]+.
Пример 17
Синтез 6-(метилсульфонил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 31)
Этап 1: Синтез 6-(метилсульфонил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
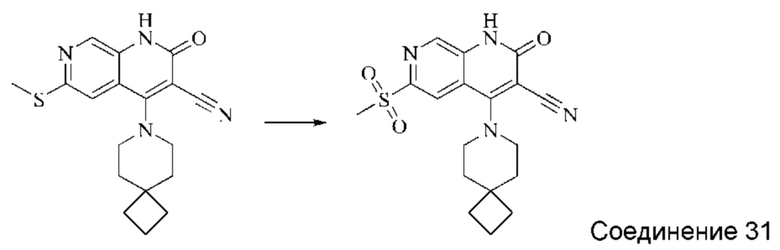
6-метилтио-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (40,0 мг, 0,12 ммоль) растворяли в дихлорметане (8 мл). Смесь охлаждали до 0°С, после чего добавляли мета-хлорпероксибензойную кислоту (59,2 мг, 0,24 ммоль), и затем медленно нагревали до комнатной температуры для начала реакции. Завершение протекания реакции определяли способами ТСХ и ЖХ-МС. Добавляли насыщенный водный раствор бикарбоната натрия (10 мл) и дихлорметан (10 мл). После разделения жидкостей водную фазу экстрагировали дихлорметаном, органические фазы объединяли, промывали водой, затем насыщенным солевым раствором, сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 100:1, 80:1), получая желтый твердый продукт (10,0 мг, выход: 20,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,44 (s, 1Н), 8,75 (s, 1Н), 8,19 (s, 1Н), 3,57 (s, 4Н), 3,26 (s, 3Н), 1,94-1,82 (m, 10Н).
Молекулярная формула: C18H20N4O3S, Молекулярная масса: 372,44, ЖХ-МС (Pos, m/z)=373,1 [М+Н]+.
Пример 18
Синтез 6-(1,2-дигидроксиэтил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 32)
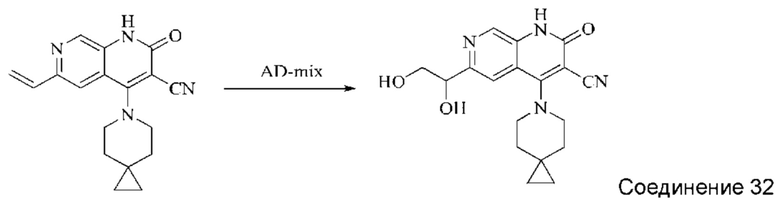
2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (700,2 мг, 2,285 ммоль, 1,0 экв.) растворяли в смеси трет-бутанола (30 мл) и воды (30 мл). Систему охлаждали до 0°С на ледяной бане, после чего добавляли метансульфонамид (217,4 мг, 2,285 ммоль, 1,0 экв.) и AD-mix-β (7,76 г), и затем энергично перемешивали при комнатной температуре в течение ночи. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор фильтровали с отсасыванием, и осадок на фильтре промывали водой (30 мл×3), сушили в вакууме, получая желтое твердое вещество (504,7 мг, выход: 64,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,97 (s, 1Н), 8,60 (s, 1Н), 7,77 (s, 1Н), 5,56 (s, 1Н), 4,69-4,65 (d, 2Н), 3,65 (s, 5Н), 3,50 (s, 1Н), 1,63 (s, 4Н), 0,45 (s, 4Н).
Молекулярная формула: C18H20N4O3, Молекулярная масса: 340,38, ЖХ-МС (Pos, m/z)=341,08 [М+Н]+.
Пример 19
Синтез 6-(1,2-дигидроксиэтил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 33)
Этап 1: Синтез 2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
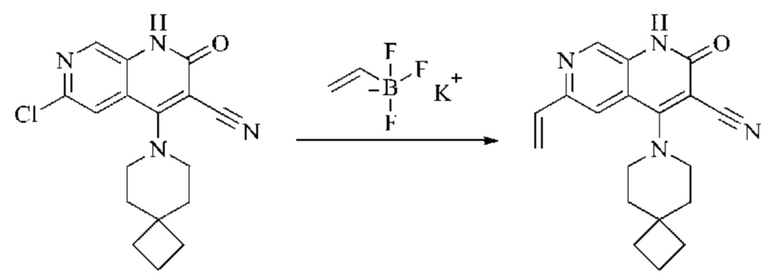
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (4,61 г, 14,02 ммоль, 1,0 экв.), трифторвинилборат калия (3,05 г, 22,77 ммоль, 3,0 экв.), карбонат цезия (13,70 г, 42,06 ммоль, 3,0 экв.), Pd(PPh3)4 (810,1 мг, 0,70 ммоль, 0,05 экв.) и Pd(dppf)Cl2 (512,9 мг, 0,70 ммоль, 0,05 экв.) растворяли в смеси 1,4-диоксана (40 мл) и воды (10 мл). Систему нагревали до 105°С и перемешивали в течение ночи в атмосфере азота. Завершение протекания реакции определяли способом ТСХ. К реакционному раствору добавляли H2O (20 мл), и систему перемешивали в течение 30 минут, охлаждали до комнатной температуры и фильтровали с отсасыванием. Осадок на фильтре промывали небольшим количеством дихлорметана, и фильтрат экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл×2), сушили безводным Na2SO4, фильтровали с отсасыванием и концентрировали при пониженном давлении. Неочищенный продукт сначала очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 80:1-30:1), а затем промывали этилацетатом (10 мл), получая желтый твердый продукт (2,3 г, выход: 51,27%).
Этап 2: Синтез 6-(1,2-дигидроксиэтил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
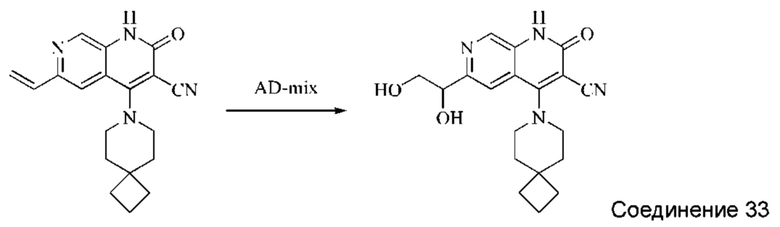
2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (2,3 г, 7,2 ммоль, 1,0 экв.) растворяли в смеси трет-бутанола (60 мл) и воды (60 мл). Систему охлаждали до 0°С на ледяной бане, затем медленно добавляли метансульфонамид (682,0 мг, 7,2 ммоль, 1,0 экв.) и AD-mix-β (10 г), после чего энергично перемешивали при комнатной температуре в течение ночи. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор фильтровали с отсасыванием, осадок на фильтре промывали водой (30 мл×3) и сушили в вакууме, получая желтый твердый продукт (857,0 мг, выход: 33,6%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,89 (s, 1Н), 8,58 (s, 1Н), 7,77 (s, 1Н), 5,56 (s, 1Н), 4,68 (m, 2Н), 3,73 (s, 1Н), 3,57 (m, 4Н), 1,84-2,01 (m, 10Н).
Молекулярная формула: C19H22N4O3, Молекулярная масса: 354,17, ЖХ-МС (Pos, m/z)=355,11 [М+Н]+.
Пример 20
Синтез 4-(4-этилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединения 34)
Этап 1: Синтез трет-бутил-4-этил-4-гидроксипиперидин-1-карбоксилата

Трет-бутил-4-оксопиперидин-1-карбоксилат (2,0 г,, 038 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (10,0 мл), затем медленно, при 0°С, в атмосфере азота добавляли раствор этилмагнийхлорида (10,04 мл, 20,075 ммоль, 2,0 экв.), разбавленный ТГФ (10,0 мл). Смесь медленно нагревали до комнатной температуры и оставляли реагировать в течение ночи. После того, как анализ ТСХ показал завершение реакции, неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА=от 10:1 до 1:1), получая продукт (800,0 мг, выход: 40%).
Этап 2: Синтез трет-бутил-4-этил-3,6-дигидропиридин-1(2H)-карбоксилата
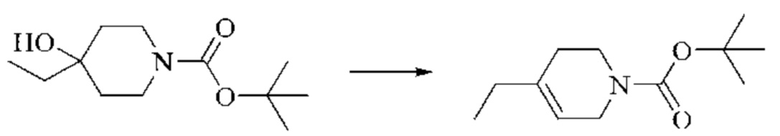
Трет-бутил-4-этил-4-гидроксипиперидин-1-карбоксилат (800,0 мг, 3,488 ммоль, 1,0 экв.) растворяли в дихлорметане (5,0 мл) и затем добавляли пиридин (827,9 мг, 10,466 ммоль, 3,0 экв.). К смеси медленно, при 0°С, в атмосфере азота добавляли дихлорсульфоксид (830,1 мг, 6,977 ммоль, 2,0 экв.). Смесь медленно нагревали до комнатной температуры и оставляли реагировать в течение ночи. После того как анализ ТСХ показал завершение реакции, реакционный раствор выливали в холодную воду и экстрагировали этилацетатом. Органическую фазу дважды промывали соляной кислотой (1 моль/л), сушили безводным Na2SO4 и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА=20:1-5:1), получая продукт в виде желтого масла (450,0 мг, выход: 56%).
Этап 3: Синтез трет-бутил-4-этилпиперидин-1-карбоксилата
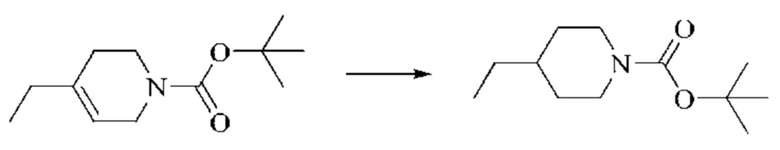
Трет-бутил-4-этил-3,6-дигидропиридин-1(2Н)-карбоксилат (450,0 мг, 2,129 ммоль, 1,0 экв.) растворяли в метаноле (10,0 мл) и затем добавляли Pd/C (225,0 мг). Воздух в системе вытесняли водородом, и реакцию проводили в течение 4 суток. После того, как анализ ТСХ показал завершение реакции, смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт непосредственно вводили в следующий этап.
Этап 4: Синтез 4-этилпиперидингидрохлорида
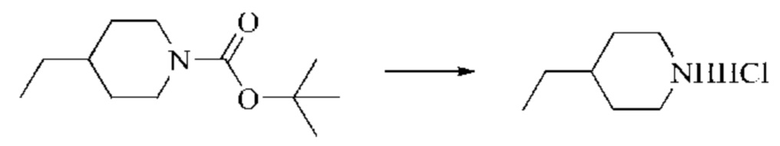
Полученный как описано выше неочищенный продукт растворяли в этаноле (3,0 мл), и в реакционный раствор добавляли по каплям хлорид водорода в этаноле (25%, 3,0 мл). Реакцию в полученной смеси оставляли протекать при комнатной температуре в течение 2 часов. После того, как анализ ТСХ показал завершение реакции, реакционный раствор концентрировали при пониженном давлении, и неочищенный продукт (102,0 мг) был непосредственно введен в следующий этап.
Этап 5: Синтез 4-(4-этилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
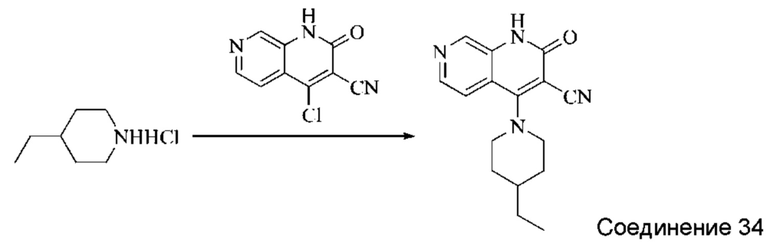
Неочищенный продукт, полученный в предыдущем этапе (102,0 мг, 0,681 ммоль, 1,0 экв.) растворяли в N,N-диметилформамиде (3,0 мл) и добавляли N,N-диизопропилэтиламин (528,1 мг, 4,086 ммоль, 6,0 экв.) и 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (140,0 мг, 0,681 ммоль, 1,0 экв.). Реакцию в полученной системе оставляли протекать при 80°С в течение 2 часов. После того, как анализ ТСХ показал завершение реакции, смесь концентрировали при пониженном давлении, суспендировали и промывали этилацетатом (1,5 мл) и простым метил-трет-бутиловым эфиром (5,0 мл) и фильтровали с отсасыванием. Осадок на фильтре промывали водой, получая желтый твердый продукт (100,0 мг, выход: 52,1%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,00 (s, 1Н), 8,65 (s, 1Н), 8,35-8,34 (m, 1Н), 7,60-7,58 (m, 1Н), 3,87-3,84 (m, 2Н), 3,42-3,39 (m, 2Н), 1,88-1,85 (m, 2Н), 1,44-1,36 (m, 5Н), 1,01-0,89 (m, 3Н).
Молекулярная формула: C16H18N4O, Молекулярная масса: 282,35, ЖХ-МС (Pos, m/z)=283,2 [М+Н]+.
Пример 21
Синтез 4-(4,4-бис(гидроксиметил)пиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединения 38)
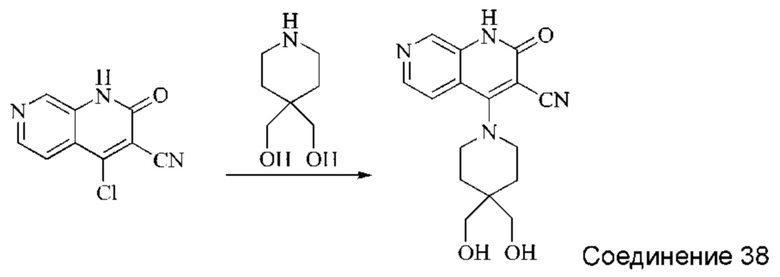
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (200,0 мг, 0,973 ммоль, 1,0 экв.), пиперидин-4,4-диилдиметанол (265,1 мг, 1,459 ммоль, 1,5 экв.) и DIPEA (1005,8 мг, 7,782 ммоль, 8,0 экв.) растворяли в ДМФА (5 мл). Смесь перемешивали при 80°С, и оставляли реагировать в течение 3 часов. Завершение протекания реакции определяли способом ТСХ. Смесь концентрировали при пониженном давлении, промывали в течение 3 часов добавлением воды (10 мл) и этилацетата (10 мл) и фильтровали с отсасыванием. Осадок на фильтре споласкивали небольшим количеством метанола, получая темно-красный твердый продукт (55,0 мг, выход: 18,0%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,94 (s, 1Н), 8,65 (s, 1Н), 8,36-8,34 (d, 1Н), 7,60-7,59 (d, 1Н), 4,55 (t, 2Н), 3,63 (t, 4Н), 3,43-3,42 (d, 4Н), 1,65 (t, 4Н).
Молекулярная формула: C16H18N4O3, Молекулярная масса: 314,35, ЖХ-МС (Pos, m/z)=315,04 [М+Н]+.
Пример 22
Синтез (R)-4-(3-(1Н-пиразол-1-ил)пирролидин-1-ил)-6-морфолино-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 39)
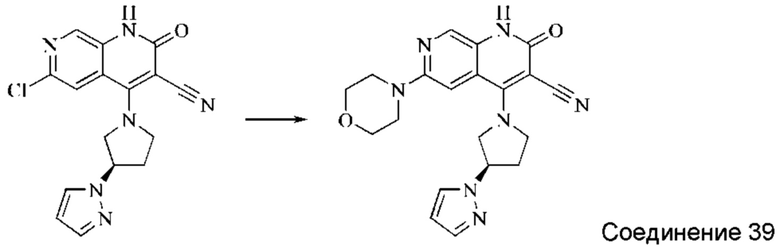
(R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-6-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (60,0 мг, 0,176 ммоль), морфолин (36,85 мг, 0,423 ммоль), Pd2(dba)3 (6,7 мг, 0,007 ммоль), трет-бутилат натрия (47,54 мг, 0,493 ммоль) и XPhos (6,7 мг, 0,014 ммоль) растворяли в 1,4-диоксане (1 мл) и ДМА (0,2 мл). Реакцию в полученной системе оставляли протекать при микроволновом нагреве до 110°С в течение 4 часов. Завершение протекания реакции определяли способом ТСХ. Смесь концентрировали при пониженном давлении, после чего добавляли воду и затем экстрагировали этилацетатом. Органические фазы объединяли, промывали водой, затем насыщенным солевым раствором, сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 80:1, 60:1, 40:1, 20:1), получая красный твердый продукт (13,0 мг, 18,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,30 (s, 1Н), 8,30 (s, 1Н), 7,89-7,88 (d, 1Н), 7,50 (d, J=1,2 Гц, 1Н), 7,15 (d, 1Н), 6,30-6,29 (m, 1Н), 5,17-5,15 (m, 1Н), 4,52-4,48 (m, 1H), 4,36-4,32 (m, 1H), 4,29-4,16 (m, 2H), 3,74-3,72 (m, 4H), 3,37 (s, 3Н), 2,46-2,33 (m, 2H).
Молекулярная формула: C20H21N7O2, Молекулярная масса: 391,44, ЖХ-МС (Pos, m/z)=392,1 [М+Н]+.
Пример 23
Синтез 4-(2-гидрокси-7-аза-спиро[3,5]нонан-7-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединения 40)
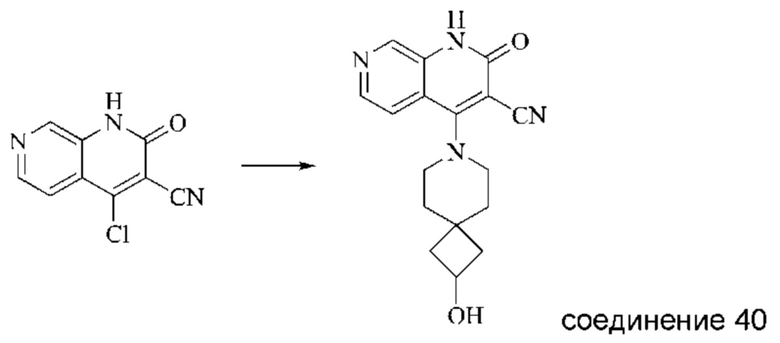
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,24 ммоль, 1,0 экв.) растворяли в ДМФА и добавляли гидрохлорид 7-аза-спиро[3,5]нонан-2-ола (60,5 мг, 0,34 ммоль, 1,4 экв.) и DIPEA (188,5 мг, 1,46 ммоль, 6,0 экв.). Реакцию в полученной системе оставляли протекать при 80°С в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Добавляли МТБЭ (2 мл) и H2O (2 мл). На смесь воздействовали вибрацией и фильтровали с отсасыванием, получая продукт (37 мг, выход: 49,7%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,97 (s, 1Н), 8,64 (s, 1Н), 8,32-8,33 (m, 1Н), 7,57-7,59 (m, 1Н), 4,97-4,98 (m, 1Н), 4,12-4,19 (m, 1Н), 3,50-3,55 (m, 4Н), 2,21-2,26 (m, 2Н), 1,74-1,75 (m, 4Н), 1,64-1,69 (m, 2Н).
Молекулярная формула: C17H18N4O2, Молекулярная масса: 310,36, ЖХ-МС (Pos, m/z)=311,02 [М+Н]+.
Пример 24
Синтез 4-(2-циано-7-аза-спиро[3,5]нонан-7-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединение 42)
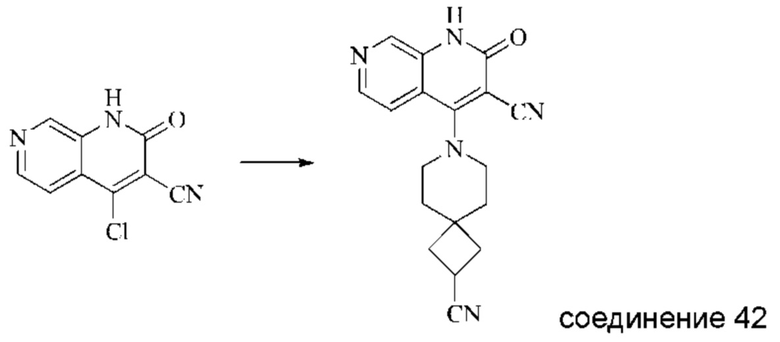
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,24 ммоль, 1,0 экв.) растворяли в ДМФА и добавляли гидрохлорид 7-аза-спиро[3,5]нонан-2-карбонитрила (51,2 мг, 0,34 ммоль, 1,4 экв.) и DIPEA (188,5 мг, 1,46 ммоль, 6,0 экв.). Реакцию в полученной системе оставляли протекать при 80°С в течение 1,5 часов, и завершение протекания реакции определяли способом ЖХ-МС. Смесь фильтровали с отсасыванием, получая продукт (19 мг, выход: 24,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,00 (s, 1Н), 8,64 (s, 1Н), 8,32-8,34 (m, 1Н), 7,57-7,58 (m, 1Н), 3,52-3,55 (m, 4Н), 3,38-3,46 (m, 1Н), 2,29-2,34 (m, 2Н), 2,15-2,20 (m, 2Н), 1,84-1,88 (m, 4Н).
Молекулярная формула: C18H17N5O, Молекулярная масса: 319,37, ЖХ-МС (Pos, m/z)=320,05 [М+Н]+.
Пример 25
Синтез 4-(2,2-дифтор-7-аза-спиро[3,5]нонан-7-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 43)
Этап 1: Синтез трет-бутил-2,2-дифтор-7-аза-спиро[3,5]нонан-7-карбоксилата
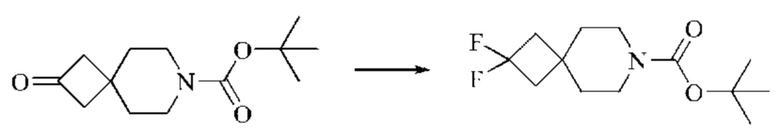
Трет-бутил-2-оксо-7-аза-спиро[3,5]нонан-7-карбоксилат (500,0 мг, 2,09 ммоль) растворяли в дихлорметане (4 мл). Систему охлаждали до 0°С на ледяной бане, после чего добавляли DAST (673,8 мг, 4,18 ммоль), медленно нагревали до комнатной температуры и перемешивали в течение ночи. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор добавляли в насыщенный водный раствор бикарбоната натрия. Водную фазу экстрагировали дихлорметаном. Органические фазы объединяли, промывали водой, затем солевым раствором и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА=30:1, 25:1, 20:1), получая продукт (170,0 мг, выход: 31,2%).
Этап 2: Синтез 2,2-дифтор-7-аза-спиро[3,5]нонана
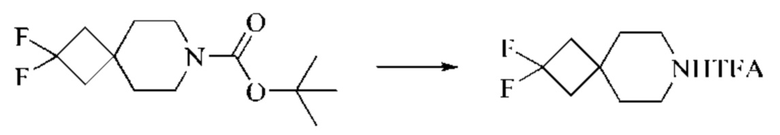
Трет-бутил-2,2-дифтор-7-аза-спиро[3,5]нонан-7-карбоксилат (170,0 мг, 0,65 ммоль) растворяли в дихлорметане (3 мл). Систему охлаждали до 0°С на ледяной бане, после чего добавляли трифторуксусную кислоту (1,5 мл), медленно нагревали до комнатной температуры и перемешивали в течение 2 часов. Завершение протекания реакции определяли способом ТСХ. Смесь концентрировали при пониженном давлении, получая продукт (115,0 мг неочищенного продукта).
Этап 3: Синтез 4-(2,2-дифтор-7-аза-спиро[3,5]нонан-7-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
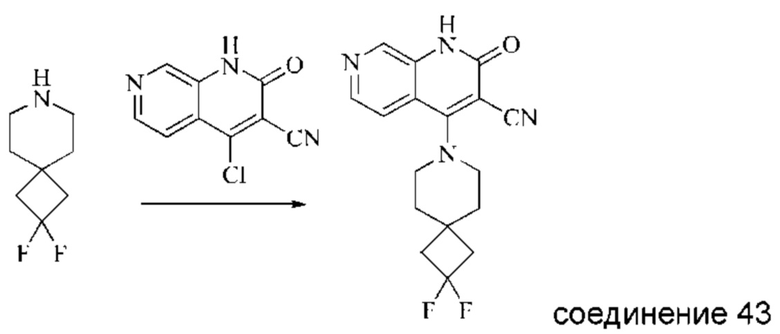
4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (95,6 мг, 0,465 ммоль) и 2,2-дифтор-7-аза-спиро[3,5]нонан (104,72 мг неочищенного продукта) растворяли в ДМФА (3 мл) и добавляли DIPEA (181.2 мг, 1,395 ммоль). Смесь нагревали до 80°С и оставляли реагировать в течение 2 часов; завершение протекания реакции определяли способом ТСХ. Реакционный раствор выливали в воду (10 мл), фильтровали с отсасыванием, и осадок на фильтре промывали этилацетатом и петролейным эфиром, получая продукт (50,0 мг).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,03 (s, 1Н), 8,65 (s, 1Н), 8,35-8,33 (d, 1Н), 3,58 (s, 4Н), 2,51 (s, 2Н), 1,99-1,88 (d, 4Н), 1,26-1,24 (d, 2Н).
Молекулярная формула: C17H16F2N4O, Молекулярная масса: 330,34, ЖХ-МС (Pos, m/z)=331,1 [М+Н]+.
Пример 26
Синтез 6-амино-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 44)
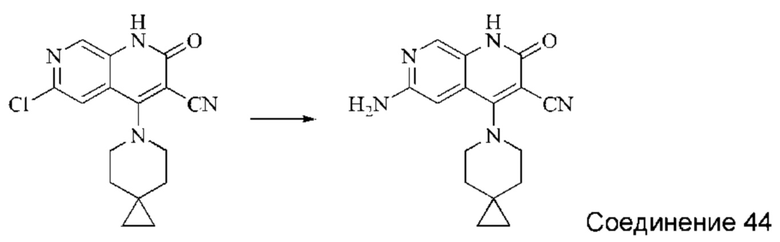
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (30 мг, 0,095 ммоль, 1,0 экв.), LiHMDS в ТГФ (1 моль/л, 1 мл), Pd2(dba)3 (9 мг, 0,0095 ммоль, 0,1 экв.) и 2-(дициклогексилфосфино)бифенил (7 мг, 0,0191 ммоль, 0,2 экв.) помещали в пробирку для микроволновой печи и нагревали под действием микроволнового излучения до 100°С в течение 2 часов. Реакционный раствор охлаждали, после чего добавляли соляную кислоту концентрацией 1 моль/л (12 мл) и перемешивали в течение 20 минут. Систему доводили до слабощелочной реакции добавлением водного раствора карбоната натрия и экстрагировали ЭА. Органическую фазу сушили безводным сульфатом натрия и дважды разделяли на пластине для препаративной тонкослойной хроматографии (ДХМ : МеОН = 30:1,15:1), получая продукт (1,03 мг, выход: 3,7%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,55 (s, 1Н), 8,13 (s, 1Н), 6,71 (s, 1Н), 5,93 (s, 1Н), 7,32-7,34 (t, 1Н), 3,54-3,56 (t, 4Н), 1,96-2,03 (m, 4Н), 1,63 (m, 2Н), 0,86 (m, 2Н).
Молекулярная формула: C16H17N5O, Молекулярная масса: 295,35, ЖХ-МС (Pos, m/z)=296,05 [М+Н]+.
Пример 27
Синтез 6-метиламино-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединение 45)
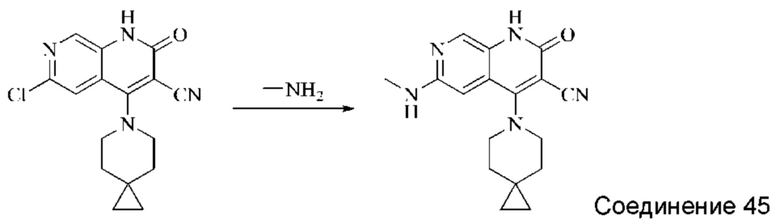
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,159 ммоль, 1,0 экв.), метиламин в ТГФ (1 моль/л, 0,8 мл, 0,794 ммоль, 5 экв.), Pd2(dba)3 (6 мг, 0,006 ммоль, 0,04 экв.), трет-бутилат натрия (46 мг, 0,476 ммоль, 2,8 экв.) и Xphos (6 мг, 0,013 ммоль, 0,08 экв.) растворяли в 1,4-диоксане (1 мл) и ДМАА (0,2 мл). Реакционный раствор нагревали под действием микроволнового излучения до 110°С в течение 4 часов, охлаждали, после чего добавляли воду и экстрагировали ЭА. Органическую фазу сушили безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 100:1-50:1), получая продукт (5,69 мг, выход: 11,6%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,54 (s, 1Н), 8,21 (s, 1Н), 6,54-6,55 (d, 1Н), 6,61 (s, 1Н), 3,57 (m, 4Н), 2,77-2,78 (d, 3Н), 1,62 (m, 4Н), 0,43 (m, 4Н).
Молекулярная формула: C17H19N5O, Молекулярная масса: 309,37, ЖХ-МС (Pos, m/z)=310,17 [М+Н]+.
Пример 28
Синтез 6-диметиламино-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 46)
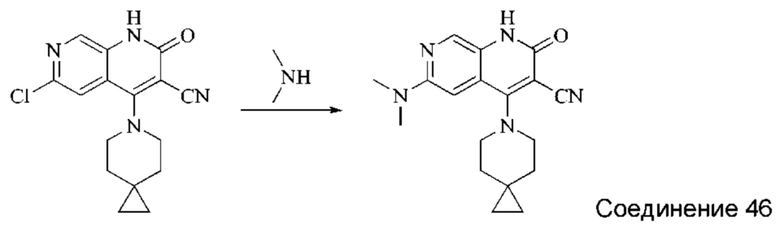
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,159 ммоль, 1,0 экв.), диметиламин в ТГФ (2 моль/л, 0,4 мл, 0,794 ммоль, 5 экв.), Pd2(dba)3 (6 мг, 0,006 ммоль, 0,04 экв.), трет-бутанол натрия (43 мг, 0,445 ммоль, 2,8 экв.) и Xphos (6 мг, 0,013 ммоль, 0,08 экв.) растворяли в 1,4-диоксане (1 мл) и ДМАА (0,2 мл). Реакционный раствор нагревали под действием микроволнового излучения до 110°С в течение 4 часов, охлаждали, после чего добавляли воду и экстрагировали ЭА. Органическую фазу сушили безводным сульфатом натрия. Неочищенный продукт выделяли способом препаративной тонкослойной хроматографии (ДХМ : МеОН = 20:1), получая продукт (26,63 мг, выход: 51,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,58 (s, 1Н), 8,34 (s, 1Н), 6,63 (s, 1Н), 3,62 (s, 4Н), 3,05 (s, 6Н), 1,62 (m, 4Н), 0,44 (m, 4Н).
Молекулярная формула: C18H21N5O, Молекулярная масса: 323,40, ЖХ-МС (Pos, m/z)=324,1 [М+Н]+.
Пример 29
Синтез 6-изопропил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 47)
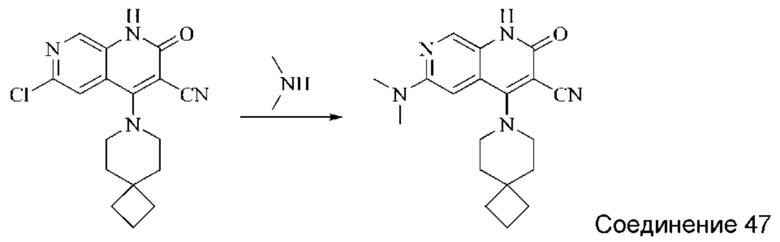
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,152 ммоль, 1,0 экв.), диметиламин в ТГФ (2 моль/л, 0,38 мл, 0,76 ммоль, 5 экв.), Pd2(dba)3 (5,7 мг, 0,006 ммоль, 0,04 экв.), трет-бутилат натрия (41,5 мг, 0,43 ммоль, 2,8 экв.) и Xphos (5,72 мг, 0,012 ммоль, 0.08 экв.) растворяли в 1,4-диоксане (1 мл) и ДМАА (0,2 мл). Реакционный раствор нагревали под действием микроволнового излучения до 110°С в течение 4 часов, охлаждали, после чего добавляли воду и экстрагировали ЭА. Органическую фазу сушили безводным сульфатом натрия. Неочищенный продукт выделяли способом препаративной тонкослойной хроматографии (проявляющий агент: ДХМ : МеОН = 20:1), получая 6-изопропил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (11,56 мг, выход: 22,5%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,57 (s, 1Н), 8,30 (s, 1Н), 6,59 (s, 1Н), 3,51 (s, 4Н), 3,03 (s, 6Н), 1,79-2,07 (m, 10Н).
Молекулярная формула: C19H23N5O, Молекулярная масса: 337,43, ЖХ-МС (Pos, m/z)=338,15 [М+Н]+.
Пример 30
Синтез 6-((диметиламино)метил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 48)
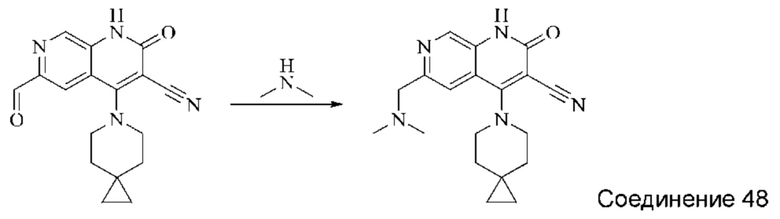
6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (10 мг, 0,03 ммоль, 1,0 экв.) растворяли в 1,2-дихлорэтане (1 мл) и затем добавляли раствор диметиламина в тетрагидрофуране (2 моль/л, 0,06 мл, 0,13 ммоль, 4,0 экв.). Смесь перемешивали при комнатной температуре в течение 1 часа. При охлаждении ледяной баней добавляли триацетоксиборгидрид натрия (21 мг, 0,10 ммоль, 3,0 экв.). Систему перемешивали при комнатной температуре в течение 2 часов и тушили, добавляя воду (1 мл). Смесь экстрагировали дихлорметаном (5 мл×2). Органические фазы объединяли и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан : метанол = 15:1), получая беловатый твердый продукт (1,6 мг, выход: 14,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,03 (s, 1Н), 8,62 (s, 1Н), 7,66 (s, 1Н), 3,64 (m, 6Н), 2,26 (m, 6Н), 1,62 (m, 4Н), 0,45 (m, 4Н).
Молекулярная формула: C19H23N5O, Молекулярная масса: 337,19, ЖХ-МС (Pos, m/z)=336,17 [М+Н]+.
Пример 31
Синтез 6-(азетидин-1-ил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 49)
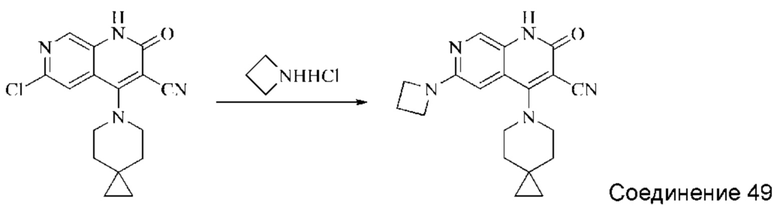
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,159 ммоль, 1,0 экв.), азетидина гидрохлорид (74 мг, 0,794 ммоль, 5 экв.), Pd2(dba)3 (6 мг, 0,006 ммоль, 0,04 экв.), трет-бутилат натрия (119 мг, 1,239 ммоль, 7,8 экв.) и Xphos (6 мг, 0,013 ммоль, 0,08 экв.) растворяли в 1,4-диоксане (1 мл) и ДМАА (0,2 мл). Реакционный раствор нагревали под действием микроволнового излучения до 110°С в течение 4 часов, охлаждали, после чего добавляли воду и экстрагировали ЭА. Органическую фазу сушили безводным сульфатом натрия. Неочищенный продукт очищали способом препаративной тонкослойной хроматографии (ДХМ : МеОН = 20:1), получая продукт (1,25 мг, выход: 2,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,65 (s, 1Н), 8,28 (s, 1Н), 6,42 (s, 1Н), 3,92-3,95 (t, 4Н), 3,59-3,62 (t, 4Н), 2,29-2,36 (m, 2Н), 1,60 (m, 4Н), 0,432 (m, 4Н).
Молекулярная формула: C19H21N5O, Молекулярная масса: 335,41, ЖХ-МС (Pos, m/z)=336,15 [М+Н]+.
Пример 32
Синтез 6-(3-гидроксиазетидин-1-ил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 50)
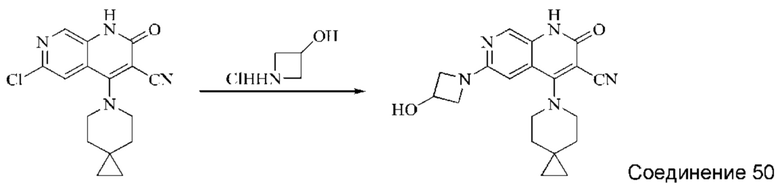
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (200 мг, 0,64 ммоль, 1,0 экв.) и азетидин-3-ол гидрохлорид (278 мг, 2,54 ммоль, 4,0 экв.) растворяли в 1,4-диоксане (5 мл). Добавляли бутилат натрия (478 мг, 4,96 ммоль, 7,8 экв.), 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (24 мг, 0,051 ммоль, 0,08 экв.) и Pd2(dba)3 (24 мг, 0,025 ммоль, 0,04 экв.). Смесь нагревали до 120°С в атмосфере азота и оставляли реагировать в течение 3 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 20:1), затем суспендировали и промывали ЭА и фильтровали с отсасыванием, получая продукт (74 мг, выход: 33%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,64 (s, 1Н), 8,27 (s, 1Н), 6,44 (s, 1Н), 5,64-5,66 (t, 1Н), 4,56-4,59 (m, 1Н), 4,14-4,18 (t, 2Н), 3,60-3,66 (m, 6Н), 1,59 (m, 6Н), 0,43 (m, 4Н).
Молекулярная формула: C19H21N5O2, Молекулярная масса: 351,41, ЖХ-МС (Pos, m/z)=352,17 [М+Н]+.
Пример 33
Синтез 6-(3-гидроксиазетидин-1-ил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 51)
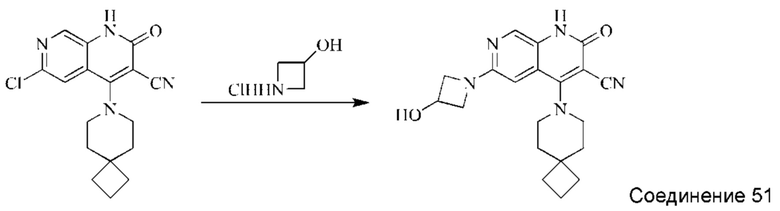
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (80 мг, 0,243 ммоль, 1,0 экв.) и гидрохлорид азетидин-3-ола (89 мг, 0,81 ммоль, 3,3 экв.) растворяли в 1,4-диоксане (5 мл). Добавляли бутилат натрия (70 мг, 0,73 ммоль, 3,0 экв.), 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (11 мг, 0,024 ммоль, 0,1 экв.) и Pd2(dba)3 (12 мг, 0,012 ммоль, 0,05 экв.). Смесь нагревали до 120°С в атмосфере азота и оставляли реагировать в течение 3 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 10:1), получая желтый твердый продукт (9 мг, выход: 10%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,59 (s, 1Н), 8,27 (s, 1Н), 6,41 (s, 1Н), 5,63-5,65 (d, 1Н), 4,56-4,63 (m, 1Н), 4,14-4,18 (m, 2Н), 3,63-3,67 (m, 2Н), 3,49 (s, 4Н), 1,74-1,94 (m, 10Н).
Молекулярная формула: C20H23N5O2, Молекулярная масса: 365,44, ЖХ-МС (Pos, m/z)=366,14 [М+Н]+.
Пример 34
Синтез 6-(3-аминоазетидин-1-ил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 52)
Этап 1: Синтез трет-бутил-(1-(3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-ил)азетидин-3-ил)карбамата
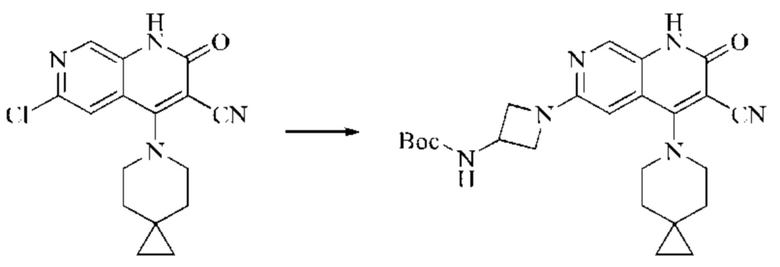
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (300 мг, 0,95 ммоль, 1,0 экв.) и трет-бутилазетидин-3-илкарбамат (795,5 мг, 3,81 ммоль, 4,0 экв.) растворяли в 1,4-диоксане, после чего добавляли Pd2(dba)3 (36,1 мг, 0,04 ммоль, 0,04 экв.), t-BuONa (643,3 мг, 6,67 ммоль, 7,0 экв.) и Xphos (36,3 мг, 0,08 ммоль, 0,08 экв.). Реакцию в полученной смеси оставляли протекать при 120°С в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Смесь концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 50:1-20:1), получая продукт (177 мг, выход: 41,2%).
Этап 2: Синтез 6-(3-аминоазетидин-1-ил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
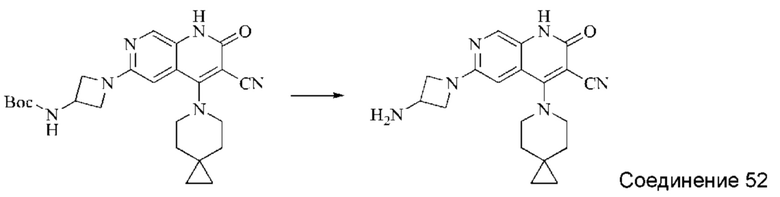
Трет-бутил-(1-(3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-ил)азетидин-3-ил)карбамат (177 мг, 0,39 ммоль, 1,0 экв.) растворяли в ДХМ и добавляли трифторуксусную кислоту (1 мл). Реакцию в полученной смеси оставляли протекать при 25°С в течение получаса. Завершение протекания реакции определяли способом ЖХ-МС. Смесь концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 10:1), получая продукт (25,5 мг, выход: 18,7%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 8,29 (s, 1Н), 6,46 (s, 1Н), 3,94-4,18 (m, 3Н), 3,67-3,70 (m, 2Н), 3,60 (m, 4Н), 1,60 (s, 4Н), 1,09-1,13 (m, 2Н), 0,81-0,85 (m, 4Н).
Молекулярная формула: C20H22N6O, Молекулярная масса: 350,43, ЖХ-МС (Pos, m/z)=351,15 [М+Н]+.
Пример 35
Синтез 6-(3-аминоазетидин-1-ил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 53)
Этап 1: Синтез (1-(3-циано-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-6-ил)азетидин-3-ил)карбамата
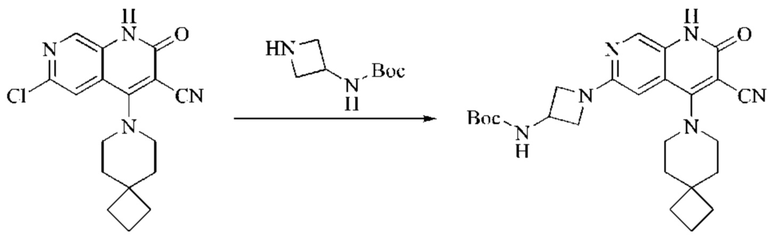
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (200 мг, 0,61 ммоль, 1,0 экв.) и трет-бутил азетидин-3-ил карбамат (105 мг, 0,61 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (5 мл), после чего добавляли трет-бутилат натрия (175 мг, 1,82 ммоль, 3,0 экв.), 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (25 мг, 0,061 ммоль, 0,1 экв.) и Pd2(dba)3 (30 мг, 0,031 ммоль, 0,05 экв.). Температуру повышали до 120°С, и реакцию в полученной системе оставляли протекать в течение 3 часов в атмосфере азота. Завершение реакции определяли способом ЖХ-МС. Реакционный раствор фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 20:1), получая желтый твердый продукт (140 мг, выход: 49%).
Этап 2: Синтез 6-(3-аминоазетидин-1-ил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
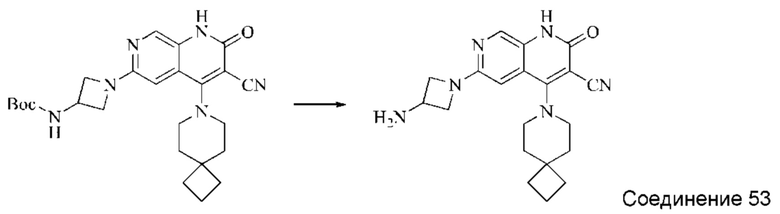
(1-(3-циано-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-6-ил)азетидин-3-ил)карбамат (140 мг, 0,30 ммоль, 1,0 экв.) растворяли в дихлорметане (4 мл), после чего добавляли трифторацетат (1 мл) и оставляли реагировать в течение 2 часов при комнатной температуре. Завершение реакции определяли способом ЖХ-МС. Образовывался твердый осадок. Смесь фильтровали, и осадок на фильтре споласкивали дихлорметаном (5 мл×2) и сушили при 45°С, получая продукт (19 мг, выход: 17%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 8,26 (s, 1Н), 6,39 (s, 1Н), 4,10-4,14 (m, 2Н), 3,79-3,85 (m, 1Н), 3,50-3,53 (m, 6Н), 1,79-1,93 (m, 10Н).
Молекулярная формула: C20H24N6O, Молекулярная масса: 364,45, ЖХ-МС (Pos, m/z)=365,16 [М+Н]+.
Пример 36
Синтез 6-(азетидин-1-ил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 54)
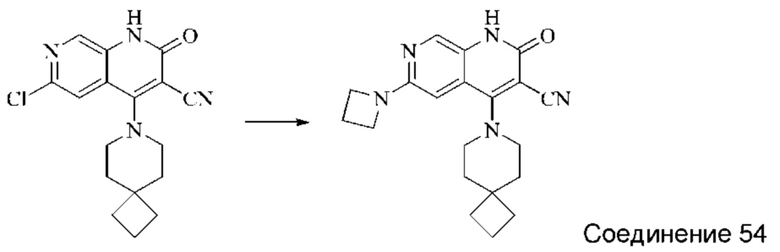
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (150 мг, 0,46 ммоль, 1,0 экв.) и азетидин (130 мг, 2,28 ммоль, 5,0 экв.) растворяли в тетрагидрофуране (5 мл) и ДМАА (1 мл), после чего добавляли трет-бутилат натрия (343 мг, 3,56 ммоль, 7,8 экв.), 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (17 мг, 0,036 ммоль, 0,08 экв.) и Pd2(dba)3 (17 мг, 0,018 ммоль, 0,04 экв.). Температуру повышали до 110°С, и реакцию в полученной системе оставляли протекать в течение 8 часов в атмосфере азота. Завершение реакции определяли способом ЖХ-МС. Реакционный раствор выливали в воду и экстрагировали ЭА. Органическую фазу сушили и концентрировали. Неочищенный продукт очищали способом препаративной тонкослойной хроматографии (ДХМ : МеОН = 20:1), получая продукт (61 мг, выход: 38%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,60 (s, 1Н), 8,27 (s, 1Н), 6,37 (s, 1Н), 3,90-3,94 (t, 4Н), 3,48 (s, 4Н), 2,30-2,36 (m, 2Н), 1,78-1,92 (m, 10Н).
Молекулярная формула: C20H23N5O, Молекулярная масса: 349,44, ЖХ-МС (Pos, m/z)=350,12 [М+Н]+.
Пример 37
Синтез 6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 55)
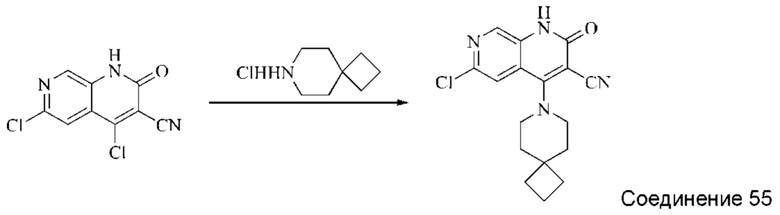
4,6-дихлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (500,0 мг, 2,08 ммоль, 1,0 экв.), гидрохлорид 7-аза-спиро[3,5]нонана (505,1 мг, 3,12 ммоль, 1,5 экв.) и DIPEA (2153,6 мг, 16,66 ммоль, 8,0 экв.) растворяли в ДМФА (10 мл), перемешивали для протекания реакции в течение 3 часов при 80°С. Завершение реакции определяли способом ТСХ. Систему концентрировали при пониженном давлении, после чего добавляли воду (10 мл) и этилацетат (10 мл) до образования суспензии и промывали в течение 3 часов, затем фильтровали с отсасыванием. Осадок на фильтре споласкивали небольшим количеством метанола, получая темно-красный твердый продукт (67,0 мг, выход: 9,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,11 (s, 1Н), 8,45 (s, 1Н), 3,54 (s, 4Н), 1,86-1,80 (d, 10Н).
Молекулярная формула: C17H17ClN4O, Молекулярная масса: 328,80, ЖХ-МС (Pos, m/z)=329,15 [М+Н]+.
Пример 38
Синтез 6-морфолино-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 56)
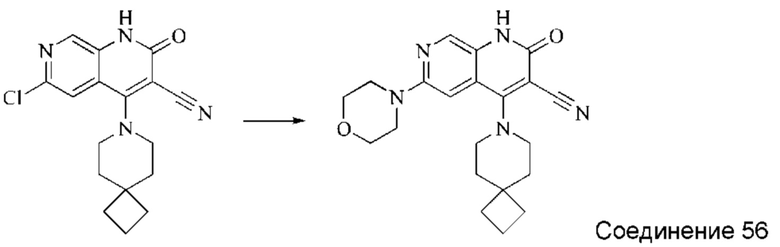
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50,0 мг, 0,152 ммоль), морфолин (32,23 мг, 0,37 ммоль), Pd2(dba)3 (5,7 мг, 0,006 ммоль), трет-бутилат натрия (41,5 мг, 0,43 ммоль) и XPhos (5,72 мг, 0,012 ммоль) растворяли в 1,4-диоксане (1 мл) и DMA (0,2 мл). В защитной атмосфере азота реакцию в полученной системе оставляли протекать при микроволновом нагреве до 110°С в течение 4 часов. Завершение реакции определяли способом ТСХ. Систему концентрировали при пониженном давлении, после чего добавляли воду и экстрагировали этилацетатом. Органические фазы объединяли, промывали водой, промывали насыщенным солевым раствором, сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 80:1-20:1), получая продукт (15,0 мг).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,67 (s, 1Н), 8,35 (s, 1Н), 6,78 (d, 1Н), 3,76-3,74 (t, 4Н), 3,38-3,35 (t, 4Н), 1,99-1,80 (m, 10Н).
Молекулярная формула: C21H25N5O2, Молекулярная масса: 379,46, ЖХ-МС (Pos, m/z)=380,17 [М+Н]+.
Пример 39
Синтез 6-(4-метилпиперазин-1-ил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 57)
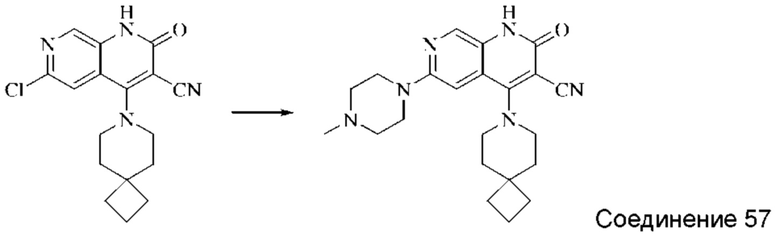
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (80,0 мг, 0,243 ммоль, 1,0 экв.), N-метилпиперазин (58,4 мг, 0,584 ммоль, 2,4 экв.), трет-бутилат натрия (65,7 мг, 0.681 ммоль, 2,8 экв.), трис(дибензилиденацетон)дипалладий (9,2 мг, 0,00972 ммоль, 0,04 экв.) и 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (9,3 мг, 0,0194 ммоль, 0,08 экв.) растворяли в 1,4-диоксане (2,0 мл) и N,N-диметилацетамиде (0,4 мл) и перемешивали при 120°С в течение ночи в атмосфере азота. Завершение реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, после чего добавляли воду (5,0 мл), экстрагировали этилацетатом (5,0 мл), сушили безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 80:1-20:1), получая желтый твердый продукт (20,0 мг, выход: 25%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,64 (s, 1Н), 8,33 (s, 1Н), 6,77 (s, 1Н), 3,52-3,32 (m, 6Н), 2,47 (s, 3Н), 2,24 (m, 2Н), 1,91-1,78 (m, 8Н), 1,36 (m, 2Н), 1,34-1,24 (m, 4Н).
Молекулярная формула: C22H28N6O, Молекулярная масса: 392,51, ЖХ-МС (Pos, m/z)=393,21 [М+Н]+.
Пример 40
Синтез 2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 58)
Этап 1: Синтез 6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
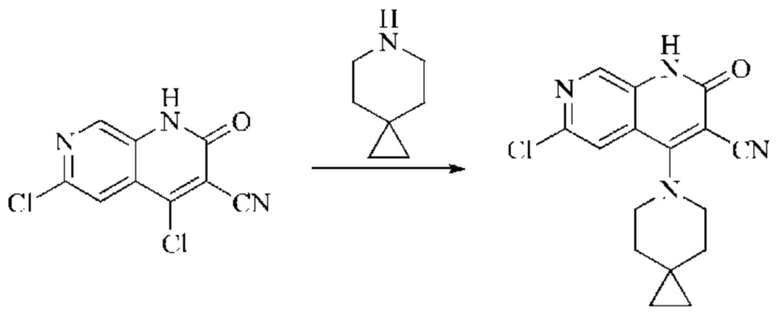
4,6-дихлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (4,00 г, 16,66 ммоль, 1,0 экв.), 6-аза-спиро[2,5]октан (2,22 г, 20,00 ммоль, 1,2 экв.) и DIPEA (8,61 г, 66,66 ммоль, 4,0 экв.) растворяли в ДМФА (40 мл), перемешивали в течение 3 часов при 80°С. Завершение реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, после чего добавляли воду (30 мл) и этилацетат (30 мл) до образования суспензии, промывали в течение 3 часов и фильтровали с отсасыванием. Осадок на фильтре споласкивали небольшим количеством метанола, получая желтый твердый продукт (3,63 г, выход: 69,2%).
Этап 2: Синтез 2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
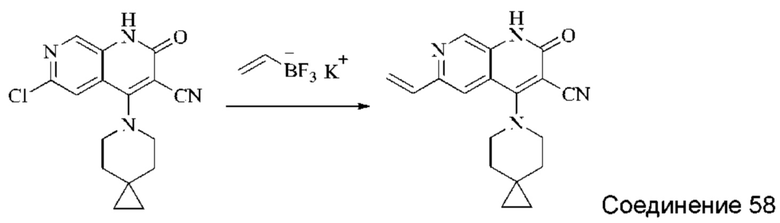
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (4,63 г, 14,71 ммоль, 1,0 экв.), трифторвинилборат калия (5,91 г, 44,13 ммоль, 3,0 экв.), карбонат цезия (14,38 г, 44,13 ммоль, 3,0 экв.), Pd(PPh3)4 (849,9 мг, 0,74 ммоль, 0,05 экв.) и Pd(dppf)Cl2 (538,1 мг, 0,74 ммоль, 0,05 экв.) растворяли в смеси 1,4-диоксана (40 мл) и воды (10 мл). Температуру повышали до 105°С, и систему перемешивали для протекания реакции в течение ночи в атмосфере азота. Завершение реакции определяли способом ТСХ. Добавляли H2O (20 мл). Реакционный раствор перемешивали в течение 30 минут, охлаждали до комнатной температуры и фильтровали с отсасыванием, и осадок на фильтре промывали небольшим количеством дихлорметана. Фильтрат экстрагировали дихлорметаном (20 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл×2), сушили безводным Na2SO4, фильтровали с отсасыванием и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 80:1-30:1) и промывали этилацетатом (10 мл), получая желтый твердый продукт (1,28 г, выход: 28,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,09 (s, 1Н), 8,65 (s, 1Н), 7,590 (s, 1Н), 6,96-6,89 (m, 1Н), 6,17 (d, J=16 Гц, 1Н), 5,40 (d, J=12 Гц, 1Н), 3,68 (m, 4Н), 1,64 (m, 4Н), 0,44 (m, 4Н).
Молекулярная формула: C18H18N4O, Молекулярная масса: 306,15, ЖХ-МС (Neg, m/z)=305,06 [М-Н]-.
Пример 41
Синтез 6-циклопропил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 59)
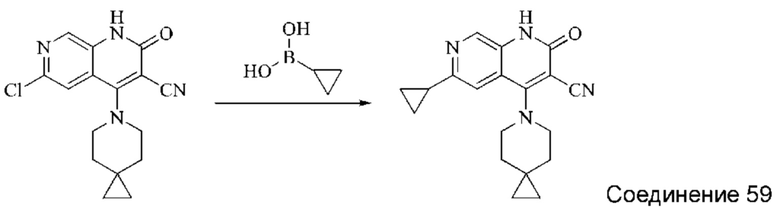
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,16 ммоль, 1,0 экв.) и циклопропилборную кислоту (55 мг, 0,64 ммоль, 4,0 экв.) растворяли в 1,4-диоксане (3 мл), после чего добавляли карбонат цезия (155 мг, 0,48 ммоль, 3,0 экв.), фосфат калия (34 мг, 0,16 ммоль, 1,0 экв.) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (12 мг, 0,016 ммоль, 0,1 экв.). Температуру повышали до 120°С, и реакцию в полученной системе оставляли протекать в течение 3 часов в атмосфере азота. Завершение реакции определяли способом ЖХ-МС. Полученную смесь фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 20:1), получая продукт (2,5 мг, выход: 4,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,89 (s, 1Н), 8,52 (s, 1Н), 7,47 (s, 1Н), 3,65 (m, 4Н), 2,16-2,25 (m, 1Н), 1,63 (s, 4Н), 0,93-0,95 (m, 2Н), 0,86-0,87 (m, 2Н), 0,44 (m, 4Н).
Молекулярная формула: C19H20N4O, Молекулярная масса: 320,40, ЖХ-МС (Pos, m/z)=321,92 [М+Н]+.
Пример 42
Синтез 6-циклопропил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 60)
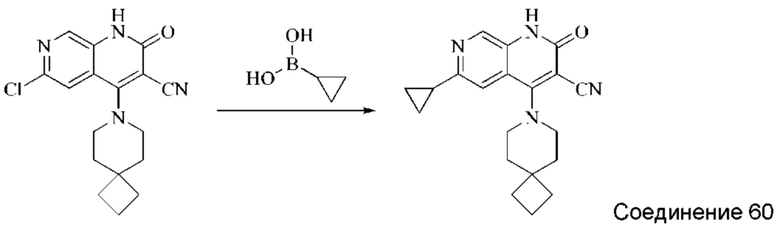
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,15 ммоль, 1,0 экв.) и циклопропилборную кислоту (53 мг, 0,61 ммоль, 4,0 экв.) растворяли в 1,4-диоксане (3 мл), после чего добавляли карбонат цезия (149 мг, 0,46 ммоль, 3,0 экв.), фосфат калия (33 мг, 0,15 ммоль, 1,0 экв.) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (11 мг, 0,015 ммоль, 0,1 экв.). В защитной атмосфере азота реакцию в полученной системе оставляли протекать при микроволновом нагреве до 120°С в течение 3 часов. Завершение реакции определяли способом ЖХ-МС. Реакционный раствор фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 20:1), получая продукт (4 мг, выход: 8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,83 (s, 1Н), 8,51 (s, 1Н), 7,43 (s, 1Н), 3,54 (s, 4Н), 2,23-2,25 (m, 1Н), 2,17-2,22 (m, 10Н), 0,93-0,95 (m, 2Н), 0,87-0,88 (m, 2Н).
Молекулярная формула: C20H22N4O, Молекулярная масса: 334,42, ЖХ-МС (Pos, m/z)=335,07 [М+Н]+.
Пример 43
Синтез 6-((2-(диметиламино)этил)(метил)амино)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 61)
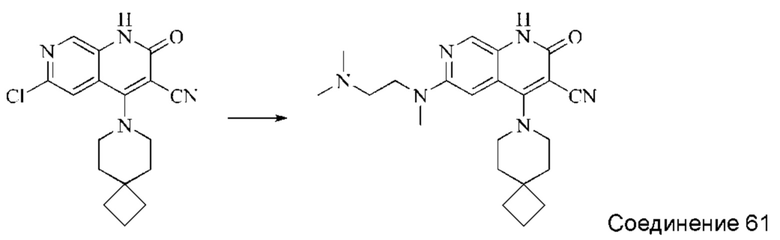
6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,15 ммоль, 1,0 экв.) и N1,N1,N2-триметилэтан-1,2-диамин (78 мг, 0,76 ммоль, 5,0 экв.) растворяли в тетрагидрофуране (2 мл) и ДМАА (0,2 мл), после чего добавляли трет-бутилат натрия (114 мг, 1,19 ммоль, 7,8 экв.), 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (6 мг, 0,012 ммоль, 0,08 экв.) и Pd2(dba)3 (6 мг, 0,006 ммоль, 0,04 экв.). Температуру повышали до 120°С, и реакцию в полученной системе оставляли протекать в течение 8 часов в атмосфере азота. Завершение реакции определяли способом ЖХ-МС. Реакционный раствор выливали в воду (10 мл) и экстрагировали ЭА. Органическую фазу сушили и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 10:1), получая продукт (14 мг, выход: 23%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,65 (s, 1Н), 8,28-8,32 (d, 1Н), 6,57-9,69 (d, 1Н), 3,80 (s, 3Н), 3,51 (s, 3Н), 2,96-3,02 (t, 3Н), 2,85 (s, 4Н), 1,80-2,03 (d, 10Н), 1,24-1,47 (t, 4Н).
Молекулярная формула: C22H30N6O, Молекулярная масса: 394,52, ЖХ-МС (Pos, m/z)=395,24 [М+Н]+.
Пример 44
Синтез 6-(гидроксиметил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 62)
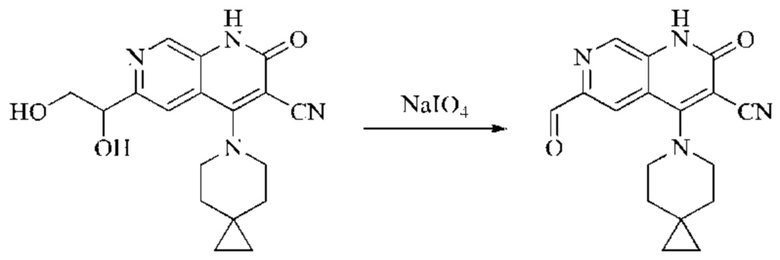
Перйодат натрия (634,3 мг, 2,966 ммоль, 1,0 экв.) растворяли в воде (10 мл), затем добавляли тетрагидрофуран (10 мл), охлаждали до 0°С на ледяной бане, после чего медленно добавляли 6-(1,2-дигидроксиэтил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (504,7 мг, 1,483 ммоль, 0,5 экв.) и перемешивали для протекания реакции в течение 3 часов при комнатной температуре. Завершение реакции определяли способом ТСХ. Смесь экстрагировали дихлорметаном (10 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл×2), сушили безводным сульфатом натрия и фильтровали с отсасыванием. Фильтрат концентрировали при пониженном давлении, получая желтый твердый продукт (350,0 мг, выход: 38,4%).
Этап 2: Синтез 6-(гидроксиметил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
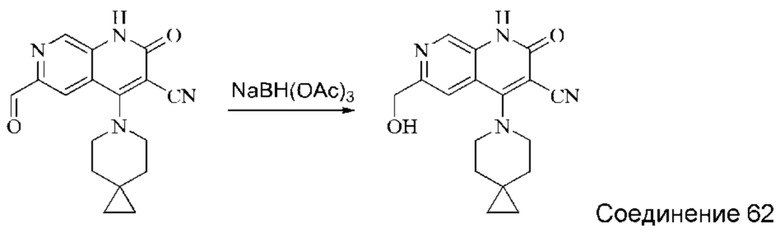
6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (100,0 мг, 0,324 ммоль, 1,0 экв.) растворяли в метаноле (15 мл), охлаждали до 0°С на ледяной бане, после чего добавляли триацетилборгидрид натрия (206,2 мг, 0,973 ммоль, 3,0 экв.) и перемешивали в течение ночи при комнатной температуре. Завершение реакции определяли способом ТСХ. Реакционный раствор тушили, добавляя H2O (10 мл), перемешивали в течение 30 минут и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 50:1-20:1), получая желтый твердый продукт (96,9 мг, выход: 96,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,97 (s, 1Н), 8,59 (s, 1Н), 7,72 (s, 1Н), 5,58-5,55 (t, 1Н), 4,62-4,60 (d, 2Н), 3,66-3,63 (t, 4Н), 1,63 (m, 4Н), 0,45 (m, 4Н).
Молекулярная формула: C17H18N4O2, Молекулярная масса: 310,36, ЖХ-МС (Pos, m/z)=311,08 [М+Н]+.
Пример 45
Синтез 6-метил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 63)
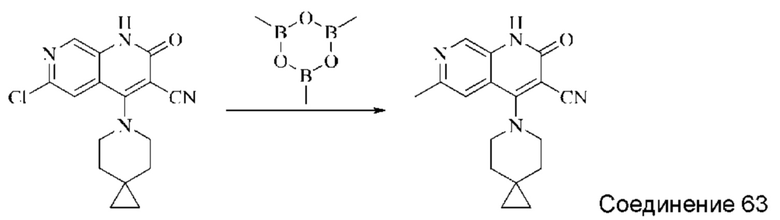
6-хлор-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,16 ммоль, 1,0 экв.) и триметилциклотриборан (160 мг, 0,64 ммоль, 4,0 экв., 50%) растворяли в 1,4-диоксане (3 мл), после чего добавляли карбонат цезия (155 мг, 0,48 ммоль, 3,0 экв.), фосфат калия (34 мг, 0,16 ммоль, 1,0 экв.) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (12 мг, 0,016 ммоль, 0,1 экв.). Температуру повышали до 120°С, и реакцию в полученной системе оставляли протекать в течение 3 часов в атмосфере азота. Завершение реакции определяли способом ЖХ-МС. Реакционный раствор фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 20:1), получая светло-желтый твердый продукт (10 мг, выход: 21%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,92 (s, 1Н), 8,55 (s, 1Н), 7,45 (s, 1Н), 3,63 (m, 4Н), 2,53 (s, 3Н), 1,63 (m, 4Н), 0,44 (m, 4Н).
Молекулярная формула: C17H18N4O, Молекулярная масса: 294,36, ЖХ-МС (Pos, m/z)=295,09 [М+Н]+.
Пример 46
Синтез 6-метокси-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 64)
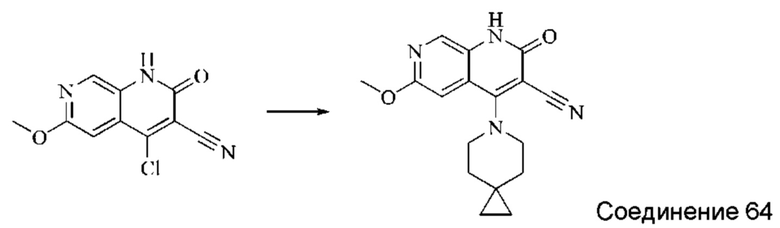
4-хлор-6-метокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (139,4 мг неочищенного продукта) растворяли в ДМФА (2 мл), после чего добавляли гидрохлорид 6-аза-спиро[2,5]октан (122,5 мг, 0,83 ммоль) и DIPEA (231,1 мг, 1,78 ммоль). Температуру повышали до 80°С, и реакцию в полученной системе оставляли протекать в течение 2 часов. Завершение реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, после чего добавляли этилацетат, промывали водой, промывали насыщенным солевым раствором и сушили безводным сульфатом натрия. Неочищенный продукт очищали способом препаративной тонкослойной хроматографии (ДХМ : МеОН = 20:1), получая продукт (2,28 мг, выход: 1,23%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,82 (s, 1Н), 8,32 (s, 1Н), 6,98 (s, 1Н), 3,88 (s, 3Н), 3,63-3,61 (t, 4Н), 1,60 (m, 4Н), 0,43 (m, 4Н).
Молекулярная формула: C17H18N4O2, Молекулярная масса: 310,36, ЖХ-МС (Pos, m/z)=311,1 [М+Н]+.
Пример 47
Синтез 6-метил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 65)
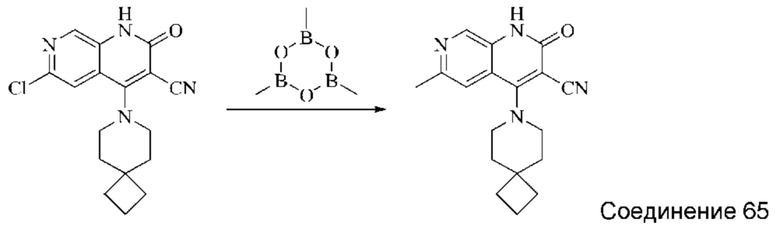
Материал 6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50 мг, 0,15 ммоль, 1,0 экв.) и триметилциклотриборан (153 мг, 0,61 ммоль, 4,0 экв., 50%) растворяли в 1,4-диоксане (3 мл), после чего добавляли карбонат цезия (148 мг, 0,47 ммоль, 3,0 экв.), фосфат калия (32 мг, 0,15 ммоль, 1,0 экв.) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (11 мг, 0,015 ммоль, 0,1 экв.). Температуру повышали до 120°С, и реакцию в полученной системе оставляли протекать в течение 3 часов в атмосфере азота. Завершение реакции определяли способом ЖХ-МС. Реакционный раствор фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 20:1), получая светло-желтый твердый продукт (15 мг, выход: 32%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,88 (s, 1Н), 8,55 (s, 1Н), 7,41 (s, 1Н), 3,52 (s, 4Н), 2,52 (s, 3Н), 1,81-1,86 (m, 10Н).
Молекулярная формула: C18H20N4O, Молекулярная масса: 308,39, ЖХ-МС (Pos, m/z)=309,07 [М+Н]+.
Пример 48
Синтез 6-метокси-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 66)
Этап 1: Синтез 6-метокси-2Н-пиридо[3,4-d][1,3]оксазин-2,4(1H)-диона
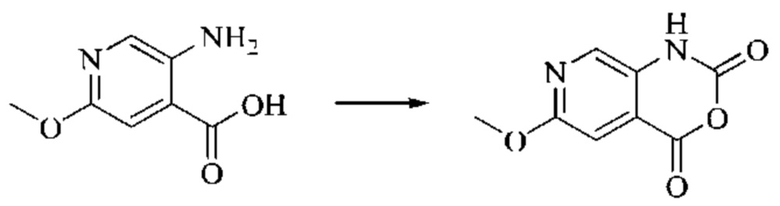
5-амино-2-метоксиизоникотиновую кислоту (500,0 мг, 2,97 ммоль) растворяли в ДМФА (3,5 мл). Температуру понижали до 0°С с помощью ледяной бани и добавляли CDI (819,7 мг, 5,05 ммоль). Температуру медленно повышали до комнатной температуры, и систему перемешивали в течение ночи. Завершение реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, получая продукт (700,0 мг неочищенного продукта), который непосредственно использовали в следующем этапе без очистки.
Этап 2: Синтез 4-гидрокси-6-метокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
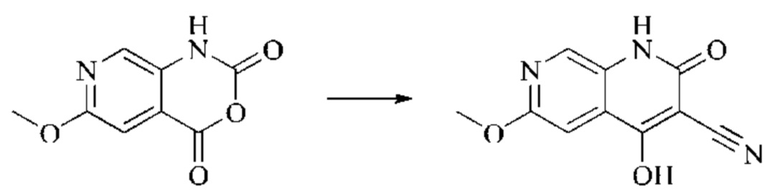
В колбу (50 мл) помещали 6-метокси-2Н-пиридо[3,4-d][1,3]оксазин-2,4(1Н)-дион (576,3 мг неочищенного продукта), после чего добавляли этилцианоацетат (353,0 мг, 3,12 ммоль) и триэтиламин (601,1 мг, 5,94 ммоль), нагревали до 150°С и оставляли реагировать в течение ночи. Завершение реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, после чего добавляли воду (10 мл), доводили до рН 1 соляной кислотой, что приводило к осаждению твердого вещества, и фильтровали с отсасыванием, получая продукт (500,0 мг неочищенного продукта).
Этап 3: Синтез 2,4-дихлор-6-метокси-1,7-нафтиридин-3-карбонитрила
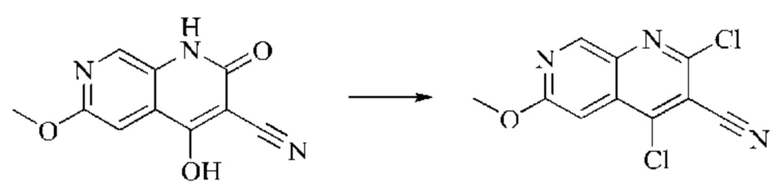
4-гидрокси-6-метокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (420,0 мг неочищенного продукта) растворяли в оксихлориде фосфора (1,34 г, 8,73 ммоль), после чего добавляли фосфопентат (phosphopentate) (807,9 мг, 3,88 ммоль), нагревали до 100°С и оставляли реагировать в течение ночи. Завершение реакции определяли способом ЖХ-МС. Реакционный раствор выливали в ледяную воду (15 мл), доводили до рН 1 добавлением насыщенного водного раствора бикарбоната натрия и фильтровали с отсасыванием, получая продукт (260,0 мг неочищенного продукта).
Этап 4: Синтез 4-хлор-6-метокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
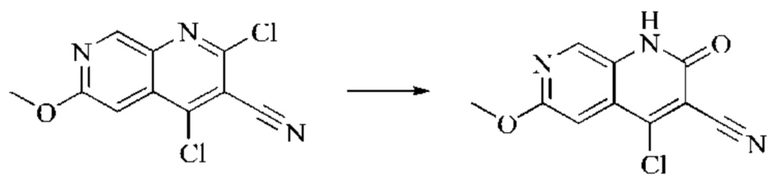
2,4-дихлор-6-метокси-1,7-нафтиридин-3-карбонитрил (100,0 мг неочищенного продукта) растворяли в ТФУК (2 мл) и воде (1 мл), нагревали до 100°С и оставляли реагировать в течение 2 часов. Поскольку по данным ТСХ реакция не завершилась, вновь добавляли ТФУК (2 мл), и реакцию в полученной системе оставляли протекать в течение 2 часов при 100°С. По завершении реакции, которое определяли способом ТСХ, реакционный раствор концентрировали при пониженном давлении, получая продукт (50,0 мг неочищенного продукта), который непосредственно использовали в следующем этапе без очистки.
Этап 5: Синтез 6-метокси-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
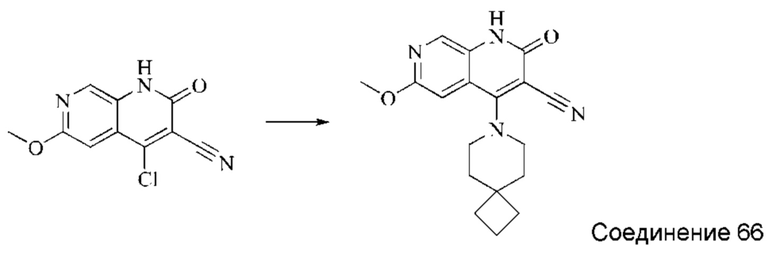
4-хлор-6-метокси-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50,0 мг неочищенного продукта) растворяли в ДМФА (2 мл), после чего добавляли гидрохлорид 7-аза-спиро[3,5]нонана (48,2 мг, 0,298 ммоль) и DIPEA (83,0 мг, 0,639 ммоль), нагревали до 80°С и оставляли реагировать в течение ночи. Завершение реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 80:1-40:1), получая продукт (4,1 мг, выход по завершении пяти этапов: 0,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,76 (s, 1Н), 8,31 (s, 1Н), 6,95 (d, 1Н), 3,88 (s, 3Н), 3,51-3,50 (t, 4Н), 1,93-1,80 (m, 10Н).
Молекулярная формула: C18H20N4O2, Молекулярная масса: 324,38, ЖХ-МС (Pos, m/z)=325,1 [М+Н]+.
Пример 49
Синтез 6-этил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 76)
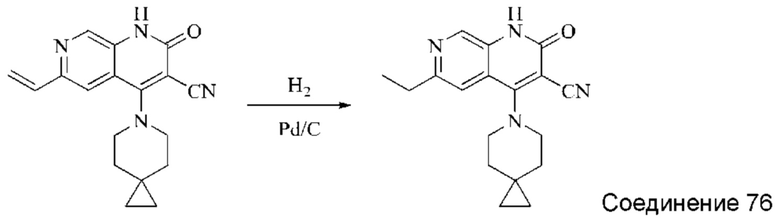
2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (50,0 мг, 0,163 ммоль) растворяли в метаноле (10 мл), после чего добавляли палладий на углеродном носителе (10,0 мг) и перемешивали в течение 30 минут при комнатной температуре в атмосфере водорода. Завершение реакции определяли способом ТСХ. Реакционный раствор фильтровали с отсасыванием. Остаток на фильтре промывали небольшим количеством метанола. Фильтрат концентрировали при пониженном давлении, получая продукт (48,9 мг, выход: 97,2%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,94 (s, 1Н), 8,60 (s, 1Н), 7,44 (s, 1Н), 3,65 (t, 4Н), 2,85-2,79 (m, 2Н), 1,63 (s, 4Н), 1,23-1,27 (t, 3Н), 0,44 (m, 4Н).
Молекулярная формула: C18H20N4O, Молекулярная масса: 308,39, ЖХ-МС (Pos, m/z)=309,08 [М+Н]+.
Пример 50
Синтез 2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3,6-дикарбонитрила (Соединение 77)
Этап 1: Синтез 6-((гидроксиимино)метил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
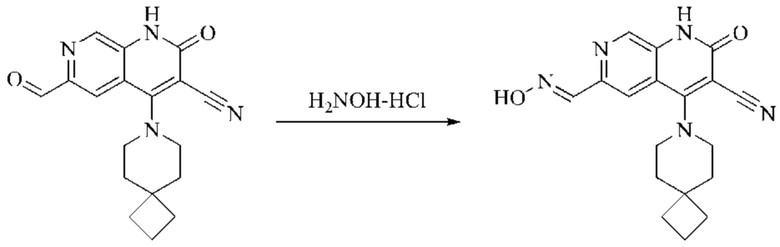
6-формил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (200 мг, 0,62 ммоль, 1,0 экв.), гидрохлорид гидроксиламина (66 мг, 0,93 ммоль, 1,5 экв.) и карбонат калия (112 мг, 0,8 ммоль, 1,3 экв.) растворяли в метаноле (4,5 мл) и воде (1,5 мл), перемешивали в течение 2 часов при 65°С, охлаждали и фильтровали. Осадок на фильтре промывали водой (2 мл × 2), получая белый твердый продукт (170 мг, выход: 81,36%).
Этап 2: Синтез 2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3,6-динитрила
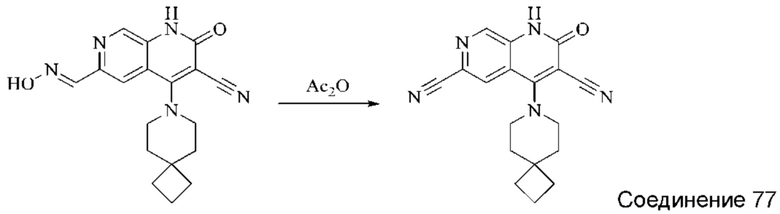
6-((гидроксиимино)метил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (60 мг, 0,18 ммоль, 1,0 экв.) растворяли в уксусном ангидриде (3 мл) и перемешивали в течение 2 часов при 120°С. Реакционный раствор охлаждали и фильтровали, получая желтый твердый продукт (18 мг, выход: 31,3%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,25 (s, 1Н), 8,70 (s, 1Н), 8,08 (s, 1Н), 3,55 (m, 4Н), 1,82-1,91 (m, 10Н).
Молекулярная формула: C18H17N5O, Молекулярная масса: 319,14, ЖХ-МС (Neg, m/z) = 318,10 [М-Н]-.
Пример 51
Синтез трифторацетата 6-(аминометил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 78)
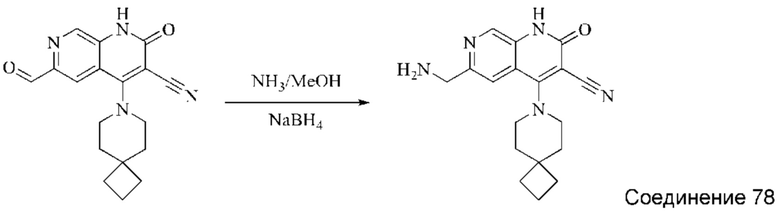
6-формил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (100 мг, 0,31 ммоль, 1,0 экв.) и тетраизопропилтитанат (440,2 мг, 1,55 ммоль, 5,0 экв.) растворяли в метанольном растворе аммиака (10 мл), перемешивали в течение 18 часов при комнатной температуре, охлаждали до 0°С на ледяной бане, затем медленно добавляли боргидрид натрия (350 мг, 1,24 ммоль, 4,0 экв.) и перемешивали в течение 1 часа при комнатной температуре. Завершение реакции определяли способом ТСХ. Для прекращения реакции добавляли воду (1 мл). Неочищенный продукт очищали колоночной хроматографией с обращенной фазой (ацетонитрил : вода : трифторуксусная кислота = 30:100:0,05%), получая желтый твердый продукт (16,27 мг, выход: 16,2%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,09 (s, 1Н), 8,67 (s, 1Н), 8,29 (br. s, 1Н), 7,66 (s, 1Н), 4,25 (m, 2Н), 3,63 (m, 4Н), 1,88-1,83 (s, 10Н).
Молекулярная формула: C18H21N50, Молекулярная масса: 323,17, ЖХ-МС (Neg, m/z) = 324,14 [М-Н]-.
Пример 52
Синтез 6-(метилтио)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 79)
Этап 1: Синтез 4-хлор-6-(метилтио)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
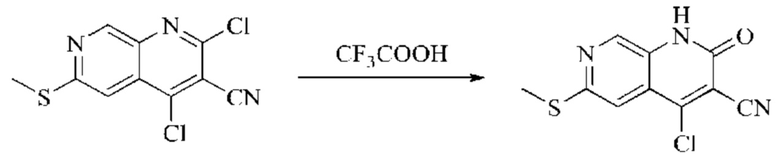
2,4-дихлор-6-(метилтио)-1,7-нафтиридин-3-карбонитрил (180,0 мг, 0,666 ммоль) растворяли в смеси трифторацетата (4,0 мл) и воды (1,0 мл) и перемешивали в течение 1 часа при 100°С. Завершение реакции определяли способом ТСХ. Смесь концентрировали при пониженном давлении, получая темно-красное твердое вещество (180 мг неочищенного продукта), которое непосредственно использовали в следующем этапе.
Этап 2: Синтез 6-(метилтио)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
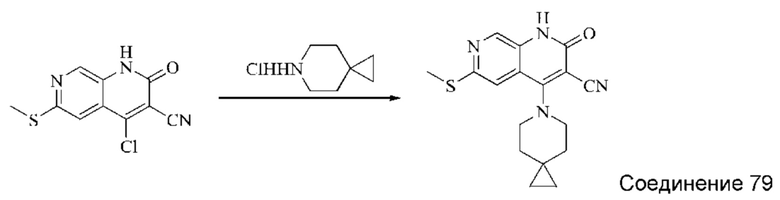
4-хлор-6-(метилтио)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (167,7 мг, 0,666 ммоль, 1,0 экв.), гидрохлорид 6-аза-спиро[2,5]октана (118,1 мг, 0,800 ммоль, 1,2 экв.) и DIPEA (688,9 мг, 5,331 ммоль, 8,0 экв.) растворяли в ДМФА (5 мл) и перемешивали в течение 3 часов при 80°С. Завершение реакции определяли способом ТСХ. Смесь концентрировали при пониженном давлении для удаления большей части ДМФА, растворяли в дихлорметане (5 мл) и концентрировали при пониженном давлении. Этот процесс повторяли 3 раза. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 100:1-60:1), получая желтый твердый продукт (21,0 мг, выход: 9,66%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,95 (s, 1Н), 8,56 (s, 1Н), 7,37 (s, 1Н), 3,65-3,63 (t, 4Н), 2,56 (s, 4Н), 1,62 (m, 4Н), 0,44 (m, 3Н).
Молекулярная формула: C17H18N4OS, Молекулярная масса: 326,42, ЖХ-МС (Pos, m/z) = 327,20 [М+Н]+.
Пример 53
Синтез 2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,5-нафтиридин-3-карбонитрила (Соединение 80)
Этап 1: Синтез 2Н-пиридо[3,2-d][1,3]оксазин-2,4(1H)-диона
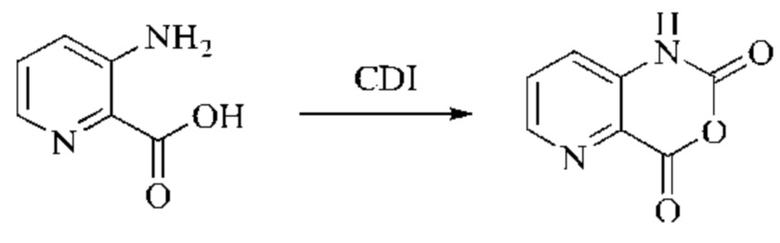
3-аминопиколиновую кислоту (460 мг, 3,3 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (8 мл), затем порциями при охлаждении на ледяной бане добавляли N,N-карбонилдиимидазол (920 мг, 5,7 ммоль, 1,7 экв.), перемешивали при комнатной температуре в течение 24 часов и фильтровали. Фильтрат концентрировали, получая желтый твердый продукт (235 мг, неочищенный продукт).
Этап 2: Синтез 4-гидрокси-2-оксо-1,2-дигидро-1,5-нафтиридин-3-карбонитрила
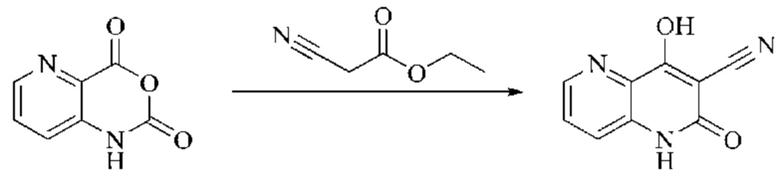
Этилцианоацетат (172 мг, 1,5 ммоль, 1,05 экв.) растворяли в тетрагидрофуране (4 мл), затем порциями при охлаждении ледяной баней добавляли гидрид натрия (128 мг, 3,2 ммоль, 2,2 экв.) и кипятили с обратным холодильником в течение 30 минут. Во время кипячения добавляли 2Н-пиридо[3,2-d][1,3]оксазин-2,4-(1Н)-дион (235 мг, 1,43 ммоль, 1,0 экв.), и реакцию продолжали в течение 18 часов. Реакционный раствор охлаждали, выливали в ледяную баню, и доводили до рН, составляющего от 3 до 4, в результате чего выпадал твердый осадок, и фильтровали, получая черный твердый продукт (130 мг, выход: 48,5%).
Этап 3: Синтез 4-хлор-2-оксо-1,2-дигидро-1,5-нафтиридин-3-карбонитрила
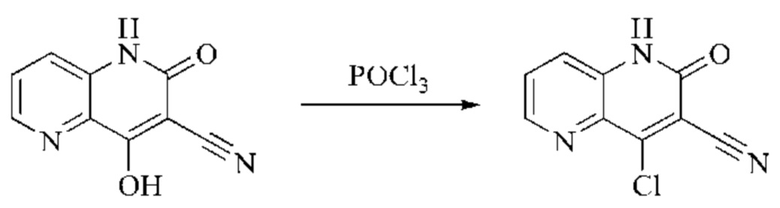
4-гидрокси-2-оксо-1,2-дигидро-1,5-нафтиридин-3-карбонитрил (130 мг, 0,69 ммоль, 1,0 экв.) растворяли в оксихлориде фосфора (3 мл) и перемешивали при 80°С в течение 2 часов. Реакционный раствор охлаждали, выливали в ледяную баню, в результате чего образовывался черный твердый осадок, и фильтровали, получая продукт в виде твердого черного вещества (60 мг, неочищенный продукт).
Этап 4: Синтез 2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,5-нафтиридин-3-карбонитрила
4-хлор-2-оксо-1,2-дигидро-1,5-нафтиридин-3-карбонитрил (38 мг, 0,18 ммоль, 1,0 экв.) и гидрохлорид 6-аза-спиро[2,5]октана (30 мг, 0,20 ммоль, 1,1 экв.) растворяли в ДМФА (2 мл), после чего добавляли триэтиламин (73 мг, 0,72 ммоль, 4,0 экв.). Реакционный раствор нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали и очищали способом колоночной хроматографии с обращенной фазой (ацетонитрил : вода : раствор аммиака = 30:100:0,05%), получая белый твердый продукт (15,1 мг, выход: 30,0%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,66 (s, 1Н), 8,49 (d, J=4 Гц, 1Н), 7,67-7,64 (m, 1Н), 7,60-7,58 (m, 1Н), 3,88 (m, 4Н), 1,60 (m, 4Н), 0,41 (m, 4Н).
Молекулярная формула: C16H16N40, Молекулярная масса: 280,33, ЖХ-МС (Pos, m/z) = 281,35 [М+Н]+.
Пример 54
Синтез 4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединение 81)
Неочищенный материал, 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (300 мг, 1,46 ммоль, 1,0 экв.), растворяли в ДМФА (3 мл) и добавляли DIPEA (1,13 г, 8,76 ммоль, 6,0 экв.) и трифторацетат 4-метокси-4-метилпиперидина (496 мг, 2,04 ммоль, 1,4 экв.). Реакцию проводили при 80°С в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор экстрагировали, добавляя воду (10 мл) и дихлорметан (10 мл × 3). Органическую фазу промывали водой (10 мл × 3), сушили безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 20:1), получая желтый твердый продукт (210 мг, выход: 48%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,99 (s, 1Н), 8,65 (s, 1Н), 8,33-8,34 (d, 1Н), 7,61-7,62 (d, 1Н), 3,59-3,62 (m, 4Н), 3,19 (s, 3Н), 1,90-1,92 (d, 2Н), 1,77-1,82 (m, 2Н), 1,21 (s, 3Н).
Молекулярная формула: C16H18N4O2, Молекулярная масса: 298,35, ЖХ-МС (Pos, m/z) = 299,14 [М+Н]+.
Пример 55
Синтез 6-(2-гидроксипропан-2-ил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединение 82)
Этап 1: Синтез 6-ацетил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
6-(1-гидроксиэтил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (220 мг, 0,65 ммоль, 1,0 экв.) растворяли в дихлорметане (4 мл), после чего добавляли окислитель Десса-Мартина (552 мг, 1,3 ммоль, 2,0 экв.) и перемешивали при комнатной температуре в течение 18 часов. Добавляли воду (10 мл) и дихлорметан (30 мл). Органическую фазу отделяли, промывали насыщенным солевым раствором, сушили и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан : метанол = 30:1), получая продукт (170 мг, выход: 77,8%).
Этап 2: Синтез 6-ацетил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
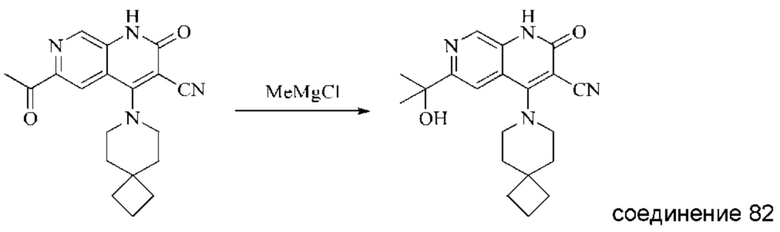
6-ацетил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (60 мг, 0,18 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (2 мл). В защитной атмосфере азота при -10°С добавляли метилмагнийхлорид (3,0 моль/л раствор в тетрагидрофуране, 0,12 мл, 0,37 ммоль, 2,0 экв.), и смесь нагревали до 0°С и перемешивали в течение 2 часов, тушили насыщенным водным раствором хлорида аммония (5 мл) при охлаждении ледяной баней, после чего добавляли этилацетат (20 мл). После разделения жидкостей органическую фазу сушили и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан : метанол = 60:1), получая продукт (17,4 мг, выход: 27,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 8,74 (s, 1Н), 7,73 (s, 1Н), 3,65-3,84 (m, 4Н), 1,84-2,07 (m, 10Н), 1,47-1,77 (m, 6Н).
Молекулярная формула: C20H24N4O2, Молекулярная масса: 352,19, ЖХ-МС (Neg, m/z) = 351,15 [М-Н]-.
Пример 56
Синтез 3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-карбоновой кислоты (соединение 83)
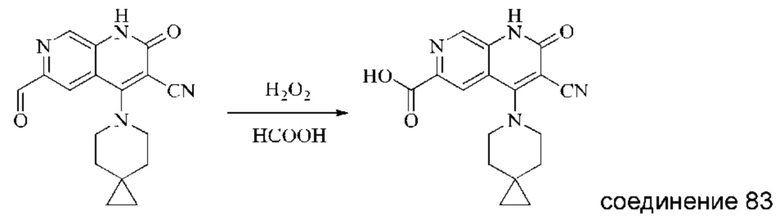
6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (92,8 мг, 0,301 ммоль, 1,0 экв.) растворяли в муравьиной кислоте (10 мл) и охлаждали до 0°C с помощью ледяной бани. Затем добавляли по каплям пероксид водорода (30%, 2,4 мл, 24,08 ммоль, 80,0 экв.). По окончании добавления по каплям, реакционную смесь перемешивали в течение ночи при охлаждении ледяной баней. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор фильтровали, осадок на фильтре промывали водой (3 мл × 3) и сушили при 40°С в течение 2 часов, получая желтый твердый продукт (46,6 мг, выход: 47,7%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 13,24 (s, 1Н), 12,32 (s, 1Н), 8,70 (s, 1Н), 8,34 (s, 1Н), 3,68 (t, 4Н), 1,63 (s, 4Н), 0,46 (s, 4Н).
Молекулярная формула: C17H16N4O3, Молекулярная масса: 324,34, ЖХ-МС (Pos, m/z) = 325,10 [М+Н]+.
Пример 57
Синтез 6-(2-гидроксипропан-2-ил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединение 84)
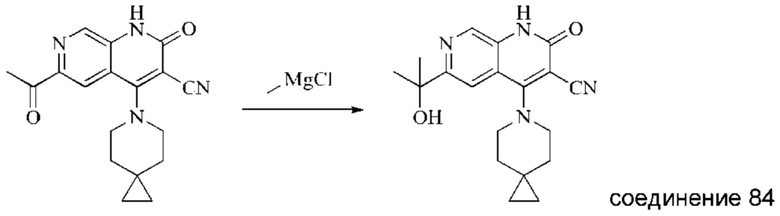
6-ацетил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (277,6 мг, 0,86 ммоль, 1,0 экв.) растворяли в безводном тетрагидрофуране (20 мл) и охлаждали до -10°С в атмосфере азота. Затем добавляли по каплям раствор метилмагнийхлорида в тетрагидрофуране (3 моль/л, 1,2 мл, 3,44 ммоль, 4,0 экв.). По окончании добавления по каплям смесь перемешивали для протекания реакции в течение времени, составляющего от 4 до 5 часов, при охлаждении ледяной баней (0°С). Завершение протекания реакции определяли способом ТСХ. Реакцию останавливали добавлением насыщенного водного раствора NH4Cl (10 мл). Смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу отделяли от реакционной смеси, промывали насыщенным солевым раствором (10 мл × 2), сушили безводным сульфатом натрия, фильтровали с отсасыванием и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 40:1-15:1), получая желтый твердый продукт (257,4 мг, выход: 88,3%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,93 (s, 1Н), 8,60 (s, 1Н), 7,95 (s, 1Н), 5,35 (s, 1Н), 3,66-3,64 (t, 4Н), 1,63 (s, 4Н), 1,45 (s, 6Н), 0,45 (s, 4Н).
Молекулярная формула: C19H22N4O2, Молекулярная масса: 338,41, ЖХ-МС (Pos, m/z) = 339,20 [М+Н]+.
Пример 58
Синтез 6-(гидроксиметил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединение 85)
Этап 1: Синтез 6-формил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
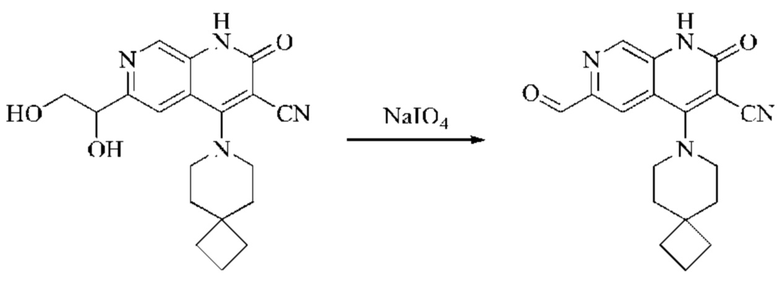
Перйодат натрия (1,63 г, 7,6 ммоль, 1,0 экв.) растворяли в воде (50 мл), после чего добавляли тетрагидрофуран (50 мл), охлаждали до 0°С ледяной баней, затем медленно добавляли 6-(1,2-дигидроксиэтил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (2,7 г, 7,6 ммоль, 0,5 экв.) и перемешивали при комнатной температуре в течение 3 часов. Завершение протекания реакции определяли способом ТСХ. Смесь экстрагировали дихлорметаном (100 мл × 3). Органическую фазу промывали насыщенным солевым раствором (40 мл × 2), сушили безводным сульфатом натрия, фильтровали с отсасыванием, концентрировали при пониженном давлении, получая продукт (857,0 мг, выход: 34,89%).
Этап 2: Синтез 6-(гидроксиметил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
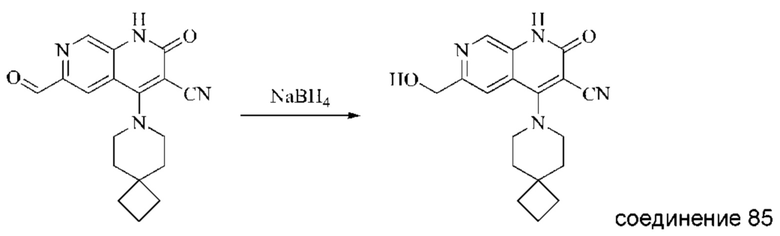
6-формил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (100,0 мг, 0,324 ммоль, 1,0 экв.) растворяли в метаноле (15 мл), охлаждали до 0°С с помощью ледяной бани, после чего добавляли боргидрид натрия (36,8 мг, 0,973 ммоль, 3,0 экв.) и перемешивали при комнатной температуре в течение ночи. Завершение протекания реакции определяли способом ТСХ, и реакцию останавливали добавлением H2O (10 мл). Реакционный раствор концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 50:1-20:1), получая продукт (96,9 мг, выход: 96,3%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,97 (s, 1Н), 8,58 (s, 1Н), 7,68 (s, 1Н), 5,58-5,55 (m, 1Н), 4,60 (d, 2Н), 3,66-3,63 (t, 4Н), 1,94-1,81 (s, 10Н).
Молекулярная формула: C18H20N4O2, Молекулярная масса: 324,16, ЖХ-МС (Neg, m/z) = 323,15 [М-Н]-.
Пример 59
Синтез 6-(1-гидрокси-2-метилпропан-2-ил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (соединение 86)
Этап 1: Синтез 1-трет-бутил-3-этил-2-(4-(метоксикарбонил)-5-нитропиридин-2-ил)малоната
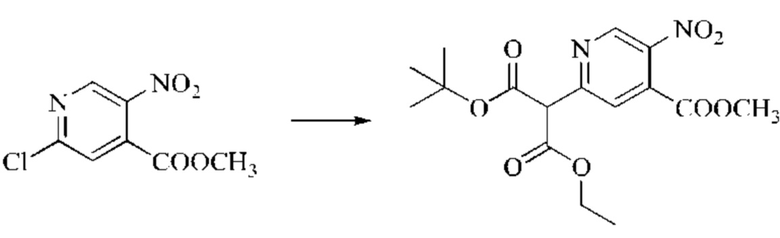
Исходное вещество, трет-бутил-этилмалонат (41,7 г, 222 ммоль, 1,2 экв.), растворяли в безводном ДМФА (100 мл), охлаждали до 0°C с помощью ледяной бани, перемешивали в течение получаса, после чего добавляли NaH (14,8 г, 370 ммоль, 2 экв.), перемешивали в течение получаса, затем медленно добавляли метил-2-хлор-5-нитроизоникотинат (40,0 г, 185 ммоль, 1,0 экв.) и перемешивали при комнатной температуре в течение 5 часов. Завершение протекания реакции определяли способом ТСХ, и реакцию останавливали добавлением H2O при 0°С. Реакционный раствор концентрировали и экстрагировали ЭА (3×500 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 10:1), получая продукт (35 г, выход: 51,4%).
Этап 2: Синтез метил-2-(2-этокси-2-оксоэтил)-5-нитроизоникотината
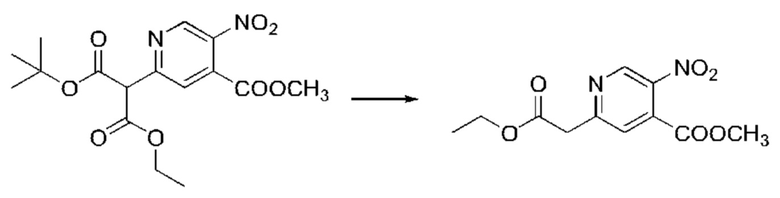
Промежуточный 1-трет-бутил-3-этил-2-(4-(метоксикарбонил)-5-нитропиридин-2-ил)малонат (36,0 г, 97,8 ммоль, 1,0 экв.) растворяли в трифторуксусной кислоте (55,7 г, 0,5 моль, 5 экв.) при 0°С, перемешивали при комнатной температуре в течение получаса, и завершение протекания реакции определяли способом ТСХ. Насыщенный водный раствор бикарбоната натрия добавляли по каплям при 0°С, перемешивали в течение получаса и экстрагировали ЭА (3×500 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 10:1), получая продукт (23 г, выход: 87,7%).
Этап 3: Синтез метил-2-(1-этокси-2-метил-1-оксопропан-2-ил)-5-нитроизоникотината
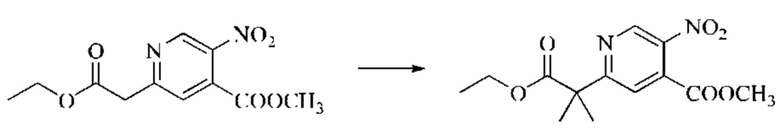
Промежуточный метил-2-(2-этокси-2-оксоэтил)-5-нитроизоникотинат (10,7 г, 40,2 ммоль, 1,0 экв.) растворяли в ДМФА (100 мл), перемешивали при 0°С в течение получаса, после чего добавляли NaH (1,1 г, 44,2 ммоль, 1,1 экв.), медленно нагревали до комнатной температуры, перемешивали в течение получаса, затем охлаждали до 0°С, затем медленно по каплям добавляли CH3I (6,2 г, 44,2 ммоль, 1,1 экв.), нагревали до комнатной температуры и перемешивали в течение 1 часа. Завершение реакции исходных материалов отслеживали с помощью ТСХ, и температуру понижали до 0°С. Повторяли описанную выше операцию. Завершение протекания реакции определяли способом ЖХ-МС. Реакцию останавливали добавлением H2O при 0°С. Реакционный раствор перемешивали в течение получаса, концентрировали и экстрагировали ЭА (3×100 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 10:1), получая продукт (5,3 г, выход: 44,5%).
Этап 4: Синтез метил-5-амино-2-(1-этокси-2-метил-1-оксопропан-2-ил)изоникотината
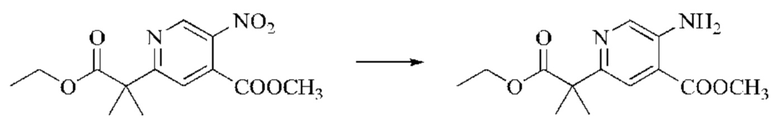
Промежуточный метил-2-(1-этокси-2-метил-1-оксопропан-2-ил)-5-нитроизоникотинат (5,0 г, 16,9 ммоль, 1,0 экв.) растворяли в метаноле (30 мл) и к нему добавляли палладий на углеродном носителе (500,0 мг). Атмосферу вытесняли из системы водородом, и реакцию проводили при комнатной температуре. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор фильтровали через целит. Фильтрат концентрировали, после чего добавляли этилацетат (30 мл), сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении, получая продукт (4,3 г, выход: 95,6%).
Этап 5: Синтез метил-5-(2-цианоацетамидо)-2-(1-этокси-2-метил-1-оксопропан-2-ил)изоникотината
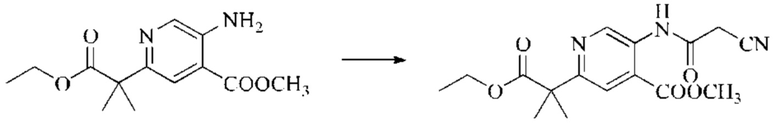
Промежуточный метил-5-амино-2-(1-этокси-2-метил-1-оксопропан-2-ил)изоникотинат (4,0 г, 15,0 ммоль, 1,0 экв.) и цианоуксусную кислоту (2,6 г, 30,0 ммоль, 2,0 экв.) растворяли в ДМФА (20 мл), после чего при 0°С добавляли EDCI (8,6 г, 45,0 ммоль, 3,0 экв.) и оставляли реагировать при комнатной температуре. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор концентрировали, затем добавляли воду и экстрагировали ЭА (3×100 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 2:1), получая продукт (3,4 г, выход: 68,1%).
Этап 6: Синтез этил 2-(3-циано-4-гидрокси-2-оксо-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионата
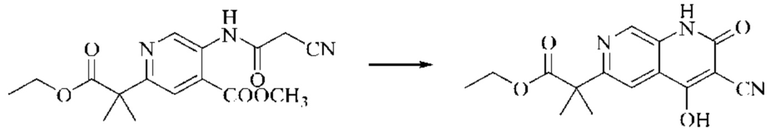
Промежуточный метил-5-(2-цианоацетамидо)-2-(1-этокси-2-метил-1-оксопропан-2-ил)изоникотинат (4,0 г, 12,0 ммоль, 1,0 экв.) и этилат натрия (1,8 г, 26,4 ммоль, 2,2 экв.) последовательно добавляли в этанол (30 мл) при 0°С, и перемешивали при комнатной температуре. Завершение протекания реакции определяли способом ЖХ-МС. Полученный как указано выше реакционный раствор добавляли по каплям в ледяную воду (150 мл), доводили до рН 2, что приводило к осаждению большого количества твердого вещества, затем фильтровали, и осадок на фильтре сушили, получая продукт (3,4 г, выход: 94,1%).
Этап 7: Синтез этил-2-(4-хлор-3-циано-2-оксо-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионата
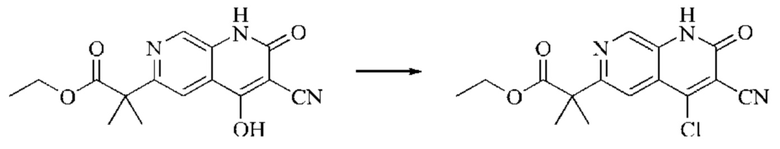
Промежуточный этил-2-(3-циано-4-гидрокси-2-оксо-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионат (2,0 г, 6,6 ммоль, 1,0 экв.) растворяли в оксихлориде фосфора (5 мл) при 0°С, и проводили реакцию при 100°С в течение получаса. Завершение протекания реакции определяли способом ЖХ-МС, и температуру понижали до 0°С. Реакционный раствор тушили, медленно добавляя воду, и экстрагировали ЭА (3×80 мл). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали. К неочищенному продукту добавляли трифторуксусную кислоту (3 мл) и кипятили с обратным холодильником. Завершение реакции дихлорзамещенного продукта отслеживали способом ЖХ-МС. Температуру понижали до 0°С. К реакционному раствору медленно по каплям добавляли воду. Смесь экстрагировали ЭА (3×20 мл). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали, получая продукт (1,3 г, выход: 61,7%).
Этап 8: Синтез этил-2-(3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионата
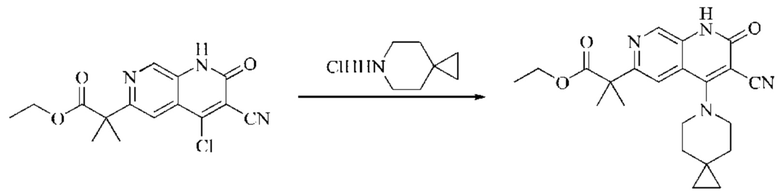
Промежуточный этил-2-(4-хлор-3-циано-2-оксо-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионат (800,0 мг, 2,5 ммоль, 1,0 экв.) растворяли в ДМФА (5 мл), затем последовательно добавляли гидрохлорид 6-аза-спиро[2,5]октана (404,2 мг, 2,75 ммоль, 1,1 экв.) и DIPEA (1,9 г, 15,0 ммоль, 6,0 экв.) и оставляли реагировать при 80°С в течение получаса. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали, после чего добавляли воду и экстрагировали ЭА (3×60 мл). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 3:2), получая продукт (500,0 мг, выход: 50,7%).
Этап 9: Синтез 6-(1-гидрокси-2-метилпропан-2-ил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
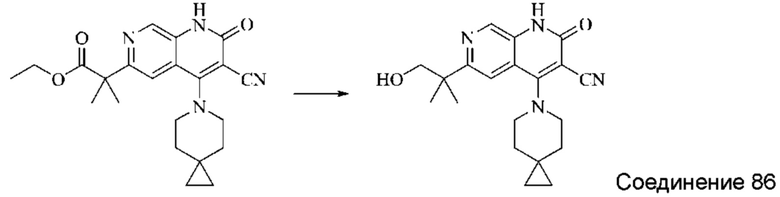
Промежуточный этил-2-(3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионат (400,0 мг, 1,01 ммоль, 1,0 экв.) растворяли в безводном 2-метилтетрагидрофуране (5 мл), при -60°С в атмосфере азота добавляли по каплям DIBAL-H (1,5 моль/л раствор в толуоле, 3,4 мл, 5,05 ммоль, 5,0 экв.), затем постепенно нагревали до комнатной температуры, перемешивали в течение 6 часов, и тушили, добавляя по каплям воду при 0°С, затем концентрировали и экстрагировали ЭА (3×60 мл). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 3:2), получая желтый твердый продукт (150,0 мг, выход: 42,2%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,93 (s, 1Н), 8,64 (s, 1Н), 7,58 (s, 1Н), 4,65 (t, 1Н), 3,63-3,66 (m, 4Н), 3,53 (d, 2Н), 1,63 (s, 4Н), 1,28 (s, 6Н), 0,45 (s, 4Н).
Молекулярная формула: C20H24N4O2, Молекулярная масса: 352,44, ЖХ-МС (Pos, m/z) = 353 [М+Н]+.
Пример 60
Синтез 6-(1-гидроксиэтил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 87)
Этап 1: Синтез 6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
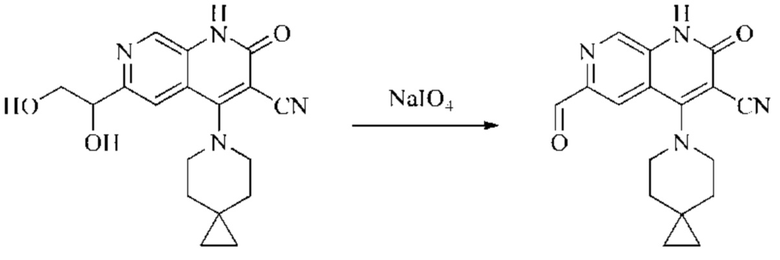
Перйодат натрия (4,00 г, 18,70 ммоль, 2,0 экв.) растворяли в воде (10 мл), после чего добавляли тетрагидрофуран (100 мл), охлаждали до 0°С на ледяной бане, затем медленно добавляли 6-(1,2-дигидроксиэтил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (3,18 г, 9,35 ммоль, 1,0 экв.) и перемешивали при комнатной температуре в течение 3 часов. Завершение протекания реакции определяли способом ТСХ, и реакционный раствор экстрагировали дихлорметаном (20 мл × 3). Органическую фазу промывали насыщенным солевым раствором (20 мл × 2), сушили безводным сульфатом натрия и фильтровали с отсасыванием, затем фильтрат концентрировали при пониженном давлении, получая желтый твердый продукт (1,50 г, выход: 52,0%).
Этап 2: синтез 6-(1-гидроксиэтил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
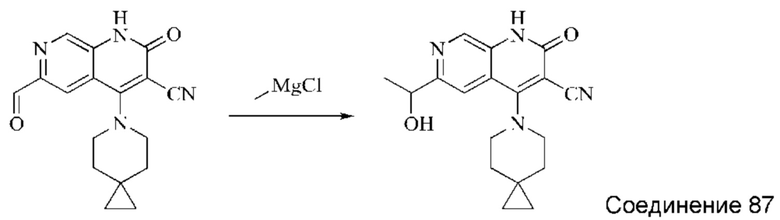
6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (1,00 г, 3,24 ммоль, 1,0 экв.) растворяли в безводном тетрагидрофуране (50 мл), охлаждали до -10°С в атмосфере азота, после чего по каплям добавляли раствор метилмагнийхлорида в тетрагидрофуране (3 моль/л, 4,3 мл, 12,97 ммоль, 4,0 экв.) и затем перемешивали на ледяной бане для протекания реакции в течение периода, составляющего от 4 до 5 часов. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор тушили добавлением насыщенного водного раствора NH4Cl (20 мл) и экстрагировали дихлорметаном (20 мл × 3). Органическую фазу промывали насыщенным солевым раствором (20 мл × 2), сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ : МеОН = 40:1-15:1), получая желтый твердый продукт (0,95 г, выход: 90,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,96 (s, 1Н), 8,59 (s, 1Н), 7,78 (s, 1Н), 5,50-5,49 (d, 1Н), 4,81-4,77 (m, 1Н), 3,65 (s, 4Н), 1,63 (s, 4Н), 1,40-1,38 (d, 3Н), 0,45 (s, 4Н).
Молекулярная формула: C18H20N4O2, Молекулярная масса: 324,38, ЖХ-МС (Pos, m/z) = 325,10 [М+Н]+.
Пример 61
Синтез 6-(циклопропил(гидрокси)метил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 90)
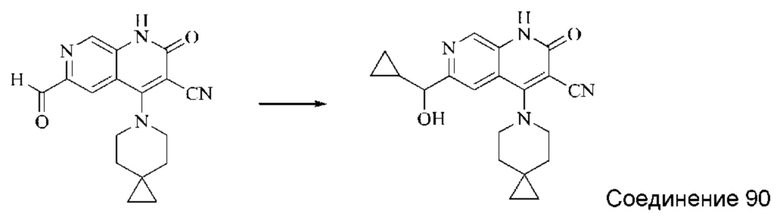
Промежуточный 6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (200,0 мг, 0,65 ммоль, 1,0 экв.) растворяли в сухом ТГФ (5 мл) и к полученному таким образом реакционному раствору при -10°С, в атмосфере азота, добавляли циклопропилмагнийбромид (1,95 мл, 1,95 ммоль, 3,0 экв.). Реакционнуюю смесь медленно нагревали до 0°С и перемешивали в течение 1 часа. По завершении реакции, реакционный раствор тушили добавлением насыщенного водного раствора хлорида аммония, концентрировали и экстрагировали этилацетатом (3×30 мл). Органические фазы объединяли и сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (DCE (дихлорэтан): МеОН = 100:1-75:1), получая беловатый твердый продукт (92 мг, выход: 40,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,96 (br, 1Н), 8,60 (s, 1Н), 7,74 (s, 1Н), 5,44 (s, 1Н), 4,24-4,27 (m, 1Н), 3,65 (t, 4Н), 1,63 (s, 4Н), 1,12-1,18 (m, 1Н), 0,45 (s, 4Н), 0,40-0,43 (m, 4Н).
Молекулярная формула: C20H22N4O2, Молекулярная масса: 350,42, ЖХ-МС (m/z) = 351 [М+Н]+.
Пример 62
Синтез промежуточного трифторацетата 4-метокси-4-метилпиперидина
Этап 1: Синтез трет-бутил-4-гидрокси-4-метилпиперидин-1-карбоксилата
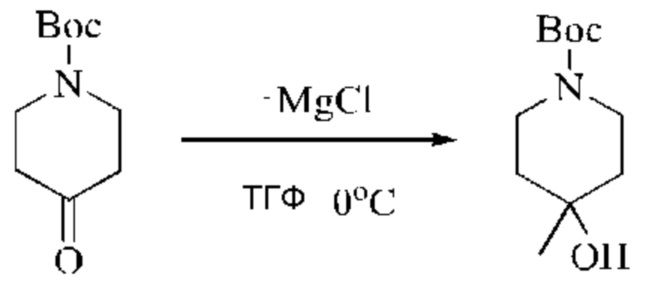
Неочищенный материал трет-бутил-4-оксопиперидин-1-карбоксилат (5,0 г, 25 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (25 мл), после чего при 0°С, в атмосфере азота, добавляли метилмагнийхлорид (9 мл, 27 ммоль, 1,1 экв.). Реакцию проводили в течение 2 часов, и завершение протекания реакции определяли способом ТСХ. Величину рН реакционного раствора доводили до 4, добавляя разбавленную соляную кислоту, после чего добавляли воду (30 мл) и экстрагировали этилацетатом (30 мл × 3). Органическую фазу сушили, фильтровали и концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 5:1), получая продукт (5,2 г, выход: 96%).
Этап 2: Синтез трет-бутил-4-метокси-4-метилпиперидин-1-карбоксилата
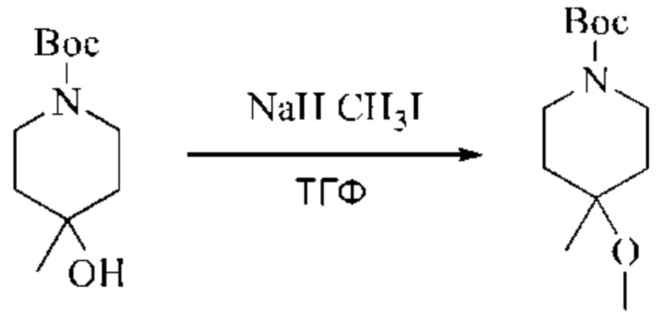
Промежуточный трет-бутил-4-гидрокси-4-метилпиперидин-1-карбоксилат (500 мг, 2,32 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (5 мл), после чего добавляли гидрид натрия (186 мг, 4,64 ммоль, 2,0 экв.), и реакция протекала в течение 1 часа, после чего добавляли йодметан (659 мг, 4,64 ммоль, 2,0 экв.), оставляли реагировать в течение 8 часов. Завершение протекания реакции определяли способом ТСХ. В реакционную колбу добавляли воду (10 мл), и реакционный раствор экстрагировали этилацетатом (20 мл × 3). Органическую фазу сушили, фильтровали и концентрировали при пониженном давлении; неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 20:1), получая продукт (306 мг, выход: 57%).
Этап 3: Синтез трифторацетата 4-метокси-4-метилпиперидина
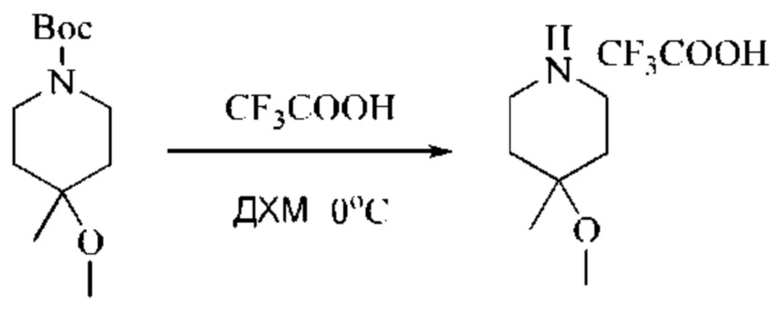
Промежуточный трет-бутил-4-метокси-4-метилпиперидин-1-карбоксилат (500 мг, 2,18 ммоль, 1,0 экв.) растворяли в дихлорметане (4 мл), после чего при 0°С добавляли трифторуксусную кислоту (3 мл), и оставляли реагировать в течение 1 часа. Завершение протекания реакции определяли способом ТСХ; реакционный раствор концентрировали при пониженном давлении, получая продукт (530 мг, выход: 100%).
Пример 63
Синтез 6-(1-гидроксипроп-2-ин-1-ил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 91)
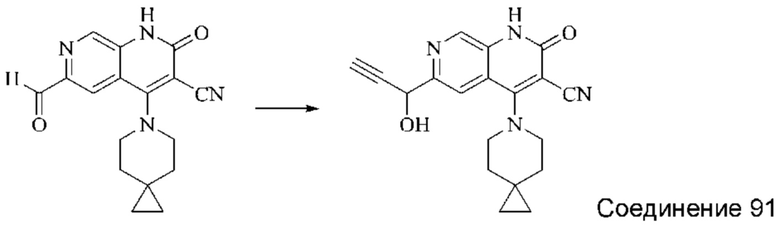
Промежуточный 6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-ацетонитрил (200,0 мг, 0,65 ммоль, 1,0 экв.) растворяли в безводном ТГФ (5 мл), и к полученному как указано выше реакционному раствору в атмосфере азота при -10°С добавляли этинилмагнийбромид (6,48 мл, 3,25 ммоль, 5,0 экв.). Реакционный раствор медленно нагревали до 0°С и перемешивали в течение 1 часа. По завершении реакции, раствор тушили добавлением насыщенного водного раствора хлорида аммония, концентрировали и экстрагировали этилацетатом (3×30 мл). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали; неочищенный продукт очищали колоночной хроматографией на силикагеле (DCE:МеОН = 100:1-75:1), получая белый твердый продукт (40 мг, выход: 18,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,05 (br. s, 1Н), 8,61 (s, 1Н), 7,86 (s, 1Н), 6,36 (d, 1Н), 5,39-5,42 (m, 1Н), 3,64 (s, 4Н), 3,51 (d, 1Н), 1,64 (s, 4Н), 0,45 (m, 4Н).
Молекулярная формула: C19H18N4O2, Молекулярная масса: 334,38, ЖХ-МС (m/z) = 335 [М+Н]+.
Пример 64
Синтез 2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-6-(2,2,2-трифтор-1-гидроксиэтил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 92)
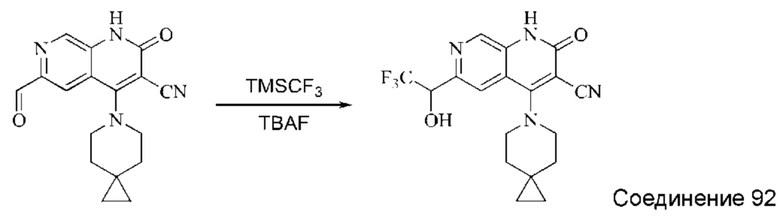
6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (300 мг, 0,973 ммоль, 1,0 экв.) растворяли в безводном N,N-диметилацетамиде (20 мл), охлаждали до -10°С в атмосфере азота, после чего добавляли TMSCF3 (1,44 мл, 9,730 ммоль, 10,0 экв.), добавляли по каплям безводный TBAF в тетрагидрофуране (0,96 мл, 0,96 ммоль) и затем перемешивали при -10°С в течение ночи для протекания реакции. Завершение протекания реакции определяли способом ТСХ. Затем в реакционный раствор добавляли дихлорметан (20 мл), промывали насыщенным солевым раствором (20 мл × 2) и водой (20 мл × 4), сушили безводным сульфатом магния, фильтровали с отсасыванием и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (DCE (дихлорэтан): МеОН = 30:1-15:1), получая желтый твердый продукт (116 мг, выход: 31,5%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,11 (s, 1Н), 8,65 (s, 1Н), 7,87 (s, 1Н), 7,13-7,12 (d, 1Н), 5,24-5,22 (t, 1Н), 3,64 (s, 4Н), 1,63 (s, 4Н), 0,45 (m, 4Н).
Молекулярная формула: C18H17F3N4O2, Молекулярная масса: 378,36, ЖХ-МС (Pos, m/z) = 379,20 [М+Н]+.
Пример 65
Синтез этил 1-(3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-ил)ацетата (Соединение 93)
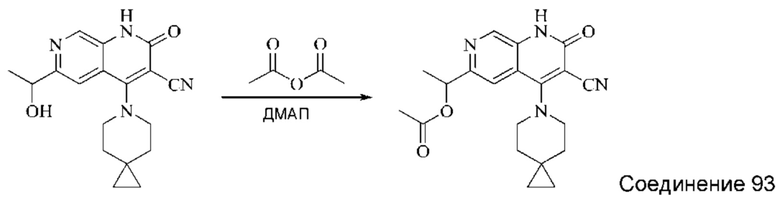
6-(1-гидроксиэтил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (100 мг, 0,308 ммоль, 1,0 экв.) растворяли в дихлорметане (15 мл), охлаждали до 0°С на ледяной бане, после чего добавляли уксусный ангидрид (63 мг, 0,616 ммоль, 2,0 экв.), триэтиламин (125 мг, 1,233 ммоль, 4,0 экв.) и ДМАП (19 мг, 0,154 ммоль, 0,5 экв.), затем перемешивали для протекания реакции в течение 5 минут на ледяной бане (0°С) и затем нагревали до комнатной температуры и перемешивали в течение 2 часов. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор гасили добавлением насыщенного водного раствора NaHCO3 (10 мл) и экстрагировали дихлорметаном (10 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл × 2), сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали способом препаративной тонкослойной хроматографии (ДХМ:МеОН = 10:1), получая оранжево-желтый твердый продукт (98 мг, выход: 86,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,05 (s, 1Н), 8,64 (s, 1Н), 7,62 (s, 1Н), 5,90-5,85 (q, 1Н), 3,64 (s, 4Н), 2,06 (s, 3Н), 1,63 (s, 4Н), 1,55-1,53 (d, 3Н), 0,45 (s, 4Н).
Молекулярная формула: C20H22N4O3, Молекулярная масса: 366,42, ЖХ-МС (Pos, m/z) = 367,20 [М+Н]+.
Пример 66
Синтез 6-(1-гидроксиэтил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 94)
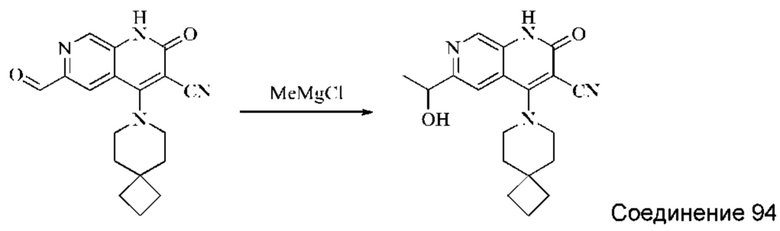
6-формил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (530,0 мг, 1,65 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (10 мл), охлаждали до -10°С, затем медленно добавляли метилмагнийхлорид концентрацией 3 моль/л (1,7 мл, 5,1 ммоль, 3,0 экв.) и перемешивали в течение 2 часов на ледяной бане. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор тушили добавлением насыщенного водного раствора хлорида аммония (10 мл) и экстрагировали этилацетатом (20 мл × 2). Органические фазы объединяли, и концентрировали при пониженном давлении; неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 50:1-20:1), получая продукт (56,1 мг, выход: 10,1%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,92 (s, 1Н), 8,58 (s, 1Н), 7,73 (s, 1Н), 5,49 (d, 1Н, J=4,64 Гц), 4,71-4,82 (m, 1Н), 3,50-3,61 (m, 4Н), 1,79-1,98 (m, 10Н), 1,34 (d, 3Н, J=6,48 Гц).
Молекулярная формула: C19H22NO2, Молекулярная масса: 338,17, ЖХ-МС (Neg, m/z) = 337,15 [М-Н]-.
Пример 67
Синтез 6-ацетил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединения 95)
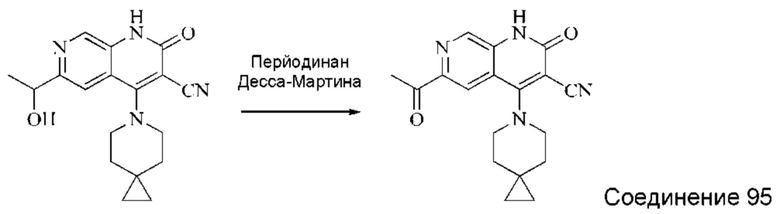
6-(1-гидроксиэтил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (0,85 г, 2,62 ммоль, 1,0 экв.) растворяли в безводном дихлорметане (60 мл), охлаждали до 0°С в атмосфере азота, после чего добавляли окислитель Десса-Мартина (2,22 г, 5,24 ммоль, 2,0 экв.) и перемешивали при комнатной температуре в течение ночи. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор тушили насыщенным водным раствором NaHCO3 (20 мл) и фильтровали с отсасыванием через целит. Фильтрат экстрагировали дихлорметаном (20 мл × 3). После разделения жидкостей органическую фазу промывали насыщенным солевым раствором (20 мл × 2), сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 80:1-50:1), получая продукт в виде желтого твердого вещества (0,35 г, выход: 41,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,35 (s, 1Н), 8,72 (s, 1Н), 8,25 (s, 1Н), 3,68 (t, 4Н), 2,64 (s, 3Н), 1,64 (s, 4Н), 0,46 (s, 4Н).
Молекулярная формула: C18H18N4O2, Молекулярная масса: 322,37, ЖХ-МС (Pos, m/z) = 323,10 [М+Н]+.
Пример 68
Синтез 3-циано-N-метил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-формамида (Соединение 96)
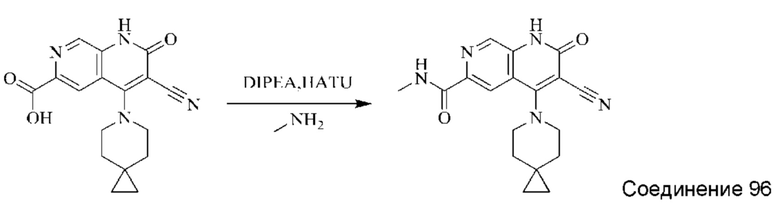
Промежуточную 3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (130 мг, 0,4 ммоль, 1,0 экв.) растворяли в N,N-диметилформамиде (2 мл), после чего добавляли N,N-диизопропилэтиламин (155 мг, 1,2 ммоль, 3,0 экв.) и HATU (228 мг, 0,6 ммоль, 1,5 экв.) и оставляли реагировать при комнатной температуре в течение 1 часа, после чего добавляли метиламин в метаноле (0,5 мл) и оставляли реагировать при комнатной температуре в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ: МеОН = 15:1), получая продукт (50 мг, выход: 37%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,19 (s, 1Н), 8,72-8,73 (m, 1Н), 8,32 (s, 1Н), 3,66-3,68 (m, 4Н), 2,83-2,84 (d, 3Н), 1,64 (m, 4Н), 0,46 (s, 4Н).
Молекулярная формула: C18H19N5O2, Молекулярная масса: 337,38 ЖХ-МС (Pos, m/z) = 338,16 [М+Н]+.
Пример 69
Синтез 3-циано-N-(2-гидроксиэтил)-2-оксо-4-(6-аза-спиро[2,5]oct-6-ил)-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 97)
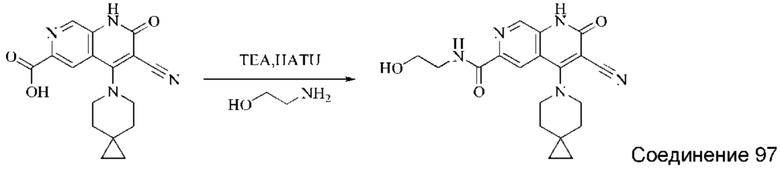
Промежуточную 3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (162 мг, 0,5 ммоль, 1,0 экв.) растворяли в N,N-диметилформамиде (2 мл), после чего добавляли триэтиламин (152 мг, 1,5 ммоль, 3,0 экв.) и HATU (285 мг, 0,75 ммоль, 1,5 экв.) и оставляли реагировать при комнатной температуре в течение 1 часа, после чего добавляли этаноламин (31 мг, 0,5 ммоль, 1,0 экв.) и оставляли реагировать при комнатной температуре в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ: МеОН = 15:1), получая продукт (60 мг, выход: 33%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,24 (s, 1Н), 8,61-8,65 (m, 2Н), 8,33 (s, 1Н), 4,77-4,80 (m, 1Н), 3,66-3,69 (m, 4Н), 3,51-3,56 (m, 2Н), 3,38-3,42 (m, 2Н), 1,65 (m, 4Н), 0,46 (s, 4Н).
Молекулярная формула: C19H21N5O3, Молекулярная масса: 367,41 ЖХ-МС (Pos, m/z) = 368,28 [М+Н]+.
Пример 70
Синтез 4-(4-гидрокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 106)
Этап 1: Синтез трифторацетата 4-метилпиперидин-4-ола
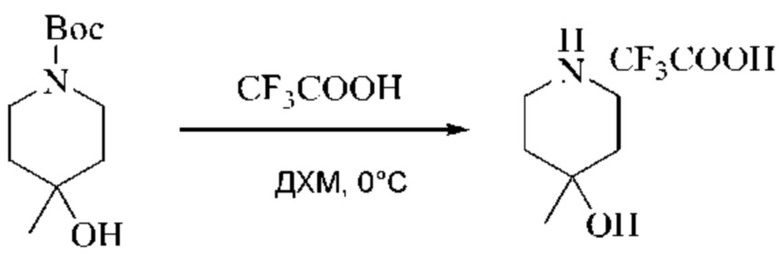
Неочищенное вещество, трет-бутил-4-гидрокси-4-метилпиперидин-1-карбоксилат (380 мг, 1,77 ммоль, 1,0 экв.), растворяли в дихлорметане (5 мл), после чего при 0°С добавляли трифторуксусную кислоту (3 мл) и оставляли реагировать в течение 1 часа. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, получая продукт (404 мг, выход: 100%).
Этап 2: Синтез 4-(4-гидрокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
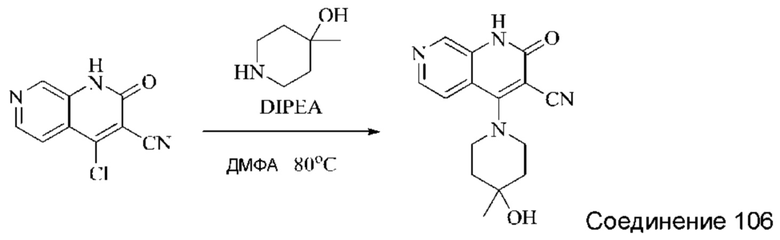
Промежуточный 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (200 мг, 0,97 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), после чего добавляли N,N-диизопропилэтиламин (753 мг, 5,87 ммоль, 6,0 экв.) и трифторацетат 4-метилпиперидин-4-ола (312 мг, 1,36 ммоль, 1,4 экв.), и реакцию проводили при 80°С в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор добавляли по каплям в воду (5 мл), перемешивали в течение 10 минут и фильтровали с отсасыванием. Осадок на фильтре затем суспендировали в дихлорметане (5 мл), фильтровали с отсасыванием и сушили при 50°С, получая продукт в виде желтого твердого вещества (136 мг, выход: 49%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,95 (s, 1Н), 8,62-8,65 (d, 1Н), 8,33-8,34 (d, 1Н), 7,59-7,61 (d, 1Н), 4,56-4,61 (m, 1Н), 3,68-3,79 (m, 2Н), 3,58-3,62 (d, 2Н), 1,77-1,83 (m, 2Н), 1,66-1,76 (m, 2Н), 1,24 (s, 3Н).
Молекулярная формула: C15H16N4O2, Молекулярная масса: 284,32 ЖХ-МС (Pos, m/z) = 285,13 [М+Н]+.
Пример 71
Синтез 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 107)
Этап 1: Синтез 6-хлор-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
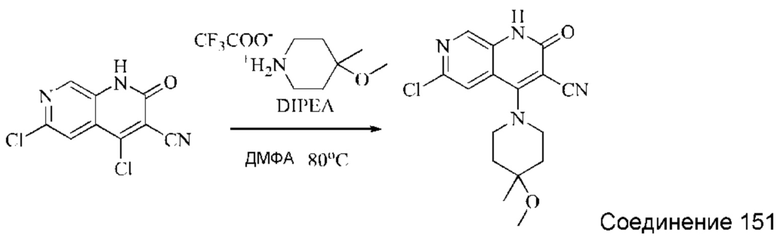
Промежуточный 4,6-дихлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (2,0 г, 8,33 ммоль, 1,0 экв.) растворяли в ДМФА (10 мл), после чего добавляли DIPEA (6,45 г, 50 ммоль, 6,0 экв.) и трифторацетат 4-метокси-4-метилпиперидина (2,2 г, 9,16 ммоль, 1,1 экв.) и оставляли реагировать при 80°С в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Затем в реакционный раствор добавляли воду (10 мл) и экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали водой (10 мл × 3), сушили безводным сульфатом натрия, фильтровали, и концентрировали при пониженном давлении, получая продукт в виде желтого твердого вещества (2,7 г, неочищенный продукт).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,11 (s, 1Н), 8,45 (s, 1Н), 7,61 (s, 1Н), 3,61-3,59 (m, 4Н), 3,18 (s, 3Н), 1,91-1,88 (m, 2Н), 1,81-1,76 (m, 2Н), 1,21 (s, 3Н).
Молекулярная формула: C16H17N4O2Cl, Молекулярная масса: 332,79 ЖХ-МС (Pos, m/z) = 333,7 [М+Н]+.
Этап 2: Синтез 4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
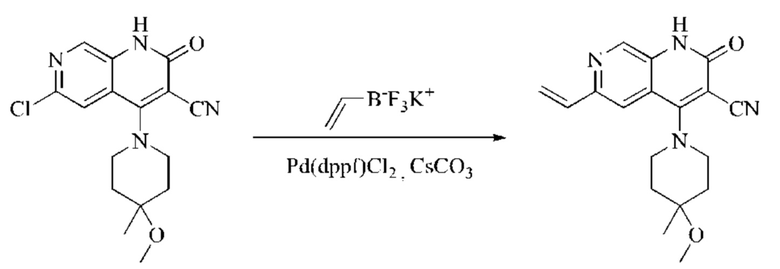
Промежуточный 6-хлор-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (2,7 г неочищенный продукт, 8,11 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (20 мл) и H2O (5 мл), после чего добавляли винилтрифторборат калия (1,63 г, 12,17 ммоль, 1,5 экв.), карбонат цезия (3,965 г, 12,17 ммоль, 1,5 экв.) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (297 мг, 0,41 ммоль, 0,05 экв.) и оставляли реагировать при 100°С в течение 8 часов в атмосфере азота. Завершение протекания реакции определяли способом ЖХ-МС. Затем в реакционный раствор добавляли воду (20 мл) и экстрагировали дихлорметаном (30 мл × 3). Органическую фазу сушили безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлениим, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 70:1), получая продукт в виде желтого твердого вещества (1,15 г, выход: 43%).
Этап 3: Синтез 6-этил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
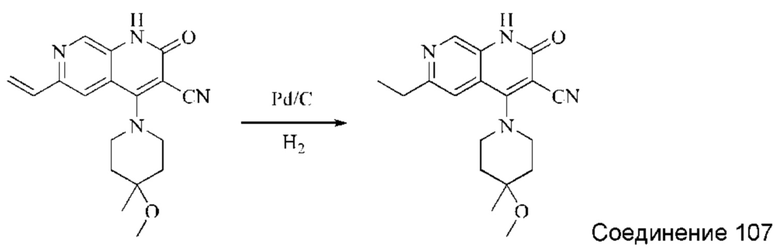
Промежуточный 4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (150 мг, 0,46 ммоль, 1,0 экв.) растворяли в метаноле (5 мл). В реакционный раствор добавляли Pd/C (100 мг). Воздух в системе вытесняли водородом три раза, и реакцию проводили в течение 1 часа в атмосфере водорода. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении, получая продукт (120 мг, выход: 80%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,89 (s, 1Н), 8,59 (s, 1Н), 7,41 (s, 1Н), 3,60-3,62 (m, 4Н), 3,19 (s, 3Н), 2,79-2,84 (m, 2Н), 1,89-1,93 (m, 2Н), 1,75-1,82 (m, 2Н), 1,22-1,27 (m, 6Н).
Молекулярная формула: C18H22N4O2, Молекулярная масса: 326,40 ЖХ-МС (Pos, m/z) = 327,26 [М+Н]+.
Пример 72
Синтез 6-(1-гидроксиэтил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 108)
Этап 1: Синтез 6-(1,2-дигидроксиэтил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
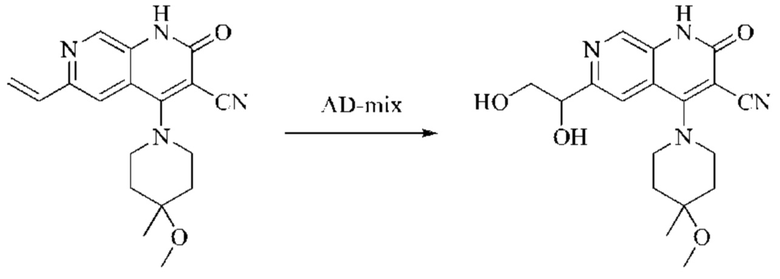
Промежуточный 4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (500 мг, 1,542 ммоль, 1,0 экв.) растворяли в трет-бутаноле (10 мл) и воде (10 мл), после чего добавляли метансульфонамид (147 мг, 1,542 ммоль, 1,0 экв.) и AD-mix-β (6,0 г) и оставляли реагировать при комнатной температуре в течение 12 часов. Завершение протекания реакции определяли способом ЖХ-МС. Затем в реакционный раствор добавляли воду (10 мл) и экстрагировали дихлорметаном (30 мл × 3). Органическую фазу сушили безводным сульфатом натрия, фильтровали, и концентрировали при пониженном давлении, получая продукт (552 мг, выход: 100%).
Этап 2: Синтез 6-формил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
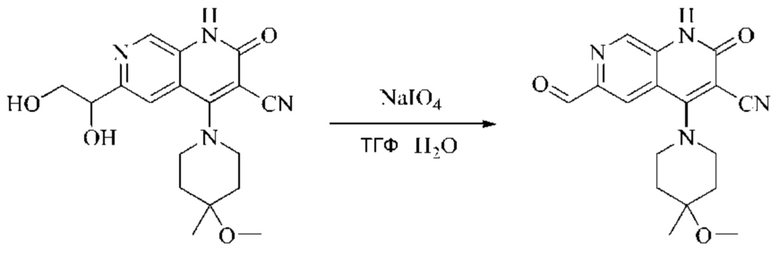
Промежуточный 6-(1,2-дигидроксиэтил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (552 мг, 1,542 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (10 мл) и воде (2 мл), после чего добавляли перйодат натрия (650 мг, 3,084 ммоль, 2,0 экв.) и оставляли реагировать в течение 4 часов. Завершение протекания реакции определяли способом ЖХ-МС. Затем в реакционный раствор добавляли воду (10 мл) и экстрагировали этилацетатом (20 мл × 3). Органическую фазу сушили безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 60:1), получая продукт в виде желтого твердого вещества (160 г, выход в результате двух этапов синтеза: 32%).
Этап 3: Синтез 6-(1-гидроксиэтил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
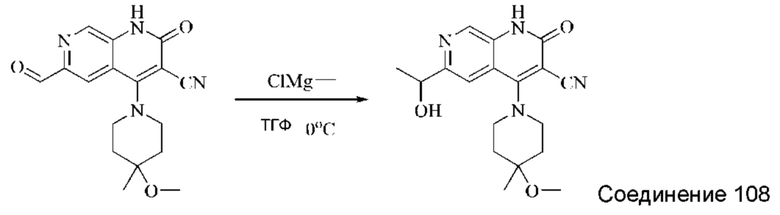
Промежуточный 6-формил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (160 мг, 0,49 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (5 мл), затем при 0°С добавляли по каплям метилмагнийхлорид (1 мл) и оставляли реагировать в течение 1 часа. Завершение протекания реакции определяли способом ЖХ-МС. Затем в реакционный раствор добавляли воду (10 мл) и экстрагировали этилацетатом (20 мл × 3). Органическую фазу сушили безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 40:1), получая продукт (108 мг, выход: 64%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,93 (s, 1Н), 8,58 (s, 1Н), 7,72 (s, 1Н), 5,48-5,49 (d, 1Н), 4,75-4,81 (m, 1Н), 3,56-3,65 (m, 4Н), 3,20 (s, 3Н), 1,91-1,95 (m, 2Н), 1,73-1,79 (m, 2Н), 1,38-1,40 (d, 3Н), 1,23 (s, 3Н).
Молекулярная формула: C18H22N4O3, Молекулярная масса: 342,40 ЖХ-МС (Pos, m/z) = 343,17 [М+Н]+.
Пример 73
Синтез 6-(2-гидроксипропан-2-ил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 109)
Этап 1: Синтез 6-ацетил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
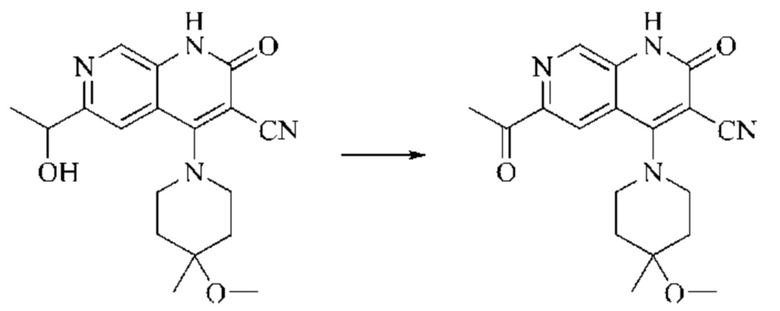
Промежуточный 6-(1-гидроксиэтил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (187 мг, 0,55 ммоль, 1,0 экв.) растворяли в сухом дихлорметане (5 мл). Температуру понижали до 0-5°С и добавляли окислитель Десса-Мартина (463,5 мг, 1,10 ммоль, 2,0 экв.). По завершении добавления температуру смеси естественным образом оставили подниматься до комнатной температуры и оставляли реагировать в течение 2 часов. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (МеОН:ДХМ = 1:100-1:50), получая продукт (185,8 мг, выход: 100%).
Этап 2: Синтез 6-(2-гидроксипропан-2-ил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
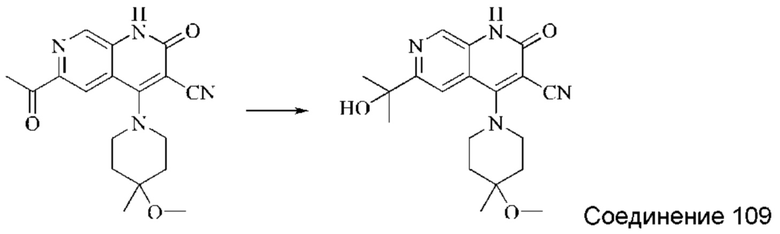
Промежуточный 6-ацетил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (185,8 мг, 0,55 ммоль, 1,0 экв.) растворяли в N,N-диметилацетамиде (3 мл), и температуру понижали до -10-0°С. В полученную смесь по каплям в атмосфере азота добавляли раствор метилмагнийхлорида в тетрагидрофуране концентрацией 3 моль/л (0,6 мл, 3,0 экв.). По завершении добавления смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Анализ ТСХ показал наличие большого количества оставшихся исходных материалов. В реакционный раствор добавляли раствор метилмагнийхлорида в тетрагидрофуране концентрацией 3 моль/л (0,6 мл, 3,0 экв.), и реакцию оставляли протекать в течение 3 часов, снова добавили раствор метилмагнийхлорида в тетрагидрофуране концентрацией 3 моль/л (0,6 мл, 3,0 экв.), реакцию оставляли протекать в течение 2 часов, охлаждали до 0-10°С, доводили до рН 5-6 добавлением уксусной кислоты и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (МеОН:ДХМ = 1:100-1:70), получая продукт (63,9 мг, выход: 32,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,91 (s, 1Н), 8,59 (s, 1Н), 7,89 (s, 1Н), 5,35 (s, 1Н), 3,62-3,60 (m, 4Н), 3,20 (s, 3Н), 1,95-1,92 (m, 2Н), 1,80-1,73 (m, 2Н), 1,45 (s, 6Н), 1,23 (s, 3Н).
Молекулярная формула: C19H24N4O3, Молекулярная масса: 356,43 ЖХ-МС (Pos, m/z) = 357,25 [М+Н]+.
Пример 74
Синтез (S)-4-(3-(1Н-1,2,4-триазол-1-ил)пирролидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 112)
Этап 1: Синтез трет-бутил-(S)-3-(1H-1,2,4-триазол-1-ил)пирролидин-1-карбоксилата
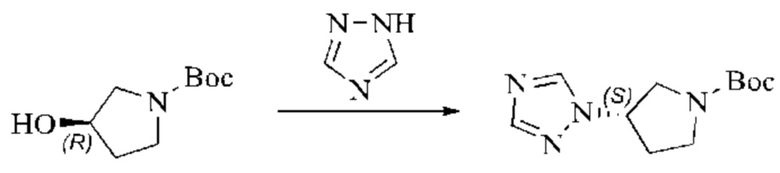
Исходное вещество, трет-бутил-(R)-3-гидроксипирролидин-1-карбоксилат (1,0 г, 5,34 ммоль, 1,0 экв.), растворяли в безводном тетрагидрофуране (20 мл) и добавляли 1Н-1,2,4-триазол (368 мг, 5,34 ммоль, 1,0 экв.) и трифенилфосфин (2,8 г, 10,68 ммоль, 2.0 экв.). По завершении добавления температуру понижали до 0°С и по каплям добавляли диэтилазодикарбоксилат (1,86 г, 10,68 ммоль, 2,0 экв.). По завершении добавления реакцию проводили при комнатной температуре в течение 12 часов. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ЭА:ПЭ = 1:15-1:2), получая продукт (559 мг, выход: 44%).
Этап 2: Синтез гидрохлорида (S)-1-(пирролидин-3-ил)-1H-1,2,4-триазола

Промежуточный трет-бутил-(S)-3-(1H-1,2,4-триазол-1-ил)пирролидин-1-карбоксилат (559 мг, 2,34 ммоль, 1,0 экв.) растворяли в безводном метаноле (5 мл), после чего добавляли 30%-ный раствор хлорида водорода в этаноле (5 мл) и оставляли реагировать при комнатной температуре в течение 2 часов до выпадения белого твердого вещества. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали при пониженном давлении, после чего добавляли воду (10 мл) и экстрагировали ЭА (3×5 мл). Водную фазу подвергали лиофилизации, получая беловатый твердый продукт (410 мг, выход: 100%).
Этап 3: Синтез (S)-4-(3-(1H-1,2,4-триазол-1-ил)пирролидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
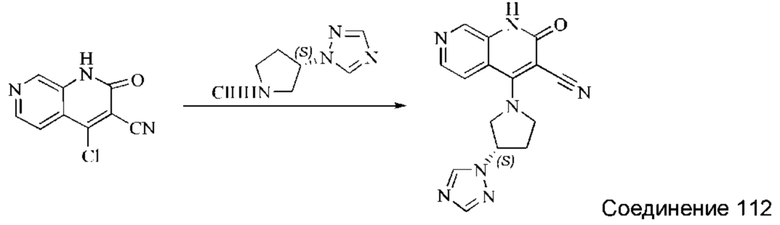
Промежуточный 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (346,2 мг, 1,68 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл) и добавляли гидрохлорид (S)-1-(пирролидин-3-ил)-1H-1,2,4-триазола (410 мг, 2,34 ммоль, 1,4 экв.) и DIPEA (1,1 г, 8,4 ммоль, 5,0 экв.). По завершении добавления температуру повышали до 80°С и реакцию проводили в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС, и температуру понижали до комнатной температуры. Реакционный раствор разделяли способом колоночной хроматографии с обращенной фазой (0,1% водный раствор соляной кислоты : ацетонитрил = 70:30), получая продукт (226 мг, выход: 43,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,68 (s, 1Н), 8,70 (s, 1Н), 8,62 (s, 1Н), 8,28-8,27 (d, 1Н), 8,04 (s, 1Н), 7,98-7,97 (s, 1Н), 5,33-5,30 (m, 1Н), 4,61-4,57 (m, 1Н), 4,35-4,28 (m, 2Н), 4,23-4,17 (m, 1Н), 2,55-2,51 (m, 1Н), 2,50-2,47 (m, 1Н).
Молекулярная формула: C15H13N7O, Молекулярная масса: 307,21 ЖХ-МС (Pos, m/z) = 307,98 [М+Н]+.
Пример 75
Синтез 2-оксо-4-(3-(тиазол-2-ил)пирролидин-1-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 113)
Этап 1: Синтез трет-бутил-3-(тиазол-2-ил)-2,5-дигидро-1Н-пиррол-1-карбоксилата
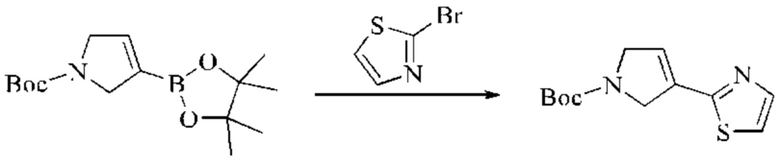
Исходное вещество, трет-бутил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат (1,0 г, 3,39 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (20 мл) и добавляли 2-бромтиазол (666,6 мг, 4,06 ммоль, 1,2 экв.), безводный карбонат натрия (897,5 мг, 8.47 ммоль, 2,5 экв.) и воду (4 мл). Воздух в системе трижды вытесняли азотом и в реакцию добавляли дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (247,8 мг, 0,34 ммоль, 0,1 экв.). Воздух в системе трижды вытесняли азотом, температуру повышали до 80°С и оставляли реагировать в течение 2 часов. Завершение протекания реакции определяли способом ТСХ. Температуру понижали до комнатной температуры. В смесь добавляли воду (10 мл) и экстрагировали этилацетатом (50 мл х 3). Смесь разделяли, органические фазы объединяли, сушили безводным сульфатом натрия и фильтровали. Маточный раствор концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ЭА:ПЭ = 1:30-1:20), получая бледно-желтый маслянистый продукт (719 мг, выход: 84,2%).
Этап 2: Синтез трет-бутил-3-(тиазол-2-ил)пирролидин-1-карбоксилата
Промежуточный трет-бутил-3-(тиазол-2-ил)-2,5-дигидро-1H-пиррол-1-карбоксилат (719 мг, 2,84 ммоль, 1,0 экв.) растворяли в безводном этаноле (20 мл), после чего добавляли 10% палладий на углеродном носителе (0,5 г). По завершении добавления воздух в системе вытесняли водородом три раза, температуру повышали до 50°С, и реакция протекала в течение 12 часов. Завершение протекания реакции определяли способом ТСХ, и температуру понижали до комнатной температуры. Реакционный раствор фильтровали, и осадок на фильтре споласкивали этанолом. Маточный раствор концентрировали при пониженном давлении, получая бледно-желтый маслянистый продукт (653 мг, выход: 90,5%).
Этап 3: Синтез гидрохлорида 2-(пирролидин-3-ил)тиазола

Промежуточный трет-бутил-3-(тиазол-2-ил)пирролидин-1-карбоксилат (653 мг, 2,57 ммоль, 1,0 экв.) растворяли в безводном метаноле (2 мл), после чего добавляли 30%-ный раствор хлорида водорода в этаноле (5 мл) и оставляли реагировать при комнатной температуре в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор концентрировали при пониженном давлении, получая продукт (880 мг неочищенного продукта), который непосредственно вводили в следующий этап без очистки.
Этап 4: Синтез 2-оксо-4-(3-(тиазол-2-ил)пирролидин-1-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
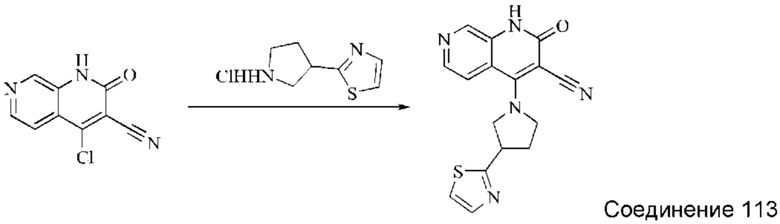
Промежуточный 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (210 мг, 1,02 ммоль, 1,0 экв.) растворяли в ДМФА (2 мл) и в реакционную смесь добавляли гидрохлорид 2-(пирролидин-3-ил)тиазола (195 мг, 1,02 ммоль, 1,0 экв.) и DIPEA (792,7 мг, 6,14 ммоль, 6,0 экв.). По завершении добавления температуру повышали до 80°С, и смесь оставляли реагировать в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС, и температуру понижали до комнатной температуры. Смесь очищали препаративной ВЭЖХ (высокоэффективной жидкостной хроматографией, также называемой жидкостной хроматографией высокого давления) (0,1% водный раствор трифторуксусной кислоты : ацетонитрил = 70:30), подвергали лиофилизации, растворяли, добавляя воду (1 мл), доводили до приблизительно рН 8 добавлением насыщенного водного раствора карбоната натрия до тех пор, пока не выпадал твердый осадок, и затем фильтровали. Осадок на фильтре сушили, получая продукт (92 мг, выход: 27,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,63 (s, 1Н), 8,61 (s, 1Н), 8,28-8,27 (d, 1Н), 7,99-7,98 (d, 1Н), 7,79-7,78 (d, 1Н), 7,70-7,69 (d, 1Н), 4,48-4,44 (m, 1Н), 4,34-4,30 (m, 1Н), 4,27-4,15 (m, 2Н), 4,06-4,00 (m, 1Н), 2,50 (m, 1Н), 2,30-2,27 (m, 1Н).
Молекулярная формула: C16H13N5OS, Молекулярная масса: 323,37 ЖХ-МС (Pos, m/z) = 346,14 [М+Н]+.
Пример 76
Синтез 2-(метилтио)-6-оксо-8-(6-аза-спиро[2,5]октан-6-ил)-5,6-дигидропиридо[3,2-d]пиримидин-7-карбонитрила (Соединение 129)
Этап 1: Синтез 6-(метилтио)-2H-пиримидо[5,4-d][1,3]оксазин-2,4-(1H)-диона
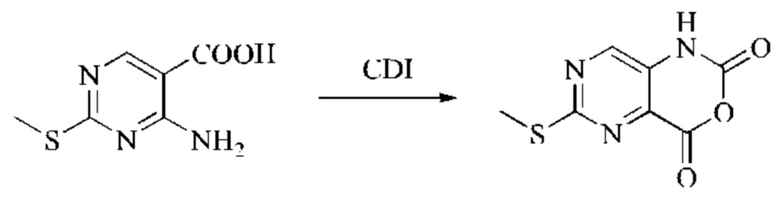
4-амино-2-(метилтио)пиримидин-5-карбоновую кислоту (2,00 г, 10,80 ммоль, 1,0 экв.) растворяли в безводном тетрагидрофуране (100,0 мл), после чего при охлаждении ледяной баней добавляли N,N'-карбонилдиимидазол (3,50 г, 21,60 ммоль, 2,0 экв.), перемешивали при комнатной температуре, проводя реакцию в течение 16 часов. Завершение протекания реакции определяли способом ТСХ. Твердое вещество отфильтровывали, и фильтрат вводили непосредственно в следующую реакцию.
Этап 2: Синтез 8-гидрокси-2-(метилтио)-6-оксо-5,6-дигидропиридо[3,2-d]пиримидин-7-карбонитрила
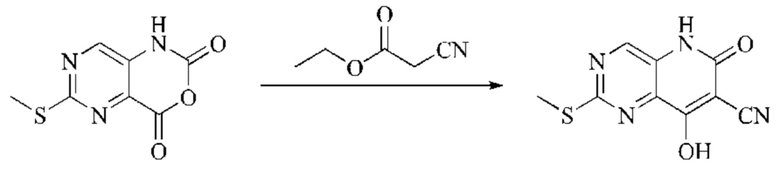
Гидрид натрия (60%, 1,88 г, 46,96 ммоль, 4,35 экв.) медленно добавляли в безводный тетрагидрофуран (100,0 мл), перемешивали в течение 10 минут и медленно, при охлаждении ледяной баней, добавляли по каплям этилцианоацетат (3,50 г, 30,95 ммоль, 2,86 экв.). Смесь перемешивали в атмосфере азота при 75°С в течение 20 минут, затем медленно по каплям добавляли 6-(метилтио)-2Н-пиримидо[5,4-d][1,3]оксазин-2,4-(1Н)-дион в ТГФ и затем перемешивали при 75°С в течение ночи в атмосфере азота. Завершение протекания реакции определяли способом ТСХ. Затем в реакционный раствор добавляли ледяную воду (30 мл), доводили до рН 3-4 концентрированной соляной кислотой и фильтровали с отсасыванием, получая желтый твердый продукт (880,0 мг, выход в результате двух этапов синтеза: 34,8%).
Этап 3: Синтез 8-хлор-2-(метилтио)-6-оксо-5,6-дигидропиридо[3,2-d]пиримидин-7-карбонитрила
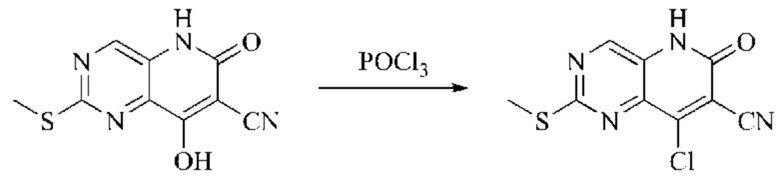
8-гидрокси-2-(метилтио)-6-оксо-5,6-дигидропиридо[3,2-d]пиримидин-7-карбонитрил (705 мг, 3,01 ммоль) добавляли к смешанному растворителю, состоящему из POCl3 (10 мл) и 1,2-дихлорэтана (20 мл), и перемешивали при 75°С; реакция протекала в течение 3-4 часов в атмосфере азота. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор охлаждали на ледяной бане, после чего добавляли ледяную воду (10 мл), затем перемешивали и фильтровали с отсасыванием, получая продукт (400,0 мг, выход: 52,6%).
Этап 4: Синтез 2-(метилтио)-6-оксо-8-(6-аза-спиро[2,5]октан-6-ил)-5,6-дигидропиридо[3,2-d]пиримидин-7-карбонитрила
8-хлор-2-(метилтио)-6-оксо-5,6-дигидропиридо[3,2-d]пиримидин-7-карбонитрил (400 мг, 1,58 ммоль, 1,0 экв.), гидрохлорид 6-аза-спиро[2,5]октана (280,5 мг, 1,90 ммоль, 1,2 экв.) и DIPEA (1636,7 мг, 12,66 ммоль, 8,0 экв.) добавляли к ДМФА (20 мл) и перемешивали при 80°С, проводя реакцию в течение 3 часов. По завершении реакции по данным ТСХ смесь концентрировали при пониженном давлении, после чего добавляли ЭА (5 мл) и воду (10 мл), перемешивали в течение 2 часов и фильтровали с отсасыванием. Осадок на фильтре суспендировали повторным добавлением ЭА (5 мл) в течение 1 часа, и продукт получали фильтрованием (59,0 мг, выход: 11,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,58 (s, 1Н), 8,69 (s, 1Н), 3,90 (t, 4Н), 2,53 (s, 3Н), 1,61 (t, 4Н), 0,42 (s, 4Н).
Молекулярная формула: C16H17N5OS, Молекулярная масса: 327,41 ЖХ-МС (Pos, m/z) = 328,10 [М+Н]+.
Пример 77
Синтез 6-(циклопропил(гидрокси)метил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 130)
Промежуточный 6-формил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (500 мг, 1,53 ммоль, 1,0 экв.) растворяли в безводном тетрагидрофуране (20 мл), и температуру понижали в атмосфере азота до -10°С. В реакционную смесь по каплям добавляли раствор циклопропилмагнийбромида в тетрагидрофуране концентрацией 1 моль/л (4,6 мл, 4,60 ммоль, 3 экв.). По завершении добавления реакцию проводили при 0°С в течение 3 часов. После этого, по данным ЖХ-МС в смеси оставалось 20% исходных материалов, поэтому вновь добавляли раствор циклопропилмагнийбромида в тетрагидрофуране концентрацией 1 моль/л (3 мл, 3 ммоль, 2 экв.), и реакцию продолжали в течение периода, составляющего от 2 до 3 часов. После этого, по данным ЖХ-МС в смеси оставалось 10% исходных материалов, поэтому рН смеси доводили до приблизительно 5-6, добавляя уксусную кислоту, и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (МеОН:ДХМ = 1:100-1:40), получая продукт (225,7 мг, выход: 40,0%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,93 (s, 1Н), 8,59 (s, 1Н), 7,68 (s, 1Н), 5,42-5,40 (d, 1Н), 4,24-4,22 (m, 1Н), 3,63-3,60 (m, 4Н), 3,19 (s, 3Н), 1,95-1,91 (m, 2Н), 1,79-1,72 (m, 2Н), 1,23 (s, 3Н), 1,23 (s, 1Н), 0,42 (m, 4Н).
Молекулярная формула: C20H24N4O3, Молекулярная масса: 368,44 ЖХ-МС (Pos, m/z) = 369,40 [М+Н]+.
Пример 78
Синтез 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-N-метил-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 131)
Этап 1: Синтез 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоновой кислоты
Промежуточный 6-формил-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (681 мг, 2,09 ммоль, 1,0 экв.) растворяли в муравьиной кислоте (5 мл), и температуру понижали до величины от -5 до 0°С. В реакционную смесь добавляли 30% пероксид водорода (1,32 мл, 10,44 ммоль, 5 экв.). По завершении добавления реакцию проводили при 0°С в течение 12 часов, затем добавляли 30% пероксид водорода (1,32 мл, 10,44 ммоль, 5 экв.) и оставляли реагировать при комнатной температуре в течение 2-3 часов. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор выливали в метил-трет-бутиловый эфир (50 мл), что приводило к осаждению бледно-желтого твердого вещества, которое отфильтровывали. Осадок на фильтре сушили, получая продукт (300 мг, выход: 42,0%).
Этап 2: Синтез 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-N-метил-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида
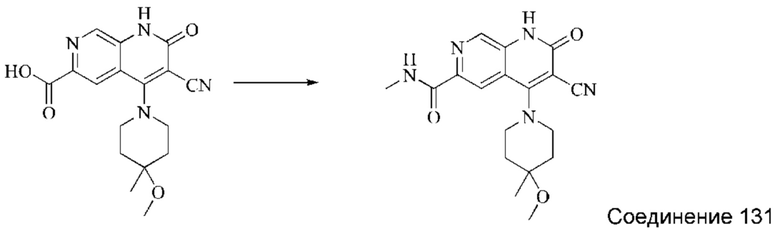
Промежуточную 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (300 мг, 0,88 ммоль, 1,0 экв.) растворяли в безводном N,N-диметилацетамиде (3 мл) и затем добавляли DIPEA (565,8 мг, 4,38 ммоль, 5,0 экв.). По завершении добавления реакционную смесь охлаждали до 0°С, затем добавляли HATU (499,7 мг, 1,31 ммоль, 1,5 экв.), перемешивали при комнатной температуре в течение времени, составляющего от получаса до 1 часа, после чего добавляли гидрохлорид метиламина (118,2 мг, 1,75 ммоль, 2,0 экв.) и оставляли реагировать при комнатной температуре в течение 1 часа, в результате чего образовывался твердый осадок. Завершение протекания реакции определяли способом ТСХ. К реакционному раствору добавляли воду (50 мл), перемешивали в течение 5 минут и фильтровали. Осадок на фильтре споласкивали водой, добавляли к этилацетату (10 мл) и кипятили с обратным холодильником в течение 1 часа, фильтровали горячим и сушили, получая продукт (199 мг, выход: 63,8%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,21 (s, 1Н), 8,74-8,73 (s, 1Н), 8,64 (s, 1Н), 8,27 (s, 1Н), 3,64-3,62 (m, 4Н), 3,20 (s, 3Н), 2,84-2,83 (d, 3Н), 1,96 (m, 1Н), 1,79-1,77 (m, 2Н), 1,24 (s, 3Н).
Молекулярная формула: C18H21N5O3, Молекулярная масса: 355,40 ЖХ-МС (Pos, m/z) = 356,26 [М+Н]+.
Пример 79
Синтез 4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 132)
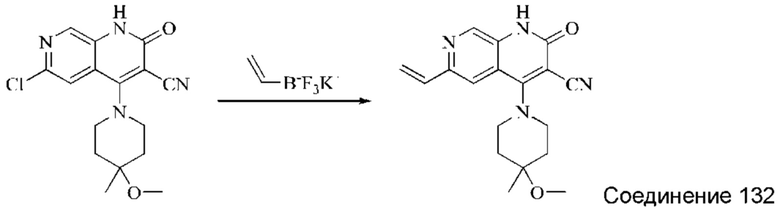
Промежуточный 6-хлор-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (20,1 г, 60,40 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (600 мл) и H2O (150 мл), после чего добавляли трифторвинилборат калия (12,14 г, 90,6 ммоль, 1,5 экв.), карбонат цезия (58 г, 181,2 ммоль, 3,0 экв.) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (4,4 г, 6,04 ммоль, 1,0 экв.) и оставляли реагировать при 100°С в течение 8 часов в атмосфере азота. Завершение протекания реакции определяли способом ЖХ-МС. Затем в реакционный раствор добавляли воду (20 мл) и экстрагировали дихлорметаном (30 мл × 3). Органическую фазу промывали водой (10 мл*3), сушили безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении; неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 50:1), получая продукт (14,63 г, выход: 74%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,03 (s, 1Н), 8,64 (s, 1Н), 7,56 (s, 1Н), 6,89-6,96 (m, 1Н), 6,15-6,19 (m, 1Н), 5,39-5,42 (m, 1Н), 3,61-3,64 (m, 4Н), 3,19 (s, 3Н), 1,77-1,93 (m, 4Н), 1,21 (s, 3Н).
Молекулярная формула: C18H20N4O2, Молекулярная масса: 324,38 ЖХ-МС (Pos, m/z) = 325,16 [М+Н]+.
Пример 80
Синтез 6-(1-гидрокси-2-метилпропил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 134)
Этап 1: Синтез 6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
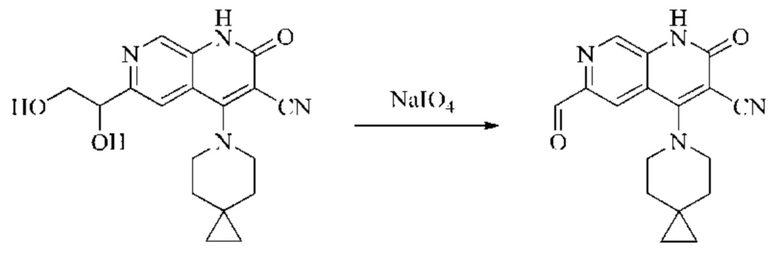
Перйодат натрия (4,00 г, 18,70 ммоль, 2,0 экв.) растворяли в воде (10 мл), после чего добавляли тетрагидрофуран (100 мл), охлаждали до 0°С на ледяной бане, затем медленно добавляли 6-(1,2-дигидроксиэтил)-2-оксо-4-(6-аза-спиро [2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (3,18 г, 9,35 ммоль, 1,0 экв.) и перемешивали при комнатной температуре в течение 3 часов. По завершении реакции, как показала ТСХ, смесь экстрагировали дихлорметаном (20 мл × 3). Органическую фазу отделяли, промывали насыщенным солевым раствором (20 мл × 2), сушили безводным Na2SO4 (0,8 г), фильтровали с отсасыванием и концентрировали при пониженном давлении, получая продукт в виде желтого твердого вещества (1,50 г, выход: 52,0%).
Этап 2: Синтез 6-(1-гидрокси-2-метилпропил)-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
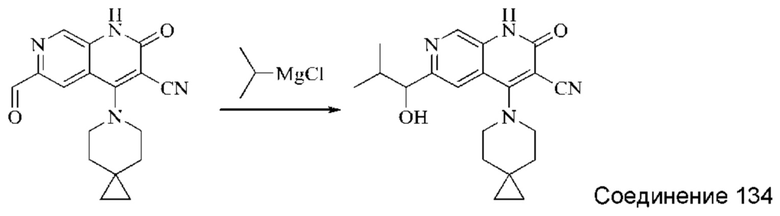
6-формил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (968,0 мг, 3,14 ммоль, 1,0 экв.) растворяли в свежеперегнанном тетрагидрофуране (100 мл), охлаждали до -30°С в атмосфере азота на бане в смеси сухого льда и этанол и затем быстро добавляли по каплям раствор изопропилмагнийхлорида в тетрагидрофуране (2 моль/л, 7,85 мл, 15,70 ммоль, 5,0 экв.). По завершении добавления по каплям смесь перемешивали при -30°С и оставляли реагировать в течение 30 минут. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор тушили насыщенным водным NH4Cl (30 мл) и экстрагировали дихлорметаном (30 мл × 3). Органическую фазу отделяли, промывали насыщенным солевым раствором (30 мл × 2), сушили безводным Na2SO4 (0,8 г), фильтровали с отсасыванием и концентрировали при пониженном давлении; неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 40:1-20:1), получая желтый твердый продукт (564,0 мг, выход: 50,9%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,96 (s, 1Н), 8,59 (s, 1Н), 7,71 (s, 1Н), 5,36-5,35 (d, 1Н), 4,47-4,45 (t, 1Н), 3,66-3,63 (t, 4Н), 2,09-2,01 (m, 1Н), 1,63 (s, 4Н), 0,90-0,88 (d, 3Н), 0,75-0,73 (d, 3Н), 0,45 (m, 4Н).
Молекулярная формула: C20H24N4O2, Молекулярная масса: 352,44 ЖХ-МС (Pos, m/z) = 353,26 [М+Н]+.
Пример 81
Синтез гидрохлорида N-(2-аминоэтил)-3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (соединение 138)
Этап 1: Синтез трет-бутил (2-(3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин)-6-формиламино)этил)карбамата
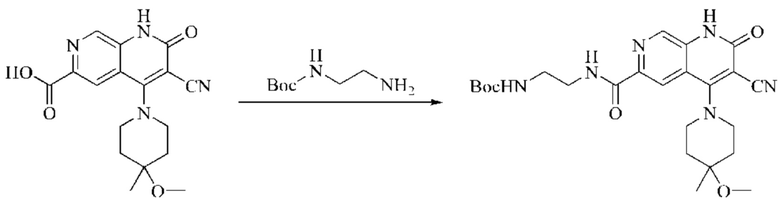
Промежуточную 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин- 6-карбоновую кислоту (200 мг, 0,58 ммоль, 1,0 экв.), HATU (333 мг, 0,88 ммоль, 1,5 экв.) и DIPEA (376 мг, 1,76 ммоль, 3,0 экв.) растворяли в ДМАА (2 мл), перемешивали при комнатной температуре в течение 30 минут, затем добавляли трет-бутил-(2-аминоэтил)карбамат (281 мг, 1,76 ммоль, 2,0 экв.) и оставляли реагировать при комнатной температуре в течение 1 часа. Завершение протекания реакции определяли способом ЖХ-МС. Затем в реакционный раствор добавляли воду (10 мл) и экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали водой (10 мл × 3), сушили безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 30:1), получая продукт (220 мг, выход: 78,3%).
Этап 2: Синтез гидрохлорида N-(2-аминоэтил)-3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида
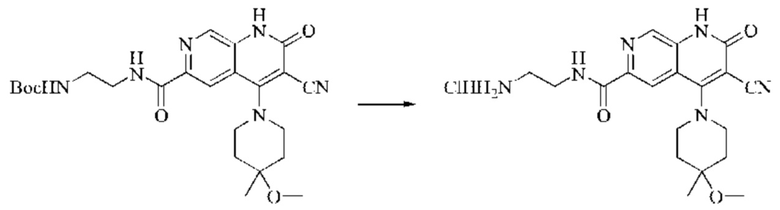
Гидрохлорид соединения 138 Промежуточный трет-бутил-(2-(3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-формиламино)этил)карбамат (220 мг, 0,45 ммоль, 1,0 экв.) растворяли в метаноле (3 мл), затем добавляли раствор хлорида водорода в этаноле (25%, 2 мл) и оставляли реагировать при комнатной температуре в течение 2 часов. Завершение протекания реакции определяли способом ТСХ. Из раствора выпадал твердый осадок, и полученную смесь фильтровали. Осадок на фильтре сушили, получая продукт (150 мг, выход: 79%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,28 (s, 1Н), 9,01-9,03 (m, 1Н), 8,68 (s, 1Н), 8,29 (s, 1Н), 7,91 (s, 3Н), 3,55-3,64 (m, 6Н), 3,21 (s, 3Н), 3,00-3,02 (m, 2Н), 1,94-1,97 (d, 2Н), 1,73-1,80 (d, 2Н), 1,24 (s, 3Н).
Молекулярная формула: C19H24N6O3, Молекулярная масса: 384,44 ЖХ-МС (Pos, m/z) = 385,19 [М+Н]+.
Пример 82
Синтез 3-циано-N-(2-(диметиламино)этил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 139)
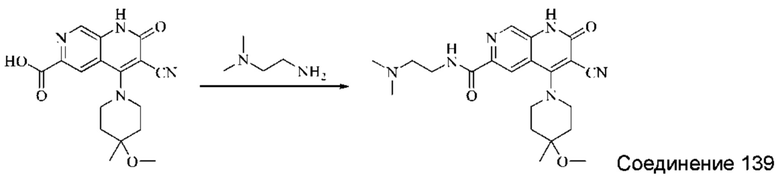
Промежуточную 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (200 мг, 0,58 ммоль, 1,0 экв.), HATU (333 мг, 0,88 ммоль, 1,5 экв.) и DIPEA (226 мг, 1,76 ммоль, 3,0 экв.) растворяли в ДМАА (2 мл), перемешивали при комнатной температуре в течение 30 минут, после чего добавляли N,N-диметилэтилендиамин (103 мг, 1,16 ммоль, 2,0 экв.) и оставляли реагировать при комнатной температуре в течение 1 часа. Завершение протекания реакции определяли способом ЖХ-МС. К реакционному раствору добавляли воду (10 мл), и реакционный раствор экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали водой (10 мл × 3), сушили безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении; неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 20:1), получая продукт (86 мг, выход: 36%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,20 (s, 1Н), 8,79 (s, 1Н), 8,66 (s, 1Н), 8,28 (s, 1Н), 3,62-3,64 (d, 4Н), 3,50-3,51 (d, 2Н), 3,21 (s, 3Н), 2,76 (s, 2Н), 2,44 (s, 6Н), 1,94-1,97 (d, 2Н), 1,74-1,81 (m, 2Н), 1,25 (s, 3Н).
Молекулярная формула: C21H28N6O3, Молекулярная масса: 412,49 ЖХ-МС (Pos, m/z) = 413,22 [М+Н]+.
Пример 83
Синтез 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-N-(2-(пирролидин-1-ил)этил)-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 140)
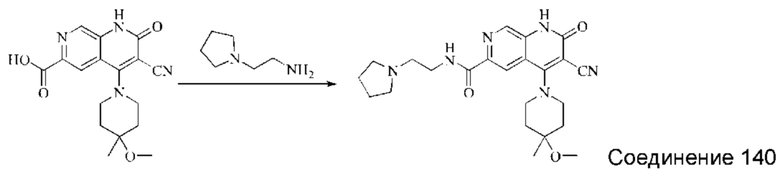
Промежуточную 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (200 мг, 0,58 ммоль, 1,0 экв.), HATU (333 мг, 0,88 ммоль, 1,5 экв.) и DIPEA (226 мг, 1,76 ммоль, 3,0 экв.) растворяли в ДМАА (2 мл), перемешивали при комнатной температуре в течение 30 минут, после чего добавляли 2-(пирролидин-1-ил)этан-1-амин (134 мг, 1,16 ммоль, 2,0 экв.) и оставляли реагировать при комнатной температуре в течение 1 часа. Завершение протекания реакции определяли способом ЖХ-МС. Затем в реакционный раствор добавляли воду (10 мл) и экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали водой (10 мл × 3), сушили безводным сульфатом натрия, фильтровали, и концентрировали при пониженном давлении; неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 20:1), получая продукт (106 мг, выход: 41%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,26 (s, 1Н), 9,08-9,11 (m, 1Н), 8,67 (s, 1Н), 8,30 (s, 1Н), 3,63-3,64 (d, 8Н), 3,06-3,20 (m, 6Н), 1,73-1,98 (m, 9Н), 1,25 (s, 3Н).
Молекулярная формула: C23H30N6O3, Молекулярная масса: 438,53 ЖХ-МС (Pos, m/z) = 439,24 [М+Н]+.
Пример 84
Синтез трифторацетата 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-N-((1-метилпиперидин-4-ил)метил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 141)
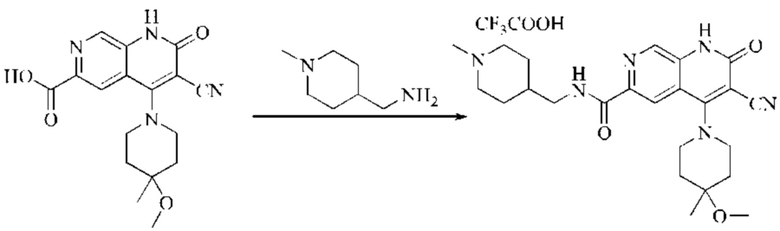
Трифторацетат соединения 141
Промежуточную 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (200 мг, 0,58 ммоль, 1,0 экв.) растворяли в безводном N,N-диметилацетамиде (2 мл), после чего добавляли DIPEA (226,3 мг, 1,75 ммоль, 3,0 экв.) и HATU (333,1 мг, 0,88 ммоль, 1,5 экв.), перемешивали при комнатной температуре в течение 0,5-1 часа, после чего добавляли (1-метилпиперидин-4-ил)метанамин (150 мг, 1,17 ммоль, 2,0 экв.) и оставляли реагировать в течение 1 часа при комнатной температуре. После того, как исходные материалы все еще обнаруживались способом ЖХ-МС, вновь добавляли (1-метилпиперидин-4-ил)метанамин (150 мг, 1,17 ммоль, 2,0 экв.) и снова оставляли реагировать в течение 2 часов. Смесь очищали препаративной ВЭЖХ (0,1% водная трифторуксусная кислота : ацетонитрил = 70:30), получая продукт (68,8 мг, выход: 20,7%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,22 (s, 1Н), 9,00-8,99 (s, 1Н), 8,95-8,94 (s, 1Н), 8,65 (s, 1Н), 8,28 (s, 1Н), 3,63-3,62 (m, 4Н), 3,43-3,40 (m, 2Н), 3,23 (s, 3Н), 2,95-2,83 (m, 2Н), 2,75-2,74 (m, 2Н), 1,97-1,94 (m, 2Н), 1,85-1,80 (m, 2Н), 1,78-1,75 (m, 3Н), 1,24 (s, 3Н).
Молекулярная формула: C24H32N6O3, Молекулярная масса: 452,56 ЖХ-МС (Pos, m/z) = 453,45 [М+Н]+.
Пример 85
Синтез трифторацетата 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-N-(1-метилазетидин-3-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида
(Соединение 142)
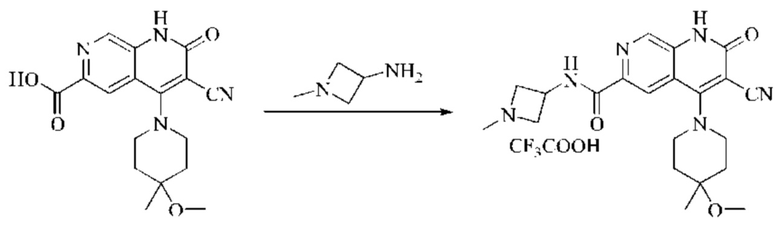
Трифторацетат соединения 142 Промежуточную 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин- 6-карбонов кислот (200 мг, 0,58 ммоль, 1,0 экв.) растворяли в безводном N,N-диметилацетамиде (2 мл), после чего добавляли DIPEA (226,3 мг, 1,75 ммоль, 3,0 экв.) и HATU (333,1 мг, 0,88 ммоль, 1,5 экв.), перемешивали при комнатной температуре в течение времени, составляющего от 0, до 1 часа, и затем добавляли 1-метилазетидин-3-амин (100,6 мг, 1,17 ммоль, 2,0 экв.) и оставляли реагировать при комнатной температуре в течение 12 часов. Неочищенный продукт очищали препаративной ВЭЖХ (0,1% водная трифторуксусная кислота : ацетонитрил = 70:30), получая продукт (113,13 мг, выход: 37,1%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,27 (s, 1Н), 9,59-9,53 (s, 2Н), 8,68 (s, 1Н), 8,27 (s, 1Н), 4,90-4,86 (m, 1Н), 4,45 (m, 2Н), 4,16 (m, 2Н), 3,63-3,62 (m, 4Н), 3,20 (s, 3Н), 2,91 (s, 3Н), 1,96-1,93 (m, 2Н), 1,79-1,72 (m, 2Н), 1,24 (s, 3Н).
Молекулярная формула: C21H26N6O3, Молекулярная масса: 410,48 ЖХ-МС (Pos, m/z) = 411,40 [М+Н]+.
Пример 86
Синтез 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-N-(1-метилпиперидин-4-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 143)
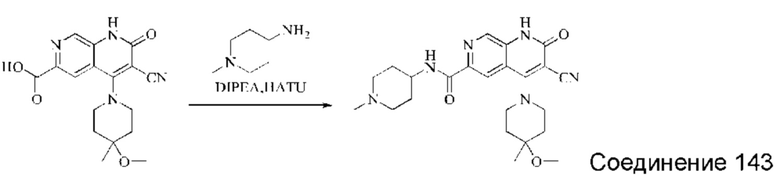
Исходное вещество, 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (200 мг, 0,58 ммоль, 1,0 экв.), растворяли в N,N-диметилформамиде (2 мл), после чего добавляли N,N-диизопропилэтиламин (226 мг, 1,75 ммоль, 3,0 экв.) и HATU (333 мг, 0,87 ммоль, 1,5 экв.), оставляли реагировать при комнатной температуре в течение 1 часа, затем добавляли 1-метилпиперидин-4-амин (67 мг, 0,58 ммоль, 1,0 экв.) и оставляли реагировать при комнатной температуре в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор очищали препаративной ВЭЖХ (0,1% водная трифторуксусная кислота : ацетонитрил = 70:30), получая продукт (49 мг, выход: 19%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,22 (s, 1Н), 9,37 (s, 1Н), 8,90 (m, 1Н), 8,67 (s, 1Н), 8,27 (s, 1Н), 4,02-4,04 (m, 1Н), 3,62-3,64 (m, 4Н), 3,46-3,48 (m, 2Н), 3,21 (s, 3Н), 3,09 (m, 1Н), 2,78 (s, 3Н), 1,88-2,01 (m, 6Н), 1,73-1,80 (m, 2Н), 1,24 (s, 3Н).
Молекулярная формула: C23H30N6O3, Молекулярная масса: 438,53 ЖХ-МС (Pos, m/z) = 439,37 [М+Н]+.
Пример 87
Синтез 3-циано-N-(2,3-дигидроксипропил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 144)
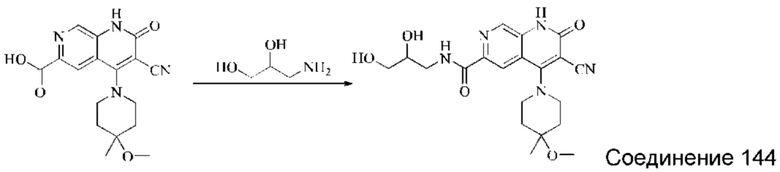
Промежуточную 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (200 мг, 0,58 ммоль, 1,0 экв.) растворяли в безводном N,N-диметилацетамиде (2 мл), после чего добавляли DIPEA (226,3 мг, 1,75 ммоль, 3,0 экв.) и HATU (333,1 мг, 0,88 ммоль, 1,5 экв.), перемешивали при комнатной температуре в течение времени, составляющего от 0,5 до 1 часа, затем добавляли 3-аминопропан-1,2-диол (106,4 мг, 1,17 ммоль, 2,0 экв.) и оставляли реагировать при комнатной температуре в течение 12 часов. Неочищенный продукт очищали препаративной ВЭЖХ (0,1% водная трифторуксусная кислота : ацетонитрил = 70:30) и подвергали лиофилизации, получая образец (93,79 мг). Образец растворяли в воде, доводили до рН 8 добавлением водного раствора бикарбоната натрия, экстрагировали н-бутанолом (20 мл × 5), и органическую фазу концентрировали, получая продукт (47,2 мг, выход: 19,4%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 8,42-8,39 (s, 1Н), 8,36 (s, 1Н), 8,08 (s, 1Н), 4,96 (s, 1Н), 4,66 (s, 1Н), 3,49 (m, 1Н), 3,47-3,45 (m, 6Н), 3,25-3,23 (m, 2Н), 3,19 (s, 3Н), 1,91-1,88 (m, 2Н), 1,75-1,70 (m, 2Н), 1,23 (s, 3Н).
Молекулярная формула: C20H25N5O5, Молекулярная масса: 415,45 ЖХ-МС (Neg, m/z) = 414,34 [М-Н]-.
Пример 88
Синтез 4-(4-метокси-4-метилпиперидин-1-ил)-6-(2-метоксиэтокси)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 145)
Этап 1: Синтез метил-2-(2-метоксиэтокси)-5-нитроизоникотината
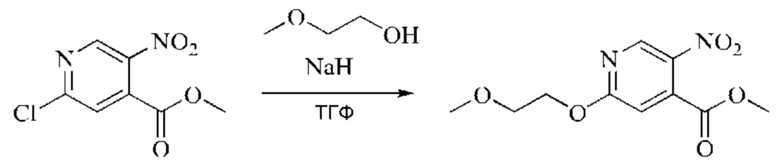
Неочищенный материал, простой монометиловый эфир этиленгликоля (3,5 г, 46,17 ммоль, 1,0 экв.), растворяли в тетрагидрофуране (50 мл), охлаждали до 0°С, после чего добавляли гидрид натрия (3,7 г, 92,34 ммоль, 2,0 экв.). Реакция протекала в течение 1 часа, после чего добавляли метил-2-хлор-5-нитроизоникотинат (10,0 г, 46,17 ммоль, 1,0 экв.). Завершение протекания реакции определяли способом ТСХ (ПЭ:ЭА = 5:1). Реакционный раствор выливали в ледяную воду (100 мл) и тушили. Водную фазу экстрагировали этилацетатом (100 мл × 2), органические фазы объединяли и сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 5:1), получая продукт в виде бледно-желтого масла (1,8 г, выход: 15%).
Этап 2: Синтез метил-5-амино-2-(2-метоксиэтокси)изоникотината
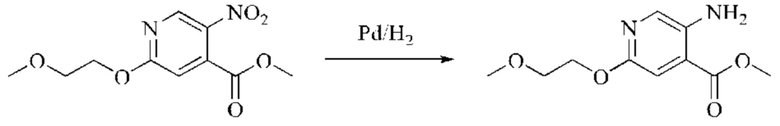
Промежуточный метил-2-(2-метоксиэтокси)-5-нитроизоникотинат (1,8 г, 7,02 ммоль, 1,0 экв.) растворяли в метаноле (10 мл), после чего добавляли 10% палладий на углеродном носителе (500 мг), подавали водород и оставляли реагировать в течение ночи при комнатной температуре. Завершение протекания реакции определяли способом ТСХ (ПЭ:ЭА = 3:1). Реакционный раствор фильтровали и концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 5:1), получая продукт в виде бледно-желтого твердого вещества (1,2 г, выход: 75%).
Этап 3: Синтез метил-5-(2-цианоацетамидо)-2-(2-метоксиэтокси)изоникотината
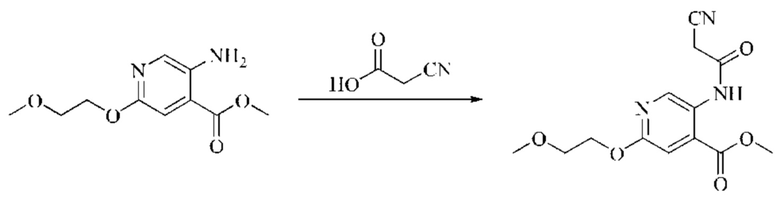
Промежуточный метил-5-амино-2-(2-метоксиэтокси)изоникотинат (1,2 г, 5,3 ммоль, 1,0 экв.) и цианоуксусную кислоту (901 мг, 10,6 ммоль, 2,0 экв.) растворяли в дихлорметане (20 мл), после чего добавляли EDCI (3,04 г, 15,9 ммоль, 3,0 экв.) и оставляли реагировать при комнатной температуре в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор выливали в ледяную воду (30 мл) и тушили. Водную фазу экстрагировали дихлорметаном (30 мл × 2), органические фазы объединяли и сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт суспендировали в простом метил-трет-бутиловом эфире, получая продукт в виде бледно-желтого твердого вещества (1,2 г, выход: 77%).
Этап 4: Синтез 4-гидрокси-6-(2-метоксиэтокси)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
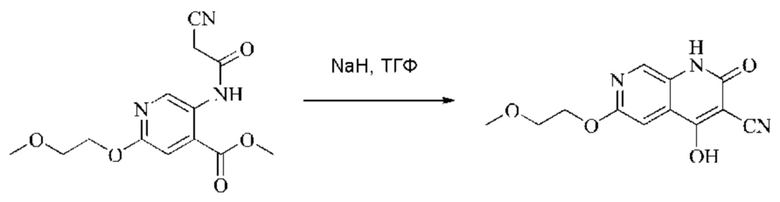
Промежуточный метил-5-(2-цианоацетамидо)-2-(2-метоксиэтокси)изоникотинат (1,2 г, 4,09 ммоль, 1,0 экв.) растворяли в ТГФ (20 мл), после чего добавляли гидрид натрия (327 мг, 8,18 ммоль, 2,0 экв.), затем нагревали до 80°С и оставляли реагировать в течение 4 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали до приблизительно 0°С, доводили до рН 2 водным раствором соляной кислоты концентрацией 2 моль/л, в результате чего не выпадал твердый осадок, и затем фильтровали. Осадок на фильтре сушили при 50°С при обычном давлении, получая продукт в виде желтого твердого вещества (800 мг, выход: 75%).
Этап 5: Синтез 4-хлор-6-(2-метоксиэтокси)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
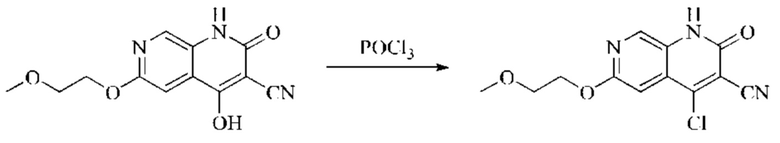
Промежуточный 4-гидрокси-6-(2-метоксиэтокси)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (800 мг, 3,06 ммоль, 1,0 экв.) растворяли в оксихлориде фосфора (8 мл), нагревали до 100°С и оставляли реагировать в течение 1 часа. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор выливали в ледяную воду (20 мл) и тушили. Водную фазу экстрагировали дихлорметаном (30 мл × 3), органические фазы объединяли и сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт суспендировали в простом метил-трет-бутиловом эфире, получая продукт в виде бледно-желтого твердого вещества (180 мг, выход: 21%).
Этап 6: Синтез 4-(4-метокси-4-метилпиперидин-1-ил)-6-(2-метоксиэтокси)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
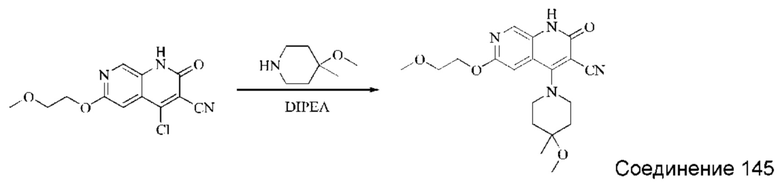
Промежуточный 4-хлор-6-(2-метоксиэтокси)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (180 мг, 0,64 ммоль, 1,0 экв.) растворяли в N,N-диметилформамиде (2 мл), после чего добавляли N,N-диизопропилэтиламин (332 мг, 2,58 ммоль, 4,0 экв.) и 4-метокси-4-метилпиперидин (110 мг, 0,97 ммоль, 1,0 экв.), нагревали до 80°С и затем оставляли реагировать в течение двух часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор выливали в ледяную воду (20 мл) и тушили. Водную фазу экстрагировали дихлорметаном (30 мл × 3), органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 15:1), получая продукт в виде бледно-желтого масла (60 мг, выход: 25%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,78 (s, 1Н), 8,28 (s, 1Н), 6,94 (s, 1Н), 4,37 (m, 2Н), 3,67 (m, 2Н), 3,56-3,58 (m, 4Н), 3,30 (s, 3Н), 3,18 (s, 3Н), 1,88-1,91 (m, 2Н), 1,75-1,80 (m, 2Н), 1,21 (s, 3Н).
Молекулярная формула: C19H24N4O4, Молекулярная масса: 372,43 ЖХ-МС (Pos, m/z) = 373,3 [М+Н]+.
Пример 89
Синтез 3-циано-N,N-диметил-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 149)
3-циано-2-оксо-4-(6-аза-спиро[2,5]октан-6-ил)-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (256,4 мг, 0,790 ммоль, 1,0 экв.) растворяли в ДМФА (20 мл), охлаждали до 0°С на ледяной бане, после чего добавляли HATU (450,8 мг, 1,186 ммоль, 1,5 экв.) и затем добавляли DIPEA (613,0 мг, 4,743 ммоль, 6,0 экв.) и гидрохлорид диметиламина (193,3 мг, 2,371 ммоль, 3,0 экв.). По завершении добавления смесь перемешивали при комнатной температуре в течение 30 минут. Завершение протекания реакции определяли способом ТСХ. В смесь добавляли воду (100 мл) и экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором (10 мл×4), сушили безводным сульфатом натрия, фильтровали с отсасыванием, концентрировали при пониженном давлении и очищали способом препаративной тонкослойной хроматографии (ДХМ:МеОН = 8:1), получая продукт в виде желтого твердого вещества (91,0 мг, выход: 32,8%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,19 (s, 1Н), 8,61 (s, 1Н), 7,89 (s, 1H), 3,68-3,65 (t, 4H), 3,06 (s, 3Н), 3,02 (s, 3H), 1,61 (s, 4H), 0,44 (s, 4H).
Молекулярная формула: C19H21N5O2, Молекулярная масса: 351,41 ЖХ-МС (Pos, m/z)=352,20 [М+Н]+.
Пример 90
Синтез 4-(4-(2-гидроксиэтил)пиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 153)
Промежуточный 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (60 мг, 0,29 ммоль, 1,0 экв.) растворяли в N,N-диметилформамиде (1 мл), и в реакционную смесь добавляли N,N-диизопропилэтиламин (112 мг, 0,87 ммоль, 3,0 экв.) и затем добавляли 2-(пиперидин-4-ил)этан-1-ол (38 мг, 0,29 ммоль, 1,0 экв.). Температуру повышали до 80°С, и реакцию проводили в течение 2 часов. Смесь охлаждали до комнатной температуры, в результате чего выпадал твердый осадок, который затем фильтровали. Осадок на фильтре последовательно споласкивали ТГФ (1 мл) и петролейным эфиром (1 мл) и сушили при 45°С, получая желтый твердый продукт (16 мг, выход: 18%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,98 (s, 1H), 8,65 (s, 1H), 8,33-8,35 (d, 1H), 7,58-7,80 (d, 1H), 4,40-4,42 (m, 1H), 3,83-3,86 (m, 2H), 3,48-3,53 (m, 2H), 3,37-3,43 (m, 2H), 1,84-1,87 (m, 2H), 1,74-1,80 (m, 1H), 1,45-1,50 (m, 4H).
Молекулярная формула: C16H18N4O2, Молекулярная масса: 298,35 ЖХ-МС (Neg, m/z)=297,15[M-H]-.
Пример 91
Синтез (R)-4-(3-(1Н-1,2,4-триазол-1-ил)пирролидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 154)
Этап 1: Синтез трет-бутил (R)-3-(1H-1,2,4-триазол-1-ил)пирролидин-1-карбоксилата
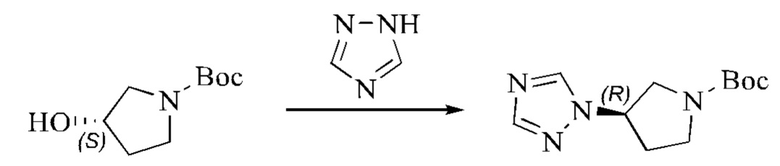
Исходное вещество, трет-бутил-(S)-3-гидроксипирролидин-1-карбоксилат (1,0 г, 5,34 ммоль, 1,0 экв.), растворяли в безводном тетрагидрофуране (20 мл), и в реакционную смесь добавляли 1H-1,2,4-триазол (552,7 мг, 8,01 ммоль, 1,5 экв.) и трифенилфосфин (2,8 г, 10,68 ммоль, 2,0 экв.). По завершении добавления температуру понижали до 0°С и по каплям добавляли диэтилазодикарбоксилат (1,86 г, 10,68 ммоль, 2,0 экв.). По завершении добавления реакцию проводили при комнатной температуре в течение 12 часов. Завершение протекания реакции определяли способом ТСХ. К полученному раствору добавляли насыщенный водный раствор карбоната натрия (10 мл), раствор смесь экстрагировали этилацетатом (50 мл×2), и слои разделяли. Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ЭА:ПЭ = 1:10-1:2), получая продукт (1,05 г, выход: 83,3%).
Этап 2: Синтез гидрохлорида (R)-1-(пирролидин-3-ил)-1H-1,2,4-триазола

Промежуточный трет-бутил-(R)-3-(1H-1,2,4-триазол-1-ил)пирролидин-1-карбоксилат (1,05 г, 4,41 ммоль, 1,0 экв.) растворяли в безводном метаноле (4 мл), после чего добавляли 30%-ный раствор хлорида водорода в этаноле (10 мл) и оставляли реагировать при комнатной температуре в течение 2 часов до осаждения белого твердого вещества. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор фильтровали, осадок на фильтре споласкивали этилацетатом и сушили, получая продукт (770 мг, выход: 100%).
Этап 3: Синтез (R)-4-(3-(1H-1,2,4-триазол-1-ил)пирролидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
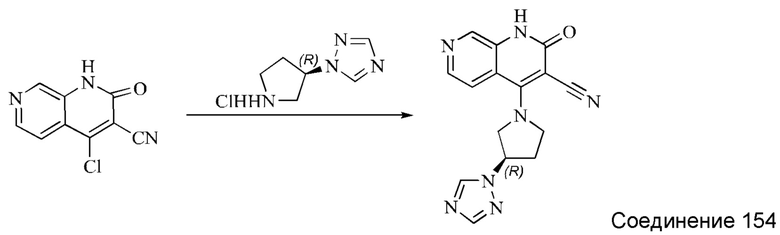
Промежуточный 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (200 мг, 0,97 ммоль, 1,0 экв.) растворяли в ДМФА (2 мл), и в реакционную смесь добавляли гидрохлорид (R)-1-(пирролидин-3-ил)-1H-1,2,4-триазола (238 мг, 1,36 ммоль, 1,4 экв.) и DIPEA (753,7 мг, 5,84 ммоль, 6,0 экв.). По завершении добавления температуру повышали до 80°С, и реакцию проводили в течение 2 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор охлаждали до комнатной температуры и очищали препаративной ВЭЖХ (0,1% водная трифторуксусная кислота : ацетонитрил = 70:30), получая продукт (98,6 мг, выход: 33%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,67 (s, 1Н), 8,69 (s, 1Н), 8,62 (s, 1H), 8,28-8,27 (d, 1Н), 8,03 (s, 1Н), 7,98-7,96 (s, 1Н), 5,33-5,28 (m, 1Н), 4,61-4,57 (m, 1Н), 4,35-4,29 (m, 2H), 4,23-4,17 (m, 1Н), 2,56-2,53 (m, 1Н), 2,48-2,47 (m, 1Н).
Молекулярная формула: C15H13N7O, Молекулярная масса: 307,21 ЖХ-МС (Pos, m/z)=307,98 [M+H]+.
Пример 92
Синтез 4-(R)-3-(1Н-пиразол-1-ил)пирролидин-1-ил)-6-(1-гидроксиэтил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 156)
Этап 1: Синтез гидрохлорида (R)-1-(пирролидин-3-ил)-1H-пиразола
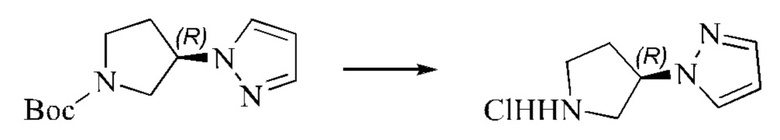
Исходное вещество, трет-бутил-(R)-3-(1H-пиразол-1-ил)пирролидин-1-карбоксилат (2,7 г, 11,37 ммоль), растворяли в безводном метаноле (4 мл), и в реакционную смесь добавляли 30%-ный раствор хлорида водорода в этаноле (10 мл). По завершении добавления реакцию проводили при комнатной температуре в течение 2 часов. Завершение протекания реакции определяли способом ТСХ. Смесь концентрировали при пониженном давлении, получая маслянистый продукт (3,29 г неочищенный продукт).
Этап 2: Синтез (R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-6-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
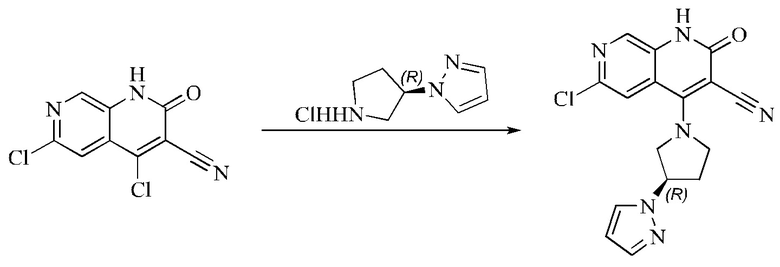
Промежуточный 4-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (2,27 г, 9,46 ммоль, 1,0 экв.) растворяли в ДМФА (8 мл), и в реакционную смесь добавляли гидрохлорид (R)-1-(пирролидин-3-ил)-1H-пиразола (3,29 г неочищенного продукта) и DIPEA (7,3 г, 56,76 ммоль, 6,0 экв.). По завершении добавления температуру повышали до 80°С, и реакцию проводили в течение 2 часов.
Завершение протекания реакции определяли способом ЖХ-МС, и температуру понижали до комнатной температуры. Реакционный раствор выливали в воду (50 мл), перемешивали в течение получаса, фильтровали. Осадок на фильтре споласкивали водой, затем суспендировали в простом метил-трет-бутиловом эфире и фильтровали; осадок на фильтре сушили, получая продукт (2,35 г, выход в результате двух этапов синтеза: 73,4%).
Этап 3: Синтез (R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-2-оксо-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
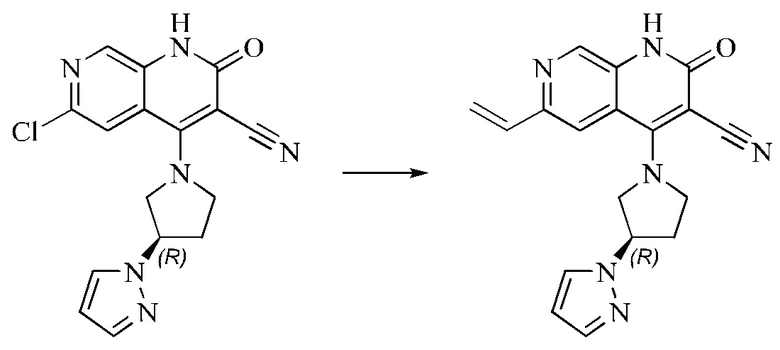
Промежуточный (R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-6-хлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (2,3 г, 6,75 ммоль, 1,0 экв.), карбонат цезия (6,6 г, 20,25 ммоль, 3,0 экв.) и винилтрифторборат калия (1,3 г, 10,12 ммоль, 1,5 экв.) растворяли в смеси растворителей 1,4-диоксана (50 мл) и воды (50 мл). Воздух в системе трижды вытесняли азотом и затем в реакционную смесь добавляли дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (493 мг, 0,68 ммоль, 0,1 экв.). Воздух в системе трижды вытесняли азотом, смесь кипятили с обратным холодильником и оставляли реагировать в течение 12 часов. Завершение протекания реакции определяли способом ТСХ, и температуру понижали до комнатной температуры. Добавляли этилацетат (50 мл) и воду (10 мл), перемешивали в течение 10 минут и фильтровали. Осадок на фильтре споласкивали этилацетатом, и жидкость отделяли. Водную фазу экстрагировали дихлорметаном (50 мл×3), органические фазы объединяли, сушили безводным сульфатом натрия и фильтровали. Маточный раствор концентрировали при пониженном давлении, суспендировали в метил-трет-бутиловом эфире, что приводило к осаждению бледно-желтого твердого вещества, и затем фильтровали. Осадок на фильтре сушили, получая продукт (1,45 г, выход: 65,9%).
Этап 4: Синтез 4-((R)-3-(1H-пиразол-1-ил)пирролидин-1-ил)-6-(1,2-дигидроксиэтил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
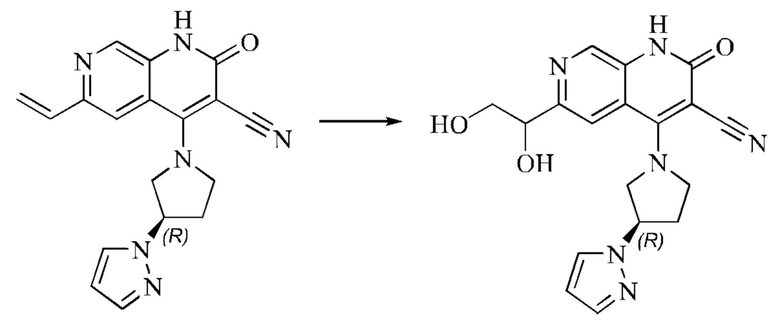
Промежуточный (R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-2-оксо-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (453 мг, 1,36 ммоль, 1,0 экв.) растворяли в смеси растворителей трет-бутанола (7 мл) и воды (7 мл), после чего добавляли метансульфонамид (129,5 мг, 1,36 ммоль, 1,0 экв.) и AD-mix-β (5,4 г). По завершении добавления, реакцию в полученной смеси оставляли протекать при комнатной температуре в течение 12 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор непосредственно вводили в следующий этап.
Этап 5: Синтез (R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-6-формил-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
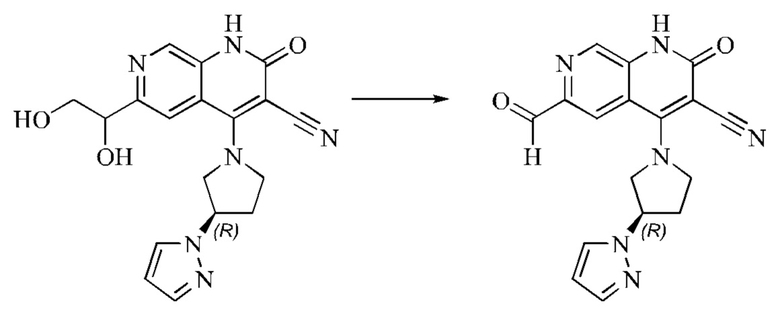
Перйодат натрия (582,6 мг, 2,72 ммоль, 2,0 экв.) и тетрагидрофуран (5 мл) добавляли к реакционному раствору, полученному в предыдущем этапе. По завершении добавления, реакцию проводили в течение 4 часов при комнатной температуре, и завершение протекания реакции определяли способом ЖХ-МС. Добавляли воду (20 мл), и жидкость отделяли. Водную фазу экстрагировали дихлорметаном (50 мл×4), органические фазы объединяли, сушили и фильтровали. Фильтрат концентрировали при пониженном давлении, получая бледно-желтое твердое вещество. Твердое вещество суспендировали в простом метил-трет-бутиловом эфире, фильтровали, и осадок на фильтре сушили, получая продукт (215 мг, выход: 47,2%).
Этап 6: Синтез 4-(R)-3-(1H-пиразол-1-ил)пирролидин-1-ил)-6-(1-гидроксиэтил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
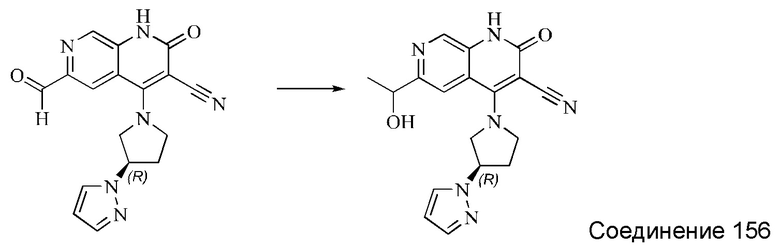
Промежуточный (R)-4-(3-(1H-пиразол-1-ил)пирролидин-1-ил)-6-формил-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (215 мг, 0,64 ммоль, 1,0 экв.) растворяли в безводном тетрагидрофуране (3 мл), охлаждали до -10 ~ 0°С в атмосфере азота, затем по каплям добавляли раствор метилмагнийхлорида в тетрагидрофуране (3 моль/л, 0,32 мл, 0,96 ммоль, 1,5 экв.). По завершении добавления реакцию проводили в течение 12 часов при комнатной температуре. Завершение протекания реакции определяли способом ЖХ-МС. рН реакционного раствора доводили до приблизительно 8, добавляя по каплям насыщенный водный раствор хлорида аммония, и экстрагировали дихлорметаном (50 мл×4). Органические фазы объединяли, сушили, фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (МеОН:ДХМ = от 1:100 до 1:40), получая продукт (44,6 мг, выход: 19,8%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,59 (s, 1Н), 8,54 (s, 1Н), 8,00 (s, 1H), 7,90-7,89 (d, 1Н), 7,58-7,56 (d, 1Н), 7,51 (d, 1Н), 6,31-6,30 (d, 1Н), 6,29 (d, 1Н), 5,46-5,45 (m, 1Н), 5,20 (d, 1H), 4,79-4,73 (m, 1H), 4,57-4,50 (m, 1H), 4,37-4,20 (m, 1H), 2,48 (m, 2H), 1,38-1,37 (d, 3H).
Молекулярная формула: C18H18N6O2, Молекулярная масса: 350,38 ЖХ-МС (Pos, m/z)=351,22[M+H]+.
Пример 93
Синтез 4-(4-метокси-4-метилпиперидин-1-ил)-6-метил-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 158)
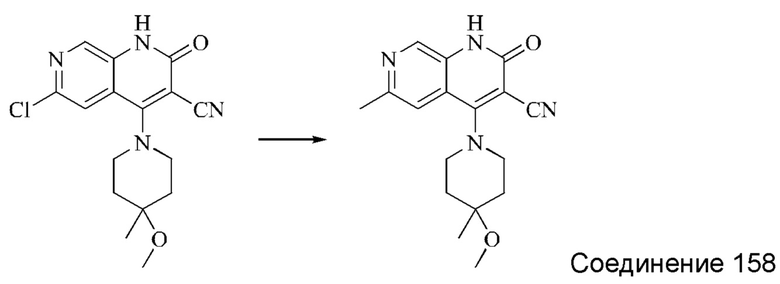
Промежуточный 6-хлор-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (1,3 г, 3,91 ммоль, 1,0 экв.), карбонат цезия (3,8 г, 11,73 ммоль, 3,0 экв.) и триметилциклотриборан (50% в ТГФ, 3,9 г, 15,62 ммоль, 4,0 экв.) растворяли в 1,4-диоксане (20 мл). По завершении добавления воздух в системе трижды вытесняли азотом и затем в реакционную смесь добавляли дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (286 мг, 0,39 ммоль, 0,1 экв.). По завершении добавления воздух в системе трижды вытесняли азотом, и смесь кипятили с обратным холодильником в течение 12 часов. Когда по данным ТСХ в системе все еще оставались исходные материалы, добавляли дополнительное количество триметилциклотриборана (50%-ный раствор в ТГФ, 3,9 г, 15,62 ммоль, 4,0 экв.) и дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия (286 мг, 0,39 ммоль, 0,1 экв.), и кипячение продолжали в течение 4 часов. Когда по данным ТСХ в системе не осталось исходного материала, систему охлаждали до комнатной температуры, после чего добавляли воду (50 мл) и дихлорметан (100 мл), перемешивали в течение 5 минут, что приводило к появлению твердого осадка, который фильтровали. Осадок на фильтре споласкивали дихлорметаном. Жидкость отделяли, водную фазу экстрагировали дихлорметаном (100 мл×3), органические фазы объединяли, сушили безводным сульфатом натрия и фильтровали. Маточный раствор концентрировали при пониженном давлении, и неочищенный продукт очищали колоночной хроматографией на силикагеле (МеОН:ДХМ = 1:100-1:50), получая продукт (309,9 мг, выход: 25,4%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,88 (s, 1Н), 8,55 (s, 1Н), 7,42 (s, 1H), 3,61-3,59 (m, 4H), 3,19 (s, 3Н), 2,53 (s, 3H), 1,92-1,89 (m, 2H), 1,82-1,75 (m, 2H), 1,22 (s, 3H).
Молекулярная формула: C17H20N4O2, Молекулярная масса: 312,37 ЖХ-МС (Pos, m/z)=313,25 [M+H]+.
Пример 94
Синтез 6-(циклопропил(гидрокси)метил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 160)
Этап 1: Синтез 6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
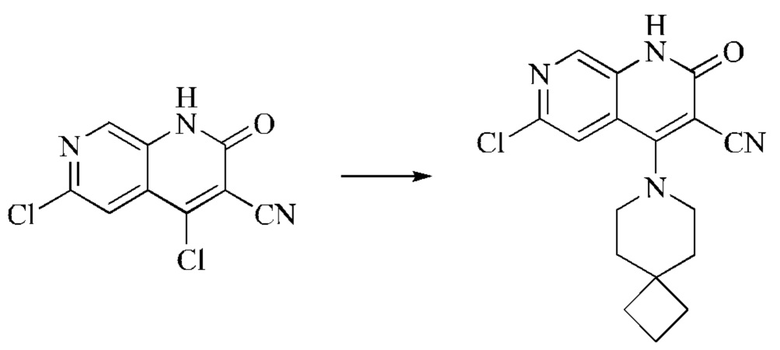
Промежуточный 4,6-дихлор-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (8,0 г, 33,3 ммоль, 1,0 экв.) растворяли в ДМФА (100 мл), после чего последовательно добавляли исходное вещество, гидрохлорид 7-аза-спиро[3,5]нонана (6,5 г, 40,0 ммоль, 1,2 экв.), и DIPEA (17,2 г, 133,2 ммоль, 4,0 экв.), и оставляли реагировать при 80°С в течение получаса. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали, после чего добавляли воду и этилацетат (40 мл/40 мл), перемешивали в течение ночи и фильтровали, получая желтый твердый продукт (10,0 г, выход: 91,5%).
Этап 2: Синтез 2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
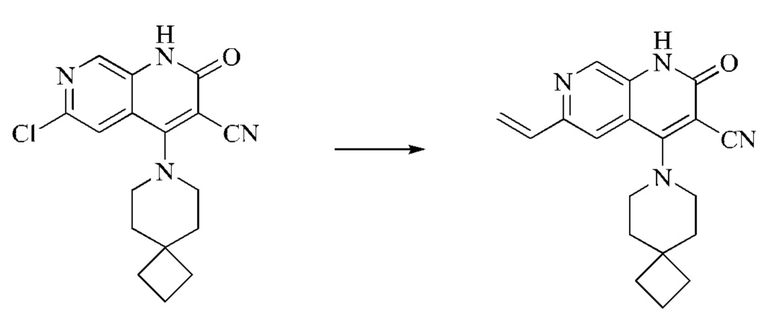
Промежуточный 6-хлор-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (5,0 г, 15,2 ммоль, 1,0 экв.) растворяли в 1,4-диоксане (50 мл). Исходные материалы, винилфторборат калия (6,1 г, 45,6 ммоль, 3,0 экв.), карбонат цезия (14,8 г, 45,6 ммоль, 3,0 экв.) и дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (1,1 г, 15,2 ммоль, 1,0 экв.), растворяли в воде (10 мл) и добавляли к полученному как указано выше реакционному раствору. Реакционную смесь кипятили в течение ночи в атмосфере азота. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор концентрировали и экстрагировали ЭА (3×100 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали с отсасыванием и концентрировали при пониженном давлении, получая продукт (4,5 г, выход: 92,5%).
Этап 3: Синтез 6-формил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
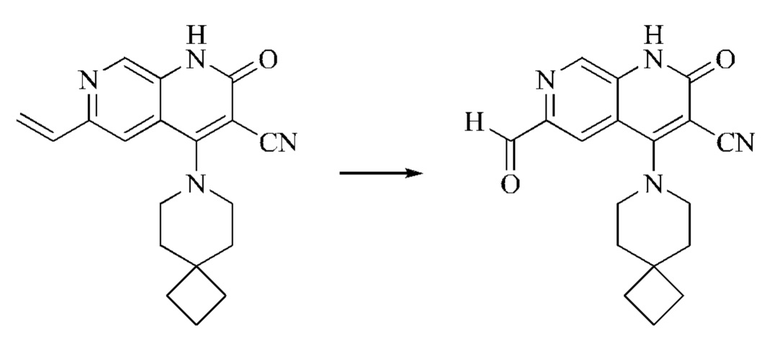
Промежуточный 2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-6-винил-1,2-дигидро-1,7-нафтиридин-3-карбонитрил (2,0 г, 6,2 ммоль, 1,0 экв.) растворяли в трет-бутаноле (50 мл) и воде (40 мл), после чего последовательно при 0°С добавляли Ad-mix-β (20 г, 10 экв.) и метансульфонамид (1,2 г, 12,5 ммоль, 2,0 экв.) и перемешивали при комнатной температуре в течение 48 часов. Завершение протекания реакции определяли способом ЖХ-МС. Перйодат натрия (4,0 г, 18,6 ммоль, 3,0 экв.) растворяли в воде (10 мл) при 0°С и добавляли по каплям к полученному как указано выше реакционному раствору. Реакционный раствор перемешивали при комнатной температуре в течение 12 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор экстрагировали ЭА (3×100 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 40:1), получая продукт в виде желтого твердого вещества (1,0 г, выход: 50,4%).
Этап 4: Синтез 6-(циклопропил(гидрокси)метил)-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
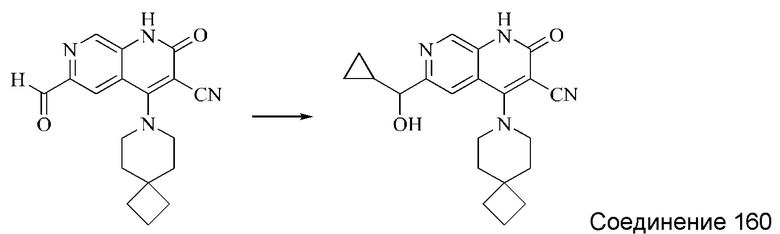
Промежуточный 6-формил-2-оксо-4-(7-аза-спиро[3,5]нонан-7-ил)-1,2-дигид PC-1,7-нафтиридин-3-карбонитрил (500,0 мг, 1,6 ммоль, 1,0 экв.) растворяли в 2-метилтетрагидрофуране (5 мл), затем медленно, в атмосфере азота, при -20°С добавляли циклопропилмагнийбромид (7,5 мл, 7,75 ммоль, 5 экв.) и перемешивали при -10°С в течение 12 часов. Завершение протекания реакции определяли способом ЖХ-МС. Реакционный раствор тушили насыщенным водным раствором хлорида аммония при 0°С и экстрагировали ЭА (3×30 мл). Органическую фазу сушили безводным сульфатом натрия, фильтровали с отсасыванием, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 30:1-20:1), получая продукт (80,0 мг, выход: 13,7%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,92 (br, 1Н), 8,58 (s, 1Н), 7,69 (s, 1H), 5,41 (d, 1Н), 4,23-4,26 (m, 1Н), 3,54 (s, 4H), 1,80-1,99 (m, 9H), 1,11-1,24 (m, 2H), 0,42 (s, 4H).
Молекулярная формула: C21H24N4O2, Молекулярная масса: 364,45 ЖХ-МС (m/z)=365 [М+Н]+.
Пример 95
Синтез 6-(1-гидрокси-2-метилпропан-2-ил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила (Соединение 161)
Этап 1: Синтез этил-2-(3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионат
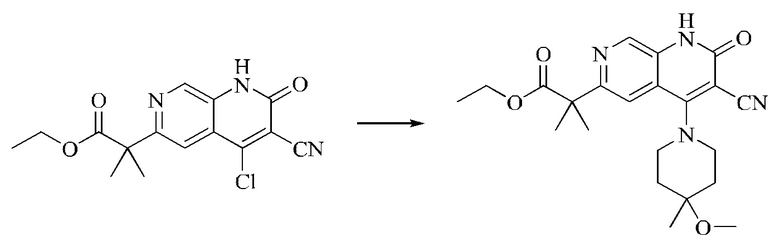
Промежуточный этил-2-(4-хлор-3-циано-2-оксо-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионат (600,0 мг, 1,87 ммоль, 1,0 экв.) растворяли в ДМФА (5 мл), после чего последовательно добавляли исходное вещество, гидрохлорид 4-метил-4-метоксипиперидина (339,4 мг, 2,05 ммоль, 1,1 экв.), и DIPEA (1,4 г, 11,2 ммоль, 6,0 экв.) и оставляли реагировать при 80°С в течение получаса. Завершение протекания реакции определяли способом ТСХ. Реакционный раствор концентрировали, после чего добавляли воду и экстрагировали ЭА (3×60 мл). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали, и концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 3:2), получая продукт (300,0 мг, выход: 38,9%).
Этап 2: Синтез 6-(1-гидрокси-2-метилпропан-2-ил)-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-3-карбонитрила
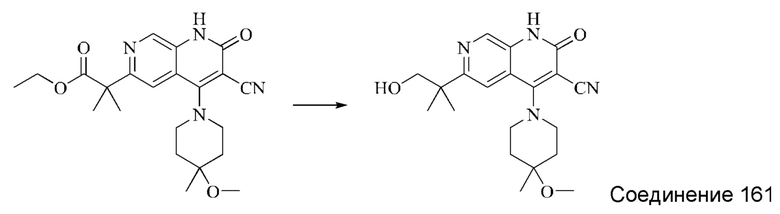
Промежуточный этил-2-(3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-ил)-2-метилпропионат (300,0 мг, 0,73 ммоль, 1,0 экв.) растворяли в безводном 2-метилтетрагидрофуране (5 мл), затем при -60°С в атмосфере азота добавляли по каплям DIBAL-H (раствор концентрацией 1,5 моль/л в толуоле, 2,4 мл, 3,65 ммоль, 5,0 экв.), после чего нагревали до комнатной температуры и перемешивали в течение 6 часов. Реакцию останавливали, добавляя по каплям воду при 0°С, концентрировали и экстрагировали ЭА (3×60 мл). Органические фазы объединяли, сушили безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ПЭ:ЭА = 3:2), получая продукт (60,0 мг, выход: 22,2%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 11,88 (s, 1Н), 8,63 (s, 1Н), 7,53 (s, 1H), 4,65 (t, 1Н), 3,59-3,62 (m, 4H), 3,54 (d, 2H), 3,19 (s, 3H), 1,91-1,95 (m, 2H), 1,73-1,78 (m, 2H), 1,28 (s, 6H), 1,23 (s, 3H).
Молекулярная формула: C20H26N4O3, Молекулярная масса: 370,45 ЖХ-МС (Pos, m/z)=371 [М+Н]+.
Пример 96
Синтез 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-N,N-диметил-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоксамида (Соединение 162)
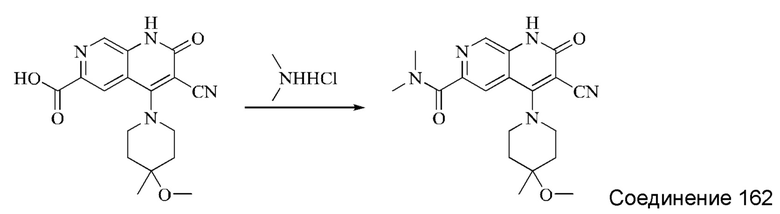
Промежуточную 3-циано-4-(4-метокси-4-метилпиперидин-1-ил)-2-оксо-1,2-дигидро-1,7-нафтиридин-6-карбоновую кислоту (200 мг, 0,58 ммоль, 1,0 экв.), HATU (333 мг, 0,88 ммоль, 1,5 экв.) и DIPEA (376 мг, 1,76 ммоль, 3,0 экв.) растворяли в ДМАА (2 мл), перемешивали при комнатной температуре в течение 30 минут, после чего добавляли гидрохлорид диметиламина (95 мг, 1,16 ммоль, 2,0 экв.), и реакцию оставляли протекать при комнатной температуре в течение 1 часа. Завершение протекания реакции определяли способом ЖХ-МС. Добавляли воду (10 мл), и смесь экстрагировали дихлорметаном (10 мл×3). Органическую фазу промывали водой (10 мл×3), сушили безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН = 50:1), получая продукт (120 мг, выход: 55%).
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.): 12,16 (s, 1Н), 8,61 (s, 1Н), 7,83 (s, 1H), 3,61-3,63 (m, 4H), 3,19 (s, 3Н), 3,03-3,06 (d, 6H), 1,90-1,93 (d, 2H), 1,71-1,78 (m, 2H), 1,22 (s, 3H).
Молекулярная формула: C19H23N5O3, Молекулярная масса: 369,18 ЖХ-МС (Pos, m/z)=370,43 [М+Н]+.
Описанными выше способами могут быть получены другие соединения. Характеристические данные некоторых других соединений согласно настоящему изобретению представлены ниже в Таблице 2.
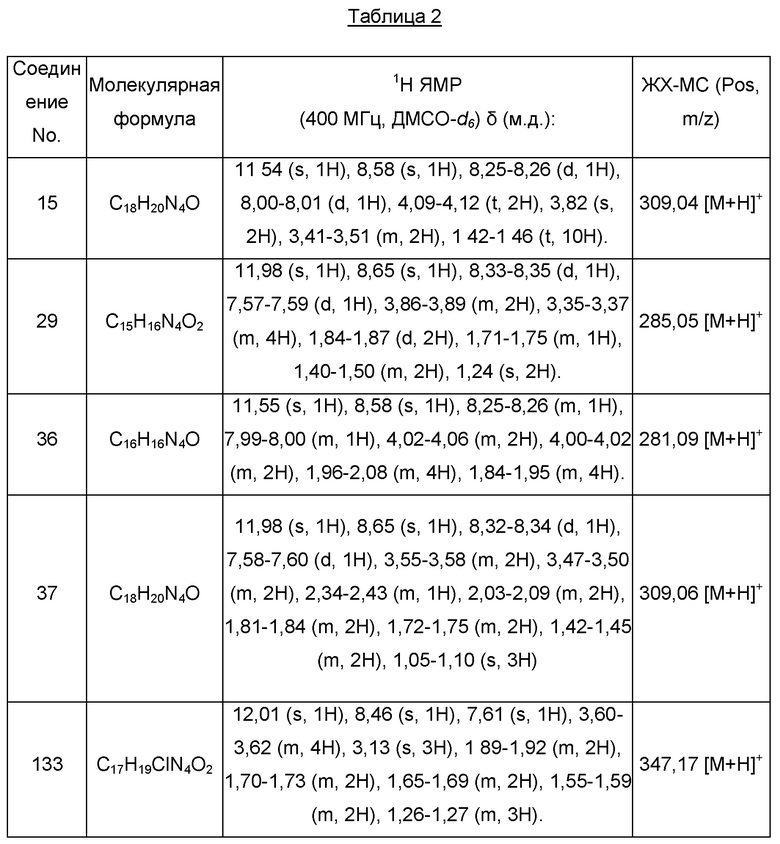
Настоящее изобретение станет более понятным после рассмотрения приведенных ниже экспериментальных примеров. Однако специалист в данной области техники должен понимать, что приведенное содержание экспериментальных примеров дано для иллюстрации изобретения и не должно ограничивать и не ограничивает объем изобретения, раскрытый в прилагаемых пунктах формулы изобретения.
Экспериментальный Пример 1
Способ оценки ферментативных свойств PDE9
Испытуемое вещество: Соединения согласно изобретению, полученные согласно соответствующим примерам осуществления изобретения
1. Экспериментальные материалы и оборудование Фермент PDE9A2 (BPS, No. по каталогу: 60090) 384-луночный планшет (Perkin Elmer, No. по каталогу: 6007279)
2. Процедура эксперимента
Приготовление соединений: соединения были приготовлены для длительного хранения в виде основного (базового) раствора соединений концентрацией 10 мМ в ДМСО (диметилсульфоксиде). Полученный основной раствор соединения разбавляли в 100 раз ДМСО, получая рабочий маточный раствор соединения концентрацией 100 мкМ, и затем рабочий маточный раствор соединения разбавляли в 3 раза ДМСО, получая 8-10 градиентов концентрации разбавленного маточного раствора соединения (100×).
Инкубация с соединениями: Для внесения разбавленного маточного раствора соединения в 384-луночный планшет применяли систему Echo для пипетирования очень малых количеств жидкости. В каждую лунку для соединения добавляли 200 панолитров разбавленного маточного раствора соединения и 10 мкл раствора фермента PDE9A2. После центрифугирования со скоростью 1000 об/мин в течение 1 минуты смесь инкубировали в течение 15 минут при комнатной температуре. Затем добавляли 10 мкл смеси субстрата. После центрифугирования со скоростью 1000 об/мин в течение 1 минуты смесь инкубировали с применением шока (with shocking) в течение 30 минут при комнатной температуре. Наконец, для остановки реакции в системе добавляли стоп-раствор. Смесь инкубировали с применением шока в течение 60 минут при комнатной температуре. При максимальном считывании (Мах) соединение было замещено растворителем. При минимальном считывании (Min), и соединение, и раствор фермента были замещены растворителем.
Измерения: Величины флуоресценции (F) при 480 нм/535 нм определяли с помощью считывающего устройства для микропланшетов.
Вычисление: Скорость ингибирования вычисляли по нижеследующей формуле, и величины IC50 аппроксимировали с помощью программы GraphPad Prism 5.0:
3. Результаты испытаний представлены в Таблице 3:
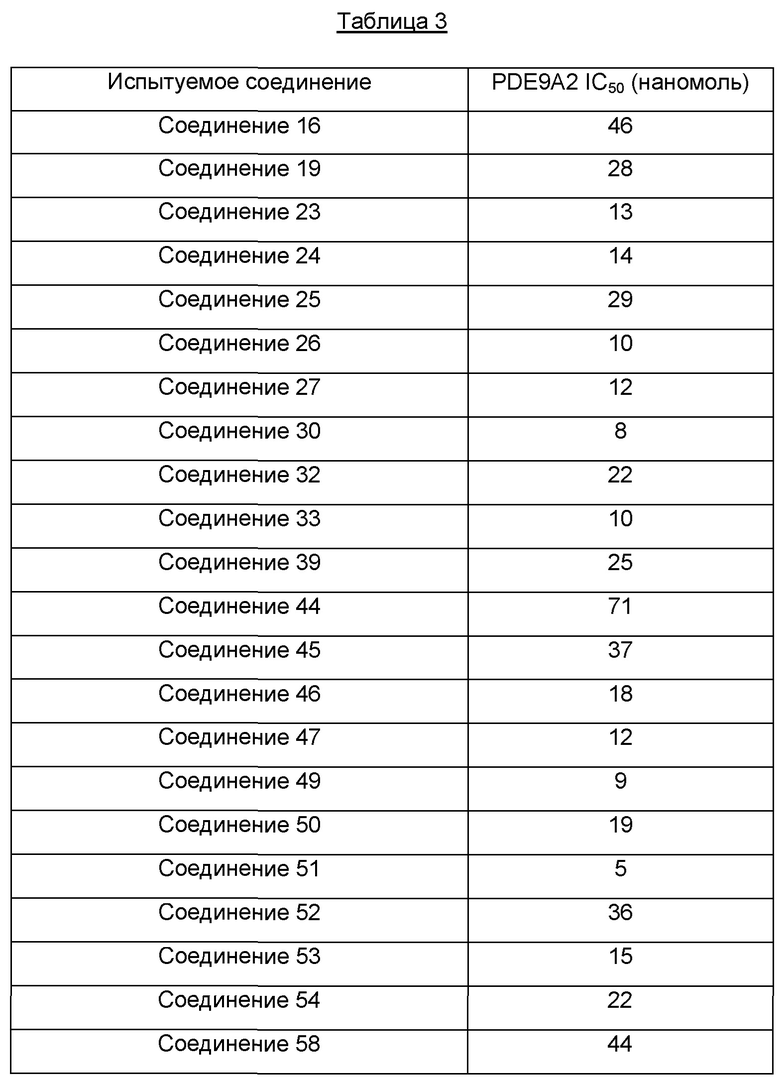
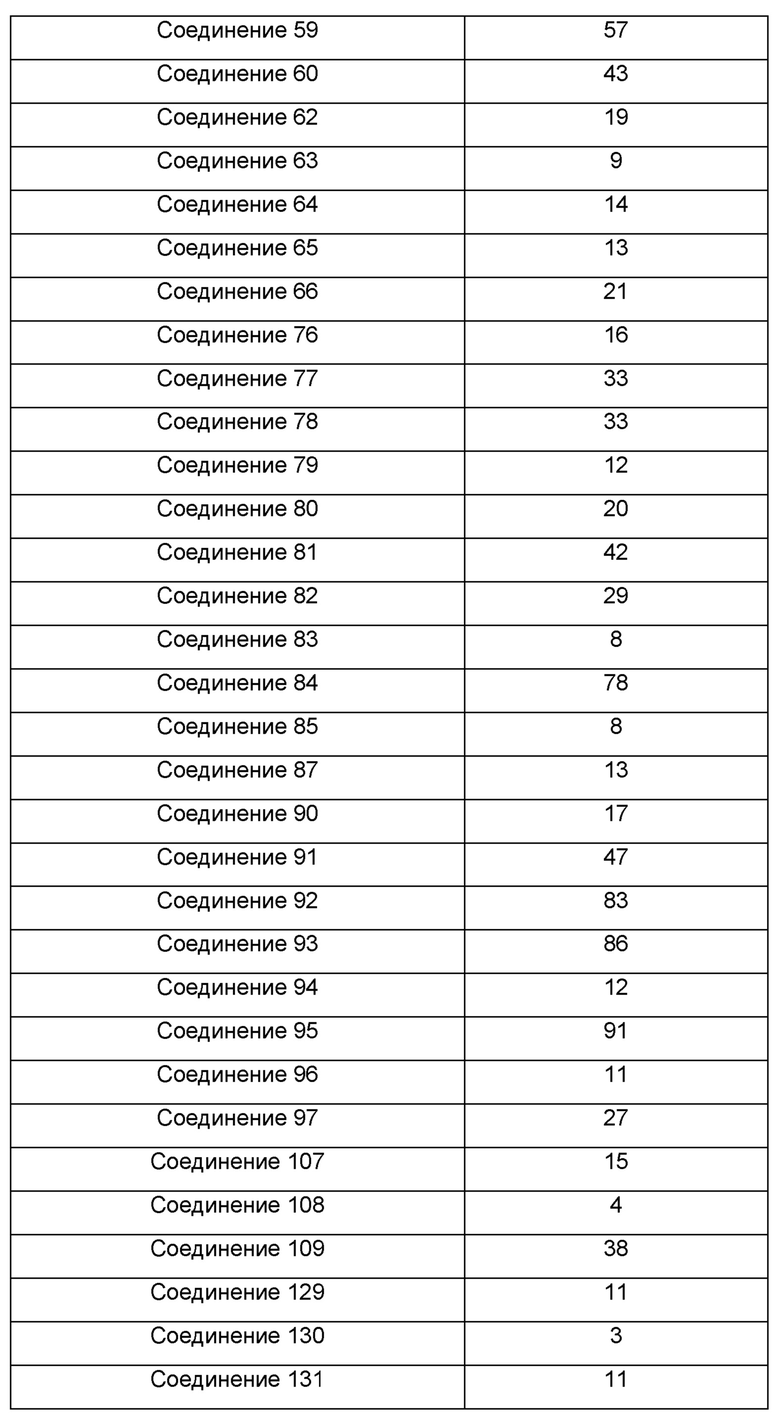
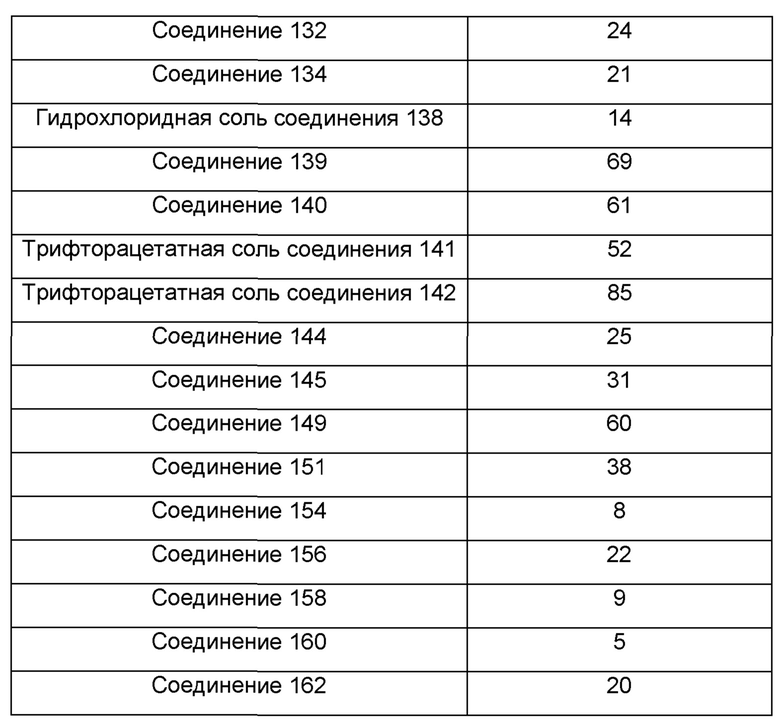
Данные, представленные в Таблице 3, показывают, что соединения согласно настоящему изобретению обладают весьма удовлетворительной ингибиторной активностью в отношении фермента PDE9, которая имеет величину, подходящую для потенциального клинического применения.
Экспериментальный Пример 2
Фармакокинетическая оценка соединений согласно изобретению в опытах на собаках
Испытуемое соединение: Соединения согласно изобретению, полученные согласно соответствующим примерам осуществления изобретения
1. Введение животным и отбор образцов
Для приготовления раствора испытуемое соединение 25 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD (сокр. от англ. 2-hydroxylpropyl-beta-cyclodextrin, русс.2-гидроксипропил-бета-декстрин)) физиологического солевого раствора. Раствор соединения 25 вводили внутрижелудочно (перорально) собакам породы бигль в дозировке 2,0 мг/кг. Отбор образцов крови осуществляли в следующие моменты времени: 0 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 25 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединения 25 вводили собакам породы бигль в дозировке 1 мг/кг посредством внутривенного болюсного введения. Отбор образцов крови осуществляли в следующие моменты времени: 0 минут, 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 65 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединения 65 вводили внутрижелудочно собакам породы бигль в дозировке 2,0 мг/кг. Отбор образцов крови осуществляли в следующие моменты времени: 0 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 65 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединения 65 вводили собакам породы бигль в дозировке 1 мг/кг посредством внутривенного болюсного введения. Отбор образцов крови осуществляли в следующие моменты времени: 0 минут, 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 107 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединения 107 вводили внутрижелудочно собакам породы бигль в дозировке 1,0 мг/кг. Отбор образцов крови осуществляли в следующие моменты времени: 0 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 107 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединения 107 вводили собакам породы бигль в дозировке 1,0 мг/кг посредством внутривенного болюсного введения. Отбор образцов крови осуществляли в следующие моменты времени: 0 минут, 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 158 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединения 158 вводили внутрижелудочно собакам породы бигль в дозировке 1,0 мг/кг. Отбор образцов крови осуществляли в следующие моменты времени: 0 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 158 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединения 158 вводили собакам породы бигль в дозировке 1,0 мг/кг посредством внутривенного болюсного введения. Отбор образцов крови осуществляли в следующие моменты времени: 0 минут, 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов и 24 часа после введения.
Отбирали приблизительно 1 мл крови из вены верхней конечности.
Собранную кровь помещали в антикоагуляционную пробирку, содержащую ЭДТУ-K2 (этилендиаминтетрауксусную кислоту-K2). Образцы крови центрифугировали при 1800 g в течение 5 мин при 4°С, получая образцы плазмы. Плазму готовили в течение 30 минут после отбора крови и до испытаний хранили в морозильной камере при -80°С.
2. Способ анализа образцов
Испытуемый образец плазмы извлекали из морозильной камеры, в которой поддерживали температуру -80°С, оставляли нагреваться при комнатной температуре и перемешивали на вортексе в течение 5 минут. Двадцать (20) мкл образца плазмы аккуратно помещали пипеткой в центрифужную пробирку объемом 1,5 мл, после чего добавляли 200 мкл рабочего раствора внутреннего стандарта (толбутамид в метаноле) в концентрации 100 нг/мл, смешивали, перемешивали на вортексе в течение 5 минут и затем центрифугировали при 12000 об/мин в течение 5 минут. Отбирали 50 мкл жидкости над осадком и аккуратно помещали ее пипеткой в 96-луночные планшеты, в которые предварительно помещали воду в количестве 150 мкл/лунка; перемешивали на вортексе в течение 5 минут и анализировали способом ЖХ-МС/МС (жидкостной хроматографии с тандемной масс-спектрометрией). Объем впрыска соединения 25, соединения 65, соединения 107 и соединения 158 составлял 20, 10, 20 и 10 мкл, соответственно.
3. Способ обработки данных
Концентрацию соединений определяли с помощью программы Analyst 1.6.3, поставляемой АВ SCIEX. Для расчета среднего значения, стандартного отклонения, коэффициента вариации и других параметров (непосредственно выдаваемых Analyst 1.6.3 и не вычисляемых) применяли программу Microsoft Excel.
Фармакокинетические параметры вычисляли с помощью программного обеспечения Pharsight Phoenix 6.1 NCA (Tmax представляет собой среднее значение).
4. Результаты
Экспериментальные результаты представлены в Таблице 4:
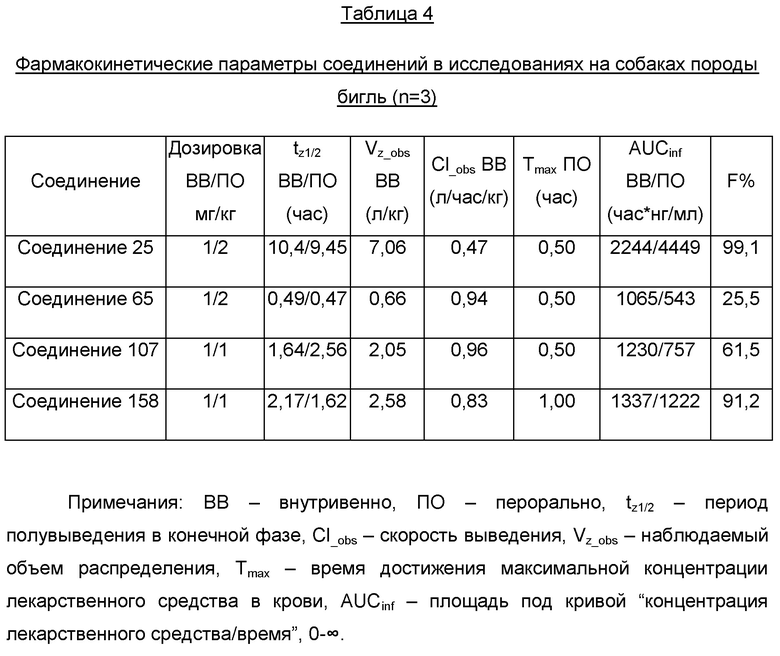
Как видно из данных, приведенных выше в Таблице, соединения согласно изобретению имеют очень хорошие фармакокинетические характеристики.
Экспериментальный Пример 3
Оценка стабильности соединений согласно изобретению в микросоме печени человека
Задача: Оценка стабильности соединений согласно изобретению в микросоме печени человека.
Испытуемое вещество: Соединения согласно настоящему изобретению и соединение 1-8, рассмотренное в патенте WO2017019723A1 и имеющее следующую структурную формулу:
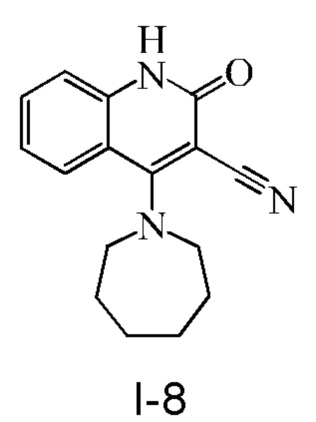
Состав инкубируемой системы представлен в Таблице 5:
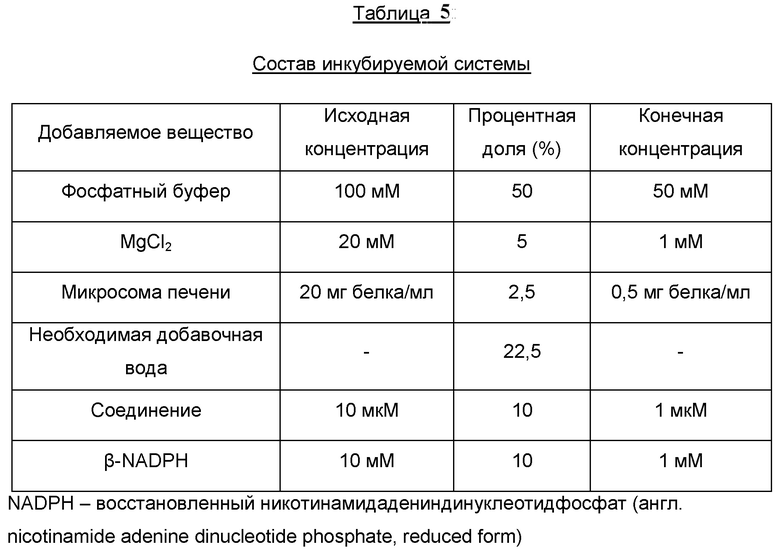
Процедура эксперимента:
(1) Микросомы печени (20 мг белка/мл) извлекали из морозильной камеры, в которой поддерживали температуру -80°С, помещали на термостатируемую водяную баню с температурой 37°С для предварительной инкубации в течение 3 минут и размораживали для дальнейшего использования.
(2) Состав смешанного раствора инкубируемой системы (без соединения и β-NADPH) готовили в соответствии с соотношениями, указанными в Таблице 4 "Состав инкубируемой системы", и предварительно инкубировали в течение 2 минут на термостатируемой водяной бане с температурой 37°С.
(3) Контрольная группа (без β-NADPH): 30 мкл воды и 30 мкл рабочего раствора соединения (10 мкМ, растворитель: 1%-ный водный раствор ДМСО) добавляли к 240 мкл смешанного раствора инкубируемой системы, полученной в этапе (2), перемешивали на вортексе в течение 30 секунд и смешивали; общий объем реакционной смеси составлял 300 мкл. Образец дублировали. Смесь инкубировали на термостатируемой водяной бане с температурой 37°С и начинали отсчет времени. Образцы отбирали в моменты времени 0 минут и 60 минут.
(4) Группа образцов: 70 мкл раствора β-NADPH (10 мМ) и 70 мкл рабочего раствора соединения (10 мкМ) добавляли к 560 мкл смешанного раствора инкубируемой системы, полученного в этапе (2). Общий объем реакционной смеси составил 700 мкл. Смесь перемешивали на вортексе в течение 30 секунд, перемешивали, и лунку дублировали. Смесь инкубировали на термостатируемой водяной бане с температурой 37°С, и начинали отсчет времени. Образцы отбирали в моменты времени 0 минут, 5 минут, 10 минут, 20 минут, 30 минут, 60 минут от начала отсчета.
(5) После перемешивания на вортексе в течение 3 минут образец центрифугировали при 12000 об/мин в течение 5 минут.
(6) Отбирали 50 мкл жидкости над осадком, туда добавляли 150 мкл воды, перемешивали на вортексе, смешивали и проводили анализ ЖХ-МС/МС.
Анализ данных:
Период полувыведения (t1/2) и скорость выведения (CI) вычисляли в соответствии с приведенной ниже формулой динамики первого порядка:
Ct=С0*e-kt
t1/2=ln2/k=0,693/k
Clint=Vd*k
Vd=1/(содержание белка в микросомах печени).
Примечания: k наклон логарифмической зависимости оставшегося количества соединения от времени, и Vd - наблюдаемый объем распределения.
Результаты представлены в Таблице 6.
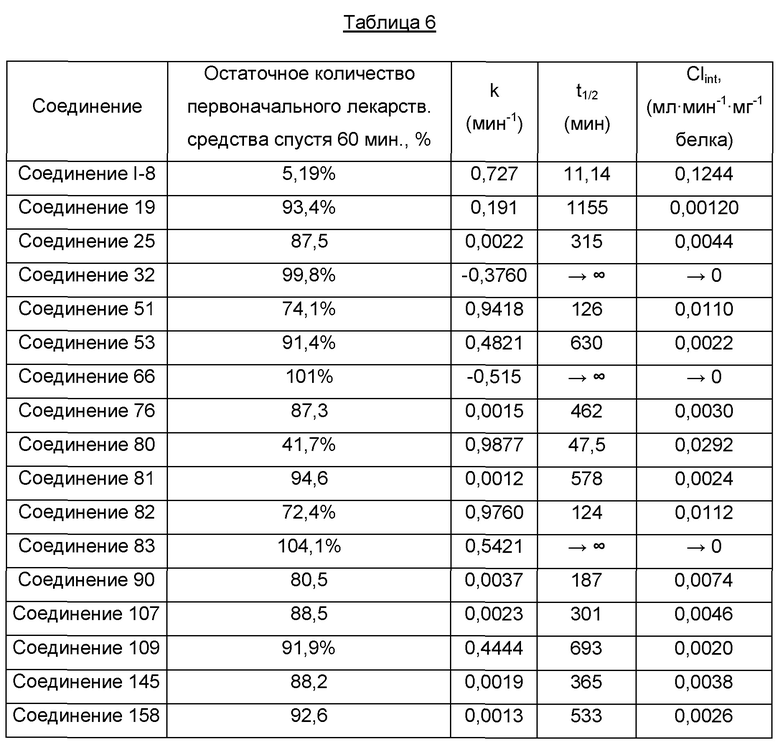
Приведенные выше результаты показывают, что соединения согласно настоящему изобретению имеют меньшую скорость выведения из микросомы печени человека, чем соединения согласно предшествующему уровню техники.
Экспериментальный Пример 4
Фармакокинетическая оценка соединений согласно изобретению в опытах с крысами
Задача: Оценка фармакокинетических параметров соединений in vivo в организмах самцов крыс SD (крыс Спрег-Доули (англ. Sprague Dawley)) (приобретенных у Beijing Vital River Animal Technology Co., Ltd.) и оценка биодоступности соединений.
Введение животным и отбор образцов:
Для приготовления раствора испытуемые соединения 65, 90, 107, 158 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединений 65, 90, 107, 158 вводили крысам SD внутрижелудочно в дозировке 5,0 мг/кг. Отбор образцов крови осуществляли в следующие моменты времени: 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения.
Для приготовления раствора испытуемые соединения 65, 90, 107, 158 растворяли в смеси 2% ДМСО + 10% ПЭГ400 + 88% (28% HP-β-CD) физиологического солевого раствора. Раствор соединений 65, 90, 107, 158 вводили крысам SD в дозировке 1 мг/кг посредством внутривенного болюсного введения. Отбор образцов крови осуществляли в следующие моменты времени: 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 76 растворяли в смеси 30% ДМФА + 30% ПЭГ400 + 40% солевого раствора. Раствор соединения 76 вводили крысам SD внутрижелудочно в дозировке 5,0 мг/кг. Отбор образцов крови осуществляли в следующие моменты времени: 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 76 растворяли в смеси 30% ДМФА + 30% ПЭГ400 + 40% солевого раствора. Раствор соединения 76 вводили крысам SD в дозировке 1 мг/кг посредством внутривенного болюсного введения. Отбор образцов крови осуществляли в следующие моменты времени: 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 25 растворяли в смеси 5% ДМСО + 20% (30% солютола) + 2% 1М NaOH + 73% солевого раствора. Раствор соединения 25 вводили крысам SD внутрижелудочно в дозировке 5,0 мг/кг. Отбор образцов крови осуществляли в следующие моменты времени: 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения.
Для приготовления раствора испытуемое соединение 25 растворяли в смеси 5% ДМСО + 20% (30% солютола) + 2% 1М NaOH + 73% солевого раствора. Раствор соединения 25 вводили крысам SD в дозировке 1 мг/кг посредством внутривенного болюсного введения. Отбор образцов крови осуществляли в следующие моменты времени: 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения.
Для приготовления раствора испытуемые соединения 84 и 96 растворяли в смеси 5% ДМСО + 10% ПЭГ400 + 85% (28% HP-β-CD) солевого раствора. Раствор соединений 84 и 96 вводили крысам SD внутрижелудочно в дозировке 5,0 мг/кг. Отбор образцов крови осуществляли в следующие моменты времени: 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения.
Для приготовления раствора испытуемые соединения 84 и 96 растворяли в смеси 5% ДМСО + 10% ПЭГ400 + 85% (28% HP-β-CD) солевого раствора. Раствор соединений 84 и 96 вводили крысам SD в дозировке 1 мг/кг посредством внутривенного болюсного введения. Отбор образцов крови осуществляли в следующие моменты времени: 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа после введения.
Животных фиксировали. Хвост животного нагревали на водяной бане в течение 10 минут перед каждым моментом отбора. Отбирали приблизительно 100 мкл крови из хвостовой вены, которую после отбора помещали в антикоагуляционную пробирку, содержащую ЭДТУ-K2. Образцы крови центрифугировали со скоростью 8000 об/мин при 4°С в течение 6 мин, получая образцы плазмы. Плазму готовили в течение 30 минут после отбора крови и до испытаний хранили в морозильной камере при -80°С.
Способ анализа образцов
Испытуемый образец плазмы извлекали из морозильной камеры, в которой поддерживали температуру -80°С, оставляли нагреваться при комнатной температуре и перемешивали на вортексе в течение 5 минут. Двадцать (20) мкл образца плазмы аккуратно помещали пипеткой в центрифужную пробирку объемом 1,5 мл, после чего добавляли 200 мкл рабочего раствора внутреннего стандарта (толбутамид в метаноле) в концентрации 100 нг/мл, смешивали, перемешивали на вортексе в течение 5 минут и затем центрифугировали при 12000 об/мин в течение 5 минут. Отбирали 50 мкл жидкости над осадком и аккуратно помещали ее пипеткой в 96-луночные планшеты, в которые предварительно помещали воду в количестве 150 мкл/лунка; перемешивали на вортексе в течение 5 минут и анализировали способом ЖХ-МС/МС. Объем впрыска соединений 25, 65, 76, 84, 90, 96, 107, 154 и 158 составлял 20, 20, 5, 10 мкл, соответственно.
Способ обработки данных
Концентрацию испытуемых образцов определяли с помощью программы Analyst 1.6.1, поставляемой АВ SCIEX. Для расчета среднего значения, стандартного отклонения, коэффициента вариации и других параметров (непосредственно выдаваемых Analyst 1.6.1 и не вычисляемых) применяли программу Microsoft Excel. Фармакокинетические параметры вычисляли с помощью программного обеспечения Pharsight Phoenix 6.1 NCA (Tmax представляет собой среднее значение).
Результаты представлены в Таблице 7:
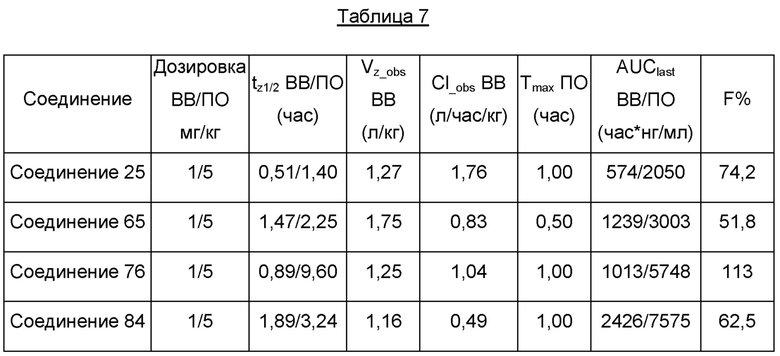
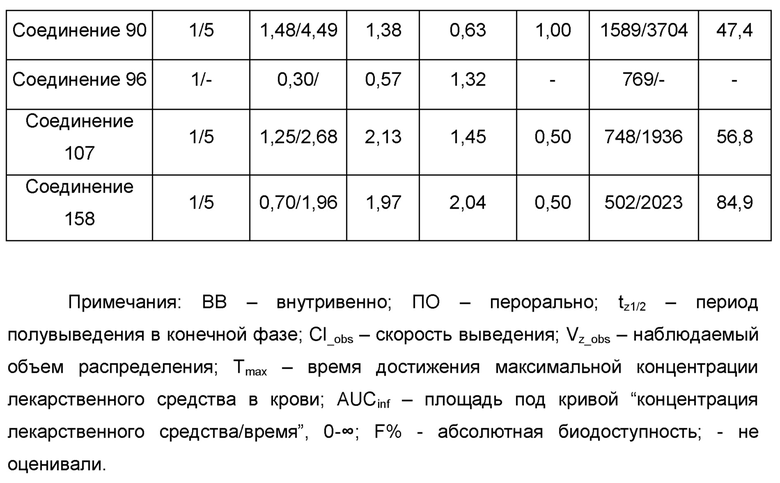
Как видно из данных, приведенных выше в Таблице, соединения согласно изобретению имеют очень хорошие фармакокинетические характеристики.
Экспериментальный Пример 5
Способ оценки ферментативных свойств PDE
Испытуемое вещество: Соединения согласно изобретению, полученные согласно соответствующим примерам осуществления изобретения
1. Экспериментальные материалы и оборудование
Фермент PDE1A1 (BPS, No. по каталогу: 60010)
Фермент PDE3A (BPS, No. по каталогу: 60030)
Фермент PDE5A1 (BPS, No. по каталогу: 60050)
Фермент PDE8A1 (BPS, No. по каталогу: 60080)
384-луночный планшет (Perkin Elmer, No. по каталогу: 6007279)
2. Процедура эксперимента
Подготовка соединений: соединения были приготовлены для длительного хранения в виде основного раствора соединений концентрацией 10 мМ в ДМСО.
Полученный основной раствор соединения разбавляли в 100 раз ДМСО, получая рабочий маточный раствор соединения концентрацией 100 мкМ, и затем рабочий маточный раствор соединения разбавляли в 3 раза ДМСО, получая 10 градиентов концентрации разбавленного маточного раствора соединения (100×).
Инкубация с соединениями: Для внесения разбавленного маточного раствора соединения в 384-луночный планшет применяли систему Echo для пипетирования очень малых количеств жидкости. В каждую лунку для соединения добавляли 200 панолитров разбавленного маточного раствора соединения и 10 мкл раствора фермента PDE1A1, PDE3A, PDE5A1 и PDE8A1. После центрифугирования со скоростью 1000 об/мин в течение 1 минуты смесь инкубировали в течение 15 минут при комнатной температуре. Затем добавляли 10 мкл смеси субстрата. После центрифугирования со скоростью 1000 об/мин в течение 1 минуты смесь инкубировали с применением шока (with shocking) в течение 30 минут при комнатной температуре. Наконец, для остановки реакции в системе добавляли стоп-раствор. Смесь инкубировали с применением шока в течение 60 минут при комнатной температуре. При максимальном считывании (Мах) соединение было замещено растворителем. При минимальном считывании (Min), и соединение, и раствор фермента были замещены растворителем.
Измерения: Величины флуоресценции (F) при 480 нм/535 нм определяли с помощью считывающего устройства для микропланшетов.
Вычисление: Скорость ингибирования вычисляли по нижеследующей формуле, и величины IC50 аппроксимировали с помощью программы GraphPad Prism 5.0:
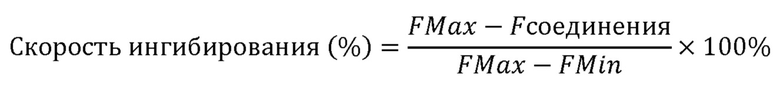
3. Результаты испытаний представлены в Таблице 8:
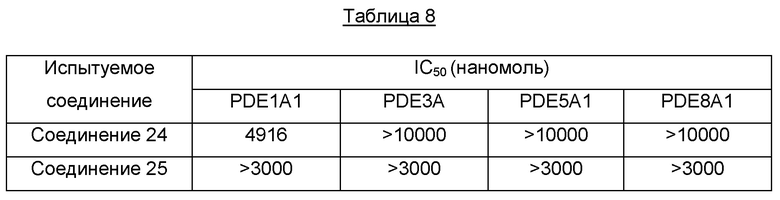
Экспериментальное заключение: Соединения согласно настоящему изобретению не оказывают существенного ингибиторного действия на ферменты PDE1A1, PDE3A, PDE5A15 или PDE8A1 и имеют относительно высокую селективность в отношении PDE9.
Следует отметить, что, если не указано иное, каждый из рассмотренных выше экспериментальных примеров может быть воплощен традиционным способом, известным в данной области техники, и для выполнения каждого из экспериментальных примеров также может быть привлечена сторонняя организация; например, рассмотренные выше экспериментальные примеры 1 и 5 выполнены сторонней организацией.
Выше приведены лишь предпочтительные примеры осуществления настоящего изобретения, и настоящее изобретение не ограничивается ими. Любые модификации, эквиваленты, усовершенствования и подобные изменения, соответствующие предмету и объему настоящего изобретения должны быть включены в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2830113C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| ТЕТРАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2010 |
|
RU2725140C2 |
| ТЕТРАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2010 |
|
RU2585622C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ПРОИЗВОДНЫЕ ТИЕНОПИРРОЛА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ НАЦЕЛИВАНИЯ НА БЕЛКИ, КОМПОЗИЦИИ С УКАЗАННЫМИ ПРОИЗВОДНЫМИ, СПОСОБЫ И ПРИМЕНЕНИЯ | 2017 |
|
RU2771166C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2840815C2 |
| АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2833035C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2800292C2 |
| Фармацевтически эффективные соединения, селективно ингибирующие изоформы миозина 2 | 2019 |
|
RU2817643C2 |
Группа изобретений относится к области техники, связанной с медициной, и включает соединение формулы (I), или его фармацевтически приемлемые соли, или стереоизомеры, фармацевтическую композицию и применение. Где в формуле (I) каждый из X1, Х2, Х3 и X4 независимо представляет собой CR3 или N, и Х1 Х2, Х3 и Х4 одновременно не представляют собой CR3; R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, карбоксила, циано, галогена, C1-6 алкила, C1-6 алкокси, С1-6 алкиламино, (С-1-6 алкил)2амино, С2-8 алкенила, С2-8 алкинила, C1-6 алкилсульфонила, C1-6 алкилтио, С3-6 циклоалкила, 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пиперазинила и морфолинила, С1-6 алкилкарбонила, аминокарбонила, C1-6 алкиламинокарбонила, (С1-6 алкил)2аминокарбонила, 4-6-членного гетероциклилкарбонила, выбранного из пиперазинилкарбонила и азетидинилкарбонила, и пиримидинилокси, где С1-6 алкил, C1-6 алкокси, (C1-6 алкил)2амино, С2-8 алкинил, 4-6-членный насыщенный азотсодержащий гетероциклил, выбранный из азетидинила, пиперазинила и морфолинила, аминокарбонил, C1-6 алкиламинокарбонил и 4-6-членный гетероциклилкарбонил, выбранный из пиперазинилкарбонила и азетидинилкарбонила, являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, C1-6 алкила, С1-6 алкокси, (C1-6 алкил)2амино, C1-6 алкилкарбонилокси, С3-6 циклоалкила, галогенированного C1-6 алкила и 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пирролидинила и пиперидинила, который является незамещенным или необязательно замещен C1-6 алкилом; L представляет собой связь, -NH-(CH2)t-, и t равен 0, 1, 2 или 3; кольцо А выбрано из группы, состоящей из 5-7-членного насыщенного моногетероциклила, имеющего атом N в качестве гетероатома, 7-11-членного насыщенного спирогетероциклила, имеющего 1 или 2 гетероатома, выбранных из О и N, и арила, имеющего от 6 до 14 атомов углерода; каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, C1-6 алкила, возможно замещенного гидроксигруппой, C1-6 алкокси, и 5-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из S и N; m равен 0, 1, 2 или 3; и R2 представляет собой водород; при условии, что: когда X1 представляет собой N и каждый из Х2, Х3 и Х4 независимо представляет собой CR3, А не является фенилом; когда каждый из X1 и Х3 независимо представляет собой N и каждый из Х2 и Х4 независимо представляет собой CR3, А не является фенилом; и когда Х3 представляет собой N и каждый из X1, Х2 и Х4 независимо представляет собой CR3, А не является фенилом. Технический результат - соединение формулы (I), проявляющее свойства ингибитора фосфодиэстеразы 9 (PDE9), которое может быть использовано для получения лекарственного средства для лечения или профилактики заболеваний, опосредуемых PDE9. 3 н. и 15 з.п. ф-лы, 8 табл., 101 пр.
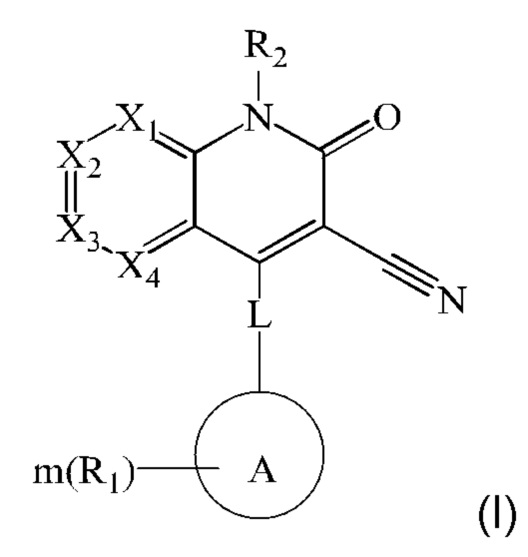
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль или стереоизомер,
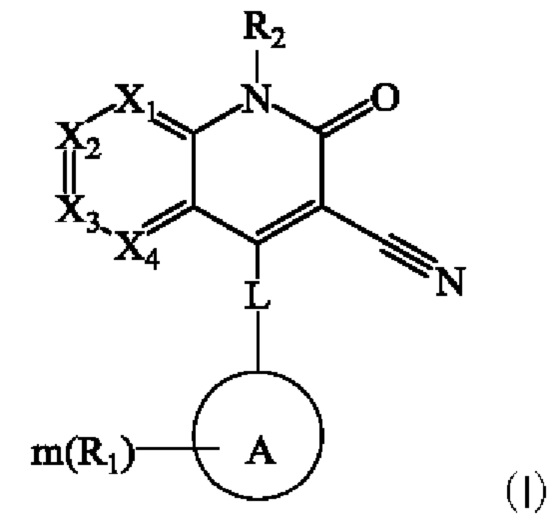
где
каждый из X1, Х2, Х3 и X4 независимо представляет собой CR3 или N, и Х1 Х2, Х3 и Х4 одновременно не представляют собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, карбоксила, циано, галогена, C1-6 алкила, C1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2амино, С2-8 алкенила, С2-8 алкинила, C1-6 алкилсульфонила, C1-6 алкилтио, С3-6 циклоалкила, 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пиперазинила и морфолинила, С1-6 алкилкарбонила, аминокарбонила, C1-6 алкиламинокарбонила, (С1-6 алкил)2аминокарбонила, 4-6-членного гетероциклилкарбонила, выбранного из пиперазинилкарбонила и азетидинилкарбонила, и пиримидинилокси, где С1-6 алкил, C1-6 алкокси, (C1-6 алкил)2амино, С2-8 алкинил, 4-6-членный насыщенный азотсодержащий гетероциклил, выбранный из азетидинила, пиперазинила и морфолинила, аминокарбонил, C1-6 алкиламинокарбонил и 4-6-членный гетероциклилкарбонил, выбранный из пиперазинилкарбонила и азетидинилкарбонила, являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, C1-6 алкила, С1-6 алкокси, (C1-6 алкил)2амино, C1-6 алкилкарбонилокси, С3-6 циклоалкила, галогенированного C1-6 алкила и 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пирролидинила и пиперидинила, который является незамещенным или необязательно замещен C1-6 алкилом;
L представляет собой связь, -NH-(CH2)t-, и t равен 0, 1, 2 или 3;
кольцо А выбрано из группы, состоящей из 5-7-членного насыщенного моногетероциклила, имеющего атом N в качестве гетероатома, 7-11-членного насыщенного спирогетероциклила, имеющего 1 или 2 гетероатома, выбранных из О и N, и арила, имеющего от 6 до 14 атомов углерода;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, C1-6 алкила, возможно замещенного гидроксигруппой, C1-6 алкокси, и 5-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из S и N;
m равен 0,1, 2 или 3; и
R2 представляет собой водород;
при условии, что:
когда X1 представляет собой N и каждый из Х2, Х3 и Х4 независимо представляет собой CR3, А не является фенилом;
когда каждый из X1 и Х3 независимо представляет собой N и каждый из Х2 и Х4 независимо представляет собой CR3, А не является фенилом; и
когда Х3 представляет собой N и каждый из X1, Х2 и Х4 независимо представляет собой CR3, А не является фенилом.
2. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 1,
где
каждый из X1, Х2, Х3 и X4 независимо представляет собой CR3 или N, и Х1, Х2, Х3 и Х4 одновременно не представляют собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, карбоксила, циано, галогена, C1-6 алкила, C1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2амино, С2-8 алкенила, С2-8 алкинила, C1-6 алкилсульфонила, C1-6 алкилтио, С3-6 циклоалкила, 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пиперазинила и морфолинила, C1-6 алкилкарбонила, C1-6 алкиламинокарбонила, (C1-6 алкил)2аминокарбонила и аминокарбонила, где C1-6 алкил, С1-6 алкокси, (С1-6 алкил)2амино, С2-8 алкинил, 4-6-членный насыщенный азотсодержащий гетероциклил, выбранный из азетидинила, пиперазинила и морфолинила, С1-6 алкиламинокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, C1-6 алкила, С1-6 алкокси, (C1-6 алкил)2амино, С3-6 циклоалкила, C1-6 алкилкарбонилокси и незамещенного или замещенного C1-6 алкилом 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пирролидинида и пиперидинила;
L представляет собой связь, -NH-(CH2)t, и t равен 0, 1, 2 или 3;
кольцо А выбрано из группы, состоящей из 5-7-членного насыщенного моногетероциклила, имеющего атом N в качестве гетероатома, 7-11-членного насыщенного спирогетероциклила, имеющего 1 или 2 гетероатома, выбранных из О и N, и арила, имеющего от 6 до 14 атомов углерода;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, C1-6 алкила, возможно замещенного гидроксигруппой, C1-6 алкокси, и 5-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из S и N;
m равен 0,1, 2 или 3; и
R2 представляет собой водород,
при условии, что:
когда X1 представляет собой N и каждый из Х2, Х3 и X4 независимо представляет собой CR3, А не является фенилом;
когда каждый из X1 и Х3 независимо представляет собой N и каждый из Х2 и X4 независимо представляет собой CR3, А не является фенилом; и
когда Х3 представляет собой N и каждый из Х1, Х2 и X4 независимо представляет собой CR3, А не является фенилом.
3. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 2, где соединение имеет структуру, представленную формулой (II):
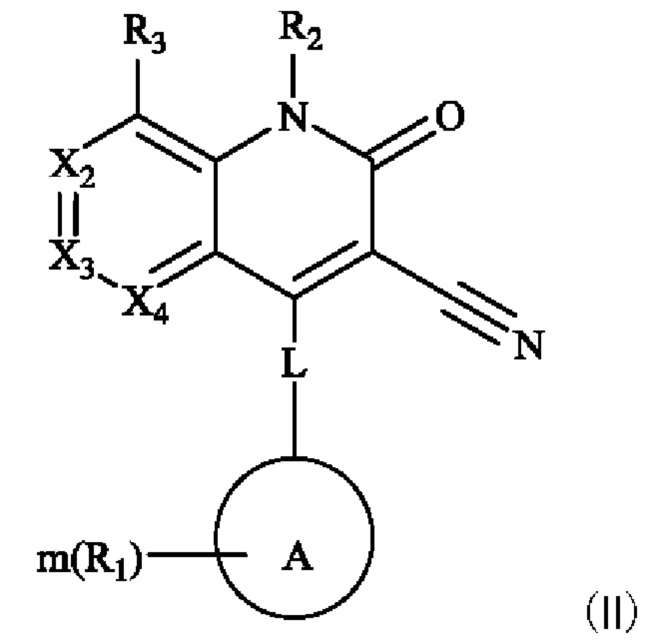
где
Х2, Х3, Х4, R1, R2, R3, кольцо A, L и m являются такими, как определено в п. 2, и Х2, Х3 и Х4 одновременно не представляют собой CR3;
при условии, что
когда Х3 представляет собой N и каждый из Х2 и Х4 независимо представляет собой CR3, А не является фенилом.
4. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 3,
где
каждый из Х2, Х3 и Х4 независимо представляет собой CR3 или N, и Х2, Х3 и X4 одновременно не представляют собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, карбоксила, циано, галогена, C1-6 алкила, C1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2амино, С2-8 алкенила, С2-8 алкинила, C1-6 алкилсульфонила, C1-6 алкилтио, С3-6 циклоалкила, 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пиперазинила и морфолинила, C1-6 алкилкарбонила, C1-6 алкиламинокарбонила, (C1-6 алкил)2аминокарбонила и аминокарбонила, где C1-6 алкил, C1-6 алкокси, (С1-6 алкил)2амино, С2-8 алкинил, 4-6-членный насыщенный азотсодержащий гетероциклил, выбранный из азетидинила, пиперазинила и морфолинила, C1-6 алкиламинокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, С1-6 алкила, C1-6 алкокси, (C1-6 алкил)2амино, С1-6 алкилкарбонилокси, С3-6 циклоалкила и незамещенного или замещенного C1-6 алкилом 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пирролидинила и пиперидинила;
L представляет собой связь;
кольцо А представляет собой 5-7-членный насыщенный моногетероциклил, имеющий атом N в качестве гетероатома, или 7-11-членный насыщенный спирогетероциклил, имеющий 1 или 2 гетероатома, выбранных из О и N;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, C1-6 алкила, возможно замещенного гидроксигруппой, C1-6 алкокси и 5-членного гетероарила, имеющего от 1 до 3 гетероатомов, выбранных из S и N;
m равен 0, 1 или 2; и
R2 представляет собой водород.
5. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 4,
где
Х2 представляет собой N и каждый из Х3 и Х4 независимо представляет собой CR3 или N;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, циано, галогена, карбоксила, С1-4 алкила, С1-4 алкокси, С1-4 алкиламино, (C1-4 алкил)2амино, С2-6 алкенила, С2-6 алкинила, C1-4 алкилкарбонилa, C1-4 алкиламинокарбонила, (С1-6 алкил)2аминокарбонила, C1-4 алкилсульфонила, С1-4 алкилтио, аминокарбонила, циклопропила, азетидинила, морфолинила и пиперазинила, где С1-4 алкил, С1-4 алкокси, (C1-4 алкил)2амино, С2-6 алкинил, C1-4 алкиламинокарбонил, аминокарбонил, циклопропил, азетидинил, морфолинил и пиперазинил являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, амино, циано, С1-4 алкила, С1-4 алкокси, (С1-4 алкил)2амино, циклопропила, C1-4 алкилкарбонилокси и незамещенного или замещенного С1-4 алкилом 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пирролидинила и пиперидинила;
L представляет собой связь;
кольцо А представляет собой 5-7-членный насыщенный моногетероциклил, имеющий атом N в качестве гетероатома, или 7-11-членный насыщенный спирогетероциклил, имеющий 1 или 2 гетероатома, выбранных из О и N, причем 7-11-членный насыщенный спирогетероциклил содержит по меньшей мере один N, и кольцо А соединено с L через атом N;
каждый R1 независимо выбран из группы, состоящей из водорода, гидрокси, циано, галогена, С1-4 алкила, возможно замещенного гидроксигруппой, C1-4 алкокси, пиразолила, тиазолила и триазолила; и
m равен 0, 1 или 2.
6. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 4 или 5,
где
Х2 представляет собой N, Х3 представляет собой CR3 и X4 представляет собой CR3 или N;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, галогена, С1-4 алкила, С1-4 алкокси, морфолинила, С2-6 алкенила, С1-4 алкиламинокарбонила, (С1-4 алкил)2аминокарбонила и аминокарбонила, где С1-4 алкил, С1-4 алкокси, морфолинил, С1-4 алкиламинокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, С1-4 алкокси, циклопропила, амино (С1-4 алкил)2амино и незамещенного или замещенного C1-4 алкилом 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пирролидинила и пиперидинила;
L представляет собой связь;
кольцо А представляет собой 5-7-членный насыщенный моногетероциклил, имеющий атом N в качестве гетероатома, кольцо А соединено с L через атом N;
каждый R1 независимо выбран из группы, состоящей из водорода, галогена, С1-4 алкила, возможно замещенного гидрокси, С1-4 алкокси, пиразолила, тиазолила и триазолила; и
m равен 0, 1 или 2.
7. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 6, где кольцо А выбрано из группы, состоящей из
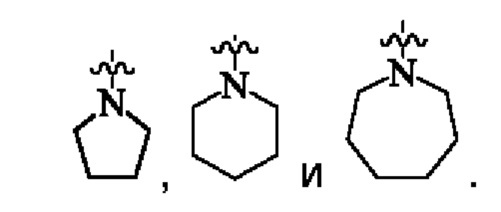
8. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 6,
где
Х2 представляет собой N и каждый из Х3 и X4 независимо представляет собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, галогена, С1-4 алкила, С1-4 алкокси, С2-6 алкенила, С1-4 алкиламинокарбонила и аминокарбонила, где С1-4 алкил, С1-4 алкокси, С1-4 алкиламинокарбонил и аминокарбонил являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, амино, С1-4 алкила, С1-4 алкокси, циклопропила, (C1-4 алкил)2амино и незамещенного или замещенного С1-4 алкилом 4-6-членного насыщенного азотсодержащего гетероциклила, выбранного из азетидинила, пирролидинила и пиперидинила;
L представляет собой связь; кольцо А представляет собой 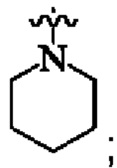
каждый R1 независимо выбран из группы, состоящей из водорода, С1-4 алкила, возможно замещенного гидроксигруппой, и С1-4 алкокси; и
m равен 0, 1 или 2.
9. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 6,
где
Х2 представляет собой N и каждый из Х3 и Х4 независимо представляет собой CR3;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, галогена, С1-4 алкила и морфолинила, где С1-4 алкил является незамещенным или замещен одной или двумя гидроксигруппами;
L представляет собой связь;
кольцо А представляет собой 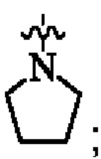 и
и
каждый R1 независимо выбран из группы, состоящей из пиразолила, тиазолила и триазолила.
10. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 4 или 5,
где
R3 в каждом случае независимо выбран из группы, состоящей из водорода, амино, циано, галогена, карбоксила, С1-4 алкила, С1-4 алкокси, С1-4 алкилкарбонила, С2-6 алкинила, С1-4 алкиламинокарбонила, (С1-4 алкил)2аминокарбонила, С1-4 алкилтио, С1-4 алкилсульфонила, С1-4 алкиламино, (С1-4 алкил)2амино, азетидинила, морфолинила, пиперазинила, С2-6 алкенила и циклопропила, где С1-4 алкил, С1-4 алкокси, С2-6 алкинил, C1-4 алкиламинокарбонил, (С1-4 алкил)2амино, азетидинил, морфолинил, пиперазинил и циклопропил являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, амино, С1-4 алкила, (С1-4 алкил)2амино, циклопропила и С1-4 алкилкарбонилокси;
L представляет собой связь;
кольцо А представляет собой 7-11-членный насыщенный спирогетероциклил, имеющий 1 или 2 гетероатома, выбранных из О и N, где 7-11-членный насыщенный спирогетероциклил содержит по меньшей мере один N, и кольцо А соединено с L через атом N;
каждый R1 независимо выбран из группы, состоящей из водорода, циано, галогена, гидрокси и незамещенного или замещенного гидроксигруппой С1-4 алкила; и
m равен 0, 1 или 2.
11. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 10, где кольцо А выбрано из группы, состоящей из
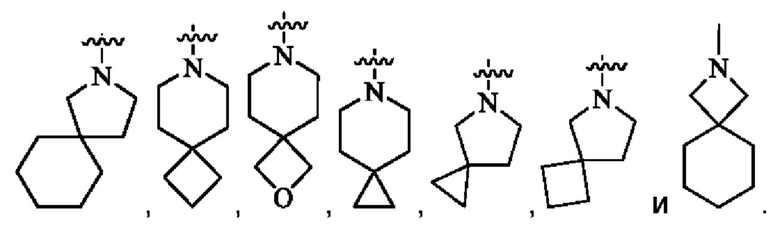
12. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 10,
где
Х2 представляет собой N и каждый из Х3 и X4 независимо представляет собой CR3 или N;
R3 в каждом случае независимо выбран из группы, состоящей из водорода, циано, амино, галогена, карбоксила, С1-4 алкила, С1-4 алкокси, С2-6 алкенила, С1-4 алкилкарбонила, С2-6 алкинила, С1-4 алкиламино, (С1-4 алкил)2амино, С1-4 алкиламинокарбонила, С1-4 алкилтио, С1-4 алкилсульфонила, циклопропила, азетидинила, морфолинила и пиперазинила, где С1-4 алкил, С1-4 алкокси, С2-6 алкинил, (С1-4 алкил)2амино, С1-4 алкиламинокарбонил, циклопропил, азетидинил, морфолинил и пиперазинил являются незамещенными или необязательно замещены одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, амино, С1-4 алкила, (С1-4 алкил)2амино, циклопропила и С1-4 алкилкарбонилокси;
L представляет собой связь;
кольцо А выбрано из группы, состоящей из 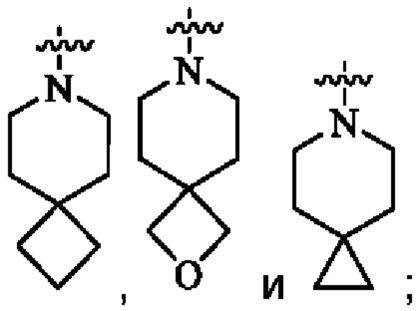 и
и
m равен 0.
13. Соединение или его фармацевтически приемлемая соль или стереоизомер по п. 4 или 5,
где
L представляет собой связь;
Х2 представляет собой N и каждый из Х3 и Х4 независимо представляет собой CR3 или N; и
кольцо А выбрано из группы, состоящей из:
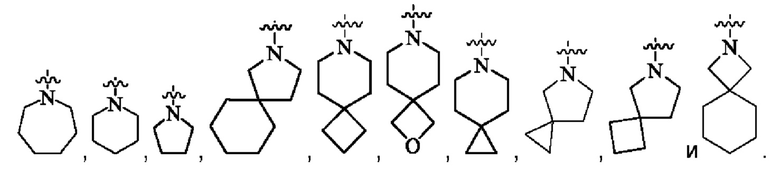
14. Соединение или его фармацевтически приемлемая соль или стереоизомер по любому из пп. 1-4, где соединение выбрано из группы, состоящей из:
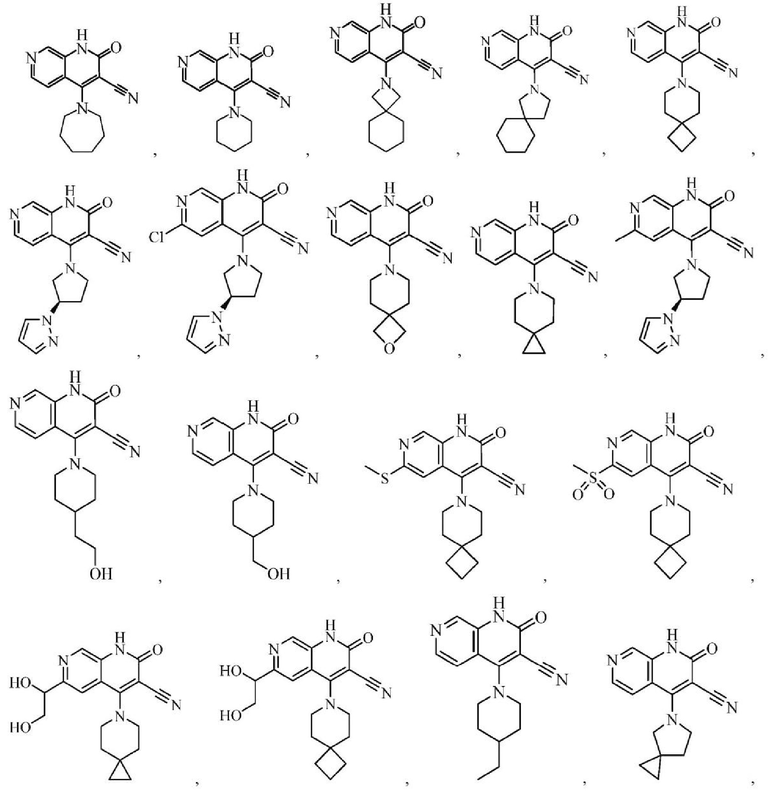
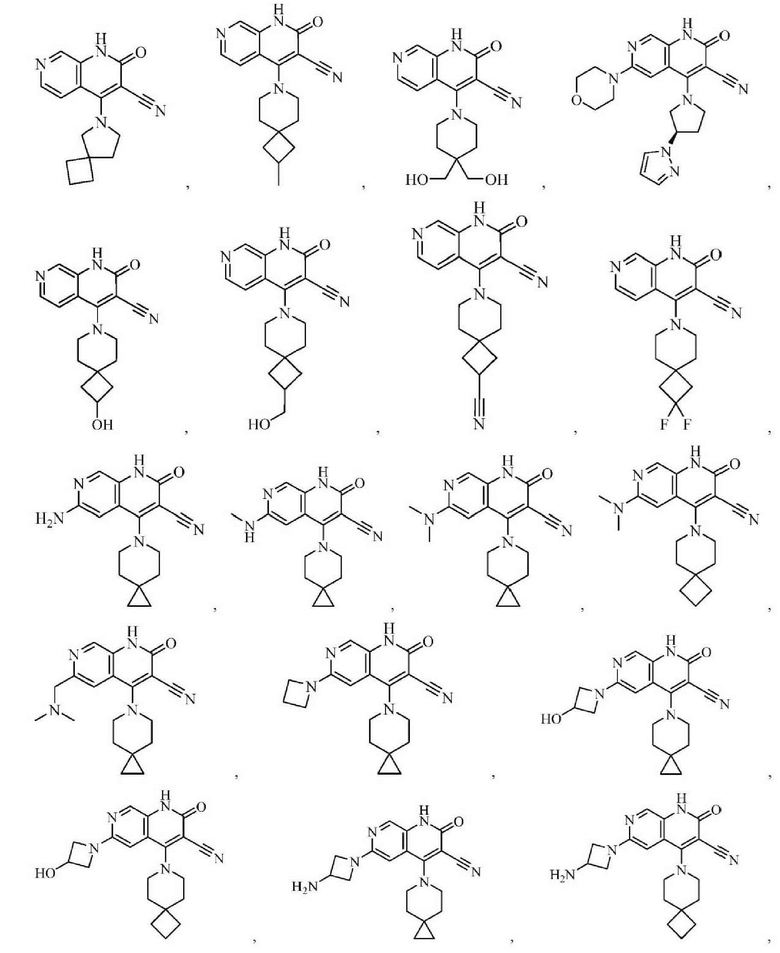
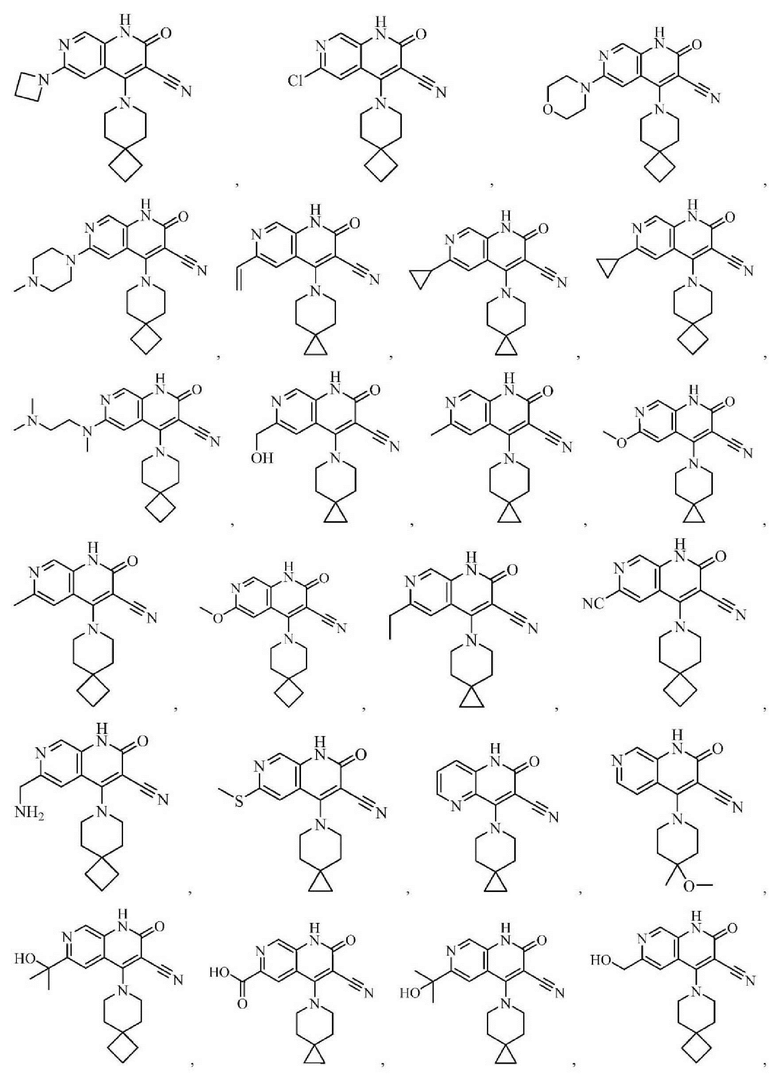
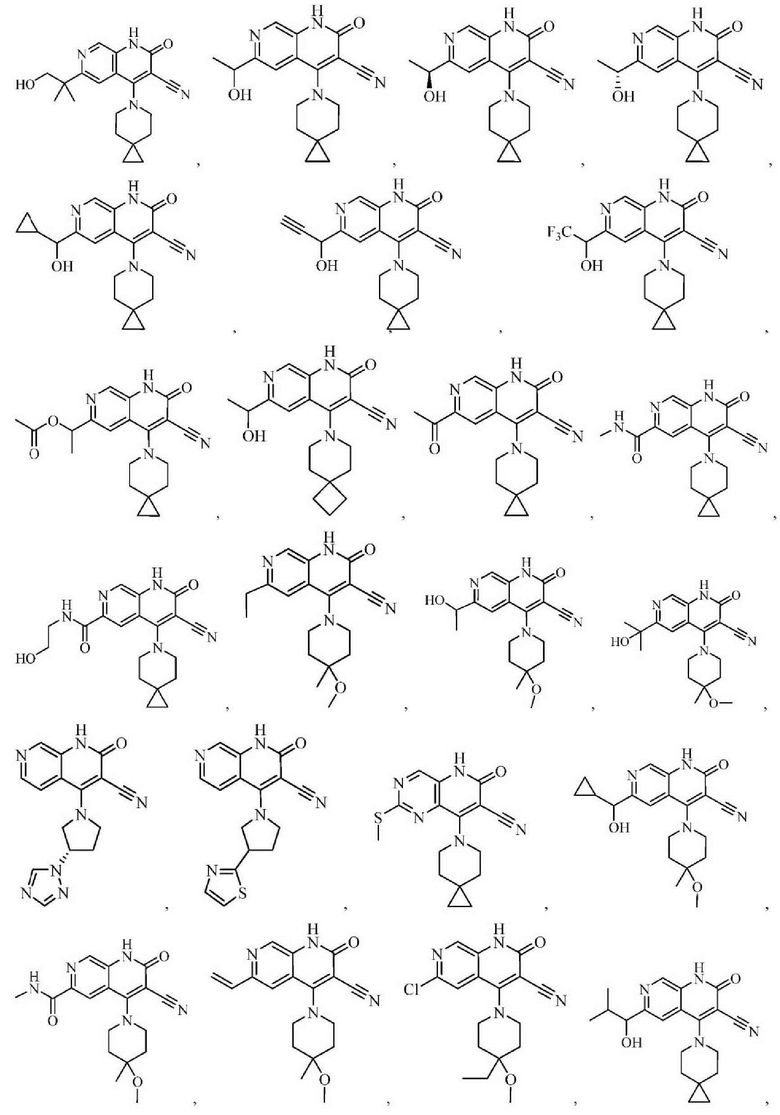
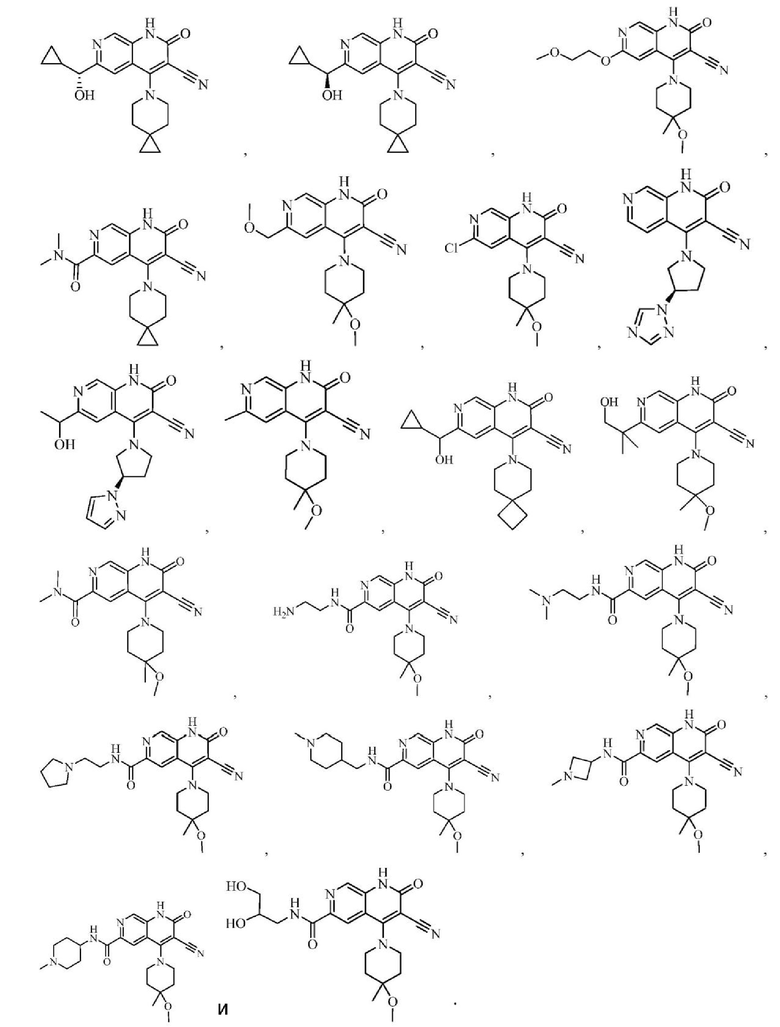
15. Фармацевтический состав, ингибирующий PDE9, включающий терапевтически эффективное количество соединения или его фармацевтически приемлемой соли или стереоизомера по любому из пп. 1-14 и фармацевтически приемлемый носитель.
16. Применение соединения или его фармацевтически приемлемой соли или стереоизомера по любому из пп. 1-14 или фармацевтического состава по п. 15 для получения лекарственного средства для лечения или профилактики заболевания, опосредованного PDE9,
17. Применение по п. 16, где заболевание, опосредованное PDE9, представляет собой когнитивное расстройство, вызванное расстройством центральной нервной системы.
18. Применение по п. 17, где когнитивное расстройство выбрано из группы, состоящей из нарушения восприятия, внимания, памяти и способности к обучению.
| ПРОИЗВОДНЫЕ НАФТИРИДИНА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2240322C2 |
| NASR M | |||
| N | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ARCHIV DER PHARMAZIE, WILEY VERLAG, WEINHEIM, 2002, vol | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| РЕЛЬСОВАЯ ПЕДАЛЬ | 1920 |
|
SU289A1 |
| WANG XIANG-SHAN et al | |||
| Three-Component, One-Pot Synthesis of Pyrido[2,3-d]pyrimidine Derivatives | |||
