Область изобретения
В настоящем изобретении предложены новые производные феникола, их применение для лечения инфекций у млекопитающих, фармацевтическая композиция, содержащая эти новые соединения, и способы получения этих соединений.
Предшествующий уровень техники
Возрастает необходимость в новых антибиотических агентах для лечения бактериальных инфекций у животных, и в частности, имеется необходимость в новых агентах, которые преодолевают растущую бактериальную устойчивость к существующим антибиотикам.
Флорфеникол представляет собой антибиотик широкого спектра действия, используемый исключительно в ветеринарной медицине. Фениколовые антибиотики как класс представляют собой сильнодействующие ингибиторы биосинтеза бактериальных белков. Флорфеникол обладает широким спектром активности против многих грамотрицательных и грамположительных бактерий и является полезным в предупреждении и лечении бактериальных инфекций, вызванных чувствительными патогенами у птиц, рептилий, рыб, моллюсков и млекопитающих. Важное применение флорфеникола заключается в лечении респираторных инфекций у крупного рогатого скота, таких как инфекции, вызванные, например, Mannheimia haemolytica, Pasteurella multocida и Haemophilus somnus. Эффективное лечение респираторной инфекции крупного рогатого скота (BRD) играет значительную роль в снижении того, что другими словами является одной из ведущих причин экономических потерь как в молочной, так и в мясной промышленности во всем мире.
Сообщения последних лет указывают на то, что развивается бактериальная устойчивость к флорфениколу, и она наблюдалась во многих бактериальных родах и видах, таких как Salmonella (Bolton, L. F., et al., Clin. Microbiol., 1999, 37, 1348), E.coli (Keyes, K., et al., Antimicrob. Agents Chemother., 2000, 44, 421), Klebsiella pneumoniae (Cloeckaert, A., et al., Antimicrob. Agents Chemother., 2001, 45, 2381), и в патогенах аквакультур, Photobacterium damselae подвид piscicida (ранее Pasteurella piscicida) (Kim, E., et al., Microbiol. Immunol., 1996, 40, 665). В свете возрастающей опасности развития устойчивости к флорфениколу и явной способности генов устойчивости перемещаться среди бактериальных видов и животных-хозяев (Cloeckaert, A., et al., Antimicrob. Agents Chemother., 2000, 44, 2858), существует важная необходимость в новых антибиотиках, которые сохраняют или превосходят активность флорфеникола, в то же время преодолевая проблемы устойчивости к флорфениколу. Соединения по настоящему изобретению представляют такое улучшение.
Краткое изложение сущности изобретения
В настоящем изобретении предложено соединение формулы I
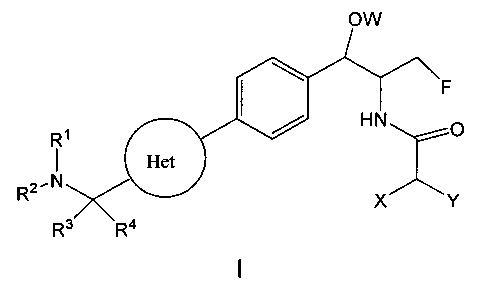
или его фармацевтически приемлемые соли или пролекарства, где:
группировка Het представляет собой 4-14-членную циклическую или бициклическую кольцевую систему, имеющую от одного до пяти гетероатомов, выбранных из N, О и S, возможно замещенную группами R6 в количестве от одного до трех;
каждый из R1 и R2 независимо представляет собой:
а. Н,
b. -C1-8алкил, возможно замещенный одной или более чем одной группой ОН, -SH, -CN, -NO2, галоген, -NHR5, -NC1-4алкилR5, -ОС1-4алкил, -SC1-4алкил, -S(C=O)С1-4алкил, -C(=O)NR5R5, -SO2R5, -SO2NR5R5 или -C3-6циклоалкил,
c. -C3-8циклоалкил, возможно замещенный группами R6 в количестве от одной до трех,
d. -SO2R5, -C(=O)NR5R5, -SO2NR5R5, -C(=O)OR5 или -C(=O)R5,
e. 4-6-членную гетероциклическую кольцевую группировку, возможно имеющую от одного до четырех гетероатомов, выбранных из группы, состоящей из N, S и О, где кольцо или атом возможно замещены группами R6 в количестве от одной до трех, или
f. R1 и R2, взятые вместе с азотом, к которому они присоединены, образуют 4-11-членную циклическую или бициклическую кольцевую группировку, возможно имеющую дополнительный(е) один или два гетероатома, выбранные из группы, состоящей из N, S и О, где указанные кольцо или атом возможно замещены группами R6 в количестве от одной до трех;
каждый из R3 и R4 независимо представляет собой:
a. -Н,
b. -С1-8алкил, возможно замещенный группами ОН, -SH, галоген, -CF3, -CN, -NO2, NH2, -NHR5, -NHR5-ОС1-4алкил, -СН2-O-СН3, -SC1-4алкил, -S(C=O)C1-4алкил, -C(=O)NR5R5, -С(=O)ОН, -SO2NR5 или -SO2R5,
c. -C3-8циклоалкил, возможно замещенный группами R6 в количестве от одной до трех,
d. -С(=O)С1-8алкил, где алкил возможно замещен группами -S(=O2)R5, -SO2NR5 или -C(=O)R5,
e. 4-6-членную гетероциклическую кольцевую группировку, возможно имеющую от одного до трех гетероатомов, выбранных из группы, состоящей из N, S и О, где гетероциклическое кольцо возможно замещено группами R6 в количестве от одной до трех,
f. R3 и R4, взятые вместе, образуют C3-8циклоалкил, возможно замещенный группами R6 в количестве от одной до трех; или
g. R3 и R4 взяты вместе с одним или двумя гетероатомами, выбранными из группы, состоящей из N, S и О, с образованием оксогруппы (=O) или с образованием 4-6-членной гетероциклической кольцевой группировки, где гетероциклическое кольцо возможно замещено группами R6 в количестве от одной до трех; или
R1 и R3, R2 и R4, R1 и R4 или R2 и R3, взятые вместе с атомом азота, к которому они присоединены, образуют 4-6-членную гетероциклическую кольцевую группировку, возможно имеющую от одного до двух гетероатомов, выбранных из группы, состоящей из N, S и О, где гетероциклическое кольцо возможно замещено группами R6 в количестве от одной до трех;
в каждом случае, R5 независимо представляет собой водород, C1-6алкил, -C3-6циклоалкил, NH2 или тетрагидро-2Н-пиранил, где указанный алкил возможно замещен одной, двумя или тремя группами R6;
в каждом случае, R6 представляет собой Н, C1-6алкил, галоген, -CN, -NO2, -CF3, -C3-6циклоалкил, оксо (=O), -NH2, -NHC1-4алкил, -N(С1-4алкил)2, -ОС1-4алкил, оксо, -SH, -SC1-4алкил, -S(C=O)С1-4алкил, -SO2R5, -SONC1-4алкил, -C(=O)C1-4алкил, -C(=O)NH2, -С(=O)NHC1-4алкил, -С(=O)N(С1-4алкил)2, -NC(=O)NH2, -NC(=O)NHC1-4алкил, NC(=O)N(С1-4алкил)2, CF3 или 4-6-членную гетероциклическую кольцевую группировку, возможно имеющую от одного до четырех гетероатомов, выбранных из группы, состоящей из N, S и О;
W представляет собой -Н, -РО(ОН)2, -РО(ОН)галоген, -СН2ОРО(ОН)2, -С(=O)С1-4алкил или -СН2ОС(=O)С1-4алкил, где С1-4алкил возможно замещен группами -OCO2H, -ОСО2С1-4алкил или -OC(=O)NHC1-4алкил;
и каждый из X, Y и Z независимо представляет собой Н, галоген, C1-4алкил, C3-6циклоалкил, -ОН, CF3, -NH2, -CN, N3 или -S-CF3;
при условии, что когда R3 и R4 взяты вместе с образованием оксогруппы (=O), тогда R1 и R2 оба не представляют собой водород.
В другом аспекте настоящего изобретения также предложены:
фармацевтические композиции, содержащие фармацевтически приемлемый носитель и соединение формулы I,
способы контролирования или лечения инфекций у млекопитающих, включающие введение нуждающемуся в этом млекопитающему терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли;
способы контролирования или лечения инфекций у домашнего скота и питомцев, включающие введение нуждающемуся в этом животному терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли; и
способы получения соединений по настоящему изобретению.
Подробное описание изобретения
В отношении вышеуказанного соединения и по всему описанию и формуле изобретения, следующие термины имеют значения, определенные ниже.
Термин «галоген» относится к хлору, брому, фтору и йоду.
Содержание атомов углерода в различных углеводород-содержащих группировках указано в префиксе, обозначающем минимальное и максимальное количество атомов углерода в группировке, то есть префикс Ci-j указывает на группировку с количеством атомов углерода от целого числа «i» до целого числе «j» включительно. Так, например, С1-4алкил относится к алкилу с 1-4 атомами углерода включительно; С1-6алкил относится к алкилу с 1-6 атомами углерода включительно; и С1-8алкил относится к алкилу с 1-8 атомами углерода включительно.
Термин «алкил» относится к прямоцепочечным, разветвленным и циклическим насыщенным моновалентным углеводородным группам, однако ссылка на индивидуальный радикал, такой как «пропил», охватывает только прямоцепочечный радикал, в то время как на изомер с разветвленной цепью, такой как «изопропил», или циклический изомер, такой как циклопропилметил или циклопентил, ссылаются специально.
Термин «циклоалкил» относится к монокольцу, такому как циклопропил, циклобутил, циклопентил или циклогексил.
Термин «Het» относится к насыщенной или ненасыщенной моноциклической или бициклической гетероциклической группе, содержащей по меньшей мере один гетероатом, выбранный из N, О и S. Бициклические гетероциклические кольца могут быть конденсированными, представлять собой спиросистемы или системы колец, связанных мостиковой связью. Моноциклические гетероциклические кольца содержат от 4 до 10 кольцевых атомов, предпочтительно от 5 до 6 представителей атомов в кольце. Бициклические гетероциклические группы содержат от 7 до 14 представителей атомов, предпочтительно от 9 до 12 представителей атомов в кольце. Примеры гетероциклических групп включают, без ограничения, замещенные или незамещенные тетрагидрофуран, диоксан, пирролидин, пиперидин, пиперазин, тетрагидротриазин, тетрагидропиразол, тетрагидротиофен, дигидро-1,3-дитиол-2-ил, гексагидротиепин-4-ил, тиенил, фуранил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тетразолил, пиридинил, пиразинил, пиридазинил, пиримидинил, пиперидинил, пирролидинил, пиперазинил, азетидинил, азиридинил, морфолинил, тиетанил, оксетарил, тиофенил, тиадиазолил, оксадизолил. Примеры подходящих бициклических гетероциклических групп включают, без ограничения, 1-, 2-, 3-, 5-, 6-, 7- или 8-индолизинил, 1-, 3-, 4-, 5-, 6- или 7-изоиндолил, 2-, 3-, 4-, 5-, 6- или 7-индолил, 2-, 3-, 4-, 5-, 6- или 7-индазолил, 2-, 4-, 5-, 6-, 7- или 8-пуринил, 1-, 2-, 3-, 4-, 6-, 7-, 8- или 9-хинолизинил, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолил, 1-, 4-, 5-, 6-, 7- или 8-фталазинил, 2-, 3-, 4-, 5- или 6- нафтиридинил, 2-, 3-, 5-, 6-, 7- или 8-хиназолинил, 3-, 4-, 5-, 6-, 7- или 8-циннолинил, 2-, 4-, 6- или 7-птеридинил, 1-, 2-, 3-, 4-, 5-, 6-, 7- или 8-4аН-карбазолил, 1-, 2-, 3-, 4-, 5-, 6-, 7- или 8-карбазолил, 1-, 3-, 4-, 5-, 6-, 7-, 8- или 9-карболинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-фенантридинил, 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8- или 9-акридинил, 1-, 2-, 4-, 5-, 6-, 7-, 8- или 9-перимидинил, 2-, 3-, 4-, 5-, 6-, 8-, 9- или 10-фенатролинил, 1-, 2-, 3-, 4-, 6-, 7-, 8- или 9-феназинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-фенотиазинил, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9- или 10-феноксазинил, 2-, 3-, 4-, 5-, 6- или 1-, 3-, 4-, 5-, 6-, 7-, 8-, 9- или 10-бензизохинолинил, 2-, 3-, 4- или тиено[2,3-b]фуранил, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10- или 11-7Н-пиразино[2,3-с]карбазолил, 2-, 3-, 5-, 6- или 7-2Н-фуро[3,2-b]-пиранил, 2-, 3-, 4-, 5-, 7- или 8-5Н-пиридо[2,3-d]-о-оксазинил, 1-, 3- или 5-1Н-пиразоло[4,3-d]-оксазолил, 2-, 4- или 5-4H-имидазо[4,5-d]тиазолил, 3-, 5- или 8-пиразино[2,3-d]пиридазинил, 2-, 3-, 5- или 6-имидазо[2,1-b]тиазолил, имидазо[1,2-а]пиридинил, тиазоло[5,4-b]пиридинил, 1-, 3-, 6-, 7-, 8- или 9-фуро[3,4-с]циннолинил, 1-, 2-, 3-, 4-, 5-, 6-, 8-, 9-, 10 или 11-4H-пиридо[2,3-c]карбазолил, 2-, 3-, 6- или 7-имидазо[1,2-b][1,2,4]триазинил, 7-бензо[b]тиенил, 2-, 4-, 5-, 6- или 7-бензоксазолил, 2-, 4-, 5-, 6- или 7-бензимидазолил, 2-, 4-, 5-, 6- или 7-бензотиазолил, 1-, 2-, 4-, 5-, 6-, 7-, 8- или 9-бензоксапинил, 2-, 4-, 5-, 6-, 7- или 8-бензоксазинил, 1-, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10- или 11-1H-пирроло[1,2-b][2]-бензазапинил. Типичные конденсированные гетероарильные группы включают, без ограничения, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолинил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолинил, 2-, 3-, 4-, 5-, 6- или 7-индолил, 2-, 3-, 4-, 5-, 6- или 7-бензо[b]тиенил, 2-, 4-, 5-, 6- или 7-бензоксазолил, 2-, 4-, 5-, 6- или 7-бензимидазолил, 2-, 4-, 5-, 6- или 7-бензотиазолил.
Для гетероциклических групп, содержащих серу, окисленная сера, такая как группы SO или SO2, также включена.
Для гетероциклических групп, содержащих азот, также включены азотные группы, такие как N→O или NH.
В каждом случае, Het возможно замещен группами ОН, галоген, -CN, -NO2, C1-6алкил, -C3-6циклоалкил, оксо (=O), -NH2, -NHC1-4алкил, -N(С1-4алкил)2, -ОС1-4алкил, -SH, -SC1-4алкил, -S(C=O)С1-4алкил, -SONC1-4алкил, -C(=O)C1-4алкил, -C(=O)NH2, -С(=O)NHC1-4алкил, -С(=O)N(С1-4алкил)2, -NC(=O)NH2, -NC(=O)NHC1-4алкил или NC(=O)N(С1-4алкил)2, в количестве от одной до трех.
Термин «млекопитающее» относится к человеку или животным, включая домашний скот и питомцев. Фраза «питомец» или «животное-компаньон» относится к животным, которых содержат как домашних любимцев. Примеры питомцев включают кошек, собак и лошадей. Термин «домашний скот» относится к животным, которых разводят или выращивают в сельскохозяйственных учреждениях для получения продуктов, таких как пищевые продукты или волокна, или для его разведения. В некоторых воплощениях домашний скот подходит для употребления млекопитающими, например людьми. Примеры домашнего скота включают млекопитающих, таких как крупный рогатый скот, козы, лошади, свиньи, овцы, включая молодых барашек, и кролики, а также птиц, таких как курицы, утки и индейки. Конкретно, домашний скот по настоящему изобретению относится к крупному рогатому скоту и свиньям. Соединения по настоящему изобретению могут также быть полезны в аквакультурах, таких как рыбы.
Термин «контролирование», «излечение» или «лечение» заболевания включает: (1) предупреждение заболевания, то есть чтобы возникновение клинических симптомов или признаков заболевания не развивалось у млекопитающего, который может подвергнуться или быть предрасположенным к заболеванию, но пока не испытывает или не проявляет симптомов/признаков заболевания; (2) подавление заболевания, то есть остановка развития или замедление развития заболевания или его клинических симптомов/признаков; или (3) облегчение заболевания, то есть индуцирование регрессии заболевания или его клинических симптомов/признаков.
Термин «терапевтически эффективное количество» означает количество соединения, которое при введении млекопитающему для лечения заболевания является достаточным для воздействия такого лечения на заболевание. «Терапевтически эффективное количество» будет варьировать в зависимости от соединения, заболевания и его тяжести и возраста, массы тела и так далее, млекопитающего, подлежащего лечению.
Термин «фармацевтически приемлемый» означает подходящий для использования млекопитающими, домашними питомцами или домашним скотом.
Термин «пролекарство» относится к биообратимой производной молекулы, то есть соединения формулы I по настоящему изобретению. Пролекарства могут изменять растворимость, липофильность и распределение лекарств in-vivo. Путем преднамеренного изменения этих ключевых свойств можно улучшить всасывание, увеличить время начала действия, уменьшить эффект метаболизма первого прохождения, обеспечить разработку водных препаратов для внутривенного введения (IV) и достичь направленной доставки. В дополнение, пролекарства полезны в улучшении трансдермальной доставки, маскировке вкуса, минимизации боли при инъекции, улучшении стабильности и так далее. В ситуациях, где фармакофор как таковой приводит к свойствам плохой доставки, пролекарства представляют собой одну из нескольких стратегий, которые могут быть использованы для восстановления высокоактивного соединения. В объем настоящего изобретения также включены все пролекарства соединений формулы I, которые могут быть получены стандартными способами, известными специалисту в данной области техники. Пролекарства соединений формулы I могут быть получены следующими способами, описанными в "Prodrugs of phosphates, phosphonates, and phosphinates," Krise JP, Stella VJ, Advanced Drug Delivery Reviews, 19: (2) 287-310 MAY 22 1996; "Targeted Prodrug Design to Optimize Drug Delivery". Hyo-Kyung Han and Gordon Amidon, AAPS PharmSci 2000; 2 (1) article 6; "Prodrugs", L. Prokai and K. Prokai-Tatrai, Chapter 12 in Injectable Drug Development: Techniques to Reduce Pain and Irritation, Interpharm Press, Buffalo Grove, IN, 1999; "Improved oral drug delivery: Solubility limitations overcome by the use of prodrugs", Fleisher D, Bong R, Stewart BH, Advanced Drug Delivery Reviews, 19: (2) 115-130 MAY 22 1996; или "Preparation and hydrolysis of water soluble, non-irritating prodrugs of pharmaceuticals with oxaalkanoic acids", Crooks, Peter Anthony; Cynkowski, Tadeusz; Cynkowska, Grazyna; Guo, Hong; Ashton, Paul, PCT Int. Appl. (2000), 65 pp. Примеры иллюстративных пролекарств включают фосфаты, фосфонаты, фосфинаты, сложные эфиры карбоновых кислот и карбаматы.
Соединения, которые имеют одну и ту же молекулярную формулу, но отличаются по природе или последовательности связывания их атомов или расположению их атомов в пространстве называют «изомерами».
В объем описанных соединений включены все изомеры (например цис-, транс-, энантиомеры или диастереомеры) описанных здесь соединений, самих по себе, а также любых смесей. Все эти формы, включая энантиомеры, диастереомеры, цис, транс, син, анти, сольваты (включая гидраты), таутомеры, и их смеси, включены в описанные соединения.
Конкретное значение для W представляет собой Н, -РО(ОН)2 или -СН2ОРО(ОН)2.
Конкретное значение для W представляет собой Н.
Конкретное значение для X и Y представляет собой хлорид; и Z представляет собой Н.
Конкретное значение для X и Y представляет собой фторид; и Z представляет собой Н.
Конкретное значение для группировки Het представляет собой 5- или 6-членную циклическую кольцевую систему, имеющую от одного до трех гетероатомов, выбранных из N, О и S, включая гетероатомные группы, такие как S-O, -SO2, N→O и -NH. Группировка Het возможно замещена группами R6.
Конкретное значение для группировки Het представляет собой пиридинил, тиофенил, тиазолил, тиадиазолил, имидазолил, оксадиазолил, пиримидинил, пиразинил, изоксазол, изотиазол или пиридазин.
Конкретное значение для группировки Het представляет собой пиридинил или тиазолил.
Конкретные значения для R1 и R2 независимо представляют собой Н, или R1 и R2, взятые вместе с азотом, к которому они присоединены, образуют 4-6-членную гетероциклическую кольцевую группировку, возможно имеющую дополнительные 1-2 гетероатома, выбранные из группы, состоящей из N, S и О, где гетероциклическое кольцо возможно замещено группами R6.
Конкретные значения для каждого из R1 и R2 представляют собой Н.
Конкретные значения для R3 и R4 независимо представляют собой Н или С1-4алкил, или R3 и R4, взятые вместе, образуют C3-6циклоалкил.
Конкретные значения для R3 и R4, взятых вместе, представляют циклопропил.
Конкретные значения для соединений по настоящему изобретению включают значения, где W представляет собой Н, -РО(ОН)2 или -CH2OPO(ОН)2; группировка Het представляет собой 5- или 6-членную циклическую кольцевую систему, имеющую од одного до трех гетероатомов, выбранных из N, О и S, возможно замещенных группами R6; каждый из R1 и R2 представляет собой Н; -R3 и R4 независимо представляют собой Н или C1-4алкил, или R3 и R4, взятые вместе, образуют циклопропил; и X, Y и Z независимо представляют собой Н, хлорид или фторид.
Конкретные значения для R3 и R4, взятых вместе с одним или двумя гетероатомами, выбранными из группы, состоящей из N, S и О, представляют собой 4-6-членную гетероциклическую кольцевую группировку, где гетероциклическое кольцо возможно замещено группами R6 в количестве от одной до трех.
Конкретные значения для R3 и R4, взятых вместе с атомом кислорода, представляют собой оксетанил.
Конкретные значения для соединений по настоящему изобретению включают значения, где W представляет собой Н или -РО(ОН)2; группировка Het представляет собой 5- или 6-членную циклическую кольцевую систему, имеющую от одного до трех гетероатомов, выбранных из N, О и S, возможно замещенных группами R6; каждый из R1 и R2 представляет собой Н; R3 и R4 взяты вместе с атомом кислорода с образованием оксетанила; и X, Y и Z независимо представляют собой Н, хлорид или фторид.
Конкретные значения для R2 и R3, взятых вместе с атомом азота, к которому они присоединены, представляют собой азетидинил.
Конкретные значения для соединений по настоящему изобретению включают значения, где W представляет собой Н или -РО(ОН)2; группировка Het представляет собой 5- или 6-членную циклическую кольцевую систему, имеющую от одного до трех гетероатомов, выбранных из N, О и S, возможно замещенных группами R6; каждый из R1 и R4 представляет собой Н; R2 и R3 взяты вместе с атомом азота, к которому они присоединены, с образованием азетидинила; и X, Y и Z независимо представляют собой Н, хлорид или фторид.
Конкретные значения для R1 и R3, взятых вместе с атомом азота, к которому они присоединены, представляют собой пирролидинил.
Конкретные значения для соединений по настоящему изобретению включают значения, где W представляет собой Н или -РО(ОН)2; группировка Het представляет собой 5- или 6-членную циклическую кольцевую систему, имеющую от одного до трех гетероатомов, выбранных из N, О и S, возможно замещенную группами R6; каждый из R2 и R4 представляет собой Н; R1 и R3 взяты вместе с атомом азота, к которому они присоединены, с образованием пирролидинила; и X, Y и Z независимо представляет собой Н, хлорид или фторид.
Примеры соединений по настоящему изобретению включают следующие:
N-((1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид;
N-((1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамид;
(1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-2-(2,2-дихлорацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-2-(2,2-дихлорацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-дигидрофосфат;
N-((1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид;
N-((1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамид;
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-дигидрофосфат;
N-((1R,2S)-1-(4-(6-((RS)-1-аминоэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид;
N-((1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамид;
(1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дихлорацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дихлорацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-дигидрофосфат;
N-((1R,2S)-1-(4-(6-(азетидин-2-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид;
N-((1R,2S)-1-(4-(6-(азетидин-2-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамид;
(1R,2S)-1-(4-(6-(азетидин-2-ил)пиридин-3-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(6-(азетидин-2-ил)пиридин-3-ил)фенил)-2-(2,2-дихлорацетамидо)-3-фторпропил-дигидрофосфат;
N-((1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид;
(1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-дигидрофосфат;
N-((1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-3-фтор-1-гидрокси-пропан-2-ил)-2,2-дихлорацетамид;
2,2-дихлор-N-{(1S,2R)-1-(фторметил)-2-гидрокси-2-[4-(6-пирролидин-2-илпиридин-3-ил)фенил]этил}ацетамид;
(1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-(4-(6-(пирролидин-2-ил)-пиридин-3-ил)фенил)пропил-дигидрофосфат;
2,2-дифтор-N-{(1S,2R)-1-фторметил-2-гидрокси-2-[4-(6-пирролидин-2-ил-пиридин-3-ил)-фенил]-этил}-ацетамид; и
(1R,2S)-2-(2,2-дифторацетамидо)-3-фтор-1-(4-(6-(пирролидин-2-ил)-пиридин-3-ил)-фенил)пропил-дигидрофосфат.
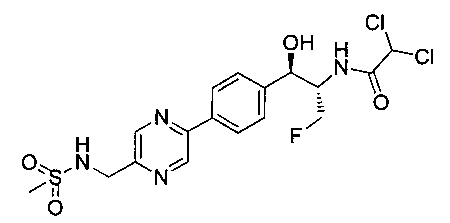
Также примером соединения по настоящему изобретению является N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)-фенил)пропан-2-ил)метансульфонамид.
Следующие схемы реакций иллюстрируют общие методики синтеза соединений по настоящему изобретению. Все исходные материалы получают согласно методикам, описанным в этих схемах, или методикам, известным специалисту в данной области техники.
Схема I
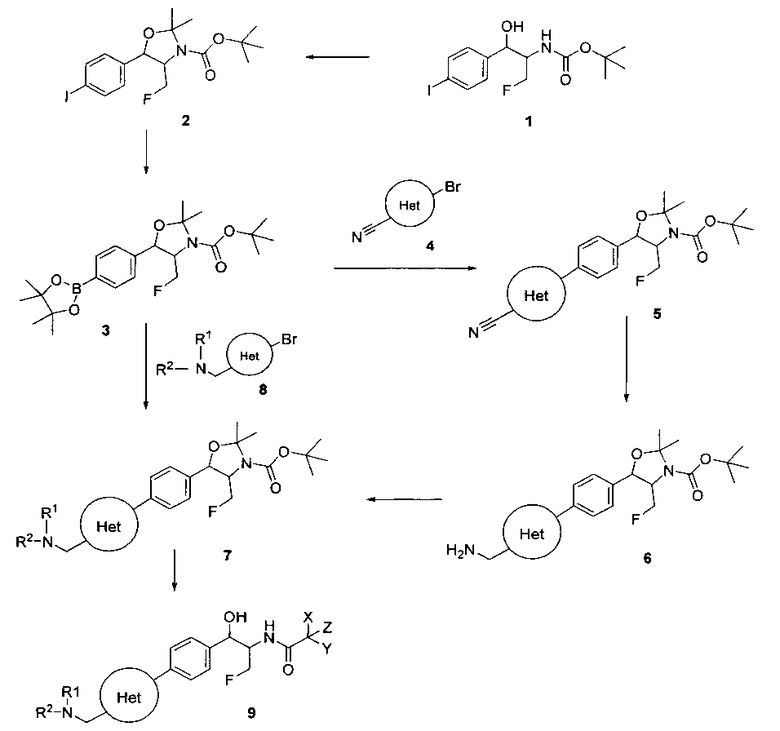
Как показано на Схеме I, соединение структуры (2) может быть получено из трет-бутил-3-фтор-1-гидрокси-1-(4-йодфенил)пропан-2-ил)карбамата (1) в присутствии подходящего агента ацетализации, такого как 2-метоксипропен, и слабой органической кислоты, такой как пара-толуолсульфоновая кислота, при температуре в интервале от 0°C до температуры дефлегмации, в полярных органических растворителях, таких как дихлорметан. Соединение структуры (3) может быть получено путем сочетания подходящего борирующего агента, такого как бис(пинаколато)дибор, с использованием каталитических количеств палладиевого катализатора, такого как хлорид бис(трифенилфосфин)палладия(II) или тетракистрифенилфосфинпалладий, в присутствии подходящего основания, такого как ацетат калия, в полярных апротонных растворителях, таких как 1,4-диоксан или тетрагидрофуран (THF), при температурах в интервале от комнатной температуры до температуры дефлегмации. Соединение структуры (5) может быть получено путем использования катализируемого палладием процесса сочетания, такого как сочетание Сузуки, между подходящим гетероарилгалогенидом (4) и бороновым сложным эфиром (3) с использованием палладиевого катализатора, такого как тетракистрифенилфосфинпалладий, в присутствии подходящего основания, такого как карбонат калия или бикарбонат натрия, в подходящей двухфазной смеси растворителей, такой как толуол и вода, при температурах от комнатной температуры до температуры дефлегмации. Соединение структуры (6) может быть получено из соединения структуры (5) путем взаимодействия с подходящим восстановителем, таким как палладий на углероде или смесь борогидрида натрия с хлоридом никеля, в подходящем протонном растворителе, таком как метанол или изопропиловый спирт, при температуре в интервале от 0°C до температуры дефлегмации. Соединение структуры (7) может быть получено путем конденсации с соответствующим агентом, таким как сульфонирующий агент, например мезилхлорид или этансульфонилхлорид или метансульфоновый ангидрид, в присутствии подходящего органического основания, такого как диизопропилэтиламин (DIPEA) или триэтиламин, в подходящем растворителе, таком как дихлорметан или THF, при температурах в интервале от -78°C до комнатной температуры. Альтернативно, соединение структуры (7) может быть получено путем прямого сочетания соответствующего агента, такого как сульфонамид (8), с бороновым эфиром структуры (3) с использованием палладиевого катализатора, такого как тетракистрифенилфосфинпалладий, в присутствии подходящего основания, такого как карбонат калия или бикарбонат натрия, в подходящей двухфазной смеси растворителей, такой как толуол и вода, при температурах в интервале от комнатной температуры до температуры дефлегмации. Использование альтернативного способа также дает соединение структуры (6), где R1 и R2 независимо представляют собой водород. Соединение структуры (9) может быть получено путем обработки соединения структуры (7) подходящей органической кислотой, такой как трифторуксусная кислота, в подходящем полярном растворителе, таком как дихлорметан или 1,2-дихлорэтан, при температурах в интервале от 0°C до комнатной температуры.
Схема II
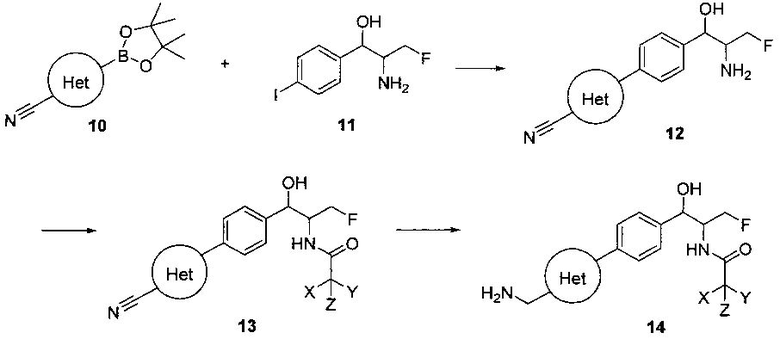
На схеме II соединение структуры (12) может быть получено из арилгалогенида (11) и подходящего боронового сложного эфира (10) с использованием палладиевого катализатора, такого как тетракистрифенилфосфинпалладий, в присутствии подходящего основания, такого как карбонат калия или бикарбонат натрия, в подходящей двухфазной смеси растворителей, такой как толуол и вода, при температурах в интервале от комнатной температуры до температуры дефлегмации. Соединение структуры (13) может быть получено путем конденсации амина (12) с подходящим ацилирующим агентом, таким как дихлорацетилхлорид или этилдифторацетат, в присутствии подходящего основания, такого как триэтиламин или DIPEA, в подходящем полярном протонном растворителе, таком как метанол, или подходящем полярном растворителе, таком как дихлорметан. Соединение структуры (14) может быть получено из соединения структуры (13) путем взаимодействия с подходящим восстановителем, таким как палладий на углероде или смесь борогидрида натрия с хлоридом никеля, в подходящем протонном растворителе, таком как метанол или изопропиловый спирт, при температуре в интервале от 0°C до температуры дефлегмации.
Схема III
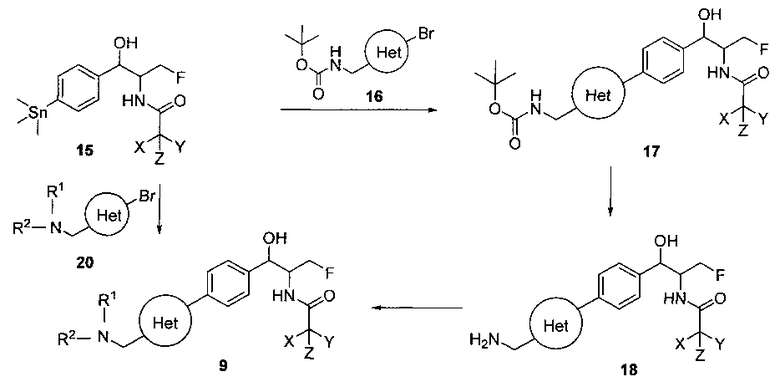
На Схеме III соединение структуры (17) может быть получено путем сочетания Стилла станнановой структуры (15) с арилгалогенидом структуры (16) с использованием палладиевого катализатора, такого как трис(дибензилиденацетон)дипалладий или хлорид бис(трифенилфосфин)-палладия(II), и возможно с подходящим фосфиновым лигандом, таким как трис(2-фурил)фосфин, вместе с подходящей добавкой галогенида металла, такого как хлорид лития или фторид цезия, в подходящем полярном апротонном растворителе, таком как N-метилпирролидон (NMP) или диметилформамид (DMF), при температурах в интервале от комнатной температуры до 120°C. Соединение структуры (18) может быть получено путем обработки соединения структуры (17) подходящей органической кислотой, такой как трифторуксусная кислота, в подходящем растворителе, таком как DCM или 1,2-дихлорэтан, при температурах в интервале от 0°C до комнатной температуры. Соединение структуры (9) может быть получено из соединения структуры (18) путем конденсации с подходящим агентом, таким как сульфонирующий агент, например мезилхлорид или этансульфонилхлорид или метансульфоновый ангидрид, в присутствии подходящего органического основания, такого как DIPEA или триэтиламин, в подходящем растворителе, таком как дихлорметан или THF, при температурах в интервале от -78°C до комнатной температуры. Альтернативно, соединение структуры (9) может быть получено путем прямого сочетания арилсульфонамида (20) с бороновым сложным эфиром станнана (15) с использованием палладиевого катализатора, такого как трис(дибензилиденацетон)дипалладий или хлорид бис(трифенилфосфин)-палладия(II), и возможно подходящего фосфинового лиганда, такого как трис(2-фурил)фосфин, вместе с подходящей добавкой галогенида металла, такого как хлорид лития или фторид цезия, в подходящем полярном апротонном растворителе, таком как NMP или DMF, при температурах в интервале от комнатной температуры до 120°C.
Фармацевтические соли
Соединение формулы I может быть использовано в его нативной форме или в виде соли. В случаях, когда желательно образование стабильных нетоксичных солей кислот или оснований, введение соединения в виде фармацевтически приемлемой соли может быть подходящим. Фармацевтически приемлемые соли соединений формулы I включают соли ацетат, аскорбат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, эдисилат, этоглутарат, эзилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, глицерофосфат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изэтионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, сахарат, стеарат, сукцинат, тартрат, тозилат и трифторацетат.
Композиция/Препарат
Фармацевтические композиции по настоящему изобретению могут быть изготовлены способами, хорошо известными в данной области техники, например, посредством традиционного смешивания, растворения, гранулирования, дражирования, измельчения, эмульгирования, инкапсулирования, включения, процессов лиофилизации или распылительной сушки.
Фармацевтическая композиции для применения в соответствии с настоящим изобретением может быть изготовлена общепринятым образом с использованием одного или более фармацевтически приемлемых носителей, включающих эксципиенты и вспомогательные агенты, которые облегчают обработку соединения и его приготовление в виде препаратов, которые могут быть использованы фармацевтически. Подходящий препарат зависит от пути выбранного введения. Фармацевтически приемлемые эксципиенты и носители в общем известны специалистам в данной области техники и, таким образом, включены в настоящее изобретение. Такие эксципиенты и носители описаны, например, в "Remington's Pharmaceutical Sciences", Mack Pub. Co., New Jersey (1991).
Препараты по изобретению могут быть разработаны для быстрого действия, быстрого высвобождения, длительного действия, продолжительного высвобождения или контролируемого высвобождения. Конкретно, препарат по изобретению может быть представлен в форме пролонгированного высвобождения. Так, фармацевтические препараты могут также быть изготовлены для контролируемого высвобождения или для медленного высвобождения.
Дозировка
Фармацевтические композиции, подходящие для применения в настоящем изобретении, включают композиции, где активные ингредиенты содержатся в количестве, достаточном для достижения намеченного назначения, например контролирования или лечения инфекций. Более конкретно, терапевтически эффективное количество означает количество соединения, эффективное для предупреждения, ослабления или облегчения симптомов/признаков инфекций или продления выживания субъекта, которого лечат.
Количество активного компонента, представляющего собой соединение по данному изобретению, в фармацевтической композиции и ее стандартной лекарственной форме, может варьировать и широко регулируется в зависимости от пути введения, активности конкретного соединения и желаемой концентрации. Определение терапевтически эффективного количества находится в компетенции специалистов в данной области. В общем, количество активного компонента находится в интервале между 0,01% до 99% по массе композиции.
В общем, терапевтически эффективное количество дозировки активного компонента находится в интервале от примерно 0,1 мг до примерно 100 мг/кг массы тела/сутки; например, от примерно 0,1 до примерно 50 мг/кг массы тела/сутки; и, например, от примерно 5 до примерно 50 мг/кг массы тела/сутки; и, например, от примерно 20 до примерно 50 мг/кг массы тела/сутки. Следует понимать, что дозировки могут варьировать в зависимости от потребностей каждого субъекта и тяжести инфекций.
Желаемая доза может удобным образом быть представлена в однократной дозе или в виде разделенных доз, вводимых через соответствующие интервалы, например, в виде двух, трех, четырех или более суб-доз в сутки. Также следует понимать, что начальная вводимая дозировка может быть увеличена за пределы верхнего уровня с целью быстрого достижения желаемой концентрации в плазме. С другой стороны, начальная дозировка может быть меньше оптимальной, и суточная дозировка может быть прогрессивно увеличена во время курса лечения в зависимости от конкретной ситуации. При желании суточная доза также может быть разделена на несколько доз для введения, например, от двух до четырех раз в сутки.
Медицинские и ветеринарные применения
Соединения по настоящему изобретению обеспечивают новые фениколовые противобактериальные агенты для лечения бычьих респираторных инфекций у крупного рогатого скота, вызванных грамотрицательными респираторными патогенами, такими как М. haemolytica, P. multocida, Н. somnus и М. bovis.
Противобактериальные анализы
Соединения по настоящему изобретение тестированы против ряда грамотрицательных и грамположительных организмов с использованием промышленных стандартных техник, описанных в М31-А3. Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals; Clinical and Laboratory Standards Institute, Approved Standard-Third Edition. Соединения по настоящему изобретению демонстрируют очень сильную противобактериальную активность против BRD патогенов, например, М. haemolytica, P. multo., H. somnus и М. bovis.
Примеры
Синтез соединений по настоящему изобретению далее проиллюстрирован следующими примерами. Исходные материалы и разнообразные промежуточные соединения, используемые в примерах, могут быть получены от коммерческих поставщиков, или их легко получить из имеющихся в продаже органических соединений с использованием способов, хорошо известных в данной области техники.
Пример 1. Получение N-((1R,2S)-1-(4-(6-(аминометил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение 5-(4-((1R,2S)-2-амино-3-фтор-1-гидроксипропил)-фенил)пиколинонитрила
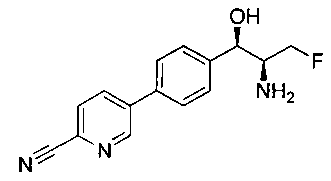
К раствору имеющегося в продаже 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиколинонитрила (0,5 г, 2,17 ммоль) в дегазированном диметоксиэтане (10 мл) и воде (3 мл) добавляли (1R,2S)-2-амино-3-фтор-1-(4-йодфенил)-пропан-1-ол (0,65 г, 2,20 ммоль) и Cs2CO3 (2,15 г, 6,6 ммоль). Добавляли Pd(PPh3)4 (0,25 г, 0,21 ммоль) и реакционную смесь нагревали до 90°C в течение 1,5 часов. Растворитель выпаривали в вакууме и неочищенный материал очищали колоночной хроматографией на силикагеле, элюируя в смеси метанол/CHCl3 с получением указанного в заголовке соединения (206 мг): 1ЯМР (400 МГц, CDCl3) δ: 3.09-3.18 (m, 1Н), 3.48 (s, 1Н), 4.24-4.28 (m, 0.5Н), 4.36-4.41 (m, 1Н), 4.49-4.52 (m, 0.5Н), 4.64 (d, J=6,16 Гц, 1Н), 7.53 (d, J=8,16 Гц, 2Н), 7.59 (d, J=8,24 Гц, 2Н), 7.76 (d, J=8,2, 1Н), 8.0 (dd, J1=8,16 Гц, J2=2,32 Гц, 1Н), 8.93 (d, J=1,76 Гц, 1Н). m/z (Cl) 272 [М+Н].
Стадия 2. Получение 2,2-дихлор-N-((1R,2S)-1-(4-(6-цианопиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)ацетамида
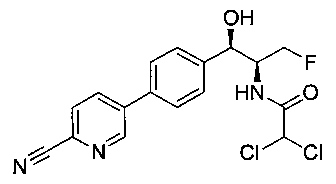
К раствору 5-(4-((1R,2S)-2-амино-3-фтор-1-гидроксипропил)фенил)-пиколинонитрила (0,5 г, 1,1 ммоль) в метаноле (5 мл) добавляли триэтиламин (0,22 г, 2,2 ммоль) и этилдихлорацетат (0,34 г, 2,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали в вакууме и неочищенный материал очищали колоночной хроматографией на силикагеле с использованием смеси метанол/CH2Cl2 с получением указанного в заголовке соединения (264 мг): 1Н ЯМР (400 МГц, CDCl3) δ: 4.26-4.35 (m, 1Н), 4.36-4.37 (m, 0.5Н), 4.45-4.51 (m, 1Н), 4.59-4.63 (m, 0.5Н), 5.02 (d, J=3,28 Гц, 1Н), 5.84 (s, 1Н), 7.47 (d, J=8,2 Гц, 2Н), 7.54 (d, J=8,24 Гц, 2Н), 7.76 (d, J=8,04 Гц, 1Н), 7.96 (dd, J1=8,08 Гц, J2=2,2 Гц, 1Н), 8.85 (d, J=1,64 Гц, 1Н). m/z (Cl) 380 [М+Н].
Стадия 3. Получение N-((1R,2S)-1-(4-(6-(аминометил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
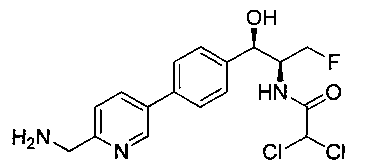
К охлажденному на льду раствору алюмогидрида лития (0,048 г, 1,33 ммоль, 4,0 экв.) в тетрагидрофуране (10 мл) до -40°C добавляли раствор 2,2-дихлор-N-((1R,2S)-1-(4-(6-цианопиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)ацетамида (0,13 г, 0,34 ммоль) в тетрагидрофуране (5 мл) при -40°C. Добавляли дополнительное количество алюмогидрида лития (0,012 г, 0,33 ммоль) три раза и реакционную смесь перемешивали при -40°C в течение 4 часов. Реакционную смесь гасили насыщенным водным сульфатом натрия и перемешивали в течение 15 минут с последующим фильтрованием. Фильтрат упаривали в вакууме и неочищенный материал очищали колоночной хроматографией на силикагеле с использованием смеси метанол/CH2Cl2 и аммиака. К неочищенному раствору в CH2Cl2 (5 мл) добавляли трифторуксусную кислоту (0,5 мл) и перемешивали реакционную смесь при комнатной температуре в течение 15 мин. Растворитель отгоняли под вакуумом и остаток промывали диэтиловым эфиром и этот остаток растворяли в 10% метанола в CH2Cl2 и упаривали досуха. Промывали н-пентаном и сушили под вакуумом с получением указанного в заголовке соединения (20 мг): 1Н ЯМР (400 МГц, CDCl3) δ: 4.24-4.25 (m, 2Н), 4.28-4.32 (m, 0.5Н), 4.40-4.44 (m, 0.5Н), 4.56-4.60 (m, 0.5Н), 4.68-4.72 (m, 0.5Н), 4.92 (bs, 1Н), 6.02 (bs, 1Н), 6.52 (s, 1Н), 7.48 (d, J=8,28 Гц, 2Н), 7.57 (d, J=10,8 Гц, 1Н,), 7.73 (d, J=8,04 Гц, 2Н), 8.17 (dd, J1=2,28 Гц, J2=8,08 Гц, 1Н), 8.28 (bs, 2Н), 8.65 (d, J=8,84 Гц, 1Н), 8.93 (d, 1Н, J=2,2 Гц), m/z (Cl) 386 [М+Н].
Пример 2. Получение N-((1R,2S)-1-(4-(5-(аминометил)тиофен-2-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение трет-бутил-((5-(4-((1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-гидроксипропил)фенил)тиофен-2-ил)метил)-карбамата
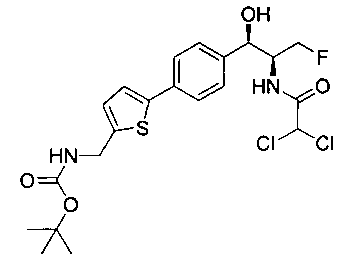
К раствору трет-бутил-((5-бромтиофен-2-ил)метил)карбамата (0,508 г, 1,74 ммоль) и 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметил-станнил)фенил)пропан-2-ил)ацетамида (0,7 г, 1,58 ммоль) в толуоле (10 мл) добавляли фторид цезия (0,478 г, 3,16 ммоль) и йодид меди (30 мг, 0,158 ммоль) и дегазировали азотом в течение 30 минут. К этой смеси добавляли Pd(PPh3)2Cl2 (0,11 г, 0,16 ммоль) и смесь нагревали до 90°C в течение 26 часов. Растворитель выпаривали в вакууме с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле с использованием метанола в CH2Cl2 с получением указанного в заголовке соединения (490 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.39 (s, 9Н), 4.17-4.29 (m, 3.5Н), 4.37-4.41 (m, 0.5Н), 4.54-4.57 (m, 0.5H), 4.65-4.69 (m, 0.5H), 4.84 (t, J=3,56 Гц, 1H), 5.95 (d, J=4,12 Гц, 1H), 6.50 (s, 1H), 6.88 (d, J=3,48 Гц, 1H), 7.29-7.37 (dd, J=8,2 Гц, J=3,6 Гц, 3H), 7.51-7.56 (d, J=8,04 Гц, 3H). m/z (Cl) 489 [M-Н].
Стадия 2. Получение N-((1R,2S)-1-(4-(5-(аминометил)тиофен-2-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
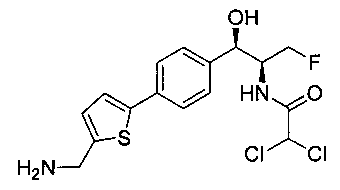
К раствору трет-бутил-((5-(4-((1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-гидроксипропил)фенил)тиофен-2-ил)метил)карбамата (140 мг, 0,238 ммоль) в CH2Cl2 (10 мл) добавляли трифторуксусную кислоту (1,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении и остаток промывали диэтиловым эфиром, затем растворяли в 10% метаноле в CH2Cl2 и упаривали досуха. Промывали н-пентаном и сушили под вакуумом с получением указанного в заголовке соединения (56 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.19-4.21 (m, 1Н), 4.24-4.26 (m, 2Н), 4.29-4.31 (m, 0.5Н), 4.39-4.43 (m, 0.5Н), 4.55-4.59 (m, 0.5Н), 4.67-4.70 (m, 0.5Н), 4.87 (bs, 1Н), 5.90 (bs, 1Н) 6.50 (s, 1Н), 7.20 (d, J=3,64 Гц, 1Н), 7.39 (d, J=8,24 Гц, 2Н), 7.43 (d, J=3,68 Гц, 1Н), 7.57 (d, J=8,16 Гц, 2Н), 8.20 (bs, 3H), 8.59-8.63 (m, 1Н). m/z (Cl) 388 [M-Н].
Пример 3. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(5-(метилсульфонамидометил)тиофен-2-ил)фенил)пропан-2-ил)ацетамида
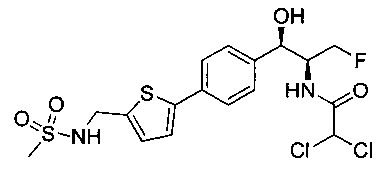
К суспензии N-((1R,2S)-1-(4-(5-(аминометил)тиофен-2-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида (150 мг, 0,383 ммоль) в дихлорметане добавляли триэтиламин (160 мкл, 1,15 ммоль) с последующим добавлением метансульфонилхлорида (30 мкл, 0,383 ммоль). Полученный желтый раствор перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали, затем очищали с использованием высокоэффективной жидкостной хроматографии (ВЭЖХ) с получением указанного в заголовке соединения (99 мг): 1Н ЯМР (400 МГц, DMSO-d6): δ 8.58 (d, 1Н), 7.71 (t, 1Н), 7.55 (d, 2Н), 7.37 (d, 2Н), 7.33 (d, 2Н), 7.01 (d, 1Н), 6.51 (s, 1Н), 5.95 (brs, 1Н), 4.85 (d, 1Н), 4.71-4.52 (m, 1Н), 4.44-4.13 (m, 4Н), 2.89 (s, 3H).
Пример 4. Получение N-((1R,2S)-1-(4-(2-(аминометил)тиазол-5-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение трет-бутил-((5-(4-((1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-гидроксипропил)фенил)тиазол-2-ил)метил)-карбамата
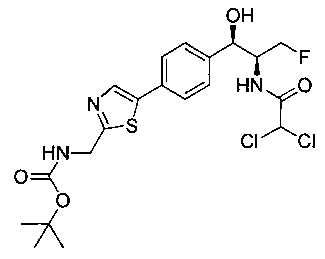
Следуя общей методике стадии 1 Примера 2 и осуществляя незначительные модификации, но используя 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметил-станнил)фенил)пропан-2-ил)ацетамид и трет-бутил-((5-бромтиазол-2-ил)метил)карбамат в качестве исходных материалов, получили указанное в заголовке соединение (346 мг): 1Н ЯМР (400 МГц, DMSO-d6) 1.41 (s, 9Н), 4.19-4.22 (m, 1Н), 4.26-4.30 (m, 0.5Н), 4.36-4.42 (m, 2.5Н), 4.54-4.58 (m, 0.5Н), 4.66-4.70 (m, 0.5Н), 4.86 (t, J=3,4 Гц, 1Н), 5.98 (d, J=4,16 Гц, 1Н), 6.50 (s, 1Н), 7.39 (d, J=8,2 Гц, 2Н), 7.60 (d, J=8,0 Гц, 2Н), 7.77-7.82 (m, 1Н), 8.05 (s, 1Н), 8.60 (d, J=8,8 Гц, 1Н) m/z (Cl) 492 [М+Н].
Стадия 2. Получение N-((1R,2S)-1-(4-(2-(аминометил)тиазол-5-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
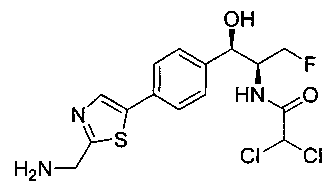
К перемешиваемому раствору продукта стадии 1 Примера 4 (0,248 г, 0,50 ммоль) в CHCl3 (10 мл) добавляли трифторуксусную кислоту (1,1 мл) и перемешивали эту реакционную смесь при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали в вакууме и остаток промывали диэтиловым эфиром. Остаток растворяли в 10% метаноле в CH2Cl2 и выпаривали досуха, затем сушили под вакуумом с получением указанного в заголовке соединения. 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.20-4.22 (m, 1Н), 4.28-32 (m, 0.5Н), 4.40-4.49 (m, 2.5Н), 4.56-4.60 (m, 0.5H), 467-4.71 (m, 0.5H), 4.89 (t, J=3,8 Гц, 1H), 6.02 (d, J=4,28 Гц, 1H), 6.50 (s, 1H), 7.43 (d, J=8,2 Гц, 2H), 7.62 (d, J=8,2 Гц, 2H), 8.28 (s, 1H), 8.48 (bs, 2H), 8.63 (d, J=9 Гц, 1H). m/z (Cl) 392 [M-Н].
Пример 5. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
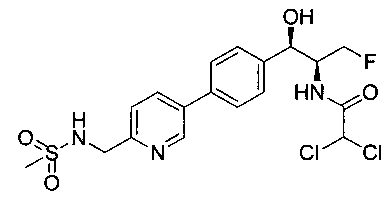
N-((5-Бромпиридин-2-ил)метил)метансульфонамид (ранее описанный в WO 9528400) (240 мг, 0,90 ммоль) и 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)фенил)пропан-2-ил)ацетамид (400 мг, 0,90 ммоль) в 2-метилпирролидиноне (5 мл) обрабатывали хлоридом лития (115 мг, 2,7 ммоль), дегазировали и продували азотом. Добавляли хлорид бис(трифенилфосфин)-палладия(II) и смесь нагревали при 100°C в течение 3 часов. Смесь охлаждали, разбавляли водой и экстрагировали этилацетатом, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали хроматографией с обращенной фазой с получением указанного в заголовке соединения (324 мг): 1Н ЯМР (400 МГц, CDCl3)δ: 2.96 (s, 3H), 4.2-4.3 (m, 3H), 4.4 (m, 0.5Н), 4.57 (m, 0.5 Н), 4.58 (m, 0.5Н), 4.70 (m, 0.5H), 4.90 (bs, 1H), 6.00 (s, 1H), 6.52 (s, 1H), 7.47 (d, 2H), 7.52, (d, 1H), 7.69 (d, 3H), 8.1 (m, 1H), 8.85 (m, 1H), 8.82 (bs, 1H). m/z (Cl) 464.
Пример 6. Получение N-((1R,2S)-1-(4-(5-(аминометил)-1,3,4-тиадиазол-2-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
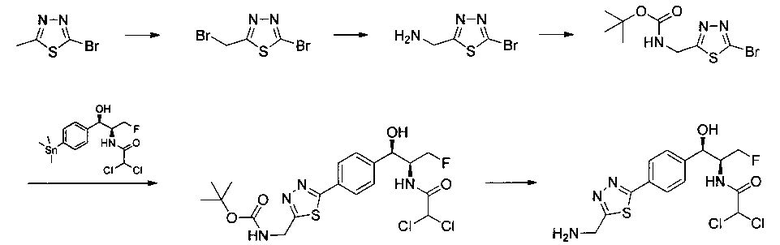
Стадия 1. Получение 2-бром-5-(бромметил)-1,3,4-тиадиазола
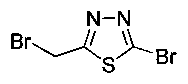
К раствору 2-бром-5-метил-1,3,4-тиадиазола (0,50 г, 2,79 ммоль) в CCl4 (8 мл) добавляли N-бромсукцинамид (0,543 г, 3,067 ммоль) и азобис-изобутиронитрил (0,022 г, 0,139 ммоль) и реакционную смесь нагревали до 70°C в течение 3 часов. Реакционную смесь охлаждали до 0°C и растворитель выпаривали в вакууме с получением неочищенного материала, который очищали колоночной хроматографией, элюируя 5%-ным этилацетатом в гексане с получением указанного в заголовке соединения (0,150 г): 1Н ЯМР (400 МГц, CDCl3) δ 4.75 (s, 2Н). ЖХ-MC (m/z): [М+Н]=260,8.
Стадия 2. Получение (5-бром-1,3,4-тиадиазол-2-ил)метанамина
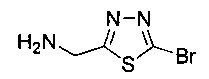
Газообразный аммиак барботировали через раствор 2-бром-5-(бромметил)-1,3,4-тиадиазола (0,6 г, 2,352 ммоль) в метаноле (15 мл) в течение 15 минут. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь концентрировали в вакууме и сушили с получением указанного в заголовке соединения (0,47 г): 1Н ЯМР (400 МГц, DMSO-d6) δ 5.14 (s, 2Н). ЖХ-MC (m/z): [М+Н]=193,90.
Стадия 3. Получение трет-бутил-((5-бром-1,3,4-тиадиазол-2-ил)метил)-карбамата
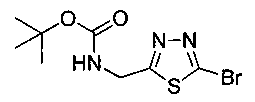
К перемешиваемому раствору (5-бром-1,3,4-тиадиазол-2-ил)метанамина (0,425 г, 2,19 ммоль) в 1,4-диоксане (10 мл) добавляли 10%-ный водный раствор К2СО3 (0,392 г, 2,84 ммоль). Смесь охлаждали до 0°C и добавляли ди-трет-бутилдикарбонат (0,525 г, 2,40 ммоль). Реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали в вакууме. Неочищенный материал разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, концентрировали и очищали колоночной хроматографией на силикагеле, элюируя 20%-ным этилацетатом в гексане с получением указанного в заголовке соединения (0,30 г): 1Н ЯМР (400 МГц, CDCl3) δ 1.45 (s, 9Н), 4.34-4.40 (bs, 1Н), 4.66 (d, J=6,28 Гц, 2Н), ЖХ-MC (m/z): [М+Н]=294.
Стадия 4. Получение трет-бутил-((5-(4-((1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-гидроксипропил)фенил)-1,3,4-тиадиазол-2-ил)-метил)карбамата
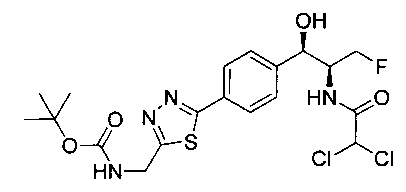
К перемешиваемому раствору трет-бутил-((5-бром-1,3,4-тиадиазол-2-ил)метил)-карбамата (0,878 г, 2,98 ммоль) и 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)фенил)пропан-2-ил)ацетамида (1,20 г, 2,71 ммоль) в диметилформамиде (20 мл) добавляли CsF (0,818 г, 5,42 ммоль) с последующим добавлением Cul (0,051 г, 0,271 ммоль). Полученную реакционную смесь дегазировали азотом в течение 30 минут и добавляли Pd(PPh3)4 (0,313 г, 0,271 ммоль). Реакционную смесь нагревали до 90°C в течение 5 часов. Реакционную смесь концентрировали в вакууме с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле, элюируя 1%-ным метанолом в CH2Cl2 с получением указанного в заголовке соединения (100 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ 1.40 (s, 9Н), 4.24-4.26 (m, 1Н), 4.29-4.33 (m, 0.5Н), 4.41-4.45 (m, 0.5Н), 4.52 (d, J=5,96 Гц, 2H), 4.57-4.61 (m, 0.5H), 4.69-4.72 (m, 0.5H), 4.93 (t, J=3,76 Гц 1H), 6.08 (d, J=4,28 Гц, 1H), 6.49 (s, 1H), 7.51 (d, J=8,24 Гц, 2H), 7.86-7.90 (m, 3H), 8.63 (d, J=8,96 Гц, 1H). LC-Ms (m/z): [M-H]=490,80.
Стадия 5. Получение N-((1R,2S)-1-(4-(5-(аминометил)-1,3,4-тиадиазол-2-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
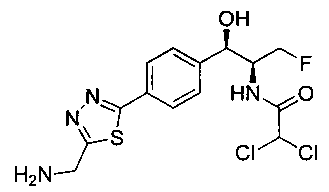
К перемешиваемому раствору трет-бутил-((5-(4-((1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-гидроксипропил)фенил)-1,3,4-тиадиазол-2-ил)-метил)карбамата (100 мг, 0,203 ммоль) в CH2Cl2 (10 мл) добавляли трифторуксусную кислоту (1,0 мл). Через 2 часа реакционную смесь концентрировали в вакууме и остаток промывали диэтиловым эфиром. Остаток растворяли в 10% метаноле в CH2Cl2 и упаривали досуха, промывали н-пентаном, сушили под вакуумом с получением указанного в заголовке соединения (108 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ 4.27-4.28 (m, 1Н), 4.31-4.35 (m, 0.5Н), 4.43-4.47 (m, 0.5Н), 4.59-4.61 (m, 0.5H), 4.64 (s, 2H), 4.70-4.74 (m, 0.5H), 4.96 (t, J=2,76 Гц, 1H), 6.10 (d, J=4,28 Гц, 1H), 6.48 (s, 1H), 7.54 (d, J=8,32 Гц, 2H), 7.94 (d, J=8,32 Гц, 2H), 8.60 (bs, 2H), 8.64 (d, J=9,12 Гц, 1H). LC-Ms (m/z): [M+H]=393,10.
Пример 7. Получение N-((1R,2S)-1-(4-(6-((1Н-имидазол-1-ил)метил)-пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
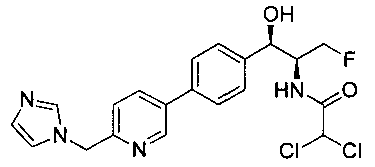
Следуя общей методике стадии 4 Примера 6 и осуществляя незначительные модификации, но используя 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметил-станнил)фенил)пропан-2-ил)ацетамид и имеющийся в продаже 2-((1Н-имидазол-1-ил)метил)-5-бромпиридин, получили указанное в заголовке соединение (20 мг): 1Н ЯМР (400 МГц, DMSO-d6) 4.21-4.24 (m, 1Н), 4.27-40.31 (m, 0.5Н), 4.38-4.42 (m, 0.5Н), 4.55-4.59 (m, 0.5H), 4.67-4.70 (m, 0.5H), 4.89 (t, J=3,6 Гц, 1H), 5.33 (s, 2H), 5.99 (d, J=4,16 Гц, 1H), 6.51 (s, 1H), 6.92 (s, 1H), 7.22 (t, J=3,84 Гц, 2H), 7.46 (d, J=8,2 Гц, 2H), 7.67 (d, J=8,28 Гц, 2H), 7.77 (s, 1H), 8.06-8.08 (d,d J=2,36 Гц, J=8,08 Гц, 1H), 8.63 (d, J=8,84 Гц, 1H), 8.84 (d, J=2,2 Гц, 1H). LC-Ms (m/z): [M+H]=437,1.
Пример 8. Получение N-((1R,2S)-1-(4-(3-(аминометил)-1,2,4-оксадиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение метил-4-((4S,5R)-3-(2,2-дихлорацетил)-4-(фтор-метил)-2,2-диметилоксазолидин-5-ил)бензоата
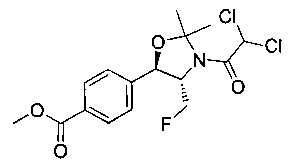
В колбу, содержащую 2,2-дихлор-1-((4S,5R)-4-(фторметил)-5-(4-йодфенил)-2,2-диметилоксазолидин-3-ил)этанон (2,0 г, 4,48 ммоль), добавляли триэтиламин (5 мл) и метанол (5 мл). Газообразный монооксид углерода барботировали через этот раствор при перемешивании в течение 30 мин. Затем добавляли Pd(OAc)2 (51 мг, 0,22 ммоль) и Xantphos (132 мг, 0,22 ммоль) и баллон, содержащий монооксид углерода, подсоединяли к выходному отверстию колбы. Реакционную смесь нагревали в течение 2 часов при 60°C и затем охлаждали до комнатной температуры. Затем реакционную смесь разбавляли водой, экстрагировали этилацетатом, сушили над Na2SO4 и концентрировали под вакуумом. Остаток подвергали хроматографии на силикагеле, элюируя от 100%-ных гексанов до смеси 50:50 этилацетат: гексаны с получением указанного в заголовке соединения (1,11 г): 1Н ЯМР (400 МГц, CDCl3) 8.10 (d, 2Н, J=8,0 Гц), 7.55 (d, 2Н, J=8,0 Гц), 6.34 (m, 1Н), 5.30-4.42 (m, 4Н), 3.95 (s, 3H), 1.97-1.53 (m, 6Н). m/z (Cl) 320 [N-(СН3)2СО].
Стадия 2. Получение 4-((4S,5R)-3-(2,2-дихлорацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)бензойной кислоты
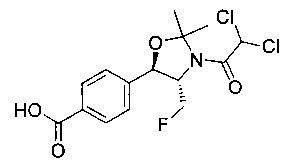
К раствору продукта стадии 1 Примера 8 (1,1 г, 2,9 ммоль) в смеси 5:1 диоксан: вода (60 мл) добавляли гидроксид лития (212 мг, 8,9 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 18 часов. Затем реакционную смесь охлаждали до 0°C и добавляли 1 н. HCl (7,5 мл) для нейтрализации (рН примерно 7). Реакционную смесь распределяли между водой (50 мл) и CH2Cl2 (150 мл). Органическую фазу собирали, сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения (979 мг): 1Н ЯМР (400 МГц, DMSO-d6) 7.98 (d, 2Н, J=8,0 Гц), 7.60 (d, 2Н, J=8,0 Гц), 5.28 (m, 1Н), 4.96-4.81 (m, 2.5 Н), 4.75-4.70 (m, 0.5Н), 4.67-4.62 (m, 0.5Н) 4.55-4.50 (m, 0.5H), 1.61 (s, 3H), 1.46 (s, 3H).
Стадия 3. Получение трет-бутил-(5-(4-((4S,5R)-3-(2,2-дихлорацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)-1,2,4-оксадиазол-3-ил)-метил-карбамата
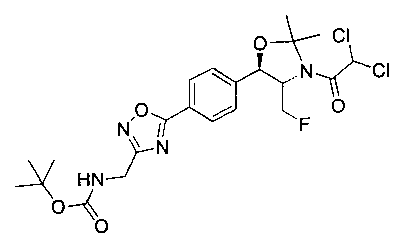
К раствору продукта стадии 2 Примера 8 (525 мг, 1,4 ммоль) в диметилформамиде (15 мл) добавляли 1,1′-карбонилдиимидазол (286 мг, 1,7 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут, во время которых добавляли ацетат натрия (140 мг, 1,7 ммоль) и имеющийся в продаже трет-бутил-2-(гидроксиамино)-2-иминоэтилкарбамат (325 мг, 1,7 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение 72 часов. Далее эту реакционную смесь разбавляли этилацетатом (75 мл) и промывали водой (3×75 мл). Органическую фазу сушили (Na2SO4) и концентрировали под вакуумом. К остатку добавляли толуол (15 мл) и ацетат натрия (140 мг, 1,7 ммоль). Полученную смесь нагревали до температуры дефлегмации в течение 18 часов при перемешивании, затем охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток подвергали хроматографии на силикагеле, элюируя от 100%-ных гексанов до смеси 50:50 EtOAc: гексаны с получением указанного в заголовке соединения (280 мг): 1Н ЯМР (400 МГц, CDCl3) 8.19 (d, 2Н, J=8,0 Гц), 7.64 (d, 2Н, J=8,0 Гц), 6.33 (m, 1Н), 5.16 (m, 2Н) 4.88-4.52 (m, 4Н), 1.75 (s, 3H), 1.59 (s, 3H), 1.50 (s, 9Н). m/z (Cl) 461 [N-(СН3)2СО].
Стадия 4. Получение N-((1R,2S)-1-(4-(3-(аминометил)-1,2,4-оксадиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
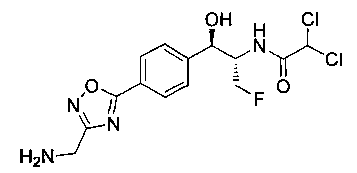
Следуя общей методике стадии 2 Примера 2 и осуществляя незначительные модификации, но используя продукт стадии 3 Примера 8, получили указанное в заголовке соединение (280 мг): 1Н ЯМР (400 МГц, DMSO-d6) 8.73-8.57 (m, 4Н), 8.07 (d, 2Н, 7=8,0 Гц), 7.65 (d, 2Н, J=8,0 Гц), 6.16 (m, 1Н), 5.02 (m, 1H) 4.76-4.72 (m, 0.5 H), 4.64-4.60 (m, 0.5H), 4.50-4.24 (m, 4H). m/z (Cl) 377 [М+Н].
Пример 9. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(1-(метилсульфонил)пирролидин-2-ил)пиридин-3-ил)фенил)пропан-2-ил)-ацетамида
Стадия 1. Получение 5-бром-2-(1-(метилсульфонил)пирролидин-2-ил)-пиридина
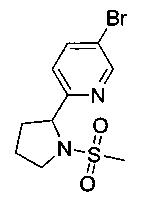
Следуя общей методике Примера 3 и осуществляя незначительные модификации, но используя 5-бром-2-(пирролидин-2-ил)пиридин (ранее описанный в WO 200853319), получили указанное в заголовке соединение (400 мг): 1Н ЯМР (400 МГц, CDCl3) 2.0-2.1 (m, 2Н), 2.15-2.25 (m, 1Н), 2.35-2.45 (m 1Н), 2.8 (s, 3H), 3.5-3.6 (m, 1Н), 3.6-3.7 (m, 1Н), 4.9-5.0 (m, 1Н), 7.45 (d, 1Н), 7.85 (dd, 1Н), 8.6 (d, 1Н). m/z (Cl) М+Н 305+307.
Стадия 2. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(1-(метилсульфонил)пирролидин-2-ил)пиридин-3-ил)фенил)пропан-2-ил)-ацетамида
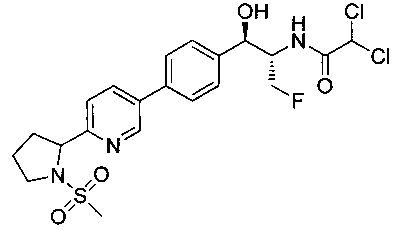
Смесь 5-бром-2-(1-(метилсульфонил)пирролидин-2-ил)пиридина (100 мг, 0,328 ммоль), 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)-фенил)-пропан-2-ил)ацетамида (145 мг, 0,328 ммоль) и трис(2-фурил)фосфина (15,5 мг, 0,066 ммоль) растворяли в N-метилпирролидиноне (1,6 мл) и затем добавляли дезоксигенированный трис(дибензилиденацетон)дипалладий(0) (30,5 мг, 0,033 ммоль) и смесь нагревали до 80°C в течение ночи. Смесь затем охлаждали и очищали препаративной ВЭЖХ (преп. ВЭЖХ Waters, колонка Gemini NX C18 21×150 мм, 5 мкм, подвижная фаза (MP) А: 0,1%-ная трифторуксусная кислота в воде, MP В: ацетонитрил, градиент от 10% В до 50% в течение 10 мин, удерживая в течение 2 мин, 20 мл/мин.) Фракции сушили на роторном испарителе и лиофилизировали с 1,4-диоксаном с получением указанного в заголовке соединения (33 мг): 1Н ЯМР (400 МГц, DMSO-d6) 1.9-2.0 (m, 2Н), 2.0-2.1 (m, 1Н), 2.3-2.4 (m, 1Н), 3.0 (s, 3H), 3.5-3.6 (m, 2Н), 4.15-4.35 (m, 1.5Н), 4.45 (t, 0.5Н), 4.55-4.65 (m, 0.5H), 4.7-4.75 (m, 0.5H), 4.9-5.0 (m, 2H), 6.5 (s, 1H), 7.5 (d, 2H), 7.65 (d, 1H), 7.75 (d, 2H), 8.2 (dd, 1H), 8.6 (d, 1H), 8.85 (d, 1H). m/z (Cl) M+H 504+506.
Пример 10. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(5-(метилсульфонамидометил)-1,3,4-тиадиазол-2-ил)фенил)пропан-2-ил)-ацетамида
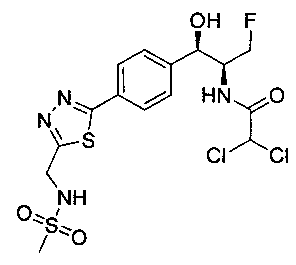
Следуя общей методике стадии 2 Примера 9 и осуществляя незначительные модификации, но используя N-((5-бром-1,3,4-тиадиазол-2-ил)-метил)метан-сульфонамид и 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметил-станнил)фенил)-пропан-2-ил)ацетамид, получили указанное в заголовке соединение (11,8 мг): 1Н ЯМР (400 МГц, DMSO-d6) 3.0 (s, 3H), 4.2-4.4 (m, 1.5Н), 4.45 (t, 1Н), 4.55-4.6 (m, 0.5Н), 4.65 (d, 2H), 4.7-4.75 (m, 0.5H), 4.95 (bt, 0.5H), 6.1 (d, 1H), 6.5 (s, 1H), 7.5 (d, 2H), 7.95 (d, 2H), 8.15 (t, 1H), 8.6 (d, 1H). m/z (Cl) M+H 471.
Пример 11. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(1-(метилсульфонамидо)этил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение (R,S)-N-(1-(5-бромпиридин-2-ил)этил)метан-сульфонамида
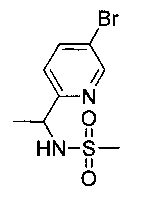
Следуя общей методике Примера 3 и осуществляя незначительные модификации, но используя имеющийся в продаже (R,S)-1-(5-бромпиридин-2-ил), получили указанное в заголовке соединение (570 мг): 1Н ЯМР (400 МГц, DMSO-d6) 1-55 (t, 3H), 2.8 (s, 3H), 4.7 (pent, 1Н), 5.6 (d, 1Н), 7.2 (d, 1Н), 7.85 (dd, 1Н), 8.65 (d, 1Н). m/z (Cl) М+Н 279+281.
Стадия 2. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(1-(метилсульфонамидо)этил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
Следуя общей методике стадии 2 Примера 9 и осуществляя незначительные модификации, но используя (R,S)-N-(1-(5-бромпиридин-2-ил)этил) и 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)-фенил)-пропан-2-ил)-ацетамид, получили указанное в заголовке соединение (17 мг): 1Н ЯМР (400 МГц, DMSO-d6) 1.5 (t, 3H), 2.8 (s, 3H), 4.15-4.35 (m, 1.5Н), 4.4 (t, 0.5Н), 4.55-4.65 (m, 1.5H), 4.65-4.75 (m, 0.5H), 4.9 (t, 1H), 6.0 (d, 1H), 6.5 (s, 1H), 7.45 (d, 2H), 7.55 (d, 1H), 7.7 (d, 2H), 8.1 (d, 1H), 8.6 (dd, 1H), 8.8 (dd, 1H). m/z (Cl) M+H 478+480.
Пример 12. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-(метилсульфонамидометил)пиримидин-5-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение (4S,5R)-трет-бутил-4-(фторметил)-5-(4-йодфенил)-2,2-диметилоксазолидин-3-карбоксилата
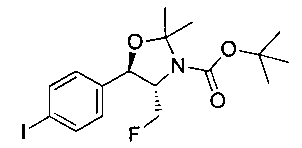
В круглодонную колбу емкостью 1 л, содержащую трет-бутил-((1R,2S)-3-фтор-1-гидрокси-1-(4-йодфенил)пропан-2-ил)карбамат (24,4 г, 61,8 ммоль), загружали CH2Cl2 (250 мл), а затем 2-метоксипропен (9,0 мл, 92,8 ммоль). Раствор охлаждали на ледяной бане и добавляли пара-толуолсульфоновую кислоту (59 мг, 0,31 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, затем гасили насыщенным раствором бикарбоната натрия (200 мл). Органическую фазу отделяли, сушили над MgSO4 и концентрировали с получением указанного в заголовке соединения (26,5 г): 1Н ЯМР (400 МГц, CDCl2) 7.73 (d, 2Н), 7.20 (d, 2Н), 5.08 (d, 1Н), 5.03-4.69 (m, 1Н), 4.54-4.31 (m, 1Н), 3.91-3.68 (m, 1Н), 1.70 (s, 3H), 1.58 (s, 3H), 1.50 (s, 9Н).
Стадия 2. Получение (4S,5R)-трет-бутил-4-(фторметил)-2,2-диметил-5-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксазолидин-3-карбоксилата
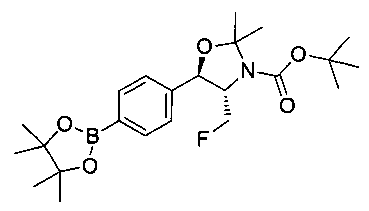
К раствору продукта стадии 1 Примера 12 (13,9 г, 32,0 ммоль) в диоксане (160 мл) добавляли последовательно бис(пинаколато)диборан (9,1 г, 35,2 ммоль), хлорид бис(трифенил-фосфин)палладия(II) (454 мг, 0,64 ммоль) и ацетат калия (9,6 г, 95,9 ммоль). Объединенную смесь нагревали до температуры дефлегмации и перемешивали в течение ночи. После охлаждения до комнатной температуры реакционную смесь распределяли между водой и этилацетатом. Органическую фазу отделяли, сушили над MgSO4, фильтровали и упаривали с получением смолы, которую очищали с использованием колоночной хроматографии, элюируя от чистого гептана до чистого этилацетата с получением указанного в заголовке соединения (8,92 г): 1Н ЯМР (400 МГц, CDCl3) 7.85 (d, 2Н), 7.45 (d, 2Н), 5.15 (d, 1Н), 4.52-4.37 (m, 1Н), 3.95-3.75 (m, 1Н), 1.72 (brs, 3H), 1.60 (brs, 3H), 1.51 (brs, 9Н), 1.37 (brs, 12Н).
Стадия 3. Получение трет-бутилового эфира 5-[4-(2-циано-пиримидин-5-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты
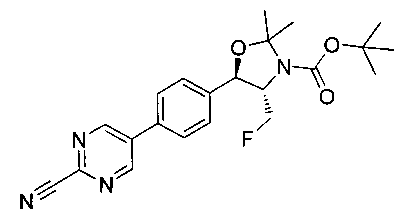
К раствору трет-бутилового эфира 4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-карбоновой кислоты (1 г, 2,298 ммоль, 1,0 экв.) в смеси толуол/вода (3:1, 24 мл) добавляли 5-бром-пиримидин-2-карбонитрил (0,42 г, 2,282 ммоль, 1,20 экв.), Na2CO3 (0,48 г, 4,528 ммоль) и дегазировали азотом в течение 15 минут с последующим добавлением тетракис(трифенилфосфин)палладия(0) (0,132 г, 0,114 ммоль) и нагревали реакционную смесь до 80°C в течение 16 часов. Растворитель выпаривали в вакууме с получением неочищенного материала, который очищали колоночной хроматографией, элюируя от 10% этилацетата/гексана с получением указанного в заголовке соединения (0,45 г): 1Н ЯМР (400 МГц, DMSO-d6) 1.43 (s, 9Н), 1.51 (s, 3H), 1.63 (s, 3H), 3.85-3.92 (m, 1Н), 4.47-4.59 (m, 1Н), 4.76-4.96 (m, 1Н), 5.17 (d, 1Н, J=7,16), 7.67 (d, 2Н, J=8,24 Гц), 7.96 (d, 2Н, J=8,24 Гц), 9.40 (s, 2Н). m/z M-Н 410,8.
Стадия 4. Получение трет-бутилового эфира 5-[4-(2-аминометил-пиримидин-5-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты
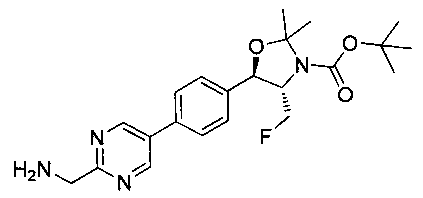
Раствор продукта стадии 3 Примера 12 (0,4 г, 0,97 ммоль, 1,0 экв.) в метаноле (20 мл) дегазировали азотом в течение 15 минут с последующим добавлением 10%-ного палладия на углероде (40 мг, 10% масс/масс.) и выдерживали в атмосфере водорода (1 атм.) при комнатной температуре в течение 16 часов. Растворитель выпаривали в вакууме с получением неочищенного материала, который очищали колоночной хроматографией, элюируя с использованием 10-13%-ного метанола/CH2Cl2 с получением указанного в заголовке соединения (0,22 г): 1Н ЯМР (400 МГц, DMSO-d6) 1.43 (s, 9Н), 1.51 (s, 3H), 1.63 (s, 3H), 3.85-3.90 (m, 1H), 3.96 (s, 2H), 4.47-4.58 (m, 1H), 4.8-4.9 (m, 2H), 5.14 (d, 1H, J=7,16 Гц), 7.62 (d, 2H, J=8,2 Гц), 7.83 (d, 2H, J=8,08 Гц), 9.11 (s, 2H). m/z M+H 417,1.
Стадия 5. Получение трет-бутилового эфира 4-фторметил-5-{4-[2-(метансульфониламино-метил)-пиримидин-5-ил]-фенил}-2,2-диметил-оксазолидин-3-карбоновой кислоты
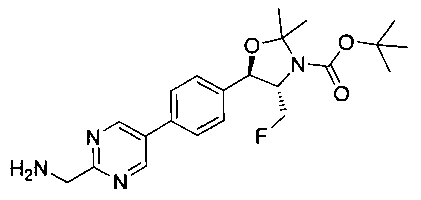
Раствор продукта стадии 4 Примера 12 (0,1 г, 0,240 ммоль, 1,0 экв.) в CH2Cl2 (10 мл) охлаждали до 0°C и добавляли триэтиламин (0,048 г, 0,48 ммоль) с последующим добавлением мезилхлорида (0,041 г, 0,36 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали в вакууме с получением неочищенного материала, который очищали колоночной хроматографией, элюируя из 30%-ного этилацетата/гексана с получением указанного в заголовке соединения (80 мг). m/z М+Н 495,1.
Стадия 6. Получение N-{5-[4-(2-амино-3-фтор-1-гидрокси-пропил)-фенил]-пиримидин-2-илметил}-метансульфонамида
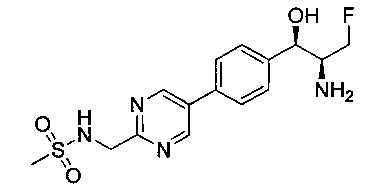
К раствору продукта стадии 5 Примера 12 (0,08 г, 0,16 ммоль, 1,0 экв.) в CH2Cl2 (2 мл) добавляли трифторуксусную кислоту (0,3 мл, 3,91 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали в вакууме с получением неочищенного указанного в заголовке соединения (60 мг). m/z М+Н 354,9.
Стадия 7. Получение 2,2-дихлор-N-(1-фторметил-2-гидрокси-2-{4-[2-(метансульфониламино-метил)-пиримидин-5-ил]-фенил}-этил)-ацетамида
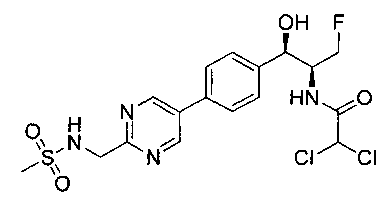
К раствору продукта стадии 6 Примера 12 (0,06 г, 0,169 ммоль, 1,0 экв.) в метаноле (1,1 мл) добавляли триэтиламин (0,034 г, 0,33 ммоль, 2,0 экв.) и этилдихлорацетат (0,053 г, 0,33 ммоль, 2,0 экв.) и перемешивали реакционную смесь при комнатной температуре в течение 24 часов. Растворитель выпаривали в вакууме с получением неочищенного материала, который очищали колоночной хроматографией, элюируя из 5%-ного метанола/CH2Cl2 с получением указанного в заголовке соединения (0,012 г). 1Н ЯМР (400 МГц, DMSO-d6) 2.97 (s, 3H), 4.28-4.30 (m, 1Н), 4.32-4.36 (m, 0.5Н), 4.43 (s, 2Н), 4.56-4.60 (m, 0.5Н), 4.68-4.71 (m, 0.5H), 4.92 (m, 1H), 6.03 (d, 1H, J=4,16 Гц), 6.52 (s, 1H), 7.50 (d, 2H, J=8,16 Гц), 7.68 (t, 1H, J=5,76 Гц), 7.78 (d, 2H, J=8,24 Гц), 8.65 (d, 1H, J=8,72 Гц), 9.13 (s, 2H). m/z M+H 464,8.
Пример 13. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(((R)-5-метил-2-оксооксазолидин-3-ил)метил)пиридин-3-ил)фенил)пропан-2-ил)-ацетамида
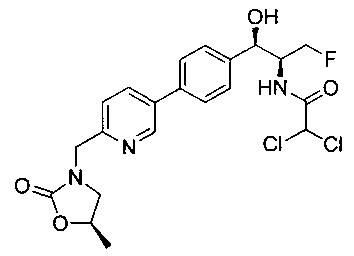
Следуя общей методике стадии 2 Примера 9 и осуществляя незначительные модификации, но используя (R)-3-(5-бром-пиридин-2-илметил)-5-метил-оксазолидин-2-он и 2,2-дихлор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-триметил-станнанил-фенил)-этил]-ацетамид, получили указанное в заголовке соединение (21 мг): 1Н ЯМР (400 МГц, DMSO-d6) 1.33 (d, 3H, J=6,24 Гц), 3.13-3.17 (m, 1Н), 3.69 (t, 1Н, J=8,4 Гц), 4.21-4.23 (m, 1.5Н), 4.27-4.29 (m, 1Н), 4.39-4.43 (m, 1Н), 4.48 (d, 2Н, J=4,96 Гц), 4.56-4.58 (m, 1Н), 4.68-4.70 (m, 2Н), 4.90 (m, 1Н), 6.0 (s, 1Н), 6.5 (s, 1Н), 7.39 (d, 1Н, J=8,04 Гц), 7.47 (d, 2Н, J=8,24 Гц), 7.68 (d, 2Н, J=8,24 Гц), 8.09 (dd, 1Н, J1=2,32 Гц, J2=8,16 Гц), 8.65 (d, 1Н, J=8,92 Гц), 8.84 (d, 1Н, J=2,12 Гц). LC-Ms (m/z): [M-Н] 468,0.
Пример 14. Получение (1R,2S)-2-(2,2-дифторацетамидо)-3-фтор-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропил-фторгидрофосфата
Стадия 1. Получение (1R,2S)-2-(2,2-дифторацетамидо)-3-фтор-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропилдифенилфосфата
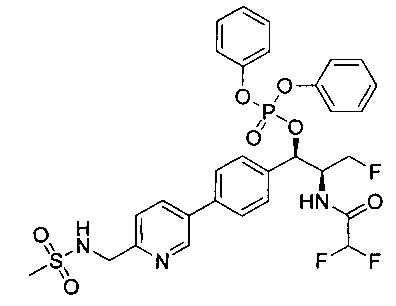
В суспензию продукта стадии 4 Примера 16 (1,0 г, 2,3 ммоль) в пиридине (2 мл) и CHCl3 (2 мл) загружали диметиламинопиридин (215 мг, 1,74 ммоль). Суспензию охлаждали, используя ледяную баню, и добавляли по каплям дифенилхлорфосфонат (1,04 мл, 4,87 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2,5 часов. Реакционную смесь разбавляли CH2Cl2 и вливали в насыщенный бикарбонат натрия (10 мл). Органическую фазу отделяли, а водную фазу подвергали обратной экстракции в CH2Cl2. Органические фазы объединяли, промывали лимонной кислотой, сушили над MgSO4, фильтровали и упаривали с получением указанного в заголовке соединения (1,57 г): 1Н ЯМР (600 МГц, CDCl3) δ: 2.95 (3H, s), 4.10-4.30 (1Н, dd), 4.38-4.70 (4Н, m), 5.62-5.83 (1Н, t), 5.76-5.85 (1Н, m), 5.95 (1Н, bs), 6.97 (2Н, d), 7.70-7.26 (7Н, m), 7.27-7.33 (1Н, m), 7.37 (2Н, t), 7.45-7.59 (4Н, m), 7.88-8.07 (1Н, dd), 8.68-8.83 (1Н, m). m/z М+Н 664,2.
Стадия 2. Получение (1R,2S)-2-(2,2-дифторацетамидо)-3-фтор-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропил-фторгидрофосфата
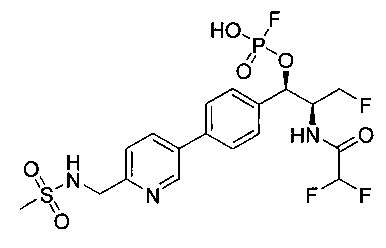
К раствору продукта стадии 1 Примера 14 (200 мг, 0,3 ммоль) в тетрагидрофуране (1,2 мл) добавляли фосфат тетрабутиламмония (102 мг, 0,39 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Растворитель выпаривали и остаток очищали хроматографией с обращенной фазой с получением указанного в заголовке соединения (19 мг): 1Н ЯМР (600 МГц, DMSO-d6) δ: 2.95 (3H, s), 4.25-4.46 (4Н, m), 4.58-4.74 (1Н, m), 5.34-5.52 (1Н, m), 5.97-6.35 (1Н, t), 7.43-7.52 (2Н, m), 7.54-7.66 (1Н, d), 7.69-7.80 (3H, m), 8.10-8.28 (1Н, m), 8.83-8.90 (1Н, m), 9.15-9.24 (1Н, m). m/zM+1 514,1.
Пример 15. Получение 2,2,2-трифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
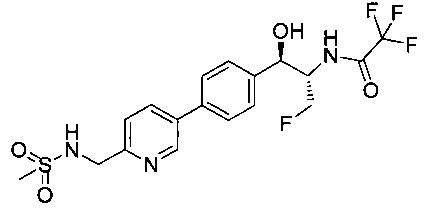
Следуя общей методике Примера 14 и осуществляя незначительные модификации, но используя трифторуксусный ангидрид на стадии 2, получили указанное в заголовке соединение: 1Н ЯМР (400 МГц, DMSO-d6) 2.95 (s, 3H), 4.30-4.45 (m, 1.5Н), 4.32 (m, 2Н), 4.50-4.60 (m, 1.5Н), 4.65-4.70 (m, 1H), 4.89 (m, 1H), 5.85 (m, 1H), 7.46 (d, 2H, J=8,08 Гц), 7.55 (d, 1H, J=8,08 Гц), 7.65-7.75 (m, 3H), 8.11 (m, 1H), 8.84 (s, 1H), 9.48 (m, 1H). m/z (Cl) M+H 450.
Пример 16. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение N-((5-бромпиридин-2-ил)метил)-метансульфонамида
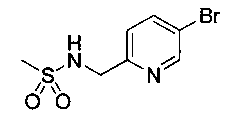
Следуя общей методике Примера 3 и осуществляя незначительные модификации, но используя имеющийся в продаже (5-бромпиридин-2-ил)-метанамин, получили указанное в заголовке соединение (1,95 г): m/z (Cl) М+Н 266.
Стадия 2. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-йодфенил)-пропан-2-ил)ацетамида
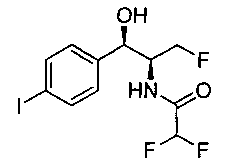
Следуя общей методике стадии 2 Примера 1 и осуществляя незначительные модификации, но используя этилдифторацетан и (1R,2S)-2-амино-3-фтор-1-(4-йодфенил)пропан-1-ол, получили указанное в заголовке соединение (18,3 г): 1Н ЯМР (400 МГц, CDCl3) 7.72 (2Н, d), 7.13 (2Н, d), 6.78 (1Н, d), 5.85 (1Н, t), 5.06 (1Н, s), 4.67-4.28 (3H, m), 2.58 (1Н, s).
Стадия 3. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)фенил)пропан-2-ил)ацетамида
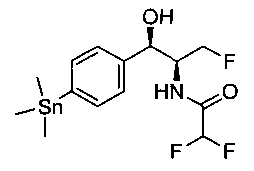
Гексаметилдиолово (9,9 г, 29,9 ммоль) добавляли к дезоксигенированному раствору продукта стадии 2 Примера 17 (10,6 г, 28,5 ммоль), дихлорбис(трифенил-фосфин)палладию (490 мг, 0,68 ммоль) в диоксане (143 мл) и смесь нагревали до 80°C в течение 1 часа. После охлаждения до комнатной температуры смесь очищали с использованием колоночной хроматографии, элюируя от чистых гептанов до чистого этилацетата с получением указанного в заголовке соединения (9,3 г): 1Н ЯМР (400 МГц, CDCl3) 7.27 (2Н, d), 7.09 (2Н, d), 6.59 (1Н, d), 5.62 (1Н, t), 4.81-4.79 (1Н, t), 4.44-4.08 (3H, m), 2.20 (1Н, d), 0.14-0.00 (9Н, m).
Стадия 4. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
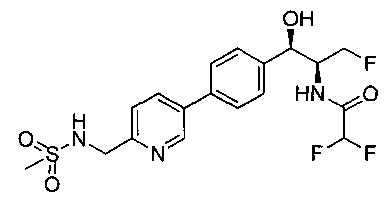
Следуя общей методике стадии 2 Примера 9 и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 17 и стадии 3 Примера 17, получили указанное в заголовке соединение (200 мг): 1Н ЯМР (400 МГц, DMSO-d6) 8.86-8.83 (2Н, m), 8.13 (1Н, d), 7.74-7.69 (3H, m), 7.55 (1Н, d), 7.49 (2Н, d), 6.23 (1Н, t), 5.91 (1Н, d), 4.91 (1Н, t), 4.70-4.56 (1Н, m), 4.47-4.29 (4Н, m), 2.97 (3H, s). m/z (Cl) М+Н 431.
Пример 17. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-(4-(5-фтор-6-(метилсульфонамидометил)пиридин-3-ил)фенил)-1-гидроксипропан-2-ил)-ацетамида
Стадия 1. Получение (4S,5R)-трет-бутил-5-(4-(6-циано-5-фторпиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилата
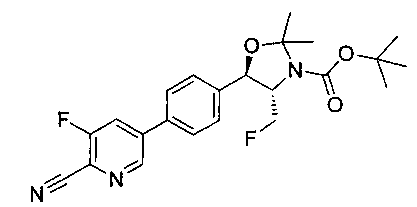
К раствору имеющегося в продаже 5-бром-3-фторпиколинонитрила (800 мг, 3,98 ммоль) в смеси 1,4-диоксан: вода (32:8 мл) добавляли продукт стадии 2 Примера 12 (1730 мг, 3,98 ммоль) и Cs2CO3 (2800 мг, 8,6 ммоль) и полученный раствор барботировали газообразным азотом в течение 30 минут. К этой реакционной смеси добавляли Pd(PPh3)4 (460 мг, 0,4 ммоль) и полученную реакционную смесь нагревали до 90°C в течение 3 часов. Полученную реакционную смесь охлаждали, разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия и концентрировали и очищали с использованием колоночной хроматографии на силикагеле, элюируя этилацетатом в гептане с получением указанного в заголовке соединения: 1Н ЯМР (400 МГц, CDCl3) 1.52 (s, 9Н), 1.63 (s, 3H), 1.75 (s, 3H), 3.80-4.00 (m, 1Н), 4.40-4.60 (m, 1Н), 4.7-5.2 (m, 1Н), 5.22 (d, 1Н, J=7,33 Гц), 7.65 (s, 4Н), 7.78 (d, 2Н, J=9,35 Гц), 8.80 (s, 1Н). m/z (Cl) М+Н 430.
Стадия 2. Получение (4S,5R)-трет-бутил-5-(4-(5-фтор-6-(метил-сульфонамидо-метил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметил-оксазолидин-3-карбоксилата
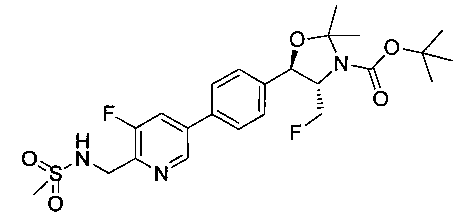
(4S,5R)-трет-Бутил-5-(4-(6-циано-5-фторпиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилат (1440 мг, 3,4 ммоль) в метаноле (25 мл) при 5°C обрабатывали NiCl2 (80 мг, 0,34 ммоль) и затем добавляли порциями борогидрид натрия (380 мг, 10 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре, концентрировали для удаления метанола, гасили водным бикарбонатом натрия и экстрагировали этилацетатом. Органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением масла. Это масло растворяли в дихлорметане (5 мл), охлаждали до 5°C и обрабатывали диизопропилэтиламином (0,88 мл, 5,0 ммоль) и метансульфонилхлоридом (0,30 мл, 3,7 ммоль) и перемешивали в течение 2 часов при комнатной температуре. Смесь концентрировали и подвергали хроматографии на диоксиде кремния, элюируя этилацетатом в гептане с получением указанного в заголовке соединения. 1Н ЯМР (400 МГц, CDCl3) 1.53 (s, 9Н), 1.63 (s, 3H), 1.75 (s, 3H), 3.80-4.00 (m, 1Н), 4.40-4.50 (m, 0.5Н), 4.55-4.65 (m, 2.5Н), 5.22 (d, 1H, J=7,33 Гц), 5.72 (bs, 1H), 7.60-7.70 (m, 5H), 8.63 (s, 1H). m/z (Cl) M+H 512.
Стадия 3. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-(4-(5-фтор-6-(метилсульфонамидометил)пиридин-3-ил)фенил)-1-гидроксипропан-2-ил)-ацетамида
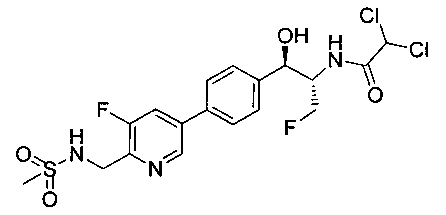
(4S,5R)-трет-Бутил-5-(4-(5-фтор-6-(метилсульфонамидометил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилат (890 мг, 1,74 ммоль) растворяли в CH2Cl2 (20 мл), охлаждали до 5°C и обрабатывали трифторуксусной кислотой (4 мл), перемешивали в течение 1 ч при комнатной температуре, разбавляли толуолом, концентрировали с получением масла. Это масло подщелачивали водным бикарбонатом натрия, экстрагировали этилацетатом, сушили, фильтровали, концентрировали с получением масла. Это масло растворяли в метаноле (8 мл) и обрабатывали диизопропилэтиламином (0,455 мл, 2,6 ммоль) и метилдихлорацетатом (305 мг, 2,1 ммоль), перемешивали при 60°C в течение 18 часов, затем охлаждали и подвергали хроматографии на диоксиде кремния, элюируя этилацетатом в гептане с получением указанного в заголовке соединения. 1Н ЯМР (400 МГц, DMSO-d6) 2.94 (s, 3H), 4.15-4.45 (m, 4Н), 4.55-4.75 (m, 1Н), 4.92 (t, 1Н, J=3,54 Гц), 6.01 (d, 1Н, J=4,29 Гц), 6.52 (s, 1Н), 7.49 (d, 2Н, J=8,08 Гц), 7.59 (t, 1Н, J=5,94 Гц), 7.77 (d, 2Н, J=8,34 Гц), 8.07 (m, 1Н), 8.63 (d, 2Н, J=8,84 Гц), 8.77 (s, 1Н). m/z (Cl) M+H 465.
Пример 18. Получение 2,2-дихлор-N-((1R,2S)-3-хлор-1-(4-(5-фтор-6-(метилсульфонамидометил)пиридин-3-ил)фенил)-1-гидроксипропан-2-ил)-ацетамида
Стадия 1. Получение (4S,5R) трет-бутил-5-(4-(6-циано-5-хлорпиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилата
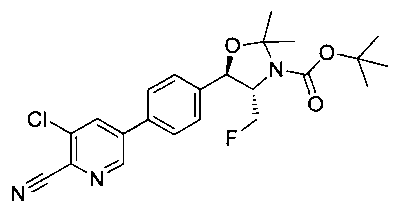
Следуя общей методике стадии 1 Примера 17 и осуществляя незначительные модификации, но используя продукт стадии 2 Примера 12, и имеющийся в продаже 5-бром-3-хлорпиколинонитрил, получили указанное в заголовке соединение. 1Н ЯМР (400 МГц, CDCl3) δ: 1.52 (s, 9Н), 1.63 (s, 3H), 1.75 (s, 3H), 3.80-4.00 (m, 1Н), 4.40-4.60 (m, 1Н), 4.7-5.2 (m, 1Н), 5.22 (d, 1Н, J=7,33 Гц), 7.64 (s, 4Н), 8.04 (d, 2Н, J=2,02 Гц), 8.84 (d, 2Н, J=2,02 Гц), m/z (Cl) М+Н 446.
Стадия 2. Получение (4S,5R)-трет-бутил-5-(4-(5-хлор-6-(метил-сульфонамидо-метил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметил-оксазолидин-3-карбоксилата
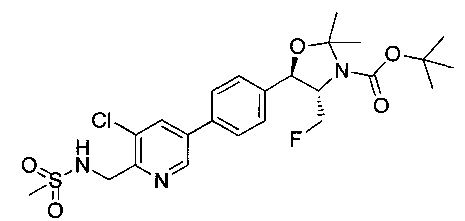
Следуя общей методике стадии 2 Примера 17 и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 19, получили указанное в заголовке соединение. 1Н ЯМР (400 МГц, CDCl3) δ: 1.53 (s, 9Н), 1.63 (s, 3H), 1.75 (s, 3H), 3.02 (s, 3H), 3.80-4.00 (m, 1Н), 4.40-4.50 (m, 0.5Н), 4.55-4.65 (m, 2.5Н), 5.21 (d, 1Н, J=7,33 Гц), 5.97 (bs, 1Н), 7.61 (s, 5Н), 7.61 (s, 1Н), 8.70 (s, 1Н). m/z (Cl) М+Н 528.
Стадия 3. Получение 2,2-дихлор-N-((1R,2S)-1-(4-(5-хлор-6-(метилсульфонамидометил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-ацетамида
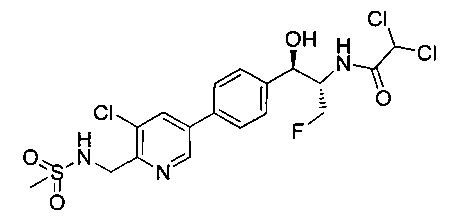
Следуя общей методике стадии 3 Примера 17 и осуществляя незначительные модификации, но используя продукт стадии 2 Примера 19, получили указанное в заголовке соединение: 1Н ЯМР (400 МГц, DMSO-d6) 2.96 (s, 3H), 4.15-4.35 (m, 1.5Н), 4.35-4.55 (m, 2.5Н), 4.55-4.75 (m, 1H), 4.92 (7, 1H, J=3,41 Гц), 6.01 (d, 1Н, J=4,04 Гц), 6.52 (m, 1Н), 6.53 (s, 1Н), 7.45-7.60 (m, 3H), 7.77 (d, 1Н, J=8,34 Гц), 8.26 (d, 2Н, J=1,77 Гц), 8.63 (d, 2Н, J=8,84 Гц), 8.77 (d, 1Н, J=1,77 Гц), m/z (Cl) M+H 498.
Пример 19. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(5-(метилсульфонамидометил)пиразин-2-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение (4S,5R)-трет-бутил-5-(4-(5-цианопиразин-2-ил)-фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилата
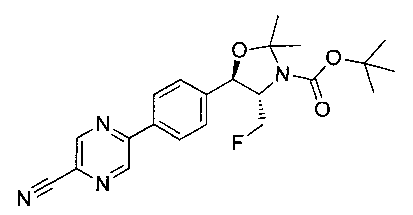
Следуя общей методике стадии 1 Примера 17 и осуществляя незначительные модификации, но используя продукт стадии 2 Примера 12, и имеющийся в продаже 5-бромпиразин-2-карбонитрил, получили указанное в заголовке соединение: 1Н ЯМР (400 МГц, CDCl3) 1.52 (s, 9Н), 1.63 (s, 3H), 1.75 (s, 3H), 3.80-4.00 (m, 1Н), 4.40-4.60 (m, 1Н), 5.23 (d, 1Н, J=7,33 Гц), 7.66 (d, 2Н, J=8,34 Гц), 8.14 (d, 2Н, J=8,34 Гц), 8.97 (d, 1Н), 9.16 (s, 1Н). m/z (Cl) М+Н 413.
Стадия 2. Получение (4S,5R)-трет-бутил-4-(фторметил)-2,2-диметил-5-(4-(5-(метилсульфонамидометил)пиразин-2-ил)фенил)оксазолидин-3-карбоксилата
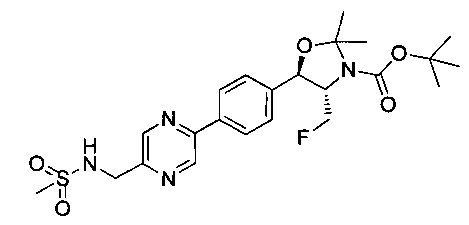
Следуя общей методике стадии 2 Примера 17 и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 20, получили указанное в заголовке соединение: 1Н ЯМР (400 МГц, CDCl3) 1.52 (s, 9Н), 1.63 (s, 3H), 1.75 (s, 3H), 3.01 (s, 3H), 3.80-4.00 (m, 1Н), 4.40-4.50 (m, 0.5Н), 4.50-4.60 (m, 2.5Н), 5.21 (d, 1Н, J=7,33 Гц), 5.61 (m, 1Н), 7.61 (d, 2Н, J=8,34 Гц), 8.05 (d, 2Н, J=8,34 Гц), 8.69 (s, 1Н), 98.98 (s, 1Н). m/z (Cl) М+Н 495.
Стадия 3. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(5-(метилсульфонамидометил)пиразин-2-ил)фенил)пропан-2-ил)ацетамида
Следуя общей методике стадии 3 Примера 17 и осуществляя незначительные модификации, но используя продукт стадии 2 Примера 20, получили указанное в заголовке соединение: 1Н ЯМР (400 МГц, DMSO-d6) 2.99 (s, 3H), 4.20-4.50 (m, 4Н), 4.50-4.75 (m, 1Н), 4.93 (t, 1Н, J=3,28 Гц), 6.04 (d, 1Н, J=4,29 Гц), 6.52 (s, 1Н), 7.51 (d, 1Н, J=8,34 Гц), 7.76 (t, 2Н, J=6,06 Гц), 8.10 (d, 2Н, J=8,34 Гц), 8.62 (d, 2Н, J=8,84 Гц), 8.73 (s, 1Н), 9.19 (s, 1Н). m/z (Cl) M-Н 483.
Пример 20. Получение N-((1R,2S)-1-(4-(6-(азетидин-1-илметил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение 2,2-дихлор-1-((4S,5R)-4-(фторметил)-5-(4-(6-(гидроксиметил)пиридин-3-ил)фенил)-2,2-диметилоксазолидин-3-ил)этанона
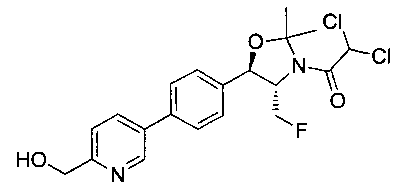
Следуя общей методике стадии 1 Примера 1 и осуществляя незначительные модификации, но используя 2,2-дихлор-1-((4S,5R)-4-(фторметил)-5-(4-йодфенил)-2,2-диметилоксазолидин-3-ил)этанон и (6-(гидроксиметил)пиридин-3-ил)бороновую кислоту в качестве исходных материалов, получили указанное в заголовке соединение (800 мг): m/z (Cl) M-Н 426,1.
Стадия 2. Получение (5-(4-((4S,5R)-3-(2,2-дихлорацетил)-4-(фтор-метил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)метил-метансульфоната
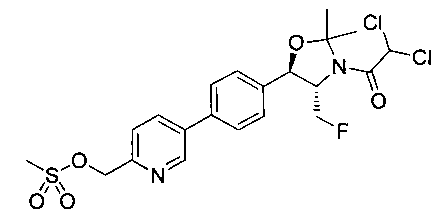
Следуя общей методике Примера 3 и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 21, получили указанное в заголовке соединение (940 мг): m/z (Cl) M-Н 504,1.
Стадия 3. Получение N-((1R,2S)-1-(4-(6-(азетидин-1-илметил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
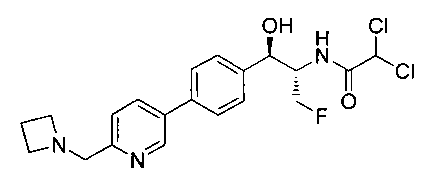
Диизопропилэтиламин (296 мкл, 1,68 ммоль) добавляли к продукту стадии 2 Примера 20 (50 мг, 0,11 ммоль) и HCl соли азетидина (146 мг, 1,12 ммоль) в диметилформамиде (1 мл) и эту смесь нагревали до 60°C в течение 4 часов. Растворитель удаляли при пониженном давлении и остаток растворяли в CH2Cl2 (2 мл). Смесь охлаждали до 0°C и добавляли трифторуксусную кислоту (2 мл). Через 1 час растворитель удаляли при пониженном давлении и неочищенный продукт очищали с использованием ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения (10 мг): m/z (Cl) M-Н 425,1.
Пример 21. Получение 2,2-дихлор-N-((1R,2S)-1-(4-(6-(этилсульфонамидо-метил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)ацетамида
Стадия 1. Получение N-((5-бромпиридин-2-ил)метил)этансульфонамида
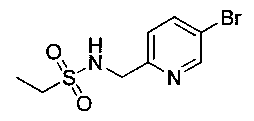
К раствору (5-бромпиридин-2-ил)метанамина (250 мг, 1,34 ммоль) в CH2Cl2 (3 мл) добавляли этилсульфонилхлорид (95 мг, 0,74 ммоль) с последующим добавлением триэтиламина (0,3 мл, 2,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь сразу же инжектировали на колонку силикагеля 12g и подвергали хроматографии, элюируя от 100%-ных гексанов до смеси 50:50 этилацетатгексаны с получением указанного в заголовке соединения (158 мг): m/z (Cl) М+Н 279+281.
Стадия 2. Получение N-((5-(4-((4S,5R)-3-(2,2-дихлорацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)метил)-этансульфонамида
2,2-Дихлор-1-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(триметилстаннил)-фенил)-оксазолидин-3-ил)этанон (281 мг, 0,66 ммоль) и N-((5-бромпиридин-2-ил)метил)этансульфонамид (155 мг, 0,56 ммоль) растворяли в N-метил-2-пирролидоне (40 мл) и продували газообразным азотом. Добавляли Pd2(dba)3 (51 мг, 0,055 ммоль) и три-2-фурилфосфин (27 мг, 0,11 ммоль); полученную смесь нагревали до 80°C в течение 18 часов. Реакционную смесь охлаждали, разбавляли этилацетатом и промывали водой. Органическую фазу сушили (Na2SO4) и концентрировали под вакуумом. Неочищенный материал подвергали хроматографии (колонка 24g Redi-Sep), элюируя от 100%-ных гексанов до смеси 90:10 этилацетат: гексаны с получением указанного в заголовке соединения (125 мг): m/z (Cl) М+Н 518.
Стадия 3. Получение 2,2-дихлор-N-((1R,2S)-1-(4-(6-(этилсульфон-амидометил)пиридинг3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)ацетамида
Следуя общей методике стадии 2 Примера 2 и осуществляя незначительные модификации, но используя продукт стадии 2 Примера 21, получили указанное в заголовке соединение (23 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.19 (t, 3H), 3.04, (q, 2Н), 4.22-4.32 (m, 3.5Н), 4.40-4.44 (m, 0.5Н), 4.56-4.60 (m, 0.5H), 4.68-4.72 (m, 0.5H), 4.91 (t, 1H), 5.98 (d, 1H), 6.53 (s, 1H), 7.46-7.54 (m, 3H), 7.68-7.73 (m, 3H), 8.10 (dd, 1H), 8.62 (d, 1H), 8.82 (d, 1H); m/z (Cl) 478 [M+H].
Пример 22. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((2-метилпропилсульфонамидо)-етил)пиридин-3-ил)фенил)пропан-2-ил)ацетамид
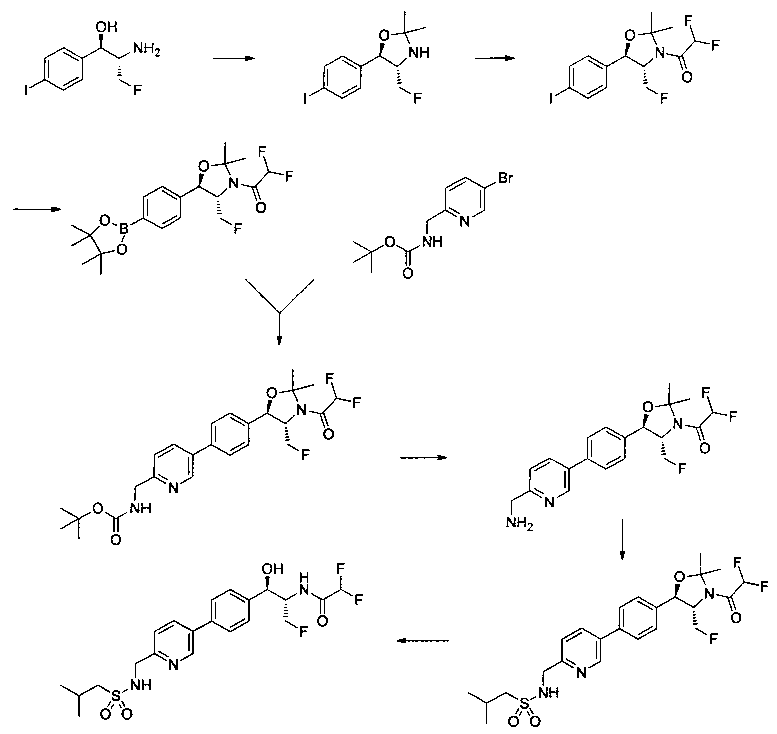
Стадия 1. Получение (4S,5R)-4-(фторметил)-5-(4-йодфенил)-2,2-диметилоксазолидина
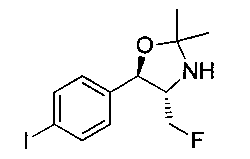
Ацетон (150 мл) добавляли к имеющемуся в продаже (1R,2S)-2-амино-3-фтор-1-(4-йодфенил)пропан-1-олу (15,0 г, 50,8 ммоль). После перемешивания в течение ночи при комнатной температуре растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения (17,6 г): m/z (Cl) М+Н 335.
Стадия 2. Получение 2,2-дифтор-1-((4S,5R)-4-(фторметил)-5-(4-йодфенил)-2,2-диметилоксазолидин-3-ил)этанона
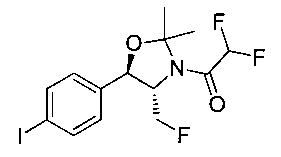
Перемешиваемый раствор продукта стадии 2 Примера 8 (3,0 г, 8,9 ммоль) в CH2Cl2 (50 мл) при 0°C добавляли в триэтиламин (6,2 мл, 44,8 ммоль) с последующим добавлением по каплям дифторацетилхлорида (2,2 мл, 27,0 ммоль). Реакционную смесь медленно оставляли нагреваться до комнатной температуры. Через 1 час реакционную смесь разбавляли водой (75 мл) и экстрагировали CH2Cl2 (2×75 мл). Объединенную органическую фазу сушили над MgSO4 и концентрировали под вакуумом. Неочищенный материал подвергали хроматографии (колонка 80g Redi-Sep), элюируя от 100%-ных гексанов до смеси 25:75 EtOAc: гексаны с получением указанного в заголовке соединения (3,54 г): m/z (Cl) М+Н 413,0.
Стадия 3. Получение 2,2-дифтор-1-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксазолидин-3-ил)-этанона
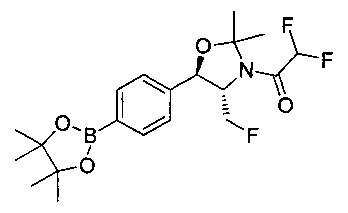
К раствору продукта стадии 3 Примера 8 (3,5 г, 8,4 ммоль) в диоксане (100 мл) добавляли бис(пинаколато)дибор (2,4 г, 9,3 ммоль), ацетат калия (2,5 г, 25,4 ммоль) и Pd(PPh3)2Cl2 (300 мг, 0,4 ммоль). Реакционную смесь нагревали до 90°C под азотом в течение 22 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали под вакуумом для удаления диоксана до объема примерно 50 мл. Остаток разбавляли водой (150 мл) и экстрагировали CH2Cl2 (2×125 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали под вакуумом. Неочищенный материал очищали хроматографией (колонка 120g Redi-Sep), элюируя от 100%-ных гексанов до смеси 25:75 EtOAc: гексаны с получением указанного в заголовке соединения (2,06 г): m/z (Cl) М+Н 413,2.
Стадия 4. Получение трет-бутил-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)-метилкарбамата
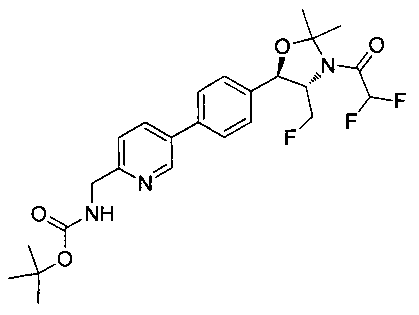
К раствору продукта стадии 3 Примера 22 (1000 мг, 2,4 ммоль) в смеси толуол/этанол (30:20 мл) добавляли имеющийся в продаже трет-бутил-(5-бромпиридин-2-ил)метилкарбамат (695 мг, 2,42 ммоль), бикарбонат натрия (5 мл насыщенного раствора) и Pd(dppf)2Cl2 (90 мг, 0,12 ммоль). Реакционную смесь нагревали до 80°C при перемешивании под азотом в течение 4 часов. Реакционную смесь охлаждали и разбавляли водой. Содержимое экстрагировали этилацетатом (2×75 мл) и объединенную органическую фазу сушили (Na2SO4) и концентрировали под вакуумом. Неочищенный материал подвергали хроматографии (колонка 80g Redi-Sep), элюируя от 100%-ных гексанов до смеси 90:10 этилацетат: гексаны с получением указанного в заголовке соединения (912 мг): m/z (Cl) М+Н 494.
Стадия 5. Получение 1-((4S,5R)-5-(4-(6-(аминометил)пиридин-3-ил)-фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-ил)-2,2-дифторэтанона
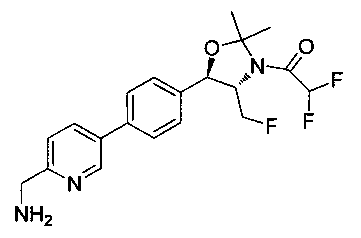
К раствору трет-бутил-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)-метилкарбамата (970 мг, 4,5 ммоль) в CH2Cl2 (25 мл) при 0°C добавляли 2,6-лутидин (460 мкл, 3,9 ммоль) с последующим добавлением трет-бутилдиметилсилилтрифторметансульфоната (700 мкл, 2,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Добавляли дополнительное количество лутидина (230 мкл, 1,9 ммоль) и трет-бутилдиметилсилилтрифторметансульфоната (350 мкл, 1,4 ммоль) и содержимое нагревали до 35°C в течение 1 часа. Добавляли насыщенный NH4Cl и реакционную смесь перемешивали в течение 15 минут. Содержимое разбавляли CH2Cl2 и разделяли. Органическую фазу промывали насыщенным NH4Cl, насыщенным NaHCO3, рассолом, сушили (Na2SO4) и концентрировали под вакуумом. Неочищенный силилкарбамат растворяли в тетрагидрофуране (20 мл) и добавляли фторид тетрабутиламмония (0,6 мл, 2,0 ммоль) и перемешивали в течение 20 минут. Добавляли насыщенный NH4Cl и содержимое экстрагировали этилацетатом. Органическую фазу промывали последовательно насыщенным гидрокарбонатом натрия, рассолом, сушили с помощью Na2SO4 и концентрировали под вакуумом с получением указанного в заголовке соединения (123 мг): m/z (Cl) М+Н 394.
Стадия 6. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((2-метилпропилсульфонамидо)метил)пиридин-3-ил)фенил)пропан-2-ил)-ацетамида
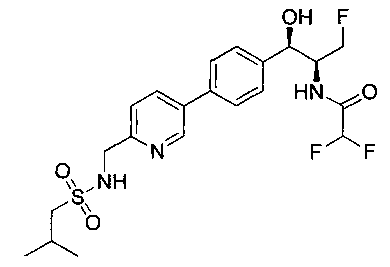
К раствору 1-((4S,5R)-5-(4-(6-(аминометил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-ил)-2,2-дифторэтанона (80 мг, 0,2 ммоль) в CH2Cl2 (15 мл) при 0°C добавляли триэтиламин (80 мкл, 0,5 ммоль) с последующим добавлением изобутилсульфонилхлорида (29 мг, 0,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при комнатной температуре в течение еще 1 часа. Добавляли толуол (20 мл) и реакционную смесь концентрировали под вакуумом. Неочищенную реакционную смесь очищали с использованием хроматографии с обращенной фазой, подщелачивали насыщенным NaHCO3, экстрагировали этилацетатом и концентрировали под вакуумом с получением указанного в заголовке соединения (17 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 0.99 (d, 6Н), 2.05-2.11 (m, 1Н), 2.94 (d, 2Н), 4.28-4.36 (m, 3.5Н), 4.41-4.45 (m, 0.5Н), 4.52-4.58 (m, 0.5Н), 4.65-4.68 (m, 0.5H), 4.89 (m, 1H), 5.90 (d, 1H), 6.21 (t, 1H), 7.47 (d, 2H), 7.54 (d, 1H), 7.69-7.74 (m, 3H), 8.12 (dd, 1H), 8.81-8.83 (m, 2H); m/z (Cl) 474 [М+Н].
Пример 23. Получение метил-(5-(4-((1R,2S)-2-(2,2-дифторацетамидо)-3-фтор-1-гидроксипропил)фенил)пиридин-2-ил)метилкарбамата
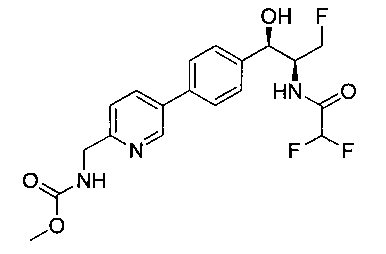
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя метилхлорформиат, получили указанное в заголовке соединение (21 мг): m/z (Cl) М+Н 412.
Пример 24. Получение N-((1R,2S)-1-(4-(6(циклопропансульфонамидо-метил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
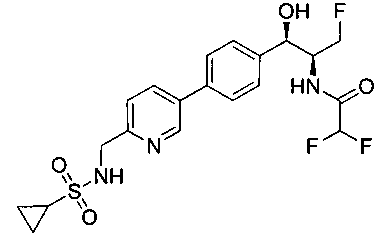
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя циклопропансульфонилхлорид, получили указанное в заголовке соединение (27 мг): m/z (Cl) М+Н 458.
Пример 25. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((2-метоксиэтилсульфонамидо)метил)пиридин-3-ил)фенил)пропан-2-ил)-ацетамид
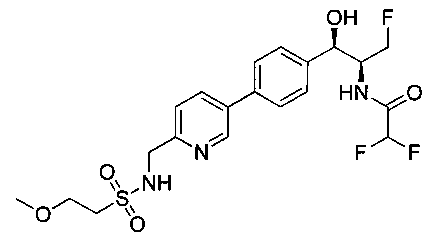
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя 2-метоксиэтансульфонилхлорид, получили указанное в заголовке соединение (27 мг): m/z (Cl) М+Н 476.
Пример 26. Получение N-((5-(4-((1R,2S)-2-(2,2-дифторацетамидо)-3-фтор-1-гидроксипропил)фенил)пиридин-2-ил)метил)циклопропанкарбоксамида
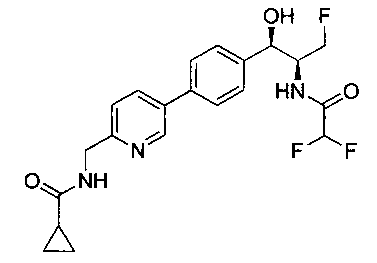
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя циклопропанкарбонилхлорид, получили указанное в заголовке соединение (21 мг): m/z (Cl) М+Н 422.
Пример 27. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((метилсульфонилметилсульфонамидо)метил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
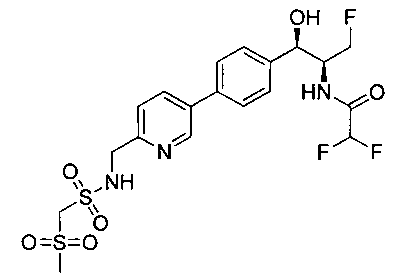
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя метилсульфонилметансульфонил-хлорид, получили указанное в заголовке соединение (14 мг): m/z (Cl) М+Н 510.
Пример 28. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((сульфамоиламино)метил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
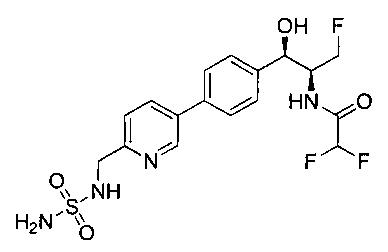
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя сульфамоилхлорид, получили указанное в заголовке соединение (12 мг): m/z (Cl) М+Н 433.
Пример 29. Получение N-((1R,2S)-1-(4-(6-(аминометил)пиридин-3-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
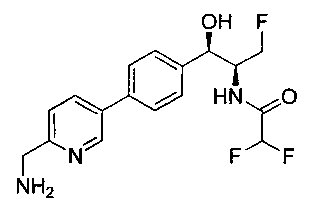
Следуя общей методике Примера 22 и осуществляя незначительные модификации, получили указанное в заголовке соединение (27 мг): m/z (Cl) М+Н 354.
Пример 30. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((2,2,2-трифторэтилсульфонамидо)метил)пиридин-3-ил)фенил)пропан-2-ил)ацетамид
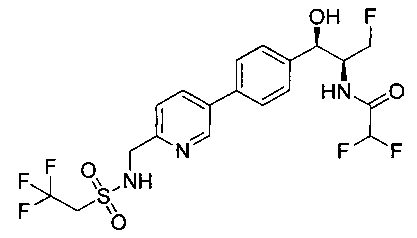
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя 2,2,2-трифторэтансульфонил-хлорид, получили указанное в заголовке соединение (21 мг): m/z (Cl) М+Н 500.
Пример 31. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((трифторметилсульфонамидо)метил)пиридин-3-ил)фенил)пропан-2-ил)-ацетамид
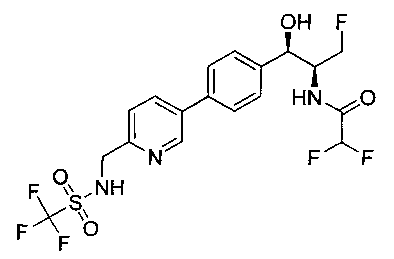
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя трифторметансульфонилхлорид, получили указанное в заголовке соединение (13 мг): m/z (Cl) М+Н 486.
Пример 32. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((тетрагидро-2Н-пиран-4-сульфонамидо)метил)пиридин-3-ил)фенил)пропан-2-ил)ацетамид
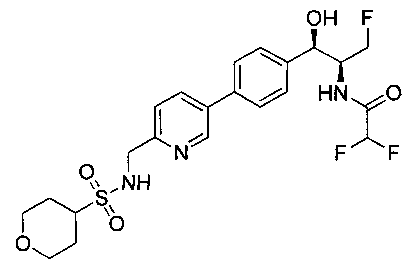
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя тетрагидро-2Н-пиран-4-сульфонилхлорид, получили указанное в заголовке соединение (43 мг): m/z (Cl) М+Н 502.
Пример 33. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((1-метилэтилсульфонамидо)метил)пиридин-3-ил)фенил)пропан-2-ил)ацетамид
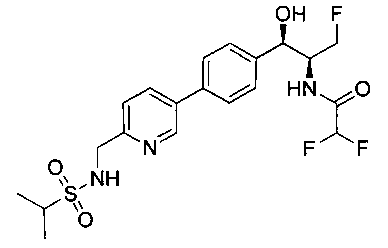
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя пропан-2-сульфонилхлорид, получили указанное в заголовке соединение (11 мг): m/z (Cl) М+Н 460.
Пример 34. N-((1R,2S)-1-(4-(6-((Цианометилсульфонамидо)метил)-пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
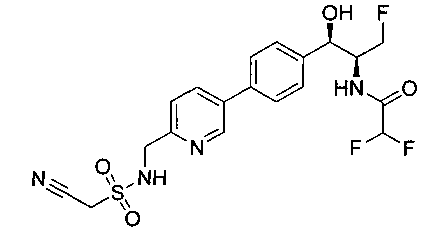
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя цианометансульфонилхлорид, получили указанное в заголовке соединение (24 мг): m/z (Cl) М+Н 457.
Пример 35. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((3-метилуреидо)метил)пиридин-3-ил)фенил)пропан-2-ил)ацетамид
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя метилизоцианат, получили указанное в заголовке соединение (14 мг): m/z (Cl) М+Н 411.
Пример 36. N-((1R,2S)-1-(4-(6-((3-Этилуреидо)метил)пиридин-3-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя этилизоцианат, получили указанное в заголовке соединение (18 мг): m/z (Cl) М+Н 425.
Пример 37. N-((1R,2S)-1-(4-(6-((Дифторметилсульфонамидо)метил)-пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
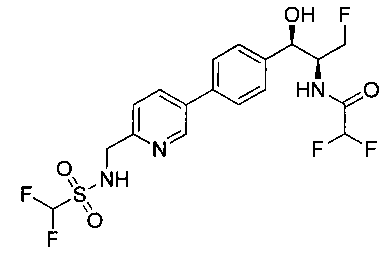
Следуя общей методике стадии 3 Примера 22 и осуществляя незначительные модификации, но используя дифторметансульфонилхлорид, получили указанное в заголовке соединение (6 мг): m/z (Cl) М+Н 468.
Пример 38. Получение N-((1R,2S)-1-(4-(6-(этилсульфонамидометил)-пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
Стадия 1. Получение N-((5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)метил)-этансульфонамида
Следуя общей методике стадии 1 Примера 22 и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 21, получили указанное в заголовке соединение (660 мг): m/z (Cl) М+Н 486.
Стадия 2. N-((1R,2S)-1-(4-(6-(Этилсульфонамидометил)пиридин-3-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
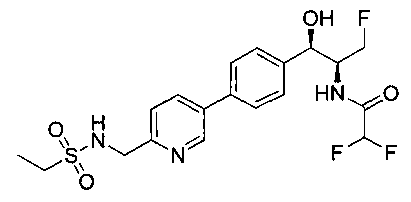
Следуя общей методике стадии 2 Примера 2 и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 38, получили указанное в заголовке соединение (157 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.20 (t, 3H), 3.03 (q, 2Н), 4.28-4.36 (m, 3.5Н), 4.41-4.45 (m, 0.5Н), 4.52-4.58 (m, 0.5H), 4.65-4.68 (m, 0.5H), 4.89 (t, 1H), 5.90 (d, 1H), 6.21 (t, 1H), 7.47 (d, 2H), 7.54 (d, 1H), 7.70-7.73 (m, 3H), 8.11 (dd, 1H), 8.81-8.83 (m, 2H); m/z (Cl) 446 [М+Н].
Пример 39. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-(4-(6-((фтор-метилсульфонамидо)метил)пиридин-3-ил)фенил)-1-гидроксипропан-2-ил)-ацетамида
Стадия 1. Получение N-((5-бромпиридин-2-ил)метил)-1-фторметансульфонамида
Следуя общей методике стадии 1 Примера 21 и осуществляя незначительные модификации, но используя фторметансульфонилхлорид, получили указанное в заголовке соединение (245 мг): m/z (Cl) М+Н 283+285.
Стадия 2. Получение N-((5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)метил)-1-фторметансульфонамида
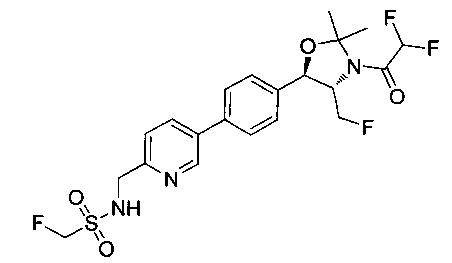
Следуя общей методике стадии 1 Примера 22 и осуществляя незначительные модификации, но используя N-((5-бромпиридин-2-ил)метил)-1-фторметан-сульфонамид, получили указанное в заголовке соединение (175 мг): m/z (Cl) М+Н 490.
Стадия 3. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-(4-(6-((фторметил-сульфонамидо)метил)пиридин-3-ил)фенил)-1-гидроксипропан-2-ил)ацетамид
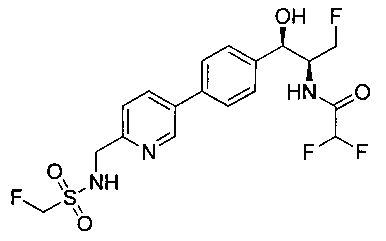
Следуя общей методике стадии 2 Примера 2 и осуществляя незначительные модификации, но используя продукт стадии 2 Примера 39, получили указанное в заголовке соединение (43 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.29-4.37 (m, 3.5Н), 4.41-4.45 (m, 0.5Н), 4.52-4.58 (m, 0.5H), 4.65-4.69 (m, 0.5H), 4.89 (t, 1H), 5.43 (d, 2H), 5.90 (d, 1H), 6.21 (t, 1H), 7.47 (d, 2H), 7.52 (d, 1H), 7.71 (d, 2H), 8.12 (dd, 1H) 8.45 (t, 1H), 8.81-8.85 (m, 2H); m/z (Cl) 450 [М+Н].
Пример 40. N-((1R,2S)-1-(4-(6-(2-Аминопропан-2-ил)пиридин-3-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
Стадия 1. Получение 2-(5-бромпиридин-2-ил)пропан-2-амина
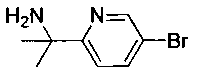
В круглодонную колбу емкостью 100 мл, содержащую 40 мл безводного тетрагидрофурана, добавляли CeCl3 (5 г, 20,28 ммоль). Полученную суспензию перемешивали при комнатной температуре в течение 18 часов. Смесь охлаждали до -78°C, добавляли по каплям раствор метиллития (12,7 мл, 1,6М) и перемешивали при -78°C в течение 30 минут. Добавляли раствор 5-бром-2-цианопиридина (1,2 г, 6,8 ммоль) в тетрагидрофуране (10 мл) и реакционную смесь перемешивали при -78°C в течение 4 часов, затем нагревали до 0°C. Добавляли насыщенный NH4Cl и реакционную смесь оставляли нагреваться до комнатной температуры при перемешивании. Добавляли этилацетат (100 мл) и органическую фазу отделяли, сушили (Na2SO4) и концентрировали под вакуумом с получением указанного в заголовке соединения (805 мг): m/z (Cl) М+Н 215+217.
Стадия 2. Получение трет-бутил-2-(5-бромпиридин-2-ил)пропан-2-ил-карбамата
К раствору 2-(5-бромпиридин-2-ил)пропан-2-амина (800 мг, 3,7 ммоль) в CH2Cl2 (25 мл) добавляли ди-трет-бутилдикарбонат (830 мг, 3,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Содержимое разбавляли NaHCO3 (50 мл) и экстрагировали CH2Cl2 (50 мл). Органическую фазу сушили (Na2SO4) и концентрировали под вакуумом. Неочищенный материал подвергали хроматографии, элюируя от 100%-ных гексанов до смеси 35:65 этилацетат: гексаны с получением указанного в заголовке соединения (635 мг): m/z (Cl) М+Н 315+317.
Стадия 3. Получение трет-бутил-2-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)пропан-2-ил-карбамата
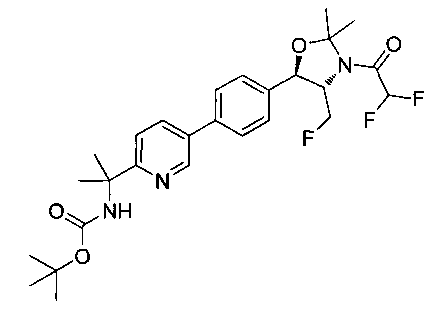
Следуя общей методике стадии 1 Примера 22 и осуществляя незначительные модификации, но используя трет-бутил-2-(5-бромпиридин-2-ил)пропан-2-илкарбамат, получили указанное в заголовке соединение (448 мг): m/z (Cl) М+Н 522.
Стадия 4. N-((1R,2S)-1-(4-(6-(2-Аминопропан-2-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
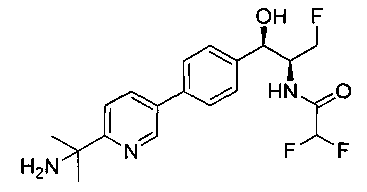
Следуя общей методике стадии 2 Примера 2 и осуществляя незначительные модификации, но используя продукт стадии 3 Примера 40, получили указанное в заголовке соединение (178 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.44 (s, 6Н), 4.29-4.37 (m, 1.5Н), 4.41-4.45 (m, 0.5Н), 4.54-4.58 (m, 0.5Н), 4.65-4.68 (m, 0.5Н), 4.88 (t, 1Н), 5.90 (d, 1Н), 6.21 (t, 1Н), 7.46 (d, 2Н), 7.68-7.73 (m, 3H), 8.04 (dd, 1Н), 8.82-8.84 (m, 2Н); m/z (Cl) 382 [М+Н].
Пример 41. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-((метиламино)метил)тиазол-5-ил)фенил)пропан-2-ил)ацетамид
Стадия 1. Получение 5-бром-2-((триизопропилсилилокси)метил)тиазола
В круглодонную колбу емкостью 100 мл загружали (5-бромтиазол-2-ил)-метанол (2,5 г, 12,9 ммоль), имидазол (1,75 г, 25,8 ммоль), диметилформамид (25 мл) и хлортриизопропилсилан (3,4 мл, 15,5 ммоль). Реакционную смесь перемешивали при комнатной температуре под азотом в течение 72 часов. Содержимое разбавляли этилацетатом (150 мл), промывали 0,5М соляной кислотой (2×100 мл) и рассолом (100 мл), сушили (Na2SO4) и концентрировали под вакуумом с получением указанного в заголовке соединения (4,7 г): m/z (Cl) М+Н 350+352.
Стадия 2. Получение 2,2-дифтор-1-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(2-((триизопропилсилилокси)метил)тиазол-5-ил)фенил)оксазолидин-3-ил)-этанона
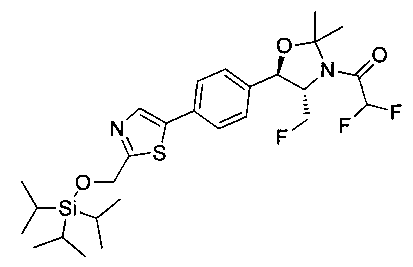
Следуя общей методике стадии 1 Примера 22 и осуществляя незначительные модификации, но используя 5-бром-2-((триизопропилсилилокси)метил)тиазол, получили указанное в заголовке соединение (2,7 г): m/z (Cl) М+Н 557.
Стадия 3. Получение 2,2-дифтор-1-((4S,5R)-4-(фторметил)-5-(4-(2-(гидроксиметил)тиазол-5-ил)фенил)-2,2-диметилоксазолидин-3-ил)этанона
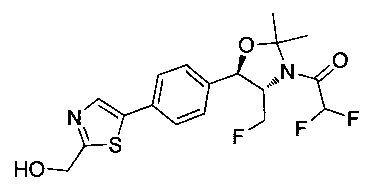
К раствору 2,2-дифтор-1-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(2-((триизопропилсилилокси)метил)тиазол-5-ил)фенил)оксазолидин-3-ил)этанона (2,6 г, 4,8 ммоль) в тетрагидрофуране (50 мл) добавляли фторид тетрабутиламмония (5,2 мл 1М раствора). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавляли этилацетатом (100 мл) и промывали водой и рассолом. Органическую фазу сушили (Na2SO4) и концентрировали под вакуумом. Неочищенный материал подвергали хроматографии, элюируя от 100%-ных гексанов до смеси 75:25 этилацетат: гексаны с получением указанного в заголовке соединения (1,8 г): m/z (Cl) М+Н 401.
Стадия 4. Получение 2,2-дифтор-1-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(2-((метиламино)метил)тиазол-5-ил)фенил)оксазолидин-3-ил)этанона
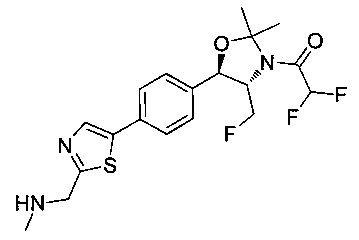
Мезилангидрид (860 мг, 4,9 ммоль) добавляли к 2,2-дифтор-1-((4S,5R)-4-(фторметил)-5-(4-(2-(гидроксиметил)тиазол-5-ил)фенил)-2,2-диметил-оксазолидин-3-ил)этанону (1,4 г, 3,5 ммоль) в CH2Cl2 (35 мл). Добавляли пиридин (0,5 мл, 5,9 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавляли CH2Cl2 (35 мл) и промывали водой и рассолом. Органическую фазу сушили (Na2SO4) и концентрировали под вакуумом. Неочищенный мезилат растворяли в ацетонитриле (10 мл) и переносили в сцинтилляционный флакон. Добавляли диизопропилэтиламин (0,65 мл, 3,7 ммоль) и метиламин HCl (130 мг, 1,9 ммоль) и реакционную смесь нагревали до 55°C в течение 18 часов. Реакционную смесь охлаждали и разбавляли насыщенным NaHCO3 (75 мл). Реакционную смесь экстрагировали CH2Cl2 (2×75 мл) и объединенную органическую фазу сушили (Na2SO4) и концентрировали под вакуумом с получением указанного в заголовке соединения (475 мг): m/z (Cl) М+Н 414.
Стадия 5. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-((метиламино)метил)тиазол-5-ил)фенил)пропан-2-ил)ацетамид
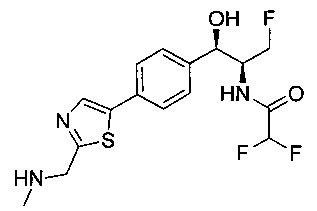
Следуя общей методике стадии 2 Примера 2 и осуществляя незначительные модификации, но используя продукт стадии 4 Примера 41, получили указанное в заголовке соединение (64 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.37 (s, 3H), 2.78 (m, 1Н), 3.92 (s, 2Н), 4.26-4.35 (m, 1.5Н), 4.40-4.44 (m, 0.5Н), 4.53-4.56 (m, 0.5H), 4.64-4.67 (m, 0.5H), 4.85 (t, 1H), 5.89 (d, 1H), 6.19 (t, 1H), 7.38 (d, 2H), 7.59 (d, 2H), 8.06 (s, 1H), 8.80 (d, 1H); m/z (Cl) 374 [М+Н].
Пример 42. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-(4-(2-((3-фторазетидин-1-ил)метил)тиазол-5-ил)фенил)-1-гидроксипропан-2-ил)ацетамид
Следуя общей методике стадий 4-5 Примера 41 и осуществляя незначительные модификации, но используя 3-фторазетидин, получили указанное в заголовке соединение (35 мг): m/z (Cl) М+Н 418.
Пример 43. N-((1R,2S)-1-(4-(2-((3-Цианоазетидин-1-ил)метил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
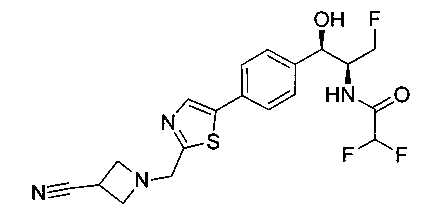
Следуя общей методике стадий 4-5 Примера 41 и осуществляя незначительные модификации, но используя 3-цианоазетидин, получили указанное в заголовке соединение (22 мг): m/z (Cl) М+Н 425.
Пример 44. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-((3-гидроксиазетидин-1-ил)метил)тиазол-5-ил)фенил)пропан-2-ил)ацетамид
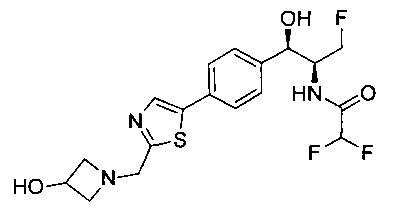
Следуя общей методике стадий 4-5 Примера 41 и осуществляя незначительные модификации, но используя 3-гидроксиазетидин, получили указанное в заголовке соединение (18 мг): m/z (Cl) М+Н 416.
Пример 45. N-((1R,2S)-1-(4-(2-((Циклопропиламино)метил)тиазол-5-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
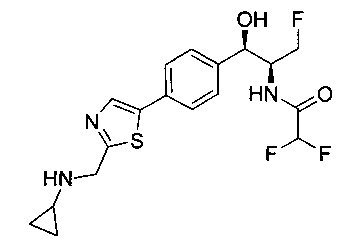
Следуя общей методике стадий 4-5 Примера 41 и осуществляя незначительные модификации, но используя циклопропанамин, получили указанное в заголовке соединение (23 мг): m/z (Cl) М+Н 400.
Пример 46. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-((2-гидроксиэтиламино)метил)тиазол-5-ил)фенил)пропан-2-ил)ацетамид
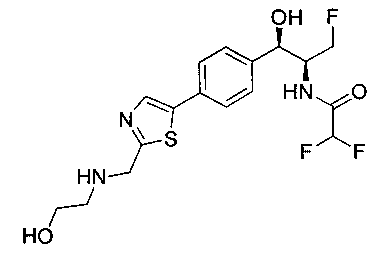
Следуя общей методике стадий 4-5 Примера 41 и осуществляя незначительные модификации, но используя 2-аминоэтанол, получили указанное в заголовке соединение (18 мг): m/z (Cl) М+Н 404.
Пример 47. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-(морфолинометил)тиазол-5-ил)фенил)пропан-2-ил)ацетамид
Следуя общей методике стадий 4-5 Примера 41 и осуществляя незначительные модификации, но используя морфолин, получили указанное в заголовке соединение (37 мг): m/z (Cl) М+Н 430.
Пример 48. 2,2-Дифтор-N-((1R,2S)-3-фтор-1-(4-(2-((2-фторэтиламино)-метил)тиазол-5-ил)фенил)-1-гидроксипропан-2-ил)ацетамид
Следуя общей методике стадий 4-5 Примера 41 и осуществляя незначительные модификации, но используя 2-фторэтанамин, получили указанное в заголовке соединение (28 мг): m/z (Cl) М+Н 406.
Пример 49. N-((1R,2S)-1-(4-(2-(1-Аминоэтил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
Стадия 1. N-(1-(5-Бромтиазол-2-ил)этил)-2-метилпропан-2-сульфинамид
Следуя методике, ранее описанной в WO 2011053542 (pp. 47-49), но используя рацемический 2-метил-2-пропансульфинамид, получили указанное в заголовке соединение (2,06 г): m/z (Cl) М+Н 311+313.
Стадия 2. Получение N-(1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)тиазол-2-ил)этил)-2-метилпропан-2-сульфинамида
Следуя общей методике стадии 1 Примера 22 и осуществляя незначительные модификации, но используя N-(1-(5-бромтиазол-2-ил)этил)-2-метилпропан-2-сульфинамид, получили указанное в заголовке соединение (323 мг): m/z (Cl) М+Н 518.
Стадия 3. N-((1R,2S)-1-(4-(2-(1-Аминоэтил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид
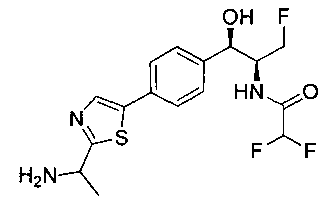
К раствору N-(1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)тиазол-2-ил)этил)-2-метилпропан-2-сульфинамида (315 мг, 0,6 ммоль) в CH2Cl2 (25 мл) при 0°C добавляли трифторуксусную кислоту (5 мл) и 5 капель воды. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 часа. Добавляли толуол (20 мл) и содержимое концентрировали под вакуумом. Добавление толуола/концентрирование повторяли и содержимое помещали в глубокий вакуум на 5 минут. В реакционную колбу добавляли метанол (25 мл) и раствор охлаждали в ледяной бане. Добавляли HCl в метаноле (4 мл в 1,25 мл раствора) и реакционную смесь оставляли нагреваться до комнатной температуры при перемешивании. Через 2 часа реакционную смесь концентрировали под вакуумом до удаления примерно 3 мл растворителя и очищали непосредственно с использованием хроматографии с обращенной фазой, элюируя от смеси 95:5 вода/ацетонитрил/0,1% трифторуксусная кислота до смеси 95:5 ацетонитрил/вода/0,1% трифторуксусная кислота. Очищенные фракции концентрировали под вакуумом для удаления ацетонитрила. Водную фазу разбавляли насыщенным NaHCO3 (100 мл) и экстрагировали этилацетатом (2×100 мл). Объединенную органическую фазу сушили (Na2SO4) и концентрировали под вакуумом с получением указанного в заголовке соединения (93 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.39 (d, 3H), 2.38 (m, 2Н), 4.18-4.33 (m, 2.5Н), 4.40-4.44 (m, 0.5Н), 4.52-4.56 (m, 0.5H), 4.64-4.67 (m, 0.5H), 4.85 (t, 1H), 5.88 (d, 1H), 6.19 (t, 1H), 7.38 (d, 2H), 7.58 (d, 2H), 8.03 (s, 1H), 8.80 (d, 1H); m/z (Cl) 374 [М+Н].
Следующие производные указанного в заголовке соединения Примера 49 могут быть получены способами, известными в данной области техники, включая описанные ниже в Примере 95А:
N-((1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамид;
(1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-2-(2,2-дихлорацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-2-(2,2-дихлорацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-гидрофосфат натрия; и
(1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-дигидрофосфат.
Пример 50. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[2-(метансульфониламино-метил)-бензотиазол-5-ил]-фенил}-этил)-ацетамида
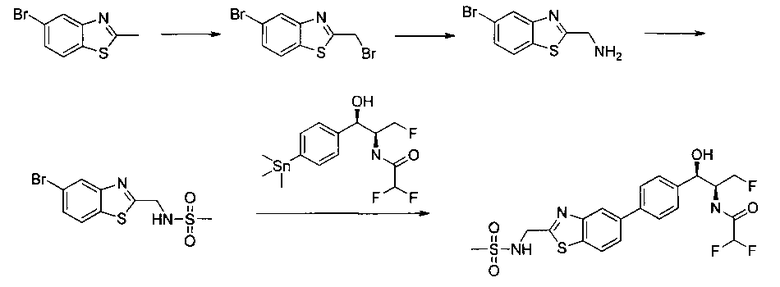
Стадия 1. Получение 5-бром-2-бромметил-бензотиазола
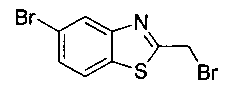
К раствору 5-бром-2-метил-бензотиазола (1,0 г, 4,385 ммоль) в CCl4 (15 мл) добавляли N-бромсукцинамид (1,1 г, 6,140 ммоль) и азобисизобутиронитрил (0,029 г, 0,175 ммоль) при комнатной температуре реакции. Реакционную смесь нагревали до 75°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и растворитель выпаривали в вакууме с получением неочищенного материала, который затем очищали флэш-хроматографией с использованием 3,5%-ного этилацетата в гексане в качестве элюента с получением указанного в заголовке соединения (0,85 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 5.12 (s, 2Н), 7.68-7.71 (dd, J1=1,92 Гц, J2=8,76 Гц, 1Н), 7.95 (d, J=8,6 Гц, 1Н), 8.43 (d, J=1,92 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=308,2.
Стадия 2. Получение С-(5-бром-бензотиазол-2-ил)-метиламина
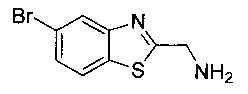
Газообразный аммиак барботировали через раствор 5-бром-2-бромметил-бензотиазола (0,850 г, 2,768 ммоль) в метаноле (16 мл) в течение 10 минут при 0°C, затем перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь сушили над сульфатом магния, фильтровали и растворитель выпаривали в вакууме. Неочищенный материал промывали н-пентаном и гексаном с получением указанного в заголовке соединения (0,78 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.52 (s, 2Н), 7.69-7.72 (dd, J1=1,96 Гц, J2=8,68 Гц, 1Н), 7.95 (d, J=8,64 Гц, 1Н), 8.46 (d, J=1,96 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=245,0.
Стадия 3. Получение N-(5-бром-бензотиазол-2-илметил)-метансульфонамида
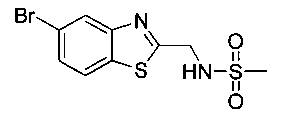
К перемешиваемому раствору С-(5-бром-бензотиазол-2-ил)-метиламина (0,780 г, 3,209 ммоль) в CH2Cl2 (12 мл) добавляли триэтиламин (1,2 мл, 8,024 ммоль). К вышеуказанному раствору добавляли по каплям метансульфонилхлорид (0,5 мл, 6,419 ммоль) при 0°C, затем перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали в вакууме и неочищенный материал разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, концентрировали и очищали CombiFlash хроматографией с использованием 50%-ного этилацетата в гексане в качестве элюента с получением указанного в заголовке соединения (0,45 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 3.03 (s, 3H), 4.60 (d, J=5,72 Гц, 2Н), 7.64-7.67 (dd, J1=1,88 Гц, J2=8,72 Гц, 1Н), 7.88 (d, J=8,68 Гц, 1Н), 8.21 (t, J=6 Гц, 1Н), 8.40 (d, J=1,88 Гц, 1Н). ЖХ-MC (m/z): [М-Н]=320,8.
Стадия 4. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[2-(метансульфониламино-метил)-бензотиазол-5-ил]-фенил}-этил)-ацетамида
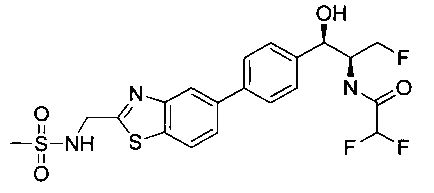
К перемешиваемому раствору N-(5-бром-бензотиазол-2-илметил)-метансульфонамида (0,21 г, 0,656 ммоль) и 2,2-дифтор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-триметилстаннанил-фенил)-этил]-ацетамида (0,404 г, 0,984 ммоль) в N-метил-2-пирролидоне (9 мл) добавляли хлорид лития (0,084 г, 1,968 ммоль) при комнатной температуре. Полученную реакционную смесь дегазировали с помощью азота в течение 15 минут, затем добавляли Pd(PPh3)2Cl2 (0,046 г, 0,065 ммоль). Реакционную смесь нагревали до 90°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме с получением неочищенного материала, который очищали флэш-колоночной хроматографией с использованием 2,4%-ного метанола в CH2Cl2 в качестве элюента с получением неочищенного соединения. Это неочищенное соединение снова очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (5 мг). 1Н ЯМР (400 МГц, DMSO) δ: 3.04 (s, 3H), 4.26-4.33 (m, 1Н), 4.41-4.43 (m, 0.5Н), 4.44-4.50 (m, 0.5Н), 4.62 (s, 2Н), 4.65-4.68 (m, 0.5Н), 4.75-4.79 (m, 0.5H), 4.87-4.88 (m, 1H), 5.9 (d, J=4,56 Гц, 1H), 6.20 (d, J=56,36 Гц, 1H), 7.45 (d, J=8,24 Гц, 2H), 7.63, (d, J=8,16 Гц, 1H), 7.73 (d, J=8,28 Гц, 2H). 7.81 (d, J1=1,8 Гц, J2=8,52 Гц, 1H), 8.42 (d, J=1,6 Гц, 1H), 8.82-8.83 (m, 2H). LC-Ms (m/z): [M-H]=485,8.
Пример 51. Получение 2,2-дифтор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-{6-[(метансульфонил-метокси-амино)-метил]-пиридин-3-ил}-фенил)-этил]-ацетамида
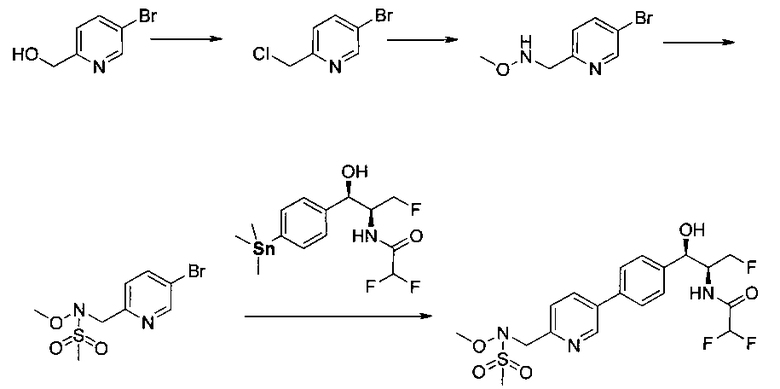
Стадия 1. Получение 5-бром-2-хлорметил-пиридина
К перемешиваемому раствору (5-бром-пиридин-2-ил)-метанола (1 г, 5,319 ммоль) в CH2Cl2 (20 мл) добавляли по каплям тионилхлорид (0,57 мл, 7,979 ммоль) при 0°C, затем перемешивали в течение еще 4 часов. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия и экстрагировали CH2Cl2. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали колоночной хроматографией с использованием 7%-ного этилацетата в гексане в качестве элюента с получением указанного в заголовке соединения (0,8 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.7 (s, 2Н), 7.53 (d, 8.36 Гц, 1Н), 8.09-8.11 (dd, J1=2,44 Гц, J2=8,23 Гц, 1Н), 8.69 (d, J=2,4 Гц, 1Н), ЖХ-MC (m/z): [М+Н]=206,0.
Стадия 2. Получение N-(5-бром-пиридин-2-илметил)-O-метил-гидроксиламина
К перемешиваемому раствору 5-бром-2-хлорметил-пиридина (2 г, 9,709 ммоль) в диметилформамиде (30 мл) добавляли соль гидрохлорид метоксиамина (0,973 г, 11,65 ммоль) и К2СО3 (3,35 г, 24,27 ммоль) при комнатной температуре, затем нагревали до 60°C в течение 16 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали CombiFlash хроматографией с использованием 30%-ного этилацетата в гексане в качестве элюента с получением указанного в заголовке соединения (0,6 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 3.36 (s, 3H), 3.99 (d, J=5,88 ГЦ, 2Н), 7.10 (t, 1Н), 7.44-7.46 (dd, J1=8,32 Гц, J2=0,32 Гц, 1Н), 7.99-8.02 (dd, J1=2,44 Гц, J2=8,4 Гц, 1Н), 8.60-8.61 (dd, J1=2,4 Гц, J2=0,4 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=217,1.
Стадия 3. Получение N-(5-бром-пиридин-2-илметил)-N-метокси-метан-сульфонамида
К перемешиваемому раствору N-(5-бром-пиридин-2-илметил)-O-метил-гидроксиламина (0,6 г, 2,765 ммоль) в тетрагидрофуране (10 мл) добавляли бис(триметилсилил)амид натрия (0,6 г, 3,318 ммоль) и мезилхлорид (0,37 г, 3,318 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли насыщенным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали флэш-хроматографией с использованием 20%-ного этилацетата в гексане в качестве элюента с получением указанного в заголовке соединения (0,3 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 3.14 (s, 3H), 3.47 (s, 3H), 4.40 (s, 2Н), 7.50 (d, J=8,28 Гц, 1Н), 8.09-8.12 (dd, J1=2,8 Гц, J2=8,4 Гц, 1Н), 8.71 (d, J=2,36 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=297,1.
Стадия 4. Получение 2,2-дифтор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-{6-[(метансульфонил-метокси-амино)-метил]-пиридин-3-ил}-фенил)-этил]-ацетамида
К перемешиваемому раствору N-(5-бром-пиридин-2-илметил)-N-метокси-метансульфонамида (0,2 г, 0,678 ммоль) в N-метил-2-пирролидоне (8 мл) добавляли 2,2-дифтор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-триметил-станнанил-фенил)этил]-ацетамид (0,27 г, 0,678 ммоль). Реакционную смесь дегазировали с помощью азота в течение 15 минут, затем добавляли одновременно трис(дибензилиденацетон)дипалладий(0) (0,06 мг, 0,068 ммоль) и три-2-фурилфосфин (0,03 г, 0,136 ммоль). Полученную реакционную смесь перемешивали при 80°C в течение 5 часов. Разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали CombiFlash хроматографией с использованием 50%-ного этилацетата в гексане в качестве элюента с получением указанного в заголовке соединения (0,05 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 3.16 (s, 3H), 3.49 (s, 3H), 4.30-4.37 (m, 1.5Н), 4.41-4.46 (m, 0.5Н), 4.46 (s, 2H), 4.54-4.56 (m, 0.5H), 4.65-4.68 (m, 0.5H), 4.89 (m, 1H), 5.94 (d, J=4,2 Гц, 1H), 6.20 (t, J=53,76 Гц, 1H), 7.47 (d, J=8,2 Гц, 2H), 7.59 (d, J=8,16 Гц, 1H), 7.74 (d, J=8,2 Гц, 2H), 8.14-8.16 (dd, J1=2,32 Гц, J2=8,16 Гц, 1H), 8.86 (d, J=8,72 Гц, 1H), 8.91 (d, J=2,08 Гц, 1H). ЖХ-MC (m/z): [M+H]=461,9.
Пример 52. Получение 2,2-дифтор-N-[(1R,2R)-1-фторметил-2-(4-{6-[(3-фтор-пропиламино)-метил]-пиридин-3-ил}-фенил)-2-гидрокси-этил]-ацетамида
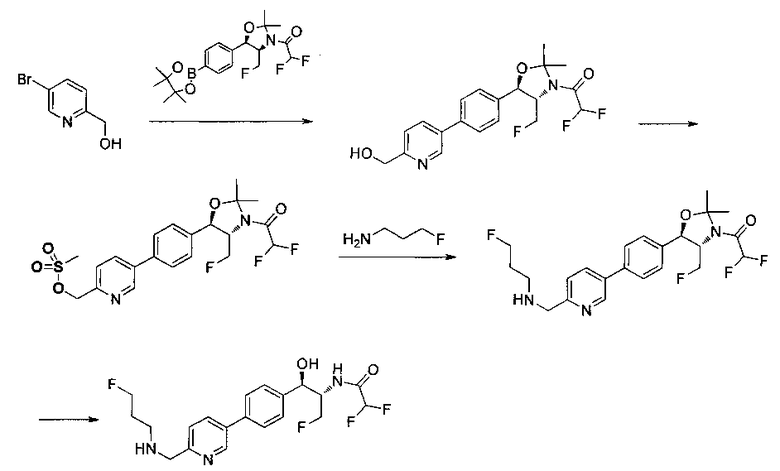
Стадия 1. Получение 2,2-дифтор-1-{(4S,5R)-4-фторметил-5-[4-(6-гидрокси-метил-пиридин-3-ил)-фенил]-2,2-диметил-оксазолидин-3-ил}-этанона
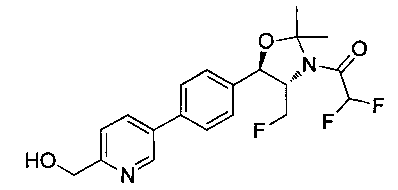
К перемешиваемому раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (2,0 г, 4,843 ммоль) в толуоле (40 мл), этаноле (10 мл), воде (10 мл) и (5-бром-пиридин-2-ил)-метаноле (0,91 г, 4,843 ммоль) добавляли Na2CO3 (1,54 г, 14,528 ммоль) при комнатной температуре. Полученную реакционную смесь дегазировали с помощью азота в течение 30 минут, затем добавляли Pd(PPh3)4 (0,559 г, 0,484 ммоль). Реакционную смесь нагревали до 90°C в течение 6 часов. Растворитель выпаривали в вакууме, затем разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали CombiFlash хроматографией с использованием 80%-ного этилацетата в гексане в качестве элюента с получением указанного в заголовке соединения (0,67 г): 1Н ЯМР (400 МГц, CDCl3) δ: 1.53 (s, 3H), 1.60 (S, 3H), 4.54-4.58 (m, 0.5Н), 4.60 (d, J=5,84 Гц, 2Н), 4.66-4.72 (m, 1Н), 4.81-4.84 (m, 0.5Н), 4.90-4.94 (m, 1Н), 5.26 (d, J=3,72 Гц, 1H), 5.48 (t, J=5,8 Гц, 1H), 6.64 (t, J=52,4 Гц, 1H), 7.55-7.64 (m, 3H), 7.78 (d, J=8,2 Гц, 2H), 8.09-8.12 (dd, J=2,32 Гц, J=8,16 Гц, 1H), 8.81 (d, 2,04 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=395,0.
Стадия 2. Получение 5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-илметилового эфира метансульфоновой кислоты
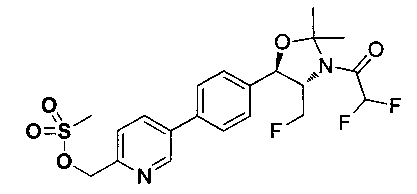
К раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-5-[4-(6-гидроксиметил-пиридин-3-ил)-фенил]-2,2-диметил-оксазолидин-3-ил}-этанона (1,1 г, 2,792 ммоль) в CH2Cl2 (50 мл) добавляли триэтиламин (0,564 г, 5,584 ммоль) с последующим добавлением метансульфонилхлорида (0,365 г, 3,071 ммоль) при 0°C, затем перемешивали в течение 30 минут при 0°C. Реакционную смесь разбавляли насыщенным водным раствором бикарбоната и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали CombiFlash хроматографией с использованием 30%-ного этилацетата в гексане в качестве элюента с получением указанного в заголовке соединения (0,875 г). 1Н ЯМР (400 МГц, DMSO) δ: 1.53 (s, 3H), 1.60 (S, 3H), 3.30 (s, 3H), 4.54-4.58 (m, 0.5Н), 4.60-4.70 (m, 1Н), 4.82-4.83 (m, 0.5Н), 4.90-4.94 (m, 1Н), 5.28 (d, J=3,68 Гц, 1Н), 5.36 (s, 2Н), 6.64 (t, J=52,4 Гц, 1Н), 7.61-7.64 (m, 3H), 7.82 (d, J=8,08 Гц, 2Н), 8.19-8.21 (dd, J1=2,4 Гц, J2=8,16 Гц, 1Н), 8.94 (d, 2,2 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=473,2.
Стадия 3. Получение 2,2-дифтор-1-[(4S,5R)-4-фторметил-5-(4-{6-[(3-фтор-пропиламино)-метил]-пиридин-3-ил}-фенил)-2,2-диметил-оксазолидин-3-ил]-этанона
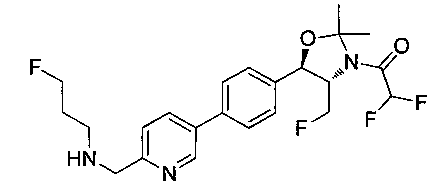
К раствору гидрохлорида 3-фтор-пропиламина (0,072 г, 0,636 ммоль) в ацетонитриле (5 мл) добавляли диизопропилэтиламин (0,082 мл, 0,636 ммоль) при комнатной температуре. После перемешивания в течение 30 минут добавляли 5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-илметиловый эфир метансульфоновой кислоты (0,1 г, 0,211 ммоль), затем полученную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали при пониженном давлении и неочищенный материал очищали CombiFlash хроматографией с использованием 12%-ного метанола в CH2Cl2 в качестве элюента с получением указанного в заголовке соединения (0,093 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.52 (s, 3H), 1.60 (S, 3H), 2.06-2.14 (m, 2Н), 3.57-3.60 (m, 2Н), 4.35 (s, 2Н), 4.49-4.51 (m, 1Н), 4.52-4.56 (m, 0.5Н), 4.57-4.63 (m, 1Н), 4.64-4.70 (m, 1Н), 4.83-4.86 (m, 0.5Н), 4.93-4.97 (m, 1Н), 5.28 (d, J=3,48 Гц, 1Н), 6.66 (t, J=52,3 Гц, 1Н), 7.61-7.65 (m, 3H), 7.84 (d, J=8,2 Гц, 2Н), 8.21-8.23 (dd, J1=3,04 Гц, J2=8,16 Гц, 1Н), 8.97 (d, J=1,96 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=454,1.
Стадия 4. Получение 2,2-дифтор-N-[(1R,2R)-1-фторметил-2-(4-{6-[(3-фтор-пропиламино)-метил]-пиридин-3-ил}-фенил)-2-гидрокси-этил]-ацетамида
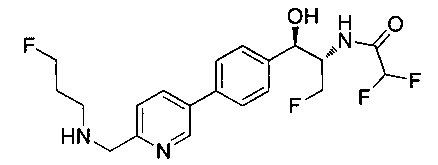
К раствору 2,2-дифтор-1-[(4S,5R)-4-фторметил-5-(4-{6-[(3-фтор-пропиламино)-метил]-пиридин-3-ил}-фенил)-2,2-диметил-оксазолидин-3-ил]-этанона (0,113 г, 0,249 ммоль) в CH2Cl2 (5 мл) добавляли трифторуксусную кислоту (1,0 мл) при 0°C. Полученную реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Растворитель выпаривали при пониженном давлении и неочищенный остаток разбавляли водным раствором бикарбоната и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали CombiFlash хроматографией с использованием 15%-ного метанола в CH2Cl2 в качестве элюента. Полученное твердое вещество дополнительно промывали н-пентаном и диэтиловым эфиром с получением указанного в заголовке соединения (0,034 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.84-1.93 (m, 2Н), 2.50-2.54 (m, 2Н), 2.76 (bs, 1Н), 3.99 (bs, 2H), 4.29-4.31 (m, 1.5H), 4.40-4.48 (m, 1.5H), 4.54-4.60 (m, 1.5H), 4.65-4.68 (m, 0.5H), 4.87 (bs, 1H), 5.92 (d, J=4,52 Гц, 1H), 6.20 (t, J=53,64 Гц, 1H), 7.46 (d, J=8,24 Гц, 2H), 7.52 (d, J=8,12 Гц, 1Н), 7.70 (d, J=8,24 Гц, 2H), 8.08-8.10 (dd, J1=2,36 Гц, J2=8,2 Гц, 1H), 8.85 (m, 2H). ЖХ-MC (m/z): [M+H]=414,1.
Пример 53. Получение N-{(1R,2R)-2-[4-(6-диметиламинометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дифтор-ацетамида
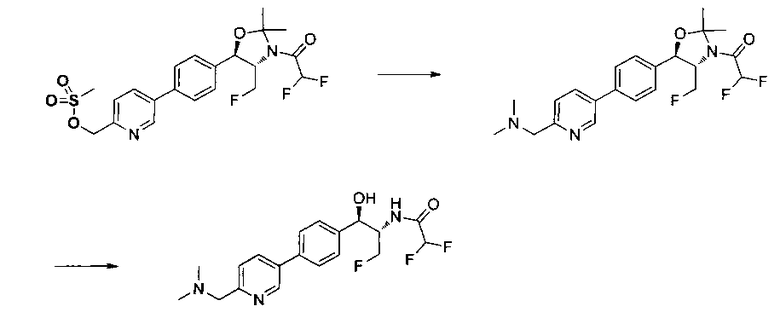
Стадия 1. Получение 1-{(4S,5R)-5-[4-(6-диметиламинометил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-ил}-2,2-дифтор-этанона
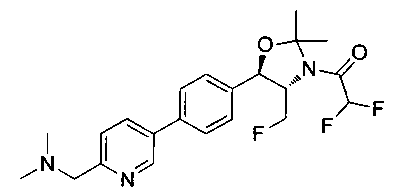
К перемешиваемому раствору диметиламина (2,0М раствор в тетрагидрофуране) (0,31 мл, 0,028 ммоль) в тетрагидрофуране (5 мл) добавляли 5-{4-[(4R,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-илметиловый эфир метансульфоновой кислоты (0,1 г, 0,212 ммоль) и перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали в вакууме и неочищенный материал очищали CombiFlash хроматографией с использованием 10%-ного метанола в CH2Cl2 в качестве элюента с получением указанного в заголовке соединения (0,072 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.53 (s, 3H), 1.60 (S, 3H), 2.21 (s, 6Н), 3.56 (s, 2Н), 4.56-4.58 (m, 0.5Н), 4.66-4.72 (m, 1Н), 4.80-4.84 (m, 0.5Н), 4.90-4.94 (m, 1Н), 5.26 (d, J=3,76 Гц, 1Н), 6.64 (t, J=52,36 Гц, 1Н), 7.50 (d, J=8,08 Гц, 1Н), 7.59 (d, J=8,08 Гц, 2H), 7.78 (d, J=8,12 Гц, 2H), 8.07-8.09 (m, 1H), 8.81 (s, 1H). ЖХ-MC (m/z): [М+Н]=422.
Стадия 2. Получение N-{(1R,2R)-2-[4-(6-диметиламинометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дифтор-ацетамида
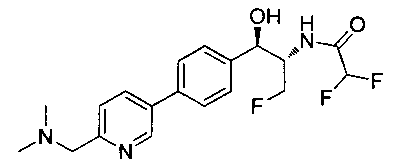
К раствору 1-{(4S,5R)-5-[4-(6-диметиламинометил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-ил}-2,2-дифтор-этанона (0,070 г, 0,166 ммоль) в CH2Cl2 (5 мл) добавляли трифторуксусную кислоту (1,0 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Концентрировали с получением неочищенного остатка и разбавляли водным раствором бикарбоната и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали CombiFlash хроматографией с использованием 15%-ного метанола в CH2Cl2 в качестве элюента. Полученное твердое вещество промывали н-пентаном и диэтиловым эфиром с получением указанного в заголовке соединения (0,030 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.44 (s, 6Н), 3.90 (bs, 2Н), 4.28-4.30 (m, 1 Н), 4.31-4.33 (m, 0.5Н), 4.41-4.45 (m, 0.5Н), 4.54-4.55 (m, 0.5H), 4.65-4.68 (m, 0.5H), 4.89 (t, J=4,0 Гц, 1H), 5.93 (d, J=4,56 Гц, 1H), 6.20 (t, J=53,76 Гц, 1H), 7.47 (d, J=8,24 Гц, 2H), 7.53 (d, J=8,16 Гц,1Н), 7.72 (d, J=8,24 Гц, 2H), 8.11-8.13 (m, 1H), 8.84-8.87 (m, 2H). ЖХ-MC (m/z): [M+H]=382,1.
Пример 54. Получение N-{(1R,2R)-2-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дифтор-ацетамида
Стадия 1. Получение 1-{(4S,5R)-5-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-ил}-2,2-дифтор-этанона
К перемешиваемому раствору этиламина (2,0М раствор в тетрагидрофуране; 0,31 мл, 0,027 ммоль) в тетрагидрофуране (5 мл) добавляли 5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-илметиловый эфир метансульфоновой кислоты (0,1 г, 0,212 ммоль) при комнатной температуре, затем перемешивали в течение ночи. Растворитель выпаривали в вакууме. Неочищенный материал очищали CombiFlash хроматографией с использованием 10%-ного метанола в CH2Cl2 в качестве элюента с получением указанного в заголовке соединения (0,062 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.09 (t, J=7,12 Гц, 3H), 1.52 (s, 3H), 1.60 (S, 3H), 2.66-2.71 (q, 2Н), 3.96 (s, 2Н), 4.51-4.56 (m, 0.5Н), 4.57-4.58 (m, 1Н), 4.59-4.69 (m, 0.5Н), 4.70-4.82 (m, 1Н), 5.27 (d, J=3,56 Гц, 1Н), 6.64 (t, J=52,4 Гц, 1Н), 7.53 (d, J=8,2 Гц, 1Н), 7.60 (d, J=8,12 Гц, 2Н), 7.79 (d, J=8,2 Гц, 2Н), 8.09-8.11 (dd, J1=2,28 Гц, J2=8,08 Гц, 1Н), 8.86 (d, J=2,08 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=422.
Стадия 2. Получение N-{(1R,2R)-2-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дифтор-ацетамида
К раствору 1-{(4S,5R)-5-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-ил}-2,2-дифтор-этанона (0,062 г, 0,147 ммоль) в CH2Cl2 (5 мл) добавляли трифторуксусную кислоту (1,0 мл) при комнатной температуре. Полученную реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Растворитель выпаривали в вакууме с получением неочищенного остатка и разбавляли водным раствором бикарбоната и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, концентрировали и очищали CombiFlash хроматографией с использованием 15%-ного метанола в CH2Cl2 в качестве элюента. Полученное твердое вещество промывали н-пентаном и диэтиловым эфиром с получением указанного в заголовке соединения (0,035 г) после сушки в лиофилизаторе. 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.16 (t, J=7,2 Гц, 3H), 2.83-2.88 (q, 2Н), 4.14 (s, 2Н), 4.30-4.33 (m, 1.5Н), 4.41-4.45 (m, 0.5Н), 4.55-4.56 (m, 0.5Н), 4.65-4.68 (m, 0.5Н), 4.89 (t, J=4,08 Гц, 1Н), 5.93 (d, J=4,55 Гц, 1Н), 6.20 (t, J=53,76 Гц, 1Н), 7.47 (d, J=8,24 Гц, 2Н), 7.54 (d, J=8,2 Гц, 1Н), 7.72 (d, J=8,24 Гц, 2Н), 8.12-8.15 (dd, J1=2,32 Гц, J2=8,16 Гц, 1H), 8.85 (d, J=8,8 Гц, 1H), 8.90 (d, J=2,12 Гц, 1H). ЖХ-MC (m/z): [М+Н]=382,1.
Пример 55. Получение N-{(1S,2R)-2-[4-(2-аминометил-оксазол-5-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дихлор-ацетамида
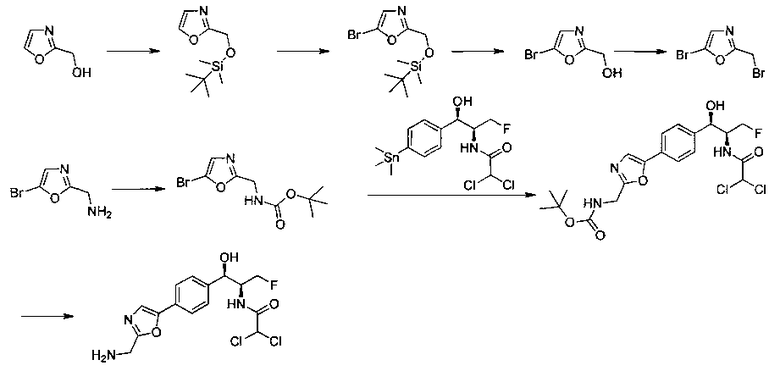
Стадия 1. Получение 2-(трет-бутил-диметил-силанилоксиметил)-оксазола
К раствору оксазол-2-ил-метанола (1,0 г, 0,010 ммоль) в безводном тетрагидрофуране (15 мл) добавляли имидазол (1,37 г, 0,020 ммоль) и трет-бутилдиметилсилилхлорид (1,97 г, 0,0131 ммоль) при 0°C. Реакционную смесь затем перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали колоночной хроматографией, элюируя 5%-ным этилацетатом в гексане с получением указанного в заголовке соединения (0,7 г). 1Н ЯМР (400 МГц, CDCl3) δ: 0.09 (s, 6Н), 0.89 (s, 9Н), 4.75 (s, 2Н), 7.06(s, 1Н), 7.62 (s, 1Н). LC-Ms (m/z) [М+Н]=214,2.
Стадия 2. Получение 5-бром-2-(трет-бутил-диметил-силанилоксиметил)-оксазола
К раствору 2-(трет-бутил-диметил-силанилоксиметил)-оксазола (0,35 г, 1,643 ммоль) в безводном тетрагидрофуране (8 мл) добавляли н-бутиллитий (0,26 г, 4,107 ммоль) по каплям в атмосфере азота при -78°C, затем перемешивали при -40°C в течение 2 часов. Реакционную смесь снова охлаждали до -78°C, затем добавляли тетрабромид углерода (1,36 г, 4,107 ммоль) и перемешивали до комнатной температуры в течение 14 часов. Реакционную смесь разбавляли насыщенным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали колоночной хроматографией, элюируя 2%-ным этилацетатом в гексане с получением неочищенного указанного в заголовке соединения (0,18 г), которое использовали на следующей стадии без очистки. ЖХ-MC (m/z): [М+Н]=294,2.
Стадия 3. Получение (5-бром-оксазол-2-ил)-метанола
Раствор 5-бром-2-(трет-бутил-диметил-силанилоксиметил)-оксазола (0,05 г, 0,171 ммоль) в метаноле (1,0 мл) продували избытком газообразного хлороводорода при 0°C в течение 30 минут. Реакционную смесь перемешивали до комнатной температуры в течение 3 часов, затем концентрировали в вакууме с получением указанного в заголовке соединения (0,03 г), которое использовали на следующей реакционной стадии без очистки. 1Н ЯМР (400 МГц, DMSO-d6) δ: 3.83 (bs, 1Н), 4.47 (s, 2Н), 7.26 (s, 1Н). ЖХ-MC (m/z): [М+Н]=180,1.
Стадия 4. Получение 5-бром-2-бромметил-оксазола
К перемешиваемому раствору (5-бром-оксазол-2-ил)-метанола (0,15 г, 0,842 ммоль) в безводном CH2Cl2 (6,0 мл) добавляли тетрабромид углерода (0,38 г, 1,011 ммоль) и трифенилфосфин (0,24 г, 0,962 ммоль) при 0°C, затем перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь концентрировали в вакууме и неочищенный материал очищали колоночной хроматографией, элюируя 5%-ным этилацетатом в гексане с получением указанного в заголовке соединения (0,05 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.42 (s, 2Н), 6.99 (s, 1Н). LC-Ms (m/z) [М+Н] 240,86.
Стадия 5. Получение С-(5-бром-оксазол-2-ил)-метиламина
К раствору аммиака в метаноле (4,0 мл) [полученному путем продувки избытка газообразного аммиака в метаноле в течение 15 минут при комнатной температуре] добавляли 5-бром-2-бромметил-оксазол (0,05 г, 0,207 ммоль) при комнатной температуре, затем перемешивали в течение 14 часов при комнатной температуре. Реакционную смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде соли гидрохлорида (0,052 г), которое использовали на следующей стадии без очистки. 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.11 (s, 2Н), 7.04 (bs, 3H), 7.36 (s, 1Н). LC-Ms (m/z): [М+Н]=178,9.
Стадия 6. Получение трет-бутилового эфира 5-бром-оксазол-2-илметил)-карбаминовой кислоты
К перемешиваемому раствору С-(5-бром-оксазол-2-ил)-метиламина (0,12 г, 0,468 ммоль) в смеси диоксан: вода 1:1 (4,0 мл) добавляли K2CO3 (0,129 г, 0,937 ммоль) с последующим добавлением ди-трет-бутилдикарбоната (0,112 г, 0,515 ммоль) при 0°C, затем перемешивали до комнатной температуры в течение 2 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали колоночной хроматографией, элюируя 30%-ным этилацетатом в гексане с получением указанного в заголовке соединения (0,52 г), которое использовали на следующей стадии без очистки. ЖХ-MC (m/z): [М+Н]=279,0.
Стадия 7. Получение трет-бутилового эфира (5-{4-[(1R,2S)-2-(2,2-дихлор-ацетиламино)-3-фтор-1-гидрокси-пропил]-фенил}-оксазол-2-илметил)-карбаминовой кислоты
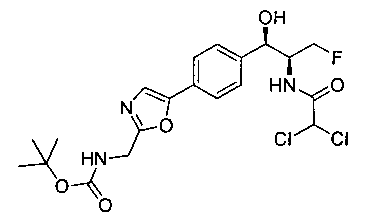
К перемешиваемому раствору трет-бутилового эфира 5-бром-оксазол-2-илметил)-карбаминовой кислоты (0,035 г, 0,13 ммоль) и 2,2-дихлор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-триметилстаннанил-фенил)-этил]-ацетамида (0,05 г, 0,11 ммоль) в безводном толуоле (2,0 мл) добавляли CsF (0,034 г, 0,22 ммоль) с последующим добавлением Cul (0,002 г, 0,011 ммоль) при комнатной температуре. Полученную реакционную смесь дегазировали с помощью азота в течение 30 минут, затем добавляли Pd2(PPh3)Cl2 (0,008 г, 0,011 ммоль) и нагревали в микроволновой печи при 100°C в течение 30 мин. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и очищали колоночной хроматографией, элюируя 10%-ным метанолом в CH2Cl2, с последующей вторичной очисткой с использованием препаративной ВЭЖХ с получением указанного в заголовке соединения (0,006 г, чистое). LC-Ms (m/z): [М-Н]=476,0.
Стадия 8. Получение N-{(1S,2R)-2-[4-(2-аминометил-оксазол-5-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дихлор-ацетамида
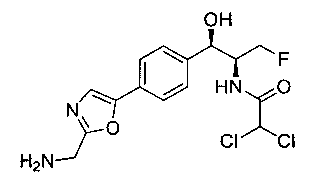
К перемешиваемому раствору трет-бутилового эфира (5-{4-[(1R,2S)-2-(2,2-дихлор-ацетиламино)-3-фтор-1-гидрокси-пропил]-фенил}-оксазол-2-илметил)-карбаминовой кислоты (0,006 г, 0,012 ммоль) в CH2Cl2 (1,0 мл) добавляли трифторуксусную кислоту (0,1 мл) при 0°C и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали в вакууме и остаток промывали н-пентаном, сушили под вакуумом с получением указанного в заголовке соединения (0,005 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.22-4.28 (m, 1.5Н), 4.31 (s, 2Н), 4.40-4.44 (m, 0.5Н), 4.56-4.60 (m, 0.5H), 4.68-4.71 (m, 0.5H), 4.89 (m, 1H), 6.02 (d, J=4,28 Гц, 1H), 6.49 (s, 1H), 7.47 (d, J=8,16 Гц, 2H), 7.67 (d, J=8,2 Гц, 2H), 7.72 (s, 1H), 8.21 (bs, 3H), 8.60 (d, J=8,88 Гц, 1H). ЖХ-MC (m/z): [M+H]=376,1.
Пример 56. Получение 2,2-дихлор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[6-(изопропиламино-метил)-пиридин-3-ил]-фенил}-этил)-ацетамида
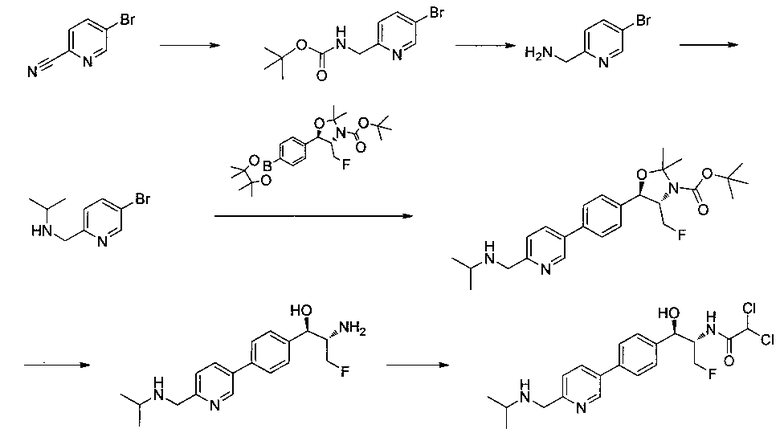
Стадия 1. Получение трет-бутилового эфира (5-бром-пиридин-2-илметил)-карбаминовой кислоты
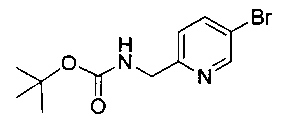
К раствору 5-бром-пиридин-2-карбонитрила (1,0 г, 5,46 ммоль) в метаноле (10 мл) при 0°C добавляли NiCl2.6H2O (0,12 г, 0,54 ммоль), ди-трет-бутилдикарбонат (2,38 г, 0,010 ммоль) и NaBH4 (0,413 г, 0,010 ммоль) при 0°C, затем перемешивали при комнатной температуре в течение 14 часов. Реакционный растворитель удаляли при пониженном давлении и неочищенный материал разбавляли водой и этилацетатом. Органический слой отделяли, сушили над сульфатом натрия и концентрировали в вакууме. Неочищенный материал очищали колоночной хроматографией, элюируя 30%-ным этилацетатом в гексане с получением указанного в заголовке соединения (650 мг). 1Н ЯМР (400 МГц, CDCl3) δ: 1.44 (s, 9Н), 4.37 (d, J=5,44 Гц, 2Н), 5.41 (bs, 1Н), 7.18 (d, J=8,28 Гц, 1Н), 7.75-7.78 (dd, J1=8,32 Гц, J2=2,28 Гц, 1Н), 8.57 (d, J=2,08 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=289,0.
Стадия 2. Получение (5-бром-пиридин-2-ил)-метиламина
К раствору трет-бутилового эфира (5-бром-пиридин-2-илметил)-карбаминовой кислоты (650 мг, 2,264 ммоль) в CH2Cl2 (8,0 мл) добавляли трифторуксусную кислоту (2,0 мл) при 0°C. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 4 часов. Летучие вещества удаляли при пониженном давлении с получением неочищенного указанного в заголовке соединения (500 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.19 (d, J=3,64 Гц, 2Н), 7.47 (d, J=8,4 Гц, 1Н), 8.14-8.16 (dd, J1=8,36 Гц, J2=2,36 Гц, 1Н), 8.32 (bs, 2Н), 8.77 (d, J=2,2 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=189,1.
Стадия 3. Получение (5-бром-пиридин-2-илметил)-изопропил-амина
К раствору (5-бром-пиридин-2-ил)-метиламина (0,350 г, 1,871 ммоль) в безводном ацетонитриле (7,0 мл) добавляли ацетон (119 мг, 2,05 ммоль) при комнатной температуре. После перемешивания в течение 1 часа при комнатной температуре добавляли триацетоксиборогидрид натрия (595 мг, 2,807 ммоль), затем перемешивали при комнатной температуре в течение 16 часов. Летучие вещества удаляли при пониженном давлении, разбавляли насыщенным водным раствором бикарбоната и этилацетатом. Органический слой отделяли и водный слой экстрагировали этилацетатом. Объединенный органический слой сушили над сульфатом натрия, растворитель выпаривали в вакууме и неочищенный материал очищали колоночной хроматографией, элюируя 6%-ным метанолом в CH2Cl2 с получением указанного в заголовке соединения (160 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 0.98 (d, J=6,24 Гц, 6Н), 2.66-2.73 (m, 1Н), 3.76 (s, 2Н), 7.44 (d, J=8,36 Гц, 1Н), 7.97-7.80 (dd, J1=8,4 Гц, J2=2,4 Гц, 1Н), 8.59 (d, J=2,32 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=230,9.
Стадия 4. Получение трет-бутилового эфира (4S,5R)-4-фторметил-5-{4-[6-(изопропиламино-метил)-пиридин-3-ил]-фенил}-2,2-диметил-оксазолидин-3-карбоновой кислоты
К раствору трет-бутилового эфира (4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-карбоновой кислоты (270 мг, 0,620 ммоль) в смеси диметоксиэтан: вода (8:2, 3,6 мл) добавляли Cs2CO3 (504 мг, 1,55 ммоль) и (5-бром-пиридин-2-илметил)-изопропил-амин (156 мг, 0,682 ммоль) при комнатной температуре. Реакционную смесь дегазировали с помощью азота в течение 30 минут, затем добавляли Pd(PPh3)4 (71 мг, 0,062 ммоль). Полученную реакционную смесь нагревали до 90°C в течение 4 часов. Реакционную смесь разбавляли водой и этилацетатом. Органический слой отделяли, сушили над сульфатом натрия и растворитель выпаривали в вакууме. Неочищенный материал очищали колоночной хроматографией, элюируя 5%-ным метанолом в CH2Cl2 с получением указанного в заголовке соединения (100 мг). ЖХ-MC (m/z): [М+Н]=458,2.
Стадия 5. Получение (1R,2S)-2-амино-3-фтор-1-{4-[6-(изопропиламино-метил)-пиридин-3-ил]-фенил}-пропан-1-ола в виде TFA соли
К раствору трет-бутилового эфира (4R,5R)-4-фторметил-5-{4-[6-(изопропиламино-метил)-пиридин-3-ил]-фенил}-2,2-диметил-оксазолидин-3-карбоновой кислоты (100 мг, 0,218 ммоль) в CH2Cl2 (0,17 мл) добавляли трифторуксусную кислоту (0,4 мл) при 0°C. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. Летучие вещества удаляли при пониженном давлении и неочищенный материал очищали колоночной хроматографией на щелочном оксиде алюминия с использованием 5%-ного метанола в CH2Cl2 в качестве элюента с получением указанного в заголовке соединения (70 мг, неочищенной). ЖХ-MC (m/z): [М+Н]=318,2.
Стадия 6. Получение 2,2-дихлор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[6-(изопропиламино-метил)-пиридин-3-ил]-фенил}-этил)-ацетамида
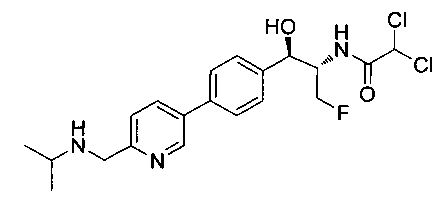
К раствору (1R,2S)-2-амино-3-фтор-1-{4-[6-(изопропиламино-метил)-пиридин-3-ил]-фенил}-пропан-1-ола в виде соли TFA (70 мг, 0,162 ммоль, неочищенный) в безводном метаноле (0,7 мл) добавляли этилдихлорацетат (51 мг, 0,324 ммоль) и триэтиламин (32 мг, 0,324 ммоль) при комнатной температуре и полученную реакционную смесь перемешивали в течение 24 часов. Летучие вещества удаляли при пониженном давлении с получением неочищенного материала и очищали колоночной хроматографией на щелочном оксиде алюминия с использованием 5%-ного метанола в CH2Cl2 с получением указанного в заголовке соединения (21 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.01 (d, J=6,2 Гц, 6Н), 2.70-2.77 (m, 1Н), 3.82 (s, 2Н), 4.19-4.24 (m, 1Н), 4.26-4.31 (m, 0.5Н), 4.39-4.43 (m, 0.5Н), 4.55-4.59 (m, 0.5H), 4.67-4.71 (m, 0.5H), 4.89 (t, J=3,64 Гц, 1H), 5.99 (d, J=4,16 Гц, 1H), 6.52 (s, 1H), 7.46 (d, J=8,08 Гц, 2H), 7.50 (d, J=7,88 Гц, 1H), 7.66 (d, J=8,2 Гц, 2H), 8.00-8.03 (dd, J1=8,24 Гц, J2=2,4 Гц, 1H), 8.64 (d, J=8,84 Гц, 1H), 8.77 (d, J=2,08 Гц, 1H). ЖХ-MC (m/z): [М+Н]=427,9.
Пример 57. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[2-(метансульфониламино-метил)-имидазо[1,2-а]пиридин-7-ил]-фенил}-этил)-ацетамида
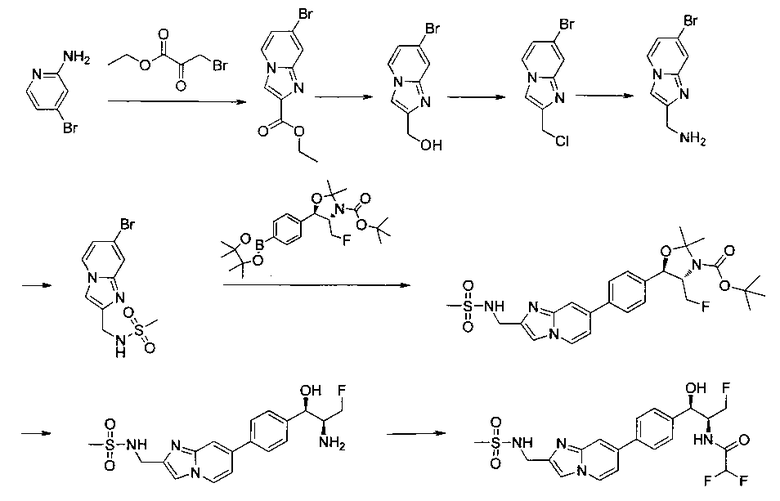
Стадия 1. Получение этилового эфира 7-бром-имидазо[1,2-а]пиридин-2-карбоновой кислоты
К перемешиваемому раствору 4-бром-пиридин-2-иламина (4 г, 23,12 ммоль) в толуоле (40 мл) добавляют этиловый эфир 3-бром-2-оксо-пропионовой кислоты (4,5 г, 23,12 ммоль). Реакционную смесь нагревают при 115°C в течение 16 часов. Реакционную смесь охлаждают до 0°C, затем разбавляют водой и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме с получением указанного в заголовке соединения (6,5 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.31 (t, J=7,2 Гц, 3H), 4.26-4.33 (q, J=7,2 Гц, 2Н), 7.18-7.20 (dd, J1=1,92 Гц, J2=7,16 Гц, 1Н), 7.98 (s, 1Н), 8.53 (d, J=7,32 Гц, 1Н), 8.57 (s, 1Н). ЖХ-MC (m/z): [М+Н]=271,0.
Стадия 2. Получение 7-бром-имидазо[1,2-а]пиридин-2-ил-метанола
К перемешиваемому раствору этилового эфира 7-бром-имидазо[1,2-а]-пиридин-2-карбоновой кислоты (3,3 г, 12,26 ммоль) в тетрагидрофуране (30 мл) добавляют алюмогидрид лития (2 М раствор в тетрагидрофуране) (5,8 мл, 12,26 ммоль). Реакционную смесь перемешивают при 0°C в течение 1 часа. Реакционную смесь разбавляют этилацетатом и смесь фильтруют через воронку Бюхнера. Остаток растворяют в насыщенном растворе бикарбоната натрия и экстрагируют 10%-ным метанолом в CH2Cl2 (30 мл). Органический слой сушат над сульфатом натрия и растворитель выпаривают в вакууме с получением неочищенного материала, который очищают колоночной хроматографией, элюируя смесью 3% MeOH:CH2Cl2, с получением указанного в заголовке соединения (0,340 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.57 (d, J=5,76 Гц, 2Н), 5.22 (t, J=5,68 Гц, 1Н), 7.00-7.02 (dd, J1=1,96 Гц, J2=7,12 Гц, 1Н), 7.76 (d, J=1,64 Гц, 1Н), 7.82 (s, 1Н), 8.48 (d, J=7,2 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=227,0.
Стадия 3. Получение 7-бром-2-хлорметил-имидазо[1,2-а]пиридина
К перемешиваемому раствору 7-бром-имидазо[1,2-а]пиридин-2-ил-метанола (0,340 г, 1,497 ммоль) в CH2Cl2 (5 мл) по каплям при 0°C добавляют SOCl2 (0,11 мл, 1,497 ммоль), затем перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь разбавляют насыщенным бикарбонатом натрия и экстрагируют CH2Cl2. Органический слой сушат над сульфатом натрия и растворитель выпаривают под вакуумом с получением указанного в заголовке соединения (0,325 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.84 (s, 2Н), 7.07-7.09 (dd, J1=1,92 Гц, J2=7,16 Гц), 7.85 (d, J=1,48 Гц, 1Н), 8.03 (s, 1Н), 8.50 (d, J=7,24 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=247,0.
Стадия 4. Получение С-(7-бром-имидазо[1,2-а]пиридин-2-ил)-метиламина
Через перемешиваемый раствор 7-бром-2-хлорметил-имидазо[1,2-а]-пиридина (0,325 г, 1,326 ммоль) в метаноле (5 мл) при 0°C в течение 30 минут продувают газообразный NH3. Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 16 часов. Растворитель концентрируют с получением указанного в заголовке соединения (0,315 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.15 (s, 2Н), 7.12-7.15 (dd J1=1,96 Гц, J2=7,24 Гц, 1Н), 7.90 (s, 1Н), 8.03 (s, 1Н), 8.32 (bs, 2Н), 8.61 (d, J=7,2 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=225,9.
Стадия 5. Получение N-(7-бром-имидазо[1,2-а]пиридин-2-илметил)-метансульфонамида
К перемешиваемому раствору С-(7-бром-имидазо[1,2-а]пиридин-2-ил)-метиламина (310 мг, 1,371 ммоль) в CH2Cl2 (5 мл) при 0°C добавляют триэтиламин (0,23 мл, 1,64 ммоль), а затем метансульфонилхлорид (0,112 мл, 1,371 ммоль), затем перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь разбавляют водой и экстрагируют CH2Cl2. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и проводят очистку посредством колоночной хроматографии с элюированием смесью 2% метанол:CH2Cl2 с получением указанного в заголовке соединения (130 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.91 (s, 3H), 4.25 (d, J=6,08 Гц, 2Н), 7.04-7.06 (dd, J1=1,88 Гц, J2=7,24 Гц, 1Н), 7.58 (t, J=6,12 Гц, 1Н), 7.81 (d, J=0,88 Гц, 1Н), 7.89 (s, 1Н), 8.51 (d, J=7,04 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=306,0.
Стадия 6. Получение трет-бутилового эфира (4S,5R)-4-фторметил-5-{4-[2-(метансульфониламино-метил)-имидазо[1,2-а]пиридин-7-ил]-фенил}-2,2-диметил-оксазолидин-3-карбоновой кислоты
К раствору N-(7-бром-имидазо[1,2-а]пиридин-2-илметил)-метансульфонамида (71 мг, 0,233 ммоль) и трет-бутилового эфира (4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-карбоновой кислоты (111 мг, 0,256 ммоль) в смеси диметоксиэтан: вода (2,2 мл:0,5 мл) в пробирке для микроволновой обработки при комнатной температуре добавляют Na2CO3 (62 мг, 0,583 ммоль). Полученную реакционную смесь дегазируют с помощью азота в течение 30 минут, затем добавляют Pd(PPh3)2Cl2 (9 мг, 0,0116 ммоль) и нагревают под воздействием микроволнового излучения при 120°C в течение 2 часов. Растворитель выпаривают и проводят очистку посредством колоночной флэш-хроматографии с элюированием 2%-ным метанолом в CH2Cl2 с получением указанного в заголовке соединения (75 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.44 (s, 9Н), 1.51 (s, 3H), 1.64 (s, 3H), 2.92 (s, 3H), 3.83-3.89 (m, 1Н), 4.28 (d, J=6,12 Гц, 2Н), 4.45-4.55 (m, 1Н), 4.65-4.75 (m, 1Н), 5.13 (d, J=7,2 Гц, 1Н), 7.27-7.29 (dd, J1=1,6 Гц, J2=7,20 Гц, 1Н), 7.56-7.58 (m, 3H), 7.82-7.88 (m, 4Н), 8.61 (d, J=7,2 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=533,1.
Стадия 7. Получение N-{7-[4-((1R,2S)-2-амино-3-фтор-1-гидрокси-пропил)-фенил]-имидазо[1,2-а]пиридин-2-илметил}-метансульфонамида
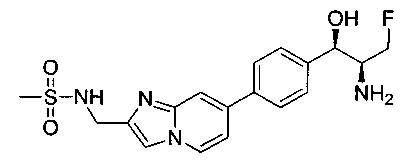
К перемешиваемому раствору трет-бутилового эфира (4S,5R)-4-фторметил-5-{4-[2-(метансульфониламино-метил)-имидазо[1,2-а]пиридин-7-ил]-фенил}-2,2-диметил-оксазолидин-3-карбоновой кислоты (75 мг, 0,14 ммоль) в CH2Cl2 (1 мл) по каплям при 0°C добавляют трифторуксусную кислоту (0,3 мл), затем перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют и обрабатывают CH2Cl2 с получением указанного в заголовке соединения (88 мг, неочищенное), которое как таковое используют на следующей стадии. ЖХ-MC (m/z): [М+Н]=393,1.
Стадия 8. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[2-(метансульфониламино-метил)-имидазо[1,2-а]пиридин-7-ил]-фенил}-этил)-ацетамида
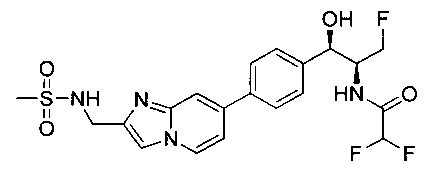
К перемешиваемому раствору N-{7-[4-((1R,2S)-2-амино-3-фтор-1-гидрокси-пропил)-фенил]-имидазо[1,2-а]пиридин-2-илметил}-метансульфонамида (88 мг, неочищенный) в метаноле (1 мл) при 0°C добавляют триэтиламин (0,05 мл, 0,346 ммоль) с последующим добавлением этилдифторацетата (0,02 мл, 0,208 ммоль), затем перемешивают при комнатной температуре в течение 20 ч. Реакционную смесь концентрируют и проводят очистку посредством колоночной флэш-хроматографии, элюируя 2,1%-ным МеОН в CH2Cl2, с последующей повторной очисткой посредством препаративной ТСХ с получением 5 мг, которые перекристаллизовывают из смеси хлороформ: гексан с получением 3 мг указанного в заголовке соединения. 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.94 (s, 3H), 4.31-4.35 (m, 3.5Н), 4.41-4.45 (m, 0.5Н), 4.54-4.57 (m, 0.5H), 4.65-4.68 (m, 0.5H), 4.89 (t, J=3,96 Гц, 1H), 5.92 (d, J=4,52 Гц, 1H), 6.20 (t, J=53,8 Гц, 1H), 7.34 (d, J=6,16 Гц, 1H), 7.46 (d, J=8,24 Гц, 2H), 7.60 (t, J=6,12, 1H), 7.78-7.83 (m, 3H), 7.90 (s, 1H), 8.63 (d, J=7,16 Гц, 1H), 8.84 (d, J=8,8 Гц, 1H), 8.93 (bs, 1H). ЖХ-MC (m/z): [M+H]=471,2.
Пример 58. Получение N-((1S,2R)-2-{4-[6-(1-амино-2,2,2-трифтор-этил)-пиридин-3-ил]-фенил}-1-фторметил-2-гидрокси-этил)-2,2-дифторацетамида
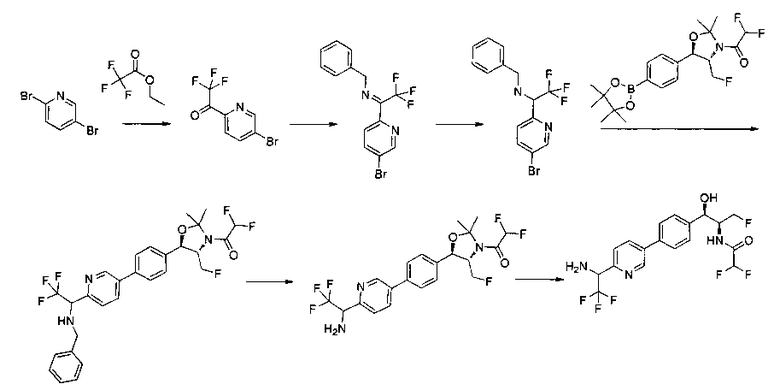
Стадия 1. Получение 1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-этанона
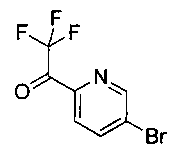
К перемешиваемому раствору 2,5-дибром-пиридина (4 г, 6,87 ммоль) в сухом тетрагидрофуране (30 мл) и сухом толуоле (40 мл) при -78°C по каплям добавляют н-бутиллитий (1,62 г, 25,31 ммоль). После перемешивания в течение 15-20 минут при -78°C добавляют этиловый эфир трифторуксусной кислоты (3,56 г, 25,31 ммоль), затем перемешивают при -78°C в течение еще 30 минут. Реакционную смесь разбавляют насыщенным раствором хлорида аммония и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и проводят очистку посредством колоночной хроматографии, элюируя 20%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (1,2 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 7.72 (d, J=8,32 Гц, 1Н), 7.85-7.88 (dd, J1=8,32 Гц, J2=2,44 Гц 1Н), 8.52 (d, J=2,36 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=255,8.
Стадия 2. Получение бензил-[1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-эт-(Е)-илиден]-амина
К раствору 1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-этанона (7,5 г, 29,52 ммоль) в сухом толуоле (30 мл) при комнатной температуре добавляют бензиламин (3,15 г, 29,52 ммоль) и этоксид титана (7,4 г, 32,48 ммоль), затем перемешивают в течение 3 часов. Реакционную смесь разбавляют насыщенным раствором бикарбоната натрия и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и проводят очистку посредством колоночной хроматографии, элюируя 20%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (0,45 г). ЖХ-MC {m/z): [М+Н]=345,0.
Стадия 3. Получение бензил-[1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-этил]-амина
К перемешиваемому раствору бензил-[1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-эт-(Е)-илиден]-амина (0,45 г, 1,31 ммоль) в метаноле (20 мл) при 0°C добавляют триацетоксиборогидрид натрия (0,55 г, 2,62 ммоль), затем перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и проводят очистку посредством колоночной хроматографии, элюируя 10%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (0,15 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 3.57-3.61 (m, 1Н), 3.64-3.73 (m, 2Н), 4.50 (m, 1Н), 7.22-7.25 (m, 1Н), 7.28-7.29 (m, 4Н), 7.73 (d, J=8,2 Гц, 1H), 7.91-7.93 (dd, J1=8,2 Гц, J2=2,24 Гц, 1Н), 8.48 (d, J=2,08 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=347,0.
Стадия 4. Получение 1-((4R,5R)-5-{4-[6-(1-бензиламино-2,2,2-трифтор-этил)-пиридин-3-ил]-фенил}-4-фторметил-2,2-диметил-оксазолидин-3-ил)-2,2-дифтор-этанона
К перемешиваемому раствору бензил-[1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-этил]-амина (0,15 г, 0,43 ммоль) и 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (0,18 г, 0,43 ммоль) в смеси толуол: этанол: вода (1:1:1; 7:7:7 мл) при комнатной температуре добавляют Na2CO3 (0,11 г, 1,08 ммоль). Полученную реакционную смесь дегазируют азотом в течение 30 минут, затем добавляют Pd(PPh3)4 (0,05 г, 0,043 ммоль) и смесь нагревают до 80°C в течение 3 часов. Реакционную смесь концентрируют в вакууме и неочищенный материал очищают посредством CombiFlash, элюируя 20%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (0,22 г). 1Н ЯМР (400 МГц, DMSO) δ: 1.52 (s, 3H), 1.60 (s, 3H), 3.59-3.69 (m, 2Н), 3.73-3.78 (m, 1Н), 4.47-4.51 (m, 0.5Н), 4.55-4.59 (m, 0.5Н), 4.66-4.72 (m, 1Н), 4.75-4.79 (m, 0.5Н), 4.80-4.93 (m, 0.5Н), 5.28 (d, J=3,68 Гц 1Н), 6.65 (t, J=51,92 Гц, 1Н), 7.23-7.24 (m, 2Н), 7.26-7.31 (m, 3H), 7.56-7.62 (m, 3H), 7.75 (d, J=8,16 Гц, 1Н), 8.06 (s, 1Н), 8.16 (d, J=8,32 Гц 1Н), 8.73 (s, 1Н). ЖХ-MC (m/z): [М+Н]=552,1.
Стадия 5. Получение 1-((4R,5R)-5-{4-[6-(1-амино-2,2,2-трифтор-этил)-пиридин-3-ил]-фенил}-4-фторметил-2,2-диметил-оксазолидин-3-ил)-2,2-дифтор-этанона
Перемешиваемый раствор 1-((4R,5R)-5-{4-[6-(1-бензиламино-2,2,2-трифтор-этил)-пиридин-3-ил]-фенил}-4-фторметил-2,2-диметил-оксазолидин-3-ил)-2,2-дифтор-этанона (0,22 г, 0,63 ммоль) в метаноле (10 мл) при комнатной температуре дегазируют азотом в течение 10 минут с последующим добавлением палладия на углероде (0,022 г, 0,0063 ммоль). Реакционную смесь перемешивают при комнатной температуре в атмосфере водорода в течение 16 часов. Реакционную смесь фильтруют через целит, концентрируют в вакууме. Неочищенный материал очищают посредством CombiFlash, элюируя 80%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (0,035 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.52 (s, 3H), 1.60 (s, 3H), 2.60-2.80 (m, 2Н), 4.54-4.58 (m, 0.5H), 4.64-4.73 (m, 2Н), 4.81-4.85 (m, 0.5Н), 4.91-4.95 (m, 1H), 5.27 (d, J=3,6 Гц, 1H), 6.65 (t, J=52,44 Гц, 1H), 7.60 (d, J=8,28 Гц, 2H), 8.03 (m, 2H), 8.14 (d, J=8,32 Гц, 2H), 8.75 (s, 1H). ЖХ-MC (m/z): [М+Н]=462,0.
Стадия 6. Получение N-((1S,2R)-2-{4-[6-(1-амино-2,2,2-трифтор-этил)-пиридин-3-ил]-фенил}-1-фторметил-2-гидрокси-этил)-2,2-дифтор-ацетамида
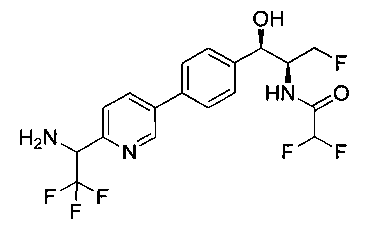
К перемешиваемому раствору 1-((4R,5R)-5-{4-[6-(1-амино-2,2,2-трифтор-этил)-пиридин-3-ил]-фенил}-4-фторметил-2,2-диметил-оксазолидин-3-ил)-2,2-дифтор-этанона (35 мг, 0,076 ммоль) в CH2Cl2 (1 мл) при 0°C добавляют трифторуксусную кислоту (1 мл), затем перемешивают до комнатной температуры в течение 3 часов. Растворитель выпаривают в вакууме и неочищенный материал разбавляют аммиачным раствором и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и проводят очистку посредством препаративной ТСХ, используя 70%-ный этилацетат в гексане, с получением указанного в заголовке соединения (0,013 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.66 (d, J=7,28 Гц, 2Н), 4.30-4.36 (m, 1.5Н), 4.40-4.44(m, 0.5Н), 4.54-4.55 (m, 0.5H), 4.61-4.68 (m, 1.5H), 4.89 (t, J=3,8 Гц, 1H), 5.93 (d, J=4,36 Гц, 1H), 6.19 (t, J=53,8 Гц, 1H), 7.45 (d, J=8,32 Гц, 2H), 8.00 (d, 1.16 Гц, 2H), 8.05 (d, J=8,28 Гц, 2H), 8.73 (s, 1H), 8.84 (d, J=8,56 Гц, 1H). ЖХ-MC (m/z): [M+H]=422,0.
Пример 59. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[6-(2,2,2-трифтор-1-метиламино-этил)-пиридин-3-ил]-фенил}-этил)-ацетамида
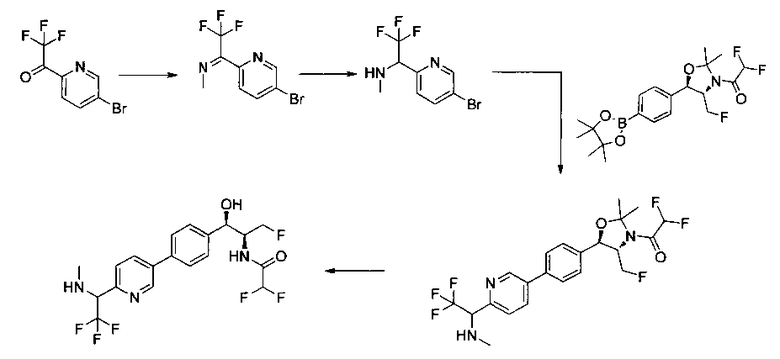
Стадия 1. Получение [1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-эт-(Е)-илиден]-метил-амина
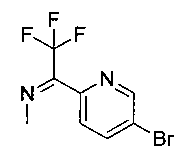
К перемешиваемому раствору 1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-этанона (1,0 г, 3,93 моль) и метиламина (20 мл, раствор в тетрагидрофуране) при 0°C добавляют этоксид титана (0,987 г, 4,33 ммоль), затем перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь разбавляют насыщенным раствором бикарбоната натрия и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, концентрируют и проводят очистку посредством колоночной хроматографии, элюируя 20%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (0,10 г). 1Н ЯМР (400 МГц, DMSO) δ: 3.25-3.26 (m, 3H), 7.85-7.90 (m, 2Н), 8.49 (s, 1Н). ЖХ-MC (m/z): [М+Н]=266,8.
Стадия 2. Получение [1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-этил]-метил-амина
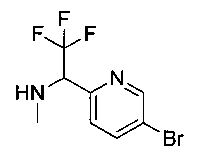
К перемешиваемому раствору [1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-эт-(Е)-илиден]-метил-амина (0,1 г, 0,375 ммоль) в метаноле (5 мл) при 0°C добавляют цианоборогидрид натрия (0,027 г, 0,449 ммоль), затем перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь разбавляют насыщенным раствором бикарбоната натрия и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и проводят очистку посредством колоночной хроматографии, элюируя 10%-ным этилацетатом в гексане, с получением неочищенного указанного в заголовке соединения (0,075 г, неочищенное). 1Н ЯМР (400 МГц, DMSO) δ: 2.21 (d, J=5,32 Гц, 3H), 3.04 (m, 1Н), 4.46 (m, 1Н), 7.74 (d, J=8,24 Гц, 1Н), 8.87 (dd, J1=2,4 Гц, J2=8,28 Гц, 1Н), 8.48 (d, J1=2,36 Гц, 1Н). ЖХ-MC (m/z): [М+Н]=271,2.
Стадия 3. Получение 2,2-дифтор-1-((4S,5R)-4-фторметил-2,2-диметил-5-{4-[6-(2,2,2-трифтор-1-метиламино-этил)-пиридин-3-ил]-фенил}-оксазолидин-3-ил)этанона
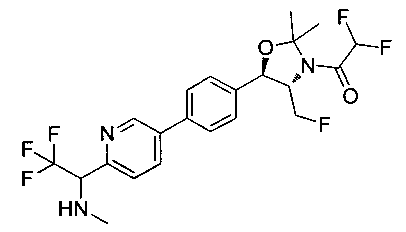
К перемешиваемому раствору [1-(5-бром-пиридин-2-ил)-2,2,2-трифтор-этил]-метил-амина (0,075 г, 0,279 ммоль) и 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (0,115 г, 0,279 ммоль) в смеси толуол: этанол: вода (3:3:3 мл) при комнатной температуре добавляют Na2CO3 (0,073 г, 0,697 ммоль). Полученную реакционную смесь дегазируют азотом в течение 30 минут, после чего добавляют Pd(PPh3)4 (0,032 г, 0,028 ммоль) и нагревают до 80°C в течение 3 ч. Реакционную смесь концентрируют в вакууме, затем разбавляют водой и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия и выпаривают в вакууме. Неочищенный материал очищают посредством CombiFlash, элюируя 20%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (0,120 г). 1Н ЯМР (400 МГц, DMSO) δ: 1.52 (s, 3H), 1.60 (s, 3H), 2.25 (d, J=5,76 Гц, 3H), 3.01-3.05 (m, 1Н), 4.44-4.48 (m, 1Н), 4.56-4.58 (m, 0.5Н), 4.67-4.72 (m, 1Н), 4.81-4.88 (m, 0.5Н), 4.90-5.00 (m, 1Н), 5.27 (d, J=3,68 Гц, 1Н), 6.65 (t, J=52,36 Гц, 1Н), 7.60 (d, J=8,24 Гц, 2Н), 7.98-8.00 (dd, J1=2,04 Гц, J2=8,2 Гц, 1Н), 8.06 (d, J=8,24 Гц, 1Н), 8.15 (d, J=8,32, 2Н), 8.73 (d, J=1,8 Гц, 1Н). ЖХ-MC (m/z): [М-Н]=476,0.
Стадия 4. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[6-(2,2,2-трифтор-1-метиламино-этил)-пиридин-3-ил]-фенил}-этил)-ацетамида
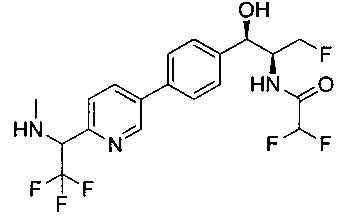
К раствору 2,2-дифтор-1-((4S,5R)-4-фторметил-2,2-диметил-5-{4-[6-(2,2,2-трифтор-1-метиламино-этил)-пиридин-3-ил]-фенил}-оксазолидин-3-ил)этанона (0,12 г, 0,235 ммоль) в CH2Cl2 (2 мл) при 0°C добавляют трифторуксусную кислоту (0,5 мл), затем перемешивают до комнатной температуры в течение 2 часов. Реакционный растворитель выпаривают в вакууме, затем проводят разбавление раствором бикарбоната натрия и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и проводят очистку посредством CombiFlash, элюируя 5%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (0,028 г). 1Н ЯМР (400 МГц, DMSO) δ: 2.25 (d, J=5,84 Гц, 3H), 3.00-3.04 (m, 1Н), 4.31-4.35 (m, 1.5Н), 4.41-4.46 (m, 1.5Н), 4.55-4.56 (m, 0.5H), 4.65-4.68 (m, 0.5H), 4.90 (m, 1H), 5.93 (d, J=4,36 Гц, 1Н), 6.19 (d, J=53,72 Гц, 1H), 7.46 (d, J=8,32 Гц, 2H), 7.95-7.98 (dd, J1=1,96 Гц, J2=8,32 Гц,1Н), 8.03 (d, J=8,32 Гц, 1Н), 8.06 (d, J=8,48 Гц, 2Н), 8.71 (d, J=1,88 Гц, 1Н), 8.84 (d, J=8,72 Гц, 1Н). ЖХ-МС (m/z): [М+Н]=436,0.
Пример 60. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[6-(1-морфлин-4-ил-этил)-пиридин-3-ил]-фенил}-этил)-ацетамида
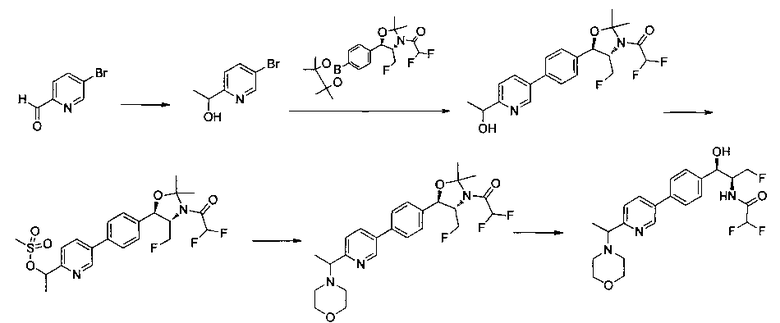
Стадия 1. Получение 1-(5-бром-пиридин-2-ил)-этанола
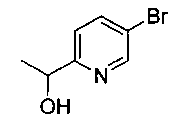
К перемешиваемому раствору 5-бром-пиридин-2-карбальдегида (10 г, 53,76 ммоль) в тетрагидрофуране (200 мл) при 0°C добавляют по каплям бромид метилмагния (45 мл, 64,51 ммоль). Реакционную смесь перемешивают при 0°C в течение 6 часов и разбавляют, используя насыщенный раствор хлорида аммония, и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, концентрируют и очищают посредством CombiFlash, используя 25%-ный этилацетат в гексане в качестве элюента, с получением указанного в заголовке соединения (8 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.34 (d, J=6,52 Гц, 3H), 4.66-4.72 (m, 1Н), 5.47(d, J=4,76 Гц, 1Н), 7.48 (d, 8.36 Гц, 1Н), 8.021 (dd, J1=2,44 Гц, J2=8,44 Гц 1Н), 8.58 (d, J=2,36 Гц, 1Н). ЖХ-МС (m/z): 203,9 [М+Н].
Стадия 2. Получение 2,2-дифтор-1-((4S,5R)-4-фторметил-5-{4-[6-(1-гидрокси-этил)-пиридин-3-ил]-фенил}-2,2-диметил-оксазолидин-3-ил)-этанона
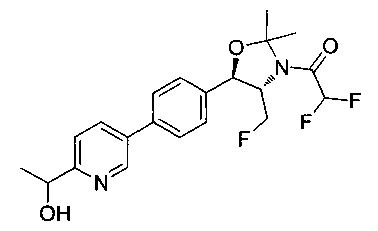
К перемешиваемому раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (3 г, 7,26 ммоль) и 1-(5-бром-пиридин-2-ил)-этанола (1,46 г, 7,26 ммоль) в смеси толуол: этанол: вода (15:15:7 мл) при комнатной температуре добавляют Na2CO3 (2,3 г, 21,79 ммоль). Полученную реакционную смесь дегазируют азотом в течение 30 минут, затем добавляют Pd(PPh3)4 (0,839 г, 0,726 ммоль). Реакционную смесь нагревают до 100°C в течение 16 часов. Растворитель выпаривают в вакууме и неочищенный материал очищают посредством CombiFlash, используя 55%-ный этилацетат в гексане в качестве элюента, с получением указанного в заголовке соединения (2,2 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.39 (d, J=6,6 Гц, 3H), 1.53 (s, 3H), 1.60 (s, 3H), 4.57-4.58 (m, 0.5Н), 4.69-4.70 (m, 1Н), 4.75-4.77 (m, 1Н), 4.78-7.80 (m, 0.5Н), 4.89-4.94 (m, 1H), 5.26 (d, J=3,88 Гц, 1H), 5.41 (d, J=4,6 Гц, 1H), 6.64 (t, J1=52,32 Гц, 1H), 7.57-7.64 (m, 3H), 7.77 (d, J=8,16 Гц, 2H), 8.08 (dd, J1=8,2 Гц, J2=2,32 Гц, 1H), 8.80 (d, J=2,04 Гц, 1Н). ЖХ-МС (m/z): 409 [М+Н].
Стадия 3. 1-(5-{4-[(4S,5R)-3-(2,2-Дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-ил)-этиловый эфир метан-сульфоновой кислоты
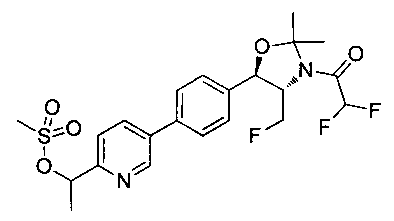
К перемешиваемому раствору 2,2-дифтор-1-((4S,5R)-4-фторметил-5-{4-[6-(1-гидрокси-этил)-пиридин-3-ил]-фенил}-2,2-диметил-оксазолидин-3-ил)-этанона (2,2 г, 5,39 ммоль) в CH2Cl2 (25 мл) добавляют триэтиламин (1,51 мл, 10,78 ммоль) и метансульфонилхлорид (0,527 мл, 6,47 ммоль). Реакционную смесь перемешивают при 0°C в течение 30 минут, затем разбавляют, используя насыщенный раствор бикарбоната, и экстрагируют CH2Cl2 (30 мл). Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и проводят очистку посредством CombiFlash, используя 45%-ный этилацетат в гексане в качестве элюента, с получением указанного в заголовке соединения (1,8 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.53 (s, 3H), 1.60 (s, 3H), 1.68 (d, J=6,56 Гц, 3H), 3.21 (s, 3H), 4.54-4.58 (m, 0.5Н), 4.66-4.71 (m, 1Н), 4.81-4.84 (m, 0.5Н), 4.90-4.95 (m, 1Н), 5.27 (d, J=3,92 Гц, 1Н), 5.79-5.84 (m, 1Н), 6.64 (t, J=52,24 Гц, 1Н), 7.60-7.63 (m, 3H), 7.81 (d, J=8,2 Гц, 2Н), 8.17-8.20 (dd, J1=8,08 Гц, J2=2,24 Гц, 1Н), 8.92 (d, J=2,00 Гц, 1Н). ЖХ-МС (m/z): 487,1 [М+Н].
Стадия 4. 2,2-Дифтор-1-((4S,5R)-4-фторметил-2,2-диметил-5-{4-[6-(1-морфолин-4-ил-этил)-пиридин-3-ил]-фенил}-оксазолидин-3-ил)-этанон
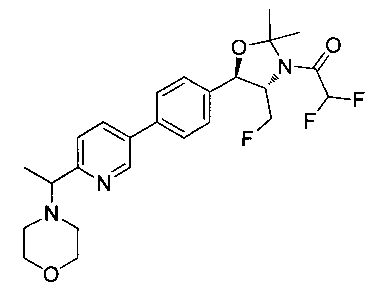
К перемешиваемому раствору 1-(5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-ил)-этилового эфира метансульфоновой кислоты (0,150 г, 0,309 ммоль) в ацетонитриле (2 мл) добавляют диизопропилэтиламин (0,161 мл, 0,926 ммоль) и морфолин (0,054 мл, 0,617 ммоль). Реакционную смесь перемешивают при 60°C в течение 16 ч. Растворитель выпаривают в вакууме и неочищенный материал очищают посредством CombiFlash, используя смесь 1% метанол: CH2Cl2 в качестве элюента, с получением указанного в заголовке соединения (0,101 г). 1Н ЯМР (400 МГц, CDCl3) δ 1.33 (d, J=6,48 Гц, 3H), 1.53 (s, 3H), 1.60 (s, 3H), 2.32-2.33 (m, 2Н), 2.45-2.50 (m, 2Н), 3.53-3.58 (m, 4Н), 3.60-3.63 (m, 1Н), 4.54-4.58 (m, 0.5Н), 4.66-4.72 (m, 1Н), 4.80-4.84 (m, 0.5Н), 4.90-4.94 (m, 1Н) 5.26 (d, J=4 Гц, 1Н), 6.64 (t, J=52,32 Гц, 1Н), 7.50 (d, J=8,2 Гц, 2Н), 7.59 (d, J=8,12 Гц, 2Н), 7.78 (d, J=8,24 Гц, 2Н), 8.06-8.10 (dd, J1=8,8 Гц, J2=6,48 Гц, 1Н), 8.83 (d, J=2,24 Гц, 1Н). ЖХ-МС - не ионизированный.
Стадия 5. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[6-(1-морфолин-4-ил-этил)-пиридин-3-ил]-фенил}-этил)-ацетамида
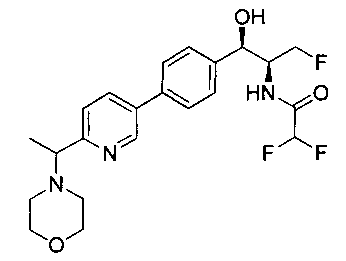
К перемешиваемому раствору 2,2-дифтор-1-((4S,5R)-4-фторметил-2,2-диметил-5-{4-[6-(1-морфолин-4-ил-этил)-пиридин-3-ил]-фенил}-оксазолидин-3-ил)-этанона (0,101 г, 0,309 ммоль) в CH2Cl2 (5 мл) при 0°C добавляют трифторуксусную кислоту (1 мл). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 4 часов. Растворитель выпаривают в вакууме и неочищенный материал разбавляют насыщенным раствором бикарбоната и экстрагируют смесью 10% MeOH:DCM. Объединенные органические слои сушат над сульфатом натрия, концентрируют и очищают посредством CombiFlash, используя смесь 5% метанол: CH2Cl2 в качестве элюента, с получением целевого соединения (0,076 г): 1Н ЯМР (400 МГц, CDCl3) δ: 1.33 (d, J=2,52 Гц, 3H), 2.32-2.35 (m, 2Н), 2.49-2.50 (m, 2Н), 3.55-3.57 (m, 4Н), 3.59-3.60 (m, 1Н), 4.29-4.30 (m, 1Н), 4.32 (m, 0.5Н), 4.40-4.44 (m, 0.5Н), 4.54-4.55 (m, 0.5H), 4.64-4.66 (m, 0.5H), 4.88 (bs, 1H), 5.92 (d, J=3,96 Гц, 1H), 6.20 (t, J=53,72 Гц, 1H), 7.45 (d, J=8,24 Гц, 2H), 7.49 (d, J=8,24 Гц, 1H), 7.69 (d, J=8,28 Гц, 2H), 8.03-8.06 (dd, J1=8,2 Гц, J2=2,4 Гц, 1H), 8.80 (d, J=2,04 Гц,1Н), 8.86 (d, J=8,56 Гц, 1H). ЖХ-МС (m/z): 438,1 [М+Н].
Пример 61. Получение 2,2-дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[6-(1-метиламино-этил)-пиридин-3-ил]-фенил}-этил)-ацетамида
Это соединение получают, используя методику, аналогичную методике из Примера 60.
2,2-Дифтор-1-((4S,5R)-4-фторметил-2,2-диметил-5-{4-[6-(1-метиламино-этил)-пиридин-3-ил]-фенил}-оксазолидин-3-ил)-этанон
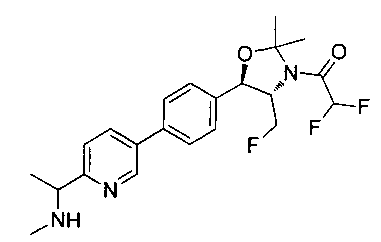
1Н ЯМР (400 МГц, DMSO-d6) δ: 1.41 (d, J=6,72 Гц, 3H), 1.52 (s, 3H), 1.60 (s, 3H), 2.36 (s, 3H), 4.14-4.16 (m, 1Н), 4.45-4.61 (m, 0.5Н), 4.69-4.72 (m, 1Н), 4.80-4.90 (m, 0.5Н), 4.91-4.96 (m, 1Н), 5.28 (d, J=3,68 Гц, 1Н), 6.65 (t, J=52,28 Гц, 1Н), 7.57 (d, J=8,12 Гц, 1Н), 7.61 (d, J=8,12 Гц, 2Н), 7.81(d, J=8,24 Гц, 2Н), 8.15-8.18 (dd, J1=8,12 Гц, J2=2,28 Гц, 1Н), 8.91(d, J=2 Гц, 1Н). ЖХ-МС (m/z): 422 [М+Н].
2,2-Дифтор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[6-(1-метиламино-этил)-пиридин-3-ил]-фенил}-этил)-ацетамид
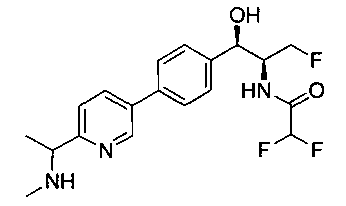
1Н ЯМР (400 МГц, DMSO-d6) δ: 1.30 (d, J=6,64 Гц, 3H), 2.21 (s, 3H), 3.80 (bs, 1Н), 4.29-4.31 (m, 1.5Н), 4.40-4.44 (m, 0.5Н), 4.54-4.55 (m, 0.5H), 4.64-4.68 (m, 0.5H), 4.87 (bs, 1H), 5.91 (d, J=4,4 Гц,1Н), 6.20 (t, J=53,84 Гц, 1H), 7.45 (d, J=8,0 Гц, 2H), 7.50 (d, J=8,16 Гц, 1H), 7.69 (d, J=8,08 Гц, 2H), 8.07 (m, 1H), 8.82-8.86 (m, 2H). ЖХ-МС (m/z): 382,2 [М+Н].
Пример 62. Получение 2,2-дифтор-N-[(1S,2R)-2-(4-{6-[1-(2-фтор-этиламино)-этил]-пиридин-3-ил}-фенил)-1-фторметил-2-гидрокси-этил]-ацетамида
Стадия 1. Получение 2,2-дифтор-1-[(4S,5R)-5-(4-{6-[1-(2-фтор-этиламино)-этил]-пиридин-3-ил}-фенил)-4-фторметил-2,2-диметил-оксазолидин-3-ил]-этанона
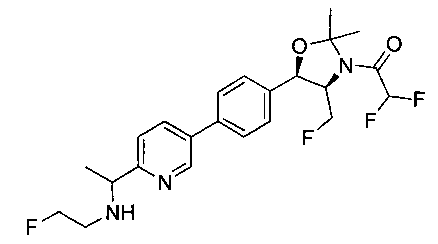
Это соединение получают, используя методику, аналогичную методике из Примера 60.
1Н ЯМР (400 МГц, DMSO-d6) δ: 1.30 (d, J=6,68 Гц, 3H), 1.53 (s, 3H), 1.60 (s, 3H), 2.54-2.67 (m, 2Н), 3.83-3.86 (m, 1Н), 4.36-4.39 (m, 1Н), 4.48-4.51 (m, 1Н), 4.56-4.58 (m, 0.5Н), 4.68-4.70 (m, 1Н), 4.80-4.83 (m, 0.5Н), 4.89-4.93 (m, 1H), 5.26 (d, J=3,96 Гц, 1H), 6.64 (t, J=52,2 Гц, 1H), 7.53 (d, J=8,12 Гц, 1H), 7.59 (d, J=8,08 Гц, 2H), 7.78 (d, J=8,12 Гц, 2H) 8.05-8.08 (dd, J1=8,04 Гц, J2=2,2 Гц, 1H), 8.2 (d, J=2,12 Гц, 1H). ЖХ-МС (m/z): 454 [М+Н].
Стадия 2. Получение 2,2-дифтор-N-[(1S,2R)-2-(4-{6-[1-(2-фтор-этиламино)-этил]-пиридин-3-ил}-фенил)-1-фторметил-2-гидрокси-этил]-ацетамида
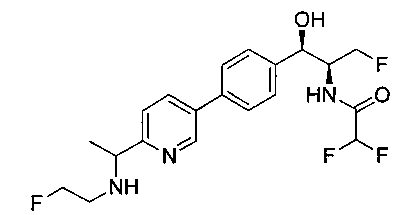
1Н ЯМР (400 МГц, DMSO-d6) δ 1.29 (d, J=6,72 Гц, 3H), 2.36-2.38 (m, 1Н), 2.60-2.61 (m, 1Н), 3.84 (bs, 1Н), 4.29-4.32 (m, 1.5Н), 4.35-4.38 (m, 1Н), 4.39-4.42 (m, 0.5Н), 4.49-4.50 (m, 1H), 4.55-4.58 (m, 0.5H), 4.65-4.70 (m, 0.5H), 4.88 (bs, 1H), 5.93 (bs, 1H), 6.20 (t, J=53,76 Гц, 1H), 7.45 (d, J=8,24 Гц, 2H), 7.51 (d, J=8,12 Гц, 1H), 7.69 (d, J=8,28 Гц, 2H), 8.03-8.06 (dd, J1=2,4 Гц, J2=8,2 Гц, 1H), 8.80 (d, J=2,2 Гц, 1H), 8.87 (d, J=8,64 Гц, 1H). ЖХ-МС (m/z): [M+H]=414,2.
Пример 63. 2,2-Дифтор-N-[(1S,2R)-2-(4-{6-[1-(3-фтор-азетидин-1-ил)-этил]-пиридин-3-ил}-фенил)-1-фторметил-2-гидрокси-этил]-ацетамида
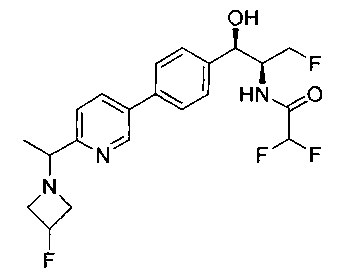
Это соединение получают, используя методику, аналогичную методике из Примера 60.
2,2-Дифтор-1-[(4S,5R)-5-(4-{6-[1-(3-фтор-азетидин-1-ил)-этил]-пиридин-3-ил}-фенил)-4-фторметил-2,2-диметил-оксазолидин-3-ил]-этанон
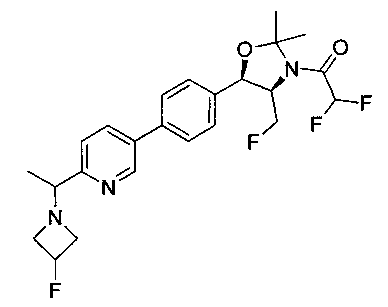
ЖХ-МС (m/z): 426 [М+Н].
2,2-Дифтор-N-[(1S,2R)-2-(4-{6-[1-(3-фтор-азетидин-1-ил)-этил]-пиридин-3-ил}-фенил)-1-фторметил-2-гидрокси-этил]-ацетамид
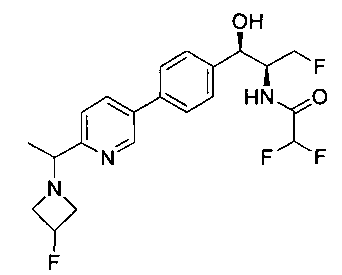
1Н ЯМР (400 МГц, DMSO-d6) δ: 1.19 (d, J=6,52 Гц, 3H), 3.03-3.31 (m, 2Н), 3.52-3.60 (m, 2Н), 4.28-4.38 (m, 1.5Н), 4.40-4.44 (m, 0.5Н), 4.54-4.55 (m, 0.5H), 4.64-4.67 (m, 0.5H), 4.87 (m, 1H), 5.08-5.10 (m, 0.5H), 5.22-5.25 (m, 0.5H), 5.93 (bs, 1H), 6.20 (t, J=53,68 Гц, 1H), 7.43-7.46 (m, 3H), 7.68 (d, J=8,24 Гц, 2H), 8.03-8.05 (dd, J1=2,36 Гц, J2=8,12 Гц, 1H), 8.79 (d, J=2,04 Гц, 2H), 8.87 (d, J=8,68 Гц, 1H). ЖХ-МС (m/z): 426,2 [М+Н].
Пример 64. Получение N-((1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
Стадия 1. Получение трет-бутил-(1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)цикло-пропил)-карбамата
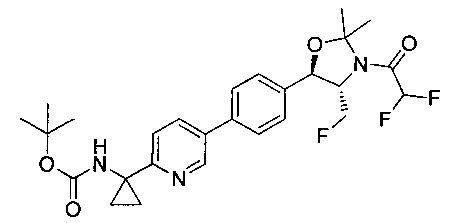
Следуя общей методике из Примера 22, стадия 1, и осуществляя незначительные модификации, но используя трет-бутил-(1-(5-бромпиридин-2-ил)циклопропил)карбамат (описан ранее в WO 2012/076063, Description 11, стр. 41), получают указанное в заголовке соединение (535 мг) m/z (ХИ) 520 [М+Н].
Стадия 2. Получение N-((1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
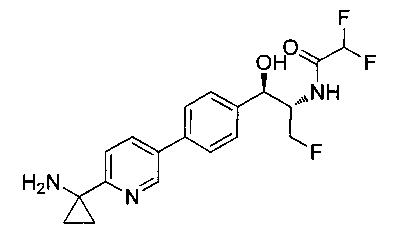
Следуя общей методике из Примера 22, стадия 2, и осуществляя незначительные модификации, но используя трет-бутил-(1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)-пиридин-2-ил)циклопропил)-карбамат, получают указанное в заголовке соединение (323 мг). 1Н ЯМР (300 МГц, DMSO-d6) δ: 0.9-1.05 (m, 2Н), 1.2-1.3 (m, 2Н), 2.45 (s, 2Н), 4.2-4.4 (m, 1.5Н), 4.4-4.6 (m, 1Н), 4.6-4.7 (m, 0.5Н), 4.85 (t, 1H), 5.9 (d, 1H), 6.2 (t, 1H), 7.45 (d, 2H), 7.65 (d, 2H), 7.8 (d, 1H), 8.0 (d, 1H), 8.70 (s, 1H), 8.8 (d, 1H). m/z (ХИ) 380 [М+Н].
Способами, известными из уровня техники, могут быть получены следующие производные указанного в заголовке соединения из Примера 64, включая соединения, описанные в Примере 95 ниже:
N-((1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамид;
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-гидрофосфат натрия и
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-дигидрофосфат.
Пример 65. Получение N-((1S,2R)-2-{4-[6-(ацетиламино-метил)-пиридин-3-ил]-фенил}-1-фторметил-2-гидрокси-этил)-2,2-дихлор-ацетамид
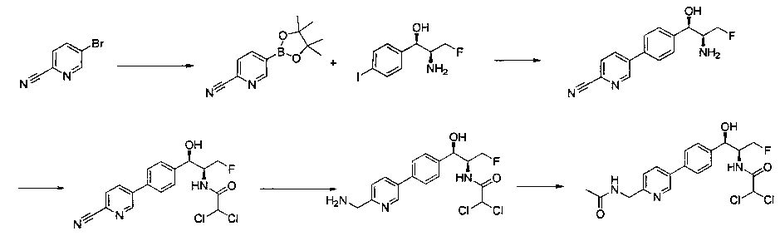
Стадия 1. Получение 5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-пиридин-2-карбонитрила
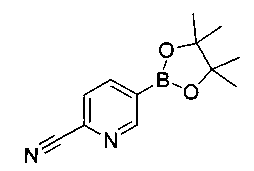
К раствору 5-бром-пиридин-2-карбонитрила (2 г, 10,92 ммоль) в диоксане (60 мл) добавляют биспинаколатодибор (3,33 г, 13,11 ммоль) и ацетат калия (1,6 г, 16,39 ммоль) и реакционную смесь дегазируют и добавляют трициклогексилфосфин (0,4 г, 1,42 ммоль) и Pd(dba)3 (0,5 г, 0,546 ммоль) и нагревают до 90°C в течение ночи. Разбавляют водой и этилацетатом. Органический слой отделяют, сушат над сульфатом натрия и растворитель выпаривают в вакууме с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 6-8%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (1,0 г): 1Н ЯМР (400 МГц, CDCl3) δ: 1.35 (s, 12H), 7.66 (d, J=7,76 Гц, 1H), 8.17 (d, J=8,88 Гц, 1H), 9.00 (s, 1Н).
Стадия 2. Получение 5-[4-((1R,2S)-2-амино-3-фтор-1-гидрокси-пропил)-фенил]-пиридин-2-карбонитрила
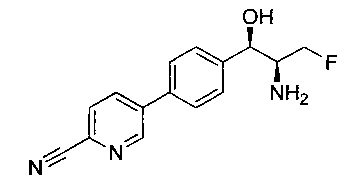
К раствору 5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-пиридин-2-карбонитрила (0,5 г, 2,17 ммоль) в диметоксиэтане (10 мл) и воде (3 мл) добавляют (1R,2S)-2-амино-3-фтор-1-(4-йод-фенил)-пропан-1-ол (0,65 г, 2,20 ммоль) и Cs2CO3 (2,15 г, 6,59 ммоль) и дегазируют азотом с последующим добавлением Pd(PPh3)4 (0,25 г, 0,21 ммоль) и нагревают реакционную смесь до 90°C в течение 1,5 часов. Растворитель выпаривают в вакууме с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 2,5-3%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (0,3 г) в виде материала с примесью. 1Н ЯМР (400 МГц, DMSO-d6) δ: 3.09-3.18 (m, 1Н), 3.48 (s, 1Н), 4.24-4.28 (m, 0.5Н), 4.36-4.41 (m, 1Н), 4.49-4.52 (m, 0.5Н), 4.64 (d, J=6,16 Гц, 1H), 7.53 (d, J=8,16 Гц, 2H), 7.59 (d, J=8,24 Гц, 2H), 7.76 (d, J=8,2 Гц, 1H), 8.0 (dd, J1=8,16 Гц, J2=2,32 Гц, 1H), 8.93 (d, J=1,76 Гц, 1H). ЖХ-МС (m/z): [M+H]=272,00.
Стадия 3. Получение 2,2-дихлор-N-{(1S,2R)-2-[4-(6-циано-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-ацетамида
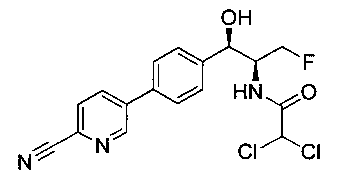
К раствору 5-(4-((1R,2S)-2-амино-3-фтор-1-гидроксипропил)фенил)-пиридин-2-карбонитрила (0,5 г, 1,1 ммоль) в метаноле (5 мл) добавляют триэтиламин (0,22 г, 2,2 ммоль) и этилдихлорацетат (0,34 г, 2,2 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Растворитель выпаривают в вакууме и неочищенный материал очищают колоночной хроматографией на силикагеле, используя смесь метанол/CH2Cl2, с получением указанного в заголовке соединения (200 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.26-4.35 (m, 1Н), 4.36-4.37 (m, 0.5Н), 4.45-4.51 (m, 1Н), 4.59-4.63 (m, 0.5Н), 5.02 (d, J=3,28 Гц, 1H), 5.84 (s, 1H), 7.47 (d, J=8,2 Гц, 2H), 7.54 (d, J=8,24 Гц, 2H), 7.76 (d, J=8,04 Гц, 1H), 7.96 (dd, J1=8,08 Гц, J2=2,2 Гц, 1Н), 8.85 (d, J=1,64 Гц, 1Н). ЖХ-МС (m/z): [М+Н]=379,70.
Стадия 4. Получение N-{(1S,2R)-2-[4-(6-аминометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дихлор-ацетамида
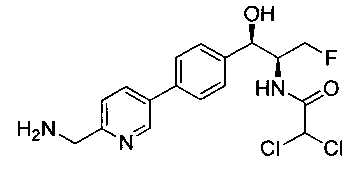
К охлажденному до -40°C на льду раствору алюмогидрида лития (0,048 г, 1,33 ммоль, 4,0 экв.) в тетрагидрофуране (10 мл) при -40°C добавляют раствор 2,2-дихлор-N-((1R,2S)-1-(4-(6-цианопиридин-3-ил)фенил)-3-фтор-1-гидрокси-пропан-2-ил)ацетамида (0,13 г, 0,34 ммоль) в тетрагидрофуране (5 мл). Три раза добавляют дополнительное количество алюмогидрида лития (0,012 г, 0,33 ммоль) и реакционную смесь перемешивают при -40°C в течение 4 часов. Реакционную смесь гасят насыщенным водным сульфатом натрия и перемешивают в течение 15 минут с последующим фильтрованием. Фильтрат выпаривают в вакууме и неочищенный материал очищают колоночной хроматографией на силикагеле, элюируя смесью метанол/CH2Cl2 и аммиаком, с получением указанного в заголовке соединения (43 мг): 1Н ЯМР (400 МГц, CDCl3) δ: 4.20-4.24 (m, 2Н), 4.27-4.29 (m, 0.5Н), 4.39-4.43 (m, 0.5Н), 4.56-4.59 (m, 1Н), 4.67-4.69 (m, 0.5Н), 4.89-4.90 (m, 1Н), 5.99 (d, J=4,2 Гц, 1Н), 6.52 (s, 1Н), 7.46-7.48 (m, 3H), 7.64-7.69 (m, 3H), 8.06-8.10 (m, 1Н), 8.63 (d, J=8,76 Гц, 1Н). ЖХ-МС (m/z): [М+Н]=386,10.
Стадия 5. Получение N-((1S,2R)-2-{4-[6-(ацетиламино-метил)-пиридин-3-ил]-фенил}-1-фторметил-2-гидрокси-этил)-2,2-дихлор-ацетамида
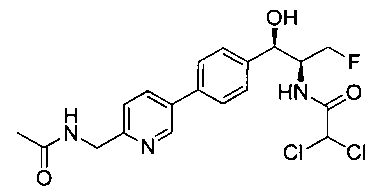
К перемешиваемому раствору неочищенного N-{(1S,2R)-2-[4-(6-аминометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дихлор-ацетамида (30 мг, 0,077 ммоль) в CH2Cl2 (2 мл) добавляют триэтиламин (8 мг, 0,078 ммоль) и уксусный ангидрид (20 мг, 0,198) и реакционную смесь перемешивают при комнатной температуре в течение 6 ч. Разбавляют водой и экстрагируют CH2Cl2, сушат над сульфатом натрия и растворитель выпаривают в вакууме с получением неочищенного материала, который очищают посредством преп. ТСХ на силикагеле, используя в качестве подвижной фазы 10%-ный метанол в CH2Cl2, с получением указанного в заголовке соединения (5 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: (s, 3H), 4.30-4.34 (m, 1Н), 4.45 (d, J=5,96 Гц, 2Н), 4.48-4.51 (m, 1Н), 4.55-4.58 (m, 1Н), 5.00 (d, 1Н), 5.95 (d, 1Н), 6.12 (s, 1Н), 6.99 (bs, 1Н), 7.34-7.36 (m, 1Н), 7.47-7.50 (m, 2Н), 7.62-7.67 (m, 2Н), 7.96 (dd, J1=8,2 Гц, J2=2,4 Гц, 1Н), 8.76 (d, J=2,2 Гц, 1Н). ЖХ-МС (m/z): [М+Н]=428,10.
Пример 66. Получение N-{(1S,2R)-2-[4-(4-аминометил-[1,2,3]триазол-1-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дихлор-ацетамида
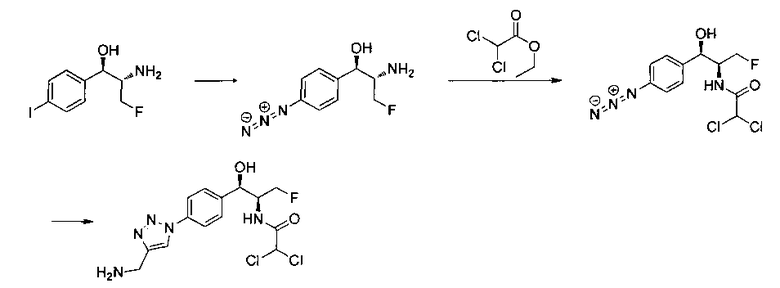
Стадия 1. Получение (1R,2R)-2-амино-1-(4-азидо-фенил)-3-фтор-пропан-1-ола
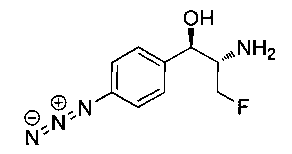
К перемешиваемому раствору (1R,2R)-2-амино-3-фтор-1-(4-йод-фенил)-пропан-1-ола (1 г, 3,38 ммоль) в смеси диметилсульфоксид: вода (9:1, 10 мл) добавляют NaN3 (0,26 г, 3,99 ммоль), Na-аскорбат (0,1 г, 0,50 ммоль), CuSO4.5H2O (0,17 г, 0,68 ммоль), L-пролин (78 мг, 0,67 ммоль), K2CO3 (93 мг, 0,67 ммоль) и полученную реакционную смесь нагревают до 60°C в течение 5 часов. Разбавляют холодной водой и экстрагируют этилацетатом и промывают избытком воды и рассола. Органический слой сушат над сульфатом натрия и растворитель выпаривают в вакууме с получением неочищенного указанного в заголовке соединения (0,6 г, неочищенное), используемого как таковое на следующей стадии. ЖХ-МС (m/z): [М+Н]=211,2.
Стадия 2. Получение ((N-[(1S,2R)-2-(4-азидо-фенил)-1-фторметил-2-гидрокси-этил]-2,2-дихлор-ацетамида
К перемешиваемому раствору (1R,2R)-2-амино-1-(4-азидо-фенил)-3-фтор-пропан-1-ола (600 мг, 2,83 ммоль) в метаноле (6,5 мл) добавляют триэтиламин (0,57 г, 5,64 ммоль) и этилдихлорацетат (0,88 г, 5,60 ммоль). Полученную реакционную смесь перемешивают под азотом при комнатной температуре в течение 16 часов. Растворитель выпаривают в вакууме с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 0,5%-ным метанолом в дихлорметане, с получением указанного в заголовке соединения (150 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.40-4.41 (m, 1Н), 4.24-4.26 (m, 0.5Н), 4.36-4.38 (m, 0.5Н), 4.52-4.54 (m, 0.5H), 4.64-4.66 (m, 0.5H), 4.84 (t, 1H, J=3,64 Гц), 5.96 (d, 1H, J=4,24 Гц), 6.49 (s, 1H), 7.05 (d, 2H, J=8,48 Гц), 7.38 (d, 2H, J=8,44 Гц), 8.58 (d, 1H, J=8,98 Гц). ЖХ-МС (m/z): [M-H]=318,8.
Стадия 3. Получение N-{(1S,2R)-2-[4-(4-аминометил-[1,2,3]триазол-1-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дихлорацетамида
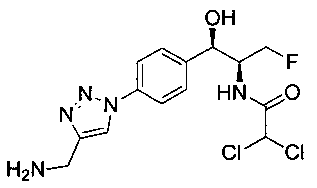
К раствору ((N-[(1S,2R)-2-(4-азидо-фенил)-1-фторметил-2-гидрокси-этил]-2,2-дихлор-ацетамида (150 мг, 0,465 ммоль) в смеси трет-бутиловый спирт: вода (1:1,3 мл) добавляют Na-аскорбат (12 мг, 0,060 ммоль), CuSO4.5H2O (3 мг, 0,012 ммоль), пропаргиламин (26 мг, 0,472 ммоль) и полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Растворитель выпаривают в вакууме с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 25-40%-ным метанолом в дихлорметане, и промывают диэтиловым эфиром и сушат под вакуумом с получением указанного в заголовке соединения (25 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 3.84 (s, 2Н), 4.23-4.29 (m, 1Н), 4.30-4.34 (m, 0.5Н), 4.42-4.46 (m, 0.5H), 4.58-4.61 (m, 0.5H), 4.69-4.73 (m, 0.5H), 4.93 (bs, 1H), 6.07 (d, 1H, J=4,28 Гц), 6.49 (s, 1H), 7.54 (d, 2H, J=8,48 Гц), 7.81 (d, 2H, J=8,52 Гц), 8.57 (s, 1H), 8.61 (d, 1H, J=5,6 Гц). ЖХ-МС (m/z): [M+H]=376,2.
Пример 67. Получение 2,2-дихлор-N-((1S,2R)-1-фторметил-2-гидрокси-2-{4-[4-(метансульфониламино-метил)-[1,2,3]триазол-1-ил]-фенил}-этил)-ацетамида
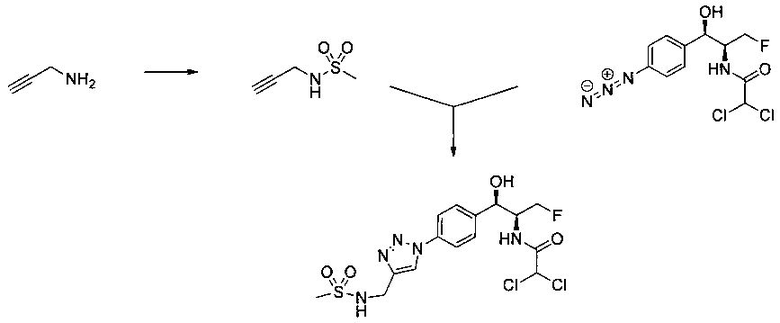
Стадия 1. Получение N-проп-2-инил-метансульфонамида
К перемешиваемому раствору проп-2-иниламина (1 г, 18,18 ммоль) в пиридине (1,5 мл) по каплям при 0°C добавляют мезилхлорид. Полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают в вакууме с получением неочищенного материала, который промывают пентаном и полностью высушивают с получением неочищенного указанного в заголовке соединения (3,0 г). 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.36 (s, 1Н), 2.95 (s, 3H), 3.32 (t, 1Н, J=2,56 Гц), 3.78 (d, J=2,48 Гц, 2Н), ЖХ-МС (m/z): [М-Н]=131,7.
Стадия 2. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(4-(метилсульфонамидометил)-1Н-1,2,3-триазол-1-ил)фенил)пропан-2-ил)-ацетамида
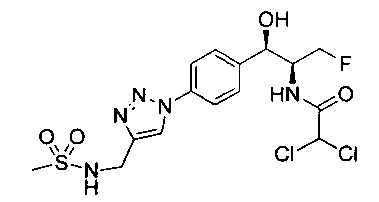
Следуя общей методике из Примера 66, стадия 3, и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 67 с продуктом Примера 66, стадия 2, получают указанное в заголовке соединение (45 мг). 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.95 (s, 3H), 4.25-4.26 (m, 1Н), 4.30 (s, 2Н), 4.33-4.34 (m, 0.5Н), 4.42-4.46 (m, 0.5Н), 4.58-4.61 (m, 0.5H), 4.69-4.73 (m, 0.5H), 4.95 (t, 1H, J=3,3 Гц), 6.08 (d, 1H, J=3,92 Гц), 6.49 (s, 1H), 7.55 (d, 2H, J=8,56 Гц), 7.84 (d, 2H, J=8,6 Гц), 8.64 (d, 1H, J=9,04 Гц), 8.70 (s, 1H). ЖХ-МС (m/z): [M+H]=453,9.
Пример 68. Получение 2,2-дихлор-N-{(1S,2R)-2-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-ацетамида
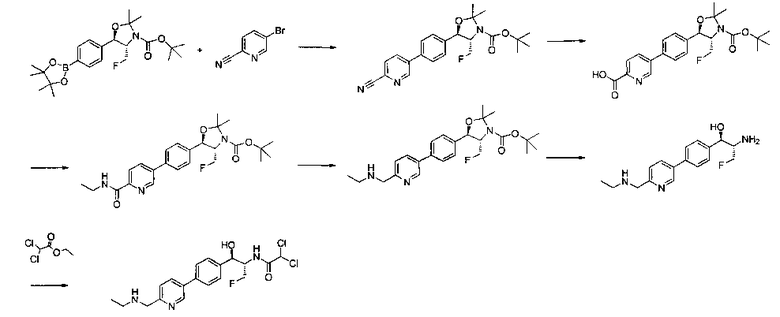
Стадия 1. трет-Бутиловый эфир (4S,5R)-5-[4-(6-циано-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты
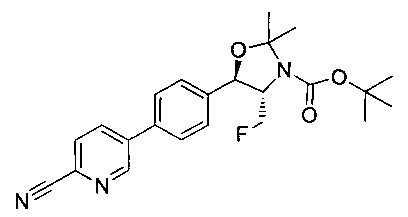
К перемешиваемому раствору трет-бутилового эфира (4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-карбоновой кислоты (0,75 г, 1,72 ммоль) в смеси диметоксиэтан: вода (8:2, 10 мл) добавляют Cs2CO3 (1,12 г, 3,44 ммоль) и 5-бром-пиридин-2-карбонитрил (0,347 г, 1,89 ммоль). Реакционную смесь дегазируют азотом в течение 30 минут с последующим добавлением Pd(PPh3)4 (0,199 г, 0,172 ммоль). Полученную реакционную смесь нагревают до 90°C в течение 3 часов. Разбавляют водой (20 мл) и этилацетатом (40 мл). Органический слой отделяют, сушат над сульфатом натрия и растворитель выпаривают в вакууме с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 15%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (0,3 г). 1Н ЯМР (400 МГц, DMSO-d6): δ: 1.43 (s, 9Н), 1.51 (s, 3H), 1.63 (s, 3H), 3.83-3.90 (m, 1Н), 4.47-4.58 (m, 2Н), 4.75-4.93 (m, 2Н), 5.15 (d, 1Н, J=7,2 Гц), 7.64 (d, 2Н, J=8,28 Гц), 7.87 (d, 2Н, J=8,24 Гц), 8.15 (d, 1Н, J=8,16 Гц), 8.36 (dd, 1Н, J=8,2 Гц, J=2,2 Гц), 9.12 (d, 1Н, J=2,24 Гц), ЖХ-МС (m/z): [М+Н]=412,1.
Стадия 2. Получение 5-[4-((4S,5R)-3-трет-бутоксикарбонил-4-фторметил-2,2-диметил-оксазолидин-5-ил)-фенил]-пиридин-2-карбоновой кислоты
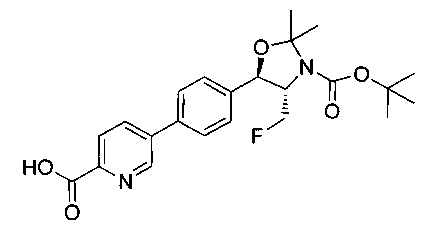
К раствору трет-бутилового эфира (4S,5R)-5-[4-(6-циано-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты (0,3 г, 0,729 ммоль) в метаноле (3 мл) при комнатной температуре добавляют КОН (0,409 г, 7,29 ммоль). Полученную реакционную смесь нагревают при 65°C в течение 48 часов. Растворитель выпаривают в вакууме, добавляют воду (10 мл) и водный слой промывают CH2Cl2. Водный слой подкисляют насыщенным раствором лимонной кислоты и экстрагируют CH2Cl2. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме с получением указанного в заголовке соединения (0,22 г): 1Н ЯМР (400 МГц, DMSO-d6): δ: 1.43 (s, 9Н), 1.51 (s, 3H), 1.64 (s, 3H), 3.84-3.90 (m, 2Н), 4.45-4.59 (m, 2Н), 4.77-5.0 (m, 2Н), 5.14 (d, 1Н, J=7,2 Гц), 7.61 (d, 2Н, J=8,08 Гц), 7.83 (d, 2Н, J=8,12 Гц), 8.08 (d, 1Н, J=8,32 Гц), 8.23 (d, 1Н, J=8,68 Гц), 9.0 (s, 1Н). ЖХ-МС (m/z): [М+Н]=431,2.
Стадия 3. Получение трет-бутилового эфира (4S,5R)-5-[4-(6-этилкарбамоил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты
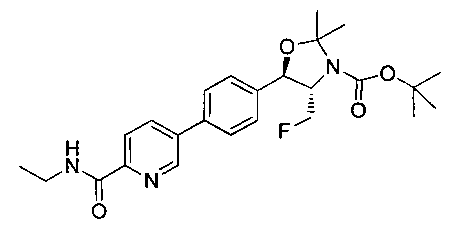
К перемешиваемому раствору 5-[4-((4S,5R)-3-трет-бутоксикарбонил-4-фторметил-2,2-диметил-оксазолидин-5-ил)-фенил]-пиридин-2-карбоновой кислоты (0,3 г, 0,697 ммоль) в сухом тетрагидрофуране (5 мл) добавляют этиламин (0,038 г, 0,837 ммоль), диизопропиламин (0,27 г, 2,092 ммоль) и 50%-ный раствор трифенилфосфина в этилацетате (0,332 г, 0,66 мл, 1,046 ммоль) и перемешивают при комнатной температуре в течение 14 часов. Разбавляют водой и этилацетатом, органический слой отделяют и водный слой экстрагируют этилацетатом. Объединенный органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и полученный неочищенный материал очищают колоночной хроматографией, элюируя 3%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (0,21 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.13 (t, 3H, J=6,98 Гц), 1.44 (s,9H), 1.51 (s, 3H), 1.64 (s, 3H), 3.34-3.37 (m, 2H), 3.84-3.91 (m, 1H), 4.47-4.59 (m, 1H), 4.76-4.95 (m, 2H), 5.15 (d, 1H, J=7,24 Гц), 7.61 (d, 2H, J=8,28 Гц), 7.83 (d, 2H, J=8,24 Гц), 8.09 (d, 1H, J=8,20 Гц), 8.29 (dd, 1H, J=8,08 Гц, J=2,2 Гц), 8.84 (t, 1H, J=5,68 Гц), 8.93 (d, 1H, J=1,88 Гц) ЖХ-МС (m/z): [М+Н]=458,1.
Стадия 4. Получение трет-бутилового эфира (4S,5R)-5-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты
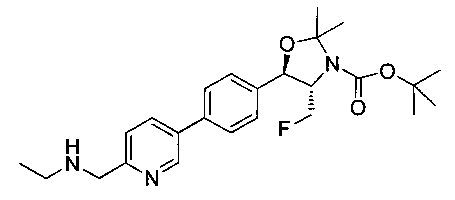
К перемешиваемому раствору трет-бутилового эфира (4S,5R)-5-[4-(6-этилкарбамоил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты (0,21 г, 0,459 ммоль) в сухом тетрагидрофуране (5,25 мл) при 0°C добавляют 2 М толуольный раствор ВН3.диметилсульфида (0,104 г, 1,378 ммоль). Реакционную смесь нагревают до 65°C в течение 16 часов. Гасят метанолом (2 мл). Летучие вещества удаляют при пониженном давлении с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 6%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (0,05 г): 1Н ЯМР (400 МГц, DMSO-d6): δ: 1.09 (t, 3H, J=7,15 Гц), 1.43 (s, 9Н), 1.51 (s, 3H), 1.63 (s, 3H), 3.15 (m, 2Н), 3.82-3.89 (m, 2Н), 3.95 (s, 2Н), 4.10 (m, 1Н), 4.45-4.55 (m, 2Н), 5.12 (d, 1Н, J=7,24 Гц), 7.52 (d, 1Н, J=8,20 Гц), 7.57 (d, 2Н,,7=8,20 Гц), 7.75 (d, 2Н, J=8,24 Гц), 8.09 (dd, 1Н, J=7,88 Гц, J=2,08 Гц), 8.84 (d, 1Н, J=1,92 Гц). ЖХ-МС (m/z): [М+Н]=444.
Стадия 5. Получение (1S,2S)-2-амино-1-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-3-фтор-пропан-1-ола
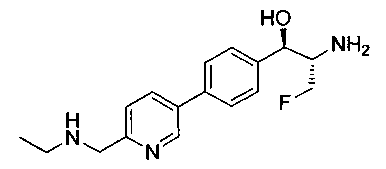
К перемешиваемому раствору трет-бутилового эфира (4S,5R)-5-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты (0,05 г, 0,112 ммоль) в CH2Cl2 (0,2 мл) при 0°C добавляют трифторуксусную кислоту (0,2 мл). Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 2 часов. Летучие вещества удаляют при пониженном давлении с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 10%-ным метанолом в CH2Cl2, с получением неочищенного указанного в заголовке соединения (0,04 г, неочищенное). ЖХ-МС (m/z): [М+Н]=304,2.
Стадия 6. Получение 2,2-дихлор-N-{(1S,2R)-2-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-ацетамида
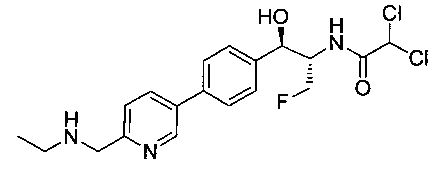
К перемешиваемому раствору (1S,2S)-2-амино-1-[4-(6-этиламинометил-пиридин-3-ил)-фенил]-3-фтор-пропан-1-ола (0,04 г, 0,095 ммоль, неочищенный) в сухом метаноле (0,17 мл) при комнатной температуре добавляют этилдихлорацетат (0,03 г, 0,191 ммоль) и триэтиламин (0,0194 г, 0,191 ммоль) и полученную реакционную смесь перемешивают в течение 16 часов. Летучие вещества удаляют при пониженном давлении с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 7%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (0,016 г): 1Н ЯМР (400 МГц, DMSO-d6) δ 1.04 (t, 3H, J=7,09 Гц), 2.54-2.58 (m, 2Н), 3.30 (s, 2Н), 4.19-4.22 (m, 1Н), 4.27-4.31 (m, 0.5Н), 4.39-4.43 (m, 0.5Н), 4.56-4.59 (m, 0.5H), 4.67-4.71 (m, 0.5H), 4.89-4.90 (m, 1H), 5.99 (d, 1H, J=4,2 Гц), 6.52 (s, 1H), 7.45-7.50 (m, 3H), 7.65 (d, 2H, J=8,2 Гц), 8.02 (dd, 1H, J=8,2 Гц, J=2,4 Гц), 8.62 (d, 1H, J=8,72 Гц), 8.77 (d, 1H, J=2,2 Гц). ЖХ-МС (m/z): [M+H]=414.
Пример 69. Получение N-((1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
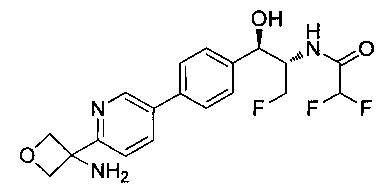
Стадия 1. Получение N-(3-(5-бромпиридин-2-ил)оксетан-3-ил)-2-метилпропан-2-сульфинамида
К толуольному раствору (20 мл) 2,5-дибромпиридина (1,93 г, 8,16 ммоль), который охлаждают до -78°C, в атмосфере азота по каплям через шприц в течение периода времени пять минут добавляют н-бутиллитий (2,5 М в гексанах, 3,6 мл, 8,98 ммоль). Реакционную смесь перемешивают при -78°C в течение еще десяти минут, после чего сразу одной порцией добавляют 2-метил-N-(оксетан-3-илиден)пропан-2-супьфинамид (1,43 г, 8,16 ммоль) в виде концентрированного раствора в толуоле. Реакционную смесь перемешивают в течение 30 минут при -78°C и в течение 20 минут при 0°C, после чего ее гасят насыщенным водным хлоридом аммония (5 мл). Летучие вещества удаляют посредством выпаривания на роторном испарителе при пониженном давлении. Оставшийся материал распределяют между водой (20 мл) и метиленхлоридом (50 мл). Водный слой экстрагируют еще один раз метиленхлоридом (30 мл). Объединенные экстракты концентрируют и оставшийся материал подвергают колоночной флэш-хроматографии, используя градиент ацетона в гексанах (от 20% до 75% за 5 объемов колонки), с получением N-(3-(5-бромпиридин-2-ил)оксетан-3-ил)-2-метилпропан-2-сульфинамида (850 мг): 1Н ЯМР (400 МГц, CDCl3) 1.32 (s, 9 Н), 4.91-4.97 (m, 2 Н), 5.13 (d, 1 Н), 5.34 (d, 1 Н), 7.78 (d, 1 Н), 8.04 (m, 1 Н), 8.68 (d, 1 Н); m/z (ХИ) 333, 335 [М+Н]+.
Стадия 2. Получение хлорида 3-(5-бромпиридин-2-ил)оксетан-3-аминия
К метанольному раствору (15 мл) N-(3-(5-бромпиридин-2-ил)оксетан-3-ил)-2-метилпропан-2-сульфинамида (850 мг, 2,55 ммоль), который был охлажден до 0°C, добавляют 4 н. HCl в диоксане (1,3 мл, 5,2 ммоль). Раствор перемешивают в течение 1 часа при 0°C. Летучие вещества удаляют посредством выпаривания на роторном испарителе при пониженном давлении. После нескольких циклов выпаривания с использованием ацетонитрила (3×10 мл) получают указанный в заголовке продукт (584 мг): m/z (ХИ) 229, 231 [М+Н]+.
Стадия 3. Получение 1-((4S,5R)-5-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-ил)-2,2-дифторэтанона
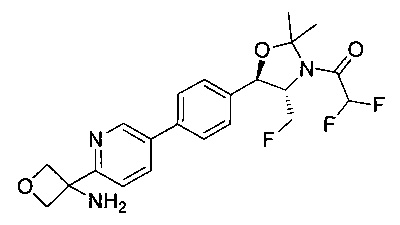
К раствору в смеси толуол/этанол (15 мл толуола, 12 мл этанола) 2,2-дифтор-1-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксазолидин-3-ил)этанона (594 мг, 1,44 ммоль) добавляют хлорид 3-(5-бромпиридин-2-ил)-3-аминия (434 мг, 1,44 ммоль), бикарбонат натрия (3 мл 2 М раствора) и Pd(dppf)2Cl2 (150 мг, 0,2 ммоль). Реакционную смесь нагревают до 80°C с перемешиванием под азотом в течение двух часов. Реакционную смесь охлаждают до комнатной температуры и разбавляют этилацетатом (60 мл). Смесь промывают водой (2×10 мл). Органическую фазу сушат над сульфатом натрия и концентрируют. Оставшийся материал очищают на силикагеле (подвижная фаза-6%-ный метанол в метиленхлориде) с получением указанного в заголовке соединения (436 мг, 70%): m/z (ХИ) 436 [М+Н]+.
Стадия 4. Получение N-((1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
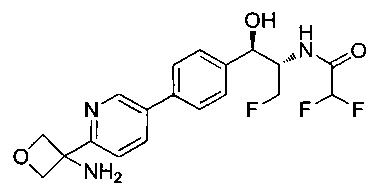
Следуя общей методике из Примера 2, стадия 2, и осуществляя незначительные модификации, но используя продукт стадии 3, Пример 69, получают указанное в заголовке соединение (132 мг): 1Н ЯМР (400 МГц, метанол-d4) 4.30-4.37 (m, 0.5 Н), 4.38-4.49 (m, 1.5 Н), 4.55 (0.5 Н), 4.62-4.70 (m, 0.5 Н), 4.82 (d, J=6,57 Гц, 2 Н), 5.00 (d, J=4,29 Гц, 1 Н), 5.04 (d, J=6,57 Гц, 2 Н), 5.86-6.16 (m, 1 Н), 7.54 (d, J=8,08 Гц, 2 Н), 7.68 (d, J=8,34 Гц, 2 Н), 7.78 (d, J=8,84 Гц, 1 Н), 8.11 (dd, J=8,21, 2,40 Гц, 1 Н), 8.85 (d, J=1,77 Гц, 1 Н); m/z (ХИ) 396 [М+Н]+.
Способами, известными из уровня техники, могут быть получены следующие производные указанного в заголовке соединения из Примера 69:
(1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-дигидрофосфат и
N-((1R,2S)-1-(4-(6-(3-аминооксетан-3-ил)пиридин-3-ил)фенил)-3-фтор-1-гидрокси-пропан-2-ил)-2,2-дихлорацетамид.
Пример 70. Получение 2,2-дихлор-N-{(1S,2R)-2-[4-(6-диметиламино-метил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-ацетамида
Стадия 1. Получение трет-бутилового эфира (4S,5R)-5-[4-(6-диметилкарбамоил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазо-лидин-3-карбоновой кислоты
К перемешиваемому раствору 5-[4-((4S,5R)-3-трет-бутоксикарбонил-4-фторметил-2,2-диметил-оксазолидин-5-ил)-фенил]-пиридин-2-карбоновой кислоты (220 мг, 0,511 ммоль) в сухом тетрагидрофуране (6 мл) при комнатной температуре добавляют раствор N,N-диметиламина, 2 M в тетрагидрофуране (28 мг, 0,31 мл, 0,61 ммоль), диизопропиламин (198 мг, 1,534 ммоль) и 50%-ный раствор трифенилфосфина в этилацетате (244 мг, 0,49 мл, 0,767 ммоль). Полученную реакционную смесь перемешивают при той же температуре в течение 14 часов. Разбавляют водой и этилацетатом, органический слой отделяют и водный слой экстрагируют этилацетатом. Объединенный органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме при пониженном давлении с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 3%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (100 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.44 (s, 9Н), 1.51 (s, 3H), 1.64 (s, 3H), 3.00 (s, 3H), 3.03 (s, 3H), 3.60-3.61 (m, 0.5Н), 3.84-3.90 (m, 1.5Н), 4.00-4.05 (m, 0.5H), 4.08-4.12 (m, 0.5H), 5.13 (d, 1H, J=7,24 Гц), 7.60 (d, 2H, J=8,24 Гц), 7.64 (d, 1H, J=8,20 Гц), 7.81 (d, 2H, J=8,32 Гц), 8.22 (dd, 1H, J=8,16 Гц, J=2,32 Гц), 8.91 (d, 1H, J=1,88 Гц), ЖХ-МС (m/z): [M+H]=458,2.
Стадия 2. Получение трет-бутилового эфира (4S,5R)-5-[4-(6-диметиламинометил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты
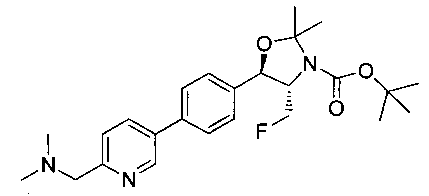
К перемешиваемому раствору трет-бутилового эфира (4S,5R)-5-[4-(6-диметилкарбамоил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты (260 мг, 0,569 ммоль) в сухом тетрагидрофуране (6,5 мл) при 0°C добавляют 2 М толуольный раствор BH3.DMS (130 мг, 1,708 ммоль). Реакционную смесь нагревают до 65°C в течение 16 часов. К реакционной смеси добавляют метанол и нагревают при 65°C в течение 2 часов. Летучие вещества удаляют при пониженном давлении с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 5%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (50 мг): ЖХ-МС (m/z): [М+Н]=444.
Стадия 3. Получение (1S,2S)-2-амино-1-[4-(6-диметиламинометил-пиридин-3-ил)-фенил]-3-фтор-пропан-1-ола
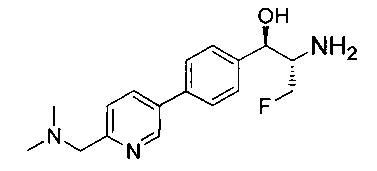
К перемешиваемому раствору трет-бутилового эфира (4S,5R)-5-[4-(6-диметиламинометил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-карбоновой кислоты (80 мг, 0,180 ммоль) в CH2Cl2 (0,32 мл) добавляют трифторуксусную кислоту (0,32 мл). Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 2 часов. Летучие вещества удаляют при пониженном давлении с получением неочищенного материала, который очищают колоночной хроматографией, элюируя 40%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (60 мг). ЖХ-МС (m/z): [М+Н]=304,1.
Стадия 4. Получение 2,2-дихлор-N-{(1S,2R)-2-[4-(6-диметиламино-метил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-ацетамида
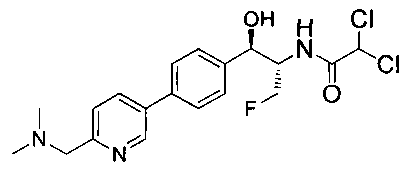
К перемешиваемому раствору (1S,2S)-2-амино-1-[4-(6-диметил-аминометил-пиридин-3-ил)-фенил]-3-фтор-пропан-1-ола (60 мг, 0,143 ммоль) в сухом метаноле (0,26 мл) при комнатной температуре добавляют этилдихлорацетат (45 мг, 0,287 ммоль) и триэтиламин (29 мг, 0,287 ммоль) и реакционную смесь перемешивают в течение 24 часов. Растворитель выпаривают в вакууме и неочищенный материал очищают колоночной хроматографией, элюируя 6%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (13 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.20 (s, 6Н), 3.54 (s, 2Н), 4.17-4.24 (m, 1Н), 4.27-4.31 (m, 0.5Н), 4.39-4.41 (m, 0.5Н), 4.56-4.58 (m, 0.5Н), 4.59-4.71 (m, 0.5Н), 4.89 (m, 1Н), 4.99 (d, 1Н, J=4 Гц), 6.52 (s, 1Н), 7.45-7.49 (m, 3H), 7.68 (d, 2Н, J=8,24 Гц), 8.03 (dd, 1Н, J1=8,08 Гц, J2=2,4 Гц), 8.64 (d, 1Н, J=8,8 Гц), 8.77 (d, 1Н, J=2,04 Гц). ЖХ-МС (m/z): [М+Н]=414.
Пример 71. Получение 2, 2-дихлор-N-{(1S,2R)-1-фторметил-2-гидрокси-2-[4-(6-уреидометил-пиридин-3-ил)-фенил]-этил}-ацетамида
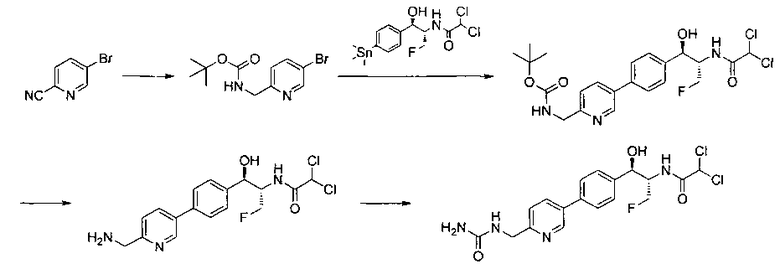
Стадия 1. Получение трет-бутилового эфира (5-бром-пиридин-2-илметил)-карбаминовой кислоты
К раствору 5-бром-пиридин-2-карбонитрила (2 г, 10,92 ммоль, 1 экв.) в метаноле (20,0 мл) добавляют NiCl2.6H2O (0,259 г, 1,09 ммоль, 0,1 экв.) и ди-трет-бутилдикарбонат (4,76 г, 21,85 ммоль, 2 экв.). К полученной смеси при 0°C добавляют NaBH4 (0,830 г, 21,57 ммоль, 2 экв.) (NaBH4 добавляют порциями). Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Растворитель выпаривают в вакууме и остаток разбавляют водой и экстрагируют этилацетатом. Органический слой сушат над Na2SO4 и растворитель выпаривают в вакууме с получением неочищенного материала, который очищают колоночной хроматографией, элюируя смесью 10% этилацетат/н-гексан, с получением указанного в заголовке соединения (1,2 г): 1Н ЯМР (400 МГц, CDCl3) δ: 1.44 (s, 9Н), 4.37 (d, 2Н, J=5,52 Гц), 5.41-5.44 (bs, 1Н), 7.18 (d, 1Н, J=8,32 Гц), 7.76 (dd, 1Н, J1=2,26 Гц, J2=8,30 Гц), 8.58 (d, 1Н, J=4 Гц). ЖХ-МС (m/z): [М+Н]=28,9.
Стадия 2. Получение трет-бутилового эфира (5-{4-[(1R,2S)-2-(2,2-дихлор-ацетиламино)-3-фтор-1-гидрокси-пропил]-фенил}-пиридин-2-илметил)-карбаминовой кислоты
К раствору продукта стадии 1 Примера 71 (1,4 г, 3,16 ммоль) в N-метил-2-пирролидоне (70,0 мл) добавляют трет-бутиловый эфир (5-бром-пиридин-2-илметил)-карбаминовой кислоты (0,905 г, 3,16 ммоль) и хлорид лития (0,399 г, 9,501 ммоль). Через полученный раствор в течение 15 минут барботируют газообразный азот и добавляют Pd(PPh3)2Cl2 (0,222 г, 0,316 ммоль). Полученную реакционную смесь нагревают до 60°C в течение 9 часов. Реакционную смесь охлаждают, разбавляют водой и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия и растворитель выпаривают в вакууме с получением неочищенного материала, который дополнительно очищают, используя колоночную хроматографию, элюируя 10%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (220 мг): МС-ЖХ (m/z): [М+Н]=483,8.
Стадия 3. Получение соли трифторуксусной кислоты и N-{(1S,2R)-2-[4-(6-аминометил-пиридин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дихлор-ацетамида
К раствору трет-бутилового эфира (5-{4-[(1R,2S)-2-(2,2-дихлор-ацетиламино)-3-фтор-1-гидрокси-пропил]-фенил}-пиридин-2-илметил)-карбаминовой кислоты (0,212 г, 0,437 ммоль) в CH2Cl2 (2 мл) по каплям при 0°C добавляют триэтиламин (1,0 мл). Полученную реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Растворитель выпаривают в вакууме с получением остатка, который обрабатывают толуолом (2×5 мл) с получением указанного в заголовке соединения (0,250 г): ЖХ-МС (m/z): М+Н=386,1.
Стадия 4. Получение 2,2-дихлор-N-{(1S,2R)-1-фторметил-2-гидрокси-2-[4-(6-уреидометил-пиридин-3-ил)-фенил]-этил}-ацетамида
К раствору продукта стадии 3 Примера 71 (0,250 г, 0,649 ммоль) в смеси 1,4-диоксана (16,0 мл) и воды (4,0 мл) добавляют изоцианат калия (0,057 г, 0,714 ммоль). Полученную реакционную смесь нагревают до 90°C в течение 2 часов. Реакционную смесь охлаждают и разбавляют водой и экстрагируют этилацетатом. Органический слой промывают водой и рассолом, сушат над сульфатом натрия и растворитель выпаривают в вакууме с получением неочищенного материала, который очищают, используя колоночную хроматографию, элюируя 2%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (0,018 г): 1Н ЯМР (400 МГЦ, DMSO-d6) δ: 4.20-4.22 (m, 1Н), 4.22-4.24 (m, 0.5Н), 4.30 (d, 2Н, J=6 Гц), 4.39-4.43 (m, 0.5Н), 4.56-4.59 (m, 0.5H), 4.67-4.71 (m, 0.5H), 4.89 (t, 1H, J=3,6 Гц), 5.65 (s, 2H), 6.00 (d, 1H, J=3,1 Гц), 6.52 (s, 1H), 6.56 (t, 1H, J=5,68 Гц), 7.35 (d, 1H, J=8,16 Гц), 7.46 (d, 2H, J=8,2 Гц), 7.66 (d, 2H, J=8,24 Гц), 8.02-8.05 (dd, 1H, J1=2,32 Гц, J2=8,16 Гц), 8.65 (d, 1H, J=8,84 Гц), 8.78 (d, 1H, J=2,08 Гц). ЖХ-МС (m/z): [M+H]=429,0.
Пример 72. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(1-(метилсульфонамидо)циклопропил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение N-(1-(5-бромпиридин-2-ил)циклопропил)метан-сульфонамида
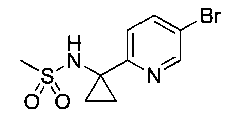
Имеющуюся в продаже соль дигидрохлорид 1-(5-бромпиридин-2-ил)циклопропанамина (494 мг, 1,73 ммоль) растворяют в CH2Cl2 (11,5 мл, 0,15 М) и добавляют 3,1 экв. триэтиламина (543 мг, 5,36 ммоль). Затем добавляют 1,1 экв. метансульфонилхлорида (218 мг, 1,9 ммоль) и смесь перемешивают в течение 16 часов. Добавляют дихлорметан (20 мл) и промывают NaHCO3 (насыщ., водн). Растворитель выпаривают с получением указанного в заголовке соединения (310 мг): m/z (ХИ) 293 [М+Н].
Стадия 2. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(1-(метилсульфонамидо)циклопропил)пиридин-3-ил)фенил)пропан-2-ил)-ацетамида
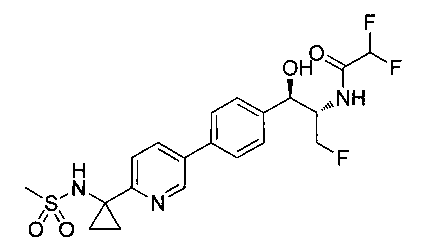
Следуя общей методике из Примера 9, стадия 2, и осуществляя незначительные модификации, но используя продукт из Примера 72, стадия 1, и Примера 17, стадия 3, получают указанное в заголовке соединение (310 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.4-1.45 (m, 2Н), 1.45-1.5 (m, 2Н), 2.9 (s, 3H), 4.2-4.35 (m, 1.5Н), 4.35-4.45 (m, 0.5Н), 4.45-4.6 (m, 0.5H), 4.6-4.7 (m, 0.5H), 4.85 (t, 1H), 5.9 (d, 1H), 6.2 (t, 1H), 7.45 (d, 2H), 7.65 (d, 2H), 7.8 (d, 1H), 8.05 (d, 1H), 8.30 (s, 1H), 8.70 (s, 1H), 8.8 (d, 1H). m/z (ХИ) 458 [М+Н].
Пример 73. Получение 2,2-дихлор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-{6-[(3-метил-бутиламино)-метил]-пиридин-3-ил}-фенил)-этил]-ацетамида
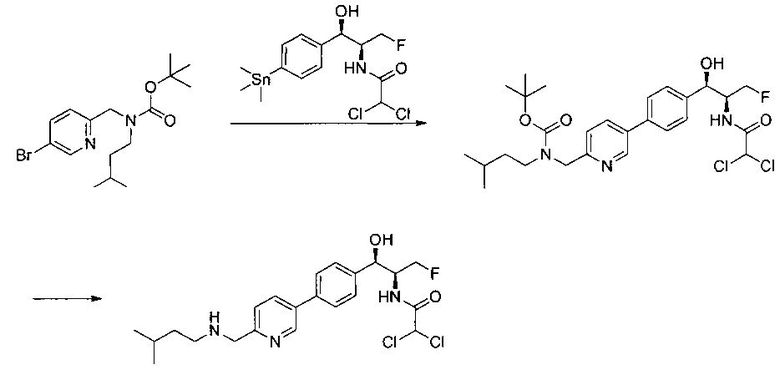
Стадия 1. Получение трет-бутилового эфира (5-{4-[(1R,2S)-2-(2,2-дихлор-ацетиламино)-3-фтор-1-гидрокси-пропил]-фенил}-пиридин-2-илметил)-(3-метил-бутил)-карбаминовой кислоты
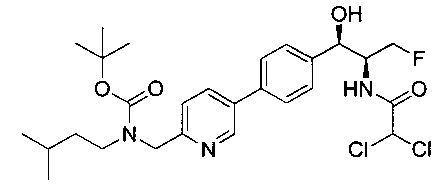
К раствору 2,2-дихлор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-триметил-станнил-фенил)-этил]-ацетамида (100 мг, 0,226 ммоль) в N-метил-2-пирролидоне (1,5 мл) добавляют трет-бутиловый эфир (5-бром-пиридин-2-илметил)-(3-метил-бутил)-карбаминовой кислоты (81 мг, 0,226 ммоль) и через полученный раствор в течение 15 минут барботируют газообразный азот. К этой реакционной смеси в атмосфере азота добавляют Pd(dba)3 (21 мг, 0,022 ммоль) и P(2-fur)3 (11 мг, 0,045 ммоль). Полученную реакционную смесь нагревают до 80°C в течение 18 часов. Охлаждают, разбавляют водой и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия и растворитель выпаривают в вакууме с получением неочищенного материала, который очищают, используя колоночную хроматографию, элюируя 2,4%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (10 мг). ЖХ-МС (m/z): [М+Н]=556,1.
Стадия 2. Получение 2,2-дихлор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-{6-[(3-метил-бутиламино)-метил]-пиридин-3-ил}-фенил)-этил]-ацетамида
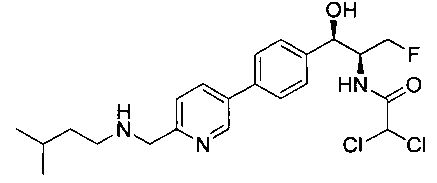
К раствору трет-бутилового эфира (5-{4-[(1R,2S)-2-(2,2-дихлор-ацетиламино)-3-фтор-1-гидрокси-пропил]-фенил}-пиридин-2-илметил)-(3-метил-бутил)-карбаминовой кислоты (8 мг, 0,014 ммоль) в сухом CH2Cl2 (0,04 мл) добавляют трифторуксусную кислоту (0,04 мл) и полученную реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают в вакууме с получением коричневого неочищенного материала, который промывают смесью диэтиловый эфир: пентан (1:9), с получением указанного в заголовке соединения (8 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ 0.89 (d, 6Н, J=6,40 Гц), 1.52-1.58 (m, 2Н), 1.60-1.65 (m, 1Н), 3.01 (bs, 1Н), 4.20-4.23 (m, 2Н), 4.29-4.31 (m, 0.5Н), 4.36-4.38 (m, 2Н), 4.40-4.44 (m, 0.5Н), 4.57-4.59 (m, 0.5H), 4.70-4.71 (m, 0.5H), 4.91-4.93 (m, 1H), 6.02 (d, 1H, J=4,28 Гц), 6.52 (s, 1H), 7.49 (d, 2H, J=8,24), 7.57 (d, 1H, J=8,16 Гц), 7.73 (d, 2H, J=8,28 Гц), 8.19 (dd, 1H, J1=8,16 Гц, J2=2,32 Гц), 8.65 (d, 1H, J=8,96 Гц), 8.96 (d, 1H, J=1,96 Гц), 9.00 (bs, 2H). ЖХ-МС (m/z): [M+H]=456,1.
Пример 74. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(5-(пирролидин-2-ил)тиофен-2-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение трет-бутил-2-(5-бромтиофен-2-ил)пирролидин-1-карбоксилата
К перемешиваемому раствору имеющегося в продаже 2-(5-бромтиофен-2-ил)-пирролидина (0,986 г, 4,25 ммоль) в 1,4-диоксане (20 мл) добавляют К2СО3 (1,21 г, 8,49 ммоль) и воду (10 мл). Смесь охлаждают до 0°C и добавляют ди-трет-бутилдикарбонат (1,02 г, 2,40 ммоль). Реакционную смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение 16 ч. Реакционную смесь концентрируют в вакууме. Неочищенный материал разбавляют водой и экстрагируют этилацетатом. Органический слой фильтруют через мелкодисперсный силикагель и концентрируют с получением указанного в заголовке соединения (1,48 г): 1Н ЯМР (400 МГц, CDCl3) δ 1.32-1.52 (m, 9Н), 1.86-2.06 (m, 3H), 2.16-2.31 (m, 1Н), 3.35-3.58 (m, 2Н), 4.91-5.15 (m, 1Н), 6.61 (bs, 1Н), 6.85 (d, J=3,5 Гц, 1Н). m/z (ХИ) М+Н 332,0.
Стадия 2. Получение трет-бутил-2-(5-(4-((1R,2S)-2-(2,2-дихлор-ацетамидо)-3-фтор-1-гидроксипропил)фенил)тиофен-2-ил)пирролидин-1-карбоксилата
К перемешиваемому раствору трет-бутил-2-(5-бромтиофен-2-ил)пирролидин-1-карбоксилата (0,750 г, 2,26 ммоль), 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)фенил)пропан-2-ил)ацетамида (1,0 г, 2,26 ммоль) и три(фуран-2-ил)фосфина (105 мг, 0,452 ммоль) в N-метил-2-пирролидоне (10 мл), дегазированному азотом, добавляют Pd2(dba)3 (207 мг, 0,226 ммоль). Реакционную смесь нагревают до 80°C в течение 5 часов, затем оставляют на 70 часов при температуре окружающей среды. Реакционную смесь распределяют между этилацетатом (100 мл) и водой (100 мл). Органическую фазу выделяют и концентрируют в вакууме с получением неочищенного материала, который очищают колоночной хроматографией на силикагеле, элюируя 0-100% этилацетата в гептане, с получением указанного в заголовке соединения (559 мг): 1Н ЯМР (400 МГц, CDCl3) δ 1.32-1.52 (m, 9Н), 1.89-2.15 (m, 3H), 2.21-2.37 (m, 1Н), 2.59-2.78 (m, 1Н), 3.40-3.66 (m, 2Н), 4.25-4.38 (m, 1Н), 4.43-4.49 (m, 0.5Н), 4.54-4.62 (m, 1Н), 4.66-4.73 (m, 0.5Н), 5.01-5.25 (m, 2H), 5.89 (s, 1H), 6.83 (bs, 1H), 7.06 (d, J=8,6 Гц, 1H), 7.14 (d, J=3,5 Гц, 1H), 7.38 (d, J=8,1 Гц, 2H), 7.56 (d, J=8,1 Гц, 2H). m/z (ХИ) M-boc+H 431,0.
Стадия 3. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(5-(пирролидин-2-ил)тиофен-2-ил)фенил)пропан-2-ил)ацетамида
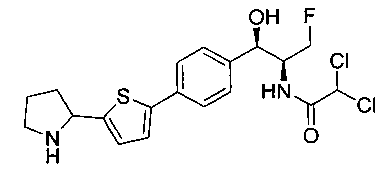
Следуя общей методике из Примера 2, стадия 2, и осуществляя незначительные модификации, но используя продукт стадии 3 Примера 74, получают указанное в заголовке соединение (257 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.60-2.01 (m, 3H), 2.18-2.33 (m, 1Н), 2.89-3.21 (m, 2Н), 3.31-3.6 (m, 2Н), 4.09-4.33 (m, 2Н), 4.37-4.44 (m, 0.5Н), 4.52-4.62 (m, 1Н), 4.64-4.71 (m, 0.5Н), 4.83-4.89 (m, 1H), 5.96 (bs, 1H), 6.52 (s, 1H), 6.92-7.12 (m, 1H), 7.26-7.41 (m, 2H), 7.50-7.59 (m, 2H), 8.55-8.62 (m, 1H): m/z (ХИ) M+H 431,0.
Пример 75. Получение N-((1R,2S)-1-(4-(5-(1-аминоэтил)тиофен-2-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение трет-бутил-(1-(5-бромтиофен-2-ил)этил)карбамата
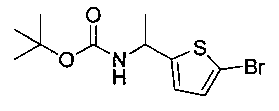
Соединение получают из имеющегося в продаже 1-(5-бромтиофен-2-ил)этанамина (992 мг, 4,09 ммоль) аналогично Примеру 74, стадия 1, что дает указанное в заголовке соединение (1,25 г): 1Н ЯМР (400 МГц, CDCl3) δ: 1.46 (s, 9Н), 1.52 (d, J=6,8 Гц, 3H), 4.78 (m, 1Н), 4.96 (m, 1Н), 6.70 (dd, J=3,8 Гц, 1Н), 6.88 (d, J=3,5 Гц, 1Н).
Стадия 2. Получение трет-бутил-(1-(5-(4-((1R,2S)-2-(2,2-дихлор-ацетамидо)-3-фтор-1-гидроксипропил)фенил)тиофен-2-ил)этил)карбамата
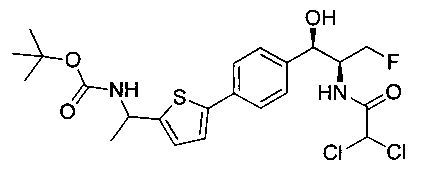
Соединение получают из трет-бутил-(1-(5-бромтиофен-2-ил)этил)-карбамата (138 мг, 0,45 ммоль) и 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)фенил)пропан-2-ил)ацетамида (200 мг, 0,45 ммоль) способом, аналогичным Примеру 74, стадия 2, что дает указанное в заголовке соединение (70 мг): 1Н ЯМР (400 МГц, CDCl3) δ: 1.47 (s, 9Н), 1.57 (d, J=6,8 Гц, 3H), 2.68 (bs, 1Н), 4.25-4.38 (m, 1Н), 4.42-4.48 (m, 0.5Н), 4.53-4.60 (m, 1Н), 4.65-4.72 (m, 0.5Н), 4.77-4.89 (m, 1H), 4.98-5.09 (m, 1H) 5.11 (d, J=4,0 Гц, 1H), 5.87 (s, 1H), 6.90 (d, J=3,5 Гц, 1H), 7.04 (d, J=8,6 Гц, 1H), 7.14 (d, J=3,8 Гц, 1H), 7.37 (d, J=8,1 Гц, 2H), 7.55 (m, 2H).
Стадия 3. Получение N-((1R,2S)-1-(4-(5-(1-аминоэтил)тиофен-2-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
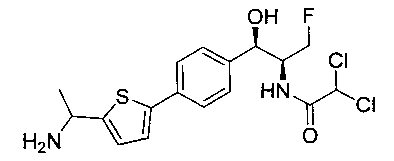
Следуя общей методике из Примера 2, стадия 2, и осуществляя незначительные модификации, но используя продукт стадии 2 Примера 75, получают указанное в заголовке соединение (8,3 мг): m/z (ХИ) М+Н 406,0.
Пример 76. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-(метилсульфонамидометил)тиазол-5-ил)фенил)пропан-2-ил)ацетамида
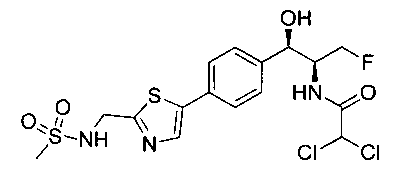
Соединение получают из N-((1R,2S)-1-(4-(2-(аминометил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида (30 мг, 0,06 ммоль) способом, аналогичным Примеру 3, что дает указанное в заголовке соединение (6,5 мг): m/z (ХИ) М+470.
Пример 77. Получение N-((1R,2S)-1-(4-(5-(аминометил)тиофен-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение трет-бутил-((4-(4-((1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-гидроксипропил)фенил)тиофен-2-ил)метил)-карбамата
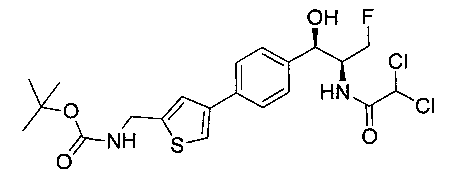
Соединение получают из трет-бутил-((4-бромтиофен-2-ил)метил)-карбамата (132 мг, 0,45 ммоль) и 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)фенил)пропан-2-ил)ацетамида (200 мг, 0,45 ммоль) способом, аналогичным Примеру 2, что дает указанное в заголовке соединение (45 мг): 1Н ЯМР (400 МГц, CDCl3) 1.47 (s, 9Н), 2.73 (bs, 1Н), 4.26-4.38 (m, 1Н), 4.39-4.52 (m, 2.5Н), 4.53-4.61 (m, 1Н), 4.65-4.72 (m, 0.5Н), 4.87-5.03 (m, 1H), 5.12 (d, J=4,0 Гц, 1H), 5.87 (s, 1H), 7.06 (d, J=8,6 Гц, 1H), 7.20 (s, 1H), 7.32 (d, J=1,5 Гц, 1H), 7.39 (d, J=8,1 Гц, 2Н), 7.54 (m, 2Н).
Стадия 2. Получение N-((1R,2S)-1-(4-(5-(аминометил)тиофен-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
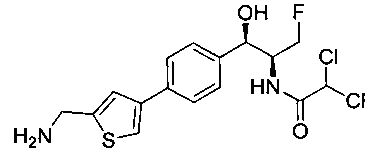
Следуя общей методике из Примера 2, стадия 2, и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 77, получают указанное в заголовке соединение (25,8 мг): m/z (ХИ) М-ОН+Н 374,0.
Пример 78. 2,2-Дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(5-(морфолино-метил)тиофен-2-ил)фенил)пропан-2-ил)ацетамида
Следуя общей методике из Примера 22, стадия 1, и осуществляя незначительные модификации, но используя 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-йодфенил)пропан-2-ил)ацетамид (150 мг, 0,34 ммоль) и 4-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиофен-2-ил)метил)-морфолин (114 мг, 0,37 ммоль), а затем общей методике из Примера 2, стадия 2, получают указанное в заголовке соединение (50 мг): 1Н ЯМР (400 МГц, CDCl3) δ: 1.66 (bs, 1Н), 2.53 (t, 4Н), 3.71 (s, 2Н,), 3.75 (t, 4Н), 4.25-4.38 (m, 1Н), 4.42-4.48 (m, 0.5Н), 4.53-4.60 (m, 1Н), 4.65-4.71 (m, 0.5Н), 5.11 (d, 1Н), 5.87 (s, 1Н), 6.89 (d, J=3,5 Гц, 1Н), 7.04 (d, J=8,8 Гц, 1Н), 7.16 (d, J=3,5 Гц, 1Н), 7.39 (m, 2Н), 7.59 (m, 2Н).
Пример 79. Получение N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метил-сульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)циклопропан-карбоксамида
Стадия 1. Получение N-((5-(4-((1R,2S)-2-амино-3-фтор-1-гидроксипропил)фенил)пиридин-2-ил)метил)метансульфонамида
(4S,5R)-трет-Бутил-4-(фторметил)-2,2-диметил-5-(4-(6-(метил-сульфонамидо-метил)пиридин-3-ил)фенил)оксазолидин-3-карбоксилат (1800 мг, 3,64 ммоль) растворяют в дихлорметане (20 мл) и в течение 4 ч при комнатной температуре обрабатывают трифторуксусной кислотой (5,4 мл). Реакционную смесь разбавляют толуолом (30 мл) и концентрируют до сиропа. Полученное неочищенное соединение подщелачивают водным бикарбонатом натрия, экстрагируют этилацетатом. Экстракты сушат над Na2SO4, фильтруют и концентрируют с получением указанного в заголовке соединения (1110 мг): m/z (Cl) М+Н 354.
Стадия 2. Получение N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метил-сульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)циклопропанкарбоксамида
Смесь аминоспирта (100 мг, 0,28 ммоль), циклопропанкарбоновой кислоты (25 мг, 0,28 ммоль), гексафторфосфата O-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилурония (118 мг, 0,31 ммоль) и диизопропилэтиламина (0,150 мл, 0,85 ммоль) в диметилформамиде (1 мл) перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь фильтруют и очищают с помощью ВЭЖХ с получением указанного в заголовке соединения (30 мг): 1Н ЯМР (400 МГц, DMSO-d6 0.5-0.7 (m, 4Н), 1.67-1.80 (m, 1Н), 2.95 (s, 3H), 4.16-4.41 (m, 4Н), 4.45-4.70 (m, 1Н), 4.81-4.94 (m, 1Н), 5.84 (dd, 1Н), 7.43-7.62 (m, 3H), 7.66-7.79 (m, 3H), 8.05-8.19 (m, 2Н), 8.85 (d, 1Н). m/z (ХИ) М+Н 422.
Пример 80. Получение 3,3,3-трифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)пропанамида
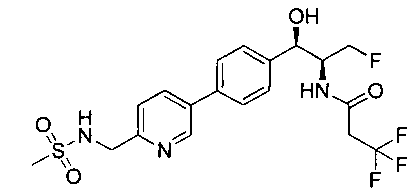
Следуя общей методике из Примера 1, стадия 2, и осуществляя незначительные модификации, за исключением использования 3,3,3-трифторпропановой кислоты, получают указанное в заголовке соединение (40 мг). 1Н ЯМР (400 МГц, DMSO-d6) 2.95 (s, 3H), 4.19-4.39 (m, 3.5Н), 4.49-4.57 (m, 0.5Н), 4.60-4.69 (m, 0.5H), 4.82-4.91 (m, 1H), 5.90 (d, 1H), 7.47 (d, 2H), 7.53 (d, 1H), 7.66-7.79 (m, 3H), 8.10 (dd, 1H), 8.34 (d, 1H), 8.82 (d, 1H). m/z (Cl) M+H 578.
Пример 81. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)пропанамида
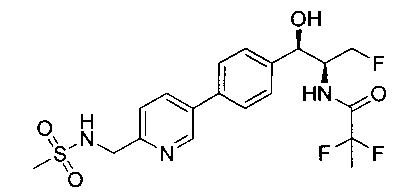
Следуя общей методике из Примера 1, стадия 2, и осуществляя незначительные модификации, но используя 2,2-дифторпропановую кислоту, получают указанное в заголовке соединение (40 мг). 1Н ЯМР (400 МГц, DMSO-d6) 1.63 (t, 3H), 2.95 (s, 3H), 4.27-4.39 (m, 3.5Н), 4.44-4.51 (m, 0.5Н), 4.52-4.59 (m, 0.5H), 4.64-4.70 (m, 0.5H), 4.86-4.92 (m, 1H), 5.80 (d, 1H), 7.44 (d, 2H), 7.53 (d, 1H), 7.66-7.74 (m, 3H), 8.11 (dd, 1H), 8.42 (d, 1H), 8.83 (d, 1H). m/z (ХИ) M+H 446.
Пример 82. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)пропанамида
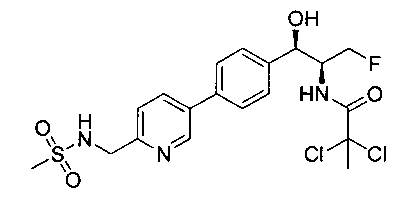
Следуя общей методике из Примера 1, стадия 2, и осуществляя незначительные модификации, но используя 2,2-дихлорпропановую кислоту, получают указанное в заголовке соединение (33 мг): 1Н ЯМР (400 МГц, DMSO-d6) 2.95 (s, 3H), 4.23-4.36 (m, 3.5Н), 4.37-4.44 (m, 0.5Н), 4.48-4.63 (m, 1Н), 4.67-4.75 (m, 0.5Н), 4.90-4.97 (m, 1Н), 5.90-5.97 (m, 1Н), 7.47 (d, 2Н), 7.53 (d, 1Н), 7.66-7.74 (m, 3H), 8.06-8.16 (m, 2Н), 8.42 (d, 1Н), 8.83 (d, 1Н). m/z (ХИ) М+Н 478.
Пример 83. Получение N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метил-сульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)-2-((трифторметил)-тио)ацетамида
Следуя общей методике из Примера 1, стадия 2, и осуществляя незначительные модификации, но используя 2-((трифторметил)тио)уксусную кислоту, получают указанное в заголовке соединение (35 мг): 1Н ЯМР (400 МГц, DMSO-d6) 2.95 (s, 3H), 3.72-3.83 (ABq, 2Н), 4.18-4.38 (m, 4Н), 4.48-4.55 (m, 0.5Н), 4.60-4.67 (m, 0.5Н), 4.85-4.92 (m, 1H), 5.90-5.92 (m, 1H), 7.48 (d, 2H), 7.53 (d, 1H), 7.64-7.74 (m, 3H), 8.10 (dd, 1H), 8.37 (d, 1H), 8.82 (d, 1H). m/z (ХИ) M+H 496.
Пример 84. Получение 2-азидо-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
К суспензии N-((5-(4-((1R,2S)-2-амино-3-фтор-1-гидроксипропил)фенил)-пиридин-2-ил)метил)метансульфонамида (800 мг, 2,26 ммоль) в смеси этилацетат/водный NaHCO3 (1:1) (10 мл) медленно в течение периода времени 20 минут добавляют бромацетилбромид (900 мг, 4,5 ммоль). Органический слой отделяют и водный слой экстрагируют этилацетатом (3×10 мл). Объединенный экстракт сушат над Na2SO4 и концентрируют с получением неочищенного продукта, m/z (ХИ) М+Н 474. Смесь неочищенного бромида (500 мг) и азида натрия (470 мг, 7,0 ммоль) в диметилформамиде (6 мл) нагревают в течение 30 минут при 50°C. Реакционную смесь охлаждают, разбавляют водой (10 мл) и экстрагируют этилацетатом (3×15 мл). Объединенный экстракт сушат над Na2SO4, концентрируют под вакуумом и затем полученный неочищенный продукт очищают на силикагелевой колонке, используя смесь 0-5% метанол/CH2Cl2, с получением указанного в заголовке соединения (300 мг): 1Н ЯМР (400 МГц, DMSO-d6) 2.95 (s, 3H), 3.71-3.87 (m, 2Н), 4.22-4.42 (m, 4Н), 4.47-4.58 (m, 0.5Н), 4.50-4.70 (m, 0.5Н), 4.83-4.93 (m, 1H), 5.83-5.93 (m, 1H), 7.47 (d, 2H), 7.54 (d, 1H), 7.64-7.77 (m, 3H), 8.06-8.20 (m, 2H), 8.83 (d, 1H). m/z (ХИ) M+H 437.
Пример 85. Получение N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метил-сульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)метансульфонамида
К суспензии N-((5-(4-((1R,2S)-2-амино-3-фтор-1-гидроксипропил)фенил)-пиридин-2-ил)метил)метансульфонамида (100 мг, 0,28 ммоль) в смеси этилацетат/водный NaHCO3 (1:1) (2 мл) медленно в течение периода времени 20 минут добавляют метансульфонилхлорид (50 мг, 0,42 ммоль). Реакционную смесь разбавляют водой (2 мл) и экстрагируют этилацетатом (3×5 мл). Объединенный органический раствор сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают, используя ВЭЖХ, с получением указанного в заголовке соединения (60 мг): 1Н ЯМР (400 МГц, DMSO-d6) 2.55 (s, 3H), 2.98 (s, 3H), 3.67-3.80 (m, 1Н), 4.18-4.26 (m, 0.5Н), 4.31-4.40 (m, 2.5Н), 4.48-4.55 (m, 0.5H), 4.60-4.67 (m, 0.5H), 4.80-4.87 (m, 1H), 5.83-5.93 (m, 1H), 7.27 (d, 1H), 7.54 (d, 2H), 7.54 (d, 1H), 7.62 (d, 1H), 7.70-7.79 (m, 3H), 8.20-8.26 (m, 1H), 8.90 (d, 1H). m/z (ХИ) M+H 432.
Пример 86. Получение N-((1R,2S)-1-(4-(2-(аминометил)тиазоло[5,4-b]-пиридин-6-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение трет-бутил((6-бромтиазоло[5,4-b]пиридин-2-ил)метил)карбамата
К раствору 6-бром-2-(бромметил)тиазоло[5,4-b]пиридина (800 мг, 2,60 ммоль) (Journal of Medicinal Chemtry, 53(10), 3927-3936; 2010) в тетрагидрофуране (10 мл) добавляют гидроксид аммония (2 мл) и перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь концентрируют, разбавляют водой (5 мл) и экстрагируют, используя этилацетат (3×10 мл). Экстракты сушат над Na2SO4 и концентрируют с получением неочищенного продукта (650 мг). m/z (ХИ) М+244. К раствору неочищенного амина (350 мг, 1,43 ммоль) в тетрагидрофуране (10 мл) и водном NaHCO3 (2 мл) добавляют boc-ангидрид и полученную смесь перемешивают при комнатной температуре в течение ночи. Органический раствор отделяют и водный слой экстрагируют этилацетатом (5 мл). Объединенный органический раствор сушат над Na2SO4 и концентрируют под вакуумом с получением указанного в заголовке соединения (400 мг): 1Н ЯМР (400 МГц, CDCl3) 1.50 (s, 9Н), 4.74 (d, 2Н), 8.36 (s, 1Н), 8.62 (s, 1Н). m/z (ХИ) М+2 346.
Стадия 2. Получение трет-бутил-((6-(4-((1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-гидроксипропил)фенил)тиазоло[5,4-b]пиридин-2-ил)метил)карбамата
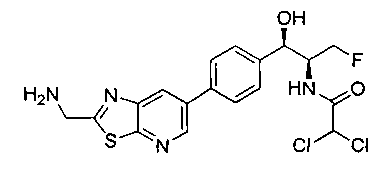
Смесь трет-бутил-((6-бромтиазоло[5,4-b]пиридин-2-ил)метил)карбамата (156 мг, 0,45 ммоль), 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)фенил)-пропан-2-ил)ацетамида (200 мг, 0,45 ммоль) и трис(2-фурил)фосфина (21 мг, 0,090 ммоль), трис(дибензилиденацетон)-дипалладия(0) (41 мг, 0,045 ммоль) в диметилформамиде (2,5 мл) нагревают под азотом при 65°C в течение 3 часов. Смесь затем охлаждают, разбавляют водой (10 мл) и экстрагируют этилацетатом (3×10 мл). Экстракты сушат над Na2SO4 и концентрируют с получением неочищенного указанного в заголовке соединения, m/z (ХИ) М+1 543. К раствору неочищенного трет-бутилкарбамата (200 мг, 0,452 ммоль) в DCM (3 мл) добавляют трифторуксусную кислоту (0,5 мл) и перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь разбавляют толуолом (10 мл), концентрируют под вакуумом, подщелачивают насыщенным водн. NaHCO3 и экстрагируют, используя этилацетат (3×10 мл). Экстракты сушат над Na2SO4, концентрируют и неочищенное соединение адсорбируют на целите и очищают на силикагелевой колонке, используя 0-20%-ный метанол в этилацетате, с получением указанного в заголовке соединения (40 мг). 1Н ЯМР (DMSO-d6): 4.17 (m, 2Н), 4.27 (m, 1.5Н), 4.43 (m, 0.5Н), 4.59 (m, 0.5H), 4.70 (m, 0.5H), 4.93 (m, 1H), 6.01 (m, 1H), 6.54 (1H), 7.50 (d, 2H), 7.80 (d, 2H), 8.51 (m, 1H), 8.64 (d, 1H), 8.87 (d, 1H), m/z(XH) M+H 443.
Пример 87. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-(метилсульфонамидометил)тиазоло[5,4-b]пиридин-6-ил)фенил)пропан-2-ил)-ацетамида
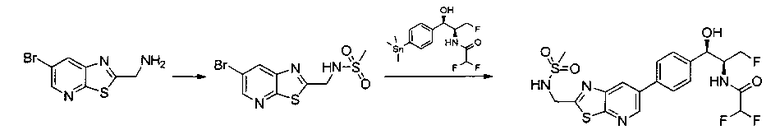
Стадия 1. Получение N-((6-бромтиазоло[5,4-b]пиридин-2-ил)метил)-метансульфонамида
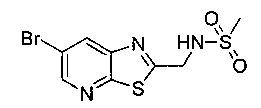
К охлажденному (лед-вода) раствору (6-бромтиазоло[5,4-b]пиридин-2-ил)метанамина (800 мг, 3,28 ммоль) и пиридина (800 мг, 9,85 ммоль) в CH2Cl2 (10 мл) медленно добавляют метансульфонилхлорид (380 мг, 3,2S) и перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь промывают водой (2×10 мл), сушат над Na2SO4 и концентрируют. Полученный неочищенный продукт очищают на силикагелевой колонке, используя смесь 0-5% метанол/CH2Cl2, с получением указанного в заголовке соединения (800 мг): m/z (ХИ) М+2 324.
Стадия 2. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(2-(метилсульфонамидометил)тиазоло[5,4-b]пиридин-6-ил)фенил)пропан-2-ил)-ацетамида
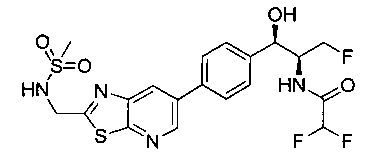
Смесь N-((6-бромтиазоло[5,4-b]пиридин-2-ил)метил)метансульфонамида (236 мг, 0,732 ммоль), 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(триметилстаннил)фенил)пропан-2-ил)ацетамида (300 мг, 0,732 ммоль) и трис(2-фурил)фосфина (34 мг, 0,146 ммоль) и трис(дибензилиденацетон)-дипалладия(0) (68 мг, 0,0732 ммоль) в диметилформамиде (3 мл) нагревают под азотом при 70°C в течение 5 часов. Смесь затем охлаждают, разбавляют водой (10 мл) и экстрагируют этилацетатом (3×15 мл). Экстракты сушат над Na2SO4 и концентрируют с получением неочищенного продукта. Полученный продукт очищают посредством ВЭЖХ с получением указанного в заголовке соединения (90 мг): 1Н ЯМР (DMSO-d6): 3.07 (s, 3H), 4.27-4.39 (m, 1.5Н), 4.40-4.48 (m, 0.5Н), 4.52-4.60 (m, 0.5H), 4.64-4.71 (m, 2.5H) 4.88-4.94 (m, 1H), 6.22 (t, 1H), 7.50 (d, 2H), 7.83(d, 2H), 8.22-8.33(m, 1H), 8.62 (bs, 1H), 8.85 (d, 1H), 8.94 (s, 1H). m/z(XH) M+H 489.
Пример 88. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-(4-(6-((3-фторазетидин-1-ил)метил)пиридин-3-ил)фенил)-1-гидроксипропан-2-ил)-ацетамида
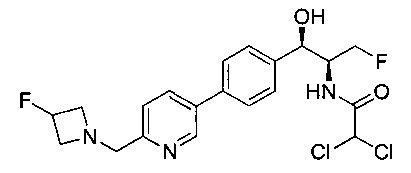
Следуя общей методике из Примера 20, стадии 2 и 3, и осуществляя незначительные модификации, но используя соль HCl 3-фторазетидина, получают указанное в заголовке соединение (5 мг). m/z (ХИ) М+Н 444.
Пример 89. Получение N-((1R,2S)-1-(4-(6-((3-аминоазетидин-1-ил)метил)-пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
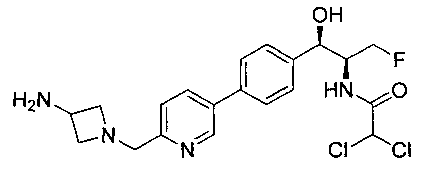
Следуя общей методике из Примера 20, стадии 2 и 3, и осуществляя незначительные модификации, но используя трет-бутил-азетидин-3-илкарбамат, получают указанное в заголовке соединение (24 мг): m/z (ХИ) М+Н 441.
Пример 90. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((3-гидроксиазетидин-1-ил)метил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
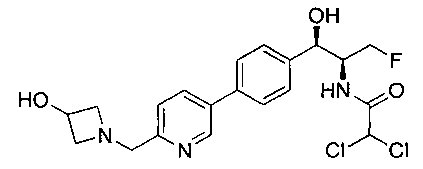
Следуя общей методике из Примера 20, стадии 2 и 3, и осуществляя незначительные модификации, но используя соль HCl азетидин-3-ола, получают указанное в заголовке соединение (15 мг): m/z (ХИ) М+Н 442.
Пример 91. Получение 2-циано-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение 3-((4S,5R)-4-(фторметил)-5-(4-йодфенил)-2,2-диметилоксазолидин-3-ил)-3-оксопропаннитрила
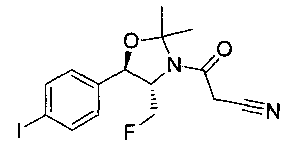
К смеси (4S,5R)-4-(фторметил)-5-(4-йодфенил)-2,2-диметил-оксазолидина (3,5 г, 10,50 ммоль), 2-цианоуксусной кислоты (1,34 г, 15,8 ммоль) и гексафторфосфата O-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилурония (6,11 г, 15,8 ммоль) в диметилформамиде (21 мл) добавляют триэтиламин (2,12 г, 21 ммоль) и перемешивают при комнатной температуре в течение ночи. Смесь распределяют между этилацетатом и рассолом. Органический раствор отделяют, сушат над MgSO4, фильтруют и выпаривают с получением желтого остатка, который очищают на силикагелевой колонке, используя градиент от гептана до чистого этилацетата, с получением указанного в заголовке соединения (2,75 г): 1Н ЯМР (DMSO-d6): 1.48 (s, 3H), 1.53 (s, 3H), 3.96-4.24 (m, 2Н), 4.47-4.84 (m, 3H), 5.11 (d, 1Н), 7.31 (d, 2Н), 7.77 (d, 2Н).
Стадия 2. Получение 3-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(триметилстаннил)фенил)оксазолидин-3-ил)-3-оксопропаннитрила
К раствору 3-((4S,5R)-4-(фторметил)-5-(4-йодфенил)-2,2-диметил-оксазолидин-3-ил)-3-оксопропаннитрила (2,57 г, 6,39 ммоль) в диоксане добавляют гексаметилолово (2,24 г, 6,84 ммоль) и затем смесь продувают азотом. Добавляют дихлорид бис(трифенилфосфин)палладия (II) (90 мг, 0,128 ммоль)) и затем содержимое реакционной массы нагревают под азотом До 80°C. Через 1,5 часа реакционную смесь охлаждают, растворитель удаляют и черное масло фильтруют через диоксид кремния (элюируя этилацетатом) и выпаривают. Неочищенное соединение очищают на силикагелевой колонке, используя 0-100%-ный этилацетат в гептане, с получением указанного в заголовке соединения (2,48 г): 1Н ЯМР (DMSO-d6): 0.7 (s, 9Н), 1.31 (s, 3H), 1.33 (s, 3H), 3.78-4.05 (m, 2Н), 4.30-4.60 (m, 3H), 5.11 (d, 1Н), 7.31 (d, 2Н), 7.77 (d, 2Н).
Стадия 3. Получение 2-циано-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(метилсульфонамидометил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
Смесь 3-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(триметилстаннил)-фенил)оксазолидин-3-ил)-3-оксопропаннитрила (150 мг, 0,342 ммоль), N-((5-бромпиридин-2-ил)метил)метансульфонамида (90 мг, 0,342 ммоль), трис(2-фурил)фосфина (16 мг, 0,068 ммоль) и трис(дибензилиденацетон)-дипалладия(0) (32 мг, 0,034 ммоль) в диметилформамиде (3 мл) нагревают под азотом при 70°C в течение 4 ч. Смесь затем охлаждают, разбавляют водой (10 мл) и экстрагируют этилацетатом (3×15 мл). Экстракты сушат над Na2SO4 и концентрируют с получением неочищенного продукта, m/z (ХИ) М+Н 461. Полученный продукт растворяют в CH2Cl2 (3 мл) и обрабатывают трифторуксусной кислотой (0,5 мл) в течение 3 часов при комнатной температуре. Реакционную смесь разбавляют толуолом (10 мл) и концентрируют до сиропа. Полученный неочищенный продукт растворяют в диметилформамиде (2 мл) и очищают, используя ВЭЖХ, с получением указанного в заголовке продукта (4 мг): m/z (ХИ) М+Н 421.
Пример 92. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(3-(метилсульфонамидометил)изоксазол-5-ил)фенил)пропан-2-ил)ацетамида
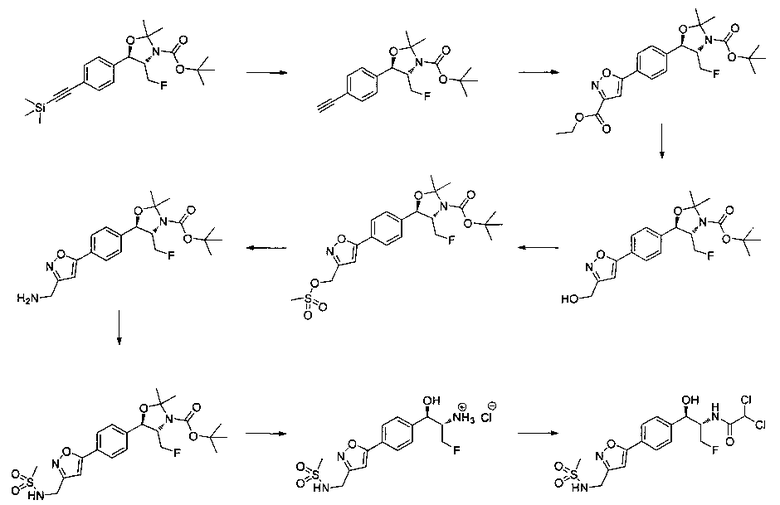
Стадия 1. Получение (4S,5R)-трет-бутил-4-(фторметил)-2,2-диметил-5-(4-((триметилсилил)этинил)фенил)оксазолидин-3-карбоксилата
К толуольному раствору (40 мл) (4S,5R)-трет-бутил-4-(фторметил)-5-(4-йодфенил)-2,2-диметилоксазолидин-3-карбоксилата (2,0 г, 4,6 ммоль) добавляют этинилтриметилсилан (451 мг, 4,6 ммоль), йодид меди (I) (88 мг, 0,46 ммоль, 0,1 эквив.), дихлорид бис(трифенилфосфин)палладия(II) (165 мг, 0,23 ммоль, 0,05 эквив.) и пиперидин (782 мг, 9,2 ммоль, 2 эквив.). Смесь нагревают в атмосфере азота при 35°C в течение шести часов. Смесь фильтруют через набивку целлита. Набивку целита промывают этилацетатом (2×15 мл). Объединенные органические фильтраты концентрировали посредством выпаривания с помощью роторного испарителя при пониженном давлении с получением неочищенного вязкого масла. Масло очищают посредством колоночной флэш-хроматографии (градиент от 100% гексанов до 15% EtOAc) с получением указанного в заголовке соединения (1,65 г): m/z (ХИ) 306 ([М+Н]+-Вос).
Стадия 2. Получение (4S,5R)-трет-бутил-5-(4-этинилфенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилата
К раствору (4S,5R)-трет-бутил-4-(фторметил)-2,2-диметил-5-(4-((триметилсилил)этинил)фенил)оксазолидин-3-карбоксилата (1,65 г, 4,1 ммоль) в THF (20 мл), который был охлажден до -78°C (сухой лед/ацетон) добавляют фторид тетра-н-бутил-аммония (5 мл 1 М раствора в тетрагидрофуране, 5 ммоль). Реакционную смесь перемешивают при -78°C в течение 1 часа. Реакционную смесь гасят (еще при -78°C) посредством добавления насыщенного водного раствора хлорида аммония (2 мл). Реакционную смесь нагревают до комнатной температуры и разбавляют этилацетатом (50 мл). Органическую фазу промывают водой (3×25 мл), сушат над сульфатом натрия и летучие вещества удаляют путем выпаривания с получением указанного в заголовке продукта (1,35 г).
Стадия 3. Получение этил-5-(4-((4S,5R)-3-(трет-бутоксикарбонил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)изоксазол-3-карбоксилата
К раствору (4S,5R)-трет-бутил-5-(4-этинилфенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилата (1,35 г, 4,0 ммоль) в этилацетате (20 мл) добавляют диметилформамидный раствор (Е)-этил-2-хлор-2-(гидроксиимино)-ацетата (12,5 мл, 1,5 г, 2,5 эквив.). К раствору добавляют гидрокарбонат натрия (3,5 г) и его оставляют перемешиваться в течение ночи при комнатной температуре. Смесь фильтруют и разбавляют этилацетатом (100 мл). После промывания водой (4×25 мл) органическую фазу концентрируют с получением неочищенного продукта, который очищают на силикагеле, используя градиент этилацетата в гексанах (от 0% до 20% этилацетата), с получением указанного в заголовке соединения (567 мг): 1Н ЯМР (400 МГц, CDCl3) 1.43-1.56 (m, 14 Н), 1.62 (s, 3 Н), 1.74 (br. s., 3 Н), 4.50 (q, J=7,24 Гц, 3 Н), 5.20 (d, J=7,33 Гц, 1 Н), 6.96 (s, 1 Н), 7.60 (d, J=8,34 Гц, 2 Н), 7.86 (d, J=8,59 Гц, 2 Н), m/z (ХИ) 449 ([М+Н]+.
Стадия 4. Получение (4S,5R)-трет-бутил-4-(фторметил)-5-(4-(3-(гидроксиметил)изоксазол-5-ил)фенил)-2,2-диметилоксазолидин-3-карбоксилата
К охлажденному (0°C) раствору этил-5-(4-((4S,5R)-3-(трет-бутоксикарбонил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)изокса-зол-3-карбоксилата (1,0 г, 2,2 ммоль) в THF (25 мл) добавляют борогидрид натрия (0,25 г, 6,7 ммоль), а затем метанол (3 мл, добавляют медленно). Реакционную смесь перемешивают в течение 30 минут при 0°C и в течение одного часа при комнатной температуре. Избыток борогидрида натрия гасят посредством добавления насыщенного водного хлорида аммония (5 мл). Реакционную смесь разбавляют этилацетатом (50 мл) и водой (25 мл). Слои смешивают и оставляют разделяться. Органическую фазу собирают, сушат над сульфатом натрия и концентрируют с получением продукта, (4S,5R)-трет-бутил-4-(фторметил)-5-(4-(3-(гидроксиметил)изоксазол-5-ил)фенил)-2,2-диметил-оксазолидин-3-карбоксилата (875 мг, 96%), в виде вязкого масла. 1Н ЯМР (400 МГц, CDCl3) 1.52 (s, 9 Н), 1.59 (s, 3 Н), 1.74 (br. s., 3 Н), 2.03-2.08 (m, 1 Н), 3.83-3.96 (m, 1 Н), 4.40-4.60 (m, 2 Н), 4.85 (d, J=6,06 Гц, 2 Н), 5.19 (d, J=7,58 Гц, 1 Н), 6.63 (s, 1 Н), 7.57 (d, J=8,34 Гц, 2 Н), 7.82 (d, J=8,34 Гц, 2 Н); m/z (ХИ) 407 ([М+Н]+.
Стадия 5. Получение (4S,5R)-трет-бутил-4-(фторметил)-2,2-диметил-5-(4-(3-((метилсульфонилокси)метил)изоксазол-5-ил)фенил)оксазолидин-3-карбоксилата
Раствор (4S,5R)-трет-бутил-4-(фторметил)-5-(4-(3-(гидроксиметил)-изоксазол-5-ил)фенил)-2,2-диметилоксазолидин-3-карбоксилата (876 мг, 2,16 ммоль) в метиленхлориде (20 мл) охлаждают до 0°C и добавляют метансульфонилхлорид (250 мг, 2,16 ммоль), а затем диизопропилэтиламин (278 мг, 2,16 ммоль). Реакционную смесь перемешивают в течение одного часа при 0°C. Реакционную смесь разбавляют метиленхлоридом (20 мл) и промывают водой (2×10 мл). Органическую фазу сушат над сульфатом натрия и концентрируют с получением указанного в заголовке соединения (987 мг): m/z (ХИ) 485 ([М+Н]+.
Стадия 6. Получение (4S,5R)-трет-бутил-5-(4-(3-(аминометил)-изоксазол-5-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилата
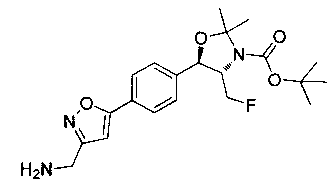
К раствору (4S,5R)-трет-бутил-4-(фторметил)-2,2-диметил-5-(4-(3-((метилсульфонилокси)метил)изоксазол-5-ил)фенил)оксазолидин-3-карбоксилата (1,0 г, 2,1 ммоль) в диоксане (10 мл) добавляют 30%-ный водный раствор гидроксида аммония (5 мл). Смесь перемешивают при 35°C в течение 12 часов. Продукт распределяют между водой (50 мл) и метиленхлоридом (25 мл). Органическую фазу сушат (сульфат натрия) и концентрируют с получением указанного в заголовке соединения (685 мг): m/z (ХИ) 406 ([М+Н]+.
Стадия 7. Получение (4S,5R)-трет-бутил-4-(фторметил)-2,2-диметил-5-(4-(3-(метилсульфонамидометил)изоксазол-5-ил)фенил)оксазолидин-3-карбоксилата
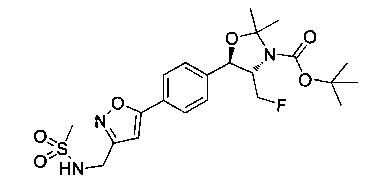
К раствору (4S,5R)-трет-бутил-5-(4-(3-(аминометил)изоксазол-5-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-карбоксилата (270 мг, 0,66 ммоль) в метиленхлориде (10 мл), который был охлажден до 0°C, последовательно добавляют мезилхлорид (83 мг, 0,73 ммоль) и диизопропилэтиламин (94 мг, 0,73 ммоль). Реакционную смесь перемешивают при 0°C в течение тридцати минут, затем гасят водой (2 мл). Погашенную реакционную смесь перемешивают в течение тридцати минут при комнатной температуре. Органическую фазу сушат над сульфатом натрия и концентрируют с получением указанного в заголовке соединения (310 мг): m/z (ХИ) 484 ([М+Н]+.
Стадия 8. Получение хлорида (1R,2S)-3-фтор-1-гидрокси-1-(4-(3-(метилсульфонамидометил)изоксазол-5-ил)фенил)пропан-2-аминия
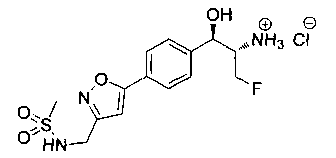
К (4S,5R)-трет-бутил-4-(фторметил)-2,2-диметил-5-(4-(3-(метилсульфон-амидо-метил)изоксазол-5-ил)фенил)оксазолидин-3-карбоксилату (300 мг, 0,62 ммоль) при комнатной температуре добавляют 4 н. HCl в диоксане (4 мл). Раствор затем охлаждают до 0°C и добавляют воду (0,5 мл). Ледяную баню удаляют спустя десять минут и реакционную смесь перемешивают при комнатной температуре в течение одного часа. Летучие вещества затем удаляют с помощью роторного испарителя. Осуществляют несколько циклов выпаривания, используя ацетонитрил для обеспечения полного удаления воды и избытка HCl, с получением указанного в заголовке соединения (234 мг): m/z (ХИ) 344 ([М+Н]+.
Стадия 9. Получение 2,2-дихлор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(3-(метилсульфонамидометил)изоксазол-5-ил)фенил)пропан-2-ил)ацетамида
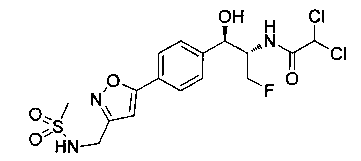
К раствору хлорида (1R,2S)-3-фтор-1-гидрокси-1-(4-(3-(метилсульфон-амидометил)изоксазол-5-ил)фенил)пропан-2-аминия (150 мг, 0,40 ммоль) в диметилформамиде (3 мл) добавляют диизопропилэтиламин (0,255 г, 2 ммоль). После охлаждения реакционной смеси до 0°C добавляют дихлорацетилхлорид (62 мг, 0,42 ммоль). Реакционную смесь перемешивают при 0°C в течение двадцати минут, затем гасят посредством добавления воды (1 мл). Реакционную смесь концентрируют до объема приблизительно два миллилитра и подвергают очистке посредством ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения (95 мг): 1Н ЯМР (400 МГц, CDCl3) 3.06 (s, 3 Н), 4.30-4.43 (m, 1 Н), 4.49 (d, J=6,32 Гц, 2 Н), 4.56-4.67 (m, 2 Н), 4.93 (m, br, 1 Н), 5.19-5.24 (m, 1 Н), 5.87 (s, 1 Н), 6.61 (s, 1 Н), 7.03 (d, J=9,09 Гц, 1 Н), 7.52 (d, J=8,08 Гц, 2 Н), 7.79 (d, J=8,59 Гц, 2 Н); m/z (ХИ) 454, 456, 458 ([М+Н]+.
Пример 93. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(3-(метилсульфонамидометил)изоксазол-5-ил)фенил)пропан-2-ил)ацетамида
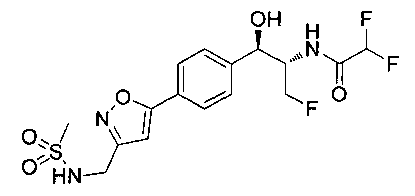
Получают, как описано на стадии 9 Примера 92, но используя дифторацетилхлорид.
1Н ЯМР (400 МГц, CDCl3) 3.06 (s, 3 Н), 4.35-4.75 (m, 6 Н), 4.93 (m, br, 1 Н), 5.20 (d, 1 Н), 5.87 (t, 1 Н), 6.61 (s, 1 Н), 7.52 (d, 2 Н), 7.79 (d, 2 Н); m/z (ХИ) 422 [М+Н]+.
Пример 94. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((RS)-1-(метилсульфонамидо)этил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение N-(1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)этил)(RS)-метансульфонамида
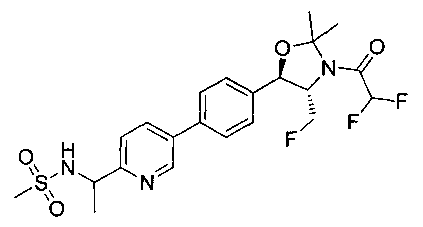
Следуя общей методике из Примера 11 и осуществляя незначительные модификации, но используя продукт стадии 1 Примера 11 и 2,2-дифтор-1-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксазолидин-3-ил)этанон, получали указанное в заголовке соединение (919 мг): 1Н ЯМР (400 МГц, CDCl3) δ: 1.6-1.9 (m, 9Н), 2.8 (s, 3H), 4.5-4.9 (m, 3H), 5.2-5.3 (m, 1Н), 5.8-5.9 (m, 1Н), 6.15 (t, 1Н), 7.4 (d, 1Н), 7.55 (d, 2Н), 7.65 (d, 2Н), 7.9 (d, 1Н), 8.8 (s, 1Н).
Стадия 2. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((RS)-1-(метилсульфонамидо)этил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
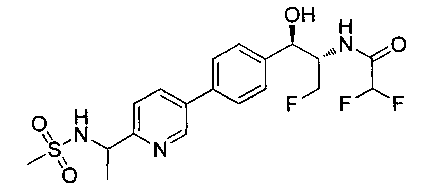
Следуя общей методике из Примера 2, стадия 2, и используя продукт стадии 1 Примера 94, получают указанное в заголовке соединение (782 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.45 (d, 3H), 2.8 (s, 3H), 4.25-4.35 (m, 1.5Н), 4.4-4.5 (m, 0.5Н), 4.5-4.7 (m, 2Н), 4.9 (m, 1Н), 5.9 (s, 1Н), 6.2 (t, 1Н), 7.45 (d, 2Н), 7.55 (d, 1Н), 7.65-7.75 (m, 3H), 8.1 (d, 1Н), 8.8 (s, 2Н). m/z (ХИ) 446 [М+Н].
Пример 95. Получение N-((1R,2S)-1-(4-(6-((RS)-1-аминоэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
Стадия 1. Получение трет-бутил-(1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)этил)-карбамата
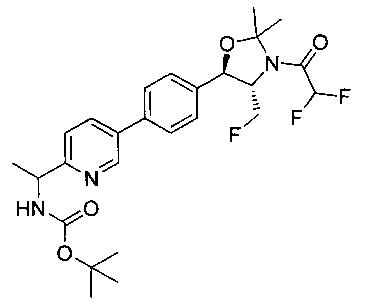
Следуя общей методике из Примера 22 и осуществляя незначительные модификации, но используя трет-бутил-(1-(5-бромпиридин-2-ил)этил)карбамат (описан ранее в WO 2011/138751), получали указанное в заголовке соединение (910 мг): m/z (ХИ) 508 [М+Н].
Стадия 2. Получение N-((1R,2S)-1-(4-(6-((RS)-1-аминоэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
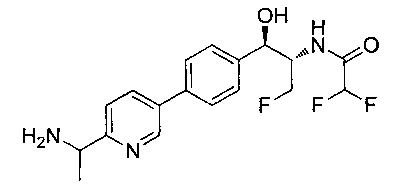
Следуя общей методике из Примера 2, стадия 2, и используя продукт со стадии 1 Примера 95, получают указанное в заголовке соединение d (970 мг): Н ЯМР (300 МГц, DMSO-d6) δ: 1.3 (d, 3H), 4.0-4.15 (m, 1Н), 4.25-4.4 (m, 1.5Н), 4.4-4.6 (m, 1Н), 4.65-4.75 (m, 0.5Н), 4.9 (t, 1H), 5.9 (d, 1H), 6.2 (t, 1H), 7.45 (d, 2H), 7.5-7.6 (m, 1H), 7.70 (d, 2H), 8.05 (d, 1H), 8.80 (d, 2H). m/z (ХИ) 368 [М+Н].
Способами, известными в уровне техники, могут быть получены следующие производные указанного в заголовке соединения из Примера 97:
N-((1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамид;
(1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-гидрофосфат натрия;
(1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-гидрофосфат натрия и
(1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-дигидрофосфат. Это производное было получено следующим образом.
Пример 95А
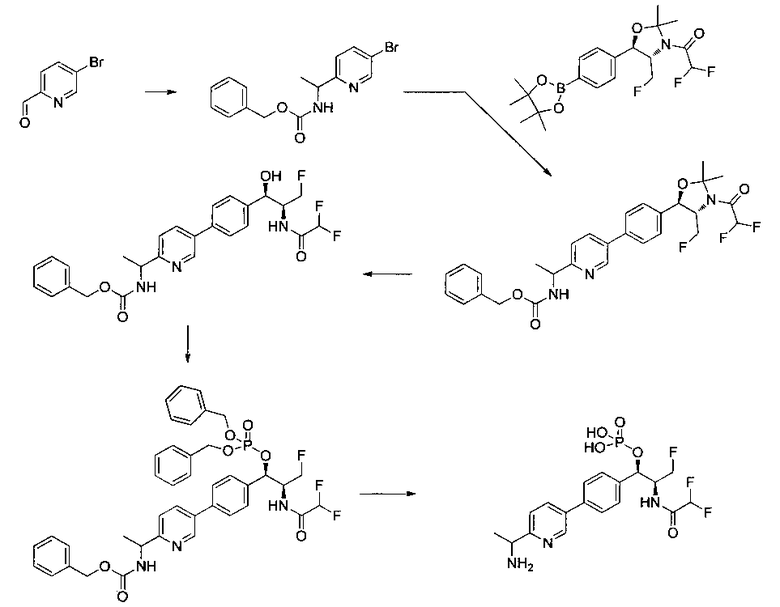
Стадия 1. Получение бензил-(1-(5-бромпиридин-2-ил)этил)карбамата
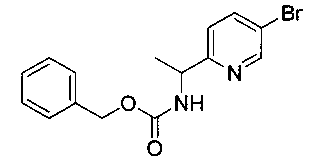
К имеющемуся в продаже 5-бром-пиридин-2-карбальдегиду (2,00 г, 10,8 ммоль) в THF (20 мл) при -20°C добавляют гексаметилдисилазид лития. Спустя 30 минут смесь охлаждают до -70°C и добавляют бромид метилмагния (4,8 мл, 4,5 ммоль). Смесь нагревают до комнатной температуры и гасят насыщенным водным хлоридом аммония (15 мл). Смесь распределяют между водой и этил ацетатом. Органический слой отделяют, промывают рассолом, сушат над MgSO4, фильтруют и выпаривают с получением промежуточного соединения в виде остатка (1,90 г). Остаток растворяют в CH2Cl2 (40 мл) и добавляют 1,0 М гидроксид натрия (24 мл, 24 ммоль). Смесь охлаждают до 0°C и по каплям добавляют бензилхлорформиат (1,69 мл, 11,8 ммоль). После перемешивания в течение 4 часов органическую фазу отделяют, промывают водой (30 мл), затем рассолом (20 мл), сушат над сульфатом натрия и растворитель удаляют при пониженном давлении с получением неочищенного продукта, который очищают хроматографией на диоксиде кремния, элюируя смесью 20% этилацетат/гептаны, с получением указанного в заголовке продукта (1,17 г): m/z: 334,03.
Стадия 2. Получение бензил-(1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)этил)-карбамата
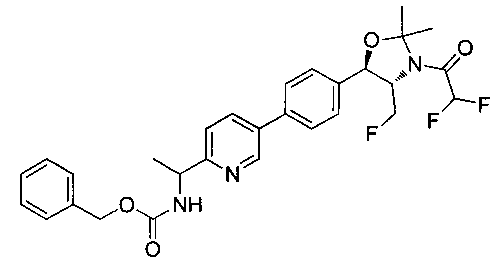
Смесь продукта из Примера 22, стадия 3, бензил-(1-(5-бромпиридин-2-ил)этил)карбамата, 2,0 М карбоната натрия в воде (3,1 мл, 6,2 ммоль) в диоксане дегазируют азотом. Добавляют комплекс 1,1′-бис(дифенилфосфино)ферроцен дихлорпалладия(II) с дихлорметаном (169 мг, 0,21 ммоль) и смесь нагревают до 80°C в течение ночи. После охлаждения до комнатной температуры реакционную смесь разбавляют водой (25 мл) и экстрагируют этилацетатом. Органическую фазу отделяют, сушат над MgSO4 и концентрируют при пониженном давлении. Неочищенный материал очищают, используя хроматографию на диоксиде кремния, элюируя градиентом от 25% этилацетат/гептан до 35% этилацетат/гептан, с получением указанного в заголовке продукта (1,00 г): m/z: 541,22.
Стадия 3. Получение бензил-(1-(5-(4-((1R,2S)-2-(2,2-дифторацетамидо)-3-фтор-1-гидроксипропил)фенил)пиридин-2-ил)этил)карбамата
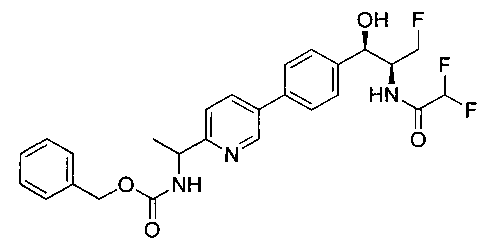
Трифторуксусную кислоту (2,2 мл) при 0°C добавляют к продукту стадии 2 Примера 95А (1,05 г, 1,94 ммоль) в CH2Cl2 (39 мл). Спустя 30 минут ледяную баню удаляют и смесь перемешивают при комнатной температуре в течение 8 часов. Добавляют насыщенный NaHCO3 (60 мл) и смесь экстрагируют CH2Cl2 (40 мл). Объединенную органическую фазу сушат над MgSO4, фильтруют и выпаривают с получением указанного в заголовке продукта (850 мг): m/z: 501,19.
Стадия 4. Получение бензил-(1-(5-(4-((1R,2S)-1-((бис(бензилокси)-фосфорил)окси)-2-(2,2-дифторацетамидо)-3-фторпропил)фенил)пиридин-2-ил)этил)карбамата
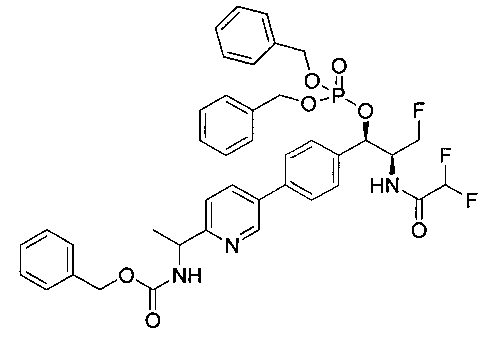
Трифторуксусную кислоту (51,6 мкл) при 0°C добавляют к продукту со стадии 3 Примера 95А (0,168 г, 0,335 ммоль) и пиридину (54,2 мкл, 0,67 ммоль) в THF (4,4 мл, 54 ммоль). Через 5 минут добавляют бис(бензилокси)(диизопропиламино)фосфин (0,219 мл, 0,586 ммоль). После ее нагрева до комнатной температуры смесь перемешивают в течение 1 часа. Добавляют смесь 30%-ная перекись водорода/вода (30:70, 60 мкл, 0,586 ммоль). Через 1 час добавляют бисульфат натрия (4 мл) и добавляют воду (10 мл). Реакционную смесь экстрагируют этилацетатом (2×15 мл). Объединенную органическую фазу сушат над MgSO4, фильтруют и выпаривают с получением неочищенного продукта, который очищают хроматографией на диоксиде кремния, элюируя градиентом от 25% этилацетат/гептаны до чистого этилацетата, с получением указанного в заголовке продукта (241 мг): m/z: 761,25.
Стадия 5. Получение (1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-дигидрофосфата
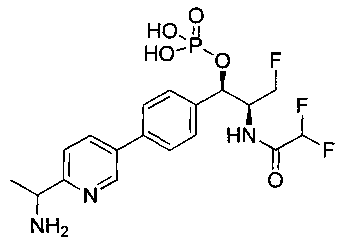
Палладий на углероде (1:9, 23 мг, 0,022 ммоль) добавляют к продукту стадии 4 Примера 95А (235 мг, 0,308 ммоль) в этаноле (5 мл) и воде (1 мл). Смесь вакуумируют три раза с помощью азота и перемешивают под водородом в течение 5 часов. Реакционную смесь фильтруют через 4 мл набивку Solka Flok и десять раз промывают чередующимися промывками по 4 мл этанола и воды. Растворитель удаляют при пониженном давлении с получением неочищенного продукта, который переносят в воду (5 мл) и фильтруют через 0,45 мкм фильтровальный диск, промывая водой (2 мл). Фильтрат сушат вымораживанием с получением указанного в заголовке продукта (126 мг): 1Н ЯМР δ 8.75 (1Н, s), 8.07 (1Н, d), 7.63 (2Н, d), 7.52-7.48 (4Н, m), 6.00 (1Н, t), 5.42 (1Н, d), 4.82 (1Н, dd), 4.64-4.43 (4Н, m), 1.62 (3H, d); m/z: 447,12.
Пример 96. Получение 2,2-дифтор-N-{(1S,2R)-1-фторметил-2-гидрокси-2-[4-(6-пирролидин-2-ил-пиридин-3-ил)-фенил]-этил}-ацетамида
Стадия 1. Получение трет-бутилового эфира 2-(5-бром-пиридин-2-ил)-пирролидин-1-карбоновой кислоты
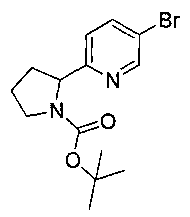
Раствор 5-бром-2-пирролидин-2-ил-пиридина (611 мг, 2,69 ммоль) (описан ранее в WO 200853319) в CH2Cl2 (20 мл) обрабатывают ди-трет-бутилдикарбонатом (881 мг, 4,04 ммоль) и триэтиламином (0,562 мл, 4,04 ммоль) и перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь промывают 10%-ным водным раствором лимонной кислоты (25 мл). Органическую фазу концентрируют на силикагеле (5 g) и очищают колоночной хроматографией (силикагель 40 g, этилацетат/гептан 0-50%) с получением указанного в заголовке соединения (500 мг): 1Н ЯМР (400 МГц, CDCl3) 1.23 (s, 5H), 1.45 (s, 4H), 1.82-2.09 (m, 3H), 2.24-2.43 (m, 1Н), 3.47-3.74 (m, 2Н), 4.67-4.99 (m, 1Н), 7.05-7.14 (m, 1Н), 7.71-7.78 (m, 1Н), 8.59 (dd, 1Н). m/z М+Н 327.
Стадия 2. Получение трет-бутилового эфира 2-(5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-ил)-пирролидин-1-карбоновой кислоты
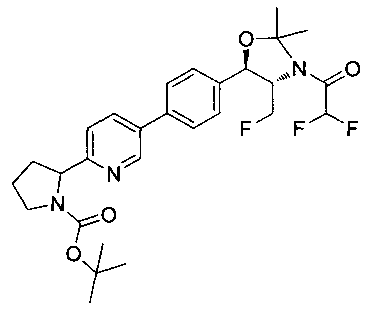
К раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (634 мг, 1,53 ммоль) в смеси толуола (12 мл) и этанола (9 мл) добавляют трет-бутиловый эфир 2-(5-бром-пиридин-2-ил)-пирролидин-1-карбоновой кислоты (500 мг, 1,53 ммоль), водный раствор бикарбоната натрия (2 М, 6 ммоль, 3 мл) и 1,1′бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (57 мг, 0,08 ммоль). Перемешанную реакционную смесь нагревают при 80°C в атмосфере азота в течение 1 часа. Реакционную смесь концентрируют до 1/3 ее объема и распределяют между этилацетатом (25 мл) и водой (25 мл). Органическую фазу промывают насыщенным рассолом (25 мл) и концентрируют на силикагеле (3 g). В результате очистки колоночной хроматографией (силикагель 40 g, этилацетат/гептан 0-100%) получают указанное в заголовке соединение (500 мг): 1Н ЯМР (400 МГц, CDCl3) 1.25 (s, 5Н) 1.48 (s, 4Н), 1.64 (s, 3H), 1.70 (s, 3H), 1.85-1.98 (m, 2Н), 1.99-2.1 (1Н), 2.3-2.48 (m, 1Н), 3.5-3.74 (m, 2Н), 4.54-5.1 (m, 4Н), 5.23-5.33 (m, 1Н), 6.13 (t, 1Н), 7.23-7.31 (m, 1Н) 7.5-7.57 (m, 2Н), 7.57-7.67 (m, 2Н), 7.78-7.87 (m, 1Н), 8.77 (s, 1Н). m/z М+Н 534,2.
Стадия 3. 2,2-Дифтор-N-{(1S,2R)-1-фторметил-2-гидрокси-2-[4-(6-пирролидин-2-ил-пиридин-3-ил)-фенил]-этил}-ацетамид
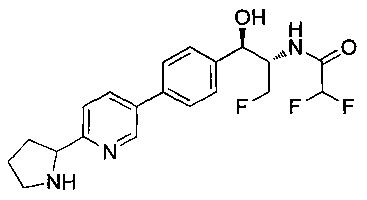
К перемешиваемому раствору трет-бутилового эфира 2-(5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-ил)-пирролидин-1-карбоновой кислоты (500 мг, 0,94 ммоль) и CH2Cl2 (20 мл), охлажденному до 0°C, добавляют трифторуксусную кислоту (3 мл) и воду (100 мкл). Реакционную смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение дополнительных 2 часов. Добавляют толуол (20 мл) и реакционную смесь концентрируют под вакуумом с получением неочищенного продукта. В результате очистки препаративной ВЭЖХ получают указанное в заголовке соединение (217 мг): 1Н ЯМР (400 МГц, CDCl3), 1.88-2.06 (m, 3H), 2.38-2.48 (m, 1Н), 3.22-3.40 (m, 3H), 4.25-4.37 (m, 1.5Н), 4.4-4.47 (m, 0.5Н), 4.52-4.59 (m, 0.5Н), 4.63-4.70 (m, 0.5Н), 4.71-4.78 (m, 1Н), 4.86-4.94 (m, 1Н), 5.89-6.01 (m, 1Н), 6.20 (t, 1Н), 7.43-7.53 (m, 2Н), 7.58-7.63 (1Н, m), 7.71-7.77 (m, 2Н), 8.15-8.24 (m, 1Н), 8.82-8.88 (m, 1Н), 8.9-8.94 (m, 1Н). m/z М+Н 394.
Способами, известными из уровня техники, может быть получено следующее производное указанного в заголовке соединения из Примера 96:
(1R,2S)-2-(2,2-дифторацетамидо)-3-фтор-1-(4-(6-(пирролидин-2-ил)пиридин-3-ил)фенил)пропил-дигидрофосфат.
Пример 97. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(морфолин-3-ил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
Стадия 1. Получение трет-бутил-3-(5-бромпиридин-2-ил)морфолин-4-карбоксилата
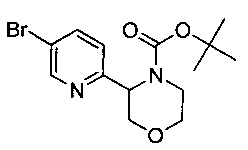
К перемешиваемой суспензии имеющегося в продаже 3-(5-бромпиридин-2-ил)-морфолина, соли ди-гидрохлорид (615 мг, 1,95 ммоль), в 1,4-диоксане (10 мл, 0,2 М) добавляют 10%-ный водный раствор К2СО3 (11,1 мл, 7,78 моль, 4 экв.). Добавляют ди-трет-бутилдикарбонат (637 мг, 2,92 ммоль, 1,5 экв.) и оставляют перемешиваться при комнатной температуре в течение ночи. Реакционную смесь оставляют еще на дополнительные 3 часа, после чего ее разбавляют водой и экстрагируют DCM (50 мл). Органический слой отделяют и концентрируют с получением светло-желтого масла (580 мг, 87%). m/z (ХИ) 343 [М+Н].
Стадия 2. Получение трет)-бутил-3-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)морфолин-4-карбоксилата
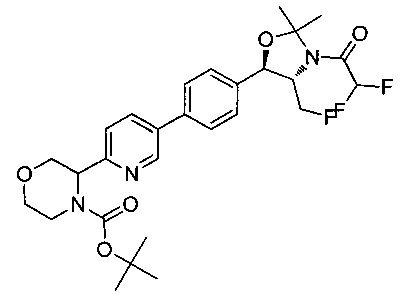
Следуя общей методике из Примера 22, стадия 1, и осуществляя незначительные модификации, но используя трет-бутил-3-(5-бромпиридин-2-ил)морфолин-4-карбоксилат, получают указанное в заголовке соединение (510 мг): m/z (ХИ) 550 [М+Н].
Стадия 3. Получение 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(морфолин-3-ил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
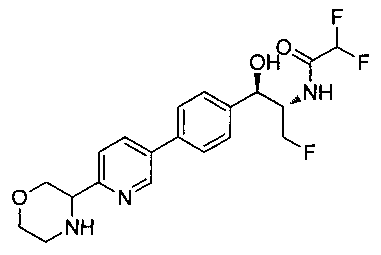
Следуя общей методике из Примера 2, стадия 2, и осуществляя незначительные модификации, но используя продукт стадии 2 Примера 97, получают указанное в заголовке соединение (251 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ 2.95 (d, 2Н), 3.35 (d, 2Н), 3.4-3.6 (m, 1Н), 3.8 (d, 1Н), 4.0 (dd, 1Н), 4.05 (dd, 1Н), 4.25-4.4 (m, 1.5Н), 4.4-4.5 (t, 0.5Н), 4.5-4.6 (m, 0.5H), 4.6-4.7 (m, 0.5H), 4.9 (t, 1H), 5.9 (d, 1H), 6.2 (t, 1H), 7.45 (d, 2H), 7.55 (d, 1H), 7.65 (d, 2H), 8.1 (dd, 1H), 8.8-8.9 (m, 2H). m/z (ХИ) 410 [М+Н].
Пример 98. Получение М-((1R,2S)-1-(4-(6-(1-амино-2-метоксиэтил)-пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
Стадия 1. Получение 1-((4S,5R)-5-(4-(6-(1-амино-2-метоксиэтил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-ил)-2,2-дифторэтанона
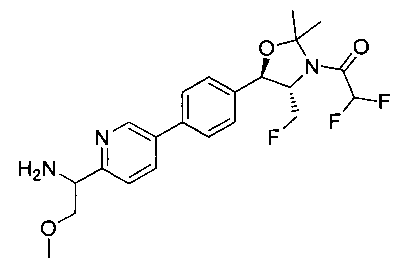
К раствору толуол/этанол (8 мл:6 мл, соответственно) добавляют последовательно 2,2-дифтор-1-((4S,5R)-4-(фторметил)-2,2-диметил-5-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)оксазолидин-3-ил)этанон (300 мг, 0,73 ммоль), 1-(5-бромпиридин-2-ил)-2-метоксиэтанамин (168 мг, 0,73 ммоль), 1,1′-бис(дифенилфосфино)ферроцендихлорпалладий (II) (53 мг, 0,07 ммоль) и гидрокарбонат натрия (2 мл, 2,0 М (водн.), 4 ммоль). Реакционную смесь нагревают с перемешиванием в атмосфере азота в течение двух часов. Темно-красную реакционную смесь концентрируют досуха посредством выпаривания при низком давлении с помощью роторного испарителя. Остаточный материал суспендируют с помощью этанола (2×20 мл). Этанол каждый раз декантируют. Декантированные растворы объединяют и концентрируют посредством выпаривания при низком давлении с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт, 1-((4S,5R)-5-(4-(6-(1-амино-2-метоксиэтил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазоютдин-3-ил)-2,2-дифторэтанон, смесь диатереомеров 1:1, использовали непосредственно на следующей стадии.
Стадия 2. Получение N-((1R,2S)-1-(4-(6-(1-амино-2-метоксиэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
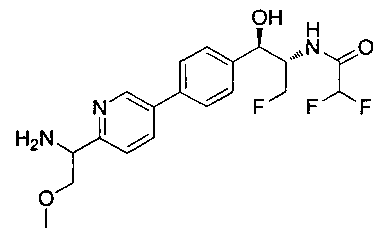
Раствор 7:3 CH2Cl2/трифторуксусная кислота (общий объем 10 мл), соответственно, добавляют к чистому 1-((4S,5R)-5-(4-(6-(1-амино-2-метоксиэтил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-ил)-2,2-дифторэтанону. К реакционной смеси добавляют воду (0,2 мл). Раствор перемешивают в течение двух часов при комнатной температуре. Летучие вещества удаляют посредством выпаривания при низком давлении с помощью роторного испарителя с получением неочищенного продукта, который очищают посредством ВЭЖХ с обращенной фазой (вода с 0,1% TFA/ацетонитрил) с получением после лиофилизации TFA соли N-((1R,2S)-1-(4-(6-(1-амино-2-метоксиэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида (17 мг): m/z (ХИ) 398 [М+Н]+.
Пример 99. Получение N-[(1S,2R)-2-{4-[6-(1-аминоэтил)пиридин-3-ил]фенил}-1-(фторметил)-2-гидроксиэтил]-2,2-дихлорацетамида
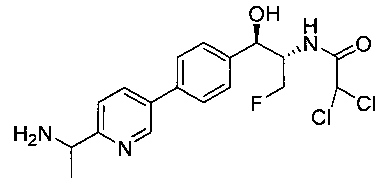
Следуя общим методикам из Примера 22 и Примера 2, и осуществляя незначительные модификации, но используя трет-бутил-[1-(5-бромпиридин-2-ил)этил]карбамат, получают указанное в заголовке соединение (140 мг): 1Н ЯМР (300 МГц, DMSOd-6) δ 1.31 (d, 3H), 2.16 (s, br, 2Н), 4.02 (q, 1Н), 4.20-4.28 (m, 1.5Н), 4.41-4.46 (m, 0.5H), 4.54-4.57 (m, 0.5H), 4.69-4.72 (m, 0.5H), 4.72-4.91 (m, 1H), 6.00 (d, 1H), 6.53 (s, 1H), 7.46 (d, 2H), 7.54 (d, 1H), 7.67 (d, 2H), 8.00-8.03 (dd, 1H), 8.65 (d, 1H), 8.77 (m, 1H). MC (ИЭР+) m/z 400,1 /402,0 [М+Н].
Пример 100. Получение N-[(1S,2R)-2-{4-[6-(1-аминоциклопропил)пиридин-3-ил]фенил}-1-(фторметил)-2-гидроксиэтил]-2,2-дихлорацетамида
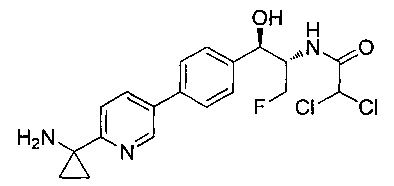
Следуя общей методике из Примера 22 и Примера 2 и осуществляя незначительные модификации, но используя трет-бутил-[1-(5-бромпиридин-2-ил)этил]карбамат, получают указанное в заголовке соединение (60 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ 0.99 (m, 2Н), 1.25 (m, 2Н), 4.25 (m, 1.5Н), 4.41 (m, 0.5Н), 4.58 (m, 0.5H), 4.70 (m, 0.5H), 4.90 (m, 1H), 6.00 (d, 1H), 6.54 (s, 1H), 7.45 (d, 2H), 7.65 (d, 2H), 7.81 (d, 1H), 8.01 (d, 1H), 8.67 (bd, 1H), 8.71 (m, 1H). MC (ИЭР+) m/z 412 [М+Н].
Пример 101. Получение бромида 4-({(1R,2S)-2-[(дифторацетил)амино]-3-фтор-1-[4-(6-{[(метилсульфонил)амино]метил}пиридин-3-ил)фенил]пропил}-окси)-N,N,N-триметил-4-оксобутан-1-аминия
Стадия 1. Получение (1R,2S)-2-[(дифторацетил)амино]-3-фтор-1-[4-(6-{[(метилсульфонил)амино]метил}пиридин-3-ил)фенил]пропил-4-бромбутаноата
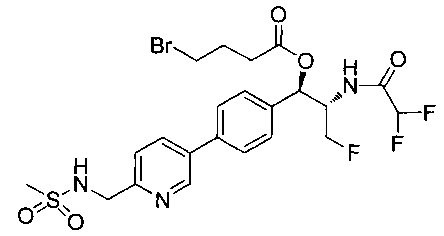
4-Бромбутаноилхлорид (29,6 мкл, 0,255 ммоль) при КТ добавляют по каплям к перемешиваемому раствору 2,2-дифтор-N-{(1S,2R)-1-(фторметил)-2-гидрокси-2-[4-(6-{[(метилсульфонил)амино]метил}пиридин-3-ил)фенил]этил}-ацетамида (100,0 мг, 0,232 ммоль), N,N-диизопропилэтиламина (48,4 мкл, 0,278 ммоль) и 4-диметиламинопиридина (11,3 мг, 0,093 ммоль) в N,N-диметилформамиде (1,00 мл). Реакционную смесь перемешивают в течение 1 часа, после чего концентрируют. Остаток очищали с помощью CombiFlash (колонка 4 g), элюируя 0-100% этилацетата, с получением указанного в заголовке соединения (80,0 мг, 59%) в виде желтой пены. МС (ИЭР+) m/z 581,7 [М+Н].
Стадия 2. Получение бромида 4-({(1R,2S)-2-[(дифторацетил)амино]-3-фтор-1-[4-(6-{[(метилсульфонил)амино]метил}пиридин-3-ил)фенил]пропил}-окси)-N,N,N-триметил-4-оксобутан-1-аминия
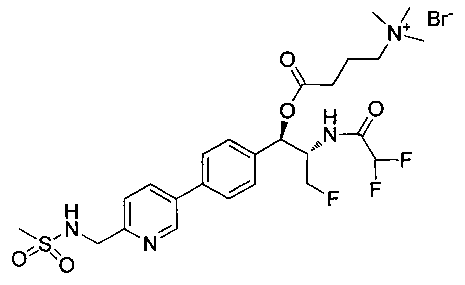
В герметизируемой пробирке при комнатной температуре к перемешиваемому раствору (1R,2S)-2-[(дифторацетил)амино]-3-фтор-1-[4-(6-{[(метилсульфонил)-амино]метил}-пиридин-3-ил)фенил]пропил-4-бромбутано-ата (стадия 1, 570,0 мг, 0,982 ммоль) в тетрагидрофуране (11,0 мл) добавляют 4,2 М триметиламин в этаноле (1,42 мл, 5,98 ммоль) и реакционную смесь нагревали при 50°C в течение ночи. После перемешивания в течение ночи реакционную смесь концентрировали и остаток переносили в воду. Водный слой экстрагировали метиленхлоридом (3х) для удаления всех примесей. Водный слой затем лиофилизировали в течение ночи с получением указанного в заголовке соединения (375 мг): 1Н ЯМР (300 МГц, DMSO-d6) δ 1.94-1.97 (m, 2Н), 2.73-2.77 (t, 2Н), 3.05 (s, 9Н), 3.23-3.31 (m, 2Н), 3.48 (s, 3H), 4.27-4.33 (m, 1.5Н), 4.43-4.53 (m, 1H), 4.67-4.71 (m, 0.5H), 4.89-4.91 (m, 1H), 5.13 (s, 2H), 5.91 (d, 1H), 6.20 (t, 1H), 7.45-7.48 (m, 3H), 7.71 (d, 2H), 8.10-8.14 (dd, 1H), 8.82 (d, 1H), 8.86 (d, 1H). MC (ИЭР+) m/z 559,1 [М].
Пример 102. Получение 2,2-дихлор-N-{(1S,2R)-1-(фторметил)-2-гидрокси-2-[4-(6-пирролидин-2-илпиридин-3-ил)фенил]этил}ацетамида
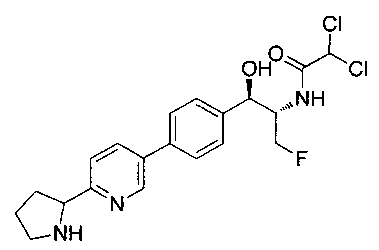
Следуя общей методике из Примера 96, но с использованием трет-бутил-2-(5-бромпиридин-2-ил)пирролидин-1-карбоксилата, получают указанное в заголовке соединение (380 мг): 1Н ЯМР (300 МГц, DMSO-d6) δ 1.66-1.79 (m, 3H), 2.12-2.18 (m, 1Н), 2.86-2.92 (m, 1Н), 3.00-3.05 (m, 1Н), 3.32 (s, br, 1Н), 4.17-4.28 (m, 2.5Н), 4.42-4.44 (m, 0.5H), 4.54-4.57 (m, 0.5H), 4.69-4.74 (m, 0.5H), 4.89-4.91 (m, 1H), 6.00 (d, 1H), 6.53 (s, 1H), 7.46 (d, 2H), 7.54 (d, 1H), 7.67 (d, 2H), 7.99-8.02 (dd, 1H), 8.65 (d, 1H), 8.77 (d, 1H). MC (ИЭР+) m/z 426,0, 428,0 (M+H).
Способами, известными из уровня техники, может быть получено следующее производное указанного в заголовке соединения из Примера 102:
(1R,2S)-2-(2,2-дихлорацетамидо)-3-фтор-1-(4-(6-(пирролидин-2-ил)пиридин-3-ил)фенил)пропилдигидрофосфат.
Пример 103. Получение N-{(1S,2R)-2-[4-(2-аминометил-пиримидин-5-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дифтор-ацетамида, соли с трифторуксусной кислотой
Стадия 1. Получение трет-бутилового эфира (5-хлор-пиримидин-2-илметил)-карбаминовой кислоты
К перемешиваемому раствору С-(5-хлор-пиримидин-2-ил)-метиламина (0,1 г, 0,532 ммоль) в CH2Cl2 (2 мл) при комнатной температуре добавляют триэтиламин (0,153 мл, 1,064 ммоль) с последующим добавлением ди-трет-бутилдикарбоната (0,134 мл, 84,50 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают и неочищенный материал очищают посредством CombiFlash, элюируя 12%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (0,1 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.37(s, 9Н), 4.27 (d, 6.16 Гц, 2Н), 7.33 (t, J1=5,92 Гц, 1Н), 8.94 (s, 2Н). ЖХ-МС (m/z): [М+Н]=288,2.
Стадия 2. Получение трет-бутилового эфира (5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиримидин-2-илметил)-карбаминовой кислоты
К перемешиваемому раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (0,286 г, 0,694 ммоль) и трет-бутилового эфира (5-хлор-пиримидин-2-илметил)-карбаминовой кислоты (0,2 г, 0,694 ммоль) в смеси изопропиловый спирт: вода (2:1, 6 мл) при комнатной температуре добавляют К2СО3 (0,287 г, 2,083 ммоль). Полученную реакционную смесь дегазируют азотом в течение 20 минут, после чего добавляют Pd(dppf)Cl2.CH2Cl2 (0,028 г, 0,035 ммоль). Полученную реакционную смесь нагревают до 140°C в течение 20 минут под действием микроволнового излучения. Растворитель выпаривают в вакууме и неочищенный материал разбавляют водой и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, концентрируют и очищают колоночной хроматографией, используя CombiFlash, элюируя 75%-ным этилацетатом в гексане, с получением указанного в заголовке соединения (0,125 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.40 (s, 9Н), 1.52 (s, 3H), 1.60 (s, 3H), 4.36 (d, J=6,16 Гц, 2Н), 4.54-4.58 (m, 0.5Н), 4.66-4.73 (m, 1Н), 4.82-4.85 (m, 0.5Н), 4.91-4.95 (m, 1H), 5.28 (d, J=3.52 Гц, 1H), 6.64 (t, J=52,44 Гц, 1H), 7.32 (t, J=6,04 Гц, 1H), 7.62 (d, J=8,04 Гц, 2H), 7.86 (d, J=8,04 Гц, 2H), 9.10 (s, 2H). ЖХ-МС (m/z): [М+Н]=495.
Стадия 3. Получение N-{(1S,2R)-2-[4-(2-аминометил-пиримидин-5-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дифтор-ацетамида, соли с трифторуксусной кислотой
К перемешиваемому раствору трет-бутилового эфира 5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиримидин-2-илметил)-карбаминовой кислоты (0,125 г, 0,253 ммоль) в CH2Cl2 (10 мл) при комнатной температуре добавляют трифторуксусную кислоту (1,0 мл). Полученную реакционную смесь оставляют перемешиваться при комнатной температуре в течение 5 часов. Растворитель выпаривают в вакууме и неочищенный материал обрабатывают CH2Cl2 с последующей промывкой диэтиловым эфиром и сушат в Lipholiser с получением указанного в заголовке соединения (0,1 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 4.31-4.33 (m, 1.5Н), 4.36 (d, J=8,84 Гц, 2H), 4.42-4.46 (m, 0.5Н), 4.56-4.57 (m, 0.5H), 4.66-4.70 (m, 0.5H), 4.92 (t, J=3,76 Гц, 1H), 5.98 (d, J=4,6 Гц, 1H), 6.20 (t, J=53,72 Гц, 1H), 7.52 (d, J=8,24 Гц, 2H), 7.84 (d, J=8,24 Гц, 2H), 8.36 (bs, 2H), 7.86 (d, J=8,84 Гц, 1H), 9.24 (s, 2H). ЖХ-МС (m/z): [M+H]=354,9.
Пример 104. Получение N-((1S,2R)-2-{4-[6-(1-амино-2-циано-этил)-пиридин-3-ил]-фенил}-1-фторметил-2-гидрокси-этил)-2,2-дифтор-ацетамида
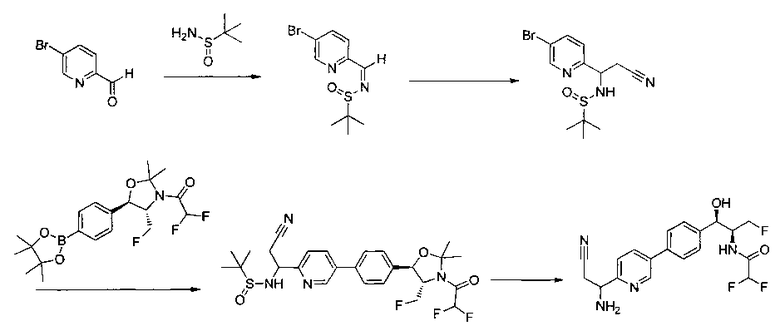
Стадия 1. Получение 5-бром-пиридин-2-илметиленамида 2-метил-пропан-2-сульфиновой кислоты
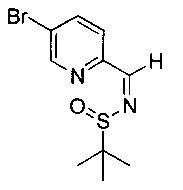
К перемешиваемому раствору 5-бром-пиридин-2-карбальдегида (2,0 г, 10,753 ммоль) в CH2Cl2 (20 мл) и амида 2-метил-пропан-2-сульфиновой кислоты (2,602 г, 21,505 ммоль) при комнатной температуре добавляют CuSO4 (5,589 г, 21,505 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь фильтруют через целит. Разбавляют водой и экстрагируют CH2Cl2. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и очищают посредством хроматографии CombiFlash, используя 5%-ный этилацетат в гексане, с получением указанного в заголовке соединения (2,5 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.19 (s, 9Н), 8.03 (d, J=8,52 Гц, 1Н), 8.25-8.27 (dd, J=2,32 Гц, J=8,32 Гц, 1Н), 8.44 (s, 1Н), 8.90 (d, J=1,84 Гц, 1Н). ЖХ-МС (m/z): [М+Н]=291,1.
Стадия 2. Получение [1-(5-бром-пиридин-2-ил)-2-циано-этил]-амида 2-метил-пропан-2-сульфиновой кислоты
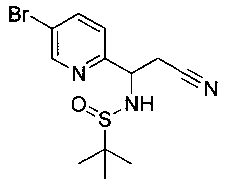
К раствору ацетонитрила (0,142 г, 3,472 ммоль) в сухом тетрагидрофуране (10 мл) при -78°C добавляют н-бутиллитий (0,222 г, 3,472 ммоль) и перемешивают в течение 30 минут при той же самой температуре с последующим добавлением по каплям раствора 5-бром-пиридин-2-илметиленамида 2-метил-пропан-2-сульфиновой кислоты (0,5 г, 1,736 ммоль) в сухом тетрагидрофуране (10 мл) и перемешивают реакционную смесь при -78°C в течение 1 ч. Реакционную смесь разбавляют водным раствором хлорида аммония и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и очищают посредством хроматографии CombiFlash, используя 80%-ный этилацетат в гексане, с получением указанного в заголовке соединения (0,25 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.17 (s, 9Н), 2.93-2.99 (m, 1Н), 3.11-3.16 (m, 1Н), 4.72-4.78 (m, 1Н), 6.09 (d, J=9,24, 1Н), 7.64 (d, J=8,44 Гц, 1H), 8.12-8.14 (dd, J1=2,36 Гц, J2=8,4 Гц, 1Н), 8.674 (d, 2.24 Гц, 1Н). ЖХ-МС (m/z): [М+Н]=330,0.
Стадия 3. Получение [2-циано-1-(5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-ил)-этил]-амида 2-метил-пропан-2-сульфиновой кислоты
К перемешиваемому раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (0,312 г, 0,758 ммоль) и [1-(5-бром-пиридин-2-ил)-2-циано-этил]-амида 2-метил-пропан-2-сульфиновой кислоты (0,25 г, 0,758 ммоль) в смеси толуол: этанол: вода (10:10:10 мл) при комнатной температуре добавляют Na2CO3 (0,16 г, 1,515 ммоль). Полученную реакционную смесь дегазируют азотом в течение15 минут с последующим добавлением Pd(PPh3)4 (0,087 г, 0,076 ммоль). Полученную реакционную смесь нагревают до 80°C в течение 3 часов. Растворитель выпаривают в вакууме, затем разбавляют водой и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, растворитель выпаривают в вакууме и очищают посредством хроматографии CombiFlash, используя 50%-ный этилацетат в гексане, с получением указанного в заголовке соединения (0,21 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.19 (s, 9Н), 1.53 (s, 3H), 1.60 (S, 3H), 2.98-3.04 (m, 1Н), 3.17-3.22 (m, 1Н), 4.54-4.58 (m, 0.5Н), 4.66-4.70 (m, 1Н), 4.78-4.84 (m, 1.5Н), 4.89-4.95 (m, 1Н), 5.26-5.27 (m, 1Н), 6.12 (d, J=9 Гц, 1Н), 6.64 (t, J=52,4 Гц, 1Н), 7.53-7.64 (m, 2Н), 7.76 (d, J=8,2 Гц, 1Н), 7.82 (d, J=8,12 Гц, 2Н), 8.89 (d, J=1,84 Гц, 1Н), 8.86 (d, J=2,08 Гц, 1Н). ЖХ-МС (m/z): [М+Н]=537,2.
Стадия 4. Получение N-((1S,2R)-2-{4-[6-(1-амино-2-циано-этил)-пиридин-3-ил]-фенил}-1-фторметил-2-гидрокси-этил)-2,2-дифтор-ацетамида
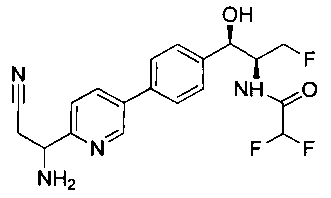
К раствору [2-циано-1-(5-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридин-2-ил)-этил]-амида 2-метил-пропан-2-сульфиновой кислоты (0,21 г, 0,392 ммоль) в этилацетате (2 мл) при 0°C добавляют HCl в диоксане (2,0 мл). Полученную реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Растворитель выпаривают в вакууме с получением неочищенного остатка и разбавляют его водным раствором аммиака и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, концентрируют и очищают посредством хроматографии CombiFlash, используя 7-8%-ный метанол в CH2Cl2, с получением указанного в заголовке соединения (0,032 г): 1Н ЯМР (400 МГц, DMSO-d6) δ: 2.85-2.99 (m, 2Н), 4.24-4.35 (m, 2.5Н), 4.40-4.44 (m, 0.5Н), 4.54-4.55 (m, 0.5H), 4.64-4.67 (m, 0.5H), 4.87-4.88 (m, 2H), 5.92 (d, J=4,44 Гц, 1H), 6.20 (t, J=53,8 Гц, 1H), 7.46 (d, J=8,16 Гц, 2H), 7.62 (d, J=8,16 Гц, 1H), 7.71 (d, J=8,22 Гц, 2H), 8.09-8.12 (dd, J1=2,32 Гц, J2=8,2 Гц, 1H), 8.84-8.87 (m, 2H). ЖХ-МС (m/z): [M+H]=393,0.
Пример 105. Получение N-{(1S,2R)-2-[4-(6-аминометил-пиридазин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дифтор-ацетамида, соли с трифторуксусной кислотой
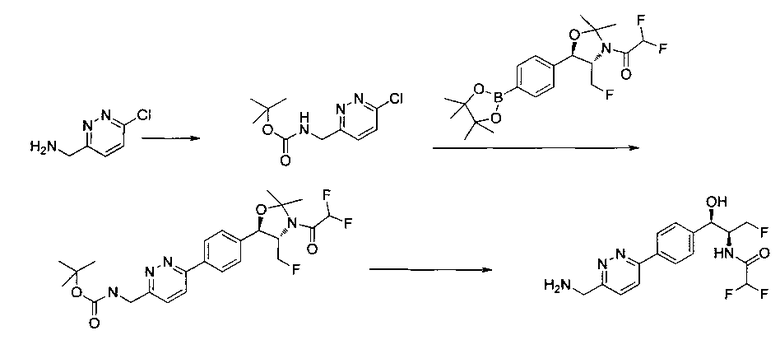
Стадия 1. Получение трет-бутилового эфира (6-хлор-пиридазин-3-илметил)-карбаминовой кислоты
К перемешиваемому раствору соли гидрохлорида С-(6-хлор-пиридазин-3-ил)-метиламина (0,05 г, 0,279 ммоль) в ацетонитриле (2 мл) при комнатной температуре добавляют триэтиламин (0,08 мл, 0,557 ммоль) с последующим добавлением ди-трет-бутилдикарбоната (0,076 мл, 0,334 ммоль). Реакционную смесь нагревают до 60°C в течение 2 часов. Растворитель выпаривают и неочищенный материал очищают посредством CombiFlash, элюируя 14%-ным метанолом в CH2Cl2, с получением указанного в заголовке соединения (0,025 г, 1Н ЯМР (400 МГц, DMSO-d6) 1.39 (s, 9Н), 4.40 (d, 6.08 Гц, 2Н), 7.59 (m, 1Н), 7.62 (d, J=9,04 Гц, 1Н), 7.89 (d, J=8,88 Гц, 2Н). ЖХ-МС (m/z): [М+Н]=244,2.
Стадия 2. Получение трет-бутилового эфира (6-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фенил}-пиридазин-3-илметил)-карбаминовой кислоты
К перемешиваемому раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (0,849 г, 2,058 ммоль) и трет-бутилового эфира (6-хлор-пиридазин-3-илметил)-карбаминовой кислоты (0,5 г, 2,058 ммоль) в смеси толуол: этанол: вода (20:10:5 мл) при комнатной температуре добавляют Na2CO3 (0,436 г, 4,115 ммоль). Полученную реакционную смесь дегазируют азотом в течение 20 минут с последующим добавлением Pd(PPh3)4 (0,237 г, 0,206 ммоль). Полученную реакционную смесь нагревают до 80°C в течение 16 ч. Растворитель выпаривают в вакууме и неочищенный материал разбавляют с помощью воды и экстрагируют этилацетатом. Органический слой сушат над сульфатом натрия, концентрируют и очищают с помощью CombiFlash, элюируя 20%-ным МеОН в DCM, с получением указанного в заголовке соединения (0,250 г): 1Н ЯМР (400 МГц, DMSO-d6) 1.40 (s, 9Н), 1.53 (s, 3H), 1.61 (s, 3H), 3.94 (s, 1Н), 4.47 (d, J=6,08 Гц, 2Н), 4.58-4.59 (m, 0.5Н), 4.70-4.71 (m, 1Н), 4.83-4.84 (m, 0.5Н), 4.94-4.97 (m, 1H), 5.30 (d, J=3,32 Гц, 1H), 6.65 (t, J=52,32 Гц, 1H), 7.61-7.67 (m, 4H), 8.18 (d, J=8,12 Гц, 2H), 8.24 (d, J=8,84 Гц, 1H). ЖХ-МС (m/z): [М+Н]=495,2.
Стадия 3. Получение соли трифторуксусной кислоты и N-{(1S,2R)-2-[4-(6-аминометил-пиридазин-3-ил)-фенил]-1-фторметил-2-гидрокси-этил}-2,2-дифтор-ацетамида
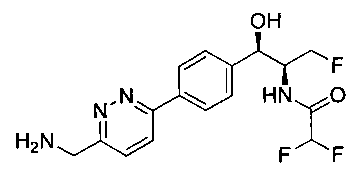
К перемешиваемому раствору трет-бутилового эфира (6-{4-[(4S,5R)-3-(2,2-дифтор-ацетил)-4-фторметил-2,2-диметил-оксазолидин-5-ил]-фен ил}-пиридазин-3-илметил)-карбаминовой кислоты (0,25 г, 0,506 ммоль) в CH2Cl2 (10 мл) при комнатной температуре добавляют трифторуксусную кислоту (1,5 мл). Полученную реакционную смесь оставляют перемешиваться при комнатной температуре в течение 5 ч. Растворитель выпаривают в вакууме и неочищенный материал обрабатывают CH2Cl2 с последующей промывкой н-пентаном и диэтиловым эфиром и сушат с получением указанного в заголовке соединения (0,181 г): 1Н ЯМР (400 МГц, DMSO-d6) 4.32-4.39 (m, 1.5Н), 4.46 (m, 2.5Н), 4.55-4.58 (m, 0.5H), 4.68-4.71 (m, 0.5H), 4.94 (bs, 1H), 5.99 (bs, 1H), 6.19 (t, J=53,76 Гц, 1H), 7.54 (d, J=8,28 Гц, 2H), 7.85 (d, J=8,92 Гц, 1H), 8.13 (d, J=8,32 Гц, 2H), 8.36 (d, J=10 Гц, 1H), 8.51 (bs, 2Н), 8.87 (d, J=8,72 Гц, 1H). ЖХ-МС (m/z): [М+Н]=355.
Пример 106. Получение N-((1R,2S)-1-(4-(6-((S)-1-амино-2-гидроксиэтил)-пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
Стадия 1. Получение (S)-трет-бутил-1-(5-бромпиридин-2-ил)-2-гидроксиэтилкарбамата
Раствор (S)-2-амино-2-(5-бромпиридин-2-ил)этанола (250 мг, 0,99) в CH2Cl2 (10 мл) обрабатывают ди-трет-бутилдикарбонатом (237 мг, 1,08 ммоль) и триэтиламином (0,275 мл, 1,97) и перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь промывают 10%-ным водным раствором лимонной кислоты (25 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и концентрируют с получением указанного в заголовке соединения (262 мг): m/z М+Н 317. Время удерживания 2,50 мин.
Стадия 2. Получение трет-бутил-(S)-1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)-2-гидрокси-этилкарбамата
К раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (339 мг, 0,820 ммоль) в смеси толуола (5 мл) и этанола (3 мл) добавляют (S)-трет-бутил-1-(5-бромпиридин-2-ил)-2-гидроксиэтилкарбамат (260 мг, 0,82 ммоль), водный раствор бикарбоната натрия (2 М, 3,28 ммоль, 1,64 мл) и [1,1′бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (30 мг, 0,04 ммоль). Перемешиваемую реакционную смесь нагревают при 80°C под слоем азота в течение 1 часа. Реакционную смесь концентрируют досуха и распределяют между метиленхлоридом (25 мл) и водой (25 мл). Органическую фазу промывают насыщенным рассолом (25 мл) и концентрируют на силикагеле (1 г). В результате очистки колоночной хроматографией (силикагель 12 g, смесь этилацетат/гептан 0-100%, 16 объемов колонки) получают указанное в заголовке соединение (260 мг): m/z М+Н 524,2. Время удерживания 2,73 мин.
Стадия 3. Получение N-((1R,2S)-1-(4-(6-((S)-1-амино-2-гидроксиэтил)-пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
К перемешиваемому раствору трет-бутил-(S)-1-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)-2-гидрокси-этилкарбамата (260 мг, 0,49 ммоль) и метиленхлорида (25 мл), охлажденному до 0°C, добавляют трифторуксусную кислоту (2,5 мл) и воду (50 мкл). Реакционную смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение дополнительного часа. Добавляют толуол (20 мл) и реакционную смесь концентрируют под вакуумом с получением неочищенного продукта в виде соли бис-трифторацетата, 400 мг. m/z М+Н 384,1. Время удерживания 1,69 мин.
Пример 107. Получение N-((1R,2S)-1-(4-(6-(2-амино-1-гидроксипропан-2-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
Стадия 1. Получение трет-бутил-2-(5-бромпиридин-2-ил)-1-гидроксипропан-2-илкарбамата
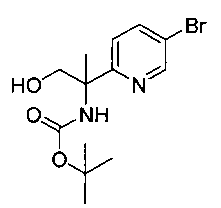
Раствор 2-амино-2-(5-бромпиридин-2-ил)пропан-1-ола (264 мг, 0,99 ммоль) в DCM (10 мл) обрабатывают ди-трет-бутилдикарбонатом (237 мг, 1,08 ммоль) и триэтиламином (0,275 мл, 1,97) и перемешивают при температуре окружающей среды в течение 16 ч. Реакционную смесь промывают 10%-ным водным раствором лимонной кислоты (25 мл). Органическую фазу сушат над безводным сульфатом магния, фильтруют и концентрируют с получением указанного в заголовке соединения (165 мг): m/z М+Н 332,1. Время удерживания 2,61 мин.
Стадия 2. Получение трет-бутил-2-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)-1-гидроксипропан-2-илкарбамата
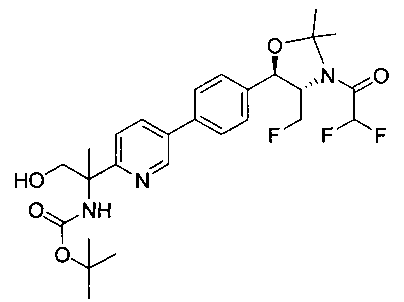
К раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (200 мг, 0,48 ммоль) в смеси толуола (3 мл) и этанола (2 мл) добавляют трет-бутил-2-(5-бромпиридин-2-ил)-1-гидроксипропан-2-илкарбамат (160 мг, 0,48 ммоль), водный раствор бикарбоната натрия (2 М, 2 ммоль, 1 мл) и [1,1′бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (18 мг, 0,024 ммоль). Перемешиваемую реакционную смесь нагревают при 80°C под слоем азота в течение 1 часа. Реакционную смесь концентрируют досуха и распределяют между метиленхлоридом (25 мл) и водой (25 мл). Органическую фазу промывают насыщенным рассолом (25 мл) и концентрируют на силикагеле (1 g). В результате очистки колоночной хроматографией (силикагель 12 g, смесь этилацетат/гептан 0-100%, 16 объемов колонки) получают указанное в заголовке соединение (62 мг): m/z М+Н 538,2. Время удерживания 2,75 мин.
Стадия 3. Получение N-((1R,2S)-1-(4-(6-(2-амино-1-гидроксипропан-2-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
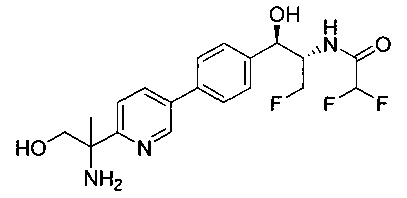
К перемешиваемому раствору трет-бутил-2-(5-(4-((4S,5R)-3-(2,2-дифторацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)пиридин-2-ил)-1-гидроксипропан-2-илкарбамата (62 мг, 0,11 ммоль) и метиленхлорида (10 мл), охлажденному до 0°C, добавляют трифторуксусную кислоту (1 мл) и воду (20 мкл). Реакционную смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение дополнительного часа. Добавляют толуол (10 мл) и реакционную смесь концентрируют под вакуумом с получением неочищенного продукта в виде соли бис-трифторацетата, 100 мг. m/z М+Н 398,1. Время удерживания 1,83 мин.
Пример 108. Получение N-((1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
Стадия 1. Получение трет-бутил-1-(5-бромтиазол-2-ил)этилкарбамата
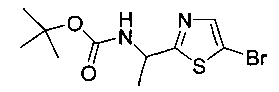
Следуя методике, описанной ранее в WO 2011053542 (стр. 47-49), но используя рацемический 2-метил-2-пропансульфинамид, получают указанное в заголовке соединение (2,06 г): m/z (ХИ) М+Н 311+313.
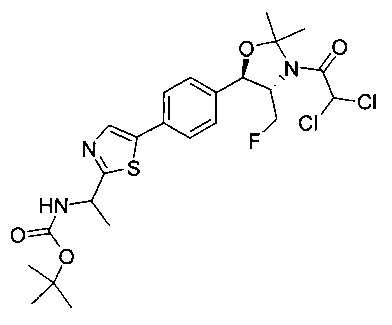
Следуя общей методике из Примера 21, стадия 2, и осуществляя незначительные модификации, но используя трет-бутил-1-(5-бромтиазол-2-ил)этилкарбамат, получают указанное в заголовке соединение (1030 мг): m/z (ХИ) 546 [М+Н].
Стадия 3. Получение N-((1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)-фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамида
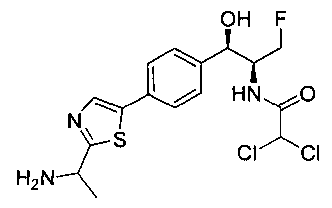
Следуя общей методике из Примера 2, стадия 2, и осуществляя незначительные модификации, но используя трет-бутил-1-(5-(4-((4S,5R)-3-(2,2-дихлорацетил)-4-(фторметил)-2,2-диметилоксазолидин-5-ил)фенил)тиазол-2-ил)этилкарбамат, получают указанное в заголовке соединение (55 мг): 1Н ЯМР (400 МГц, DMSO-d6) δ: 1.42 (d, 3H), 3.75 (m, 2Н), 4.15-4.35 (m, 2.5Н), 4.39-4.43 (m, 0.5Н), 4.55-4.59 (m, 0.5H), 4.67-4.70 (m, 0.5H), 4.87 (t, 1H), 5.98 (d, 1H), 6.51 (s, 1H), 7.40 (d, 2H), 7.58 (d, 2H), 8.06 (s, 1H), 8.59 (d, 1H); m/z (ХИ) 406 [М+Н].
Пример 109. Получение 2,2-дифтор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-{6[(1-метил-1Н-имидазол-2-иламино)-метил]-пиридин-3-ил}-фенил)-этил]-ацетамида
Стадия 1. Получение 2,2-дифтор-1-{(4S,5R)-4-фторметил-5-[4-(6-гидроксиметил-пиридин-3-ил)-фенил]-2,2-диметил-оксазолидин-3-ил}-этанона
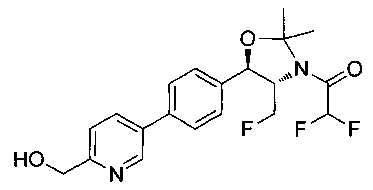
К раствору 2,2-дифтор-1-{(4S,5R)-4-фторметил-2,2-диметил-5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-оксазолидин-3-ил}-этанона (200 мг, 0,48 ммоль) в толуоле (3,2 мл) и этаноле (2,4 мл) добавляют (5-бром-пиридин-2-ил)-метанол (91 мг, 0,48 ммоль), водный раствор бикарбоната натрия (1,94 ммоль, 2 М, 1 мл) и [1,1′бис(дифенилфосфино)ферроцен]дихлор-палладий (II) (18 мг, 0,02 ммоль). Реакционную смесь под азотом нагревают при 80°C в течение 45 минут. Реакционную смесь концентрируют при пониженном давлении и очищают колоночной хроматографией (силикагель 40 g, смесь 0-100% этилацетат/гептан) с получением указанного в заголовке соединения (92 мг): 1Н ЯМР (400 МГц, CDCl3) 1.61-1.79 (m, 6Н), 3.7-3.81 (m, 1Н), 4.50-4.88 (m, 5Н), 5.24-5.31 (m, 1Н), 6.13 (t, 1Н), 7.36 (d, 1Н), 7.55 (d, 2Н), 7.63 (d, 2Н), 7.87-7.92 (m, 1Н), 8.78-8.82 (m, 1Н). m/z М+Н 395.
Стадия 2. Получение 1-{(4S,5R)-5-[4-(6-хлорметил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-ил}-2,2-дифтор-этанона
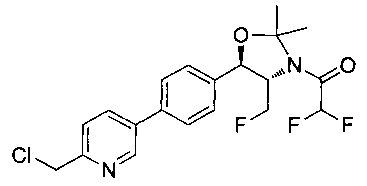
Перемешиваемый раствор 2,2-дифтор-1-{(4S,5R)-4-фторметил-5-[4-(6-гидрокси-метил-пиридин-3-ил)-фенил]-2,2-диметил-оксазолидин-3-ил}-этанона (1660 мг, 4,2 ммоль) и триэтиламина (0,88 мл, 6,31 ммоль) в CH2Cl2 (30 мл) обрабатывают метансульфонилхлоридом (0,49 мл, 6,31 ммоль) и перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь концентрируют досуха с получением указанного в заголовке соединения в виде красного масла (1,53 г): 1Н ЯМР (400 МГц, CDCl3) 1.60-1.77 (m, 6Н), 4.53-4.84 (m, 5Н), 5.25-5.31 (m, 1Н), 6.12 (t, 1Н), 7.58 (d, 2Н), 7.65 (d, 2Н), 7.68 (d, 1Н), 8.03-8.07 (m, 1Н), 8.86-8.90 (m, 1Н). m/z М+Н 413.
Стадия 3. Получение 2,2-дифтор-N-[(1S,2R)-1-фторметил-2-гидрокси-2-(4-{6-[(1-метил-1Н-имидазол-2-иламино)-метил]-пиридин-3-ил}-фенил)-этил]-ацетамида
Раствор 1-{(4S,5R)-5-[4-(6-хлорметил-пиридин-3-ил)-фенил]-4-фтор-метил-2,2-диметил-оксазолидин-3-ил}-2,2-дифтор-этанона (25 мг, 0,06 ммоль) и 1-метил-1Н-имидазол-2-иламина (30 мг, 0,3 ммоль) в диметилформамиде (2 мл) нагревают при 75°C в течение 16 ч. Реакционную смесь концентрируют досуха, растворяют в CH2Cl2 (1 мл) и обрабатывают трифторуксусной кислотой (0,5 мл) и водой (0,1 мл). Смесь встряхивают при температуре окружающей среды в течение 2 часов и затем концентрируют досуха. Неочищенный продукт очищают препаративной ВЭЖХ с получением указанного в заголовке соединения (7 мг): 1Н ЯМР (400 МГц, DMSO-d6) 2.50 (s, 3H), 3.51 (s, 2Н), 4.24-4.36 (m, 1.5Н), 4.37-4.46 (m, 0.5Н), 4.51-4.59 (m, 0.5H), 4.63-4.71 (m, 0.5H), 4.87-4.92 (m, 1H), 5.31 (s, 2H), 6.22 (t, 1H), 7.05-7.10 (m, 2H), 7.40-7.52 (m, 3H), 7.66-7.74 (m, 2H), 8.09-8.16 (m, 1H), 8.86 (s, 1H), 9.63-9.69 (m, 1H). m/z M+H 434.
В следующей далее Таблице 1 представлены другие примеры, полученные этим способом с помощью разных аминов вместо тиазол-5-иламина в Примере 109, стадия 3.
Время удерживания относится к следующему методу ВЭЖХ: колонка-Phenomenex Luna Cl8 4,6×20 мм, 5 мкм, подвижная фаза А - 20 мМ NH4OAc в H2O, подвижная фаза B-ACN, линейный градиент от 10% В до 60% за 5 минут, 1 мл/мин.
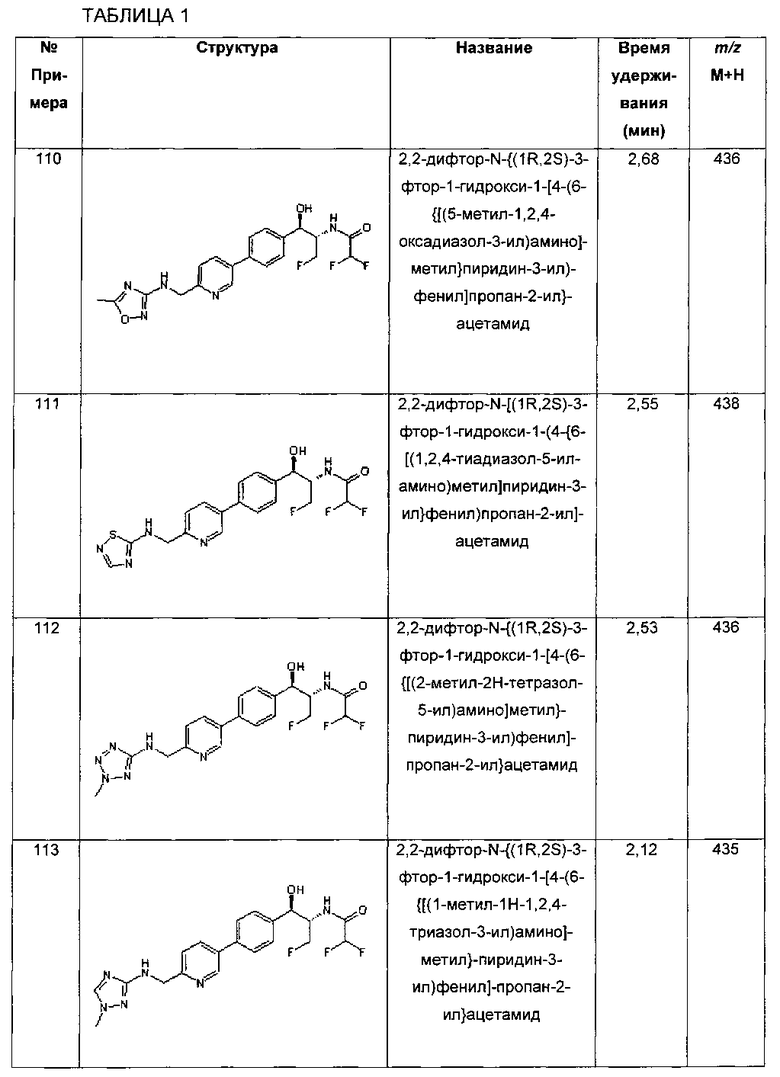
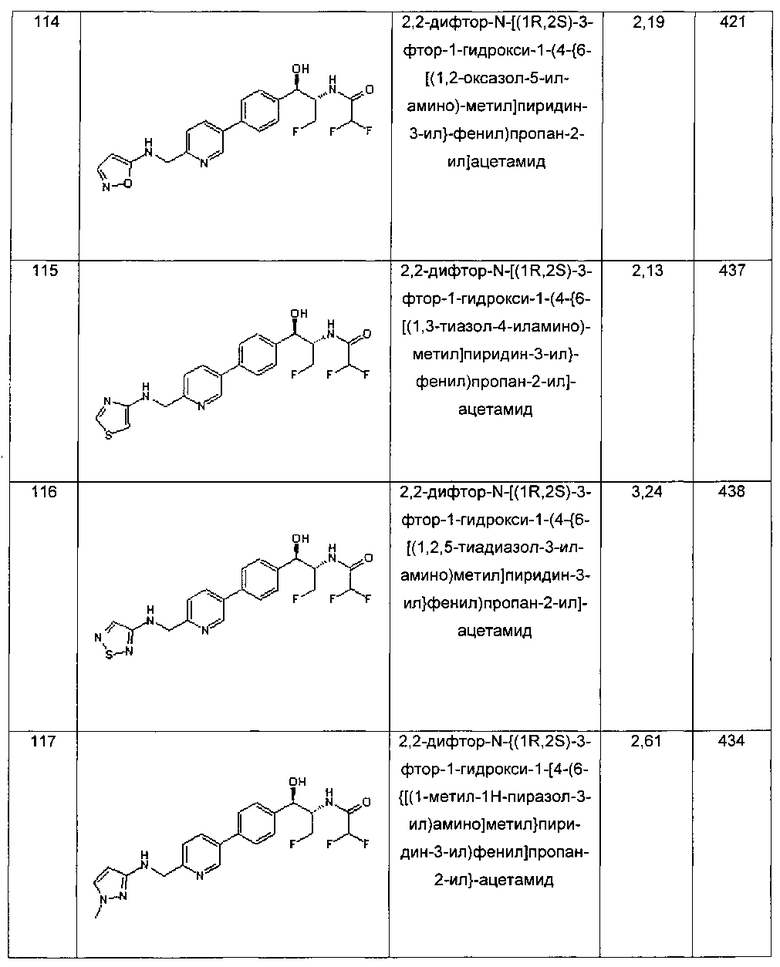
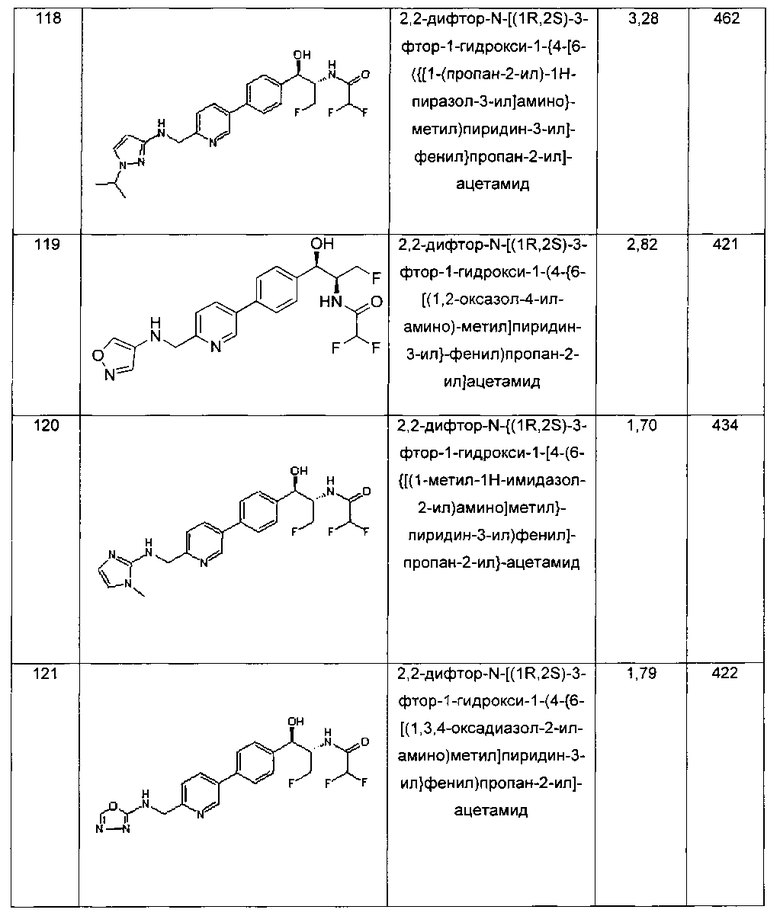
Пример 128. Получение N-((1R,2S)-1-(4-(6-(2-окса-5-азаспиро[3.4]октан-5-илметил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифтор-ацетамида
Стадия 1. Получение 1-((4S,5R)-5-(4-(6-(2-окса-5-азаспиро[3.4]октан-5-илметил)пиридин-3-ил)фенил)-4-(фторметил)-2,2-диметилоксазолидин-3-ил)-2,2-дифторэтанона
Смесь 2-окса-5-азаспиро[3.4]октана (0,1 ммоль, 15,8 мг), 1-{(4S,5R)-5-[4-(6-хлорметил-пиридин-3-ил)-фенил]-4-фторметил-2,2-диметил-оксазолидин-3-ил}-2,2-дифтор-этанона (Пример 99, стадия 2, 0,09 ммоль, 37 мг) и карбоната цезия (0,25 ммоль, 81 мг) и ацетонитрила (1 мл) нагревали до 55°C в течение 5 часов. Неочищенную смесь фильтровали на набивке целита и осадок на фильтре промывали CH2Cl2 (2 мл). Летучие вещества удаляли. Неочищенные смеси использовали "как есть" на стадии 2.
Стадия 2. Получение N-((1R,2S)-1-(4-(6-(2-окса-5-азаспиро[3.4]октан-5-илметил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифтор-ацетамида
Неочищенную смесь растворяли в 0,75 мл смеси TFA/CH2Cl2/H2O (25/10/1) и перемешивали при КТ в течение 2 часов. Затем добавляли 1 мл смеси МеОН/толуол (50/50) и смесь концентрировали досуха. Неочищенный продукт очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (12 мг). Время удерживания (RT) и МС относятся к следующему методу ВЭЖХ-МС: колонка-Waters ACQUITY UPLC ВЕН С8 1,7 мкм, 2,1×50 мм, подвижная фаза А - 0,1%-ная TFA в H2O, подвижная фаза В - 0,1%-ная TFA в ACN, линейный градиент от 10% В до 100% В за 5 мин, 0,8 мл/мин.
RT=3,270 мин, MC m/z=450,2 (М+Н).
В следующей далее Таблице 2 представлены другие примеры, полученные с помощью этого способа, с разными аминами вместо 2-окса-5-азаспиро[3.4]октана на стадии 1 Примера 128. Время удерживания относится к следующему методу ВЭЖХ: колонка-Waters ACQUITY UPLC ВЕН С8 1,7 мкм, 2,1×50 мм, подвижная фаза А - 0,1%-ная TFA в H2O, подвижная фаза В - 0,1%-ная TFA в ACN, линейный градиент от 10% В до 100% за 5 мин, 0,8 мл/мин.
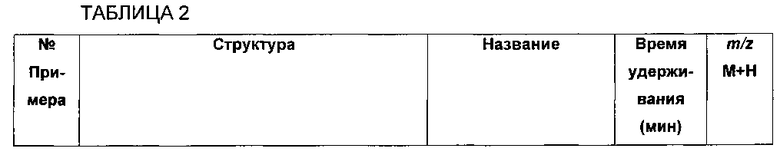
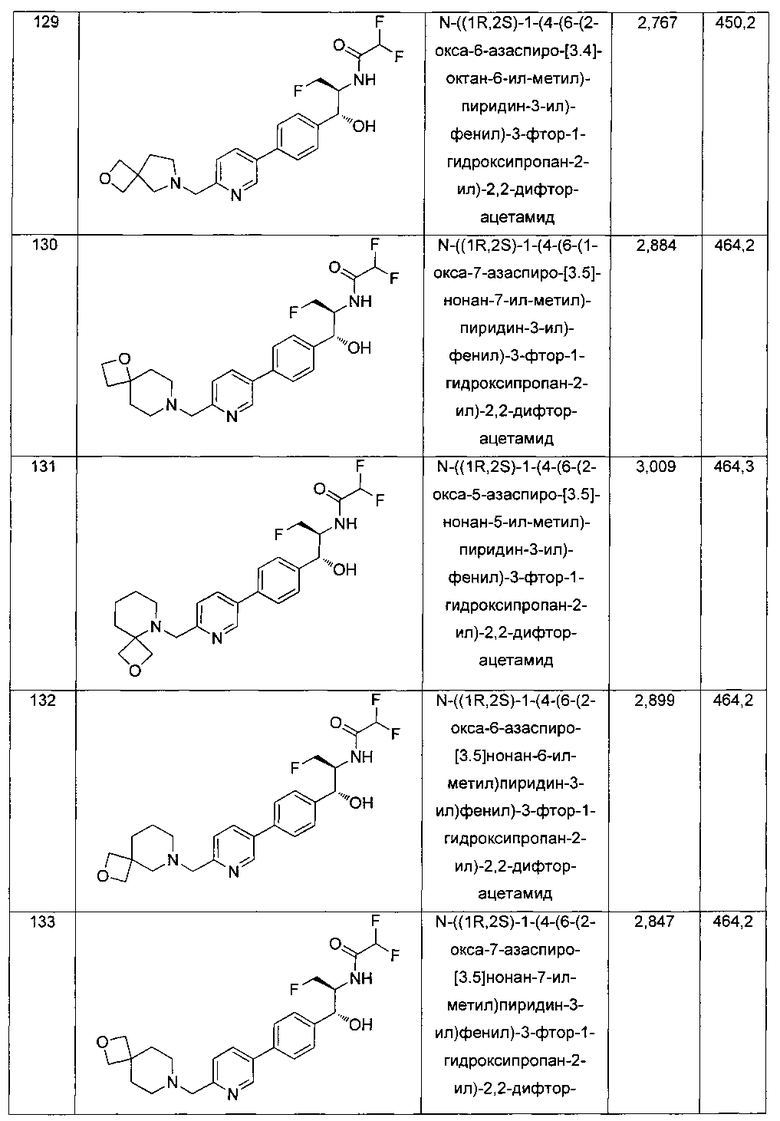
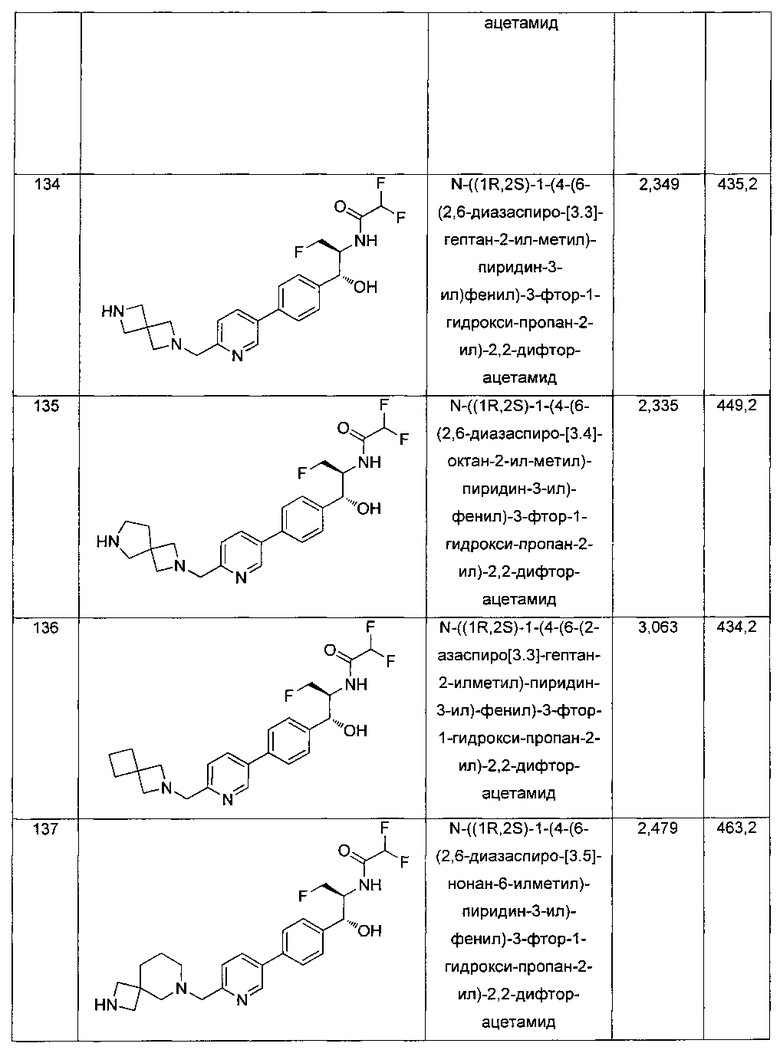
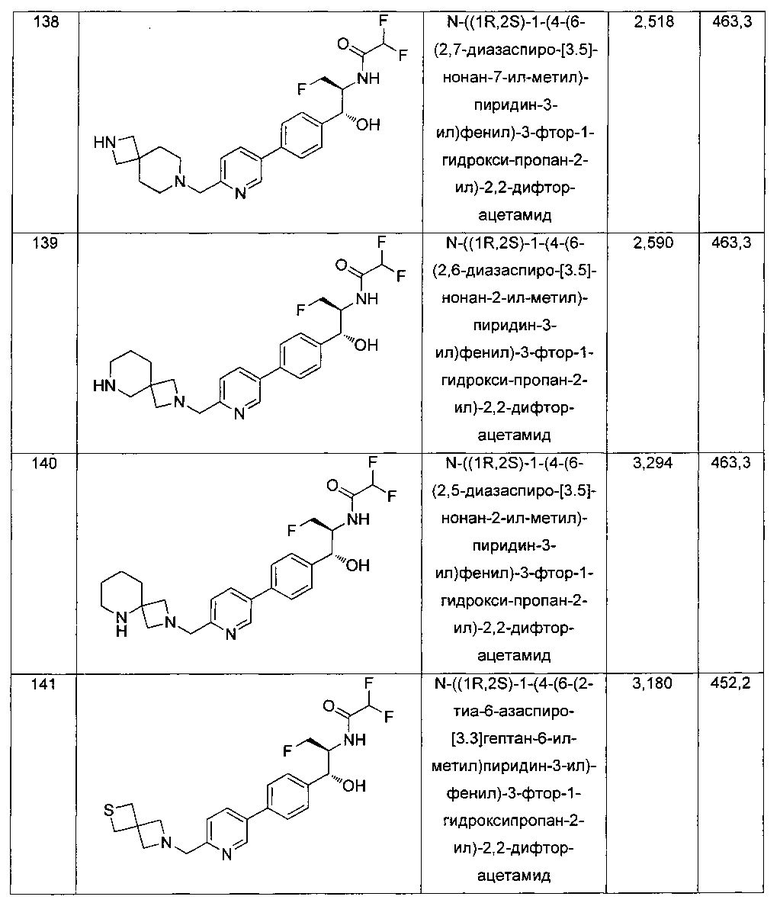
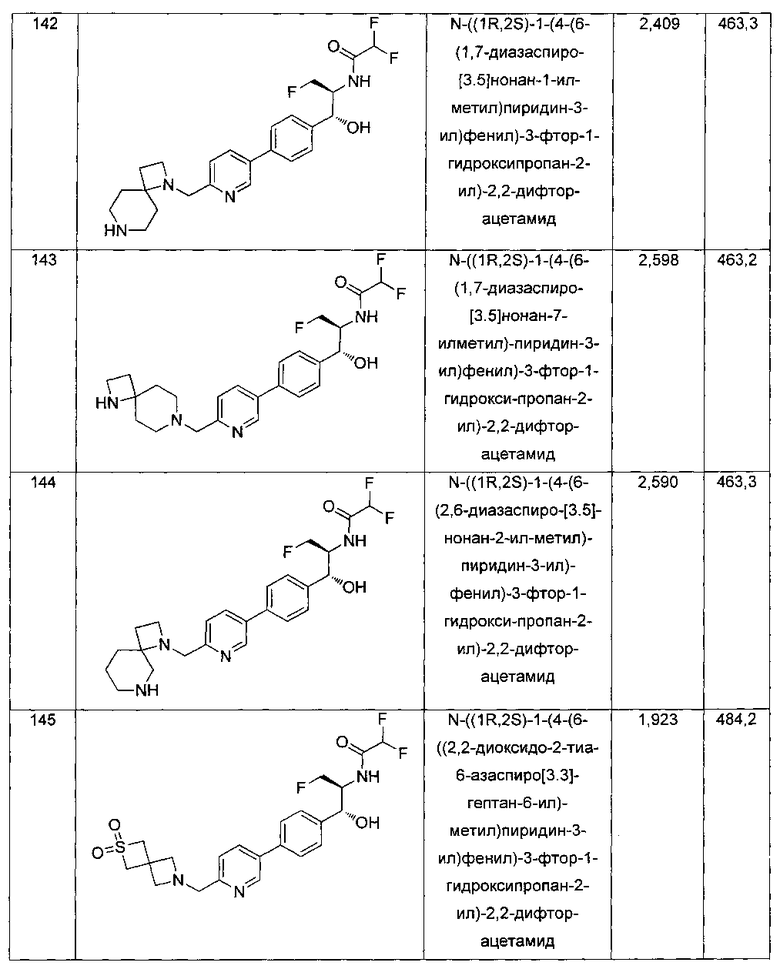
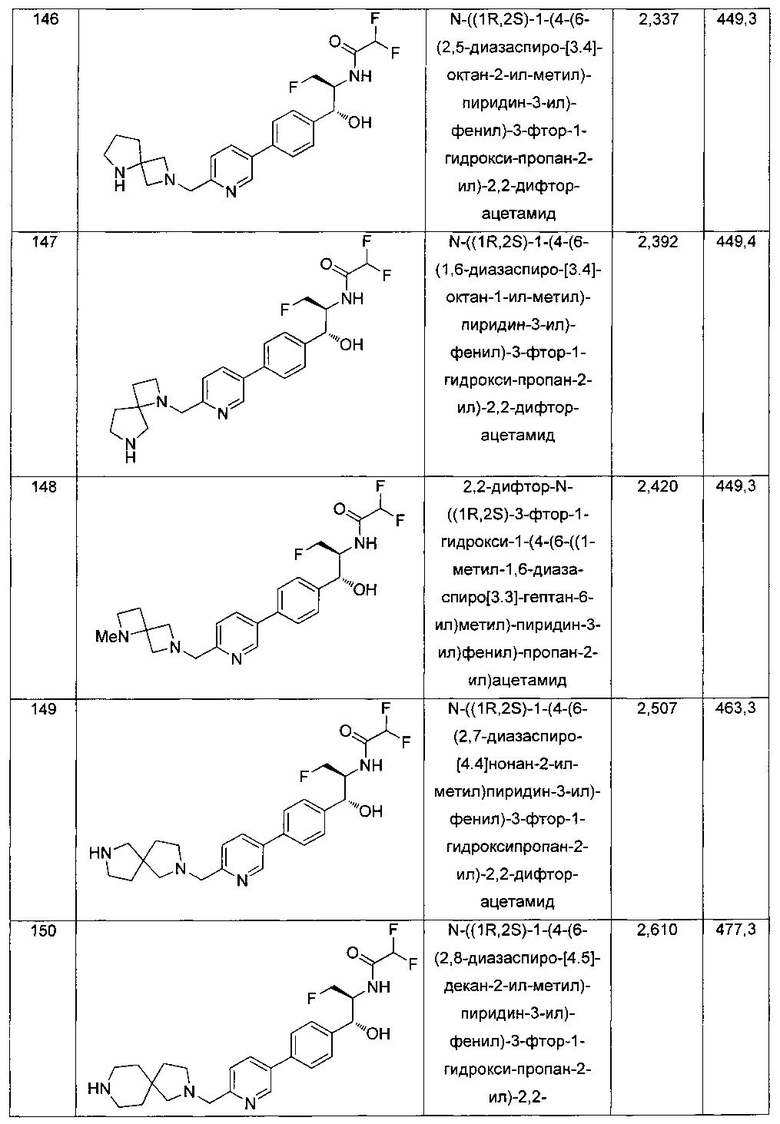
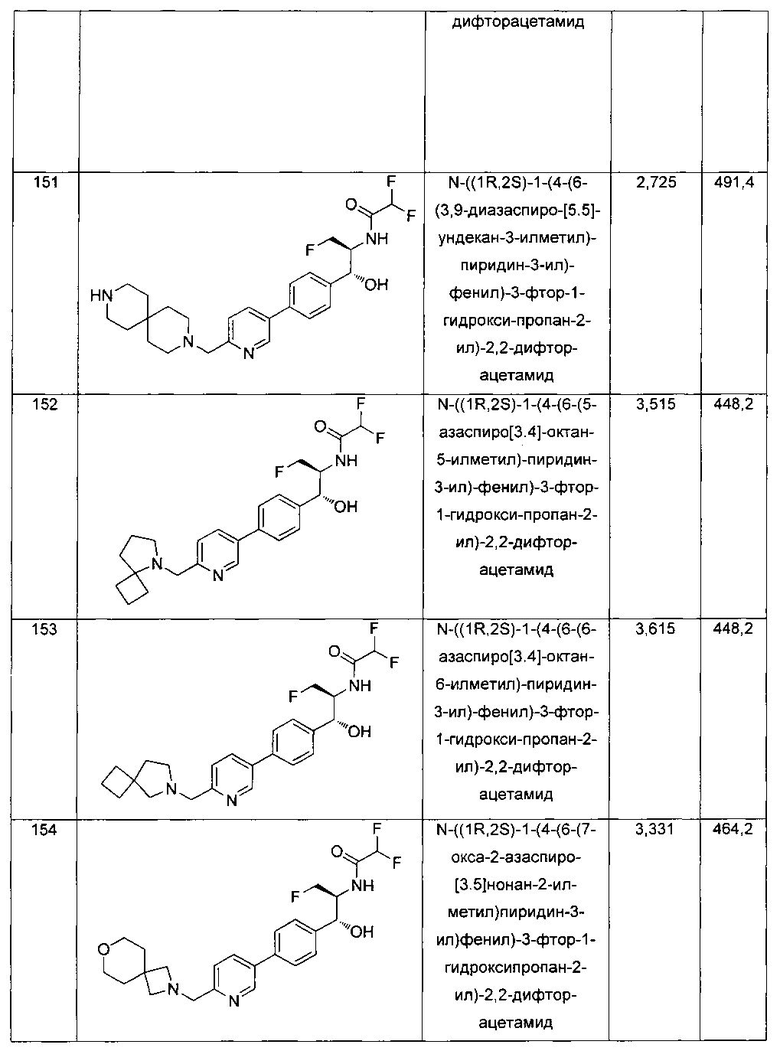
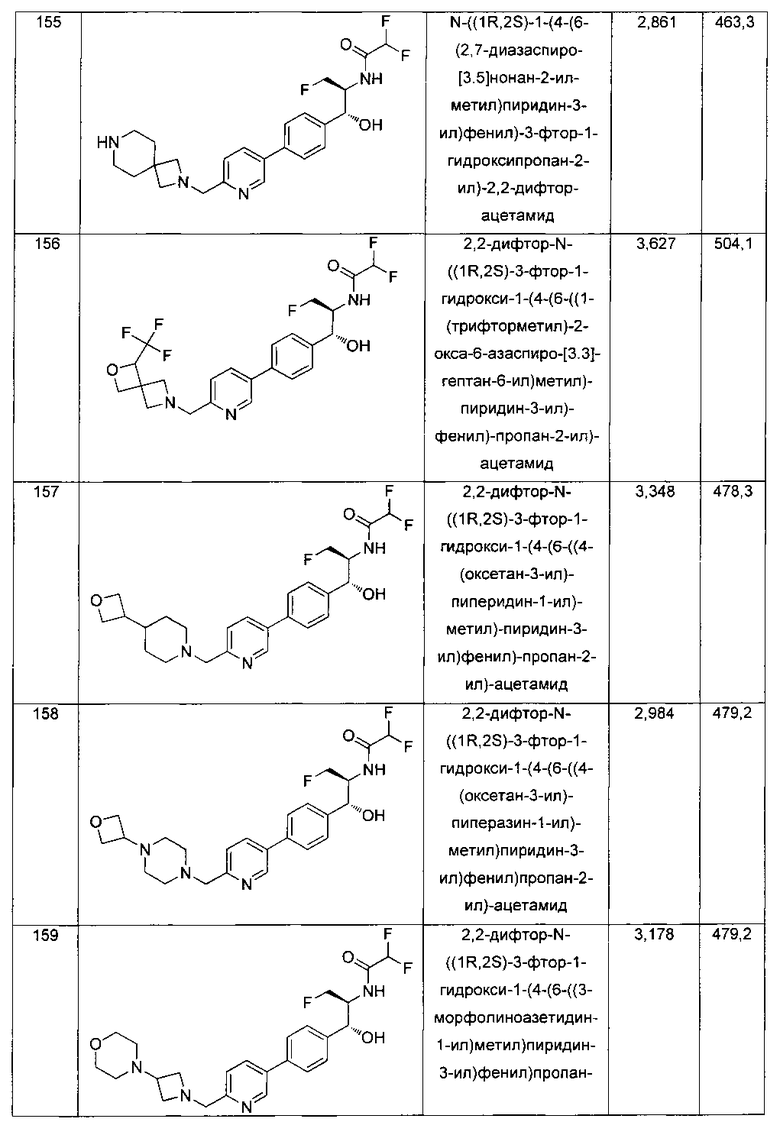
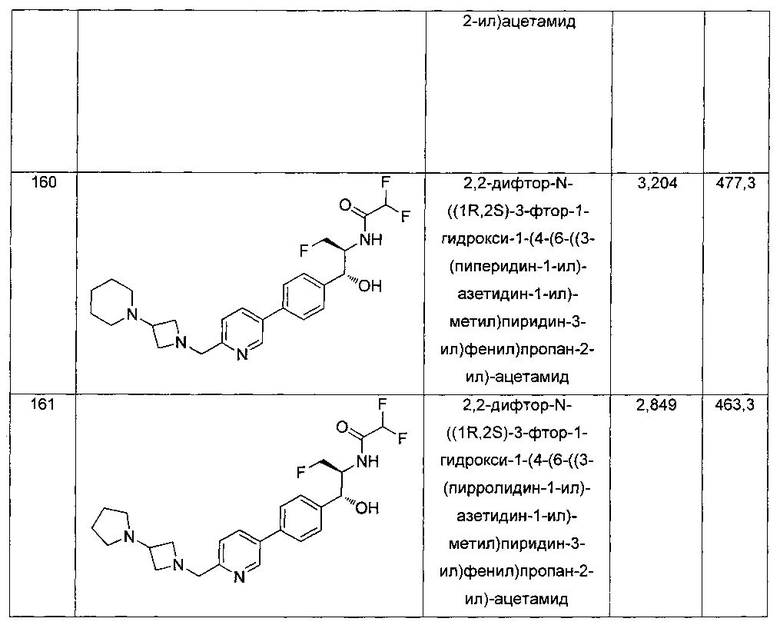
Следующие соединения из Таблицы 3 могут быть получены, используя схемы, показанные ниже.
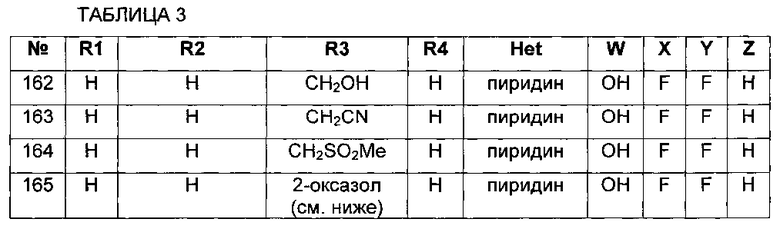
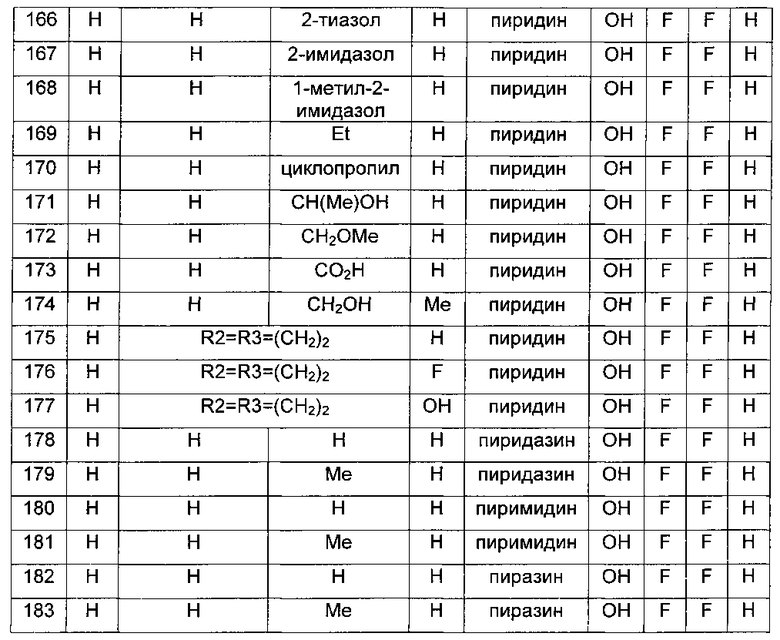
Например, соединение, указанное в заголовке Примера 175, N-((1R,2S)-1-(4-(6-(азетидин-2-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид, соединения из Примеров 162, 171-174 и 177-183 могут быть синтезированы из имеющихся в продаже бромпиридинов, и поэтому для их получения могут быть использованы методики из Примера 22, стадия 3. Соединения из Примеров 163-170 могут быть синтезированы, используя показанную ниже схему:
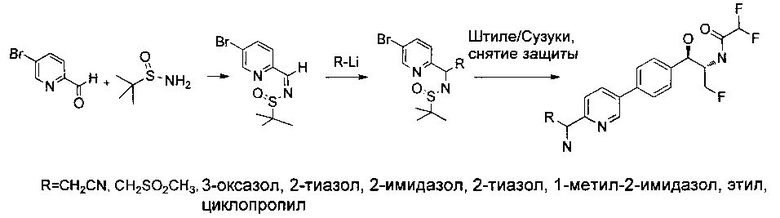
Соединения из Примеров 175, 176 и 177 могут быть синтезированы, используя показанную ниже схему:
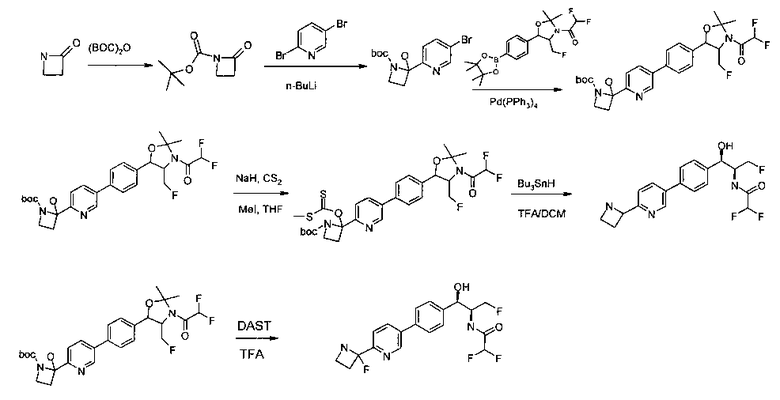
Известными из уровня техники способами могут быть получены следующие производные соединения, указанного в заголовке Примера 175:
N-((1R,2S)-1-(4-(6-(азетидин-2-ил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дихлорацетамид; и
(1R,2S)-1-(4-(6-(азетидин-2-ил)пиридин-3-ил)фенил)-2-(2,2-дифтор-ацетамидо)-3-фторпропил-дигидрофосфат;
(1R,2S)-1-(4-(6-(азетидин-2-ил)пиридин-3-ил)фенил)-2-(2,2-дихлор-ацетамидо)-3-фторпропил-дигидрофосфат.
Посредством методик, сходных с описанными выше, могут быть получены дополнительные соединения, содержащие другие группировки Het, такие как тиазолил, тиофенил, пиридазинил и пиримидинил.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| АНИЛИДЫ АМИНОКИСЛОТ В КАЧЕСТВЕ НИЗКОМОЛЕКУЛЯРНЫХ МОДУЛЯТОРОВ IL-17 | 2019 |
|
RU2815505C2 |
| АРИЛ-ЗАМЕЩЕННЫЕ КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ БЛОКАТОРОВ КАЛЬЦИЕВЫХ ИЛИ НАТРИЕВЫХ КАНАЛОВ | 2010 |
|
RU2575168C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ГЕТЕРОАРОМАТИЧЕСКИЕ МОДУЛЯТОРЫ РЕТИНОЛ-СВЯЗАННОГО ОРФАННОГО РЕЦЕПТОРА ГАММА | 2017 |
|
RU2771280C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2019 |
|
RU2812128C2 |
| НЕСТЕРОИДНЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИОДИДНЫХ РЕЦЕПТОРОВ ДЛЯ МЕСТНОЙ ДОСТАВКИ ЛЕКАРСТВ | 2016 |
|
RU2731618C2 |
Изобретение относится к соединению формулы I или его фармацевтически приемлемым солям, где группировка Het представляет собой пиридинил или тиазолил; каждый из R1 и R2 представляет собой Н; каждый из R3 и R4 независимо представляет собой Н, -С1-8алкил или R3 и R4, взятые вместе, образуют С3-6циклоакил; W представляет собой -Н, -РО(ОН)2 или -СН2ОРО(ОН)2; каждый из X и Y представляет собой хлор или каждый из X и Y представляет собой фтор, и Z представляет собой Н. Соединения формулы I используют в способе контролирования или лечения инфекций у домашнего скота, включающем введение нуждающемуся в этом животному терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. 2 н. и 6 з.п. ф-лы, 3 табл., 161 пр.
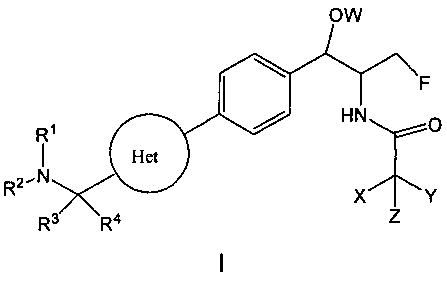
1. Соединение формулы I
или его фармацевтически приемлемые соли, где
группировка Het представляет собой пиридинил или тиазолил;
каждый из R1 и R2 представляет собой Н;
каждый из R3 и R4 независимо представляет собой Н, -С1-8алкил или R3 и R4, взятые вместе, образуют С3-6циклоакил;
W представляет собой -Н, -РО(ОН)2 или -СН2ОРО(ОН)2;
каждый из X и Y представляет собой хлор или каждый из X и Y представляет собой фтор, и
Z представляет собой Н.
2. Соединение по п. 1, где W представляет собой Н.
3. Соединение по п. 1 или 2, где X и Y представляют собой хлорид.
4. Соединение по п. 1 или 2, где X и Y представляют собой фторид.
5. Соединение по п. 1, где R3 и R4 взяты вместе с образованием циклопропила.
6. Соединение по п. 1, выбранное из группы, состоящей из:
N-((1R,2S)-1-(4-(2-(1-аминоэтил)тиазол-5-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида;
N-((1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-3-фтор-1-гидрокси-пропан-2-ил)-2,2-дифторацетамида;
(1R,2S)-1-(4-(6-(1-аминоциклопропил)пиридин-3-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропилдигидрофосфата и
N-((1R,2S)-1-(4-(6-((RS)-1-аминоэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида.
7. Соединение по п. 6, представляющее собой N-((1R,2S)-1-(4-(6-((RS)-1-аминоэтил)пиридин-3-ил)фенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид или (1R,2S)-1-(4-(6-(1-аминоэтил)пиридин-3-ил)фенил)-2-(2,2-дифторацетамидо)-3-фторпропил-дигидрофосфат.
8. Способ контролирования или лечения инфекций у домашнего скота, включающий введение нуждающемуся в этом животному терапевтически эффективного количества соединения по п. 1 или его фармацевтически приемлемой соли.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| КАРБОНАТЫ АНТИБИОТИКОВ ФЕНИКОЛОВ | 2006 |
|
RU2432352C2 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |

