Область техники
Настоящее изобретение относится к новым соединениям конденсированных гетероциклил-карбоногидразоноилдицианидов и их применению.
Предшествующий уровень техники
Тау-белок (tau (τ) белок), представляющий собой белок, ассоциированный с микротрубочками (MAP), в основном экспрессируемый в аксонах нервных клеток с молекулярной массой от 50000 до 70000, служит для стабилизации микротрубочек и представляет молекулярное разнообразие посредством фосфорилирования. У человека тау-белок формируется в шесть изоформ путем вставки 29 или 58 аминокислотных остатков на N-конце и альтернативного сплайсинга мРНК из 3 или 4 повторяющихся структур (называемых доменом связывания микротрубочек) на С-конце.
В здоровых нервах тау-белок стабилизирует микротрубочки, способствуя росту аксонов и поляризации нервных клеток. При патологическом гиперфосфорилировании тау-белок отделяется от микротрубочек, что приводит к нерастворимой агрегации. Кроме того, был предложен структурный скелет, индуцирующий агрегацию тау-белка, и были представлены доказательства того, что нерастворимые филаменты формируются из 10 растворимых мономеров, и что эти филаменты связываются в высокоразмерные структуры, называемые нейрофибриллярными клубками (NFT). Полноразмерный тау-белок человека включает домен связывания микротрубочек, состоящий из четырех повторяющихся консервативных последовательностей. Среди этих повторяющихся последовательностей положительно заряженные остатки играют важную роль в связывании с сильно отрицательно заряженными микротрубочками (от 20 до 30 электронов на димер αβ-тубулина). Аффинность связывания с тау-микротрубочками также активно регулируется фосфорилированием тау-белка, и это фосфорилирование вызывает динамическую перестройку сетей микротрубочек. При аномально чрезмерном фосфорилировании тау-белка баланс этой динамической перестройки нарушается, и сродство к микротрубочкам быстро снижается.
Гиперфосфорилирование и/или агрегация тау-белков вызывают аномальное накопление этих тау-белков в нервных клетках, на которое указывают как на причину различных нейродегенеративных заболеваний и т.п. Агрегаты тау-белков в основном находятся в телах и дендритах нервных клеток, и эти агрегаты тау-белков называют нейрофибриллярными клубками (NFT) и нейропилевыми нитями. Изучение микроструктур нейрофибриллярных клубков (NFT) показывает, что такие их микроструктуры состоят из парных спиральных филаментов (NFT), в которых тау-белки запутаны в виде тонких нитей, агрегированы и гиперфосфорилированы, в отличие от нормального тау-белка. Феномен аномальной агрегации тау-белка проявляется также при таупатии. В этом случае, хотя точно не известно, какую роль играет агрегация тау-белка в развитии таупатии, этот феномен агрегации тау-белка оказывается сходным с феноменом агрегации, характерным для общих нейродегенеративных заболеваний.
Таким образом, хотя известно, что гиперфосфорилирование и/или агрегация тау-белка вызывают различные нейродегенеративные заболевания, включая болезнь Альцгеймера и таупатию, конкретный механизм того, каким образом эти аномальные виды тау вызывают изменения в сигнальном пути и вызывают нейротоксичность, еще не подтверждены, и пока не существует эффективных способов лечения или терапевтических агентов для лечения этих заболеваний.
Раскрытие изобретения
Техническая задача
В результате интенсивных усилий по разработке новых низкомолекулярных соединений, способных ингибировать агрегацию и/или гиперфосфорилирование тау-белка, авторы настоящего изобретения обнаружили, что ряд новых соединений конденсированных гетероциклил-карбогидразоноилдицианидов эффективно ингибирует агрегацию тау-белка, не проявляя цитотоксичности в эффективных концентрациях, тем самым создав настоящее изобретение.
Техническое решение
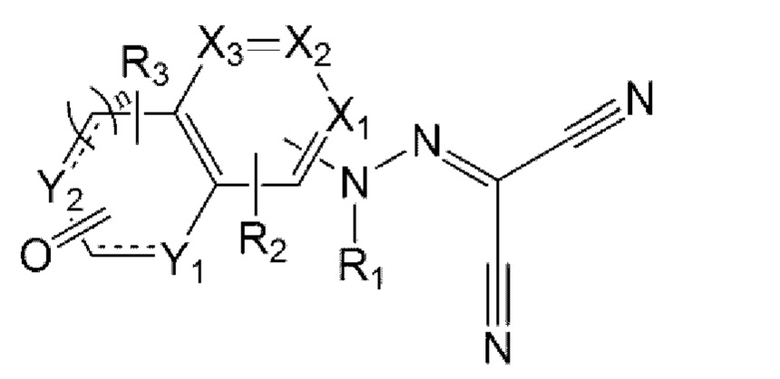
Задача настоящего изобретения заключается в предложении соединения, представленного формулой 1 ниже, или его фармацевтически приемлемой соли:
[Формула 1]
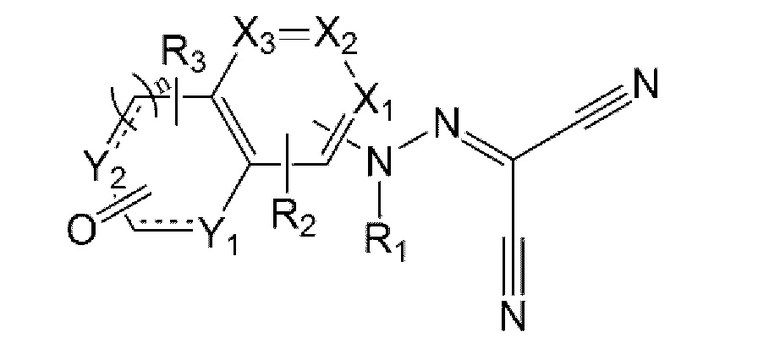
в формуле 1 выше:
X1 - Х3 каждый независимо представляет собой N или С(Н);
Y1 и Y2 каждый независимо представляет собой N(R4), С(Н), О или S;
n равен 0 или 1;
 представляет собой
представляет собой  или
или  с образованием ароматического или неароматического конденсированного гетероциклического кольца;
с образованием ароматического или неароматического конденсированного гетероциклического кольца;
R1 представляет собой водород, C1-6алкил или C1-6алкилкарбонил;
R2 и R3 каждый независимо представляет собой водород, C1-6алкил, галоген, циано, C1-6галогеналкил, C1-6алкокси, C1-6галогеналкокси, C1-6алкиламино, ди(C1-6алкил)амино, C1-6алкиламинокарбонил или ди(C1-6алкил)аминокарбонил; и
R4 представляет собой водород, C1-6алкил, C1-6алкокси, C1-6алкокси- C1-6алкил, C1-6галогеналкил, С3-6циклоалкил-С0-6алкил, 5- или 6-членный гетероцикл-С0-6алкил, С6-10арил или 5-10-членный гетероарил,
где указанные 5- или 6-членный гетероцикл, C1-10арил и 5-10-членный гетероарил являются незамещенными или замещены C1-6алкокси или C1-6алкоксикарбонилом.
Другая задача настоящего изобретения заключается в предложении способа получения соединения, описанного выше.
Еще одна задача настоящего изобретения заключается в предложении композиции для ингибирования агрегации тау-белка, включающей соединение, описанное выше, в качеств активного ингредиента.
Еще одна задача настоящего изобретения заключается в предложении композиции для ингибирования гиперфосфорилирования тау-белка, включающей соединение, описанное выше, в качестве активного ингредиента.
Еще одна задача настоящего изобретения заключается в предложении фармацевтической композиции для предупреждения или лечения заболевания, вызванного агрегацией или гиперфосфорилированием тау-белка, включающей соединение, описанное выше, в качестве активного ингредиента.
Еще одна задача настоящего изобретения заключается в предложении способа предупреждения или лечения заболевания, вызванного агрегацией или гиперфосфорилированием тау-белка, включающего введение субъекту, нуждающемуся в этом, фармацевтической композиции, описанной выше.
Полезные эффекты
Новые соединения конденсированных гетероциклил-карбоногидразоноилдицианидов по настоящему изобретению могут эффективно ингибировать агрегацию и/или гиперфосфорилирование тау-белка и, таким образом, могут быть эффективно использованы для предупреждения или лечения вызываемых этим заболеваний, таких как болезнь Альцгеймера и различные таупатии.
Наилучший способ осуществления изобретения
В первом аспекте настоящего изобретения предложено соединение, представленное формулой 1 ниже, или его фармацевтически приемлемая соль:
[Формула 1]
в формуле 1 выше:
X1 - Х3 каждый независимо представляет собой N или С(Н);
Y1 и Y2 каждый независимо представляет собой N(R4), С(Н), О или S;
n равен 0 или 1;
 представляет собой
представляет собой  или
или  с образованием ароматического или неароматического конденсированного гетероциклического кольца;
с образованием ароматического или неароматического конденсированного гетероциклического кольца;
R1 представляет собой водород, C1-6алкил или C1-6алкилкарбонил;
R2 и R3 каждый независимо представляет собой водород, C1-6алкил, галоген, циано, C1-6галогеналкил, C1-6алкокси, C1-6галогеналкокси, C1-6алкиламино, ди(C1-6алкил)амино, C1-6алкиламинокарбонил или ди(C1-6алкил)аминокарбонил; и
R4 представляет собой водород, C1-6алкил, C1-6алкокси, C1-6алкокси-С0-6алкил, C1-6галогеналкил, С3-6циклоалкил- C0-6алкил, 5- или 6-членный гетероцикл- C0-6алкил, C6-60арил или 5-10-членный гетероарил,
где указанные 5- или 6-членный гетероцикл, С6-10арил и 5-10-членный гетероарил являются незамещенными или замещены C1-6алкокси или C1-6алкоксикарбонилом.
Например, в соединении по настоящему изобретению:
R1 представляет собой водород, C1-6алкил или C1-6алкилкарбонил;
R2 представляет собой водород, галоген, C1-6алкил или C1-6алкокси;
R3 представляет собой водород или C1-6алкил; и
R4 представляет собой водород, C1-6алкил, C1-6галогеналкил, С3-6циклоалкил-C0-6алкил или C1-10арил, без ограничения этим.
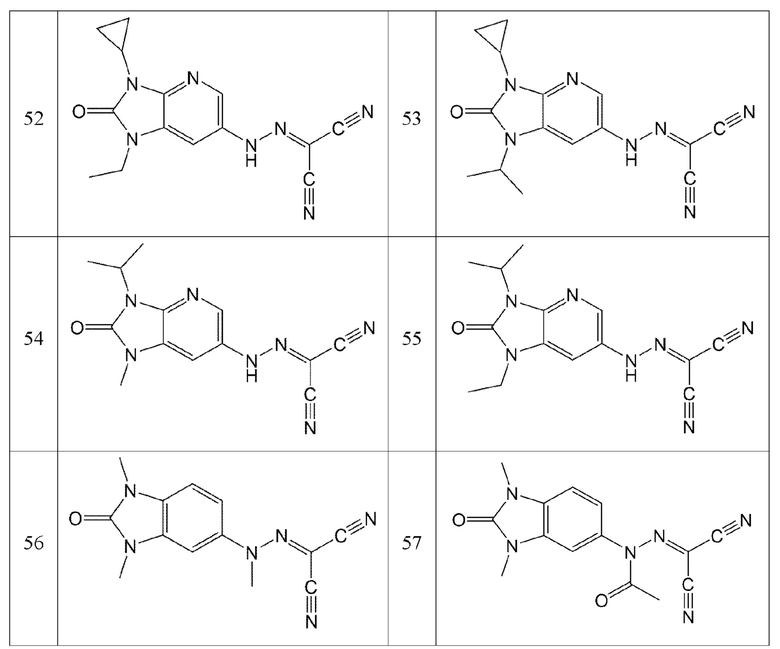
В частности, в соединении по настоящему изобретению:
R1 представляет собой водород, метил или ацетил;
R2 представляет собой водород, хлор, фтор, метил или метокси;
R3 представляет собой водород или метил; и
R4 представляет собой водород, метил, этил, изопропил, дифторметил, циклопропил или фенил, без ограничения этим.
Например, соединение по настоящему изобретению может представлять собой соединение, представленное формулой 2 ниже:
[Формула 2]
в формуле 2 выше:
один из Y1 и Y2 представляет собой СН, а другой представляет собой NR4;
 при связи с СН представляет собой
при связи с СН представляет собой  а
а  при связи с NR4 представляет собой
при связи с NR4 представляет собой 
R3 представляет собой водород или метил; и
R4 представляет собой водород, метил или изопропил.
Альтернативно, соединение по настоящему изобретению может представлять собой соединение, представленное формулой 3 ниже:
[Формула 3]
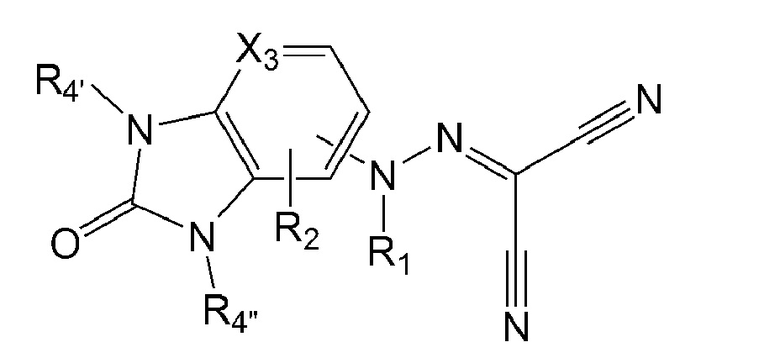
в формуле 3 выше:
Х3 представляет собой С(Н) или N;
R1 представляет собой водород, метил или ацетил;
R2 представляет собой водород, хлор, фтор, метил или метокси; и
R4' и R4'' каждый независимо представляет собой водород, метил, этил, изопропил, дифторметил, циклопропил или фенил.
Кроме того, соединение по настоящему изобретению может представлять собой соединение, представленное формулой 4 ниже:
[Формула 4]
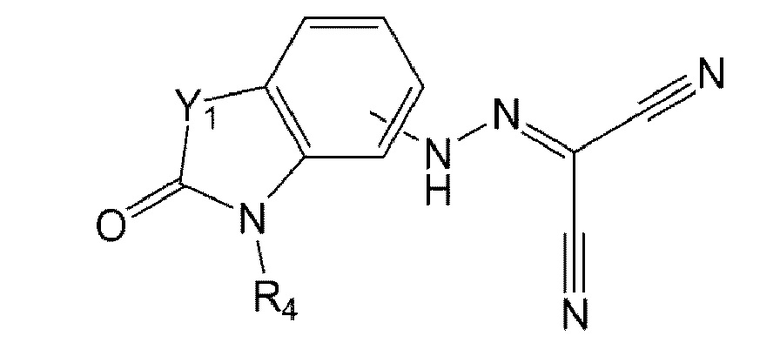
в формуле 4 выше:
Y1 представляет собой S или О; и
R4 представляет собой водород, метил, изопропил или дифторметил.
Более конкретно, соединение, представленное формулой 1, может представлять
собой:
1) (1-оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноилдицианид,
2) (2-метил-1 -оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноилдицианид,
3) (2-изопропил-1 -оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноилдицианид,
4) (1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианид,
5) (2-метил-1 -оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианид,
6) (2-изопропил-1 -оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианид,
7) (2,3 -диметил-1 -оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианид,
8) (2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианид,
9) (6-фтор-1,3-диметил-2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)карбо ногидр аз оноилд ицианид,
10) (6-хлор-1,3-Диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбо ногидр аз оноилд ицианид,
11) (7-хлор-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбо ногидр аз оноилд ицианид,
12) (1,3-Диметил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
13) (2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
14) (3-метил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
15) (2-оксо-2,3-дигидробензо[d|оксазол-6-ил)карбоногидразоноилдицианид,
16) (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианид,
17) (1-метил-2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
18) (3-изопропил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
19) (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
20) (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианид,
21) (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианид,
22) (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
23) (1,3-Диизопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
24) (1,3-бис(дифторметил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
25) (1-метил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианид,
26) (4-метил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианид,
27) (1,4-диметил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилд ицианид,
28) (3-метил-2-оксо-2,3-Дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианид,
29) (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианид,
30) (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианид,
31) (1,3,6-триметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
32) (6-метокси-1,3-Диметил-2-оксо-2,3-Дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
33) (7-метокси-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
34) (1,3-Диизопропил-2-оксо-2,3-Дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
35) (3-этил-1-метил-2-оксо-2,3-Дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
36) (3-изопропил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
37) (3-(дифторметил)-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
38) (1-этил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d|имидазол-5-ил)карбоногидразоноилдицианид,
39) (3-(дифторметил)-1-этил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
40) (1-циклопропил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
41) (1-циклопропил-3-этил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
42) (1-циклопропил-3-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
43) (3-(дифторметил)-1-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
44) (1-этил-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбо ногидр аз оноилд ицианид,
45) (1-изопропил-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
46) (1-(дифторметил)-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
47) (3-метил-2-оксо-1-фенил-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
48) (3-этил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
49) (3-этил-1-изопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
50) (1-(дифторметил)-3-этил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
51) (3-циютопропил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
52) (3-циклопропил-1-этил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
53) (3-циклопропил-1-изопропил-2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-6 -ил)карбоногидразоноилдицианид,
54) (3-изопропил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
55) (1-этил-3-изопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
56) (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)(метил)карбоногидразоноилдицианид или
57) ацетил(1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид, без ограничения ими.
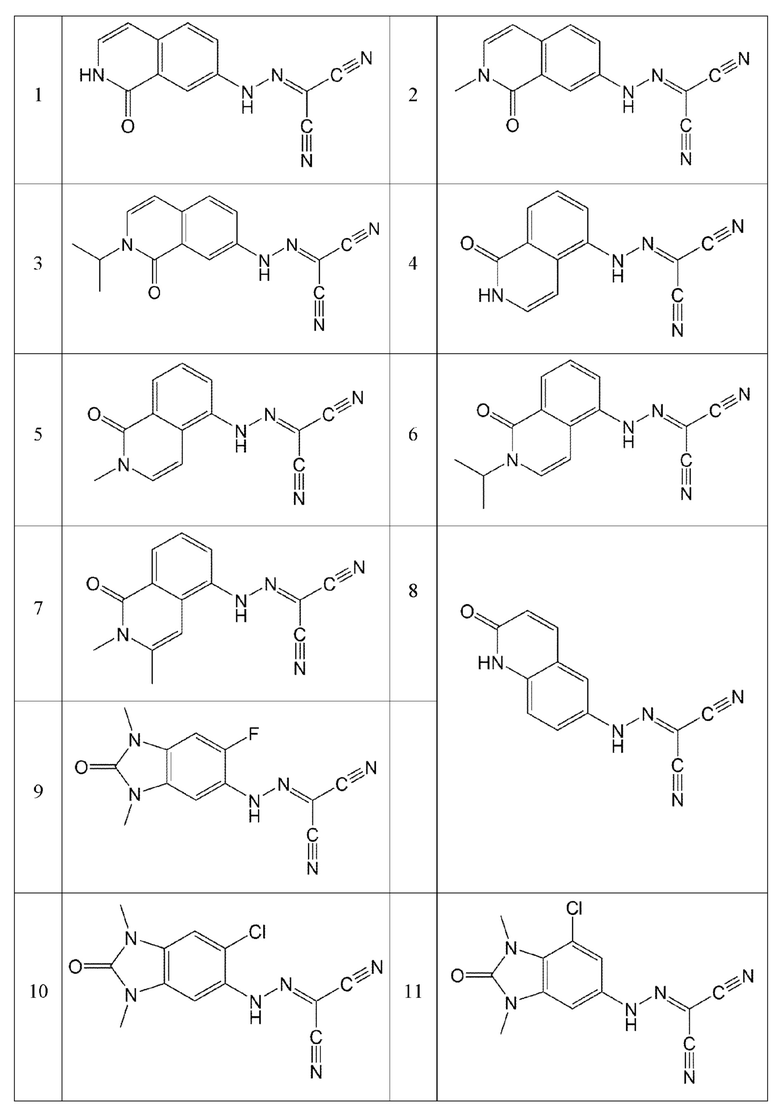
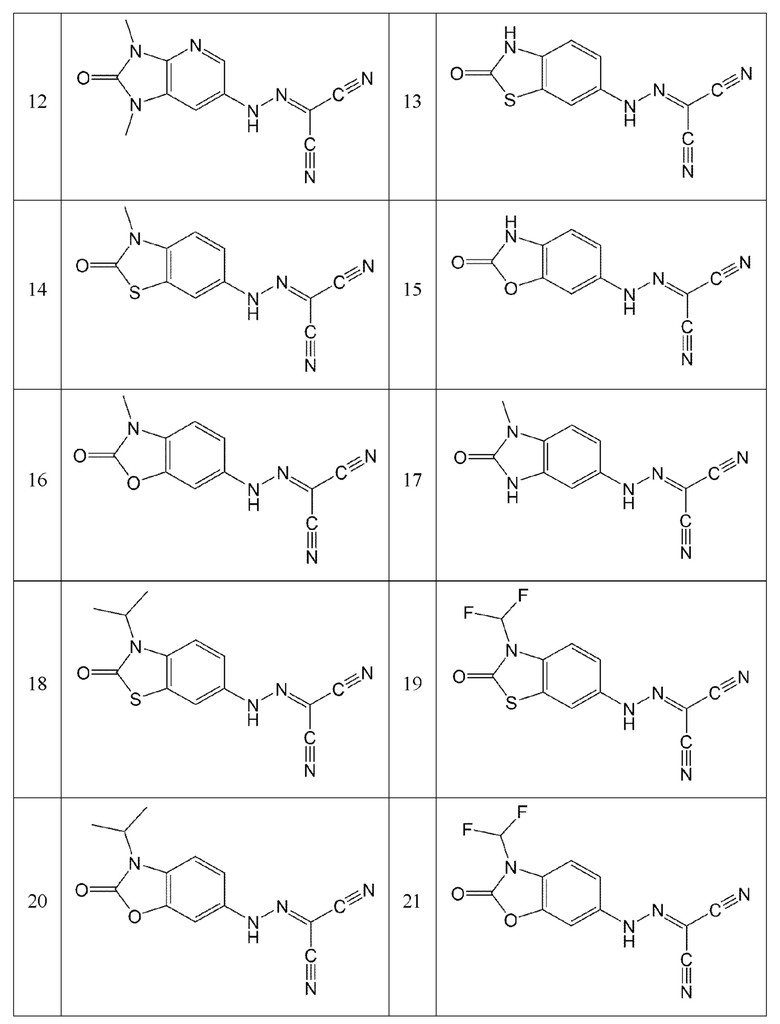
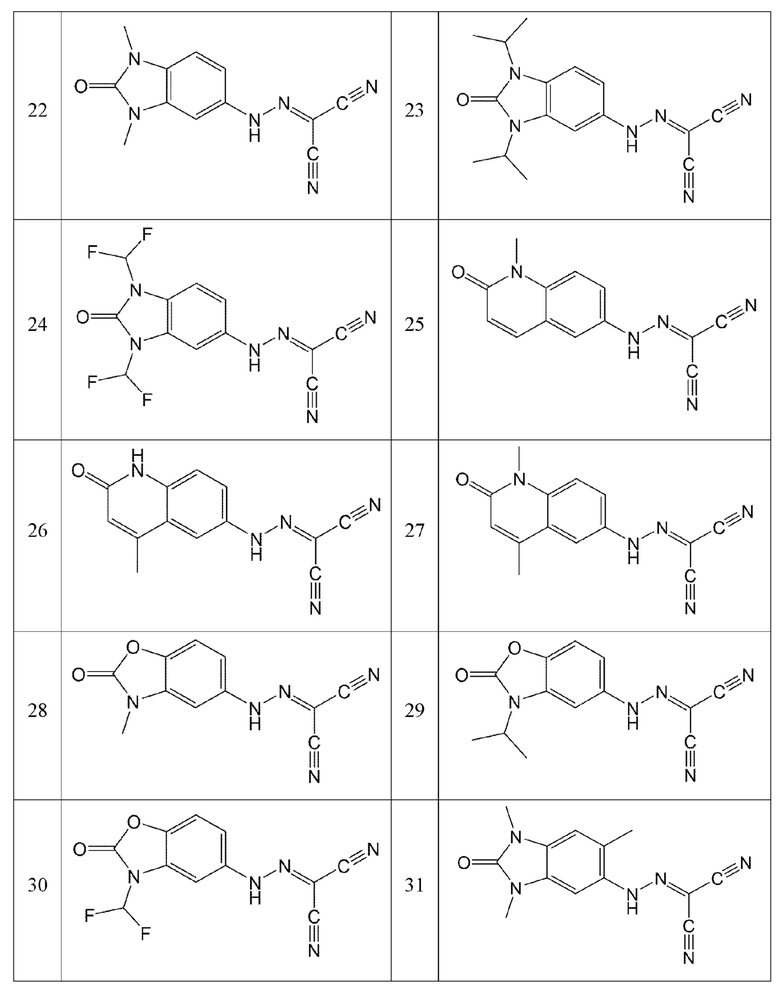
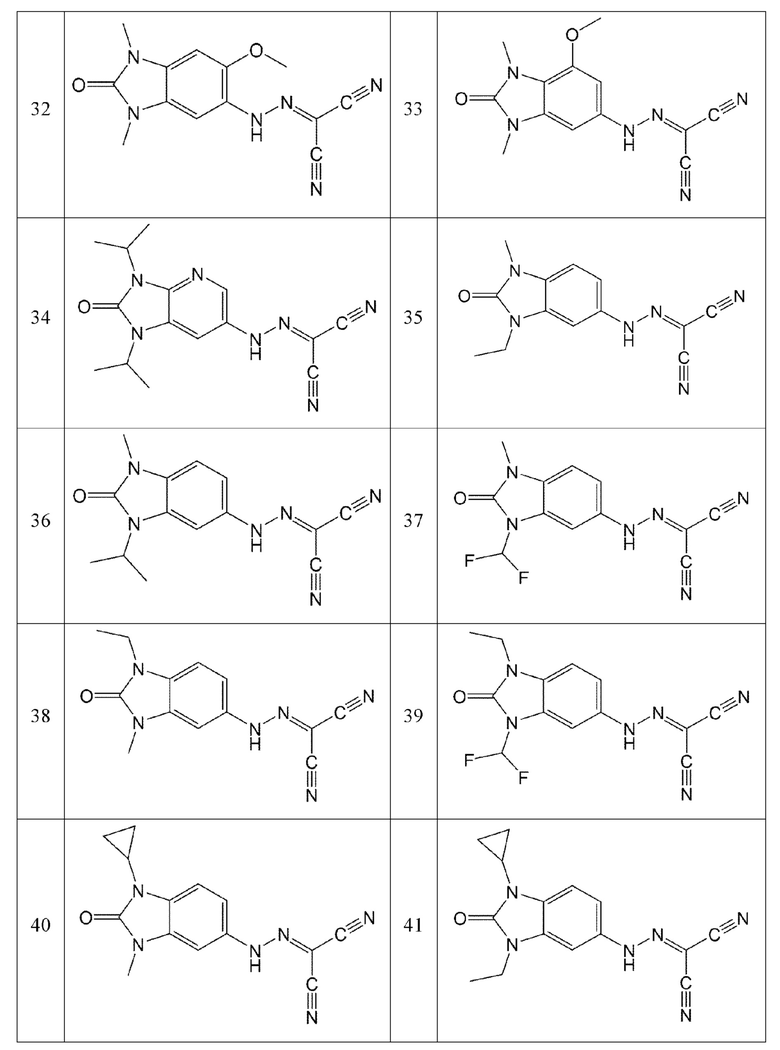
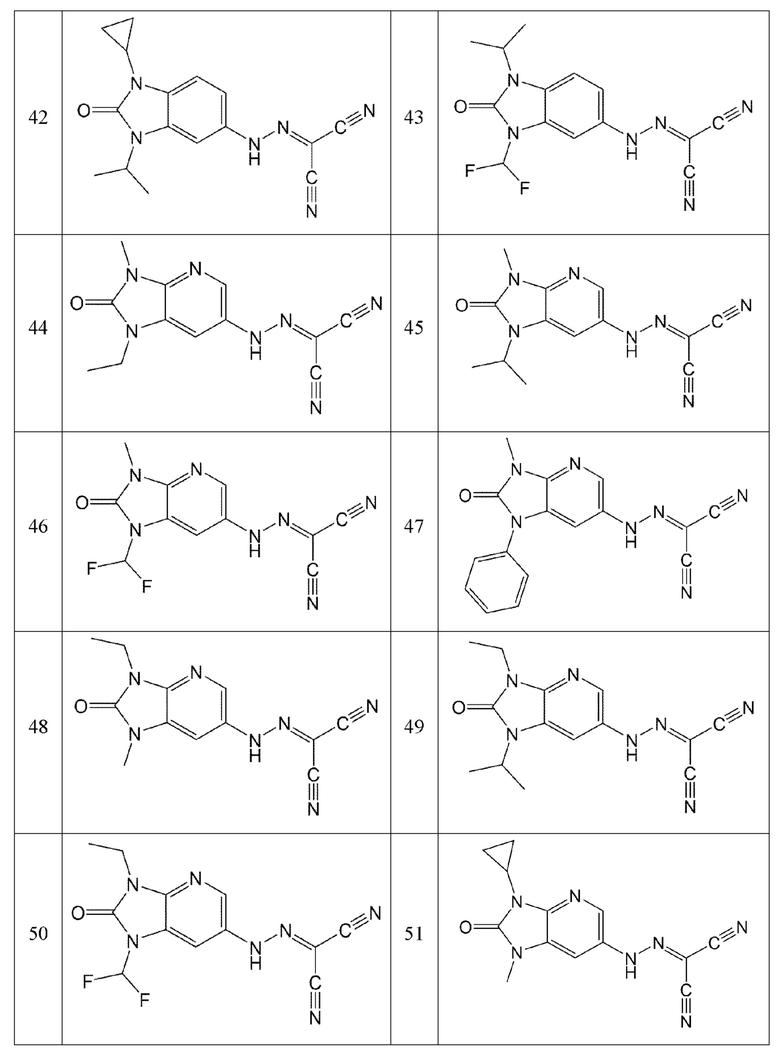
Кроме того, эти соединения могут представлять собой соединения, представленные формулами, показанными в Таблице 1 ниже.
При этом соединения по настоящему изобретению могут существовать в форме фармацевтически приемлемой соли. В качестве соли полезна кислая соль, образованная фармацевтически приемлемой свободной кислотой. Используемый здесь термин «фармацевтически приемлемая соль» относится к любой соли присоединения органической или неорганической кислоты соединений, представленных формулами 1 -4, которая является относительно нетоксичной и безвредной для пациентов, а побочные эффекты, вызываемые этой солью, не ухудшают благоприятные эффекты этого соединения.
Соль присоединения кислоты получают обычным способом, например, путем растворения соединения в избыточном количестве водного раствора кислоты и осаждения этого раствора с использованием смешивающегося с водой органического растворителя, такого как метанол, этанол, ацетон или ацетонитрил. Одинаковые молярные количества соединения и кислоты или спирта (например, моноэтилового эфира гликоля) в воде нагревают, а затем смесь упаривают и сушат, или осажденную соль фильтруют отсасыванием.
В этом случае в качестве свободной кислоты может быть использована органическая кислота или неорганическая кислота. В качестве неорганической кислоты может быть использована соляная кислота, фосфорная кислота, серная кислота, азотная кислота, винная кислота и т.п. В качестве органической кислоты может быть использована метансульфоновая кислота, пара-толуолсульфоновая кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, янтарная кислота, щавелевая кислота, бензойная кислота, винная кислота, фумаровая кислота, миндальная кислота, пропионовая кислота, лимонная кислота, молочная кислота, гликолевая кислота, глюконовая кислота, галактуроновая кислота, глутаминовая кислота, глутаровая кислота, глюкуроновая кислота, аспарагиновая кислота, аскорбиновая кислота, карбоновая кислота, ванилиновая кислота, йодистоводородная кислота и т.п. Однако настоящее изобретение не ограничивается ими.
Кроме того, фармацевтически приемлемая соль металла может быть получена с использованием основания. Соль щелочного металла или соль щелочноземельного металла получают путем растворения соединения в избыточном количестве раствора гидроксида щелочного металла или гидроксида щелочноземельного металла, отфильтровывания нерастворимой соли соединения, а затем упаривания и сушки фильтрата. В этом случае для фармацевтического применения подходит получение соли натрия, калия или кальция в качестве соли металла, но настоящее изобретение этим не ограничивается. Кроме того, соответствующая соль серебра может быть получена путем взаимодействия соли щелочного металла или щелочноземельного металла с подходящей солью серебра (например нитратом серебра).
Фармацевтически приемлемые соли соединений по настоящему изобретению включают соли кислых или основных групп, которые могут присутствовать в соединениях формул 1-4, если не указано иное. Например, фармацевтически приемлемые соли могут включать натриевые, кальциевые и калиевые соли гидроксильных групп, а другие фармацевтически приемлемые соли аминогрупп могут включать гидробромид, сульфат, гидросульфат, фосфат, гидрофосфат, дигидрофосфат, ацетат, сукцинат, цитрат, тартрат, лактат, манделат, метансульфонат (мезилат) и пара-толуолсульфонат (тозилат). Эти фармацевтически приемлемые соли могут быть получены способами получения солей, известными в данной области техники.
В качестве солей соединений формул 1-4 по настоящему изобретению можно использовать без ограничений любую соль в качестве фармацевтически приемлемой соли, при условии что она проявляет фармакологическую активность, эквивалентную соединениям формул 1-4, например, она ингибирует агрегацию и/или гиперфосфорилирование тау-белка.
Кроме того, соединения, представленные формулами 1-4 по настоящему изобретению, включают, без ограничения, их фармацевтически приемлемые соли, а также сольваты, такие как возможные гидраты, которые могут быть получены из них, и все возможные стереоизомеры. Сольваты и стереоизомеры соединений, представленных формулами 1-4, могут быть получены из соединений, представленных формулами 1-4, с использованием любого способа, известного в данной области техники.
Более того, соединения, представленные формулами 1-4 по настоящему изобретению, могут быть получены в кристаллической или аморфной форме и могут быть возможно гидратированы или сольватированы, если получены в кристаллической форме. В настоящем изобретении могут быть предложены соединения, содержащие различные количества воды, а также стехиометрические гидраты соединений, представленных формулами 1-4. Сольваты соединений, представленных формулами 1-4, по настоящему изобретению включают как стехиометрические сольваты, так и нестехиометрические сольваты.
Во втором аспекте настоящего изобретения предложен способ получения соединения формулы 1.
Например, соединение по настоящему изобретению может быть получено способом, включающим:
первую стадию взаимодействия соединения, представленного формулой 5 ниже, включающего реакционноспособную аминогруппу на одном конце, с нитритом натрия и малононитрилом в присутствии кислоты с образованием иминной связи; и
возможно, вторую стадию введения заместителя Ri в продукт, полученный на предыдущей стадии, когда Ri является заместителем, отличным от водорода:
[Формула 5]
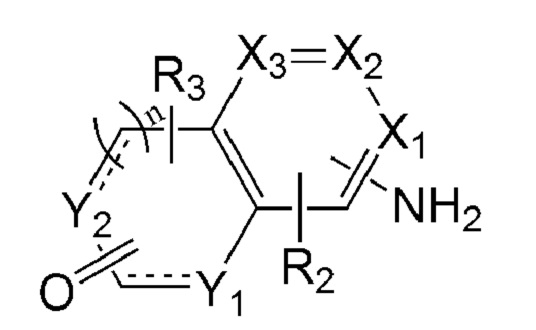
в формуле 5 выше:
X1 - Х3, Y1, Y2, n,  R2 и R3 являются такими, как определено выше, и
R2 и R3 являются такими, как определено выше, и
когда Y1 и Y2 представляют собой N(H), тогда N(H) может быть защищен трет-бутоксикарбонилом.
В этом отношении, когда N(H) в Y1 и Y2 защищен трет-бутоксикарбонилом, указанный способ может дополнительно включать стадию снятия защиты после взаимодействия.
В частности, первая стадия способа может быть выполнена в виде серии процессов, включающих стадии:
1-1) растворение соединения формулы 5 и нитрита натрия в растворителе C1-4 низший спирт и добавление к нему водного раствора кислоты при температуре от -5°С до 5°С с образованием соли диазония,
1-2) добавление малононитрила к реакционному раствору, включающему соль диазония, полученную на стадии 1-1), и проведение реакции при температуре от 15°С до 40°С, и
1-3) нейтрализация реакционного раствора со стадии 1-2) добавлением к нему водного раствора основания, без ограничения этим.
Например, первая стадия может быть проведена путем осуществления взаимодействия стадии 1-1) при низкой температуре около 0°С в течение от 2 минут до 1 часа с использованием 1 М раствора соляной кислоты, а затем осуществления взаимодействия стадии 1-2) при комнатной температуре от 2 минут до 1 часа, без ограничения этим.
Например, вторая стадия может быть осуществлена путем взаимодействия карбоногидразоноилдицианида соединения, полученного на предыдущей стадии, в которой R1 не замещен, с галогенированным производным R1 в органическом растворителе. В частности, когда R1 представляет собой С1-6алкил, вторую стадию можно проводить путем растворения карбоногидразоноилдицианида соединения, полученного на предыдущей стадии, в котором R1 не замещен, в органическом растворителе, таком как DMF, и добавления галогеналкана, такого как йодид алкана, соответствующего R1, к реакционному раствору, и проведения реакции при температуре от 50°С до 70°С, где реакционный раствор может дополнительно включать трет-бутоксид калия. Однако настоящее изобретение не ограничивается этим. В частности, когда R1 представляет собой C1-6алкилкарбонил, вторую стадию можно проводить путем растворения карбоногидразоноилдицианида соединения, полученного на предыдущей стадии, в которой R1 не замещен, в низшем спирте, таком как метанол, с последующей реакцией с основанием, таким как гидроксид калия, и отвердеванием с получением продукта, и взаимодействия данного продукта с галогенированным алкилкарбонилом, таким как ацетилхлорид, соответствующий C1-6алкилкарбонилу, в органическом растворителе, таком как ацетонитрил, в присутствии триэтиламина. Однако настоящее изобретение не ограничивается этим.
Например, соединение формулы 5, используемое в получении соединения по настоящему изобретению, может быть получено из соединения, представленного одной из формул от 5-а до 5-с ниже:
[Формула 5-а]
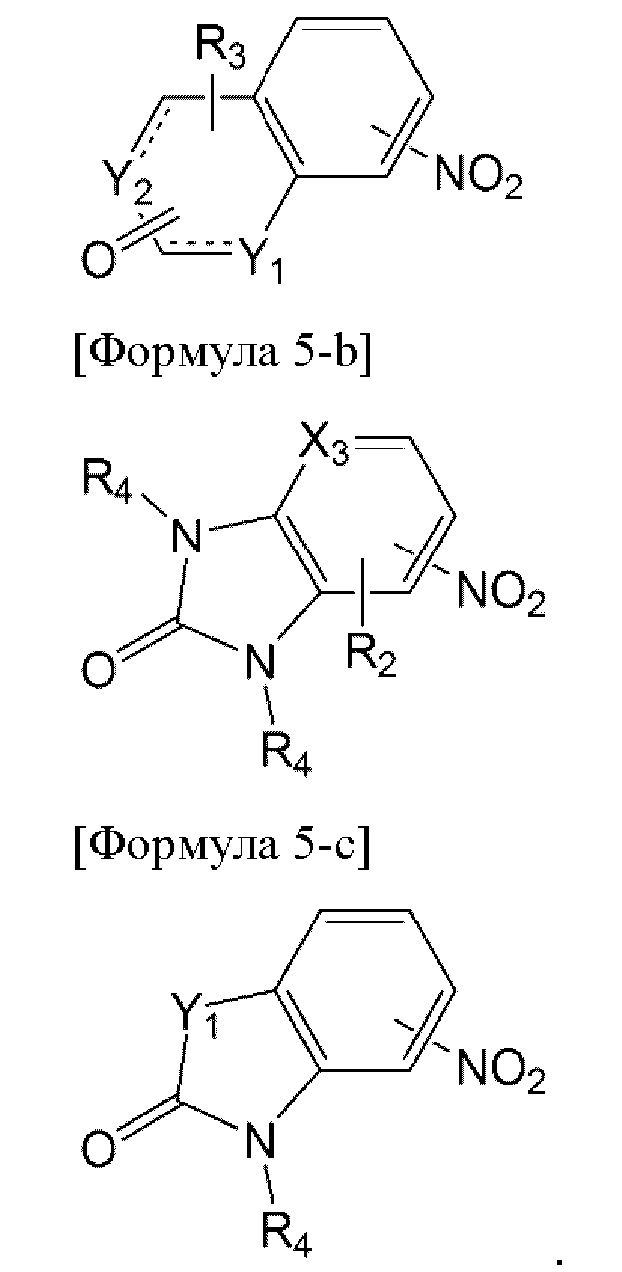
В формуле 5-b каждый из двух R4 может быть выбран из ряда заместителей, определенных выше, и оба R4 могут быть одинаковыми, или один R4 может отличаться от другого R4.
В частности, способ по настоящему изобретению может дополнительно включать перед первой стадией стадию восстановления нитрогруппы соединения, представленного одной из формул от 5-а до 5-с, до аминогруппы.
Например, восстановление может быть осуществлено посредством реакции в органическом растворителе, таком как 1,4-диоксан или метанол, в присутствии катализатора Pd/C, посредством реакции с АсОН в присутствии Fe или посредством реакции с хлоридом аммония в присутствии Fe, без ограничения этим.
В частности, соединение, представленное формулой 5-а, может быть получено путем взаимодействия его предшественника, в котором один из Y1 и Y2 представляет собой СН, а другой представляет собой О, с соединением на основе амина NH2R4 в состоянии растворения в органическом растворителе, так чтобы заместить О-сайт группой NR4, но соединение этим не ограничивается. Например, реакцию можно проводить путем перемешивания предшественника и аммиака или Бд-замещенного амина в органическом растворителе, таком как THF или метанол, при температуре от 70°С до 130°С в течение от 30 минут до 5 часов, но не ограничивается этим. В связи с этим для перемешивания можно использовать микроволновую печь, без ограничения этим.
Кроме того, предшественник соединения, представленного формулой 5-а выше, в котором один из Y1 и Y2 представляет собой СН, а другой представляет собой О, может быть получен, необязательно, путем циклизации между производным нитробензойной кислоты и диметилацеталем N,N-диметилформамида или ацеталем ацетона, без ограничения этим. Например, циклизацию можно проводить посредством реакции с диметилацеталем N,N-диметилформамида в растворителе DMF или реакции с ацеталем ацетона в трет-бутиловом спирте в присутствии Cu. Реакцию можно проводить при температуре от 100°С до 130°С в течение от 2 до 24 часов при перемешивании, но не ограничиваясь этим. В связи с этим для перемешивания можно использовать микроволновую печь, без ограничения этим.
В частности, в способе по настоящему изобретению соединение, представленное формулой 5-b или 5-с выше, может быть получено, необязательно, путем дополнительного проведения стадии введения заместителя R4 посредством реакции с предшественником соединения, включающим R4, и реакционноспособным галогенидом. Например, реакцию с DBU (1,8-диазабицикло[5.4.0]ундец-7-ен) и R4-галогенидом или R4-галогенацетатом натрия можно проводить в растворителе DMF при перемешивании при комнатной температуре в течение от 6 до 24 часов, но не ограничивается этим.
Кроме того, в способе по настоящему изобретению соединение, представленное формулой 5-b выше, может быть получено путем циклизации между незамещенным или R2-замещенным 1,2-диаминнитрофенилом и карбонилдиимидазолом (CDI). Например, реакцию можно проводить с использованием растворителя DMF при комнатной температуре в течение от 6 до 24 часов, без ограничения этим.
Например, имеющиеся в продаже соединения можно использовать в виде приобретенных в качестве реагентов и промежуточных соединений, используемых на каждой стадии способа по настоящему изобретению, или реагенты и промежуточные соединения, используемые на каждой стадии, можно синтезировать с использованием имеющихся в продаже соединений посредством реакций, хорошо известных в данной области техники, самих по себе или в комбинации, но настоящее изобретение не ограничивается этим.
Кроме того, при необходимости способ может дополнительно включать процессы выделения и/или очистки продукта после каждой реакции, и эти процессы можно проводить с использованием различных способов, хорошо известных в данной области техники.
Третий аспект настоящего изобретения заключается в предложении композиции для ингибирования агрегации тау-белка, включающей соединение по настоящему изобретению в качестве активного ингредиента.
Четвертый аспект настоящего изобретения заключается в предложении композиции для ингибирования гиперфосфорилирования тау-белка, включающей соединение по настоящему изобретению в качестве активного ингредиента.
Пятый аспект настоящего изобретения заключается в предложении фармацевтической композиции для предупреждения или лечения заболевания, вызванного агрегацией или гиперфосфорилированием тау-белка, включающей соединение по настоящему изобретению в качестве активного ингредиента.
Шестой аспект настоящего изобретения заключается в предложении способа предупреждения или лечения заболевания, вызванного агрегацией или гиперфосфорилированием тау-белка, включающего введение соединения по настоящему изобретению субъекту, нуждающемуся в этом.
В конкретных воплощениях настоящего изобретения было вновь синтезировано в общей сложности 57 соединений, пронумерованных от 1 до 57 и представленных формулой 1, и было подтверждено их влияние на ингибирование агрегации и гиперфосфорилирования тау-белка. Более того, для подтверждения возможности использования в качестве фармацевтической композиции было подтверждено, что данные соединения не проявляют цитотоксичности.
Используемый здесь термин «предупреждение» относится к любому действию, которое ингибирует или задерживает возникновение, распространение и рецидив заболевания, вызванного агрегацией или гиперфосфорилированием тау-белка при введении фармацевтической композиции по настоящему изобретению, и термин «лечение» относится к любому действию, при котором симптомы заболевания улучшаются или благотворно изменяются при введении фармацевтической композиции по настоящему изобретению.
Как описано выше, поскольку соединение по настоящему изобретению не только ингибирует агрегацию или гиперфосфорилирование тау-белка, но также не проявляет токсичности по отношению к клеткам, фармацевтическая композиция, содержащая это соединение в качестве активного ингредиента, может быть использована для предупреждения или лечения заболеваний, вызванных агрегацией или гиперфосфорилированием тау-белка. Заболевание, вызванное агрегацией или гиперфосфорилированием тау-белка, для лечения которого можно применять фармацевтическую композицию по настоящему изобретению, может представлять собой болезнь Альцгеймера, болезнь Паркинсона, сосудистую деменцию, острый инсульт, травму, цереброваскулярное заболевание, травму головного мозга, травму спинного мозга, периферическую невропатию, ретинопатию, глаукому или таупатию. Неограничивающие примеры таупатии могут включать хроническую травматическую энцефалопатию (СТЕ), первичную возрастную таупатию, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию, болезнь Пика, аргирофильную болезнь зерна (AGD), лобно-височную деменцию (FTD), паркинсонизм, связанный с хромосомой 17, литико-бодигскую болезнь (комплекс Гуама паркинсонизм-деменция), ганглиоглиому, ганглиоцитому, менингиоангиоматоз, постэнцефалитический паркинсонизм, подострый склерозирующий панэнцефалит, свинцовую энцефалопатию, туберозный склероз, нейродегенерацию, ассоциированную с пантотенаткиназой, липофусциноз, посттравматическое стрессовое расстройство и травматическое повреждение головного мозга.
Например, композиция по настоящему изобретению может дополнительно включать фармацевтически приемлемый носитель, разбавитель или эксципиент, может быть изготовлена и использована в различных формах, таких как пероральные формы, такие как порошки, гранулы, таблетки, капсулы, суспензии, эмульсии, сиропы, аэрозоли, и инъекционные препараты в виде стерильных растворов для инъекций в соответствии с общим способом для каждой цели применения, и могут быть введены перорально или могут быть введены различными путями, включая внутривенное, внутрибрюшинное, подкожное, ректальное и местное введение. Примеры подходящего носителя, эксципиента или разбавителя, включенного в эту композицию, могут включать лактозу, декстрозу, сахарозу, сорбит, маннит, ксилит, эритрит, мальтит, крахмал, аравийскую камедь, альгинат, желатин, фосфат кальция, силикат кальция, целлюлозу, метилцеллюлозу, микрокристаллическую целлюлозу, поливинилпирролидон, воду, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния и минеральное масло. Композиция по настоящему изобретению может дополнительно включать наполнитель, антиагрегант, смазывающий агент, увлажнитель, ароматизатор, эмульгатор, консервант и т.п.
Твердые препараты для перорального введения включают таблетки, пилюли, порошки, гранулы, капсулы и т.п., и такой твердый препарат готовят путем смешивания одного или более эксципиентов, таких как крахмал, карбонат кальция, сахароза, лактоза и желатин, с композицией. При этом в дополнение к простому эксципиенту можно использовать смазывающее вещество, такое как стеарат магния или тальк.
В качестве примера перорального жидкого препарата можно привести суспензию, раствор для внутреннего применения, эмульсию, сироп и т.п., и пероральный жидкий препарат может включать различные эксципиенты, такие как увлажнитель, подсластитель, отдушка и консервант, в дополнение к воде и жидкому парафину, которые обычно используют в качестве простого разбавителя.
Препараты для парентерального введения включают водный растворитель, неводный растворитель, суспензионный агент, эмульгатор, лиофилизированный препарат и суппозиторий, которые стерилизуют.В качестве неводного растворителя или суспендирующего агента можно использовать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, сложный эфир для инъекций, такой как этилолеат, или тому подобное. В качестве основы суппозитория можно использовать витепсол, макрогол, твин 61, масло какао, лауриновое масло, глицерожелатин и т.п. При этом, инъекционные препараты могут включать обычные добавки, такие как солюбилизирующий агент, изотонический агент, суспендирующий агент, эмульгатор, стабилизатор и консервант.
Композиция может быть приготовлена обычным способом смешивания, гранулирования или нанесения покрытия и может содержать активный ингредиент в количестве от примерно 0,1 до 75% масс., предпочтительно от примерно 0,1% до 50% масс. Стандартная композиция для млекопитающего весом от 50 до 70 кг содержит от 10 до 200 мг активного ингредиента.
В данном случае композицию по настоящему изобретению вводят в фармацевтически эффективном количестве. Используемый здесь термин «фармацевтически эффективное количество» относится к количеству, достаточному для лечения заболевания с разумным соотношением польза/риск, применимым к медикаментозному лечению и не вызывающим побочных эффектов, и уровень эффективного количества может быть определен в зависимости от состояния здоровья пациента, типа заболевания, тяжести, активности лекарственного средства, чувствительности к лекарственному средству, способа введения, времени введения, пути введения, скорости выведения, периода лечения, факторов, включающих лекарственные препараты, применяемые в комбинации или одновременно, и других факторов, хорошо известных в области медицины. Композицию по настоящему изобретению можно вводить в виде индивидуального терапевтического агента или вводить в комбинации с другими терапевтическими агентами, можно вводить последовательно или одновременно с традиционным терапевтическим агентом, а также можно вводить в виде одной дозы или нескольких доз. Важно вводить минимальное количество, способное обеспечить максимальный эффект без побочных эффектов, принимая во внимание все вышеперечисленные факторы, которые могут быть легко определены специалистами в данной области техники.
Например, поскольку дозировка может увеличиваться или уменьшаться в зависимости от пути введения, тяжести заболевания, пола, массы тела, возраста и т.п., дозировка никоим образом не ограничивает объем настоящего изобретения.
Предпочтительная доза соединения по настоящему изобретению варьирует в зависимости от состояния и массы тела пациента, тяжести заболевания, формы лекарственного средства, способа и продолжительности введения, но может быть надлежащим образом выбрана специалистами в данной области техники. Однако для желаемого эффекта соединение по настоящему изобретению можно вводить в количестве от 0,0001 мг/кг до 100 мг/кг (массы тела), предпочтительно от 0,001 мг/кг до 100 мг/кг (массы тела) в сутки. Соединение можно вводить один раз в сутки или несколько раз в сутки в разделенных дозах пероральным или парентеральным путем.
Седьмой аспект настоящего изобретения заключается в предложении способа предупреждения или лечения заболевания, вызванного агрегацией или гиперфосфорилированием тау-белка, включающего введение фармацевтической композиции по настоящему изобретению нуждающемуся в этом субъекту.
Используемый здесь термин «субъект» относится к любому животному, включая обезьян, коров, лошадей, овец, свиней, кур, индеек, перепелов, кошек, собак, мышей, кроликов и морских свинок, помимо людей, у которых развились или может развиться заболевание, вызванное агрегацией или гиперфосфорилированием тау-белка. Заболевания можно эффективно предупреждать или лечить путем введения субъекту фармацевтической композиции по настоящему изобретению. Кроме того, поскольку фармацевтическая композиция по настоящему изобретению проявляет терапевтический эффект путем ингибирования агрегации или гиперфосфорилирования тау-белка, синергический эффект может проявляться при введении этой композиции в комбинации с традиционным терапевтическим агентом.
Используемый здесь термин «введение» относится к предоставлению заранее определенного вещества пациенту любым подходящим способом, и путь введения композиции по настоящему изобретению может быть любым общим путем, при условии что вещество способно достигнуть ткани-мишени. Композицию можно вводить посредством внутрибрюшинного введения, внутривенного введения, внутримышечного введения, подкожного введения, внутрикожного введения, перорального введения, местного введения, интраназального введения, внутрилегочного введения или ректального введения, но настоящее изобретение не ограничивается этим. Кроме того, фармацевтическую композицию по настоящему изобретению можно вводить с помощью любого устройства, способного доставлять активное вещество к клетке-мишени. Предпочтительные способы введения и композиции включают внутривенные инъекции, подкожные инъекции, внутрикожные инъекции, внутримышечные инъекции и капельные инъекции лекарственных средств. Инъекционные препараты могут быть приготовлены с использованием водного растворителя, такого как физиологический раствор или раствор Рингера, или неводного растворителя, такого как растительное масло, сложный эфир высшей жирной кислоты (например, этилолеат) или спирт (например, этанол, бензиловый спирт, пропиленгликоль или глицерин), и могут включать фармацевтический носитель, такой как стабилизирующий агент для предотвращения денатурации (например, аскорбиновая кислота, гидросульфит натрия, пиросульфит натрия, ВНА, токоферол или EDTA), эмульгатор, буферный агент для контроля рН или консервант для ингибирования роста микроорганизмов (например, нитрат фенилртути, тимеросал, хлорид бензалкония, фенол, крезол или бензиловый спирт).
Осуществление изобретения
Далее настоящее изобретение будет описано более подробно со ссылкой на примеры и экспериментальные примеры. Однако эти примеры и экспериментальные примеры являются только иллюстрацией настоящего изобретения, и объем настоящего изобретения не ограничивается этими примерами и экспериментальными примерами.
Пример 1: Получение (1-оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноилдицианида (Соединение 1)
Стадия 1-1: Получение 7-нитроизохинолин-1(2H)-она
2,0 М раствор аммиака (3,4 мл, 6,80 ммоль) добавляли к 7-нитро-1H-изохромен-1-ону (130 мг, 0,68 ммоль) с получением реакционной смеси и эту реакционную смесь перемешивали при 80°С в течение 2 часов в микроволновой печи. После завершения взаимодействия реакционный продукт отверждали с использованием дистиллированной воды и отфильтровывали с получением 73 мг (выход: 56%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.76 (s, 1H), 8.90 (d, J=2,5 Гц, 1H), 8.44 (dd, J=8,8 Гц, 2,5 Гц, 1H), 7.91 (d, J=8,7 Гц, 1Н), 7.46 (d, J=7,1 Гц, 1Н), 6.73 (d, J=7,1 Гц, 1H).
Стадия 1-2: Получение 7-аминоизохинолин-1(2H)-она
7-Нитроизохинолин-1(2H)-он (70 мг, 0,37 ммоль), полученный на Стадии 1-1, растворяли в 10% Pd/C (78 мг, 0,07 ммоль) и 1,4-диоксане и реакционную смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере водорода. После завершения взаимодействия реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением 27 мг (выход: 46%) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) δ 10.82 (s, 1H), 7.40-7.20 (m, 2Н), 6.97 (dd, J=8,4 Гц, 2,5 Гц, 1H), 6.80 (dd, J=7,0 Гц, 5,5 Гц, 1H), 6.34 (d, J=7,0 Гц, 1H), 5.47 (s, 2Н).
Стадия 1-3: Получение (1-оксо-1,2-дигидроизохинолин-7-ил)карбогидразоноилдииианида
7-Аминоизохинолин-1(2H)-он (20 мг, 0,12 ммоль), полученный на Стадии 1-2, и нитрит натрия (13 мг, 0,19 ммоль) растворяли в этаноле в атмосфере азота и 1,0 М водный раствор соляной кислоты (0,4 мл, 0,37 ммоль) добавляли к нему при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 минут с образованием соли диазония. Малононитрил (16 мг, 0,25 ммоль) добавляли к реакционной смеси, включающей соль диазония, и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Значение рН реакционной смеси доводили до 6,0 с использованием водного раствора гидроксида натрия и затем дополнительно перемешивали при комнатной температуре в течение 1 часа. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 20 мг (выход: 67%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.22 (s, 1H), 11.33 (d, J=5,7 Гц, 1H), 8.24 (d,J=2,4 Гц, 1H), 7.81 (dd, J=8,7 Гц, 2,4 Гц, 1H), 7.72 (d, J=8,7 Гц, 1H), 7.16 (dd, J=7,1 Гц, 5,8 Гц, 1Н), 6.56(d, J=7,0 Гц, 1H).
Пример 2: Получение (2-метил-1-оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноилдицианида (Соединение 2)
Стадия 2-1: Получение 7-нитро-1H-изохромен-1-она
2-Метил-5-нитробензойную кислоту (500 мг, 2,76 ммоль) растворяли в диметилформамиде (DMF, 10 мл) и добавляли к нему диметилацеталь N,N-диметилформамида (1,10 мл, 8,23 ммоль) и реакционную смесь перемешивали при 115°С в течение 12 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 65 мг (выход: 12%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.79 (d, J=2,5 Гц, 1H), 8.61 (dd, J=8,6 Гц, 2,5 Гц, 1H), 7.93 (d, J=8,7 Гц, 1H), 7.80 (d, J =5,6 Гц, 1H), 7.01 (d,J=5,6 Гц, 1H).
Стадия 2-2: Получение 2-метил-7-нитроизохинолин-1(2H)-она
1,0 М метиламин (10,5 мл, 20,93 ммоль) добавляли к 7-нитро-1H-изохромен-1-ону (200 мг, 1,05 ммоль), полученному на Стадии 2-1, и реакционную смесь перемешивали при 120°С в течение 12 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и дихлорметана с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 126 мг (выход: 59%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.90 (d, J=2,5 Гц, 1Н), 8.43 (dd, J=8,8 Гц, 2,5 Гц, 1H), 7.89 (d, J=8,8 Гц, 1H), 7.75 (d, J=7,3 Гц, 1H), 6.78 (d, J=7,3 Гц, 1Н), 3.55 (s, 3Н).
Стадия 2-3: Получение 7-амино-2-метилизохинолин-1(2H)-она
49 мг (выход: 57%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 2-метил-7-нитроизохинолин-1(2H)-он (100 мг, 0,49 ммоль), полученный на Стадии 2-2 выше, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.36-7.29 (m, 2Н), 7.08 (d, J=7,2 Гц, 1H), 6.97 (dd, J=8,4 Гц, 2,5 Гц, 1H), 6.40 (d, J=7,2 Гц, 1Н), 5.51 (s, 2Н), 3.44 (s, 3Н).
Стадия 2-4: Получение (2-метил-1-оксо-1,2-дигидроизохинолин-7-ил)карбогидразоноилдицианида
49 мг (выход: 85%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 7-амино-2-метилизохинолин-1(2H)-он (40 мг, 0,23 ммоль), полученный на Стадии 2-3 выше, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.25 (s, 1H), 8.28 (d,J=2,4 Гц, 1H), 7.81 (dd, J=8,7 Гц, 2,4 Гц, 1H), 7.72 (d, J=8,7 Гц, 1H), 7.46 (d, J=7,3 Гц, 1H), 6.63 (d, J=7,3 Гц, 1H), 3.52 (s, 3Н).
Пример 3: Получение (2-изопропил-1-оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноилдицианида (Соединение 3)
Стадия 3-1: Получение 2-изопропил-7-нитроизохинолин-1(2H)-она
Изопропиламин (0,27 мл, 3,14 ммоль) и метанол (3 мл) добавляли к 7-нитро-1H-изохромен-1-ону (30 мг, 0,16 ммоль), полученному на Стадии 2-1 Примера 2 выше, и реакционную смесь перемешивали при 80°С в течение 12 часов в микроволновой печи. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и отверждали с использованием эфира и гексана и отфильтровывали с получением 20 мг (выход: 54%) указанного в заголовке соединения.
1H ЯМР (400 МГц, Хлороформ-d) δ 9.31 (d, J=2,4 Гц, 1H), 8.41 (dd, J=8,7 Гц, 2,4 Гц, 1H), 7.62 (d, J=8,7 Гц, 1H), 7.34 (d, J=7,6 Гц, 1H), 6.62 (d, J=7,5 Гц, 1H), 5.38 (р, J=6,8 Гц, 1H), 1.42 (d, J=6,8 Гц, 6Н).
Стадия 3-2: Получение 7-амино-2-изопропилизохинолин-1(2H)-она
54 мг (выход: 89%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 2-изопропил-7-нитроизохинолин-1(2H)-он (70 мг, 0,30 ммоль), полученный на Стадии 3-1 выше, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.31 (d, J=2,4 Гц, 1Н), 8.41 (dd, J=8,7 Гц, 2,4 Гц, 1H), 7.62 (d, J=8,7 Гц, 1H), 7.34 (d, J=7,6 Гц, 1H), 6.62 (d, J=7,5 Гц, 1H), 5.38 (p, J=6,8 Гц, 1H), 1.42 (d, J=6,8 Гц, 6H).
Стадия 3-3: Получение (2-изопропил-1-оксо-1,2-дигидроизохинолин-7-ил)карбогидразоноилдицианида
30 мг (выход: 72%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 7-амино-2-изопропилизохинолин-1(2H)-он (30 мг, 0,15 ммоль), полученный на Стадии 3-2 выше, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.25 (s, 1H), 8.30 (d, J=2,3 Гц, 1H), 7.81 (dd, J=8,7 Гц, 2,3 Гц, 1H), 7.72 (d, J=8,7 Гц, 1H), 7.52 (d, J=7,5 Гц, 1H), 6.69 (d, J=7,5 Гц, 1H), 5.19 (р, J=6,8 Гц, 1H), 1.33 (d, J=6,8 Гц, 6Н).
Пример 4: Получение (1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианида (Соединение 4)
Стадия 4-1: Получение 5-нитро-1H-изохромен-1-она
88 мг (выход: 42%) указанного в заголовке соединения было получено таким же способом, как на Стадии 2-1 Примера 2 выше, за исключением того, что 2-метил-3-нитробензойную кислоту (200 мг, 1,10 ммоль) использовали вместо 2-метил-5-нитробензойной кислоты.
1Н ЯМР (400 МГц, Хлороформ-d) δ 8.69-8.61 (m, 1H), 8.49 (dd, J=8,1 Гц, 1,4 Гц, 1Н), 7.68 (t,J=8,0 Гц, 1H), 7.44 (d, J=6,l Гц, 1Н), 7.38 (d, J=6,0 Гц, 1H).
Стадия 4-2: Получение 5-нитроизохинолин-1(2H)-она
45 мг (выход: 45%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-1 Примера 1 выше, за исключением того, что 5-нитро-1H-изохромен-1-он (100 мг, 0,52 ммоль), полученный на Стадии 4-1 выше, использовали вместо 7-нитро-1H-изохромен-1-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.75 (s, 1H), 8.58 (d, J=8,0 Гц, 1H), 8.46 (dd, J=7,9 Гц, 1,4 Гц, 1H), 7.67 (t, J=8,0 Гц, 1H), 7.45 (d, J=7,6 Гц, 1H), 6.97 (d, J=7,6 Гц, 1H).
Стадия 4-3: Получение 5-амино-2-изохинолин-1(2H)-она
18 мг (выход: 42%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 5-нитроизохинолин-1(2H)-он (50 мг, 0,26 ммоль), полученный на Стадии 4-2 выше, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.05 (s, 1Н), 7.39 (d, J=7,9 Гц, 1H), 7.14 (t, J=7,8 Гц, 1H), 7.02 (dd, J=7,4 Гц, 4,8 Гц, 1H), 6.86 (dd, J=7,7 Гц, 1,2 Гц, 1H), 6.67 (d, J=7,4 Гц, 1H), 5.61 (s, 2H).
Стадия 4-4: Получение (1-оксо-1,2-дигидроизохинолин-5-ил)карбогидразоноилдицианида
6 мг (выход: 38%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-2-изохинолин-1(2H)-он (30 мг, 0,15 ммоль), полученный на Стадии 4-3, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.42 (s, 1H), 8.10 (d, J=7,9 Гц, 1H), 7.69 (dd, J=7,8 Гц, 1,3 Гц, 1H), 7.50 (t, J=7,9 Гц, 1H), 7.27 (dd, J=7,3 Гц, 5,8 Гц, 1H), 6.82 (d, J=7,4 Гц, 1H).
Пример 5: Получение (2-метил-1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианида (Соединение 5)
Стадия 5-1: Получение 2-метил-5-нитроизохинолин-1(2H)-она
37 мг (выход: 47%) указанного в заголовке соединения было получено таким же способом, как на Стадии 2-2 Примера 2 выше, за исключением того, что 5-нитро-1H-изохромен-1-он (74 мг, 0,39 ммоль) использовали вместо 7-нитро-1H-изохромен-1-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.61 (d, J=7,9 Гц, 1H), 8.47 (dd, J=7,9 Гц, 1,3 Гц, 1H), 7.74 (d, J=7,8 Гц, 1H), 7.68 (t, J=8,0 Гц, 1H), 7.03 (dd, J=7,7 Гц, 0,8 Гц, 1H), 3.55 (s, 3Н).
Стадия 5-2: Получение 5-амино-2-метилизохинолин-1(2H)-она
30 мг (выход: 69%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 2-метил-5-нитроизохинолин-1(2H)-он (50 мг, 0,24 ммоль), полученный на Стадии 5-1, использовали вместо 7-нитроизохинолин-1 (2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.42 (d, J=7,9 Гц, 1H), 7.32 (d, J=7,5 Гц, 1H), 7.16 (t, J=7,8 Гц, 1H), 6.84 (dd, J=7,8 Гц, 1,2 Гц, 1H), 6.73 (d, J=7,6 Гц, 1H), 5.64 (s, 2H), 3.46 (s, 3Н).
Стадия 5-3: Получение (2-метил-1-оксо-1,2-дигидроизохинолин-5-ил)карбогидразоноилдицианида
31 мг (выход: 86%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-2-метилизохинолин-1(2H)-он (25 мг, 0,14 ммоль), полученный на Стадии 5-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, Хлороформ-d6) δ9.87 (s, 1H), 8.38 (d, J=8,0 Гц, 1H), 7.81 (dd, J=8,0 Гц, 1,2 Гц, 1Н), 7.55 (t, J=8,0 Гц, 1Н), 7.24 (d, J=7,7 Гц, 1Н), 6.46 (d, J=7,6 Гц, 1H), 3.64 (s, 3Н).
Пример 6: Получение (2-изопропил-1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианида (Соединение 6)
Стадия 6-1: Получение 2-изопропил-5-нитроизохинолин-1(2H)-она
11 мг (выход: 30%) указанного в заголовке соединения было получено таким же способом, как на Стадии 3-1 Примера 3 выше, за исключением того, что 5-нитро-1H-изохромен-1-он (30 мг, 0,16 ммоль) использовали вместо 7-нитро-1H-изохромен-1-она.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 8.80 (dd, J=8,0 Гц, 1,4 Гц, 1H), 8.40 (dd, J=8,0 Гц, 1,4 Гц, 1H), 7.56 (t, J=8,0 Гц, 1Н), 7.35 (s, 2Н), 5.35 (р, J=6,6 Гц, 1H), 1.42 (d, J=6,8 Гц, 6Н).
Стадия 6-2: Получение 5-амино-2-изопропилизохинолин-1(2H)-она
34 мг (выход: 99%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 2-изопропил-5-нитроизохинолин-1(2H)-он (40 мг, 0,17 ммоль), полученный на Стадии 6-1 выше, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, Хлороформ-d) δ 7.92 (d, J=8,l Гц, 1Н), 7.29 (d, J=7,9 Гц, 1H), 7.13 (d, J=7,7 Гц, 1Н), 6.94 (dd, J=7,6 Гц, 1,2 Гц, 1H), 6.55-6.42 (m, 1H), 5.40 (р, J=6,9 Гц, 1H), 3.95 (s, 2Н), 1.38 (d, J=6,9 Гц, 6Н).
Стадия 6-3: Получение (2-изопропил-1-оксо-1,2-дигидроизохинолин-5-ил)карбогидразоноилдицианида
22 мг (выход: 79%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-2-изопропилизохинолин-1(2H)-он (20 мг, 0,10 ммоль), полученный на Стадии 6-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.14 (d, J=8,0 Гц, 1Н), 7.72-7.58 (m, 2H), 7.51 (t, J=7,9 Гц, 1H), 6.96 (d, J=7,8 Гц, 1H), 5.18 (p, J=6,8 Гц, 1H), 1.34 (d, J=6,8 Гц, 6H).
Пример 7: Получение (2,3-диметил-1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианида (Соединение 7)
Стадия 7-1: Получение 3-метил-5-нитро-1H-изохромен-1-она
Ацетилацетон (1,04 мл, 10,16 ммоль), медь (13 мг, 0,20 ммоль), трет-бутоксид калия (456 мг, 4,06 ммоль) и трет-бутиловый спирт (10 мл) добавляли к 2-бром-3-нитробензойной кислоте (500 мг, 2,03 ммоль) и реакционную смесь перемешивали при 110°С в течение 5 часов в микроволновой печи. После завершения взаимодействия реакционную смесь подкисляли добавлением к этой реакционной смеси водного раствора соляной кислоты, реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 68 мг (выход: 16%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 8.58 (dt, J=7,8 Гц, 1,1 Гц, 1H), 8.44 (dd, J=8,2 Гц, 1,4 Гц, 1Н), 7.57 (t, J=8,0 Гц, 1H), 7.16 (t, J=1,0 Гц, 1Н), 2.38 (d, J=1,0 Гц, 3Н).
Стадия 7-2: Получение 2,3-Диметил-5-нитроизохинолин-1(2H)-она
56 мг (выход: 88%) указанного в заголовке соединения было получено таким же способом, как на Стадии 2-2 Примера 2 выше, за исключением того, что 3-метил-5-нитро-1H-изохромен-1-он (60 мг, 0,29 ммоль), полученный на Стадии 7-1, использовали вместо 7-нитро-1H-изохромен-1-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.26 (d, J=7,8 Гц, 1H), 8.18 (d, J=8,2 Гц, 1H), 7.62 (t, J=7,9 Гц, 1H), 6.23 (d, J=1,0 Гц, 1H), 3.02 (s, 3H), 1.46 (s, 3H).
Стадия 7-3: Получение 5-амино-2,3-диметилизохинолин-1(2H)-она
13 мг (выход: 29%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 2,3-диметил-5-нитроизохинолин-1(2H)-он (50 мг, 0,23 ммоль), полученный на Стадии 7-2, использовали вместо 7-нитроизохинолин-1 (2)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.38 (d, J=7,9 Гц, 1H), 7.08 (t, J=7,8 Гц, 1H), 6.81 (dd, J=7,7 Гц, 1,2 Гц, 1H), 6.64 (s, 1H), 5.51 (s, 2H), 3.48 (s, 3H), 2.39 (d, J=1,0 Гц, 3Н).
Стадия 7-4: Получение (2,3-диметил-1-оксо-1,2-дигидроизохинолин-5-ил)карбогидразоноилдицианида
13 мг (выход: 92%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-2,3-диметилизохинолин-1(2H)-он (10 мг, 0,05 ммоль), полученный на Стадии 7-3, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.11 (d, J=8,0 Гц, 1H), 7.65 (dd, J=7,8 Гц, 1,3 Гц, 1H), 7.44 (t, J=7,9 Гц, 1H), 6.78 (s, 1H), 3.54 (s, 3H), 2.47 (s, 3H).
Пример 8: Получение (2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианида (Соединение 8)
Стадия 8-1: Получение 6-аминоизохинолин-2(1H)-она
34 мг (выход: 13%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 6-нитроизохинолин-2(1H)-он (300 мг, 1,58 ммоль) использовали вместо 7-нитроизохинолин-1 (2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.36 (s, 1H), 7.66 (d, J=9,5 Гц, 1H), 7.02 (s, 1H), 6.83 (dd, J=8,7 Гц, 2,5 Гц, 1H), 6.71 (d, J=2,5 Гц, 1H), 6.36 (d, J=9,5 Гц, 1H), 4.98 (s, 2Н).
Стадия 8-2: Получение (2-оксо-1,2-дигидроизохинолин-6-ил)карбогидразоноилдицианида
17 мг (выход: 59%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-аминоизохинолин-2(1H)-он (20 мг, 0,12 ммоль), полученный на Стадии 8-1, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.13 (s, 1H, 11.87 (s, 1H), 7.97 (d, J=9,6 Гц, 1H), 7.74 (s, 1H), 7.66 (dd, J=8,9 Гц, 2,4 Гц, 1H), 7.34 (d, J=8,9 Гц, 1H), 6.55 (d, J=9,6 Гц, 1H).
Пример 9: Получение (6-фтор-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 9)
Стадия 9-1: Получение 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она
4-Фтор-5-нитробензол-1,2-диамин (500 мг, 2,92 ммоль) и карбонилдиимидазол (CDI, 1,44 г, 8,77 ммоль) растворяли в DMF в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 512 мг (выход: 89%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 10.38 (s, 1H), 10.08 (s, 1H), 7.75 (d, J=6,4 Гц, 1H), 7.09 (d, J=11,2 Гц, 1H).
Стадия 9-2: Получение 5-фтор-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-она
Затем 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-он (450 мг, 2,28 ммоль), полученный на Стадии 9-1, растворяли в DMF в атмосфере азота, добавляли к нему 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU, 1,02 мл, 6,84 ммоль) и метилйодид (0,42 мл, 6,84 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение от 1 до 5 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 328 мг (выход: 64%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.85 (d, J=6,4 Гц, 1H), 7.26 (d, J=1 1,6 Гц, 1H), 3.49 (s, 3Н), 3.46 (s, 3Н).
Стадия 9-3: Получение 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она
5-Фтор-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(37H)-он (250 мг, 1,11 ммоль), полученный на Стадии 9-2, и железо (496 мг, 8,88 ммоль) растворяли в уксусной кислоте в атмосфере азота и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 190 мг (выход: 88%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 6.84 (d, J=10,8 Гц, 1H), 6.59 (d, J=7,6 Гц, 1H), 4.36 (s, 2Н), 3.28 (s, 3Н), 3.26 (s, 3Н).
Стадия 9-4: Получение (6-фтор-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
5-Амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-он (100 мг, 0,51 ммоль), полученный на Стадии 9-3, и 35% водный раствор соляной кислоты (0,2 мл) растворяли в дистиллированной воде в атмосфере азота и добавляли к нему нитрит натрия (65 мг, 0,77 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 минут с образованием соли диазония. Малононитрил (51 мг, 0,77 ммоль) добавляли к этой реакционной смеси, включающей соль диазония, и реакционную смесь перемешивали при комнатной температуре в течение 11 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 28 мг (выход: 20%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.36 (d, J=6,8 Гц, 1H), 7.15 (d, J=10,8 Гц, 1H), 3.43 (s, 3Н), 3.39 (s, 3Н).
Пример 10: Получение (6-хлор-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 10)
Стадия 10-1: Получение 5-хлор-6-нитро-1H-бензо[d]имидазол-2(3H)-она
882 мг (выход: 77%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что 4-хлор-5-нитробензол-1,2-диамин (1 г, 5,33 ммоль) использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, Ацетон-d6) δ 10.30 (s, 1H), 10.19 (s, 1H), 7.71 (s, 1H), 7.26 (s,1Н).
Стадия 10-2: Получение 5-хлор-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-она
410 мг (выход: 73%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 5-хлор-6-нитро-1H-бензо[d]имидазол-2(3H)-он (500 мг, 2,34 ммоль), полученный на Стадии 10-1, использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.83 (s, 1Н), 7.41 (s, 1Н), 3.48 (s, 3Н), 3.47 (s, 3Н).
Стадия 10-3: Получение 5-амино-6-хлор-1,3-Диметил-1H-бензо[d]имидазол-2(3H)-она
190 мг (выход: 88%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-3 Примера 9 выше, за исключением того, что 5-хлор-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-он (250 мг, 1,03 ммоль), полученный на Стадии 10-2, использовали вместо 5-фтор-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 6.84 (d, J=10,8 Гц, 1Н), 6.59 (d, J=7,6 Гц, 1H), 4.36 (s, 2Н), 3.28 (s, 3Н), 3.26 (s, 3Н).
Стадия 10-4: Получение (6-хлор-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
28 мг (выход: 20%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 5-амино-6-хлор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-он (100 мг, 0,51 ммоль), полученный на Стадии 10-3, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.39 (s, 1H), 7.30 (s, 1H), 3.34 (s, 3Н), 3.33 (s, 3Н).
Пример 11: Получение (7-хлор-1,3-диметил-2-оксо-2,3-дигидро-17/-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 11)
Стадия 11-1: Получение 4-хлор-6-нитро-1H-бензо[d]имидазол-2(3H)-она
720 мг (выход: 63%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что 3-хлор-5-нитробензол-1,2-диамин (2,63 г, 15,99 ммоль) использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, Ацетон-d6) δ 10.77 (s, 1H), 10.40 (s, 1H), 8.01 (d, J=2,0 Гц, 1H), 7.86 (d,J=2,0 Гц, 1H).
Стадия 11-2: Получение 4-хлор-1у3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-она
412 мг (выход: 73%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 4-хлор-6-нитро-1H-бензо[d]имидазол-2(3H)-он (500 мг, 2,34 ммоль), полученный на Стадии 11-1, использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.99 (d, J=2,0 Гц, 1H), 7.95 (d, J=2,4 Гц, 1Н), 3.75 (s, 3Н), 3.53 (s, 3Н).
Стадия 11-3: Получение 6-амино-4-хлор-1,3-Диметил-1H-бензо[d]имидазол-2(3H)-она
154 мг (выход: 71%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-3 Примера 9 выше, за исключением того, что 4-хлор-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-он (250 мг, 1,03 ммоль), полученный на Стадии 11-2, использовали вместо 5-фтор-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1H ЯМР (400 МГц, Ацетон-d6) δ 6.39 (d, J=2,0 Гц, 1H), 6.37 (d, J=2,0 Гц, 1Н), 4.64 (s, 2Н), 3.55 (s, 3Н), 3.26 (s, 3Н).
Стадия 11-4: Получение (7-хлор-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
42 мг (выход: 41%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-амино-4-хлор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-он (100 мг, 0,47 ммоль), полученный на Стадии 11-3, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она.
1H ЯМР (400 МГц, DMSO-d6) δ 7.20 (s, 2Н), 3.58 (s, 3Н).
Пример 12: Получение (1,3-диметил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 12)
Стадия 12-1: Получение 6-нитро-1H-имидазо[4,5-61пиридин-2(3H)-она
5-Нитропиридин-2,3-диамин (600 мг, 3,89 ммоль) и N,N'-дисукцинимидил-карбонат (15 г, 5,84 ммоль) растворяли в ацетонитриле в атмосфере азота и реакционную смесь перемешивали при 80°С в течение 8 часов. После завершения взаимодействия реакционный продукт фильтровали с использованием ацетонитрила с получением 570 мг (выход: 81%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 11.00 (s, 1H), 10.29 (s, 1H), 8.87 (d, J=2,4 Гц, 1H), 8.02 (d, J=2,4 Гц, 1H).
Стадия 12-2: Получение 1,3-диметил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
351 мг (выход: 76%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он (400 мг, 2,22 ммоль), полученный на Стадии 12-1, использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.94 (d, J=2,4 Гц, 1H), 8.18 (d, J=2,0 Гц, 1H), 3.54 (s, 3Н), 3.48 (s, 3Н).
Стадия 12-3: Получение 6-амино-1,3-диметил-1H-имидазо[4,5-b]пиридин-2(3H)-она
1,3-Диметил-6-нитро-1H-имидазо[4,5-6]пиридин-2(3H)-он (300 мг, 1,44 ммоль), полученный на Стадии 12-2, и 10% Pd/C (307 мг, 0,29 ммоль) растворяли в метаноле и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения взаимодействия реакционный продукт фильтровали с использованием метанола и фильтрат концентрировали с получением 241 мг (выход: 94%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.49 (d, J=2,4 Гц, 1H), 6.78 (d, J=2,4 Гц, 1H), 4.49 (s, 2Н), 3.30 (s, 3Н), 3.29 (s, 3Н).
Стадия 12-4: Получение (1,3-диметил-2-оксо-2,3-Дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида
100 мг (выход: 47%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-амино-1,3-диметил-1H-имидазо[4,5-b]пиридин-2(3H)-он (150 мг, 0,84 ммоль), полученный на Стадии 12-3, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.18 (d, J=2,0 Гц, 1H), 7.63 (d, J=2,0 Гц, 1Н), 3.47 (s, 3Н), 3.41 (s, 3Н).
Пример 13: Получение (2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 13)
Стадия 13-1: Получение 6-аминобензо[d]тиазол-2(3H)-она
6-Нитробензо[d]тиазол-2(3H)-он (200 мг, 1,02 ммоль) растворяли в смеси этанол/вода в атмосфере азота и добавляли к нему Fe (228 мг, 4,08 ммоль) и хлорид аммония (545 мг, 10,19 ммоль) и реакционную смесь перемешивали при 80°С в течение 1 часа. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 83 мг (выход: 49%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.34 (s, 1H), 6.79 (d, J=8,4 Гц, 1H), 6.69 (d, J=2,2 Гц, 1Н), 6.51 (dd, J=8,5 Гц, 2,3 Гц, 1H), 4.95 (s, 2Н).
Стадия 13-2: Получение (2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида
34 мг (выход: 47%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-аминобензо[d]тиазол-2(3H)-он (50 мг, 0,30 ммоль), полученный на Стадии 13-1, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.96 (s, 1H), 7.68 (s, 1H), 7.39 (d, J=8,8 Гц, 1H), 7.13 (d, J=8,6 Гц, 1Н).
Пример 14: Получение (3-метил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 14)
Стадия 14-1: Получение 3-метил-6-нитробензо[d]тиазол-2(3H)-она
68 мг (выход: 64%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 6-нитробензо[d]тиазол-2(3H)-он (100 мг, 0,48 ммоль) использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.61 (d, J=2,4 Гц, 1H), 8.31 (dd, J=8,9 Гц, 2,4 Гц, 1H), 7.50 (d, J=8,9 Гц, 1H), 3.58 (s, 3Н).
Стадия 14-2: Получение 6-амино-3-метилбензо[d]тиазол-2(3H)-она
79 мг (выход: 91%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-метил-6- нитробензо[d]тиазол-2(3H)-он (100 мг, 0,48 ммоль), полученный на Стадии 14-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо 1,4-диоксана.
1H ЯМР (400 МГц, Ацетон-d6) δ 6.95 (d, J=8,5 Гц, 1H), 6.87 (d, J=2,3 Гц, 1Н), 6.72 (dd, J=8,5 Гц, 2,3 Гц, 1H), 4.62 (s, 2Н), 3.35 (s, 3Н).
Стадия 14-3: Получение (3-метил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида
94 мг (выход: 83%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-амино-3-метилбензо[d]тиазол-2(3H)-он, полученный на Стадии 14-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.81 (d, J=2,2 Гц, 1H), 7.51 (dd, J=8,8 Гц, 2,3 Гц, 1H), 7.36 (d, J=8,8 Гц, 1H), 3.41 (s, 3H).
Пример 15: Получение (2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида (Соединение 15)
Стадия 15-1: Получение 6-аминобензо[d]оксазол-2(3H)-она
131 мг (выход: 78%) указанного в заголовке соединения было получено таким же способом, как на Стадии 14-2 Примера 14 выше, за исключением того, что 6-нитробензо[d]оксазол-2(3H)-он использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 6.83 (d, J=8,3 Гц, 14H), 6.61 (d, J=2,l Гц, 1H), 6.49 (dd, J=8,3 Гц, 2,1 Гц, 1H), 4.60 (s, 2Н).
Стадия 15-2: Получение (2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида
115 мг (выход: 76%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-аминобензо[d]оксазол-2(3H)-он, полученный на Стадии 15-1, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.77 (s, 1H), 7.40 (d, J=2,0 Гц, 1H), 7.30 (dd, J=8,4 Гц, 2,0 Гц, 1Н), 7.13 (d, J=8,4 Гц, 1H).
Пример 16: Получение (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида (Соединение 16)
Стадия 16-1: Получение 3-метил-6-нитробензо[d]оксазол-2(3H)-она
282 мг (выход: 87%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 6-нитробензо[d]оксазол-2(3H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1H ЯМР (400 МГц, Ацетон-d6) δ 6.87 (d, J=8,3 Гц, 1H), 6.64 (d, J=2,1 Гц, 1Н), 6.55 (dd, J=8,3 Гц, 2,1 Гц, 1H), 4.64 (s, 2Н), 3.32 (s, 3Н).
Стадия 16-2: Получение 6-амино-3-метилбензо[d]оксазол-2(3H)-она
111 мг (выход: 88%) указанного в заголовке соединения было получено таким же способом, как на Стадии 14-2 Примера 14 выше, за исключением того, что 3-метил-6-нитробензо[d]оксазол-2(3H)-он, полученный на Стадии 16-1, использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 6.95 (d, J=8,5 Гц, 1H), 6.87 (d, J =2,3 Гц, 1Н), 6.72 (dd, J=8,5 Гц, 2,3 Гц, 1Н), 4.62 (s, 2Н), 3.35 (s, 3Н).
Стадия 16-3: Получение (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида
107 мг (выход: 73%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-амино-3-метилбензо[d]оксазол-2(3H)-он, полученный на Стадии 16-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.43 (d, J=2,0 Гц, 1Н), 7.36 (dd, J=8,5 Гц, 2,0 Гц, 1H), 7.29 (d,J=8,5 Гц, 1H), 3.34 (s, 3Н).
Пример 17: Получение (1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 17)
Стадия 17-1: Получение N-метил-2,4-динитроанилина
1-Хлор-2,4-динитробензол (3 г, 14,81 ммоль) растворяли в THF в атмосфере азота, добавляли к нему 2,0 М метиламин (37,03 мл, 74,06 ммоль), растворенный в THF, и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество отверждали с использованием эфира и отфильтровывали с получением 2,81 г (выход: 96%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.99 (d, J=2,8 Гц, 1H), 8.82 (s, 1H), 8.35-8.32 (m, 1H), 7.23 (d, J=9,6 Гц, 1H), 3.24 (d, J=5,2 Гц, 3Н).
Стадия 17-2: Получение N'-метил-4-нитробензол-1,2-диамина
Затем N'-метил-2,4-динитроанилин (300 мг, 1,52 ммоль), полученный на Стадии 17-1, растворяли в метаноле, добавляли к нему сульфид натрия (356 мг, 4,57 ммоль) и гидрокарбонат натрия (384 мг, 4,57 ммоль), растворенный в дистиллированной воде, и реакционную смесь перемешивали в атмосфере азота при 80°С в течение 8 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество отверждали с использованием эфира и отфильтровывали с получением 193 мг (выход: 76%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.68 (dd, J=8,8 Гц, 2,8 Гц, 1H), 7.58 (d, J=2,8 Гц, 1H), 6.55 (d, J=8,8 Гц, 1H), 5.42 (s, 1H), 4.54 (s, 2Н), 2.96 (s, 3Н).
Стадия 17-3: Получение 1-метил-5-нитро-1,3-дигидро-2H-бензо[d]имидазол-2-она
Затем N'-метил-4-нитробензол-1,2-диамин (900 мг, 5,38 ммоль), полученный на Стадии 17-2, растворяли в DMF, добавляли к нему CDI (2,56 г, 16,15 ммоль) и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 15 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество отверждали с использованием эфира и отфильтровывали с получением 568 мг (выход: 55%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 10.27 (s, 1Н), 8.10 (dd, J=8,6 Гц, 2,2 Гц, 1Н), 7.93 (d, J=2,0 Гц, 1H), 7.29 (d, J=8,8 Гц, 1H), 3.47 (s, 3Н).
Стадия 17-4: Получение 5-амино-1-метил-1.3-Дигидро-2H-бензо[d]имидазол-2-она
306 мг (выход: 91%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-метил-5-нитро-1,3-дигидро-2H-бензо[d]имидазол-2-он (400 мг, 2,07 ммоль), полученный на Стадии 17-3, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо 1,4-диоксана.
1H ЯМР (400 МГц, Ацетон-d6) δ 9.37 (s, 1H), 6.72 (d, J=8,4 Гц, 1H), 6.44 (d, J=2,0 Гц, 1H), 6.39 (dd, J=8,4 Гц, 2,0 Гц, 1H), 4.31 (s, 2Н), 3.23 (s, 3Н).
Стадия 17-5: Получение (1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
62 мг (выход: 17%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 5-амино-1-метил-1,3-дигидро-2H-бензо[d]имидазол-2-он (250 мг, 1,53 ммоль), полученный на Стадии 17-4, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.01 (s, 1H), 10.99 (s, 1H), 7.19 (dd, J=8,4 Гц, 2,0 Гц, 1H), 7.12-7.10 (m, 2H), 3.27 (s, 3H).
Пример 18: Получение (3-изопропил-2-оксо-2,3-Дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 18)
Стадия 18-1: Получение 3-изопронил-6-нитробензо[d]тиазол-2(3H)-она
162 мг (выход: 44%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 6-нитробензо[d]тиазол-2(3H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она, и 2-йодпропан использовали вместо метилйодида.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.59 (d, J=2,4 Гц), 8.26 (dd, J=9,2 Гц, 2,4 Гц), 7.67 (d, J=9,2 Гц), 4.97-4.90 (m, 1Н), 1.61 (d, J=6,8 Гц, 6H).
Стадия 18-2: Получение 6-амино-3-изопропилбензо[d]тиазол-2(3H)-она
112 мг (выход: 85%) указанного в заголовке соединения было получено таким же способом, как на Стадии 14-2 Примера 14 выше, за исключением того, что 3-изопропил-6-нитробензо[d]тиазол-2(3H)-он, полученный на Стадии 18-1, использовали вместо 3-метил-6-нитробензо[d]тиазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) 5 7.13 (d, J=8,8 Гц, 1H), 6.87 (d, J=2,4 Гц, 1H), 6.71 (dd, J=8,8 Гц, 2,4 Гц, 1H), 4.76-4.69 (m, 1H), 4.64 (s, 2H), 1.53 (d, J=6,8 Гц, 6H).
Стадия 18-3: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида
109 мг (выход: 80%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-амино-3-изопропилбензо[d]тиазол-2(3H)-он, полученный на Стадии 18-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.79 (d, J=2,4 Гц, 1H), 7.55 (d, J=9,2 Гц, 1H), 7.49 (d,J=2,4 Гц, 1H), 4.81-4.74 (m, 1H), 1.50 (d,J=7,2 Гц, 6Н).
Пример 19: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида (Соединение 19)
Стадия 19-1: Получение 3-(дифторметил)-6-нитробензо[d]тиазол-2(3H)-она
После растворения 6-нитробензо[d]тиазол-2(3Н)-она (300 мг, 1,53 ммоль) в DMF в атмосфере азота к нему добавляли 2-хлор-2,2-дифторацетат натрия (467 мг, 3,06 ммоль) и DBU (0,46 мл, 3,06 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество отверждали с использованием метанола и отфильтровывали с получением 176 мг (выход: 47%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.76 (d, J=2,4 Гц, 1Н), 8.38 (dd, J=9,2 Гц, 2,4 Гц, 1H), 7.79 (d, J=8,8 Гц, 1H), 7.75 (t,J=57,2 Гц, 1H).
Стадия 19-2: Получение 6-амино-3-(дифторметил)бензо[d]тиазол-2(3H)-она
125 мг (выход: 95%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-(дифторметил)-6-нитробензо[d]тиазол-2(3H)-он, полученный на Стадии 19-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо 1,4-диоксана.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.55 (t, J=58,0 Гц, 1H), 7.23 (d, J=8,8 Гц, 1H), 6.93 (d,J=2,4 Гц, 1H), 6.76 (dd, J=8,6 Гц, 2,2 Гц, 1H), 4.89 (s, 2Н).
Стадия 19-3: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианида
85 мг (выход: 63%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-амино-3-(дифторметил)бензо[d]тиазол-2(3H)-он, полученный на Стадии 19-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.90 (s, 1H), 7.82 (t, J=56,8 Гц, 1H), 7.53 (s, 2Н).
Пример 20: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида (Соединение 20)
Стадия 20-1: Получение 3-изопропил-6-нитробензо[d]оксазол-2(3H)-она
201 мг (выход: 54%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 6-нитробензо[d]оксазол-2(3H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она, и 2-йодпропан использовали вместо метилйодида.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.23 (dd, J=8,8 Гц, 2,0 Гц, 1H), 8.16 (d, J=2,0 Гц, 1H), 7.61 (d, J=8,8 Гц, 1H), 4.72-1.65 (m, 1H), 1.61 (d, J=6,8 Гц, 6Н).
Стадия 20-2: Получение 6-амино-3-изопропилбензо[d]оксазол-2(3H)-она
124 мг (выход: 85%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-изопропил-6-нитробензо[d]тиазол-2(3H)-он, полученный на Стадии 20-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо 1,4-диоксана.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.00 (d, J=8,4 Гц, 1H), 6.61 (d, J=2,0 Гц, 1H), 6.51 (dd, J=8,4 Гц, 2,0 Гц, 1H), 4.62 (s, 2Н), 4.48-4.41 (m, 1H), 1.48 (d, J=6,8 Гц, 6Н).
Стадия 20-3: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида
99 мг (выход: 59%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-амино-3-изопропилбензо[d]оксазол-2(3H)-он, полученный на Стадии 20-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.47-7.43 (m, 2Н), 7.33 (dd, J=8,8 Гц, 2,0 Гц, 1H), 4.51-4.44 (m, 1H), 1.46 (d, J=6,8 Гц, 6Н).
Пример 21: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида (Соединение 21)
Стадия 21-1: Получение 3-(дифторметил)-6-нитробензо[d]оксазол-2(3H)-она
144 мг (выход: 38%) указанного в заголовке соединения было получено таким же способом, как на Стадии 19-1 Примера 19 выше, за исключением того, что 6-нитробензо[d]оксазол-2(3H)-он (300 мг, 1,67 ммоль) использовали вместо 6-нитробензо[d]тиазол-2(3Н)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.34-8.30 (m, 2Н), 7.68 (d, J=8,8 Гц, 1Н), 7.65 (t, J=57,6 Гц, 1H).
Стадия 21-2: Получение 6-амино-3-(дифторметил)бензо[d]оксазол-2(3H)-она
110 мг (выход: 98%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-(дифторметил)-6-нитробензо[d]оксазол-2(3H)-он, полученный на Стадии 21-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо 1,4-диоксана.
1H ЯМР (400 МГц, Ацетон-d6) δ 7.68 (t, J=58,4 Гц, 1H), 7.08 (d, J=8,4 Гц, 1H), 6.70 (d, J=2,4 Гц, 1H), 6.61 (dd, J=8,4 Гц, 2,0 Гц, 1H) 4.91 (s, 2Н).
Стадия 21-3: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианида
85 мг (выход: 63%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 6-амино-3-(дифторметил)бензо[d]оксазол-2(3H)-он, полученный на Стадии 21-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.73 (t, J=57,2 Гц, 1H), 7.52 (d, J=1,2 Гц, 1H), 7.45-7.40 (m, 2Н).
Пример 22: Получение (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 22)
Стадия 22-1: Получение 1,3-Диметил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
207 мг (выход: 64%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.09 (dd, J=8,6 Гц, 2,2 Гц), 7.98 (d, J=2,0 Гц), 7.30 (d, J=8,4 Гц), 3.51 (s, 3Н), 3.48 (s, 3Н).
Стадия 22-2: Получение 5-амино-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она
105 мг (выход: 82%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1,3-диметил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 22-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо 1,4-диоксана.
1Н ЯМР (400 МГц, Ацетон-d6) δ 6.76 (d, J=8,0 Гц, 1H), 6.43-6.40 (m, 2Н), 4.38 (s, 2Н), 3.27 (s, 3Н), 3.26 (s, 3Н).
Стадия 22-3: Получение (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
11 мг (выход: 8%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 5-амино-1,3-диметил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 22-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, Ацетон-d6) δ 7.18-7.14 (m, 2Н), 6.97 (d, J=8,0 Гц, 1H), 3.37 (s, 3Н), 3.35 (s, 3Н).
Пример 23: Получение (1,3-диизопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 23)
Стадия 23-1: Получение 1,3-диизопропил-5-нитро-1H-бензо[d]имидазол-2(3H)она
204 мг (выход: 46%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она, и 2-йодпропан использовали вместо метилйодида.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.05-8.02 (m, 2Н), 7.46 (d, J=8,8 Гц, 1H), 4.80-4.72 (m, 2Н), 1.58-1.54(m, 12Н).
Стадия 23-2: Получение 5-амино-1,3-диизопропил-1H-бензо[d]имидазол-2(3H)-она
118 мг (выход: 89%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1,3-диизопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 23-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо 1,4-диоксана.
1Н ЯМР (400 МГц, Ацетон-d6) δ 6.92 (d, J=8,0 Гц, 1H), 6.64 (d, J=2,0 Гц, 1H), 6.37 (dd, J=8,4 Гц, 2,4 Гц, 1Н), 4.62-4.54 (m, 2Н), 4.33 (s, 2Н), 1.46-1.43 (m, 12Н).
Стадия 23-3: Получение (1,3-Диизопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
12 мг (выход: 10%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 5-амино-1,3-диизопропил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 23-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.22-7.20 (m, 2Н), 7.04 (dd, J=8,8 Гц, 1.6 Гц, 1H), 4.61-4.55 (m, 2Н), 1.44-1.42 (m, 12Н).
Пример 24: Получение (1,3-бис(дифторметил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 24)
Стадия 24-1: Получение 1,3-бис(дифторметил)-5-нитро-1H- бензо[d]имидазол-2(3H)-она
179 мг (выход: 38%) указанного в заголовке соединения было получено таким же способом, как на Стадии 19-1 Примера 19 выше, за исключением того, что 5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 6-нитробензо[d]тиазол-2(3Н)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.33 (dd, J=8,8 Гц, 2,0 Гц, 1H), 8.22 (d, J=2,0 Гц, 1H), 7.83-7.53 (m, 2Н).
Стадия 24-2: Получение 5-амино-1,3-бис(дифторметил)-1Н-бензо[d]имидазол-2(3H)-она
69 мг (выход: 96%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того? что 1,3- бис(дифторметил)-6-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 24-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо 1,4-диоксана.
1H ЯМР (400 МГц, Ацетон-d6) δ 7.62-7.32 (m, 2Н), 7.13 (d, J=8,8 Гц, 1H), 6.82 (s, 1H), 6.63 (dd, J=8,6 Гц, 2,2 Гц, 1H), 4.90 (s, 2Н).
Стадия 24-3: Получение (1,3-бис(дифторметил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
45 мг (выход: 58%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-4 Примера 9 выше, за исключением того, что 5-амино-1,3-бис(дифторметил)-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 24-2, использовали вместо 5-амино-6-фтор-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она, и смешанный раствор этанола и воды в соотношении 1:3 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.93-7.61 (m, 2Н), 7.56 (s, 1H), 7.49-7.42 (m, 2Н).
Пример 25: Получение (1-метил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианида (Соединение 25)
Стадия 25-1: Получение 1-метил-6-нитрохинолин-2(17/)-она
123 мг (выход: 38%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 6-нитрохинолин-2(1H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.74 (d, J=2,7 Гц, 1Н), 8.40 (dd, J=9,4 Гц, 2,7 Гц, 1H), 8.15 (d, J=9,5 Гц, 1H), 7.73 (d, J=9,3 Гц, 1H), 6.80 (d, J=9,5 Гц, 1H), 3.67 (s, 3H).
Стадия 25-2: Получение 6-амино-1-метилхинолин-2(1H)-она
56 мг (выход: 59%) указанного в заголовке соединения было получено таким же способом, как на Стадии 13-2 Примера 13 выше, за исключением того, что 1-метил-6-нитрохинолин-2(1H)-он, полученный на Стадии 25-1, использовали вместо 6-нитробензо[d]тиазол-2(3H)-она, и THF и воду использовали в качестве растворителей.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.66 (d, J=9,4 Гц, 1H), 7.25 (d, J=8,9 Гц, 1Н), 6.94 (dd, J=8,9 Гц, 2,7 Гц, 1H), 6.78 (d, J=2,6 Гц, 1H), 6.48 (d, J=9,4 Гц, 1H), 5.09 (s, 2H), 3.53 (s, 3H).
Стадия 25-3: Получение (1-метил-2-оксо-1,2-дигидрохинолин-6- ил)карбоногидразоноилдицианида
55 мг (выход: 77%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-1-метилхинолин-2(1H)-он, полученный на Стадии 25-2, использовали вместо 7-аминоизохинолин- 1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.16 (s, 1H), 7.98 (d, J=9,5 Гц, 1H), 7.79 (d, J=2,5 Гц, 1H), 7.74 (dd, J=9,l Гц, 2,6 Гц, 1H), 7.59 (d, J=9,l Гц, 1H), 6.66 (d, J=9,5 Гц, 1H), 3.61 (s, 3H).
Пример 26: Получение (4-метил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианида (Соединение 26)
Стадия 26-1: Получение 4-метил-6-нитрохинолин-2(1H)-она
После растворения 4-метилхинолин-2(1H)-она (1 г, 6,28 ммоль) в серной кислоте (4 мл) к нему добавляли 65% смешанный раствор азотной кислоты (0,5 мл) и серной кислоты (0,5 мл) и реакционную смесь перемешивали при 0°С в течение 2 часов. После завершения взаимодействия к ней добавляли дистиллированную воду (30 мл) и реакционную смесь перемешивали при 0°С и фильтровали с получением 1,08 г (выход: 84%) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) δ 12.18 (s, 1H), 8.51 (d, J=2,6 Гц, 1H), 8.35 (dd, J=9,0 Гц, 2,5 Гц, 1H), 7.44 (d, J=9,0 Гц, 1H), 6.58 (s, 1H).
Стадия 26-2: Получение 6-амино-4-метилхинолин-2(1H)-она
47 мг (выход: 37%) указанного в заголовке соединения было получено таким же способом, как на Стадии 13-2 Примера 13 выше, за исключением того, что 4-метил-6-нитрохинолин-2(1H)-он, полученный на Стадии 26-1, использовали вместо 6-нитробензо[d]тиазол-2(3H)-она, и THF и воду использовали в качестве растворителей.
1H ЯМР (400 МГц, DMSO-d6) δ 11.22 (s, 1H), 7.04 (d, J=8,5 Гц, 1Н), 6.87-6.78 (m, 2H), 6.28 (s, 1H), 5.00 (s, 2H), 2.31 (s, 3Н).
Стадия 26-3: Получение (4-метил-2-оксо-1,2-Дигидрохинолин-6- ил)карбоногидразоноилдицианида
48 мг (выход: 83%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-4-метилхинолин-2(1H)-он, полученный на Стадии 26-2, использовали вместо 7-аминоизохинолин- 1(2H)-она.
1H ЯМР (400 МГц, DMSO-d6) δ 13.13 (s, 1Н), 11.73 (s, 1H), 7.78 (d, J=2,4 Гц, 1H), 7.66 (dd, J=8,9 Гц, 2,4 Гц, 1H), 7.34 (d, J=8,9 Гц, 1H), 6.47 (s, 1H), 2.41 (s, 3Н).
Пример 27: Получение (1,4-диметил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианида (Соединение 27)
Стадия 27-1: Получение 1,4-диметил-6-нитрохинолин-2(1H)-она
206 мг (выход: 38%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 4-метил-6-нитрохинолин-2(1H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1H ЯМР (400 МГц, DMSO-d6) δ 8.55 (d, J=2,6 Гц, 1Н), 8.42 (ddd, J=9,3 Гц, 2,6 Гц, 1,1 Гц, 1H), 7.73 (dd, J=9,4 Гц, 1,1 Гц, 1H), 6.72 (s, 1H), 3.65 (s, 3H), 2.53 (s, 3Н).
Стадия 27-2: Получение 6-амино-1,4-диметилхинолин-2(1H)-она
33 мг (выход: 19%) указанного в заголовке соединения было получено таким же способом, как на Стадии 13-2 Примера 13 выше, за исключением того, что 1,4-диметил-6-нитрохинолин-2(1H)-он, полученный на Стадии 27-1, использовали вместо 6-нитробензо[d]тиазол-2(3H)-она, и THF и воду использовали в качестве растворителей.
1H ЯМР (400 МГц, DMSO-d6) δ 7.25 (d, J=8,9 Гц, 1Н), 6.94 (dd, J=8,9 Гц, 2,5 Гц, 1H), 6.89 (d, J=2,5 Гц, 1H), 6.42 (s, 1H), 5.10 (s, 2H), 3.52 (s, 3Н), 2.32 (s, 3Н).
Стадия 27-3: Получение (1,4-Диметил-2-оксо-1,2-Дигидрохинолин-6- ил)карбоногидразоноилдицианида
55 мг (выход: 77%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-1,4-диметилхинолин-2(1H)-он, полученный на Стадии 27-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.14 (s, 1H), 7.82 (d,J=2,5 Гц, 1H), 7.73 (dd, J=9,2 Гц, 2,5 Гц, 1Н), 7.58 (d, J=9,2 Гц, 1H), 6.59 (s, 1Н), 3.59 (s, 3Н), 2.42 (s, 3Н).
Пример 28: Получение (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианида (Соединение 28)
Стадия 28-1: Получение 3-метил-5-нитробензо[d]оксазол-2(3H)-она
226 мг (выход: 70%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 5-нитробензо[d]диоксол-2(3H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.21 (d, J=2,3 Гц, 1Н), 8.10 (dd, J=8,8 Гц, 2,4 Гц, 1H), 7.58 (d, J=8,8 Гц, 1H), 3.43 (s, 3Н).
Стадия 28-2: Получение 5-амино-3-метилбензо[d]оксазол-2(3H)-она
150 мг (выход: 88%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-метил-5-нитробензо[d]оксазол-2(3H)-он, полученный на Стадии 28-1, использовали вместо 7-нитроизохинолин-1 (2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 6.95 (d, J=8,5 Гц, 1H), 6.37 (d, J=2,2 Гц, 1H), 6.28 (dd, J=8,5 Гц, 2,2 Гц, 1H), 5.07 (s, 2H), 3.23 (s, 3H).
Стадия 28-3: Получение (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианида
73 мг (выход: 38%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-3-метилбензо[d]оксазол-2(3H)-он, полученный на Стадии 28-2, использовали вместо 7-аминоизохинолин- 1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.11 (s, 1H), 7.36 (d, J=8,6 Гц, 1H), 7.32 (d, J=2,1 Гц, 1H), 7.24 (dd, J=8,6 Гц, 2,2 Гц, 1H).
Пример 29: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианида (Соединение 29)
Стадия 29-1: Получение 3-изопропил-5-нитробензо[d]оксазол-2(3H)-она
94 мг (выход: 25%) указанного в заголовке соединения было получено таким же способом, как на Стадии 28-1 Примера 28 выше, за исключением того, что 2-йодпропан использовали вместо метилйодида.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.22 (d, J=2,3 Гц, 1H), 8.10 (dd, J=8,8 Гц, 2,3 Гц, 1H), 7.59 (d, J=8,8 Гц, 1H), 4.62 (hept, J=6,9 Гц, 1H), 1.48 (d, J=6,9 Гц, 6H).
Стадия 29-2: Получение 5-амино-3-изопропилбензо[d]оксазол-2(3H)-она
48 мг (выход: 61%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-изопропил-5-нитробензо[d]оксазол-2(3H)-он, полученный на Стадии 29-1, использовали вместо 7-нитроизохинолин-1 (2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 6.96 (d, J=8,4 Гц, 1H), 6.57 (d, J=2,2 Гц, 1H), 6.28 (dd, J=8,4 Гц, 2,2 Гц, 1H), 5.01 (s, 2H), 4.37 (hept, J=6,9 Гц, 1H), 1.42 (d, J=7,l Гц, 6H).
Стадия 29-3: Получение (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианида
9 мг (выход: 15%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-3-изопропилбензо[d]оксазол-2(3H)-он, полученный на Стадии 29-2, использовали вместо 7-аминоизохинолин-1 (2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.00 (s, 1H), 7.46 (d, J=2,1 Гц, 1H, 7.38 (d, J=8,7 Гц, 1H), 7.24 (dd, J=8,7 Гц, 2,2 Гц, 1H), 4.51 (hept, J=6,9 Гц, 1H), 1.46 (d, J=6,9 Гц, 6H).
Пример 30: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианида (Соединение 30)
Стадия 30-1: Получение 3-(дифторметил)-5-нитробензо[d]оксазол-2(3H)-она
104 мг (выход: 27%) указанного в заголовке соединения было получено таким же способом, как на Стадии 28-1 Примера 28 выше, за исключением того, что 2-хлор-2,2-дифторацетат натрия использовали вместо метилйодида.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.05 (s, 1H), 8.71 (s, 1H), 8.38 (dd, J=9,0 Гц, 2,3 Гц, 1H), 8.06 (d, J=9,0 Гц, 1H).
Стадия 30-2: Получение 5-амино-3-(дифторметил)бензо[d]оксазол-2(3H)-она
Указанное в заголовке соединение получали таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-(дифторметил)-5-нитробензо[d]оксазол-2(3H)-он, полученный на Стадии 30-1, использовали вместо 7-нитроизохинолин-1(2H)-она и использовали на следующей стадии без дополнительной очистки.
Стадия 30-3: Получение (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианида
30 мг (выход: 22%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-3-(дифторметил)бензо[d]оксазол-2(3H)-он, полученный на Стадии 30-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.13 (s, 1H), 8.80 (s, 1H), 7.85-7.81 (m, 2Н), 7.59 (dd, J=9,0 Гц, 2,1 Гц, 1H).
Пример 31: Получение (1,31б-триметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 31)
Стадия 31-1: Получение 5-метил-6-нитро-1H-бензо[d]имидазол-2(3H)-она
67 мг (выход: 29%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что 4-метил-5-нитробензол-1,2-диамин использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.25 (s, 1H), 11.00(s, 1H), 7.59 (s, 1H), 6.96 (s, 1H), 2.55 (s, 3H).
Стадия 31-2: Получение 13,5-триметил-6-нитро-1H-бензо[d]имидазол-2(3H)она
268 мг (выход: 78%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 5-метил-6-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 31-1, использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.91 (s, 1H), 7.26 (s, 1H), 3.37 (d, J=5,5 Гц, 6Н), 2.60 (s, 3Н).
Стадия 31-3: Получение 5-амино-1,3,6-триметил-1H-бензо[d]имидазол-2(3H)-она
169 мг (выход: 78%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1,3,5-триметил-6-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 31-2, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 6.73 (s, 1Н), 6.41 (s, 1H), 4.55 (s, 2Н), 3.20 (d, J=5,0 Гц, 6Н), 2.09 (s, 3Н).
Стадия 31-4: Получение (1,3,6-триметил-2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
20 мг (выход: 10%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-1,3,6-триметил-6-нитро-1H-бензо[d]имидазол-он, полученный на Стадии 31-3, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 12.26 (s, 1H), 7.14 (s, 1H), 7.05 (s, 3H), 3.31 (d, J=2,7 Гц, 6Н), 2.38 (s, 3Н).
Пример 32: Получение (6-метокси-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 32)
Стадия 32-1: Получение 5-метокси-6-нитро-1H-бензо[d]имидазол-2(3H)-она
336 мг (выход: 98%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что 4-метокси-5-нитробензол-1,2-диамин использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.26 (s, 1H), 10.86 (s, 1H), 7.48 (s, 1H), 6.81 (s, 1H), 3.90 (s, 3H).
Стадия 32-2: Получение 5-метокси-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-она
224 мг (выход: 66%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 5-метокси-6-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 32-1, использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.81 (s, 1H), 7.19 (s, 1H), 3.95 (s, 3H), 3.39 (s, 3H), 3.34 (s, 3H).
Стадия 32-3: Получение 5-амино-6-метокси-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она
78 мг (выход: 44%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 5-метокси-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 32-2, использовали вместо 7-нитроизохинолин-1(2H)-она.
1H ЯМР (400 МГц, DMSO-d6) δ 6.78 (s, 1Н), 6.47 (s, 1Н), 4.45 (s, 2Н), 3.78 (s, 3Н), 3.25 (s, 3Н), 3.20 (s, 3Н).
Стадия 32-4: Получение (6-метокси-1,3-диметил-2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
37 мг (выход: 54%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-6-метокси-1,3-диметил-6-нитро-1H-бензо[d]имидазол-он, полученный на Стадии 32-3, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.75 (s, 1H), 7.21 (s, 1Н), 7.12 (s, 1H), 3.94 (s, 3Н), 3.34 (s, 3Н), 3.31 (s, 3Н).
Пример 33: Получение (7-метокси-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 33)
Стадия 33-1: Получение 4-метокси-6-нитро-1H-бензо[d]имидазол-2(3H)-она
509 мг (выход: 89%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что 3-метокси-5-нитробензол-1,2-диамин использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.57 (s, 1H), 11.15 (s, 1H), 7.57 (d, J=2,0 Гц, 1H), 7.48 (d, J=2,0 Гц, 1H), 3.96 (s, 3H).
Стадия 33-2: Получение 4-метокси-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-она
119 мг (выход: 70%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 4-метокси-6-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 33-1, использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.83 (d, J=2,0 Гц, 1H), 7.64 (d, J=2,0 Гц, 1H), 3.98 (s, 3Н), 3.55 (s, 3Н), 3.40 (s, 3Н).
Стадия 33-3: Получение 6-амино-4-метокси-1,3-диметил-1H-бензо[d]имидазол-2(3H)-она
15 мг (выход: 16%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 4-метокси-1,3-диметил-6-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 33-2, использовали вместо 7-нитроизохинолин-1(2H)-она.
1H ЯМР (400 МГц, DMSO-d6) δ 6.04 (d, J=1,7 Гц, 1H), 5.97 (d, J=1,7 Гц, 1Н), 4.85 (s, 2Н), 3.76 (s, 3Н), 3.38 (s, 3Н), 3.17 (s, 2Н).
Стадия 33-4: Получение (7-метокси-1,3-диметил-2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
20 мг (выход: 30%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-4-метокси-1,3-диметил-1H-бензо[d]имидазол-он, полученный на Стадии 33-3, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.00 (s, 1H), 6.96 (d, J=1,8 Гц, 1Н), 6.91 (d, J=1,8 Гц, 1H), 3.87 (s, 3Н), 3.48 (s, 3Н), 3.30 (s, 3Н).
Пример 34: Получение (1,3-Диизопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 34)
Стадия 34-1: Получение 1,3-диизопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
387 мг (выход: 30%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 6-нитро-1H- имидазо[4,5-6]пиридин-2(3H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она, и 2-йодпропан использовали вместо метилйодида.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.94 (d, J=2,3 Гц, 1H), 8.32 (d, J=2,3 Гц, 1Н), 4.72 (dhept, J=16,6 Гц, 6,9 Гц, 2Н), 1.50 (dd, J=16,1 Гц, 6,9 Гц, 12Н).
Стадия 34-2: Получение 6-амино-13-Диизопропил-1H-имидазо[4,5-b] пиридин-2(3H)-она
162 мг (выход: 42%) указанного в заголовке соединения было получено таким же способом, как на Стадии 28-2 Примера 28 выше, за исключением того, что 1,3-диизопропил-6-нитро-1H-имидазо[4,5-6]пиридин-2(3H)-он, полученный на Стадии 34-1, использовали вместо 1-метил-6-нитрохинолин-2(1H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.37 (d, J=2,2 Гц, 1H), 6.92 (d, J=2,2 Гц, 1Н), 4.84 (s, 2Н), 4.61-4.48 (m, J=7,0 Гц, 2Н), 1.41 (dd, J=19,6 Гц, 6,9 Гц, 12Н).
Стадия 34-3: Получение 6-амино-1,3-Диизопропил-1H-имидазо[4,5-Ь] пиридин-2(3H)-она
43 мг (выход: 27%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-1,3-диизопропил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 34-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.13 (d, J=2,2 Гц, 1H), 7.64 (d, J=2,2 Гц, 1H), 4.64 (dp, J=l 1,5 Гц, 6,9 Гц, 2H), 1.46 (dd, J=19,8 Гц, 6,9 Гц, 12Н).
Пример 35: Получение (3-этил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 35)
Стадия 35-1: Получение 3-этил-1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
1-Метил-5-нитро-1H-бензо[d]имидазол-2(3H)-он (150 мг, 0,78 ммоль) растворяли в DMF (3 мл) и добавляли к нему карбонат калия (214,7 мг, 1,55 ммоль) и затем реакционную смесь охлаждали до 0°С. К ней медленно добавляли этилйодид (0,156 мл, 1,94 ммоль) и реакционную смесь медленно нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 17 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество отверждали с использованием эфира и отфильтровывали с получением 133,5 мг (выход: 77%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 8.11 (dd, J=8,7 Гц, 2,2 Гц, 1H), 7.90 (d, J=2,1 Гц, 1Н), 7.02 (d, J=8,7 Гц, 1H), 3.49 (s, 3Н).
Стадия 35-2: Получение 5-амино-3-этил-1-метил-1H-бензо[d]имидазол-2(3H)-она
74,7 мг (выход: 68%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-этил-1-метил-5-нитро-1H-бензо[d]имидазол-2(37/)-он, полученный на Стадии 35-1, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 6.79 (d, J=8,3 Гц, 1H), 6.40 (d, J=2,1 Гц, 1Н), 6.31 (dd, J=8,2 Гц, 2,1 Гц, 1H), 4.76 (s, 2Н), 3.74 (q, J=7,2 Гц, 2Н), 3.21 (s, 3Н), 1.16 (t, J=7,2 Гц, 3Н).
Стадия 35-3: Получение (3-этил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
60,9 мг (выход: 58%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-3-этил-1-метил-1H-бензо[димидазол-2(3H)-он, полученный на Стадии 35-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1H ЯМР (400 МГц, DMSO-d6) δ 13.04 (s, 1H), 7.30 (d,J=2,0 Гц, 1H), 7.23 (dd, J=8,4 Гц, 2,0 Гц, 1H), 7.18 (d, J=8,4 Гц, 1H), 3.88 (q, J=7,2 Гц, 2Н), 3.32 (s, 3Н), 1.20 (t, J=7,2 Гц, 3Н).
Пример 36: Получение (3-изопропил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 36)
Стадия 36-1: Получение 3-изопропил-1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
135,9 мг (выход: 55%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-2 Примера 9 выше, за исключением того, что 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-он использовали вместо 5-фтор-6-нитро-1H-бензо[d]имидазол-2(3H)-она, и изопропилйодид использовали вместо метилйодида.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 8.10 (dd, J=8,7 Гц, 2,1 Гц, 1H), 8.01 (d, J=2,2 Гц, 1H), 7.01 (d, J=8,7 Гц, 1Н), 4.75 (hept, J=7,0 Гц, 1H), 3.47 (s, 3Н), 1.58 (d, J=7,1 Гц, 7Н).
Стадия 36-2: Получение 5-амино-3-изопропил-1-метил-1H-бензо[d]имидазол-2(3H)-она
114,2 мг (выход: 100%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-изопропил-1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 36-1, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 6.78 (d, J=8,2 Гц, 1H), 6.56 (d,J=2,0 Гц, 1Н), 6.30 (dd, J=8,3 Гц, 2,0 Гц, 1H), 4.73 (s, 2Н), 4.49 (hept, J=7,0 Гц, 1H), 3.19 (s, 3Н), 1.39 (d, J=7,0 Гц, 6Н).
Стадия 36-3: Получение (3-изопропил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
15,2 мг (выход: 9%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-3-изопропил-1-метил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 36-2, использовали вместо 7-аминоизохинолин-1(2d)-она.
1H ЯМР (400 МГц, DMSO-d6) δ 12.96 (s, 1H), 7.42 (d, J=1,9 Гц, 1H), 7.22 (dd, J=8,5 Гц, 1,9 Гц, 1H), 7.17 (d, J=8,5 Гц, 1H), 4.61 (hept, J=7,0 Гц, 1Н), 3.30 (s, 3Н), 1.44 (d, J=6,9 Гц, 6Н).
Пример 37: Получение (3-(дифторметил)-1-метил-2-оксо-2,3-дигидро-1H- бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 37)
Стадия 37-1: Получение 3-дифторметил-1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
После растворения 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она (100 мг, 0,52 ммоль) в DMF в атмосфере азота к нему добавляли 2-хлор-2,2-дифторацетат натрия (157,9 мг, 1,04 ммоль) и карбонат калия (143,1 мг, 1,04 ммоль) и реакционную смесь нагревали до 80°С и перемешивали в течение 5 часов. После завершения взаимодействия к ней добавляли воду и полученный осадок отфильтровывали и очищали колоночной хроматографией с получением 58,5 мг (выход: 46%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.23 (dd, J=8,8 Гц, 2,2 Гц, 1H), 8.05 (d, J=2,2 Гц, 1H), 7.81 (t, J=57,5 Гц, 1H), 7.52 (d, J=8,8 Гц, Ш), 3.41 (s, 3Н).
Стадия 37-2: Получение 5-амино-3-дифторметил-1-метил-1H-бензо[d]имидазол-2(3H)-она
56.1 мг (выход: 72%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-дифторметил-1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 37-1, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.56 (t, J=58,3 Гц, 1H), 6.92 (d, J=8,4 Гц, 1H), 6.65 (d, J=2,0 Гц, 1H), 6.45 (dd, J=8,4 Гц, 2,1 Гц, 1H), 5.03 (s, 2H), 3.24 (s, 3Н).
Стадия 37-3: Получение (3-дифторметил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
53.2 мг (выход: 71%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-3-дифторметил-1-метил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 37-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 11.86 (s, 1H), 7.56 (d, J=2,l Гц, 1H), 7.52 (t, J=58,4 Гц, 1H), 7.45 (dd, J=8,6 Гц, 2,1 Гц, 1H), 7.30 (d, J=8,6 Гц, 1H), 3.42 (s, 3Н).
Пример 38: Получение (1-этил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 38)
Стадия 38-1: Получение N-этил-2,4-Динитроанилина
1,95 г (выход: 93%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-1 Примера 17 выше, за исключением того, что этиламин использовали вместо метиламина.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.13 (d, J=2,7 Гц, 1Н), 8.49 (s, 1H), 8.27 (ddd, J=9,5 Гц, 2,7 Гц, 0,8 Гц, 1H), 6.92 (d, J=9,5 Гц, 1H), 3.48 (qd, J=7,2 Гц, 5,2 Гц, 2Н), 1.43 (t, J=7,2 Гц, 3Н).
Стадия 38-2: Получение N1-этил-4-нитробензол-1,2-диамина
1,46 г (выход: 88%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-2 Примера 17 выше, за исключением того, что N-этил-2,4-динитроанилин, полученный на Стадии 38-1, использовали вместо N-метил-2,4-динитроанилина.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 7.83 (dd, J=8,9 Гц, 2,5 Гц, 1H), 7.62 (d, J=2,5 Гц, 1H), 6.54 (d, J=8,9 Гц, 1H), 4.18 (s, 1H), 3.35 (s, 2Н), 3.31-3.17 (m, 2Н), 1.34 (t, J=7,2 Гц, 3Н).
Стадия 38-3: Получение 1-этил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
1,57 г (выход: 95%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что N-этил-4-нитробензол-1,2-диамин, полученный на Стадии 38-2, использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.42 (s, 1H), 8.01 (dd, J=8,7 Гц, 2,3 Гц, 1H), 7.75 (d, J=2,3 Гц, 1H), 7.36 (d, J=8,7 Гц, 1H), 3.90 (q, J=7,2 Гц, 2Н), 1.21 (t, J=7,2 Гц, 3Н).
Стадия 38-4: Получение 1-этил-3-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
172,0 мг (выход: 80%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что 1-этил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 38-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она, и метилйодид использовали вместо этилйодида.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 8.11 (dd, J=8,7 Гц, 2,2 Гц, 1H), 7.89 (d, J=2,1 Гц, 1Н), 7.04 (d,J=8,7 Гц, 1H), 4.00 (q, J=7,3 Гц, 2Н), 3.49 (s, 3Н), 1.37 (t, J=7,2 Гц, 3Н).
Стадия 38-5: Получение 5-амино-1-этил-3-метил-H-бензо[d]имидазол-2(3H)-она
117,6 мг (выход: 80%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-этил-3-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 38-4, использовали вместо 7-нитроизохинолин-1 (2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 6.82 (d, J=8,2 Гц, 1H), 6.35 (d, J=2,1 Гц, 1H), 6.30 (dd, J=8,2 Гц, 2,1 Гц, 1H), 4.78 (s, 2Н), 3.75 (q, J=7,1 Гц, 2Н), 3.20 (s, 3Н), 1.15 (t, J=7,1 Гц, 3Н).
Стадия 38-6: Получение (1-этил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
10,8 мг (выход: 6%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-1-этил-3-метил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 38-5, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.07 (s, 1H), 7.26-7.22 (m, 3Н), 3.86 (q, J=7,2 Гц, 2Н), 1.20 (t, J=7,1 Гц, 3Н).
Пример 39: Получение (3-(дифторметил)-1-этил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 39)
Стадия 39-1: Получение 3-дифторметил-1-этил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
133,0 мг (выход: 42%) указанного в заголовке соединения было получено таким же способом, как на Стадии 37-1 Примера 37 выше, за исключением того, что 1-этил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 38-3 Примера 38, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) 5 8.22 (dd, J=8,8 Гц, 2,2 Гц, 1H), 8.06 (d, J=2,2 Гц, 1H), 7.81 (t, J=57,5 Гц, 1H), 7.60 (d, J=8,9 Гц, 1H), 3.97 (q, J=7,2 Гц, 2Н), 1.26 (t, J=7,2 Гц, 3Н).
Стадия 39-2: Получение 5-амино-3-дифторметил-1-этил-1Н-бензо[d]имидазол-2(3H)-она
88,2 мг (выход: 75%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-дифторметил-1-этил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 39-1, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.56 (t, J=58,3 Гц, 1H), 6.97 (d, J=8,4 Гц, 1H), 6.65 (d, J=2,0 Гц, 1H), 6.44 (dd, J=8,4 Гц, 2,1 Гц, 1H), 5.03 (s, 2Н), 3.77 (q, J=7,2 Гц, 2H), 1.19 (t, J=7,2 Гц, 3Н).
Стадия 39-3: Получение (3-дифторметил-1-этил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
64,7 мг (выход: 56%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-3-дифторметил-1-этил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 39-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 11.85 (s, 1H), 7.57 (d, J=2,l Гц, Ш), 7.52 (t, J=58,4 Гц, 1Н), 7.44 (dd, J=8,6 Гц, 2,1 Гц, 1H), 7.37 (d, J=8,4 Гц, 1Н), 3.97 (q, J=7,3 Гц, 2Н), 1.32 (t, J=7,2 Гц, 3Н).
Пример 40: Получение (1-циклопропил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 40)
Стадия 40-1: Получение N-циклопропил-2,4-динитроанилина 179,9 мг (выход: 81%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-1 Примера 17 выше, за исключением того, что циклопропиламин использовали вместо метиламина.
1H ЯМР (400 МГц, Хлороформ-d6) δ 9.13 (d, J=2,6 Гц, 1Н), 8.56 (s, 1H), 8.31 (ddd, J=9,5 Гц, 2,6 Гц, 0,7 Гц, 1H), 7.40 (d, J=9,5 Гц, Ш), 2.70 (tdt, J=6,8 Гц, 3,8 Гц, 1,8 Гц, 1H), 1.12-0.98 (m, 2Н), 0.82-0.67 (m, 2Н).
Стадия 40-2: Получение N'-циклопропил-4-нитробензол-1,2-диамина
1,69 г (выход: 89%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-2 Примера 17 выше, за исключением того, что N-циклопропил-2,4-динитроанилин, полученный на Стадии 40-1 выше, использовали вместо N-метил-2,4-динитроанилина.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 7.85 (ddd, J=8,9 Гц, 2,5 Гц, 0.6 Гц, 1Н), 7.61 (d, J=2,5 Гц, 1H), 6.98 (d, J=8,9 Гц, 1H), 4.66 (s, 1H), 3.29 (s, 2Н), 2.53 (ttd, J=6,7 Гц, 3,6 Гц, 1,3 Гц, 1H), 0.93-0.82 (m, 2Н), 0.63-0.55 (т, 2Н).
Стадия 40-3: Получение 1-циклопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
1,83 г (выход: 96%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что N-циклопропил-4-нитробензол-1,2-диамин, полученный на Стадии 40-2, использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.30 (s, 1Н), 8.01 (dd, J=8,7 Гц, 2,3 Гц, 1H), 7.71 (d, J=2,3 Гц, 1H), 7.32 (d, J=8,7 Гц, 1H), 2.93 (tt, J=7,0 Гц, 3,7 Гц), 1.09-1.01 (m, 2H), 0.93-0.85 (m, 2H).
Стадия 40-4: Получение 1-циклопропил-3 -метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
186,6 мг (выход: 87%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 1-циклопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 40-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она, и метилйодид использовали вместо этилйодида.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 8.10 (ddd, J=8,6 Гц, 2,3 Гц, 1,0 Гц, 1H), 7.85 (d, J=2,0 Гц, 1H), 7.23 (s, 1H), 3.45 (d, J=1,0 Гц, 3Н), 2.93 (tdd, J=7,1 Гц, 4,1 Гц, 3,1 Гц, 1H), 1.22 1.11 (m, 2Н), 1.07 0.98 (m, 2Н).
Стадия 40-5: Получение 5-амино-1-циклопропил-3-метил-H-бензо[d]имидазол-2(3H)-она
41,6 мг (выход: 75%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-циклопропил-3-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 40-4, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1H ЯМР (400 МГц, Хлороформ-d6) δ 6.96 (d, J=8,2 Гц, 1H), 6.44 (dd, J=8,2 Гц, 2,1 Гц, 1H), 6.34 (d, J=2,1 Гц, 1H), 3.58 (s, 2Н), 3.31 (s, 3Н), 2.90-2.69 (m, 1H), 1.08-1.01 (m, 2Н), 1.01-0.94 (m, 2Н).
Стадия 40-6: Получение (1-циклопропил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
29,9 мг (выход: 52%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-1-циклопропил-3-метил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 40-5, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.86 (s, 1Н), 7.18 (d, J=8,3 Гц, 1Н), 7.05 (d, J=2,1 Гц, 1H), 6.95 (dd, J=8,4 Гц, 2,1 Гц, 1H), 3.41 (s, 3Н), 2.89 (tt, J=7,0 Гц, 3,7 Гц, 1H), 1.17-1.09 (m, 2Н), 1.01 (tt, J=5,3 Гц, 3,7 Гц, 2Н).
Пример 41: Получение (1-циклопропил-3-этил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 41)
Стадия 41-1: Получение 1-циклопропил-3-этил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
159,9 мг (выход: 70%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 1-циклопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 40-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.09 (d, J=2,2 Гц, 1Н), 8.07 (dd, J=8,6 Гц, 2,3 Гц, 1H), 7.38 (d, J=8,6 Гц, 1H), 3.94 (q, J=7,1 Гц, 2H), 2.98 (tt, J=7,l Гц, 3,7 Гц, 1H), 1.21 (t, J=7,2 Гц, 3Н), 1.12-1.03 (m, 2H), 0.95-0.86 (m, 2H).
Стадия 41-2: Получение 5-амино-1-циклопропил-3-этил-1H-бензо[d]имидазол-2(3H)-она
83,2 мг (выход: 61%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-циклопропил-3-этил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 41-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 6.96 (d, J=8,2 Гц, 1H), 6.43 (dd, J=8,2 Гц, 2,2 Гц, 1Н), 6.37 (d, J=2,1 Гц, 1H), 3.83 (q, J=7,2 Гц, 2Н), 3.58 (brs, 2Н), 2.90-2.72 (m, 1H), 1.28 (t, J=7,2 Гц, 3Н), 1.08-0.94 (m, 4Н).
Стадия 41-3: Получение (1-циклопропил-3-этил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
53,6 мг (выход: 49%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-1-циклопропил-3-этил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 41-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.04 (s, 1Р), 7.52-7.06 (m, 3Н), 3.83 (q, J=7,2 Гц, 2Н), 2.89 (tt, J=7,0 Гц, 3.7 Гц, 1H), 1.18 (t, 7=7,2 Гц, 3Н), 1.02 (td, J=7,3 Гц, 5,0 Гц, 2Н), 0.92-0.82 (m, 2Н).
Пример 42: Получение (1-циклопропил-3-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 42)
Стадия 42-1: Получение 1-ииклопропил-3-изопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
207,5 мг (выход: 69%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 1-циклопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 40-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она, и изопропилйодид использовали вместо этилйодида.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.17 (d, J=2,3 Гц, 1H), 8.03 (dd, J=8,8 Гц, 2,2 Гц, 1Н), 7.51 (d,J=8,8 Гц, 1H), 5.27 (hept, J=6,2 Гц, 1H), 3.18 (tt, J=7,1 Гц, 3,7 Гц, 1H), 1.43 (d, J=6,2 Гц, 6Н), 1.13 (td, J=7,4 Гц, 5,2 Гц, 2Н), 0.98-0.90 (m, 2Н).
Стадия 42-2: Получение 5-амино-1-циклопропил-3-изопропил-H-бензо[d]имидазол-2(3H)-она
114,5 мг (выход: 62%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-циклопропил-3-изопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 42-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1H ЯМР (400 МГц, Хлороформ-d6) δ 6.97 (d, J=8,2 Гц, 1Н), 6.52 (d, J=2,1 Гц, 1H), 6.43 (dd, J=8,2 Гц, 2,1 Гц, 1H), 4.65 (hept, J=7,0 Гц, 1H), 3.56 (s, 2Н), 2.84-2.71 (m, 1H), 1.48 (d, J=7,0 Гц, 6Н), 1.07-0.94 (m, 4Н).
Стадия 42-3: Получение (1-циклопропил-3-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
39,8 мг (выход: 26%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-1-циклопропил-3-изопропил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 42-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 12.96 (s, 1H), 7.39 (d, J=1,3 Гц, 1H), 7.22 (d, J=l,2 Гц, 2H), 4.58 (hept, J=6,9 Гц, 1H), 2.87 (tt,J=7,0 Гц, 3.6 Гц, 1H), 1.42 (d, J=6,9 Гц, 6H), 1.01 (td, J=7,2 Гц, 5,0 Гц, 2H), 0.90-0.82 (m, 2Н).
Пример 43: Получение (3-(дифторметил)-1-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 43)
Стадия 43-1: Получение N-изопропил-2,4-Динитроанилина
2,02 г (выход: 90%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-1 Примера 17 выше, за исключением того, что изопропиламин использовали вместо метиламина.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.14 (d, J=2,7 Гц, 1H), 8.50 (s, 1H), 8.26 (ddd, J=9,6 Гц, 2,7 Гц, 0,8 Гц, 1H), 6.93 (d, J=9,6 Гц, 1H), 4.00-3.89 (m, 1H), 1.40 (d, J=6,4 Гц, 6Н).
Стадия 43-2: Получение N1-изопропил-4-нитробензол-1,2-Диамина
1,08 г (выход: 62%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-2 Примера 17 выше, за исключением того, что N-изопропил-2,4-динитроанилин, полученный на Стадии 43-1, использовали вместо N-метил-2,4-динитроанилина, и смешанный раствор метанола и воды в соотношении 1:1 использовали в качестве растворителя.
1Н ЯМР (400 МГц, Хлороформ- d6) δ 7.84 (dd, J=8,9 Гц, 2,6 Гц, 1H), 7.63 (d, J=2,6 Гц, 1H), 6.55 (d, J=9,0 Гц, 1Н), 4.20 (s, 1Н), 3.82-3.63 (m, J=6,3 Гц, 1H), 3.29 (s, 2Н), 1.29 (d, J=6,3 Гц, 6Н).
Стадия 43-3: Получение 1-изопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
1,15 г (выход: 94%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что N-изопропил-4-нитробензол-1,2-диамин, полученный на Стадии 43-2, использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.42 (s, 1Н), 7.96 (dd, J=8,8 Гц, 2,4 Гц, 1H), 7.74 (d, j=2,3 Гц, 1H), 7.47 (d, J=8,8 Гц, 1H), 4.63 (hept, J=6,9 Гц, 1H), 1.46 (d, J=7,0 Гц, 6Н).
Стадия 43-4: Получение 3-дифторметил-1-изопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-она
89,3 мг (выход: 29%) указанного в заголовке соединения было получено таким же способом, как на Стадии 37-1 Примера 37 выше, за исключением того, что 1-изопропил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 43-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.17 (dd, J=8,9 Гц, 2,3 Гц, 1H), 8.05 (d, J=2,3 Гц, 1Н), 7.79 (t, J=57,6 Гц, 1H), 7.69 (d, J=8,9 Гц, 1H), 4.68 (hept, J=6,9 Гц, 1Н), 1.49 (d, J=6,9 Гц, 6Н).
Стадия 43-5: Получение 5-амино-3-дифторметил-1-этил-H-бензо[d]имидазол-2(3H)-она
67,2 мг (выход: 84%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-дифторметил-1-этил-5-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 43-4, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.53 (t, J=58,4 Гц, 1H), 7.07 (d, J=8,5 Гц, 1Н), 6.65 (d, J=2,1 Гц, 1H), 6.42 (dd, J=8,5 Гц, 2,1 Гц, 1H), 5.03 (s, 2Н), 4.49 (hept, J=6,9 Гц, 1H), 1.41 (d, J=6,9 Гц, 6Н).
Стадия 43-6: Получение (1-этил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
53,6 мг (выход: 60%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-3-дифторметил-1-этил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 43-5, использовали вместо 7-аминоизохинолин-1(2H)-она, и воду использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.10 (s, 1Н), 7.69 (t, J=57,8 Гц, 1H), 7.53-7.44 (m, 2H), 7.35 (dd, J=8,8 Гц, 2,1 Гц, 1H), 4.59 (р, J=6,9 Гц, 1Н), 1.46 (d, J=7,0 Гц, 6Н).
Пример 44: Получение (1-этил-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 44)
Стадия 44-1: Получение N-метил-3.5-динитропиридин 2 амина
1,62 г (выход: 83%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-1 Примера 17 выше, за исключением того, что 2-хлор-3,5-динитропиридин использовали вместо 1-хлор-2,4-динитробензола.
1Н ЯМР (400 МГц, Ацетон-d6) δ 9.27 (d, J=2,6 Гц, 1H), 9.13 (s, 1H), 9.10 (d, J=2,6 Гц, 1Н), 3.30 (d,J=1,2 Гц, 3Н).
Стадия 44-2: Получение N'-метил-5-нитропиридин-2,3-Диамина
1,14 г (выход: 83%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-2 Примера 17 выше, за исключением того, что N-метил-3,5-динитропиридин-2-амин, полученный на Стадии 44-1, использовали вместо N-метил-2,4-динитроанилина.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.40 (d, J=2,5 Гц, 1H), 7.34 (d, J=2,6 Гц, 1H), 7.12 (s, 1H), 5.32 (s, 2H), 2.97 (d, J=4,6 Гц, 3Н).
Стадия 44-3: Получение 3-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
669.7 мг (выход: 53%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что N-метил-5-нитропиридин-2,3-диамин, полученный на Стадии 44-2, использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.69 (s, 1H), 8.92 (d, J=2,3 Гц, 1H), 7.92 (d, J=2,3 Гц, 1H), 3.36 (s, 3H).
Стадия 44-4: Получение 1-этил-3-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
148.8 мг (выход: 65%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 3-метил-6-нитро-1/7-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 44-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.03 (d, J=2,2 Гц, 1H), 7.96 (d, J=2,2 Гц, 1H), 4.01 (q, J=7,3 Гц, 2H), 3.56 (s, 3Н), 1.40 (t, J=7,3 Гц, 3Н).
Стадия 44-5: Получение 6-амино-1-этил-3-метил-1H-имидазо[4,5-b]пиридин-2(3H)-она
92,8 мг (выход: 74%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-этил-3-метил-6-нитро-1/7-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 44-4, использовали вместо 7-нитроизохинолин-1(2H)-она, метанол использовали в качестве растворителя вместо диоксана и реакцию проводили при 40°С.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 7.56 (d, J=2,2 Гц, 1H), 6.65 (d, J=2,3 Гц, 1H), 3.88 (q, J=7,3 Гц, 2Н), 3.52 (s, 2Н), 3.43 (s, 3Н), 1.31 (t, J=7,2 Гц, 3Н).
Стадия 44-6: Получение (1-циклопропил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
45,8 мг (выход: 31%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-1-этил-3-метил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 44-5, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.10 (s, 1H), 8.16(d, J=2,l Гц, 1H), 7.62 (d, J=2,1 Гц, 1H), 3.92 (q, J=7,1 Гц, 2Н), 3.33 (s, 4Н), 1.20 (t, J=7,1 Гц, 3Н).
Пример 45: Получение (1-изопропил-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-А]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 45)
Стадия 45-1: Получение 1-изопропил-3-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
98,5 мг (выход: 40%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 3-метил-6-нитро-1H-имидазо[4,5-6]пиридин-2(3H)-он, полученный на Стадии 44-3 Примера 44, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.01 (d, J=2,2 Гц, 1Н), 8.05 (d, J=2,3 Гц, 1H), 4.76 (hept, J=7,0 Гц, 1H), 3.54 (s, 3Н), 1.57 (d, J=7,0 Гц, 6Н).
Стадия 45-2: Получение 6-амино-1-изопропил-3-метил-1H-имидазо[4,5-b]пиридин-2(3H)-она
64,1 мг (выход: 81%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-изопропил-3- метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 45-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и этилацетат использовали в качестве растворителя вместо диоксана.
1H ЯМР (400 МГц, Хлороформ-d6) δ 7.55 (d, J=2,3 Гц, 1Н), 6.78 (d, J=2,2 Гц, 1H), 4.71 (hept, J=7,0 Гц, 1H), 3.51 (s, 2Н), 3.41 (s, 3Н), 1.48 (d, J=7,0 Гц, 6Н).
Стадия 45-3: Получение (1-изопропил-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида
8,6 мг (выход: 9%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 5-амино-1-циклопропил-3-этил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 45-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.10 (s, 1H), 8.15 (d, J=2,1 Гц, 1Н), 7.66 (d, J=2,1 Гц, 1H), 4.64 (hept, J=6,9 Гц, 1H), 3.32 (s, 3Н), 1.44 (d, J=6,9 Гц, 6Н).
Пример 46: Получение (1-(дифторметил)-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 46)
Стадия 46-1: Получение 1-дифторметил-3-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
58,5 мг (выход: 46%) указанного в заголовке соединения было получено таким же способом, как на Стадии 37-1 Примера 37 выше, за исключением того, что 3-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 44-3 Примера 44, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.11 (d, J=2,3 Гц, 1H), 8.25 (d, J=2,3 Гц, 1H), 7.87 (t, J=57,4 Гц, 1H), 3.40 (s, 3Н).
Стадия 46-2: Получение 6-амино-1-диФторметил-3-метил-1H-имидазо[4,5-b]пиридин-2(3H)-она
121,1 мг (выход: 88%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-дифторметил-3-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 46-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 7.66 (d, J=2,4 Гц, 1Н), 7.31 (t, J=58,9 Гц, 1H), 7.02 (d, J=2,3 Гц, 1Н), 3.62 (s, 2Н), 3.42 (s, 3Н).
Стадия 46-3: Получение (1-дифторметил-3-метил-2-оксо-2.3-Дигидро-1H-имидазо [4,5-b]пиридин-6-ил)карбоногидразоноилдицианида
57,3 мг (выход: 81%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-1-дифторметил-3-метил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 46-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.29 (d, J=2,1 Гц, 1H), 7.79 (t, J=57,6 Гц, 1Н), 7.68 (d, J=2,1 Гц, 1H), 3.33 (s, 3Н).
Пример 47: Получение (3-метил-2-оксо-1-фенил-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 47)
Стадия 47-1: Получение 3-метил-6-нитро-1-фенил-1H-имидазо [4,5-b]пиридин-2(3H)-она
3-Метил-6-нитро-1H-имидазо[4,5-6]пиридин-2(3H)-он (50 мг, 0,26 ммоль), полученный на Стадии 44-3 Примера 44, карбонат калия (71,2 мг, 0,52 ммоль), йодид меди (9,8 мг, 0,052 ммоль) и транс-4-гидрокси-L-пролин (13,5 мг, 0,10 ммоль) растворяли в DMSO и к нему медленно добавляли бензолйодид (28,7 мг, 0,26 ммоль), растворенный в DMSO. Реакционную смесь нагревали до 130°С и перемешивали в течение 14 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием насыщенного водного раствора хлорида аммония и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 36,6 мг (выход: 52%) указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) δ 9.05 (d, J=2,3 Гц, 1H), 7.88 (d, J=2,3 Гц, 1Н), 7.63 (d, J=5,7 Гц, 4Н), 7.58-7.47 (m, 1H), 3.50 (s, 3Н).
Стадия 47-2: Получение 6-амино-3-метил-1-фенил-1H-имидазо[4,5-b]пиридин-2(3H)-она
18,2 мг (выход: 34%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-метил-6-нитро-1-фенил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 47-1, использовали вместо 7-нитроизохинолин-1(2H)-она, DMF использовали в качестве растворителя вместо диоксана и реакцию проводили при 60°С.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.61-7.49 (m, 4Н), 7.46 (d, J=2,3 Гц, 1H), 7.45-7.41 (m, 1H), 6.73 (d, J=2,2 Гц, 1H), 4.96 (s, 2H), 3.32 (s, 3H).
Стадия 47-3: Получение (1-дифторметил-3-метил-2-оксо-2,3-дигидро-1H-имидазо [4,5-b]пиридин-6-ил)карбоногидразоноилдицианида
14,0 мг (выход: 58%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-3-метил-1-фенил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 47-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 11.91 (s, 1H), 8.26 (d, J=2,2 Гц, 1H), 7.67-7.56 (m, 4Н), 7.53 (d, J=2,2 Гц, 1H), 7.48 (tt, J=6,l Гц, 2,2 Гц, 1H), 3.48 (s, 3Н).
Пример 48: Получение (3-этил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 48)
Стадия 48-1: Получение N-этил-3,5-динитропиридин-2-амина
1,66 г (выход: 79%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-1 Примера 17 выше, за исключением того, что 2-хлор-3,5-динитропиридин использовали вместо 1-хлор-2,4-динитробензола, и этиламин использовали вместо метиламина.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.26 (d, J=2,6 Гц, 1H), 9.22 (d, J=2,6 Гц, 1H), 8.73 (s, 1H), 3.80 (qd, J=7,2 Гц, 5,6 Гц, 2Н), 1.37 (t, J=7,3 Гц, 3Н).
Стадия 48-2: Получение N1-этил-5-нитропиридин-2,3-диамина
1,15 г (выход: 80%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-2 Примера 17 выше, за исключением того, что N-этил-3,5-динитропиридин-2-амин, полученный на Стадии 48-1, использовали вместо N-метил-2,4-динитроанилина, и смешанный раствор метанола и воды в соотношении 1:1 использовали в качестве растворителя.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 8.74 (d, J=2,4 Гц, 1H), 7.60 (d, J=2,4 Гц, 1H), 5.02 (s, 1H), 3.61 (qd, J=7,2 Гц, 5,4 Гц, 2Н), 3.30 (s, 2Н), 1.31 (t, J=7,2 Гц, 3Н).
Стадия 48-3: Получение 3-этил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
891,8 мг (выход: 68%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что N-этил-5-нитропиридин-2,3-диамин, полученный на Стадии 48-2, использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.69 (s, 1H), 8.93 (d, J=2,4 Гц, 1H), 7.93 (d, J=2,3 Гц, 1H), 3.92 (q, J=7,2 Гц, 2H), 1.27 (t, J=7,2 Гц, 3Н).
Стадия 48-4: Получение 3-этил-1-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
127,8 мг (выход: 80%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 3-этил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 48-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она, и метилйодид использовали вместо этилйодида.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.03 (d, J=2,2 Гц, 1H), 7.94 (d, J=2,2 Гц, 1H), 4.11 (q, J=7,2 Гц, 2Н), 3.50 (s, 3Н), 1.41 (t, J=7,2 Гц, 3Н).
Стадия 48-5: Получение 6-амино-3-этил-1-метил-1H-имидазо[4,5-b]пиридин-2(3H)-она
94,8 мг (выход: 87%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-этил-1-метил-6-нитро-1/7-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 48-4, использовали вместо 7-нитроизохинолин-1(2H)-она, и этилацетат использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, Хлороформ-d) 5 7.55 (d, J=2,3 Гц, 1H), 6.61 (d, J=2,3 Гц, 1H), 3.98 (q, J=7,2 Гц, 2Н), 3.53 (s, 2Н), 3.35 (s, 3Н), 1.34 (t, J=7,2 Гц, 3Н).
Стадия 48-6: Получение (3-этил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-и л)карбоногидразоноилдицианида
61,6 мг (выход: 46%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-3-этил-1-метил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 48-5, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, Ацетон-d6) δ 8.15 (d, J=2,2 Гц, 1H), δ.61 (d, J=2,2 Гц, 1H), 3.96 (q, J=7,2 Гц, 2Н), 3.45 (s, 3Н), 1.31 (t, J=7,2 Гц, 3Н).
Пример 49: Получение (3-этил-1-изопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 49)
Стадия 49-1: Получение 3-этил-1-изопропил-6-нитро-1H-имидазо[4,5-b] пиридин-2(3H)-она
108,3 мг (выход: 45%) указанного в заголовке соединения было получено таким же способом, как на Стадии 48-4 Примера 48 выше, за исключением того, что изопропилйодид использовали вместо метилйодида.
1H ЯМР (400 МГц, Хлороформ-d6) δ 9.01 (d, J=2,2 Гц, 1Н), 8.05 (d, J=2,2 Гц, 1H), 4.77 (hept, J=7,0 Гц, 1H), 4.10 (q, J=7,2 Гц, 2Н), 1.57 (d, J=7,1 Гц, 6Н), 1.41 (t, J=7,2 Гц, 3Н).
Стадия 49-2: Получение 6-амино-3-этил-1-изопропил-1H-имидазо[4,5-b] пиридин-2(3H)-она
75,2 мг (выход: 82%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-этил-1-изопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 49-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и этилацетат использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 7.54 (d, J=2,3 Гц, 1H), 6.78 (d, J=2,2 Гц, 1H), 4.71 (hept, J=7,0 Гц, 1H), 3.97 (q, J=7,2 Гц, 2Н), 3.51 (s, 2Н), 1.48 (d, J=7,0 Гц, 6Н), 1.34 (t, J=7,2 Гц, 3Н).
Стадия 49-3: Получение (3-этил-1-изопропил-2-оксо-2,3-дигидро-1H-имидазо [4,5-b]пиридин-6-ил)карбоногидразоноилдицианида
43,5 мг (выход: 42%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-3-этил-1-изопропил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 49-2, использовали вместо 7-аминоизохинолин-1(2H)-она, и воду использовали в качестве растворителя.
1Н ЯМР (400 МГц, Ацетон-d6) δ 11.89 (s, 1Н), 8.16 (d, J=2,2 Гц, 1Н), 7.72 (d, J=2,2 Гц, 1H), 4.72 (hept, J=6,9 Гц, 1Н), 3.95 (q, J=7,2 Гц, 2Н), 1.52 (d, J=7,0 Гц, 6Н), 1.30 (t, J=7,2 Гц, 3Н).
Пример 50: Получение (1-(дифторметил)-3-этил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 50)
Стадия 50-1: Получение 1-дифторметил-3-этил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
129,6 мг (выход: 70%) указанного в заголовке соединения было получено таким же способом, как на Стадии 37-1 Примера 37 выше, за исключением того, что 3-этил-6- нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 48-3 Примера 48, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.12 (d,J=2,3 Гц, 1H), 8.26 (d, J=2,3 Гц, 1Н), 7.87 (t, J=57,4 Гц, 1H), 3.97 (q, J=7,2 Гц, 2H), 1.31 (t, J=7,2 Гц, 3Н).
Стадия 50-2: Получение 6-амино-1-дифторметил-3-этил-1H-имидазо[4,5-b] 1 шридин-2(3H)-она
103,7 мг (выход: 90%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-дифторметил-3-этил-6-нитро-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 50-1, использовали вместо 7-нитроизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.67 (t, J=58,2 Гц, 1H), 7.53 (d, J=2,2 Гц, 1Н), 7.02 (d, J=2,2 Гц, 1H), 5.18 (s, 2Н), 3.81 (q, J=7,2 Гц, 2Н), 1.23 (t, J=7,2 Гц, 3Н).
Стадия 50-3: Получение (3-дифторметbл-1-этил-2-оксо-2,3-дигидро-1-имидазо [4,5-b]пиридин-6-ил)карбоногидразоноилдицианида
69,3 мг (выход: 80%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-1-дифторметил-3-этил-1H-бензо[d]имидазол-2(3H)-он, полученный на Стадии 50-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.30 (d, J=2,l Гц, 1H), 7.79 (t, J=57,6 Гц, 1H), 7.70 (d, J=2,2 Гц, 1H), 3.90 (q, J=7,2 Гц, 2Н), 1.28 (t, J=7,2 Гц, 3Н).
Пример 51: Получение (3-циклопропил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 51)
Стадия 51-1: Получение N-циклопропил-3,5-динитрпиридин-2-амина
1,99 г (выход: 90%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-1 Примера 17 выше, за исключением того, что 2-хлор-3,5-динитропиридин использовали вместо 1-хлор-2,4-динитробензола, и циклопропиламин использовали вместо метиламина.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.32 (d, J=2,5 Гц, 1Н), 9.21 (d, J=2,5 Гц, 1H), 8.69 (s, 1H), 3.21 (ddt, J=11,1 Гц, 7,1 Гц, 3.9 Гц, 1H), 1.11-0.97 (m, 2Н), 0.80-0.64 (m, 2Н).
Стадия 51-2: Получение N2-циклопропил-5-нитропиридин-2,3-диамина
1,33 г (выход: 78%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-2 Примера 17 выше, за исключением того, что N-циклопропил-3,5-динитропиридин-2-амин, полученный на Стадии 51-1, использовали вместо N-метил-2,4-динитроанилина, и смешанный раствор метанола и воды в соотношении 1:1 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.41 (d, J=2,5 Гц, 1H), 7.34 (d, J=2,5 Гц, 1Н), 7.12 (d, J=3.6 Гц, 1H), 5.35 (s, 2H), 2.93 (tq, J=7,3 Гц, 3.8 Гц, 1H), 0.84-0.71 (m, 2H), 0.57-0.45 (m, 2H).
Стадия 51-3: Получение 3-1щклопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
1,16 г (выход: 77%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что N2-циклопропил-5-нитропиридин-2,3-диамин, полученный на Стадии 51-2, использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) δ 11.56 (s, 1H), 8.91 (d, J=2,4 Гц, 1Н), 7.88 (d, J=2,4 Гц, 1H), 3.07-2.92 (m, 1H), 1.17-0.78 (m, 4H).
Стадия 51-4: Получение 3-циклопропил-1-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)она
172,0 мг (выход: 80%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 3-циклопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 51-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она, и метилйодид использовали вместо этилйодида.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.95 (d, J=2,3 Гц, 1H), 8.31 (d,J=2,3 Гц, 1Н), 3.40 (s, 3Н), 3.08-2.97 (m, 1H), 1.11-0.99 (m, 4Н).
Стадия 51-5: Получение 6-амино-3-циклопропил-1-метил-1H-имидазо[4,5-b]пиридин-2(3H)-она
89,3 мг (выход: 78%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-циклопропил-1-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 51-4, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.37 (d, J=2,3 Гц, 1H), 6.69 (d, J=2,3 Гц, 1H), 4.92 (s, 2H), 3.20 (s, 3Н), 2.86 (tt, J=7,2 Гц, 3,5 Гц, 1H), 1.04-0.85 (m, 4Н).
Стадия 51-6: Получение (3-циклопропил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
25,6 мг (выход: 20%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-3-циклопропил-1-метил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 51-5, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.15 (d, J=2,2 Гц, 1H), 7.52 (d, J=2,2 Гц, 1H), 3.32 (s, 3Н), 2.94 (td, J=6,8 Гц, 3.6 Гц, 1H), 0.99 (d, J=7,5 Гц, 4H).
Пример 52: Получение (3-циклопропил-1-этил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 52)
Стадия 52-1: Получение 3-циклопропил-1-этил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
179,5 мг (выход: 79%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 3-циклопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 51-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ Хлороформ-d6) δ 9.03 (d, J=2,3 Гц, 1H), 7.92 (d, J=2,2 Гц, 1H), 3.98 (q, J=7,3 Гц, 2Н), 3.07 (tt, J=6,6 Гц, 4.1 Гц, 1Н), 1.38 (t, J=7,3 Гц, 3Н), 1.29-1.09 (m, 4Н).
Стадия 52-2: Получение 6-амино-3-никлопропил-1-этил-1H-имидазо[4,5-b] пиридин-2(3H)-она
112,0 мг (выход: 71%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-циклопропил-1-этил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 52-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, Хлороформ-d) δ 7.58 (d, J=2,3 Гц, 1Н), 6.62 (d, J=2,3 Гц, 1H), 3.84 (q, J=7,3 Гц, 2Н), 3.52 (s, 2Н), 2.93 (tt, J=6,4 Гц, 4.1 Гц, 1H), 1.30 (t, J=7,2 Гц, 3Н), 1.10 (dddt, J=7,0 Гц, 4.5 Гц, 2,8 Гц, 1,2 Гц, 4Н).
Стадия 52-3: Получение (3-циклопропил-1-этил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
66,2 мг (выход: 44%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-3-циклопропил-1-этил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 52-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.11 (s, 1H), 8.16 (d, J=2,2 Гц, 1H), 7.58 (d, J=2,2 Гц, 1H), 3.88 (q, J=7,1 Гц, 2H), 2.95 (td, J=7,0 Гц, 3,6 Гц, 1H), 1.19 (t, J=7,l Гц, 3Н), 1.00 (ddt, J=9,9 Гц, 5,2 Гц, 2,7 Гц, 4Н).
Пример 53: Получение (3-циклопропил-1-изопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 53)
Стадия 53-1: Получение 3-циклопропил-1-изопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
187,7 мг (выход: 76%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 3-циклопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 51-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она, и изопропилйодид использовали вместо этилйодида.
1Н ЯМР (400 МГц, DMSO-d6) δ Хлороформ-d6) δ 9.02 (d, J=2,2 Гц, 1H), 8.02 (d, J=2,3 Гц, 1H), 4.74 (hept, J=7,0 Гц, 1H), 3.06 (tt, J=6,7 Гц, 4.1 Гц, 1H), 1.56 (d, J=7,l Гц, 6Н), 1.24-1.12 (m, 4Н).
Стадия 53-2: Получение 6-амино-3-циклопропил-1-изопропил-1H-имидазо[4,5-b]пиридин-2(3H)-она
130,9 мг (выход: 79%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-циклопропил-1-изопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 53-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 7.58 (d, J=2,3 Гц, 1H), 6.75 (d, J=2,3 Гц, 1H), 4.69 (hept, J=7,0 Гц, 1Н), 3.50 (s, 2Н), 2.92 (tt, J=6,5 Гц, 4,1 Гц, 1Н), 1.47 (d, J=7,0 Гц, 6Н), 1.10 (dddd, J=9,7 Гц, 4.2 Гц, 2,6 Гц, 1,2 Гц, 4Н).
Стадия 53-3: Получение (3-циклопропил-1-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдинианида
22,9 мг (выход: 34%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-3-циклопропил-1-изопропил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 53-2, использовали вместо 7-аминоизохинолин-1(2H)-она, и воду использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.13 (s, 1H), 8.14 (d, J=2,l Гц, 1H), 7.62 (d,J=2,2 Гц, 1H), 4.61 (hept, J=6,9 Гц, 1H), 3.03-2.79 (m, 1H), 1.42 (d,J=6,9 Гц, 6H), 1.08-0.91 (m, 4H).
Пример 54: Получение (3-изопропил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 54)
Стадия 54-1: Получение N-изопропил-3,5-динитропиридин-2-амина
2,07 г (выход: 93%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-1 Примера 17 выше, за исключением того, что 2-хлор-3,5-динитропиридин использовали вместо 1-хлор-2,4-динитробензола, и изопропиламин использовали вместо метиламина.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.25 (d, J=2,6 Гц, 1Н), 9.22 (d, J=2,5 Гц, 1H), 8.60 (s, 1H), 4.62 (dh, J=7,6 Гц, 6,6 Гц, 1H), 1.37 (d, J=6,5 Гц, 6Н).
Стадия 54-2: Получение N2-изопропил-5-нитропиридин-2,3-диамина
1.07 г (выход: 59%) указанного в заголовке соединения было получено таким же способом, как на Стадии 17-2 Примера 17 выше, за исключением того, что N-изопропил-3,5-динитропиридин-2-амин, полученный на Стадии 54-1, использовали вместо N-метил-2,4-динитроанилина, и смешанный раствор метанола и воды в соотношении 1:1 использовали в качестве растворителя.
1Н ЯМР (400 МГц, DMSO-d6) δ 8.37 (d, J=2,5 Гц, 1H), 7.32 (d, J=2,6 Гц, 1H), 6.74 (d, J=7,2 Гц, 1H), 5.41 (s, 2Н), 4.39-4.26 (m, J=6,6 Гц, 1Н), 1.21 (d, J=6,5 Гц, 6H).
Стадия 54-3: Получение 3-изопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
1.08 г (выход: 91%) указанного в заголовке соединения было получено таким же способом, как на Стадии 9-1 Примера 9 выше, за исключением того, что N2-изопропил-5-нитропиридин-2,3-диамин, полученный на Стадии 54-2, использовали вместо 4-фтор-5-нитробензол-1,2-диамина.
1Н ЯМР (400 МГц, DMSO-d6) 8,91 (d, J=2,4 Гц, 1H), 7.91 (d, J=2,4 Гц, 1H), 4.69 (hept, J=6,9 Гц, 1H), 1.51 (d, J=6,9 Гц, 6H).
Стадия 54-4: Получение 3-изопропил-1-метил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
143,5 мг (выход: 67%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 3-изопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 54-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она, и метилйодид использовали вместо этилйодида.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.01 (d, J=2,3 Гц, 1Н), 7.92 (d, J=2,3 Гц, 1H), 4.88 (hept, J=6,9 Гц, 1H), 3.48 (s, 3Н), 1.61 (d, J=6,9 Гц, 6Н).
Стадия 54-5: Получение 6-амино-3-изопропил-1-метил-1H-имидазо[4,5-b]пиридин-2(3H)-она
97,2 мг (выход: 78%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 3-изопропил-1-метил-6-нитро-1Р-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 54-4, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.37 (d, J=2,3 Гц, 1H), 6.71 (d, J=2,2 Гц, 1Н), 4.90 (s, 2H), 4.55 (hept, J=6,9 Гц, 1H), 3.22 (s, 3Н), 1.44 (d, J=6,9 Гц, 6H).
Стадия 54-6: Получение (3-изопропил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
103,5 мг (выход: 77%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-3-изопропил-1-метил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 54-5, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.18 (s, 1H), 8.16 (d, J=2,2 Гц, 1Н), 7.56 (d, J=2,2 Гц, 1H), 4.66 (hept, J=6,9 Гц, 1H), 3.35 (s, 3Н), 1.49 (d, J=6,9 Гц, 6H).
Пример 55: Получение (1-этил-3-изопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианида (Соединение 55)
Стадия 55-1: Получение 1-этил-3-изопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-она
194,1 мг (выход: 86%) указанного в заголовке соединения было получено таким же способом, как на Стадии 35-1 Примера 35 выше, за исключением того, что 3-изопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 54-3, использовали вместо 1-метил-5-нитро-1H-бензо[d]имидазол-2(3H)-она.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 9.00 (d, J=2,3 Гц, 1H), 7.92 (d, J=2,3 Гц, 1H), 4.87 (hept, J=6,9 Гц, 1H), 3.98 (q, J=7,2 Гц, 2Н), 1.61 (d, J=6,9 Гц, 6Н), 1.39 (t, J=7,3 Гц, 3Н).
Стадия 55-2: Получение 6-амино-1-этил-3-изопропил-1H-имидазо[4,5-b] пиридин-2(3H)-она
27,4 мг (выход: 16%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-2 Примера 1 выше, за исключением того, что 1-этил-3-изопропил-6-нитро-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 55-1, использовали вместо 7-нитроизохинолин-1(2H)-она, и метанол использовали в качестве растворителя вместо диоксана.
1H ЯМР (400 МГц, DMSO-d6) δ 7.37 (d, J=2,2 Гц, 1H), 6.77 (d,J=2,3 Гц, 1Н), 4.88 (s, 2H), 4.55 (hept, J=6,9 Гц, 1H), 3.75 (q, J=7,2 Гц, 2H), 1.44 (d, J=6,9 Гц, 6H), 1.17 (t, J=7,2 Гц, 3Н).
Стадия 55-3: Получение (1-этил-3-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида
5,7 мг (выход: 15%) указанного в заголовке соединения было получено таким же способом, как на Стадии 1-3 Примера 1 выше, за исключением того, что 6-амино-1-этил-3-изопропил-1H-имидазо[4,5-b]пиридин-2(3H)-он, полученный на Стадии 55-2, использовали вместо 7-аминоизохинолин-1(2H)-она.
1Н ЯМР (400 МГц, DMSO-d6) δ 13.17 (s, 1H), 8.16 (d, J=2,2 Гц, 1Н), 7.61 (d, J=2,2 Гц, 1H), 4.65 (h, J=7,0 Гц, 1H), 3.90 (q, J=7,2 Гц, 2H), 1.49 (d, J=6,9 Гц, 6H), 1.21 (t, J=7,0 Гц, 3Н).
Пример 56: Получение (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)(метил)карбоногидразоноилдицианида (Соединение 56)
(1,3-Диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил) карбоногидразоноилдицианид (100 мг, 0,39 ммоль), полученный в Примере 22, растворяли в диметилформамиде в атмосфере азота и добавляли к нему при комнатной температуре трет-бутоксид калия (66 мг, 0,59 ммоль), а затем метанйодид (122 мкл, 1,97 ммоль). Реакционную смесь перемешивали при 60°С в течение 4 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенное вещество очищали колоночной хроматографией с получением 41 мг (выход: 38%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, DMSO-d6) δ 7.48-7.15 (m, 3Н), 4.10-3.84 (m, 3Н), 3.35 (s, 6Н).
Пример 57: Получение ацетил(1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианида (Соединение 57)
(1,3-Диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил) карбоногидразоноилдицианид (150 мг, 0,59 ммоль), полученный в Примере 22, и KОН (36 мг, 0,65 ммоль) растворяли в метаноле и этот раствор перемешивали при комнатной температуре в течение 3 часов. После завершения взаимодействия растворитель удаляли при пониженном давлении и остаток отверждали с использованием эфира. Затем полученное твердое вещество (170 мг, 0,58 ммоль) и триэтиламин (40 мкл, 0,29 ммоль) растворяли в ацетонитриле, добавляли к нему ацетилхлорид (103 мкл, 1,45 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения взаимодействия реакционный продукт экстрагировали с использованием дистиллированной воды и этилацетата с получением органического слоя и этот органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и отверждали с использованием эфира и затем фильтровали. Фильтрат снова концентрировали при пониженном давлении и отверждали с использованием гексана с получением 43 мг (выход: 25%) указанного в заголовке соединения.
1Н ЯМР (400 МГц, Хлороформ-d6) δ 7.07 (d, J=8,5 Гц, 1H), 6.91 (dd, J=8,2 Гц, 1,7 Гц, 1H), 6.77 (s, 1H), 3.46 (s, 3Н), 3.42 (s, 3Н), 2.61 (s, 3Н).
[Примеры препаратов]
При этом, новое соединение, представленное формулой 1 по настоящему изобретению, может быть приготовлено в виде препарата в различных формах. Следующие примеры иллюстративно описывают несколько способов приготовления препаратов, включающих соединение, представленное формулой 1, по настоящему изобретению в качестве активного ингредиента, и настоящее изобретение не ограничивается этим.
Пример Препарата 1: Изготовление таблетки прямым прессованием
5,0 мг каждого из активных ингредиентов, полученных в Примерах 1-57, просеивали, смешивали с 14,1 мг лактозы, 0,8 мг кросповидона USNF и 0,1 мг стеарата магния и затем прессовали в таблетки.
Пример Препарата 2: Изготовление таблетки влажным гранулированием
5,0 мг каждого из активных ингредиентов, полученных в Примерах 1-57, просеивали и смешивали с 16,0 мг лактозы и 4,0 мг крахмала. 0,3 мг полисорбата 80 растворяли в чистой воде и этот раствор добавляли к смеси в подходящем количестве с последующим распылением с получением мелкодисперсных частиц. После сушки мелкодисперсные частицы просеивали, смешивали с 2,7 мг коллоидного диоксида кремния и 2,0 мг стеарата магния и прессовали в таблетки.
Пример Препарата 3: Изготовление порошка и капсулы 5,0 мг каждого из активных ингредиентов, полученных в Примерах 1-57, просеивали и затем смешивали с 14,8 мг лактозы, 10,0 мг поливинилпирролидона и 0,2 мг стеарата магния. Смесью заполняли твердые желатиновые капсулы No.5 с использованием подходящего устройства для приготовления капсул.
Пример Препарата 4: Изготовление инъекционного лекарственного средства
100 мг каждого из активных ингредиентов, полученных в Примерах 1-57, смешивали с 180 мг маннита, 26 мг Na2HPO4⋅12H2O и 2974 мг дистиллированной воды с получением инъекционных препаратов.
Экспериментальный Пример 1: Выбор вещества, ингибирующего агрегацию тау-белка, с использованием клеточной модели
Для выбора новых веществ, ингибирующих агрегацию тау-белков, использовали клеточную модель tau-BiFC, в которой легко наблюдается образование тау-олигомеров в живых клетках. Аликвоты клеток Tau-BiFC помещали в 384-луночный планшет. На следующие сутки клетки обрабатывали каждым из соединений, полученных согласно Примерам 1-57, в концентрациях 1 мкМ, 3 мкМ и 10 мкМ вместе с форсколином (в концентрации для обработки 30 мкМ), который является соединением, индуцирующим агрегацию тау-белка путем активации тау-фосфорилазы РКА. Через 48 часов ядра в клетках окрашивали с помощью Hoechst (в концентрации для обработки 2 мкг/мл), а интенсивность флуоресценции BiFC автоматически измеряли с помощью Operetta (PerkinElmer) для подсчета окрашенных ядер в каждой лунке из всего луночного планшета. Группа, получавшая только форсколин, который индуцирует агрегацию тау-белка, была установлена в качестве контрольной точки 100% агрегированного состояния тау-белка, а эффекты соединений подтверждали с помощью уравнения «Интенсивность флуоресценции BiFC, вызванной соединением, синтезированным в соответствии с воплощением настоящего изобретения / (интенсивность флуоресценции контрольной группы, получавшей только форсколин, индуцирующий агрегацию тау-белка -интенсивность флуоресценции необработанной контрольной группы) х 100». Кроме того, степень цитотоксичности, индуцированной новым синтезированным соединением, также измеряли из расчета 100% жизнеспособности клеток в группе, получавшей только форсколин, в качестве эталона, а значение цитотоксичности каждого соединения рассчитывали по уравнению «(количество окрашенных ядер в группе, получавшей соединение / количество окрашенных ядер в группе, получавшей форсколин) × 100». На основании этих результатов обработки, вещества, ингибирующие внутриклеточную агрегацию тау-белка, были выбраны из ряда групп кандидатов, демонстрирующих скорость ингибирования агрегации тау-белка 70% и более и жизнеспособность клеток 100% при концентрации соединения для обработки 10 мкМ и более.
Экспериментальный Пример 2: Подтверждение зависимого от концентрации ингибирующего действия нового соединения на агрегацию тау-белка
Чтобы оценить дозозависимое ингибирование агрегации тау-белка соединениями, выбранными в соответствии с Экспериментальным Примером 1, на агрегацию тау-белка, клетки тау-BiFC обрабатывали выбранными соединениями в концентрациях 0,03 мкМ, 0,01 мкМ, 0,3 мкМ, 1 мкМ, 3 мкМ, 10 мкМ и 30 мкМ, соответственно, вместе с форсколином (в концентрации для обработки 30 мкМ), который является веществом, индуцирующим агрегацию тау-белка. Через 48 часов анализировали реакцию агрегации тау-белка и степень цитотоксичности путем наблюдения за изображениями клеток. IC50 и токсичность соединений анализировали с помощью нелинейного регрессионного анализа программного обеспечения prism (Graph Pad). Результаты расчетов репрезентативных соединений показаны в Таблице 2 ниже.
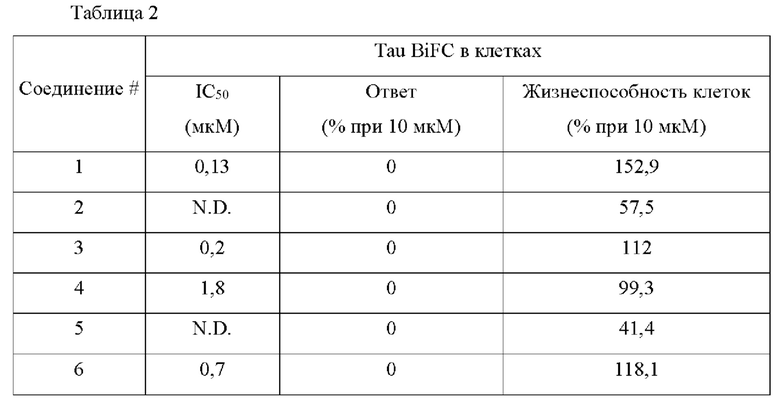
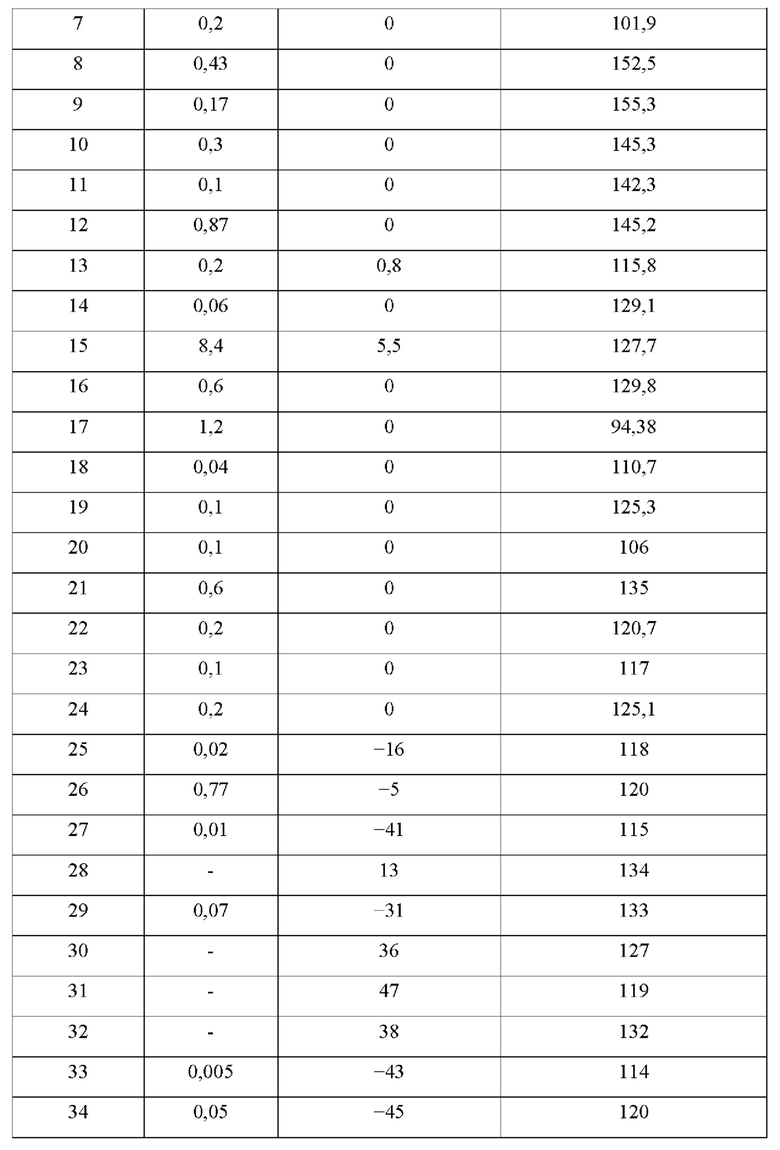
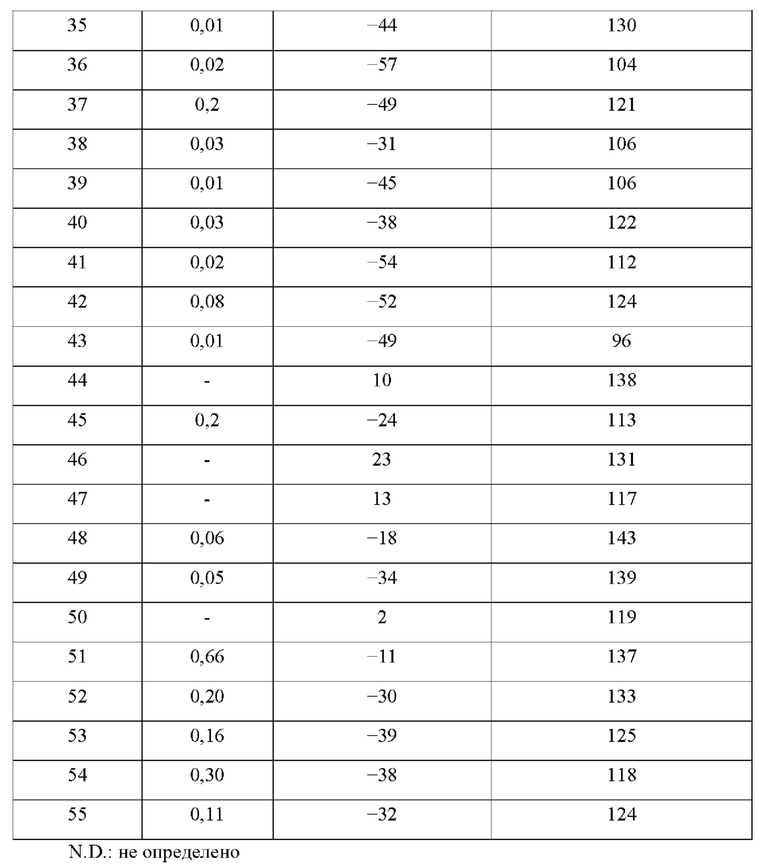
Экспериментальный Пример 3: Ингибирующее действие нового соединения на активность кофермента CYP
Было выявлено ингибирующее действие соединений, полученных в соответствии с Примерами 1-57, на активность кофермента CYP. В частности, микросомы печени человека (0,25 мг/мл), 0,1 М фосфатный буферный раствор (рН 7,4), коктейль лекарств-субстратов из пяти типов ферментов метаболизма лекарств (50 мкМ фенацетина, 10 мкМ диклофенака, 100 мкМ S-мефенитоина, 5 мкМ декстрометорфана и 2,5 мкМ мидазолама) и соединение в концентрации 0 мкМ или 10 мкМ смешивали и предварительно культивировали при 37°С в течение 5 минут, а затем дополнительно культивировали при 37°С в течение 15 минут вместе с добавленным к нему раствором системы генерации НАДФН. После этого реакцию останавливали добавлением раствора внутреннего стандарта (терфенадин) в ацетонитриле и центрифугировали в течение 5 минут (14000 об/мин, 4°С), а затем супернатант вводили в систему ЖХ-МС/МС для одновременного анализа метаболитов лекарств-субстратов, чтобы таким образом оценить ингибирующее действие на метаболизм лекарств.
Метаболиты каждого лекарства-индикатора кофермента CYP, образующиеся в результате реакции, анализировали с использованием системы Shimadzu Nexera XR и TSQ Vantage (Thermo). В ВЭЖХ использовали колонку Kinetex С18 (2,1 мм × 100 мм, 2,6 мкм, размер частиц; Phenomenex, США), подвижными фазами служили (А) дистиллированная вода, содержащая 0,1% муравьиной кислоты и (В) ацетонитрил, содержащий 0,1% муравьиной кислоты, и к ним применяли программу градиента, показанную в Таблице 3.
Продуцированные метаболиты были количественно определены с использованием режима количественного определения мониторинга множественных реакций (MRM), и Xcalibur (версия 1.6.1) использовали для анализа данных. Для отображения ингибирующего действия нового соединения, полученного в соответствии с примерами по настоящему изобретению, на активность кофермента CYP, активность кофермента CYP (%) по отношению к контрольной группе, не получавшей соединение, показана в Таблице 4 ниже. 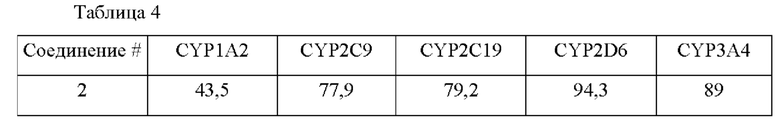
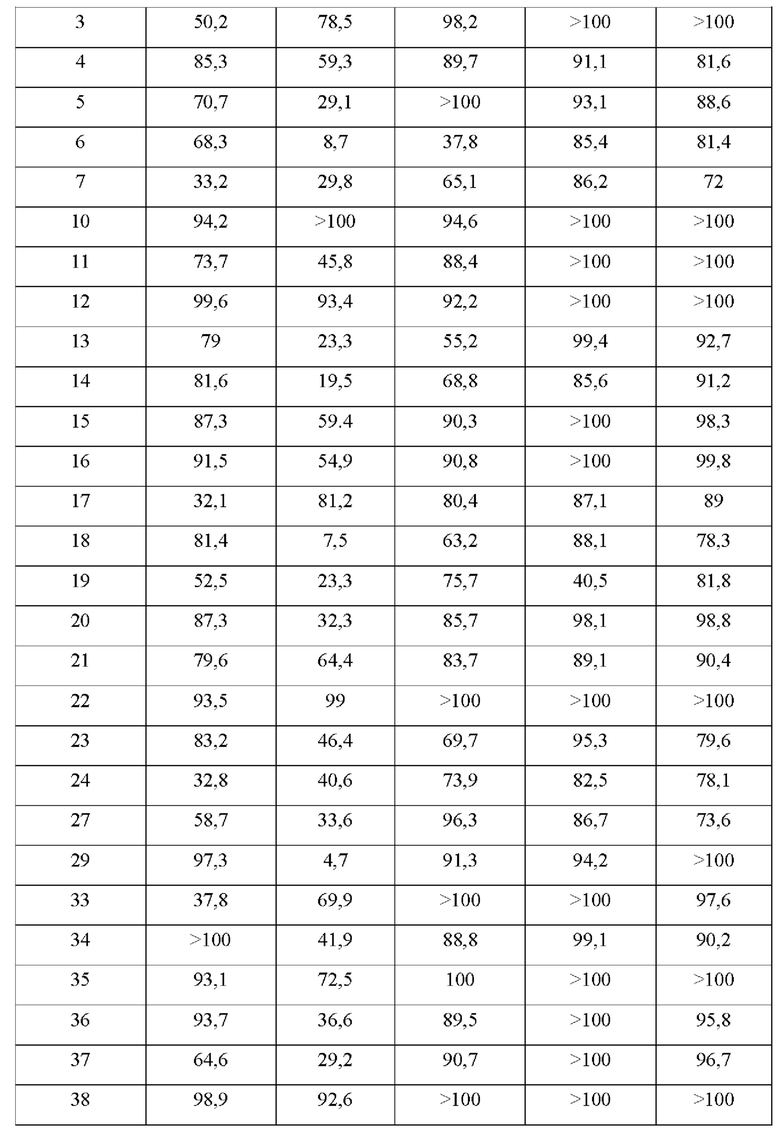
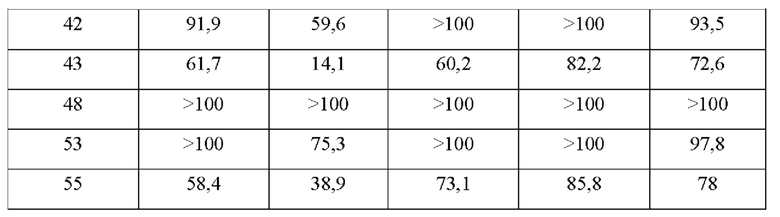
Экспериментальный Пример 4: Идентификация стабильности микросом печени, обеспечиваемой новым соединением
Подтверждали стабильность микросом печени, обеспечиваемую соединениями, полученными согласно Примерам 1-57. В частности, четыре типа микросом печени (человека, собаки, крысы и мыши, каждого по 0,25 мг/мл), 0,1 М фосфатный буферный раствор (рН 7,4) и каждое из соединений в концентрации 1 мкМ смешивали и предварительно культивировали при 37°С в течение 5 минут и затем культивировали при 37°С в течение 30 минут вместе с добавлением раствора системы генерации НАДФН. После этого реакцию останавливали добавлением раствора внутреннего стандарта (хлорпропамид) в ацетонитриле и центрифугировали в течение 5 минут (14000 об/мин, 4°С), а затем супернатант вводили в систему ЖХ-МС/МС для анализа лекарств-субстратов, чтобы таким образом оценить метаболическую стабильность для 8 типов соединений.
Количество субстрата, оставшегося после реакции, анализировали с помощью системы Shimadzu Nexera XR и TSQ Vantage (Thermo). В ВЭЖХ использовали колонку Kinetex ХВ-С18 (2,1 мм × 100 мм, размер частиц 2,6 мкм; Phenomenex, США), подвижными фазами служили (А) дистиллированная вода, содержащая 0,1% муравьиной кислоты и (В) ацетонитрил, содержащий 0,1% муравьиной кислоты. Для анализа данных использовали программы Analyst (версия 1.6.3) и Xcalibur (версия 1.6.1). Результаты расчетов представлены в Таблице 5 ниже.
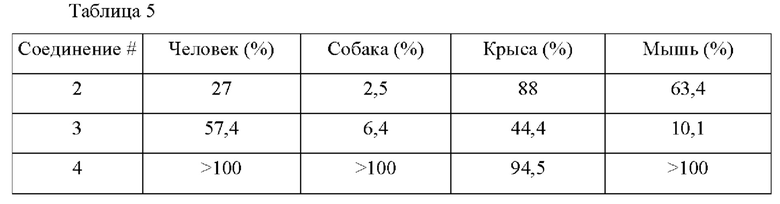
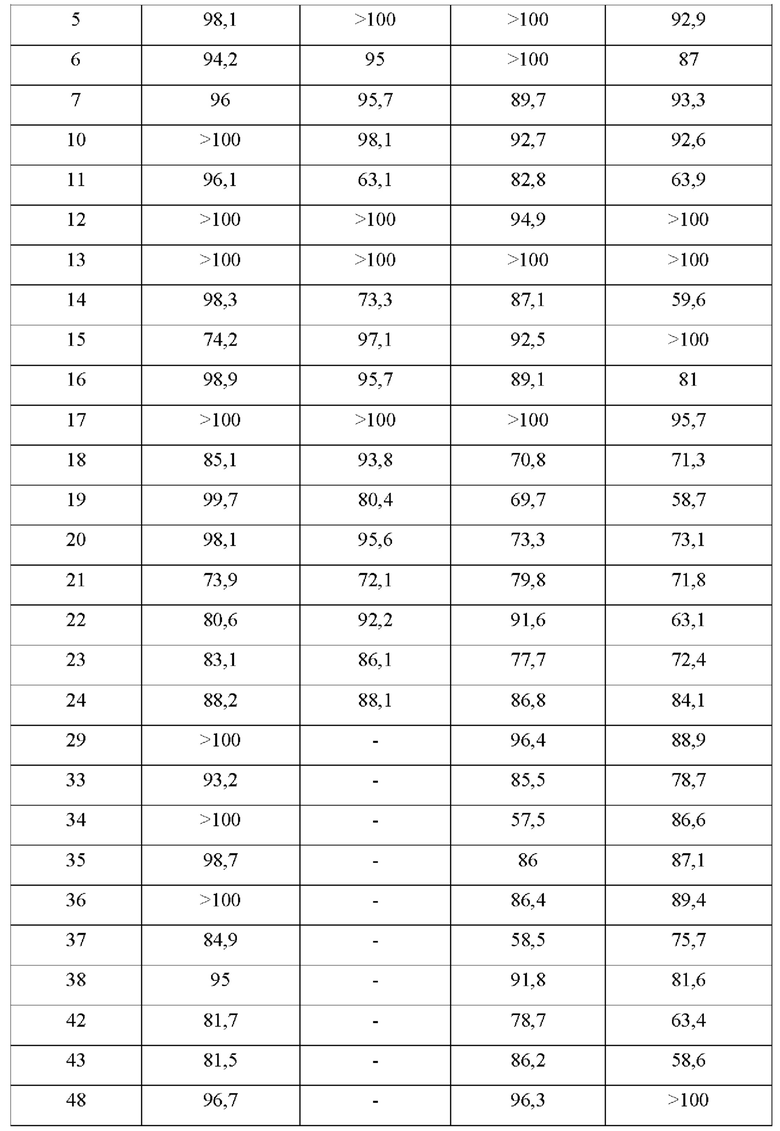
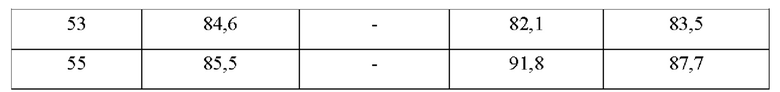
Приведенное выше описание настоящего изобретения предоставлено с целью иллюстрации, и специалистам в данной области техники будет понятно, что различные изменения и модификации могут быть выполнены без изменения технической концепции и существенных признаков настоящего изобретения. Таким образом, понятно, что вышеописанные воплощения являются иллюстративными во всех аспектах и не ограничивают настоящее изобретение. Различные воплощения, раскрытые в данном документе, не предназначены для ограничения, а истинный объем и сущность указаны в следующей формуле изобретения. Настоящее изобретение должно быть ограничено только терминами прилагаемой формулы изобретения, а также полным объемом эквивалентов, на которые распространяется такая формула изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2827641C1 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2733384C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| 4-(ИМИДАЗО[1,2-а]ПИРИДИН-3-ИЛ)-ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2822388C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ IV, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ НА ИХ ОСНОВЕ | 2003 |
|
RU2297418C9 |
| Новые производные пиразола | 2022 |
|
RU2816835C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ 637: ПИРИДОПИРИМИДИНДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2008 |
|
RU2479584C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
Группа изобретений относится к фармацевтической химии и включает производные конденсированных гетероциклил-карбоногидразоноилдицианидов формулы 1, их фармацевтически приемлемые соли, способ их получения и фармацевтические композиции на их основе. В формуле 1 X1 - Х3 каждый независимо представляет собой N или С(Н); Y1 и Y2 каждый независимо представляет собой N(R4), С(Н), О или S, и по меньшей мере один из Y1 и Y2 представляет собой N(R4); n равен 0 или 1;  представляет собой
представляет собой  или
или  с образованием ароматического или неароматического конденсированного гетероциклического кольца; R1 представляет собой водород, C1-6алкил или C1-6алкилкарбонил; R2 и R3 каждый независимо представляет собой водород, C1-6алкил, галоген или C1-6алкокси; и R4 представляет собой водород, C1-6алкил, C1-6галогеналкил, С3-6циклоалкил-С0-6алкил или С6-10арил. Технический результат – соединения формулы 1, ингибирующие агрегацию тау-белка. 4 н. и 16 з.п. ф-лы, 5 табл., 65 пр.
с образованием ароматического или неароматического конденсированного гетероциклического кольца; R1 представляет собой водород, C1-6алкил или C1-6алкилкарбонил; R2 и R3 каждый независимо представляет собой водород, C1-6алкил, галоген или C1-6алкокси; и R4 представляет собой водород, C1-6алкил, C1-6галогеналкил, С3-6циклоалкил-С0-6алкил или С6-10арил. Технический результат – соединения формулы 1, ингибирующие агрегацию тау-белка. 4 н. и 16 з.п. ф-лы, 5 табл., 65 пр.
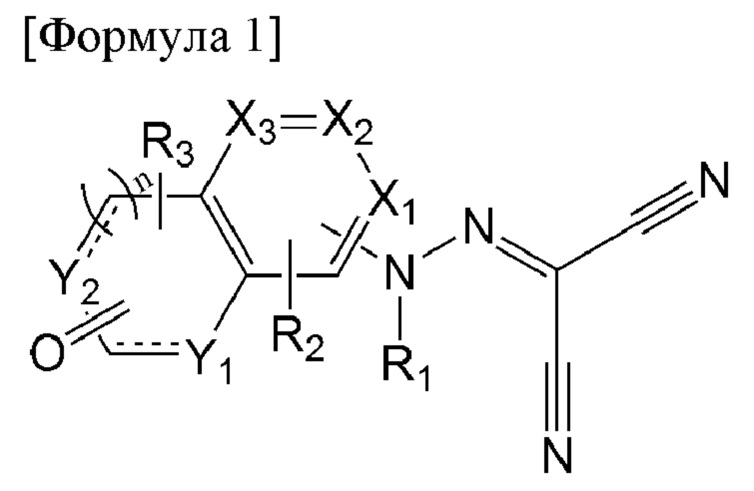
1. Соединение, представленное формулой 1 ниже, или его фармацевтически приемлемая соль:
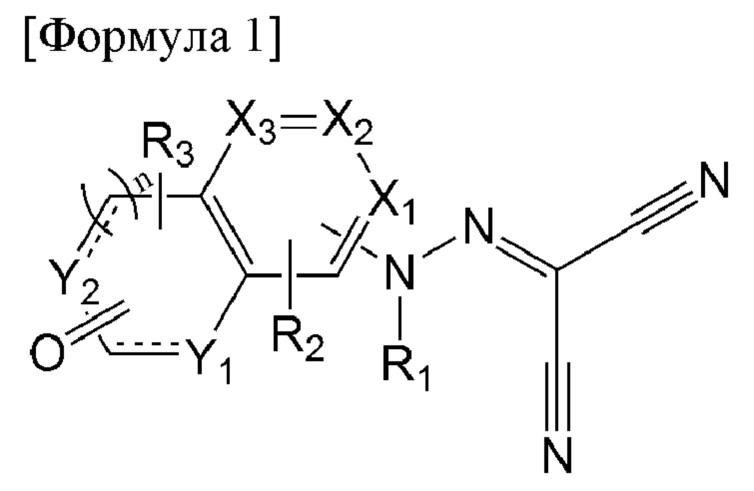
где в формуле 1:
X1 - Х3 каждый независимо представляет собой N или С(Н);
Y1 и Y2 каждый независимо представляет собой N(R4), С(Н), О или S, и по меньшей мере один из Y1 и Y2 представляет собой N(R4);
n равен 0 или 1;
 представляет собой
представляет собой  или
или  с образованием ароматического или неароматического конденсированного гетероциклического кольца;
с образованием ароматического или неароматического конденсированного гетероциклического кольца;
R1 представляет собой водород, C1-6алкил или C1-6алкилкарбонил;
R2 и R3 каждый независимо представляет собой водород, C1-6алкил, галоген или C1-6алкокси; и
R4 представляет собой водород, C1-6алкил, C1-6галогеналкил, С3-6циклоалкил-С0-6алкил или С6-10арил.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где
R1 представляет собой водород, C1-6алкил или C1-6алкилкарбонил;
R2 представляет собой водород, галоген, C1-6алкил или C1-6алкокси;
R3 представляет собой водород или C1-6алкил; и
R4 представляет собой водород, C1-6алкил, C1-6галогеналкил, С3-6циклоалкил-С0-6алкил или С6-10арил.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где
R1 представляет собой водород, метил или ацетил;
R2 представляет собой водород, хлор, фтор, метил или метокси;
R3 представляет собой водород или метил; и
R4 представляет собой водород, метил, этил, изопропил, дифторметил, циклопропил или фенил.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение представлено формулой 2 ниже:
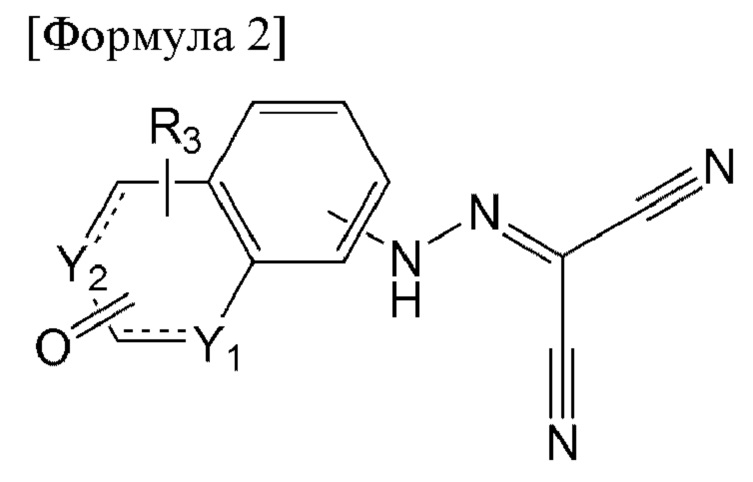
где в формуле 2 выше:
один из Y1 и Y2 представляет собой СН, а другой представляет собой NR4;
 при связи с СН представляет собой
при связи с СН представляет собой  , а
, а  при связи с NR4 представляет собой
при связи с NR4 представляет собой  ;
;
R3 представляет собой водород или метил; и
R4 представляет собой водород, метил или изопропил.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение представлено формулой 3 ниже:
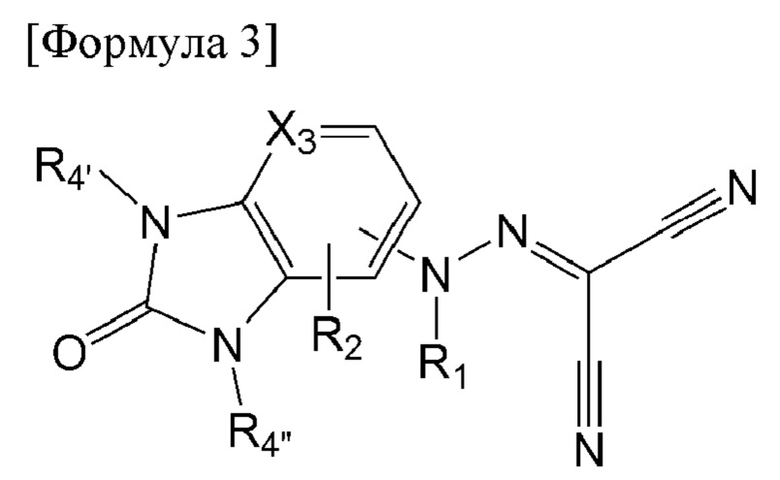
где в формуле 3 выше:
Х3 представляет собой С(Н) или N;
R1 представляет собой водород, метил или ацетил;
R2 представляет собой водород, хлор, фтор, метил или метокси; и
R4' и R4ʺ каждый независимо представляет собой водород, метил, этил, изопропил, дифторметил, циклопропил или фенил.
6. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение представлено формулой 4 ниже:
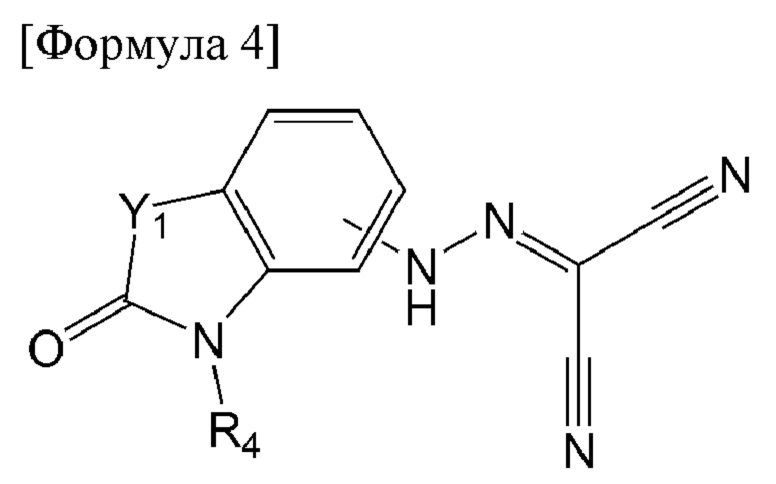
где в формуле 4 выше:
Y1 представляет собой S или О; и
R4 представляет собой водород, метил, изопропил или дифторметил.
7. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение представляет собой
1) (1-оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноилдицианид,
2) (2-метил-1-оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноилдицианид,
3) (2-изопропил-1-оксо-1,2-дигидроизохинолин-7-ил)карбоногидразоноил-дицианид,
4) (1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианид,
5) (2-метил-1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноилдицианид,
6) (2-изопропил-1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноил-дицианид,
7) (2,3-диметил-1-оксо-1,2-дигидроизохинолин-5-ил)карбоногидразоноил-дицианид,
8) (2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианид,
9) (6-фтор-1,3-диметил-2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
10) (6-хлор-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
11) (7-хлор-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
12) (1,3-диметил-2-оксо-2,3-дигидро-1Н-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
13) (2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
14) (3-метил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноил-дицианид,
15) (2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноилдицианид,
16) (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноил-дицианид,
17) (1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
18) (3-изопропил-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноил-дицианид,
19) (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]тиазол-6-ил)карбоногидразоноилдицианид,
20) (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-6-ил)карбоногидразоноил-дицианид,
21) (3-(дифторметил)-2-оксо-2,3-дигидробензо[d|оксазол-6-ил)карбоногидразоноилдицианид,
22) (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
23) (1,3-диизопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
24) (1,3-бис(дифторметил)-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
25) (1-метил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианид,
26) (4-метил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианид,
27) (1,4-диметил-2-оксо-1,2-дигидрохинолин-6-ил)карбоногидразоноилдицианид,
28) (3-метил-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианид,
29) (3-изопропил-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианид,
30) (3-(дифторметил)-2-оксо-2,3-дигидробензо[d]оксазол-5-ил)карбоногидразоноилдицианид,
31) (1,3,6-триметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
32) (6-метокси-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
33) (7-метокси-1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
34) (1,3-диизопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
35) (3-этил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
36) (3-изопропил-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
37) (3-(дифторметил)-1-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
38) (1-этил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
39) (3-(дифторметил)-1-этил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
40) (1-циклопропил-3-метил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
41) (1-циклопропил-3-этил-2-оксо-2,3-Дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
42) (1-циклопропил-3-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
43) (3-(дифторметил)-1-изопропил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид,
44) (1-этил-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
45) (1-изопропил-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
46) (1-(дифторметил)-3-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
47) (3-метил-2-оксо-1-фенил-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
48) (3-этил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
49) (3-этил-1-изопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
50) (1-(дифторметил)-3-этил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
51) (3-циютопропил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
52) (3-циклопропил-1-этил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
53) (3-циклопропил-1-изопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
54) (3-изопропил-1-метил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
55) (1-этил-3-изопропил-2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-6-ил)карбоногидразоноилдицианид,
56) (1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)(метил)карбоногидразоноилдицианид или
57) ацетил(1,3-диметил-2-оксо-2,3-дигидро-1H-бензо[d]имидазол-5-ил)карбоногидразоноилдицианид.
8. Способ получения соединения по любому из пп. 1-7, включающий:
первую стадию взаимодействия соединения, представленного формулой 5 ниже, включающего реакционноспособную аминогруппу на одном конце, с нитритом натрия и малононитрилом в присутствии кислоты с образованием иминной связи; и
необязательно, вторую стадию введения заместителя R1 в продукт, полученный на предыдущей стадии, когда R1 является заместителем, отличным от водорода: 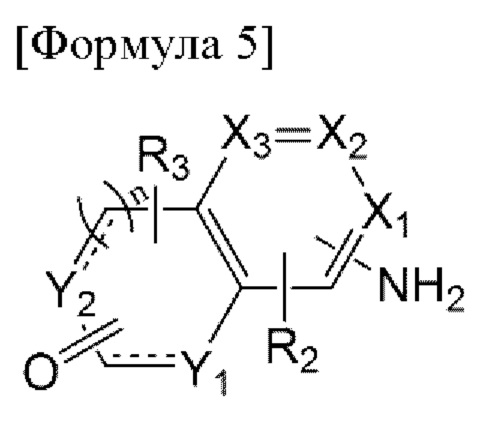
где в формуле 5 выше:
X1 - Х3, Y1, Y2, n,  , R2 и R3 являются такими, как определено в п. 1, и
, R2 и R3 являются такими, как определено в п. 1, и
когда Y1 и Y2 представляют собой N(H), тогда N(H) защищен трет-бутоксикарбонилом.
9. Способ по п. 8, где когда N(H) в Y1 и Y2 защищен трет-бутоксикарбонилом, способ дополнительно включает стадию снятия защиты после указанного взаимодействия.
10. Способ по п. 8, где первую стадию выполняют в виде серии процессов, включающих стадии:
1-1) растворение соединения формулы 5 и нитрита натрия в растворителе C1-4 низший спирт и добавление к нему водного раствора кислоты при температуре от -5°С до 5°С с образованием соли диазония,
1-2) добавление малононитрила к реакционному раствору, включающему соль диазония, полученную на стадии 1-1), и проведение реакции при температуре от 15°С до 40°С, и
1-3) нейтрализация реакционного раствора со стадии 1-2) путем добавления к нему водного раствора основания.
11. Способ по п. 8, где соединение формулы 5 получают из соединения, представленного любой из формул от 5-а до 5-с:
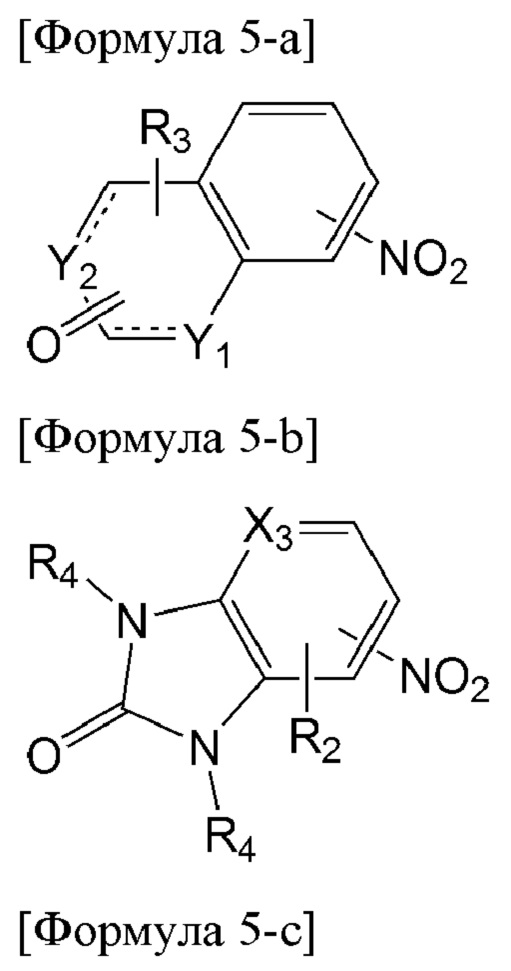
12. Способ по п. 11, дополнительно включающий перед первой стадией стадию восстановления нитрогруппы соединения, представленного одной из формул от 5-а до 5-с, до аминогруппы.
13. Способ по п. 12, где соединение, представленное формулой 5-а выше, получают путем взаимодействия предшественника соединения, в котором один из Y1 и Y2 представляет собой СН, а другой представляет собой О, с соединением на основе амина NH2R4 в состоянии растворения в органическом растворителе, так чтобы О-сайт заместить NR4.
14. Способ по п. 13, где предшественник соединения, представленного формулой 5-а выше, в котором один из Y1 и Y2 представляет собой СН, а другой представляет собой О, необязательно получают путем циклизации между производным нитробензойной кислоты и диметилацеталем N,N-диметилформамида или ацеталем ацетона.
15. Способ по п. 11, где соединение, представленное формулой 5-b или 5-с выше, необязательно получают путем дополнительного проведения стадии введения заместителя R4 посредством реакции с предшественником соединения, включающим R4, и реакционноспособным галогенидом.
16. Способ по п. 11, где соединение, представленное формулой 5-b выше, необязательно получают путем циклизации между незамещенным или R2-замещенным 1,2-диаминнитрофенилом и карбонилдиимидазолом (CDI).
17. Фармацевтическая композиция для ингибирования агрегации тау-белка, содержащая эффективное количество соединения по п. 1 в качестве активного ингредиента и фармацевтически приемлемый носитель.
18. Фармацевтическая композиция для предупреждения или лечения заболевания, вызванного агрегацией тау-белка, содержащая эффективное количество соединения по п. 1 в качестве активного ингредиента и фармацевтически приемлемый носитель.
19. Фармацевтическая композиция по п. 18, где заболевание, вызванное агрегацией тау-белка, выбрано из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, сосудистой деменции, острого инсульта, травмы, цереброваскулярного заболевания, травмы головного мозга, травмы спинного мозга, периферической невропатии, ретинопатии, глаукомы и таупатии.
20. Фармацевтическая композиция по п. 19, где таупатия выбрана из группы, состоящей из хронической травматической энцефалопатии (СТЕ), первичной возрастной таупатии, прогрессирующего надъядерного паралича, кортикобазальной дегенерации, болезни Пика, аргирофильной болезни зерна (AGD), лобно-височной деменции (FTD), паркинсонизма, связанного с хромосомой 17, литико-бодигской болезни (комплекс Гуама паркинсонизм-деменция), ганглиоглиомы, ганглиоцитомы, менингиоангиоматоза, постэнцефалитического паркинсонизма, подострого склерозирующего панэнцефалита, свинцовой энцефалопатии, туберозного склероза, нейродегенерации, ассоциированной с пантотенаткиназой, липофусциноза, посттравматического стрессового расстройства и травматического повреждения головного мозга.
| US 9962384 B1, 08.05.2018 | |||
| EP 3424908 А1, 09.01.2019 | |||
| Crowe Alex et al., Compound screening in cell-based models of tau inclusion formation: Comparison of primary neuron and HEK293 cell assays | |||
| Journal of Biological Chemistry, 2020, vol.295, no.12, p.4001-4013 | |||
| US 2012270836 A1, 25.10.2012 | |||
| WO 03045379 A1, 05.06.2003 | |||
| ЖАРОСТОЙКАЯ ХРОМОНИКЕЛЕВАЯ СТАЛЬ | 0 |
|
SU177080A1 |

