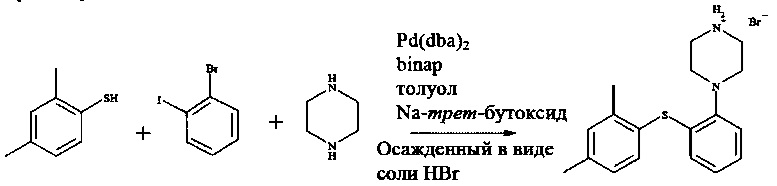
Область изобретения
Изобретение относится к способу получения соединения 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина и его фармацевтически приемлемых солей.
Предпосылки изобретения
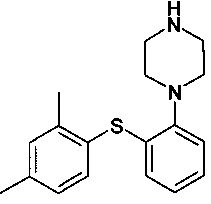
В международных патентных заявках WO 03/029232 и WO 2007/144005 раскрывается соединение 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин, в том числе различные пути получения. В остальной части данного документа 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин и его фармацевтически приемлемые соли обозначают как соединение I или соединение I XX, если требуется обозначить соль XX.
Соединение I является антагонистом 5-HT3, 5-HT7 и 5-HT1D-рецепторов, агонистом 5-HT1A-рецептора, частичным агонистом 5-HT1B-рецептора и ингибитором транспортера серотонина. Дополнительно, соединение I, как было показано, повышает уровни нейротрансмиттеров серотонина, норадреналина, дофамина, ацетилхолина и гистамина в специфических участках головного мозга. Все эти активности считаются клинически значимыми и потенциально связанными с механизмом действия соединения [J. Med. Chem., 54, 3206-3221, 2011; Eur. Neuropshycopharmacol., 18 (suppl 4), S321, 2008; Eur. Neuropshycopharmacol., 21 (suppl 4), S407-408, 2011; Int. J. Psychiatry Clin Pract. 5, 47, 2012].
Как было показано в клинических испытаниях, соединение I является безопасным и эффективным средством для лечения депрессии. Документ, в котором приведены результаты экспериментального подтверждения концепции с целью оценки эффективности и переносимости соединения у пациентов с большим депрессивным расстройством (MDD), под авторством Alvares et al стал доступен в Интернете в Int. J. Neuropsychopharm. 18 июля 2011 г. Результаты этого шестинедельного рандомизированного плацебо-контролируемого исследования с приблизительно 100 пациентами в каждой группе демонстрируют, что соединение I статистически значимо отличается от плацебо при лечении депрессивных и тревожных симптомов у пациентов с MDD. Также сообщается, что не наблюдалось клинически значимых изменений результатов клинических лабораторных исследований, показателей жизненно важных функций, веса или параметров ЭКГ. Результаты долгосрочного исследования также демонстрируют, что соединение I является эффективным при предупреждении рецидива у пациентов, страдающих MDD [Eur. Neuropsychopharmacol. 21 (suppl 3), S396-397, 2011].
В WO 2007/144005 раскрывается способ получения, при котором соединения
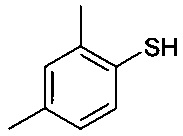 ,
, 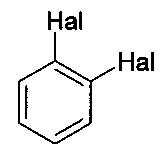 и
и 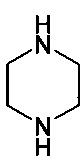
смешивают в присутствии основания и палладиевого катализатора, состоящего из источника палладия и фосфинового лиганда. Палладиевый катализатор катализирует образование связи C-N, как раскрыто в US 5573460. В приведенном выше способе к пиперазину необязательно можно присоединить защитную группу по одному из атомов азота. Этот способ приводит к получению необходимого соединения с высоким выходом и при относительно высокой чистоте. Несмотря на это, образуются примеси, которые затем необходимо удалять. Особенно трудно удалять примеси, которые, подобно соединению I, содержат вторичный амин, т.е. пиперазиновый фрагмент. Такие соединения обычно имеют сходные свойства растворимости, например, в том числе pH-зависимые свойства растворимости, и, следовательно, их трудно отделить от соединения I при помощи способов, в которых используется разница в растворимости, например, кристаллизации. В WO 2010/094285 раскрывается способ очистки, при помощи которого эффективно удаляют такие примеси, например, 1-[2-(2,4-диметилфенилсульфанил)фенил]-4-(2-пиперазин-1-илфенил)пиперазин, который образуется, если оба атома азота в пиперазиновой группе образуют связь C-N. Способ очистки включает осаждение изопропанольного сольвата соединения I, HBr.
Настоящее изобретение предоставляет новый путь получения, при котором устранены или уменьшены вышеупомянутые трудноудаляемые примеси.
Краткое описание изобретения
Автор настоящего изобретения обнаружил, что реакция между 1-галоген-2,4-диметилфенилом, 2-галогентиофенолом и необязательно защищенным пиперазином в присутствии основания и палладиевого катализатора предоставляет реакцию с высоким выходом и чистотой, и при этом примеси уменьшены или устранены. Соответственно, в одном варианте осуществления настоящее изобретение относится к способу получения
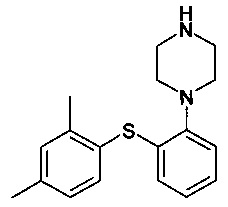
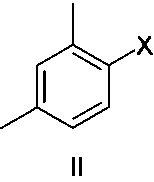
или его фармацевтически приемлемых солей (соединения I), причем указанный способ включает осуществление реакции соединения формулы II
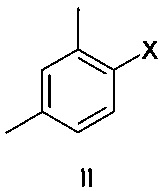
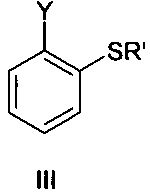
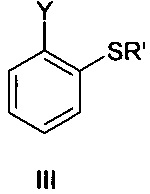
где X представляет собой Br или I, с соединением формулы III
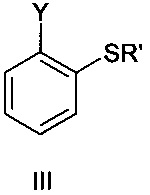
где Y представляет собой Cl или Br и Rʹ представляет собой водород или ион металла, и соединением формулы IV
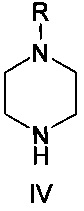
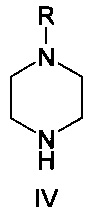
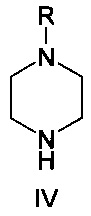
где R представляет собой водород или защитную группу, в присутствии растворителя, основания и палладиевого катализатора, состоящего из источника палладия и фосфинового лиганда, при температуре от 50 до 130°C.
Подробное описание изобретения
В одном варианте осуществления на первом этапе можно осуществлять реакцию соединения формулы II и соединения формулы III с получением промежуточного соединения,
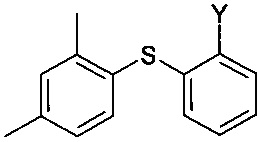 .
.
Данное промежуточное соединение затем можно выделять перед последующей реакцией с соединением формулы IV с получением соединения I. В альтернативном случае, эта дополнительная реакция может происходить без выделения промежуточного соединения. Синтез в одном реакционном сосуде, т.е. пути синтеза, при которых все реагенты загружают в реактор в начале реакции без выделения или очистки промежуточных соединений, как правило, является предпочтительными путями из-за присущей им простоты. С другой стороны, при синтезе в одном реакционном сосуде также увеличивается количество возможных нежелательных побочных реакций, причем с соответствующим увеличением побочных продуктов и уменьшением выхода. Что касается способа по настоящему изобретению, известно, что пиперазин содержит два идентичных вторичных амина, оба из которых потенциально могут принимать участие в образовании связи С-N. Несмотря на это, было обнаружено, что данный путь получения очень эффективен и позволяет избежать таких побочных реакций. В частности, было обнаружено, что данный способ получения позволяет эффективно избежать или уменьшить образование примесей, образующихся при образовании связи C-N при участии вторичного амина в соединении I. Примеры таких примесей включают 1-(2,4-диметилфенил)-4-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин.
В WO 2010/094285 раскрывается способ очистки, который можно применять для удаления примесей, образующихся при участии второго амина в пиперазиновом фрагменте в образовании связи C-N. Способ очистки, раскрытый в WO 2010/094285, применяется в способе получения соединения I, при котором осуществляют реакцию 2,4-диметилтиофенола, 1,2-дигалогенбензола и необязательно замещенного пиперазина в присутствии палладиевого катализатора. Этот способ был впервые раскрыт в WO 2005/144005. Данные, представленные в WO 2010/094285, по всей видимости, указывают на то, что примеси, образующиеся при участии второго амина в образовании связи C-N в этой реакции, например 1-[2-(2,4-диметилфенилсульфанил)фенил]-4-(2-пиперазин-1-илфенил)пиперазин, образуются в количестве от 0,5 до 4,8%. Это согласуется с данными, показанными в Примере 4 в данной заявке, где образуется 1% этой примеси. Как обсуждается ниже, палладиевый катализатор также катализирует образование связей C-S. Кроме того, следует отметить, что в способе получения по настоящему изобретению примеси, образующиеся при реакции двух (или более) тиоловых соединений (соединение формулы III), не образуются в сколько-нибудь существенном количестве.
Как показано в примерах, настоящее изобретение предоставляет альтернативный способ получения для соединения I, при котором образуется мало примесей или они не образуются путем образования связи C-N по второму атому азота пиперазина. В то же время, уровень всех примесей также снижается по сравнению со способом, раскрытым в WO 2005/144005, и общий выход сохраняется на высоком уровне. Кроме того, способ получения по настоящему изобретению предоставляет более простой общий способ, характеризующийся тем, что можно избежать этапов очистки для удаления примесей, образующихся путем образования связи C-N по второму атому азота пиперазина, например, раскрытых в WO 2010/094285. Данные, показанные в примерах 1-4, демонстрируют, что примеси, образующиеся при участии второго амина в образовании связи C-N, например примесь А и В, с практической точки зрения не образуются. Это выгодно отличается от уровня таких примесей, составляющего приблизительно 1%, при применении способа из WO 2005/144005, как продемонстрировано в примере 4, и даже больших количеств, приведенных в WO 2010/094285. Также следует отметить, что общий уровень примесей значительно ниже, т.е. на приблизительно 50% ниже, при применении способа получения по настоящему изобретению, что опять же означает, что чистота соединения I, полученного в способе получения по настоящему изобретению, намного выше.
Соединение формулы II представляет собой 1-галоген-2,4-диметилфенил, где указанный галоген выбран из Br и I. В одном варианте осуществления соединение формулы II представляет собой 1-йод-2,4-диметилфенил.
Соединение формулы III представляет собой 2-галогентиофенол, где указанный галоген выбран из Cl и Br. В одном варианте осуществления соединение формулы III представляет собой 2-бромтиофенол.
Соединение формулы III представляет собой тиол или соответствующий тиолат. Из-за основных условий реакции участвующая в реакции частица представляет собой тиолат. С точки зрения гигиены труда может быть целесообразным использовать тиолат, такой как тиолат Li+, Na+, K+ или Ca++, во избежание проблем с запахом, присущим тиолам. При этом в одном варианте осуществления Rʹ представляет собой водород.
Соединения формулы II и III обычно добавляют в эквимолярных количествах, и эти соединения также обычно добавляют в лимитирующем количестве.
Соединение формулы IV представляет собой пиперазиновое соединение. Пиперазин имеет два атома азота, причем необходимо, чтобы только один из них участвовал в образовании связи C-N. В одном варианте осуществления образования связей со вторым атомом азота избегают путем применения монозащищенного пиперазина, т.е. вариант осуществления, где R представляет собой защитную группу. Много защитных групп известно в данной области техники, и пригодные примеры включают -C(=O)O-W, -C(=O)-W, boc, Bn и Cbz, и, в частности, boc. W представляет собой алкил или арил; Bn является сокращением бензила; boc является сокращением t-бутилоксикарбонила; и cbz является сокращением бензилоксикарбонила. Если в реакциях используется защищенный пиперазин, защитная группа должна быть удалена на следующем этапе, обычно после добавления водной кислоты. Способ по настоящему изобретению, как было обнаружено, приводит в результате только к низким уровням примесей, образующихся путем образования связи C-N по второму амину пиперазина. Это дает возможность использовать незащищенный пиперазин (т.е. R представляет собой водород). Применение незащищенного пиперазина обеспечивает по существу более простой способ, характеризующийся тем, что можно избежать этапа снятия защитной группы.
Соединение формулы IV обычно добавляют в количестве 1-100 эквивалентов, например 1-10 эквивалентов, обычно 1-3 эквивалентов. В альтернативном случае, пиперазин можно применять в качестве растворителя, т.е. вариант осуществления, где пиперазин представляет собой как реагент, так и растворитель.
Растворитель, применяемый в способе по настоящему изобретению, можно выбрать из апротонных органических растворителей или смесей таких растворителей с температурой кипения в пределах диапазона температуры реакции, т.е. 50-130°C. Обычно растворитель выбирают из толуола, ксилола, триэтиламина, трибутиламина, диоксана, N-метилпирролидона, пиридина или из любой их смеси. В особенности следует упомянуть толуол в качестве растворителя.
Самым важным в способе по настоящему изобретению является применение палладиевого катализатора, без которого соединение I не образуется. Как обсуждалось выше, палладиевый катализатор катализирует образование связи C-N, но также образование связи C-S [Bull. Chem. Soc. Jpn., 53, 1385-1389, 1980]. Палладиевый катализатор состоит из источника палладия и фосфинового лиганда. Пригодные источники палладия включают палладий в различных степенях окисления, таких как, например, 0 и II. Примеры источников палладия, которые можно применять в способе по настоящему изобретению, представляют собой Pd2(dba)3, Pd(dba)2 и Pd(OAc)2. Dba является сокращением дибензилиденацетона. В особенности следует упомянуть Pd(dba)2 и Pd(OAc)2. Источник палладия обычно применяют в количестве от 0,1 до 6 мол. %, например от 0,5 до 2 мол. %, обычно приблизительно 1 мол. %. Во всей данной заявке мол. % и эквивалент рассчитаны относительно лимитирующего реагента.
Известны многочисленные фосфиновые лиганды, как монодентатные, так и бидентатные. Пригодные фосфиновые лиганды включают рацемический 2,2ʹ-бис-дифенилфосфанил-[1,1ʹ]бинафталенил (rac-BINAP), (S)-BINAP, (R)-BINAP, 1,1ʹ-бис(дифенилфосфин)ферроцен (DPPF), бис-(2-дифенилфосфинфенил)эфир (DPEphos), три-t-бутилфосфин (соль Фу),
бифенил-2-илди-t-бутилфосфин,
бифенил-2-илдицикпогексилфосфин,
(2ʹ-дициклогексилфосфанилбифенил-2-ил)диметиламин,
[2ʹ-(ди-t-бутилфосфанил)бифенил-2-ил]диметиламин и
дициклогексил(2ʹ,4ʹ,6ʹ-трипропилбифенил-2-ил)фосфан.
Более того, карбеновые лиганды, такие как, например, хлорид 1,3-бис-(2,6-диизопропилфенил)-3H-имидазол-1-ия можно применять вместо фосфиновых лигандов. В одном варианте осуществления фосфиновый лиганд представляет собой rac-BINAP, DPPF или DPEphos и, в частности, rac-BINAP. Фосфиновый лиганд обычно применяют в количестве от 0,2 до 12 мол. %, например от 0,5 до 4 мол. %, обычно приблизительно 2 мол. %.
Основание добавляют к реакционной смеси для повышения значения pH. В частности, основания, выбранные из NaOt-Bu, KOt-Bu, Na2CO3, K2CO3 и Cs2CO3, являются пригодными. Органические основания, например 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) и 1,4-диазабицикло[2.2.2]октан (DABCO), также можно применять. В особенности следует упомянуть NaO(t-Bu) и KO(t-Bu). Обычно основание добавляют в количестве приблизительно 2-10 эквивалентов, например 2-5 эквивалентов, например 2,5-3,5 эквивалента.
В некоторых ситуациях может требоваться получение соли присоединения кислоты 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина, а не свободного основания. Соли присоединения кислоты можно получать на дополнительном этапе способа, на котором осуществляют реакцию свободного основания с соответствующей кислотой, такой как, например, молочная кислота, соляная кислота или бромистоводородная кислота. Кислоту можно добавлять непосредственно в реакционную смесь или, в альтернативном случае, можно предварительно очищать свободное основание до любой подходящей степени перед этим этапом. Если свободное основание было выделено в виде твердого соединения, может быть необходимо применять растворитель для введения свободного основания в раствор перед реакцией с кислотой. В одном варианте осуществления водную бромистоводородную кислоту добавляют непосредственно в реакционную смесь без любой предварительной очистки свободного основания. В альтернативном случае, HBr можно добавлять в спиртовом растворе.
В способах, где применяют защищенный пиперазин, защитная группа должна быть удалена, например, при помощи добавления водной кислоты, как объяснено выше. В одном варианте осуществления указанную водную кислоту можно выбрать для осуществления двух превращений, т.е. снятия защитной группы защищенного пиперазина и образования соли присоединения кислоты. В частности, водную бромистоводородную кислоту можно применять для снятия защитной группы защищенного пиперазина и для получения соли присоединения бромистоводородной кислоты на одном этапе способа.
В одном варианте осуществления способ по настоящему изобретению осуществляют при 75-120°С или при 80-120°С.
Применительно ко всем реакциям и реакционным смесям, упомянутым в данном документе, может быть преимущественным продувать их инертным газом или работать с ними под слоем инертного газа. Азот является дешевым и легко доступным примером инертного газа.
В одном варианте осуществления соединение I получают в способе, который включает осуществление реакции
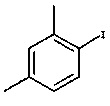
с
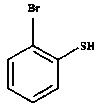
и
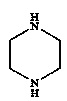
в присутствии растворителя, основания и палладиевого катализатора, состоящего из источника палладия и фосфинового лиганда, при температуре от 50 до 130°C. В дополнительном варианте осуществления осуществляют реакцию полученного соединения с кислотой с получением необходимой фармацевтически приемлемой соли 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина.
В одном варианте осуществления настоящее изобретение предоставляет способ получения соединения I, который включает этапы
a) растворения или диспергирования 0,1 мол. %-3 мол. % бис(дибензилиденацетон)палладия(0), 0,5 мол. %-4 мол. % рацемического 2,2ʹ-бис-дифенилфосфанил-[1,1ʹ]бинафталенила и 2-6 эквивалентов основания в толуоле с получением смеси А;
b) добавления 1 эквивалента 1-йод-2,4-диметилбензола, 0,8-1,2 эквивалента 2-бромтиофенола и 1-10 эквивалентов пиперазина к смеси А с получением смеси В; и
c) нагревания смеси В до 80-120°C или 75-120°С с получением
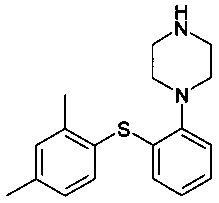 ; и
; и
d) необязательно добавления соответствующей кислоты к продукту, полученному на этапе с), с получением соответствующей соли. В особенности следует упомянуть добавление водной HBr.
В одном варианте осуществления настоящее изобретение относится к соединению I и, в частности, соединению I HBr, полученному в способе, включающем этапы a)-d), приведенные выше, или состоящем из них.
В одном варианте осуществления настоящее изобретение предоставляет способ получения соединения I, который включает этапы
a) растворения или диспергирования 0,1 мол. %-3 мол. % бис(дибензилиденацетон)палладия(0) и 0,5 мол. %-4 мол. % рацемического 2,2ʹ-бис-дифенилфосфанил-[1,1ʹ]бинафталенила, 1-10 эквивалентов пиперазина и 2-6 эквивалентов основания в толуоле с получением смеси А;
b) добавления 1 эквивалента 1-йод-2,4-диметилбензола и 0,8-1,2 эквивалента 2-бромтиофенола к смеси А с получением смеси В; и
c) нагревания смеси В до 80-120°C или 75-120°С с получением
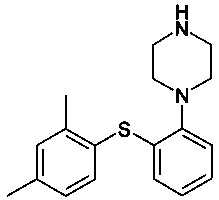 ; и
; и
d) необязательно добавления соответствующей кислоты к продукту, полученному на этапе с), с получением соответствующей соли. В особенности следует упомянуть добавление водной HBr.
В одном варианте осуществления настоящее изобретение относится к соединению I и, в частности, соединению I HBr, полученному в способе, включающем этапы a)-d), приведенные выше, или состоящем из них.
Все ссылки, включая публикации, патентные заявки и патенты, приведенные в данном документе, включены при помощи ссылки в данный документ во всей своей полноте и в той же степени, как если бы каждая ссылка была индивидуально и конкретно обозначена для включения при помощи ссылки и приведена во всей своей полноте в данном документе (в максимальной степени, допускаемой законом), вне зависимости от любого другого отдельно сделанного в любом месте данного документа включения конкретных документов.
Использование в контексте описания настоящего изобретения форм единственного и множественного числа следует понимать как взаимозаменяемое, если в данном документе не указано иное или это однозначно не противоречит контексту. Например, выражение "соединение" следует понимать как относящееся к различным соединениям по настоящему изобретению или конкретному описанному аспекту, если не указано иное.
Описание в данном документе любого аспекта или аспекта настоящего изобретения с использованием терминов, таких как "включающий", "имеющий", "в том числе" или "содержащий" по отношению к элементу или элементам предназначено для подтверждения сходного аспекта или аспекта настоящего изобретения, который "состоит из", "практически состоит из" или "по существу включает" этот конкретный элемент или элементы, если не указано иное или это однозначно не противоречит контексту (например, композиция, описанная в данном документе как включающая определенный элемент, должна также пониматься как описывающая композицию, состоящую из этого элемента, если не указано иное или это однозначно не противоречит контексту).
Примеры
Пример 1
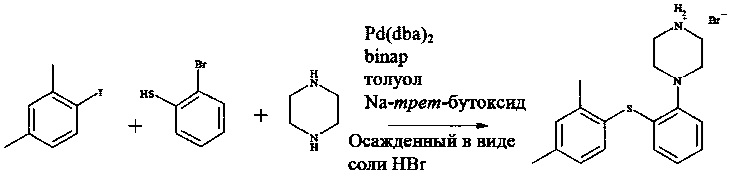
Смешивали бис(дибензилиденацетон)палладий(0) (0,610 г, 1,06 ммоль), рацемический 2,2ʹ-бис(дифенилфосфин)-1,1ʹ-бинафтил (1,34 г, 2,15 ммоль), трет-бутоксид натрия (31,0 г, 323 ммоль) и толуол (150 мл, дегазированный). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре (23°C) в атмосфере азота.
К этой смеси добавляли пиперазин (23,0 г, 267 ммоль), 1-йод-2,4-диметилбензол (25,0 г, 108 ммоль) и 2-бромтиофенол (20,5 г, 108 ммоль). Реакционную смесь затем нагревали при 100°C в течение 5 часов, после чего ее охлаждали до комнатной температуры. К реакционной смеси добавляли воду (80 мл) (в этот момент отбирали образцы для IPC (технологический контроль)) и затем целит 545 (8,0 г). Реакционную смесь перемешивали в течение 20 минут перед фильтрацией. Фазы разделяли и толуольную фазу промывали 2 раза водой (2×80 мл).
Толуольную фазу нагревали при 60°C. К теплой толуольной фазе добавляли бромистоводородную кислоту (8,9 М, 13,0 мл, 116 ммоль), добавляли затравочные кристаллы ((соль HBr) титульного соединения, 10 мг) и раствору давали возможность охладиться до комнатной температуры.
1-[2-(2,4-Диметилфенилсульфанил)фенил]пиперазин, HBr выделяли путем фильтрации и осадок на фильтре промывали 2 раза толуолом (2×30 мл).
Выход 80,1% 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина, HBr (33,5 г, 88,3 ммоль).
Чистота 97,9% (ВЭЖХ, УФ-детектирование при 254 нм).
1Н ЯМР (DMSO-d6; 500 МГц): 8,83 (bs, 2Н), 7,34 (d, 1Н), 7,26 (s, 1Н), 7,15 (m, 3H), 6,98 (dd, 1Н), 6,43 (d, 1Н), 3,26 (bm, 4Н), 3,21 (bm, 4Н), 2,33 (s, 3H), 2,25 (s, 3H).
Пример 2
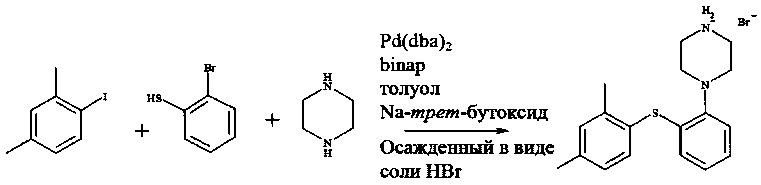
Смешивали бис(дибензилиденацетон)палладий(0) (665 мг, 1,15 ммоль), рацемический 2,2ʹ-бис(дифенилфосфин)-1,1ʹ-бинафтил (1,44 г, 2,30 ммоль), трет-бутоксид натрия (22,2 г, 231 ммоль), пиперазин (16,6 г, 193 ммоль) и толуол (110 мл, дегазированный). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре (23°C) в атмосфере азота.
К этой смеси добавляли 1-йод-2,4-диметилбензол (15,1 г, 65,0 ммоль) и 2-бромтиофенол (11,8 г, 62,4 ммоль). Реакционную смесь затем нагревали при 100°C в течение 4 часов, после чего ее охлаждали до комнатной температуры. К реакционной смеси добавляли воду (60 мл) (в этот момент отбирали образцы для IPC) и затем целит 545 (9,5 г). Реакционную смесь перемешивали в течение 20 минут перед фильтрацией. Фазы разделяли и толуольную фазу промывали 2 раза водой (2×60 мл).
Толуольную фазу нагревали при 60°C. К теплой толуольной фазе добавляли бромистоводородную кислоту (8,9 М, 9,3 мл, 82,8 ммоль), добавляли затравочные кристаллы (соль HBr титульного соединения, 10 мг) и раствору давали возможность охладиться до комнатной температуры.
1-[2-(2,4-Диметилфенилсульфанил)фенил]пиперазин, HBr выделяли путем фильтрации и осадок на фильтре промывали 3 раза толуолом (3×25 мл).
Выход 77,1% 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина, HBr (18,6 г, 49,0 ммоль).
Чистота 98,2% (ВЭЖХ, УФ-детектирование при 254 нм).
1Н ЯМР (DMSO-d6): 8,83 (bs, 2Н), 7,34 (d, 1Н), 7,26 (s, 1Н), 7,14 (m, 3H), 6,97 (dd, 1Н), 6,42 (d, 1Н), 3,26 (bm, 4Н), 3,21 (bm, 4Н), 2,33 (s, 3H), 2,25 (s, 3H).
Пример 3
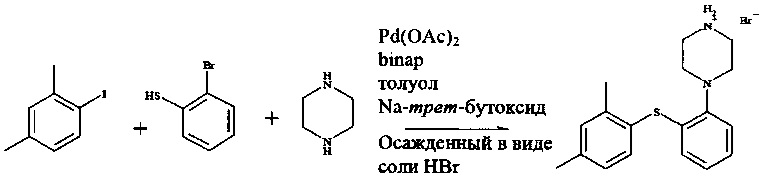
Смешивали ацетат палладия(II) (580 мг, 2,58 ммоль), рацемический 2,2ʹ-бис(дифенилфосфин)-1,1ʹ-бинафтил (3,20 г, 5,16 ммоль), трет-бутоксид натрия (49,5 г, 516 ммоль), пиперазин (37,0 г, 430 ммоль) и толуол (250 мл, дегазированный). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре (23°C) в атмосфере азота.
К этой смеси добавляли 1-йод-2,4-диметилбензол (40,0 г, 172 ммоль) и 2-бромтиофенол (32,6 г, 172 ммоль). Реакционную смесь затем нагревали при 100°C в течение 1 часа, после чего ее охлаждали до комнатной температуры. К реакционной смеси добавляли воду (80 мл) (в этот момент отбирали образцы для IPC) и затем целит 545 (12 г). Реакционную смесь перемешивали в течение 20 минут перед фильтрацией. Фазы разделяли и толуольную фазу промывали 2 раза водой (2×80 мл).
Толуольную фазу нагревали при 60°C. К теплой толуольной фазе добавляли бромистоводородную кислоту (8,9 М, 20,8 мл, 185 ммоль), добавляли затравочные кристаллы (соль HBr титульного соединения, 10 мг) и раствору давали возможность охладиться до комнатной температуры.
1-[2-(2,4-Диметилфенилсульфанил)фенил]пиперазин, HBr выделяли путем фильтрации и осадок на фильтре промывали 3 раза толуолом (3×40 мл).
Выход 84,3% 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина, HBr, содержащего 0,60 экв. трет-бутанола (62,1 г, 147 ммоль).
Чистота 98,6% (ВЭЖХ, УФ-детектирование при 254 нм).
1Н ЯМР (DMSO-d6): 8,82 (bs, 2Н), 7,34 (d, 1Н), 7,26 (s, 1Н), 7,15 (mp, 3Н), 6,98 (dd, 1Н), 6,43 (d, 1Н), 3,27 (bm, 4Н), 3,21 (bm, 4Н), 2,34 (s, 3Н), 2,25 (s, 3Н), 1,12 (s, 5,4Н).
Пример 4
Смешивали бис(дибензилиденацетон)палладий(0) (307 мг, 0,530 ммоль), рацемический 2,2ʹ-бис(дифенилфосфин)-1,1ʹ-бинафтил (0,66 г, 1,06 ммоль), трет-бутоксид натрия (44,7 г, 466 ммоль), пиперазин (40,3 г, 469 ммоль) и толуол (200 мл, дегазированный). Реакционную смесь перемешивали в течение 35 минут при комнатной температуре (23°C) в атмосфере азота.
К этой смеси добавляли 1-бром-2-йодбензол (39,2 г, 135 ммоль) и 2,4-диметилбензолтиол (18,3 г, 133 ммоль). Реакционную смесь затем нагревали с обратным холодильником в течение 6 часов, после чего ее охлаждали до комнатной температуры. К реакционной смеси добавляли воду (60 мл) (в этот момент отбирали образцы для IPC) и затем целит 545 (9,0 г). Реакционную смесь перемешивали в течение 20 минут перед фильтрацией. Фазы разделяли и толуольную фазу промывали 2 раза водой (2×60 мл).
Толуольную фазу нагревали при 60°C. К теплой толуольной фазе добавляли бромистоводородную кислоту (8,9 М, 16,8 мл, 150 ммоль), добавляли затравочные кристаллы (соль HBr титульного соединения, 10 мг) и раствору давали возможность охладиться до комнатной температуры.
1-[2-(2,4-Диметилфенилсульфанил)фенил]пиперазин, HBr выделяли путем фильтрации и осадок на фильтре промывали 3 раза толуолом (3×25 мл).
Выход 81,5% 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина, HBr, содержащего 0,70 экв. трет-бутанола (51,2 г, 118 ммоль).
Чистота 96,0% (ВЭЖХ, УФ-детектирование при 254 нм).
1Н ЯМР (DMSO-d6): 8,78 (bs, 2Н), 7,34 (d, 1Н), 7,26 (s, 1Н), 7,15 (m, 3H), 6,98 (dd, 1Н), 6,43 (d, 1Н), 3,27 (bm, 4Н), 3,20 (bm, 4Н), 2,34 (s, 3H), 2,26 (s, 3H), 1,12 (s, 6,3H).
В таблице ниже показаны данные анализа примесей (площадь, % при помощи ВЭЖХ) для IPC и конечного продукта применительно к примерам 1-4.
Способ ВЭЖХ
- Тип колонки: Acquity UPLC BEH С18 1,7 мкм; 2,1×150 мм.
- Температура колонки: 60°C.
- Детектирование при 254 нм.
- Расход: 0,6 мл/мин.
- Растворители
А: Вода, содержащая 0,05% TFA (трифторуксусной кислоты).
В: Ацетонитрил, содержащий 5% воды и 0,035% TFA.
Примесь А представляет собой 1,2-бис(1ʹ-пиперазинил)бензол.
Примесь В представляет собой 1-фенилпиперазин.
Примесь С представляет собой 1-(2-бромфенил)пиперазин.
Примесь D представляет собой 1-(2,4-диметилфенил)пиперазин.
Примесь Е представляет собой 1-[2-(2,4-диметилфенилсульфанил)фенил]-4-(2-пиперазин-1-илфенил)пиперазин.
Примесь F представляет собой 1-(2,4-диметилфенил)-4-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин.
Примесь G представляет собой 2,4-диметил-1-фенилсульфанилбензол.

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВОРТИОКСЕТИНА | 2014 |
|
RU2652265C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО ПРОДУКТА ДЛЯ СИНТЕЗА АНТАГОНИСТОВ мю-РЕЦЕПТОРА ОПИОИДОВ | 2008 |
|
RU2469035C2 |
| ПОДГОТОВКА И ИСПОЛЬЗОВАНИЕ ИНГИБИТОРА КИНАЗЫ | 2016 |
|
RU2691401C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА АКТИВИН-РЕЦЕПТОРОПОДОБНОЙ КИНАЗЫ | 2020 |
|
RU2826600C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА И ЕГО АНАЛОГОВ | 2013 |
|
RU2619928C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-( 4'- ИЗОБУТИЛФЕНИЛ)ПРОПИОНОВОЙ КИСЛОТЫ (ИБУПРОФЕНА) (ЕГО ВАРИАНТЫ) | 1988 |
|
RU2005715C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ АВЕРМЕКТИНА | 1998 |
|
RU2211221C2 |
| ДИФОСФИНЫ И МЕТАЛЛОКОМПЛЕКСЫ | 2006 |
|
RU2408600C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ БИФЕНИЛИМИДАЗОЛА | 2010 |
|
RU2552350C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ 5-(2,6-ДИ-4-МОРФОЛИНИЛ-4-ПИРИМИДИНИЛ)-4-ТРИФТОРМЕТИЛПИРИДИН-2-АМИНА | 2013 |
|
RU2646760C2 |
Изобретение относится к способу получения 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина или его фармацевтически приемлемых солей
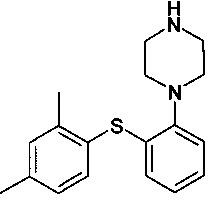 ,
, 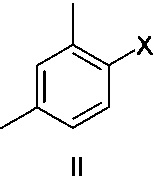 ,
,  ,
, 
при этом способ включает осуществление реакции соединения формулы II, где X представляет собой Br или I, с соединением формулы III, где Υ представляет собой Cl или Br, и R’ представляет собой водород или ион металла, и соединением формулы IV, где R представляет собой водород или защитную группу, в присутствии растворителя, основания и палладиевого катализатора, состоящего из источника палладия и фосфинового лиганда, при температуре от 50 до 130°C. Технический результат: предложен новый способ получения 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазина с высоким выходом и чистотой, который позволяет избежать или уменьшить образование примесей. 17 з.п. ф-лы, 2 табл., 4 пр.
1. Способ получения
или его фармацевтически приемлемых солей (соединения I), при этом способ включает осуществление реакции соединения формулы II,
где X представляет собой Br или I, с соединением формулы III,
где Υ представляет собой Cl или Br и R’ представляет собой водород или ион металла, и соединением формулы IV,
где R представляет собой водород или защитную группу, в присутствии растворителя, основания и палладиевого катализатора, состоящего из источника палладия и фосфинового лиганда, при температуре от 50 до 130°C.
2. Способ по п. 1, отличающийся тем, что X представляет собой I.
3. Способ по п. 1 или 2, отличающийся тем, что Υ представляет собой Br, a R’ представляет собой H.
4. Способ по п. 2, отличающийся тем, что R представляет собой H.
5. Способ по п. 2, отличающийся тем, что R представляет собой защитную группу, выбранную из -C(=O)O-W, -C(=O)-W, boc, Bn и Cbz, где W представляет собой алкил или арил.
6. Способ по п. 2, отличающийся тем, что указанный растворитель является апротонным растворителем.
7. Способ по п. 6, отличающийся тем, что указанный растворитель представляет собой толуол.
8. Способ по п. 2, отличающийся тем, что указанный источник палладия выбран из Pd(dba)2, Pd(OAc)2 и Pd2(dba)3.
9. Способ по п. 8, отличающийся тем, что указанный источник палладия представляет собой Pd(dba)2 или Pd(OAc)2.
10. Способ по п. 2, отличающийся тем, что указанный фосфиновый лиганд выбран из:
рацемического 2,2’-бис-дифенилфосфанил-[1,1’]бинафталенила (rac-BINAP),
1,1’-бис(дифенилфосфин)ферроцена (DPPF),
бис-(2-дифенилфосфинфенил)эфира (DPEphos),
три-t-бутилфосфина (соль Фу),
бифенил-2-илди-t-бутилфосфина,
бифенил-2-илдициклогексилфосфина,
(2’-дициклогексилфосфанилбифенил-2-ил)диметиламина,
[2’-(ди-t-бутилфосфанил)бифенил-2-ил]диметиламина и
дициклогексил(2’,4’,6’-трипропилбифенил-2-ил)фосфана.
11. Способ по п. 10, отличающийся тем, что указанный фосфиновый лиганд представляет собой 2,2’-бис-дифенилфосфанил-[1,1’]бинафталенил (rac-BINAP).
12. Способ по п. 2, отличающийся тем, что указанное основание выбрано из NaO(t-Bu), KO(t-Bu), K2CO3, Na2CO3, Cs2CO3, DBU и DABCO.
13. Способ по п. 12, отличающийся тем, что указанное основание представляет собой NaO(t-Bu).
14. Способ по п. 1, дополнительно включающий осуществление реакции продукта, полученного согласно указанным пунктам, с соответствующей кислотой с удалением защитной группы (если R представляет собой защитную группу) и/или с получением необходимой фармацевтически приемлемой соли.
15. Способ по п. 1, включающий осуществление реакции
с
и
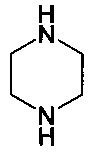
в присутствии растворителя, основания и палладиевого катализатора, состоящего из источника палладия и фосфинового лиганда, при температуре от 50 до 130°C.
16. Способ по п. 15, дополнительно включающий осуществление реакции соединения, полученного согласно указанному пункту, с кислотой с получением необходимой фармацевтически приемлемой соли указанного соединения.
17. Способ по п. 1, включающий этапы:
a) растворения или диспергирования 0,1-3 мол.% бис(дибензилиденацетон)палладия(0), и 0,5-4 мол.% рацемического 2,2’-бис-дифенилфосфанил-[1,1’]бинафталенила, и 2-6 эквивалентов основания в толуоле с получением смеси A;
b) добавления 1 эквивалента 1-йод-2,4-диметилбензола, 0,8-1,2 эквивалента 2-бромтиофенола и 1-10 эквивалентов пиперазина к смеси A с получением смеси В; и
c) нагревания смеси В до 80-120°C с получением
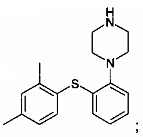 и
и
d) необязательно добавления соответствующей кислоты к продукту, полученному на этапе c), с получением соответствующей соли.
18. Способ по п. 1, включающий этапы:
a) растворения или диспергирования 0,1-3 мол.% бис(дибензилиденацетон)палладия(0) и 0,5-4 мол.% рацемического 2,2’-бис-дифенилфосфанил-[1,1’]бинафталенила, 1-10 эквивалентов пиперазина и 2-6 эквивалентов основания в толуоле с получением смеси А;
b) добавления 1 эквивалента 1-йод-2,4-диметилбензола и 0,8-1,2 эквивалента 2-бромтиофенола к смеси A с получением смеси В; и
c) нагревания смеси B до 80-120°C с получением
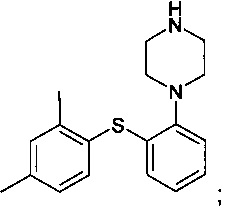 и
и
d) необязательно добавления соответствующей кислоты к продукту, полученному на этапе c), с получением соответствующей соли.
| Видоизменение охарактеризованного в патенте по заяв. свид. № 15997 кузнечного горна | 1927 |
|
SU15287A1 |
| МАШИНА ДЛЯ ОТЛИВКИ ПЛОМБ | 1926 |
|
SU7537A1 |
| WO 03053942 A1, 03.07.2003. | |||

