ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к новым фосфатам и боронатам пиридинон- и пиримидинон-гидроксамовой кислоты. Изобретение также относится к способам применения таких соединений для лечения бактериальных инфекций (в особенности инфекций, вызываемых грамотрицательными бактериями) и к фармацевтическим композициям, содержащим такие соединения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Инфекция, вызываемая грамотрицательными бактериями, такими как Pseudomonas aeruginosa, энтеробактерии, продуцирующие β-лактамазу расширенного спектра (ESBL), и Acinetobacter baumannii, представляет собой серьезную проблему для здоровья, особенно в случае внутрибольничных инфекций. Кроме того, возрастает уровень устойчивости к современным видам антибиотикотерапии, что серьезно ограничивает возможности лечения. Например, в 2002 г. 33% инфекций, вызванных Pseudomonas aeruginosa в отделениях интенсивной терапии, оказались устойчивы к фторхинолонам, при этом устойчивость к имипенему составляла 22% (CID, 42: 657-68, 2006). Помимо этого также растет число инфекций с множественной лекарственной устойчивостью (MDR); в случае Pseudomonas aeruginosa число инфекций с MDR увеличилось с 4% в 1992 г. до 14% в 2002 г. (Biochem. Pharm., 71: 991, 2006).
Грамотрицательные бактерии уникальны тем, что их наружная мембрана содержит липополисахарид (LPS), который принципиально важен для поддержания целостности мембраны и играет существенную роль в плане жизнеспособности бактерий (обзор в Ann. Rev. Biochem., 76: 295-329, 2007). Основным липидным компонентом LPS является липид А, и ингибирование биосинтеза липида А вызывает гибель бактерий. Липид А синтезируется на обращенной в цитоплазму поверхности внутренней мембраны бактерий в ходе пути, в котором участвуют девять разных ферментов. Эти ферменты высоко консервативны у большинства грамотрицательных бактерий. LpxC (деацетилаза УДФ(уридин-5'-дифосфат)-3-О-(R-3-гидроксимиристоил)-N-ацетилглюкозамина) представляет собой фермент, который катализирует осуществляемую первой стадию пути биосинтеза липида А, удаление N-ацетильной группы с УДФ-3-O-(R-3-гидроксимиристоил)-N-ацетилглюкозамина. LpxC является Zn2+ -зависимым ферментом, не имеющим гомолога среди млекопитающих, что делает его хорошей мишенью в плане разработки новых антибиотиков. Сообщалось о нескольких ингибиторах LpxC с низкой аффинностью (наномолярного (нМ) уровня) (Biochemistry, 45: 7940-48, 2006).
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к некоторым новым фосфатам и боронатам пиридинон- и пиримидинон-гидроксамовой кислоты, фармацевтическим композициям, содержащим эти соединения, и способам ингибирования LpxC и лечения бактериальных инфекций этими соединениями.
В одном из воплощений настоящего изобретения предложено новое LpxC-ингибирующее соединение формулы (1), представляющее собой фосфат пиридинон-или пиримидинон-гидроксамовой кислоты, и его стереоизомеры
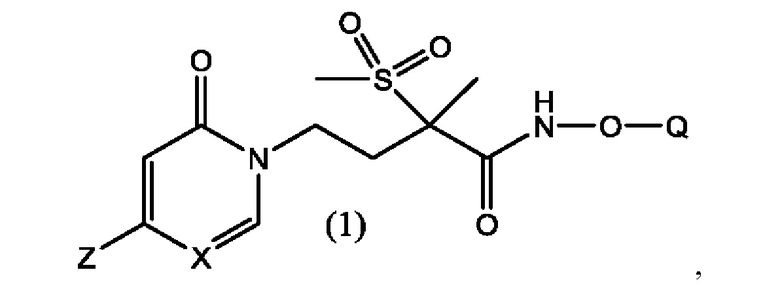
где Q выбран из группы, состоящей из -Р(O)(ОН)2, -Р(O)(ОН)(О-М+), -Р(O)(О-М+)2 и -Р(O)(O)2М2+;
X представляет собой СН или N;
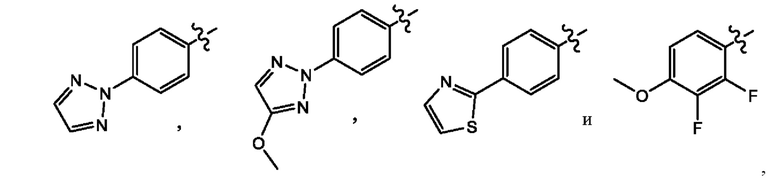
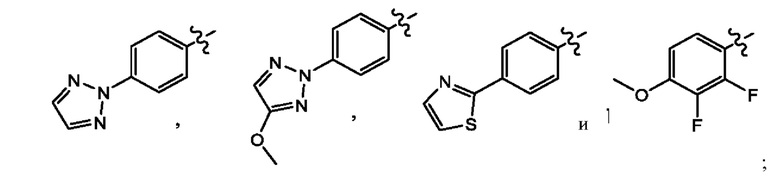
Z выбран из группы, состоящей из
М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион; и
М2+ представляет собой фармацевтически приемлемый двухвалентный катион.
В другом воплощении настоящего изобретения предложено соединение формулы (1а)
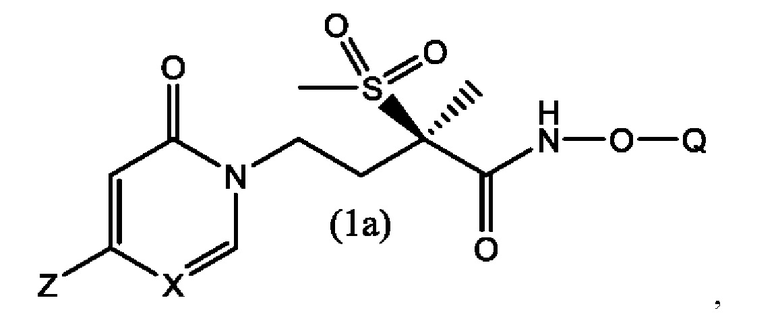
где Q выбран из группы, состоящей из -Р(O)(ОН)2, -Р(O)(ОН)(О-М+), -Р(O)(O-М+)2 и -Р(O)(O)2М2+;
X представляет собой СН или N;
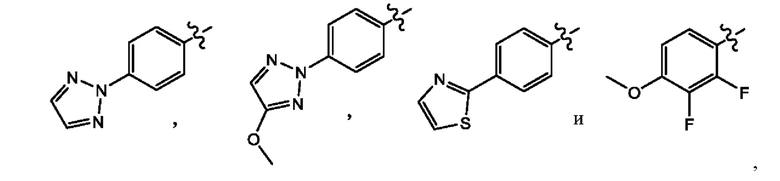
Z выбран из группы, состоящей из
М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион; и
М2+ представляет собой фармацевтически приемлемый двухвалентный катион.
В другом воплощении настоящего изобретения предложено соединение формулы (1а), где X представляет собой СН; Z представляет собой
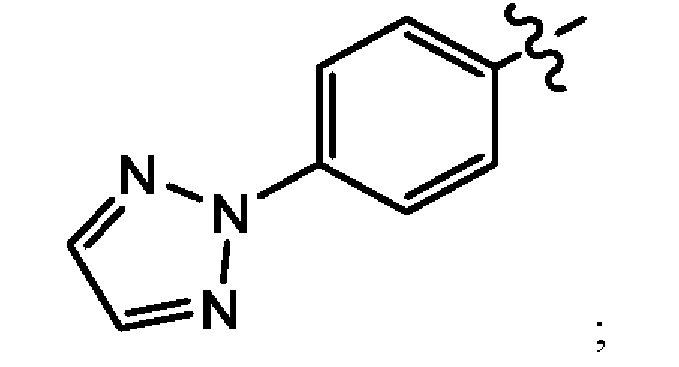
Q выбран из группы, состоящей из -Р(O)(ОН)2, -Р(O)(ОН)(O-М+), -Р(O)(O-М+)2 и -Р(O)(O-)2М2+; М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион; и М2+ представляет собой фармацевтически приемлемый двухвалентный катион.
В еще одном воплощении настоящего изобретения предложено соединение формулы (1а), где X представляет собой СН; Z представляет собой
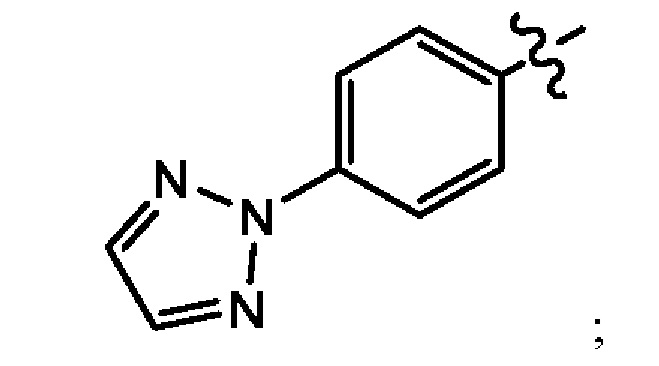
Q представляет собой -P(O)(OH)2; -P(O)(OH)(O-M+); -Р(O)(O-М+)2 или -Р(O)(O-)2М2+; и М+ в каждом случае независимо выбран из группы, состоящей из Li+, K+, Na+, NH4+, NH3+C(CH2OH)3, NH2+(CH2CH3)2, NH2+(CH2CH3)2, пирролидиния и глициния; и где М2+ выбран из группы, состоящей из Са2+, Mg2+ и Zn2+. В другом воплощении М+ в каждом случае независимо выбран из группы, состоящей из Li+, K+ и Na+; или М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион, независимо выбранный из NH4+, NH3+C(CH2OH)3, NH2+(CH2CH3)2, NH2+(CH2CH3)2, пирролидиния и глициния; и где М2+ выбран из группы, состоящей из Са2+, Mg2+ и Zn2+.
В еще одном воплощении настоящего изобретения предложено соединение формулы (1а), выбранное из группы, состоящей из:
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, динатриевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, диаммониевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, дикалиевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, дилитиевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, кальциевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, магниевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, цинковой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, пирролидиновой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, трис-(гидроксиметил)метиламиновой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, диэтиламиновой соли и
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, глициновой соли, и других его фармацевтически приемлемых солей.
В другом воплощении настоящего изобретения предложено соединение формулы (1а), где X представляет собой N; Z представляет собой
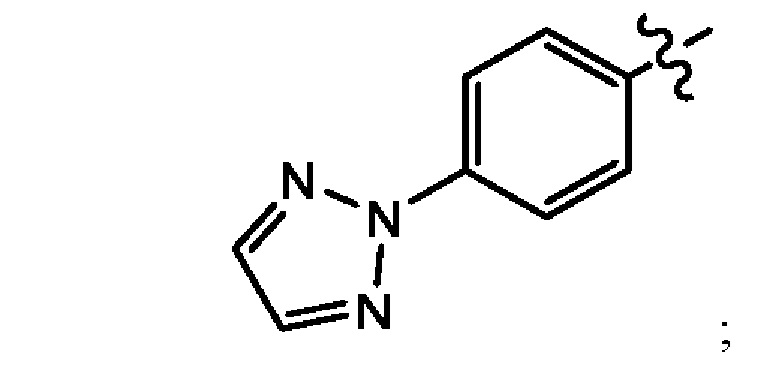
Q выбран из группы, состоящей из -Р(O)(ОН)2, -Р(O)(ОН)(O-М+), -Р(O)(O-М+)2 и -Р(O)(O-)2М2+; М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион; и М2+ представляет собой фармацевтически приемлемый двухвалентный катион.
В еще одном воплощении настоящего изобретения предложено соединение формулы (1а), где X представляет собой N; Z представляет собой
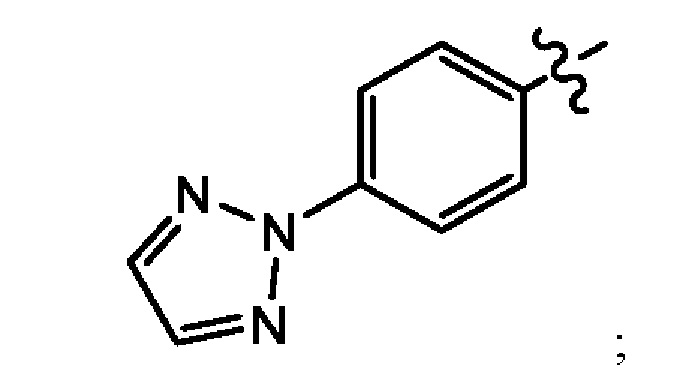
Q представляет собой -Р(O)(ОН)2; -Р(O)(ОН)(O-М+); -Р(O)(O-М+)2 или -Р(O)(O-)2М2+; М+ в каждом случае независимо выбран из группы, состоящей из Li+, K+ и Na+, либо М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион, независимо выбранный из NH4+, NH3+C(CH2OH)3, NH2+(CH2CH3)2, NH2+(CH2CH3)2, пирролидиния и глициния; и где М2+ выбран из группы, состоящей из Са2+, Mg2+ и Zn2+.
В еще одном воплощении настоящего изобретения предложены боронатные пролекарства соединений формулы (1) и формулы (1а), которые представляют собой соединения формулы (2) и формулы (2а), соответственно,
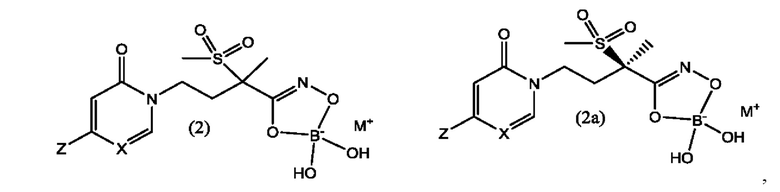
где X представляет собой CH или N; и Z выбран из группы, состоящей из
и М+ представляет собой фармацевтически приемлемый одновалентный катион.
В еще одном воплощении настоящего изобретения предложено соединение формулы (2а), где X представляет собой CH; Z представляет собой
М+ представляет собой фармацевтически приемлемый одновалентный катион, выбранный из группы, состоящей из Li+, K+ и Na+; или М+ представляет собой фармацевтически приемлемый одновалентный катион, независимо выбранный из NH4+, NH3+C(CH2OH)3, NH2+(CH2CH3)2; NH2+(CH2CH3)2; пирролидиния и глициния.
В еще одном воплощении настоящего изобретения предложено соединение формулы (2а), где X представляет собой N; Z представляет собой
где М+ представляет собой фармацевтически приемлемый одновалентный катион, выбранный из группы, состоящей из Li+, K+ и Na+; или М+ представляет собой фармацевтически приемлемый одновалентный катион, независимо выбранный из NH4+, NH3+C(CH2OH)3, NH2+(CH2CH3)2; NH2+(CH2CH3)2; пирролидиния и глициния.
В еще одном воплощении настоящего изобретения предложено соединение формулы (2а), представляющее собой боронатное пролекарство (R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)-бутанамида и его фармацевтически приемлемые соли. В еще одном воплощении настоящего изобретения предложено соединение формулы (2а), представляющее собой
(R)-5-(4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уид натрия, и другие его фармацевтически приемлемые соли.
В еще одном воплощении настоящего изобретения предложено соединение формулы (1а), выбранное из группы, состоящей из:
(2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, динатриевой соли;
(R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, диаммониевой соли;
(2R)-Н-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфата, динатриевой соли;
(2R)-Н-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфата, динатриевой соли;
(R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, динатриевой соли;
(2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2H)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, диаммониевой соли;
(2R)-N-гидрокси-4-{4-[4-(4-метокси-2H-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфата, диаммониевой соли;
(2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфата, аммониевой соли;
(2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, дикалиевой соли;
(2R)-N-гидрокси-4-{4-[4-(4-метокси-2H-l,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфата, дикалиевой соли;
(2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфата, дикалиевой соли;
(R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, дикалиевой соли;
(2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, дилитиевой соли;
(2R)-N-гидрокси-4-{4-[4-(4-метокси-2H-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфата, дилитиевой соли;
(2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(l,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфата, дилитиевой соли и
(R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, дилитиевой соли, и других его фармацевтически приемлемых солей.
В еще одном воплощении настоящего изобретения предложено соединение формулы (2а), выбранное из группы, состоящей из:
(R)-5-(4-(4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уида натрия;
(R)-2,2-дигидрокси-5-(4-(4-(4-(4-метокси-2H-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-1,3,4,2-диоксазаборол-2-уида натрия;
(R)-2,2-дигидрокси-5-(2-(метилсульфонил)-4-(2-оксо-4-(4-(тиазол-2-ил)фенил)пиридин-1(2Н)-ил)бутан-2-ил)-1,3,4,2-диоксазаборол-2-уида натрия и
(R)-5-(4-(4-(4-(2H-l,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6H)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уида натрия; и других его фармацевтически приемлемых солей.
В еще одном воплощении настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (1), формулы (1а), формулы (2) или формулы (2а) в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом, разбавителем или носителем.
В еще одном воплощении настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (1), формулы (1а), формулы (2) или формулы (2а) либо его фармацевтически приемлемую соль в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом, разбавителем или носителем; для введения пациенту посредством перорального, местного или инъекционного введения.
В еще одном воплощении настоящего изобретения предложен способ лечения бактериальной инфекции у пациента, включающий введение терапевтически эффективного количества соединения формулы (1), формулы (1а), формулы (2) или формулы (2а) либо его фармацевтически приемлемой соли пациенту, нуждающемуся в этом. В еще одном воплощении настоящего изобретения предложен способ лечения бактериальной инфекции у пациента, включающий введение терапевтически эффективного количества соединения формулы (1), формулы (1а), формулы (2) или формулы (2а) либо его фармацевтически приемлемой соли пациенту, нуждающемуся в этом, посредством перорального, местного или инъекционного введения.
В еще одном воплощении настоящего изобретения предложено применение соединения формулы (1), формулы (1а), формулы (2) или формулы (2а) либо его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения бактериальной инфекции у пациента.
В еще одном воплощении бактериальная инфекция представляет собой вызываемую грамотрицательными бактериями инфекцию. В еще одном воплощении вызываемая грамотрицательными бактериями инфекция вызвана грамотрицательными бактериями, выбранными из группы, состоящей из Mannheimia haemolytica, Pasteurella multocida, Histophilus somni, Actinobacillus pleuropneumoniae, Salmonella enteritidis, Salmonella gallinarium, Lawsonia intracellular!s, Brachyspira hyodysenteriae, Brachyspira pilosicoli, Acinetobacter baumannii, Acinetobacter spp., Citrobacter spp., Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Serratia marcescens, Stenotrophomonas maltophilia и Pseudomonas aeruginosa. В еще одном воплощении вызываемая грамотрицательными бактериями инфекция выбрана из группы, состоящей из респираторной инфекции, желудочно-кишечной инфекции, нозокомиальной пневмонии, инфекции мочевыводящих путей, бактериемии, сепсиса, кожной инфекции, инфекции мягких тканей, инфекции брюшной полости, легочной инфекции, эндокардита, синдрома диабетической стопы, остеомиелита и инфекции центральной нервной системы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Следующие далее термины, которые использованы по всей данной заявке, включая формулу изобретения, имеют определенные ниже значения, если конкретно не указано иное. Множественное и единственное число следует рассматривать как взаимозаменяемые, за исключением случаев указания числа.
«Алкил» относится к гидрокарбильному заместителю с линейной или разветвленной цепью (т.е. заместителю, получаемому в результате удаления водорода из углеводорода); в одном из воплощений, к заместителю, содержащему от одного (C1) до двенадцати (C12) атомов углерода, т.е. C1-C12. Неограничивающие примеры таких заместителей включают метил, этил (С2), пропил (включая н-пропил и изопропил), бутил (включая н-бутил, изобутил, втор-бутил и трет-бутил), пентил, изоамил, гексил, гептил, октил, нонил, децил, ундецил, додецил и тому подобное.
«Циклоалкил» относится к карбоциклическому заместителю, получаемому в результате удаления водорода из насыщенной карбоциклической молекулы, например, молекулы, имеющей от трех до шести атомов углерода. Термин «С3.6циклоалкил» означает радикал трех-шести-членного кольца, который включает группы циклопропил, циклобутил, циклопентил и циклогексил.
В некоторых случаях число атомов углерода в гидрокарбильном заместителе (т.е. алкиле, циклоалкиле и т.д.) указывается префиксом «Сх-Су-» или «Сх-у», где х представляет собой минимальное, а у представляет собой максимальное число атомов углерода в этом заместителе. Так, например, «С1-С12-алкил» или «С1-12алкил» относится к алкильному заместителю, содержащему от 1 до 12 атомов углерода, а «C1-С6-алкил» или «С1-6алкил» относится к алкильному заместителю, содержащему от 1 до 6 атомов углерода. В качестве другой иллюстрации, С3-С6циклоалкил или С3-6-циклоалкил относится к насыщенной циклоалкильной группе, содержащей от 3 до 6 атомов углерода в кольце.
Термин «соединения по настоящему изобретению» означает соединения формулы (1), формулы (1а), формулы (2) и формулы (2а), их стереоизомеры и их фармацевтически приемлемые соли.
«Двухвалентный катион», определенный в данном описании как М2+, представляет собой катион с валентностью 2 и включает катионы металлов: Mg2+, Са2+ и Zn2+.
«Геометрический изомер» означает любой из двух или более стереоизомеров, которые отличаются расположением атомов или групп атомов относительно структурно жесткой связи, такой как двойная связь, или относительно кольца и определяются как цис- (с расположением на одной и той же стороне) и транс-изомеры (с расположением на противоположных сторонах) относительно этой связи или этого кольца.
«Изомер» означает «стереоизомер» и «геометрический изомер», как он определен в данном описании.
«Одновалентный катион», определенный в данном описании как М+, включает аммоний (NH4+), моно-, ди-, три- и тетра-(С1-С12алкил)аммоний (т.е. (С1-С12алкил)МН3+, (С1-С12алкил)2NH2+, (С1-С12алкил)3NH+ и (С1-С12алкил)4N+), где алкильная(ые) группа(ы) может быть замещена/могут быть замещены так, как указано, моно-, ди-, три- и тетра-(С3-С6циклоалкил)аммоний (т.е. (С3-С6циклоалкил)NH3+, (С3-С6циклоалкил)2NH2+, (С3-С6циклоалкил)3NH+ и (С3-С6циклоалкил)4N+), ионы щелочных металлов, такие как ионы натрия, лития и калия, ионы органических аминов, таких как пирролидин, пиперидин или пиридин, и ионы аминокислот, такие как ионы глицина, аланина, β-аланина, валина, лизина, изолейцина, лейцина, метионина, треонина, аспарагина, глутамина, гистидина, аргинина, орнитина, триптофана, пролина, глутамина, цистеина, фенилаланина, тирозина и серина. Когда органический амин или аминокислота находится в своей протонированной форме, это может быть указано путем использования окончания «ий». Например, протонированный пирролидин означает пирролидиний, протонированный пиперидин означает пиперидиний, протонированный пиридин означает пиридиний, а протонированный глицин означает глициний.
«Исходное соединение» относится к биологически активной структуре, которая высвобождается под действием ферментов в ходе метаболического или катаболического процесса или в результате химического процесса после введения соединений формулы (1) или формулы (1а), представляющих собой фосфатную соль, или соединений формулы (2) или формулы (2а), представляющих собой боронат.
Термин «пациент» относится к теплокровным животным, таким как, например, люди и не являющиеся человеком субъекты. Термин «не являющиеся человеком субъекты» относится к таким животным, как сельскохозяйственные животные (т.е. крупный рогатый скот, свиньи, овцы и козы) и домашние животные (т.е. кошка, собака и лошадь); и также включает других не являющихся человеком животных, например, морских свинок, мышей, крыс, карликовых песчанок, кроликов, обезьян, шимпанзе и тому подобных.
«Фармацевтически приемлемый» указывает на то, что данные вещество или композиция должны быть совместимы с точки зрения химии и/или токсикологии с другими ингредиентами, входящими в состав композиции, и/или с пациентом, которого лечат ими. Данный термин является синонимом термину «ветеринарно приемлемый» (т.е. эти ингредиенты совместимы с не являющимся человеком пациентом).
Термин «пролекарство» относится к соединениям, являющимся предшественниками лекарственных средств, которые, после их введения и всасывания, высвобождают лекарственное средство in vivo в результате протекания некоторого метаболического, катаболического или химического процесса; например, посредством гидролитического отщепления фосфата в соединениях формулы (1) и формулы (1а) или бороната в соединениях формулы (2) и формулы (2а).
В данной заявке термины «пиридон» и «пиридинон» использованы взаимозаменяемо. Никакой разницы или отличительной особенности не подразумевается, если не указано иное.
«Стереоизомер» означает соединения, имеющие один или более хиральных центров, и каждый центр может находиться в R- или S-конфигурации. Стереоизомеры включают все диастереомерные, энантиомерные и эпимерные формы, а также рацематы и их смеси.
«Терапевтически эффективное количество» относится к количеству соединения по изобретению (т.е. соединения формулы I, Ia, II или IIa), которое при введении пациенту обеспечивает желаемый эффект; например, ослабление тяжести симптомов, ассоциированных с бактериальной инфекцией, уменьшение числа бактерий в пораженной ткани и/или предупреждение увеличения числа бактерий в пораженной ткани (локально или системно).
Термин «лечить», «подвергание лечению», «лечение» и тому подобный относится к способности соединений по настоящему изобретению ослаблять, облегчать или замедлять развитие бактериальной инфекции (или вызываемого ей состояния) у пациента или любого повреждения ткани, ассоциированного с данным заболеванием.
Соединения по настоящему изобретению являются ингибиторами LpxC, полезными для лечения пациентов с бактериальной инфекцией, вызываемой грамотрицательными бактериями.
Первым воплощением первого аспекта настоящего изобретения является новое LpxC-ингибирующее соединение формулы (1), представляющее собой фосфат пиридинон- или пиримидинон-гидроксамовой кислоты
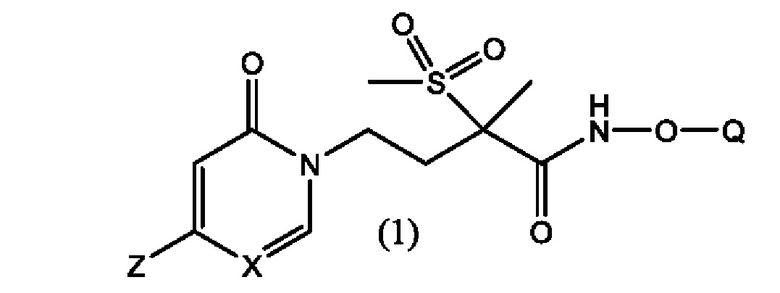
или его фармацевтически приемлемая соль; их стереоизомеры и их фармацевтически приемлемые соли; где Q выбран из группы, состоящей из -Р(O)(ОН)2, -Р(O)(ОН)(O-М+), -Р(O)(O-М+)2 и -Р(O)(O-)2М2+; X представляет собой СН или N; и где Z выбран из группы, состоящей из
М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион; и
М2+ представляет собой фармацевтически приемлемый двухвалентный катион.
Первым воплощением второго аспекта настоящего изобретения является новое LpxC-ингибирующее соединение формулы (2), представляющее собой боронат
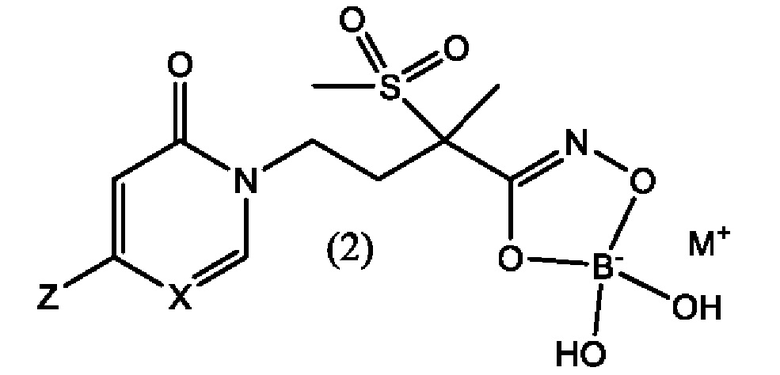
где X представляет собой СН или N; М+ представляет собой фармацевтически приемлемый одновалентный катион; и Z выбран из группы, состоящей из
Соединения формулы (1) и формулы (2), будучи введенными пациенту, нуждающемуся в этом, проявляют антибактериальную активность, в особенности против грамотрицательных микроорганизмов. Эти соединения можно использовать для лечения бактериальных инфекций у млекопитающих, особенно у людей. Соединения также можно использовать для приложений в области ветеринарии, например, для лечения инфекций у сельскохозяйственных животных и домашних животных.
Соединения формулы (1) и формулы (2) полезны для лечения ряда инфекций; особенно инфекций, вызываемых грамотрицательными бактериями, включая нозокомиальную пневмонию, инфекции мочевыводящих путей, системные инфекции (бактериемию и сепсис), инфекции кожи и мягких тканей, хирургические инфекции, инфекции брюшной полости, легочные инфекции (в том числе инфекции у пациентов с кистозным фиброзом), инфекции Helicobacter pylori (и для облегчения сопутствующих желудочных осложнений, таких как язвенная болезнь, канцерогенез в желудке и т.д.), эндокардит, синдромы диабетической стопы, остеомиелит и инфекции центральной нервной системы.
Чтобы упростить процедуру введения, соединения обычно смешивают по меньшей мере с одним эксципиентом и на их основе готовят фармацевтическую лекарственную форму. Примеры таких лекарственных форм включают таблетки, капсулы, растворы/суспензии для инъекций, аэрозоли для ингаляции, кремы/мази для местного применения, для лечения ушей и глаз, растворы/суспензии для перорального приема внутрь и формы, используемые в качестве лечебных кормовых добавок. Соединения по настоящему изобретению обладают повышенной растворимостью в воде по сравнению с исходным соединением гидроксамовой кислоты, из которого они получены, и поэтому соединения по настоящему изобретению могут предпочтительно применяться в лекарственных формах для инъекций.
Вторым воплощением первого аспекта настоящего изобретения является соединение по первому воплощению первого аспекта формулы 1а
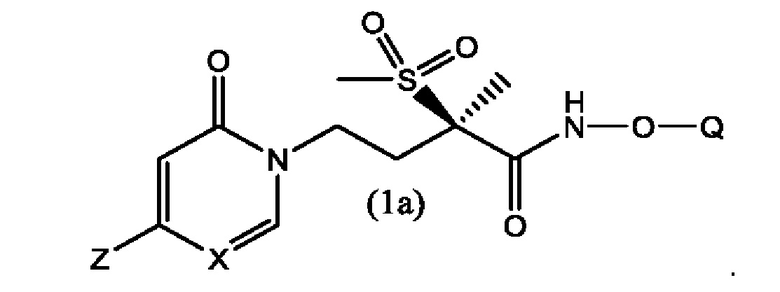
Третьим воплощением первого аспекта настоящего изобретения является соединение по второму воплощению первого аспекта, где X представляет собой СН.
Четвертым воплощением первого аспекта настоящего изобретения является соединение по третьему воплощению первого аспекта, где Z представляет собой
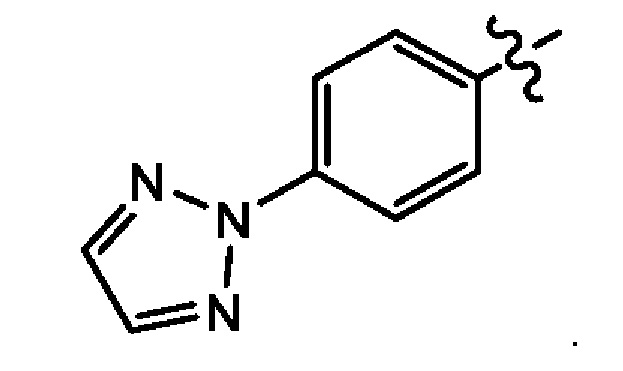
Пятым воплощением первого аспекта настоящего изобретения является соединение по третьему воплощению первого аспекта, где Z представляет собой
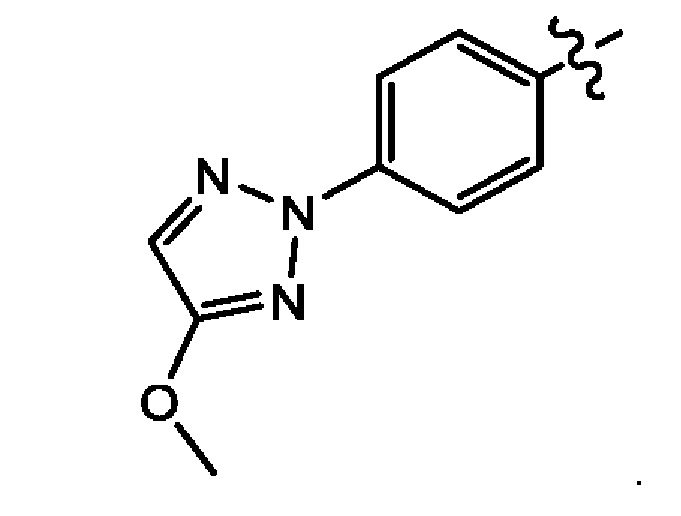
Шестым воплощением первого аспекта настоящего изобретения является соединение по третьему воплощению первого аспекта, где Z представляет собой
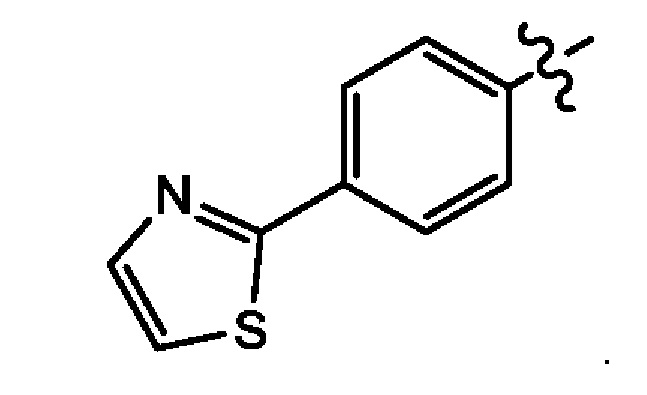
Седьмым воплощением первого аспекта настоящего изобретения является соединение по третьему воплощению первого аспекта, где Z представляет собой
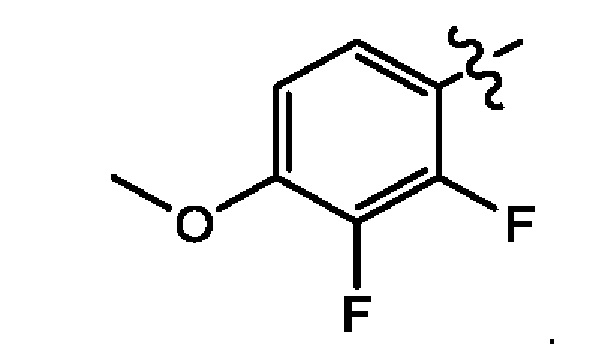
Восьмым воплощением первого аспекта настоящего изобретения является соединение по второму воплощению первого аспекта, где X представляет собой N; и Z представляет собой
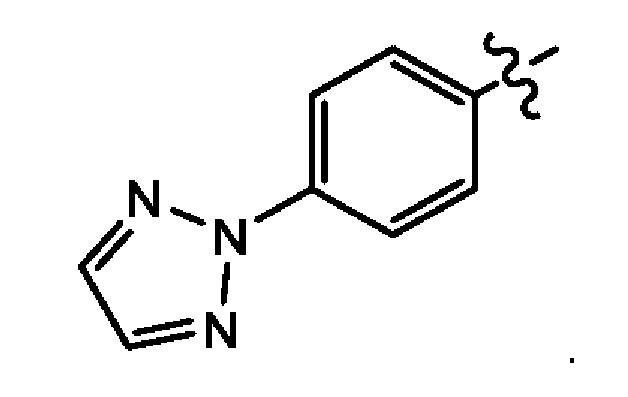
Девятым воплощением первого аспекта настоящего изобретения является соединение по второму воплощению первого аспекта, где Q представляет собой -Р(O)(ОН)2. Десятым воплощением первого аспекта настоящего изобретения является соединение по второму воплощению первого аспекта, где Q представляет собой -Р(O)(ОН)(O-М+) или -Р(O)(O-М+)2. Одиннадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где Q представляет собой -Р(O)(O-М+)2. Двенадцатым воплощением первого аспекта настоящего изобретения является соединение по второму воплощению первого аспекта, где Q представляет собой -Р(O)(O-)2М2+. Тринадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ в каждом случае независимо выбран из группы, состоящей из Li+, K+ и Na+.
Четырнадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион, независимо выбранный из аммония, (С1-С12алкил)аммония, (С1-С12алкил)2аммония, (C1-С12алкил)3аммония, (С1-С12алкил)4аммония, (С3-С6циклоалкил)аммония, (С3-С6циклоалкил)2аммония, (С3-С6циклоалкил)3аммония, (С3-С6циклоалкил)4аммония, пирролидиния, пиперидиния и пиридиния; при этом каждая из группировок (C1-С12алкил) или (С3-С6циклоалкил) возможно замещена одной-тремя группами гидрокси или одним-тремя атомами галогена.
Пятнадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ в каждом случае представляет собой фармацевтически приемлемый одновалентный катион, независимо выбранный из группы, состоящей из глициния, аланиния, β-аланиния, валиния, лизиния, изолейциния, лейциния, метиониния, треониния, аспарагиния, глутаминия, гистидиния, аргининия, орнитиния, триптофания, пролиния, глутаминия, цистеиния, фенилаланиния, тирозиния и сериния.
Шестнадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ представляет собой Na+. Семнадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ представляет собой К+. Восемнадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ представляет собой Li+.
Девятнадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ представляет собой NH4+. Двадцатым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ представляет собой NH3+С(CH2OH)3. Двадцать первым воплощением первого аспекта настоящего изобретения является соединение по десятому воплощению первого аспекта, где М+ представляет собой NH2+(CH2CH3)2. Двадцать вторым воплощением первого аспекта настоящего изобретения является соединение по двенадцатому воплощению первого аспекта, где М2+ выбран из группы, состоящей из Са2+, Mg2+ и Zn2+.
Двадцать третьим воплощением первого аспекта настоящего изобретения является соединение по третьему воплощению первого аспекта, выбранное из группы, состоящей из:
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, динатриевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, диаммониевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, дикалиевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, дилитиевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, кальциевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, магниевой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, цинковой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, пирролидиновой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, трис-(гидроксиметил)метиламиновой соли;
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, диэтиламиновой соли и
(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфата, глициновой соли, и других его фармацевтически приемлемых солей.
Двадцать четвертым воплощением первого аспекта настоящего изобретения является соединение по второму воплощению первого аспекта, выбранное из группы, состоящей из:
(2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, динатриевой соли;
(R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6H)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, диаммониевой соли;
(2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфата, динатриевой соли;
(2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфата, динатриевой соли;
(R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6H)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, динатриевой соли;
(2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, диаммониевой соли;
(2R)-Н-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфата, диаммониевой соли;
(2R)-Н-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфата, аммониевой соли;
(2R)-4-[4-(2,3-Дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, дикалиевой соли;
(2R)-Н-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфата, дикалиевой соли;
(2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфата, дикалиевой соли;
(R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, дикалиевой соли;
(2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-Н-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, дилитиевой соли;
(2R)-N-гидрокси-4-{4-[4-(4-метокси-2H-l,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфата, дилитиевой соли;
(2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(l,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфата, дилитиевой соли и
(R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6H)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфата, дилитиевой соли, и других его фармацевтически приемлемых солей.
Вторым воплощением второго аспекта настоящего изобретения является соединение по первому воплощению второго аспекта формулы (2а)
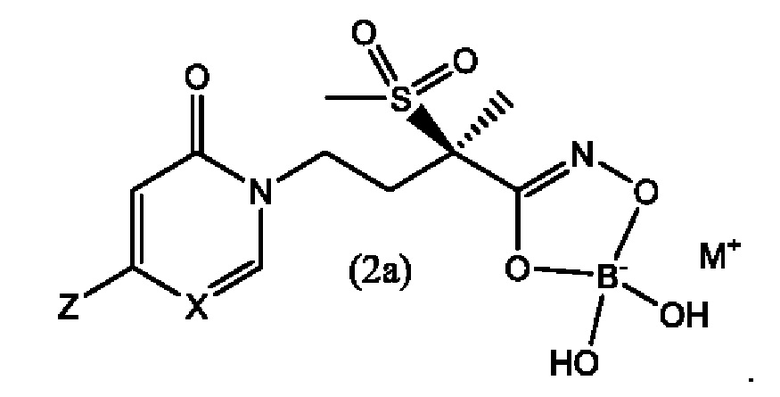
Третьим воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где X представляет собой СН.
Четвертым воплощением второго аспекта настоящего изобретения является соединение по третьему воплощению второго аспекта, где Z представляет собой
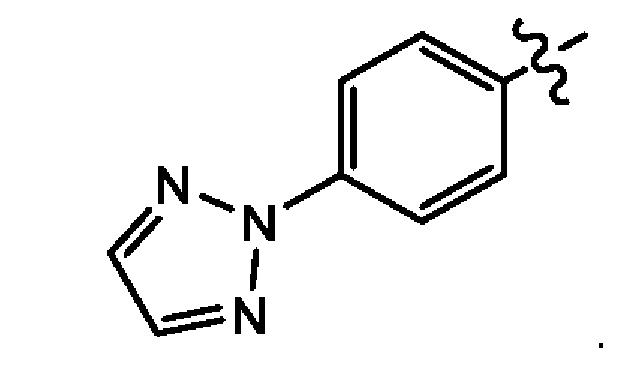
Пятым воплощением второго аспекта настоящего изобретения является соединение по третьему воплощению второго аспекта, где Z представляет собой
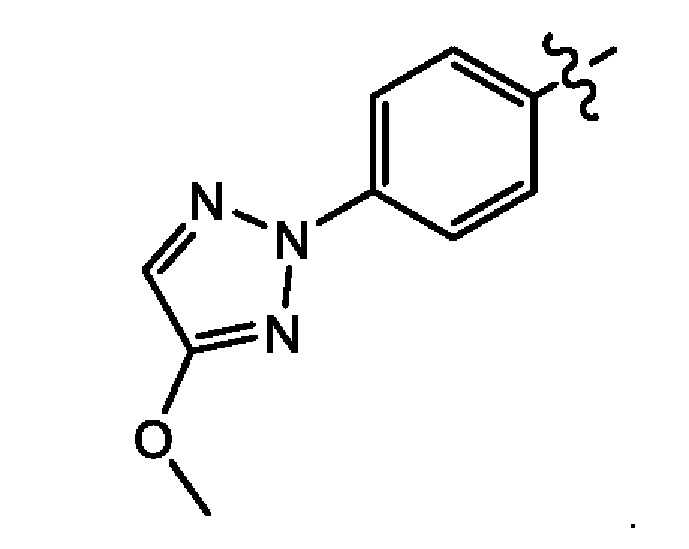
Шестым воплощением второго аспекта настоящего изобретения является соединение по третьему воплощению второго аспекта, где Z представляет собой
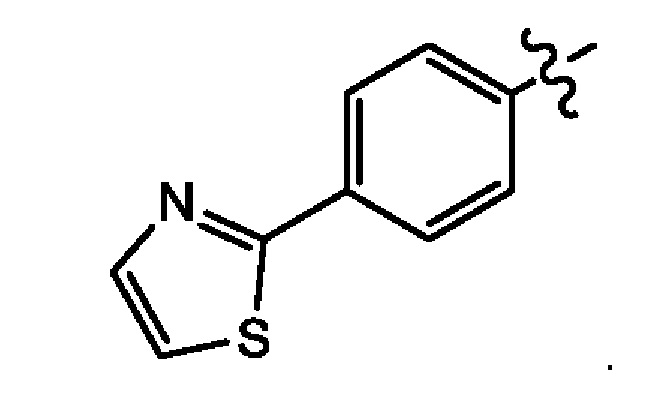
Седьмым воплощением второго аспекта настоящего изобретения является соединение по третьему воплощению второго аспекта, где Z представляет собой
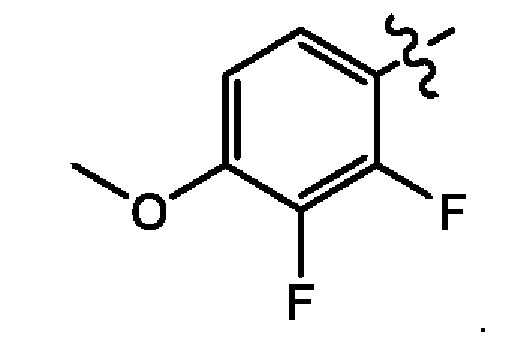
Восьмым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где X представляет собой N; и Z представляет собой
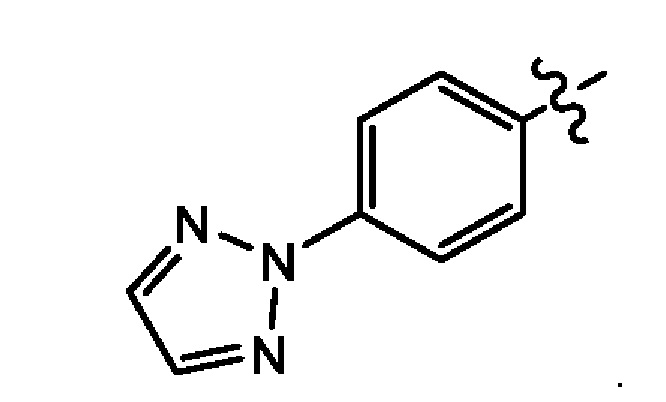
Девятым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ выбран из группы, состоящей из Li+, К+ и Na+.
Десятым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ выбран из группы, состоящей из аммония, (С1-С12алкил)аммония, (С1-С12алкил)2аммония, (C1-С12алкил)3аммония, (С1-С12алкил)4аммония, (С3-С6циклоалкил)аммония, (С3-С6циклоалкил)2аммония, (С3-С6циклоалкил)3аммония, (С3-С6циклоалкил)4аммония, пирролидиния, пиперидиния и пиридиния; при этом каждая из группировок (C1-С12алкил) или (С3-С6циклоалкил) возможно замещена одной-тремя группами гидрокси или одним-тремя атомами галогена.
Одиннадцатым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ выбран из группы, состоящей из глициния, аланиния, β-аланиния, валиния, лизиния, изолейциния, лейциния, метиониния, треониния, аспарагиния, глутаминия, гистидиния, аргининия, орнитиния, триптофания, пролиния, глутаминия, цистеиния, фенилаланиния, тирозиния и сериния.
Двенадцатым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ представляет собой Na+. Тринадцатым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ представляет собой K+. Четырнадцатым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ представляет собой Li+. Пятнадцатым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ представляет собой NH4+. Шестнадцатым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ представляет собой NH3+С(СН2ОН)3. Семнадцатым воплощением второго аспекта настоящего изобретения является соединение по второму воплощению второго аспекта, где М+ представляет собой NH2+(СН2СН3)2.
Восемнадцатым воплощением второго аспекта настоящего изобретения является второе воплощение второго аспекта, представляющее собой боронатное пролекарство (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида и его фармацевтически приемлемые соли.
Девятнадцатым воплощением второго аспекта настоящего изобретения является второе воплощение второго аспекта, представляющее собой боронатное пролекарство (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида, то есть (R)-5-(4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уид натрия и другие его фармацевтически приемлемые соли.
Двадцатым воплощением второго аспекта настоящего изобретения является второе воплощение второго аспекта, представляющее собой боронатное пролекарство, выбранное из группы, состоящей из:
(R)-5-(4-(4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уида натрия;
(R)-2,2-дигидрокси-5-(4-(4-(4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-1,3,4,2-диоксазаборол-2-уида натрия;
(R)-2,2-дигидрокси-5-(2-(метилсульфонил)-4-(2-оксо-4-(4-(тиазол-2-ил)фенил)пиридин-1(2Н)-ил)бутан-2-ил)-1,3,4,2-диоксазаборол-2-уида натрия и
(R)-5-(4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уида натрия; и других его фармацевтически приемлемых солей.
Первым воплощением третьего аспекта настоящего изобретения является фармацевтическая композиция, содержащая соединение по любому из воплощений первого или второго аспектов в смеси по меньшей мере с одним фармацевтически приемлемым эксципиентом, разбавителем или носителем.
Первым воплощением четвертого аспекта настоящего изобретения является способ лечения вызываемой грамотрицательными бактериями инфекции у пациента, включающий введение терапевтически эффективного количества соединения по любому из воплощений первого или второго аспектов пациенту, нуждающемуся в этом.
Вторым воплощением четвертого аспекта настоящего изобретения является способ по первому воплощению четвертого аспекта, где вызываемая грамотрицательными бактериями инфекция вызвана грамотрицательными бактериями, выбранными из группы, состоящей из Mannheimia haemolytica, Pasteurella multocida, Histophilus somni, Actinobacillus pleuropneumoniae, Salmonella enteritidis, Salmonella gallinarium, Lawsonia intracellular!s, Brachyspira hyodysenteriae, Brachyspira pilosicoli, Acinetobacter baumannii, Acinetobacter spp., Citrobacter spp., Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Serratia marcescens, Stenotrophomonas maltophilia и Pseudomonas aeruginosa.
Третьим воплощением четвертого аспекта настоящего изобретения является способ по первому воплощению четвертого аспекта, где вызываемая грамотрицательными бактериями инфекция выбрана из группы, состоящей из респираторной инфекции, желудочно-кишечной инфекции, нозокомиальной пневмонии, инфекции мочевыводящих путей, бактериемии, сепсиса, кожной инфекции, инфекции мягких тканей, инфекции брюшной полости, легочной инфекции, эндокардита, синдрома диабетической стопы, остеомиелита и инфекции центральной нервной системы.
Изобретение относится к солям присоединения основания соединений по настоящему изобретению. Химическими основаниями, которые могут быть использованы в качестве реагентов для получения таких фармацевтически приемлемых солей с основаниями, являются основания, которые образуют нетоксичные соли присоединения основания с такими соединениями. Такие нетоксичные соли с основаниями включают, но не ограничиваются этим, соли, полученные из таких фармакологически приемлемых катионов (М+ или М2+), как катионы щелочных металлов (например, лития, калия и натрия) и катионы щелочноземельных металлов (например, кальция, магния и цинка), аммоний, катионы алкиламина, диалкиламина, триалкиламина, тетралкиламмоний, пиридиний, или водорастворимые соли присоединения аминов, таких как N-метилглюкамин (меглумин) и (низший спирт)аммоний, и другие соли с основаниями фармацевтически приемлемых органических аминов, таких как пиперидин, N-метилпиперидин, морфолин, N-метилморфолин, аминокислоты и другие амины, которые используют для образования солей карбоновых кислот и фосфорных кислот.
Подходящие соли с основаниями получают из оснований, которые образуют нетоксичные соли. Неограничивающие примеры подходящих солей с основаниями включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Также могут быть образованы гемисоли кислот и оснований, например гемисульфатные и гемикальциевые соли. Для обзора подходящих солей см. Handbook of Pharmaceutical Salts: Properties, Selection, and Use под ред. Stahl и Wermuth (Wiley-VCH, 2002). Помимо способов, описанных в данной заявке, специалисту в данной области техники известны способы получения фармацевтически приемлемых солей фосфатов и боронатов.
Соединения формулы (1), где Q представляет собой Р(O)(ОН)(O-М+), -Р(O)(O- М+)2 или -Р(O)(O-)2М2+, могут быть получены общепринятым образом путем смешивания соединения формулы (1), где Q представляет собой -Р(O)(ОН)2, с соответствующим выбранным основанием, предпочтительно путем приведения их в контакт в растворе с применением избытка обычно используемых инертных растворителей, таких как вода, эфир, ацетонитрил, диоксан, метилен хлорид, изопропанол, метанол, этанол и этилацетат. Соединения формулы (1), где Q представляет собой Р(O)(ОН)(O-М+), -Р(O)(O-М+)2 или -Р(O)(O-)2М2+, также могут быть получены в результате обмена или в результате обработки с использованием ионообменной смолы в условиях, в которых одновалентный катион, М+, или двухвалентный катион, М2+, в соединении формулы 1 заменяют на другой одновалентный катион, М+, или двухвалентный катион, М2+, при необходимости, в условиях, которые позволяют разделить желаемые соединения, как например, путем осаждения из раствора, или экстракции в растворитель, или элюирования из ионообменной смолы либо удерживания на ней. Аналогичным образом, соединения формулы (2) также могут быть получены в результате обмена или в результате обработки с использованием ионообменной смолы в условиях, при которых одновалентный катион, М+, в соединении формулы (2) заменяют на другой одновалентный катион, М+, в условиях, которые позволяют разделить желаемые соединения, как например, путем осаждения из раствора, или экстракции в растворитель, или элюирования из ионообменной смолы либо удерживания на ней.
Соединения формулы (1) имеют асимметрический центр, тем самым существуя в виде двух стереоизомерных форм. Настоящее изобретение включает все индивидуальные стереоизомеры соединений формулы (1) и их смеси. Индивидуальные энантиомеры могут быть получены посредством хирального разделения или с использованием при их синтезе релевантного энантиомера. Например, индивидуальные (R)- и (S)-энантиомеры соединения формулы (1) могут быть получены посредством хирального разделения из энантиомерной смеси, или они могут быть получены по отдельности с использованием метода хирального синтеза. Предпочтительным воплощением является соединение формулы 1а, при этом соединение имеет (R)-стереохимическую конфигурацию относительно атома углерода как хирального центра. Аналогичным образом, соединения формулы (2) также имеют асимметричный центр, и предпочтительными воплощениями являются соединения формулы IIa, которые имеют изображенную стереохимическую конфигурацию.
Кроме того, соединения по настоящему изобретению могут существовать в несольватированной, а также в сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное. Обычно считается, что в рамках задач настоящего изобретения сольватированные формы эквивалентны несольватированным формам. Соединения также могут существовать в одном или более кристаллических состояниях, т.е. являться полиморфами, или они могут существовать в виде аморфных твердых веществ. Все такие формы включены в объем настоящего изобретения и в формулу изобретения.
Соединения по настоящему изобретению действуют как пролекарства (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида; (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида; (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамида; (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамида и (R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида или рацематов этих соединений. Сами эти соединения могут обладать незначительной или не обладать никакой фармакологической активностью, но после введения внутрь организм или нанесения на тело могут превращаться в исходное соединение, имеющее желаемую активность, например, в результате гидролитического отщепления фосфатной в соединениях формулы (1) или боронатной группировки в соединении формулы (2).
Данное изобретение также охватывает соединения, содержащие защитные группы. Например, некоторые промежуточные соединения, используемые для получения соединений формулы (1) или формулы (2), могут содержать защитные группы. Специалисту в данной области техники также будет очевидно, что соединения по настоящему изобретению также могут быть получены с определенными защитными группами, присутствие которых полезно с точки зрения очистки или хранения и которые могут быть удалены перед введением пациенту. Введение и удаление защитных функциональных групп описано в "Protective Groups in Organic Chemistry", под редакцией J.W.F. McOmie, Plenum Press (1973) и "Protective Groups in Organic Synthesis", 3-е издание, T.W. Greene and P.G.M. Wuts, Wiley-Interscience (1999).
Настоящее изобретение также включает меченные изотопом соединения, которые идентичны соединениям, описанным формулой (1) или формулой (2), если не считать, что один или более атомов заменены на атом(ы), имеющий(ие) атомную массу или массовое число, отличную(ое) от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть инкорпорированы в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как, но не ограничиваясь этим, 2Н, 3Н, 13С, 14С, 15N, 17O, 18O, 31Р, 32Р, 35S, 18F и 36Cl, соответственно. Соединения по настоящему изобретению, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов, включены в объем данного изобретения. Некоторые меченные изотопом соединения по настоящему изобретению, например те, в которые инкорпорированы такие радиоактивные изотопы, как 3Н и 14С, применимы в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы трития, т.е. 3Н, и углерода-14, т.е. 14С, особенно предпочтительны ввиду простоты их получения и детекции. Кроме того, замена на более тяжелые изотопы, такие как дейтерий, т.е. Н, может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный in vivo период полувыведения или снижение дозировки, и поэтому в некоторых обстоятельствах может быть предпочтительной. В общем случае, меченные изотопом соединения по данному изобретению могут быть получены путем осуществления методик, описанных ниже на схемах и/или в разделе Примеры, с заменой не меченного изотопом реагента легко доступным меченным изотопом реагентом.
Каждое из соединений формулы (1) содержит сульфонильную группировку, которая изображена ниже:
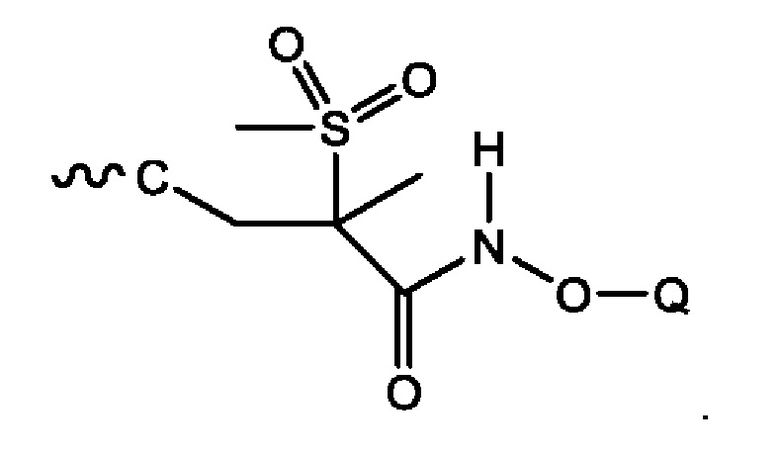
Специалисту в данной области техники совершенно очевидно, что атом углерода, примыкающий к сульфонильной группировке, представляет собой хиральный центр. Ввиду этого, соединения могут существовать в виде рацемата, в виде S-энантиомера или в виде R-энантиомера либо в виде их смесей. В следующем воплощении соединения формулы (1) могут быть получены и введены в виде R-энантиомера (т.е. в виде соединения формулы (1а), которое изображено ниже):
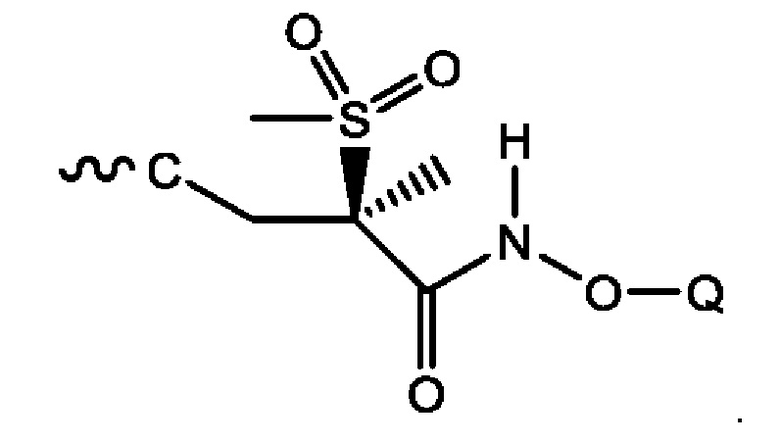
Показанные соединения формулы (1) и формулы (2) могут представлять собой рацемические соединения, индивидуальные изомеры или их смеси, в то время как соединения формулы (1а) и формулы (2а) имеют стереохимическую конфигурацию, изображенную на этих формулах, соответственно. Специалисту в данной области техники совершенно очевидно, что синтезированные соединения в редких случаях будут присутствовать исключительно в виде единственного энантиомера. Энантиомер с противоположной конфигурацией (т.е. S-энантиомер) может присутствовать в незначительных количествах (т.е. «по существу чистое»). Такое незначительное количество может составлять до 10 масс./масс. % включительно, в более типичном случае не выше 5 масс/масс. %, в следующем воплощении не выше 1 масс/масс. % или, более конкретно, не выше 0,5 масс/масс. %.
Экспериментальный синтез
Соединения формулы (1) и формулы (2) могут быть получены рядом способов, аналогичных известным в данной области техники. На реакционных схемах А и В, представленных ниже, иллюстрируются два альтернативных способа получения промежуточных соединений формулы I' или I''. Другие, в том числе их модификации, будут совершенно очевидны специалисту в данной области техники. Затем в синтезе соединений формулы (1) и формулы (2) могут быть использованы соединения формулы I' или I''.
Синтез соединений формулы I' или I'' показан на приведенных ниже схемах А и В. Первая стадия заключается в осуществлении N-алкилирования, показанного на стадии А. Пиридинон/пиримидинон (где X представляет собой СН или N, соответственно) структуры 1 взаимодействует с сульфонильным производным структуры 2, образуя промежуточное соединение структуры 3. Для получения соединений формулы (1) структура 3 может быть модифицирована далее. Показаны два альтернативных варианта синтеза (вариант А или В), тем не менее читатель легко отметит, что они являются вариациями одного и того же синтеза. Единственным различием является порядок, согласно которому выполняются стадии.
Первоначально согласно варианту А соответствующую уходящую группу, такую как галогенид, обозначенную как Lg, в положении 4 пиридинона/пиримидинона структуры 3 заменяют на желаемую группировку Z посредством взаимодействия с Z-М1, где М1 представляет собой металл-содержащую структуру, такую как бор-содержащее производное, подходящее для проведения типичного перекрестного сочетания, такого как реакция Сузуки-Мияуры. После гидролиза или удаления этильной защитной группы (или других подходящих защитных групп) на стадии С получают соединение структуры 5. Затем путем модификации концевой группы карбоновой кислоты структуру 5 превращают в защищенное производное гидроксамовой кислоты, обозначенное как структура 8 (где Pr представляет собой соответствующую защитную группу). После удаления защиты с защищенного производного гидроксамовой кислоты структуры 8, как показано на стадии Н, получают промежуточное соединение формулы I'. Несмотря на то, что эти реакции хорошо известны специалисту в данной области техники, их более подробное обсуждение приводится ниже.
Первоначально, согласно варианту В схемы А, этильную защитную группу (или другие традиционные защитные группы) удаляют из пиридинона/пиримидинона структуры 3 с образованием соединения структуры 6, как показано на стадии Е. На стадии F путем модификации концевой группы карбоновой кислоты структуру 6 превращают в защищенное производное гидроксамовой кислоты структуры 7, применяя условия амидирования. Затем на стадии G уходящую группу Lg, такую как галогенидная функциональная группа на пиридиноновой/пиримидиноновой группировке, заменяют непосредственно на желаемую группировку Z путем взаимодействия с Z-M1 в реакции сочетания с получением защищенных производных гидроксамовой кислоты структуры 8. Как и ранее, после удаления защиты с защищенных производных гидроксамовой кислоты, как показано на стадии Н, получают соединения формулы I'.
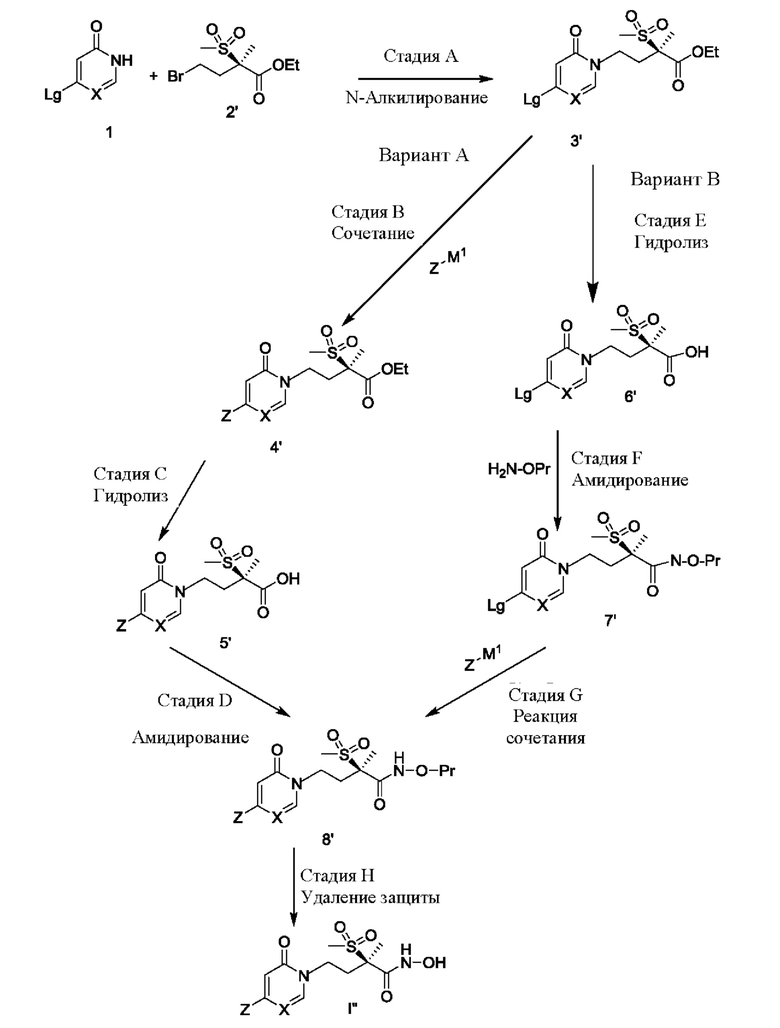
Схема В, показанная ниже, аналогична схеме А за исключением того, что пиридинон/пиримидинон структуры 1 взаимодействует с сульфонильным производным структуры 2' с образованием промежуточного соединения структуры 3'. Для получения соединений формулы I'' структура 3' может быть модифицирована далее. Первоначально, согласно варианту А, соответствующую уходящую группу, такую как галогенид, обозначенную как Lg, на 2-пиридиноне/пиримидиноне структуры 3' заменяют на желаемую группировку Z посредством взаимодействия с Z-M1, где М1 представляет собой металл-содержащую структуру, такую как бор-содержащее производное, подходящее для проведения типичного перекрестного сочетания, такого как реакция Сузуки-Мияуры. После гидролиза или удаления этильной защитной группы (или других подходящих защитных групп) на стадии С получают соединение структуры 5'. Затем путем модификации концевой группы карбоновой кислоты структуру 5' превращают в защищенное производное гидроксамовой кислоты, обозначенное как структура 8' (где Pr представляет собой соответствующую защитную группу). После удаления защиты с защищенного производного гидроксамовой кислоты структуры 8', как показано на стадии Н, получают промежуточное соединение формулы I''. Несмотря на то, что эти реакции хорошо известны специалисту в данной области техники, их более подробное обсуждение приводится ниже.
Первоначально, согласно варианту В схемы В этильную защитную группу (или другие традиционные защитные группы) удаляют из пиридинона/пиримидинона структуры 3' с образованием соединения структуры 6', как показано на стадии Е. На стадии F путем модификации концевой группы карбоновой кислоты структуру 6' превращают в защищенное производное гидроксамовой кислоты структуры 7', применяя условия амидирования. Затем на стадии G соответствующую уходящую группу Lg, такую как галогенидная функциональная группа на пиридиноновой/пиримидиноновой группировке, заменяют непосредственно на желаемую группировку Z путем взаимодействия с Z-M1 в реакции сочетания с получением защищенных производных гидроксамовой кислоты структуры 8'. Как и ранее, после удаления защиты с защищенных производных гидроксамовой кислоты, как показано на стадии Н, получают соединения формулы I''.
СХЕМА А
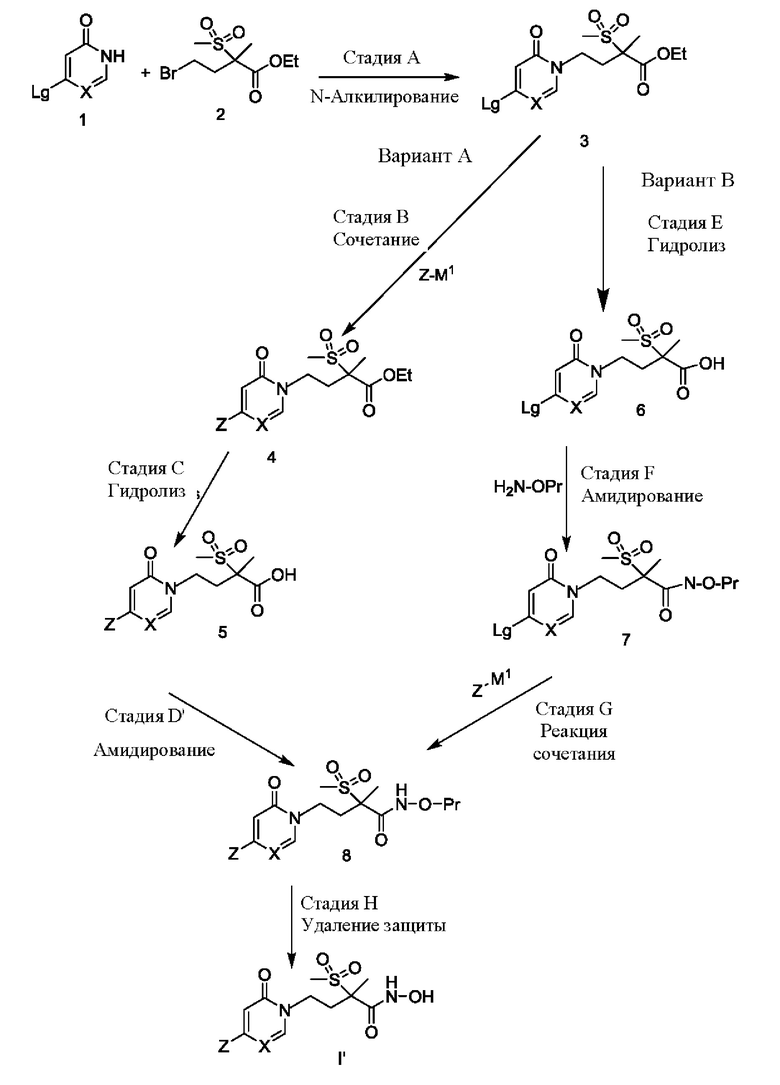
СХЕМА В
Следующее далее описание относится к стадиям синтеза, использованным на схемах А и В. N-Алкилирование, показанное выше на стадии А схемы А и схемы В, может быть выполнено с использованием методов, хорошо известных специалисту в данной области техники. Одним из исходных веществ является производное 2-пиридинонов или пиримидинонов структуры 1. В этом пиридиноне или пиримидиноне Lg представляет собой соответствующую уходящую группу, такую как галогенид. Многие из таких производных пиридинонов или пиримидинонов известны в данной области техники, а остальные можно получить, используя методы синтеза, аналогичные известным в данной области техники. Для ознакомления с описанием таких методов внимание читателя обращается на Tet. Lett. (2005) Vol. 46, 7917. Приведенный ниже подготовительный пример 2 также иллюстрирует их получение.
Другим реагентом при N-алкилировании, показанном на стадии А, является защищенный алкилсульфонат структуры 2 или 2'. В структуре 2 или 2' изображена этильная защитная группа (т.е. защита карбоновой кислоты в виде ее сложного этилового эфира), но она может быть заменена на любую стандартную защитную группу карбоновой кислоты. Эти алкилсульфонаты также известны в данной области техники. Для ознакомления с описанием их получения внимание читателя обращается на Journal of Organic Chemistry, (1980) Vol. 45, 8, 1486-1489. Приведенный ниже подготовительный пример 1 также иллюстрирует их получение.
Такое N-алкилирование может быть проведено так, как известно в данной области техники. Обычно, эквивалентные количества соединений структуры 1 и 2 или 2' приводят в контакт друг с другом в смеси апротонных и протонных растворителей, таких как тетрагидрофуран и трет-бутшол, в присутствии основания, такого как карбонат калия, карбонат цезия, карбонат натрия, гидрид натрия и так далее. При желании можно использовать агент переноса, такой как бромид тетрабутиламмония. Обычно реагенты нагревают и дают возможность завершиться реакции. Желаемый продукт структуры 3 или 3' может быть выделен способами, известными в данной области техники. При желании продукт структуры 3 или 3' может быть очищен, или, альтернативно, на следующей стадии реакции можно использовать неочищенный продукт. Приведенный ниже подготовительный пример 2 иллюстрирует такое N-алкилирование.
На схеме А иллюстрируется, как осуществить введение группировки гидроксамовой кислоты в молекулы. Сначала с карбоновой кислоты удаляют защитную группу, в результате чего образуется промежуточное соединение структуры 5 или 5' и 6 или 6', которое показано на стадии С (варианта А) и стадии Е (варианта В), соответственно. То, каким образом это осуществляется, будет варьировать в зависимости от природы самой защитной группы, и хорошо известно специалистам в данной области техники. Для обсуждения возможных защитных групп и способов их удаления внимание читателя обращается на работу McOmie или Greene, выше. В приведенном ниже подготовительном примере 2 описывается, как удалить этильную группировку, которая показана на схемах А и В.
На стадиях F и D показано, как группировка гидроксамовой кислоты включается в молекулу. Можно использовать защищенный источник гидроксиламина, после чего провести реакцию удаления защиты (альтернативно, чтобы исключить стадии удаления защиты, гидроксиламин может быть введен непосредственно). В любом случае гидроксамовую кислоту вводят в молекулу с использованием стандартных реакций амидирования. Например, соединение структуры 5 или 5' (варианта А) либо 6 или 6' (варианта В) могут быть приведены в контакт с избытком оксалилхлорида в апротонном растворителе, таком как дихлорметан, в течение периода времени, достаточного для осуществления образования соответствующего хлорангидрида кислоты, с последующим добавлением избытка либо гидроксиламина, либо защищенного гидроксиламина. Затем дают возможность завершиться реакции и из реакционной среды выделяют защищенные промежуточные соединения структуры 7 или 7' (варианта В) либо 8 или 8' (варианта А) и очищают так, как известно в данной области техники. Как упомянуто выше, любое удаление защиты может быть выполнено так, как известно в данной области техники (см. Greene или McOmie, выше). Альтернативно, амид может быть образован с использованием реагента амидного сочетания, 1,1'-карбонилдиимидазола (CDI), 2-хлор-4,6-диметокси-1,3,5-триазина (CDMT) или 1-этил-3-(3-диметиламинопропил)-карбодиимида (EDCI), так, как известно в данной области техники.
На схемах А и В также показано как осуществить введение в молекулу концевой группировки Z. Независимо от того, выбран ли вариант А или вариант В, в конечном итоге проводят реакцию сочетания для присоединения концевой группировки Z к промежуточному пиридинону/пиримидинону. На обеих схемах А и В этот сореагент показан как Z-M1, где М1 представляет собой металл (или металлоид), такой как магний, медь, олово, сложный эфир бороновой кислоты/бороновая кислота и так далее, в желаемом месте присоединения к промежуточному пиридинону/пиримидинону структуры 3 или 3' либо 7 или 7' (т.е. другой реагент).
Реакция сочетания может быть осуществлена разнообразными методами. Для образования углерод-углеродной связи можно использовать стратегию Сузуки-Мияуры. В такой реакции М1 будет представлен бороновой кислотой/сложным эфиром бороновой кислоты. Эквивалентные молярные количества реагентов будут приведены в контакт друг с другом в растворителе, таком как тетрагидрофуран, 2-метилтетрагидрофуран, 1,4-диоксан, вода, толуол или их смесь, в присутствии катализатора переходных металлов, такого как свободные или связанные со смолой палладий- или никель-содержащие структуры, вместе с основанием, таким как карбонат натрия, карбонат калия, фторид цезия, карбонат цезия и так далее. Реакционную смесь можно нагревать с использованием микроволн или других традиционных методов до достижения адекватного превращения. После завершения желаемый продукт можно выделить и извлечь из реакционной смеси и далее очистить так, как известно в данной области техники. Аналогично, для проведения реакции сочетания можно использовать другие способы образования углерод-углеродных связей, известные в данной области техники. В такой реакции М1 может быть представлен образуемым in situ купратом или группировкой триалкилолова, такой как триметилстаннил, трибутилстаннил или три-трет-бутилстаннил. В контакт будут приведены эквивалентные молярные количества реагентов в растворителе, таком как тетрагидрофуран, 2-метилтетрагидрофуран, диметилформамид или их смесь, в присутствии катализатора переходных металлов, такого как свободный или связанный со смолой палладий или никель, вместе с соответствующим основанием, таким как подходящее органическое основание, например, N,N-диизопропилэтиламин. Реакционную смесь можно нагревать с использованием микроволн или других традиционных методов до достижения адекватного превращения. После завершения желаемый продукт можно выделить и извлечь из реакционной смеси и далее очистить так, как известно в данной области техники.
СХЕМА С
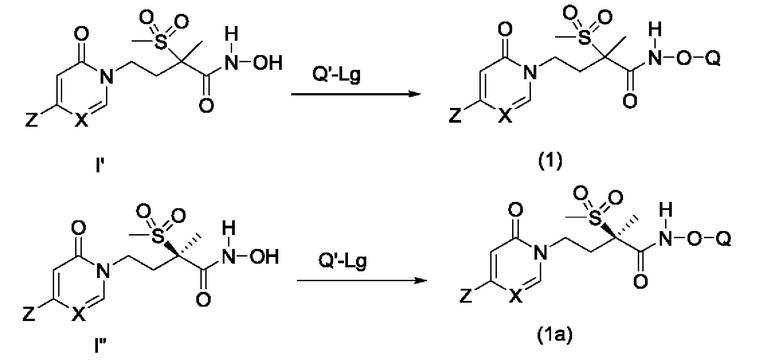
На схеме С показано получение соединений формулы (1) и формулы (1а) из соединений I' и I'', соответственно. Соединение формулы I' или I'' приводят во взаимодействие с соответствующим соединением-предшественником фосфата, Q'-Lg, где Lg представляет собой соответствующую уходящую группу, a Q' представляет собой фосфор-содержащую группу, которая может быть преобразована в соответствующую фосфатную группу Q. Примеры соединений-предшественников фосфата Q'-Lg включают оксихлорид фосфора (POCl3) или фосфорамидитный реагент (PgO)2P-NR'2. В соответствующих условиях реакции группировка Q' превращается в группу Q, как указано в формуле (1) или формуле (1а). Более подробное описание таких превращений Q' в Q приведено ниже на схемах D и Е.
СХЕМА D
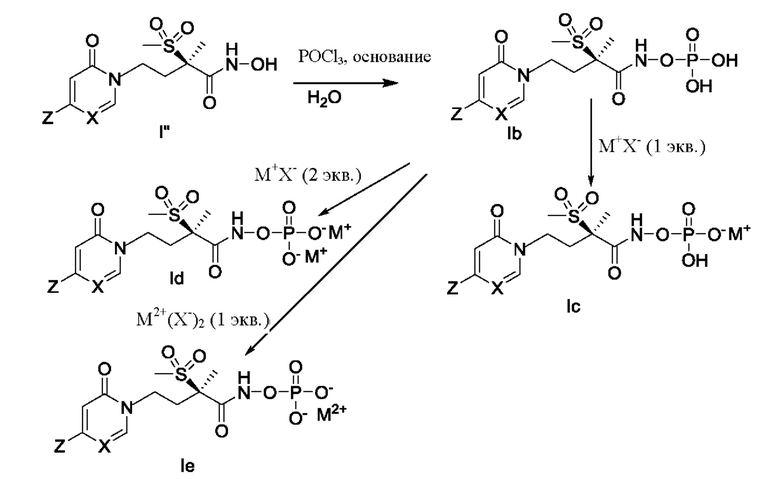
На схеме D показано получение новых фосфатов в объеме формулы (1) (т.е. соединений формулы Ib, Ic, Id и Ie). Гидроксамовую кислоту, соединение формулы I'', растворяют в соответствующем растворителе, таком как ацетонитрил, и обрабатывают соответствующим основанием, таким как N-метилморфолин, при пониженной температуре, например, от 0°С до -10°С. Затем полученную смесь приводят во взаимодействие с оксихлоридом фосфора, и после этого ее можно погасить водой с получением фосфата формулы Ib. Затем соединение формулы Ilb может быть приведено во взаимодействие, как показано, с соответствующим основанием (т.е. с М+Х- или М2+(Х-)2, где X- представляет собой анионный противоион) с получением соединений формулы Ic, Id или Ie. Альтернативно, соединение формулы Ib можно обработать с использованием соответствующей ионообменной смолы, такой как ионообменная смола Dowex, в водном растворе с получением соединения формулы Id.
СХЕМА Е
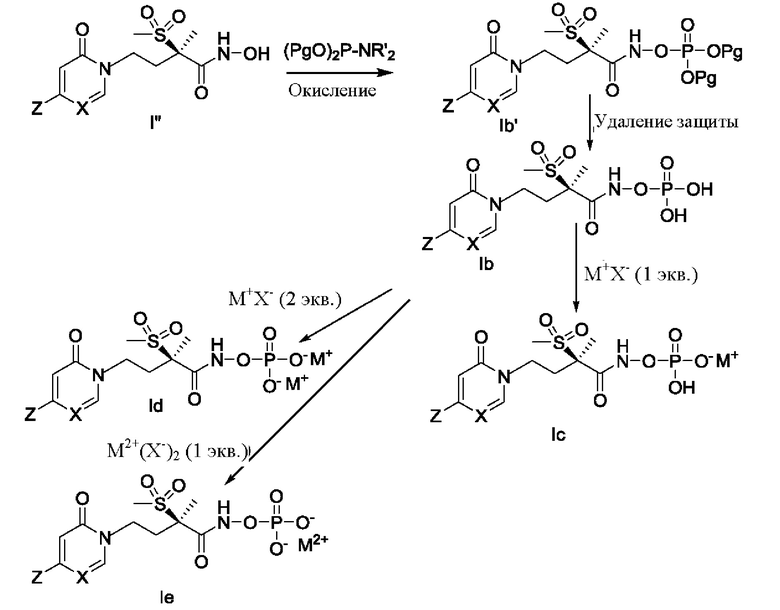
На схеме Е показан альтернативный способ получения соединений формулы Ib-Ie. Соединение формулы I'' приводят во взаимодействие с подходящим фосфорамидитным реагентом, (PgO)2P-NR'2, в котором группа Pg представляет собой соответствующую защитную группу, такую как трет-бутил или бензил, а группа R' представляет собой низшую алкильную группу, такую как этил или изопропил. Обычно эту реакцию проводят при температуре, приблизительно равной температуре окружающей среды, в соответствующем растворителе, таком как ацетонитрил, дихлорметан или их смесь, в присутствии активирующего агента, такого как тетразол, в течение периода времени от одного до восьми часов. Затем реакционную смесь можно охладить и провести окисление in situ, используя обработку соответствующим окисляющим агентом, таким как перекись водорода, трет-бутилгидропероксид или m-СРВА, с получением соединения формулы Ib'. Далее с использованием стандартной методологии с соединения формулы Ib' удаляют защитную группу, получая соединения формулы Ib. Например, если Pg представляет собой трет-бутил, то защитная группа с соединения формулы Ib' может быть удалена путем обработки сильной кислотой, такой как соляная кислота или трифторуксусная кислота. Альтернативно, если Pg представляет собой бензил, то защитная группа с соединения формулы Ib' может быть удалена путем каталитического гидрирования. Затем соединение формулы Ib можно использовать для получения соединений формулы Ic, Id или Ie, как описано ранее для реакционной схемы D.
СХЕМА F
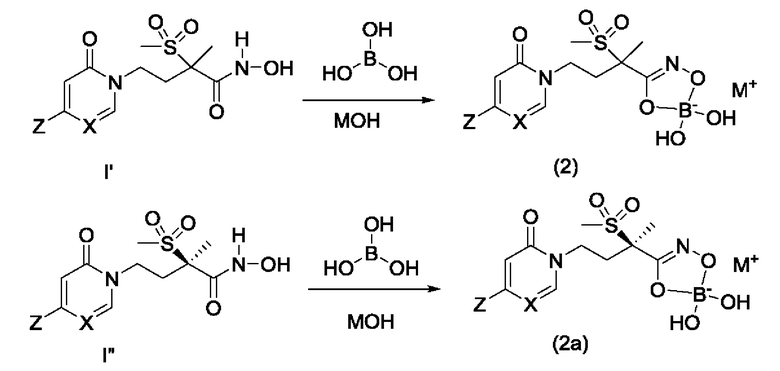
На схеме F показано получение мономерных боратных соединений формулы (2) и формулы (2а). Один эквивалент гидроксамовой кислоты формулы I' или I'' объединяют с одним эквивалентом борной кислоты в воде в присутствии одного эквивалента соответствующего основания, такого как гидроксид натрия, гидроксид калия или гидроксид лития (МОН). Смесь перемешивают при температуре окружающей среды в течение от 30 минут до четырех часов, затем смесь можно либо сконцентрировать в вакууме, либо заморозить и лиофилизировать, чтобы получить моноборонатное соединение формулы (2) или формулы (2а).
Показанные выше реакционные схемы для получения соединений по настоящему изобретению являются главным образом иллюстративными. Специалисту в данной области техники совершенно очевидно, что они могут быть модифицированы в зависимости от конкретного соединения, доступности реагентов и так далее.
Применения в медицине и ветеринарии
Соединения по настоящему изобретению могут быть использованы для лечения или предупреждения инфекционных расстройств, в особенности расстройств, вызываемых восприимчивыми и обладающими множественной лекарственной устойчивостью (MDR) грамотрицательными бактериями. Примеры таких грамотрицательных бактерий включают Acinetobacter baumannii, Acinetobacter spp., Achromobacter spp., Aeromonas spp., Bacteroides fragilis, Bordetella spp., Borrelia spp., Brucella spp., Campylobacter spp., Citrobacter diversus (koseri), Citrobacter freundii, Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Francisella tularensis, Fusobacterium spp., Haemophilus influenzae (β-лактамаза-позитивные и -негативные), Helicobacter pylori, Klebsiella oxytoca, Klebsiella pneumoniae (в том числе таковые, кодирующие р-лактамазы расширенного спектра (далее «ESBL»)), Legionella pneumophila, Moraxella catarrhalis β-лактамаза-позитивные и -негативные), Morganella morganii, Neisseria gonorrhoeae, Neisseria meningitidis, Proteus vulgaris, Porphyromonas spp., Prevotella spp., Mannheimia haemolyticus, Pasteurella spp., Proteus mirabilis, Providencia spp., Pseudomonas aeruginosa, Pseudomonas spp., Salmonella sppro, Shigella spp., Serratia marcescens, Treponema spp., Burkholderia cepacia, Vibrio spp., Yersinia spp.и Stenotrophomonas mulophilia. Примеры других грамотрицательных микроорганизмов включают представителей энтеробактерий, которые экспрессируют ESBL; КРС (карбапенемаза из Klebsiella pneumoniae), СТХ-М, металло-β-лактамазы (такие как NDM-1 (металло-бета-лактамаза из Нью-Дели), например) и бета-лактамазы AmpC-типа, которые придают устойчивость к имеющимся в настоящее время цефалоспоринам, цефамицинам, карбапенемам и комбинациям бета-лактам/ингибитор бета-лактамазы.
В более конкретном воплощении грамотрицательные бактерии выбраны из группы, состоящей из Acinetobacter baumannii, Acinetobacter spp., Citrobacter spp., Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Serratia marcescens, Stenotrophomonas maltophilia, Pseudomonas aeruginosa и представителей энтеробактерий и Pseudomonas, которые экспрессируют ESBL, КРС, СТХ-М, металло-β-лактамазы и бета-лактамазы AmpC-типа, которые придают устойчивость к имеющимся в настоящее время цефалоспоринам, цефамицинам, карбапенемам и комбинациям бета-лактам/ингибитор бета-лактамазы.
Примеры инфекций, которые могут подлежать лечению соединениями формулы (1), включают нозокомиальную пневмонию, инфекции мочевыводящих путей, системные инфекции (бактериемию и сепсис), инфекции кожи и мягких тканей, хирургические инфекции, инфекции брюшной полости, легочные инфекции у пациентов с кистозным фиброзом, у пациентов, страдающих от легочных инфекций, эндокардит, синдромы диабетической стопы, остеомиелит и инфекции центральной нервной системы.
Помимо этого, данные соединения можно использовать для лечения инфекций, вызываемых Helicobacter pylori в желудочно-кишечном (ЖК) тракте людей (и других млекопитающих). Устранение этих бактерий связывается с улучшенными показателями состояния здоровья, включая уменьшение количества диспепсических симптомов, уменьшение рецидивов пептической язвы и повторного кровотечения, снижение риска рака желудка и так далее. Более подробное обсуждение искоренения Н. pylori и ее влияния на желудочно-кишечное заболевание можно найти во всемирной сети по адресу: informahealthcare.com, Expert Opin. Drug Saf. (2008) 7(3).
Чтобы соединения проявили эту противоинфекционную активность, их необходимо вводить в терапевтически эффективном количестве. Подразумевается, что термин «терапевтически эффективное количество» описывает количество соединения, достаточное для лечения инфекции, при разумном соотношении польза/риск, принятом для любого такого медицинского лечения. Однако будет понятно, что лечащий врач назначит общую суточную дозу соединения на основании тщательной медицинской оценки. Конкретный терапевтически эффективный уровень доз для любого конкретного пациента будет зависеть от ряда факторов, включая подвергаемое лечению расстройство и тяжесть этого расстройства; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и питание пациента; время введения, путь введения и скорость экскреции конкретного применяемого соединения; продолжительность лечения; лекарственные средства, используемые в комбинации или совместно с конкретным применяемым соединением; и подобные факторы, хорошо известные в области медицины. Однако, как правило, общая суточная доза обычно будет изменяться в диапазоне от примерно 0,1 мг/кг/сутки до примерно 5000 мг/кг/сутки в однократной или в разделенных дозах. В типичном случае дозировки для людей будут изменяться в диапазоне от примерно 10 мг до примерно 3000 мг в сутки, в однократной или многократных дозах.
Для введения соединений можно применять любой путь, обычно используемый для лечения инфекционных болезней, в том числе пероральный, парентеральный, местный, ректальный, трансмукозальный и через кишечник. Парентеральные введения включают инъекции для достижения системного эффекта или инъекции непосредственно в пораженный участок. Примерами парентеральных введений являются методы подкожной, внутривенной, внутримышечной, интрадермальной, интратекальной и интраокулярной, интраназальной, интравентрикулярной инъекций или инфузий. Методы местного введения включают обработку участков, легко доступных для локального нанесения, таких как, например, глаза, уши, в том числе в случае инфекций наружного и среднего уха, вагина, открытая рана, кожа, включая поверхностный слой кожи и нижележащие дермальные структуры, или нижний отдел кишечника. Трансмукозальное введение включает методы нанесения с использованием назального аэрозоля или ингаляции. Пероральное введение включает прием таблеток, капсул, растворов, суспензий, смеси с водой и/или пищей, саше и тому подобное.
Композиции
На основе соединений по настоящему изобретению могут быть приготовлены композиции для введения тем или иным способом с целью применения в медицине или ветеринарии по аналогии с другими биологически активными агентами, такими как антибиотики. Такие способы известны в данной области техники и кратко изложены ниже.
Композиция может быть приготовлена для введения любым путем, известным в данной области техники, таким как подкожный, ингаляционный, пероральный, местный или парентеральный. Композиции могут быть представлены в любой форме, известной в данной области техники, включая, но не ограничиваясь этим, таблетки, капсулы, порошки, гранулы, пастилки, кремы или препараты в жидкой форме, такие как пероральные или стерильные парентеральные растворы или суспензии.
Композиции для местного применения по настоящему изобретению могут быть представлены в виде, например, мазей, кремов или лосьонов, глазных мазей/капель и ушных капель, пропитанных повязок и аэрозолей, и могут содержать соответствующие традиционные вспомогательные вещества, такие как консерванты, растворители для содействия проникновению лекарственного средства и смягчающие вещества и так далее. Такие композиции для местного применения также могут содержать традиционные носители, такие как основы для крема или мази и этанол или олеиловый спирт для лосьонов. Такие носители могут быть представлены в композиции в количестве, например, от примерно 1% до примерно 98%.
Формой представления таблеток и капсул для перорального введения могут быть стандартные дозы, и они могут содержать такие традиционные эксципиенты, как связующие вещества, например, аравийскую камедь, желатин, сорбит, трагакант или поливинилпирролидон; наполнители, например, лактозу, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающие вещества для таблетирования, например, стеарат магния, тальк, полиэтиленгликоль или диоксид кремния; разрыхлители, например, картофельный крахмал; или приемлемые увлажняющие агенты, такие как лаурилсульфат натрия. Таблетки могут быть покрыты оболочкой способами, хорошо известными в обычной фармацевтической практике.
Жидкие препараты для перорального применения могут быть в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров, или могут быть представлены в виде сухого продукта для повторного разведения водой или другим подходящим разбавителем перед применением. Такие препараты в жидкой форме могут содержать традиционные вспомогательные вещества, такие как суспендирующие агенты, например, сорбит, метилцеллюлозу, глюкозный сироп, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, стеарат алюминия в виде геля или гидрогенизированные пищевые жиры, эмульгирующие агенты, например, лецитин, сорбитана моноолеат или аравийскую камедь; неводные разбавители (которые могут включать пищевые масла), например, миндальное масло, сложные эфиры жирных кислот, такие как глицериновый, пропиленгликолевый или из этилового спирта; консерванты, например, метил- или пропил-и-гидроксибензоат или сорбиновую кислоту, и, при желании, традиционные ароматизаторы или красители.
В случае парентерального введения готовят жидкие стандартные лекарственные формы, используя соединение и стерильный разбавитель, обычно воду. Соединение, в зависимости от используемого разбавителя и применяемой концентрации, может быть либо суспендировано, либо растворено в разбавителе или другом подходящем растворителе. При приготовлении растворов соединение может быть растворено в воде для инъекций и стерилизовано фильтрованием перед заполнением подходящего(ей) флакона или ампулы и герметичным закрыванием. Предпочтительно, чтобы такие агенты, как местный анестетик, консервирующие и буферные агенты, могли быть растворены в данном разбавителе. Для повышения стабильности композицию можно заморозить после заполнения флакона, а воду удалить под вакуумом. Затем флакон с сухим лиофилизированным порошком герметично закрывают, и для повторного приготовления жидкой формы перед применением к нему может быть приложен флакон с водой для инъекций. Суспензии для парентерального введения готовят по существу таким же образом, за исключением того, что вместо раствора готовят суспензию соединения в разбавителе, и стерилизацию невозможно осуществить путем фильтрования. Перед суспендированием в стерильном разбавителе можно провести стерилизацию соединения под действием этиленоксида. Предпочтительно, в композицию включают поверхностно-активное вещество или увлажняющий агент для облегчения равномерного распределения соединения.
Композиции могут содержать активное вещество, например, от приблизительно 0,1% по массе до приблизительно 100% по массе в зависимости от способа введения. Если композиции представляют собой стандартные дозировки, то каждая стандартная доза будет содержать, например, примерно 0,5-1000 мг активного ингредиента. Дозировка, которую применяют для лечения взрослого человека, будет находиться в диапазоне, например, от приблизительно 10 до 3000 мг в сутки, в зависимости от пути и частоты введения.
При желании соединения по настоящему изобретению можно вводить в комбинации с одним или более чем одним дополнительным антибактериальным агентом («дополнительным активным агентом»). Такое применение соединений по настоящему изобретению в комбинации с дополнительным активным агентом может быть предназначено для совместного, раздельного или последовательного использования.
В разделах Примеры и Подготовительные примеры, приведенных ниже, дополнительно иллюстрируются и разъясняются на примерах соединения по настоящему изобретению и способы получения таких соединений. Следует понимать, что объем настоящего изобретения никоим образом не ограничивается объемом следующих далее примеров и подготовительных примеров. В следующих далее примерах молекулы с единственным хиральным центром, если не указано иное, существуют в виде рацемической смеси. Те молекулы, которые имеют два или более хиральных центров, если не указано иное, существуют в виде рацемической смеси диастереомеров. Индивидуальные энантиомеры/диастереомеры могут быть получены способами, известными специалистам в данной области техники.
ПРИМЕРЫ
Экспериментальные методики
Обычно эксперименты проводили в атмосфере инертного газа (азота или аргона), особенно в случаях, когда использовали чувствительные к кислороду или влаге реагенты или промежуточные соединения. Как правило, имеющиеся в продаже растворители и реагенты, включая безводные растворители, использовали без дополнительной очистки там, где это целесообразно (обычно продукты SureSeal™ от Aldrich Chemical Company, Milwaukee, Wisconsin). Данные масс-спектрометрии приведены на основании результатов либо жидкостной хроматографии-масс-спектрометрии (LCMS), либо масс-спектрометрии с химической ионизацией при атмосферном давлении (APCI). Химические сдвиги для данных ядерного магнитного резонанса (ЯМР) выражены в частях на миллион (млн-1, δ) относительно остаточных пиков от используемых дейтерированных растворителей. Точки плавления не скорректированы. Масс-спектры низкого разрешения (LRMS) регистрировали либо на приборе Hewlett Packard 5989®, использующем химическую ионизацию (аммоний), либо на платформе Fisons (или MicroMass) с химической ионизацией при атмосферном давлении (APCI), для которой используют смесь 50/50 ацетонитрил/вода с 0,1% муравьиной кислоты в качестве ионизирующего агента. Значение комнатной температуры или температуры окружающей среды составляет 20-25°С.
Что касается процедур синтеза, ссылающихся на методики в других примерах, то условия реакции (продолжительность реакции и температура) могут варьировать. В общем случае ход реакций отслеживали по тонкослойной хроматографии или масс-спектрометрии и при необходимости подвергали дополнительной обработке. Процедуры очистки могут варьировать от эксперимента к эксперименту: обычно растворители и соотношения растворителей, используемые для элюентов/градиентов, выбирали для получения соответствующих значений фактора удерживания (Rf) или времени удерживания.
В приведенном выше обсуждении и в приведенных ниже примерах следующие сокращения имеют следующие значения. Если сокращение не определено, то оно имеет свое общепринятое значение: химическая ионизация при атмосферном давлении (APCI); водный (водн.); дейтерированный хлороформ (CDCl3); 2-хлор-4,6-диметокси-1,3,5-триазин (CDMT); дейтерированный метанол (CD3OD); дихлорметан (DCM); диметилформамид (DMF); диметилсульфоксид (DMSO); этилацетат (EtOAc); граммы (г); часы (ч); соляная кислота (HCl); жидкостная хроматография высокого давления (HPLC); гидроксид калия (КОН); жидкостная хроматография/масс-спектрометрия (LCMS); уходящая группа (Lg); гидроксид лития (LiOH); мета-хлорпербензойная кислота (mCPBA); сульфат магния (MgSO4); минуты (мин); гидроксид натрия (NaOH); палладий (Pd); ацетат палладия и BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил), микроинкапсулированные в полимочевинную матрицу с загрузкой по Pd 0,39 ммоль/г, BINAP 0,25, Pd 1,0 (Pd EnCat™); хлорид бис(дифенилфосфино)ферроценпалладия(II) (Pd(dppf)Cl2); фактор удерживания (Rf); время удерживания (Rt); комнатная температура (КТ); трифторуксусная кислота (TFA); тетрагидрофуран (THF); тетрагидропиранил (ТНР); тетраметилсилан (TMS); теоретический выход (TY) и уридин-5'-дифосфат (УДФ).
ПОЛУЧЕНИЕ ИСХОДНЫХ ВЕЩЕСТВ
Подготовительный пример 1 и подготовительный пример 1А
(+/-)-Этил-4-бром-2-метил-2-(метилсульфонил)бутаноат и индивидуальные (R)- и (S)-энантиомеры
Стадия А). Этил-2-(метилсульфонил)пропаноат
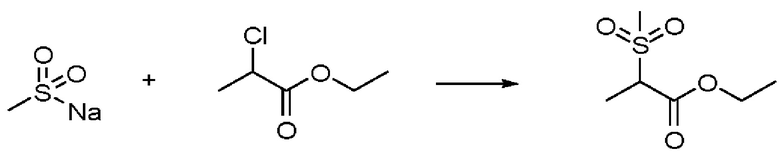
Метилсульфинат натрия (103 г; 937 ммоль) объединяли с этил-2-хлорпропионатом (109 г; 892 ммоль) в этаноле (350 мл) в одногорлой круглодонной колбе емкостью 500 мл. Реакционную смесь нагревали до 77°С в течение 20 часов и затем оставляли охлаждаться до комнатной температуры. Твердые вещества удаляли фильтрованием через целит, фильтровальную набивку промывали этанолом и объединенные фильтраты концентрировали в вакууме. Неочищенный продукт суспендировали в диэтиловом эфире (250 мл) и твердые вещества удаляли фильтрованием. Фильтрат концентрировали в вакууме, получая указанное в заголовке соединение в виде бледно-желтого масла (51 г; 73%). 1Н ЯМР (CDCl3, 400 МГц) δ млн-1 1.32 (t, J=7,05 Гц, 3Н); 1.67 (d, J=7,47 Гц, 3Н); 3.05 (s, 3Н); 3.83-3.92 (m, 1H); 4.18-4.37 (m, 2Н).
Стадия В). (+/-)-Этил-4-бром-2-метил-2-(метилсульфонил)бутаноат
Гидрид натрия (60%-ную дисперсию в минеральном масле; 2,33 г; 58,3 ммоль) промывали гексаном (2×10 мл) в двухгорлой круглодонной колбе емкостью 100 мл в атмосфере азота, затем суспендировали в DMF (30 мл). Суспензию обрабатывали этил-2-(метилсульфонил)пропаноатом (10,0 г; 55,49 ммоль) в DMF (10 мл), добавляя его по каплям. Смесь перемешивали в течение 30 мин при КТ, охлаждали до 0°С и обрабатывали 1,2-дибромэтаном (5,17 мл; 58,8), добавляя его по каплям. Смесь оставляли нагреваться до комнатной температуры с перемешиванием в течение ночи. Смесь гасили насыщенным хлоридом аммония (100 мл) и смесь экстрагировали диэтиловым эфиром (4×50 мл). Объединенные органические экстракты промывали 50%-ным насыщенным хлоридом аммония (4×50 мл), сушили (MgSO4), фильтровали и фильтрат концентрировали в вакууме. Неочищенное вещество хроматографировали через силикагель (350 г; 230-400 меш) с элюированием смесью 10-20% EtOAc/гексан, получая указанное в заголовке соединение в виде бледно-желтого масла (7,9 г; 50%). 1Н ЯМР (CDCl3, 400 МГц) δ млн-1 1.33 (t, J=7,05 Гц, 3Н); 1.64 (s, 3Н) 2.49-2.59 (m, 1Н); 2.78 (ddd, J=13,89; 10,16; 6,64 Гц, 1H); 3.05 (s, 3Н); 3.33-3.41 (m, 1Н); 3.46-3.54 (m, 1Н); 4.22-4.37 (m, 2Н).
Стадия С). Хиральное разделение (+/-)-этил-4-бром-2-метил-2-(метилсульфонил)бутаноата
Неочищенный (+/-)-этил-4-бром-2-метил-2-(метилсульфонил)бутаноат (1,82 кг) очищали посредством флэш-хроматографии с использованием колонки LP-600 и толуола в качестве элюента, получая чистый (+/-)-этил-4-бром-2-метил-2-(метилсульфонил)бутаноат (1,63 кг). Очищенное вещество растворяли в этаноле (75 г/л) и разделяли хиральной мультикол о ночной хроматографией (условия перечислены в Таблице 1) на МСС-2, получая энантиомер 1 (738,4 г; Rt=4,719 мин, [α]58920=+14,1°) с энантиомерной чистотой 99% и энантиомер №2 (763,8 г; Rt=4,040 мин) с энантиомерной чистотой 95%. Чистоту энантиомеров определяли хиральной HPLC; колонка Chiralpak AD 4,6×250 мм, 10 мкм; длина волны 215 нм; подвижная фаза: этанол; изократическое элюирование со скоростью 1 мл/мин при температуре окружающей среды.
Установлено, что энантиомер 1 представлял собой этил-(2R)-4-бром-2-метил-2-(метилсульфонил)бутаноат.
Подготовительный пример 1В
Бензил-(+/-)-4-бром-2-метил-2-(метилсульфонил)бутаноат и индивидуальные (R)- и (S)-энантиомеры.
Стадия А). Бензил-2-хлорпропаноат
Бензиловый спирт (242 мл; 253 г; 2,34 моль) и пиридин (204 мл; 204 г; 2,57 моль) растворяли в метиленхлориде (2,5 л) и охлаждали до 0°С.По каплям добавляли 2-хлорпропаноилхлорид (250 мл; 327 г; 2,57 моль), поддерживая температуру от 0°С до 5°С. После добавления смесь оставляли нагреваться до КТ в течение ночи. Смесь промывали 20%-ным водным раствором лимонной кислоты (2,5 л), насыщенным водным NaHCO3 (2,5 л), рассолом (2,5 л), сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученную коричневую жидкость (450 г) разбавляли небольшим количеством метиленхлорида и фильтровали через короткую набивку силикагеля. После концентрирования неочищенное вещество очищали посредством дистилляции на приборе, в конструкцию которого входит набранный из круглодонных сосудов каскад (bulb-to-bulb distillation) (2*10-2 мбар (2Па), 90-95°С), получая указанное в заголовке соединение в виде бледно- желтой жидкости (420 г; 90%). 1Н ЯМР (CDCl3, 300 МГц) δ млн-1 1.75 (d, 3Н, СН3), 4.45 (q, 1H, CHCl), 5.25 (s, 2Н, CH2Ar), 7.40 (m, 5Н, ArH).
Стадия В). Бензил-2-(метилсульфонил)пропаноат
Бензил-2-хлорпропаноат превращали в указанное в заголовке соединение, следуя общей методике, изложенной для этил-2-(метилсульфонил)пропаноата в подготовительном примере 1А. Указанное в заголовке соединение получали в виде желтой жидкости (389 г; 70%). 1Н ЯМР (CDCl3, 300 МГц) δ млн-1 1.65 (dt, 3Н, СНСН3), 3.00 (s, 3Н, SO2CH3), 3.95 (q, 1H, СН), 5.25 (m, 2Н, CO2CH2Ar), 7.40 (m, 5Н, ArH).
Стадия С). Бензил-(+/-)-4-бром-2-метил-2-(метилсульфонил)бутаноат
Бензил-2-(метилсульфонил)пропаноат превращали в указанное в заголовке соединение, следуя общей методике, изложенной для этил-(+/-)-4-бром-2-метил-2-(метилсульфонил)бутаноата в подготовительном примере 1А. Указанное в заголовке соединение получали в виде бледно-желтой жидкости (300 г; 58%). 1Н ЯМР (CDCl3 , 300 МГц) δ млн-1 1.70 (s, 3Н, СН3), 2.60 (m, 1H, CH2CH2Br), 2.80 (m, 1H, CH2CH2Br), 3.00 (s, 3Н, SO2CH3), 3.35 (m, 1Н, CH2CH2Br), 3.50 (m, 1H, CH2CH2Br), 5.30 (m, 2Н, CO2CH2Ar), 7.40 (m, 5Н, ArH).
Стадия D). Хиральное разделение бензил-(+/-)-4-бром-2-метил-2-(метилсульфонил)бутаноата
Бензил-(+/-)-4-бром-2-метил-2-(метилсульфонил)бутаноат (275 г) растворяли в смеси изопропанол/ацетонитрил (900 мл) и разделяли с использованием аналитического прибора SFC-4, колонки AS-H (30×250), подвижной фазы: CO2/пропанол (90/10) со скоростью потока 120 г/мин, получая энантиомер 1 (98 г; Rt=3,09 мин, [α]58920=-13,9°) с энантиомерной чистотой 99,94% и энантиомер 2 (101,5 г; время удерживания = 4,18 мин, [α]589=+11,61°) с энантиомерной чистотой 97,77%.
(S)-Бензил-4-бром-2-метил-2-(метилсульфонил)бутаноат
1Н ЯМР (CDCl3, 400 МГц) δ млн-1 1.65 (s, 3Н); 2.48-2.60 (m, 1H); 2.74 2.86 (m, 1Н); 2.95 (s, 3Н); 3.25-3.37 (m, 1Н); 3.40-3.52 (m, 1Н); 5.16-5.31 (m, 2Н); 7.31-7.40 (m, 5Н). [α]58920=-13,9°.
(R)-Бензил-4-бром-2-метил-2-(метилсульфонил)бутаноат
1H ЯМР (CDCl3, 400 МГц) δ млн-1 1.67 (s, 3Н); 2.51-2.61 (m, 1H); 2.75-2.87 (m, 1Н); 2.97 (s, 3Н); 3.28-3.37 (m, 1Н); 3.40-3.60 (m, 1Н); 5.15-5.36 (m, 2Н); 7.30-7.48 (m, 5Н). [α]58920=+11,61°.
Подготовительный пример 2
На приведенной ниже реакционной схеме показано получение 4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамида и его соответствующего R-энантиомера. Последовательность реакций в подготовительном примере 2 В является такой же, за исключением того, что в качестве исходного вещества для получения желаемого изомера используется бензил-(2R)-4-бром-2-метил-2-(метилсульфонил)бутаноат.
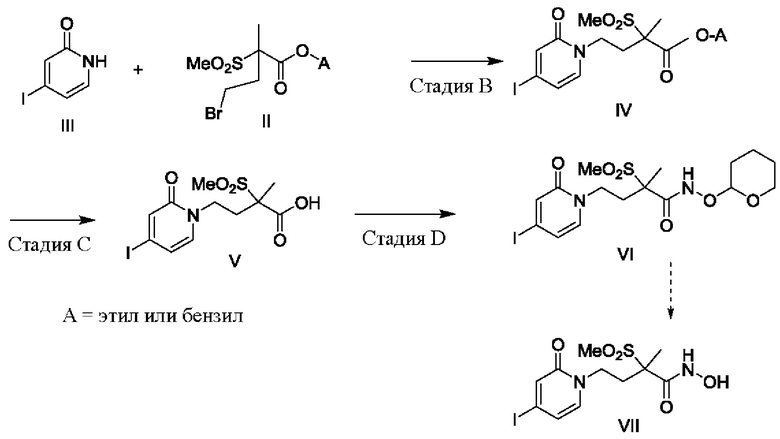
Синтез соединения VI (Т3): 4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамида в виде смеси диастереоизомеров
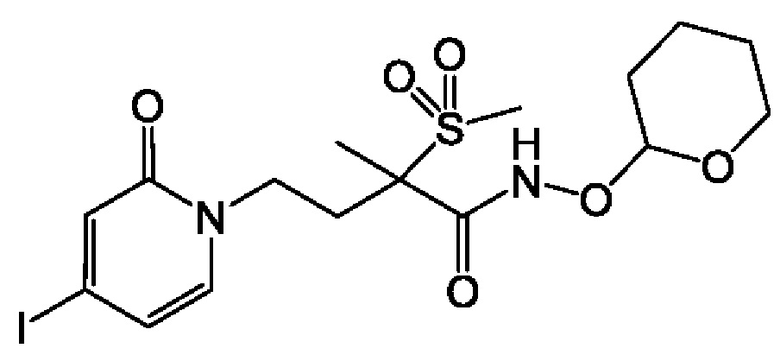
Стадия А). 4-Иодпиридин-2(1Н)-он (соединение III)
2-Фтор-4-иодпиридин (2,21 кг; 9,91 моль) суспендировали в смеси уксусной кислоты (7 л) и Н2О (3,5 л) с механическим перемешиванием. Смесь нагревали при температуре дефлегмации в течение ночи. После охлаждения до комнатной температуры твердое вещество отфильтровывали и концентрировали в вакууме. Остаток перемешивали в Et2O (3 л), указанное в заголовке соединение (1,72 кг; 7,78 моль) собирали фильтрованием в виде бледно-желтого твердого вещества. 1Н ЯМР (DMSO-d6, 300 МГц) δ млн-1 6.50 (d, 1H), 6.85 (s, 1H), 7.15 (d, 1H), 11.80 (s, 1Н).
Стадия В). Соединение IV (Т1): этил-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутаноат (А представляет собой Et)
К смеси 4-иодпиридин-2(1Н)-она (3,9 г; 18 ммоль), который может быть получен на приведенной выше стадии А, и карбоната цезия (11,9 г; 35,3 ммоль) в тетрагидрофуране (176 мл) при температуре окружающей среды добавляли этил-4-бром-2-метил-2-(метилсульфонил)бутаноат (6,08 г; 21,2 ммоль) (соединение II). Смесь нагревали до 50°С и перемешивали в течение ночи. Смесь оставляли охлаждаться до температуры окружающей среды и фильтровали через набивку целита. Набивку промывали метиленхлоридом и фильтрат концентрировали в вакууме. Неочищенное масло очищали посредством хроматографии на силикагеле, элюируя смесью гептаны/этилацетат. Желаемые фракции выделяли, растворитель удаляли посредством упаривания на роторном испарителе, получая этил-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутаноат в виде твердого вещества. 4,73 г. LCMS: (М+1) 428,2.
Стадия С). Соединение V (Т2): 4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутановая кислота
К раствору этил-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутаноата (3,26 г; 7,63 ммоль), который может быть получен на приведенной выше стадии В, в смеси тетрагидрофуран/метанол (4:1; 60 мл) при температуре окружающей среды добавляли раствор гидроксида лития моногидрата (0,9 М в воде; 15,3 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 3 часов. Смесь подкисляли водным раствором соляной кислоты (1 н.; 16 мл) и три раза экстрагировали метиленхлоридом. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая 4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутановую кислоту в виде твердого вещества. 3,05 г. LCMS: (М+1) 400,1.
Стадия D). Соединение VI (Т3): 4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамид
К раствору 4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутановой кислоты (3,01 г; 7,54 ммоль), которая может быть получена на приведенной выше стадии С, в метиленхлориде (75 мл) при температуре окружающей среды добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (2,02 г; 10,6 ммоль), 1-гидроксибензотриазола моногидрат (2,08 г; 13,6 ммоль), триэтиламин (1,89 мл; 13,6 ммоль) и O-тетрагидро-2Н-пиран-2-ил-гидроксиламин (1,33 г; 11,3 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение ночи. Смесь разбавляли метиленхлоридом и водой. Проводили разделение фаз и водный слой два раза экстрагировали метиленхлоридом. Органические экстракты объединяли и сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая неочищенный остаток. Неочищенный остаток очищали посредством хроматографии на силикагеле, элюируя метиленхлоридом и метанолом. Фракции, содержащие желаемый продукт, объединяли и концентрировали, получая 4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамид в виде твердого вещества. 3,62 г. LCMS: (М-1)497.
Подготовительный пример 2В
Синтез Т6: (2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамида
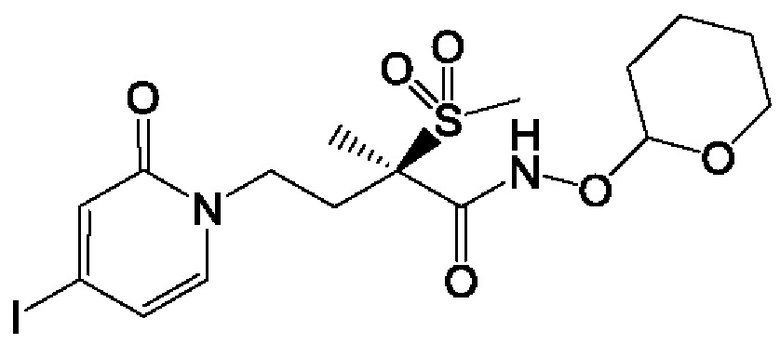
Стадия А). Т4: бензил-(2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутаноат
К смеси 4-иодпиридин-2(1Н)-она, который может быть получен так, как на стадии А подготовительного примера 2 (32,9 г; 149 ммоль), и карбоната цезия (102 г; 312 ммоль) в тетрагидрофуране (400 мл) при температуре окружающей среды добавляли бензил-(2R)-4-бром-2-метил-2-(метилсульфонил)бутаноат (62,3 г; 178,4 ммоль). Смесь нагревали до 60°С и перемешивали в течение ночи. Смесь оставляли охлаждаться до температуры окружающей среды и фильтровали через набивку целита. Набивку промывали этилацетатом (500 мл), фильтраты объединяли и концентрировали в вакууме, получая оранжевое масло. Неочищенное масло очищали путем фильтрования через набивку силикагеля, элюируя смесью гептаны/этилацетат. Желаемые фракции выделяли и растворитель удаляли посредством упаривания на роторном испарителе, получая бензил-(2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутаноат в виде белого твердого вещества. 44,91 г. 1ЯМР (CDCl3) δ млн-1 7.39-7.36 (5Н, m), 7.03 (1Н, d, J=1,76 Гц), 6.77 (1H, d, J=7,03 Гц), 6.41 (1H, dd, J=1,76 Гц, J=7,03 Гц), 5.21 (2Н, d, J=1,56 Гц), 4.19-4.12 (1H, m), 3.82-3.75 (1H, m), 2.97 (3Н, s), 2.47-2.42 (2Н, m), 1.73 (3Н, s).
Стадия В). Т5: (2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутановая кислота
К раствору бензил-(2R)-4-(4-иод-2-оксопиридин-1 (2Н)-ил)-2-метил-2-(метилсульфонил)бутаноата (44,91 г; 91,7 ммоль), который может быть получен на приведенной выше стадии А, в тетрагидрофуране (300 мл) и метаноле (300 мл) при температуре окружающей среды добавляли гидроксид калия (3,76 М раствор в воде; 564 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 16 часов. Растворитель удаляли посредством упаривания на роторном испарителе и остаток растворяли в воде. Водный слой промывали диэтиловым эфиром и затем подкисляли концентрированной соляной кислотой (приблизительно до рН 2), что позволило получить белый осадок. Осадок собирали посредством фильтрования, промывали водой и сушили в вакууме до постоянной массы, получая (2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутановую кислоту в виде белого твердого вещества. 33,2 г. LCMS: (М+1) 400,4. 1ЯМР (CD3OD) δ млн-1 7.34 (1H, d, J=7,23 Гц), 7.03 (1Н, d, J=1,76 Гц), 6.69 (1H, dd, J=1,95 Гц, J=7,23 Гц), 4.24-4.16 (1Н, m), 4.05-3.98 (1H, m), 3.14 (3Н, s), 2.57-2.50 (1Н, m), 2.35-2.28 (1H, m), 1.68 (3Н, s).
Стадия С). Т6: (2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамид
К раствору (2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутановой кислоты, которая может быть получена на приведенной выше стадии В (33,18 г; 83,12 ммоль), в метиленхлориде (400 мл) при температуре окружающей среды добавляли 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (22,3 г; 116 ммоль), 1-гидроксибензотриазола моногидрат (22,9 г; 150 ммоль), триэтиламин (20,9 мл; 150 ммоль) и 0-тетрагидро-2Н-пиран-2-ил-гидроксиламин (14,6 г; 125 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение ночи. Смесь разбавляли метиленхлоридом и водой. Проводили разделение фаз и водную фазу два раза экстрагировали метиленхлоридом. Органические экстракты объединяли и сушили над сульфатом магния, фильтровали и концентрировали, получая неочищенный остаток. Неочищенный остаток растворяли в метиленхлориде (приблизительно в 150 мл) с минимальным количеством метанола. К этому раствору добавляли гептаны (450 мл), смесь концентрировали в вакууме до объема 150 мл и фильтровали. Твердое вещество промывали гептанами и сушили в вакууме, получая (2R)-4-(4-иод-2-оксопиридин-1 (2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамид. 26,1 г. LCMS: (М-1) 497,6.
Подготовительный пример 3А: (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(2Н-1,2,3-триазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамид
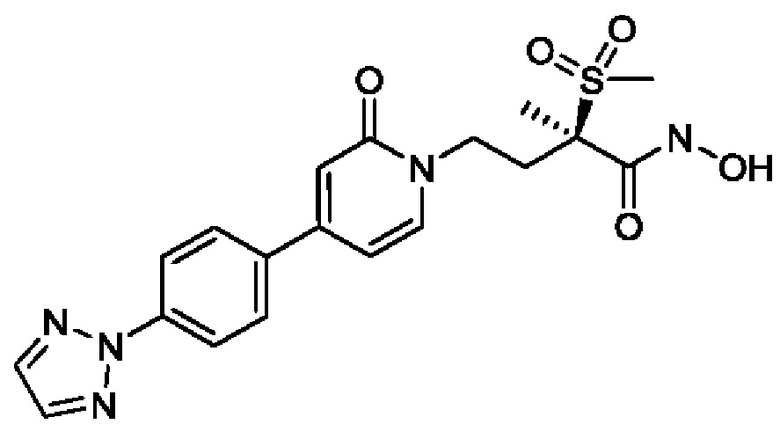
Стадия А). Получение 2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-2Н-1,2,3-триазола
Ацетат калия (391 мг; 3,98 ммоль) добавляли к раствору 2-(4-бромфенил)-2Н-1,2,3-триазола (1,0 эквивалента), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-1,3,2-диоксаборолана (1,20 эквивалента) и комплекса [1,1'-бис-(дифенилфосфино)ферроцен]-дихлорпалладия(II) и DCM (0,30 эквивалента) в 1,4-диоксане во флаконе. Флакон закрывали, нагревали до 80°С и содержимое перемешивали при этой температуре в течение ночи. К реакционной смеси добавляли комплекс [1,1'-бис-(дифенилфосфино)ферроцен]-дихлорпалладия(II) и DCM (0,30 эквивалента), смесь повторно нагревали до 80°С и перемешивание продолжали при этой температуре в течение ночи. Реакционную смесь охлаждали, разбавляли этилацетатом и водой, фильтровали через целит и фильтровальную набивку промывали этилацетатом. Органический слой отделяли и водный слой экстрагировали этилацетатом. Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали. Неочищенное вещество очищали посредством флэш-хроматографии с использованием колонки Analogix SF15-24g и этилацетата в гептане (30-80%) в качестве элюента, получая указанное в заголовке соединение. Указанное в заголовке соединение получали в виде оранжевого твердого вещества (240,6 мг; 78%) LC-MS m/z 272,4 (М+1). 1H ЯМР (CDCl3 , 400 МГц) δ млн-1 1.37 (s, 12Н), 7.83 (s, 2Н), 7.94 (d, J=8,59 Гц, 2Н), 8.10 (d, J=8,59 Гц, 2Н).
Стадия В). (2R)-2-Метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(2Н-1,2,3-триазол-2-ил)фенил]пиридин-1(2Н)-ил}-Н-(тетрагидро-2Н-пиран-2-илокси)бутанамид
Pd EnCat™ (0,08 эквивалента) добавляли к смеси карбоната калия (2,54 эквивалента), 2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-2Н-1,2,3-триазола (1,5 эквивалента) и 4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамида (1,0 эквивалента) в смеси диоксан : вода (4:1) во флаконе для микроволнового реактора и реакционную смесь нагревали при 90°С в течение ночи. Реакционную смесь фильтровали и смолу промывали этилацетатом и водой. Фильтрат концентрировали досуха, неочищенное вещество очищали посредством флэш-хроматографии с использованием колонки Analogix SF15-12g и элюировали этилацетатом в гептане (0-80%), получая указанное в заголовке соединение. Указанное в заголовке соединение получали в виде белого твердого вещества (101 мг; 48,8%). LC-MS m/z 514,7 (М-1).
Стадия С). (2R)-N-Гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(2Н-1,2,3-триазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамид
4,0 М раствор HCl в 1,4-диоксане медленно добавляли к раствору (2R)-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(2Н-1,2,3-триазол-2-ил)фенил]пиридин-1(2Н)-ил}-N-(тетрагидро-2Н-пиран-2-илокси)бутанамида в смеси дихлорметана с водой (5:1) при 0°С. Ледяную баню удаляли и реакционную смесь оставляли нагреваться до КТ. Через 30 мин (реакция завершилась по данным тонкослойной хроматографии (TLC)) реакционную смесь концентрировали, получая неочищенное твердое вещество. Это неочищенное вещество растирали в изопропаноле в течение ночи. Твердое вещество собирали посредством фильтрования, промывали изопропанолом, смесью изопропанолтептан (1:1), гептаном и эфиром. Указанное в заголовке соединение получали в виде беловатого твердого вещества (63,7 мг; 74%). LC-MS m/z 432,5 (М+1). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 1.58 (s, 3Н), 2.09-2.25 (m, 1H), 2.34-2.47 (m, 1H), 3.11 (s, 3Н), 3.70-3.82 (m, 1H), 4.04-4.19 (m, 1H), 6.68-6.73 (m, 1Н), 6.78 (d, J=2,15 Гц, 1H), 7.79 (d, J=7,22 Гц, 1H), 7.95 (d, J=8,78 Гц, 2Н), 8.12 (d, J=8,59 Гц, 2Н), 8.17 (s, 2Н), 11.15 (br. s., 1H).
Подготовительный пример 3В: (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамид
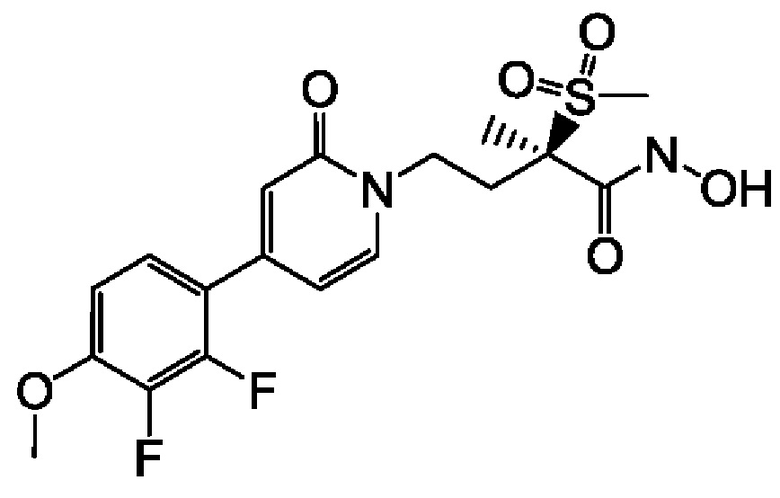
Стадия А). (2R)-4-[4-(2,3-Дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамид
Pd EnCat™ (200 мг; 0,06 ммоль) добавляли к смеси карбоната калия (250 мг; 1,81 ммоль), (2,3-дифтор-4-метоксифенил)бороновой кислоты (113 мг; 0,602 ммоль) и (2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамида (300 мг; 0,602 ммоль) в смеси диоксан : вода (5,5 мл; смесь 10:1) в круглодонной колбе емкостью 25 мл. Колбу нагревали в течение ночи при 80°С. Реакционную смесь охлаждали до температуры окружающей среды, фильтровали через целит и промывали этилацетатом (20 мл). Неочищенное вещество концентрировали, получая неочищенный продукт. Полученное неочищенное вещество очищали хроматографией на силикагеле (растворитель для элюирования: этилацетат), получая указанное в заголовке соединение в виде вязкого пенообразного масла. Выход: 132 мг; 42,6%. MS (APCI) m/z 515,5 (М+Н). 1H ЯМР (CDCl3 , 400 МГц) δ млн-1 1.54-1.66 (m, 3Н), 1.68 (d, J=2,34 Гц, 3Н), 1.71-1.97 (m, 3Н), 2.30-2.44 (m, 1H), 2.45-2.58 (m, 1H), 3.18 (d, J=3,12 Гц, 3Н), 3.54-3.68 (m, 1H), 3.92 (s, 3Н) 3.99-4.08 (m, 1H), 4.11-4.23 (m, 1H), 4.26-4.40 (m, 1H), 5.10-5.21 (m, 1H), 6.42-6.53 (m, 1H), 6.75 (s, 1H), 6.77-6.86 (m, 1Н), 7.05-7.17 (m, 1H), 7.37 (d, J=7,02 Гц, 1H), 12.10 (d, J=7,61 Гц, 1H).
Стадия В). (2R)-4-[4-(2,3-Дифтор-4-метоксифенил)-2-оксопиридин-1(2H)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамид
1,0 М водный раствор HCl (2,76 мл) медленно добавляли к раствору (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамида (132 мг; 0,26 ммоль) в 1,4-диоксане (15 мл) при комнатной температуре. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Через 18 часов реакционную смесь концентрировали до 25% от начального объема, получая белый осадок. Осадок отфильтровывали через воронку Бюхнера и промывали гексанами (20 мл), получая белое твердое вещество. Выход 45 мг; 41%. MS (APCI) m/z 431,1. (М+Н). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 1.55 (s, 3Н), 2.14 (td, J=12,20; 4,88 Гц, 1Н), 2.35-2.45 (m, 1H), 3.08 (s, 3Н), 3.72 (td, J=12,05; 4,78 Гц, 1H), 3.90 (s, 3Н), 4.09 (td, J=11,90; 5,27 Гц, 1H), 6.46 (dt, J=7,02; 1,85 Гц, 1H), 6.54 (s, 1H), 7.03-7.17 (m, 1H), 7.37 (td, J=8,63; 2,24 Гц, 1H), 7.72 (d, J=7,22 Гц, 1H), 9.22 (br. s., 1H), 11.10 (s, 1H).
Подготовительный пример 3С: (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамид
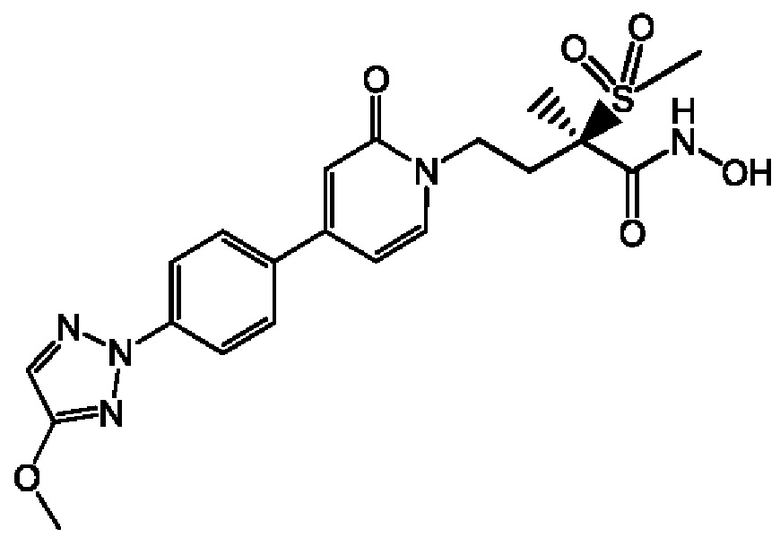
Стадия А). 2-(4-Бромфенил)-2Н-1,2,3-триазол-1-оксид
В колбу, содержащую глиоксаль (2,0 г; 14 ммоль), добавляли воду (20 мл). В колбу с глиоксалем добавляли в виде одной порции гидроксиламин HCl (958 мг; 13,8 ммоль) и карбонат натрия (1,53 г; 14,5 ммоль) (наблюдали выделение С02). Реакционную смесь перемешивали при КТ в течение 20 минут (реакционная смесь становилась желтой). К реакционной смеси добавляли метанол (40 мл) и порциями с охлаждением во льду добавляли 4-бромфенилгидразин HCl (3,1 г; 13,8 ммоль). Затем реакционную смесь перемешивали при КТ в течение 30 мин. К реакционной смеси добавляли гексагидрат сульфата меди(II) (20 г; 78 ммоль). Добавляли смесь вода : пиридин (1:1) (200 мл), затем нагревали при 90°С в течение 16 часов. Реакционную смесь охлаждали и значение рН подводили до 3, используя 6 н. раствор HCl (приблизительно 200 мл). Смесь фильтровали через целит для удаления нерастворе иных веществ. Целит промывали дополнительным количеством этилацетата (1000 мл). Органический слой отделяли и продукт дополнительно экстрагировали из водного слоя, используя EtOAc (3×250 мл). Органические фазы объединяли, сушили над карбонатом калия, фильтровали и концентрировали приблизительно до половины объема. Затем это вещество фильтровали через набивку диоксида кремния (приблизительно 6 дюймов (15 см)). Диоксид кремния промывали еще 300 мл этилацетата. Затем растворитель упаривали в вакууме. Неочищенное вещество очищали хроматографией на силикагеле (от смеси 4:1 гептан : EtOAc до смеси 3:1 гептан : EtOAc). После концентрирования фракций получали светлое рыжевато-коричневое твердое вещество (1,0 г; TY 30%). MS (LC/MS) m/z 240,1 (М+1). 1H ЯМР (CDCl3 , 400 МГц) δ млн-1 7.47 (d, J=0,98 Гц, 1H), 7.65-7.69 (m, 2Н), 7.73 (d, J=0,78 Гц, 1H), 7.86-7.90 (m, 2Н).
Стадия В). 2-(4-Бромфенил)-2Н-1,2,3-триазол-4-илацетат
В колбу, содержащую 2-(4-бромфенил)-2Н-1,2,3-триазол-1-оксид (500 мг; 2,08 ммоль), добавляли ацетилхлорид (4,71 мл; 63 ммоль) и перемешивали при КТ в течение 16 часов. Ацетилхлорид удаляли в вакууме, добавляли этилацетат (30 мл) и концентрировали (2х), получая светло-коричневое твердое вещество (520 мг; 90%). MS (LC/MS) m/z 282,1 (М+1). 1H ЯМР (CDCl3 , 400 МГц) δ млн-1 2.39 (s, 3Н), 7.57-7.63 (m, 2Н), 7.84 (s, 1H), 7.87-7.93 (m, 2Н).
Стадия С). 2-(4-Бромфенил)-2Н-1,2,3-триазол-4-ол
2-(4-Бромфенил)-2Н-1,2,3-триазол-4-илацетат (520 мг; 1,84 ммоль) обрабатывали метанолом (10 мл) и водой (10 мл), затем 1,4-диоксаном (5 мл). Полученный раствор обрабатывали гидроксидом лития (265 мг; 11,1 ммоль). Реакционную смесь перемешивали при КТ в течение 36 часов. К реакционной смеси добавляли 1 н. раствор HCl (40 мл) и продукт экстрагировали этилацетатом (3×100 мл). Объединенные органические фазы сушили над карбонатом калия, фильтровали и концентрировали. Неочищенное вещество очищали хроматографией на силикагеле (от смеси 4:1 гептан : EtOAc до смеси 1:4 гептан : EtOAc), получая светлое рыжевато-коричневое твердое вещество (440 мг; TY 98%). MS (LC/MS) m/z 240,21 (М+1). 1H ЯМР (CDCl3 , 400 МГц) δ млн-1 7.33 (s, 1H), 7.58 (d, J=8,98 Гц, 2Н), 7.78 (d, J=8,98 Гц, 2Н).
Стадия D). 2-(4-Бромфенил)-4-метокси-2Н-1,2,3-триазол
Во флакон емкостью 20 мл, снабженный крышкой с резиновой уплотнительной мембраной, отвешивали 2-(4-бромфенил)-2Н-1,2,3-триазол-4-ол (200 мг; 0,833 ммоль). Добавляли THF (10,0 мл). В этот флакон добавляли карбонат цезия (814 мг; 2,5 ммоль), затем добавляли метилиодид (65,8 мкл; 1,04 ммоль) через шприц. Реакционную смесь нагревали при 60°С в течение 16 часов. Добавляли воду (20 мл) и продукт экстрагировали этилацетатом (2×75 мл). Органические фазы объединяли, сушили над карбонатом калия, фильтровали и концентрировали, получая светлое рыжевато-коричневое твердое вещество (190 мг; TY 89%). 1H ЯМР (CDCl3 , 400 МГц) δ млн-1 4.04 (s, 3Н), 7.30 (s, 1H), 7.56 (d, J=8,98 Гц, 2H), 7.84 (d, J=8,98 Гц, 2H).
Стадия E). 4-Метокси-2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-2H-1,2,3-триазол
Ацетат калия (220 мг; 2,24 ммоль) добавляли к 2-(4-бромфенил)-4-метокси-2Н-1,2,3-триазолу (190 мг; 0,748 ммоль), бис(пинаколато)дибору (228 мг; 0,898 ммоль) и комплексу Pd(dppf)Cl2 DCM (185 мг; 0,224 ммоль) во флаконе емкостью 20 мл, снабженном крышкой с резиновой уплотнительной мембраной. Флакон вакуумировали и 3 раза заполняли азотом. В этот флакон добавляли 1,4-диоксан (8 мл). Реакционную смесь нагревали при 80°С в течение 16 часов. Реакционную смесь фильтровали через целит (приблизительно 2 дюйма (5 см)). Целит промывали дополнительным количеством этилацетата (150 мл). Фильтрат концентрировали в вакууме и неочищенное вещество очищали хроматографией на силикагеле (от смеси 9:1 гептан : EtOAc до смеси 2:4 гептан : EtOAc), получая светлое рыжевато-коричневое твердое вещество (145 мг; TY 65%). MS (LC/MS) m/z 302,3 (М+1). 1Н ЯМР (CDCl3, 400 МГц) δ млн-1 1.37 (s, 12Н), 4.06 (s, 3Н), 7.31 (s, 1Н), 7.90 (s, 2H), 7.95 (s, 2H).
Стадия F). (2R)-4-{4-[4-(4-Метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамид
Pd EnCat™ (98 мг; 0,03 ммоль) добавляли к смеси карбоната калия (171 мг; 1,24 моль), 4-метокси-2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-2Н-1,2,3-триазола (138 мг; 0,457 ммоль) и (2R)-4-(4-иод-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамида (190 мг; 0,381 ммоль) в смеси диоксан : вода (6 мл; 5:1) во флаконе емкостью 20 мл. Реакционную смесь охлаждали и фильтровали через целит (приблизительно 1 дюйм (2,5 см)). Целит промывали дополнительным количеством метанола (100 мл). Фильтрат концентрировали в вакууме и неочищенное вещество очищали хроматографией на силикагеле (от смеси 4:1 гептан : EtOAc к 100%-ному EtOAc, затем до смеси 85% EtOAc : 15% метанола), получая светлое рыжевато-коричневое смолообразное вещество (120 мг; TY 58%). MS (LC/MS) m/z 546,2 (М+1). 1Н ЯМР (CD3OD, 400 МГц) δ млн-1 1.28 (s, 1Н), 1.57-1.70 (m, 2Н), 1.68-1.81 (m, 3Н), 1.78-1.92 (m, 3Н), 2.36-2.50 (m, 1H), 2.55-2.72 (m, 1H), 3.09-3.21 (m, 3Н), 3.56-3.70 (m, 1H), 4.07 (s, 3Н), 4.12 (d, J=7,22 Гц, 2Н), 4.15-4.25 (m, 1H), 4.25-4.42 (m, 1H), 5.01-5.14 (m, 1H), 6.76-6.85 (m, 1H), 6.87 (s, 1Н), 7.49 (s, 1H), 7.68-7.80 (m, 1H), 7.85 (d, J=9,17 Гц, 2Н), 8.08 (d, J=8,98 Гц, 2Н).
Стадия G). (2R)-N-Гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилеульфонил)бутанамид
К (2R)-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)-N-(тетрагидро-2Н-пиран-2-илокси)бутанамиду (120 мг; 0,22 ммоль) добавляли диоксан (2 мл), дихлорметан (2 мл) и воду (1 мл). Содержимое реакционной колбы охлаждали снаружи с помощью льда, затем обрабатывали 4,0 М раствором HCl в диоксане (0,55 мл). Реакционную смесь перемешивали в течение 15 минут, затем концентрировали при пониженном давлении. Добавляли изопропанол (10 мл) и концентрировали путем удаления любой оставшейся воды азеотропной перегонкой, получая рыжевато-коричневое твердое вещество (80 мг; TY 80%). MS (LC/MS) m/z 462,3 (М+1). 1H ЯМР (CD3OD, 400 МГц) δ млн-1 1.74 (s, 3Н), 2.34-2.51 (m, 1H), 2.55-2.81 (m, 1H), 3.13 (s, 3Н), 3.96-4.06 (m, 1H), 4.07 (s, 3Н), 4.26-4.45 (m, 1H), 6.84-7.00 (m, 2Н), 7.49 (s, 1H), 7.75-7.93 (m, 3Н), 8.09 (d, J=8,78 Гц, 2Н).
Подготовительный пример 3D: (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамид
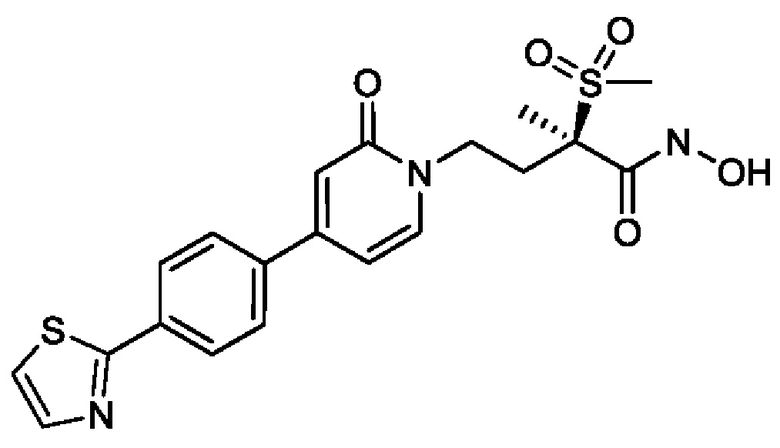
Указанное в заголовке соединение может быть получено способом, аналогичным методикам, описанным выше. Обычно продукт может быть получен в результате проведения реакции перекрестного сочетания Сузуки-Мияуры с возможным удалением концевой защитной группы гидроксамовой кислоты. Способы, используемые для описания синтеза предшественников или партнеров по сочетанию, таких как бороновые кислоты или сложные эфиры, известны специалистам в данной области техники. Время удерживания: 0,48. Масса иона 448. 'Н ЯМР (400 МГц, DMSO-d6) δ млн-1 1.58 (s, 3Н), 2.18 (td, J=12,05; 4,98 Гц, 1H), 2.40-2.48 (m, 1H), 3.11 (s, 3Н), 3.77 (td, J=12,15; 5,37 Гц, 1H), 4.08-4.19 (m, 1H), 6.72 (dd, J=7,22; 2,15 Гц, 1H), 6.79 (d, J=2,15 Гц, 1H), 7.80 (d, J=7,22 Гц, 1Н) 7.84.
Подготовительный пример 4А: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамид
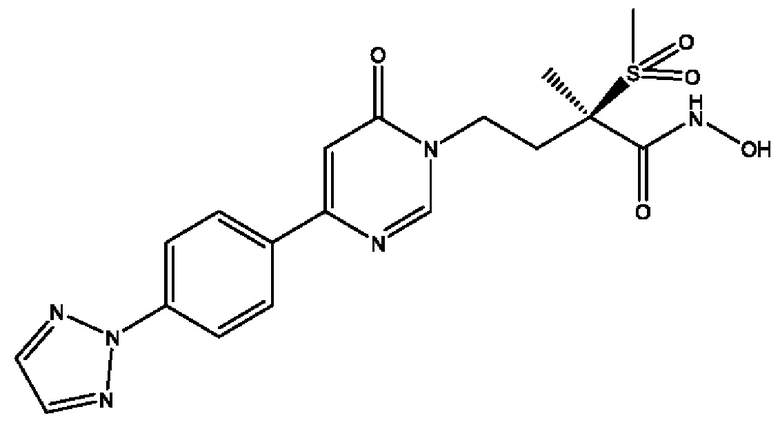
Стадия А. Получение 4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-метоксипиримидина
Приведенную далее реакцию проводили в том же масштабе за две отдельные загрузки, при этом различие между этими загрузками заключалось в способе нагревания и времени нагревания. К смеси 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-2Н-1,2,3-триазола (619 мг; 2,28 ммоль) и 4-хлор-6-метоксипиримидина (300 мг; 2,08 ммоль) добавляли хлорид бис(трифенилфосфин)палладия(П) (150 мг; 0,21 ммоль), затем 1,2-диметоксиэтан (6 мл), этанол (2 мл) и 2,0 М водный раствор карбоната натрия (3,1 мл). Реакционную смесь либо нагревали при 120°С в микроволновом реакторе в течение 15 минут, либо альтернативно нагревали в масляной бане при 120°С в течение 1 часа. Реакционную смесь очищали флэш-хроматографией на силикагеле с использованием градиентного элюирования (гептан : EtOAc; 0-100%). Фракции, содержащие продукт, концентрировали в вакууме, получая указанное в заголовке соединение (130 мг; выход 25% для случая нагревания микроволновым излучением; 80 мг; выход 15% для случая нагревания в масляной бане). 1H ЯМР (400 МГц, CDCl3 ) δ млн-1 8.86-8.90 (m, 1H), 8.21 (m, 4 Н), 7.87 (s, 2Н), 7.15-7.18 (m, 1H), 4.06 (s, 3Н).
Стадия В. Получение 6-(4-(2Н-1,2,3-триазол-2-ил)фенил)пиримидин-4(3Н)-она
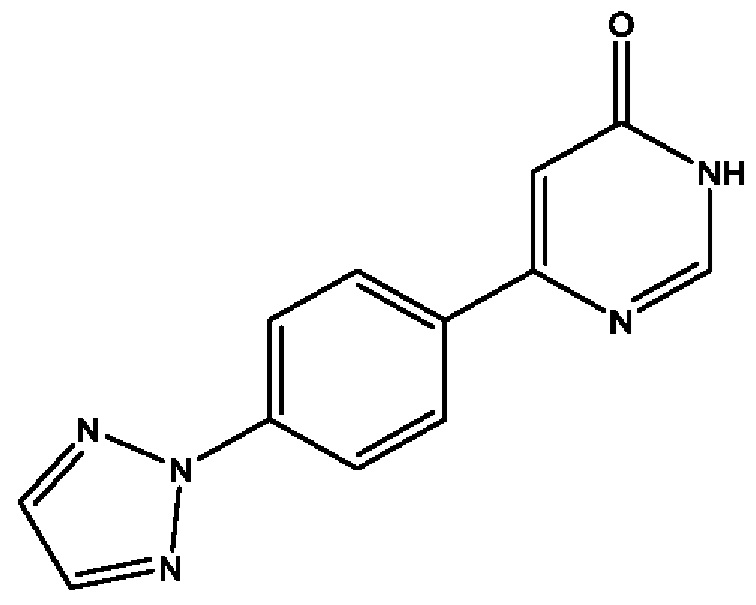
К раствору 4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-метоксипиримидина (210 мг; 0,829 ммоль) в уксусной кислоте (6 мл) добавляли бромистоводородную кислоту (0,533 мл). Реакционную смесь нагревали в течение ночи при 85°С, затем концентрировали в вакууме. К остатку добавляли EtOAc, затем смесь концентрировали в вакууме, получая указанное в заголовке соединение. Продукт использовали на следующей стадии.
Стадия С. Получение этил-(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-2-метил-2-(метилсульфонил)бутаноата
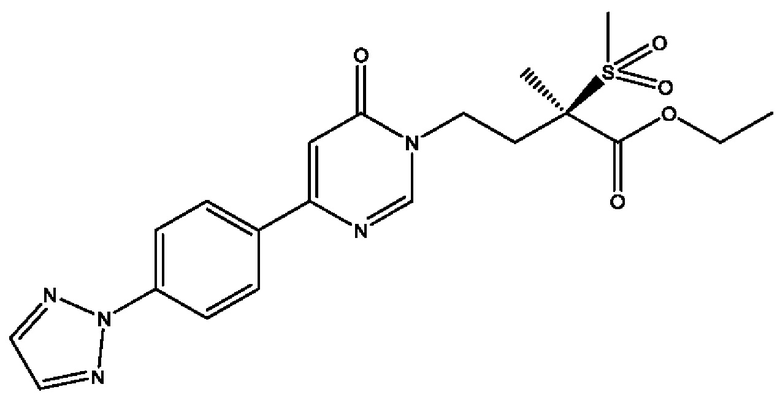
Суспензию 6-(4-(2Н-1,2,3-триазол-2-ил)фенил)пиримидин-4(3Н)-она (260 мг; 1,09 ммоль), этил-(И)-4-бром-2-метил-2-(метилсульфонил)бутаноата (344 мг; 1,20 ммоль), карбоната калия (451 мг; 3,26 ммоль) и бромида тетрабутиламмония (35,9 мг; 0,11 ммоль) в ацетонитриле (10 мл) кипятили с обратным холодильником в течение 1 часа. Образовывался белый осадок, а данные LC/MS указывали на отсутствие продукта, поэтому добавляли дополнительное количество ацетонитрила (10 мл). Реакционную смесь кипятили с обратным холодильником в течение ночи. Данные LC/MS указывали на то, что образовывалась смесь двух продуктов (продуктов О-алкилирования и N-алкилирования). Реакционную смесь оставляли охлаждаться, затем концентрировали в вакууме. Остаток фильтровали через небольшую колонку с силикагелем, элюировали метиленхлоридом и фильтрат концентрировали в вакууме. Затем полученный остаток очищали флэш-хроматографией на силикагеле с использованием градиентного элюирования (гептан: EtOAc; 40-100% по EtOAc). Первый продукт (О-алкилированный) элюировался в смеси 50% гептана/EtOAc, тогда как второй продукт (желаемый N-алкилированный) элюировался в смеси 20% гептана/80% EtOAc. Фракции, содержащие продукт, концентрировали в вакууме, получая указанное в заголовке соединение (160 мг; выход 33%).
Стадия D. Получение (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-2-метил-2-(метилсульфонил)бутановой кислоты
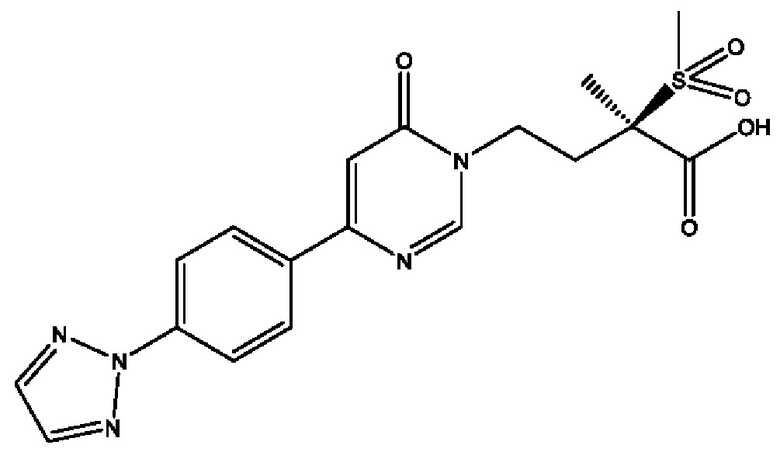
К раствору этил-(R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-2-метил-2-(метилсульфонил)бутаноата (160 мг; 0,359 ммоль) в 2-метилтетрагидрофуране (5 мл) добавляли раствор гидроксида лития (43,0 мг; 1,80 ммоль). Реакционную смесь нагревали при 50°С в течение ночи, и данные LC/MS указывали на образование продукта. Смесь оставляли охлаждаться, затем проводили разделение слоев. Органический слой обрабатывали 1 н. раствором гидроксида натрия (4 мл). Объединенный водный слой подкисляли до рН 2, используя 3 н. раствор соляной кислоты. Образовывалось белое кремообразное твердое вещество, и его собирали фильтрованием и сушили, получая указанное в заголовке соединение (100 мг; выход 67%).
Стадия Е. Получение (2R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-2-метил-2-(метилсульфонил)-Н-((тетрагидро-2Н-пиран-2-ил)окси)бутанамида
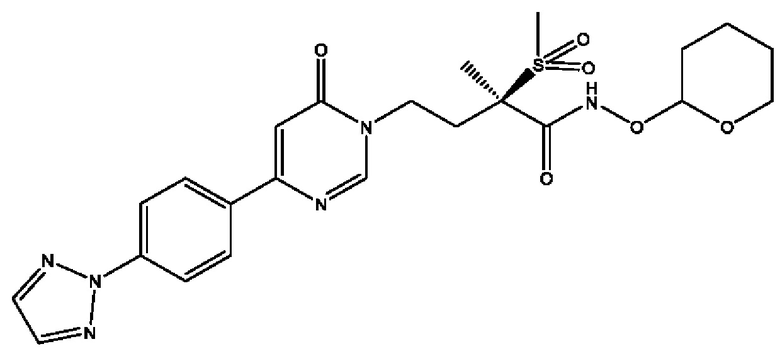
К суспензии (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-2-метил-2-(метилсульфонил)бутановой кислоты (100 мг; 0,24 ммоль) в 2-метилтетрагидрофуране (5 мл) добавляли N-метилморфолин (0,04 мл; 0,36 ммоль) и 2-хлор-4,6-диметокси-1,3,5-триазин (56,5 мг; 0,312 ммоль). Реакционную смесь перемешивали в течение одного часа при КТ, затем добавляли O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (36,6 мг; 0,312 ммоль) и реакционную смесь перемешивали в течение 1 часа при КТ. Затем реакционную смесь фильтровали и концентрировали в вакууме. Остаток растворяли в дихлорметане и полученный раствор затем очищали флэш-хроматографией на силикагеле с использованием градиентного элюирования (гептан : EtOAc, 40-100% по EtOAc). Фракции, содержащие продукт, которые элюировались в смеси 50% EtOAc/50% гептана, концентрировали в вакууме, получая указанное в заголовке соединение (50 мг; 40%).
Стадия F. Получение (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида
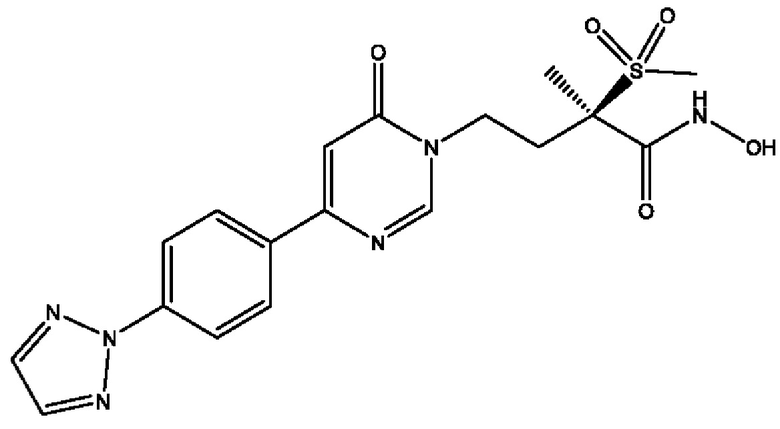
К раствору (2R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-2-метил-2-(метилсульфонил)-N-((тетрагидро-2Н-пиран-2-ил)окси)бутанамида (50,0 мг; 0,10 ммоль) в диоксане (5 мл) добавляли хлористый водород (0,50 ммоль; 0,125 мл 4,0 М раствора в диэтиловом эфире). Реакционную смесь перемешивали в течение одного часа, затем концентрировали в вакууме и остаток промывали этилацетатом и этанолом, получая указанное в заголовке соединение (40 мг; 93%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 1.59 (s, 3Н), 2.20 (ddd, J=13,22; 11,07; 4,98 Гц, 1H), 2.52-2.58 (m, 1H), 3.10 (s, 3Н), 3.84 (ddd, J=12,93; 10,88; 5,27 Гц, 1H), 4.09 (ddd, J=12,93; 10,88; 4,68 Гц, 1Н), 7.04-7.07 (m, 1H), 8.11-8.16 (m, 2Н), 8.19 (s, 2Н), 8.26-8.31 (m, 2Н), 8.52-8.64 (m, 1Н).
Пример 1
(R)-4-(4-(4-(2Н-1,2,3-Триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, динатриевая соль
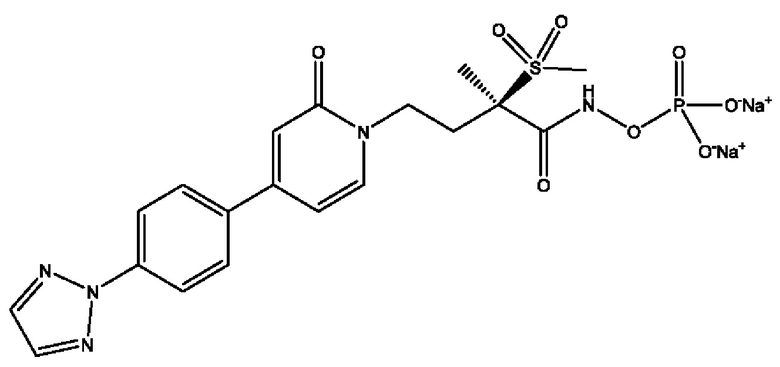
(R)-4-(4-(4-(2Н-1,2,3-Триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-М-гидрокси-2-метил-2-(метилсульфонил)бутанамид (500 мг; 1,16 ммоль) нагревали в тетрагидрофуране (150 мл) до его растворения, после чего его охлаждали до КТ и добавляли триэтиламин (3,9 мл; 28 ммоль). Затем смесь охлаждали до -40°С, добавляли оксихлорид фосфора (0,32 мл; 3,3 ммоль), смесь нагревали до -12°С и добавляли воду (20 мл). Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Затем раствор экстрагировали этилацетатом и объединенные органические экстракты экстрагировали водой. Объединенные водные слои затем частично упаривали, добавляли 4 М раствор NaOH до рН 13 и водный слой упаривали, получая беловатое твердое вещество. Добавляли смесь 1:1 DMSO и воды и декантировали, после чего белое твердое вещество растирали с водой (10 мл), получая белое твердое вещество. 1Н-ЯМР (400 МГц, D2O) δ 1.6 (s, 3Н), 2.25 (dt, 1H), 2.6 (dt, 1H), 3.25 (s, 3Н), 4.00 (dt, 1H), 4.25 (dt, 1H), 6.75 (s, 1H), 6.85 (d, 1H), 7.75 (d, 2H), 7.85 (d, 1H), 7.9 (d, 2H), 8.00 (s, 2H). m/z (CI) 512 (M-2Na+3H).
Пример 2
(2R)-4-[4-(2,3-Дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, динатриевая соль
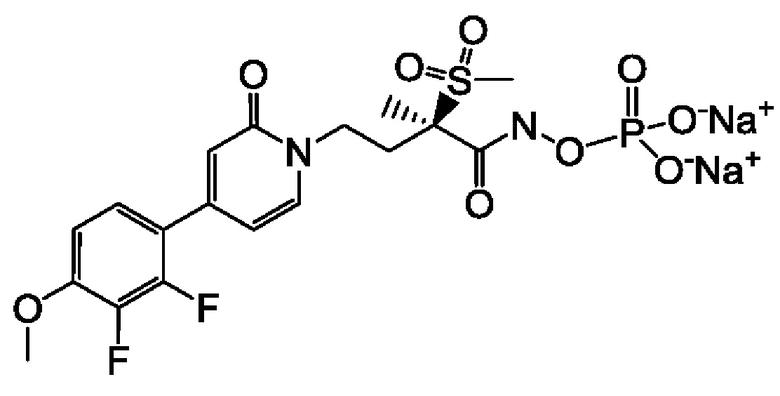
Указанное в заголовке соединение можно получить с применением методики, описанной для примера 1, используя (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамид в качестве исходного вещества.
Пример 3
(2R)-N-Гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфат, динатриевая соль
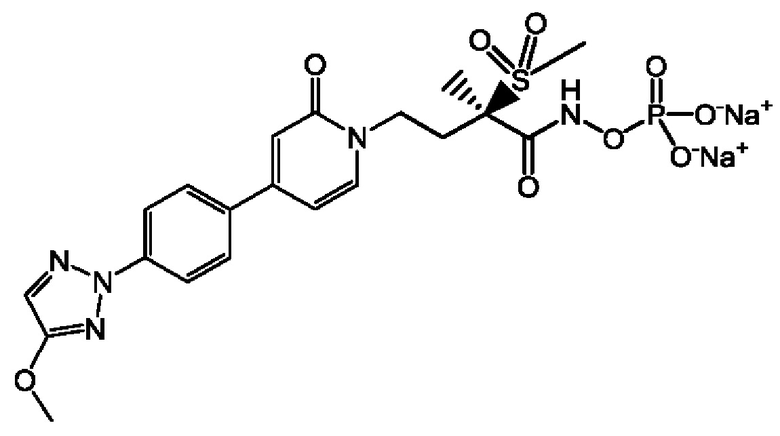
Указанное в заголовке соединение можно получить с применением методики, описанной для примера 1, используя (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамид в качестве исходного вещества.
Пример 4
(2R)-Н-Гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфат, динатриевая соль
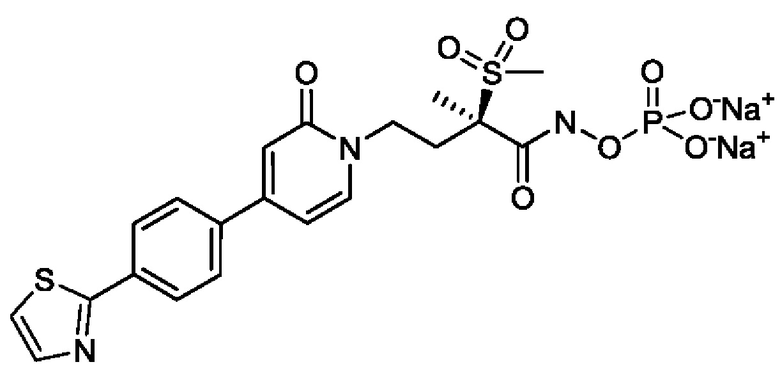
Указанное в заголовке соединение можно получить с применением методики, описанной для примера 1, используя (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамид в качестве исходного вещества.
Пример 5
(R)-4-(4-(4-(2H-1,2,3-Триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, динатриевая соль
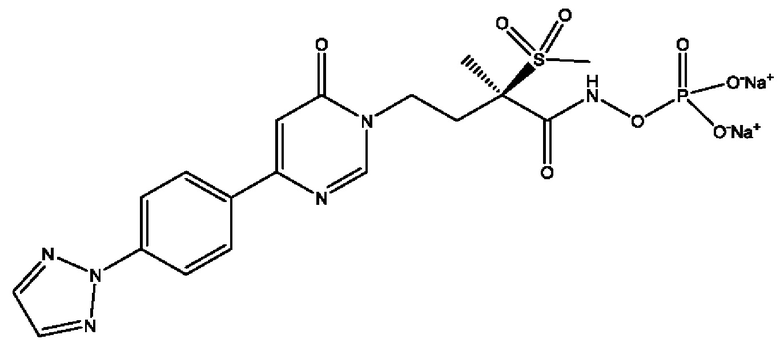
Указанное в заголовке соединение можно получить с применением методики, описанной для примера 1, используя (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамид в качестве исходного вещества.
Пример 6
(R)-4-(4-(4-(2Н-1,2,3-Триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, диаммониевая соль
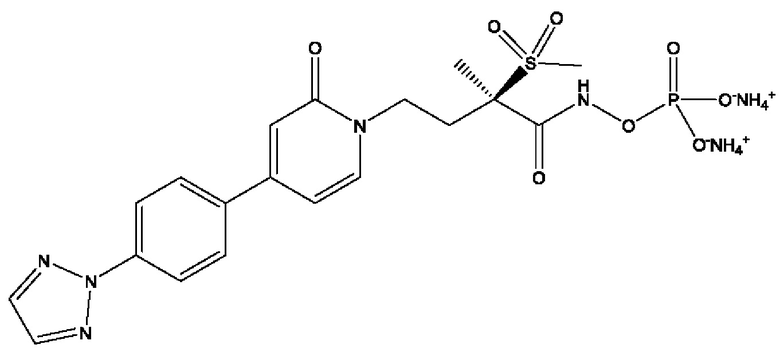
Указанное в заголовке соединение можно получить способом, аналогичным таковому для соединения из примера 1, используя концентрированный водный раствор гидроксида аммония вместо 4 М раствора NaOH.
Пример 7
(2R)-4-[4-(2,3-Дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, диаммониевая соль
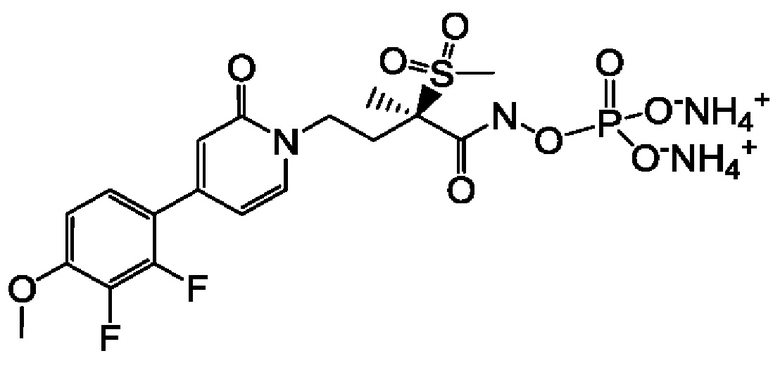
Указанное в заголовке соединение можно получить способом, аналогичным таковому для соединения из примера 2, используя концентрированный водный раствор гидроксида аммония вместо 4 М раствора NaOH.
Пример 8
(2R)-N-Гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфат, диаммониевая соль
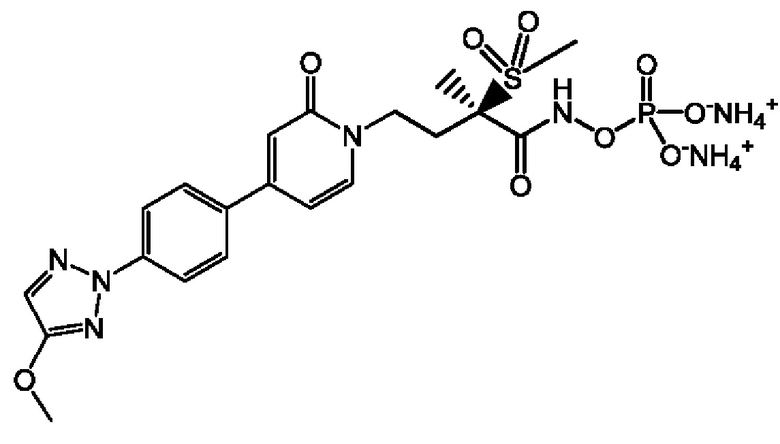
Указанное в заголовке соединение можно получить способом, аналогичным таковому для соединения из примера 3, используя концентрированный водный раствор гидроксида аммония вместо 4 М раствора NaOH.
Пример 9
(2R)-N-Гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфат, аммониевая соль
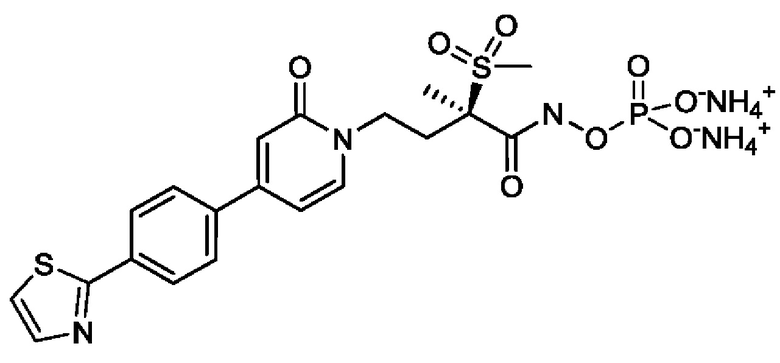
Указанное в заголовке соединение можно получить способом, аналогичным таковому для соединения из примера 3, используя концентрированный водный раствор гидроксида аммония вместо 4 М раствора NaOH.
Пример 10
(R)-4-(4-(4-(2H-1,2,3-Триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, диаммониевая соль
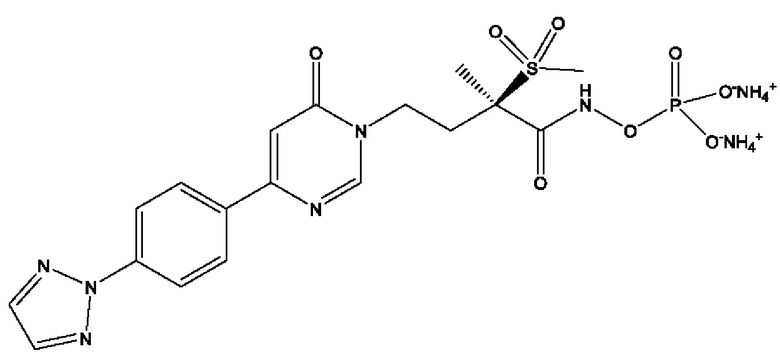
Примеры 11-15
Соединения из примеров 11-15 можно получить способами, аналогичными таковым для соответствующих соединений из примеров 1-5, используя 4 М раствор КОН вместо 4 М раствора NaOH.
Пример 11: (R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, дикалиевая соль.
Пример 12: (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, дикалиевая соль.
Пример 13: (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфат, дикалиевая соль.
Пример 14: (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфат, дикалиевая соль.
Пример 15: (R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, дикалиевая соль.
Примеры 16-20
Соединения из примеров 16-20 можно получить способами, аналогичными таковым для соответствующих соединений из примеров 1-5, используя 4 М раствор LiOH вместо 4 М раствора NaOH.
Пример 16: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, дилитиевая соль.
Пример 17: (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, дилитиевая соль.
Пример 18: (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфат, дилитиевая соль.
Пример 19: (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамидофосфат, дилитиевая соль.
Пример 20: (R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, дилитиевая соль.
Общая методика I для получения солей
Катионообменную смолу Dowex-50wx8-100 промывают водой, метанолом и еще раз водой. Затем смолу подщелачивают, обрабатывая соответствующим раствором гидроксида металла (такого как гидроксид лития, гидроксид калия, гидроксид натрия), гидроксида аммония, аминокислоты или органического амина и затем промывают водой. Готовую к применению смолу делят на три части. К раствору соответствующей соли фосфата пиридинон- или пиримидинон-гидроксамовой кислоты (такой как аммониевая или диаммониевая соль либо натриевая или динатриевая соль (например, соединению из примеров 1-10 или соответствующей моносоли)) в воде добавляют одну порцию смолы. Смесь перемешивают в течение 10 минут, затем ее фильтруют и твердое вещество промывают водой. Добавляют еще одну часть смолы к объединенному фильтрату и перемешивают в течение 10 минут, фильтруют и твердое вещество промывают водой. Добавляют заключительную часть смолы, перемешивают в течение 10 минут, фильтруют и твердое вещество промывают водой. Фильтрат концентрируют в вакууме, остаток растворяют в ацетонитриле, фильтруют и фильтрат концентрируют в вакууме. Остаток растворяют в метиленхлориде, добавляют гексан и концентрируют в вакууме, получая соответствующий фосфат в виде моно- или двойной соли.
Общая методика II для получения солей двухвалентных катионов
Один эквивалент соответствующего фосфата пиридинон- или пиримидинон-гидроксамовой кислоты (такого как (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфат, (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил] пиридин-1(2Н)-ил}бутанамидофосфат или (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат) переносят в соответствующий растворитель, такой как метанол, до концентрации приблизительно 10 мг/мл и обрабатывают одним эквивалентом соответствующего ацетата металла (такого как ацетат кальция, ацетат цинка или ацетат магния). Полученную смесь перемешивают при температуре окружающей среды в течение нескольких суток, затем концентрируют в вакууме. Полученный остаток промывают небольшим количеством метанола и осуществляют сушку продукта.
Приведенные далее соединения из примеров 21-23 могут быть получены в соответствии с общей методикой П.
Пример 21: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, кальциевая соль.
Пример 22: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, магниевая соль.
Пример 23: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, цинковая соль.
Общая методика III для получения солей одновалентных катионов
Один эквивалент соответствующего фосфата пиридинон- или пиримидинон-гидроксамовой кислоты (такого как (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат, (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамидофосфат, (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил] пиридин-1(2Н)-ил}бутанамидофосфат или (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамидофосфат) переносят в соответствующий растворитель, такой как метанол, до концентрации приблизительно 10 мг/мл и обрабатывают надлежащим соответствующим амином (таким как пирролидин, пиперидин, пиридин, морфолин, пиперазин, трис-(гидроксиметил)метиламин, диэтиламин, глицин) в количестве 1,0-1,1 эквивалента. Полученную смесь перемешивают при температуре окружающей среды в течение нескольких суток, затем концентрируют в вакууме. Полученный остаток промывают небольшим количеством метанола и осуществляют сушку продукта.
Приведенные далее соединения из примеров 24-27 могут быть получены в соответствии с общей методикой III.
Пример 24: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, пирролидиновая соль.
Пример 25: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, трис-(гидроксиметил)метиламиновая соль.
Пример 26: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, диэтиламиновая соль.
Пример 27: (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-метил-2-(метилсульфонил)бутанамидофосфат, глициновая соль.
Общая методика IV для получения боронатов
Один эквивалент соответствующей гидроксамовой кислоты (например, (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида, (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида, (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамида, (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамида или (R)-4-(4-(4-(2H-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамида) суспендируют в воде (в концентрации приблизительно 1,5 М). Добавляют борную кислоту (1,0 эквивалента), затем соответствующее основание (например, гидроксид натрия, гидроксид калия или гидроксид лития (1,0 эквивалента)). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение приблизительно 30 минут. Реакционный раствор фильтруют через тефлоновый фильтр. Фильтрат переносят в круглодонную колбу емкостью 250 мл, в которой его замораживают при -78°С. Замороженное твердое вещество помещают в установку для сублимационной сушки и оставляют сушиться в течение ночи (вакуум = 0,2 мбар (20 Па)), получая желаемый продукт.
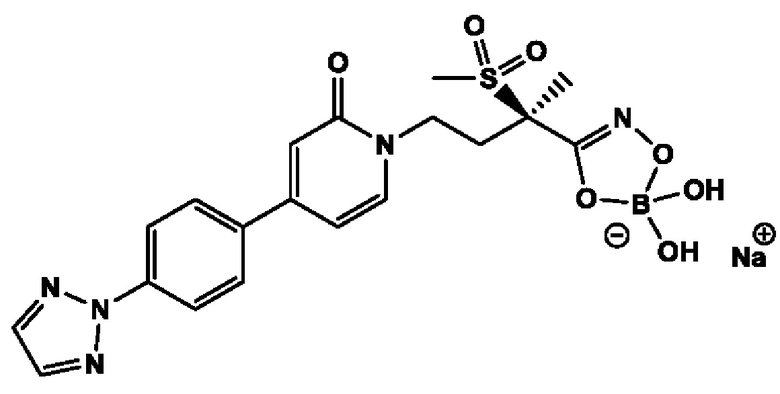
Показанное выше соединение, (R)-5-(4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уид натрия, можно получить в соответствии с общей методикой IV, используя (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-Н-гидрокси-2-метил-2-(метилсульфонил)бутанамид в качестве исходного вещества и гидроксид натрия в качестве основания.
Пример 29
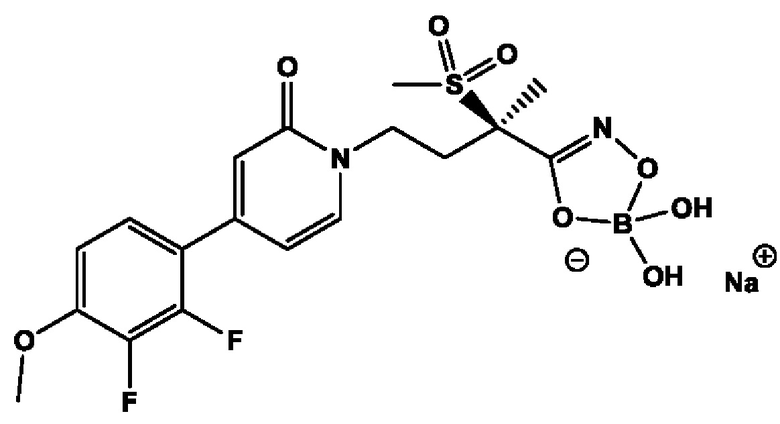
Показанное выше соединение, (R)-5-(4-(4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уид натрия, можно получить в соответствии с общей методикой IV, используя (2R)-4-[4-(2,3-дифтор-4-метоксифенил)-2-оксопиридин-1(2Н)-ил]-N-гидрокси-2-метил-2-(метилсульфонил)бутанамид в качестве исходного вещества и гидроксид натрия в качестве основания.
Пример 30
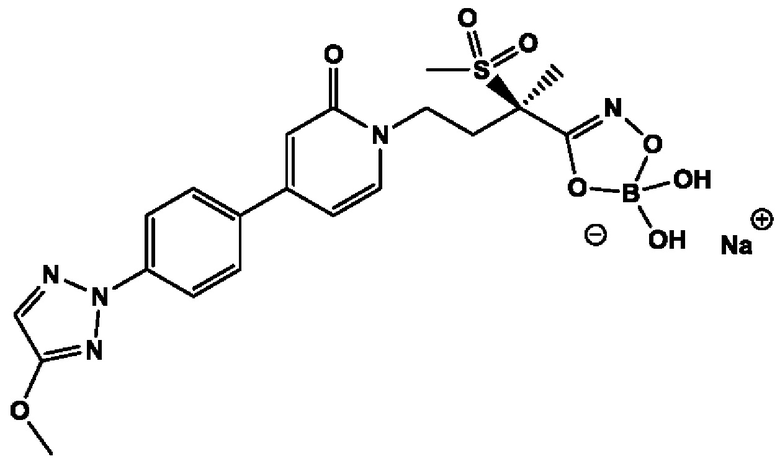
Показанное выше соединение, (R)-2,2-дигидрокси-5-(4-(4-(4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-1,3,4,2-диоксазаборол-2-уид натрия, можно получить в соответствии с общей методикой IV, используя (2R)-N-гидрокси-4-{4-[4-(4-метокси-2Н-1,2,3-триазол-2-ил)фенил]-2-оксопиридин-1(2Н)-ил}-2-метил-2-(метилсульфонил)бутанамид в качестве исходного вещества и гидроксид натрия в качестве основания.
Пример 31
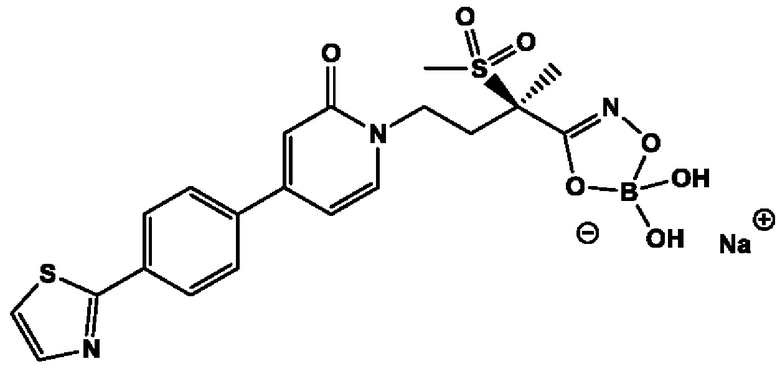
Показанное выше соединение, (R)-2,2-дигидрокси-5-(2-(метилсульфонил)-4-(2-оксо-4-(4-(тиазол-2-ил)фенил)пиридин-1(2Н)-ил)бутан-2-ил)-1,3,4,2-диоксазаборол-2-уид натрия, можно получить в соответствии с общей методикой IV, используя (2R)-N-гидрокси-2-метил-2-(метилсульфонил)-4-{2-оксо-4-[4-(1,3-тиазол-2-ил)фенил]пиридин-1(2Н)-ил}бутанамид в качестве исходного вещества и гидроксид натрия в качестве основания.
Пример 32
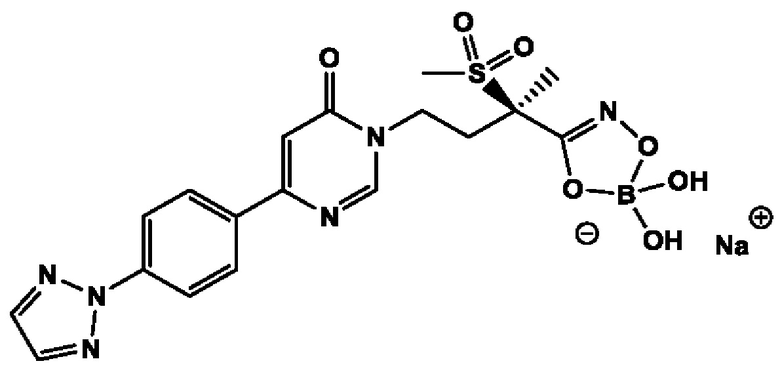
Показанное выше соединение, (R)-5-(4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уид натрия, можно получить в соответствии с общей методикой IV, используя (R)-4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-6-оксопиримидин-1(6Н)-ил)-N-гидрокси-2-метил-2-(метилсульфонил)бутанамид в качестве исходного вещества и гидроксид натрия в качестве основания.
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Для того, чтобы оценить биологическую активность соединений, с отобранными соединениями проводили отдельные анализы in vitro. В одном из этих анализов измеряли способность соединений нарушать синтез липополисахарида, LPS, представляющего собой компонент наружной мембраны грамотрицательных бактерий. Нарушение этого синтеза приводит к гибели бактерий. В этом анализе определяли способность соединений ингибировать LpxC, который представляет собой первый фермент в пути биосинтеза LPS (которую измеряли в виде IC50). Кроме того, для некоторых бактерий определяли MIC (минимальные ингибирующие концентрации). Конкретные протоколы описаны ниже.
А) Анализ IC50, с применением фермента LpxC из P. aeruginosa (обозначено как IC50 для фермента LpxC из РА): определение IC50 в анализе с использованием фермента LpxC проводили аналогично тому, как описано Malikzay и др. в постере 2006 г. Screening LpxC (UDP-3-O-(R-3-hydroxymyristoyl)-GlcNAc deacetylase) using BioTrove RapidFire HTS Mass Spectrometry (aNew Lead Discovery and ^Inflammation and Infectious Disease, cStructural Chemistry, Schering-Plough Research Institute, Kenilworth, NJ 07033 (BioTrove, Inc. 12 Gill St., Suite 4000, Woburn, MA 01801). Кратко, фермент LpxC из Pseudomonas aeruginosa (0,1 нМ), очищенный из сверхэкспрессирующих бактерий Е. coli, инкубировали при 25°С в конечном объеме 50 мкл, содержащем 0,5 мкМ УДФ-3-O-(R-3-гидроксидеканоил)-N-ацетилглюкозамин, BSA (бычий сывороточный альбумин) в концентрации 1 мг/мл и 50 мМ буферный раствор фосфата натрия, рН 8,0, в присутствии и в отсутствие ингибирующего соединения. По окончании 1 часа добавляли 5 мкл 1 н. раствора HCl для остановки ферментативной реакции, планшеты центрифугировали и затем обрабатывали, используя систему для высокопроизводительной масс-спектрометрии (HTMS) BioTrove Rapidfire. Использовали не содержащий фермента контроль при расчете значений IC50 исходя из значений процента превращения.
В) Определения MIC: оценку антибактериальной активности исходных соединений in vitro среди соединений, описанных в примерах, проводили по минимальной ингибирующей концентрации (MIC), выполняя тестирование согласно протоколу Института клинических и лабораторных стандартов (CLSI). См.: Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard-Eighth Edition. CLSI document M7-A8 [ISBN 1-56238-689-1]. Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006; также Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; Twentieth Informational Supplement. CLSI document M100-S20 [ISBN1-56238-716-2].Clinical and Laboratory Standards Institute.
Определение MIC представляет собой стандартный лабораторный метод оценки антибактериальной активности соединения. MIC представляет собой самую низкую концентрацию лекарственного средства, при которой происходит ингибирование видимого роста бактерий после инкубирования в течение ночи. Чтобы определить значение MIC, инкубирование с определенным штаммом бактерий выполняют в диапазоне концентраций лекарственного средства (например, от 0,06 мкг/мл до 64 мкг/мл). Обычно, диапазон концентраций лекарственного средства разбивают для получения 2-кратных приращений концентрации (например, 0,06 мкг/мл; 0,12 мкг/мл; 0,25 мкг/мл; 0,50 мкг/мл; 1,0 мкг/мл и т.д.), и лекарственное средство во всех различных концентрациях инкубируют по отдельности в течение ночи приблизительно с одним и тем же числом бактерий. Затем определяют MIC, визуально фиксируя эффект лекарственного средства для каждой концентрации и определяя самую низкую концентрацию лекарственного средства, при которой наблюдалось ингибирование бактериального роста, по сравнению с не содержащим лекарственного средства контролем. Обычно бактерии продолжают расти при концентрации лекарственного средства более низкой, чем MIC, и не растут при концентрации равной и превышающей MIC.
Значения MIC, описанные ниже в Таблице 2, получали на основании анализов, при этом каждое тестируемое соединение оценивали в двух повторах. В случаях, когда значения для повторов различались в 0-2 раза, ниже указывали меньшее из этих двух значений. Вообще говоря, если значения для повторов различались более чем в 2 раза, результаты анализа считались недостоверными, и анализ повторяли до тех пор, пока разброс между измерениями для этих повторов не становился ≤2-кратным. В соответствии с указаниями CLSI, упомянутыми выше, для обеспечения надлежащего контроля качества в каждом анализе MIC использовали как контрольные микроорганизмы, так и референсные соединения. Значения MIC, полученные с использованием этих контрольных микроорганизмов и референсных соединений, должны были находиться в пределах определенного диапазона, чтобы результаты анализа считались достоверными и были включены в данное описание. Специалистам в данной области техники будет известно, что значения MIC могут варьировать и действительно варьируют от эксперимента к эксперименту. Вообще говоря, следует признать, что значения MIC часто варьируют +/- в 2 раза от эксперимента к эксперименту. Хотя для каждого соединения и каждого микроорганизма указывается одно значение MIC, читателю не следует делать заключение, что каждое соединение тестировали только один раз. Некоторые соединения подвергали многократным тестированиям. Данные, представленные в Таблице 2, отражают относительную активность соединений, и в этих случаях в соответствии с указаниями, описанными выше, могли быть получены разные значения MIC.
В таких определениях MIC использовали следующие бактериальные штаммы:
1) Pseudomonas aeruginosa UI-18: дикий тип, обозначенный как РА-7 в Таблице 2;
2) Acinetobacter baumannii/haemolyticus: клинический изолят с множественной лекарственной устойчивостью, обозначенный как АВ-3167 в Таблице 2;
3) Escherichia coli ЕС-1: VOGEL, вирулентный для мышей, обозначенный как ЕС-1 в Таблице 2;
4) Klebsiella pneumoniae: ципрофлоксацин-резистентный изолят, экспрессирует (3-лактамазы расширенного спектра (ESBL), клинический изолят, обозначенный как КР-3700 в Таблице 2.
В приведенной ниже Таблице 2 показаны результаты, полученные для исходных соединений, использованных для получения соединений в примерах 1-32. Если конкретная позиция таблицы оставлена пустой, то данные в настоящее время не установлены.
Колонка 1 соответствует исходному соединению, привязанному к номерам примеров, в колонке 2 приведены названия по IUPAC (Международный союз по теоретической и прикладной химии), в колонке 3 приведены результаты анализа с использованием фермента LpxC, описанного выше, а в колонках 4-7 приведены данные для MIC, как описано выше.
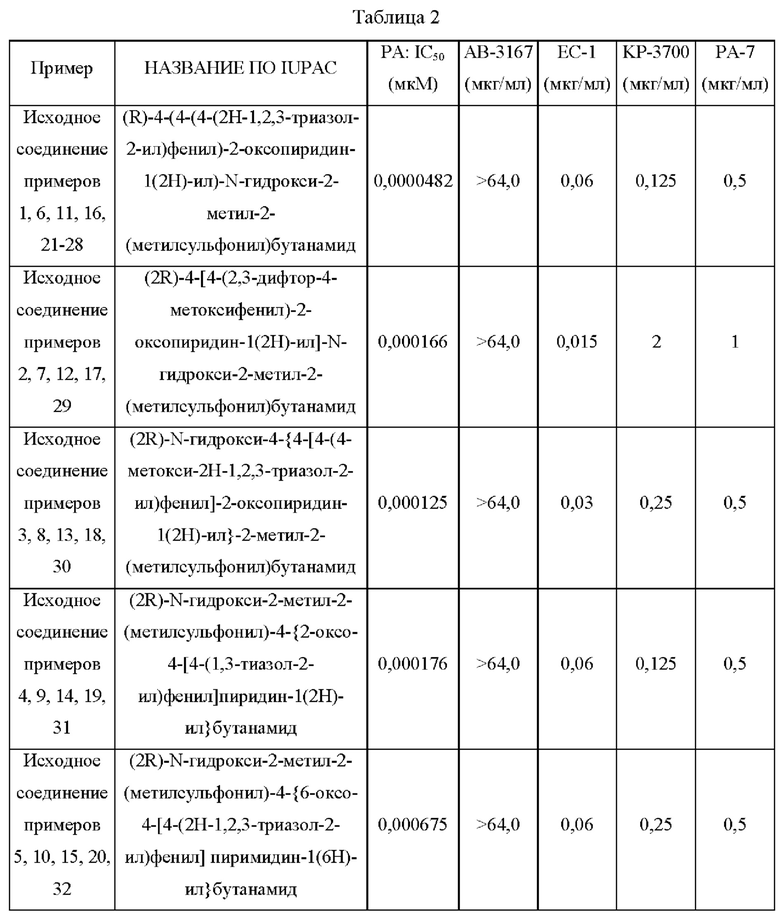
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДВУХ 4-{ [(2S)-2-{ 4-[5-ХЛОР-2-(1H-1,2,3-ТРИАЗОЛ-1-ИЛ)ФЕНИЛ]-5-МЕТОКСИ-2-ОКСОПИРИДИН-1(2H)-ИЛ} БУТАНОИЛ]АМИНО} -2-ФТОРБЕНЗАМИДНЫХ ПРОИЗВОДНЫХ | 2019 |
|
RU2804663C2 |
| ПРОИЗВОДНОЕ ОКСОПИКОЛИНАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2742771C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| Новые производные имидазо[4,5-c] хинолинов и имидазо[4,5-c][1,5] нафтиридинов в качестве ингибиторов LRRK2 | 2016 |
|
RU2722149C1 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА И ИХ ПРИМЕНЕНИЕ ПРИ НЕВРОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЯХ | 2011 |
|
RU2581504C2 |
| СОЕДИНЕНИЯ ПИРИМИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2807277C2 |
| Соединения циклопентана | 2019 |
|
RU2794997C2 |
| ТИЛОЗИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2714482C2 |
Изобретение относится к соединению формулы (2a), которое может найти применение в качестве пролекарства для ингибирования деацетилазы УДФ(уридин-5'-дифосфат)-3-О-(R-3-гидроксимиристоил)-N-ацетилглюкозамина (LpxC). В формуле (2a) X представляет собой СН; Z представляет собой  и M+ представляет собой фармацевтически приемлемый одновалентный катион, выбранный из группы, состоящей из Li+, K+, Na+, NH4+, NH3+С(СН2ОН)3 или NH2+(CH2CH3)2. Изобретение относится также к фармацевтической композиции для использования в качестве пролекарства для ингибирования деацетилазы УДФ(уридин-5'-дифосфат)-3-O-(R-3-гидроксимиристоил)-N-ацетилглюкозамина (LpxC), содержащей эффективное количество указанного соединения в смеси с по меньшей мере одним фармацевтически приемлемым эксципиентом, разбавителем или носителем, и к способу лечения вызываемой грамотрицательными бактериями инфекции у нуждающегося в этом животного, не являющегося человеком, включающему введение нуждающемуся в этом животному, не являющемуся человеком, терапевтически эффективного количества указанного соединения. 5 н. и 5 з.п. ф-лы, 2 табл., 32 пр.
и M+ представляет собой фармацевтически приемлемый одновалентный катион, выбранный из группы, состоящей из Li+, K+, Na+, NH4+, NH3+С(СН2ОН)3 или NH2+(CH2CH3)2. Изобретение относится также к фармацевтической композиции для использования в качестве пролекарства для ингибирования деацетилазы УДФ(уридин-5'-дифосфат)-3-O-(R-3-гидроксимиристоил)-N-ацетилглюкозамина (LpxC), содержащей эффективное количество указанного соединения в смеси с по меньшей мере одним фармацевтически приемлемым эксципиентом, разбавителем или носителем, и к способу лечения вызываемой грамотрицательными бактериями инфекции у нуждающегося в этом животного, не являющегося человеком, включающему введение нуждающемуся в этом животному, не являющемуся человеком, терапевтически эффективного количества указанного соединения. 5 н. и 5 з.п. ф-лы, 2 табл., 32 пр.
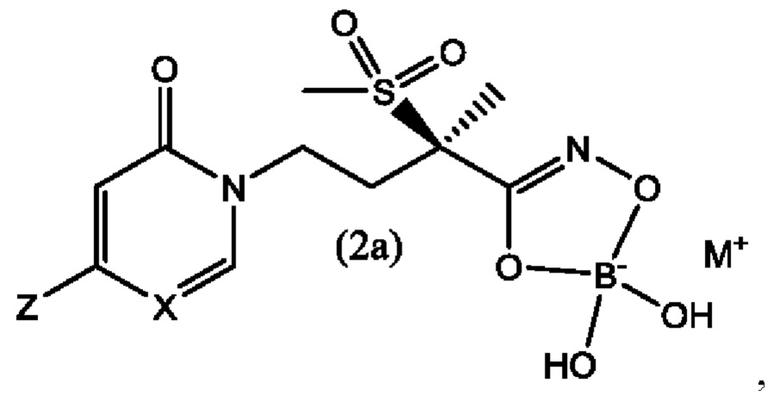
1. Соединение формулы (2а)
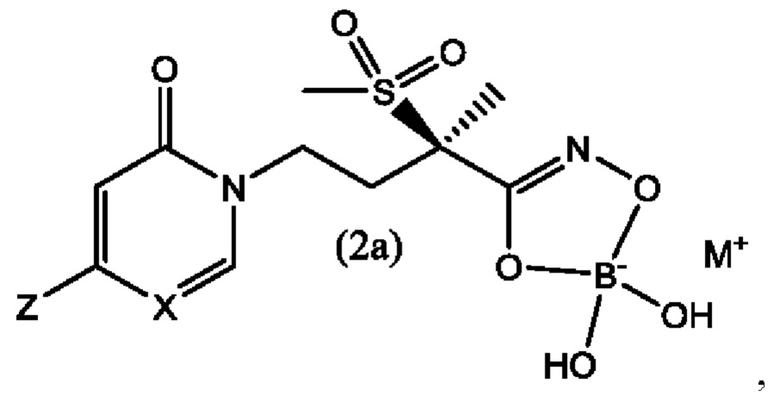
где X представляет собой СН; Z представляет собой
и M+ представляет собой фармацевтически приемлемый одновалентный катион, выбранный из группы, состоящей из Li+, K+, Na+, NH4+, NH3+С(СН2ОН)3 или NH2+(CH2CH3)2.
2. Соединение формулы (2а) по п. 1, где M+ выбран из группы, состоящей из Li+, K+ и Na+.
3. Фармацевтическая композиция для использования в качестве пролекарства для ингибирования деацетилазы УДФ(уридин-5'-дифосфат)-3-O-(R-3-гидроксимиристоил)-N-ацетилглюкозамина (LpxC), содержащая эффективное количество соединения по п. 1 или 2 в смеси с по меньшей мере одним фармацевтически приемлемым эксципиентом, разбавителем или носителем.
4. Способ лечения вызываемой грамотрицательными бактериями инфекции у нуждающегося в этом животного, не являющегося человеком, включающий введение нуждающемуся в этом животному, не являющемуся человеком, терапевтически эффективного количества соединения по п. 1 или 2.
5. Способ по п. 4, где указанное животное, не являющееся человеком, представляет собой сельскохозяйственное животное.
6. Способ по п. 4, где вызываемая грамотрицательными бактериями инфекция вызвана грамотрицательными бактериями, выбранными из группы, состоящей из Mannheimia haemolytica, Pasteurella multocida, Histophilus somni, Actinobacillus pleuropneumoniae, Salmonella enteritidis, Salmonella gallinarium, Lawsonia intracellularis, Brachyspira hyodysenteriae, Brachyspira pilosicoli, Citrobacter spp., Enterobacter aerogenes, Enterobacter cloacae, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Serratia marcescens, Stenotrophomonas maltophilia и Pseudomonas aeruginosa.
7. Способ по п. 4, где вызываемая грамотрицательными бактериями инфекция выбрана из группы, состоящей из респираторной инфекции, желудочно-кишечной инфекции, нозокомиальной пневмонии, инфекции мочевыводящих путей, бактериемии, сепсиса, кожной инфекции, инфекции мягких тканей, инфекции брюшной полости, легочной инфекции, эндокардита, синдрома диабетической стопы, остеомиелита и инфекции центральной нервной системы.
8. Способ по п. 4, где терапевтически эффективное количество соединения вводят перорально, местно или путем инъекции.
9. Соединение, представляющее собой (R)-5-(4-(4-(4-(2Н-1,2,3-триазол-2-ил)фенил)-2-оксопиридин-1(2Н)-ил)-2-(метилсульфонил)бутан-2-ил)-2,2-дигидрокси-1,3,4,2-диоксазаборол-2-уид натрия.
10. Водная фармацевтическая композиция для использования в качестве пролекарства для ингибирования деацетилазы УДФ(уридин-5'-дифосфат)-3-O-(R-3-гидроксимиристоил)-N-ацетилглюкозамина (LpxC), содержащая эффективное количество соединения по пп. 1, 2 или 9 в смеси с водой.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| J.I.MONTGOMERY et al., Pyridone methylsulfone hydroxamate LpxC inhibitors for the treatment of serious gram-negative infections, J.MED.CHEM., 2012, V.55, N.4, p.1662-1670 | |||
| L.A.MCALLISTER et al., Heterocyclic methylsulfone hydroxamic acid LpxC inhibitors as Gram-negative antibacterial agents, BIOORG.MED.CHEM., 2012, | |||

