Изобретение относится к области синтеза органических соединений, более конкретно - к получению действующих веществ пестицидов, в частности - к способу получения моносульфурона (1-(2-нитрофенилсульфонил)-3-(4-метилпиримидин-2-ил)мочевины (N-[(4-метилпиримидин-2-ил)карбамоил]-2-нитробензолсульфонамида) (I), регистрационный номер CAS [155860-63-2]). Это соединение является высокоселективным системным до- и послевсходовым гербицидом из класса сульфонилмочевин, обладающим низкой токсичностью для млекопитающих, и применяется в сельском хозяйстве для эффективной борьбы с широколиственными сорняками и травами при выращивании пшеницы, проса, кукурузы, риса и др. (см., напр., Modern Crop Protection Compounds, Edited by P. Jeschke et al., 3rd Ed., Vol. 1: Herbicides. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2019, p. 56, 71-72; Zh. Fan et al., J. Environ. Sci., 2005, 17(3), 399-403; J. Wang et al., J. Comput. Aided Mol. Des., 2005, 19(11), 801-820). Моносульфурон был разработан в Национальном исследовательском центре пестицидов при Нанькайском университете (КНР) в 1993 году (патент CN №1080116A) и выведен на рынок в 2012 году.
Известные промышленные методы получения моносульфурона основаны на использовании 2-нитробензолсульфонамида (II) (далее по тексту - сульфонамид II), доступного в 2-3 стадии из 1-хлор-2-нитробензола или о-нитроанилина, в качестве исходного сырья (Схема 1). Для получения целевой сульфонилмочевины I этот сульфонамид II а) фосгенируют с помощью фосгена в присутствии бутилизоцианата (BuCNO) и каталитических количеств третичных аминов (см., напр., патент US №4379769А) или оксалилхлорида в присутствии триэтиламина или 1,4-диазабицикло[2.2.2]октана (DABCO) в качестве катализатора при нагревании с генерированием в реакционном растворе соответствующего сульфонилизоцианата III, который после выделения его из реакционного раствора в чистом виде перегонкой при пониженном давлении или без такого выделения сразу используют в реакции с 2-амино-4-метилпиримидином (V) (далее по тексту - аминопиримидин V) в отсутствие или в присутствии третичных аминов в качестве катализатора при нагревании или б) вводят в реакцию с этилхлорформиатом в присутствии карбоната калия в ацетоне с промежуточным получением соответствующего карбамата IV, который выделяют в чистом виде и далее также используют в реакции с аминопиримидином V при нагревании. В результате, все эти подходы [а) или б); Схема 1] приводят к целевому моносульфурону с выходом до 81% (см., напр., патенты CN №1080116A, CN №1106393A, CN №105439970A, CN №115108997A, CN №116023338A; Zh. Yu et al., Chin. Sci. Bull., 2007, 52(14), 1929-1941; J. Wang et al., Youji Huaxue (Chin. J. Org. Chem.), 2006, 26(5), 648-652).
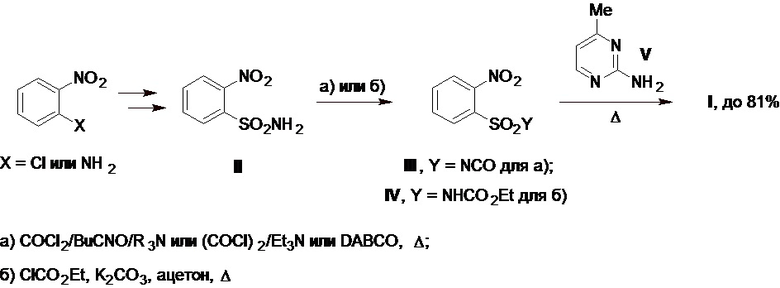 Схема 1.
Схема 1.
К основным недостаткам вышеперечисленных методов получения моносульфурона можно отнести а) необходимость работы с токсичными, нестабильными или опасными при обращении исходными соединениями, полупродуктами и (или) реагентами, такими как фосген, оксалилхлорид, бутилизоцианат, этилхлорформиат, сульфонилизоцианат III и др.; б) использование повышенных температур (до 140-150°С) на стадиях фосгенирования исходного сульфонамида II или превращения полупродуктов III и IV в целевой продукт I; в) стадийность и продолжительность таких процессов; г) необходимость использования специального коррозионно-стойкого оборудования с его защитой от попадания влаги и воздуха, специальных мер промышленной безопасности и др. Все эти факторы негативно отражаются на безопасности и технико-экономических характеристиках процесса промышленного производства моносульфурона, в основе которого лежит один из таких подходов (Схема 1).
Другим методом получения моносульфурона служит двухстадийная схема, предложенная в патенте WO №1996022284 A1, в которой исходный аминопиримидин V первоначально превращают в один из стабильных бис-карбаматов VI с помощью избытка соответствующего хлорформиата (метил-, этил-, фенилхлорформиат и др.) в присутствии неорганических или органических оснований в органических растворителях, среди которых наиболее предпочтительными являются ацето-, пропио- или бутиронитрил. Эти бис-карбаматы VI выделяют в чистом виде и далее используют в реакции с сульфонамидом II в присутствии сильных оснований, самыми эффективными из которых являются гидриды или алкоголяты натрия, калия, кальция, магния и др., приводящей к получению целевого продукта I, выход которого не указан (Схема 2).
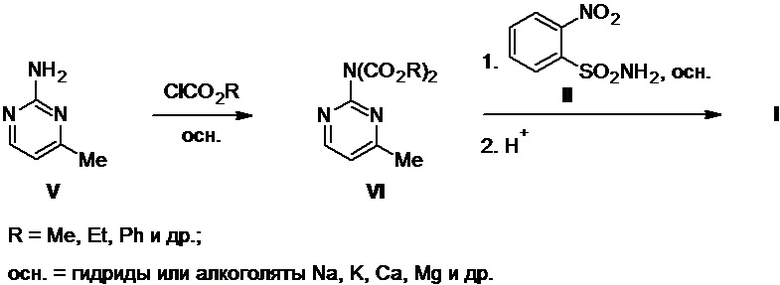
Схема 2.
К недостаткам такого подхода, негативно влияющим на масштабируемость и безопасность процесса в целом, можно вновь отнести необходимость использования в нем хлорформиатов (метил-, этил-, фенилхлорформиат и др.), являющихся чувствительными к влаге и кислороду воздуха и температурному режиму хранения высокотоксичными жидкостями, а также предпочтительное использование опасных в работе гидридов или алкоголятов щелочных или щелочноземельных металлов на второй его стадии, что автоматически требует применения безводных условий в ходе ее осуществления.
Также известны двухстадийные схемы получения моносульфурона, в которых в качестве исходного сырья используют 2-(феноксикарбонил)амино-4-метилпиримидин (VII) (далее по тексту - фенилкарбамат VII) в виде индивидуального соединения или в виде раствора в органическом растворителе, генерируемого в процессе его синтеза из аминопиримидина V (Схемы 3, 4).
Например, взаимодействие аминопиримидина V с дифенилкарбонатом в присутствии сильных оснований (алкоголяты натрия или калия) в среде безводного ацетонитрила, тетрагидрофурана (ТГФ), ацетона, N,N-диметилацетамида (ДМАА), N,N-диметилформамида (ДМФА), диметилсульфоксида (ДМСО) и др. приводит к генерированию раствора смеси фенилкарбамата VII и фенолята натрия или калия в используемом органическом растворителе (Схема 3), которую без выделения из этого раствора используют в реакции с сульфонамидом II, завершающейся получением целевого моносульфурона, выход которого не указан (патент RU №2177002C2).
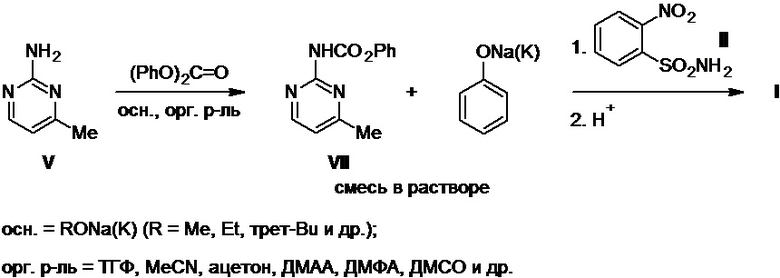
Схема 3.
В патенте US №5886176A раскрыто, что фенилкарбамат VII может быть удобным исходным сырьем в синтезе солей моносульфурона с щелочными или щелочноземельными металлами I-M (M = Na, K, Mg, Ca и др.), которые образуются при его взаимодействии с предварительно полученными из сульфонамида II и алкоголятов или гидроксидов щелочных или щелочноземельных металлов солями сульфонамида II (II-M, где M = Na, K, Mg, Ca и др.) в таких растворителях как метанол, вода, ацетонитрил, ацетон, ТГФ, ДМФА и др.; при этом выход этих солей I-M в источнике не указан (Схема 4).
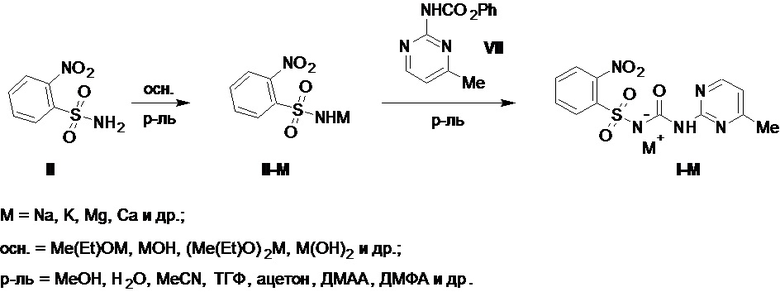 Схема 4.
Схема 4.
Среди основных недостатков таких бесфосгенных схем получения моносульфурона, основанных на применении фенилкарбамата VII в том или ином виде в качестве исходного сырья, при их возможном промышленном применении можно выделить а) их стадийность, составляющая не менее двух стадий из аминопиримидина V или сульфонамида II, и как следствие этого - продолжительность; б) наличие проблем с безопасностью осуществления таких процессов из-за необходимости использования опасных в обращении алкоголятов щелочных или щелочноземельных металлов; в) применение безводных органических растворителей для проведения реакций, кроме случаев, где допустимо применение растворов гидроксидов щелочных или щелочноземельных металлов в воде (напр., трансформация II в II-M, Схема 4), при этом эту воду все равно необходимо удалять тем или иным способом из реакционной массы после получения соли II-M для ее применения на второй стадии процесса, для реализации которой требуются безводные условия, и др.
Задачей предлагаемого технического решения является улучшение технико-экономических показателей технологического процесса производства моносульфурона за счет использования новой одностадийной технологии его синтеза исходя из сульфонамида II и фенилкарбамата VII.
Техническим результатом является а) улучшение технико-экономических показателей технологического процесса производства моносульфурона за счет упрощения его аппаратурного оформления, сокращения продолжительности, исключения использования в нем нестабильных, опасных и токсичных исходных соединений, полупродуктов и (или) реагентов и повышенной температуры; б) повышение безопасности и общей эффективности процесса; в) получение целевого продукта высокого качества с хорошим выходом.
Технический результат достигается при использовании способа получения моносульфурона, включающего взаимодействие 2-нитробензолсульфонамида с 2-(феноксикарбонил)амино-4-метилпиримидином в полярном органическом растворителе в присутствии органического основания при температуре 20-25°С в течение 1-2 ч с последующей обработкой полученной реакционной массы водой и кислотой с получением целевого продукта, его выделением и очисткой.
В качестве полярного органического растворителя используют ацетонитрил, пропионитрил, ТГФ или 1,4-диоксан; в качестве органического основания используют 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), 1,5-диазабицикло[4.3.0]нон-5-ен (DBN) или триэтиламин; в качестве кислоты используют соляную, серную, муравьиную или уксусную кислоту.
Изобретение иллюстрируется следующими примерами (Схема 5).
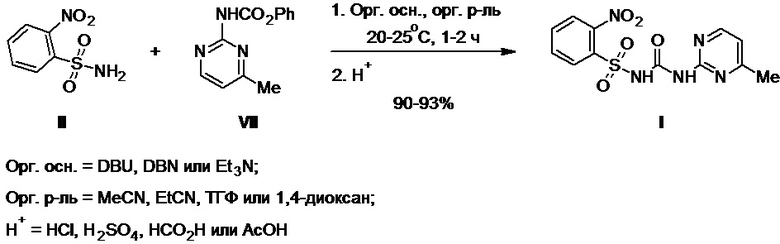
Схема 5.
Пример 1.
В реактор синтеза помещают сульфонамид II (7,0 г, 0,03 моль), фенилкарбамат VII (7,9 г, 0,03 моль) и ацетонитрил и к полученной суспензии при перемешивании добавляют DBU (5,3 г, 0,03 моль), поддерживая температуру 20-25°С. Содержимое реактора выдерживают при этой температуре в течение 1-2 ч (ТСХ-контроль), затем разбавляют водой и подкисляют конц. соляной кислотой до pH 2-3. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают. Получают 10,9 г (93%) моносульфурона в виде белого порошка с т.пл. 191-192°С (разл.) и чистотой не менее 98,0% (по данным ВЭЖХ). Спектральные характеристики продукта: 1Н ЯМР-спектр (500 МГц, DMSO-d6, δ, м.д.): 13,62 (уш. с, 1Н, NH), 10,91 (с, 1Н, NH), 8,56 (д, 1Н, J = 5,1 Гц), 8,24-8,26 (м, 1Н), 8,01-8,04 (м, 1Н), 7,90-7,97 (м, 2Н), 7,15 (д, 1Н, J = 5,1 Гц), 2,49 (с, 3H, CH3); ИК-спектр (в порошке, ν, см-1): 3154-2828 уш. сл, 1724 с, 1602 ср, 1578 с, 1537 с, 1460 с, 1415 с, 1363 с, 1352 с, 1306 ср, 1259 ср, 1202 ср, 1169 с, 1122 с, 1078 ср, 1054 ср, 950 сл, 881 ср, 856 с, 842 ср, 793 с, 744 с, 729 с, 692 с, 671 с; масс-спектр (APCI-): [M-H]-: 336,1.
Пример 2.
В реактор синтеза помещают сульфонамид II (5,0 г, 0,02 моль), фенилкарбамат VII (5,7 г, 0,02 моль) и пропионитрил и к полученной суспензии при перемешивании добавляют триэтиламин (2,5 г, 0,02 моль), поддерживая температуру 20-25°С. Содержимое реактора выдерживают при этой температуре в течение 1-2 ч (ТСХ-контроль), затем разбавляют водой и подкисляют 70%-ным водным раствором уксусной кислоты до pH 2-3. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают. Получают 7,6 г (91%) моносульфурона в виде белого порошка.
Пример 3.
В реактор синтеза помещают сульфонамид II (2,5 г, 0,01 моль), фенилкарбамат VII (2,8 г, 0,01 моль) и тетрагидрофуран и к полученной суспензии при перемешивании добавляют DBN (1,5 г, 0,01 моль), поддерживая температуру 20-25°С. Содержимое реактора выдерживают при этой температуре в течение 1-2 ч (ТСХ-контроль), затем разбавляют водой и подкисляют муравьиной кислотой до pH 2-3. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают. Получают 3,8 г (90%) моносульфурона в виде белого порошка.
Пример 4.
В реактор синтеза помещают сульфонамид II (5,7 г, 0,03 моль), фенилкарбамат VII (6,5 г, 0,03 моль) и 1,4-диоксан и к полученной суспензии при перемешивании добавляют DBU (4,3 г, 0,03 моль), поддерживая температуру 20-25°С. Содержимое реактора выдерживают при этой температуре в течение 1-2 ч (ТСХ-контроль), затем разбавляют водой и подкисляют 40%-ным водным раствором серной кислоты до pH 2-3. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают. Получают 8,7 г (91%) моносульфурона в виде белого порошка.
В результате использования предлагаемого способа получения моносульфурона удается а) улучшить технико-экономические показатели технологического процесса его производства за счет упрощения аппаратурного оформления процесса, сокращения его продолжительности, исключения использования в нем нестабильных, опасных и токсичных исходных соединений, полупродуктов и (или) реагентов и повышенной температуры; б) повысить безопасность и общую эффективность процесса; в) получить целевой продукт высокого качества с хорошим выходом.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения моносульфурон-метила | 2024 |
|
RU2834839C1 |
| Способ получения пиразосульфурон-этила | 2024 |
|
RU2828085C1 |
| Способ получения сульфонилмочевинных гербицидов, содержащих 4-метил- или 4,6-диметилпиримидин-2-ильный заместитель | 2024 |
|
RU2837390C1 |
| Способ получения метсульфурон-метила | 2024 |
|
RU2824626C1 |
| Способ получения бенсульфурон-метила | 2024 |
|
RU2826376C1 |
| Способ получения хлорсульфурона | 2023 |
|
RU2813093C1 |
| Способ получения тифенсульфурон-метила | 2023 |
|
RU2802004C1 |
| Способ получения сульфометурон-метила | 2023 |
|
RU2805743C1 |
| Способ получения сульфонилмочевинных гербицидов, содержащих 4,6-диметоксипиримидин-2-ильный заместитель | 2024 |
|
RU2834461C1 |
| Способ получения гербицида никосульфурона | 2024 |
|
RU2827153C1 |
Изобретение относится к области синтеза органических соединений, а именно к способу получения моносульфурона формулы (I). Способ включает взаимодействие 2-нитробензолсульфонамида с 2-(феноксикарбонил)амино-4-метилпиримидином в полярном органическом растворителе в присутствии органического основания при температуре 20-25°С в течение 1-2 ч с последующей обработкой полученной реакционной массы водой и кислотой с получением целевого продукта, его выделением и очисткой. Технический результат – улучшение технико-экономических показателей технологического процесса производства моносульфурона за счет упрощения его аппаратурного оформления, сокращения продолжительности, исключения использования в нем нестабильных, опасных и токсичных исходных соединений, полупродуктов и (или) реагентов и повышенной температуры, повышение безопасности и общей эффективности процесса, получение целевого продукта высокого качества с хорошим выходом. 3 з.п. ф-лы, 4 пр.
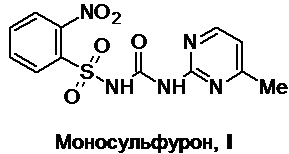 (I)
(I)
1. Способ получения моносульфурона, включающий взаимодействие 2-нитробензолсульфонамида с 2-(феноксикарбонил)амино-4-метилпиримидином в полярном органическом растворителе в присутствии органического основания при температуре 20-25°С в течение 1-2 ч с последующей обработкой полученной реакционной массы водой и кислотой с получением целевого продукта, его выделением и очисткой.
2. Способ по п. 1, характеризующийся тем, что в качестве полярного органического растворителя используют ацетонитрил, пропионитрил, тетрагидрофуран или 1,4-диоксан.
3. Способ по п. 1, характеризующийся тем, что в качестве органического основания используют 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен или триэтиламин.
4. Способ по п. 1, характеризующийся тем, что в качестве кислоты используют соляную, серную, муравьиную или уксусную кислоту.
| WO 1996022284 A1, 25.07.1996 | |||
| US 5886176 A1, 23.03.1999 | |||
| Способ получения тифенсульфурон-метила | 2023 |
|
RU2802004C1 |
| Способ получения сульфометурон-метила | 2023 |
|
RU2805743C1 |
| Способ получения никосульфурона | 2023 |
|
RU2807708C1 |

