Изобретение относится к области получения пестицидов, более конкретно - к способу получения никосульфурона (2-[(4,6-диметоксипиримидин-2-илкарбамоил)сульфамоил]-N,N-диметилникотинамида (I), регистрационный номер CAS [111991-09-4]). Это соединение является селективным системным послевсходовым гербицидом из класса сульфонилмочевин и применяется в сельском хозяйстве для борьбы с широким кругом одно- и многолетних сорняков на посевах кукурузы и др. (см., напр., Modern Crop Protection Compounds, Edited by P. Jeschke et al., 3rd ed., Vol. 1: Herbicides. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2019, p. 60). Никосульфурон был разработан и выведен на рынок компаниями «Ishihara Sangyo Kaisha, Ltd.» и «E. I. du Pont de Nemours and Company» в начале 90-х годов XX века (патенты EP № 0232067 A2 и US № 5008393 А соответственно).
Все известные методы получения никосульфурона являются сложными и включают первоначальный многостадийный синтез двух его ключевых предшественников, которые являются производными 2-Х-сульфонил-N,N-диметилникотинамида (А) [Х = Cl, NH2, NCO, NHCO2R (R = Me, Et, Ph), NHC(O)Cl или NHC(O)NH2] и 2-Y-4,6-диметоксипиримидина (Б) [Y = NH2, NCO, NHCO2R или N(CO2R)2 (R = Me, Et, Ph), NHC(O)Cl или NHC(O)NH2] (Схема 1). На заключительной стадии синтеза осуществляют формирование сульфонилмочевинного фрагмента никосульфурона, для чего используют реакцию одной из возможных пар соответствующих предшественников А и Б между собой в подходящих условиях (патенты JPH № 04139170 A, DE № 19501174 A1, WO № 2009128512 A1, CN № 113461667 А, CN № 110878084 А, CN № 113387928 А, CN № 104803982 А, CN № 103483318 А, CN № 112645930 А, CN № 111269214 А, CN № 107759569 А, CN № 106749183 А, CN № 103450155 А, CN № 101671328 А, CN № 103524493 А, CN № 101659642 А, CN № 102993176 А, CN № 101503403 А, CN № 101671327 А, CN № 108558830 А; Y. Lei, Q. Lin, Xiandai Nongyao, 2013, 12(5), 9-11, 14; X. Li et al., Xiandai Nongyao, 2006, 5(3), 13-14, 16).
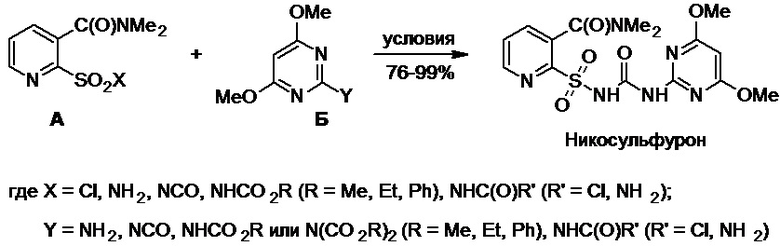
Схема 1.
Несмотря на то, что известно множество способов построения структуры целевого никосульфурона, в промышленной практике наиболее удобным, технологичным, масштабируемым и безопасным подходом к осуществлению финальной стадии его синтеза является реакция 2-аминосульфонил-N,N-диметилникотинамида (II) (далее по тексту - сульфонамид II) [предшественник А, где Х = NH2, Схема 1] с 2-феноксикарбониламино-4,6-диметоксипиримидином (III) (далее по тексту - фенилкарбамат III) [предшественник Б, где Y = NHCO2Ph, Схема 1] (Схема 2). Это взаимодействие проводят в присутствии сильных органических оснований, в качестве которых чаще всего используют бициклические амидины (1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) или 1,5-диазабицикло[4.3.0]нон-5-ен (DBN)) в безводных полярных органических растворителях, в качестве которых применяют ацетонитрил, пропионитрил, 1,4-диоксан и др. После реакции никосульфурон выделяют, подкисляя образовавшуюся реакционную смесь соляной кислотой и разбавляя ее водой, с последующей фильтрацией полученного осадка продукта, его промывкой и высушиванием на воздухе или при пониженном давлении. При этом получают технический никосульфурон с выходом до 84% (см., напр., патенты EP № 0232067 A2, US № 5008393 А, WO № 1990005728 А1).
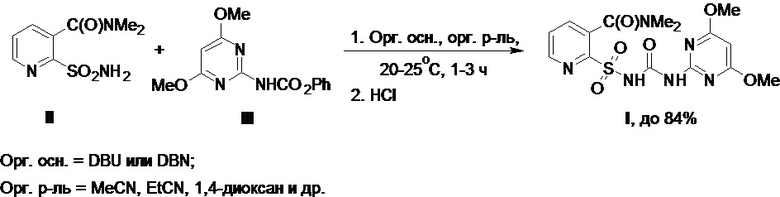
Схема 2.
Данная схема получения никосульфурона исключает использование в ходе процесса повышенной или пониженной температуры и/или давления, токсичного фосгена или низших хлорформиатов, а также нестабильных при нормальных условиях и в присутствии влаги изоцианатов и карбомоилхлоридов в качестве исходных соединений (см. схему 1), что является ее преимуществом. Выделение и очистка целевого продукта, образующегося после реакции в виде осадка, включает лишь его фильтрацию и промывку водой, осуществляемые различными методами.
Среди главных недостатков этой бесфосгенной схемы синтеза никосульфурона, снижающих общую эффективность и экономические показатели процесса, можно выделить а) проведение процесса в безводной среде, что требует отсутствия остаточной влаги в используемых органических растворителях, технологическом оборудовании до и в ходе реакции и в исходном сырье и б) использование дорогостоящих амидинов (DBU и DBN) в качестве органических оснований, при этом их регенерация из водно-органических маточных растворов, где они содержатся в виде гидрохлоридов, после проведения реакции и выделения целевого продукта является отдельной проблемой в виду их нестабильности в присутствии воды.
Задачей предлагаемого технического решения является улучшение технико-экономических показателей процесса производства никосульфурона за счет упрощения технологии его получения из 2-аминосульфонил-N,N-диметилникотинамида и 2-феноксикарбониламино-4,6-диметоксипиримидина.
Техническим результатом является улучшение технико-экономических показателей технологического процесса производства никосульфурона из 2-аминосульфонил-N,N-диметилникотинамида и 2-феноксикарбониламино-4,6-диметоксипиримидина за счет отказа от использования в нем органических оснований и безводных органических растворителей, что приводит к а) упрощению и удешевлению подготовки необходимого емкостного оборудования за счет ликвидации требования к отсутствию остаточной влаги на всех этапах процесса; б) снижению общего количества используемых органических веществ (основание, растворитель) в ходе процесса и, как следствие, снижению содержания органических компонентов в водно-органических отходах производства, что выгодно с точки зрения дальнейшей переработки таких отходов и охраны окружающей среды; в) снижению стоимости конечного продукта за счет исключения из процесса дорогостоящих органических оснований; г) получению целевого продукта высокого качества с выходом 92-99% в зависимости от условий проведения процесса.
Технический результат достигается при использовании способа получения никосульфурона, включающего взаимодействие 2-аминосульфонил-N,N-диметилникотинамида с 2-феноксикарбониламино-4,6-диметоксипиримидином в водно-органической среде, представляющей собой смесь воды и от 10 до 90 об.% органического компонента, в качестве растворителя в присутствии неорганического основания с последующей обработкой полученной реакционной массы, содержащей соответствующую соль никосульфурона, минеральной кислотой, выбранной из соляной, серной или фосфорной кислоты, или органической кислотой, выбранной из муравьиной, уксусной или пропионовой кислоты, с получением целевого продукта, его выделением и очисткой.
В качестве органического компонента водно-органической среды предпочтительно используют водорастворимый полярный протонный органический растворитель, выбранный из одноатомных спиртов С1-С3 (метанол, этанол, н-пропанол или изопропанол), многоатомных спиртов С2-С3 (этиленгликоль, пропиленгликоль или глицерин) или целлозольвов С1-С3 (2-метоксиэтанол, 2-этоксиэтанол или 2-пропоксиэтанол), или водорастворимый полярный апротонный органический растворитель, выбранный из кетонов (ацетон или метилэтилкетон), нитрилов (ацетонитрил или пропионитрил), циклических или ациклических простых эфиров (1,4-диоксан, тетрагидрофуран или 1,2-диметоксиэтан), сульфоксидов (диметилсульфоксид или диэтилсульфоксид), амидов (N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон или N-этил-2-пирролидон) или мочевин (N,N,N',N'-тетраметилмочевина, N,N'-диметилэтиленмочевина или N,N'-диметилпропиленмочевина).
В качестве неорганического основания предпочтительно используют оксид, гидроксид, карбонат, гидрокарбонат, фосфат или гидрофосфат щелочного металла, выбранного из лития, натрия, калия или цезия, или щелочноземельного металла, выбранного из магния, кальция, стронция или бария. При этом неорганическое основание добавляют к раствору или суспензии смеси сульфонамида II и фенилкарбамата III в водно-органической среде или неорганическое основание в виде водного раствора или суспензии добавляют к раствору или суспензии смеси сульфонамида II и фенилкарбамата III в водно-органической среде или в органическом компоненте водно-органической среды.
В ходе реакции между сульфонамидом II и фенилкарбаматом III в водно-органической среде в присутствии неорганического основания получают раствор или суспензию соли никосульфурона I x M (Схема 3) с катионом соответствующего щелочного (M = Li, Na, K или Cs) или щелочноземельного (M = Mg, Ca, Sr или Ba) металла, первоначально входящим в состав исходной неорганической соли, используемой в качестве основания. Из полученного в ходе реакции раствора или суспензии соли I x M в используемой водно-органической среде целевой никосульфурон получают подкислением реакционной массы минеральной или органической кислотой, при этом на данном этапе синтеза соль I x M может быть также выделена в индивидуальном виде, если это необходимо.
Предпочтительным является использование в процессе: а) от 1,0 до 1,3 мол. экв. фенилкарбамата III по отношению к сульфонамиду II; б) от 0,5 до 2,5 мол. экв. неорганического основания по отношению к сульфонамиду II; в) от 1,0 до 5,0 мол. экв. минеральной или органической кислоты по отношению к сульфонамиду II; г) температуры в ходе реакции от 15 до 50°С; д) времени реакции от 0,5 до 5,0 ч.
Изобретение иллюстрируется следующими примерами (Схема 3).
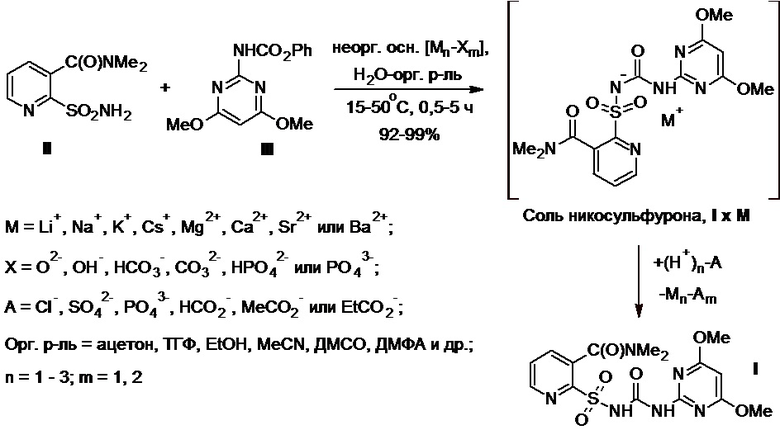
Схема 3.
Пример 1.
В реактор синтеза никосульфурона помещают сульфонамид II (45,9 г, 0,2 моль), фенилкарбамат III (60,6 г, 0,22 моль) и водный раствор этанола (20 об.%). К полученной суспензии при перемешивании добавляют карбонат натрия (25,4 г, 0,24 моль), поддерживая температуру реакционной массы не выше 25°С. По окончании прибавления основания содержимое реактора выдерживают до полного срабатывания сульфонамида II при температуре 15-20°С (контроль ТСХ или ВЭЖХ), после чего разбавляют водой и подкисляют концентрированной соляной кислотой до pH 2-3. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают. Получают 78,0 г (95%) никосульфурона в виде белого порошка с т.пл. 171-173°С и чистотой не менее 98,0% (по данным ВЭЖХ).
Пример 2.
В реактор синтеза никосульфурона помещают сульфонамид II (45,9 г, 0,2 моль), фенилкарбамат III (66,1 г, 0,24 моль) и пропиленгликоль. К полученной суспензии при перемешивании добавляют воду (образуется водно-органическая смесь, содержащая 40 об.% пропиленгликоля), после чего добавляют тригидрат фосфата калия (47,9 г, 0,18 моль), поддерживая температуру реакционной массы не выше 25°С. По окончании прибавления основания содержимое реактора выдерживают до полного срабатывания сульфонамида II при температуре 30-35°С (контроль ТСХ или ВЭЖХ), после чего разбавляют водой и подкисляют 70%-ной водной уксусной кислотой до pH 2-3. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают. Получают 76,2 г (93%) никосульфурона в виде белого порошка с т.пл. 171-173°С и чистотой не менее 98,0% (по данным ВЭЖХ).
Пример 3.
В реактор синтеза никосульфурона помещают сульфонамид II (45,9 г, 0,2 моль), фенилкарбамат III (55,1 г, 0,2 моль) и 2-метоксиэтанол. К полученной суспензии при перемешивании добавляют 10%-ный водный раствор гидроксида натрия (120,0 г, 0,3 моль), поддерживая температуру реакционной массы не выше 25°С (образуется водно-органическая смесь, содержащая 25 об.% 2-метоксиэтанола). По окончании прибавления основания содержимое реактора выдерживают до полного срабатывания сульфонамида II при температуре 20-25°С (контроль ТСХ или ВЭЖХ), после чего разбавляют водой и подкисляют 85%-ной фосфорной кислотой до pH 2-3. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают. Получают 77,2 г (94%) никосульфурона в виде белого порошка с т.пл. 171-173°С и чистотой не менее 98,0% (по данным ВЭЖХ).
Пример 4.
В реактор синтеза никосульфурона помещают сульфонамид II (45,9 г, 0,2 моль), фенилкарбамат III (71,6 г, 0,26 моль) и ацетон. К полученному раствору при перемешивании дозируют суспензию гидроксида кальция в воде (29,6 г, 0,4 моль), поддерживая температуру реакционной массы не выше 25°С (образуется водно-органическая смесь, содержащая 70 об.% ацетона). По окончании прибавления основания содержимое реактора выдерживают до полного срабатывания сульфонамида II при температуре 35-40°С (контроль ТСХ или ВЭЖХ), после чего разбавляют водой и подкисляют пропионовой кислотой до pH 2-3. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают. Получают 81,2 г (99%) никосульфурона в виде белого порошка с т.пл. 171-173°С и чистотой не менее 98,0% (по данным ВЭЖХ).
Пример 5.
Способ получения никосульфурона осуществлялся аналогично примеру 1 с загрузкой 91,7 г (0,4 моль) сульфонамида II и 115,6 г (0,42 моль) фенилкарбамата III. Отличие состояло в использовании водного раствора ацетонитрила (50 об.%) вместо водного раствора этанола (20 об.%), карбоната цезия (130,3 г, 0,4 моль) вместо карбоната натрия, температуры выдержки реакционной массы, равной 20-25°С вместо 15-20°С, и 40%-ной серной кислоты вместо концентрированной соляной кислоты. Получают 160,7 г (98%) никосульфурона в виде белого порошка.
Пример 6.
Способ получения никосульфурона осуществлялся аналогично примеру 1 с загрузкой 91,7 г (0,4 моль) сульфонамида II и 110,1 г (0,4 моль) фенилкарбамата III. Отличие состояло в использовании водного раствора 1,4-диоксана (90 об.%) вместо водного раствора этанола (20 об.%), гидроксида лития (23,9 г, 1,0 моль) вместо карбоната натрия. Получают 159,2 г (97%) никосульфурона в виде белого порошка.
Пример 7.
Способ получения никосульфурона осуществлялся аналогично примеру 2 с загрузкой 91,7 г (0,4 моль) сульфонамида II и 121,1 г (0,44 моль) фенилкарбамата III. Отличие состояло в использовании водного раствора диметилсульфоксида (20 об.%) вместо водного раствора пропиленгликоля (40 об.%), оксида магния (24,2 г, 0,6 моль) вместо тригидрата фосфата калия, температуры выдержки реакционной массы равной 45-50°С вместо 30-35°С и 50%-ной водной муравьиной кислоты вместо 70%-ной водной уксусной кислоты. Получают 156,0 г (95%) никосульфурона в виде белого порошка.
Пример 8.
Способ получения никосульфурона осуществлялся аналогично примеру 2 с загрузкой 91,7 г (0,4 моль) сульфонамида II и 132,1 г (0,48 моль) фенилкарбамата III. Отличие состояло в использовании водного раствора диметилформамида (60 об.%) вместо водного раствора пропиленгликоля (40 об.%), карбоната стронция (29,5 г, 0,2 моль) вместо тригидрата фосфата калия и температуры выдержки реакционной массы, равной 20-25°С вместо 30-35°С. Получают 154,3 г (94%) никосульфурона в виде белого порошка.
Пример 9.
Способ получения никосульфурона осуществлялся аналогично примеру 3 с загрузкой 91,7 г (0,4 моль) сульфонамида II и 110,1 г (0,4 моль) фенилкарбамата III. Отличие состояло в использовании водного раствора N,N,N',N'-тетраметилмочевины (35 об.%) вместо водного раствора 2-метоксиэтанола (25 об.%), 20%-ного водного раствора гидрокарбоната калия (401 г, 0,8 моль) вместо 10%-ного водного раствора гидроксида натрия, температуры выдержки реакционной массы, равной 35-40°С вместо 20-25°С, и концентрированной соляной кислоты вместо 85%-ной фосфорной кислоты. Получают 152,7 г (93%) никосульфурона в виде белого порошка.
Пример 10.
Способ получения никосульфурона осуществлялся аналогично примеру 3 с загрузкой 91,7 г (0,4 моль) сульфонамида II и 132,1 г (0,48 моль) фенилкарбамата III. Отличие состояло в использовании водного раствора N,N-диметилацетамида (40 об.%) вместо водного раствора 2-метоксиэтанола (25 об.%) и 40%-ного водного раствора гидрофосфата калия (304,8 г, 0,7 моль) вместо 10%-ного водного раствора гидроксида натрия и температуры выдержки реакционной массы, равной 40-45°С вместо 20-25°С. Получают 151,0 г (92%) никосульфурона в виде белого порошка.
Пример 11.
Способ получения никосульфурона осуществлялся аналогично примеру 4 с загрузкой 91,7 г (0,4 моль) сульфонамида II и 115,6 г (0,42 моль) фенилкарбамата III. Отличие состояло в использовании водного раствора изопропанола (50 об.%) вместо водного раствора ацетона (70 об.%), суспензии карбоната бария в воде (55,3 г, 0,28 моль) вместо суспензии гидроксида кальция в воде и 50%-ной водной муравьиной кислоты вместо пропионовой кислоты. Получают 156,0 г (95%) никосульфурона в виде белого порошка.
В результате использования предлагаемого способа получения никосульфурона удается улучшить технико-экономические показатели технологического процесса его производства из 2-аминосульфонил-N,N-диметилникотинамида и 2-феноксикарбониламино-4,6-диметоксипиримидина, за счет отказа от использования в нем органических оснований и безводных органических растворителей, что приводит к а) упрощению и удешевлению подготовки необходимого емкостного оборудования, за счет ликвидации требования к отсутствию остаточной влаги на всех этапах процесса; б) снижению общего количества используемых органических веществ (основание, растворитель) в ходе процесса и, как следствие, снижению содержания органических компонентов в водно-органических отходах производства, что выгодно с точки зрения дальнейшей переработки таких отходов и охраны окружающей среды; в) снижению стоимости конечного продукта за счет исключения из процесса дорогостоящих органических оснований; г) получению целевого продукта высокого качества с выходом 92-99% в зависимости от условий проведения процесса.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения гербицида никосульфурона | 2024 |
|
RU2827153C1 |
| Способ получения R-N-[[3-[(диметиламино)карбонил]пиридин-2-ил]сульфонил]карбаматов, в которых заместителем R является метил или этил | 2023 |
|
RU2816572C1 |
| Способ получения пиразосульфурон-этила | 2024 |
|
RU2828085C1 |
| Способ получения сульфонилмочевинных гербицидов, содержащих 4,6-диметоксипиримидин-2-ильный заместитель | 2024 |
|
RU2834461C1 |
| Способ получения бенсульфурон-метила | 2024 |
|
RU2826376C1 |
| Способ получения моносульфурон-метила | 2024 |
|
RU2834839C1 |
| Способ получения метсульфурон-метила | 2024 |
|
RU2824626C1 |
| Способ получения сульфометурон-метила | 2023 |
|
RU2805743C1 |
| Способ получения моносульфурона | 2024 |
|
RU2834000C1 |
| АЦИЛИРОВАННЫЕ АМИНОФЕНИЛСУЛЬФОНИЛМОЧЕВИНЫ ИЛИ ИХ СОЛИ, ПРОМЕЖУТОЧНЫЕ ФЕНИЛСУЛЬФОНИЛЫ, ГЕРБИЦИДНОЕ СРЕДСТВО И СПОСОБ БОРЬБЫ С СОРНОЙ РАСТИТЕЛЬНОСТЬЮ | 1995 |
|
RU2171253C2 |
Изобретение относится к области получения пестицидов, более конкретно - к способу получения никосульфурона. Способ получения никосульфурона осуществляют взаимодействием 2-аминосульфонил-N,N-диметилникотинамида с 2-феноксикарбониламино-4,6-диметоксипиримидином в водно-органической среде, представляющей собой смесь воды и от 10 до 90 об.% органического компонента, в качестве растворителя в присутствии неорганического основания. Далее полученную реакционную массу, содержащую соответствующую соль никосульфурона, обрабатывают минеральной кислотой, выбранной из соляной, серной или фосфорной кислоты, или органической кислотой, выбранной из муравьиной, уксусной или пропионовой кислоты, с получением целевого продукта, его выделением и очисткой. В качестве органического компонента водно-органической среды используют водорастворимый полярный протонный органический растворитель, выбранный из одноатомных спиртов, многоатомных спиртов или целлозольвов, или водорастворимый полярный апротонный органический растворитель, выбранный из кетонов, нитрилов, циклических или ациклических простых эфиров, сульфоксидов, амидов или мочевин. В качестве неорганического основания используют оксид, гидроксид, карбонат, гидрокарбонат, фосфат или гидрофосфат щелочного металла, выбранного из лития, натрия, калия или цезия, или щелочноземельного металла, выбранного из магния, кальция, стронция или бария. Неорганическое основание добавляют к раствору или суспензии смеси 2-аминосульфонил-N,N-диметилникотинамида и 2-феноксикарбониламино-4,6-диметоксипиримидина в водно-органической среде или неорганическое основание в виде водного раствора или суспензии добавляют к раствору или суспензии смеси 2-аминосульфонил-N,N-диметилникотинамида и 2-феноксикарбониламино-4,6-диметоксипиримидина в водно-органической среде или в органическом компоненте водно-органической среды. Предлагаемый способ получения никосульфурона обеспечивает улучшение технико-экономических показателей технологического процесса за счет отказа от использования в нем органических оснований и безводных органических растворителей, упрощение процесса, снижение общего количества используемых органических веществ, снижение содержания органических компонентов в водно-органических отходах производства, получение целевого продукта высокого качества с выходом 92-99%. 4 з.п. ф-лы, 11 пр.
1. Способ получения никосульфурона, характеризующийся тем, что осуществляют взаимодействие 2-аминосульфонил-N,N-диметилникотинамида с 2-феноксикарбониламино-4,6-диметоксипиримидином в водно-органической среде, представляющей собой смесь воды и от 10 до 90 об.% органического компонента, в качестве растворителя в присутствии неорганического основания с последующей обработкой полученной реакционной массы, содержащей соответствующую соль никосульфурона, минеральной кислотой, выбранной из соляной, серной или фосфорной кислоты, или органической кислотой, выбранной из муравьиной, уксусной или пропионовой кислоты, с получением целевого продукта, его выделением и очисткой.
2. Способ по п. 1, характеризующийся тем, что в качестве органического компонента водно-органической среды используют водорастворимый полярный протонный органический растворитель, выбранный из одноатомных спиртов, многоатомных спиртов или целлозольвов, или водорастворимый полярный апротонный органический растворитель, выбранный из кетонов, нитрилов, циклических или ациклических простых эфиров, сульфоксидов, амидов или мочевин.
3. Способ по п. 1, характеризующийся тем, что в качестве неорганического основания используют оксид, гидроксид, карбонат, гидрокарбонат, фосфат или гидрофосфат щелочного металла, выбранного из лития, натрия, калия или цезия, или щелочноземельного металла, выбранного из магния, кальция, стронция или бария.
4. Способ по п. 1, характеризующийся тем, что неорганическое основание добавляют к раствору или суспензии смеси 2-аминосульфонил-N,N-диметилникотинамида и 2-феноксикарбониламино-4,6-диметоксипиримидина в водно-органической среде.
5. Способ по п. 1, характеризующийся тем, что неорганическое основание в виде водного раствора или суспензии добавляют к раствору или суспензии смеси 2-аминосульфонил-N,N-диметилникотинамида и 2-феноксикарбониламино-4,6-диметоксипиримидина в водно-органической среде или в органическом компоненте водно-органической среды.
| АРИЛСУЛЬФОНИЛМОЧЕВИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ГЕРБИЦИДОВ | 1992 |
|
RU2314291C2 |
| Способ получения замещенной арилсульфонилмочевины | 1972 |
|
SU503513A3 |
| WO 1990005728 A1, 31.05.1990 | |||
| DE 3750633 T2, 23.02.1995 | |||
| CN 108558830 B, 20.09.2019. | |||

