ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет и преимущество заявки на патент Китая №201811533849.X, поданной в Национальное управление интеллектуальной собственности Китая 14 декабря 2018 г., раскрытие которой включено в данный документ посредством ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ
Настоящая заявка относится к области медицинской химии, относится к соли ингибитора Syk и ее кристаллической форме, и более конкретно относится к гидрохлориду 5-фтор-1-метил-3-((5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-ил)амино)-6-(1H-пиразол-3-ил)хинолин-2(1H)-она и его кристаллической форме, и способу его получения, фармацевтической композиции на его основе и его применению.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Тирозинкиназа селезенки (Syk) является внутриклеточной тирозинпротеинкиназой и относится к семейству протеинкиназ ZAP70. Syk играет важную роль в раннем развитии B-клеток, онтогенезе лимфоцитов и функционировании зрелых B-клеток. Во время этого процесса она вовлечена в различные пути передачи сигнала и функционирует без фосфорилирования Src-киназой. Syk, помимо того, что является повсеместно экспрессируемой в гемопоэтических стволовых клетках, экспрессируется в клетках, отличных от гемопоэтических, таких как эпителиальные клетки, гепатоциты, фибробласты, нейроны и ткань молочной железы, и она имеет различные функции.
Нарушение функции PTK Syk обнаруживается при многих заболеваниях у людей, таких как аллергические реакции, астма, воспаление и аутоиммунные заболевания, и многочисленные исследования показали, что Syk является важным медиатором при остром или хроническом воспалении. Активация Syk обнаруживается в некоторых обычных B-клеточных злокачественных опухолях. Например, антигеннезависимое фосфорилирование Syk можно выявить при фолликулярной лимфоме, диффузной В-крупноклеточной лимфоме, лимфоме из клеток мантийной зоны и B-клеточном хроническом лимфоцитарном лейкозе. Исследователи обнаружили, что ингибирование Syk в клетках фолликулярной лимфомы и диффузной В-крупноклеточной лимфомы может снижать уровень фосфорилирования нижеследующих сигнальных молекул, за счет этого подавляя пролиферацию и выживаемость опухолевых клеток. Кроме того, транслокации Syk были обнаружены при миелодиспластическом синдроме и периферической Т-клеточной лимфоме, дополнительно указывая на то, что киназа может выступать в качестве протоонкогена. Ингибирование активности Syk может, таким образом, использоваться для лечения конкретных типов видов рака, включая В-клеточную лимфому и лейкоз.
КРАТКОЕ ОПИСАНИЕ
Предусмотрено соединение формулы I, имеющее следующее химическое название: 5-фтор-1-метил-3-((5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-ил)амино)-6-(1H-пиразол-3-ил)хинолин-2(1H)-он,
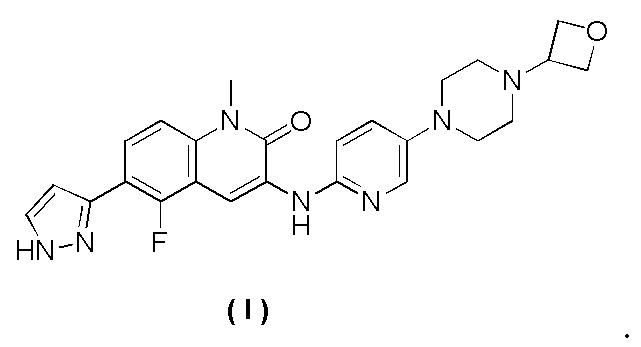
В одном аспекте в настоящей заявке предусмотрен гидрохлорид соединения формулы I.
В некоторых вариантах осуществления гидрохлорид соединения формулы I представляет собой гидрохлорид соединения формулы I (формула II) 1:1,
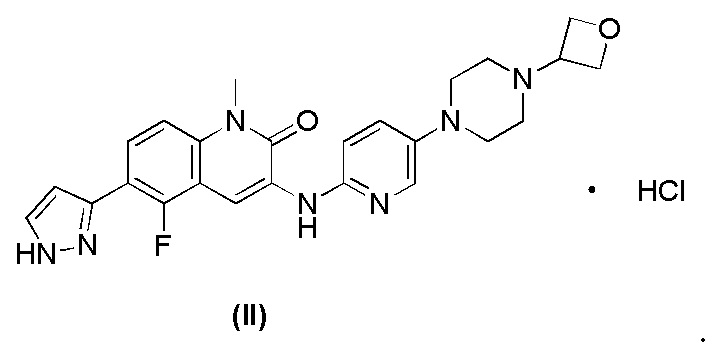
Гидрохлорид соединения формулы I, описанный в данном документе, может находиться в кристаллической форме или аморфной форме, предпочтительно в кристаллической форме.
Кристаллическая форма гидрохлорида соединения формулы I характеризуется высокой стабильностью, небольшой гигроскопичностью, лучшим уровнем метаболизма in vivo, более длительным периодом полувыведения, лучшей активностью в отношении ингибирования тирозинкиназы селезенки, лучшими физическими свойствами, безопасностью и метаболической стабильностью и лучшими фармацевтическими свойствами.
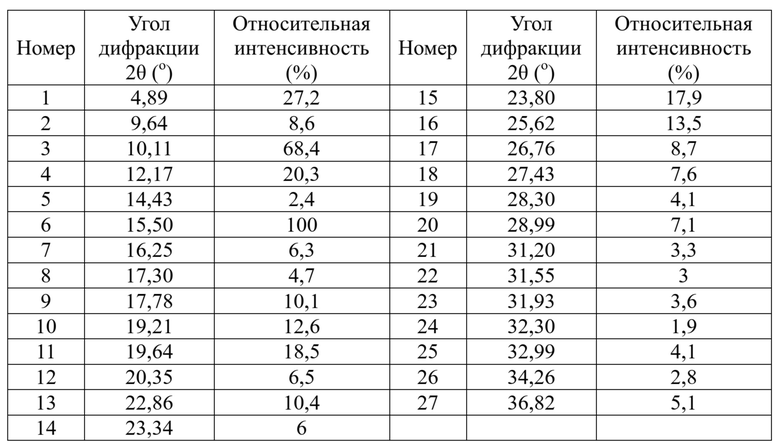
В некоторых вариантах осуществления гидрохлорид соединения формулы I, раскрытый в данном документе, представляет собой кристалл формы A, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при приблизительно 4,9°, 10,1°, 12,2°, 15,5°, 19,6° и 23,8° на спектре порошковой рентгеновской дифракции; обычно наличием дифракционных пиков, представленных с помощью значений 2θ, при приблизительно 4,9°, 10,1°, 12,2°, 15,5°, 17,8°, 19,2°, 19,6°, 22,9°, 23,8° и 25,6° на спектре порошковой рентгеновской дифракции; обычно наличием дифракционных пиков, представленных с помощью значений 2θ, при приблизительно 4,9°, 9,6°, 10,1°, 12,2°, 15,5°, 16,3°, 17,8°, 19,2°, 19,6°, 20,4°, 22,9°, 23,3°, 23,8°, 25,6°, 26,8°, 27,4°, 29,0° и 36,8 на спектре порошковой рентгеновской дифракции; и обычно наличием дифракционных пиков, представленных с помощью значений 2θ, при приблизительно 4,9°, 9,6°, 10,1°, 12,2°, 14,4°, 15,5°, 16,3°, 17,3°, 17,8°, 19,2°, 19,6°, 20,4°, 22,9°, 23,3°, 23,8°, 25,6°, 26,8°, 27,4°, 28,3°, 29,0°, 31,2°, 31,6°, 31,9°, 32,3°, 33,0°, 34,3° и 36,8° на спектре порошковой рентгеновской дифракции.
В качестве одного варианта осуществления настоящей заявки положения пиков и интенсивности характеристических пиков на спектре порошковой рентгеновской дифракции для кристалла формы A гидрохлорида соединения формулы I, раскрытого в данном документе, показаны в таблице 1.
Таблица 1. Данные определения характеристик с помощью рентгенограммы XRPD для кристалла формы A
В одном варианте осуществления настоящей заявки предусмотрен кристалл формы A гидрохлорида соединения формулы I, который характеризуется порошковой рентгеновской дифрактограммой, показанной на фиг. 1.
В одном варианте осуществления настоящей заявки предусмотрен кристалл формы A гидрохлорида соединения формулы I, который характеризуется пиком поглощения при приблизительно 272°C на термограмме дифференциальной сканирующей калориметрии (DSC).
В одном варианте осуществления настоящей заявки предусмотрен кристалл формы A гидрохлорида соединения формулы I, который характеризуется термограммой DSC, показанной на фиг. 2.
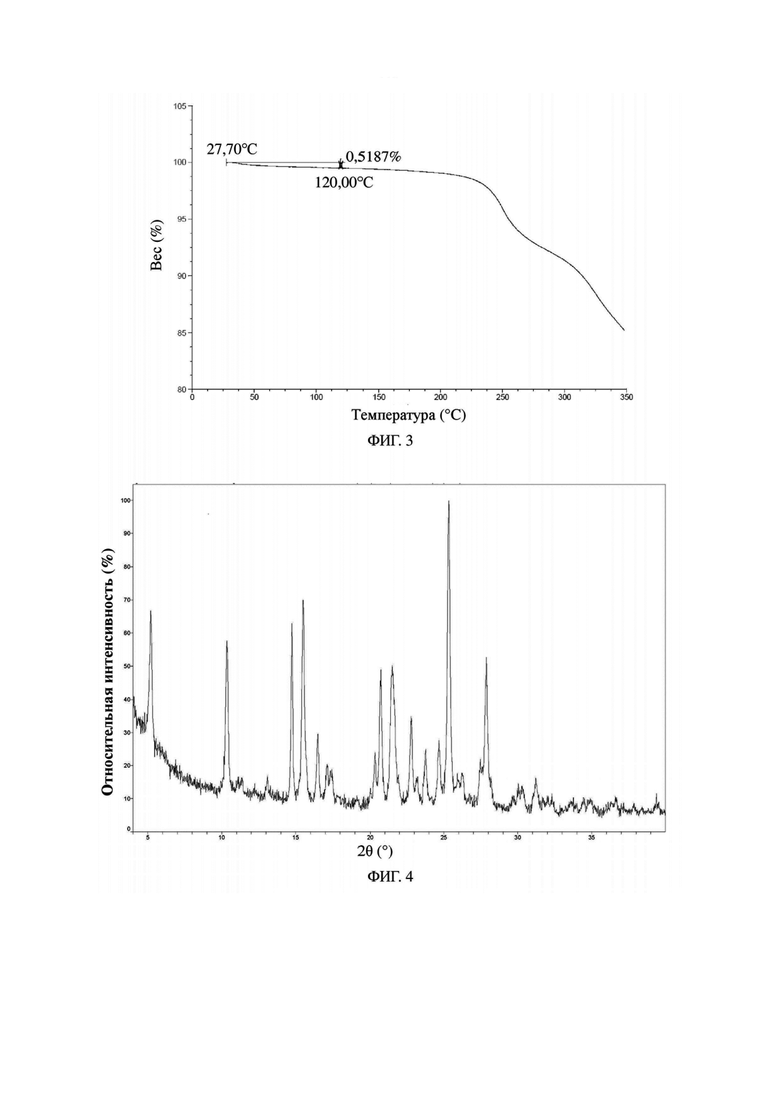
В одном варианте осуществления настоящей заявки предусмотрен кристалл формы A гидрохлорида соединения формулы I, который характеризуется термограммой термогравиметрического анализа (TGA), показанной на фиг. 3.
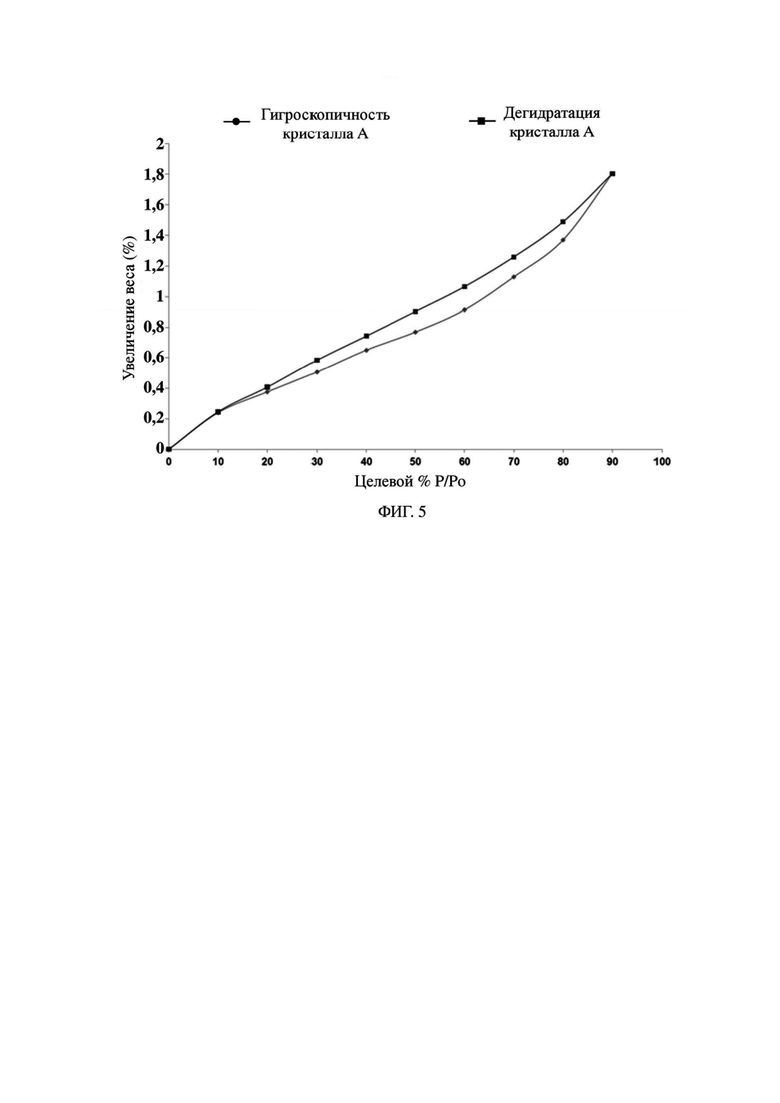
В некоторых вариантах осуществления гидрохлорид соединения формулы I, раскрытый в данном документе, представляет собой кристалл формы B, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при приблизительно 5,2°, 10,4°, 14,7°, 15,5° и 25,3° на спектре порошковой рентгеновской дифракции; обычно наличием дифракционных пиков, представленных с помощью значений 2θ, при приблизительно 5,2°, 10,4°, 14,7°, 15,5°, 16,5°, 20,7°, 21,5°, 22,8°, 25,3° и 27,9° на спектре порошковой рентгеновской дифракции; обычно наличием дифракционных пиков, представленных с помощью значений 2θ, при приблизительно 5,2°, 10,4°, 14,7°, 15,5°, 16,5°, 17,1°, 17,5°, 20,3°, 20,7°, 21,5°, 22,8°, 23,8°, 24,7°, 25,3°, 27,5°, 27,9° и 31,2° на спектре порошковой рентгеновской дифракции; и обычно наличием дифракционных пиков, представленных с помощью значений 2θ, при приблизительно 5,2°, 10,4°, 13,1°, 14,7°, 15,5°, 16,5°, 17,1°, 17,5°, 20,0°, 20,3°, 20,7°, 21,5°, 22,8°, 23,2°, 23,8°, 24,7°, 25,3°, 25,9°, 26,2°, 27,5°, 27,9°, 28,2°, 29,7°, 30,0°, 30,3°, 31,2°, 31,7°, 32,3°, 34,5°, 34,9° и 36,6° на спектре порошковой рентгеновской дифракции.
В качестве одного варианта осуществления настоящей заявки положения пиков и интенсивности характеристических пиков на спектре порошковой рентгеновской дифракции для кристалла формы В гидрохлорида соединения формулы I, раскрытого в данном документе, показаны в таблице 2.
Таблица 2. Данные определения характеристик с помощью рентгенограммы XRPD для кристалла формы В

В одном варианте осуществления настоящей заявки предусмотрен кристалл формы В гидрохлорида соединения формулы I, который характеризуется порошковой рентгеновской дифрактограммой, показанной на фиг. 4.
В другом аспекте в настоящей заявке предусмотрен способ получения кристалла формы A гидрохлорида соединения формулы I, включающий: (1) добавление соединения формулы I в предварительно нагретый растворитель, затем добавление по каплям другого растворителя, пока раствор не станет прозрачным, и перемешивание при поддержании температуры; (2) добавление по каплям разбавленной хлористоводородной кислоты в раствор из стадии (1), и перемешивание в течение ночи при поддержании температуры, и (3) медленное добавление по каплям растворителя в раствор из стадии (2), перемешивание с осаждением твердого вещества, фильтрование и высушивание с получением кристалла формы A гидрохлорида соединения формулы I.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I предварительно нагретый растворитель на стадии (1) выбран из группы, состоящей из воды, этилацетата, дихлорметана, этанола, ацетона, ацетонитрила, тетрагидрофурана и метил-трет-бутилового эфира; предпочтительно предварительно нагретый растворитель представляет собой воду или этилацетат.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I предварительно нагретый растворитель на стадии (1) находится при температуре 30-40°C, предпочтительно при 35-38°C.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I другой растворитель на стадии (1) выбран из группы, состоящей из муравьиной кислоты, уксусной кислоты, пропионовой кислоты и щавелевой кислоты; предпочтительно другой растворитель представляет собой муравьиную кислоту.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I мольно-объемное отношение соединения формулы I к другому растворителю на стадии (1) составляет от 1 ммоль:0,8 мл до 1 ммоль:1,5 мл, предпочтительно от 1 ммоль:0,8 мл до 1 ммоль:1,2 мл и более предпочтительно от 1 ммоль:0,8 мл до 1 ммоль:1,0 мл.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I объемное отношение предварительно нагретого растворителя к другому растворителю на стадии (1) составляет от 0,5:1 до 3:1, предпочтительно от 0,5:1 до 2:1.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I мольное отношение хлористоводородной кислоты в разбавленной хлористоводородной кислоте на стадии (2) к соединению формулы на стадии (1) составляет от 1:1 до 3:1, предпочтительно от 1:1 до 2:1.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I концентрация разбавленной хлористоводородной кислоты на стадии (2) составляет 1-2 моль/л, предпочтительно 1-1,2 моль/л; разбавленная хлористоводородная кислоты получена путем разбавления коммерчески доступной концентрированной хлористоводородной кислоты водой, или разбавленная хлористоводородная кислота относится к смешанному раствору коммерчески доступной концентрированной хлористоводородной кислоты и другого растворителя, подобного предварительно нагретому растворителю на стадии (1).
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I растворитель на стадии (3) выбран из группы, состоящей из метанола, этанола, ацетона, изопропанола, ацетонитрила, тетрагидрофурана, этиленгликоля или пропиленгликоля; предпочтительно растворитель представляет собой этанол.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I мольно-объемное отношение растворителя на стадии (3) к соединению формулы I на стадии (1) составляет от 2 мл:1 ммоль до 10 мл:1 ммоль, предпочтительно от 4 мл:1 ммоль до 9 мл:1 ммоль.
При вышеуказанном получении кристалла формы A гидрохлорида соединения формулы I «поддержание температуры» на стадии (1) и стадии (2) означает, что температура перемешивания поддерживается на таком же уровне температуры, что и предварительно нагретый растворитель на стадии (1).
В другом аспекте в настоящей заявке дополнительно предусмотрен способ получения кристалла формы B гидрохлорида соединения формулы I, включающий: (1) добавление соединения формулы I в растворитель и перемешивание с растворением; (2) добавление разбавленной хлористоводородной кислоты в раствор из стадии (1), и перемешивание в течение ночи, и (3) центрифугирование раствора из стадии (2), и высушивание твердого вещества с получением кристалла формы B гидрохлорида соединения формулы I.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы B гидрохлорида соединения формулы I растворитель на стадии (1) выбран из группы, состоящей из метанола, этанола, изопропанола, уксусной кислоты, ацетона, ацетонитрила или смешанного растворителя из любых двух из них; предпочтительно растворитель представляет собой этанол, уксусную кислоту, ацетон или смешанный растворитель из любых двух из них и более предпочтительно растворитель представляет собой этанол, смешанный растворитель из этанола и уксусной кислоты или смешанный растворитель из ацетона и уксусной кислоты.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы B гидрохлорида соединения формулы I мольно-объемное отношение соединения формулы I к другому растворителю на стадии (1) составляет от 1 ммоль:10 мл до 1 ммоль:40 мл, предпочтительно от 1 ммоль:20 мл до 1 ммоль:35 мл.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы B гидрохлорида соединения формулы I растворитель на стадии (1) представляет собой смешанный растворитель из уксусной кислоты и этанола или смешанный растворитель из уксусной кислоты и ацетона, причем объемное отношение уксусной кислоты к этанолу или уксусной кислоты к ацетону составляет от 1:5 до 1:10, предпочтительно от 1:5 до 1:8.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы B гидрохлорида соединения формулы I мольное отношение соединения формулы I на стадии (1) к хлористоводородной кислоте на стадии (2) составляет от 1:1 до 1:5, предпочтительно от 1:1 до 1:3,5.
В одном варианте осуществления настоящей заявки при вышеуказанном получении кристалла формы B гидрохлорида соединения формулы I концентрация разбавленной хлористоводородной кислоты на стадии (2) составляет 1-2 моль/л, предпочтительно 1-1,2 моль/л; разбавленная хлористоводородная кислота получена путем разбавления коммерчески доступной концентрированной хлористоводородной кислоты водой.
В другом аспекте в настоящей заявке предусмотрен гидрохлорид соединения формулы I, причем кристалл формы A гидрохлорида соединения формулы I составляет 50% или больше, предпочтительно 75% или больше, более предпочтительно 90% или больше и наиболее предпочтительно 95% или больше веса гидрохлорида соединения формулы I. Гидрохлорид соединения формулы I может также содержать небольшое количество других кристаллических или аморфных форм гидрохлорида соединения формулы I, например, небольшое количество кристалла формы B гидрохлорида соединения формулы I.
В другом аспекте в настоящей заявке предусмотрен гидрохлорид соединения формулы I, причем кристалл формы B гидрохлорида соединения формулы I составляет 50% или больше, предпочтительно 75% или больше, более предпочтительно 90% или больше и наиболее предпочтительно 95% или больше веса гидрохлорида соединения формулы I.
В другом аспекте в настоящей заявке предусмотрена фармацевтическая композиция, содержащая терапевтически эффективное количество гидрохлорида соединения формулы I, описанного выше; при этом фармацевтическая композиция может содержать по меньшей мере один фармацевтически приемлемый носитель или другое вспомогательное вещество.
В другом аспекте в настоящей заявке предусмотрено применение гидрохлорида соединения формулы I или фармацевтической композиции, описанных выше, при получении лекарственного препарата для лечения заболевания, связанного с рецептором Syk.
В другом аспекте в настоящей заявке предусмотрен способ лечения заболевания, связанного с рецептором Syk, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества гидрохлорида соединения формулы I или фармацевтической композиции, описанных выше.
В другом аспекте в настоящей заявке предусмотрены гидрохлорид соединения формулы I или фармацевтическая композиция, описанные выше, для применения в лечении заболевания, связанного с рецептором Syk, у млекопитающего.
В некоторых вариантах осуществления настоящей заявки млекопитающее представляет собой человека.
В настоящей заявке фармацевтическая композиция может быть составлена в определенной лекарственной форме, и путь введения предпочтительно представляет собой пероральное введение, парентеральное введением (включая подкожное, внутримышечное и внутривенное введение), ректальное введение и т.п. Например, подходящие лекарственные формы для перорального введения включают таблетки, капсулы, гранулы, порошкообразные препараты, пилюли, порошки, пастилки, сиропы или суспензии; подходящие лекарственные формы для парентерального введения включают водные или неводные растворы или эмульсии для инъекции; подходящие лекарственные формы для ректального введения включают суппозитории с гидрофильными или гидрофобными носителями. Лекарственные формы также при необходимости могут быть составлены для быстрого, замедленного или модифицированного высвобождения активного ингредиента.
В некоторых вариантах осуществления настоящей заявки заболевание, связанное с рецептором Syk, выбрано из рака и воспалительных заболеваний.
В некоторых вариантах осуществления настоящей заявки, заболевание связанное с рецептором Syk, выбрано из В-клеточной лимфомы, лимфомы Ходжкина, неходжкинской лимфомы, волосатоклеточного лейкоза, множественной миеломы, хронического гранулоцитарного лейкоза, острого гранулоцитарного лейкоза, хронического лимфолейкоза, острого лимфолейкоза, ревматоидного артрита, аллергического ринита, хронического обструктивного заболевания легких (COPD), синдрома расстройства дыхания у взрослых (ARD), вызванных аллергией воспалительных заболеваний, рассеянного склероза, аутоиммунных заболеваний, острых воспалительных реакций, аллергических нарушений и поликистозных заболеваний почек.
В настоящей заявке спектр порошковой рентгеновской дифракции образца измеряют при следующих условиях: прибор: рентгеновский дифрактометр Bruker D8 ADVANCE; мишень: Cu:Kα; длина волны λ = 1,54056 Å; диапазон угла 2θ: 4°-40°; скорость сканирования: 10°/мин; скорость вращения образца: 15 об/мин; давление и ток трубки-мишени Cu: 40 кВ, 40 мA.
В настоящей заявке спектр DSC измеряют при следующих условиях: прибор: дифференциальный сканирующий калориметр TA Q2000; диапазон температур: 30-300°C; скорость нагревания: 10°C/мин.
В настоящей заявке термогравиметрический анализ (TGA) осуществляют при следующих условиях: прибор: термогравиметрический анализатор TA Q5000; диапазон температур: 25-300°C; скорость нагревания: 10°C/мин.
Следует отметить, что если речь идет о спектре порошковой рентгеновской дифракции, дифракционная рентгенограмма, полученная для кристаллического соединения, обычно является характеристической для конкретного кристалла, при этом значения относительной интенсивности полос (особенно под малыми углами) могут варьировать вследствие эффектов доминирующей ориентации, возникающих из-за различий в условиях кристаллизации, размере частиц и других условиях измерения. Таким образом, значения относительной интенсивности дифракционных пиков не являются характеристическими для рассматриваемого кристалла, и важно рассматривать относительные положения пиков, а не их значения относительной интенсивности для определения того, является ли он таким же, как известный кристалл. Кроме того, для любого конкретного кристалла могут иметь место небольшие погрешности в положениях пиков, что также хорошо известно в области кристаллографии. Например, положение пика может сдвигаться из-за изменений температуры, перемещения образца или калибровки прибора при анализе образца, и погрешность измерения значения 2θ иногда составляет приблизительно ±0,2°. Таким образом, эта погрешность должна учитываться при определении кристаллической структуры. На рентгенограмме XRD положение пика обычно представлено с помощью угла 2θ или межплоскостного расстояния d, и существует простая формула преобразования между ними двумя: d = λ/2sinθ, где d представляет собой межплоскостное расстояние, λ представляет собой длину волны падающих рентгеновских лучей, и θ представляет собой угол дифракции. Для одного и того же кристалла одного и того же соединения положения пиков на рентгенограммах XRD в целом имеют сходство, хотя погрешность значений относительной интенсивности может быть большой. Следует также отметить, что при идентификации смесей часть дифракционных линий может отсутствовать из-за, например, снижения содержания, в случае чего нет необходимости полагаться на все полосы, наблюдаемые в образце высокой чистоты, и даже одна полоса может быть характеристической для данного кристалла.
С помощью DSC измеряют температуру фазового перехода, когда кристаллическая форма поглощает или высвобождает тепло из-за изменения в кристаллической структуре или плавления кристаллической формы. Для одного и того же кристалла одного и того же соединения погрешность температуры теплового перехода и температуры плавления в последовательных анализах обычно находится в пределах приблизительно 5°C, обычно в пределах приблизительно 3°C или в пределах приблизительно 2°C, и заданный пик DSC или температура плавления соединения, когда на нее ссылаются, означает пик DSC или температуру плавления ± 5°C. DSC представляет собой вспомогательный способ для идентификации различных кристаллов. Различные кристаллические морфологии могут быть идентифицированы по их различным характеристическим температурам фазового перехода. Следует отметить, что для смеси ее пик DSC или температура плавления может варьировать в широком диапазоне. Кроме того, температура плавления зависит от скорости нагревания.
Определения и описание
При использовании в описании и формуле изобретения настоящей заявки следующие термины, если не указано иное, имеют указанные значения.
«Млекопитающее» включает человека, домашних животных, таких как лабораторные млекопитающие и домашние питомцы (например, кошка, собака, свинья, корова, овца, коза, лошадь, кролик), и неодомашненные млекопитающие, такие как дикие млекопитающие.
Термин «фармацевтическая композиция» относится к составу соединения, раскрытого в данном документе, со средой-носителем, обычно признаваемой в данной области для доставки биологически активного соединения млекопитающему, такому как человек. Среда-носитель включает все фармацевтически приемлемые носители для ее применения. Фармацевтическая композиция способствует введению соединения в организм.
Термин «терапевтически эффективное количество» относится к количеству лекарственного средства или лекарственного препарата, которое является достаточным для обеспечения необходимого эффекта, но является нетоксичным. Определение эффективного количества различается в зависимости от индивидуума, в зависимости от возраста и общего состояния здоровья субъекта, а также в зависимости от конкретного активного вещества. Подходящее эффективное количество в каждом случае может быть определено специалистами в данной области техники путем проведения обычных тестов.
Термин «фармацевтически приемлемые носители» относится к носителям, которые вводят вместе с активным ингредиентом, которые не вызывают значительного раздражающего воздействия на организм и не ухудшают биологическую активность и свойства активного соединения. Для дополнительной информации касательно носителей ссылка может быть сделана на Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание которого включено в данный документ посредством ссылки.
При использовании в данном документе, если не указано иное, термины «содержать», «содержит» и «содержащий» или их эквиваленты являются открытыми утверждениями и означают, что элементы, компоненты и стадии, которые не определены, могут быть включены в дополнение к перечисленным.
Все патенты, заявки на патенты и другие указанные публикации специально включены в данный документ посредством ссылки с целью описания и раскрытия. Эти публикации представлены только из-за того, что они были раскрыты до даты подачи настоящей заявки. Все утверждения относительно дат этих документов или описания касательно содержания этих документов основаны на информации, доступной заявителю, и не являются признанием в отношении правильности дат этих документов или содержания этих документов. Кроме того, в любой стране или регионе любая ссылка на эти публикации в данном документе не должна толковаться как признание того, что публикации являются частью общепризнанных знаний в данной области.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1 представлена порошковая рентгеновская дифрактограмма (XRPD) кристалла формы A гидрохлорида формулы II в примере 2.
На фиг. 2 представлена термограмма дифференциальной сканирующей калориметрии (DSC) кристалла формы A гидрохлорида формулы II в примере 2.
На фиг. 3 представлена термограмма термогравиметрического анализа (TGA) кристалла формы A гидрохлорида формулы II в примере 2.
На фиг. 4 представлена порошковая рентгеновская дифрактограмма (XRPD) кристалла формы В гидрохлорида формулы II в примере 6.
На фиг. 5 представлен график динамической сорбции паров (DVS) кристалла формы A гидрохлорида формулы II в примере 2.
ПОДРОБНОЕ ОПИСАНИЕ
Следующие конкретные примеры предоставлены для того, чтобы позволить специалистам в данной области техники более четко понять и осуществлять на практике изложенное в настоящей заявке. Эти конкретные примеры не должны рассматриваться как ограничивающие объем настоящей заявки, а только как иллюстрирующие описание и представляющие настоящую заявку.
Все операции, включающие легко окисляемые или гидролизуемые материалы, проводили в атмосфере азота. Если не указано иное, все исходные материалы, используемые в настоящей заявке, были коммерчески доступны и использовались без дополнительной очистки. Все растворители, используемые в настоящей заявке, являются коммерчески доступными и используются без специальной обработки. Соединения названы либо вручную, либо с применением программного обеспечения ChemDraw®, и для коммерчески доступных соединений приводятся названия согласно каталогу производителя.
В настоящей заявке применяют следующие сокращения: DMAP представляет собой 4-диметиламинопиридин; Pd2(dba)3 представляет собой трис(дибензилиденацетон)дипалладий; Xantphos представляет собой 4,5-бис(дифенилфосфино)-9,9-диметилксантен; Pd(dppf)Cl2 представляет собой дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия.
Пример 1. Получение соединения формулы I

Стадия 1
Гидрид натрия (19,24 г, 480,99 ммоль, чистота: 60%) добавляли в раствор 1a (59,00 г, 436,59 ммоль) в тетрагидрофуране (600 мл) при 0°C в атмосфере азота. После перемешивания в течение 30 мин в реакционную смесь добавляли метилтрифлат (93,14 г, 567,57 ммоль, 62,09 мл), а затем перемешивали при 15°C в течение 2 ч. После завершения реакции в реакционную смесь добавляли насыщенный раствор хлорида аммония (1 л) с гашением реакции, а затем добавляли этилацетат для экстракции (500 мл × 3). Органическую фазу промывали насыщенным солевым раствором (1 л) и высушивали над безводным сульфатом натрия. После фильтрации и концентрирования остаток подвергали колоночной хроматографии с получением 1b.
1H-ЯМР (400 МГц, CDCl3) δ = 7,22-7,08 (m, 2H), 7,03 (br d, J=3,0 Гц, 1H), 6,86-6,73 (m, 1H), 6,58 (d, J=2,5 Гц, 1H), 3,81 (s, 3H).
Стадия 2
N-Бромсукцинимид (65,63 г, 368,72 ммоль) добавляли в раствор 1b (55,00 г, 368,72 ммоль) в диметилсульфоксиде (400 мл) в атмосфере азота и реакционную смесь затем перемешивали при 20°C в течение 1 ч. После еще одного добавления N-бромсукцинимида (65,63 г, 368,72 ммоль) реакционную смесь нагревали до 60°C и перемешивали в течение 10 ч. После завершения реакции реакционную смесь выливали в воду (6 л) и фильтровали. Осадок на фильтре растворяли в ацетоне (2 л) и фильтровали и полученный осадок на фильтре промывали ацетоном (500 мл). После концентрирования фильтрата осадок подвергали колоночной хроматографии с получением 1c.
1H-ЯМР (400 МГц, DMSO-d6) δ = 7,72 (dt, J=5,8, 8,2 Гц, 1H), 6,99 (d, J=7,8 Гц, 1H), 6,93 (t, J=8,8 Гц, 1H), 3,15 (s, 3H).
Стадия 3
N-Бромсукцинимид (40,04 г, 224,95 ммоль) добавляли в раствор 1c (31,0 г, 173,04 ммоль) в ацетонитриле (300 мл) и воде (600 мл) в атмосфере азота и реакционную смесь затем перемешивали при 15°C в течение 16 ч. После завершения реакции реакционную смесь фильтровали и осадок на фильтре промывали водой (300 мл) и высушивали с получением 1d.
1H-ЯМР (400 МГц, DMSO-d6) δ = 7,99 (dd, J=7,3, 8,3 Гц, 1H), 6,98 (d, J=8,5 Гц, 1H), 3,14 (s, 3H).
Стадия 4
Триметилсилилдиазометан (2 моль/л, 65,11 мл) добавляли по каплям в раствор 1d (32,00 г, 124,01 ммоль) и триэтиламина (25,1 г, 248,02 ммоль) в этаноле (300 мл) при 0°C в атмосфере азота и реакционную смесь затем перемешивали при 0-15°C в течение 1 ч. После завершения реакции реакционную смесь концентрировали. Остаток суспендировали с этилацетатом (500 мл) и фильтровали, и осадок на фильтре высушивали с получением 1e.
1H-ЯМР (400 МГц, DMSO-d6) δ= 10,17 (br s, 1H), 7,65 (dd, J=7,5, 9,0 Гц, 1H), 7,30 (d, J=9,0 Гц, 1H), 7,11 (s, 1H), 3,69 (s, 3H).
Стадия 5
Ангидрид трифторметансульфоновой кислоты (13,48 г, 47,78 ммоль) добавляли по каплям в раствор 1e (10,0 г, 36,76 ммоль), пиридина (8,72 г, 110,27 ммоль) и DMAP (449,04 мг, 3,68 ммоль) в дихлорметане (200 мл) при 0°C в атмосфере азота и реакционную смесь затем перемешивали при 15°C в течение 1 ч. После завершения реакции реакционную смесь гасили водой (300 мл), а затем доводили до pH 5 с помощью 1 н. хлористоводородной кислоты. Органическую фазу промывали насыщенным раствором хлорида натрия (250 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением 1f.
1H-ЯМР (400 МГц, CDCl3) δ = 7,93 (s, 1H), 7,82-7,75 (m, 1H), 7,11 (d, J=9,0 Гц, 1H), 3,79 (s, 3H).
Стадия 6
1f (10,00 г, 24,74 ммоль), 1 г (6,38 г, 27,21 ммоль), Pd2(dba)3 (2,27 г, 2,47 ммоль), Xantphos (2,15 г, 3,71 ммоль) и карбонат цезия (16,12 г, 49,48 ммоль) добавляли в тетрагидрофуран (200 мл) в атмосфере азота и реакционную смесь затем перемешивали при 50°C в течение 16 ч. После завершения реакции реакционную смесь добавляли в воду (200 мл) и фильтровали, и осадок на фильтре суспендировали с этилацетатом (100 мл). После фильтрации твердое вещество высушивали с получением 1h.
1H-ЯМР (400 МГц, DMSO-d6) δ = 9,07-8,76 (m, 2H), 8,00 (br d, J=2,3 Гц, 1H), 7,68-7,40 (m, 2H), 7,32 (br dd, J=9,0, 13,3 Гц, 2H), 4,71-4,39 (m, 4H), 3,75 (s, 3H), 3,52-3,39 (m, 1H), 3,14 (br s, 4H), 2,42 (br s, 4H).
Стадия 7
1h (9,00 г, 18,43 ммоль), карбонат калия (6,37 г, 46,07 ммоль), Pd(dppf)Cl2 (1,08 г, 1,47 ммоль) и 1i (5,36 г, 27,64 ммоль) добавляли в 1,4-диоксан (160 мл) и воду (40 мл) в атмосфере азота и реакционную смесь затем перемешивали при 110°C в течение 16 ч. После завершения реакции реакционную смесь охлаждали для осаждения твердого вещества, а затем фильтровали, и осадок на фильтре промывали водой (200 мл) и этилацетатом (100 мл), и высушивали с получением соединения формулы I.
1H-ЯМР (400 МГц, DMSO-d6) δ = 13,08 (br s, 1H), 9,04 (br s, 1H), 8,78 (br s, 1H), 8,16-7,70 (m, 3H), 7,57-7,23 (m, 3H), 6,73 (br s, 1H), 4,74-4,37 (m, 4H), 3,79 (br s, 3H), 3,56 (br s, 2H), 3,14 (br s, 3H), 2,42 (br s, 4H).
Пример 2. Получение кристалла формы A гидрохлорида формулы II
Деионизированную воду (5440 мл) добавляли в реакционную емкость объемом 50 л, а затем перемешивали механически со скоростью 200 об/мин. После нагревания реакционной емкости до внутренней температуры 35-38°C соединение формулы I (1360 г) порциями добавляли и реакционную смесь перемешивали в течение 0,5 ч. с получением желтой суспензии. Муравьиную кислоту (2720 мл) добавляли по каплям в реакционную емкость в течение периода приблизительно 1 ч., пока раствор не становился полностью прозрачным, и полученную реакционную смесь затем перемешивали в течение 1 ч. 1 моль/л приготовленный водный раствор хлористоводородной кислоты (4290 мл) затем добавляли по каплям в реакционную емкость в течение периода приблизительно 2 ч. и реакционную смесь, которая все еще была желтой прозрачной жидкостью, перемешивали в течение ночи без изменения условий. Этанол (24900 мл) добавляли по каплям в реакционную емкость в течение периода приблизительно 5 ч., когда в нее добавляли приблизительно 7000 мл этанола, ярко-желтое твердое вещество начинало осаждаться, и добавление по каплям продолжали, пока твердое вещество полностью не осаждалось. Реакционную смесь перемешивали в течение ночи без изменения условий. Реакционную смесь затем фильтровали и осадок на фильтре высушивали под вакуумом до постоянного веса с получением кристалла формы A гидрохлорида формулы II (1220 г, чистота: 99,79%).
Пример 3. Получение кристалла формы A гидрохлорида формулы II
Деионизированную воду (2 мл) добавляли в реакционную колбу объемом 100 мл. После нагревания реакционной колбы до внутренней температуры 35-38°C соединение формулы I (0,6 г) добавляли и реакционную смесь перемешивали в течение 0,5 ч. с получением желтой суспензии. Муравьиную кислоту (1 мл) добавляли по каплям в реакционную колбу в течение периода приблизительно 5 мин, пока раствор не становился полностью прозрачным, и полученную реакционную смесь затем перемешивали в течение 1 ч. 1 моль/л приготовленный водный раствор хлористоводородной кислоты (1,26 мл) затем добавляли по каплям в реакционную колбу в течение периода приблизительно 5 мин и реакционную смесь, которая все еще была желтой прозрачной жидкостью, перемешивали в течение ночи без изменения условий. Этанол (8 мл) добавляли по каплям в реакционную колбу в течение периода приблизительно 15 мин, когда в нее добавляли приблизительно 5 мл этанола, ярко-желтое твердое вещество начинало осаждаться, и добавление по каплям продолжали, пока твердое вещество полностью не осаждалось. Реакционную смесь перемешивали в течение 4 ч. без изменения условий. Реакционную смесь затем фильтровали и осадок на фильтре высушивали под вакуумом до постоянного веса с получением кристалла формы A гидрохлорида формулы II (0,55 г).
Пример 4. Получение кристалла формы A гидрохлорида формулы II
Деионизированную воду (2 мл) добавляли в реакционную колбу объемом 100 мл. После нагревания реакционной колбы до внутренней температуры 35-38°C соединение формулы I (0,6 г) добавляли и реакционную смесь перемешивали в течение 0,5 ч. с получением желтой суспензии. Муравьиную кислоту (1 мл) добавляли по каплям в реакционную колбу в течение периода приблизительно 5 мин, пока раствор не становился полностью прозрачным, и полученную реакционную смесь затем перемешивали в течение 1 ч. 1 моль/л приготовленный водный раствор хлористоводородной кислоты (2,52 мл) затем добавляли по каплям в реакционную колбу в течение периода приблизительно 5 мин и реакционную смесь, которая все еще была желтой прозрачной жидкостью, перемешивали в течение ночи без изменения условий. Этанол (8 мл) добавляли по каплям в реакционную колбу в течение периода приблизительно 15 мин, когда в нее добавляли приблизительно 5 мл этанола, ярко-желтое твердое вещество начинало осаждаться, и добавление по каплям продолжали, пока твердое вещество полностью не осаждалось. Реакционную смесь перемешивали в течение 4 ч. без изменения условий. Реакционную смесь затем фильтровали и осадок на фильтре высушивали под вакуумом до постоянного веса с получением кристалла формы A гидрохлорида формулы II (0,55 г).
Пример 5. Получение кристалла формы A гидрохлорида формулы II
Этилацетат (2 мл) добавляли в реакционную колбу объемом 100 мл. После нагревания реакционной колбы до внутренней температуры 35-38°C соединение формулы I (1,0 г) добавляли и реакционную смесь перемешивали в течение 0,5 ч. с получением желтой суспензии. Муравьиную кислоту (3 мл) добавляли по каплям в реакционную колбу в течение периода приблизительно 15 мин, пока раствор не становился полностью прозрачным, и полученную реакционную смесь затем перемешивали в течение 1 ч. 35% водный раствор хлористоводородной кислоты (0,26 мл) и этилацетат (2,0 мл) затем добавляли по каплям в реакционную колбу в течение периода приблизительно 5 мин и реакционную смесь, которая все еще была желтой прозрачной жидкостью, перемешивали в течение ночи без изменения условий. Этанол (20 мл) добавляли по каплям в реакционную колбу в течение периода приблизительно 15 мин, когда в нее добавляли приблизительно 15 мл этанола, ярко-желтое твердое вещество начинало осаждаться, и добавление по каплям продолжали, пока твердое вещество полностью не осаждалось. Реакционную смесь перемешивали в течение 4 ч. без изменения условий. Реакционную смесь затем фильтровали и осадок на фильтре высушивали под вакуумом до постоянного веса с получением кристалла формы A гидрохлорида формулы II (0,95 г).
Пример 6. Получение кристалла формы B гидрохлорида соединения формулы I
Соединение формулы I (500 мг) добавляли в стеклянный флакон объемом 40 мл, а затем добавляли этанол (35 мл). После перемешивания в течение 2 ч. при 40°C с магнитной мешалкой, в реакционную смесь добавляли 1,2 моль/л разбавленную хлористоводородную кислоту (3 мл), 10-кратно разбавленную водой. Полученную реакционную смесь перемешивали в течение ночи при 40°C с магнитной мешалкой. Реакционную смесь центрифугировали и твердое вещество высушивали в вакуумной печи в течение ночи с получением кристалла формы B гидрохлорида формулы II.
Пример 7. Получение кристалла формы B гидрохлорида соединения формулы I
Соединение формулы I (500 мг) добавляли в стеклянный флакон объемом 40 мл, а затем добавляли смешанный раствор из этанола (29,2 мл) и уксусной кислоты (5,8 мл). После перемешивания в течение 2 ч. при 40°C с магнитной мешалкой, в реакционную смесь добавляли 1,2 моль/л разбавленную хлористоводородную кислоту (3 мл), 10-кратно разбавленную водой. Полученную реакционную смесь перемешивали в течение ночи при 40°C с магнитной мешалкой. Реакционную смесь центрифугировали и твердое вещество высушивали в вакуумной печи в течение ночи с получением кристалла формы B гидрохлорида формулы II.
Пример 8. Получение кристалла формы B гидрохлорида соединения формулы I
Соединение формулы I (500 мг) добавляли в стеклянный флакон объемом 40 мл, а затем добавляли смешанный раствор из ацетона (29,2 мл) и уксусной кислоты (5,8 мл). После перемешивания в течение 2 ч. при 40°C с магнитной мешалкой, в реакционную смесь добавляли 1,2 моль/л разбавленную хлористоводородную кислоту (3 мл), 10-кратно разбавленную водой. Полученную реакционную смесь перемешивали в течение ночи при 40°C с магнитной мешалкой. Реакционную смесь центрифугировали и твердое вещество высушивали в вакуумной печи в течение ночи с получением кристалла формы B гидрохлорида формулы II.
Экспериментальный пример 1. Исследование стабильности кристалла формы A гидрохлорида формулы II
Согласно «Guidelines for the Stability Test of APIs and Preparations» (General Principles 9001 of the Four Parts of the Chinese Pharmacopoeia, 2015 Edition) стабильность кристалла формы A соединения формулы II исследовали при высокой температуре (60°C, на воздухе), высокой влажности (комнатная температура/относительная влажность 92,5%, на воздухе) и освещении (полная освещенность 1,2×106 люкс•ч./энергия в ближней УФ-области 200 Вт•ч./м2, на воздухе).
Кристалл формы A гидрохлорида формулы II (5 мг) взвешивали, помещали на дно стеклянной бутылки для образцов и распределяли тонким слоем. Флаконы, в которые образцы помещали при высокой температуре и высокой влажности, закрывали алюминиевой фольгой, и небольшие отверстия обеспечивали в алюминиевой фольге, чтобы убедиться, что образцы контактировали в достаточной степени с атмосферным воздухом. Флакон, в который образец помещали при сильном облучении светом, помещали открытым без накрытия алюминиевой фольгой. Образцы, помещенные в различные условия, отбирали и тестировали с помощью спектра XRPD на 5 день и 10 день. Результаты теста сравнивали с результатами исходного теста в 0 день, и результаты теста показаны в таблице 3 ниже.
Таблица 3. Экспериментальные результаты касательно стабильности кристалла формы A гидрохлорида формулы II
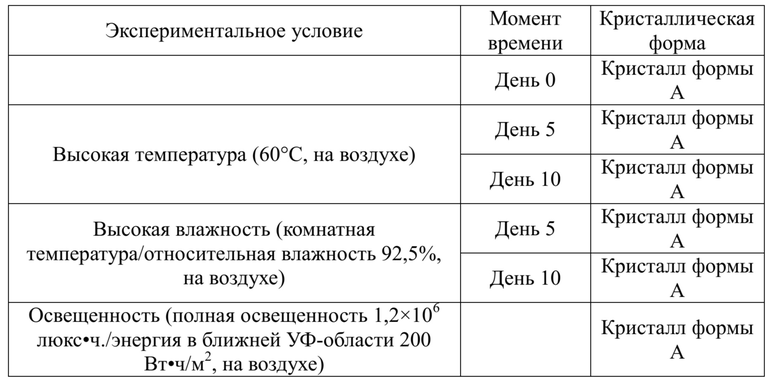
Результаты показали, что кристалл формы A гидрохлорида формулы II имеет хорошую стабильность при условиях высокой температуры, высокой влажности и освещенности.
Экспериментальный пример 2. Исследование гигроскопичности кристалла формы A гидрохлорида формулы II
Прибор: SMS DVS Advantage.
Способ: помещение 10-15 мг кристалла формы A гидрохлорида формулы II на поднос для образца для DVS для тестирования.
Параметры DVS являются следующими.
Температура: 25°C.
Критерий равновесия: dm/dt = 0,01%/мин (самый короткий: 10 мин, самый длинный: 180 мин).
Высушивание: высушивание при 0% RH в течение 120 мин.
Тестовый градиент RH (%): 10%.
Диапазон тестового градиента RH (%): 0%-90%-0%.
Результаты: график DVS кристалла формы A гидрохлорида формулы II показан на фиг. 5, где ΔW = 1,371%.
Вывод: кристалл формы A гидрохлорида формулы II является менее гигроскопичным при 25±1°C и 80±2% RH.
Экспериментальный пример 3. Исследование на стабильность кристалла формы A гидрохлорида формулы II в органическом растворителе
Кристалл формы A гидрохлорида формулы II (60 мг) взвешивали и помещали в стеклянный флакон объемом 8 мл. После добавления метанола (4 мл) стеклянный флакон помещали на магнитную мешалку и смесь перемешивали при 20°C и 50°C, в каждом случае в течение 24 ч. Суспензию центрифугировали с удалением органического растворителя, и полученное твердое вещество высушивали под вакуумом, и затем тестировали с помощью спектра XRPD, который сравнивали со спектром XRPD кристалла формы A гидрохлорида формулы II.
Согласно вышеуказанному способу этанол, этилацетат, ацетон, ацетонитрил, тетрагидрофуран, 1,4-диоксан и н-гексан использовали в качестве растворителей для проведения параллельных тестов.
Результаты показали, что спектр XRPD кристалла формы A гидрохлорида формулы II не изменяется после его суспендирования в вышеуказанном растворителе в течение 24 ч., что указывает на то, что никакого превращения кристаллической формы не происходило для кристалла формы A гидрохлорида формулы II в исследуемой системе растворителей, и его кристаллическая форма является относительно стабильной.
Экспериментальный пример 4. Тестирование in vitro ингибирования киназы Syk соединением формулы I
4.1 Цель эксперимента: обнаружить взаимодействие между субстратом и ферментом с помощью технологии гомогенной флуоресценции с временным разрешением (HTRF) и оценить ингибирование соединением тирозинкиназы (Syk) путем получения значения концентрации полумаксимального ингибирования (IC50) соединения в качестве показателя.
4.2 Экспериментальные материалы:
Тирозинкиназа (Invitrogen, PV3857)
Дитиотреитол (DTT) (Sigma № 43815)
Аденозинтрифосфат (АТФ) (Sigma № A7699)
Хлорид магния (MgCl2) (Sigma № 63020)
Хлорид марганца (MnCl2) (Sigma № M1787)
Этилендиаминтетрауксусная кислота (EDTA) (Invitrogen № 15575-020)
Буфер на основе 4-гидроксиэтилпиперазинэтансульфоновой кислоты (буфер HEPES) (Invitrogen № 15630-080)
Набор для измерения активности тирозинкиназы HTRF® KinEASE™ (Cisbio №62TK0PEC, 20000 тестов):
384-луночный белый полистирольный планшет с небольшим объемом (Greiner № 784075)
384-луночный микропланшет (Greiner № 781946)
Центрифуга (Eppendorf № 5810R)
Пипеточный дозатор (Eppendorf)
Пипетка (Greiner)
Автоматический пипеточный дозатор (Eppendorf)
Многоканальный автоматический дозатор Multidrop
POD 810 Plate Assembler, полностью автоматизированная система предварительной обработки микропланшетов
Envision Reader, мультифункциональный микропланшет-ридер
4.3 Экспериментальные процедуры и способы
a) Разведение и нанесение соединения
1) Порошок соединения формулы I взвешивали и растворяли в некотором количестве диметилсульфоксида, и исходная концентрация составляла 10 мМ.
2) Соединение разводили до концентрации 0,74 мМ и добавление образца проводили с помощью полностью автоматизированной системы предварительной обработки микропланшетов в количестве 135 нл на лунку. Исходная концентрация соединения составляла 10 мкМ, и 11 точек концентрации получали после 3-кратного градиентного разбавления со снижением концентрации.
b) Реакция фермента с субстратом
1) Подготовка для разбавления экспериментального буфера. 5x буфер HTRF в наборе разводили до 1x, и конкретное количество DTT и раствора MgCl2 добавляли согласно руководству для набора для дальнейшего использования.
2) Реакционный раствор тирозиназы готовили с 1x буфером HTRF, и конечная концентрация тирозинкиназы в реакционном растворе составляла 0,0156 нг/мкл.
3) Смешанный раствор тирозинкиназы-субстрата-биотина/ATФ готовили, и конечная концентрация субстрата составляла 0,2 мкМ, и концентрация ATФ составляла 2 мкМ.
4) Раствор тирозиназы и смешанный раствор тирозинкиназы-субстрата-биотина/ATФ добавляли в микропланшет с соединением формулы I в количестве 5 мкл на лунку с помощью автоматического дозатора Multidrop, и смесь инкубировали при 23°C в течение 1 ч.
c) Обнаружение
1) Раствор EDTA (13,33 мл) добавляли в буфер обнаружения в наборе, а затем конкретное количество меченного радиоактивным элементом (Eu) антитела и стрептавидин XL-665 добавляли согласно руководству для набора для получения раствора для обнаружения.
2) Раствор для обнаружения (10 мкл) добавляли в каждую лунку вышеуказанного микропланшета с помощью автоматического дозатора Multidrop, и полученную смесь инкубировали при 23°C в течение 1 ч. для остановки реакции фермента со смешанным раствором субстрата.
3) После центрифугирования полученную надосадочную жидкость считывали на мультифункциональном микропланшет-ридере.
d) Анализ данных
Данные анализировали с помощью XL-Fit для расчета значения IC50 для соединения формулы I.
Экспериментальный пример 5. Тестирование in vitro ингибирования фосфорилирования AKT соединением формулы I
5.1 Цель эксперимента: обнаружить фосфорилирование протеинкиназы AKT в клетках с помощью иммуноферментного твердофазного анализа (ELISA) и оценить ингибирование соединением фосфорилирования AKT путем получения значения концентрации полумаксимального ингибирования (IC50) соединения в качестве показателя.
5.2 Экспериментальные материалы
Клеточная линия: Клеточная линия Ramos
Среда для культивирования клеток (RPMI1640, Invitrogen № 22400-105; 10% эмбриональная бычья сыворотка, Gibco № 10099-141; L-глутамин 1x, Gibco № 25030-081)
Среда для эксперимента (без сыворотки, RPMI 1640, Invitrogen № 22400-105; L-глутамин 1x, Gibco № 25030)
Буфер для лизиса (Трис-HCl, Invitrogen 15567-1000 мл; NaCl, внутренний; дезоксихолат натрия, Sigma 30970-25G; октилфениловый эфир полиэтиленгликоля, Sigma T9284-100 мл; додецилсульфонат натрия, Sigma L3771; EDTA, Invitrogen 15575-038-100 мл; ультрачистая вода, MilliQ)
Ингибитор протеазы (Roche, 4693159001-30/BOX)
Смесь 2 ингибиторов фосфатазы (Sigma, P5726-5 ML)
Смесь 3 ингибиторов фосфатазы (Sigma, P0044-5 ML)
Антитело козы к иммуноглобулину M человека (F(ab')2 козы к IgM человека) (JacksonImmuno Research-109-006-129)
Набор для обнаружения фосфорилирования AKT (Phospho-AKT 1/2/3(ser473)) (TGR Bioscience, EKT002)
10x сбалансированный солевой раствор Хэнкса (Gibco № 14065-056)
96-луночный планшет для клеток (Greiner № 655090)
Планшет для разбавления соединения с V-образными лунками (Axygen № WITP02280)
Инкубатор с CO2 (Thermo № 371)
Центрифуга (Eppendorf № 5810R)
Устройство для подсчета клеток Vi-cell (Beckman Coulter)
Пипеточный дозатор (Eppendorf)
Пипетка (Greiner)
Автоматический пипеточный дозатор (Eppendorf)
Мультифункциональный микропланшет-ридер (Envision Reader)
5.3 Экспериментальные процедуры и метод
a) Высевание клеток (клетки Ramos)
1) Среду для культивирования клеток предварительно нагревали на водяной бане при 37°C, и суспендированную культуру клеток Ramos пипетировали и центрифугировали в течение 5 мин при 1000 об/мин.
2) После отбора пипеткой надосадочной жидкости после центрифугирования предварительно нагретую среду для культивирования клеток (10 мл) добавляли в центрифужную пробирку. Клетки повторно суспендировали пипетированием и затем полученную снова суспензию клеток Ramos (1 мл) пипетировали и подвергали подсчету клеток с помощью устройства для подсчета клеток Vi-cell.
3) Полученную снова суспензию клеток Ramos разводили средой для культивирования клеток до плотности клеток 5×106 клеток/мл и разбавленные клетки добавляли в 96-луночный планшет для культивирования клеток (100 мкл/лунку) с помощью автоматического пипеточного дозатора; планшет для культивирования клеток инкубировали в течение ночи в инкубаторе при 37°C, 5% CO2.
b) Клеточное истощение
После культивирования в течение ночи высеянные клетки центрифугировали при 1000 об/мин в течение 5 мин на следующий день. Исходную среду для культивирования клеток отбирали пипеткой, а затем добавляли среду без сыворотки для эксперимента. Планшет для культивирования клеток помещали в инкубатор при 37°C, 5% CO2 и клетки подвергали истощению в течение ночи.
c) Получение и нанесение тестового образца
1) Соединение формулы I в виде тестового образца растворяли в диметилсульфоксиде с получением раствора соединения формулы I с исходной концентрацией 5 мМ; трехкратное градиентное разведение раствора проводили с помощью планшета для разбавления соединения с V-образными лунками с получением 10 точек концентрации.
2) В другой новый планшет для разбавления соединения с V-образными лунками добавляли среду без сыворотки для эксперимента в количестве 198 мкл на лунку, а затем добавляли в лунки раствор с исходной концентрацией (2 мкл) и каждый раствор соединения после трехкратного градиентного разведения (2 мкл), соответственно, и смеси хорошо перемешивали с помощью автоматического пипеточного дозатора; в это время соединение разводили 100-кратно, причем максимальная концентрация разбавленного соединения формулы I в лунке составляла 50 мкМ.
3) Каждый разбавленный раствор соединения, полученный на стадии 2), добавляли в планшет для культивирования клеток, содержимое которого подвергали истощению в течение ночи на b), в количестве 25 мкл на лунку, причем среда для культивирования клеток в каждой лунке составляла 100 мкл, и каждый раствор соединения был 5-кратно разбавлен; в это время максимальная концентрация разбавленного соединения формулы I в лунке составляла 10 мкМ, и в других лунках были разбавленные растворы соединения формулы I в 10 точках концентрации, полученные 3-кратным градиентным разбавлением.
4) Содержимое планшета для культивирования клеток со стадии 3) центрифугировали при 1000 об/мин в течение 1 мин, а затем помещали в инкубатор при 37°C, 5% CO2, для обеспечения протекания реакции соединения в течение 1 ч.
d) Стимуляция стимулирующим фактором
1) Две пробирки 1x сбалансированного солевого раствора получали путем разбавления 10x сбалансированного солевого раствора до 1x сбалансированного солевого раствора с помощью дважды дистиллированной воды, и соответственно помещали в термостат при 37°C и холодильник при 4°C для дальнейшего использования.
2) Пробирку со смешанным раствором для лизиса получали и помещали в холодильник при 4°C для дальнейшего использования. Состав являлся следующим: 1 таблетка ингибитора протеазы + 100 мкл смеси 2 ингибитора фосфатазы + 100 мкл смеси 3 ингибитора фосфатазы + 10 мл буфера для лизиса.
3) Антитело козы к иммуноглобулину M человека (F(ab')2 козы к IgM человека) (1,2 мг/мл) разводили до 60 мкг/мл с помощью 1x сбалансированного солевого раствора, предварительно нагретого при 37°C.
4) Через один час обработки клеток Ramos соединением разведенное антитело козы к иммуноглобулину M человека (F(ab')2 козы к IgM человека) (25 мкл) добавляли на лунку, причем рабочая концентрация IgM в это время составляла 10 мкг/мл.
5) Клетки стимулировали с помощью IgM в течение 10 мин, а затем центрифугировали при 4000 об/мин в течение 5 мин для осаждения суспендированных клеток на дне 96-луночного планшета. Жидкость в 96-луночном планшете несколько сливали без выливания суспедированных клеток, и остаточную жидкость абсорбировали с помощью бумажного полотенца.
6) Стимуляцию клеток с помощью IgM завершали добавлением предварительно охлажденного (4°C) 1x сбалансированного солевого раствора (250 мкл) в каждую лунку и центрифугированием смеси при 4000 об/мин в течение 5 мин.
e) Получение клеточного лизата
1) Жидкость в 96-луночном планшете аккуратно выливали и остаточную жидкость абсорбировали с помощью бумажного полотенца; смешанный раствор для лизиса (100 мкл) добавляли в каждую лунку и смесь встряхивали на шейкере при 4°C в течение 1 ч. для лизиса клеток.
2) После лизиса клеток в течение 1 ч. клетки центрифугировали при 4000 об/мин в течение 5 мин при 4°C, и надосадочную жидкость аккуратно отбирали пипеткой, таким образом получая клеточный лизат.
f) Иммуноферментный твердофазный анализ (Elisa)
1) 96-луночный планшет для Elisa в наборе для обнаружения фосфорилированной AKT доводили до комнатной температуры и добавляли в него клеточный лизат в количестве 50 мкл на лунку.
2) Реагент, представляющий собой иммобилизированное антитело, и реагент, представляющий собой детекторное антитело, в наборе смешивали в отношении 1:1, а затем смесь добавляли в 96-луночный планшет для Elisa в количестве 50 мкл на лунку; полученный смешанный раствор клеточного лизата и смеси реагентов, представляющих собой антитела, встряхивали на шейкере при комнатной температуре в течение 1 ч.
3) Промывочный раствор (10x) в наборе разводили до 1x с помощью дважды дистиллированной воды; жидкость из планшета для Elisa выливали и планшет промакивали досуха на впитывающей бумаге; 1x промывочный раствор (200 мкл) добавляли в каждую лунку для промывания, а затем планшет промакивали досуха и процесс повторяли 4 раза.
4) Субстрат 10-ацетил-3,7-дигидроксифеназин (ADHP) (100x) разводили до 1x с помощью разбавителя для ADHP, а затем добавляли в 96-луночный планшет для Elisa в количестве 100 мкл на лунку и смесь встряхивали на шейкере при комнатной температуре в течение 10 мин.
5) Останавливающий раствор (10 мкл) добавляли в каждую лунку и смесь центрифугировали сразу же, встряхивая в течение 5 мин при комнатной температуре, и считывали на мультифункциональном микропланшет-ридере Envision Reader.
g) Анализ данных
Данные анализировали с помощью XL-Fit для расчета значения IC50 для соединения.
Результаты экспериментального примера 4 и экспериментального примера 5 показаны в таблице 4.
Таблица 4
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛ ЦИКЛОПЕПТИДА ВЫСОКОЙ ЧИСТОТЫ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2607083C2 |
| БОРАТНОЕ ПРОИЗВОДНОЕ АЗЕТИДИНА | 2019 |
|
RU2802287C2 |
| КРИСТАЛЛ ПИРРОЛОПИРИМИДИНА ДЛЯ ПОЛУЧЕНИЯ JAK-ИНГИБИТОРА | 2017 |
|
RU2746045C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ ТРИАЗОЛОПИРИМИДИНА | 2017 |
|
RU2754856C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТНОГО ИНГИБИТОРА JAK-КИНАЗЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2716260C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО ТИОФЕНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2022 |
|
RU2830948C1 |
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТИАЗОЛЬНОГО ПРОИЗВОДНОГО | 2016 |
|
RU2738937C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНЫХ ДИГИДРОПИРИМИДИНА | 2013 |
|
RU2646599C2 |
| КРИСТАЛЛЫ ПРОИЗВОДНЫХ 6,7-НЕНАСЫЩЕННОГО-7-КАРБАМОИЛМОРФИНАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2607084C2 |

Изобретение относится к гидрохлориду соединения формулы I. Гидрохлорид соединения формулы (I) представляет собой кристалл формы A, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 4,9°, 10,1°, 12,2°, 15,5°, 19,6° и 23,8° на спектре порошковой рентгеновской дифракции, или кристалл формы B, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 5,2°, 10,4°, 14,7°, 15,5° и 25,3° на спектре порошковой рентгеновской дифракции. Изобретение относится к фармацевтической композиции для лечения заболевания, связанного с рецептором Syk, содержащей терапевтически эффективное количество гидрохлорида соединения формулы I по изобретению. Способ получения гидрохлорида соединения формулы I формы А осуществляют путем (1) добавления соединения формулы I в предварительно нагретый растворитель, выбранный из группы, состоящей из воды и этилацетата, при температуре 30-40°C, добавления по каплям другого растворителя, выбранного из группы, состоящей из муравьиной кислоты, уксусной кислоты и пропионовой кислоты, пока раствор не станет прозрачным, и перемешивания при поддержании температуры; (2) добавления по каплям разбавленной хлористоводородной кислоты в раствор из стадии (1), и перемешивания в течение ночи при поддержании температуры, и (3) добавления по каплям растворителя, выбранного из группы, состоящей из метанола и этанола, в раствор из стадии (2), перемешивания с осаждением твердого вещества, фильтрования и высушивания с получением кристалла формы A гидрохлорида соединения формулы I. Способ получения гидрохлорида соединения формулы I формы В осуществляют путем (1) добавления соединения формулы I в растворитель, выбранный из группы, состоящей из метанола, этанола, уксусной кислоты, ацетона или смеси любых двух указанных растворителей, и перемешивания с растворением; (2) добавления разбавленной хлористоводородной кислоты в раствор из стадии (1), и перемешивания в течение ночи, и (3) центрифугирования раствора из стадии (2), и высушивания твердого вещества с получением кристалла формы B гидрохлорида соединения формулы I. Технический результат - гидрохлорид соединения формулы I в кристаллической форме, обладающий высокой стабильностью, небольшой гигроскопичностью и применяемый в получении лекарственного препарата для лечения заболевания, связанного с рецептором Syk. 6 н. и 12 з.п. ф-лы, 5 ил., 4 табл., 13 пр. 
1. Гидрохлорид соединения формулы I,

2. Гидрохлорид соединения формулы I по п. 1, где гидрохлорид представляет собой 1:1 гидрохлорид соединения формулы I.
3. Гидрохлорид соединения формулы I по п. 1 или 2, где гидрохлорид представляет собой кристалл формы A, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 4,9°, 10,1°, 12,2°, 15,5°, 19,6° и 23,8° на спектре порошковой рентгеновской дифракции.
4. Гидрохлорид соединения формулы I по п. 3, где гидрохлорид представляет собой кристалл формы A, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 4,9°, 10,1°, 12,2°, 15,5°, 17,8°, 19,2°, 19,6°, 22,9°, 23,8° и 25,6° на спектре порошковой рентгеновской дифракции.
5. Гидрохлорид соединения формулы I по п. 4, где гидрохлорид представляет собой кристалл формы A, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 4,9°, 9,6°, 10,1°, 12,2°, 15,5°, 16,3°, 17,8°, 19,2°, 19,6°, 20,4°, 22,9°, 23,3°, 23,8°, 25,6°, 26,8°, 27,4°, 29,0° и 36,8° на спектре порошковой рентгеновской дифракции.
6. Гидрохлорид соединения формулы I по п. 5, где гидрохлорид представляет собой кристалл формы A, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 4,9°, 9,6°, 10,1°, 12,2°, 14,4°, 15,5°, 16,3°, 17,3°, 17,8°, 19,2°, 19,6°, 20,4°, 22,9°, 23,3°, 23,8°, 25,6°, 26,8°, 27,4°, 28,3°, 29,0°, 31,2°, 31,6°, 31,9°, 32,3°, 33,0°, 34,3° и 36,8° на спектре порошковой рентгеновской дифракции.
7. Гидрохлорид соединения формулы I по любому из пп. 3-6, где гидрохлорид представляет собой кристалл формы A, характеризующийся наличием пика поглощения при 272°C на термограмме дифференциальной сканирующей калориметрии (DSC).
8. Гидрохлорид соединения формулы I по п. 1 или 2, где гидрохлорид представляет собой кристалл формы B, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 5,2°, 10,4°, 14,7°, 15,5° и 25,3° на спектре порошковой рентгеновской дифракции.
9. Гидрохлорид соединения формулы I по п. 8, где гидрохлорид представляет собой кристалл формы B, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 5,2°, 10,4°, 14,7°, 15,5°, 16,5°, 20,7°, 21,5°, 22,8°, 25,3° и 27,9° на спектре порошковой рентгеновской дифракции.
10. Гидрохлорид соединения формулы I по п. 9, где гидрохлорид представляет собой кристалл формы B, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 5,2°, 10,4°, 14,7°, 15,5°, 16,5°, 17,1°, 17,5°, 20,3°, 20,7°, 21,5°, 22,8°, 23,8°, 24,7°, 25,3°, 27,5°, 27,9° и 31,2° на спектре порошковой рентгеновской дифракции.
11. Гидрохлорид соединения формулы I по п. 10, где гидрохлорид представляет собой кристалл формы B, характеризующийся наличием дифракционных пиков, представленных с помощью значений 2θ, при 5,2°, 10,4°, 13,1°, 14,7°, 15,5°, 16,5°, 17,1°, 17,5°, 20,0°, 20,3°, 20,7°, 21,5°, 22,8°, 23,2°, 23,8°, 24,7°, 25,3°, 25,9°, 26,2°, 27,5°, 27,9°, 28,2°, 29,7°, 30,0°, 30,3°, 31,2°, 31,7°, 32,3°, 34,5°, 34,9° и 36,6° на спектре порошковой рентгеновской дифракции.
12. Фармацевтическая композиция для лечения заболевания, связанного с рецептором Syk, содержащая терапевтически эффективное количество гидрохлорида соединения формулы I по любому из пп. 1-11.
13. Применение гидрохлорида соединения формулы I по любому из пп. 1-11 в получении лекарственного препарата для лечения заболевания, связанного с рецептором Syk.
14. Применение фармацевтической композиции по п. 12 в получении лекарственного препарата для лечения заболевания, связанного с рецептором Syk.
15. Применение по любому из пп. 13 или 14, где заболевание, связанное с рецептором Syk, выбрано из рака и воспалительных заболеваний.
16. Применение по любому из пп. 13-15, где заболевание, связанное с рецептором Syk, выбрано из В-клеточной лимфомы, лимфомы Ходжкина, неходжкинской лимфомы, волосатоклеточного лейкоза, множественной миеломы, хронического гранулоцитарного лейкоза, острого гранулоцитарного лейкоза, хронического лимфолейкоза, острого лимфолейкоза, ревматоидного артрита, аллергического ринита, хронического обструктивного заболевания легких (COPD), синдрома расстройства дыхания у взрослых (ARD), вызванных аллергией воспалительных заболеваний, рассеянного склероза, аутоиммунных заболеваний, острых воспалительных реакций, аллергических расстройств и поликистозных заболеваний почек.
17. Способ получения гидрохлорида соединения формулы I по любому из пп. 3-7, включающий: (1) добавление соединения формулы I в предварительно нагретый растворитель, выбранный из группы, состоящей из воды и этилацетата, при температуре 30-40°C, затем добавление по каплям другого растворителя, выбранного из группы, состоящей из муравьиной кислоты, уксусной кислоты и пропионовой кислоты, пока раствор не станет прозрачным, и перемешивание при поддержании температуры; (2) добавление по каплям разбавленной хлористоводородной кислоты в раствор из стадии (1), и перемешивание в течение ночи при поддержании температуры, и (3) добавление по каплям растворителя, выбранного из группы, состоящей из метанола и этанола, в раствор из стадии (2), перемешивание с осаждением твердого вещества, фильтрование и высушивание с получением кристалла формы A гидрохлорида соединения формулы I.
18. Способ получения гидрохлорида соединения формулы I по любому из пп. 8-11, включающий: (1) добавление соединения формулы I в растворитель, выбранный из группы, состоящей из метанола, этанола, уксусной кислоты, ацетона или смеси любых двух указанных растворителей, и перемешивание с растворением; (2) добавление разбавленной хлористоводородной кислоты в раствор из стадии (1), и перемешивание в течение ночи, и (3) центрифугирование раствора из стадии (2), и высушивание твердого вещества с получением кристалла формы B гидрохлорида соединения формулы I.
| WO 2016097862 A2, 23.06.2016 | |||
| EA 201791369 A1, 31.10.2017 | |||
| WO 2018228475 A1, 20.12.2018 | |||
| WO 2004080463 A1, 23.09.2004 | |||
| WO 2017001733 A1, 05.01.2017 | |||
| O’Brien N.J., Brzozowski M., Wilson, D.J.D., Deady L.W | |||
| & Abbott B.M | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Bioorganic & Medicinal | |||
