Область техники
Настоящее изобретение относится к соли 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (соединение Z) и его полиморфу, а также фармацевтической композиции, содержащей их, к способу получения соли и полиморфа, а также применению таковых при получении фармацевтической композиции.
Предпосылки создания изобретения
В опубликованной международной заявке № WO 2010/066111 раскрывается ряд соединений, обладающих ингибиторной активностью в отношении фосфодиэстеразы типа 5 (ВДЭ5), а также соединений, демонстрирующих чрезвычайно высокую активность и избирательность в отношении фермента ВДЭ5 в скрининговых тестах вне организма, в качестве ингибиторов фермента. Соединение 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (соединение Z) также входит в него и представлено следующей структурной формулой:
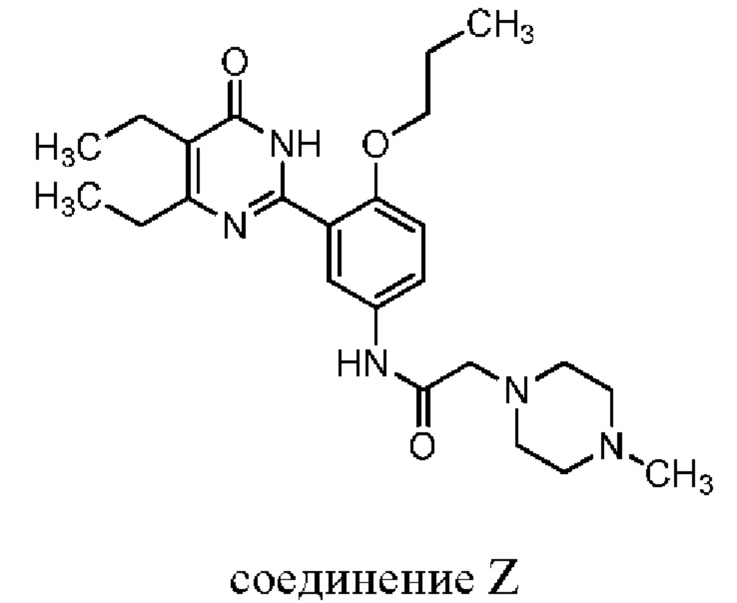
Сущность изобретения:
Авторы изобретения по настоящей заявке обнаружили, что соединение Z в свободном состоянии обладает плохой растворимостью в воде, и, таким образом, его трудно растворить, что отрицательно скажется на технологии получения фармацевтической композиции; с другой стороны, соединение само по себе обладает едким запахом, что приведет к отрицательному воздействию в случае использования в лекарственном препарате для медицинского применения. Таким образом, целью по настоящему изобретению является разработка приемлемой формы соединения Z для получения лекарственного средства, которое должно обладать следующими преимуществами: хорошая стабильность, высокая растворяемость в воде, низкая гигроскопичность, отсутствие неприятного запаха и т.п.
Физические свойства фармацевтических соединений и их солей, а также их кристаллов и аморфных форм оказывают большое влияние на биодоступность лекарственного средства, чистоту активной фармацевтической субстанции, а также рецептуру препарата. Таким образом, при разработке лекарственного средства необходимо изучить, какие виды солей, кристаллических форм и аморфных форм соединения являются предпочтительными. Поскольку вышеупомянутые физические свойства зависят от свойств различных соединений, как правило, трудно предсказать, обладают ли исходные лекарственные соли, кристаллические формы и аморфные вещества надлежащими физическими свойствами, и, следовательно, необходимо проводить различные исследования в отношении каждого соединения.
Таким образом, другой целью настоящего изобретения является обеспечение различных солей, кристаллов и аморфных форм соединения Z.
Авторы настоящего изобретения синтезировали и разделили различные соли и кристаллические формы соединения Z, изучили различные физические и химические свойства таковых, и обнаружили, что соли, кристаллические формы и сольватированные формы соединения Z обладают надлежащими физическими свойствами, которые могут быть использованы в качестве активной фармацевтической субстанции или в качестве промежуточного вещества для получения активной фармацевтической субстанции, в соответствии с сущностью настоящего изобретения.
Настоящее изобретение относится к соли и полиморфу соединения 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (соединение Z), а также фармацевтической композиции и фармацевтической стандартной лекарственной форме, содержащим их. Дополнительно, настоящее изобретение относится к со-кристаллам или комплексу соединения Z, а также к фармацевтической композиции, содержащей их. Настоящее изобретение также относится к способу получения вышеуказанных веществ.
Согласно одному из аспектов настоящего изобретения, оно позволяет получить соединение, представленное формулой (I), и его фармацевтически приемлемый полиморф, сольват, гидрат, со-кристалл, ангидрит или аморфную форму:
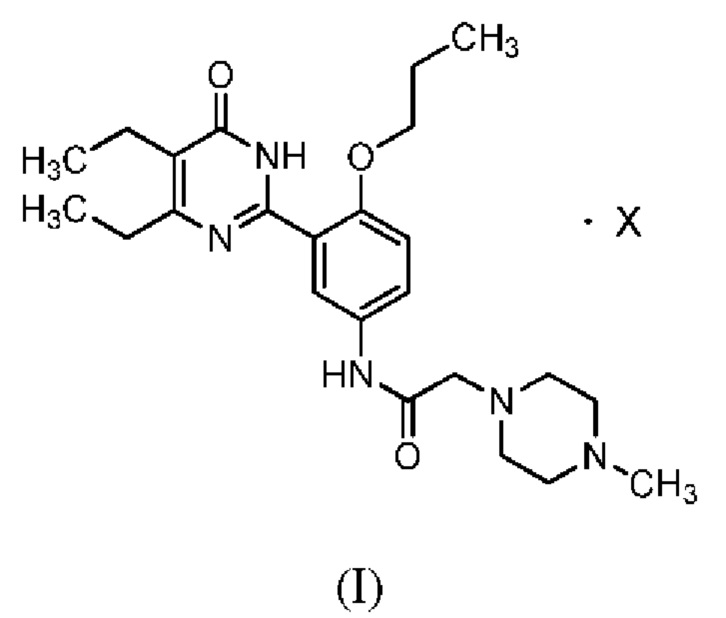
при этом X включает в себя, помимо прочего, органическую или неорганическую кислоту. Например, органическая кислота может включать в себя, помимо прочего, малеиновую кислоту, янтарную кислоту, лимонную кислоту, винную кислоту, фумаровую кислоту, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, малоновую кислоту, щавелевую кислоту, бензойную кислоту, фталевую кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, толуолсульфоновую кислоту, нафталинсульфоновую кислоту, 1,5-нафталиндисульфоновую кислоту, камфорную кислоту, камфорсульфоновую кислоту, салициловую кислоту, ацетилсалициловую кислоту, аспарагиновую кислоту, глутаминовую кислоту, молочную кислоту, глюконовую кислоту, аскорбиновую кислоту, галловую кислоту, миндальную кислоту, яблочную кислоту, сорбиновую кислоту, трифторуксусную кислоту, таурин, гомотаурин, 2-гидроксиэтансульфоновую кислоту, коричную кислоту, муциновую кислоту; а неорганическая кислота может включать в себя, в числе прочего, соляную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, перхлорную кислоту; и другие подобные протонсодержащие кислоты.
при этом, X, предпочтительно, является малеиновой кислотой, янтарной кислотой, лимонной кислотой, винной кислотой, фумаровой кислотой, муциновой кислотой, уксусной кислотой, метансульфоновой кислотой, соляной кислотой, азотной кислотой или серной кислотой.
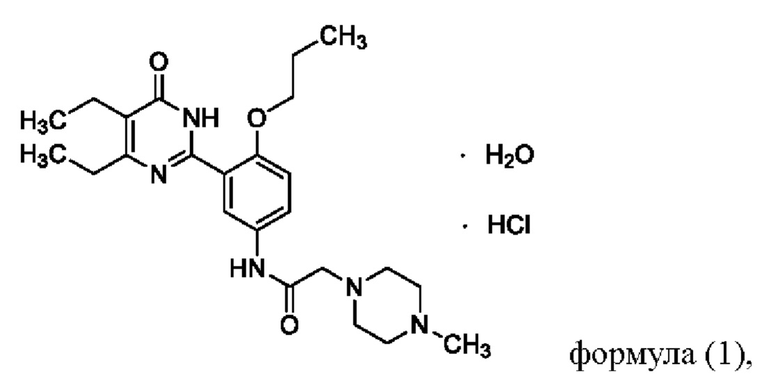
X более предпочтительно представляет собой соляную кислоту, то есть соединение формулы (I) является, предпочтительно, соединением формулы (I-A):
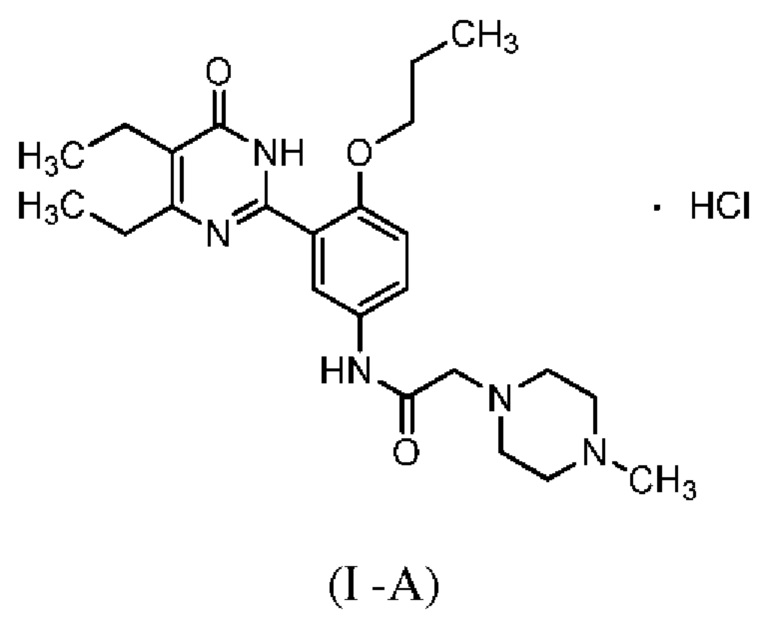
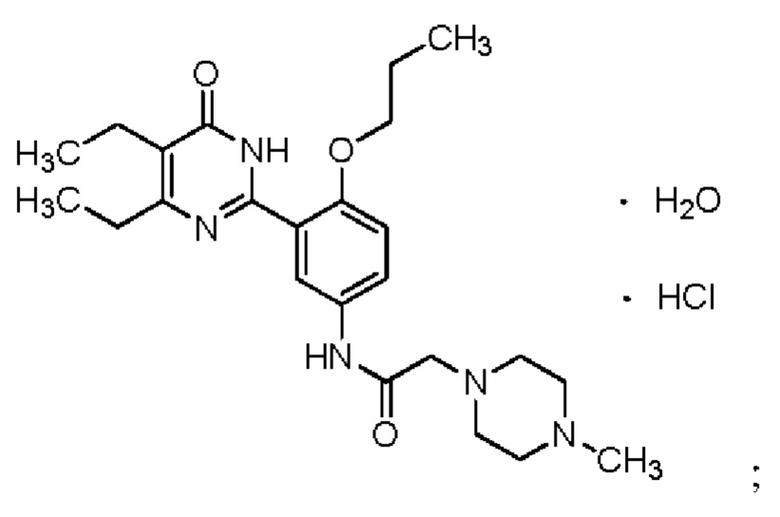
Более предпочтительно, соединение формулы (I) является кристаллической формой А или В гидрохлорида соединения Z. Для кристаллической формы А, на дифракционной рентгенограмме пики дифракции наблюдаются при угле дифракции 2θ, равным 9,3°±0,2°, 12,2°±0,2°, 16,7°+0,2°, точнее, на дифракционной рентгенограмме при угле дифракции 2θ, равным 6,1°±0,2°, 9,3°±0,2°, 11,1°±0,2°, 12,2°±0,2°, 15,3°±0,2°, 16,7°±0,2°, 21,6°±0,2°, 22,9°±0,2°. Кристаллическая форма А является моногидратом и обладает следующей структурой:
Для кристаллической формы В, на дифракционной рентгенограмме пики дифракции наблюдаются при угле дифракции 2θ, равным 12,5°±0,2°, 13,5°±0,2°, 17,9°±0,2°, 19,5°±0,2°, 19,9°±0,2°, 23,0°±0,2°, 26,5°±0,2°, 26,8°±0,2°, точнее, на дифракционной рентгенограмме при угле дифракции 2θ, равным 12,5°±0,2°, 12,8°±0,2°, 13,5°±0,2°, 14,1°±0,2°, 17,9°±0,2°, 19,5°±0,2°, 19,9°±0,2°, 23,0°±0,2°, 26,5°±0,2°, 26,8°±0,2°. Кристаллическая форма А гидрохлорида соединения Z является наиболее предпочтительной.
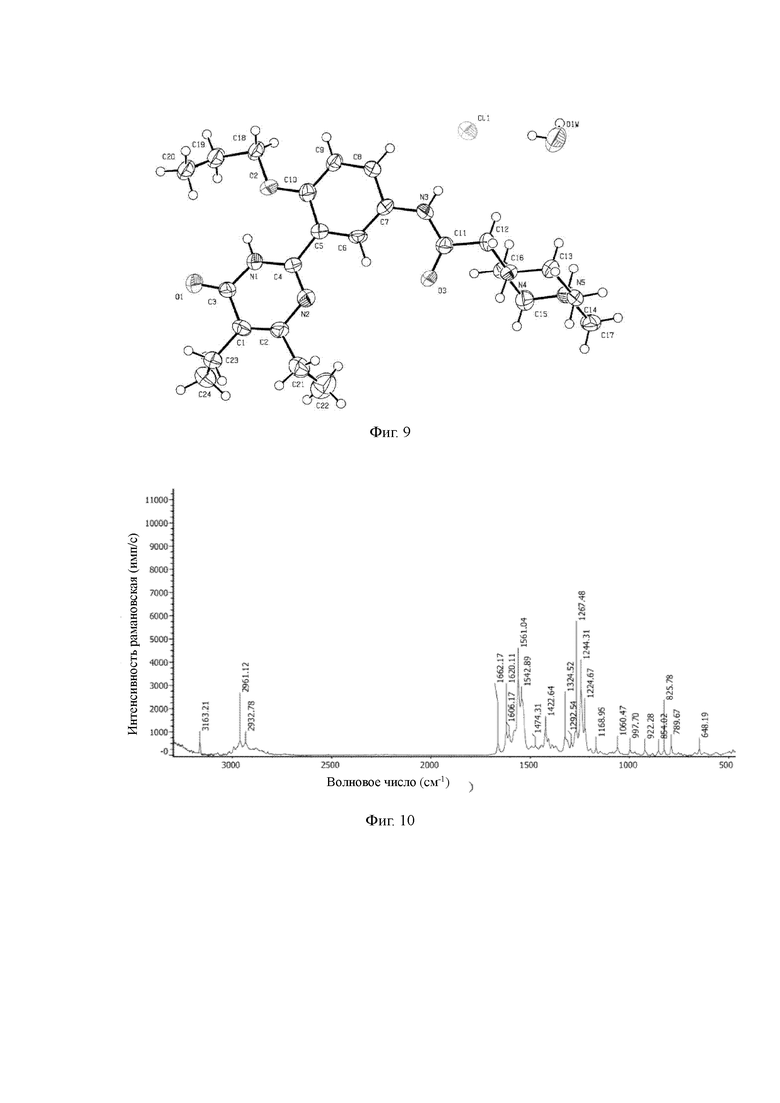
Дополнительно, настоящее изобретение позволяет получить монокристалл кристаллической формы А гидрохлорида соединения Z. Исследование монокристалла формы А проводится с помощью анализа рентгеновской дифракции монокристаллов; дифракционная рентгенограмма монокристалла представлена фигурой 9, которая показывает: кристаллическая система: моноклинная, пространственная группа: Рс, длина оси: а=9,935(4) 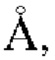 b=14,44(6)
b=14,44(6) 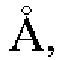 с=9,147(5)
с=9,147(5) 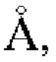 угол плоскости решетки: α=90°, β=106,47(5)°, γ=90°, объем=1260,5(11)
угол плоскости решетки: α=90°, β=106,47(5)°, γ=90°, объем=1260,5(11) 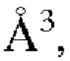 и количество молекул на элементарную ячейку составляет Z=2.
и количество молекул на элементарную ячейку составляет Z=2.
Измерение монокристаллической структуры производилось с помощью рентгеновского монокристального дифрактометра, производитель: Japanese Science (японская корпорация (Rigaku)); модель прибора: SuperNova, Dual, Cu at zero, AtlasS2; температура в ходе определения: 100K,
Кристаллическая форма А по настоящему изобретению содержит от 3,28 до 5,35 масс. % воды, а именно, от 3,28 до 3,98 масс. % воды. Содержание воды определено по методу Карла Фишера.
В данном контексте выражение «соль» или «соли» включает в себя как фармацевтически приемлемую соль, так и фармацевтически неприемлемую соль. Фармацевтически неприемлемая соль не является предпочтительной для введения пациенту, но такая соль может быть использована для получения промежуточных веществ и нефасованных лекарственных форм.
В данном контексте выражение «фармацевтически приемлемая соль» или «фармацевтически приемлемая соль присоединения кислоты» относится к соли, полученной с помощью фармацевтически приемлемой кислоты и соединения Z, включающей, помимо прочего, соль органической кислоты и соль неорганической кислоты, предпочтительно соль малеиновой кислоты, янтарной кислоты, лимонной кислоты, винной кислоты, фумаровой кислоты, муциновой кислоты, уксусной кислоты, метансульфоновой кислоты, соляной кислоты, азотной кислоты или серной кислоты, наиболее предпочтительно соль метансульфоновой кислоты, фумаровой кислоты или соляной кислоты.
Другой вариант осуществления данного изобретения относится к фармацевтической композиции и лекарственной форме, содержащим терапевтически или профилактически эффективное количество соединения формулы (I), или полиморф, сольват, гидрат, со-кристалл, ангидрит или аморфное вещество такового.
Дополнительно, при размещении соли соединения формулы (I) настоящего изобретения на воздухе или путем рекристаллизации, за счет поглощения воды, образуется гидрат с адсорбированной водой, и в настоящее изобретение также включены соли присоединения кислоты, содержащие такую адсорбированную воду.
Выражение «сольват» в настоящем изобретении не ограничивается только тем, что он образуется растворителем, применяемым для производства соли или образования кристалла, и, в частности, например, сольват может представлять собой гидрат, алкоголят, ацетоновый сольват, толуоловый сольват. Предпочтительными являются гидрат и алкоголят.
Дополнительный вариант осуществления настоящего изобретения относится к способу получения фармацевтической композиции и лекарственной формы, содержащими соединение формулы (I) или его полиморф, сольват, гидрат, со-кристалл, безводное вещество или аморфную форму.
Конкретный способ получения выглядит следующим образом.
1. Способ получения соли 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н))-она (соединение формулы I)
Соединение Z можно получить, ссылаясь на пример WO 2010/066111. Также соединение Z можно получить одним из следующих способов:
1) проведение реакции метилирования соединения Z-3 и диметилсульфата в присутствии основания в целях получения соединения Z-4;
2) проведение реакции соединения Z-4 и аммиака в целях получения соединения Z-5;
3) проведение реакции дегидратации и циклизации соединения Z-5 и метил-2-этил-3-оксопентаноата в присутствии основания в целях получения соединения Z,
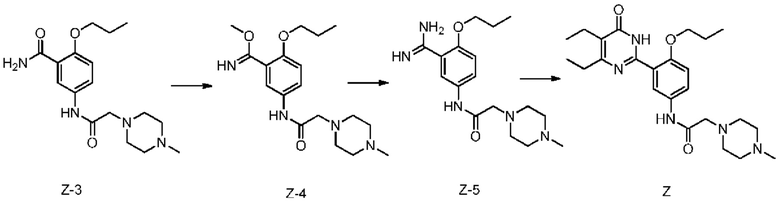
при этом, на вышеупомянутом этапе 1) температура реакции составляет, как правило, от комнатной температуры до 100°С, растворителем, предпочтительно, является дихлорметан, толуол, хлороформ или N,N-диметилформамид, а основанием является, предпочтительно, карбонат калия или карбонат натрия;
На вышеупомянутом этапе 2), температура реакции, как правило, составляет 0°С~50°С, растворителем является, предпочтительно, метанол, этанол или N,N-диметилформамид;
На вышеупомянутом этапе 3), температура реакции, как правило, составляет 50~120°С, растворителем, предпочтительно, является хлороформ, метанол, этанол, монометиловый эфир этиленгликоля, N,N-диметилформамид или 1,4-диоксан. Основанием является, предпочтительно, карбонат калия, карбонат натрия, метоксид натрия или этоксид натрия.
Соединение формулы (I) можно получить путем образования соли посредством реакции соответствующей кислоты с соединением Z, например, используя один из следующих способов:
Способ I:
1) растворение соединения Z в первом растворителе с целью образования раствора а;
2) растворение соответствующей кислоты X во втором растворителе с целью образования раствора b;
3) добавление раствора а к раствору b, или добавление раствора b к раствору а для получения смешанного раствора, и выделение соли соединения Z (т.е. соединения формулы I) из смешанного раствора;
Способ II:
1) растворение соединения Z в первом растворителе с целью образования раствора а;
2) непосредственное добавление соответствующей кислоты X к раствору а, с последующим выделением соли соединения Z (т.е. соединения формулы I) из раствора;
Способ III:
1) растворение соответствующей кислоты X во втором растворителе с целью образования раствора b;
2) непосредственное добавление соединения Z к раствору b, с последующим выделением соли соединения Z (т.е. соединения формулы I) из раствора;
Предпочтительно, соединение формулы (I-A) можно получить путем образования соли посредством реакции хлороводорода с соединением Z, например, используя один из следующих способов:
Способ I:
1) растворение соединения Z в первом растворителе с целью образования раствора а;
2) растворение хлороводорода во втором растворителе с целью образования раствора b;
3) добавление раствора а к раствору b, или добавление раствора b к раствору а для получения смешанного раствора, и выделение гидрохлорида соединения Z (т.е. соединения формулы (I-A)) из смешанного раствора;
Способ II:
1) растворение соединения Z в первом растворителе с целью образования раствора а;
2) непосредственное добавление хлороводорода к раствору а, с последующим выделением гидрохлорида соединения Z (т.е. соединения формулы (I-A)) из раствора;
Способ III:
1) растворение хлороводорода во втором растворителе с целью образования раствора b;
2) непосредственное добавление соединения Z к раствору b, с последующим выделением гидрохлорида соединения Z (т.е. соединения формулы (I-A)) из раствора.
В вышеупомянутом способе первый и второй растворители, каждый по себе независимо отобран из воды, неводного растворителя и их смешанного растворителя, а именно, отобраны из воды, спиртов, эфиров, сложных эфиров, углеводородов, кетонов и смешанных растворителей таковых и тому подобного. Более конкретно, первый растворитель и второй растворитель, каждый независимо, один или несколько, отобран из воды; таких сложных эфиров, как этилацетат, метилацетат, пропилацетат, бутилацетат, метилформиат, этиловый эфир муравьиной кислоты, формиат пропила, формиат бутила; таких спиртов, как метанол, этанол, пропанол, изопропиловый спирт, бутанол, этиленгликоль, пропиленгликоль; таких эфиров, как диэтиловый эфир, пропиловый эфир, изопропиловый эфир, нефтяной эфир, монометиловый эфир этиленгликоля, моноэтиловый эфир этиленгликоля, монопропиловый эфир этиленгликоля, диметиловый эфир этиленгликоля, диэтиловый эфир этиленгликоля, монометиловый эфир пропиленгликоля, моноэтиловый эфир пропиленгликоля, диметиловый эфир пропиленгликоля, тетрагидрофуран, диоксан, диметоксиэтан, диглим; таких кетонов, как ацетон, бутанон, N-метилпирролидон, диэтиловый кетон; таких углеводородов, как n-пентан, n-гексан, гептан; таких ароматических углеводородов, как толуол, бензол, ксилол, хлорбензол, дихлорбензол; таких галогенизировавших алканов, как дихлорметан, хлороформ или 1,2 дихлорэтан, тетрахлорид углерода; таких кислот, как уксусная кислота, пропионовая кислота; нитрилов, таких как ацетонитрил, пропионитрил;
Определение соответствующей кислоты выполняется так же, как определение X в формуле (I).
Температура реакции изменяется в зависимости от реагентов или растворителей, и, как правило, составляет от -20°С до 200°С, предпочтительно, от 0°С до 100°С.
Время реакции не ограничено и составляет, как правило, от 10 минут до 10 часов.
Хлороводород может присутствовать в газообразной форме, или в таком водном или неводном растворителе, как соляная кислота, хлорид водорода в метаноле, хлорид водорода в этаноле.
2. Способы получения различных полиморфов соли соединения Z (соединение формулы I)
Другой аспект настоящего изобретения обеспечивает полиморф соли соединения Z (т.е. соединения формулы I), а также способ получения такового. При необходимости в процесс можно добавить затравочный кристалл. Здесь и далее затравочный кристалл относится к «затравке» кристаллического вещества соединения формулы (I) или самодельному соединению формулы (I), который используется для возбуждения кристаллизации.
Полиморф соли соединения Z (т.е. соединения формулы I) может быть получен одним из следующих способов:
Способ I:
1) растворение основного соединения Z в третьем растворителе с целью образования раствора С;
2) растворение соответствующей кислоты X в четвертом растворителе с целью образования раствора D;
3) добавление раствора С к раствору D, или добавление раствора D к раствору С, или непосредственное добавление соответствующей кислоты X к раствору Сс целью получения смешанного раствора Е;
4) дополнительно, добавление пятого растворителя к смешанному раствору Е;
5) осаждение целевого соединения путем отстаивания, или перемешивания, или добавления соответствующего затравочного кристалла к раствору, полученному на этапе 4);
Способ II:
1) растворение соли соединения Z (т.е. соединения формулы (I)) в третьем растворителе с целью образования раствора F;
2) дополнительно, добавление пятого растворителя к раствору F;
3) осаждение целевого соединения путем отстаивания, или перемешивания, или добавления соответствующего затравочного кристалла к раствору, полученному на этапе 2);
Способ III:
1) растворение соли соединения Z (т.е. соединения формулы (I)) в третьем растворителе с целью образования суспензии G;
2) дополнительно, добавление пятого растворителя к раствору G;
3) нагревание, перемешивание, охлаждение с целью осаждения целевого соединения, или добавление затравочного кристалла к раствору, полученному на этапе 2) с целью осаждения.
Предпочтительно, полиморф соединения формулы (I-A) может быть получен одним из следующих способов:
Способ I:
1) растворение основного соединения Z в третьем растворителе с целью образования раствора С;
2) растворение хлороводорода в четвертом растворителе с целью образования раствора D;
3) добавление раствора С к раствору D, или добавление раствора D к раствору С, или непосредственное добавление хлороводорода к раствору С с целью получения смешанного раствора Е;
4) дополнительно, добавление пятого растворителя к смешанному раствору Е;
5) осаждение целевого соединения путем отстаивания, или перемешивания, или добавления соответствующего затравочного кристалла к раствору, полученному на этапе 4).
Способ II:
1) растворение соединения формулы (I-A) в третьем растворителе с целью образования раствора F;
2) дополнительно, добавление пятого растворителя к раствору F;
3) осаждение целевого соединения путем отстаивания, или перемешивания, или добавления соответствующего затравочного кристалла к раствору, полученному на этапе 2);
Способ III:
1) растворение соединения формулы (I-A) в третьем растворителе с целью образования суспензии G;
2) дополнительно, добавление пятого растворителя к раствору G;
3) нагревание, перемешивание, охлаждение с целью осаждения целевого соединения, или добавление затравочного кристалла к раствору, полученному на этапе 2) с целью осаждения.
При этом, третий раствор, четвертый раствор и пятый раствор могут быть одинаковыми или разными, и определение третьего раствора, четвертого раствора и пятого раствора является идентичным определению первого раствора и второго раствора, и температурный диапазон является идентичным тому, который используется для получения соли соединения Z (т.е. соединения формулы I).
В частности, предпочтительный способ получения кристаллической формы А гидрохлорида соединения Z является следующим:
Добавление соединения Z к спирту, добавление соляной кислоты, нагревание, дополнительное добавление активированного угля для обесцвечивания, добавление сложного эфира к полученному фильтрату, отстаивание или перемешивание с последующим отделением осажденной твердой части с целью получения кристаллической формы А соединения формулы (I-A).
В частности, кристаллическая форма А гидрохлорида соединения Z, предпочтительно, получается способом, как указано далее: добавление гидрохлорида соединения Z к спирту, его растворение путем нагревания, охлаждения для осаждения твердой части с последующим отделением в целях получения кристаллической формы А гидрохлорида соединения Z.
В частности, кристаллическая форма А гидрохлорида соединения Z, предпочтительно, получается способом, как указано далее: добавление гидрохлорида соединения Z к смешанной системе сложного эфира и спирта, его растворение путем нагревания, охлаждения для осаждения твердой части с последующим отделением в целях получения кристаллической формы А гидрохлорида соединения Z.
Предпочтительно, спиртом является С1-С4 линейный или разветвленный алканол, как метанол, этанол, изопропиловый спирт; более предпочтительно, этанол.
Сложный эфир включает в себя, помимо прочего, этилацетат, метилацетат, пропилацетат, бутилацетат, метилформиат, этиловый эфир муравьиной кислоты, формиат пропила, формиат бутила; более предпочтительно, этилацетат.
3. Определение характеристик соли соединения Z (соединение формулы I)
В целом, в порошковой рентгеновской дифракции может возникать отклонение угла дифракции (2θ) в диапазоне ±0,2°. Таким образом, значение угла дифракции следует понимать как включающее в себя значения в диапазоне ±0,2°. Настоящее изобретение включает в себя не только кристаллы, которые характеризуются пиками (углами дифракции), полностью совпадающими с теми, которые присутствуют в порошковой дифракционной рентгенограмме, но также и те кристаллы, которые характеризуются пиками (углом дифракции), совпадающими с пиками в диапазоне отклонения, равного ±0,2°.
(1) Определение характеристик кристаллической формы А гидрохлорида соединения Z
Настоящее изобретение обеспечивает кристаллическую форму А гидрохлорида соединения Z, что является моногидратом.
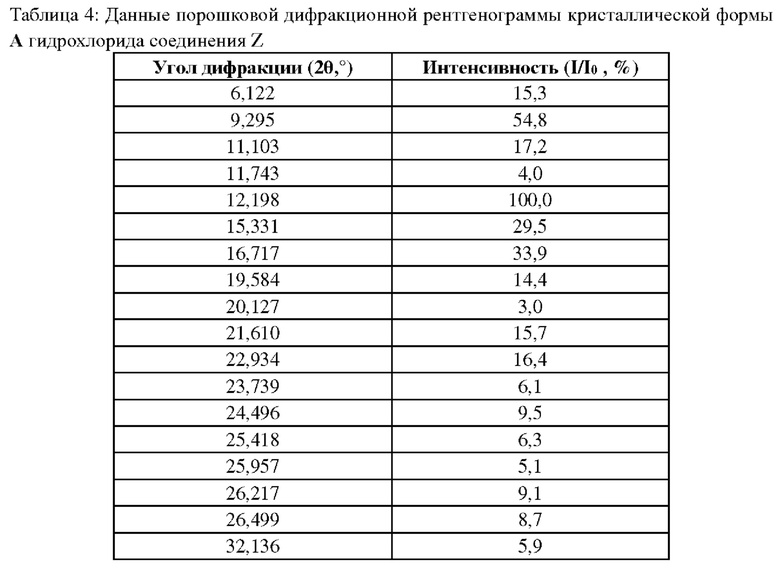
Данные порошковой рентгеновской дифракции для кристаллической формы А гидрохлорида соединения Z являются следующими: на порошковой дифракционной рентгенограмме кристаллическая форма характеризуется дифракционными пиками при углах дифракции 2θ, равными 6,1°±0,2°, 9,3°+0,2°, 11,1°±0,2°, 12,2°±0,2°, 15,3°±0,2°, 16,7°±0,2°, 21,6°±0,2°, 22,9°±0,2°.
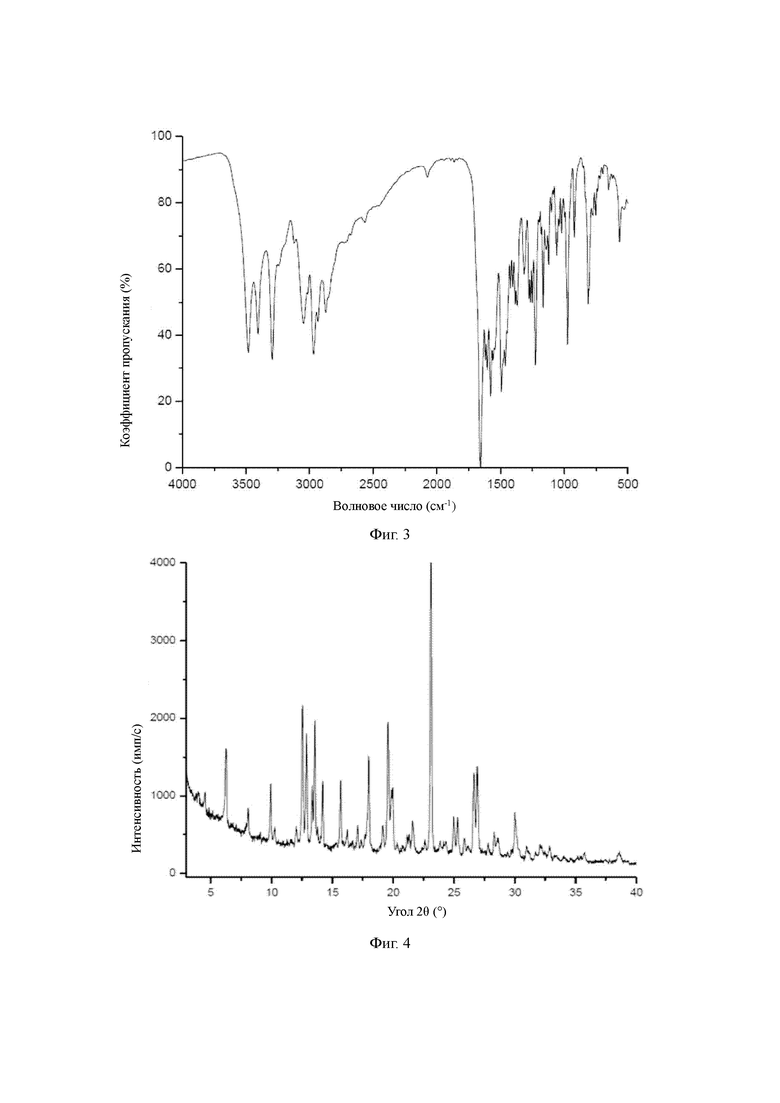
Для монокристаллов формы А проведен анализ рентгеновской дифракции монокристаллов, исходя из дифракционной рентгенограммы монокристалла он показывает, что форма А содержит в себе одну молекулу связанной воды. В инфракрасном спектре поглощения, измеренном с помощью методики гранул бромида калия, кристаллическая форма А характеризуется пиками, по меньшей мере, примерно, при 3482,81 см-1, 3405,67 см-1, 3293,82 см-1, 3046,98 см-1, 2967,91 см-1, 2871,49 см-1, 1656,55 см-1, 1604,48 см-1, 1579,41 см-1, 1494,56 см-1, 1226,50 см-1, 973,88 см-1, 813,81 см-1.
В рамановском спектре, кристаллическая форма А характеризуется пиками, по меньшей мере, примерно, при 3163,21 см-1, 2961,12 см-1, 2932,78 см-1, 1662,17 см-1, 1620,11 см-1, 1606,17 см-1, 1561,04 см-1, 1542,89 см-1, 1422,64 см-1, 1292,54 см-1, 1267,48 см-1, 1244,31 см-1, 1224,67 см-1, 854,02 см-1, 825,78 см-1.
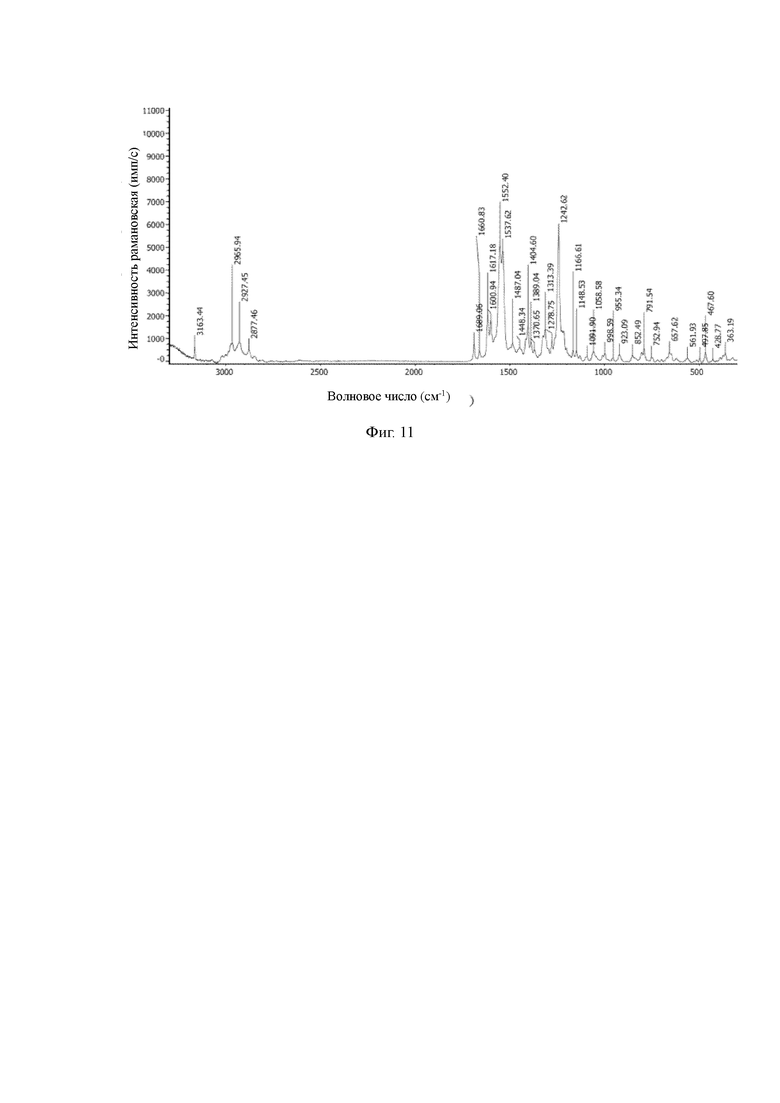
(2) Определение характеристик кристаллической формы В гидрохлорида соединения Z
Данное изобретение также обеспечивает кристаллическую форму В гидрохлорида соединения Z.
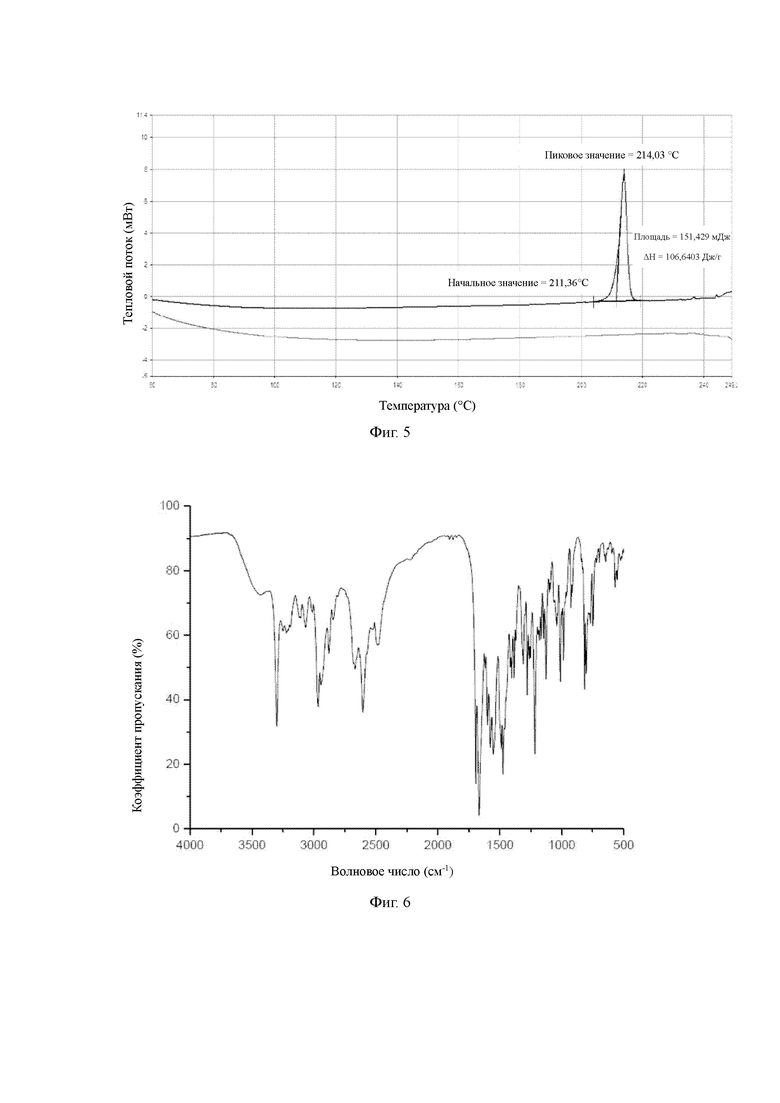
Кристаллическая форма В гидрохлорида соединения Z характеризуется эндотермическим пиком при 211,36°С как это определено ДСК, данные спектра являются следующими:
Начальное значение (начало процесса) = 211,36±3°С, пиковое значение (пик) = 214,03±3°С
Условия измерения ДСК являются следующими:
Модель прибора: Perkin Elmer DSC 8500, диапазон температур: 50-280°С, скорость сканирования: 10°С/мин, расход азота: 50 мл/мин.
Данные порошковой рентгеновской дифракции для кристаллической формы В гидрохлорида соединения Z являются следующими: на порошковой дифракционной рентгенограмме кристаллическая форма характеризуется дифракционными пиками при углах дифракции 2θ, равными 12,5°±0,2°, 12,8°±0,2°, 13,5°±0,2°, 14,1°±0,2°, 17,9°±0,2°, 19,5°±0,2°, 19,9°±0,2°, 23,0°±0,2°, 26,5°±0,2°, 26,8°±0,2°.
В рамановском спектре, кристаллическая форма В характеризуется пиками, по меньшей мере, примерно, при 3163,44 см-1, 2965,94 см-1, 2927,45 см-1, 2877,46 см-1, 1689,06 см-1, 1617,18 см-1, 1600,94 см-1, 1552,40 см-1, 1537,62 см-1, 1404,60 см-1, 1242,62 см-1, 657,62 см-1.
(3) Определение характеристик аморфной формы гидрохлорида соединения Z
Дополнительно, настоящее изобретение обеспечивает аморфную форму гидрохлорида соединения Z, порошковая дифракционная рентгенограмма которой по существу показана на фигуре 7.
Соответствующим образом, в другом аспекте, настоящее изобретение обеспечивает фармацевтическую композицию, содержащую одно или более соединений формулы (I), а также фармацевтически приемлемые вспомогательные лекарственные вещества. Предпочтительно, фармацевтическая композиция содержит одно или более, выбранных из малеата, сукцината, метансульфоната, цитрата, гидрохлорида, тартрата, фумарата, муката, ацетата и сульфатов соединения Z. Более предпочтительно, фармацевтическая композиция содержит кристаллическую форму А гидрохлорида соединения Z, порошковая дифракционная рентгенограмма которого приведена в таблице 1.
Вспомогательное средство может представлять собой наполнитель, связующее вещество, смазывающее вещество, разделяющее средство, окрашивающее вещество, ароматизирующее вещество, эмульгатор, поверхностно-активное вещество, ожижающее средство, суспендирующее вещество, изотоническое средство, буферное средство, консервант, антиокислитель, стабилизатор, ускоритель впитывания и т.п., которые обычно используются в медицинской области. Вышеупомянутое может, соответственно, использоваться в комбинации по мере необходимости.
Предпочтительно, соль соединения Z по настоящему изобретению может быть составлена, по меньшей мере, с одним фармацевтическим вспомогательным средством для пероральной фармацевтической композиции, и каждая лекарственная форма содержит 10 мг~200 мг активной фармацевтической субстанции.
При получении твердой композиции типа таблетки, основная активная фармацевтическая субстанция смешивается с одним фармацевтическим носителем, как крахмал, лактоза, стеарат магния или подобным, а также таблетку можно покрыть сахарным покрытием или иным приемлемым веществом. Или таблетки подвергаются обработке с целью обеспечения продленного или замедленного высвобождения, так чтобы таблетки могли непрерывно высвобождать предопределенное количество активной фармацевтической субстанции.
Альтернативно, активная фармацевтическая субстанция может смешиваться с разбавителем, что приводит к образованию смеси с последующим заполнением ее в капсулу.
Соль присоединения кислоты соединения Z по настоящему изобретению (т.е. соединения формулы I), при использовании в качестве терапевтического или профилактического средства для вышеупомянутых заболеваний, может использоваться как сама по себе, так и вместе с приемлемым фармацевтическим вспомогательным средством, разбавителем или подобным, и вводиться перорально в форме таблетки, капсулы, гранулы, порошка или сиропа, или неперорально, например, в форме инъекции, инъецируемого порошка, спрея или суппозиториев.
Такие составы можно получать обычным способом.
Количество лекарственного средства изменяется в зависимости от симптомов, возраста и тому подобного. Например, взрослому можно вводить от 1 до 7 раз в течение от 1 до 7 дней, вводимая доза составляет от 0,01 мг до 1000 мг. Способ введения не ограничен.
Другой аспект настоящего изобретения обеспечивает использование соединения, представленного формулой (I), при получении лекарственного средства для предупреждения или лечения заболеваний, связанных с ферментом ФДЭ5. К заболеваниям, связанным с ферментом ФДЭ5, относят следующие: эректильная дисфункция, артериальная гипертензия легочной артерии, женская половая дисфункция, преждевременные роды, дисменорея, доброкачественная гипертрофия предстательной железы, обструкция выходного отверстия мочевого пузыря, недержание, нестабильная и вариантная стенокардия, артериальная гипертензия, острая сердечная недостаточность, почечная недостаточность, атеросклероз, инсульт, периферическое сосудистое заболевание, болезнь Рейно, воспалительное заболевание, бронхит, хроническая астма, аллергическая астма, аллергический ринит, глаукома или заболевания, характеризующиеся нарушениями перистальтики кишечника.
КРАТКОЕ ОПИСАНИЕ ФИГУР:
Фигура 1: порошковая дифракционная рентгенограмма кристаллической формы А соединения формулы (I-A);
Фигура 2: график термогравиметрического анализа кристаллической формы А соединения формулы (I-А);
Фигура 3: инфракрасный спектр кристаллической формы А соединения формулы (I-А);
Фигура 4: порошковая дифракционная рентгенограмма кристаллической формы В соединения формулы (I-А);
Фигура 5: график дифференциальной сканирующей калориметрии кристаллической формы В соединения формулы (I-А);
Фигура 6: инфракрасный спектр кристаллической формы В соединения формулы (I-А);
Фигура 7: порошковая дифракционная рентгенограмма аморфной формы соединения формулы (I-А);
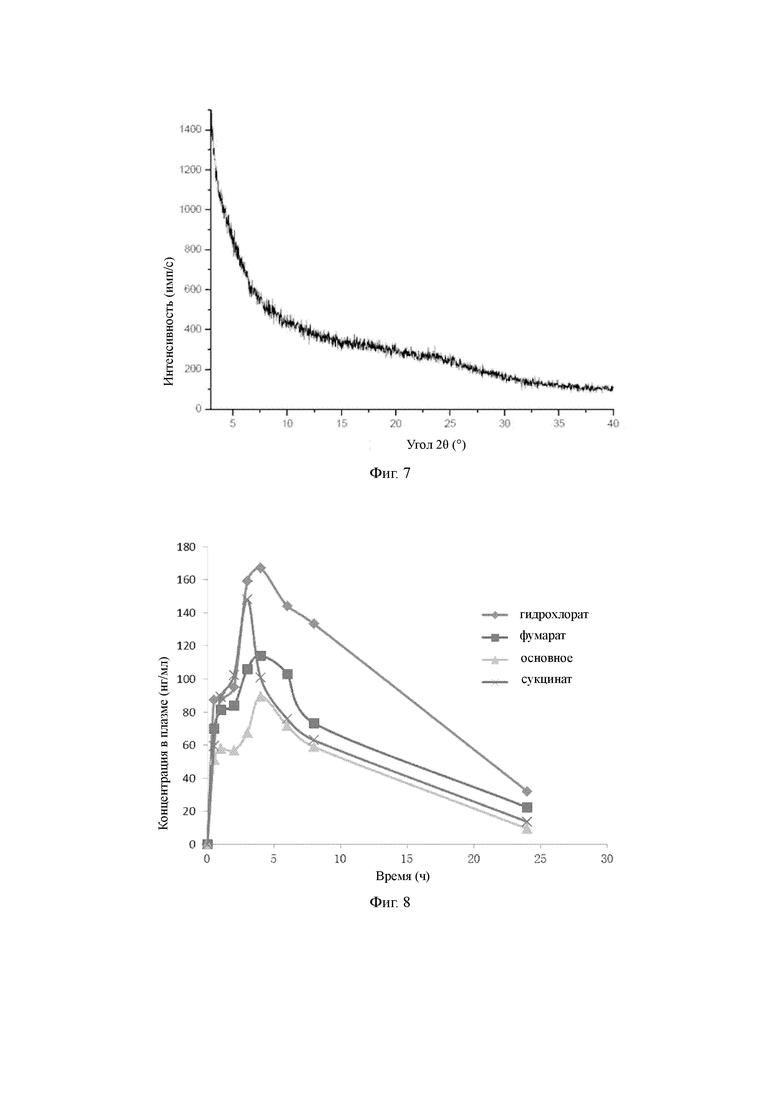
Фигура 8: кривая концентрация в плазме - время для соединения Z и его различных солей после введения;
Фигура 9: дифракционная рентгенограмма структуры монокристалла кристаллической формы А соединения формулы (I-А);
Фигура 10: рамановский спектр кристаллической формы А соединения формулы (I-А);
Фигура 11: рамановский спектр кристаллической формы В соединения формулы (I-А).
Эффект изобретения:
Соль соединения Z по настоящему изобретению обладает такими преимуществами, как высокая стабильность, улучшенная растворимость в воде, отсутствие неприятного запаха и т.п.
Различные кристаллические формы различных солей соединения Z по настоящему изобретению обладают такими преимуществами, как низкая гигроскопичность, хорошая химическая стабильность, высокая чистота, постоянный состав, простой способ получения в сочетании с хорошей воспроизводимостью, простое хранение образцов и т.п.
Подробное описание вариантов осуществления изобретения
Настоящее изобретения будет дополнительно показано при помощи следующих примеров, которые просто используются для пояснения предпочтительных вариантов осуществления настоящего изобретения, и не направлены на ограничение настоящего изобретения. Как значения температура, так и реагенты, примененные в следующих примерах, могут быть заменены соответствующими значениями температуры и реагентами, упомянутыми выше, для достижения целей настоящего изобретения,
В следующих примерах элементарный анализ выполнялся с помощью устройства ElementarVario EL; масс-спектрометрия осуществлялась с помощью масс-спектрометра МАТ-95 (производство Finnigan Corporation); спектроскопия ядерного магнитного резонанса была получена на ЯМР-спектрометрах Mercury-300 и Mercury-400 (Varian, Inc.); измерение инфракрасного спектра проводилось на контрольно-измерительном приборе: инфракрасный спектрометр American Nicolet Magna FTIR 6700 с преобразованием Фурье; спектрометр рамановского рассеяния: Thermo Scientific DXR Raman Microscope. Волновое число (см-1) инфракрасного и рамановского спектров может содержать отклонение от -0,5 до +0,5%, но такой уровень отклонения находится в настоящем изобретении в пределах приемлемого диапазона.
Пример 1: Получение малеата 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (5 г, 0,01 моль) суспендировали в 50 мл безводного этанола, затем полученную суспензию нагревали до 65°С для растворения соединения, с добавлением малеиновой кислоты (1,16 г, 0,01 моль), нагреванием при 65°С в течение 40 минут, затем в немного охлажденную систему добавляли активированный уголь и поддерживали такую температуру в течение 30 минут. Активированный уголь отфильтровывали и перемешивали полученный фильтрат при комнатной температуре в течение 30 минут для осаждения твердого вещества. Осажденное твердое вещество отфильтровывали и сушили в течение 4 часов для получения на выходе целевого соединения в виде белого твердого вещества (4 г) с чистотой 99% (ВЭЖХ).
Пример 2: Получение сукцината 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (5 г, 0,01 моль) суспендировали в 30 мл безводного этанола, затем полученную суспензию нагревали до 65°С для растворения соединения, с добавлением янтарной кислоты (1,18 г, 0,01 моль), перемешиванием при 65°С в течение 40 минут, затем в немного охлажденную систему добавляли активированный уголь для обесцвечивания. Активированный уголь отфильтровывали и перемешивали полученный фильтрат при комнатной температуре в течение 30 минут для осаждения твердого вещества. Осажденное твердое вещество отфильтровывали и сушили для получения на выходе целевого соединения в виде белого твердого вещества (3 г) с чистотой 99% (ВЭЖХ).
Пример 3: Получение фумарата 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (5 г, 0,01 моль) суспендировали в 30 мл безводного этанола, затем полученную суспензию нагревали до 65°С для растворения соединения, с добавлением фумаровой кислоты (1,16 г, 0,01 моль), перемешиванием при 65°С в течение 40 минут, затем в немного охлажденную систему добавляли активированный уголь для обесцвечивания. Активированный уголь отфильтровывали и перемешивали полученный фильтрат при комнатной температуре в течение 30 минут для осаждения твердого вещества. Осажденное твердое вещество отфильтровывали и сушили для получения на выходе целевого соединения в виде белого твердого вещества (3 г) с чистотой 99% (ВЭЖХ).
Пример 4: Получение ацетата 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (5 г, 0,01 моль) суспендировали в 30 мл безводного этанола, затем полученную суспензию нагревали до 65°С для растворения соединения, с добавлением уксусной кислоты (0,6 г, 0,01 моль), перемешиванием при 65°С в течение 40 минут, затем в немного охлажденную систему добавляли активированный уголь для обесцвечивания. Активированный уголь отфильтровывали и перемешивали полученный фильтрат при комнатной температуре в течение 30 минут для осаждения твердого вещества. Осажденное твердое вещество отфильтровывали и сушили для получения на выходе целевого соединения в виде белого твердого вещества (3 г) с чистотой 99% (ВЭЖХ).
Пример 5: Получение метансульфоната 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (5 г, 0,01 моль) суспендировали в 40 мл безводного этанола с добавлением 1 мл метансульфоновой кислоты на ледяной бане, после этого температуру повышали до 60~70°С, и перемешивали полученную смесь в течение 30 минут с добавлением активированного угля для обесцвечивания. Активированный уголь отфильтровывали и концентрировали полученный фильтрат до малого объема при пониженном давлении с добавлением 80 мл этил ацетата, перемешиванием, с последующим постепенным осаждением продукта, фильтрованием с отсасыванием и сушкой для получения на выходе 4,4 г целевого соединения.
Пример 6: Получение тартрата 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (5 г, 0,01 моль) растворяли в 25 мл безводного этанола, с добавлением к нему 1,5 г винной кислоты, после чего смесь нагревалась до 60~70°С, перемешивалась для реакции в течение 30 минут, охлаждалась до комнатной температуры, и перемешивалась в течение ночи, и после осаждения белого твердого вещества, обеспечивалась ее фильтрация и сушка для получения на выходе 4 г целевого соединения.
Пример 7: Получение муката 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (5 г, 0,01 моль) растворяли в 25 мл безводного этанола, с добавлением к нему 2,1 г муциновой кислоты, после чего смесь нагревалась до 60~70°С, перемешивалась для реакции в течение 30 минут, охлаждалась до комнатной температуры, и перемешивалась в течение ночи, и после осаждения белого твердого вещества, обеспечивалась ее фильтрация и сушка для получения на выходе 4 г целевого соединения.
Пример 8: Получение гидрохлорида 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она.
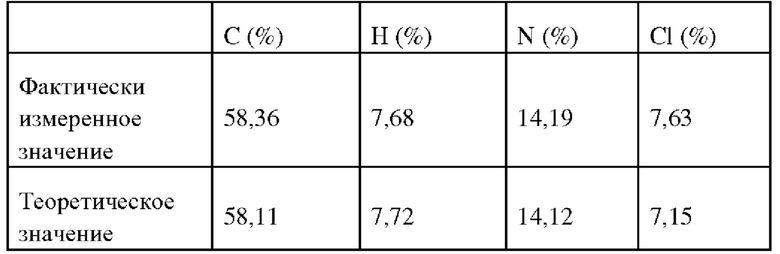
К соединению Z (51 г, 0,11 моль) добавляли ацетонитрил (200 мл), концентрированную хлороводородную кислоту (10,5 мл) добавляли капельным приливом на ледяной бане, после завершения капельного прилива смесь подвергалась нагреванию с обратным холодильником в течение 1 часа, обесцвечивалась с помощью активированного угля и отфильтровывалась. Полученный фильтрат охлаждался до комнатной температуры, концентрировался до малого объема, продукт постепенно осаждался при перемешивании, фильтровался и сушился для получения на выходе целевого соединения в виде 50 г белого твердого вещества. Было проведено высокоэффективное определение жидкой фазы, чистота >99%. ИЭР-МС: 442 (М+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ 0,96 (t, 3Н), 1,03 (t, 3Н), 1,19 (t, 3Н), 1,72 (m, 2Н), 2,45 (q, 2Н), 2,56 (q, 2Н), 2,65-2,75 (br, 5Н), 2,90-3,03 (br, 2Н), 3,03-3,17 (br, 2Н), 3,25 (s, 2Н), 3,30-3,40 (br, 2Н), 4,01 (t, 2Н), 7,13 (d, 1H), 7,79 (dd, 1H), 8,00 (d, 1H), 10,02 (s, 1H), 10,86 (br, 1H), 11,84 (s, 1H). 13С ЯМР (100 МГц, ДМСО-d6) δ 167,54, 163,31, 161,53, 152,70, 152,40, 131,75, 123,75, 123,07, 121,57, 121,35, 113,21, 70,11, 60,07, 52,27, 49,11, 42,03, 26,93, 21,95, 18,01, 13,37, 13,09, 10,37.
Элементарный анализ:
Пример 9: Получение гидрохлорида 5,6-Диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (51 г, 0,11 моль) суспендировали в смешанном растворе 400 мл безводного этанола и 10 мл воды, с добавлением 11 мл концентрированной хлороводородной кислоты капельным приливом на ледяной бане, с последующим нагревом смеси при 60°С в течение 30 минут, добавлением активированного угля и нагревом в течение 30 минут при перемешивании. Активированный уголь отфильтровывали, растворитель частично удаляли концентрированием при пониженном давлении, с добавлением 600 мл этилацетата, продукт постепенно осаждали, осуществлялась фильтрация с отсасыванием, затем отфильтрованное вещество сушили для получения на выходе 35 г целевого соединения.
Пример 10: Получение сульфата 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она
Соединение Z (5 г) суспендировали в смешанном растворе 19 мл безводного этанола и 0,5 мл воды, добавляли 0,7 мл концентрированной серной кислоты на ледяной бане, полученная смесь подвергалась нагреванию с обратным холодильником в течение 30 минут, естественным образом охлаждалась до комнатной температуры и концентрировалась до малого объема с пониженным давлением с последующим добавлением 30 мл этилацетата. Продукт постепенно осаждался, осуществлялась фильтрация с отсасыванием, затем отфильтрованное вещество сушили для получения на выходе 3,5 г продукта.
Пример 11: Получение моногидрата гидрохлорида 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (форма А)
Соединение Z (51 г, 0,11 моль) добавляли к 400 мл безводного этанола, с последующим добавлением 11 мл концентрированной соляной кислоты капельным приливом, затем, полученная смесь нагревалась при 60°С в течение 30 минут, в нее добавляли активированный уголь, и система нагревалась в течение 30 минут при перемешивании. Активированный уголь отфильтровывали, в фильтрат добавляли 600 мл этилацетата при перемешивании, продукт постепенно осаждали, осуществлялась фильтрация с отсасыванием, затем отфильтрованное вещество сушили для получения на выходе 30 г целевого соединения.
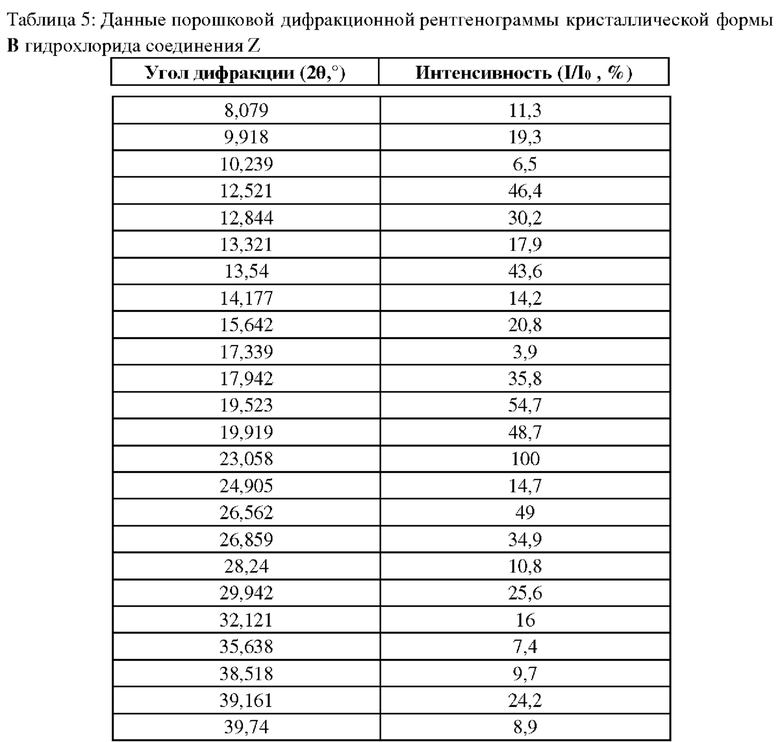
Как можно видеть на кривой ТГА, форма А потеряла в массе 3,92% в пределах 60°С~170°С, ТГ спектр был показан на фигуре 2. Условия испытания, как представлено далее: Аналитический прибор ТГ: термогравиметрический анализатор mettler toledo TGA2; условия испытания: продувочный газ: азот 50 мл/мин; тепловой поток: 200 К/мин; диапазон температур: 40°С-300°С.
Пример 12: Получение моногидрата гидрохлорида 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (форма А)
Целевое соединение (1 г) по примеру 5 или 6 добавляли к этанолу (8 мл), растворяли при нагреве с последующим охлаждением полученной смеси естественным путем до комнатной температуры и постепенным осаждением белого твердого вещества, его отфильтровали и сушили для получения 0,6 г целевого соединения в виде белого твердого вещества.
Пример 13: Получение моногидрата гидрохлорида 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (форма А)
Целевое соединение (1 г) по примеру 5 или 6 добавляли к этанолу (4 мл) и этилацетату (5 мл) с последующим охлаждением полученной смеси естественным путем до комнатной температуры и постепенным осаждением белого твердого вещества, его отфильтровывали и сушили для получения 0,85 г целевого соединения в виде белого твердого вещества.
Пример 14: Получение гидрохлорида 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (форма В)
Целевое соединение (1 г) по примеру 5 или 6 добавляли к этанолу (15 мл), растворяли при нагреве с последующим добавлением изопропанола (15 мл) и охлаждением полученной смеси естественным путем до комнатной температуры и постепенным осаждением белого твердого вещества, его отфильтровывали и сушили для получения 0,7 г целевого соединения в виде гранулированного кристалла.
Пример 15: Получение гидрохлорида 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (аморфного)
Целевое соединение по примеру 5 или 6 (200 мг) добавляли к хлороформу (10 мл), перемешивали при комнатной температуре с медленным удалением летучих соединений, белое твердое вещество осаждали, отфильтровывали и сушили для получения 0,15 г целевого соединения в виде белого пластинчатого твердого вещества.
Пример 16: Получение 5,6-диэтил-2-[2-n-пропокси-5-(2-(4-метилпиперазин-1-ил)ацетамидо)фенил]пиримидин-4(3Н)-она (соединение Z)
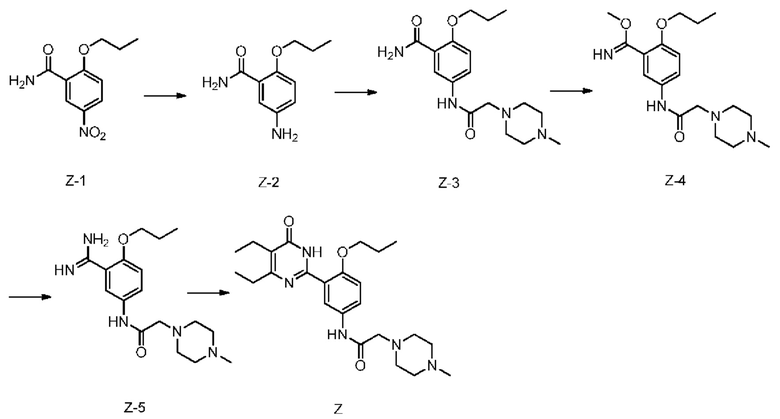
Метанол (60 мл) и соединение Z-1 (10 г) добавляли в сосуд объемом 100 мл с одной горловиной, при этом воздух замещали азотом 5 раз, после чего к ним добавлялся активный никель 0,3 г, азот замещался водородом 5 раз, смесь перемешивалась при комнатной температуре в течение 12 часов, после чего, по завершении реакции, активный никель удалялся путем фильтрации. Полученный фильтрат концентрировали при пониженном давлении с целью получения соединения Z-2 (8 г), выход 92%.
Соединение Z-2 (1,94 г, 10 ммоль) добавляли к 20 мл дихлорметана, с последующим добавлением 4-метил-1-пиперазин уксусной кислоты (1,58 г, 10 ммоль) и N,N'-карбонилдиимидазола (1,94 г, 12 ммоль), и перемешивали полученную смесь при комнатной температуре в течение 8 часов. После завершения реакции, в реакционную систему добавляли 10 мл воды, ее перемешивали, отстаивали для разделения, с последующим концентрированием органической фазы до высыхания с целью получения соединения Z-3 (2,34 г), выход 70%.
Соединение Z-3 (334 г, 1 моль) добавляли к N,N-диметилформамиду (1 л), к ним добавляли карбонат калия (276 г, 2 моль) и диметилсульфат (126 г, 1 моль) при помешивании, полученная смесь нагревалась до 40-50°С, перемешивалась в течение 12 часов с последующей концентрацией при пониженном давлении с целью получения остатка. К нему добавляли 1 л воды, полученная смесь перемешивалась при комнатной температуре и отфильтровывалась с целью получения 260 г соединения Z-4 в виде твердого вещества почти белого цвета, выход 75%. ИЭР-МС: 349 (М+1).
Соединение Z-4 (348 г, 1 моль) растворяли в метаноле (1 л), и полученная смесь перемешивалась и нагревалась до 50°С с последующим медленным введением аммиачного газа в нее. Затем систему концентрировали при пониженном давлении для получения остатка после завершения реакции путем очистки кристаллизации с метил-терт-бутиловым эфиром получали 198 г соединения Z-5, выход: 59%. ИЭР-МС: 334 (М+1).
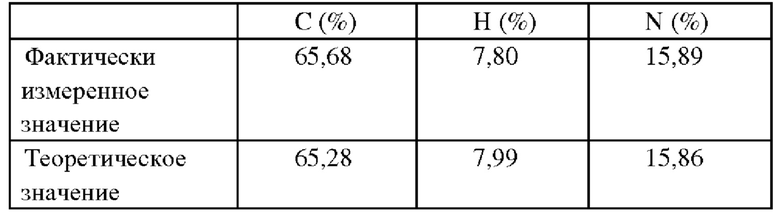
Соединение Z-5 (333 г, 1 моль) растворяли в метаноле (2 л), к ним добавляли метилат натрия (54 г, 1 моль) и метил-2-этил-3-оксопентаноат (158 г, 1 моль), полученная смесь подвергалась нагреванию с обратным холодильником и поддерживалась в таком состоянии в течение 12 часов, затем охлаждалась до комнатной температуры. Путем добавления слабоконцентрированной хлороводородной кислоты система приводилась к рН=7, затем концентрировалась при пониженном давлении с целью удаления метанола. Для экстрагирования добавляли этилацетат, оставляли раствор для разделения с последующим концентрированием органической фазы до высыхания с целью получения 410 г соединения Z, выход 93%. ИЭР-МС: 442 (М+1). 1Н ЯМР (400 МГц, CDCl3) δ 1,12 (t, 6Н), 1,30 (t, 3Н), 1,96 (m, 2Н), 2,56 (q, 2Н), 2,67 (q, 2Н), 2,31 (br, 3Н), 2,45-2,55 (br, 4Н), 2,60-2,70 (br, 4Н), 3,14 (s, 2Н), 4,13 (t, 2Н), 6,99 (d, 1H), 7,97 (dd, 1H), 8,26 (d, 1H), 9,11 (s, 1H), 11,14 (br, 1H). 13С ЯМР (100 МГц, CDCl3) δ 167,99, 163,87, 161,74, 153,41, 150,78, 131,23, 124,33, 123,93, 121,31, 119,23, 113,06, 70,95, 61,37, 54,76, 53,05, 45,57, 27,17, 21,97, 18,24, 12,93, 12,65, 10,16.
Элементарный анализ:
Экспериментальные примеры
1. Преимущества солей соединения Z (т.е. соединения формулы I)
(1) Сравнение растворимости соединений
Проводили сравнение растворимости соединения Z, полученного в примере 16, и различных солей соединения Z, полученных в примерах 1-7 и 9-10. Подходящее количество образца взвешивали в стеклянной пробирке, к нему постепенно добавляли выбранный растворитель и наблюдали его осветление. Замеряли растворимость различных солей при разных значениях рН, равных 2, 4,5, 6,8, и в дистиллированной воде, как приведено далее:
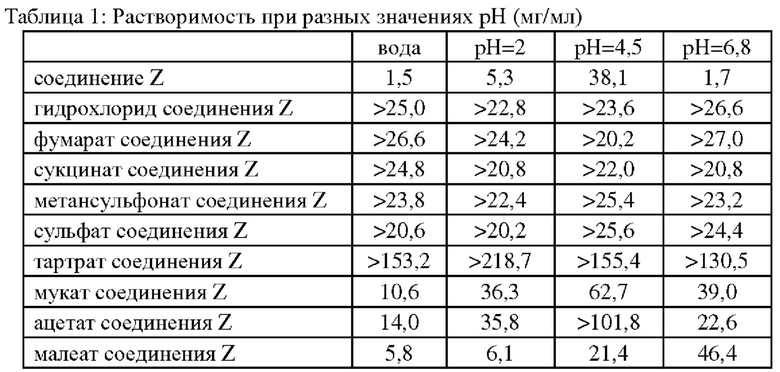
Как можно увидеть из вышеупомянутой таблицы, растворимость основного соединения Z в воде значительно ниже, чем таковая у солей соединения Z. Растворимость в воде оказывает решающее влияние на получение фармацевтической композиции, пероральную биодоступность и тому подобное. Таким образом, образование соли соединения Z является более полезным для получения лекарственного препарата для медицинского применения.
(2) Сравнение внешнего вида и признаков соединения
Наблюдали внешний вид и признаки основного соединения Z, полученного в примере 16, а также различных солей соединения Z, полученных в примерах 1-2, 5-6, 9; результаты показаны в таблице далее:
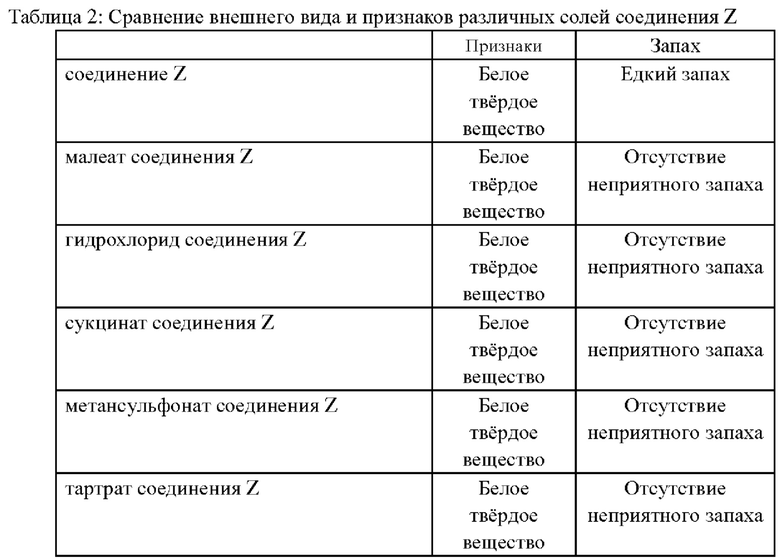
Как можно увидеть из вышеупомянутой таблицы, соли основного соединения Z могут скрывать исходный едкий запах и являются более подходящими для перорального введения.
(3) Сравнение стабильности соединений
Сравнивали химическую стабильность основного соединения Z, полученного в примере 16, а также солей соединения Z, полученных в примерах 5, 9 и 10, посредством испытания под нагрузкой; результаты показаны в таблице далее:
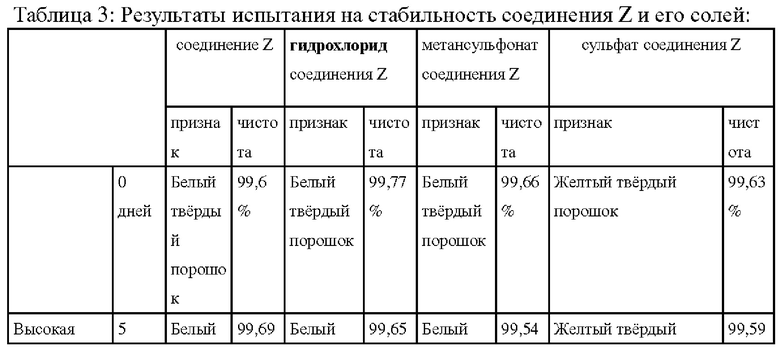
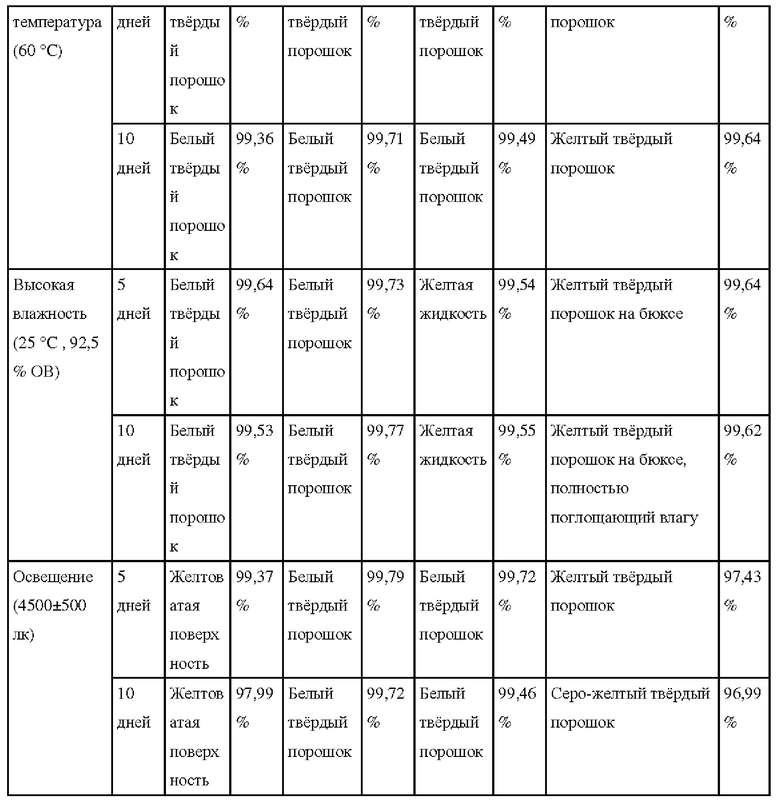
Как можно увидеть из результатов сравнительного испытания оказывающих влияние факторов, приведенных в таблице выше, соли соединения Z, особенно гидрохлорид, выказывали стабильность при воздействии высокой температуры, света и высокой влажности, при этом стабильность соединения Z является относительно слабой, особенно в условиях 10-дневного воздействия светом, и значение чистоты было значительно снижено.
(4) Сравнение фармакокинетических свойств
Для проведения фармакокинетического экспериментального исследования на живых организмах крыс использовали основное соединение Z, полученное в примере 16, гидрохлорид соединения Z, полученный в примере 9, сукцинат соединения Z, полученный в примере 2, фумарат соединения Z, полученный в примере 3 (введенный через желудочный зонд, доза 3 мг/кг); результаты приведены на фигуре 8 описания изобретения.
По результатам экспериментального исследования можно видеть, что когда соединение Z преобразовано в соль, то фармакокинетические свойства для живых организмов крыс являются более предпочтительными, чем имеющиеся у основного соединения Z, в частности, фармакокинетические свойства в живом организме у гидрохлорида соединения Z значительно лучше таковых у соединения Z.
Из результатов экспериментального исследования можно видеть, что соединения формулы I, полученные путем реакции солеобразования у соединения Z, могут улучшить растворимость соединения Z, скрыть неприятный запах соединения Z и увеличить стабильность. В частности, гидрохлорид соединения Z обладает лучшими физическими и химическими свойствами (хорошая растворимость и высокая стабильность), высокой оральной биодоступностью, выказывает наилучшую обширную применяемость в виде лекарства, и в большей степени подходит для производства фармацевтических препаратов и консервации.
2. Порошковые дифракционные рентгенограммы кристаллических форм А и В гидрохлорида соединения Z
Измеряли порошковые дифракционные рентгенограммы кристаллической формы А, полученной в примерах 11 или 12, кристаллической формы В, полученной в примере 14, а также аморфной формы, полученной в примере 15; данные показаны в таблицах 4 и 5, на фигурах 1, 4 и 7.
Определение порошковой дифракционной рентгенограммы проводилось в условиях, как это указано далее:
Модель прибора: Bruker D8 advance, цель: Cu K α (40 кВ, 40 мА), расстояние от образца до детектора: 30 см, диапазон сканирования: 3°-40° (2θ значение), шаг сканирования: 0,1 с.
3. Превосходство кристаллической формы А гидрохлорида соединения Z
Сравнивали кристаллическую форму А, полученную в примерах 11 или 12, кристаллическую форму В, полученную в примере 14, а также аморфное вещество, полученное в примере 15; результаты показаны в следующей таблице 6.
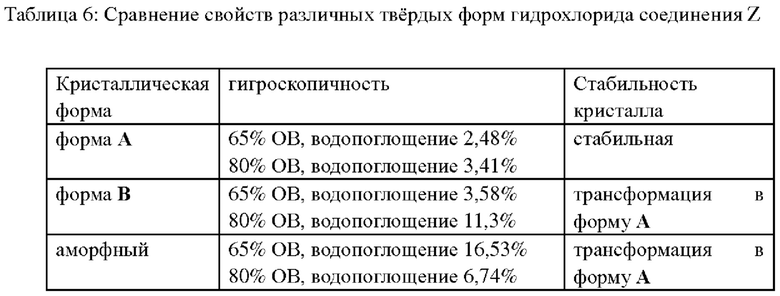
Как можно видеть из вышеприведенной таблицы, кристаллическая форма А обладает стабильными свойствами и низкой гигроскопичностью, и является предпочтительной кристаллической формой.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ С ТРЕМЯ КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ СОЛЬ ИЛИ КРИСТАЛЛИЧЕСКАЯ ФОРМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2020 |
|
RU2835105C1 |
| ПРОИЗВОДНЫЕ ФЕНИЛПИРИМИДОНА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2009 |
|
RU2522578C2 |
| Способ получения полиморфа гидрохлорида 2-[4-(метиламинометил)фенил]-5-фтор-бензофуран-7-карбоксамида | 2018 |
|
RU2783418C1 |
| ПРОИЗВОДНОЕ ГЕТЕРОАРИЛ[4,3-с]ПИРИМИДИН-5-АМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2018 |
|
RU2764655C2 |
| ПРОИЗВОДНОЕ ПИРИДОПИРИМИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2019 |
|
RU2778524C2 |
| Триазолпиримидиновые соединения и их применение в лечении рака | 2019 |
|
RU2793249C2 |
| КРИСТАЛЛ ПИРРОЛОПИРИМИДИНА ДЛЯ ПОЛУЧЕНИЯ JAK-ИНГИБИТОРА | 2017 |
|
RU2746045C2 |
| ПИРИДОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ УКАЗАННЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2662713C2 |
| ПРОИЗВОДНОЕ ФЕНИЛПРОПАНАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2738207C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
Настоящее изобретение относится к способу получения соединения формулы (1): 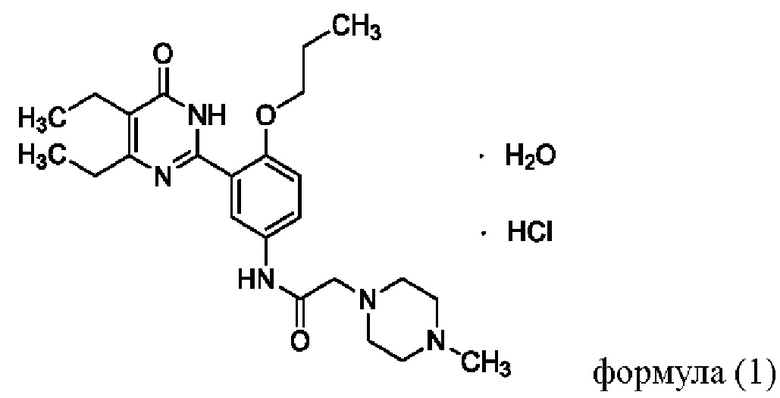 . Способ включает добавление соединения Z к спирту, добавление соляной кислоты, нагревание при температуре от 60 до 70°С, дополнительное добавление активированного угля для обесцвечивания, добавление сложного эфира к полученному фильтрату, отстаивание или перемешивание с последующим отделением осажденного твердого вещества с получением упомянутого соединения, причем соединение Z имеет следующую структурную формулу:
. Способ включает добавление соединения Z к спирту, добавление соляной кислоты, нагревание при температуре от 60 до 70°С, дополнительное добавление активированного угля для обесцвечивания, добавление сложного эфира к полученному фильтрату, отстаивание или перемешивание с последующим отделением осажденного твердого вещества с получением упомянутого соединения, причем соединение Z имеет следующую структурную формулу: 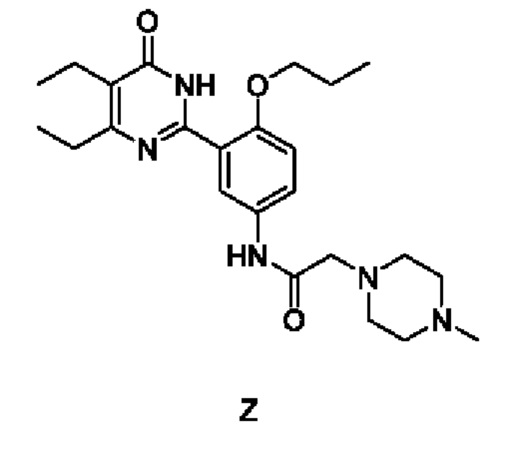 . Также предложены варианты способа получения соединения формулы (1). Технический результат изобретения заключается в осуществлении нового способа получения соединения формулы (1), которое обладает стабильными свойствами и низкой гигроскопичностью. 3 н. и 1 з.п. ф-лы, 11 ил., 6 табл., 16 пр.
. Также предложены варианты способа получения соединения формулы (1). Технический результат изобретения заключается в осуществлении нового способа получения соединения формулы (1), которое обладает стабильными свойствами и низкой гигроскопичностью. 3 н. и 1 з.п. ф-лы, 11 ил., 6 табл., 16 пр.
1. Способ получения соединения согласно формуле (1):
который осуществляется по Способу I, включающему: добавление соединения Z к спирту, добавление соляной кислоты, нагревание при температуре от 60 до 70°С, дополнительное добавление активированного угля для обесцвечивания, добавление сложного эфира к полученному фильтрату, отстаивание или перемешивание с последующим отделением осажденного твердого вещества с получением упомянутого соединения, причем соединение Z имеет следующую структурную формулу:
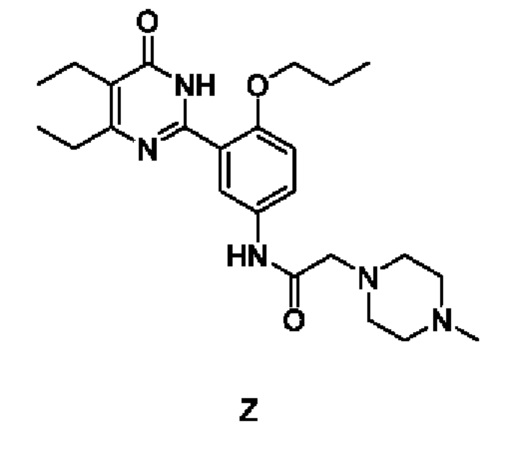
2. Способ по п. 1, в котором соединение Z получают следующим способом:
1) проведение реакции метилирования соединения Z-3 и диметилсульфата в присутствии основания в целях получения соединения Z-4;
2) проведение реакции соединения Z-4 и аммиака в целях получения соединения Z-5;
3) проведение реакции дегидратации и циклизации соединения Z-5 и метил-2-этил-3-оксопентаноата в присутствии основания в целях получения соединения Z
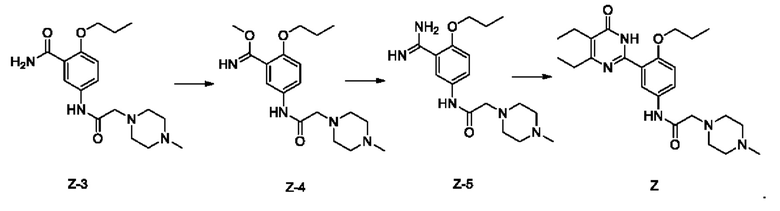
3. Способ получения соединения согласно формуле (1), который осуществляется по Способу II, включающему: добавление гидрохлорида соединения Z к спирту, его растворение путем нагревания, охлаждение для осаждения твердого вещества с последующим отделением с получением упомянутого соединения,
причем соединение Z имеет следующую структурную формулу:
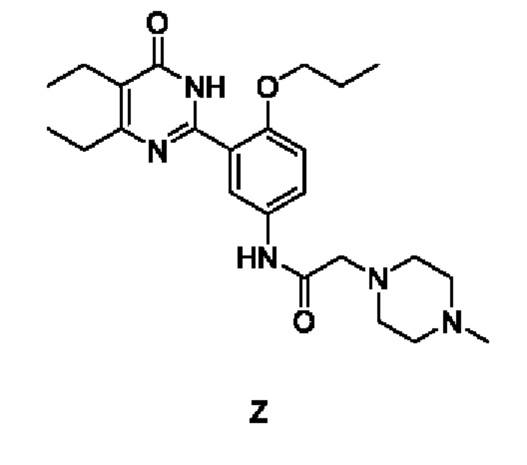
4. Способ получения соединения согласно формуле (1), который осуществляется по Способу III, включающему: добавление гидрохлорида соединения Z к смешанной системе из эфира и спирта, ее растворение путем нагревания, охлаждение для осаждения твердого вещества с последующим отделением с получением упомянутого соединения,
причем соединение Z имеет следующую структурную формулу:
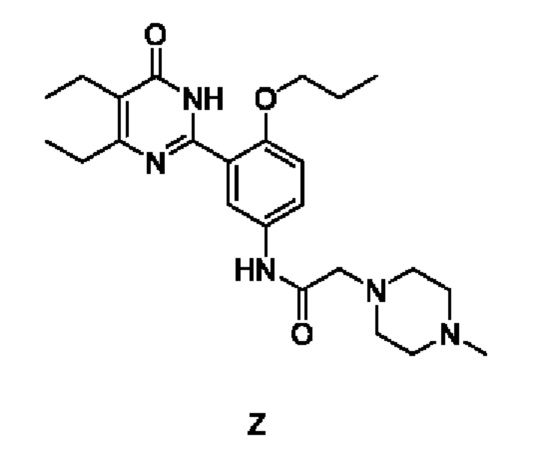
| WO 2010066111 A1, 17.06.2010 | |||
| Устройство для определения средневзвешенного значения функции | 1976 |
|
SU636626A1 |
| Сушилка для электротехнических изделий | 1989 |
|
SU1746171A1 |
| ПИРАЗИЛПИРИМИДИНОНЫ ДЛЯ ЛЕЧЕНИЯ ИМПОТЕНЦИИ | 1994 |
|
RU2373938C9 |

