ОБЛАСТЬ ТЕХНИКИ
[1] Настоящее изобретение относится к области техники лекарственных средств, и, в частности, относится к способу синтеза противоопухолевого соединения и его промежуточных соединений.
УРОВЕНЬ ТЕХНИКИ
[2] Помимо того, что рост и миграция опухолей связаны со сверхэкспрессией митотических киназ (например, киназы Aurora), они также зависят от образования большого количества новых кровеносных сосудов, причем ключевую роль в неоваскуляризации опухолей играет путь с участием VEGF/VEGFR (фактор роста эндотелия сосудов/рецептор фактора роста эндотелия сосудов).
[3] В патенте WO 2018108079 A1 раскрыт ряд соединений, способных ингибировать, модулировать и/или регулировать активность одной или более протеинкиназ, таких как киназы Aurora и киназы VEGFR, и изменять микроокружение опухоли, оказывать противоопухолевый иммунный эффект и противоопухолевый терапевтический эффект путем ингибирования роста и миграции опухолей. В данном патенте описывается способ синтеза соединения 29. Дальнейшие исследования показывают, что процесс синтеза еще нуждается в оптимизации, поэтому требуется найти способ синтеза, который отличается более простым процессом, более высоким выходом, более высокой чистотой и более низкими затратами и подходит для промышленного крупномасштабного производства.
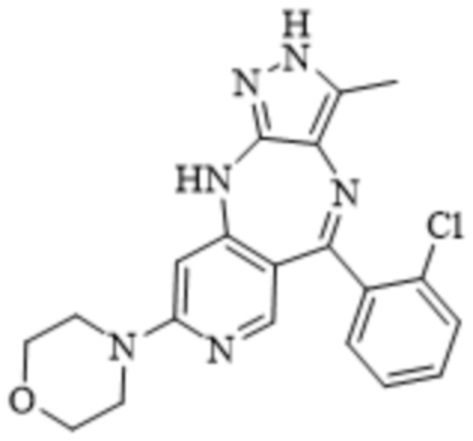
Соединение 29
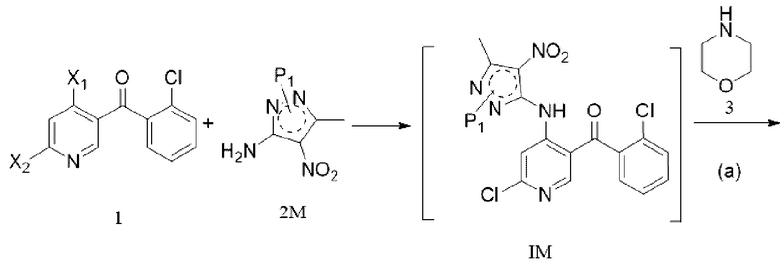
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[4] В первом аспекте в настоящем изобретении предлагаются следующие технические решения 1-15:
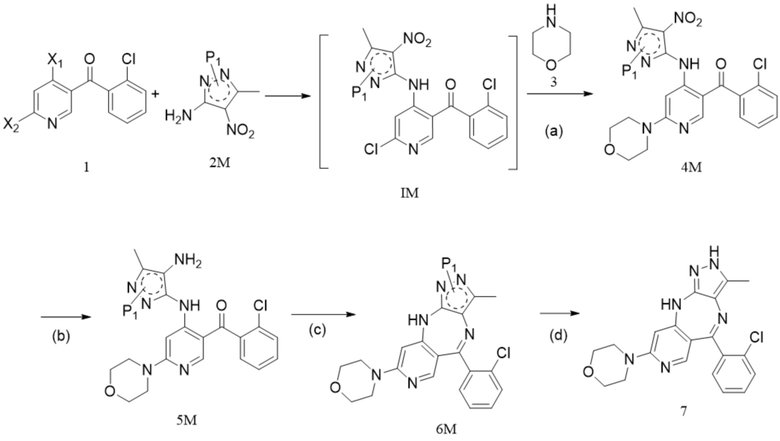
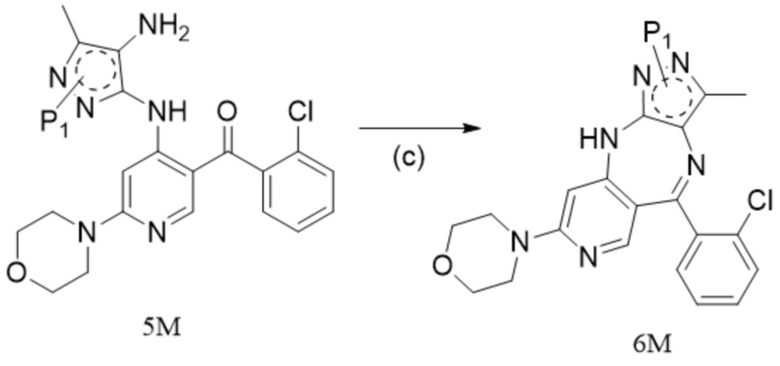
[5] 1. Способ синтеза противоопухолевого соединения формулы 7, включающий следующие стадии:
 ,
,
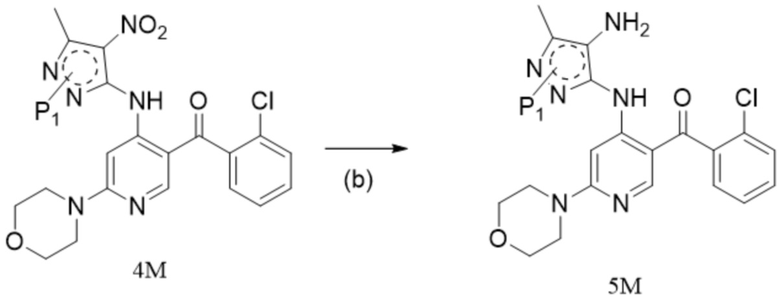
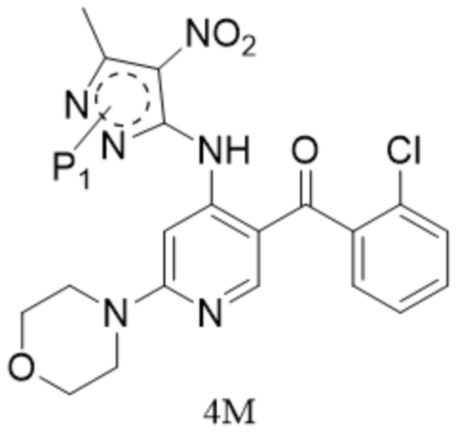
[6] (a) приведение соединения формулы 1 в реакцию ароматического нуклеофильного замещения соединением формулы 2M в органическом растворителе под действием основания cf получением соединения формулы IM, и приведение соединения формулы IM, отделенного или не отделенного, в другую реакцию ароматического нуклеофильного замещения соединением формулы 3, например, в щелочных условиях, с получением соединения формулы 4M;
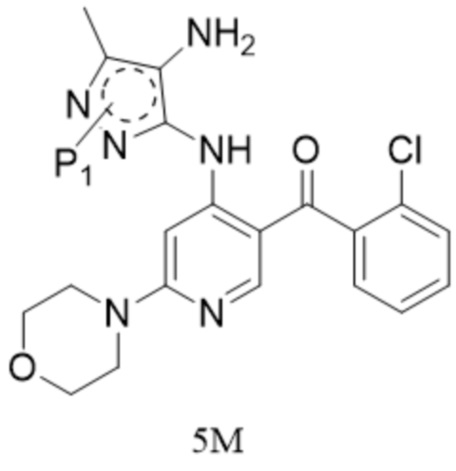
[7] (b) приведение соединения формулы 4M в реакцию восстановления в органическом растворителе под действием катализатора с получением соединения формулы 5M;
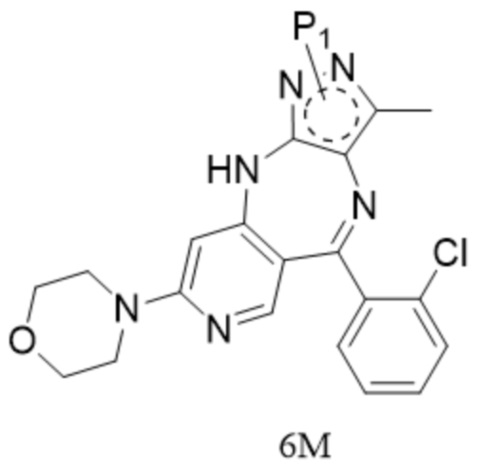
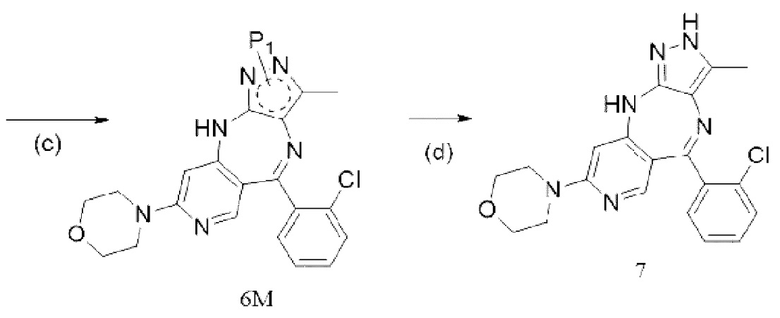
[8] (c) приведение соединения формулы 5M в реакцию замыкания кольца в органическом растворителе под действием кислоты с получением соединения формулы 6M; и
[9] (d) приведение соединения формулы 6M в реакцию в органическом растворителе для удаления аминозащитной группы с получением противоопухолевого соединения формулы 7;
[10] схема реакции является следующей:
[11] где каждый X1 и X2 выбраны из галогена; P1 представляет собой аминозащитную группу, и предпочтительно P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например, Boc, Cbz, Tos, Fmoc, PMB, Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1;
[12] 2. Способ синтеза противоопухолевого соединения формулы 7,
,
[13] включающий следующую стадию:
[14] (d) приведение соединения формулы 6M в реакцию в органическом растворителе для удаления аминозащитной группы с получением противоопухолевого соединения формулы 7;
[15] схема реакции является следующей:
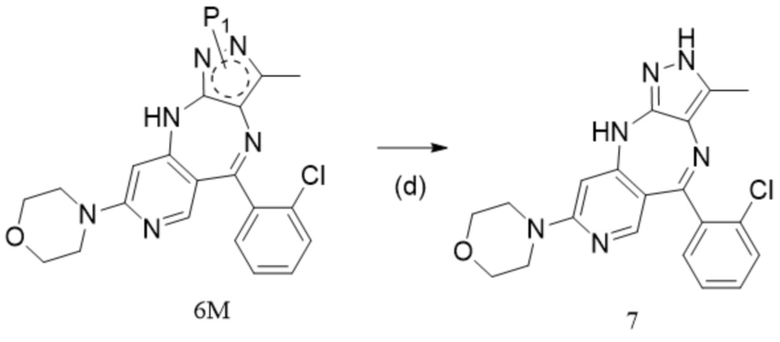
[16] где P1 представляет собой аминозащитную группу и предпочтительно P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например, Boc, Cbz, Tos, Fmoc, PMB, Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1;
[17] 3. Способ синтеза промежуточного соединения формулы 6M или способ по любому из предшествующих технических решений,
 ,
,
[18] включающий следующую стадию:
[19] (c) приведение соединения формулы 5M в реакцию замыкания кольца в органическом растворителе под действием кислоты с получением соединения формулы 6M;
[20] схема реакции является следующей:
[21] где P1 представляет собой аминозащитную группу и предпочтительно P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например, Boc, Cbz, Tos, Fmoc, PMB, Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1;
[22] 4. Способ синтеза промежуточного соединения формулы 5M или способ по любому из предшествующих технических решений,
 ,
,
[23] включающий следующую стадию:
[24] (b) приведение соединения формулы 4M в реакцию восстановления в органическом растворителе под действием катализатора с получением соединения формулы 5M;
[25] схема реакции является следующей:
[26] где P1 представляет собой аминозащитную группу и предпочтительно P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например, Boc, Cbz, Tos, Fmoc, PMB, Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1;
[27] 5. Способ синтеза промежуточного соединения формулы 4M или способ по любому из предшествующих технических решений,
[28] включающий следующую стадию:
[29] (a) приведение соединения формулы 1 в реакцию ароматического нуклеофильного замещения соединением формулы 2M в органическом растворителе под действием основания с получением соединения формулы IM, и приведение соединения формулы IM, отделенного или не отделенного, в другую реакцию ароматического нуклеофильного замещения соединением формулы 3, например, в щелочных условиях, с получением соединения формулы 4M;
[30] схема реакции является следующей:
[31] где каждый X1 и X2 выбраны из галогена; P1 представляет собой аминозащитную группу, и предпочтительно P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например, Boc, Cbz, Tos, Fmoc, PMB, Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1;
[32] 6. Способ по любому из предшествующих технических решений, в котором на стадии (a) органический растворитель выбран из одного или более из 2-метилтетрагидрофурана, ацетонитрила, тетрагидрофурана и толуола; основание выбрано из одного или более из гидрида натрия, гидроксида натрия, карбоната цезия, триэтилендиамина, трет-бутоксида натрия, трет-бутоксида калия, бис(триметилсилил)амида лития и гексаметилдисилазида натрия;
[33] необязательно на стадии (a);
[34] реакцию проводят при давлении от 0 МПа до 10 МПа (избыточное давление), например, при атмосферном давлении;
[35] реакцию проводят в течение периода времени 1-96 ч, например, 17 ч; реакцию проводят при температуре, которая может быть температурой кипения растворителя;
[36] в частности, реакцию ароматического нуклеофильного замещения соединения формулы 1 соединением формулы 2M под действием основания проводят в течение периода времени 3-18 ч, например 3-8 ч, при температуре от 40°C до 70 °C, например от 40°C до 60 °C;
[37] в частности, другую ароматическую реакцию нуклеофильного замещения соединением формулы 3 проводят в течение периода времени от 8 ч до 16 ч, например от 13 ч до 16 ч, при температуре, которая может быть температурой кипения растворителя;
[38] соотношение следующее:
[39] растворитель : соединение формулы 1 : соединение формулы 2М : соединение формулы 3 : основание=(0,5-60) л : (0,1-11) моль : 1 моль : (0,3-30) моль : (0,2-25) моль, например 5,8 л : 1,1 моль : 1 моль : 3 моль : 2,5 моль;
[40] в частности, добавляют сульфат магния в реакцию ароматического нуклеофильного замещения соединения формулы 1 соединением формулы 2М под действием основания, и добавляют уксусную кислоту в другую реакцию ароматического нуклеофильного замещения соединением формулы 3,
[41] соотношение следующее:
[42] растворитель : соединение формулы 1 : соединение формулы 2М : соединение формулы 3 : основание:сульфат магния : уксусная кислота=(0,5-60) л:(0,1-11) моль : 1 моль : (0,3-30) моль : (0,2-25) моль : (0,2-20) моль : (0,1-15) моль, например 5,8 л : 1,1 моль : 1 моль : 3 моль : 2,5 моль : 1,6 моль : 1,5 моль;
[43] 7. Способ по любому из предшествующих технических решений, в котором на стадии (b) органический растворитель выбран из одного или более из метанола, этанола, изопропанола, тетрагидрофурана, 2-метилтетрагидрофурана, уксусной кислоты и ацетонитрила; катализатор выбран из порошка железа, оксида платины, Pt/C, Pd(OH)2/C, Rh/C и Pd/C;
[44] необязательно на стадии (b), когда катализатор представляет собой порошок железа, реакцию проводят в течение периода времени 1-96 ч, например 15-24 ч, при давлении от 0 МПа до 10 МПа (избыточное давление), например при атмосферном давлении, при температуре, которая является температурой кипения растворителя;
[45] соотношение следующее:
[46] органический растворитель : соединение формулы 4M : катализатор=(1-130) л : 1 моль : (1-150) моль, например 13 л : 1 моль : (10-18) моль, например 13 л : 1 моль : (14-18) моль, например 13 л : 1 моль : 15 моль;
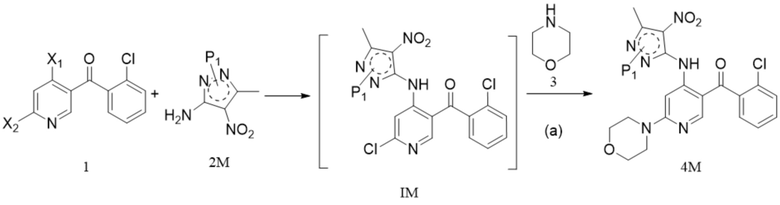
[47] в частности, когда катализатор представляет собой порошок железа: на стадии (b) добавляют также хлорид аммония в количестве 0,05-5 моль, например 0,5 моль; растворитель представляет собой смешанный растворитель из этанола и тетрагидрофурана, например в объемном соотношении этанола и тетрагидрофурана (6-10) : 10;
[48] в частности, на стадии (b), когда катализатор представляет собой оксид платины, Pt/C, Pd(OH)2/C, Rh/C или Pd/C, реакцию проводят в атмосфере водорода при температуре от 30°C до 80°C в течение 20-80 ч; например реакцию проводят в атмосфере водорода при температуре от 60°C до 80°C в течение 20-72 ч; реакцию проводят при давлении от 0 МПа до 10 МПа (избыточное давление), например 0,5-2 МПа; соотношение следующее: растворитель : соединение формулы 4М : катализатор=(1-50) мл : 1 г : (0,03-0,2) г, например 22 мл : 1 г: 0,1 г;
[49] необязательно на стадии (b) добавляют также небольшое количество воды; например объемное соотношение воды и тетрагидрофурана составляет (0,2-2) : 10;
[50] 8. Способ по любому из предшествующих технических решений, в котором на стадии (c) кислота выбрана из одной или более из трифторуксусной кислоты, метансульфоновой кислоты, п-толуолсульфоновой кислоты, муравьиной кислоты, уксусной кислоты и хлористоводородной кислоты; органический растворитель выбран из по меньшей мере одного из дихлорметана, метанола, этанола и изопропанола;
[51] необязательно на стадии (c) реакцию проводят в течение периода времени 1-96 ч, например 1,5-3 ч, при давлении от 0 МПа до 10 МПа (избыточное давление), например при атмосферном давлении, при температуре, которая является температурой кипения растворителя; соотношение следующее: растворитель : соединение формулы 5M : кислота=(0,5-70) л : 1 моль : 0,220 моль, например 6,6 л : 1 моль : 2 моль;
[52] 9. Способ по любому из предшествующих технических решений, в котором, в частности, стадия (c) представляет собой: выдерживание соединения формулы 5M при температуре кипения изопропанола под действием трифторуксусной кислоты в течение от 1 ч до 3 ч;
[53] 10. Способ по любому из предшествующих технических решений, в котором на стадии (d) органический растворитель выбран из одного или более из толуола, дихлорметана, ацетонитрила, тетрагидрофурана, метанола, этанола и изопропанола;
[54] на стадии (d) добавляют также кислоту, и кислота выбрана из по меньшей мере одной из муравьиной кислоты, уксусной кислоты, хлористоводородной кислоты, метансульфоновой кислоты и трифторуксусной кислоты;
[55] на стадии (d) в дополнение к растворителю добавляют спирт или фенол; спирт выбран из метанола и/или этанола, и фенол выбран из фенола и/или п-метоксифенола;
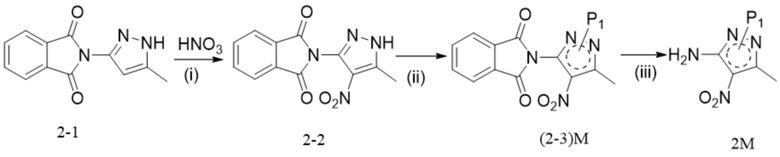
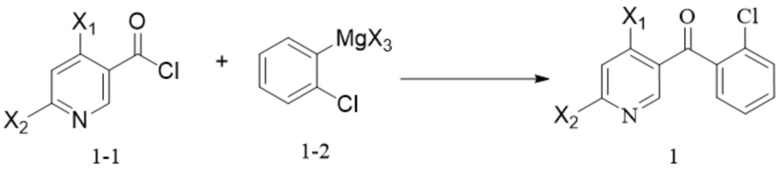
[56] необязательно на стадии (d) реакцию проводят в течение периода времени 1-96 ч, например 17 ч, при давлении от 0 МПа до 10 МПа (избыточное давление), например при атмосферном давлении, при температуре, которая является температурой кипения растворителя; соотношение следующее: растворитель : соединение формулы 6M : кислота : спирт или фенол=(10-800) л : 1 моль : (0,1-10) моль : (0,2-20) моль, например 84 л : 1 моль : 1 моль : 2 моль;
[57] 11. способ по любому из предшествующих технических решений, дополнительно включающий стадии синтеза соединения формулы 2M:
[58] (i) приведение соединения формулы 2-1 (например, 1 молярный эквивалент) в реакцию нитрования с азотной кислотой (например, 1-2 молярных эквивалента) в растворителе, таком как концентрированная серная кислота, уксусный ангидрид или уксусная кислота, в течение периода времени 0,5-2 ч при температуре 10-20°C с получением соединения формулы 2-2;
[59] (i) приведение соединения формулы 2-2 (например, 1 молярный эквивалент) в реакцию защиты аминогруппы с аминозащитным реактивом (например, 1-1,5 молярных эквивалента) в органическом растворителе, таком как тетрагидрофуран, 2-метилтетрагидрофуран, дихлорметан и ацетонитрил, в течение периода времени 1-48 ч при температуре 60-100°C с получением соединения формулы (2-3)M; и
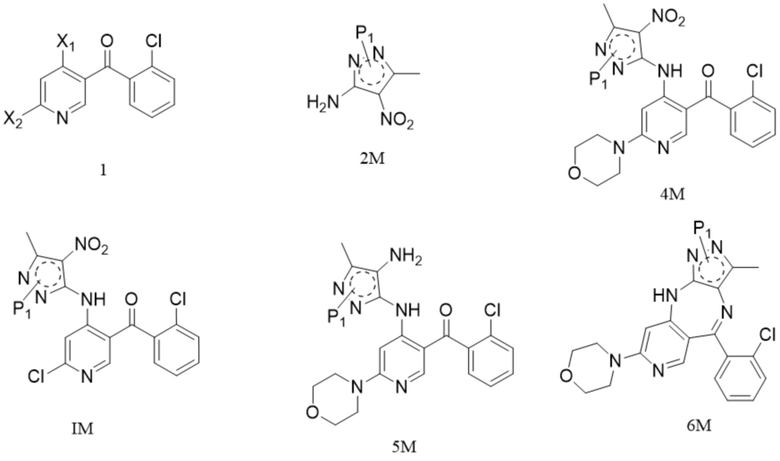
[60] (iii) приведение соединения формулы (2-3)M (например, 1 молярный эквивалент) в реакцию в органическом растворителе, таком как один или более из этанола, тетрагидрофурана и ацетонитрила, например в течение периода времени 1-3 ч при температуре, которая является температурой кипения растворителя, для удаления фталоильной группы с получением соединения формулы 2M;
[61] схема реакции является следующей:
 ;
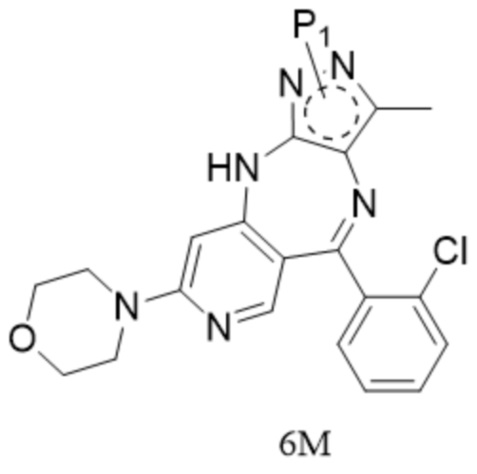
;
[62] причем аминозащитный реактив представляет собой Boc2O, CbzCl, TosCl, FmocCl, PMBBr, MOMCl, EOMCl, трет-бутанол, изобутилен, BnCl, уксусный ангидрид, SEMCl, TrtCl или DHP, например Boc2O, CbzCl, TosCl, FmocCl, PMBBr, TrtCl или DHP; P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например Boc, Cbz, Tos, Fmoc, PMB, Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1;
[63] 12. Способ по техническому решению 11, причем
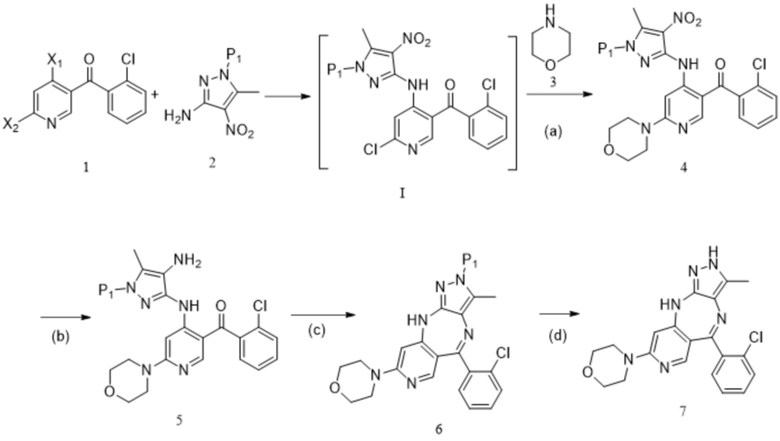
[64] когда аминозащитный реактив представляет собой TrtCl, реакцию на стадии (ii) проводят в следующих условиях: под действием основания при температуре от 60°C до 100°C в течение от 1 ч до 48 ч;
[65] когда аминозащитный реактив представляет собой DHP, реакцию на стадии (ii) проводят в следующих условиях: под действием п-толуолсульфоновой кислоты или п-толуолсульфоната пиридиния при температуре от 60°C до 100°C с обратным холодильником в течение от 3 ч до 48 ч;
[66] 13. Способ по любому из предшествующих технических решений, дополнительно включающий стадию синтеза соединения формулы 1:
[67] приведение соединения формулы 1-1 (например, 1-2 молярных эквивалента) в реакцию сочетания с соединением формулы 1-2 (например, 1-2 молярных эквивалента) в органическом растворителе, таком как THF, в течение периода времени 2-4 ч при температуре от -70°C до 60°C с получением соединения формулы 1; и
[68] схема реакции является следующей:
 ;
;
[69] где каждый X1, X2 и X3 выбраны из галогена; предпочтительно каждый X1, X2 и X3 выбраны из хлора;
[70] 14. Промежуточное соединение для получения противоопухолевого соединения формулы 7, имеющее следующие структурные формулы:
 ;
;
[71] где каждый X1 и X2 независимо выбраны из галогена;
[72] P1 выбран из Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1; и
[73] 15. Промежуточное соединение для получения противоопухолевого соединения формулы 7, имеющее следующую структурную формулу:
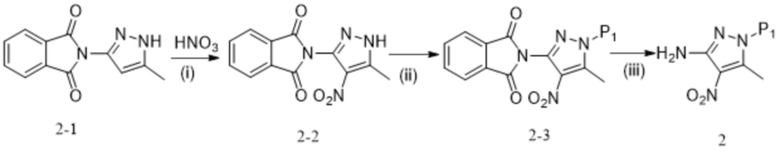
[74] где P1 выбран из Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1;
[75] формула 6М представляет собой комплекс, содержащий одну молекулу трифторуксусной кислоты и одну молекулу изопропанола.
[76] Во втором аспекте настоящее изобретение предназначено для предоставления способа синтеза противоопухолевого соединения формулы 7, который включает следующие стадии:
,
[77] (a) приведение соединения формулы 1 в реакцию ароматического нуклеофильного замещения соединением формулы 2 в органическом растворителе под действием основания, и затем в другую реакцию ароматического нуклеофильного замещения соединением формулы 3, например, в щелочных условиях, с получением соединения формулы 4;
[78] (b) приведение соединения формулы 4 в реакцию восстановления в органическом растворителе под действием катализатора с получением соединения формулы 5;
[79] (c) приведение соединения формулы 5 в реакцию замыкания кольца в органическом растворителе под действием кислоты с получением соединения формулы 6; и
[80] (d) приведение соединения формулы 6 в реакцию в органическом растворителе для удаления аминозащитной группы с получением противоопухолевого соединения формулы 7;
[81] схема реакции является следующей:
[82] где каждый X1 и X2 независимо выбраны из галогена; P1 представляет аминозащитную группу, выбранную из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например Boc, Cbz, Tos, Fmoc, PMB, Trt и THP.
[83] В одном варианте осуществления изобретения на стадии (a) органический растворитель выбран из одного или более из 2-метилтетрагидрофурана, ацетонитрила, тетрагидрофурана и толуола; основание выбрано из одного или более из гидрида натрия, гидроксида натрия, карбоната цезия, триэтилендиамина, трет-бутоксида натрия, трет-бутоксида калия, бис(триметилсилил)амида лития и гексаметилдисилазида натрия.
[84] В одном варианте осуществления изобретения на стадии (b) органический растворитель выбран из одного или более из метанола, этанола, изопропанола, тетрагидрофурана, 2-метилтетрагидрофурана и ацетонитрила; катализатор выбран из порошка железа, оксида платины, Pt/C и Pd/C.
[85] В одном варианте осуществления изобретения на стадии (c) кислота выбрана из одной или более из трифторуксусной кислоты, метансульфоновой кислоты, п-толуолсульфоновой кислоты, муравьиной кислоты, уксусной кислоты и хлористоводородной кислоты; органический растворитель выбран из по меньшей мере одного из дихлорметана, метанола, этанола и изопропанола.
[86] В одном варианте осуществления изобретения конкретно стадия (c) представляет собой: выдерживание соединения формулы 5 в изопропаноле при температуре его кипения под действием трифторуксусной кислоты в течение от 1 ч до 3 ч, например от 1,5 ч до 3 ч.
[87] В одном варианте осуществления изобретения на стадии (d) органический растворитель выбран из одного или более из толуола, дихлорметана, ацетонитрила, тетрагидрофурана, метанола, этанола и изопропанола;
[88] на стадии (d) добавляют также катализатор, и катализатор выбран из по меньшей мере одной из муравьиной кислоты, уксусной кислоты, хлористоводородной кислоты, метансульфоновой кислоты и трифторуксусной кислоты;
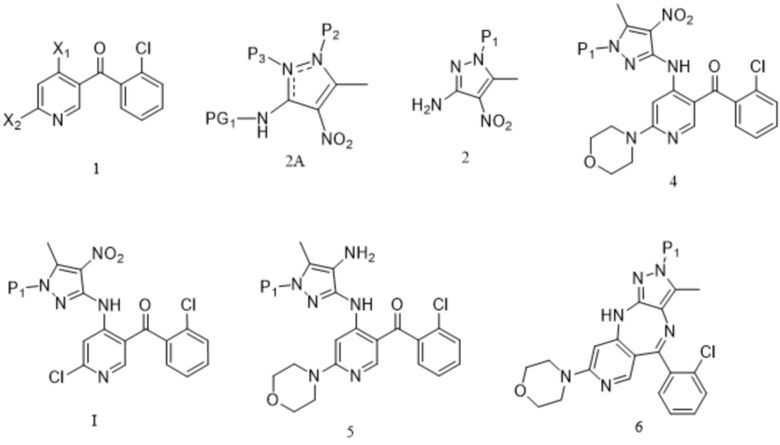
[89] на стадии (d) добавляют спирт или фенол; спирт выбран из метанола и/или этанола, и фенол выбран из фенола и/или п-метоксифенола.
[90] В одном варианте осуществления изобретения дополнительно содержатся стадии синтеза соединения формулы 2:
[91] (i) приведение соединения формулы 2-1 в реакцию нитрования с азотной кислотой в растворителе с получением соединения формулы 2-2;
[92] (ii) приведение соединения формулы 2-2 в реакцию защиты аминогруппы с аминозащитным реактивом в органическом растворителе с получением соединения формулы 2-3; и
[93] (iii) приведение соединения формулы 2-3 в реакцию в органическом растворителе для удаления фталоильной группы с получением соединения формулы 2;
[94] схема реакции является следующей:
 ;
;
[95] причем аминозащитный реактив представляет собой Boc2O, CbzCl, TosCl, FmocCl, PMBBr, MOMCl, EOMCl, трет-бутанол, изобутилен, BnCl, уксусный ангидрид, SEMCl, TrtCl или DHP, например Boc2O, CbzCl, TosCl, FmocCl, PMBBr, TrtCl или DHP; P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например Boc, Cbz, Tos, Fmoc, PMB, Trt и THP.
[96] В одном варианте осуществления изобретения, когда аминозащитный реактив представляет собой TrtCl, реакцию на стадии (ii) проводят в следующих условиях: под действием основания при температуре от 60°C до 100°C в течение от 1 ч до 48 ч;
[97] когда аминозащитный реактив представляет собой DHP, реакцию на стадии (ii) проводят в следующих условиях: под действием п-толуолсульфоновой кислоты или п-толуолсульфоната пиридиния при температуре от 60°C до 100°C с обратным холодильником в течение от 3 ч до 48 ч.
[98] В одном варианте осуществления изобретения дополнительно содержатся стадии синтеза соединения формулы 1:
[99] приведение соединения формулы 1-1 в реакцию сочетания с соединением формулы 1-2 в органическом растворителе с получением соединения формулы 1;
[100] схема реакции является следующей:
;
[101] где каждый X1, X2 и X3 выбраны из галогена; предпочтительно каждый X1, X2 и X3 выбраны из хлора;
[102] Во третьем аспекте в настоящем изобретении также предлагается промежуточное соединение для получения противоопухолевого соединения формулы 7, которое имеет следующие структурные формулы:
 ;
;
[103] где каждый X1 и X2 независимо выбраны из галогена;
[104] P1 выбран из Trt и THP;
[105] P2 выбран из Trt и THP, и отсутствует P3; или P3 выбран из Trt и THP, и отсутствует P2;
[106] PG1 представляет собой ацетил, или PG1-NH- представляет  ;
;
[107]  показывает возможную одинарную связь или двойную связь.
показывает возможную одинарную связь или двойную связь.
[108] В четвертом аспекте в настоящем изобретении предлагается промежуточное соединение для получения противоопухолевого соединения формулы 7, которое имеет следующую структурную формулу:
[109] где P1 выбран из Trt и THP;
[110] формула 6 представляет собой комплекс, содержащий одну молекулу трифторуксусной кислоты и одну молекулу изопропанола.
[111] По сравнению с известным уровнем техники, настоящее изобретение обладает преимуществами, описанными ниже.
[112] (1) Настоящее изобретение отличается от существующего процесса синтеза, в котором снятие защитной группы происходит до образования трициклического кольца, тем, что принят процесс, в котором трициклическое кольцо синтезируется до снятия защитной группы. В этом процессе промежуточное соединение (соединение формулы 2) используется в качестве исходного вещества и приводится в реакции ароматического нуклеофильного замещения и реакцию восстановления с получением соединения формулы 5, посредством чего могут достигать процесса, в котором образование трициклического кольца предшествует снятию защитной группы, тем самым позволяя избегать недостатков существующего процесса, а именно: a) при предварительном снятии защитной группы требуется большое количество трифторуксусной кислоты, что приводит к накоплению стоков; b) при последующем образовании трициклического кольца для восстановления используется хлорид олова, что приводит к проблемам с реализуемостью и сложной отработке; c) для процесса необходимо применение колоночной хроматографии, что в целом усложняет процесс.
[113] (2) Способ синтеза по настоящему изобретению обладает улучшенной реализуемостью и позволяет упростить процесс, в котором более высокой чистоты могут достигать без сложного процесса перекристаллизации синтезированного соединения формулы 7, и, таким образом, образуется меньше трех отходов, что делает данный способ более подходящим для промышленного крупномасштабного производства.
[114] (3) Когда промежуточные соединения по настоящему изобретению используют для получения противоопухолевого соединения, во время реакций эффективно снижается содержание побочных продуктов, и, таким образом, улучшается общий выход реакций, и он по меньшей мере в два раза выше, чем выход, полученный с использованием способа синтеза соединения 29 в патенте WO2018108079A1.
ПОДРОБНОЕ ОПИСАНИЕ
[115] Приведенное выше описание настоящего изобретения более подробно поясняется следующим описанием конкретных вариантов осуществления изобретения, однако не следует подразумевать, что объем настоящего изобретения ограничен следующими примерами. Все способы, реализованные на основе приведенного выше описания настоящего изобретения, входят в объем настоящего изобретения.
[116] Сокращения, используемые в данном документе, имеют следующие общепринятые в данной области техники значения:
[117] «Галоген» относится к фтору, хлору, брому, йоду и т. д.
[118] «Boc2O» относится к ди-трет-бутилдикарбонату;
[119] «Boc» относится к трет-бутилоксикарбонилу;
[120] «CbzCl» относится к бензилхлорформиату;
[121] «Cbz» относится к бензилоксикарбонилу;
[122] «TosCl» относится к п-толуолсульфонилхлориду;
[123] «Tos» относится к п-толуолсульфонилу;
[124] «FmocCl» относится к 9-флуоренилметилхлорформиату;
[125] «Fmoc» относится к 9-флуоренилметоксикарбонилу;
[126] «PMBBr» относится к п-метоксибензилбромиду;
[127] «PMB» относится к п-метоксибензилу;
[128] «MOMCl» относится к хлорметилметиловому эфиру;
[129] «MOM» относится к метоксиметилу;
[130] «EOMCl» относится к хлорметилэтиловому эфиру;
[131] «EOM» относится к этоксиметилу;
[132] «tBu» относится к трет-бутилу;
[133] «BnCl» относится к бензилхлориду;
[134] «Bn» относится к бензилу;
[135] «Ac» относится к ацетилу;
[136] «SEMCl» относится к 2-(триметилсилил)этоксиметилхлориду;
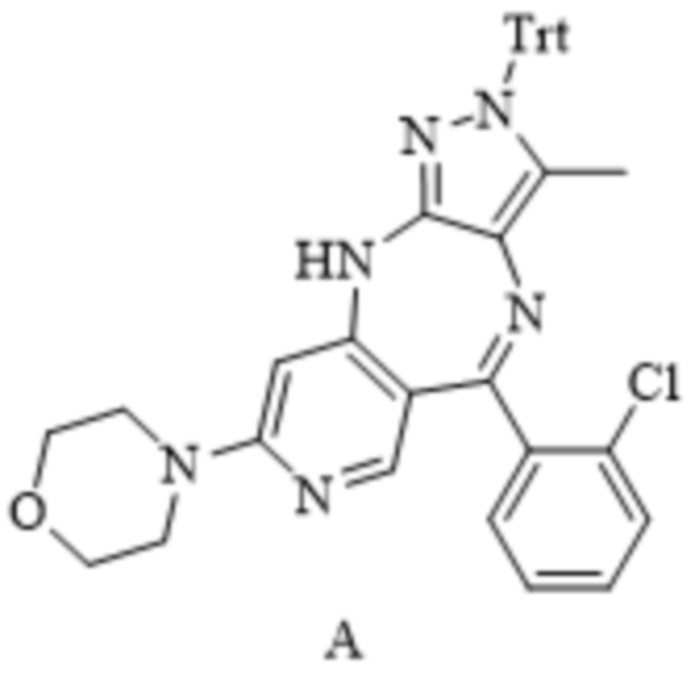
[137] «SEM» относится к 2-(триметилсилил)этоксиметилу;
[138] «TrtCl» относится к трифенилхлорметану;
[139] «Trt» относится к трифенилметилу;
[140] «DHP» относится к 3,4-дигидро-2H-пирану;
[141] «THP» относится к 2-тетрагидропиранилу;
[142] «THF» относится к тетрагидрофурану;
[143] «2-MeTHF» относится к диметилтетрагидрофурану;
[144] «MTBE» относится к метил-трет-бутиловому эфиру;
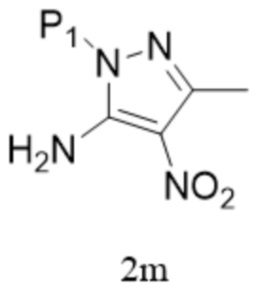
[145] «ACN» относится к ацетонитрилу;
[146] «TEA» относится к триэтиламину;
[147] «TFA» относится к трифторуксусной кислоте;
[148] «IPA» относится к изопропанолу;
[149] «TEMPO» относится к 2,2,6,6-тетраметилпиперидин-1-оксиду;
[150] «DCM» относится к дихлорметану;
[151] «ТСХ» относится к тонкослойной хроматографии;
[152] «H-ЯМР» относится к спектроскопии протонного ядерного магнитного резонанса;
[153] «DMSO» относится к диметилсульфоксиду;
[154] «ВЭЖХ» относится к высокоэффективной жидкостной хроматографии;
[155] «PE» относится к петролейному эфиру;
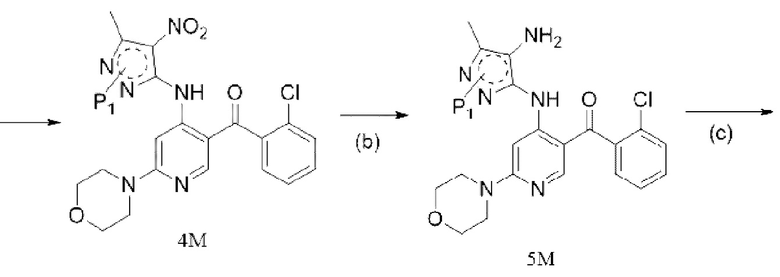
[156] «EA» относится к этилацетату;
[157] «ЖХ-МС» относится к жидкостной хроматографии - масс-спектрометрии.
[158] В настоящем изобретении катализатор включает вещества, способные изменять скорость реакции в обычных условиях, а также вещества, выполняющие в реакции окислительно-восстановительные или кислотно-основные функции.
[159] В настоящем изобретении реакцию проводят при атмосферном давлении, если не указано иное.
[160] В настоящем изобретении температура, при которой проводят реакцию, относится к самой высокой температуре, которая достигается в ходе реакции. Весь процесс реакции или часть процесса реакции проводят при этой самой высокой температуре.
[161] В настоящем изобретении, когда упоминается «соотношение», например растворитель : соединение формулы 1 : соединение формулы 2М : соединение формулы 3 : основание=5,8 л : 1,1 моль : 1 моль : 3 моль : 2,5 моль, такое выражение относится к пропорциональной взаимосвязи между добавленными в реакции количествами веществ, а именно, добавляют 1 моль соединения формулы 2М, и соответственно добавленные количества растворителя, соединения формулы 1, соединения формулы 3 и основания составляют 5,8 л, 1,1 моль, 3 моль и 2,5 моль; соответственно, если добавленное количество соединения формулы 2М увеличивается или уменьшается, добавленные количества других веществ увеличиваются или уменьшаются пропорционально. Добавленное количество каждого вещества относится к общему количеству этого вещества, добавленного в реакции. Например, если вещество добавляют одной порцией, добавленное количество относится к количеству этой порции, а если вещество добавляют несколькими порциями, добавленное количество относится к общему количеству этих нескольких порций. Вещества, указанные в соотношении, могут добавлять одновременно или по отдельности.
[162] В настоящем изобретении химическая структурная формула представляет соединение этой формулы в форме свободного основания или свободной кислоты, или гидрата, сольвата, соли кислоты или соли основания, или их комбинации. Например, приведенная ниже формула А может включать свободную форму основания формулы А, а также различные комплексы формулы А, например комплекс, состоящий из одной молекулы трифторуксусной кислоты и одной молекулы изопропанола (как показано ниже в формуле В).
 ,
, 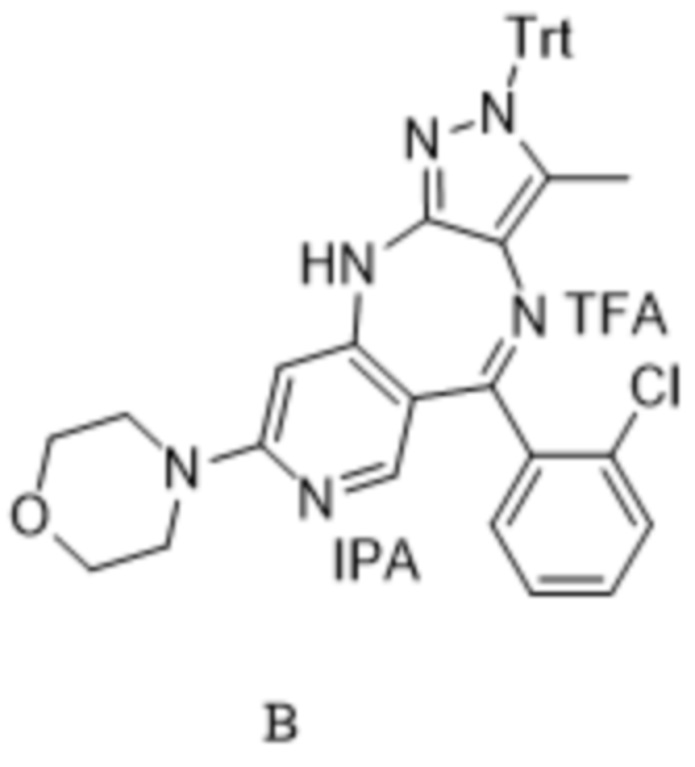 .
.
[163] В настоящем изобретении предлагается способ синтеза противоопухолевого соединения формулы 7, который включает следующие стадии:
,
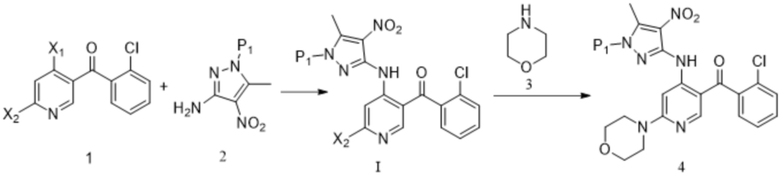
[164] (a) приведение соединения формулы 1 в реакцию ароматического нуклеофильного замещения соединением формулы 2M в органическом растворителе под действием основания с получением соединения формулы IM, и приведение соединения формулы IM, отделенного или не отделенного, в другую реакцию ароматического нуклеофильного замещения соединением формулы 3, например, в щелочных условиях, с получением соединения формулы 4M;
[165] (b) приведение соединения формулы 4M в реакцию восстановления в органическом растворителе под действием катализатора с получением соединения формулы 5M;
[166] (c) приведение соединения формулы 5M в реакцию замыкания кольца в органическом растворителе под действием кислоты с получением соединения формулы 6M; и
[167] (d) приведение соединения формулы 6M в реакцию в органическом растворителе для удаления аминозащитной группы с получением противоопухолевого соединения формулы 7;
[168] схема реакции является следующей:
[169] где каждый X1 и X2 выбраны из галогена; P1 представляет собой аминозащитную группу, и предпочтительно P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например Boc, Cbz, Tos, Fmoc, PMB, Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1.
[170] В некоторых вариантах осуществления изобретения соединение формулы 2M включает соединение формулы 2 и соединение формулы 2m, как показано ниже:
[171] 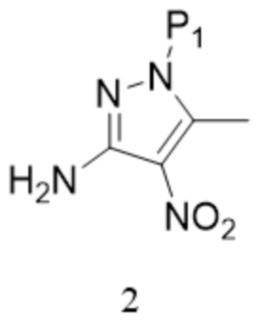 ,
,  ; предпочтительно соединение формулы 2M представляет собой соединение формулы 2.
; предпочтительно соединение формулы 2M представляет собой соединение формулы 2.
[172] Соединение формулы IM включает соединение формулы I и соединение формулы Im, как показано ниже:
[173] 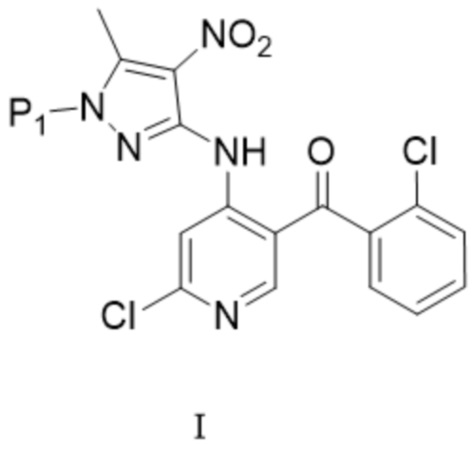 ,
, 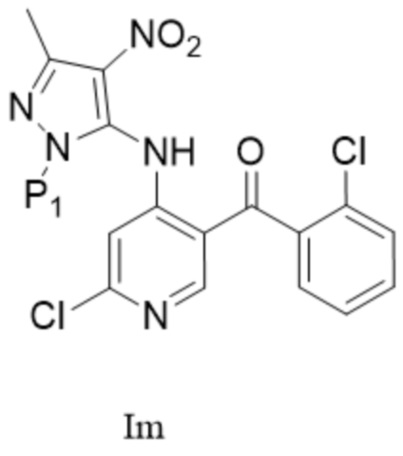 ; предпочтительно соединение формулы IM представляет собой соединение формулы I.
; предпочтительно соединение формулы IM представляет собой соединение формулы I.
[174] Соединение формулы 4M включает соединение формулы 4 и соединение формулы 4m, как показано ниже:
[175] 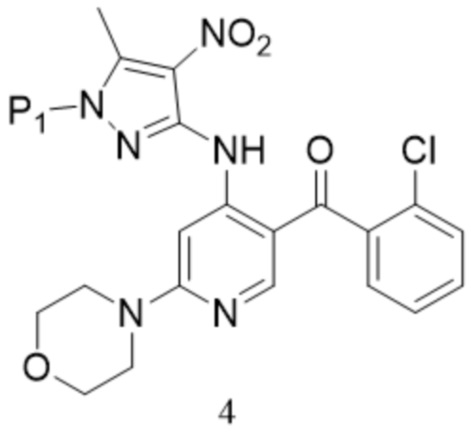 ,
, 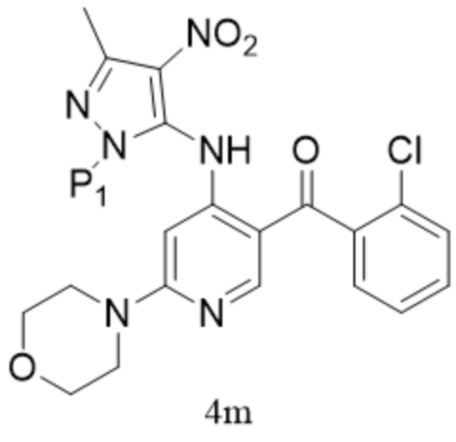 ; предпочтительно соединение формулы 4M представляет собой соединение формулы 4.
; предпочтительно соединение формулы 4M представляет собой соединение формулы 4.
[176] Соединение формулы 5M включает соединение формулы 5 и соединение формулы 5m, как показано ниже:
[177] 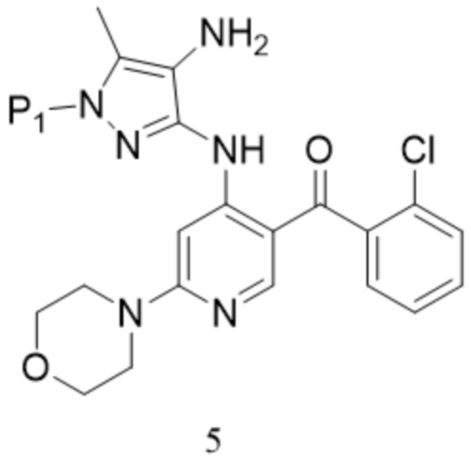 ,
, 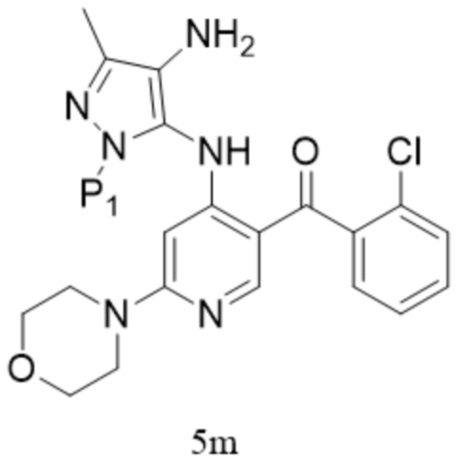 ; предпочтительно соединение формулы 5M представляет собой соединение формулы 5.
; предпочтительно соединение формулы 5M представляет собой соединение формулы 5.
[178] Соединение формулы 6M включает соединение формулы 6 и соединение формулы 6m, как показано ниже:
[179] 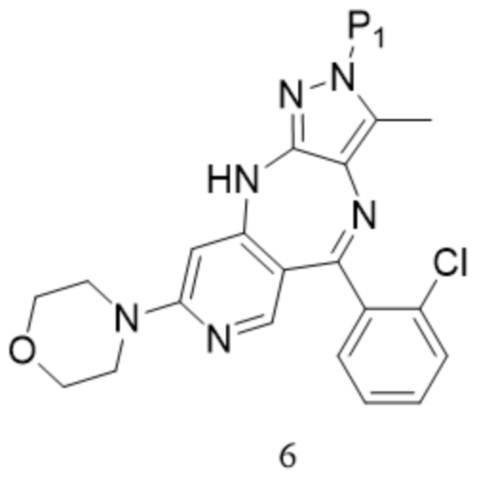 ,
, 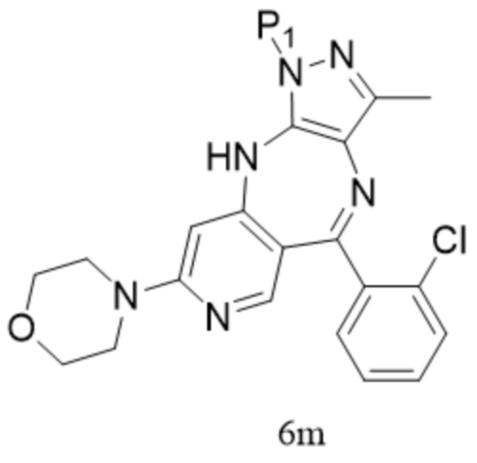 ; предпочтительно соединение формулы 6M представляет собой соединение формулы 6.
; предпочтительно соединение формулы 6M представляет собой соединение формулы 6.
[180] Предпочтительно P1 выбран из Trt и THP.
[181] В некоторых вариантах осуществления изобретения способ синтеза соединения формулы 4 содержит стадию (a): приведение соединения формулы 1 в реакцию ароматического нуклеофильного замещения соединением формулы 2 в органическом растворителе под действием основания с получением соединения формулы I, и приведение не отделенного соединения формулы IM, в другую реакцию ароматического нуклеофильного замещения соединением формулы 3, например, в щелочных условиях, с получением соединения формулы 4.
[182] В настоящем изобретении при синтезе соединения формулы 4, продукт I реакции соединения формулы 1 с соединением формулы 2 не нужно отделять, а непосредственно проводить в реакцию с соединением формулы 3, поэтому упрощается процесс и улучшается выход.
[183] В некоторых вариантах осуществления изобретения на стадии (a) органический растворитель выбран из одного или более из 2-метилтетрагидрофурана, ацетонитрила, тетрагидрофурана и толуола.
[184] Предпочтительно органический растворитель выбран из 2-метилтетрагидрофурана и ацетонитрила.
[185] В некоторых вариантах осуществления изобретения на стадии (a) из одного или более из гидрида натрия, гидроксида натрия, карбоната цезия, триэтилендиамина, трет-бутоксида натрия, трет-бутоксида калия, бис(триметилсилил)амида лития и гексаметилдисилазида натрия.
[186] В некоторых вариантах осуществления для обеспечения щелочных условий на стадии (a) в качестве основания можно использовать морфолин вместо добавления дополнительного основания или можно добавлять дополнительный основной реактив, такой как один или более из гидрида натрия, гидроксида натрия, карбоната цезия, триэтилендиамина, трет-бутоксида натрия, трет-бутоксида калия, бис(триметилсилил)амида лития и гексаметилдисилазида натрия. В некоторых вариантах осуществления изобретения на стадии (a) основание представляет собой гидрид натрия, а растворитель - 2-метилтетрагидрофуран.
[187] В некоторых вариантах осуществления изобретения на стадии (a) реакцию ароматического нуклеофильного замещения соединения формулы 1 соединением формулы 2 проводят при следующих условиях: при температуре от 40°C до 70°C в течение от 3 ч до 18 ч, например при температуре от 40°C до 60°C в течение от 3 ч до 8 ч.
[188] В некоторых вариантах осуществления изобретения на стадии (a) реакцию другого ароматического нуклеофильного замещения соединением формулы 3 проводят при следующих условиях: с обратным холодильником в течение от 8 ч до 16 ч, например с обратным холодильником в течение от 8 ч до 12 ч, или например с обратным холодильником в течение от 13 ч до 16 ч.
[189] В некоторых вариантах осуществления изобретения на стадии (a) молярное соотношение соединения формулы 1, соединения формулы 2 и соединения формулы 3 составляет (1-1,2) : 1 : 3.
[190] В некоторых вариантах осуществления изобретения на стадии (a) в качестве катализатора могут добавлять сульфат магния, молекулярное сито или активированный уголь.
[191] В частности, сульфат магния добавляют в качестве катализатора в количестве от 0,2 до 3 молярных эквивалентов соединения формулы 2, например от 2 до 3 молярных эквивалентов соединения формулы 2, или например от 1 до 3 молярных эквивалентов соединения формулы 2.
[192] Дополнительно, после завершения реакции на стадии (a), очищают соединение формулы 4, причем очистку можно проводить с использованием обычного способа, известного в данной области техники.
[193] В настоящем изобретении предлагается также способ синтеза противоопухолевого соединения формулы 4, который включает в себя следующие стадии:
[194] приведение соединения формулы 1 в реакцию ароматического нуклеофильного замещения соединением формулы 2 в органическом растворителе под действием основания с получением соединения формулы I; и
[195] приведение соединения формулы I в реакцию ароматического нуклеофильного замещения с приведенным выше соединением формулы 3 в органическом растворителе под действием основания с получением соединения формулы 4;
[196] схема реакции является следующей:
 ;
;
[197] где каждый X1 и X2 независимо выбраны из галогена; P1 представляет аминозащитную группу, предпочтительно выбранную из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например Boc, Cbz, Tos, Fmoc, PMB, Trt и THP.
[198] В данном способе органический растворитель выбран из одного или более из 2-метилтетрагидрофурана, ацетонитрила, тетрагидрофурана и толуола. Основание выбрано из одного или более из гидрида натрия, гидроксида натрия, карбоната цезия, триэтилендиамина, трет-бутоксида натрия, трет-бутоксида калия, бис(триметилсилил)амида лития и гексаметилдисилазида натрия.
[199] В некоторых вариантах осуществления изобретения стадии синтеза соединения формулы 4m и условия реакции такие же, как описанные выше в способе синтеза соединения формулы 4, за исключением того, что соединение формулы 2m используют вместо соединения формулы 2, а соединение формулы Im - вместо соединения формулы I.
[200] В некоторых вариантах осуществления изобретения способ синтеза соединения формулы 5 включает стадию (b): приведение соединения формулы 4 в реакцию восстановления в органическом растворителе под действием катализатора с получением соединения формулы 5.
[201] В некоторых вариантах осуществления изобретения на стадии (b) органический растворитель выбран из одного или более из метанола, этанола, изопропанола, тетрагидрофурана, 2-метилтетрагидрофурана, уксусной кислоты и ацетонитрила.
[202] В некоторых вариантах осуществления изобретения на стадии (b) катализатор выбран из порошка железа, оксида платины, Pt/C, Pd(OH)2/C, Rh/C и Pd/C.
[203] В некоторых вариантах осуществления изобретения массовая доля Pt/C и Rh/C составляет 5%; массовая доля Pd/C составляет 5% или 10%; массовая доля Pd(OH)2/C составляет 10% или 20%.
[204] Когда катализатор представляет собой порошок железа, его добавляют в количестве от 10 до 18 молярных эквивалентов, например от 14 до 18 молярных эквивалентов, соединения формулы 4.
[205] В частности, на стадии (b) добавляют также хлорид аммония в количестве от 0,4 до 0,6 молярных эквивалентов соединения формулы 4; растворитель представляет собой смешанный растворитель из этанола и тетрагидрофурана, например в объемном соотношении этанола и тетрагидрофурана (6-10) : 10. В частности, на стадии (b) добавляют также небольшое количество воды; и объемное соотношение воды и тетрагидрофурана составляет (0,2-0,5) : 10. Дополнительно реакцию восстановления проводят в следующих условиях: с обратным холодильником в течение от 15 ч до 24 ч.
[206] Когда катализатор представляет собой оксид платины, реакцию восстановления проводят в следующих условиях: в атмосфере водорода (например, при давлении 0,5-0,8 МПа) при температуре от 35°C до 80°C в течение от 20 ч до 70 ч. Например, реакцию восстановления проводят в следующих условиях: в атмосфере водорода при температуре от 35°C до 50°C в течение от 40 ч до 50 ч.
[207] Дополнительно, после завершения реакции на стадии (b), очищают соединение формулы 5, и очистку могут проводить с использованием обычного способа, известного в данной области техники.
[208] В некоторых вариантах осуществления изобретения стадии синтеза соединения формулы 5m и условия реакции такие же, как описанные выше в способе синтеза соединения формулы 5, за исключением того, что соединение формулы 4m используют вместо соединения формулы 4.
[209] В некоторых вариантах осуществления изобретения способ синтеза соединения формулы 6 включает стадию (c): приведение соединения формулы 5 в реакцию замыкания кольца в органическом растворителе под действием кислоты с получением соединения формулы 6.
[210] В одном варианте осуществления изобретения на стадии (c) кислота выбрана из одной или более из трифторуксусной кислоты, метансульфоновой кислоты, п-толуолсульфоновой кислоты, муравьиной кислоты, уксусной кислоты и хлористоводородной кислоты. Органический растворитель выбран из по меньшей мере одного из дихлорметана, метанола, этанола и изопропанола.
[211] В частности, кислота выбрана из трифторуксусной кислоты, а органический растворитель - из изопропанола.
[212] В некоторых вариантах осуществления изобретения на стадии (c) соотношение количества вещества соединения формулы 5 и кислоты составляет 1 : (1,5-2).
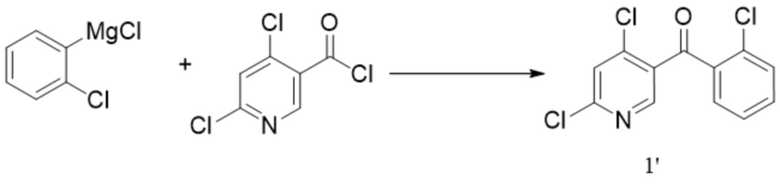
[213] В одном варианте осуществления изобретения, конкретно, стадия (c) представляет собой: выдерживание соединения формулы 5 в изопропаноле при температуре его кипения под действием трифторуксусной кислоты в течение от 1 ч до 3 ч, например в течение периода времени от 1,5 ч до 3 ч.
[214] Дополнительно, после завершения реакции на стадии (c), очищают соединение формулы 6, а именно, реакционную смесь фильтруют под вакуумом, и осадок на фильтре промывают изопропанолом и сушат с получением очищенного соединения формулы 6.
[215] В некоторых вариантах осуществления изобретения стадии синтеза соединения формулы 6m и условия реакции такие же, как описанные выше в способе синтеза соединения формулы 6, за исключением того, что соединение формулы 5m используют вместо соединения формулы 5.
[216] В некоторых вариантах осуществления изобретения способ синтеза соединения формулы 7 включает стадию (d): приведение соединения формулы 6 в реакцию в органическом растворителе для удаления аминозащитной группы с получением противоопухолевого соединения формулы 7.
[217] В некоторых вариантах осуществления изобретения на стадии (d) органический растворитель выбран из одного или более из толуола, дихлорметана, ацетонитрила, тетрагидрофурана, метанола, этанола и изопропанола. В частности, органический растворитель представляет собой толуол.
[218] В некоторых вариантах осуществления изобретения на стадии (d) могут также добавлять катализатор, и катализатор выбран из по меньшей мере одной из муравьиной кислоты, уксусной кислоты, хлористоводородной кислоты, метансульфоновой кислоты и трифторуксусной кислоты. В частности, катализатор выбран из трифторуксусной кислоты.
[219] Дополнительно на стадии (d) добавляют спирт или фенол; спирт выбран из метанола и/или этанола, и фенол выбран из фенола и/или п-метоксифенола. Дополнительно, например, добавляют спирт или фенол в количестве от 1 до 2 молярных эквивалентов соединения формулы 6.
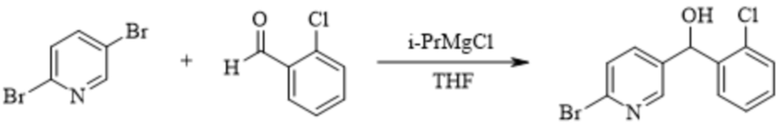
[220] Дополнительно, после завершения реакции на стадии (d), очищают соединение формулы 7, и очистку могут проводить с использованием обычного способа, известного в данной области техники.
[221] В некоторых вариантах осуществления изобретения на стадии синтеза соединения формулы 7 использовали соединение формулы 6m вместо соединения формулы 6, и условия реакции такие же, как описанные выше в способе синтеза соединения формулы 7.
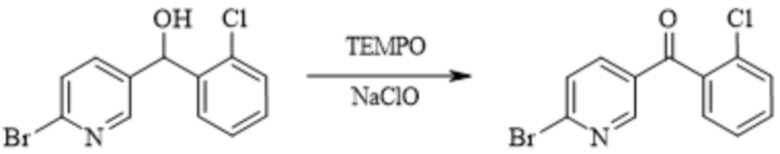
[222] В некоторых вариантах осуществления изобретения соединение формулы 7 могут также синтезировать непосредственно из соединения формулы 5M в одну стадию. Реакцию проводят в следующих условиях: приведение соединения формулы 5M в реакцию в органическом растворителе под действием кислоты с получением соединения формулы 7. Например, реакцию проводят с обратным холодильником под действием трифторуксусной кислоты в течение 20-40 ч. Органический растворитель выбран из одного или более из: толуола, дихлорметана, ацетонитрила, 2-метилтетрагидрофурана, тетрагидрофурана, метанола, этанола и изопропанола.
[223] В некоторых вариантах осуществления изобретения способ синтеза противоопухолевого соединения формулы 7 дополнительно включает стадии синтеза соединения формулы 2M:
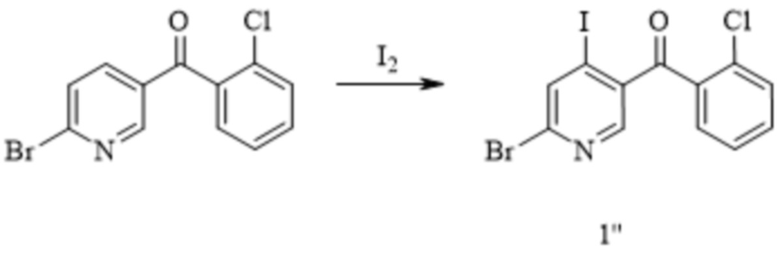
[224] (i) приведение соединения формулы 2-1 в реакцию нитрования с азотной кислотой в растворителе с получением соединения формулы 2-2;
[225] (ii) приведение соединения формулы 2-2 в реакцию защиты аминогруппы с аминозащитным реактивом в органическом растворителе с получением соединения формулы (2-3)M; и
[226] (iii) приведение соединения формулы (2-3)M в реакцию в органическом растворителе для удаления фталоильной группы с получением соединения формулы 2M;
[227] схема реакции является следующей:
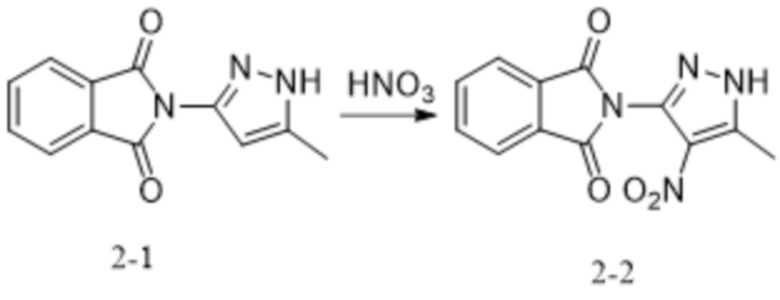
;
[228] причем аминозащитный реактив представляет собой Boc2O, CbzCl, TosCl, FmocCl, PMBBr, MOMCl, EOMCl, трет-бутанол, изобутилен, BnCl, уксусный ангидрид, SEMCl, TrtCl или DHP, например Boc2O, CbzCl, TosCl, FmocCl, PMBBr, TrtCl или DHP; P1 выбран из Boc, Cbz, Tos, Fmoc, PMB, MOM, EOM, tBu, Bn, Ac, SEM, Trt и THP, например Boc, Cbz, Tos, Fmoc, PMB, Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1. Предпочтительно аминозащитный реактив представляет собой TrtCl или DHP; P1 выбран из Trt и THP.
[229] В некоторых вариантах осуществления изобретения соединение формулы (2-3)M включает соединение формулы 2-3 и соединение формулы (2-3)m, как показано ниже:
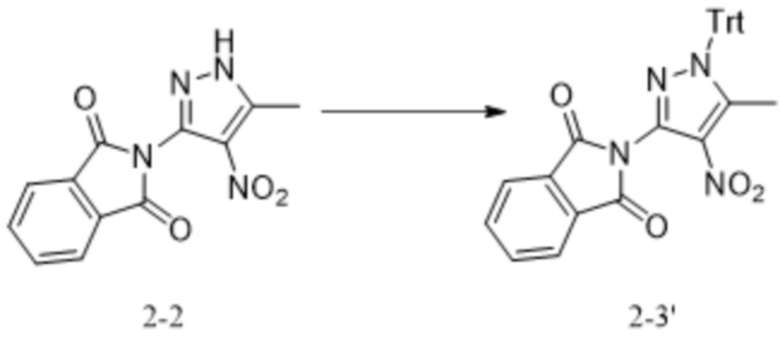
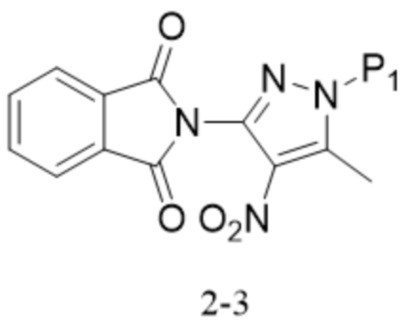 ,
, 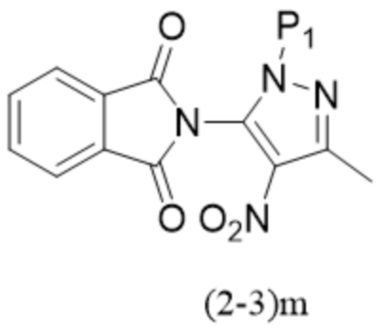 .
.
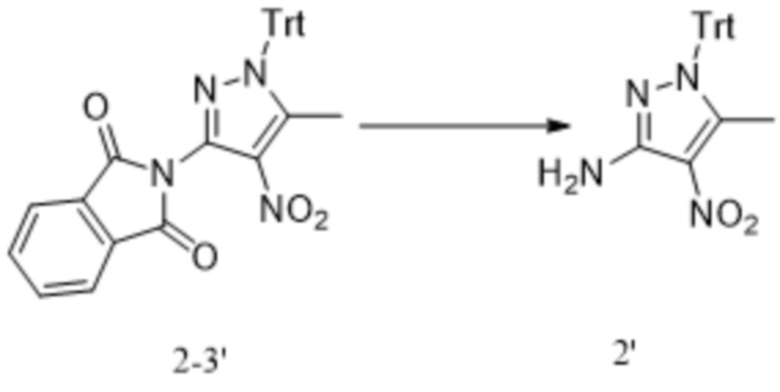
[230] В некоторых вариантах осуществления изобретения растворитель на стадии (i) представляет собой, например, концентрированную серную кислоту, уксусный ангидрид или уксусную кислоту.
[231] В некоторых вариантах осуществления изобретения реакцию нитрования на стадии (i) проводят в следующих условиях: в концентрированной серной кислоте при температуре от 10°C до 20°C в течение от 0,5 ч до 2 ч.
[232] Дополнительно соотношение массы к объему соединения формулы 2-1 и концентрированной серной кислоты составляет 100 г : (150-250) мл.
[233] Дополнительно, после завершения реакции нитрования, полученное соединение формулы 2-2 легко фильтруется и имеет хорошую растворимость в таких растворителях, как тетрагидрофуран, дихлорметан, ацетонитрил и т. д., поэтому выбор растворителей для реакции более разнообразен.
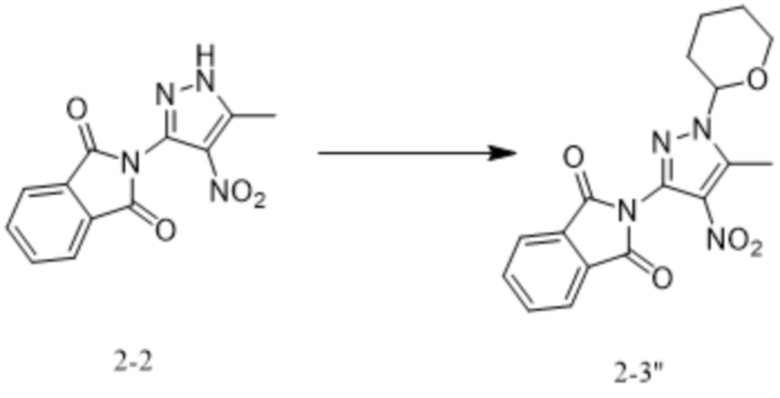
[234] В некоторых вариантах осуществления изобретения на стадии (ii) устанавливаются различные условия реакции в зависимости от природы аминозащитного реактива. В частности, когда аминозащитный реактив представляет собой TrtCl, реакцию на стадии (ii) проводят в следующих условиях: под действием основания при температуре от 60°C до 100°C в течение от 1 ч до 48 ч; когда аминозащитный реактив представляет собой DHP, реакцию на стадии (ii) проводят в следующих условиях: под действием п-толуолсульфоновой кислоты или п-толуолсульфоната пиридиния при температуре от 60°C до 100°C с обратным холодильником в течение от 3 ч до 48 ч.
[235] В некоторых вариантах осуществления изобретения органический растворитель для реакции на стадии (ii) выбран из одного или более из тетрагидрофурана, 2-метилтетрагидрофурана, дихлорметана и ацетонитрила.
[236] В частности, когда аминозащитный реактив представляет собой TrtCl, реакцию на стадии (ii) проводят в следующих условиях: под действием катализатора триэтиламина в ацетонитриле с обратным холодильником в течение от 1 ч до 3 ч
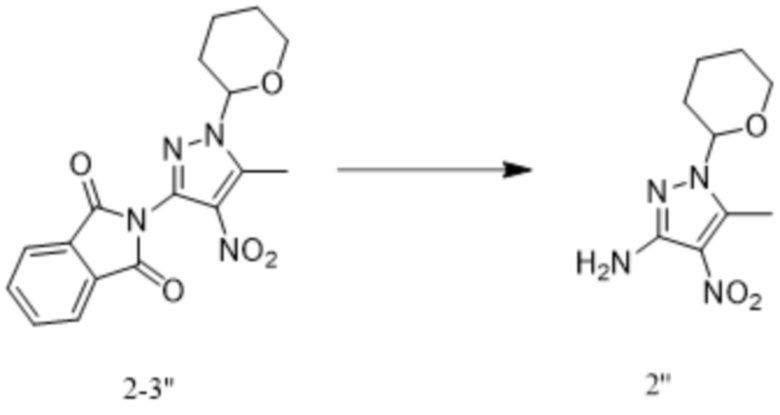
[237] В некоторых вариантах осуществления изобретения реакцию удаления фталоильной группы на стадии (iii) проводят в следующих условиях: под действием гидрата гидразина в органическом растворителе с обратным холодильником в течение от 1 ч до 3 ч, причем органический растворитель выбран из одного или более из этанола, тетрагидрофурана и ацетонитрила.
[238] В некоторых вариантах осуществления изобретения способ синтеза противоопухолевого соединения формулы 7 дополнительно включает стадию синтеза соединения формулы 1:
[239] приведение соединения формулы 1-1 в реакцию сочетания с соединением формулы 1-2 в органическом растворителе с получением соединения формулы 1;
[240] схема реакции является следующей:
;
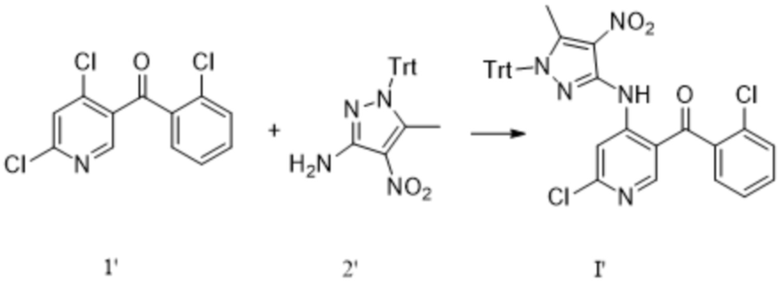
[241] где каждый X1, X2 и X3 выбраны из галогена. Предпочтительно каждый X1X2 и X3 выбраны из хлора.
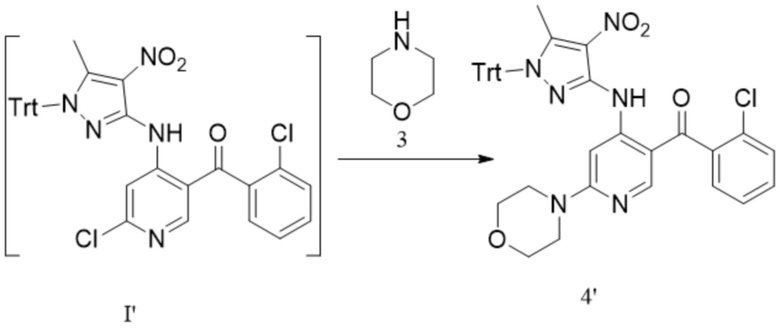
[242] Дополнительно реакцию сочетания соединения формулы 1-1 с соединением формулы 1-2 проводят в следующих условиях: под действием ацетилацетоната железа при температуре от -70°C до -60 °C.
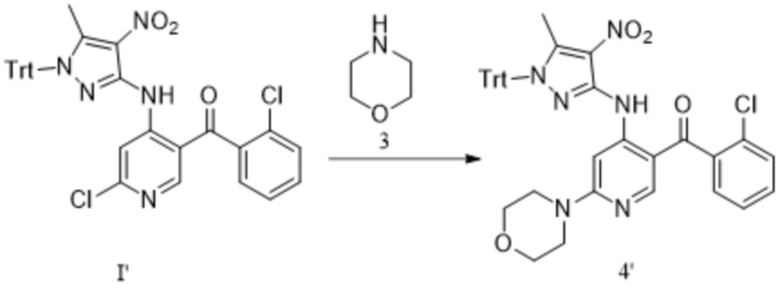
[243] В настоящем изобретении также предлагается промежуточное соединение для получения противоопухолевых соединений формулы 7, которые имеют следующие структурные формулы:
;
[244] Предпочтительно промежуточное соединение имеет следующие структуры:
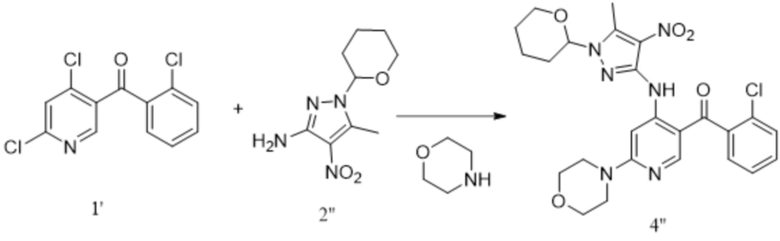
 ;
;
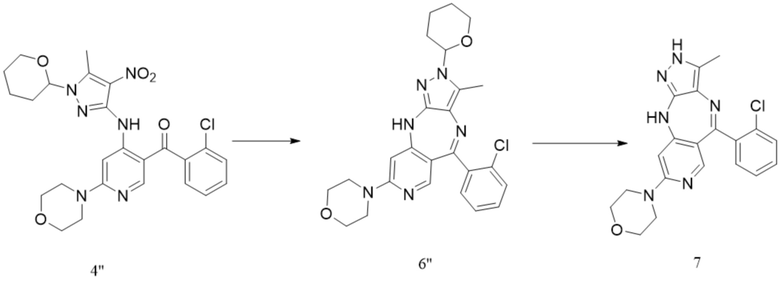
[245] где каждый X1 и X2 независимо выбраны из галогена;
[246] где P1 выбран из Trt и THP; P1 связан с любым атомом N в кольце, где расположен P1, и предпочтительно P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1.
[247] В настоящем изобретении также предлагается промежуточное соединение для получения противоопухолевого соединения формулы 7, которое имеет следующую структурную формулу:
[248] предпочтительно следующей структуры:
[249] где P1 выбран из Trt и THP;
[250] формула 6М представляет собой комплекс, содержащий одну молекулу трифторуксусной кислоты и одну молекулу изопропанола.
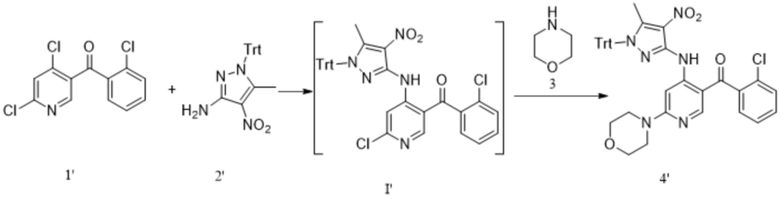
[251] Настоящее изобретение дополнительно иллюстрируется следующим описанием примеров, но объем охраны настоящего изобретения этим не ограничивается.
[252] Все исходные вещества, используемые в примерах настоящего изобретения, являются коммерчески доступными, химически чистыми продуктами.
[253] Пример 1
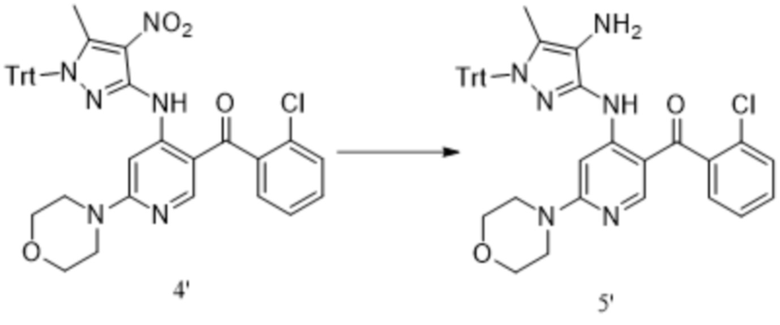
[254] Синтез соединения формулы 1'
[255] Приготовление раствора хлорида (2-хлорфенил)магния в THF: раствор хлорида (2-хлорфенил)магния (27,34 моль) готовили в THF (50,44 л, 0,542 М) и хранили при температуре от -5°C до 5°C в отсутствии кислорода для последующего использования.
[256] Приготовление раствора 4,6-дихлорникотиноилхлорида в THF: 4,6-дихлорникотиноилхлорид (3,85 кг, 18,32 моль) добавляли к THF (25,0 л) и ацетилацетонату железа (193,16 г, 0,55 моль, 0,03 экв.) для получения раствора (28,0 л, 0,651 М) для дальнейшего использования.
[257] Приготовление разбавленной хлористоводородной кислоты: в реактор объемом 100 л добавляли 2,8 л концентрированной хлористоводородной кислоты и 5,6 л воды, и температуру контролировали от 5°C до 15 °C; приготовленный раствор использовали для гашения.
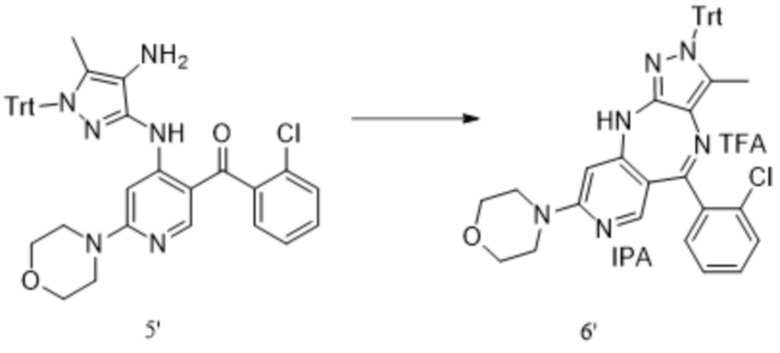
[258] В микрореакторе с заданной температурой от -70°C до -60°C раствор хлорида (2-хлорфенил)магния в THF приводили в реакцию с раствором 4,6-дихлорникотиноилхлорида в THF с получением в реакционной смеси (2-хлорфенил)(4,6-дихлорпиридин-3-ил)метанона. Реакционную смесь гасили введением в подготовленную разбавленную хлористоводородную кислоту. Отделяли и концентрировали прозрачную желтую органическую фазу. Добавляли MTBE (17,5 л) к остатку после концентрирования, органическую фазу отделяли, концентрировали для удаления растворителя и затем добавляли к 2 л н-гептана с получением неочищенного коричневато-желтого продукта. Неочищенный продукт добавляли к 3,3 л этанола и 9,9 л н-гептана, и смесь нагревали до 60 °C, после полного растворения неочищенного продукта охлаждали до от 5°C до 10°C и фильтровали. Высушивали отфильтрованный осадок с получением светло-желтого порошка (2,78 кг, выход 53,3%).
[259] 1H-ЯМР (400 МГц, DMSO-d6) δ (м. д.): 8,61-8,57 (м, 1H), 8,02 (с, 1H), 7,70-7,62 (м, 3H), 7,55-7,51 (м, 1H).
[260] Синтез соединения формулы 1''
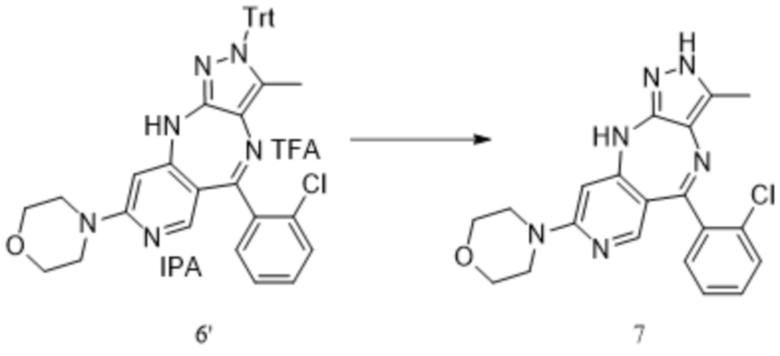
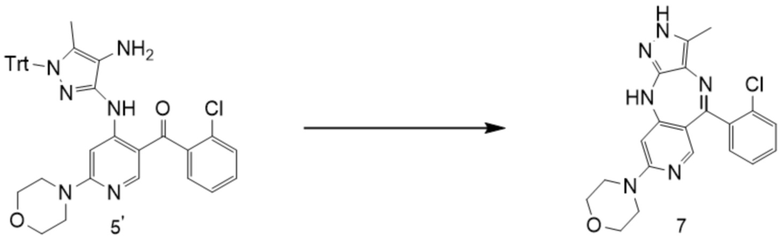
[261] Стадия 1. Синтез (6-бромпиридин-3-ил)(2-хлорфенил)метанола
[262] В четырехгорлую колбу объемом 2 л добавляли безводный тетрагидрофуран (500 мл) и 2,5-дибромпиридин (100,0 г, 0,42 моль, 1,0 экв.), и смесь охлаждали при перемешивании на водяной бане со льдом до 2 °C. Добавляли по каплям хлорид изопропилмагния (210,5 мл, 2,0 М, 0,42 моль, 1,0 экв.) в течение около 0,5 ч при контролируемой температуре не выше 10 °C. Смесь перемешивали при комнатной температуре (20 °C) в течение 1 ч, и затем охлаждали на водяной бане со льдом до 10 °C, и по каплям добавляли раствор 2-хлорбензальдегида (62,3 г, 0,443 моль, 1,05 экв.) в тетрагидрофуране (200 мл) в течение около 0,5 ч. Смесь перемешивали при 10°C в течение 2 ч, и реакцию завершали по показателям ТСХ. В реакционную систему добавляли насыщенный водный раствор хлорида аммония (300 мл). Смесь перемешивали в течение 10 мин, и затем органическую фазу отделяли и концентрировали с получением желтого масла. Водную фазу экстрагировали этилацетатом (1,0 л × 2), и экстракты объединяли с полученным ранее желтым маслом, промывали водой (500 мл) и насыщенным солевым раствором (500 мл), высушивали над безводным Na2SO4 и концентрировали с получением коричневого масла (140 г, неочищенное).
[263] Стадия 2. Синтез (6-бромпиридин-3-ил)(2-хлорфенил)метанона
[264] Растворяли (6-бромпиридин-3-ил)(2-хлорфенил)метанол (140 г, неочищенный) в DCM (1,3 л), и добавляли TEMPO (1,51 г, 9,4 ммоль) и NaBr (1,92 г, 18,8 ммоль). Смесь охлаждали на водяной бане со льдом до 3 °C, и по каплям добавляли водный раствор NaClO (1,34 моль/л, 600 л, 0,71 моль), нейтрализованный NaHCO3 (45,0 г), при этом температура не превышала 20 °C. После добавления по каплям смесь перемешивали в течение 10 мин, и реакцию завершали по показателям ТСХ. Водную фазу отделяли и экстрагировали DCM (1,0 л), органические фазы объединяли, промывали водой (1,0 л) и насыщенным солевым раствором (1,0 л), высушивали над безводным Na2SO4 и концентрировали с получением желтого масла. Неочищенный продукт суспендировали в 150 мл метил трет-бутилового эфира/500 мл петролейного эфира с получением желтого твердого вещества (50,3 г, 39,7% выход за две стадии).
[265] Стадия 3. Синтез (6-бром-4-иодпиридин-3-ил)(2-хлорфенил)метанона
[266] В атмосфере азота в четырехгорлую колбу объемом 2 л добавляли раствор тетраметилпиперидин лития/хлорида магния (281 мл, 1,5 моль/л, 0,43 моль, 2,5 экв.), охлаждали на бане с сухим льдом/этанолом до -65°C и добавляли по каплям раствор (6-бромпиридин-3-ил)(2-хлорфенил)метанона (50,0 г, 0,17 моль, 1,0 экв.) в тетрагидрофуране (50 мл) в течение около 0,5 ч. Затем смесь нагревали до -45 °C, перемешивали в течение 1 ч, и затем охлаждали до -65 °C, и добавляли по каплям в течение около 1 ч раствор I2 (129,3 г, 0,51 моль, 3,0 экв.) в тетрагидрофуране (400 мл). Смесь перемешивали в течение 20 мин, и затем реакцию завершали по показателям ТСХ. К реакционной системе добавляли насыщенный водный раствор хлорида аммония (500 мл) и насыщенный водный раствор NaHSO3 (500 мл). Смесь перемешивали в течение 15 мин и фильтровали, и нерастворимое вещество промывали этилацетатом (500 мл × 2). Фильтраты объединяли, водную фазу отделяли и экстрагировали этилацетатом (1,0 л × 2). Все органические фазы объединяли, промывали водой (800 мл) и насыщенным солевым раствором (800 мл), высушивали над безводным Na2SO4 и концентрировали с получением желтого твердого вещества. Твердое вещество суспендировали с метил-трет-бутиловым эфиром (500 мл) / петролейным эфиром (500 мл) и высушивали с получением желтого твердого вещества (30 г, выход 41,8%).
[267] Пример 2
[268] Синтез соединения 2'
[269] Стадия 1. Синтез соединения формулы 2-2
[270] Концентрированную серную кислоту (7,5 л) добавляли в четырехгорлую колбу и хорошо перемешивали, проводя перемешивание, и систему охлаждали на водяной бане со льдом до температуры ниже 20 °C. Добавляли порциями соединение формулы 2-1 (3590 г, 15,80 моль, 1,0 экв.). При контролируемой температуре системы от 10°C до 20°C в реакционную систему по каплям и медленно добавляли 63% (массовая доля) концентрированную азотную кислоту (1896,4 г, 18,96 моль, 1,2 экв.). После добавления по каплям удаляли водяную баню со льдом, и реакционной системе давали прореагировать при температуре от 10°C до 20°C в течение 1 ч. Исходные вещества полностью прореагировали по показателям ТСХ (DCM : MeOH=20 : 1). Реакционную смесь медленно выливали в 36 кг ледяной воды, и полученную смесь фильтровали под вакуумом до сухого состояния. Затем отфильтрованный осадок добавляли в емкость с широкой горловиной объемом 50 л, после чего добавляли 36 л воды. Полученную смесь перемешивали в течение 0,5 ч и фильтровали под вакуумом, и отфильтрованный осадок промывали водой (6 л). Отфильтрованный осадок сушили до постоянной массы (около 72 ч) с получением соединения формулы 2-2 в виде грязно-белого твердого вещества (4160г, выход 96,7%, чистота ВЭЖХ 95,5%).
[271] 1H-ЯМР (400 МГц, DMSO-d6) δ(м. д.): 14,23 (уш., 1H), 8,08-7,97 (м, 4H), 2,64 (с, 3H).
[272] Стадия 2. Синтез соединения формулы 2-3'
[273] В реактор добавляли ACN (38 л), с последующим добавлением при перемешивании соединения формулы 2-2 (4140 г, 15,21 моль, 1,0 экв.) и TEA (1847 г, 18,25 моль, 1,2 экв.). Смесь хорошо перемешивали и добавляли трифенилхлорметан (4664 г, 16,73 моль, 1,1 экв.). Систему нагревали с обратным холодильником (82 °C) в течение 2 ч, и завершали реакцию. Систему охлаждали до 25°C и фильтровали под вакуумом, и отфильтрованный осадок дважды промывали ACN (4 л × 2). После слива жидкости отфильтрованный осадок суспендировали 30 л воды в течение 2 ч с последующим фильтрованием под вакуумом, и отфильтрованный осадок дважды промывали водой (4 л × 2). После слива жидкости отфильтрованный осадок сушили в сушильном шкафу с принудительной подачей воздуха при температуре 50°C в течение 24 ч с получением соединения формулы 2-3' в виде белого твердого вещества (6920 г, ВЭЖХ чистота 99,7%, выход 88,4%).
[274] 1H-ЯМР (400 МГц, CDCl3) δ(м. д.): 7,97-7,92 (м, 2H), 7,82-7,78 (м, 2H), 7,41-7,33 (м, 9H), 7,22-7,19 (м, 6H), 2,08 (с, 3H).
[275] Стадия 3. Синтез соединения формулы 2'
[276] THF (55 л) в равных долях разделяли по четырем четырехгорлым колбам объемом 20 л, и в каждую колбу при перемешивании добавляли соединение формулы 2-3' (6900 г, 13,41 моль, 1,0 экв.) и 85% (массовая доля) N2H4⋅H2O (1580 г, 26,82 моль, 2,0 экв.). Смеси нагревали с обратным холодильником в течение 3 ч и завершали реакции. Смеси естественным образом охлаждали и фильтровали под вакуумом, и отфильтрованный осадок дважды промывали THF (2 л × 2). Фильтрат концентрировали на водяной бане при 50°C под пониженным давлением с удалением 24 л THF, и оставшийся фильтрат переносили в реактор объемом 50 л и нагревали с обратным холодильником. Медленно добавляли 95% этанол (36 л), и медленно осаждалось желтовато-зеленое твердое вещество. Смесь перемешивали в течение 2 ч. Смесь охлаждали до комнатной температуры и перемешивали для кристаллизации в течение ночи. Смесь фильтровали под вакуумом, и отфильтрованный осадок промывали один раз 2 л 95% этанола и сушили с получением соединения формулы 2' в виде порошкообразного желтовато-зеленого вещества (4480 г, чистота ВЭЖХ 100,0%, выход 86,9%).
[277] 1H-ЯМР (400 МГц, CDCl3) δ(м. д.): 7,38-7,36 (м, 9H), 7,25-7,22 (м, 6H), 5,43 (с, 2H), 2,46 (с, 3H).
[278] Синтез соединения 2''
[279] Стадия 1. Синтез 2-(5-метил-4-нитро-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-ил)изоиндолин-1,3-диона
[280] В реакционную колбу добавляли безводный ацетонитрил (270 моль), п-толуолсульфоновую кислоту (1,72 г, 0,01 моль, 0,1 экв.) и пиридин (0,79 г, 0,01 моль, 0,1 экв.), смесь подогревали до 50°C и перемешивали в течение 2 ч. Добавляли соединение формулы 2-2 (27,2 г, 0,1 моль, 1,0 экв.). После нагревания смеси с обратным холодильником в течение 1 ч, по каплям добавляли DHP (16,8 г, 0,2 моль, 2,0 экв.). После выдерживания реакционной смеси около 20 часов, ее концентрировали. Остаток суспендировали с этилацетатом (90 мл) в течение 1 ч и фильтровали, и отфильтрованный осадок высушивали с получением соединения формулы 2-3'' (28,6 г, выход 80,3%).
[281] 1H-ЯМР (400 МГц, DMSO-d6) δ(м. д.): 8,08-7,98 (м, 4H), 5,76-5,71 (м, 1H), 4,01-3,75 (м, 2H), 2,76 (с, 3H), 2,30-2,16 (м, 1H), 1,98-1,94 (м, 2H), 1,72-1,67 (м, 1H), 1,57-1,56 (м, 2H).
[282] Стадия 2. Синтез 5-метил-4-нитро-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-амина
[283] В реакционную колбу добавляли соединения формулы 2-3'' (569,4 г, 1,6 моль, 1,0 экв.) и THF (5,7 л). Смесь нагревали с обратным холодильником, и добавляли по каплям гидрат гидразина (85%) (141,1 г, 2,4 моль, 1,5 экв.) в течение 0,5 ч. Реакционную смесь нагревали с обратным холодильником еще 8 ч, и реакцию завершали по показателям ТСХ (PE : EA=2 : 1). Реакционную смесь охлаждали до 20-25°C и фильтровали под вакуумом, и фильтрат концентрировали с получением неочищенного продукта (303 г). Неочищенный продукт добавляли к 3 л 95% этанола, смесь нагревали с обратным холодильником, охлаждали до комнатной температуры и фильтровали с получением соединения формулы 2'' в виде желтовато-зеленого твердого вещества. Выход=62,5%.
[284] 1H-ЯМР (400 МГц, DMSO-d6) δ(м. д.): 6,15 (с, 2H), 5,42-5,38 (м, 1H), 3,91-3,87 (м, 1H), 3,69-3,65 (м, 1H), 2,58 (с, 3H), 2,17-1,98 (м, 1H), 1,98-1,93 (м, 1H), 1,81-1,76 (м, 1H), 1,66-1,52 (м, 3H).
[285] Пример 3
[286] Синтез соединения формулы 4', [(2-хлорфенил)(4-((5-метил-4-нитро-1-тритил-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)метанона]
[287] Стадия 1. Синтез (6-хлор-4-((5-метил-4-нитро-1-тритил-1H-пиразол-3-ил)амино)пиридин-3-ил)(2-хлорфенил)метанона
[288] Добавляли NaH (10,0 г) к 2-MeTHF (600 мл). После подогревания смеси до 50 °C, добавляли соединение формулы 2' (38,4 г, 0,1 моль). После перемешивания смеси в течение 1 ч, добавляли соединение формулы 1' (28,5 г, 0,1 моль). Смеси давали прореагировать в течение 6-8 ч, и завершали реакцию. Реакционную смесь охлаждали до 20-25 °C, и по каплям добавляли уксусную кислоту (9 г) с последующим добавлением 200 мл воды. Смесь перемешивали, органическую фазу отделяли и концентрировали, пока ее не осталось около 100 мл, после чего добавляли этанол (300 мл). После выдерживания смеси в течение 4-6 ч нагревание прекращали. Реакционную смесь охлаждали до 20-25°C и затем фильтровали, и отфильтрованный осадок высушивали с получением соединения формулы I' в виде желтого твердого вещества (41,4 г, выход 65,4%), которое непосредственно использовали на следующей стадии.
[289] Стадия 2. Синтез (2-хлорфенил)(4-((5-метил-4-нитро-1-тритил-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)метанона
[290] В реакционную колбу добавляли соединение формулы I' (41,4 г), морфолин (17,1 г) и 2-MeTHF (400 мл) и нагревали при температуре 80°C в течение 12-16 ч. После концентрирования реакционной смеси до остатка около 200 мл, добавляли этанол (300 мл) с использованием обратного холодильника. Реакционную смесь нагревали с обратным холодильником в течение 1 ч, затем охлаждали до 20-25°C и фильтровали, и отфильтрованный осадок высушивали с получением соединения формулы 4' в виде желтого твердого вещества (41 г, выход 91,7%).
[291] Пример 4
[292] Стадия 1. Синтез соединения формулы 4'', (2-хлорфенил)(4-((5-метил-4-нитро-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)метанона
[293] Помещали 60% (массовая доля) гидрида натрия (87,36 г, 2,18 моль) в трехгорлую колбу объемом 10 л и добавляли 2-MeTHF (4,3 л). Система становилась молочно-белой и мутной. Систему перемешивали при температуре 45-50°C в течение 1 ч, и порциями добавляли соединение формулы 2'' (197,4 г, 0,87 моль). Система меняла цвет с желтого на коричневый. После перемешивания 1 ч добавляли соединение формулы 1' (231,5 г, 0,81 моль). Реакционной смеси давали прореагировать при температуре 45-50°C в течение 2,5 ч, и реакцию завершали по данным ВЭЖХ-контроля в технологическом процессе. Температуру снижали до 12 °C, и порциями добавляли уксусную кислоту (78,61 г, 1,31 моль). Внутреннюю температуру снижали до 18 °C, и затем добавляли морфолин (228,3 г, 2,62 моль). Реакционной смеси давали прореагировать при температуре 90°C в течение 15 ч, и реакцию завершали по данным ВЭЖХ-контроля в технологическом процессе. Реакционную смесь охлаждали до 20-25 °C, и добавляли 2 л воды. Органическую фазу отделяли и концентрировали, и остаток суспендировали в 1 л этилацетата в течение 2-4 ч, после чего фильтровали, и отфильтрованный осадок сушили в сушильном шкафу с принудительной подачей воздуха с получением соединения формулы 4'' в виде желтого твердого вещества (218 г, выход 51,2%).
[294] Стадия 2. Синтез соединения формулы 7, 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо[4,3-e][1,4]диазепин-8-ил)морфолина
[295] Соединение 4'' (1 г, 1,90 ммоль) растворяли в 60 мл уксусной кислоты, и добавляли 6 мл воды и 300 мг Pd/C. Реакционной смеси давали прореагировать в атмосфере водорода в течение 24 ч и фильтровали ее, а маточный раствор концентрировали. Добавляли 50 мл воды, и в большом количестве осаждалось желтое твердое вещество. Твердое вещество собирали фильтрацией и сушили с получением соединения формулы 6'' (720 мг, выход 80%).
[296] Соединение 6'' (1 г, 2,09 ммоль) растворяли в метаноле, и добавляли п-толуолсульфоновую кислоту (0,43 г, 2,50 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч, подогревали до 55 °C, добавляли п-толуолсульфоновую кислоту (0,28 г, 1,6 ммоль), давали прореагировать в течение 3 ч, концентрировали, и остаток очищали колоночной хроматографией с получением продукта 7 в виде желтого твердого вещества.
[297] Молекулярная формула: C20H19ClN6O; молекулярная масса: 394,86 по данным ЖХ-МС (полож., m/z)=395,22[M+H]+.
[298] Пример 5
[299] Синтез соединения формулы 7
[300] Стадия 1. Синтез соединения формулы 4', (2-хлорфенил)(4-((5-метил-4-нитро-1-тритил-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)метанона
[301] В реакционную колбу добавляли 2-MeTHF (15 л), затем при перемешивании - сульфат магния (500 г, 4,15 моль) и 60% (массовая доля) гидрид натрия (260 г, 6,50 моль), и смесь перемешивали при 50°C в течение 2 ч. Соединение формулы 2' (1000 г, 2,60 моль) добавляли порциями, и система становилась коричнево-желтой. Через 1 ч перемешивания, система становилась коричневой. Добавляли соединение формулы 1' (820 г, 2,86 моль). Реакционной смеси давали прореагировать при температуре 46-60°C в течение 3,5 ч, и соединение формулы 1' полностью прореагировало по данным ВЭЖХ-контроля в технологическом процессе. Порциями добавляли уксусную кислоту (234 г, 3,90 моль), и система превращалась в коричневую суспензию. Добавляли морфолин (680 г, 7,80 моль), систему нагревали с обратном холодильником в течение 15 ч, и реакцию завершали по данным ВЭЖХ-контроля в технологическом процессе. Систему охлаждали до 15-25 °C. Добавляли 7,5 л воды, и органическую фазу отделяли, высушивали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали, пока не осталось около 1-2 л растворителя, и добавляли 5 л толуола и 15 л этанола. Проводили суспендирование при температуре 78°C в течение 2 ч, охлаждали смесь до комнатной температуры и фильтровали ее. Высушивали отфильтрованный осадок с получением желтого твердого вещества (1250 г, выход 70,3%).
[302] 1H-ЯМР (400 МГц, DMSO-d6) δ(м. д.): 12,55 (с, 1H), 7,89 (с, 1H), 7,60-7,52 (м, 2H), 7,50-7,49 (м, 2H), 7,42-7,33 (м, 9H), 7,29-7,27 (м, 6H), 7,21 (с, 1H), 3,60-3,50 (м, 4H), 3,70 (с, 4H), 2,07 (с, 3H).
[303] Стадия 2. Синтез соединения формулы 5', (4-((4-амино-5-нитро-1-тритил-1H-пиразол-3-ил)амино)-6-морфолинопиридин-3-ил)(2-хлорфенил)метанона
[304] (Реализация 1) В реакционную колбу объемом 20 л добавляли воду (300 мл), этанол (95%, 7,5 л), хлорид аммония (30,30 г, 0,57 моль) и порошок железа (950 г, 17 моль), и смесь перемешивали при температуре 55-60°C в течение 1,5 ч с последующим добавлением THF (7,5 л) и соединения формулы 4' (775 г, 1,13 моль). Реакционную смесь нагревали с обратным холодильником в течение 5 ч, добавляли порошок железа (320 г, 5,66 моль), нагревали с обратным холодильником в течение 15 ч, и реакцию завершали по данным ВЭЖХ-контроля в технологическом процессе. Реакционную смесь фильтровали в горячем состоянии через целит (1,5 кг), и отфильтрованный осадок промывали THF (3 л). Фильтрат концентрировали, пока не осталось около 5 л растворителя, и добавляли 5 л этанола. Проводили суспендирование при температуре 65°C в течение 3 ч, охлаждали смесь до комнатной температуры и фильтровали ее. Промывали отфильтрованный осадок 4 л этанола и высушивали его с получением желтого твердого вещества (590 г, выход 79,6%).
[305] 1H-ЯМР (300 МГц, CDCl3) δ(м. д.): 11,42 (с, 1H), 8,04 (с, 1H), 7,47-7,23 (м, 19H), 7,11 (с, 1H), 3,67-3,63 (м, 4H), 3,27-3,24 (м, 4H), 1,59 (с, 3H).
[306] (Реализация 2) Соединение формулы 4' (6,8 г, 10,0 ммоль) и THF (150 мл) добавляли в автоклав с последующим добавлением оксида платины (700 мг), и вводили водород при давлении 0,5-0,8 МПа. Реакционной смеси давали прореагировать при температуре 40°C в течение 46 ч, и реакцию завершали по данным ВЭЖХ. Реакционную смесь фильтровали, и фильтрат концентрировали, пока его не осталось около 50 мл, затем по каплям добавляли 100 мл н-гептана с обратным холодильником. Реакционную смесь охлаждали до комнатной температуры и фильтровали, и отфильтрованный осадок высушивали с получением желтого твердого вещества (5,5 г, выход 84,6%).
[307] Стадия 3. Синтез соединения формулы 6', комплекса изопропанола и 4-(5-(2-хлорфенил)-3-метил-2-тритил-2,10-дигидропиразоло[4,3-b]пиридо[4,3-e][1,4]диазепин-8-ил)морфолина трифторацетата
[308] В реактор объемом 50 л добавляли 20 л изопропанола с последующим добавлением соединения формулы 5' (1980 г, 3,03 моль) и TFA (690 г, 6,05 моль). После этого реакционную смесь нагревали с обратным холодильником в течение 2 ч, реакцию завершали по данным ВЭЖХ-контроля в технологическом процессе. Реакционную смесь охлаждали до 20°C и фильтровали под вакуумом, отфильтрованный осадок промывали изопропанолом до получения прозрачного фильтрата и сушили с получением желтого твердого вещества (2300 г, выход 93,8%).
[309] 1H-ЯМР (400 МГц, DMSO-d6) δ(м. д.): 9,24 (с, 1H), 7,51-7,48 (м, 1H), 7,43-7,35 (м, 9H), 7,32-7,29 (м, 3H), 7,17-7,15 (м, 6H), 6,79 (с, 1H), 6,17 (с, 1H), 3,81-3,74 (м, 1H), 3,66-3,64 (м, 4H), 3,36-3,33 (м, 4H), 1,36 (с, 3H), 1,48-1,32 (д, 6H, J=6,12 Гц).
[310] Стадия 4. Синтез соединения формулы 7, 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо[4,3-e][1,4]диазепин-8-ил)морфолина
[311] Добавляли соединение формулы 6' (58,0 г, 71,5 ммоль) и метанол (4,6 г, 143,7 ммоль) к толуолу (600 мл) с последующим добавлением трифторуксусной кислоты (8,2 г, 71,9 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 17 ч, охлаждали до 50 °C, фильтровали, и отфильтрованный осадок суспендировали в толуоле (400 мл) при температуре 50°C в течение 1 ч. Затем смесь фильтровали под вакуумом, и отфильтрованный осадок сушили в сушильном шкафу с принудительной подачей воздуха при 100°C с получением желтого твердого вещества (32 г). Полученное твердое вещество растворяли в метаноле (300 мл) и воде (100 мл). Значение pH раствора корректировали до 7-8 водным аммиаком (1-кратное разбавление), и в большом количестве осаждалось желтое твердое вещество. Смесь перемешивали при 50°C в течение 1 ч и фильтровали под вакуумом, и отфильтрованный осадок высушивали с получением желтого твердого вещества (24 г, чистота 99,7%, выход 85,4%).
[312] 1H-ЯМР (DMSO-d6, 400 МГц) 11,57 (с, 1H), 8,29 (с, 1H), 7,32-7,47 (м, 4H), 6,89 (с, 1H), 5,95 (с, 1H), 3,61 (м, 4H), 3,31 (м, 4H), 1,97 (с, 3H).
[313] Пример 6
[314] Стадия 1. Синтез соединения формулы 7, 4-(5-(2-хлорфенил)-3-метил-2,10-дигидропиразоло[4,3-b]пиридо[4,3-e][1,4]диазепин-8-ил)морфолина
[315] Соединение формулы 5' (1 г, 1,53 ммоль) растворяли в DCM (10 мл), и добавляли TFA (350 мг, 3,06 ммоль). Систему перемешивали в течение 2,5 ч, и осаждалось желтое твердое вещество. Систему нагревали с обратным холодильником, и она превращалась из желтой мутной жидкости в оранжевую прозрачную жидкость. Систему перемешивали в течение ночи, и реакцию проводили в общей сложности 24 ч. Система становилась черной и прозрачной. Добавляли EA (100 мл) и насыщенный водный раствор бикарбоната натрия, органическую фазу отделяли и концентрировали. Остаток суспендировали в 20 мл EA, и смесь фильтровали с получением соединения формулы 7.
[316] Пример 7
[317] Стадия данного примера аналогична стадии 1 примера 5. Соединение формулы 4' получали, используя толуол вместо 2-MeTHF.
[318] Пример 8
[319] Стадия данного примера аналогична стадии 1 примера 5. Соединение формулы 4' получали, используя ацетонитрил вместо 2-MeTHF.
[320] Пример 9
[321] Стадия данного примера аналогична стадии 1 примера 5. Соединение формулы 4' получали, используя тетрагидрофуран вместо 2-MeTHF.
[322] Пример 10
[323] Добавляли соединение формулы I' (1,3 г, 2 ммоль), морфолин (5 мл, 57 ммоль) и карбонат калия (2,0 г, 14,5 ммоль) в DCM (10 мл). После этого реакционную смесь нагревали при температуре 40°C в течение 2 ч, реакцию завершали по данным ВЭЖХ, и прекращали нагревание. Реакционную смесь фильтровали, и фильтрат добавляли к EtOH (10 мл). После нагревания смеси с обратным холодильником в течение 2 ч, нагревание прекращали. Реакционную смесь естественным образом охлаждали до 20-25°C и фильтровали, и отфильтрованный осадок высушивали с получением продукта (1,2 г).
[324] Пример 11
[325] В реакционную колбу добавляли 2-MeTHF (10 л), с последующим добавлением при перемешивании 60% гидрида натрия (300 г, 7,50 моль). Смесь подогревали до температуры 50 °C, и порциями добавляли соединение формулы 2' (1153,2 г, 3 моля). После перемешивания 1 ч добавляли раствор соединения формулы 1' (940,3 г, 3,3 моль) в 2-MeTHF (2 л). Реакционной смеси давали прореагировать при температуре 50°C в течение 6 ч, и соединение формулы 1' полностью прореагировало по показателям ТСХ. Реакционную смесь охлаждали до 20 °C, и по каплям и медленно добавляли уксусную кислоту (270 г, 4,5 моль). Через 10 мин перемешивания добавляли морфолин (784,8 г, 9 моль). Реакционную смесь нагревали с обратным холодильником в течение 15 ч, и реакцию завершали по данным ВЭЖХ-контроля в технологическом процессе. Нагревание останавливали, и систему охлаждали до 15-25 °C. Реакционную смесь промывали водой (10 л × 2), и органическую фазу сушили над безводным Na2SO4 и фильтровали под вакуумом. Отфильтрованный осадок промывали 2-MeTHF (2 л). К фильтрату добавляли 200 г активированного угля, и фильтрат нагревали с обратным холодильником в течение 1 ч и фильтровали в горячем состоянии. Отфильтрованный осадок промывали 2-MeTHF (1 л), и выпаривали из фильтрата 6 л растворителя при атмосферном давлении с последующим добавлением EtOH (8 л). Смесь нагревали с обратным холодильником в течение 1 ч, естественным образом охлаждали до 20-25 °C, прекращая нагревание, и фильтровали, а отфильтрованный осадок промывали EtOH (1 л). Влажный продукт переносили в DCM (15 л), и полученную смесь нагревали с обратным холодильником с последующим добавлением активированного угля (300 г). Смесь перемешивали в течение 0,5 ч и фильтровали в горячем состоянии, и отфильтрованный осадок промывали DCM (1 л). Выпаривали из фильтрата 2 л растворителя при атмосферном давлении с последующим добавлением EtOH (12 л). Смесь перемешивали с обратным холодильником в течение 4 ч, охлаждали до 20-25 °C, прекращая нагревание, и фильтровали под вакуумом, и отфильтрованный осадок высушивали с получением желтого твердого вещества (1234,5 г, выход 60,1%).
[326] Пример 12
[327] Стадия данного примера аналогична стадии 2 (реализация 2) примера 5. Используя Pt/C вместо оксида платины, посредством реакций получали соединение формулы 5' с выходом 80% или более.
[328] Пример 13
[329] Стадия данного примера аналогична стадии 2 (реализация 2) примера 5. Используя Pd/C вместо оксида платины, посредством реакций получали соединение формулы 5'.
[330] Пример 14
[331] Стадия данного примера аналогична стадии 2 (реализация 2) примера 5. Используя Pd(OH)2/C вместо оксида платины, посредством реакций получали соединение формулы 5'.
[332] Пример 15
[333] Стадия данного примера аналогична стадии 2 (реализация 2) примера 5. Используя Rh/C вместо оксида платины, посредством реакций получали соединение формулы 5'.
[334] Пример 16
[335] Стадия данного примера аналогична стадии 2 примера 5. Используя смесь THF и изопропанола вместо THF (реализация 2), посредством реакций получали соединение формулы 5'.
[336] Пример 17
[337] Стадия данного примера аналогична стадии 3 примера 5. Используя п-толуолсульфоновую кислоту вместо трифторуксусной кислоты (TFA), посредством реакций получали соединение формулы 6'.
[338] Пример 18
[339] Стадия данного примера аналогична стадии 3 примера 5. Используя метансульфоновую кислоту вместо трифторуксусной кислоты (TFA), посредством реакций получали соединение формулы 6'.
[340] Пример 19
[341] Стадия данного примера аналогична стадии 4 примера 5. Используя этанол вместо метанола, посредством реакций получали соединение формулы 7 с выходом 80% или более.
[342] Пример 20
[343] Добавляли соединение формулы 6' (385 г, 0,47 ммоль), TFA (138 г, 1,21 моль) и EtOH (55,66 г, 1,21 ммоль) в DCM (4 л). После нагревания реакционной смеси с обратным холодильником в течение 1,5 ч, нагревание прекращали, и реакционную смесь охлаждали до 20-25 °C. Добавляли раствор NaOH (66 г) в воде (2 л), и осаждалось твердое вещество. Смесь перемешивали в течение 1 ч и фильтровали под вакуумом. Отфильтрованный осадок растворяли в 12 л EtOH и 2 л воды, и добавляли 15 г активированного угля. Смесь перемешивали в течение 0,5 ч и фильтровали под вакуумом. Фильтрат концентрировали пока его не осталось 1,5 л, затем нагревали с обратным холодильником в течение 1 ч, охлаждали до 20-25 °C, и затем фильтровали под вакуумом, и отфильтрованный осадок сушили с получением желтого твердого вещества (114 г, 61,0%).
[344] Приведенное выше описание служит только для иллюстрации предпочтительного примера настоящего изобретения и не предназначено для ограничения объема настоящего изобретения. Любые модификации, эквиваленты, усовершенствования и тому подобное, выполненные без отступления от сущности и принципа настоящего изобретения, должны быть включены в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АМИНОКИСЛОТНЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2643146C2 |
| НОВЫЕ СПОСОБЫ ПОЛУЧЕНИЯ БАРУСИБАНА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2016 |
|
RU2726414C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ JAK И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2019 |
|
RU2786361C2 |
| СПОСОБ ПОЛУЧЕНИЯ F-БФА И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2019 |
|
RU2776178C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ БЕТА-МЕТИЛКАРБАПЕНЕМОВ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1993 |
|
RU2130927C1 |
| СПОСОБ ПРОИЗВОДСТВА ДЕГАРЕЛИКСА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2602042C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (ЦИКЛОПЕНТИЛ[d]ПИРИМИДИН-4-ИЛ)ПИПЕРАЗИНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2732404C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛОАЛКИЛКАРБОКСАМИДО-ИНДОЛЬНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2569678C2 |
| Способ получения пептида Ac-His-Ala-Glu-Glu-NH | 2021 |
|
RU2767030C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, ПРИМЕНИМОГО В КАЧЕСТВЕ ИНГИБИТОРА TAFIa | 2010 |
|
RU2538506C2 |
Изобретение относится к способу синтеза противоопухолевого соединения формулы 7, включающему следующие стадии:
и промежуточным соединениям. Технический результат: разработан новый способ синтеза противоопухолевого средства, который обладает улучшенной реализуемостью и позволяет упростить процесс, в котором образуется меньше отходов, что делает данный способ более подходящим для промышленного крупномасштабного производства, снижается содержание побочных продуктов и, таким образом, улучшается общий выход реакций. 3 н. и 33 з.п. ф-лы, 20 пр.
1. Способ синтеза противоопухолевого соединения формулы 7 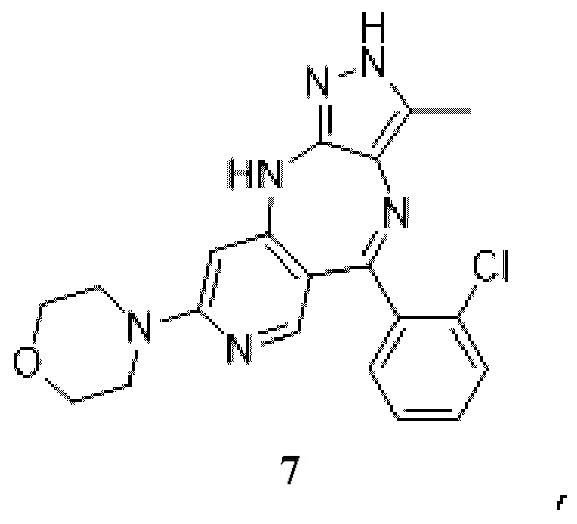
включающий следующие стадии:
(а) приведение соединения формулы 1 в реакцию ароматического нуклеофильного замещения соединением формулы 2М в органическом растворителе под действием основания с получением соединения формулы IM и приведение соединения формулы IM, отделенного или не отделенного, в другую реакцию ароматического нуклеофильного замещения соединением формулы 3, в щелочных условиях, с получением соединения формулы 4М; где
на стадии (а):
органический растворитель выбран из одного или более из 2-метилтетрагидрофурана, ацетонитрила, тетрагидрофурана и толуола;
основание выбрано из одного или более из гидрида натрия, гидроксида натрия, карбоната цезия, триэтилендиамина, трет-бутоксида натрия, трет-бутоксида калия, бис(триметилсилил)амида лития и гексаметилдисилазида натрия;
реакцию проводят при давлении от 0 до 10 МПа (избыточное давление);
реакцию ароматического нуклеофильного замещения соединения формулы 1 соединением формулы 2М под действием основания проводят в течение периода времени 3-18 ч при температуре от 40 до 70°С;
другую ароматическую реакцию нуклеофильного замещения соединением формулы 3 проводят в течение периода времени от 8 до 16 ч;
для обеспечения щелочных условий на стадии (а) вместо добавления дополнительного основания в качестве основания используют морфолин или добавляют дополнительное основание, выбранное из одного или более из гидрида натрия, гидроксида натрия, карбоната цезия, триэтилендиамина, трет-бутоксида натрия, трет-бутоксида калия, бис(триметилсилил)амида лития и гексаметилдисилазида натрия;
(b) приведение соединения формулы 4М в реакцию восстановления в органическом растворителе под действием катализатора с получением соединения формулы 5М;
на стадии (b):
органический растворитель выбран из одного или более из метанола, этанола, изопропанола, тетрагидрофурана, 2-метилтетрагидрофурана, уксусной кислоты и ацетонитрила;
катализатор выбран из порошка железа, оксида платины, Pt/C, Pd(OH)2/C, Rh/C и Pd/C;
(c) приведение соединения формулы 5М в реакцию замыкания кольца в органическом растворителе под действием кислоты с получением соединения формулы 6М;
на стадии (с):
кислота выбрана из одной или более из трифторуксусной кислоты, метансульфоновой кислоты, п-толуолсульфоновой кислоты, муравьиной кислоты, уксусной кислоты и хлористоводородной кислоты;
органический растворитель выбран из по меньшей мере одного из дихлорметана, метанола, этанола и изопропанола;
реакцию проводят в течение периода времени 1-96 ч при давлении от 0 до 10 МПа (избыточное давление); и
(d) приведение соединения формулы 6М в реакцию в органическом растворителе для удаления аминозащитной группы с получением противоопухолевого соединения формулы 7;
на стадии (d):
органический растворитель выбран из одного или более из толуола, дихлорметана, ацетонитрила, тетрагидрофурана, метанола, этанола и изопропанола;
добавляют также кислоту и кислота выбрана из по меньшей мере одной из муравьиной кислоты, уксусной кислоты, хлористоводородной кислоты, метансульфоновой кислоты и трифторуксусной кислоты;
в дополнение к растворителю добавляют спирт или фенол; спирт выбран из метанола и/или этанола и фенол выбран из фенола и/или п-метоксифенола;
реакцию проводят в течение периода времени 1-96 ч при давлении от 0 до 10 МПа (избыточное давление); и
где схема реакции является следующей:
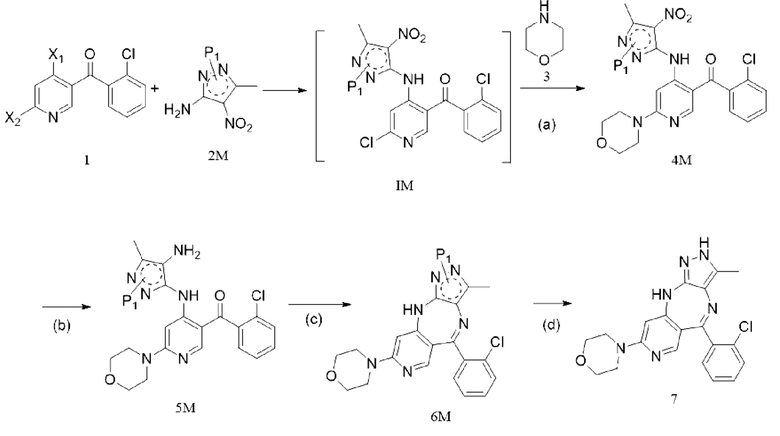
где каждый Х1 и Х2 выбран из хлора; Р1 представляет собой Trt или ТНР; P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен Р1.
2. Способ по п. 1, в котором на стадии (а):
реакцию проводят при атмосферном давлении; при соотношении растворитель : соединение формулы 1 : соединение формулы 2М : соединение формулы 3 : основание = (0,5-60) л : (0,1-11) моль : 1 моль : (0,3-30) моль : (0,2-25) моль.
3. Способ по любому из пп. 1 и 2, в котором на стадии (а):
реакцию ароматического нуклеофильного замещения соединения формулы 1 соединением формулы 2М под действием основания проводят в течение периода времени 3-8 ч при температуре от 40 до 60°С.
4. Способ по любому из пп. 1-3, в котором на стадии (а):
другую ароматическую реакцию нуклеофильного замещения соединением формулы 3 проводят в течение периода времени от 13 до 16 ч.
5. Способ по любому из пп. 1-4, в котором на стадии (а):
другую ароматическую реакцию нуклеофильного замещения соединением формулы 3 проводят при температуре, которая является температурой кипения растворителя;
при соотношении растворитель : соединение формулы 1 : соединение формулы 2М : соединение формулы 3 : основание = 5,8 л : 1,1 моль : 1 моль : 3 моль : 2,5 моль.
6. Способ по любому из пп. 1-5, в котором на стадии (а):
добавляют сульфат магния в реакцию ароматического нуклеофильного замещения соединения формулы 1 соединением формулы 2М под действием основания и добавляют уксусную кислоту в другую реакцию ароматического нуклеофильного замещения соединением формулы 3,
при соотношении растворитель : соединение формулы 1 : соединение формулы 2М : соединение формулы 3 : основание : сульфат магния : уксусная кислота = (0,5-60) л : (0,1-11) моль : 1 моль : (0,3-30) моль : (0,2-25) моль : (0,2-20) моль : (0,1-15) моль.
7. Способ по любому из пп. 1-6, в котором на стадии (а):
добавляют сульфат магния в реакцию ароматического нуклеофильного замещения соединения формулы 1 соединением формулы 2М под действием основания и добавляют уксусную кислоту в другую реакцию ароматического нуклеофильного замещения соединением формулы 3,
при соотношении растворитель : соединение формулы 1 : соединение формулы 2М : соединение формулы 3 : основание : сульфат магния : уксусная кислота = 5,8 л : 1,1 моль : 1 моль : 3 моль : 2,5 моль : 1,6 моль : 1,5 моль.
8. Способ по любому из пп. 1-7, в котором на стадии (b):
когда катализатор представляет собой порошок железа, реакцию проводят в течение периода времени 1-96 ч при давлении от 0 до 10 МПа (избыточное давление), при температуре, которая является температурой кипения растворителя;
при соотношении органический растворитель : соединение формулы 4М : катализатор = (1-130) л : 1 моль : (1-150) моль.
9. Способ по п. 7, где на стадии (b):
когда катализатор представляет собой порошок железа, реакцию проводят в течение периода времени 15-24 ч при атмосферном давлении, при температуре, которая является температурой кипения растворителя;
при соотношении органический растворитель : соединение формулы 4М : катализатор = 13 л : 1 моль : (10-18) моль.
10. Способ по п. 8, где
соотношение органический растворитель : соединение формулы 4М : катализатор = 13 л : 1 моль : (14-18) моль.
11. Способ по п. 8, где
соотношение органический растворитель : соединение формулы 4М : катализатор = 13 л : 1 моль : 15 моль.
12. Способ по п. 8, где:
на стадии (b) добавляют также хлорид аммония в количестве 0,05-5 молярных эквивалентов;
растворитель представляет собой смешанный растворитель из этанола и тетрагидрофурана.
13. Способ по п. 12, в котором
на стадии (b) хлорид аммония добавляют в количестве 0,5 молярных эквивалентов.
14. Способ по п. 12, в котором
на стадии (b) растворитель представляет собой смешанный растворитель из этанола и тетрагидрофурана в объемном соотношении этанола и тетрагидрофурана (6-10) : 10.
15. Способ по любому из пп. 1-7, в котором
на стадии (b), когда катализатор представляет собой оксид платины, Pt/C, Pd(OH)2/C, Rh/C или Pd/C, реакцию проводят в атмосфере водорода при температуре от 30 до 80°С в течение 20-80 ч, при давлении от 0 до 10 МПа (избыточное давление), при соотношении растворитель : соединение формулы 4М : катализатор = (1-50) мл : 1 г : (0,03-0,2) г.
16. Способ по п. 15, в котором
на стадии (b), когда катализатор представляет собой оксид платины, Pt/C, Pd(OH)2/C, Rh/C или Pd/C, реакцию проводят в атмосфере водорода при температуре от 60 до 80°С в течение 20-72 ч.
17. Способ по п. 15, в котором
реакцию проводят при давлении более чем 0,5-2 МПа (избыточное давление).
18. Способ по п. 15, в котором
соотношение растворитель : соединение формулы 4М : катализатор = 22 мл : 1 г : 0,1 г.
19. Способ по любому из пп. 1-18, в котором
на стадии (b) добавляют также небольшое количество воды;
растворитель содержит тетрагидрофуран; объемное соотношение воды и тетрагидрофурана составляет (0,2-2) : 10.
20. Способ по любому из пп. 1-19, в котором
на стадии (с) реакцию проводят в течение периода времени 1,5-3 ч.
21. Способ по любому из пп. 1-20, в котором
на стадии (с) реакцию проводят при температуре, которая является температурой кипения растворителя.
22. Способ по любому из пп. 1-21, в котором
на стадии (с) реакцию проводят атмосферном давлении.
23. Способ по любому из пп. 1-22, в котором
на стадии (с) реакцию проводят при соотношении растворитель : соединение формулы 5М : кислота=(0,5-70) л : 1 моль : 0,220 моль.
24. Способ по любому из пп. 1-23, в котором
на стадии (с) реакцию проводят при соотношении растворитель : соединение формулы 5М : кислота = 6,6 л : 1 моль : 2 моль.
25. Способ по любому из пп. 1-19 и 22-24, в котором
на стадии (с) соединение формулы 5М выдерживают при температуре кипения изопропанола под действием трифторуксусной кислоты в течение от 1 до 3 ч.
26. Способ по любому из пп. 1-25, в котором
на стадии (d) реакцию проводят в течение периода времени 17 ч.
27. Способ по любому из пп. 1-26, в котором
на стадии (d) реакцию проводят при атмосферном давлении.
28. Способ по любому из пп. 1-27, в котором
на стадии (d) реакцию проводят при температуре, которая является температурой кипения растворителя; при соотношении растворитель : соединение формулы 6М : кислота : спирт или фенол = (10-800) л : 1 моль : (0,1-10) моль : (0,2-20) моль.
29. Способ по любому из пп. 1-28, в котором
на стадии (d) реакцию проводят при соотношении растворитель : соединение формулы 6М : кислота : спирт или фенол = 84 л : 1 моль : 1 моль : 2 моль.
30. Способ по любому из пп. 1-29, дополнительно включающий стадии синтеза соединения формулы 2М:
(i) приведение соединения формулы 2-1 в реакцию нитрования с азотной кислотой в растворителе с получением соединения формулы 2-2;
(ii) приведение соединения формулы 2-2 в реакцию защиты аминогруппы с аминозащитным реактивом в органическом растворителе с получением соединения формулы (2-3)М; и
(iii) приведение соединения формулы (2-3)М в реакцию в органическом растворителе для удаления фталоильной группы с получением соединения формулы 2М;
схема реакции является следующей:
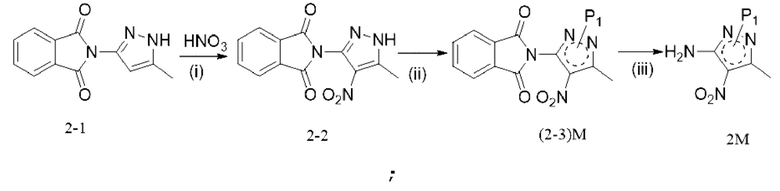
причем аминозащитный реактив представляет собой TrtCl или DHP; P1 выбран из Trt и ТНР; P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1.
31. Способ по п. 30, в котором
на стадии (i):
соединение формулы 2-1 присутствует в количестве 1 молярный эквивалент,
азотная кислота присутствует в количестве 1-2 молярных эквивалента,
растворитель выбран из концентрированной серной кислоты, уксусного ангидрида и уксусной кислоты и
реакцию стадии (i) проводят в течение периода времени 0,5-2 ч при температуре 10-20°С;
на стадии (ii):
соединение формулы 2-2 присутствует в количестве 1 молярный эквивалент,
аминозащитный реагент присутствует в количестве 1-1,5 молярных эквивалента,
органический растворитель выбран из одного или более из тетрагидрофурана, 2-метилтетрагидрофурана, дихлорметана и ацетонитрила и
реакцию стадии (ii) проводят в течение периода времени 1-48 ч при температуре 60-100°С; и
на стадии (iii):
соединение формулы (2-3)М присутствует в количестве 1 молярный эквивалент,
органический растворитель выбран из одного или более из этанола, тетрагидрофурана и ацетонитрила и
реакцию стадии (iii) проводят в течение периода времени 1-3 ч при температуре кипения растворителя.
32. Способ по п. 30, в котором:
когда аминозащитный реактив представляет собой TrtCl, реакцию на стадии (ii) проводят в следующих условиях: под действием основания при температуре от 60 до 100°С в течение от 1 до 48 ч;
когда аминозащитный реактив представляет собой DHP, реакцию на стадии (ii) проводят в следующих условиях: под действием п-толуолсульфоновой кислоты или п-толуолсульфоната пиридиния при температуре от 60 до 100°С с обратным холодильником в течение от 3 до 48 ч.
33. Способ по любому пп. 1-32, дополнительно включающий стадию синтеза соединения формулы 1:
приведение соединения формулы 1-1 в реакцию сочетания с соединением формулы 1-2 в органическом растворителе в течение периода времени 2-4 ч при температуре от -70 до 60°С с получением соединения формулы 1; и
схема реакции является следующей:
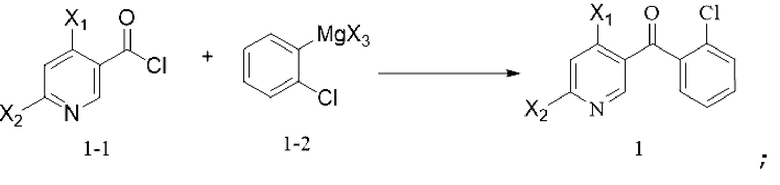
где Х3 представляет собой галоген, предпочтительно хлор; каждый X1 и Х2 выбраны из хлора.
34. Способ по п. 33, в котором
соединение формулы 1-1 присутствует в количестве 1-2 молярных эквивалента;
соединение формулы 1-2 присутствует в количестве 1-2 молярных эквивалента;
органический растворитель представляет собой тетрагидрофуран.
35. Промежуточное соединение для получения противоопухолевого соединения формулы 7, имеющее следующие структурные формулы:
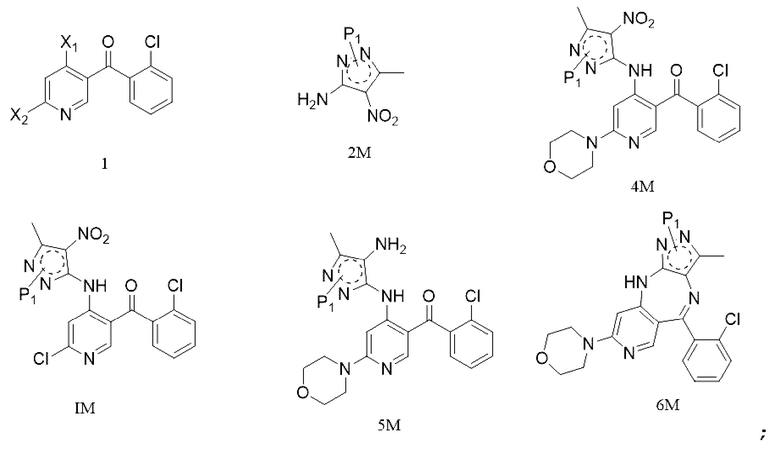
где каждый X1 и Х2 независимо выбраны из хлора;
P1 выбран из Trt и ТНР; P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1.
36. Промежуточное соединение для получения противоопухолевого соединения формулы 7, имеющее следующую структурную формулу:
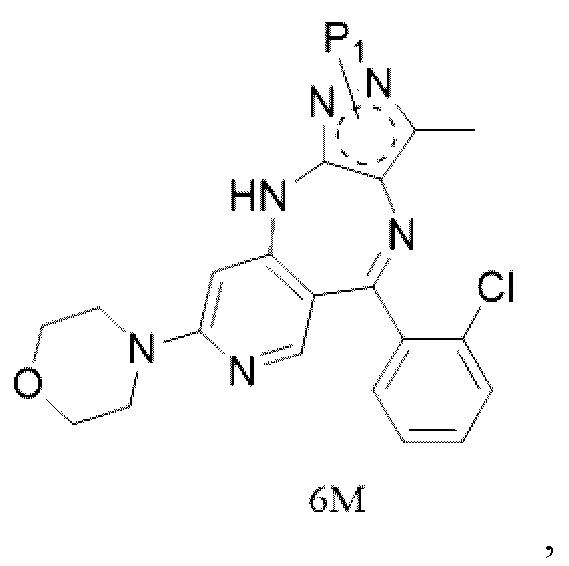
где P1 выбран из Trt и ТНР; P1 связан с атомом N в орто-положении к метильной группе в кольце, где расположен P1;
формула 6М представляет собой комплекс, содержащий одну молекулу трифторуксусной кислоты и одну молекулу изопропанола.
| US 2019040064 A1, 07.02.2019 | |||
| ПИРАЗОЛБЕНЗОДИАЗЕПИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ CDK2, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ | 2000 |
|
RU2249593C2 |
| WO 2006040036 A1, 20.04.2006. | |||

