Уровень техники изобретения
Область техники, к которой относится изобретение
Изобретение относится к способу получения соединений, которые можно использовать в качестве промежуточных соединений для получения лекарственных средств. В частности, изобретение относится к получению промежуточных соединений для ингибиторов JAK.
Существующий уровень техники
Семейство JAK состоит из четырех членов: JAK1, JAK2, JAK3 и тирозинкиназа 2 (TYK2). Связывание цитокина с JAK-зависимым цитокиновым рецептором вызывает димеризацию рецептора, что приводит к фосфорилированию остатков тирозина на киназе JAK, влияя на активацию JAK. Фосфорилированные JAK, в свою очередь, связывают и фосфорилируют различные белки STAT, которые димеризуются, интернализуются в ядре клетки и напрямую модулируют транскрипцию генов, приводя, среди прочего, к последующим эффектам, связанным с воспалительным заболеванием. JAK обычно ассоциируются с рецепторами цитокинов парами в виде гомодимеров или гетеродимеров. Каждый из четырех членов семейства JAK участвует в передаче сигналов по меньшей мере одного из цитокинов, связанных с воспалением. Следовательно, химический ингибитор с пан-активностью против всех членов семейства JAK может модулировать широкий спектр провоспалительных путей, которые способствуют воспалительным заболеваниям легких, таким как тяжелая астма, ХОБЛ и хроническая дисфункция аллотрансплантата легких (CLAD). Поэтому было бы желательно иметь эффективный процесс получения специфических ингибиторов JAK.
Сущность изобретения
Изобретение направлено на способ получения соединений, которые могут быть использованы в качестве промежуточных соединений для получения лекарственных средств, обладающих ингибирующей активностью в отношении JAK.
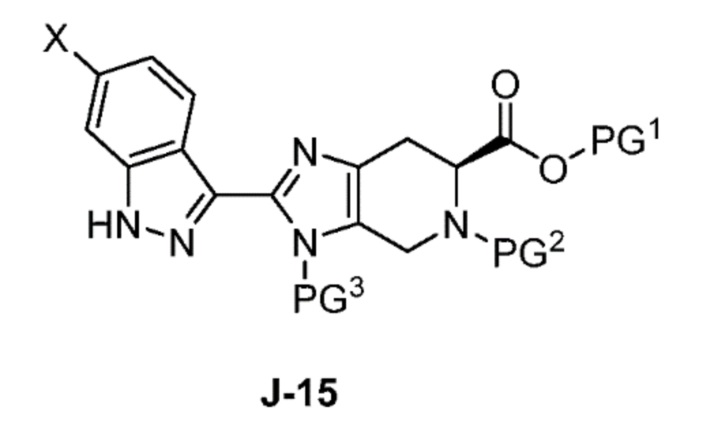
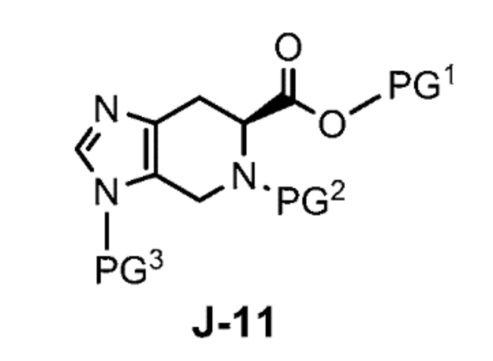
Соответственно, в одном аспекте изобретение предоставляет способ получения соединения формулы J-15 или его соли:
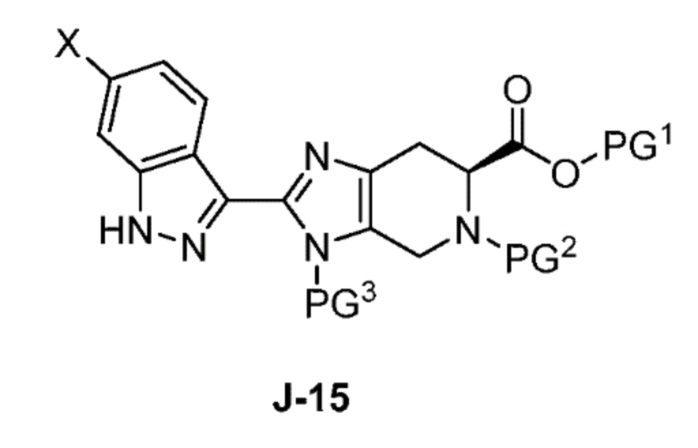
где
X выбран из группы, состоящей из Br, I и Cl;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу;
способ, включающий:
(a) взаимодействие соединения формулы J-14:
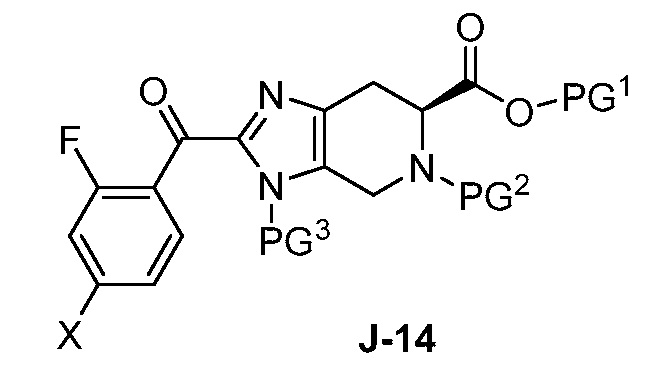
или его соли с гидразином с получением соединения формулы J-15 и
(b) необязательно образование соли соединения J-15.
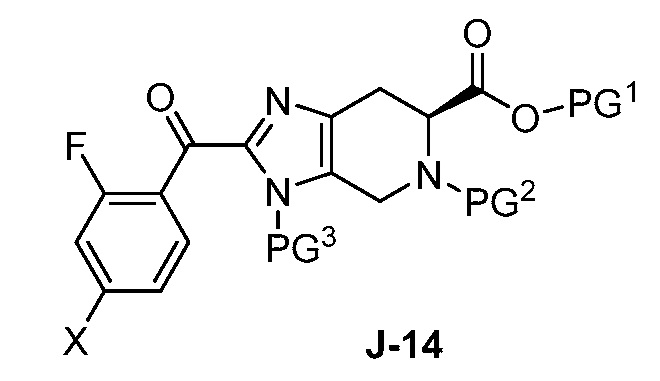
В другом аспекте изобретение относится к соединению формулы J-14:
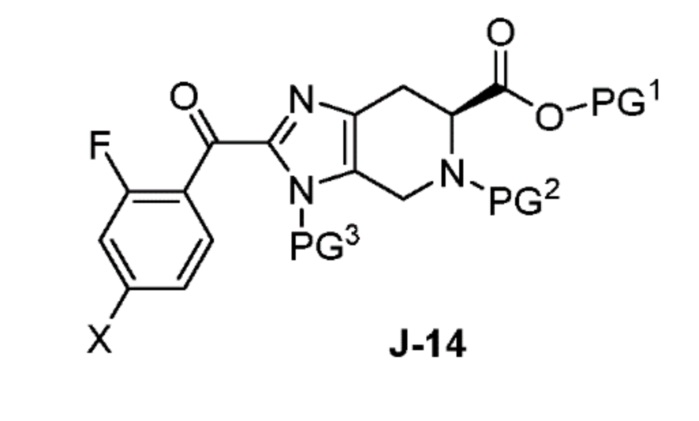
или его соли, где
X выбран из группы, состоящей из Br, I и Cl;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу.
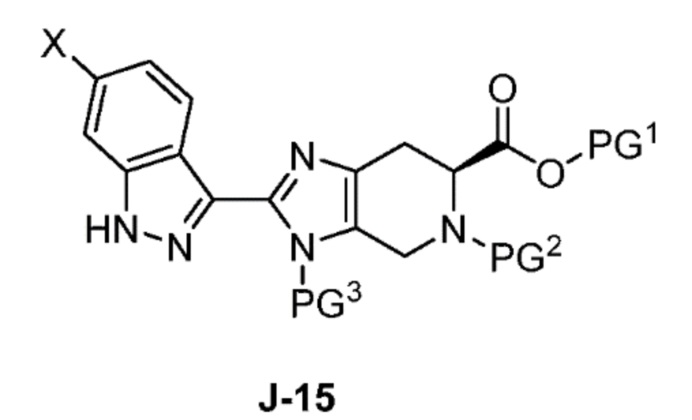
В другом аспекте изобретение относится к соединению формулы J-15:
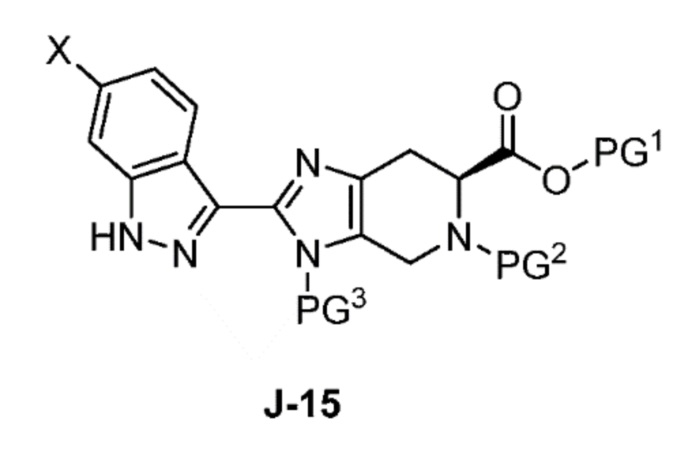
или его соли,
где
X выбран из группы, состоящей из Br, I и Cl;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу.
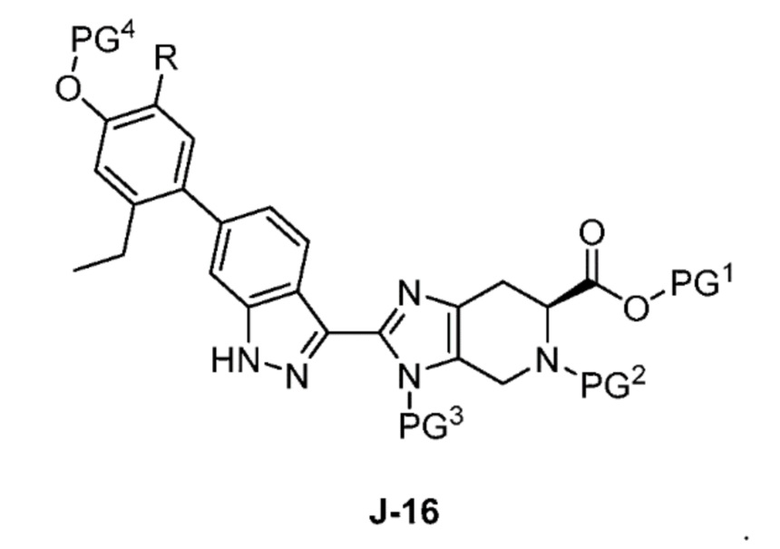
В другом аспекте изобретение относится к способу получения соединения формулы J-16, или его соли:
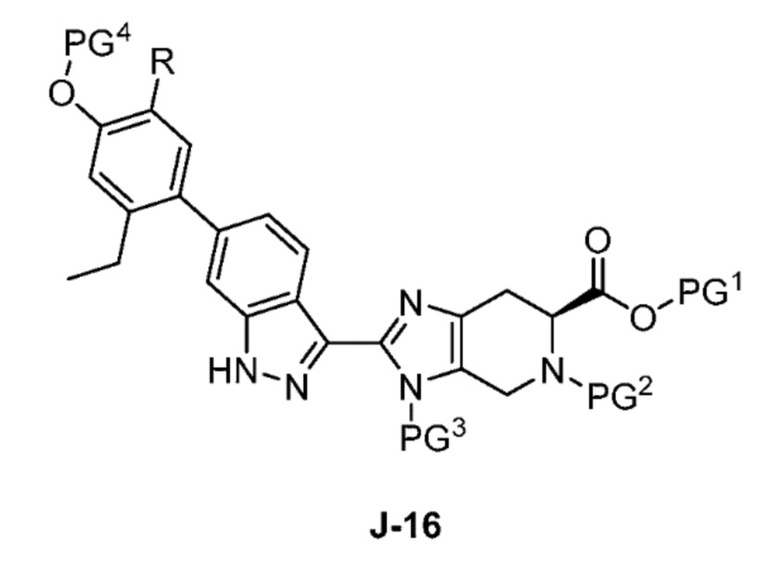
где
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу;
PG3 представляет собой аминозащитную группу;
PG4 представляет собой гидроксилзащитную группу; и
R представляет собой H или F;
включающему:
(a) взаимодействие соединения формулы J-13:
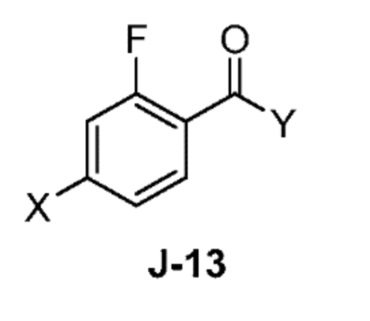
где X выбран из группы, состоящей из Br, I и Cl; и Y представляет собой уходящую группу; с соединением формулы J-11:
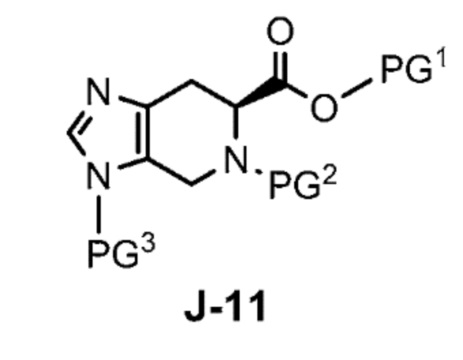
в присутствии основания, с получением соединения формулы J-14:
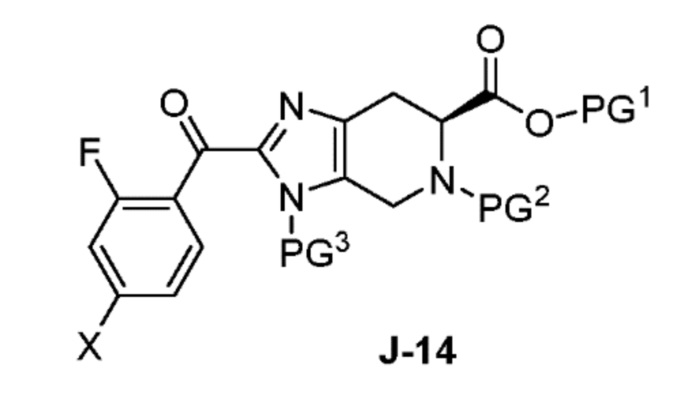
и необязательно образование соли соединения J-14;
(b) взаимодействие соединения формулы J-14 или его соли с гидразином с получением соединения формулы J-15:
и необязательно образование соли соединения J-15;
(c) взаимодействие соединения формулы J-15, или его соли, с соединением формулы J-5, J-6 или J-7:
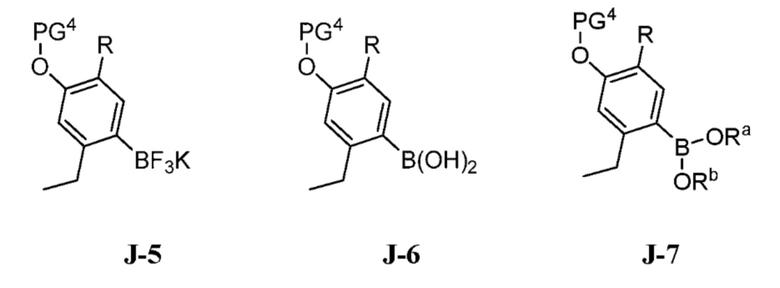
где Ra и Rb, каждый, независимо выбраны из C1-8 алкила, где Ra и Rb необязательно могут быть соединены с образованием 4-8-членного кольца; в присутствии основания, палладиевого катализатора и фосфинового лиганда с получением соединения формулы J-16, и необязательно образование соли соединения J-16.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте изобретение относится к способу получения соединения формулы J-15 или его соли:
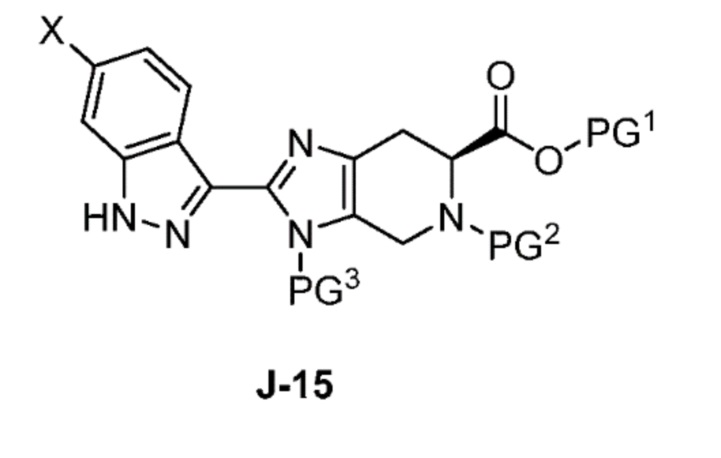
где
X выбран из группы, состоящей из Br, I и Cl;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу;
включающему:
(a) взаимодействие соединения формулы J-14:
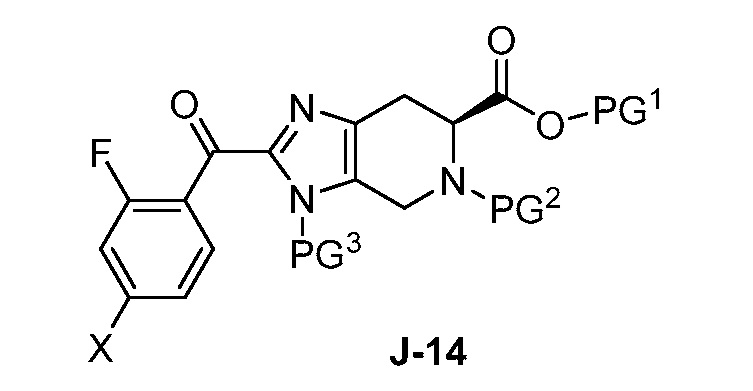
или его соли с гидразином с получением соединения формулы J-15 и
(b) необязательно образование соли соединения J-15.
В некоторых аспектах, реакцию с гидразином проводят при примерно 60°C. В некоторых аспектах, реакцию с гидразином проводят при 60°C±20°C.
В некоторых аспектах, X выбран из группы, состоящей из Br, I и Cl; PG1 представляет собой алкильную или бензильную группу, где бензильная группа является необязательно замещенной; PG2 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы; и PG3 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы.
В некоторых аспектах, X представляет собой Br. В некоторых аспектах, X представляет собой Br, PG1 представляет собой бензил, PG2 представляет собой трет-бутоксикарбонил, и PG3 представляет собой бензил.
В некоторых аспектах, соединение J-14 или его соль получают:
(a) взаимодействием соединения формулы J-13:
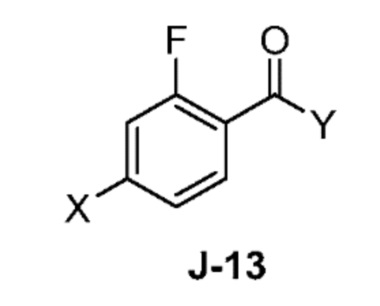
где Y представляет собой уходящую группу, с соединением формулы J-11:
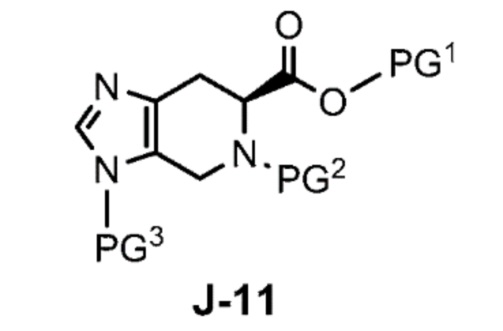
в присутствии основания, с получением J-14, и
(b) необязательно образование соли соединения J-14.
В некоторых аспектах, Y представляет собой Cl.
В другом аспекте изобретение относится к соединению формулы J-14:
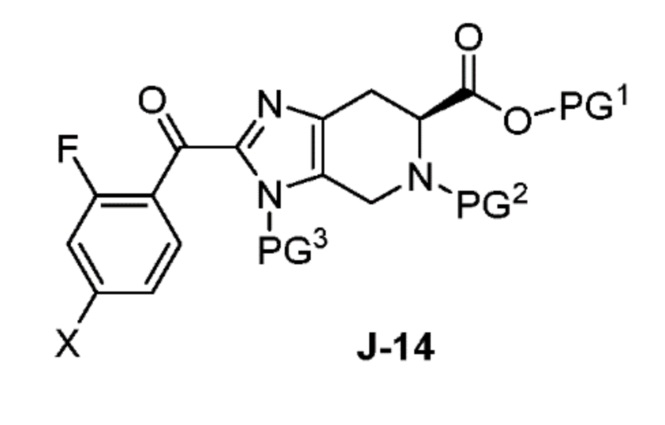
или его соли, где
X выбран из группы, состоящей из Br, I и Cl;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу.
В некоторых аспектах, X выбран из группы, состоящей из Br, I и Cl; PG1 представляет собой алкильную или бензильную группу, где бензильная группа является необязательно замещенной; PG2 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы; и PG3 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы.
В другом аспекте изобретение относится к соединению формулы I-14:
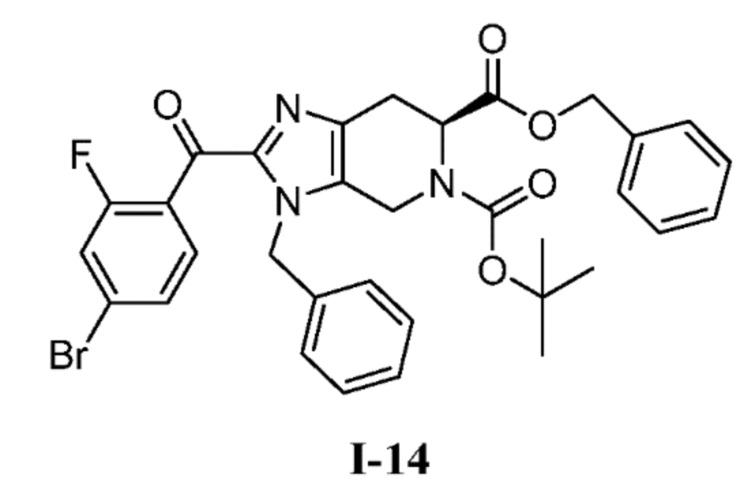
или его соли.
В другом аспекте изобретение относится к соединению формулы J-15:
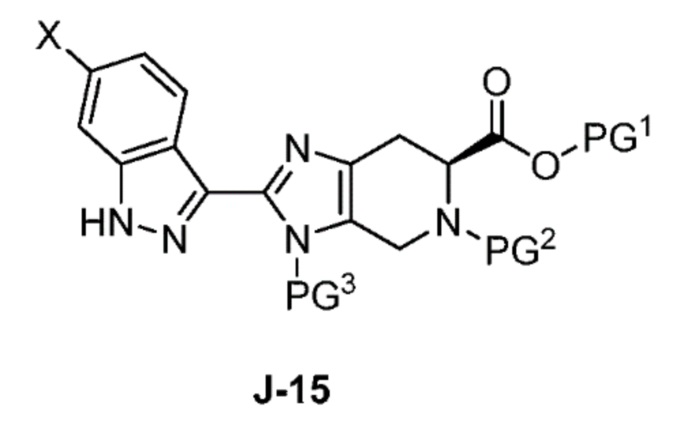
или его соли,
где
X выбран из группы, состоящей из Br, I и Cl;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу.
В некоторых аспектах, X выбран из группы, состоящей из Br, I и Cl; PG1 представляет собой алкильную или бензильную группу, где бензильная группа является необязательно замещенной; PG2 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы; и PG3 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы.
В другом аспекте изобретение относится к соединению формулы I-15:
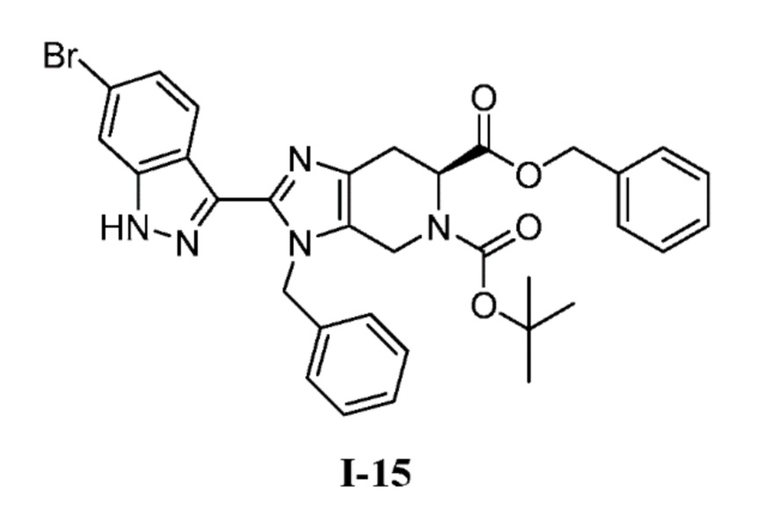
или его соли.
В другом аспекте изобретение относится к способу получения соединения формулы J-16, или его соли:
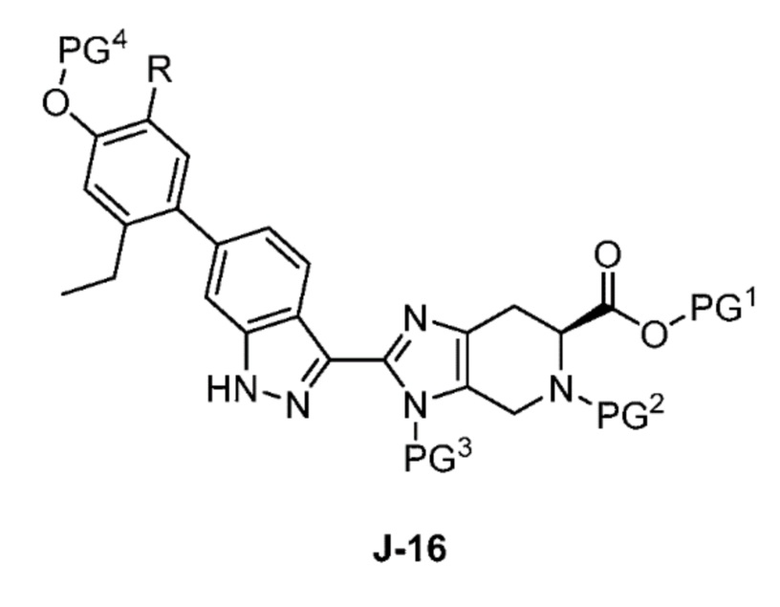
где
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу;
PG3 представляет собой аминозащитную группу;
PG4 представляет собой гидроксилзащитную группу; и
R представляет собой H или F;
включающему:
(a) взаимодействие соединения формулы J-13:
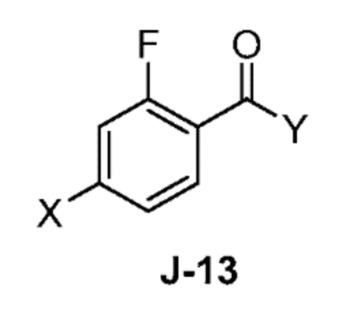
где X выбран из группы, состоящей из Br, I и Cl; и Y представляет собой уходящую группу; с соединением формулы J-11:
в присутствии основания, с получением соединения формулы J-14:
и необязательно образование соли соединения J-14;
(b) взаимодействие соединения формулы J-14 или его соли с гидразином с получением соединения формулы J-15:
и необязательно образование соли соединения J-15;
(c) взаимодействие соединения формулы J-15 или его соли с соединением формулы J-5, J-6 или J-7:
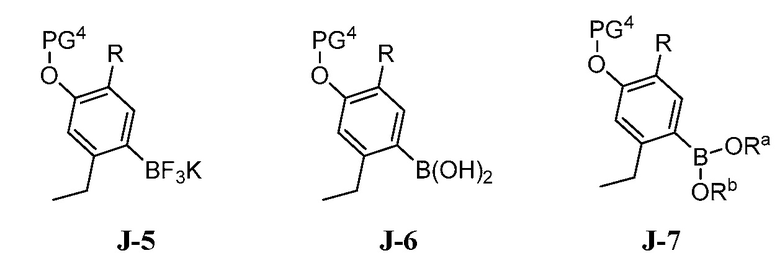
где Ra и Rb, каждый, независимо выбраны из C1-8 алкила, где Ra и Rb необязательно могут быть соединены с образованием 4-8-членного кольца; в присутствии основания, палладиевого катализатора и фосфинового лиганда с получением соединения формулы J-16, и необязательно образование соли соединения J-16.
В некоторых аспектах, стадию (c) проводят в присутствии диборонового реагента. В некоторых аспектах, стадию (c) проводят в присутствии тетрагидроксидиборона или сложного эфира дибороновой кислоты. В некоторых вариантах осуществления катализатор представляет собой продукт реакции бис(пинаколато)диборона с гидрофторидом фторида калия. В некоторых вариантах осуществления катализатор получают взаимодействием бис(пинаколато)диборона в пропан-2-оле с гидрофторидом фторида калия в воде с последующей фильтрацией и сушкой полученных твердых веществ.
В некоторых аспектах, взаимодействие с гидразином на стадии (b) проводят при 60°C±20°C.
В некоторых аспектах, X выбран из группы, состоящей из Br, I и Cl;
PG1 представляет собой алкильную или бензильную группу, где бензильная группа является необязательно замещенной; PG2 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы; PG3 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы; PG4 выбран из группы, состоящей из силильной группы, ацильной группы и арилметильной группы. В некоторых аспектах, X представляет собой Br. В некоторых аспектах, X представляет собой Br, PG1 представляет собой бензил, PG2 представляет собой трет-бутоксикарбонил, PG3 представляет собой бензил, и PG4 представляет собой бензил.
В некоторых аспектах, Y представляет собой Cl.
В некоторых аспектах, палладиевый катализатор и фосфиновый лиганд стадии (c) представляют собой бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий(II).
В настоящем способе соединение J-13:
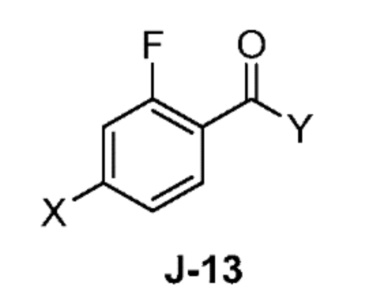
взаимодействует с соединением формулы J-11:
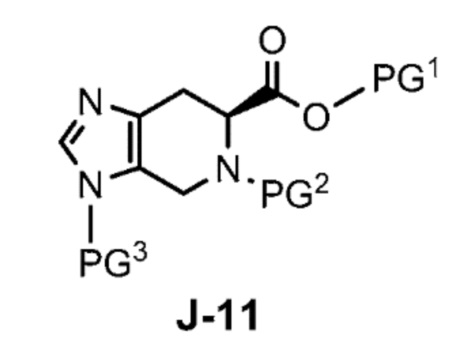
в присутствии основания, с получением соединения формулы J-14:
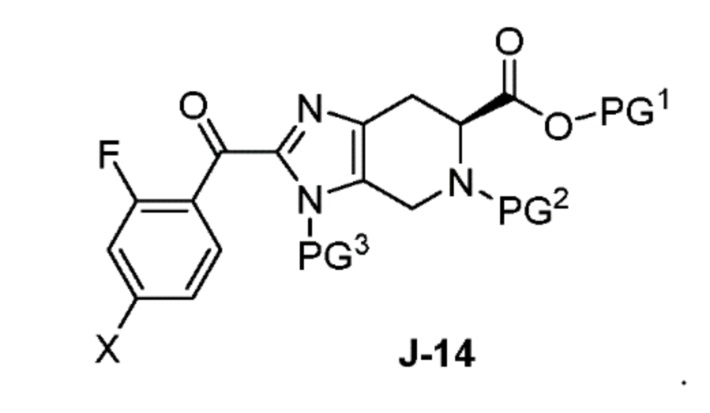
Обычно реакцию проводят в растворителе, таком как ацетонитрил, в присутствии избытка (например, от 3 до 7 эквивалентов) основания, такого как триметиламин. Обычно используется от одного до двух эквивалентов J-13.
В настоящем способе соединение J-14 или его соль взаимодействует с гидразином с получением соединения формулы J-15:
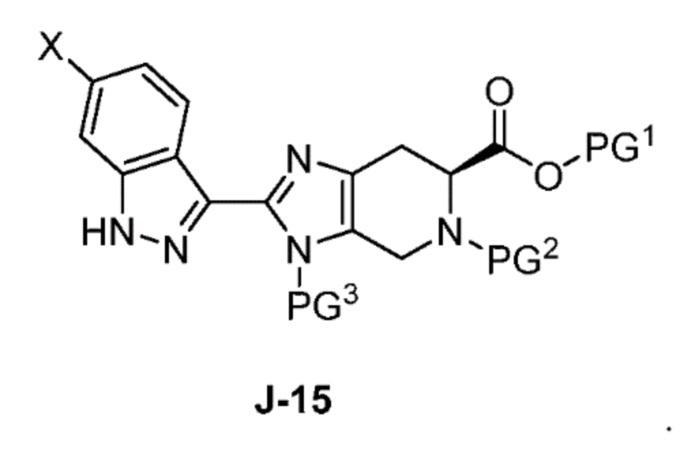
Обычно реакцию проводят в растворителе, таком как THF, и используют избыток гидразина, например от 2 до 10 эквивалентов. Реакцию нагревают при примерно 60°C до завершения, обычно от 0,5 до 6 часов.
В настоящем способе соединение J-15 или его соль взаимодействует с соединением формулы J-5, J-6 или J-7:
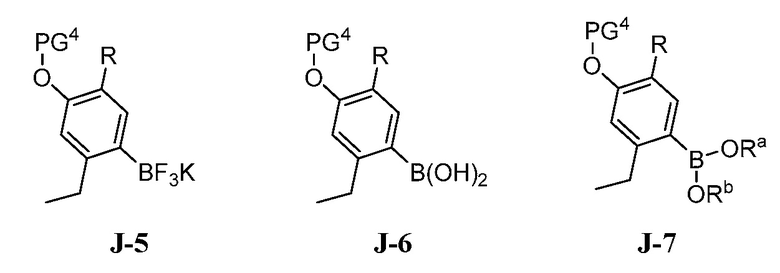
в присутствии основания, палладиевого катализатора и фосфинового лиганда с получением соединения формулы J-16:
Реакцию обычно проводят путем приведения в контакт J-16 с от примерно 1 до примерно 1,5 эквивалентов J-5, J-6 или J-7 в присутствии каталитического количества палладиевого катализатора и фосфинового лиганда (между примерно 0,005 и примерно 0,1 эквивалентов) и от примерно 2 до примерно 6 эквивалентов основания. Проведение реакции в присутствии дополнительного катализатора увеличивает выход продукта J-16. Дополнительным катализатором может быть дибороновый реагент. Дополнительным катализатором может быть тетрагидроксидиборон, сложный эфир дибороновой кислоты или продукт реакции бис(пинаколато)диборона с гидрофторидом фторида калия, как показано в получении 4.
Подходящие палладиевые катализаторы включают бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий(II), трис (дибензилиденацетон)дипалладий(0) (Pd2dba3), ацетат палладия (II) (Pd(OAc)2), дихлор(1,1’-бис(дифенилфосфино)ферроцен)дипалладий(II) (Pd(dppf)Cl2), дихлор бис(трифенилфосфин)-палладий(II) (Pd(PPh3)2Cl2), и тому подобное, где в скобках даны общепринятые сокращения. Фосфиновые лиганды, используемые в настоящей реакции, включают трициклогексилфосфин (PCy3), трициклогексилфосфинтетрафторборат (PCy3HBF4), 1,1'-бис(дифенилфосфино)ферроцен (dppf), 1,1'-бис(ди-трет-бутилфосфино)ферроцен, три(2-фурил)фосфин, 1,3-бис(дифенилфосфино)пропан (dppp), 1,5-бис(дифенилфосфино)пентан (dpppe), три-трет-бутилфосфин (P(t-Bu)3), и 99,9-диметил-4,5-бис(дифенилфосфино)ксантен (ксантфос).
Конкретные основания для реакции сочетания включают фторид калия, карбонат цезия и фторид цезия. Альтернативно, в качестве основания можно использовать карбонат натрия, карбонат калия, ацетат натрия, трет-бутоксид калия, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) или 1,4-диазабицикло[2.2.2]октан (DABCO). Реакцию обычно проводят в инертном растворителе, таком как тетрагидрофуран, N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон. Подходящие системы смешанных растворителей включают тетрагидрофуран и воду, тетрагидрофуран и N,N-диметилформамид, тетрагидрофуран и N-метилпирролидон, ацетон и воду, этанол и воду и изопропанол и воду. Реакцию обычно проводят при температуре от приблизительно 40 до приблизительно 120°С в течение от приблизительно 1 до приблизительно 20 часов или до тех пор, пока реакция по существу не завершится. Продукт J-16 выделяют в виде твердого вещества общепринятыми способами.
Определения
При описании настоящего изобретения, включая его различные аспекты и варианты осуществления, следующие термины имеют следующие значения, если не указано иное.
Термин «алкил» означает одновалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной, или их комбинациями. Если не указано иное, такие алкильные группы обычно содержат от 1 до 10 атомов углерода. Типичные алкильные группы включают, например, метил (Me), этил (Et), н-пропил (n-Pr) или (nPr), изопропил (i-Pr) или (iPr), н-бутил (n-Bu) или (nBu), втор-бутил, изобутил, трет-бутил (t-Bu) или (tBu), н-пентил, н-гексил, 2,2-диметилпропил, 2-метилбутил, 3-метилбутил, 2-этилбутил, 2,2-диметилпентил, 2-пропилпентил и тому подобное.
Когда определенное количество атомов углерода предназначено для определенного термина, количество атомов углерода указано перед термином. Например, термин «C1-3 алкил» означает алкильную группу, имеющую от 1 до 3 атомов углерода, где атомы углерода находятся в любой химически приемлемой конфигурации, включая линейные или разветвленные конфигурации.
Термин «циклоалкил» означает одновалентную насыщенную карбоциклическую группу, которая может быть моноциклической или полициклической. Если не указано иное, такие циклоалкильные группы обычно содержат от 3 до 10 атомов углерода. Типичные циклоалкильные группы включают, например, циклопропил (cPr), циклобутил (cBu), циклопентил, циклогексил, циклогептил, циклооктил, адамантил и тому подобное.
Термин «цпропил» означает циклопропил.
Термин «гетероциклил», «гетероцикл», «гетероциклический» или «гетероциклическое кольцо» означает одновалентную насыщенную или частично ненасыщенную циклическую неароматическую группу, имеющую от 3 до 10 общих кольцевых атомов, где кольцо содержит от 2 до 9 кольцевых атомов углерода и от 1 до 4 кольцевых гетероатомов, выбранных из азота, кислорода и серы. Гетероциклические группы могут быть моноциклическими или полициклическими (т.е. конденсированными или соединенными мостиковой связью). Типичные гетероциклильные группы включают, например, пирролидинил, пиперидинил, пиперазинил, имидазолидинил, морфолинил, тиоморфолил, индолин-3-ил, 2-имидазолинил, тетрагидропиранил, 1,2,3,4-тетрагидроизохинхолин-2-ил, 7-азанорборнанил, нортропанил и т.п., где точка присоединения находится у любого доступного кольцевого атома углерода или азота. Если из контекста точка присоединения гетероциклической группы очевидна, такие группы могут альтернативно называться невалентными видами, то есть пирролидином, пиперидином, пиперазином, имидазолом, тетрагидропираном и т.п.
Термин «галоген» означает фтор, хлор, бром или йод.
Термин «терапевтически эффективное количество» означает количество, достаточное для эффективного лечения при введении пациенту, нуждающемуся в лечении.
Термин «лечение» или «терапия» означает предупреждение, улучшение или подавление медицинского состояния, заболевания или расстройства, подлежащего лечению (например, респираторного заболевания) у пациента (в частности, человека); или облегчение симптомов состояния здоровья, заболевания или расстройства.
Термин «фармацевтически приемлемая соль» означает соль, приемлемую для введения пациенту или млекопитающему, например человеку (например, соли, имеющие приемлемую безопасность для млекопитающих для данного режима дозирования). Типичные фармацевтически приемлемые соли включают соли уксусной, аскорбиновой, бензолсульфоновой, бензойной, камфорсульфоновой, лимонной, этансульфоновой, эдисиловой, фумаровой, гентизиновой, глюконовой, глюкуроновой, глутаминовой, гиппуровой, бромистоводородной, хлористоводородной, изэтионовой, молочной, лактобионовой, малеиновой, яблочной, миндальной, метансульфоновой, слизевой, нафталинсульфоновой, нафталин-1,5-дисульфоновой, нафталин-2,6-дисульфоновой, никотиновой, азотной, оротовой, памоиновой, пантотеновой, фосфорной, янтарной, серной, винной, п-толуолсульфоновой и ксинафоевой кислоты и тому подобное.
Термин «его соль» означает соединение, образующееся, когда водород кислоты заменен катионом, таким как катион металла или органический катион и тому подобное. Например, катион может быть протонированной формой соединения, то есть формой, в которой одна или несколько аминогрупп были протонированы кислотой. Обычно соль представляет собой фармацевтически приемлемую соль, хотя это не требуется для солей промежуточных соединений, которые не предназначены для введения пациенту.
Термин «аминозащитная группа» означает защитную группу, подходящую для предотвращения нежелательных реакций азота аминогруппы. Типичные аминозащитные группы включают, но не ограничиваются ими, формил; ацильные группы, например алканоильные группы, такие как ацетил и трифторацетил; алкоксикарбонильные группы, такие как трет-бутоксикарбонил (Boc); арилметоксикарбонильные группы, такие как бензилоксикарбонил (Cbz), 9-флуоренилметоксикарбонил (Fmoc), п-нитробензилоксикарбонил (PNZ), 2,4-дихлорбензилоксикарбонил и 5-бензизоксазолилметоксикарбонил; арилметильные группы, такие как бензил (Bn), 4-метоксибензил, тритил (Tr) и 1,1-ди-(4’-метоксифенил)метил; силильные группы, такие как триметилсилил (TMS), трет-бутилдиметилсилил (TBDMS), [2-(триметилсилил)этокси]метил (SEM); и тому подобное.
Термин «защитная группа карбоновой кислоты» означает защитную группу, подходящую для предотвращения нежелательных реакций с карбоновой кислотой. Типичные защитные группы карбоновой кислоты включают, но не ограничиваются ими, сложные эфиры, такие как метил, этил, трет-бутил, бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm), триметилсилил (TMS), трет-бутилдиметилсилил (TBS, TBDMS), дифенилметил (бензгидрил, DPM) и тому подобное.
Термин «гидроксильная защитная группа» означает защитную группу, подходящую для предотвращения нежелательных реакций на гидроксильной группе. Типичные гидроксильные защитные группы включают, но не ограничиваются ими, силильные группы, включая три(C1-6 алкил) силильные группы, такие как триметилсилил (TMS), триэтилсилил (TES), трет-бутилдиметилсилил (TBS) и тому подобное; сложные эфиры (ацильные группы), включая C1-6 алканоильные группы, такие как формил, ацетил и тому подобное; арилметильные группы, такие как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm), дифенилметил (бензгидрил, DPM) и тому подобное.
Многочисленные защитные группы, а также их введение и удаление описаны в T. W. Greene and P.G.M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York
Примеры
Следующие примеры синтеза предоставлены для иллюстрации изобретения и не должны истолковываться никоим образом как ограничение объема изобретения. Ниже в примерах следующие аббревиатуры имеют следующие значения, если не оговорено особо. Аббревиатуры, не указанные ниже, имеют их обычно используемые значения.
ACN=ацетонитрил
DMF=N, N-диметилформамид
EtOAc=этилацетат
EtOH=этанол
NaHMDS=бис(триметилсилил)амид натрия
MeTHF=2-метилтетрагидрофуран
MTBE=трет-бутилметиловый эфир
фунт/кв. дюйм=фунты на квадратный дюйм
Rt=время удерживания
Реагенты и растворители покупали у коммерческих поставщиков (Aldrich, Strem Chemicals, Inc., etc.) и использовали без дополнительной очистки. Наблюдение за протеканием реакции проводили с помощью аналитической высокоэффективной жидкостной хроматографии и масс-спектрометрии. Эндо/экзо отношения продуктов определяли анализом ВЭЖХ с применением описанных ниже протоколов. Реакционные смеси обрабатывали, как описано конкретно в каждой реакции; обычно их очищали экстракцией и другими способами очистки, такими как кристаллизация, в зависимости от температуры и растворителя, и осаждение. Характеризацию продуктов реакции общепринятым образом проводили с помощью масс- и 1Н-ЯМР-спектрометрии. Для ЯМР измерения образцы растворяли в дейтерированном растворителе (DMSO-d6 или CDCl3) и 1Н-ЯМР-спектры регистрировали с помощью оборудования Varian Gemini 2000 (400 МГц) в стандартных условиях измерения. Масс-спектрометрическую идентификацию соединений проводили с использованием оборудования Agilent (Palo Alto, CA) model 1100 LC/MSD.
Общие условия ВЭЖХ
Колонка: Zorbax SB-Aq, 5 мкм. 4,6×250 мм
Температура колонки: 40ºC
Скорость потока: 1,0 мл/мин
Подвижные фазы: A=вода/ACN (98:2) + 0,1% TFA
B=вода/ACN (10:90) + 0,1% TFA,
Объем впрыскивания: 10 мкл
Длина волны детектора: 214 нм
Способ ВЭЖХ 1
Неочищенные соединения растворяли в смеси вода/ACN (50:50) в количестве приблизительно 1 мг/мл и анализировали с использованием следующего градиента в течение 20 мин (время (мин)/% В): 0/10, 2.5/20, 9/75, 15/90, 17/90, 18/10, 20/10.
Способ ВЭЖХ 2
Соединения растворяли в смеси вода/ACN (90:10) в количестве приблизительно 1 мг/мл и анализировали с использованием следующего градиента в течение 30 мин (время (мин)/% B): 0/10, 13/10, 23/65, 28/90, 29/90, 30/10.
Способ ВЭЖХ 3
Соединения растворяли в смеси вода/ACN (90:10) в количестве приблизительно 1 мг/мл и анализировали с использованием следующего градиента в течение 55 мин (время (мин)/% B): 0/10, 10/20, 46/75, 47/90, 50/10, 55/10.
Получение 1: 4-(бензилокси)-2-этилфенил)трифтор-λ4-боран, калиевая соль I-5
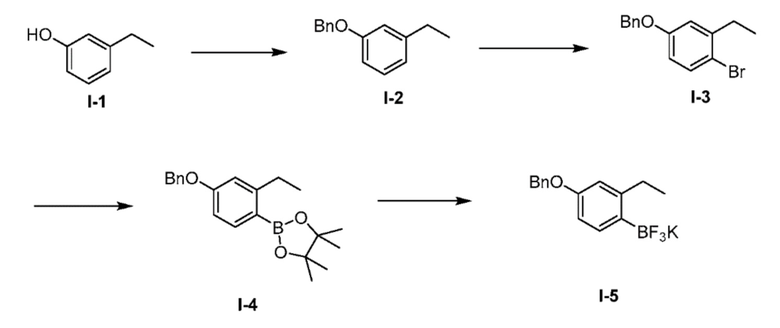
(а) 1-(бензилокси)-3-этилбензол (I-2)
К перемешиваемому раствору 3-этилфенола (I-1) (25,0 г, 204,0 ммоль) в ACN (250 мл, 10 об.) добавляли карбонат калия (42,0 г, 306 ммоль) при комнатной температуре. Полученную реакционную массу перемешивали при комнатной температуре в течение 15 минут, с последующим добавлением по каплям бензилбромида (24,0 мл, 204 ммоль). Полученную реакционную смесь перемешивали в течение 6 часов при комнатной температуре. После завершения реакции (мониторинг ТСХ), полученную реакционную массу выливали в воду (1,0 л) с последующей экстракцией соединения EtOAc (2×2 л). Объединенные органические слои промывали холодной водой, насыщенным солевым раствором и сушили над сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Затем неочищенный продукт очищали колоночной хроматографией на силикагеле (100-200 М) с использованием элюентов 2% EtOAc в гексане с получением желаемого продукта (I-2) в виде светло-желтого маслянистого соединения (35,0 г, 81%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,46-7,44 (м, 2H), 7,39 (т, J=7,6 Гц, 2H), 7,34-7,31 (м, 1H), 7,21 (т, J=7,6 Гц), 6,86-6,80 (м, 3H), 5,07 (с, 2H), 2,64 (кв, J=7,6 Гц, 2H), 1,24 (т, J=7,6 Гц, 3H).
(b) 4-(бензилокси)-1-бром-2-этилбензол (I-3)
К охлажденному льдом перемешиваемому раствору 1-(бензилокси)-3-этилбензола (I-2) (35,0 г, 164 ммоль) в ACN (525 мл, 15 об.) добавляли N-бромсукцинимид (32,0 г 181 ммоль) порциями в течение 15 минут. Полученную реакционную смесь перемешивали в течение 1 часа при комнатной температуре. После завершения реакции (мониторинг ТСХ), полученную реакционную массу выливали в ледяную воду (1,50 л) с последующей экстракцией соединения EtOAc (2×1 л). Объединенные органические слои промывали водой и сушили над сульфатом натрия, фильтровали и выпаривали при пониженном давлении с получением неочищенного продукта. К неочищенному веществу добавляли н-гексан (250 мл), получая суспензию, с последующей фильтрацией через фильтр Шотта. Маточный раствор выпаривали при пониженном давлении с получением желаемого продукта I-3 в виде светло-желтого маслянистого соединения (42,0 г, 87%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,52-7,29 (м, 7H), 6,88 (с, 1H), 6,68 (д, J=6,0 Гц, 1H), 5,04 (с, 2H), 2,69 (кв, J=7,6 Гц, 2H), 1,20 (т, J=7,5 Гц, 3H).
(с) 2-(4-(бензилокси)-2-этилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (I-4)
Перемешиваемый раствор 4-(бензилокси)-1-бром-2-этилбензола (I-3) (42,0 г, 144 ммоль), бис(пинаколато) диборона (44,0 г, 173 ммоль), и ацетата калия (28 г, 288 ммоль) в диоксане (440 мл) дегазировали продувкой N2 (г) в течение 15 мин с последующим добавлением PdCl2(dppf).DCM комплекса (11,0 г, 15 ммоль). Полученную реакционную смесь нагревали до 80°C в течение 16 ч. После завершения реакции (мониторинг ТСХ), реакционную массу фильтровали через слой целита и маточный раствор выпаривали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (100-200M) с использованием элюентов 1% EtOAc в гексане с получением желаемого продукта (I-4) в виде светло-желтого маслянистого соединения (32,0 г, 66%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,74 (д, J=8,4 Гц, 1H), 7,45-7,36 (м, 5H), 6,84-6,78 (м, 2H), 5,08 (с, 2H), 2,91 (кв, J=7,6 Гц), 1,33 (с, 12H), 1,19 (т, J=7,6 Гц, 3H).
(d) (4-(бензилокси)-2-этилфенил)трифтор-λ4-боран, калиевая соль (I-5)
К перемешиваемому раствору соединения 2-(4-(бензилокси)-2-этилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (I-4) (20 г, 59,0 ммоль), в ацетоне:метаноле (200 мл, 1:1 отношение, 10 об.), добавляли 3М раствор фтороводорода калия (23,0 г, 295 ммоль, растворенный в 98,0 мл воды). Полученную реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения реакции (мониторинг ТСХ), полученную реакционную массу выпаривали при пониженном давлении. Полученное таким образом твердое вещество помещали в воду (100 мл) и перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную массу фильтровали через фильтр Шотта воронку, промывали н-гексаном и сушили при пониженном давлении с получением желаемого продукта (I-5) в виде белого твердого вещества (14,0 г, 74%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,43 (д, J=7,2 Гц, 2H), 7,37 (т, J=7,5 Гц, 2H), 7,30 (т, J=7,1 Гц, 1H), 7,22 (д, J=8,0 Гц), 6,58 (с, 1H), 6,53 (д, J=7,9 Гц, 1H), 5,00 (с, 2H), 2,65 (кв, J=7,5 Гц, 2H), 1,07 (т, J=7,4 Гц, 3H).
Получение 2: 6-бензил 5-(трет-бутил) (S)-3-бензил-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат (I-11)
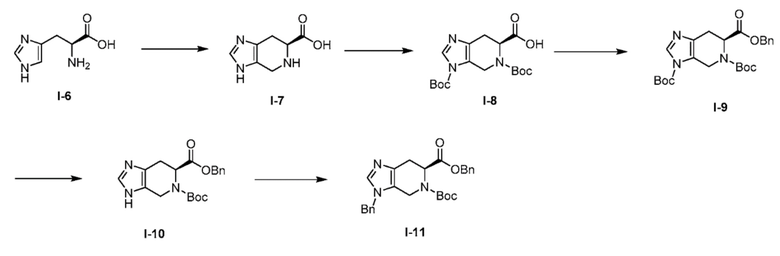
(а) (S)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота, хлористоводородная соль (I-7)
К охлажденной льдом перемешиваемой суспензии L-гистидина (I-6) (5,0 кг, 32,14 моль) в воде (40 л, 8 об.) добавляли концентрированную хлористоводородную кислоту (3,93 л, 33,75 моль), с последующим добавлением формальдегида (5,50 л, 67,5 моль, 37% водный раствор) по каплям. Полученный раствор перемешивали в течение 30 минут при той же температуре и затем нагревали при 80°C в течение 8 часов. За ходом реакции следили с помощью LCMS. Воду удаляли при пониженном давлении с получением неочищенного продукта, и полученный неочищенный продукт перемешивали в течение 2 часов в толуоле (20 л). Органические вещества удаляли при пониженном давлении для удаления избытка воды и соединение азеотропно сушили. Полученное вещество затем помещали в диэтиловый эфир (20 л) и перемешивали в течение 2 часов. Твердое вещество затем фильтровали и сушили воздухом с получением желаемого продукта (I-7) в виде не совсем белого твердого вещества (6,50 кг, 85%). 1H ЯМР (400 МГц, D2O) δ 8,69 (с, 1H), 4,56 (д, J=15,4 Гц, 1H), 4,42 (д, J=15,5 Гц, 1H), 4,20 (дд, J=5,5, 5,2 Гц, 1H), 3,42 (дд, J=5,0, 17,0 Гц, 1H), 3,11 (дд, J=10,2, 16,8 Гц, 1H).
(b) (S)-3,5-бис(трет-бутоксикарбонил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота (I-8)
К охлажденному льдом перемешиваемому раствору (S)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота ди-гидрохлорида (I-7) (6,10 кг, 25,40 моль) в 1,4-диоксане (48 л, 8 об.) и воде (48 л, 8 об.) по каплям добавляли триэтиламин (12,36 л, 89 моль) с последующим добавлением ди-трет-бутилдикарбоната (18,07 л, 78,74 моль, растворенного в 1,4-диоксане) в течение 30 мин. Полученную реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения реакции (мониторинг ТСХ & LCMS), желтоватую реакционную смесь разбавляли водой (10 л) и последовательно промывали диэтиловым эфиром (2×10 л) и EtOAc (2×7,50 л). Органическую фазу отбрасывали. Водный слой охлаждали и доводили до pH ~3 с помощью 6 н. Раствора HCl Водный слой охлаждали и доводили до pH ~ 3 с помощью 6 н. раствора HCl; водную фазу экстрагировали EtOAc (3×10 л). Объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфатом натрия, и концентрировали при пониженном давлении. Маслянистый остаток кристаллизовали из 30% EtOAc:гексаны с получением желаемого продукта (I-8) в виде не совсем белого твердого вещества (5,1 кг, 55%). (m/z): [M+H]+ вычислено для C17H25N3O6 368,18 найдено 368,21.
(с) 6-бензил 3,5-ди-трет-бутил (S)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5,6(4H)-трикарбоксилат (I-9)
К ледяному раствору (S)-3,5-бис(трет-бутоксикарбонил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновой кислоты (I-8) (5,1 кг, 13,88 моль) в DCM (51 л, 10 об.) последовательно добавляли насыщенный водный бикарбонат натрия (41,0 л, 8 об.), тетрабутиламмоний йодид (5,13 кг, 13,88 моль) и бензилбромид (2,47 л, 20,82 моль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения реакции (мониторинг ТСХ & LCMS), двухфазный раствор разделяли. Водный слой экстрагировали DCM (3×10 л). Объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией через силикагель (100-200M) с использованием элюентов 40% EtOAc в гексане с получением желаемого продукта (I-9) в виде вязкого масла (4,50 кг, 72%). (m/z): [M+H]+ вычислено для C24H31N3O6 458,22 найдено 458,60.
(d) 6-бензил 5-(трет-бутил) (S)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат (I-10)
К ледяному раствору 6-бензил 3,5-ди-трет-бутил (S)-6,7-дигидро-3H-имидазо[4,5-c]пиридин-3,5,6(4H)-трикарбоксилата (I-9) (4,50 кг, 9,84 моль) в IPA (45 л, 10 об.) по каплям добавляли гидроксид аммония (36 л, 8 об.). Полученную реакционную смесь дополнительно перемешивали при комнатной температуре в течение следующих 16 часов. После завершения реакции (мониторинг ТСХ & LCMS), полученную смесь разбавляли водой (25 л) с последующей экстракцией EtOAc (3×20 л). Объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией через силикагель (100-200M) с использованием элюентов 2% MeOH в DCM с получением желаемого продукта (I-10) в виде густого вязкого масла (2,70 кг, 77%). (m/z): [M+H]+ вычислено для C19H23N3O4 358,17 найдено 358,33.
(е) 6-бензил 5-(трет-бутил) (S)-3-бензил-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат (I-11)
К ледяному раствору 6-бензил 5-(трет-бутил) (S)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилата (I-10) (2,70 кг, 7,55 моль) в DCM (32,4 л, 12 об.) добавляли водный 1 н. гидроксид натрия (24,3 л, 9 об.) с последующим последовательным добавлением тетрабутиламмоний йодида (2,80 кг, 7,55 моль) и бензилбромида (0,99 л, 8,31 моль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения реакции (мониторинг ТСХ & LCMS), двухфазный раствор разделяли. Водный слой экстрагировали DCM (3×10 л). Объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (100-200M) с использованием элюентов 40% EtOAc в гексане с получением желаемого продукта (I-11) в виде вязкого масла (1,70 кг, 63%). (m/z): [M+H]+ вычислено для C26H29N3O4 448,22 найдено 448,20.
Получение 3: 6-бензил 5-(трет-бутил) (S)-3-бензил-2-(6-бром-1H-индазол-3-ил)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат (I-15)
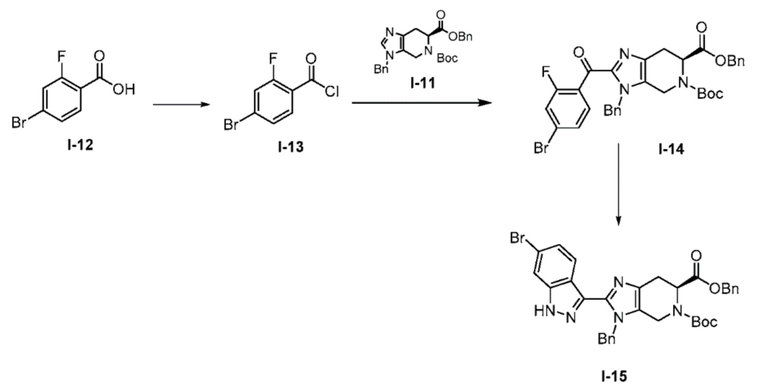
(а) 4-бром-2-фторбензоил хлорид (I-13)
К охлажденному льдом перемешиваемому раствору 4-бром-2-фторбензойной кислоты (I-12) (1,25 кг, 5,71 моль) в DCM (12,5 л, 15 об.), добавляли по каплям оксалилхлорид (0,98 л, 11,42 моль). Полученную реакционную смесь перемешивали в течение 10 мин при той же температуре. Затем к реакционной смеси по каплям добавляли DMF (150 мл). Полученной реакционной массе давали нагреться до комнатной температуры и перемешивали в течение 2 часов. После завершения реакции (мониторинг ТСХ), избыток оксалилхлорида удаляли при пониженном давлении в атмосфере азота с получением неочищенного продукта (I-13) (1,08 кг, 80%), который использовали на следующей стадии без дополнительной очистки.
(b) 6-бензил 5-(трет-бутил) (S)-3-бензил-2-(4-бром-2-фторбензоил)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат (I-14)
К перемешиваемому раствору 6-бензил 5-(трет-бутил) (S)-3-бензил-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилата (I-11) (1,70 кг, 3,80 моль) в ACN (13,6 л, 8 об.) добавляли триэтиламин (2,11 л, 15,2 моль) с последующим добавлением 4-бром-2-фторбензоил хлорида (I-13) (1,08 кг, 4,56 моль в 3,4 л ACN, 2 об.) при комнатной температуре. После завершения добавления цвет полученной реакционной смеси стал коричневым со светло-желтого. Полученную реакционную смесь перемешивали при той же температуре в течение 30 мин, и за ходом реакции следили с помощью ТСХ. Полученную реакционную смесь гасили ледяной водой (10 л), с последующей экстракцией EtOAc (3×5 л) и объединенные органические слои промывали насыщенным солевым раствором. Органические слои сушили над сульфатом натрия, фильтровали, и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (100-200M) с использованием элюентов 20% EtOAc в гексане с получением желаемого продукта (I-14) (1,74 кг, 71%). %). (m/z): [M+H]+ вычислено для C33H31BrFN3O5 648,14 найдено 648,20.
(с) 6-бензил 5-(трет-бутил) (S)-3-бензил-2-(6-бром-1H-индазол-3-ил)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат (I-15)
К перемешиваемому раствору 6-бензил 5-(трет-бутил) (S)-3-бензил-2-(4-бром-2-фторбензоил)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилата (I-14) (1,74 кг, 2,68 моль) в THF (26,0 л, 15 об.) добавляли гидразин-гидрат (0,705 л, 13,4 моль) при комнатной температуре. Полученную реакционную смесь нагревали при 60°C в течение 3 часов. После завершения реакции (мониторинг ТСХ), полученную реакционную массу выливали в ледяную воду (10 л) с последующей экстракцией соединения EtOAc (3×10 л). Объединенные органические слои промывали насыщенным солевым раствором и сушили над сульфатом натрия, фильтровали и выпаривали при пониженном давлении, с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (100-200M) с использованием элюентов 20% EtOAc в гексане с получением желаемого продукта (I-15) в виде не совсем белого твердого вещества (1,12 кг, 65%). (m/z): [M+H]+ вычислено для C33H32BrN5O4 642,16 найдено 642,21.
Получение 4: 6-бензил 5-(трет-бутил) (S)-3-бензил-2-(6-(4-(бензилокси)-2-этилфенил)-1H-индазол-3-ил)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат (I-16)
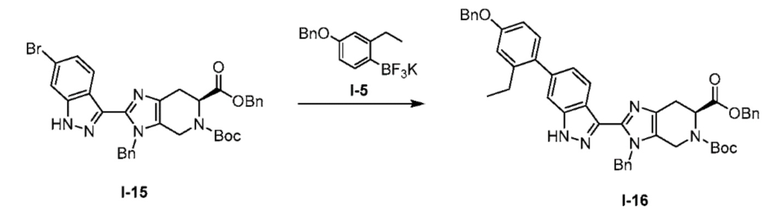
Бис(пинаколато)диборон (250 г, 984 ммоль) загружали в трехгорлую одностенную колбу объемом 5 л, предварительно протравленную с помощью химии фторидов, вместе с пропан-2-олом (1882 мл, 2,46E+04 ммоль) и смесь перемешивали до полного растворения. Растворение было эндотермическим (-4°C). В колбе Эрленмейера объемом 4 л, предварительно протравленной с использованием химии фторидов, гидрофторид фторида калия (538 г, 6891 ммоль) растворяли в воде (2306 мл, 1,28E+05 ммоль) с образованием 3M раствора. Растворение было эндотермическим (-12°C). Затем раствор фильтровали для удаления небольшого количества нерастворимого вещества из KHF2. После полного растворения обоих растворов содержимое колбы Эрленмейера загружали в одностенную колбу порциями в течение 15 минут. Наблюдался умеренный экзотермический эффект (+10°C). Раствор превратился в густую и светопроницаемую полупрозрачную серую суспензию во время добавления, и перемешивание увеличивали, для хорошего перемешивания содержимого. Смесь перемешивали в течение 1,5 ч, и затем фильтровали через грубую пористую стеклянную воронку (4 л, предварительно протравленная). Для завершения фильтрации требовалось 30-45 минут. Прозрачный двухфазный фильтрат отбрасывали. Белые твердые частицы сушили в течение 10 минут на фильтре (наблюдали растрескивание лепешки). Твердые частицы переносили обратно в очищенную трехгорлую одностенную колбу на 5 л и повторно суспендировали с водой (1330 мл, 7,38E+04 ммоль). Суспензию перемешивали в течение 2 ч по истечении этого времени она образовывала прозрачный гомогенный гидрогель. Раствор перемешивали в течение еще 1 ч, после чего твердые частицы/гель отфильтровывали с помощью грубой стеклянной воронки объемом 4 л (предварительно протравленная). Твердым веществам давали высохнуть на фильтре в течение 30 минут. Твердые вещества переносили обратно в очищенную трехгорлую одностенную колбу на 5 л и повторно суспендировали в ацетоне (1084 мл, 1,48E+04 ммоль). Белую/серую суспензию перемешивали в течение 1 ч и затем фильтровали через грубую стеклянную воронку 4 л (предварительно протравленная). Для завершения фильтрации требовалось 20 минут, и затем сушили на воронке в течение еще 1 ч. В течение этого времени твердые частицы время от времени перемешивали для обеспечения однородной сушки. После высыхания на фильтре остался легкий белый порошок. Твердые вещества сушили в течение 20 часов при 55°C под вакуумом с медленным выпуском азота, с получением рыхлого белого твердого вещества (собрали 200,3 г).
К перемешиваемому раствору 6-бензил 5-(трет-бутил) (S)-3-бензил-2-(6-бром-1H-индазол-3-ил)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилата (I-15) (10,0 г, 16,0 ммоль) в 2-метилтетрагидрофуране (100 мл, 10 об.) добавляли (4-(бензилокси)-2-этилфенил)трифтор-λ4-боран, калиевая соль (I-5) (8,0 г, 20 ммоль) и рыхлое белое твердое вещество, полученное выше (0,20 г). Полученную реакционную смесь дегазировали азотом в течение 30 минут. К этому раствору добавляли приготовленный водный раствор карбоната цезия (20,0 г, 62,0 ммоль в 60 мл воды, 6 об.). Полученную реакционную смесь дополнительно дегазировали в течение 15 минут с последующим добавлением бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладия(II) (0,66 г, 0,93 ммоль), и реакционную смесь откачивали под вакуумом и продували азотом. Полученную реакционную смесь нагревали при 110°C в течение 20 часов. После завершения реакции (мониторинг ТСХ & LCMS), полученную реакционную смесь охлаждали до комнатной температуры и фильтровали через слой целита, затем дополнительно промывали EtOAc (3×0,5 л). Объединенные органические слои промывали 1 н. раствором гидроксида натрия (3×0,5 л). Объединенные органические слои затем промывали насыщенным солевым раствором и сушили над сульфатом натрия, фильтровали, и выпаривали при пониженном давлении, с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (100-200M) с использованием элюентов 20% EtOAc в гексане с получением желаемого продукта (I-16) (в виде смеси N-бензиловых региоизомеров) в виде светло-желтого твердого вещества (8,0 г, 66%). (m/z): [M+H]+ вычислено для C48H47N5O5 774,36 найдено 774,59.
Получение 5: (S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота, гидрохлорид (I-18)
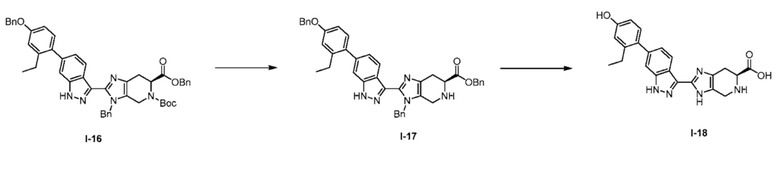
(а) бензил (S)-3-бензил-2-(6-(4-(бензилокси)-2-этилфенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоксилат, гидрохлорид (I-17)
6-Бензил 5-(трет-бутил) (S)-3-бензил-2-(6-(4-(бензилокси)-2-этилфенил)-1H-индазол-3-ил)-3,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат (I-16) (1,0 г, 1,292 ммоль) растворяли в диоксане (8 мл) и воде (1,5 мл), затем добавляли раствор хлороводорода, 4 М в диоксане (7 мл, 28,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов (ход реакции контролировали с помощью LCMS). Затем реакционную смесь замораживали и лиофилизировали, и неочищенный продукт (I-17) использовали непосредственно в следующей реакции (предполагался количественный выход). (m/z): [M+H]+ вычислено для C43H39N5O3 674,31 найдено 674,3.
(b) (S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота, гидрохлорид (I-18)
Бензил (S)-3-бензил-2-(6-(4-(бензилокси)-2-этилфенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоксилат, гидрохлорид (I-17) (0,918 г, 1,292 ммоль) растворяли в 2-пропаноле (15 мл), растворе хлористого водорода, 5 M в воде (0,258 мл, 1,292 ммоль), и воде (0,25 мл) при 50°C, затем добавляли палладий, 10% мас. на угле, 50% воды (0,138 г, 0,065 ммоль). Затем реакционную колбу продували азотом, присоединяли баллон с водородом, и реакционную смесь перемешивали при 50°C в течение 4 дней, при необходимости пополняя баллон с водородом (ход реакции контролировали с помощью LCMS). Затем все твердые вещества удаляли фильтрацией и полученный раствор концентрировали. Остаток растворяли в ACN/вода 1:1, замораживали и лиофилизировали. Полученный порошок (I-18) использовали без дополнительной очистки (предполагался количественный выход). (m/z): [M+H]+ вычислено для C22H21N5O3 404,17 найдено 404,2.
Получение 6: (S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновой кислоты (I-19)
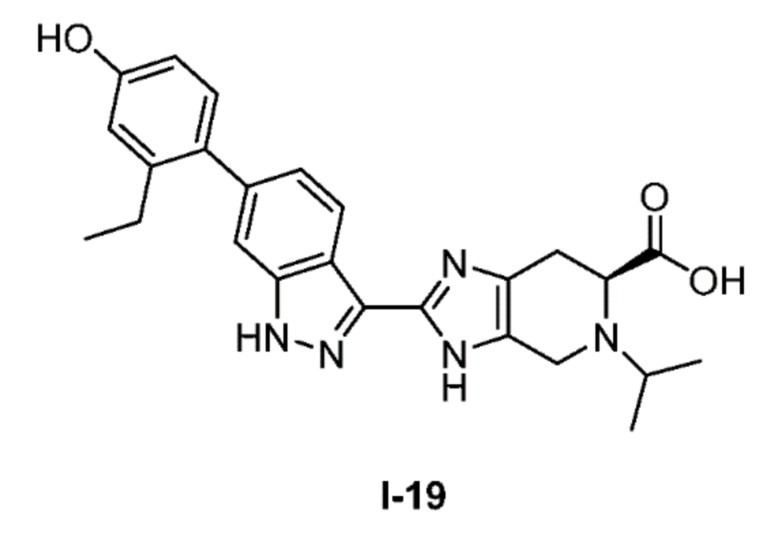
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, HCl (I-18) (0,25 г, 0,568 ммоль) суспендировали в DMF (2,5 мл) и ацетоне (2,5 мл), затем добавляли уксусную кислоту (0,098 мл, 1,705 ммоль) и цианоборгидрид натрия (0,179 г, 2,84 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 24 часов (за ходом реакции следили с помощью LCMS). Реакционную смесь концентрировали, затем неочищенный продукт очищали хроматографией с обращенной фазой (градиент 5-70% ACN/вода, колонка 50 г C18aq) с получением соли TFA указанного в заголовке соединения (149 мг, 47% выход). (m/z): [M+H]+ вычислено для C25H27N5O3 446,21 найдено 446,3.
Получение 7: (S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота (I-20)
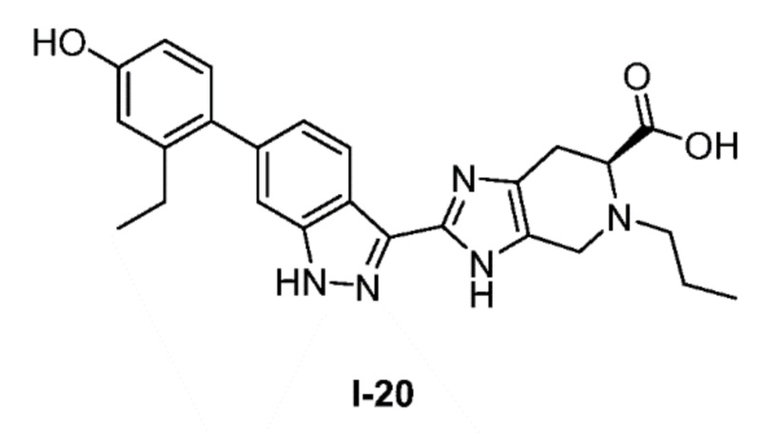
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, HCl (I-18) (0,160 г, 0,364 ммоль) и пропиональдегид (0,039 мл, 0,546 ммоль) растворяли в метаноле (3,0 мл), затем добавляли цианоборгидрид натрия (0,069 г, 1,091 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 24 часов (за ходом реакции следили с помощью LCMS). Реакционную смесь концентрировали и неочищенный продукт очищали хроматографией с обращенной фазой (градиент 5-70% ACN/вода, колонка 50 г C18) с получением соли TFA указанного в заголовке соединения (78 мг, 38% выход). (m/z): [M+H]+ вычислено для C25H27N5O3 446,21 найдено 446,3.
Получение 8: (S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота (I-21)
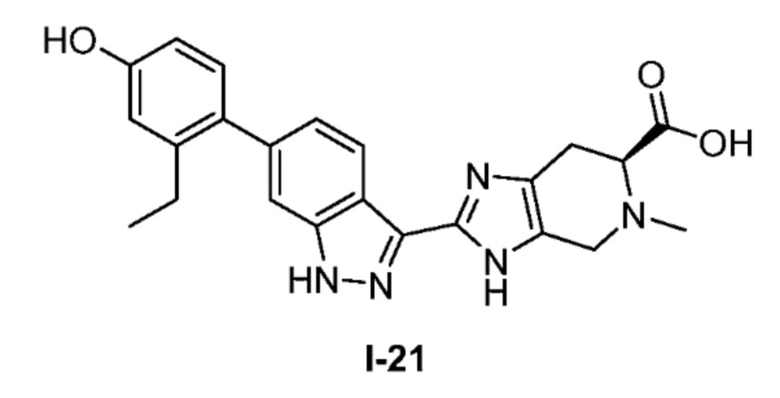
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, HCl (I-18) (0,160 г, 0,364 ммоль) и раствор формальдегида, 37 масс.% в воде (0,032 мл, 0,436 ммоль) растворяли в метаноле (3,0 мл), затем добавляли цианоборгидрид натрия (0,069 г, 1,091 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 часов (за ходом реакции следили с помощью LCMS). Боргидрид натрия (14 мг, 0,364 ммоль) добавляли для гашения избытка формальдегида, затем реакционную смесь концентрировали. Неочищенный продукт очищали хроматографией с обращенной фазой (градиент 5-70% ACN/вода, колонка 50 г C18) с получением соли TFA указанного в заголовке соединения (110 мг, 57% выход). (m/z): [M+H]+ вычислено для C23H23N5O3 418,18 найдено 418,2.
Получение 9: (R)-2-(3-метилпиперазин-1-ил)этан-1-ол, дигидрохлорид (I-24)
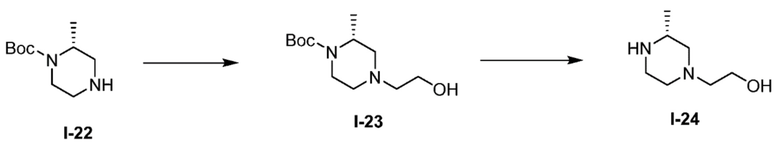
(а) трет-бутил (R)-4-(2-гидроксиэтил)-2-метилпиперазин-1-карбоксилат (I-23)
(R)-1-boc-2-метил-пиперазин (0,2 г, 0,999 ммоль), DIPEA (0,698 мл, 3,99 ммоль), и 2-бромэтанол (0,085 мл, 1,198 ммоль) растворяли в DMF (5 мл) и реакционную смесь перемешивали при 60°C в течение 16 часов (за ходом реакции следили с помощью LCMS). Реакционную смесь концентрировали, затем к остатку добавляли 10 мл диэтилового эфира. Раствор обрабатывали ультразвуком до исчезновения остатка геля и образования твердого вещества. Затем раствор эфира декантировали от твердого вещества. Затем твердое вещество использовали непосредственно в следующей реакции (предполагался количественный выход). (m/z): [M+H]+ вычислено для C12H24N2O3 245,18 найдено 245,4.
(b) (R)-2-(3-метилпиперазин-1-ил)этан-1-ол, дигидрохлорид (I-24)
Трет-бутил (R)-4-(2-гидроксиэтил)-2-метилпиперазин-1-карбоксилат (0,244 г, 0,999 ммоль) растворяли в диоксане (3,0 мл) и воде (0,5 мл), затем добавляли раствор хлороводорода, 4 М в диоксане (2,0 мл, 65,8 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов (за ходом реакции следили с помощью LCMS). Реакционную смесь замораживали и лиофилизировали, и полученный продукт использовали без очистки (предполагался количественный выход). (m/z): [M+H]+ вычислено для C7H16N2O 145,13 найдено 145,4.
Пример 1: (S)-(2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)(4-(2-гидроксиэтил)пиперазин-1-ил)метанон
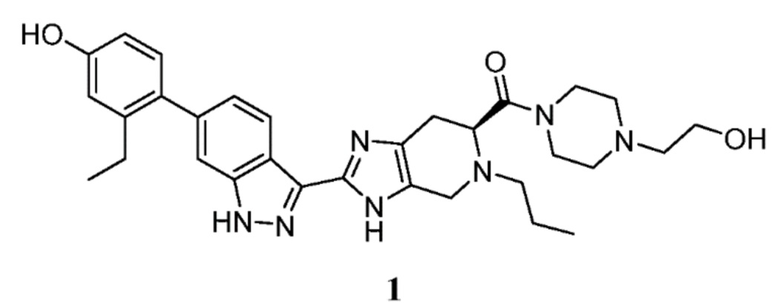
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (I-20) (40 мг, 0,071 ммоль), n-(2-гидроксиэтил)пиперазин (0,018 мл, 0,143 ммоль) и DIPEA (0,025 мл, 0,143 ммоль) растворяли в DMF (1,5 мл), затем добавляли HATU (40,8 мг, 0,107 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов (за ходом реакции следили с помощью LCMS). Гидразин (0,011 мл, 0,357 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-70% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (31 мг, 56% выход). (m/z): [M+H]+ вычислено для C31H39N7O3 558,31 найдено 558,3. 1H ЯМР (400 МГц, DMSO-d6) δ 13,10 (с, 1H), 12,27 (д, J=48,92 Гц, 1H), 9,40 (с, 1H), 8,28 (т, J=8,10 Гц, 1H), 7,30 (с, 1H), 7,04 (м, 2H), 6,71 (д, J=2,46 Гц, 1H), 6,64 (дд, J=2,50, 8,23 Гц, 1H), 4,44 (т, J=5,31 Гц, 1H), 4,11 (кв, J=5,26, 2H), 3,96 (м, 1H), 3,86-3,52 (м, 6H), 3,49 (кв, J=6,01 Гц, 2H), 2,95 (м, 2H), 2,48 (кв, J=7,48 Гц, 2H), 2,42-2,21 (м, 4H), 2,37 (т, J=6,20 Гц, 2H), 1,42 (м, 2H), 1,00 (т, J=7,52 Гц, 3H), 0,81 (м, 3H).
Пример 2: ((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)((1S,4S)-5-метил-2,5-диазабицикло[2.2.1]гептан-2-ил)метанон
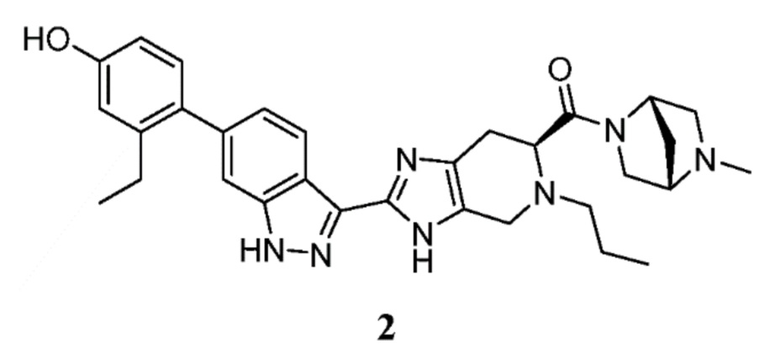
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (I-20) (40 мг, 0,071 ммоль), (1S,4S)-5-метил-2,5-диазабицикло[2.2.1]гептан дигидробромид (29,4 мг, 0,107 ммоль), и DIPEA (0,062 мл, 0,357 ммоль) растворяли в DMF (1,5 мл), затем добавляли HATU (40,8 мг, 0,107 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов (за ходом реакции следили с помощью LCMS). Гидразин (0,011 мл, 0,357 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-70% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (32 мг, 59% выход). (m/z): [M+H]+ вычислено для C31H37N7O2 540,30 найдено 540,3.
Пример 3: ((S)-2,4-диметилпиперазин-1-ил)((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон 3
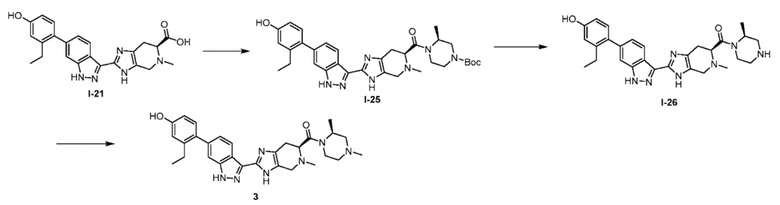
(а) трет-бутил (S)-4-((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбонил)-3-метилпиперазин-1-карбоксилат (I-25)
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (I-21) (55 мг, 0,103 ммоль), (s)-4-n-boc-2-метилпиперазин (41,5 мг, 0,207 ммоль), и DIPEA (0,036 мл, 0,207 ммоль) растворяли в DMF (1,5 мл), затем добавляли HATU (59,0 мг, 0,155 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов (за ходом реакции следили с помощью LCMS). Гидразин (0,016 мл, 0,517 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали хроматографией с обращенной фазой (градиент 5-70% ACN/вода, колонка 50 г C18) с получением соли TFA указанного в заголовке соединения (54 мг, 72% выход). (m/z): [M+H]+ вычислено для C33H41N7O4 600,33 найдено 600,3.
(b) ((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)((S)-2-метилпиперазин-1-ил)метанон (I-26)
Трет-бутил (S)-4-((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбонил)-3-метилпиперазин-1-карбоксилат, TFA (I-25) (0,126 г, 0,177 ммоль) растворяли в диоксане (1,5 мл) и воде (0,3 мл), затем добавляли раствор хлороводорода, 4 М в диоксане (1,5 мл, 6,00 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов (за ходом реакции следили с помощью LCMS). Реакционную смесь замораживали и лиофилизировали, и полученный порошок использовали непосредственно на следующей реакции (предполагался количественный выход). (m/z): [M+H]+ вычислено для C28H33N7O2 500,27 найдено 500,3.
(c) ((S)-2,4-диметилпиперазин-1-ил)((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)((S)-2-метилпиперазин-1-ил)метанон, дигидрохлорид (0,101 г, 0,176 ммоль) и раствор формальдегида, 37% масс. в воде (0,016 мл, 0,212 ммоль) растворяли в метаноле (3,0 мл), затем добавляли цианоборгидрид натрия (0,055 г, 0,882 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов (за ходом реакции следили с помощью LCMS). Боргидрид натрия (7 мг, 0,176 ммоль) добавляли, чтобы погасить оставшийся формальдегид. Реакционную смесь концентрировали, затем неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-60% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (28 мг, 21% выход). (m/z): [M+H]+ вычислено для C29H35N7O2 514,29 найдено 514,3.
Пример 4: (S)-(2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)(4-метил-1,4-диазепан-1-ил)метанон
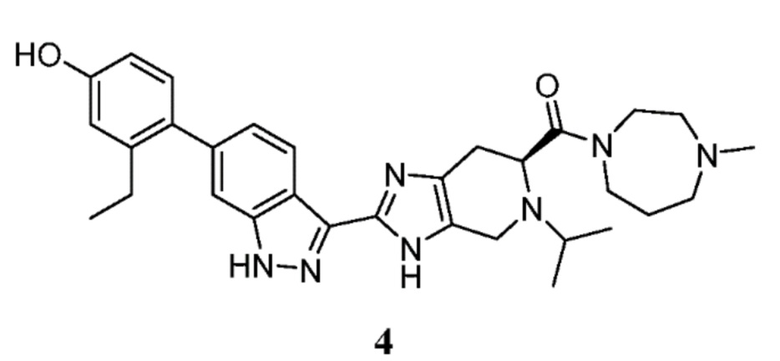
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (I-19) (50 мг, 0,089 ммоль), 1-метилгомопиперазин (0,022 мл, 0,179 ммоль), и DIPEA (0,031 мл, 0,179 ммоль) растворяли в DMF (1,5 мл), затем добавляли HATU (51,0 мг, 0,134 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов (за ходом реакции следили с помощью LCMS). Гидразин (0,014 мл, 0,447 ммоль) добавляли для расщепления нежелательных побочных продуктов, и раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 2-70% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (29 мг, 42% выход). (m/z): [M+H]+ вычислено для C31H39N7O2 542,32 найдено 542,3. 1H ЯМР (400 МГц, DMSO-d6) δ 13,08 (с, 1H), 12,21 (д, J=29,9 Гц, 1H), 9,40 (с, 1H), 8,27 (д, J=8,33 Гц, 1H), 7,30 (с, 1H), 7,04 (т, J=8,05, 2H), 6,71 (д, J=2,53 Гц, 1H), 6,64 (дд, J=2,54, 8,23 Гц, 1H), 4,11 (м, 3H), 3,91-3,52 (м, 6H), 2,97 (м, 1H), 2,91-2,53 (м, 4H), 2,49 (кв, J=7,46, 2H), 2,23 (д, J=13,9 Гц, 3H), 1,76 (м, 2H), 1,0 (м, 9H).
Пример 5: ((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)((R)-4-(2-гидроксиэтил)-2-метилпиперазин-1-ил)метанон
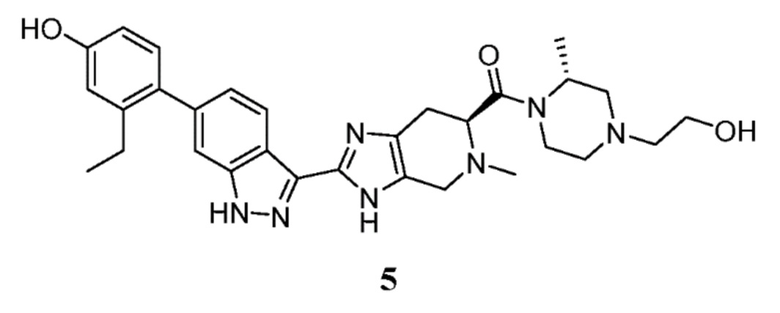
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-метил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота, TFA (I-21) (55 мг, 0,103 ммоль), (R)-2-(3-метилпиперазин-1-ил)этан-1-ол, дигидрохлорид (I-24) (33,7 мг, 0,155 ммоль), и DIPEA (0,090 мл, 0,517 ммоль) растворяли в DMF (1,5 мл), затем добавляли HATU (59,0 мг, 0,155 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов (за ходом реакции следили с помощью LCMS). Гидразин (0,016 мл, 0,517 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали, и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-70% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (22 мг, 28% выход). (m/z): [M+H]+ вычислено для C30H37N7O3 544,30 найдено 544,3.
Пример 6: ((S)-3-(диметиламино)пирролидин-1-ил)((S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
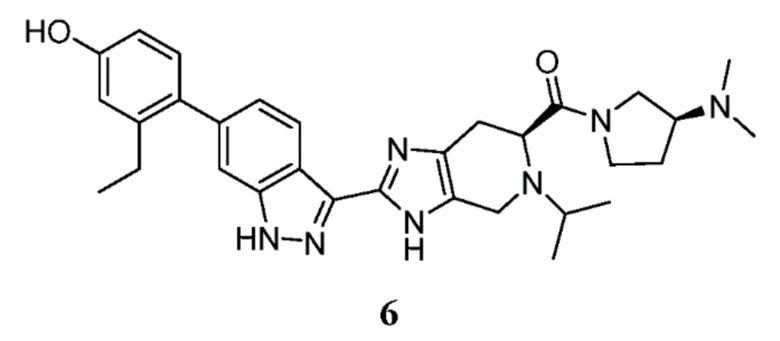
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (I-19) (50 мг, 0,089 ммоль), (S)-(-)-3-(диметиламино)пирролидин (0,023 мл, 0,179 ммоль), и DIPEA (0,031 мл, 0,179 ммоль) растворяли в DMF (1,5 мл), затем добавляли HATU (51,0 мг, 0,134 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов (за ходом реакции следили с помощью LCMS). Гидразин (0,014 мл, 0,447 ммоль) добавляли для расщепления нежелательных побочных продуктов, и раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-70% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (37 мг, 53% выход). (m/z): [M+H]+ вычислено для C31H39N7O2 542,32 найдено 542,3.
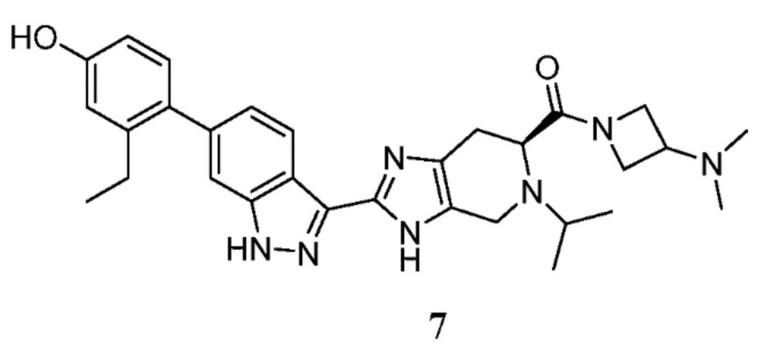
Пример 7: (S)-(3-(диметиламино)азетидин-1-ил)(2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (I-19) (50 мг, 0,089 ммоль), 3-(диметиламино)азетидин дигидрохлорид (23,20 мг, 0,134 ммоль), и DIPEA (0,078 мл, 0,447 ммоль) растворяли в DMF (1,5 мл), затем добавляли HATU (51,0 мг, 0,134 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов (за ходом реакции следили с помощью LCMS). Гидразин (0,014 мл, 0,447 ммоль) добавляли для расщепления нежелательных побочных продуктов, и раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-70% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (25 мг, 37% выход). (m/z): [M+H]+ вычислено для C30H37N7O2 528,30 найдено 528,3. 1H ЯМР (400 МГц, DMSO-d6) δ 13,09 (с, 1H), 9,40 (с, 1H), 8,27 (д, J=8,31, 1H), 7,30 (с, 1H), 7,04 (м, 2H), 6,71 (д, J=2,54, 1H), 6,64 (дд, J=2,53, 8,26, 1H), 4,26 (м, 1H), 4,06 (м, 2H), 3,82 (м, 2H), 3,64 (м, 2H), 3,03 (м, 2H), 2,74 (м, 2H), 2,47 (кв, J=7,56, 2H), 2,07 (д, J=3,69, 6H), 1,07 (м, 6H), 1,00 (т, J=7,50, 3H).
Получение 10: 6-бензил 5-(трет-бутил) (S)-1-бензил-2-(6-(4-(бензилокси)-2-этил-5-фторфенил)-1H-индазол-3-ил)-1,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилат
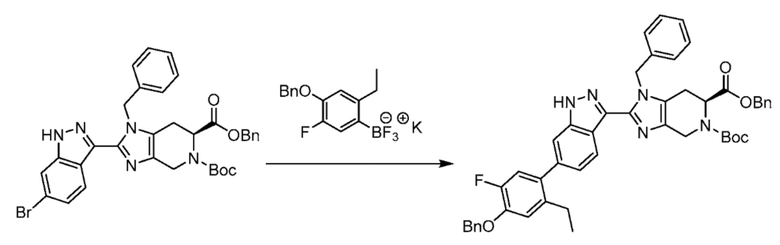
К перемешиваемому раствору 6-бензил 5-(трет-бутил) (S)-1-бензил-2-(6-бром-1H-индазол-3-ил)-1,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилата (1,12 кг, 1,74 моль) в 2-метилтетрагидрофуране (11,2 л, 10 об.) добавляли ((4-(бензилокси)-2-этил-5-фторфенил)трифторборан, калиевую соль) (0,702 кг, 2,1 моль) и рыхлое белое твердое вещество, выделенное в получении 4 (0,223 кг, 1,04 моль). Полученную реакционную смесь дегазировали азотом в течение следующих 30 мин через впускное отверстие капельницы. К этому раствору добавляли полученный водный раствор Cs2CO3 (2,27 кг, 6,96 моль в 7,30 л H2O, 6 об.). Полученную реакционную смесь дополнительно дегазировали в течение следующих 15 мин. В полученную реакционную смесь добавляли Pd(amphos) (0,74 кг, 1,04 моль) и реакционную смесь откачивали под вакуумом и продували азотом. Полученную реакционную смесь нагревали до 90°C в течение 20 часов. После завершения реакции, полученную реакционную смесь охлаждали до комнатной температуры и фильтровали через слой целита, и промывали этилацетатом (3×7,5 л). Объединенные органические слои промывали 1н раствором NaOH (3×3 л). Объединенные органические слои промывали насыщенным солевым раствором и сушили над Na2SO4, фильтровали и выпаривали при пониженном давлении с получением неочищенного продукта, который очищали с помощью колоночной хроматографии на силикагеле (100-200 М) с использованием элюентов 20% этилацетат в гексане с получением указанного в заголовке продукта в виде смеси регион-изомеров в виде не совсем белого твердого вещества (1,10 кг, 80%). (m/z): [M+H]+ вычислено для C48H46FN5O5 792,92 найдено 792,34.
Получение 11: Бензил (S)-1-бензил-2-(6-(4-(бензилокси)-2-этил-5-фторфенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридин-6-карбоксилат, 2Бензолсульфоновая кислота
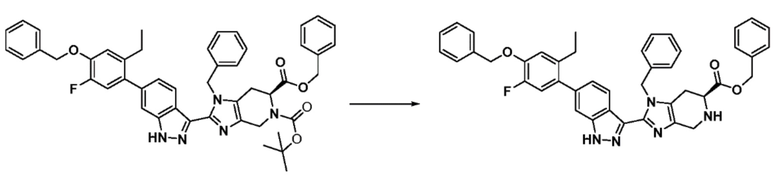
К раствору 6-бензил 5-(трет-бутил) (S)-1-бензил-2-(6-(4-(бензилокси)-2-этил-5-фторфенил)-1H-индазол-3-ил)-1,4,6,7-тетрагидро-5H-имидазо[4,5-c]пиридин-5,6-дикарбоксилата (1,005 г, 1269 ммоль) в 2-метилтетрагидрофуране (10,050 мл) и изопропилацетате (4321,5 мл) в атмосфере азота добавляли бензолсульфоновую кислоту (703 г, 4,442 ммоль). Полученную реакционную смесь перемешивали при 50ºC в течение 15 часов. После завершения реакции добавляли изопропилацетат (5728,5 мл). Суспензию охлаждали до 20ºC и выдерживали в течение 1 часа. Затем загустевшую суспензию фильтровали под давлением азота. Затем осадок промывали изопропилацетатом (5000 мл) и сушили под давлением азота при 25ºC в течение 2 часов с последующей сушкой при 60ºC в течение 16 часов в высоком вакууме с продувкой азота с получением указанного в заголовке соединения в виде не совсем белого свободно текучего твердого вещества (1210 г, 95% выход). (m/z): [M+H]+ вычислено для C43H38FN5O3 692,81 найдено 692,88.
Получение 12: Бензил (S)-1-бензил-2-(6-(4-(бензилокси)-2-этил-5-фторфенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридин-6-карбоксилат
К суспензии молекулярных сит (1,21 кг) в ацетоне (12,0 л) добавляли бензил (S)-1-бензил-2-(6-(4-(бензилокси)-2-этил-5-фторфенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридин-6-карбоксилат, 2Бензолсульфоновую кислоту (1,20 кг, 1,190 ммоль) и уксусную кислоту (26 мл, 446 ммоль). Суспензии давали перемешиваться при 25ºC в течение 30 минут с получением гетерогенной не совсем бело-желтой гетерогенной смеси. К реакционной смеси добавляли триацетоксиборгидрид натрия (492 г, 2,321 ммоль) и перемешивали при 25ºC в течение 1 часа. Реакционную смесь фильтровали через целит и осадок промывали 2-метилтетрагидрофураном (500 мл). Фильтрат разбавляли 2-метилтетрагидрофураном (8,0 л) и растворитель заменяли отгонкой ацетона. Для промывки раствора добавляли насыщенный бикарбонат натрия (3025 мл), и смесь перемешивали в течение 1 часа, перемешивание прекращали, слои разделяли в течение 15 минут, водный слой (pH=7,5) удаляли и на этот раз повторяли промывку, оставляя 2 часа перемешивания перед разделением слоев. Органический слой перегоняли до 2,0 л, добавляли изопропилацетат (8,0 л) и заменяли растворитель путем отгонки 2-метилтетрагидрофурана с получением суспензии. Суспензию перемешивали при 20ºC в течение 1 часа, затем фильтровали под давлением азота, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (795 г, 91% выход). (m/z): [M+H]+ вычислено для C46H44FN5O3 734,89 найдено 734,96.
Получение 13: (S)-2-(6-(2-этил-5-фтор-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота, 2HCl
К дегазированному перемешиваемому гомогенному раствору бензил (S)-1-бензил-2-(6-(4-(бензилокси)-2-этил-5-фторфенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридин-6-карбоксилата (760 г, 1,036 ммоль), пропан-2-ола (3800 мл) и 1M хлороводорода(вод.) (2589 мл, 2,589 ммоль) при 50oC добавляли 10% масс.Pd/C, 50% масс. H2O (76 г, 35,7 ммоль) сразу с последующим барботированием газообразного водорода через реакционную смесь в течение 4 часов. Реакционную смесь фильтровали через подушку из целита (200 грамм). К прозрачному темно-желтому фильтрату добавляли SiliaMetS Thiol (76 г, в виде белого твердого вещества) и перемешивали при 50oC в течение 1 часа для удаления остатка палладия. SiliaMetS Thiol отфильтровывали через фильтр 0,2 мкм, получая однородный фильтрат светло-желтого цвета, затем SiliaMetS Thiol стал ярко-оранжевым. К фильтрату добавляли изопропилацетат (7600 мл) и его концентрировали на роторном испарителе до приблизительно 3,0 литров. К концентрату, затем в виде густой суспензии, добавляли изопропилацетат (7600 мл), после чего суспензия становилась свободно текучей и фильтруемой. Суспензию фильтровали, и лепешку промывали изопропилацетатом (3000 мл), сушили в высоком вакууме в течение 1 часа, затем дополнительно сушили в высоком вакууме с продувкой азота при 50°C в течение 18 часов с получением указанного в заголовке соединения (472 г, 81% выход). (m/z): [M+H]+ вычислено для C25H26FN5O3 464,51 найдено 464,58.
Пример 8: (S)-(3-(диметиламино)азетидин-1-ил)(2-(6-(2-этил-5-фтор-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
К перемешиваемому раствору (S)-2-(6-(2-этил-5-фтор-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновой кислоты, 2HCl (470 г, 876 ммоль) и N, N-диметилазетидин-3-амина, 2HCl (197 г, 1139 ммоль) в N, N-диметилацетамиде (2436 мл), охлажденному до -20ºC, добавляли DIPEA (413 мл, 2366 ммоль) в течение не менее 15 минут (экзотермическое добавление вызывало повышение температуры партии до -9,1°C). Партию снова охлаждали до -15ºC и добавляли HCTU (453 г, 1095 ммоль). Смесь нагревали до 20°C в течение 1 часа и выдерживали еще час. К полученной реакционной смеси добавляли изопропилацетат (5,0 л) и 1M HCl (2,0 л) и смесь перемешивали в течение 15 минут, слои разделяли для извлечения примесей в слой изопропилацетата. Водный слой, содержащий продукт, трижды экстрагировали изопропилацетатом (5,0 л каждый). После 4-й экстракции водный слой добавляли к 2-метилтетрагидрофурану, затем насыщенный раствор бикарбоната натрия (~2,2 л, для доведения значения pH=8) перемешивали 15 минут, слои разделяли, а водный слой отбрасывали. Органический слой заменяли растворителем на ацетонитрил и перемешивали до конечного объема 2,35 л, при этом продукт выпадал в осадок из раствора в виде аморфных фильтруемых твердых веществ. Затем суспензию фильтровали под давлением азота с получением 345 граммов неочищенного продукта. Неочищенный продукт (345 г) растворяли в метаноле (1035 л) при перемешивании при 55ºC и выдерживали в течение 15 часов для кристаллизации продукта из раствора. Суспензию охлаждали до 10ºC и выдерживали при этой температуре при перемешивании в течение 2 часов. Загустевшую суспензию фильтровали под давлением азота в течение 2 часов при 20°C с последующей сушкой в высоком вакууме с продувкой азота при 65°C в течение 18 часов с получением указанного в заголовке соединения в виде свободно текучего твердого вещества от не совсем белого до белого цвета (253 г, 53% выход). (m/z): [M+H]+ вычислено для C30H36FN7O2 546,66 найдено 546,73.
Получение 14: (S)-5-этил-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновая кислота
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, HCl (0,100 г, 0,227 ммоль) (I-18) и ацетальдегид (0,019 мл, 0,341 ммоль) растворяли в метаноле (3,0 мл), затем добавляли цианоборгидрид натрия (0,057 г, 0,909 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов (за ходом реакции следили с помощью LCMS). Боргидрид натрия (9 мг, 0,227 ммоль) добавляли для гашения любого оставшегося ацетальдегида, затем реакционную смесь концентрировали. Затем неочищенный продукт очищали хроматографией с обращенной фазой (градиент 5-70% ACN/вода, колонка 40 г C18) с получением соли TFA указанного в заголовке соединения (62 мг, 50% выход). (m/z): [M+H]+ вычислено для C24H25N5O3 432,20 найдено 432,1.
Пример 9: (S)-(3-(диметиламино)азетидин-1-ил)(5-этил-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
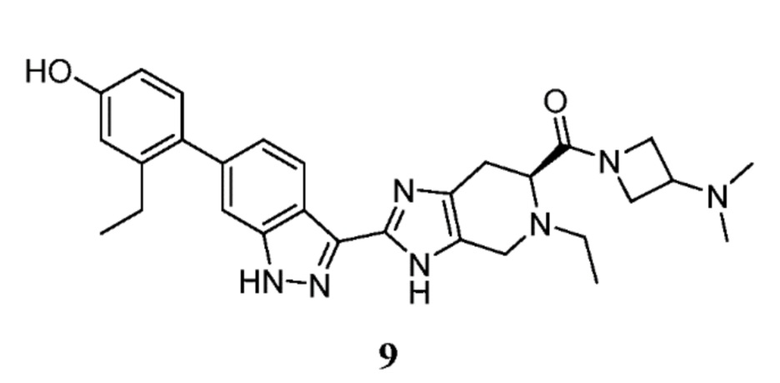
(S)-5-этил-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (30 мг, 0,055 ммоль), 3-(диметиламино)азетидин дигидрохлорид (14,28 мг, 0,082 ммоль), и DIPEA (0,048 мл, 0,275 ммоль) растворяли в DMF (1,50 мл), затем добавляли HATU (31,4 мг, 0,082 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа (за ходом реакции следили с помощью LCMS). Гидразин (5,18 мкл, 0,165 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-60% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (25 мг, 63% выход). (m/z): [M+H]+ вычислено для C29H35N7O2 514,29 найдено 514,2.
Пример 10: (S)-(3-(диметиламино)-3-метилазетидин-1-ил)(5-этил-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
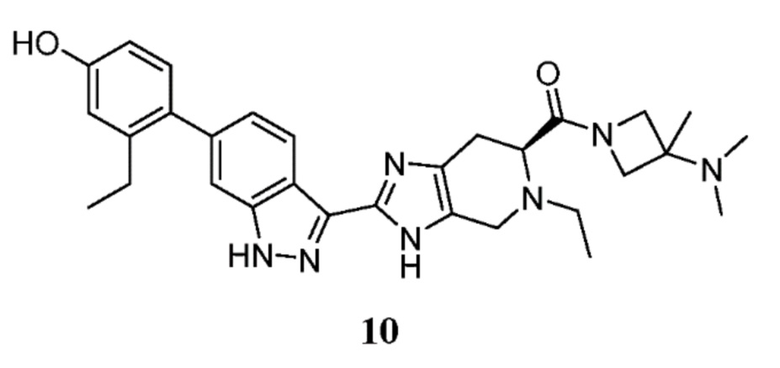
(S)-5-этил-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (30 мг, 0,055 ммоль), N, N,3-триметилазетидин-3-амин гидрохлорид (12,43 мг, 0,082 ммоль), и DIPEA (0,048 мл, 0,275 ммоль) растворяли в DMF (1,50 мл), затем добавляли HATU (31,4 мг, 0,082 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа (за ходом реакции следили с помощью LCMS). Гидразин (5,18 мкл, 0,165 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-60% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (25 мг, 62% выход). (m/z): [M+H]+ вычислено для C30H37N7O2 528,30 найдено 528,2.
Пример 11: (S)-(3-(диметиламино)-3-метилазетидин-1-ил)(2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
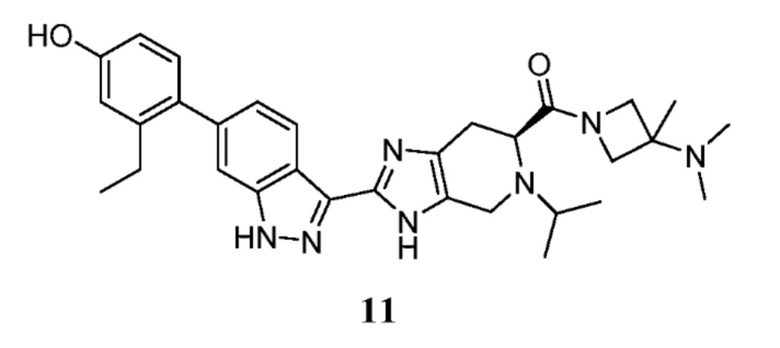
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-изопропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту (40 мг, 0,090 ммоль) (I-19) N, N,3-триметилазетидин-3-амин гидрохлорид (20,29 мг, 0,135 ммоль), и DIPEA (0,047 мл, 0,269 ммоль) растворяли в DMF (1,50 мл), затем добавляли HATU (51,2 мг, 0,135 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов (за ходом реакции следили с помощью LCMS). Гидразин (8,45 мкл, 0,269 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-60% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (26 мг, 38% выход). (m/z): [M+H]+ вычислено для C31H39N7O2 542,32 найдено 542,2.
Пример 12: (S)-(3-(диметиламино)азетидин-1-ил)(2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
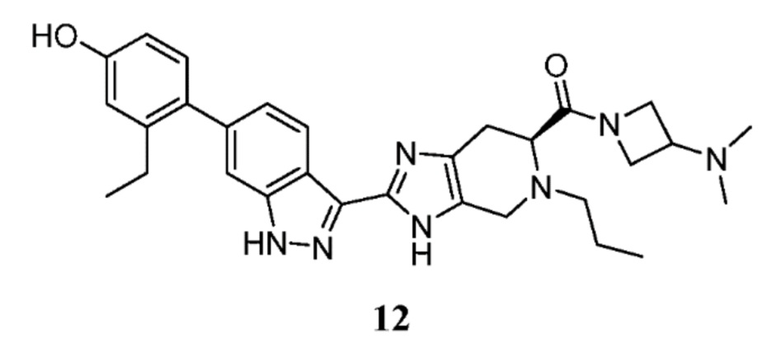
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (30 мг, 0,054 ммоль), (I-20) 3-(диметиламино)азетидин дигидрохлорид (13,92 мг, 0,080 ммоль), и DIPEA (0,047 мл, 0,268 ммоль) растворяли в DMF (1,50 мл), затем добавляли HATU (30,6 мг, 0,080 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа (за ходом реакции следили с помощью LCMS). Гидразин (5,05 мкл, 0,161 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-60% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (26 мг, 63% выход). (m/z): [M+H]+ вычислено для C30H37N7O2 528,30 найдено 528,2.
Пример 13: (S)-(3-(диметиламино)-3-метилазетидин-1-ил)(2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-ил)метанон
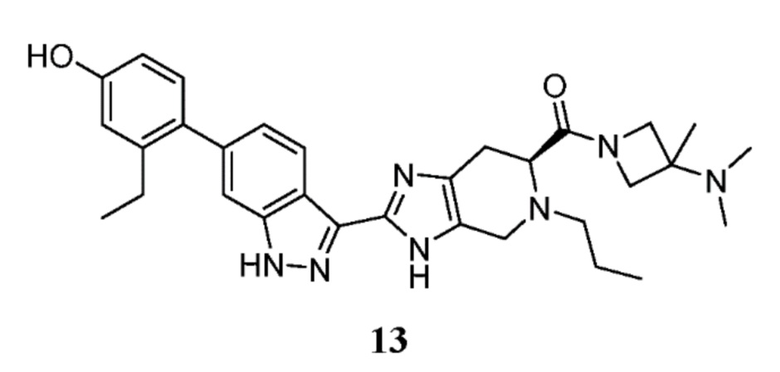
(S)-2-(6-(2-этил-4-гидроксифенил)-1H-индазол-3-ил)-5-пропил-4,5,6,7-тетрагидро-3H-имидазо[4,5-c]пиридин-6-карбоновую кислоту, TFA (30 мг, 0,054 ммоль), (I-20) N, N,3-триметилазетидин-3-амин гидрохлорид (12,12 мг, 0,080 ммоль), и DIPEA (0,047 мл, 0,268 ммоль) растворяли в DMF (1,50 мл), затем добавляли HATU (30,6 мг, 0,080 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа (за ходом реакции следили с помощью LCMS). Гидразин (5,05 мкл, 0,161 ммоль) добавляли для расщепления нежелательных побочных продуктов, затем раствор перемешивали при комнатной температуре в течение 10 минут. Затем реакционную смесь концентрировали и неочищенный продукт очищали препаративной ВЭЖХ (градиент 5-60% ACN/вода, колонка C18) с получением соли TFA указанного в заголовке соединения (18 мг, 44% выход). (m/z): [M+H]+ вычислено для C31H39N7O2 542,32 найдено 542,2.
Биологические анализы
Соединения настоящего описания охарактеризованы в одном или нескольких следующих биологических анализах.
Анализ 1: Биохимические анализы киназы JAK
Панель четырех биохимических анализов LanthaScreen JAK (JAK1, 2, 3 и Tyk2) проводили в общем киназном реакционном буфере (50 мМ HEPES, pH 7,5, 0,01% Brij-35, 10 мМ MgCl2 и 1 мМ EGTA). Рекомбинантные GST-меченные ферменты JAK и GFP-меченный пептидный субстрат STAT1 были получены от Life Technologies.
Последовательно разведенные соединения предварительно инкубировали с каждым из четырех ферментов JAK и субстратом в белых 384-луночных микропланшетах (Corning) при температуре окружающей среды в течение 1 часа. Затем добавляли АТФ для инициации киназных реакций в общем объеме 10 мкл с 1% DMSO. Конечные концентрации фермента для JAK1, 2, 3 и Tyk2 составляли 4,2 нМ, 0,1 нМ, 1 нМ и 0,25 нМ, соответственно; соответствующие используемые концентрации Km АТФ составляли 25 мкМ, 3 мкМ, 1,6 мкМ и 10 мкМ; в то время как концентрация субстрата составляли 200 нМ для всех четырех анализов. Киназным реакциям позволяли протекать в течение 1 часа при температуре окружающей среды, после чего добавляли 10 мкл препарата EDTA (конечная концентрация 10 мМ) и Tb-анти-pSTAT1 (pTyr701) антитело (Life Technologies, конечная концентрация 2 нМ) в буфере для разведения TR-FRET (Life Technologies). Планшеты оставляли для инкубации при температуре окружающей среды в течение 1 часа перед считыванием на считывателе EnVision (Perkin Elmer). Сигналы коэффициента излучения (520 нм/495 нм) записывали и использовали для расчета значений процентного ингибирования на основе DMSO и фоновых контролей.
Для анализа доза-ответ данные процентного ингибирования наносили на график в зависимости от концентраций соединения, и значения IC50 определяли с помощью 4-параметрической робастной модели подгонки с программным обеспечением Prism (GraphPad Software). Результаты выражали как pIC50 (отрицательный логарифм IC50) и затем конвертировали в pKi (отрицательный логарифм константы диссоциации Ki) с использованием уравнения Ченга-Прусоффа.
Тестируемые соединения, имеющие более низкое значение Ki или более высокое значение pKi в каждом из четырех анализов JAK, показывают большее ингибирование активности JAK.
Анализ 2: Ингибирование стимулированного IL-2 pSTAT5 в T-клетках Tall-1
Эффективность тестируемых соединений в отношении ингибирования фосфорилирования STAT5, стимулированного интерлейкином-2 (IL-2), измеряли в линии Т-клеток человека Tall-1 (DSMZ) с использованием AlphaLisa. Поскольку IL-2 передает сигнал через JAK1/3, этот анализ обеспечивает измерение клеточной активности JAK1/3.
Фосфорилированный STAT5 измеряли с помощью набора AlphaLISA SureFire Ultra pSTAT5 (Tyr694/699) (PerkinElmer).
Человеческие Т-клетки из линии клеток Tall-1 культивировали во влажной камере при 37°C, 5% CO2 в RPMI (Life Technologies), дополненной 15% инактивированной нагреванием фетальной бычьей сывороткой (FBS, Life Technologies), 2 мМ глутамакса (Life Technologies), 25 мМ HEPES (Life Technologies) и 1X Pen/Strep (Life Technologies). Соединения последовательно разбавляли в DMSO и акустически распределяли в пустые лунки. Среду для анализа (DMEM без фенолового красного (Life Technologies), дополненная 10% FBS (ATCC)) распределяли (4 мкл/лунка), и планшеты встряхивали при 900 об/мин в течение 10 минут. Клетки высевали из расчета 45000 клеток/лунку в среду для анализа (4 мкл/лунку) и инкубировали при 37°C, 5% CO2 в течение 1 часа с последующим добавлением IL-2 (R&D Systems; конечная концентрация 300 нг/мл) в предварительно нагретую среду для анализа (4 мкл) в течение 30 минут. После стимуляции цитокинов клетки лизировали 6 мкл 3x буфера для лизиса AlphaLisa (PerkinElmer), содержащего 1x таблетки PhosStop and Complete (Roche). Лизат встряхивали при 900 об/мин в течение 10 минут при комнатной температуре (комн.темп.). Фосфорилированный STAT5 измеряли с помощью набора pSTAT5 AlphaLisa (PerkinElmer). Свежеприготовленную смесь акцепторных гранул распределяли на лизат (5 мкл) при отфильтрованном зеленом свете <100 люкс. Планшеты встряхивали при 900 об/мин в течение 2 минут, непродолжительное время центрифугировали и инкубировали в течение 2 часов при комнатной температуре в темноте. Донорные гранулы распределяли (5 мкл) под фильтрованным зеленым светом <100 люкс. Планшеты встряхивали при 900 об/мин в течение 2 минут, непродолжительное время центрифугировали и инкубировали в течение ночи при комнатной температуре в темноте. Люминесценцию измеряли при возбуждении при 689 нм и испускании при 570 нм с использованием планшет-ридера EnVision (PerkinElmer) в зеленом фильтрованном свете <100 люкс.
Для определения ингибирующей способности тестируемых соединений в ответ на IL-2 измеряли среднюю интенсивность излучения шариков, связанных с pSTAT5, в линии Т-клеток человека. Значения IC50 определяли из анализа кривых ингибирования интенсивности сигнала в зависимости от концентрации соединения. Данные выражали в виде значений pIC50 (отрицательный десятичный логарифм IC50) (среднее значение± стандартное отклонение).
Результаты анализа in vitro
Соединения по изобретению тестировали в четырех ферментных анализах JAK; JAK1, JAK2, JAK3 и Tyk2, а также анализе активности клеток BEAS-2B, описанных выше.
Таблица 1
pKi
pKi
pIC50
Анализ 3: Крысиная (мышиная) модель индуцированной IL-13 индукции pSTAT6 в легочной ткани.
IL-13 представляет собой важный цитокин, лежащий в основе патофизиологии астмы (Kudlacz et al. Eur. J. Pharmacol, 2008, 582,154-161). IL-13 связывается с рецепторами клеточной поверхности, активирующими членов семейства янус киназ (JAK), которые затем фосфорилируют STAT6 и впоследствии активируют дальнейшие пути транскрипции. В описанной модели дозу IL-13 доставляли локально в легкие мышей, чтобы вызвать фосфорилирование STAT6 (pSTAT6), которое затем измеряли в качестве конечной точки.
В анализе использовали взрослых мышей Balb/c от Harlan. В день исследования животных слегка анестезировали изофлураном и вводили либо носитель, либо тестируемое соединение (1 мг/мл, общий объем 50 мкл за несколько вдохов) посредством пероральной аспирации. После введения дозы животных помещали в положение лежа на боку и наблюдали за полным восстановлением после анестезии перед возвращением в их домашнюю клетку. Через четыре часа животных снова ненадолго анестезировали и вводили либо носитель, либо IL-13 (общая доставленная доза 0,03 мкг, общий объем 50 мкл) с помощью пероральной аспирации, после чего наблюдали за восстановлением после анестезии и возвращали в их домашнюю клетку. Через час после введения носителя или IL-13 цельную кровь и легкие собирали как для обнаружения pSTAT6 в гомогенатах легких, используя набор для анализа Perkin Elmer AlphaLISA® SureFire® Ultra™ HV p-STAT6 (Tyr641), так и для анализа общей концентрации лекарственного средства как в легких, так и в плазме. Образцы крови центрифугировали (центрифуга Eppendorf, 5804R) в течение 4 минут при приблизительно 12000 об/мин при 4°C для сбора плазмы. Легкие промывали в DPBS (физиологический раствор, забуференный фосфатом Дульбекко), высушивали, замораживали, взвешивали и гомогенизировали в разведении 1:3 в 0,1% муравьиной кислоте в воде ВЭЖХ. Уровни тестируемого соединения в плазме и легких определяли с помощью анализа ЖХ-МС по аналитическим стандартам, построенным на стандартной кривой в тестовой матрице. Отношение легких к плазме определяли как отношение концентрации в легких в нг/г к концентрации в плазме в нг/мл через 5 часов.
Активность в модели подтверждается снижением уровня pSTAT6, присутствующего в легких животных, получивших лечение, через 5 часов, по сравнению с получившими носитель контрольными животными с IL-13. Разница между контрольными животными с IL-13, которым вводили носитель, и контрольными животными с носителем, которым вводили носитель, диктовала 0% и 100% ингибирующий эффект, соответственно, в любом данном эксперименте. Соединения, протестированные в этом анализе, проявляли ингибирование фосфорилирования STAT6 через 5 часов после введения IL-13, как указано ниже.
Таблица 2: Наблюдаемое ингибирование pSTAT6 и воздействие на плазму/легкие
Наблюдение за значительной концентрацией тестируемых соединений в легких мыши подтвердило, что наблюдаемое ингибирование индуцированной IL-13 индукции pSTAT6 было результатом активности тестируемого соединения. Соотношение содержания в легких и плазме через 5 часов показало, что соединения с 1 по 6 показали значительно большее воздействие в легких, чем воздействие в плазме мышей.
Анализ 4: Ингибирование вызванного TSLP высвобождения TARC в мононуклеарных клетках периферической крови человека
Тимический стромальный лимфопоэтин (TSLP) и тимусом и активацией регулируемый хемокин (TARC) сверхэкспрессируются в дыхательных путях при астме и коррелируют с тяжестью заболевания. В легких TSLP может выделяться эпителиальными клетками бронхов в ответ на аллергены и вирусные инфекции. Сигналы TSLP через гетеродимер IL-7Rα/TSLPR обнаружены в широком диапазоне тканей и типов клеток, включая эпителиальные клетки, эндотелиальные клетки, нейтрофилы, макрофаги и тучные клетки. Связывание TSLP с его рецептором вызывает конформационное изменение, которое активирует JAK1 и JAK2 для фосфорилирования различных факторов транскрипции, включая STAT3 и STAT5. В иммунных клетках это запускает каскад внутриклеточных событий, которые приводят к пролиферации клеток, антиапоптозу, миграции дендритных клеток и продукции Th2 цитокинов и хемокинов. В мононуклеарных клетках периферической крови (PBMC) TSLP оказывает провоспалительное действие, активируя миелоидные дендритные клетки для привлечения и стимуляции Т-клеток, процесс, опосредованный хемоаттрактантом TARC.
В этом анализе было показано, что стимуляция TSLP индуцирует высвобождение TARC из PBMC, и что этот ответ ослабляется дозозависимым образом при лечении соединением. Эффективность тестируемых соединений измеряли по ингибированию высвобождения TARC.
Аликвоты PBMC (ранее выделенные из цельной крови и замороженные в аликвотах при -80°C) от 3-5 доноров размораживали при 37°C и добавляли по каплям в 40 мл предварительно нагретой, стерильно фильтрованной, полной среды RPMI в 50 мл пробирки фирмы Falcon. Клетки осаждали и ресуспендировали в полной среде при 2,24×106 клеток/мл. Клетки высевали по 85 мкл (190000 клеток) на лунку в 96-луночный планшет с плоским дном, обработанным культурой ткани. Клеткам давали отдохнуть в течение 1 часа при 37°C с 5% CO2.
Соединения получали в виде 10 мМ исходных растворов в DMSO. 3,7-кратные серийные разведения были выполнены для получения 9 концентраций тестируемого соединения в DMSO при 300X конечной тестовой концентрации для анализа. 150-кратные промежуточные разведения осуществляли в полной среде для получения соединения в 2X конечной тестовой концентрации для анализа с 0,2% DMSO. После 1-часового периода отдыха в каждую лунку PBMC добавляли 95 мкл 2X соединения для конечного диапазона концентраций для анализа от 33,33 мкМ до 0,95 мкМ. 95 мкл 0,2% DMSO в полной среде добавляли в необработанные контрольные лунки. Клетки предварительно обрабатывали соединением в течение 1 часа при 37°C с 5% CO2 перед стимуляцией.
Рекомбинантный белок TSLP человека восстанавливали в концентрации 10 мкг/мл в стерильном DPBS с 0,1% BSA и хранили в аликвотах при -20°C. Непосредственно перед использованием аликвоту размораживали и получали при 20X конечной концентрации анализа в полной среде. 10 мкл 20X TSLP добавляли в каждую лунку PBMC для конечной концентрации анализа 10 нг/мл. В нестимулированные контрольные лунки добавляли 10 мкл полной среды. Клетки стимулировали в присутствии соединения в течение 48 часов при 37°C с 5% CO2.
После стимуляции собирали супернатанты клеточных культур и определяли уровни TARC с помощью твердофазного иммуноферментного анализа (ELISA) с использованием набора Human CCL17/TARC Quantikine ELISA (R&D Systems #DDN00) в соответствии с инструкциями производителя.
Для анализа эффекта дозы log [тестируемое соединение (M)] наносили на график в зависимости от значений процентного ответа для каждого донора, и значения IC50 определяли с использованием нелинейного регрессионного анализа с программным обеспечением GraphPad Prism Software с использованием 4-параметрического сигмоидального алгоритма зависимости доза-ответ с переменный наклоном. Данные выражены в виде средних значений pIC50 (отрицательный десятичный логарифм IC50), рассчитанных на основе значений pIC50 отдельных доноров и округленных до одного десятичного знака. Значения эффективности ингибирования приведены в таблице 3.
Таблица 3: Значения эффективности (pIC50) тестируемых соединений для ингибирования TSLP-вызванного высвобождения TARC в мононуклеарных клетках периферической крови человека
Хотя настоящее изобретение было описано со ссылкой на его конкретные варианты осуществления, специалисту в данной области будет понятно, что могут быть внесены различные изменения и могут быть заменены эквиваленты без отклонения от истинной сущности и объема изобретения. Кроме того, могут быть сделаны многие модификации для адаптации конкретной ситуации, вещества, рассматриваемой композиции, способа, стадии или стадий способа для задачи, существа и объема настоящего изобретения. Предполагается, что все такие модификации входят в объем прилагаемой формулы изобретения. Кроме того, все публикации, патенты и патентные документы, цитированные выше в данном контексте, включены в данное описание во всей своей полноте посредством ссылки, как если бы каждый из них был включен по отдельности посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| Новые производные пиразола | 2022 |
|
RU2816835C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ АНАБОЛИЧЕСКИХ АГЕНТОВ СКОТА | 2007 |
|
RU2419621C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ-ИНГИБИТОРА JAK | 2018 |
|
RU2790535C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2009 |
|
RU2531274C2 |
| ПИРРОЛОПИРАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2673064C2 |
Изобретение относится к получению промежуточных соединений для получения лекарственных средств. Предложен способ получения соединения формулы J-15 или его соли, где X представляет собой Br; PG1 представляет собой защитную группу для карбоновой кислоты; PG2 представляет собой аминозащитную группу и PG3 представляет собой аминозащитную группу; включающий:(a) взаимодействие соединения формулы J-14 или его соли с гидразином с получением соединения формулы J-15 и (b) необязательно образование соли соединения J-15. Также предложены соединения формул J-14, J-15 и способ получения соединения формулы J-16. Технический результат – получение промежуточных соединений для ингибиторов JAK. 4 н. и 16 з.п. ф-лы, 3 табл., 13 пр.
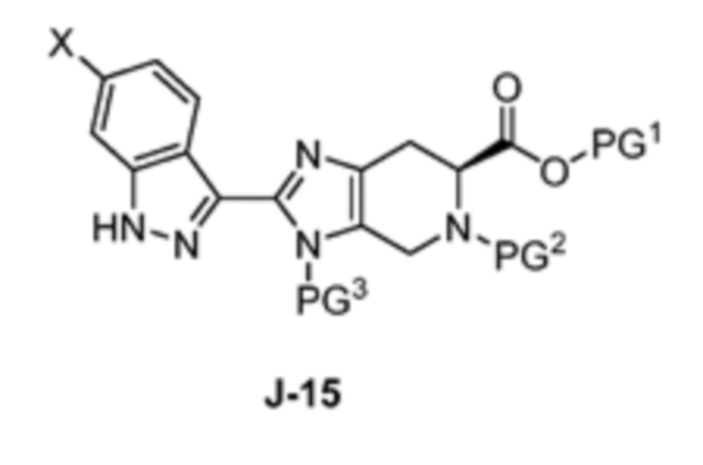
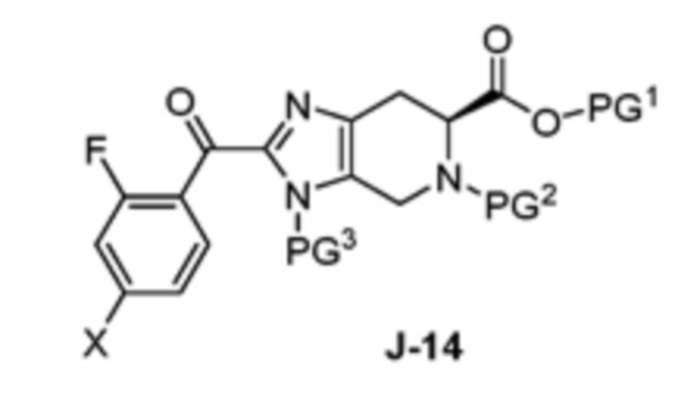
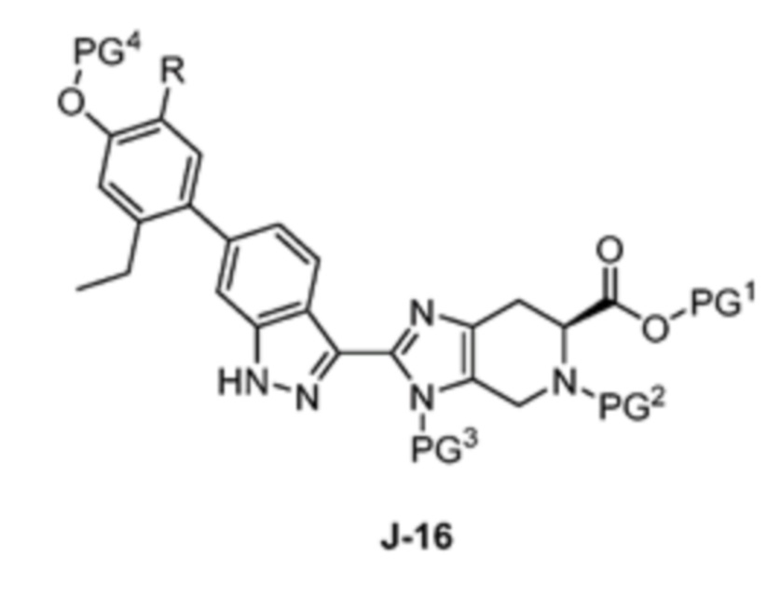
1. Способ получения соединения формулы J-15 или его соли:
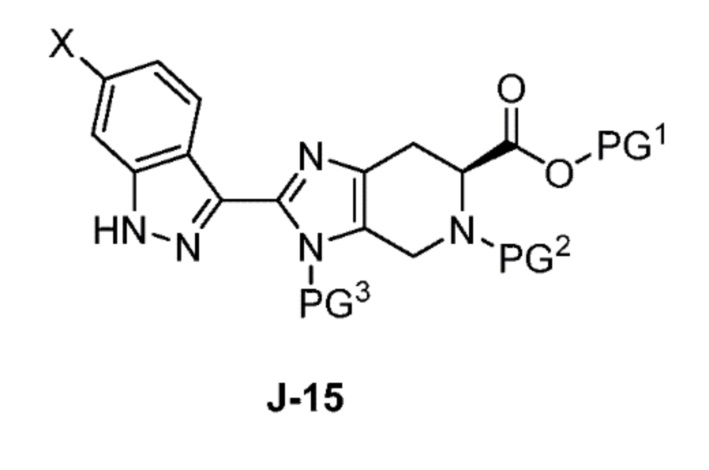
где
X представляет собой Br;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу;
включающий:
(a) взаимодействие соединения формулы J-14:
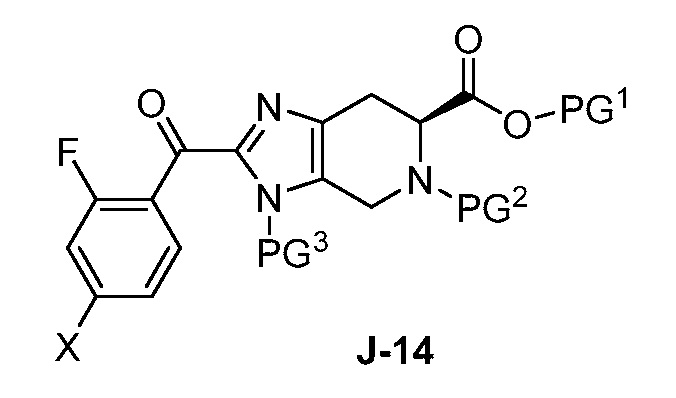
или его соли с гидразином с получением соединения формулы J-15 и
(b) необязательно образование соли соединения J-15.
2. Способ по п. 1, где реакцию с гидразином проводят при 60±20°C.
3. Способ по любому из пп. 1 или 2, где
PG1 представляет собой алкильную или бензильную группу, где бензильная группа является необязательно замещенной;
PG2 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы; и
PG3 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы.
4. Способ по любому из пп. 1 или 2, где PG1 представляет собой бензил, PG2 представляет собой трет-бутоксикарбонил и PG3 представляет собой бензил.
5. Способ по любому из пп. 1-4, где соединение J-14 или его соль получают:
(a) взаимодействием соединения формулы J-13:
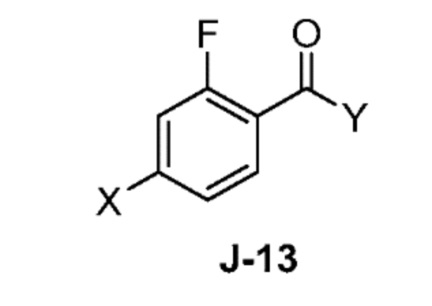
где Y представляет собой уходящую группу, с соединением формулы J-11:
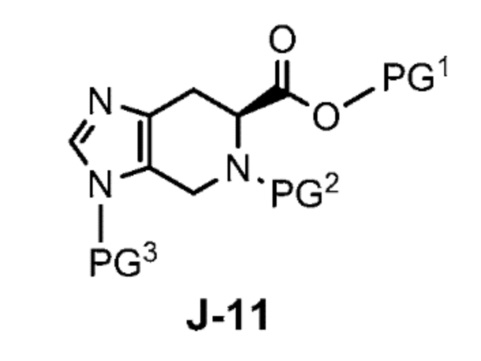
в присутствии основания с получением J-14, и
(b) необязательно образованием соли соединения J-14.
6. Способ по п. 5, где Y представляет собой Cl.
7. Соединение формулы J-14:
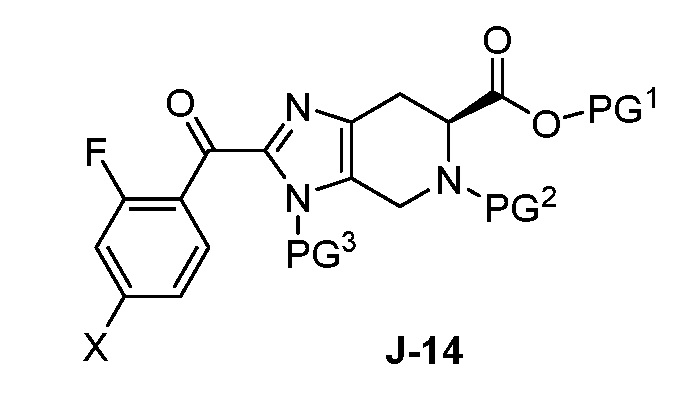
или его соль, где
X представляет собой Br;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу.
8. Соединение по п. 7 или его соль, где
PG1 представляет собой алкильную или бензильную группу, где бензильная группа является необязательно замещенной;
PG2 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы; и
PG3 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы.
9. Соединение по п. 7, имеющее формулу I-14:
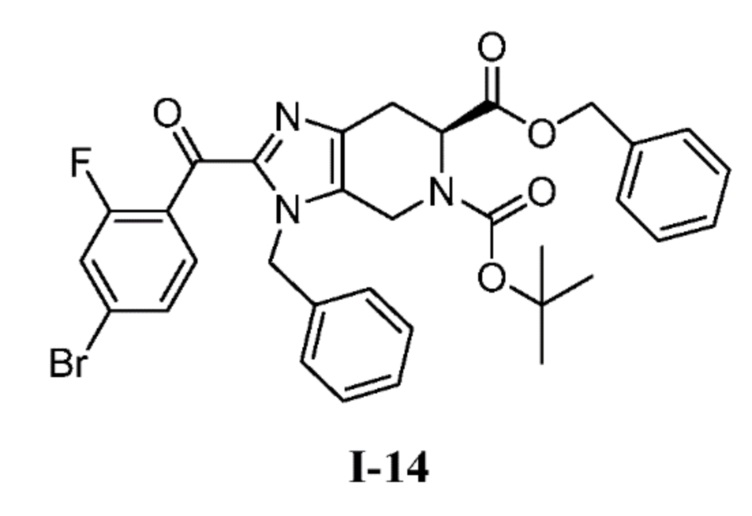
или его соль.
10. Соединение формулы J-15:
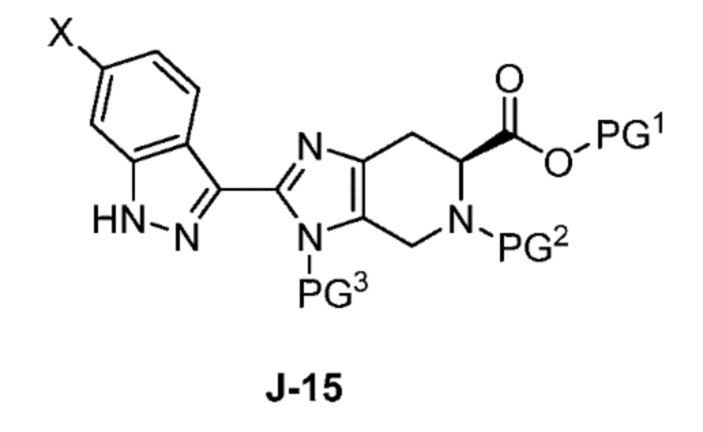
или его соль,
где
X представляет собой Br;
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу; и
PG3 представляет собой аминозащитную группу.
11. Соединение по п. 10 или его соль, где
PG1 представляет собой алкильную или бензильную группу, где бензильная группа является необязательно замещенной;
PG2 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы; и
PG3 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы.
12. Соединение по п. 10, имеющее формулу I-15:
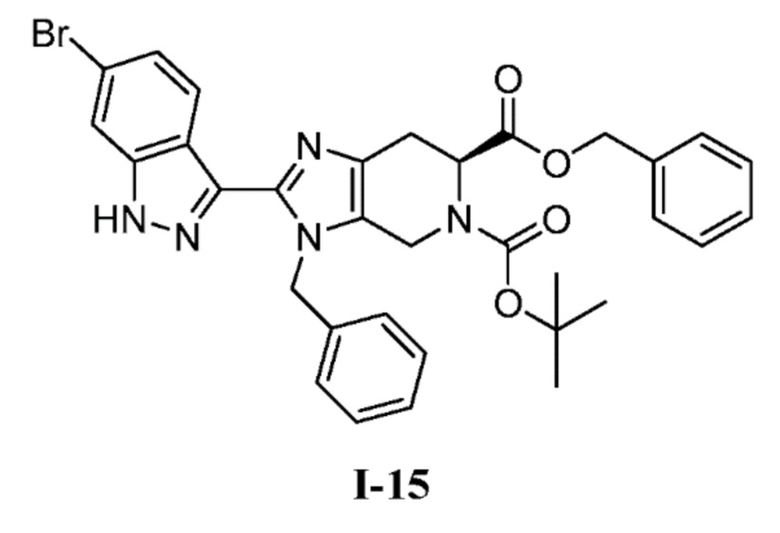
или его соль.
13. Способ получения соединения формулы J-16 или его соли:
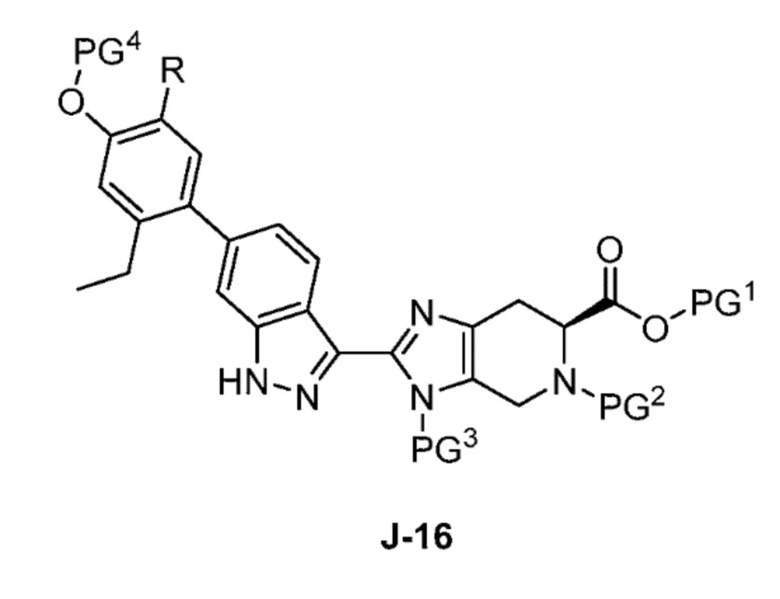
где
PG1 представляет собой защитную группу для карбоновой кислоты;
PG2 представляет собой аминозащитную группу;
PG3 представляет собой аминозащитную группу;
PG4 представляет собой гидроксилзащитную группу; и
R представляет собой H или F;
включающий:
(a) взаимодействие соединения формулы J-13:
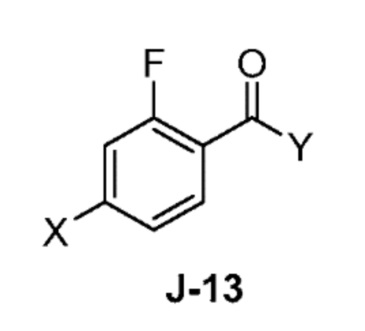
где X представляет собой Br и Y представляет собой уходящую группу; с соединением формулы J-11:
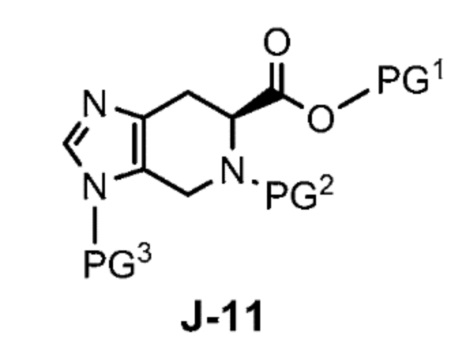
в присутствии основания, с получением соединения формулы J-14:
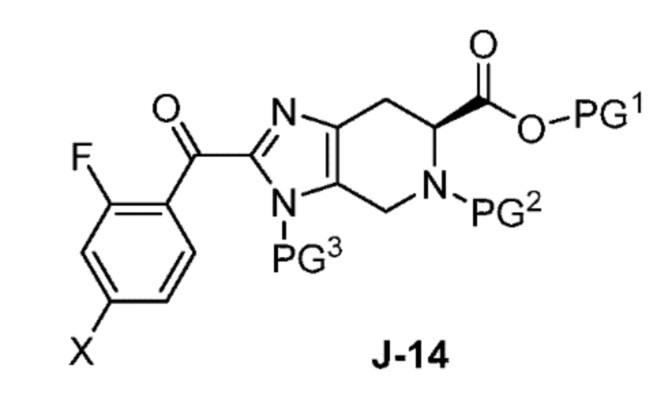
и необязательно образование соли соединения J-14;
(b) взаимодействие соединения формулы J-14 или его соли с гидразином с получением соединения формулы J-15:
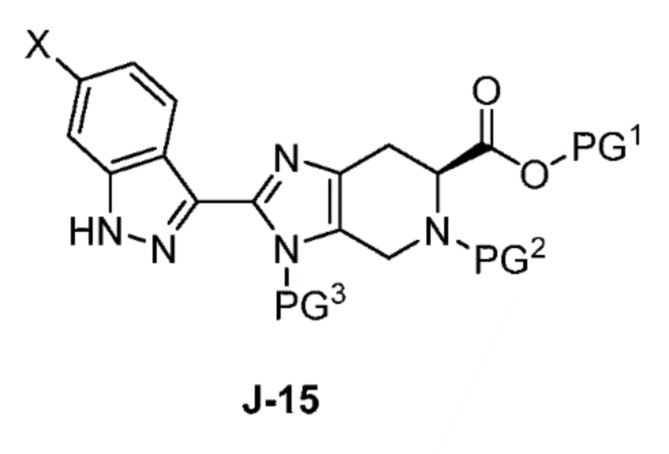
и необязательно образование соли соединения J-15;
(c) взаимодействие соединения формулы J-15 или его соли с соединением формулы J-5:
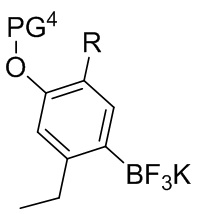
J-5
в присутствии основания, палладиевого катализатора и фосфинового лиганда с получением соединения формулы J-16 и необязательно образование соли соединения J-16.
14. Способ по п. 13, где стадию (c) проводят в присутствии диборонового реагента или катализатора.
15. Способ по п. 14, где стадию (c) проводят в присутствии тетрагидроксидиборона, сложного эфира дибороновой кислоты или продукта реакции бис(пинаколато)диборона с гидрофторидом фторида калия.
16. Способ по любому из пп. 13-15, где взаимодействие с гидразином на стадии (b) проводят при 60±20°C.
17. Способ по любому из пп. 13-16, где
PG1 представляет собой алкильную или бензильную группу, где бензильная группа является необязательно замещенной;
PG2 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы;
PG3 выбран из группы, состоящей из ацильной группы, алкоксикарбонильной группы, арилметильной группы и силильной группы;
PG4 выбран из группы, состоящей из силильной группы, ацильной группы и арилметильной группы.
18. Способ по любому из пп. 13-16, где PG1 представляет собой бензил, PG2 представляет собой трет-бутоксикарбонил, PG3 представляет собой бензил и PG4 представляет собой бензил.
19. Способ по любому из пп. 13-18, где Y представляет собой Cl.
20. Способ по любому из пп. 13-19, где палладиевый катализатор и фосфиновый лиганд стадии (c) представляют собой бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий (II).
| WO 2013014567 A1, 31.01.2013 | |||
| WO 2017079205 A1, 11.05.2017 | |||
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| 1Н-ИНДАЗОЛЫ, 1, 2-БЕНЗИЗОКСАЗОЛЫ И 1, 2-БЕНЗИЗОТИАЗОЛЫ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2389729C2 |
| Ценовая палочка | 1931 |
|
SU26641A1 |
| BAO Lei et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Прибор с двумя призмами | 1917 |
|
SU27A1 |

