Изобретение относится к способу получения 1-бета-метилкарбапенемов. 1-Бета-метилкарбапенемы являются хорошо известными антибиотиками, используемыми для лечения грам-отрицательных и грам-положительных бактериальных инфекций широкого спектра (см., например, патент США 4 962 103, выданный 9 октября 1990 г.; патент США 4 933 333, выданный 12 июня 1990 г.; патент США 4 943 569, выданный 24 июля 1990 г.; патент США 5 122 604, выданный 16 июня 1992 г.; патент США 5 034 384, выданный 23 июля 1991 г.; патенты США ('256) и 5 011 832, выданные 30 апреля 1991 г).
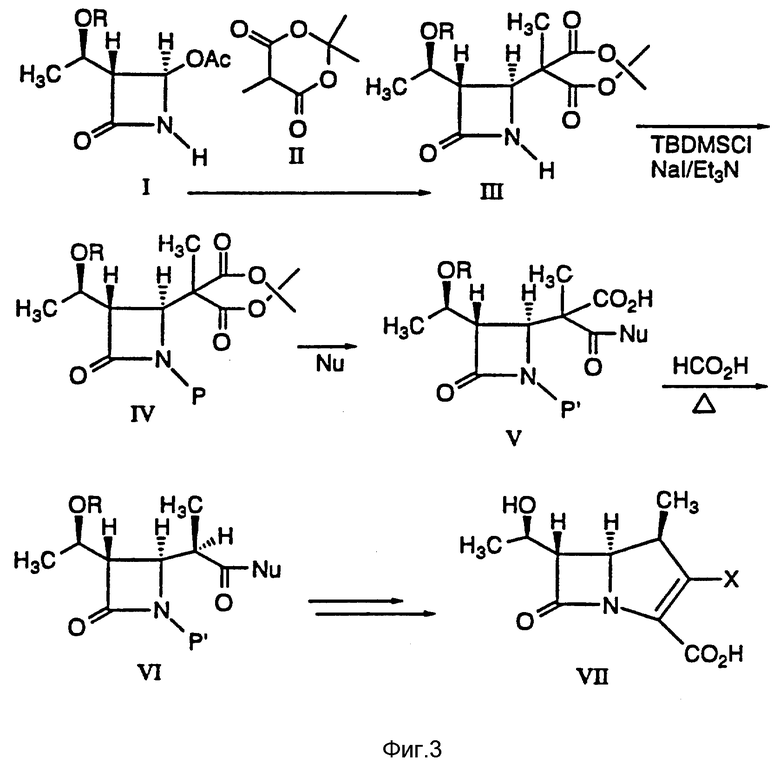
В литературе описано много способов получения промежуточных соединений бета-метилкабапенема формулы VI: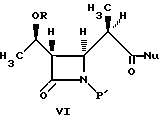
Tetrahedron Letters, Vol 26, N 39, pp 4739 - 4742, 1985; J.Am. Chem. Soc. 1986, 108, pp 4673 - 4675; Tetrahedron Letters, Vol. 27, N 19, pp 2149 - 2152, 1986; Tetrahedron Letters, Vo; 27, N 51, pp 6242 - 6244, 1986; Can. J. Chem. 65, 2140 (1987); J.Org. Chem. 1987, 52, 3174 - 3176; J.Org. Chem. 1987, 52, 2563 - 2567; J. Org. Chem. 1987, 52, 5491 - 5492; Tetrahedron Letters, Vol 28, N 1, pp 83 - 86, 1987; Tetrahedron Letters, Vpl. 28, N 5, pp 507 - 510, 1987' Tetrahedron Letters, Vol. 28, N 17, pp 1857 - 1860, 1987; Tetrahedron Letters, Vol. 28, N 52, pp 6625 - 6628, 1987; Can.J. Chem. 66, 1400 (1988); Can. J. Chem. 66, (1988); J.Chem. Soc., Chemm. Commun., 1988; J. Otg. Che, . 1988, 53, 2131 - 2132; J.Org. Chem. 1988, 53, 4154 - 4156; Tetrahedron, Vol. 44, N 8, pp 2149 - 2165, 1988; Tetrahedron Letters, Vol. 29, N 1, pp 61 - 64, 1988; Tetrahedron Letters, Vol. 29, N 49, pp 6461 - 6464, 1988; Tetrahedron Letters. Vol. 29, N 48, pp 6345 - 6348, 1988; Chemistry Letters, pp 445 - 448, 1989; J.Chem. Soc. Perkin Trans. I. 1989; J.Org. Chem. 1989, 54, 2103 - 2112; Tetrahedron Letters. Vol.30, N 1, pp 113 - 116, 1989; Tetrahedron Letters. Vol.31, N 2, pp 271 - 274, 1990; Tetrahedron Letters. Vol. 31, N 4, pp 549 - 552, 1990; Chem. Pharm. Bull. 39(9) 2225 - 2232 (1991); Tetrahedron: Asymmetry, Vol. 2, N 4, pp 255 - 256, 1991; Tetrahedron Letters. Vol.32, N 19, pp 2143 - 2144, 1991; J.Org. Chem. 1992, 57, 2144 - 2418; Tetrahedron. Vol. 48, N 1, pp 55 - 66, 1992.
Предшествующие методы стереоселективного получения β- метилкарбапенемов включают в себя:
(1) гидрогенизацию 4-(2-пропенил)замещенного азетидинона;
(2) стереоселективное протонирование иона енолята;
(3) реакцию 4-ацетоксиазетидинона с хиральным енолятом.
Указанные методы требуют трудоемкого многостадийного получения промежуточных соединений (1) и/или реагентов (3), утомительного манипулирования с высокореактивными промежуточными соединениями при низкой температуре (2); или использования дорогостоящих реагентов (2, 3).
Настоящее изобретение относится к универсальному способу получения β- метиловых промежуточных соединении (VI, схема I), осуществляемому в четыре стадии с высокой степенью стереоселактивности и с использованием легкодоступных исходных материалов.
Краткое описание изобретения.
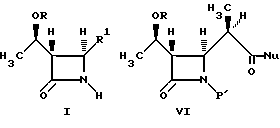
где R и P1 являются защитными группами;
R1 являются сложным эфиром метилмалоновой кислоты;
Nu является нуклеиофильной группой.
Раскрывается также способ получения промежуточных соединений.
В одном из вариантов осуществления настоящее изобретение относится к способу получения промежуточных соединений бета-метилкарбапенема формулы VI: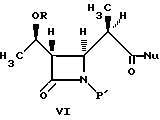
где R представляет собой: (a) водород, (b) метил или (c) гидрокси- защитную группу, такую как три-органосилил, включая три-C1-4- алкилсилил, фенил-ди-C1-4-алкилсилил и дифенилмоно-C1-4-алкилсилилокси, включая трет-бутил- диметилсилил; и изопропил-диметилсилил;
P1 представляет собой азотную защитную группу, такую как три-органосилил, включая три-C1-4-алкилсилил, фенил-ди-C1-4-алкилсилил, и дифенил-моно-C1-4-алкилсилил, включая трет-бутил-диметисилил; и изопропилдиметилсилил;
причем указанный способ включает в себя:
(a) реакцию взаимодействия соединения формулы I: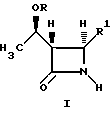
где R1 представляет собой:
(a) -O-C(O)-R'', где R'' является C1-4-алкилом, аллилом и замещенным фенилом, где заместитель представляет собой водород, C1-3-алкил, галогено, нитро, циано или C1-3-алкокси;
(b) - S(O)n - R2, где n= 1 или 2, а R2 является либо ароматической группой (такой, как фенил, бифенил, нафтил), которая может быть, но не обязательно, замещенной, например, галидом (таким как хлорид или бромид); либо C1-4-алкилом;
(c) галоген, включая Cl и Br,
в инертном растворителе с 2,2,5-триметил-1,3--диоксан-4,6- дионом и основанием с получением в результате соединения формулы III: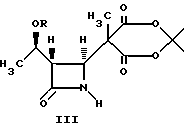
Защитными группами R и P1, которые могут быть использованы в целях настоящего изобретения, являются (но не ограничиваются ими) три-органосилилы, определенные выше, причем могут быть использованы и другие подходящие группы, описанные в Protecting Groups In Organic Suthesis, Theodaro W. Green, John Wiley abd Sons, 1981.
Для осуществления настоящего изобретения в качестве инертных растворителей может быть использован широкий ряд нереакционноспособных солюбилизирующих агентов, включая ароматические растворители, такие как бензол, толуол и ксилол; эфирные растворители, включая диэтиловый простой эфир, ди-н-бутиловый и диизопентиловый простые эфиры, анизол, циклические простые эфиры, такие как тетрагидропиран, 4-метил-1,3-диоксан, дигидропиран, тетрагидрофурфурил, метиловый эфир, этиловый эфир, фуран, 2-этокситетрагидрофуран и тетрагидрофуран (ТГФ); сложноэфирные растворители, такие как этил- и изопропилацетат; галогенуглеводородные растворители, такие как моно- или дигалоген-C1-4-алкил; спирты, такие как C1-6-алканол; C6-10-линейные, разветвленные или циклические углеводородные растворители, такие как гексан и толуол; и азотсодержащие растворители, такие как N,N-диметилацетамид, N,N-диметилформамид и ацетонитрил.
Для осуществления настоящего изобретения в качестве оснований могут быть использованы карбонаты щелочных металлов, например, K2CO3, и третичные C1-4-алкиламины, например, триэтиламин.
Молярное соотношение соединения формулы I к метил-кислоте Мелдрума (Meldrum) должно составлять приблизительно 1:1 или более. Молярное соотношение соединения формулы I к основанию должно составлять приблизительно 0,8 - 1,2:1. Вышеописанная реакция может быть проведена при температуре приблизительно 0 - 60oC, а предпочтительно при 40 - 50oC. Реакцию осуществляют в течение периода времени от 1 мин до 2 ч, а обычно в течение 14 ч, до тех пор, пока реакция не завершится.
Указанный способ также включает в себя:
(b) реакцию взаимодействия соединения формулы III в апротонном растворителе с основанием-ацептором, галидом щелочного металла и три-органогалогенсиланом с получением соединения формулы IV: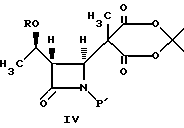
В соответствии с настоящим изобретением апротонным растворителем является N, N-ди-C1-6-алкилкарбониламид, такой как N-N- диметилформамид (ДМФ), толуол, тетрагидрофуран и дихлорметан. Основанием-акцептором является пиррол, пиридин, пирролидин, N,N-ди-C1-3-алкиламинопиридин, такой как N,N-диметиламинопиридин; три-C1-4-алкиламин, такой как триэтиламин и имидазол. Галидом щелочного металла может быть иодид, бромид или хлорид натрия, калия или лития. Три-органогалогенсиланом является три-C1-4-алкилгалогенсилан, такой как бутилдиметилсилилхлорид; фенил-ди-C1-4-алкилгалогенсилан и дифенил-С1-4-алкилгалогенсилан, где галогеном является хлорид, бромид и иодид. Отношение соединения формулы III к силану составляет приблизительно 1:1 или менее. Молярное отношение силана к основанию-акцептору составляет приблизительно 1:1 и менее. Отношение силана к галиду составляет приблизительно 1: 1 или менее.
Эту реакцию осуществляют при 0 - 70oC до тех пор, пока реакция не завершится, т.е. в течение 2 - 72 ч.
Кроме того, указанный способ включает в себя:
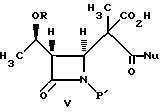
(c) реакцию взаимодействия соединения формулы IV в инертном растворителе или в C1-6-алканоле с основанием и нуклеофилом формулы NuX с получением после подкисления соединения формулы V: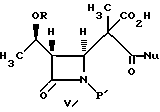
Инертным растворителем является растворитель, описанный выше.
В соответствии с настоящим изобретением C1-6-алканолом может быть метиловый, этиловый, пропиловый, изопропиловый, бутиловый и изобутиловый спирт. Основанием может быть гидроксид щелочного металла, например, гидроксид калия, лития или натрия; карбонат щелочного металла, например, карбонат натрия или калия. Подкисление может быть осуществлено любой подходящей кислотой, такой как минеральная кислота, например, HCl, H2SO4 или органическая кислота, например, уксусная или муравьиная кислота.
Отношение соединения формулы IV к основанию должно составлять приблизительно 1:1 или менее, а отношение к кислоте, используемой для подкисления, должно составлять приблизительно 1:2.
Отношение соединения формулы IV к нуклеофилу должно составлять приблизительно 1: 1 или более. Реакцию осуществляют от -20 до 25oC, в основном до полного завершения, в течение 10 - 100 мин.
При этом следует отметить, что выбор конкретного нуклеофила не имеет решающего значения для целей настоящего изобретения. В качестве нуклеофила формулы NuX могут быть использованы соли щелочных металлов, такие как алкоксиды, тиолаты и еноляты. Так, например, X может быть Na, K, Li и Сs, а Nu может быть R2O-, где R2 является водородом, C1-6-алкилом и замещенным С1-6- алкилом и фенилом.

Кроме того, в случае R2S-, R2 может иметь заместители во 2-положении карбапенема, как это имеет место в коммерчески доступных или других активных карбапенемовых антибиотиках.
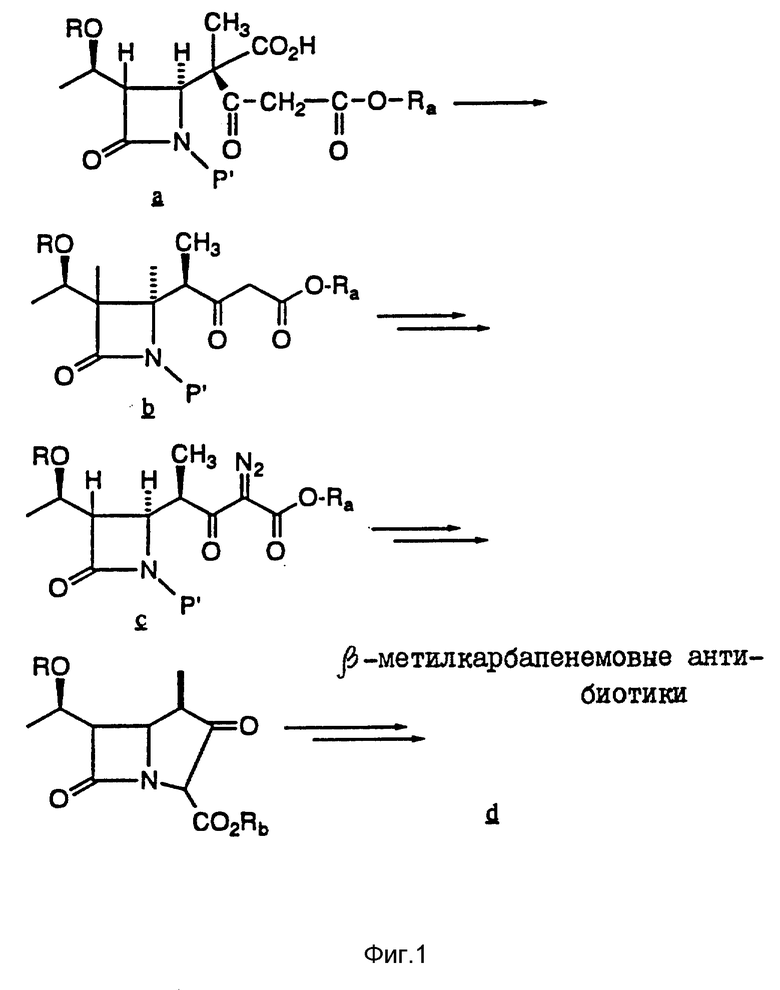
Таким образом, в одном из вариантов настоящего изобретения предусматривается использование соединений и способа, проиллюстрированных ниже на фиг. 1 и 2.
Как показано на схеме 1, соединение a подвергают стереоселективному декарбоксилипрованию, в результате чего получают соединение b. Затем соединение b превращают в активный антибиотик хорошо известным специалистам способом. См. ShihD. H. , et al., Heterocycles 1984, 21, 79. Аналогичным образом, как показано на схеме 2, соединение a подвергают стереоселективному декарбоксилированию, в результате чего получают тиокетон b', который затем превращают в активный антибиотик хорошо известным специалистам способом. См. Greenlee. et al. , Heterocycles 1989, 28, 195 и работы, указанные в настоящем описании.
В соответствии с настоящим изобретением нуклеофилом Nu являются (но не ограничиваются ими) CH2CO2t-Bu, серо- или кислородсодержащие группы - CR2, где R2 выбирают из группы, включающей в себя: водород; прямой или разветвленный низший алкил, имеющий 1 - 10 атомов углерода; алкенил, алкинил, имеющий 2 - 10 атомов углерода; циклоалкил, имеющий 3 - 6 атомов углерода; циклоалкилалкил, где циклоалкильная часть содержит 3 - 6 атомов углерода, а алкильная часть содержит 1 - 10 атомов углерода; алкилциклоалкил, где алкильная часть содержит 1 - 6 атомов углерода, а циклоалкильная часть содержит 3 - 6 атомов углдерода; арил, такой как фенил и нафтил; аралкил, такой как бензил, фенетил и т.п.; гетероциклил (насыщенный и ненасыщенный), содержащий моно- и бициклические структуры, имеющие 5 - 10 атомов на кольце, где один или несколько гетероатомов выбирают из кислорода, азота и серы, например, такой как тиофен, имидазол, тетразолил, фурил и т.п.; гетероциклилалкил, содержащий вышеуказанные гетероциклильные части и алкильную часть, имеющую 1 - 10 атомов углерода; при этом заместитель (или заместителей) для вышеупомянутых радикалов выбирают из группы, включающей в себя амино; гидроксил; циано; карбоксил; нитро; хлоро; бромо; фторо; низшую алкоксигруппу, имеющую 1 - 6 атомов углерода; меркапто; пергалогено (низший) алкил, такой как трифторметил; (низший) алкилтио; гуанидино; амидино; сульфамоил; и N-замещенные сульфамоил, амидино и гуанидино, где заместителем является низший алкил, имеющие 1 - 6 атомов углерода, или арил, имеющий 6 - 10 атомов углерода,
Например, арильные группы могут представлять собой радикалы R2 (но не ограничиваются ими), определенные в патенте США 4 962 103 (выданном 9 октября 1990 г.,) где - SR2 является:
определенные в патентах США 4 933 333, 4 943, 569 и 5 122 604, где - SR2 является: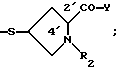
определенные в патенте США 4 866 171 (выданном 12 сент. 1989 г.), где - SR2 является: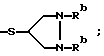
определенные в патенте США 5 034 384 (выданном 23 июля 1991 г.), где R2 является: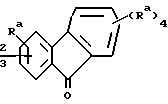
и определенные в патенте США 5 011 832 (выданном 30 апреля 1991 г.), где R2 является: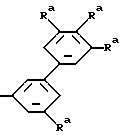
Все указанные работы вводятся в настоящее описание в качестве ссылок. Конкретные заместители на указанных арильных группах могут быть проиллюстрированы нижеследующими структурами, раскрытыми в вышеуказанных работах: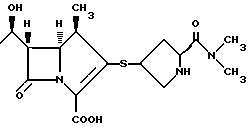
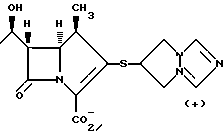
Кроме того, рассматриваемый способ включает в себя:
(d) реакцию взаимодействия соединения формулы V в эфирном или сложноэфирном растворителе со слабым основанием с получением соединения формулы VI: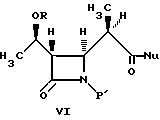
В соответствии с настоящим изобретением сложноэфирными растворителями являются этилацетат и изопропилацетат, а простыми эфирными растворителями являются растворители, указанные выше, включая метил-трет-бутиловый эфир. В качестве слабой кислоты могут быть использованы уксусная и муравьиная кислоты. Молярное отношение соединения формулы V к кислоте должно составлять 1: 1 или более. Данная реакция может протекать при температуре от 10 до 150oC в течение 10 - 120 мин, в основном до полного завершения.
Вторым объектом настоящего изобретения являются промежуточные соединения формул III, IV, и V.
Настоящее изобретение подробно проиллюстрировано на нижеприведенной схеме 3 и в последующих примерах.
4-Ацетоксиазединон I (фиг. 3) подвергают реакции с метил-кислотой Мелдрума II (2,2,5-триметил-1,3-диоксан-4,6-дион), в результате чего получают β- лактам III с использованием т-бутилдиметилсилилхлорида/триэтиламина/иодида натрия получают N- силилированный аддукт IV. В результате реакции этого аддукта IV с нуклеофилом получают производное карбоновой кислоты B, которое затем подвергают стереоселективному декарбоксидированию и получают β- метилазетидинон VI, который является предшественником, используемым для синтеза β- метилкарбапенемовых антибиотиков. Использование указанного промежуточного соединения VI проиллюстрировано на схемах 1 и 2.
Пример 1. Получение аддукта III кислоты Мелдрума.
2,2,5-триметил-1,3-диоксан-4,6-дион II (17,4 г, 110 мМ), 4-ацетокси-азетидинон I (28,7 г, 100 мМ) и K2CO3 (15,3 г, 110 мМ) смешивали в безводном ацетонитриле (150 мл, KF=5,6 мг/мл) и эту смесь выдерживали 14 ч при 45 - 50oC. После завершения перемешивания реакционную смесь охлаждали до комнатной температуры и добавляли воду (150 мл). Органический слой отделяли, а водный слой подвергали обратному экстрагированию ацетонитрилом (100 мл). После этого объединенные органические экстракты промывали солевым раствором (100 мл) и концентрировали приблизительно до объема 50 мл. Полученную смесь разводили гептаном (200 мл) и концентрировали до объема 50 мл. Затем добавляли еще 150 мл гептана и полученную смесь выдерживали при комнатной температуре для кристаллизации. Полученный продукт собирали путем фильтрации, промывали 50 мл гептана и осушали в вакууме при 40o - 50oC в течение 15 часов, в результате чего получали беловатое кристаллическое твердое вещество (30,9 г, 80,2 мМ). Второй сбор продукта получали путем концентрирования объединенного фильтрата, затем промывали приблизительно до объема 50 мл и выдерживали при комнатной температуре. Таким образом получали 2,07 г (5,4 мМ) продукта в виде белых твердых хлопьев. Объединенный выход составлял 85,6%. Т.пл. 78 - 83oC (с разложением).
1H-ЯМР (CDCl3): 6,19 (1H, шир., NH), 4,20 (1H, дкв., J=3,7 и 6,4 Гц), 4,15 (1H, д, J=2,1 Гц), 3,54 (1H, дд, J=2,1 и 3,7 Гц), 1,77 (3H, с, CH3), 1,73 (3H, с, CH3), 1,62 (3H, с, CH3), 1,17 (3H, д, J=6,4 Гц), 0,85 (9H, с, Si-t-Bu), 0,06 и 0,05 (6H, 2с, 2Si-CH3);
13C-ЯМР (CDCl3): 168,91, 168,51, 167,72, 105,47, 64,70, 61,22, 55,63, 50,99, 30,04, 28,28, 25,78, 22,82, 18,60, 17,95, -4,32, -4,94.
Пример 2. Получение соединения IV.
Азетидинон III (7,7 г, 20 мМ) растворяли в диметилформамиде (100 мл, KF= 10 мг/мл) и последовательно добавляли NaI (6,6 г, 44 мМ), триэтиламин (8,4 мл, 60 мМ) и N,N-диметиламинопиридин (0,25 г, 2 мМ). Полученную смесь перемешивали в течение 5 мин и добавляли одной порцией 6,6 г (44 мМ) т-бутилдиметилсилилхлорида. Затем эту смесь перемешивали в течение 48 ч при комнатной температуре и 15 ч при температуре 50 - 60oC. После завершения перемешивания реакционную смесь охлаждали до комнатной температуры и добавляли воду (100 мл). Полученную смесь экстрагировали гексаном (2х100 мл). Объединенные экстракты промывали 100 мл 1 H водного раствора соляной кислоты и 100 мл воды, а затем концентрировали досуха. Полученный маслообразный остаток растворяли в 2-пропаноле (40 мл) и по капле добавляли 40 мл воды. После добавления по капле еще 40 мл воды смеси выдерживали при комнатной температуре для кристаллизации. Образовавшийся продукт собирали фильтрованием и осушали в вакууме в течение 15 часов при 40 - 50oC. Таким образом получали светло-оранжевое твердое кристаллическое вещество (8,4 г, 16,8 мМ). Выход = 84,1%. Т.пл. 73 - 74oC (с разложением).
1H-ЯМР (CDCl3): 4,32 (1H, д, J=2,2 Гц), 3,96 (1H, дкв., J=5,9 и 9,4 Гц), 3,66 (1H, дд, J=2,2 и 9,4 Гц), 1,79 (6H, с, 2CH3), 1,71 (3H, с, CH3), 1,34 (3H, д, J=5,9 Гц), 0,95 (и 0,91 (18H, 2с, 2Si-t-B), 0,25, 0,14, 0,12 и 0,11 (12H, 4с, 4Si-CH3);
13C-ЯМР (CDCl3): 173,72, 168,56, 167,44, 105,43, 68,39, 63,13, 60,92, 50,45, 29,80, 28,09, 26,67, 25,95, 23,61, 23,03, 19,06, 18,02, -4,38, -4,76, -4,90.
Пример 3. Гидролиз силилированного аддукта IV.
Аддукт IV (2,5 г, 5 мМ) растворяли в ТГФ (10 мл) и раствор охлаждали до 0oC. Затем к раствору по капле добавляли 10 мл (10 мМ) 1 H водного раствора NaOH, поддерживая внутреннюю температуру ниже 5oC. После этого смесь отстаивали в течение часа при 0oC и по капле добавляли раствор NaOH (2 мл, 2 мМ). После завершения добавления смесь подкисляли путем добавления муравьиной кислоты (1,1 мл, 30 мМ) и экстрагировали этилацетатом (40 мл). Полученный раствор использовали в последующей стадии. Двухосновная кислота может быть выделена путем концентрирования растворителя с последующей кристаллизацией полученного твердого вещества из смеси метанол/вода. Температура плавления данного продукта составляет 97 - 99oC (с разложением).
1H-ЯМР (CDCl3): 4,39 (1H, с), 4,07 (1H, кв., J=6,5 Гц), 3,29 (1H, д, J= 6,2 Гц), 1,28 (3H, д, J=7,6 Гц), 0,95 и 0,90 (18H, 2с, 2Si-t-Bu), 0,28, 0,14 и 0,11 (12H, 3С, 4Si-CH3).
13C-ЯМР (CD3OD): 176,79, 173,78, 67,94, 62,15, 57,81, 27,21, 26,59, 20,14, 19,01, 17,71, -3,77, -4,46.
Пример 4. Метанолиз силилированного аддукта IV.
Аддукт IV (1,50 г, 3 мМ) растворяли в метаноле (20 мл) и раствор охлаждали до 0oC. После этого к раствору тремя порциями добавляли K2CO3 (0,86 г, 6,2 мМ) и полученную смесь выдерживали в течение часа при комнатной температуре. Эту реакционную смесь гасили путем добавления воды (10 мл) и 1 H водного раствора соляной кислоты (10 мл), а затем экстрагировали этилацетатом (20 мл). Органический слой промывали 20 мл воды и концентрировали досуха, в результате чего образовалось белое пенообразное вещество (1,18 г, 2,5 мМ), выход = 80%. Затем это пенообразное вещество очищали путем кристаллизации из смеси метанол/вода и получали белое кристаллическое твердое вещество. Т.пл. 125 - 135oC (с разложением).
1H-ЯМР (CDCl3): 8,5 (1H, шир., CO2H), 4,34 (1H, д, J=2,4 Гц) 4,09 (1H, кв., J=6,4 Гц), 3,76 (3H, с, OCH3), 3,11 (1H, дд, J=2,4 и 6,8 Гц), 1,50 (3H, с, CH3), 1,22 (3H, д, J=6,2 Гц), 0,96 и 0,89 (18H, 2с, 2Si-t-Bu), 0,29, 0,09, и 0,08 (12H, 2с, 4Si-CH3).
13C-ЯМР (CDCl3): 174,57, 174,20, 171,26, 67,37, 61,33, 56,88, 56,54, 53,05, 25,54, 26,02, 22,52, 19,32, 18,34, 18,12, -3,99, -4,43, -4,60, -4,70.
Пример 5. Декарбоксилирование двухосновной кислоты V.
Полученную на стадии основного гидролиза двухосновную кислоту в этилацетате нагревали с обратным холодильником в течение 2 ч с дополнительным количеством муравьиной кислоты (1,10 мл, 30 мМ). Анализ аликвот показал наличие смеси β- метилового продукта VI и α- метилового продукта VI (95:5). Эту смесь охлаждали до комнатной температуры и концентрировали с получением маслообразного вещества. Полученное маслообразное вещество растворяли в водном растворе NaOH (1H, 10 мл, 10 мМ) и смесь отстаивали в течение 2 ч при комнатной температуре. Затем этот раствор подкисляли 1 H водным раствором соляной кислоты (15 мл, 15 мМ), после чего экстрагировали 30 мл этилацетата. Экстракт промывали водой и концентрировали приблизительно до объема 5 мл. После добавления 60 мл гексана смесь выдерживали при комнатной температуре. Белое твердое кристаллическое вещество собирали путем фильтрации, а затем промывали гексаном 910 мл), в результате чего получали чистое β- метиловое соединение формулы VII (0,96 г 3,18 мМ). Полный выход из промежуточного соединения IV составлял 64%. Температура плавления составляла 144 -146oC (с разложением).
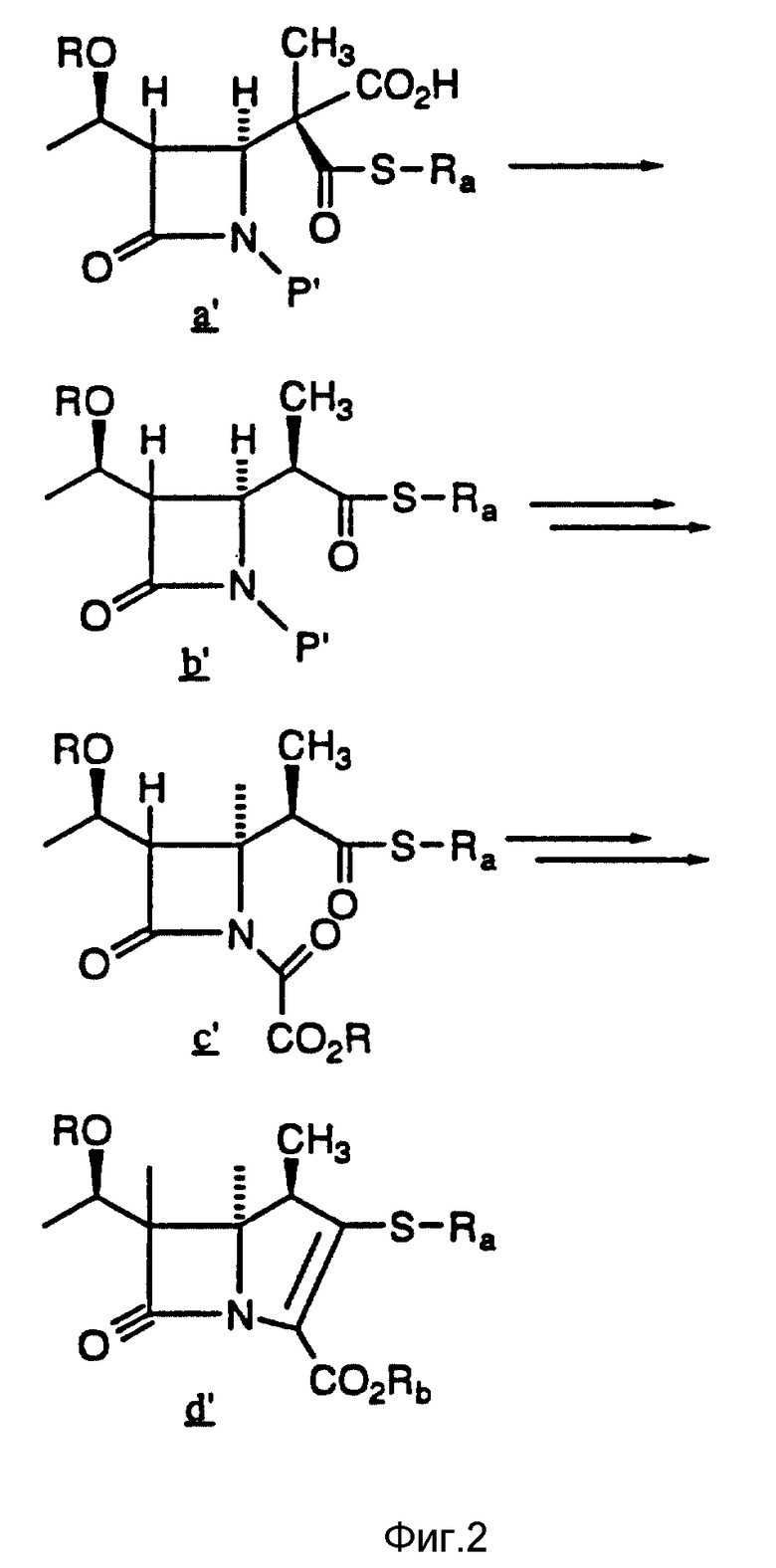
Описывается способ получения промежуточного соединения карбапенема формулы VI, где R представляет собой: (a) водород, (b) метил или (c) гидроксизащитную группу, и P1 представляет собой азотную защитную группу; Nu представляет собой нуклеофильную группу, выбранную из -CH2CO2-t-Bu и -SR2, где R2 представляет собой водород, прямо и или разветвленный C1-C10 низший алкил, прямой или разветвленный C2-C10 алкенил и алкинил; C3-C6 циклоалкил, C3-C6-циклоалкил- C1-C10 алкил, C1-C6 алкил- C3-C6-циклоалкил, арил и аралкил. Способ включает реакцию взаимодействия соединения формулы V, где R, P1 и Nu принимают значения, определенные выше, в эфирном или сложноэфирном растворителе со слабой кислотой. Технический результат - универсальность способа получения целевых продуктов. 5 с. и 3 з.п.ф-лы, 3 ил.
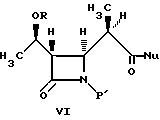
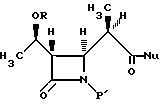
где R представляет собой (а) водород, (b) метил или (с) гидрокси-защитную группу;
P' представляет собой N-защитную группу;
Nu представляет собой нуклеофильную группу, выбранную из -CH2CO2-t-Bu и -SR2, где R2 представляет собой водород, прямой или разветвленный C1-C10 низший алкил, прямой или разветвленный C2-C10 алкенил и алкинил; C3-C6 циклоалкил, C3-C6-циклоалкил - C1-C10алкил, C1-C6 алкил-C3-C6-циклоалкил, арил и аралкил,
стереоселективным декарбоксилированием, отличающийся тем, что проводят декарбоксилирование соединения формулы V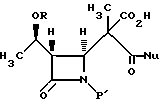
где R, P' и Nu принимают значения, определенные выше, в эфирном или сложноэфирном растворителе со слабой кислотой с получением соединения формулы VI, где R, P' и Nu определены выше.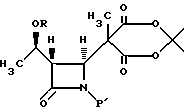
в водном инертном растворителе или в C1-C6-алканоловом растворителе с основанием и нуклеофилом с получением после подкисления соединения формулы V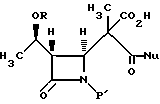
3. Способ по п.2, отличающийся тем, что предварительно проводят реакцию взаимодействия соединения формулы III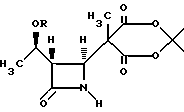
в апротонном растворителе с основанием-акцептором, галидом щелочного металла и три-органо-галогеносиланом с получением соединения формулы IV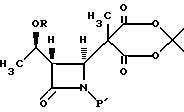
4. Способ получения промежуточного соединения бета-метилкарбапенема формулы VI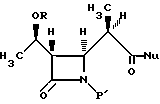
где R представляет собой (а) водород, (b) метил или (с) гидрокси-защитную группу;
P' представляет собой N-защитную группу;
Nu представляет собой нуклеофильную группу, выбранную из -CH2CO2-t-Bu и -SR2, где R2 представляет собой водород, прямой или разветвленный C1-C10 низший алкил, прямой или разветвленный C2-C10 алкенил и алкинил; C3-C6-циклоалкил, C3-C6-циклоалкил-C1-C10алкил, C1-C6 алкил-C3-C6-циклоалкил; арил и аралкил,
с использованием стереоселективного декарбоксилирования, отличающийся тем, что проводят (а) реакцию взаимодействия соединения формулы I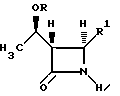
где R1 представляет собой: (а) -O-C(O)-R'', где R'' представляет собой C1-C6-алкил, аллил и замещенный фенил, в котором заместителем является водород, C1-C3-алкил, галоген, нитро-, циано или C1-C3-алкил-окси; (b) -S(O)п-R2, где п равно 1 или 2, а R2 представляет собой фенил, бифенил, нафтил, необязательно замещенный галидом, или C1-4-алкил и (с) галоген;
в инертном безводном растворителе с 2,2,5-триметил-1,3-диоксан-4,6-дионом и основанием с получением соединения формулы III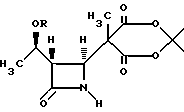
(b) реакцию взаимодействия соединения формулы III в апротонном растворителе с основанием-акцептором, галидом щелочного металла и три-органо-галогеносиланом с получением соединения формулы IV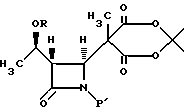
(с) реакцию взаимодействия соединения формулы IV в инертном растворителе или C1-6-алканоле с основанием и нуклеофилом формулы NuX с получением после подкисления соединения формулы V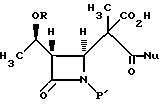
где Nu является нуклеофильной группой, выбираемой из -CH2CO2-t-Bu и -SR2, где R2 представляет собой водород, прямой или разветвленный C1-C10 низший алкил, прямой или разветвленный C2-C10 алкенил и алкинил; C3-C6-циклоалкил, C3-C6-циклоалкил-C1-C10-алкил, C1-C6-алкил-C3-C6-циклоалкил, арил и аралкил, а Х является уходящей группой;
(d) декарбоксилирование соединения формулы V в эфирном или сложноэфирном растворителе со слабой кислотой с получением соединения формулы VI.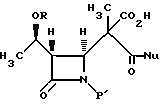
где R представляет собой (а) водород, (b) метил или (с) три-органо-силил, выбранный из три-C1-4-алкилсилила, фенил-ди-C1-4-алкилсилила и дифенил-моно-C1-4-алкилсилила;
P' представляет собой три-органо-силил, выбранный из три-C1-4-алкилсилила, фенил-ди-C1-4-алкилсилила и дифенил-моно-C1-4-алкилсилила;
Nu представляет собой нуклеофильную группу, выбранную из -CH2CO2-t-Bu и -SR2, где R2 представляет собой водород, прямой или разветвленный C1-C10 низший алкил, прямой или разветвленный C2-C10-алкенил и алкинил; C3-C6 циклоалкил, C3-C6-циклоалкил - C1-C10-алкил, C1-C6-алкил-C3-C6-циклоалкил, арил и аралкил,
отличающийся тем, что включает (с) реакцию взаимодействия соединения формулы IV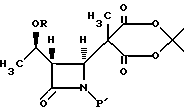
в водном инертном растворителе или C1-6-алканоле с основанием и соединением формулы NuX с получением после подкисления соединения формулы V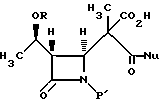
где Nu является нуклеофильной группой, выбранной из -CH2CO2-t-Bu и -SR2, где R2 представляет собой водород, прямой или разветвленный C1-C10 низший алкил, прямой или разветвленный C2-C10- алкенил и алкинил; C3-C6-циклоалкил, C3-C6-циклоалкил- C1-C10-алкил, C1-C6-алкил-C3-C6-циклоалкил, арил и аралкил, а Х является уходящей группой.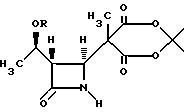
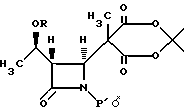
где R представляет собой (а) водород, (b) метил или (с) защитную группу;
P' представляет собой защитную группу.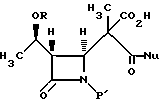
где R представляет собой: (а) водород, (b) метил или (с) защитную группу;
P' представляет собой защитную группу,
Nu представляет собой нуклеофильную группу, выбранную из -CH2CO2-t-Bu и -SR2, где R2 представляет собой водород, прямой или разветвленный C1-C10 низший алкил, прямой или разветвленный C2-C10-алкенил и алкинил; C3-C6-циклоалкил, C3-C6-циклоалкил-C1-C10-алкил, C1-C6-алкил-C3-C6-циклоалкил, арил и аралкил.
| Способ получения производных 4-ацетокси-3-оксиэтилазетидин-2-она (его варианты) | 1985 |
|
SU1442071A3 |
| US 4960880 A, 02.10.90 | |||
| US 4576746 A, 18.03.86. | |||

