Область техники
Настоящее изобретение относится к составам назального спрея, содержащим по меньшей мере один активный фармацевтический ингредиент и по меньшей мере один увлажняющий агент.
Уровень техники
Пациенты, страдающие аллергическим ринитом, часто жалуются на сухость в носу и связанный с ней дискомфорт в полости носа. В одном из исследований по меньшей мере 41% людей, страдающих аллергией, сообщили, что ощущают сухость в носу, являющуюся симптомом аллергического ринита или побочным действием наиболее распространенных лекарств для лечения симптомов аллергии, включая назальные спреи. Представленные в настоящее время на рынке назальные спреи, в том числе назальные спреи под торговым наименованием Флоназа (FLONASE®) от GlaxoSmithKline Consumer Healthcare, лечат симптомы аллергического ринита, но практически не решают проблему сухости в носу. Сухость доставляет не меньше беспокойства, чем другие симптомы у людей с аллергией.
Сухость может приводить к повреждению кожи внутри носовых ходов, что может вызывать кровотечение из носа или появление волдырей. Кроме того, может возникать чувство жжения, особенно при сморкании или вытирании носа. Сухости может быть подвержена не только кожа внутри носовых ходов. Она может проявляться даже снаружи носа и в прилегающих областях между носом и губами. На рынке существует неудовлетворенная потребность в увлажняющем назальном спрее, который помимо лечения симптомов аллергического ринита облегчал бы сухость в носу и обеспечивал ощущение увлажнения.
Одним из основных препятствий для введения увлажняющего агента в состав назального спрея является то, что введение увлажняющего агента может негативно влиять на доступность активного фармацевтического ингредиента в месте его нанесения, что снижает эффективность состава при лечении симптомов аллергического ринита. Кроме того, увлажняющий агент может влиять на осмолярность, реологию и/или рН состава, что может приводить к раздражению или влиять на стабильность состава.
С учетом вышеизложенного желательно разработать состав назального спрея, который эффективен для лечения симптомов аллергического ринита и при этом облегчает сухость в носу.
Краткое описание изобретения
Один из вариантов реализации настоящего изобретения относится к составу назального спрея, содержащему по меньшей мере один активный фармацевтический ингредиент, глицерин, полиэтиленгликоль и декстрозу, при этом указанный состав удерживает по меньшей мере примерно 3% воды под действием относительной влажности примерно 80% при температуре примерно 23°С в течение примерно 750 минут.
Другой вариант реализации настоящего изобретения относится к составу назального спрея, содержащему по меньшей мере один активный фармацевтический ингредиент, глицерин, полиэтиленгликоль и декстрозу.
В одном из вариантов реализации составы согласно настоящему изобретению имеют рН от примерно 5 до примерно 7. В одном из вариантов реализации составы имеют осмолярность от примерно 100 мОсмолей до примерно 800 мОсмолей.
В одном из вариантов реализации активный фармацевтический ингредиент в составах присутствует в количестве от примерно 0,005% масс./масс., до примерно 0,2% масс./масс. В одном из вариантов реализации активный фармацевтический ингредиент представляет собой флутиказона пропионат, флутиказона фуроат, азеластин, оксиметазолин, ксилометазолин, беклометазон, мометазон, будесонид, их соли и сложные эфиры, или их комбинацию. В одном из предпочтительных вариантов реализации активный фармацевтический ингредиент представляет собой флутиказона пропионат.
В другом варианте реализации составы согласно настоящему изобретению являются изотоническими и содержат агент, регулирующий изотоничность, представляющий собой хлорид натрия, декстрозу, хлорид калия или их комбинацию. В одном из вариантов реализации агент, регулирующий изотоничность, представляет собой хлорид натрия.
В одном из вариантов реализации глицерин присутствует в составах согласно настоящему изобретению в количестве от примерно 0,5% масс./масс., до примерно 8% масс./масс. В одном из вариантов реализации декстроза присутствует в количестве от примерно 0,3% масс./масс. до примерно 7% масс./масс. В одном из вариантов реализации полиэтиленгликоль присутствует в количестве от примерно 0,5% масс./масс. до примерно 20% масс./масс. В одном из вариантов реализации полиэтиленгликоль имеет среднюю молекулярную массу от примерно 200 до примерно 600. В одном из предпочтительных вариантов реализации полиэтиленгликоль имеет среднюю молекулярную массу примерно 400.
В одном из вариантов реализации составы согласно настоящему изобретению имеют распределение по капель размеру, при котором примерно 10% капель имеют размер менее примерно 18 мкм, примерно 50% капель имеют размер от примерно 33 мкм до примерно 51 мкм, и примерно 90% капель имеют размер менее примерно 120 мкм. В одном из вариантов реализации примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 10 мкм.
Один из вариантов реализации настоящего изобретения относится к способу лечения симптомов аллергического ринита, обеспечивающему ощущение увлажнения в полости носа, включающему введение состава согласно изобретению. Один из вариантов реализации настоящего изобретения относится к способу лечения симптомов аллергического ринита, обеспечивающему мягкое ощущение облегчения в полости носа, включающему введение состава согласно изобретению. Один из вариантов реализации настоящего изобретения относится к способу лечения симптомов аллергического ринита, обеспечивающему ощущение комфорта в полости носа, включающему введение состава согласно изобретению.
Подробное описание изобретения
Аспекты настоящего изобретения относятся к составам назального спрея, содержащим по меньшей мере один активный фармацевтический ингредиент, подходящий для назального введения, и увлажняющий агент или комбинацию увлажняющих агентов. Заявители обнаружили, что эти составы назального спрея неожиданно облегчают сухость и обеспечивают ощущение увлажнения без ухудшения стабильности, эффективности или безопасности указанных составов.
Составы согласно настоящему изобретению содержат по меньшей мере один активный фармацевтический ингредиент или комбинацию активных фармацевтических ингредиентов. В одном варианте реализации активный фармацевтический ингредиент предназначен для лечения воспалительных и аллергических состояний, включая сезонный и круглогодичный аллергический ринит. Подходящие активные фармацевтические ингредиенты включают, среди прочего, кортикостероиды, антигистамины, альфа-адренергические агонисты. Примеры активного фармацевтического ингредиента, который может входить в составы согласно настоящему изобретению, включают, но не ограничиваются ими, флутиказон, азеластин, оксиметазолин, ксилометазолин, беклометазон, мометазон, будесонид, хлорфенирамина малеат, лоратадин, азатадин, их соли и сложные эфиры, или их комбинацию. Количество активного фармацевтического ингредиента может составлять от примерно 0,001% масс./масс. до примерно 0,5% масс./масс. в расчете на общую массу состава.
В одном из предпочтительных вариантов реализации активный фармацевтический ингредиент представляет собой флутиказона пропионат. Количество флутиказона пропионата может составлять от примерно 0,005% масс./масс. до примерно 0,2% масс./масс. в расчете на общую массу состава. В одном варианте реализации количество флутиказона пропионата может составлять от примерно 0,01% масс./масс. до примерно 0,09% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество флутиказона пропионата составляет примерно 0,05% масс./масс. в расчете на общую массу состава.
В другом варианте реализации активный фармацевтический ингредиент представляет собой флутиказона фуроат. Количество флутиказона фуроата может составлять от примерно 0,005% масс, до примерно 0,2% масс./масс. в расчете на общую массу состава.
В одном варианте реализации количество флутиказона фуроата может составлять от примерно 0,01% масс./масс. до примерно 0,09% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество флутиказона фуроата составляет примерно 0,05% масс./масс. в расчете на общую массу состава.
Например, композиция согласно настоящему изобретению, предназначенная для введения путем дозирования из дозирующей емкости для назального спрея для многократного применения, может содержать флутиказона пропионат в количестве, достаточном для обеспечения 0,05 мг/впрыск; или флутиказона фуроат в количестве, достаточном для обеспечения 0,0275 мг/впрыск; или азеластин или его фармацевтически приемлемую соль (например, гидрохлорид азеластина) в количестве, достаточном для обеспечения эквивалента 0,125 мг основания на впрыск, при этом общее количество впрысков на одну емкость составляет, например, 50, 60, 120 или 144 впрыска.
В одном варианте реализации состав может быть изотоническим и может включать агент, регулирующий изотоничность, вместо активного фармацевтического ингредиента или в комбинации с активным фармацевтическим ингредиентом. Примеры агентов, регулирующих изотоничность, включают, но не ограничиваются ими, хлорид натрия, декстрозу, хлорид калия или их комбинацию.
В одном из предпочтительных вариантов реализации агент, регулирующий изотоничность, представляет собой хлорид натрия. Количество хлорида натрия может составлять от примерно 0,1% масс./масс. до примерно 1,5% масс./масс. в расчете на общую массу состава. В одном варианте реализации количество хлорида натрия может составлять от примерно 0,5% масс./масс. до примерно 1,3% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество хлорида натрия составляет примерно 0,9% масс./масс. в расчете на общую массу состава.
В другом варианте реализации агент, регулирующий изотоничность, представляет собой декстрозу. Количество декстрозы может составлять от примерно 1% масс./масс. до примерно 10% масс./масс. в расчете на общую массу состава. В одном варианте реализации количество декстрозы может составлять от примерно 3% масс./масс. до примерно 7% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество декстрозы составляет примерно 5% масс./масс. в расчете на общую массу состава.
Составы согласно настоящему изобретению включают увлажняющий агент или комбинацию увлажняющих агентов. Увлажняющий агент или комбинация увлажняющих агентов может обеспечивать ощущение увлажнения и/или облегчать сухость в полости носа и/или прилегающих областях. Обычно добавление любого увлажняющего агента может негативно сказываться, среди прочего, на стабильности состава, доступности активного фармацевтического ингредиента в составе и/или безопасности состава. Заявители обнаружили, что добавление в составы согласно настоящему изобретению конкретной комбинации увлажняющих агентов неожиданно обеспечивает увлажняющее действие с поддержанием при этом характеристик состава, включающих, но без ограничения ими, рН, распределение капель по размеру (РКР), распределение частиц по размеру (РЧР), растворимость и осмоляриость, в пределах заданных диапазонов для сохранения стабильности, эффективности и безопасности составов.
Существенным преимуществом составов согласно настоящему изобретению по сравнению с FLONASE® является то, что несмотря на то, что составы согласно настоящему изобретению содержат увлажняющие агенты, эти составы по-прежнему биоэквивалентны одобренной и представленной в настоящее время на рынке FLONASE®, не содержащей каких-либо увлажняющих агентов. Это является существенным преимуществом, поскольку составы согласно настоящему изобретению могут лечить симптомы аллергического ринита практически так же эффективно и безопасно, как и FLONASE®, при этом обеспечивая дополнительное увлажняющее действие.
В настоящем документе биоэквивалентность представляет собой отсутствие значительной разницы в скорости и степени доступности активного фармацевтического ингредиента в месте действия лекарственного средства при введении в той же дозе в аналогичных условиях в рамках надлежащим образом спланированного исследования. Лекарственный препарат, содержащий тот же активный ингредиент в том же количестве, что и другой лекарственный препарат, считается биоэквивалентным одобренному лекарственному препарату, если скорость и степень всасывания не отличается значительно от этих показателей у одобренного лекарственного препарата, или если степень всасывания не отличается значительно, а любые различия в скорости всасывания являются преднамеренными или не являются значимыми с медицинской точки зрения.
Для составов согласно настоящему изобретению биоэквивалентность может зависеть, среди прочих факторов, от РКР и РЧР активного фармацевтического ингредиента. Скорость и степень доступности активного фармацевтического ингредиента в месте действия для составов согласно настоящему изобретению по существу аналогичны FLONASE®, поэтому указанные составы биоэквивалентны FLONASE®.
Подходящие увлажняющие агенты, которые могут быть включены в составы согласно настоящему изобретению, включают, но не ограничиваются ими, глицерин, пропиленгликоль, гиалуронат натрия, DL-молочную кислоту, поливинилпирролидин, полиэтиленгликоль или их комбинацию. В одном варианте реализации увлажняющий агент может представлять собой глицерин, полиэтиленгликоль со средней молекулярной массой от примерно 200 до примерно 600, пропиленгликоль или их комбинацию.
В одном из предпочтительных вариантов реализации составы согласно настоящему изобретению содержат комбинацию увлажняющих агентов, представляющую собой комбинацию глицерина и полиэтиленгликоля со средней молекулярной массой 400 (ПЭГ 400). Без ограничения рамками какой-либо конкретной теории, полагают, что ПЭГ 400 способствует увлажнению полости носа за счет увеличения влагоудержания и обеспечения успокаивающего действия.
Кроме того, глицерин, как полагают, усиливает увлажняющее действие, притягивая воду из воздуха во внешний слой кожи и образуя защитный слой, который помогает предотвратить потерю влаги.
Количество ПЭГ 400 может составлять от примерно 0,5% масс./масс. до примерно 20% масс./масс. в расчете на общую массу состава. В одном варианте реализации ПЭГ 400 может быть включен в количестве от примерно 1% масс./масс. до примерно 5% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество ПЭГ 400 составляет примерно 2% масс./масс. в расчете на общую массу состава.
Количество глицерина может составлять от примерно 0,5% масс./масс. до примерно 8% масс./масс. в расчете на общую массу состава. В одном варианте реализации количество глицерина может составлять от примерно 1% масс./масс. до примерно 4% масс./масс. в расчете на общую массу состава. В предпочтительном варианте реализации количество глицерина составляет примерно 2,5% масс./масс. в расчете на общую массу состава.
В предпочтительном варианте реализации комбинация увлажняющих средств представляет собой комбинацию примерно 2% масс./масс. ПЭГ 400 в расчете на общую массу состава и примерно 2,5% масс./масс. глицерина в расчете на общую массу состава.
По мере увеличения содержания увлажняющих агентов в составах согласно настоящему изобретению характеристики указанных составов могут становиться отличными от FLONASE®. Было обнаружено, что включение комбинации увлажняющих агентов, в отличие от включения единственного увлажняющего агента, обеспечивает увлажняющее действие без излишнего увеличения общего количества увлажняющих агентов в составах. Кроме того, применение комбинации увлажняющих агентов обеспечивает улучшенное влагоудержание и поддержание заданных характеристик составов по сравнению с единственным увлажняющим агентом.
Полость носа является чувствительной областью, поэтому составы назального спрея предпочтительно должны иметь осмолярность, близкую к физиологическим жидкостям. Для поддержания осмолярности составов в пределах заданного диапазона в составы согласно настоящему изобретению может быть включен регулятор осмолярности. Примеры подходящих регуляторов осмолярности включают, но не ограничиваются ими, хлорид натрия, декстрозу и хлорид кальция. В предпочтительном варианте реализации регулятор осмолярности представляет собой декстрозу, наиболее предпочтительно безводную декстрозу.
Добавление увлажняющих агентов в составы согласно настоящему изобретению может влиять на осмолярность, повышая ее до такой степени, что возможно появление раздражения. Следовательно, количество декстрозы, используемой в составах согласно настоящему изобретению, должно быть подходящим для поддержания соответствующей осмолярности.
Количество декстрозы может составлять от примерно 0,03% масс./масс. до примерно 7% масс./масс. в расчете на общую массу состава. В одном варианте реализации количество декстрозы может составлять от примерно 1% масс./масс. до примерно 2,5% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество декстрозы составляет примерно 2% масс./масс. в расчете на общую массу состава.
Осмолярность составов согласно настоящему изобретению может составлять от примерно 100 мОсмолей до примерно 800 мОсмолей. В одном варианте реализации осмолярность может составлять от примерно 300 мОсмолей до примерно 600 мОсмолей. В предпочтительном варианте реализации осмолярность составляет примерно 454 мОсмолей.
Составы согласно настоящему изобретению могут иметь влагосодержание от примерно 80% масс./масс. до примерно 99% масс./масс. в расчете на общую массу состава. В одном из вариантов реализации влагосодержание может составлять от примерно 85% масс./масс. до примерно 97% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации влагосодержание может составлять от примерно 90% масс./масс. до примерно 95% масс./масс. в расчете на общую массу состава.
Для оценки способности составов удерживать воду и обеспечивать увлажняющее действие можно использовать прибор для измерения динамической сорбции паров (ДСП). Указанный прибор содержит микровесы, в лодочку которых помещают образцы в микролитровых количествах (т.е. 8-12 микролитров, например 10 мкл). Лодочка находится внутри камеры с контролируемой температурой и влажностью. В ходе воздействия на образец постоянной влажности микровесы регистрируют потерю массы с течением времени.
Исследуемые составы первоначально содержат от примерно 90% масс./масс. до примерно 95% масс./масс. воды в расчете на общую массу состава. Скорость потери воды, регистрируемая микровесами по потере массы, записывается прибором. Относительную влажность (ОВ) поддерживали равной примерно 80% в течение первоначальных 750 минут, после чего ОВ снижали до примерно 20% и поддерживали в течение последующих 750 минут. Температуру поддерживали равной примерно 23°С в течение всего испытания.
В одном варианте реализации результаты ДСП указывают на по меньшей мере примерно 3% воды, удерживаемой в течение первоначальных 750 минут. В другом варианте реализации результаты ДСП указывают на более примерно 3% воды, удерживаемой в течение первоначальных 750 минут. В другом варианте реализации результаты ДСП указывают на от примерно 3% до примерно 10% воды, удерживаемой в течение первоначальных 750 минут. В другом варианте реализации результаты ДСП указывают на от примерно 3,5% до примерно 7% воды, удерживаемой в течение первоначальных 750 минут. В предпочтительном варианте реализации влагоудержание по результатам ДСП составляет примерно 5,82% воды, удерживаемой в течение первоначальных 750 минут. Для сравнения, FLONASE® имеет влагоудержание примерно 3%, измеренное методом ДСП в течение первоначальных 750 минут.
В одном варианте реализации составы согласно настоящему изобретению удерживают по меньшей мере примерно на 50% больше воды, чем FLONASE®, в течение первоначальных 750 минут. В другом варианте реализации составы согласно настоящему изобретению удерживают по меньшей мере примерно на 75% больше воды, чем FLONASE®, в течение первоначальных 750 минут.
Относительную влажность снижали до примерно 20% для последующих 750 минут исследования методом ДСП.
В одном варианте реализации результаты ДСП указывают на от примерно 0,3% до примерно 1,5% воды, удерживаемой в течение последующих 750 минут. В другом варианте реализации результаты ДСП указывают на от примерно 0,5% до примерно 0,9% воды, удерживаемой в течение последующих 750 минут. В предпочтительном варианте реализации результаты ДСП указывают на примерно 1% воды, удерживаемой в течение последующих 750 минут. Для сравнения, FLONASE® имеет влагоудержание от примерно 0,15% до примерно 0,3%, измеренное методом ДСП в течение последующих 750 минут. В одном варианте реализации составы согласно настоящему изобретению удерживают по меньшей мере примерно на 50% больше воды, чем FLONASE®, в течение последующих 750 минут. В другом варианте реализации составы согласно настоящему изобретению удерживают по меньшей мере примерно на 75% больше воды, чем FLONASE®, в течение последующих 750 минут. В другом варианте реализации составы согласно настоящему изобретению удерживают по меньшей мере примерно на 100% больше воды, чем FLONASE®, в течение последующих 750 минут.
Предпочтительно составы согласно настоящему изобретению сохраняют рН от примерно 5 до примерно 7 в течение срока их годности. Было замечено, что добавление одного глицерина в количестве, эффективном для его применения в качестве увлажняющего агента, повышало рН до значений за пределами приемлемого диапазона. Кроме того, добавление одного ПЭГ 400 в количестве, эффективном для его применения в качестве увлажняющего агента, снижало рН до значений за пределами приемлемого диапазона. Заявители обнаружили, что включение комбинации глицерина и ПЭГ 400 обеспечивает рН в пределах приемлемого диапазона с обеспечением при этом увлажняющего действия. Некоторые варианты реализации настоящего изобретения имеют рН от примерно 5 до примерно 7 на протяжении всего срока годности.
Составы согласно настоящему изобретению предпочтительно распыляются в виде мелкодисперсного аэрозоля, и капли оседают местно. Указанные капли затем всасываются и становятся доступными местно для лечения симптомов аллергического ринита. Полагают, что доступность активного фармацевтического ингредиента в месте осаждения влияет на эффективность и безопасность составов. Доступность активного фармацевтического ингредиента является функцией РКР и РЧР активного фармацевтического ингредиента в каплях составов.
Вязкость составов может влиять на РКР. Глицерин может увеличивать вязкость составов, что приводит к потенциально большему распределению капель по размеру. Напротив, ПЭГ 400 может снижать вязкость составов, что приводит к потенциально меньшему распределению капель по размеру. Заявители обнаружили, что конкретная комбинация глицерина и ПЭГ 400 позволяет найти компромисс между вязкостью и реологическими характеристиками с обеспечением подходящего РКР. Предпочтительно РКР и РЧР составов согласно настоящему изобретению могут быть по существу аналогичны FLONASE®, и они, таким образом, обладают соответствующей безопасностью и эффективностью.
РКР составов согласно настоящему изобретению может быть следующим: примерно 10% капель имеют размер менее примерно 25 мкм, примерно 50% капель имеют размер от примерно 25 мкм до примерно 60 мкм, и примерно 90% капель имеют размер менее примерно 150 мкм. В другом варианте реализации РКР составов согласно настоящему изобретению может быть следующим: примерно 10% капель имеют размер менее примерно 20 мкм, примерно 50% капель имеют размер от примерно 30 мкм до примерно 55 мкм, и примерно 90% капель имеют размер менее примерно 130 мкм. В предпочтительном варианте реализации РКР составов согласно настоящему изобретению может быть следующим: примерно 10% капель имеют размер менее примерно 18 мкм, примерно 50% капель имеют размер от примерно 33 мкм до примерно 51 мкм, и примерно 90% капель имеют размер менее примерно 120 мкм.
В одном из вариантов реализации в нулевой момент времени (Т0) РКР является следующим: примерно 10% капель имеют размер менее примерно 18 мкм, примерно 50% капель имеют размер от примерно 33 мкм до примерно 51 мкм, и примерно 90% капель имеют размер менее примерно 120 мкм.
Т0 представляет собой момент времени, когда препарат помещают в контролируемую среду для испытания стабильности. В одном варианте реализации Т0 находится в пределах примерно 60 дней после завершения процесса изготовления. В другом варианте реализации Т0 находится в пределах примерно 7 дней после завершения процесса изготовления.
В одном из вариантов реализации после хранения в течение 1 месяца при 40°С/ОВ 75% РКР является следующим: примерно 10% капель имеют размер менее примерно 18 мкм, примерно 50% капель имеют размер от примерно 33 мкм до примерно 51 мкм, и примерно 90% капель имеют размер менее примерно 120 мкм.
В одном из вариантов реализации после хранения в течение 3 месяцев при 40°С/ОВ 75% РКР является следующим: примерно 10% капель имеют размер менее примерно 18 мкм, примерно 50% капель имеют размер от примерно 33 мкм до примерно 51 мкм, и примерно 90% капель имеют размер менее примерно 120 мкм.
В одном из вариантов реализации после хранения в течение 6 месяцев при 40°С/ОВ 75% РКР является следующим: примерно 10% капель имеют размер менее примерно 18 мкм, примерно 50% капель имеют размер от примерно 33 мкм до примерно 51 мкм, и примерно 90% капель имеют размер менее примерно 120 мкм.
В одном варианте реализации РКР составов согласно настоящему изобретению находится в пределах по меньшей мере примерно 20% от РКР FLONASE®. В одном варианте реализации РКР составов согласно настоящему изобретению находится в пределах по меньшей мере примерно 20% от РКР FLONASE® при Т0, после хранения в течение 1 месяца при 40°С/ОВ 75%, после хранения в течение 3 месяцев при 40°С/ОВ 75% и после хранения в течение 6 месяцев при 40°С/ОВ 75%.
РКР составов согласно настоящему изобретению подтверждает, что составы биоэквивалентны FLONASE®.
Составы согласно настоящему изобретению дополнительно исследовали с помощью морфологически направленной рамановской спектроскопии (Morphologically Directed Raman Spectroscopy, MDRS). MDRS представляет собой метод, позволяющий измерить РЧР частиц активного фармацевтического ингредиента. Этот метод позволяет отличить частицы активного фармацевтического ингредиента от частиц вспомогательных веществ и позволяет измерять частицы, специфичные для активного фармацевтического ингредиента.
РЧР активного фармацевтического ингредиента для составов согласно настоящему изобретению может быть следующим: от примерно 50% до примерно 75% частиц активного фармацевтического ингредиента имеют размер менее примерно 2 мкм, от примерно 85% до примерно 99% частиц активного фармацевтического ингредиента имеют размер менее примерно 3 мкм, от примерно 95% до примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 5 мкм, и от примерно 97% до примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 10 мкм.
В одном варианте реализации при Т0 РЧР активного фармацевтического ингредиента является следующим: примерно 66% частиц активного фармацевтического ингредиента имеют размер менее примерно 2 мкм, примерно 95% частиц активного фармацевтического ингредиента имеют размер менее примерно 3 мкм, примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 5 мкм, и примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 10 мкм.
РЧР для FLONASE® при Т0 является следующим: примерно 60% частиц активного фармацевтического ингредиента имеют размер менее примерно 2 мкм, примерно 95% частиц активного фармацевтического ингредиента имеют размер менее примерно 3 мкм, примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 5 мкм, и примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 10 мкм.
После хранения в течение 1 месяца при 40°С/ОВ 75% в одном варианте реализации РЧР активного фармацевтического ингредиента является следующим: примерно 61% частиц активного фармацевтического ингредиента имеют размер менее примерно 2 мкм, примерно 95% частиц активного фармацевтического ингредиента имеют размер менее примерно 3 мкм, примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 5 мкм, и примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 10 мкм.
РЧР для FLONASE® после хранения в течение 1 месяца при 40°С/ОВ 75% является следующим: примерно 49% частиц активного фармацевтического ингредиента имеют размер менее примерно 2 мкм, примерно 86% частиц активного фармацевтического ингредиента имеют размер менее примерно 3 мкм, примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 5 мкм, и примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 10 мкм.
После хранения в течение 3 месяцев при 40°С/ОВ 75% в одном варианте реализации РЧР является следующим: примерно 65% частиц активного фармацевтического ингредиента имеют размер менее примерно 2 мкм, примерно 94% частиц активного фармацевтического ингредиента имеют размер менее примерно 3 мкм, примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 5 мкм, и примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 10 мкм.
РЧР для FLONASE® после хранения в течение 3 месяцев при 40°С/ОВ 75% является следующим: примерно 50% частиц активного фармацевтического ингредиента имеют размер менее примерно 2 мкм, примерно 85% частиц активного фармацевтического ингредиента имеют размер менее примерно 3 мкм, примерно 99% частиц активного фармацевтического ингредиента имеют размер менее примерно 5 мкм, и примерно 100% частиц активного фармацевтического ингредиента имеют размер менее примерно 10 мкм.
В одном варианте реализации РЧР частиц активного фармацевтического ингредиента находится в пределах по меньшей мере примерно 20% от РЧР частиц активного фармацевтического ингредиента для FLONASE®. В одном варианте реализации РЧР частиц активного фармацевтического ингредиента находится в пределах по меньшей мере 20% от РЧР частиц активного фармацевтического ингредиента для FLONASE® при Т0, после хранения в течение 1 месяца при 40°С/ОВ 75% и после хранения в течение 3 месяцев при 40°С/ОВ 75%. РЧР частиц активного фармацевтического ингредиента составов согласно настоящему изобретению подтверждает, что составы биоэквивалентны FLONASE®.
Составы согласно настоящему изобретению дополнительно изучали на предмет скорости растворения частиц активных фармацевтических ингредиентов с применением методики растворения in vitro. РЧР может влиять на скорость растворения и всасывание, то есть более крупные частицы могут растворяться медленнее, чем более мелкие частицы, что влияет на доступность активного фармацевтического ингредиента в месте осаждения.
Испытание проводили для образцов, хранившихся при 40°С/ОВ 75%, при Т0, через 1 месяц и через 3 месяца. В одном варианте реализации профиль растворения показал, что скорость растворения активного фармацевтического ингредиента в составах согласно настоящему изобретению по существу такая же как скорость растворения активного фармацевтического ингредиента в FLONASE®.
Составы согласно настоящему изобретению могут дополнительно содержать суспендирующий агент. Примеры суспендирующих агентов включают, но не ограничиваются ими, карбоксиметилцеллюлозу, вигум, трагакант, бентонит, метилцеллюлозу и полиэтиленгликоли, или их комбинацию.
В предпочтительном варианте реализации суспендирующий агент представляет собой микрокристаллическую целлюлозу и натрийкарбоксиметилцеллюлозу, предпочтительно применяемые в виде продукта с торговым наименованием AVICEL® RC-591 или AVUCEL® CL-611. AVICEL® RC-591 обычно содержит от примерно 87% до примерно 91% микрокристаллической целлюлозы и от примерно 9% до примерно 13% натрийкарбоксиметилцеллюлозы.
Микрокристаллическая целлюлоза и натрийкарбоксиметилцеллюлоза могут присутствовать в количестве от примерно 0,5% масс./масс. до примерно 5% масс./масс. в расчете на общую массу состава. В одном варианте реализации микрокристаллическая целлюлоза и натрийкарбоксиметилцеллюлоза могут присутствовать в количестве от примерно 1% масс./масс. до примерно 3% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество микрокристаллической целлюлозы и натрийкарбоксиметилцеллюлозы составляет примерно 1,5% масс./масс. в расчете на общую массу состава.
Составы согласно настоящему изобретению могут дополнительно включать консерванты для защиты состава от загрязнения и роста микроорганизмов. Примеры консервантов включают, но не ограничиваются ими, четвертичные соединения аммония (бензалкония хлорид, бензетония хлорид, цетримид и цетилпиридиния хлорид), ртутьсодержащие агенты (например, нитрат фенилртути, ацетат фенилртути и тимеросал), спиртовые агенты (например, хлорбутанол, фенилэтиловый спирт и бензиловый спирт), сложные эфиры антибактериальных соединений (например, сложные эфиры пара-гидроксибензойной кислоты), хелатирующие агенты, такие как эдетат натрия (ЭДТА), и другие противомикробные агенты, такие как хлоргексидин, хлоркрезол, сорбиновая кислота и ее соли и полимиксин, или их комбинацию.
В одном варианте реализации консерванты представляют собой бензалкония хлорид, фенилэтиловый спирт или их комбинацию. В предпочтительном варианте реализации консерванты представляют собой комбинацию бензалкония хлорида и фенилэтилового спирта.
Количество бензалкония хлорида может составлять от примерно 0,005% масс./масс. до примерно 0,2% масс./масс. в расчете на общую массу состава. В одном варианте реализации количество бензалкония хлорида может составлять от примерно 0,01% масс./масс. до примерно 0,09% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество бензалкония хлорида составляет примерно 0,02% масс./масс. в расчете на общую массу состава.
Количество фенилэтилового спирта может составлять от примерно 0,05% масс./масс. до примерно 0,5%) масс./масс. в расчете на общую массу состава. В одном варианте реализации количество фенилэтилового спирта может составлять от примерно 0,15% масс./масс. до примерно 0,35% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество фенилэтилового спирта составляет примерно 0,25% масс./масс. в расчете на общую массу состава.
Настоящее изобретение дополнительно включает смачивающий агент. Примеры подходящих смачивающих агентов включают, но не ограничиваются ими, жирные спирты, сложные эфиры, простые эфиры или их комбинацию. В одном из предпочтительных вариантов реализации смачивающий агент представляет собой полиоксиэтилен (2) сорбитан моноолеат, предпочтительно в виде продукта с торговым наименованием POLYSORBATE® 80 (Полисорбат 80).
Количество полиоксиэтилен (2) сорбитан моноолеата может составлять от примерно 0,0005% масс./масс. до примерно 0,09% масс./масс. в расчете на общую массу состава. В одном варианте реализации количество полиоксиэтилен (2) сорбитан моноолеата может составлять от примерно 0,001% масс./масс. до примерно 0,01% масс./масс. в расчете на общую массу состава. В одном из предпочтительных вариантов реализации количество полиоксиэтилен (2) сорбитан моноолеата составляет примерно 0,005% масс./масс. в расчете на общую массу состава.
Составы согласно настоящему изобретению можно применять совместно с устройством доставки, содержащим насос, для облегчения местного введения в полость носа при помощи дозирующего распылительного насоса. Насос может быть выполнен с обеспечением возможности доставки примерно 100 мг (100 мкл) суспензии за одно срабатывание. Устройство доставки предпочтительно обеспечивает одинаковую дозу с одинаковым РКР и характером распыления за одну активацию. В предпочтительном варианте реализации для обеспечения равномерности дозирования получают гомогенную дисперсию активного ингредиента за счет применения микронизированного флутиказона пропионата, предварительно смоченного полиоксиэтилен (2) сорбитан моноолеатом, в комбинации с тиксотропным суспендирующим агентом, микрокристаллической целлюлозой и натрийкарбоксиметилцеллюлозой (AVICEL® RC591).
Составы согласно настоящему изобретению можно вводить местно в полость носа человека, нуждающегося в таком лечении, путем распыления составов согласно настоящему изобретению с помощью устройства доставки. В одном варианте реализации составы согласно настоящему изобретению можно вводить в полость носа для лечения симптомов аллергического ринита, включая, среди прочего, сезонный и круглогодичный ринит, воспалительные состояния, астму, ХОБЛ, дерматит. Примеры симптомов включают, но не ограничиваются ими, заложенность носа, чихание, слезотечение, зуд в глазах, зуд в носу, насморк или их комбинацию.
В одном варианте реализации составы согласно настоящему изобретению могут обеспечивать ощущение увлажнения, мягкого облегчения, комфорта в полости носа, или их комбинацию. Кроме того, составы согласно настоящему изобретению могут облегчать раздражение, сухость, дискомфорт в полости носа, или их комбинацию.
Примеры
Примеры 1-6 - Примеры составов назального спрея
Варианты реализации настоящего изобретения могут быть получены как указано ниже в Примерах 1-5. В одном из предпочтительных вариантов реализации состав назального спрея получен согласно Примеру 1. В Примере 6 приведен состав FLONASE®.
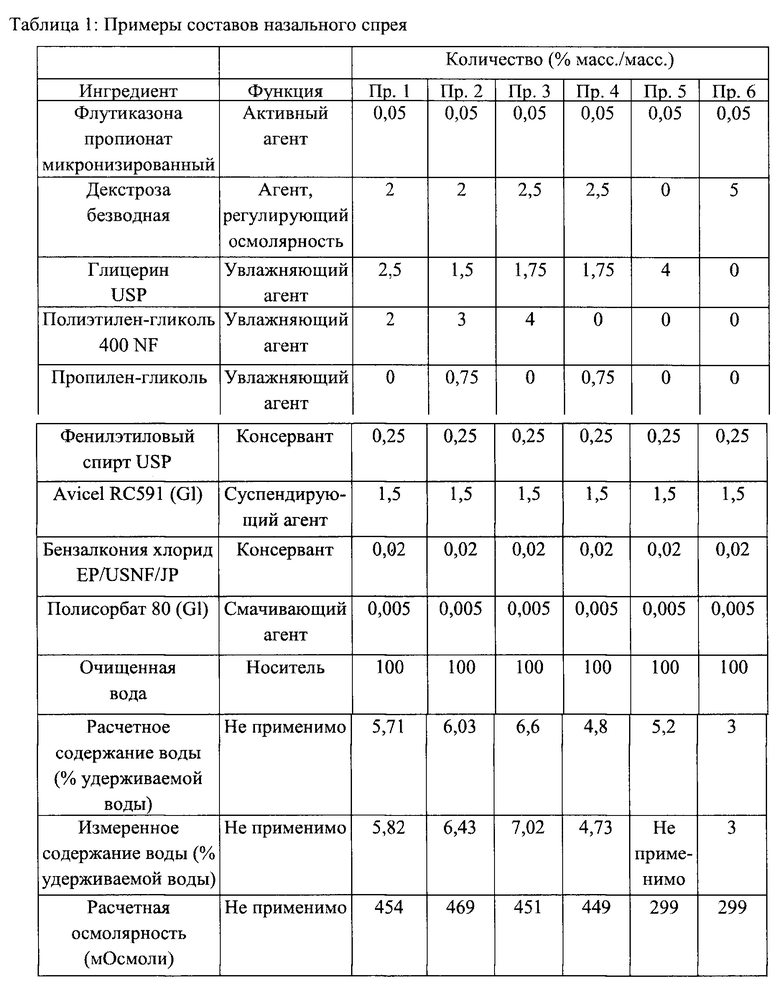
Измеренное содержание воды определяли с помощью ДСП. Расчетное содержание воды рассчитывали с помощью программного обеспечения Design-Expert® Software с получением прогностической статистической модели. Измеренные значения влагосодержания наносили на график помощью программного обеспечения Design-Expert® Software с получением проектного поля с известными концентрациями глицерина, ПЭГ 400 и декстрозы в качестве входных данных и измеренными значениями влагосодержания в качестве выходных данных.
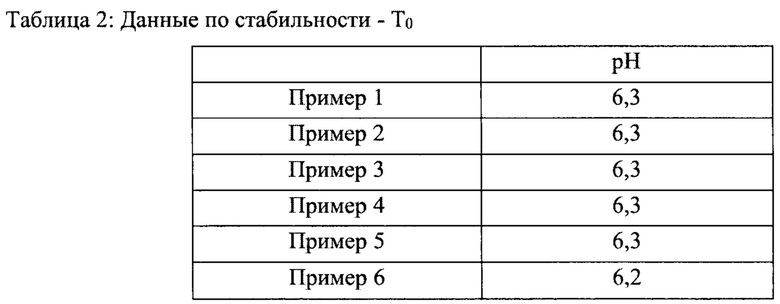
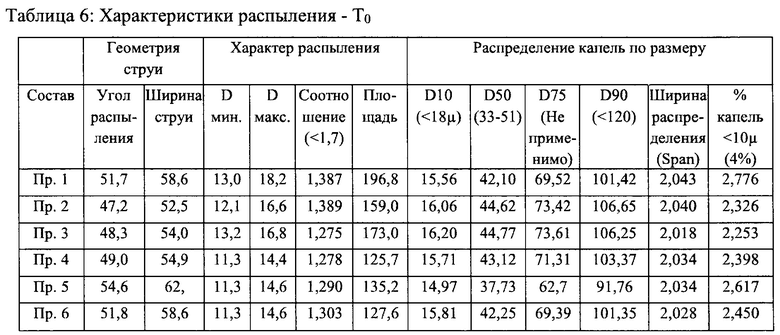
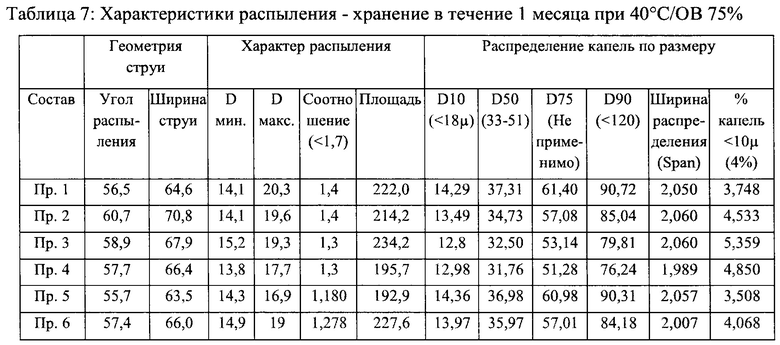
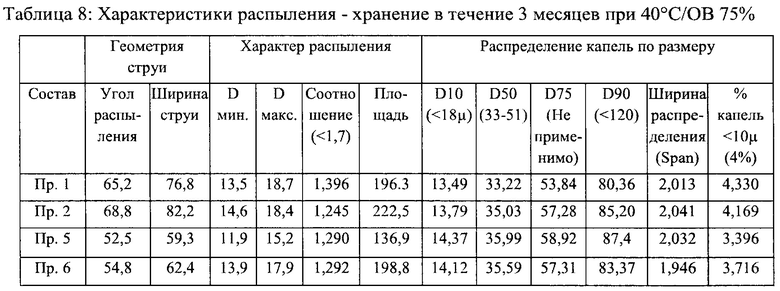
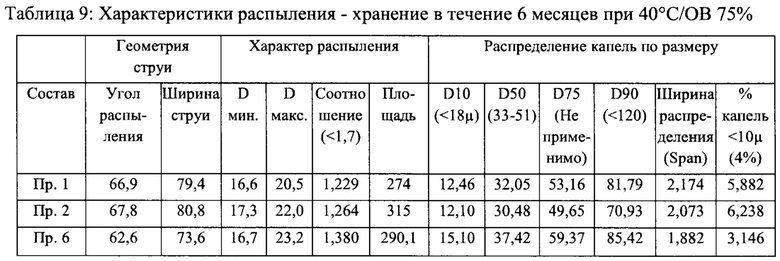
Пример 7 - Стабильность и характеристики распыления составов назального спрея
Стабильность и характеристики распыления некоторых вариантов реализации настоящего изобретения исследовали при То, после хранения в течение 3 месяцев при 40°С/ОВ 75% и после хранения в течение 6 месяцев при 40°С/ОВ 75%.
Результаты испытаний на стабильность приведены в таблицах 2-5 ниже. В результате указанного исследования в целом был сделан вывод о том, что составы согласно настоящему изобретению соответствовали заявленным значениям стабильности для всех моментов времени и условий хранения.
Результаты исследования характеристик распыления приведены в таблицах 6-9 ниже. В результате исследования характеристик распыления в целом был сделан вывод о том, что составы согласно настоящему изобретению соответствовали заявленным значениям характеристик распыления для всех моментов времени и условий хранения.



Пример 8 - Исследование сенсорных ощущений у потребителей
Предметом исследования сенсорных ощущений у потребителей был предпочтительный вариант реализации настоящего изобретения, подробно описанный в Примере 1. Методики проведения и результаты исследования сенсорных ощущений у потребителей представлены ниже.
В исследовании принимал участие 301 человек, пользующийся назальным спреем от аллергии в настоящее время или готовый пользоваться им в будущем. Респонденты тестировали состав назального спрея у себя дома, один раз в день в течение пяти дней подряд. Респонденты проходили на компьютере количественный онлайн-опрос каждый день после использования назального спрея, отвечая на вопросы о своем восприятии сенсорных свойств состава. Респонденты отвечали на вопросы каждый день сразу после использования спрея в одно и то же время (+/- 30 минут).
Свойства или вопросы, касающиеся состава назального спрея, которые представляли интерес для исследования, включали следующие: 1) назальный спрей дает мне ощущение увлажнения в носу; 2) назальный спрей дает мне мягкое ощущение облегчения в носу; и 3) назальный спрей дает мне ощущение комфорта в носу.
Дозировка составляла один впрыск в каждую ноздрю в день. Исследуемые образцы хранили при температуре окружающей среды и поставляли в простой небрендированной упаковке, имеющейся в продаже. Все респонденты страдали от аллергии и сухости в носу. Среди респондентов было 54% женщин и 46% мужчин. Распределение респондентов по возрасту было следующим: 29% были в возрасте 18-34 лет, 24% были в возрасте 35-44 лет, 25% были в возрасте 45-54 лет, и 23% были в возрасте 55-65 лет.36% респондентов пользовались назальными спреями, а 64% принимали таблетки. Все респонденты были в целом здоровы.
Статистический анализ полученных данных основан на биноминальном распределении, которое описывает вероятность получения некоторого числа удачных исходов (ЧУИ) в некотором количестве испытаний (КИ), из чего можно вычислить р (ЧУИ/КИ).
Нулевая гипотеза исследования представляла собой Н0: р ≤ 80%, что указывает на то, что процент респондентов, чувствующих данное ощущение, не будет превышать значение, заданное нулевой гипотезой (в данном случае 80%). Если полученное значение р значительно выше, чем значение, заданное нулевой гипотезой, то нулевая гипотеза отвергается, и можно сделать вывод, что наблюдаемое ощущение не обусловлено случайностью и что р>80%.
Биноминальное распределение предполагает, что каждое отдельное испытание может быть независимо описано распределением Бернулли с вероятностью удачного исхода р. В контексте данного исследования это выражается в предположении, что респонденты не влияли друг на друга в ходе исследования, и что для каждого отдельного респондента вероятность того, что он/она почувствует ощущение, составляет р. Целью исследования является определение значения р и определение того, является ли полученное значение значимым или может быть отнесено к случайным.
Результаты:
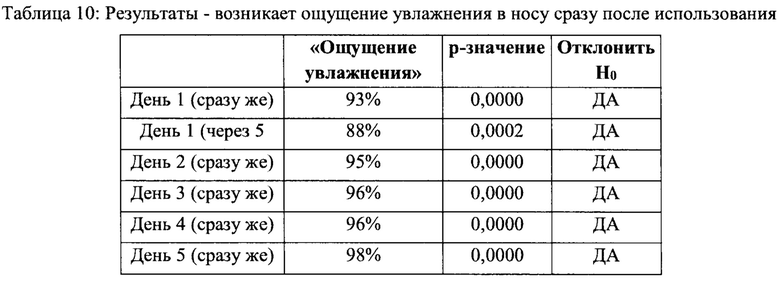
Раздел 1 - «Назальный спрей дает мне ощущение увлажнения в носу» - сразу возникающее ощущение
Нулевая гипотеза (Но) может быть отвергнута, и может быть принята альтернативная гипотеза (На). Более 80% респондентов согласны с утверждением «этот назальный спрей дает мне ощущение увлажнения в носу» сразу после использования во все дни и через 5 минут после использования в день 1 (см. Таблицу 10).
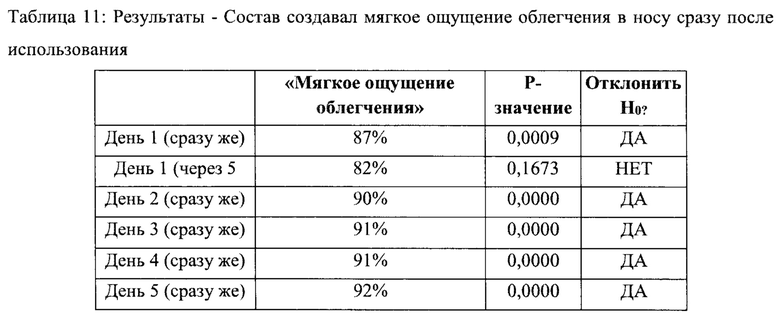
Раздел 2 - «Состав назального спрея дает мне мягкое ощущение облегчения в носу» - сразу возникающее ощущение
Нулевая гипотеза (Но) может быть отвергнута, и может быть принята альтернативная гипотеза (На). Более 80% респондентов согласны с утверждением «этот назальный спрей дает мне мягкое ощущение облегчения в носу» сразу после использования во все дни, за исключением 5 минут после использования в день 1 (см. Таблицу 11).
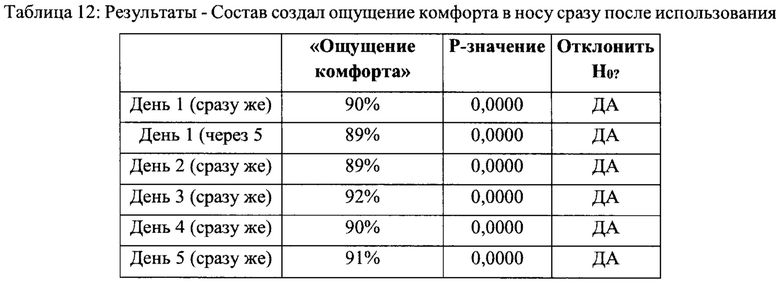
Раздел 3 - «Назальный спрей дает мне ощущение комфорта в носу» - сразу возникающее ощущение
Нулевая гипотеза (Н0) может быть отвергнута, и может быть принята альтернативная гипотеза (HA). Более 80% респондентов согласны с утверждением «этот назальный спрей дает мне ощущение комфорта в носу» сразу после использования во все дни и через 5 минут после использования в день 1 (см. Таблицу 12).
Вывод:
На основании вышеприведенного исследования можно сделать обоснованный вывод, что респонденты в целом полагали, что состав назального спрея согласно настоящему изобретению дает им ощущение увлажнения в носу сразу после использования, состав назального спрея дает им мягкое ощущение облегчения в носу сразу после использования, и что назальный спрей дает им ощущение комфорта в носу сразу после использования.
Пример 9 - Исследование растворимости и анализ подобия
Исследование проводили в соответствии с валидированным аналитическим методом. При оценке коэффициента подобия между партиями также применяли валидированный аналитический метод.
Средний профиль для каждой исследованной партии приведен в Таблице 13 и представляет собой результат усреднения 12 повторов для каждой партии.
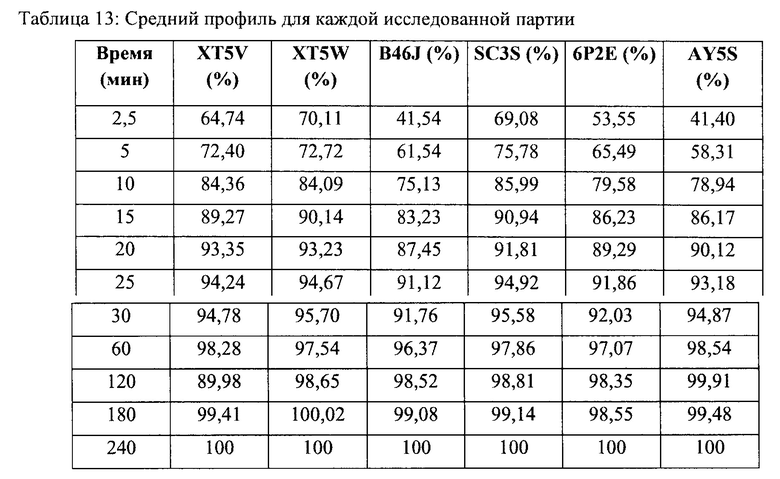
После 85% растворимости обоих продуктов учитывали только один результат измерения, поэтому в анализ были включены 4 временные точки (2,5 мин, 5 мин, 10 мин и 15 мин), поскольку кумулятивное высвобождение лекарства через 10 минут оказалось ниже 85% для 5 из 6 средних профилей растворимости. Профили растворимости сравнивали с использованием следующего уравнения, которое определяет коэффициент подобия (f2):
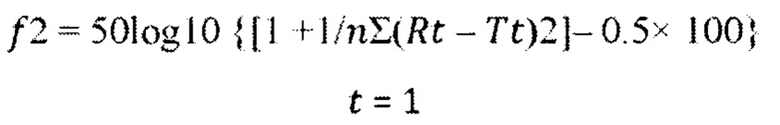
В приведенном выше уравнении Rt и Tt представляют собой процент растворенного вещества в каждой временной точке. Значение f2 от 50 до 100 указывает на то, что два данных профиля растворимости подобны. Значения 12 были получены в результате сравнения каждого референтного продукта (РП), который представляет собой FLONASE®, с исследуемым продуктом (ИП), который представляет собой предпочтительный вариант реализации настоящего изобретения, подробно описанный в Примере 1. Эти данные представлены ниже в таблице 14.
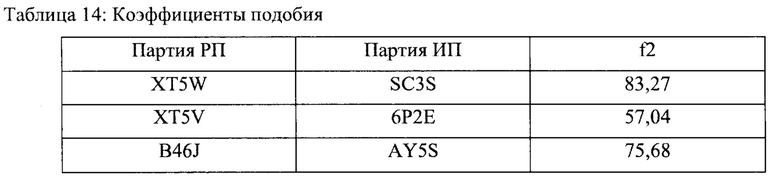
Все коэффициенты подобия, приведенные в таблице 14, свидетельствуют о том, что профили растворимости исследуемых РП и ИП сопоставимы и эквивалентны.
Пример 10 - Данные MDRS и результаты исследования
Приведенные ниже данные и результаты были получены с помощью системы Morphologi М4-ID с использованием технологии морфологически направленной рамановской спектрометрии (Morphologically Directed Raman Spectrometry, MDRS). Кроме того, ниже приведены результаты статистического анализа, необходимые для оценки того, является ли предпочтительный вариант реализации настоящего изобретения, приведенный в Примере 1, эквивалентным FLONASE® с точки зрения размера частиц активного фармацевтического ингредиента. Исследование проводили в соответствии с валидированным аналитическим методом.
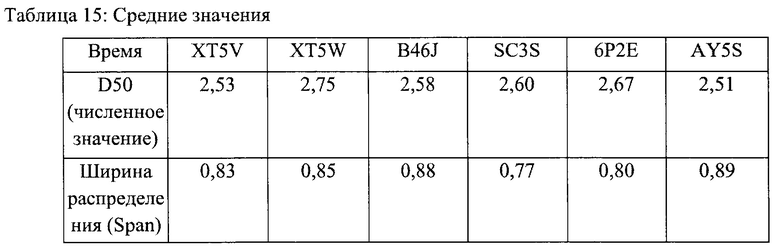
В таблице 15 приведены средние значения D50 и ширины распределения span (в количественном распределении) для каждой исследованной партии как среднее из 25 повторов для каждой партии.
Span рассчитывали из процентилей РЧР по следующей формуле:
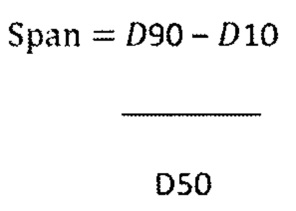 .
.
И span, и D50 являются переменными, предложенными в Проекте руководства по флутиказона пропионату (Draft Guidance on Fluticasone Propionate) в качестве основы для анализа популяционной биоэквивалентности (ПБЭ) в рамках альтернативного подхода к сравнительному исследованию биоэквивалентности (БЭ) по клиническим конечным точкам.
Статистический анализ популяционной биоэквивалентности проводили в соответствии с Проектом руководства по будесониду (Draft Guidance on Budesonide), опубликованным Управлением по контролю за продуктами и лекарствами США (FDA). Критерий популяционной биоэквивалентности определяется как:
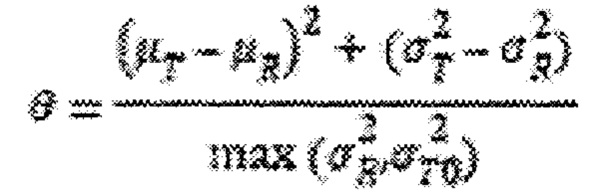 ,
,
где μT и μR - средние значения для исследуемого и контрольного продукта в логарифмическом выражении, 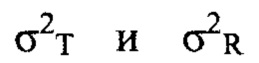 - дисперсии для исследуемого и контрольного состава в логарифмическом выражении, и
- дисперсии для исследуемого и контрольного состава в логарифмическом выражении, и  представляет собой нормативную константу со значением 0,01. Популяционная биоэквивалентность определяется как:
представляет собой нормативную константу со значением 0,01. Популяционная биоэквивалентность определяется как:

Вышеприведенное описывает среднее значение для исследуемого продукта, равное 90% или 100% от среднего значения для референтного продукта, и дисперсию для исследуемого продукта, которая в два раза больше дисперсии для референтного продукта. Опубликованный Проект руководства по будесониду включает метод расчета доверительных интервалов ПБЭ с использованием следующей линеаризованной формы критерия:
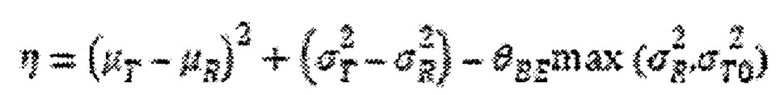 .
.
В вышеприведенной формуле популяционную биоэквивалентность определяют при верхнем 95% доверительном интервале меньше 0.
В Таблицах 16 и 17 представлены результаты по популяционной биоэквивалентности, как предложено в Проекте руководства по будесониду. Среднее геометрическое и общая дисперсия для исследуемого и референтного образцов приведены в Таблице 16.
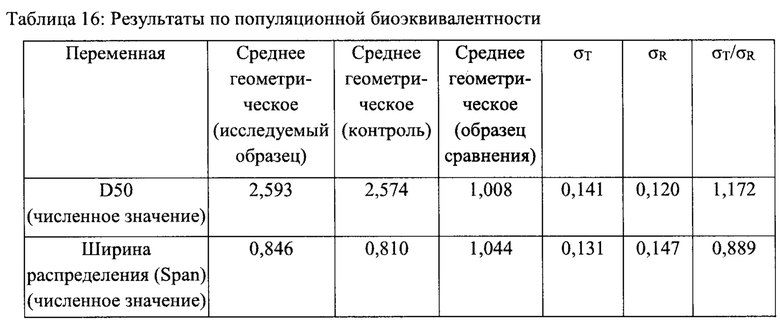
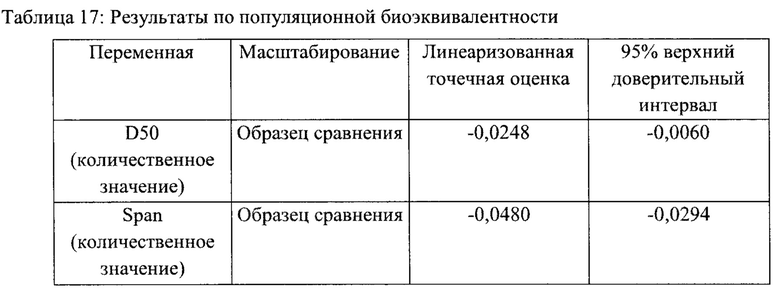
Линеаризованная точечная оценка и 95% верхний доверительный интервал, полученные путем ПБЭ сравнения результатов РЧР для исследуемого и контрольного образцов, приведены в Таблице 17. Все верхние 95% доверительные интервалы меньше 0, поэтому можно сделать вывод о наличии популяционной биоэквивалентности для всех показателей.
Вывод:
Таким образом, сравнительное исследование распределения частиц активного фармацевтического ингредиента по размеру и анализ данных по популяционной биоэквивалентности, проведенный в этом исследовании, показывают, что наличие популяционной биоэквивалентности может быть признано для всех оцениваемых показателей (span и D50) при сравнении исследуемого состава (предпочтительный вариант реализации настоящего изобретения) и контрольного состава (FLONASE®).
Аспекты настоящего изобретения относятся к составу назального спрея, содержащему по меньшей мере один активный фармацевтический ингредиент, глицерин, полиэтиленгликоль и декстрозу. Составы, согласно настоящему изобретению, обеспечивают увлажняющее действие и удерживают по меньшей мере примерно 3% воды под действием относительной влажности примерно 80% при температуре примерно 23°С в течение примерно 750 минут в исследовании методом динамической сорбции паров (ДСП). 2 н. и 6 з.п. ф-лы, 17 табл., 10 пр.
1. Состав назального спрея, содержащий:
флутиказона пропионат или флутиказона фуроат в количестве от примерно 0,005% масс./масс. до примерно 0,2% масс./масс.;
глицерин в количестве от примерно 0,5% масс./масс. до примерно 8% масс./масс.;
полиэтиленгликоль, имеющий среднюю молекулярную массу примерно 400, присутствующий в количестве от примерно 0,5% масс./масс. до примерно 20% масс./масс.;
декстрозу, присутствующую в количестве от примерно 0,3% масс./масс. до примерно 7% масс./масс.;
полиоксиэтилен (2) сорбитан моноолеат, присутствующий в количестве от примерно 0,0005% масс./масс. до примерно 0,09% масс./масс.;
микрокристаллическую целлюлозу и натрий карбоксиметилцеллюлозу, в количестве от примерно 0, 5% масс./масс. до примерно 5% масс./масс.; и
воду, в количестве от примерно 80% масс./масс. до примерно 99% масс./масс.,
отличающийся тем, что указанный состав имеет рН от примерно 5 до примерно 7 и имеет осмолярность от примерно 100 мОсмолей до примерно 800 мОсмолей.
2. Состав назального спрея по п. 1, отличающийся тем, что активный фармацевтический ингредиент представляет собой флутиказона пропионат.
3. Состав назального спрея по п. 1, отличающийся тем, что указанный состав является изотоническим.
4. Состав назального спрея по п. 3, отличающийся тем, что указанный состав содержит агент, регулирующий изотоничность, представляющий собой хлорид натрия, хлорид калия или их комбинацию.
5. Состав назального спрея по п. 4, отличающийся тем, что указанный агент, регулирующий изотоничность, представляет собой хлорид натрия.
6. Состав назального спрея по п. 1, отличающийся тем, что указанный состав имеет распределение по капель размеру, при котором в нулевой момент времени Т0 примерно 10% капель имеют размер менее примерно 18 мкм, примерно 50% капель имеют размер от примерно 33 мкм до примерно 51 мкм, и примерно 90% капель имеют размер менее примерно 120 мкм.
7. Состав назального спрея по п. 1, отличающийся тем, что примерно 100% частиц активного фармацевтического ингредиента в нулевой момент времени Т0 имеют размер менее примерно 10 мкм.
8. Способ лечения симптомов аллергического ринита, включающий введение состава назального спрея по п. 1.
| US 2004208830 A1, 2004.10.21 | |||
| WO 9746243 A1, 1997.12.11 | |||
| WO 2017139765 A1, 2017.08.17 | |||
| US 4782047 A, 1988.11.01 | |||
| Jingshun Sun, The Use of Dynamic Vapor Sorption Method in the Determination of Water Sorption Limit and Setting Specification for Drug Substance, 2011, American Pharmaceutical Review | |||
| Флутиказон (Fluticasone) инструкция по применению, |

