В настоящей заявке испрашивается преимущество и приоритет китайской патентной заявки №202010042865.Х, поданной в Национальное управление интеллектуальной собственности Китая 15 января 2020 г., которая полностью включена в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
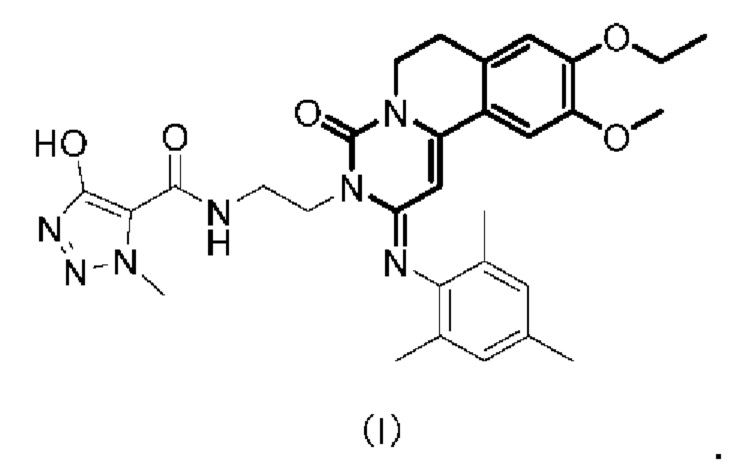
Настоящая заявка относится к технической области фармацевтики, а именно к фармацевтической композиции трициклического соединения двойного ингибитора ФДЭ3/ФДЭ4, способу его получения и его применения, и, более конкретно, к фармацевтической композиции соединения Формулы (I) или его фармацевтически приемлемой соли, способу ее получения и ее применения.
УРОВЕНЬ ТЕХНИКИ
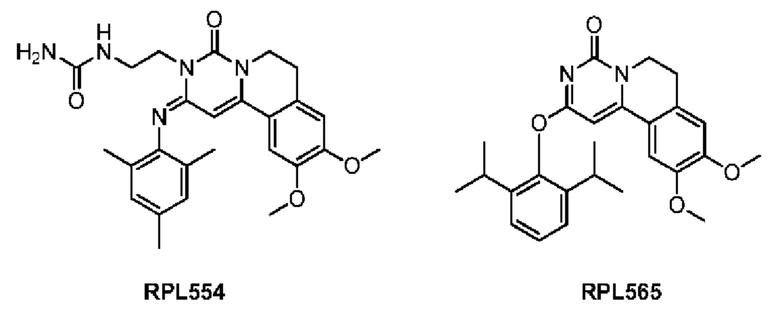
Фосфодиэстеразы (ФДЭ) представляют собой надсемейство ферментных систем, включающее 11 членов, которые участвуют в различных сигнальных путях и регулируют различные физиологические процессы. Среди них ФДЭ3 представляет собой основную фосфодиэстеразу в гладкой мускулатуре дыхательных путей человека (ГМДП), а ингибирование ФДЭ3 увеличивает внутриклеточную концентрацию цАМФ и, таким образом, ослабляет гладкую мускулатуру бронхов. ФДЭ4 играет главную регулирующую роль в экспрессии провоспалительных и противовоспалительных медиаторов, а ингибитор ФДЭ4 может ингибировать высвобождение вредных медиаторов из воспалительных клеток. Таким образом, теоретически ингибитор, который ингибирует как ФДЭ3, так и ФДЭ4, будет иметь как бронхолитическое действие агониста бета-адренорецепторов, так и противовоспалительное действие ингаляционного глюкокортикоида. Функциональное дополнение двойного нацеливания теоретически является более эффективным, чем однонаправленное, обеспечивая терапевтический эффект, который может быть достигнут только комбинацией, в настоящее время достигается за счет монотерапии и, таким образом, устраняет недостаток, заключающийся в том, что физико-химические свойства ингредиентов медикаментов, используемых в комбинации, не могут быть полностью согласованы. Таким образом, введение упрощается и является удобным для режима фиксированной дозы.
Victoria Boswell et al., J. Pharmaco. Experi. Therap., 2006, 318:840-848 и WO 200005830 сообщают, что соединения RPL554 и RPL565 обладают длительно действующим бронхолитическим и противовоспалительным действием, а также плохой растворимостью, высоким плазменным клиренсом и другими физико-химическими свойствами и подходят для ингаляционного введения. Но данные также показали, что ингибирующая активность ФДЭ4 неудовлетворительна, а противовоспалительный эффект недостаточен. Таким образом, все еще существует потребность в разработке соединения, обладающего хорошей ингибирующей активностью в отношении ФДЭ3/4.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение Формулы (I) или его фармацевтически приемлемую соль и поверхностно-активное вещество.
В некоторых вариантах реализации изобретения фармацевтическая композиция дополнительно содержит хелатирующий металл агент (или агент, образующий комплекс с металлом).
В некоторых вариантах реализации изобретения фармацевтическая композиция дополнительно содержит буферный агент.
В некоторых вариантах реализации изобретения фармацевтическая композиция дополнительно содержит разбавитель.
В некоторых вариантах реализации изобретения фармацевтическая композиция дополнительно содержит регулятор осмотического давления.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I) или его фармацевтически приемлемую соль, поверхностно-активное вещество и буферный агент.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I) или его фармацевтически приемлемую соль, поверхностно-активное вещество, буферный агент и регулятор осмотического давления.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I) или его фармацевтически приемлемую соль, поверхностно-активное вещество, буферный агент и хелатирующий металл агент.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I) или его фармацевтически приемлемую соль, поверхностно-активное вещество, буферный агент, регулятор осмотического давления и хелатирующий металл агент.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I) или его фармацевтически приемлемую соль, поверхностно-активное вещество, буферный агент, регулятор осмотического давления, хелатирующий металл агент и разбавитель.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I) или его фармацевтически приемлемую соль, поверхностно-активное вещество, по меньшей мере один из буферного агента, регулятора осмотического давления, хелатирующего металл агента и разбавителя.
В некоторых вариантах реализации изобретения фармацевтически приемлемая соль выбрана из группы, состоящей из малеата, сульфата, метансульфоната или n-толуолсульфоната.
В некоторых вариантах реализации изобретения в фармацевтически приемлемой соли соединения Формулы (I) молярное отношение соединения Формулы (I) к ионному радикалу кислоты, образующему фармацевтически приемлемую соль, может составлять от 1:1 до 1:2, например, 1:1 или 1:2.
В некоторых вариантах реализации изобретения в фармацевтической композиции «соединение Формулы (I) или его фармацевтически приемлемая соль» может использоваться взаимозаменяемо с «соединением Формулы (I)».
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), поверхностно-активное вещество, буферный агент, регулятор осмотического давления, хелатирующий металл агент и разбавитель.
В некоторых вариантах реализации изобретения в фармацевтической композиции соединение Формулы (I) или его фармацевтически приемлемая соль на основе соединения Формулы (I) имеет концентрацию от около 0,001 мг/мл до около 80 мг/мл, предпочтительно от 0,002 мг/мл до 50 мг/мл, и более предпочтительно от 0,1 мг/мл до 20 мг/мл.
Поверхностно-активное вещество по настоящей заявке представляет собой фармацевтически приемлемое поверхностно-активное вещество, например, смачивающий агент. Поверхностно-активное вещество может представлять собой неионогенное поверхностно-активное вещество, анионное поверхностно-активное вещество, катионное поверхностно-активное вещество или цвиттер-ионное поверхностно-активное вещество. Предпочтительно одно или более поверхностно-активных веществ представляют собой неионогенные поверхностно-активные вещества.
В некоторых вариантах реализации изобретения поверхностно-активное вещество представляет собой одно или более, выбранных из группы, состоящей из полиоксиэтиленгликоля, алкилового эфира полипропиленгликоля, алкилполиглюкозида, октилфенолполиоксиэтиленового эфира, алкилфенолполиоксиэтиленового эфира, глицериналкилового эфира, полиоксиэтиленсорбитанового эфира жирной кислоты (полисорбата), сорбитаналкилового сложного эфира, эфира сорбитана и жирной кислоты, кокамида МЭА, кокамида ДЭА, оксида додецилдиметиламина, блок-сополимера полиэтиленгликоля и полипропиленгликоля (полоксамера) и полиэтоксилированного таллового амина (ПОЭА).
Предпочтительно поверхностно-активное вещество представляет собой одно или более, выбранных из группы, состоящей из сложного эфира полиоксиэтиленсорбитана и жирной кислоты (например, Tween) и сложного эфира сорбитана и жирной кислоты (например, Span).
В некоторых вариантах реализации изобретения сложный эфир полиоксиэтиленсорбитана и жирной кислоты представляет собой один или более, выбранных из группы, состоящей из полисорбата 20 (лаурата полиоксиэтиленсорбитана, Tween 20), полисорбата 40 (монопальмитата полиоксиэтиленсорбитана), полисорбата 60 (стеарата полиоксиэтиленсорбитана) и полисорбата 80 (моноолеата полиоксиэтиленсорбитана; Tween 80).
В некоторых вариантах реализации изобретения сложный эфир сорбитана и жирной кислоты представляет собой один или более, выбранных из группы, состоящей из монолаурата сорбитана (Span 20), монопальмитата сорбитана, моностеарата сорбитана, тристеарата сорбитана и моноолеата сорбитана.
Более предпочтительно поверхностно-активное вещество представляет собой одно или более, выбранных из группы, состоящей из Tween и Span.
В некоторых конкретных вариантах реализации изобретения поверхностно-активное вещество представляет собой одно или более, выбранных из группы, состоящей из Tween 20, Tween 80 и Span 20.
В некоторых вариантах реализации изобретения поверхностно-активное вещество имеет концентрацию от около 0,01 мг/мл до около 8 мг/мл. Более типично концентрация поверхностно-активного вещества в фармацевтической композиции составляет от около 0,01 мг/мл до 5 мг/мл, предпочтительно от около 0,02 мг/мл до 3 мг/мл, более предпочтительно от около 0,05 мг/мл до 2 мг/мл, и еще более предпочтительно от около 0,1 мг/мл до 1 мг/мл.
В некоторых вариантах реализации изобретения в фармацевтической композиции массовое отношение соединения Формулы (I) или его фармацевтически приемлемой соли (на основе соединения Формулы (I)) к поверхностно-активному веществу составляет от около 1:200 до 100:1, предпочтительно от около 1:150 до 50:1, более предпочтительно от около 1:50 до 25:1, и еще более предпочтительно от около 1:1 до 15:1, например, около 10:1.
В некоторых вариантах реализации изобретения буферный агент представляет собой фармацевтически приемлемый буферный агент. Буферный агент может представлять собой любой буфер, пригодный для использования в жидкой фармацевтической композиции, подходящей для ингаляции. Буферный агент, как правило, представляет собой один или более, выбранных из группы, состоящей из серной кислоты, соляной кислоты, гидроксида натрия, лимонной кислоты, цитрата натрия, молочной кислоты, лактата натрия, уксусной кислоты, ацетата натрия, тринатрийфосфата, дигидрофосфата натрия, гидрофосфата динатрия, дигидрофосфата калия, винной кислоты, тартрата натрия, глицина, борной кислоты и фталевой кислоты. Предпочтительное количество буферных агентов составляет 2 или более 2, и предпочтительным типом буферного агента является цитратно-солевой буфер или фосфатно-солевой буфер, более предпочтительно натриевая соль лимонной кислоты или фосфорной кислоты. Цитратно-солевой буфер содержит лимонную кислоту, цитрат натрия и их смесь. Фосфатно-солевой буфер содержит фосфорную кислоту, монофосфат натрия (т.е. дигидрофосфат натрия), гидрофосфат динатрия и их смесь.
В некоторых вариантах реализации изобретения буферный агент выбран из группы, состоящей из лимонной кислоты, цитрата (например, цитрата натрия), винной кислоты, тартрата (например, тартрата натрия), фосфорной кислоты и фосфата (например, дигидрофосфата натрия и гидрофосфата динатрия).
В некоторых вариантах реализации изобретения буферный агент выбран из группы, состоящей из цитрата (например, цитрата натрия), тартрата (например, тартрата натрия) и фосфата (например, дигидрофосфата натрия и гидрофосфата динатрия).
В некоторых вариантах реализации изобретения буферный агент имеет концентрацию от около 0,01 мг/мл до около 50 мг/мл, предпочтительно от около 0,05 мг/мл до около 40 мг/мл, более предпочтительно от 0,1 мг/мл до около 25 мг/мл, и еще более предпочтительно от 0,5 мг/мл до около 6 мг/мл.
В некоторых вариантах реализации изобретения буферный агент используется для контроля рН фармацевтической композиции на уровне от около 3,0 до около 8,5, предпочтительно от около 5 до около 7.
В некоторых вариантах реализации изобретения регулятор осмотического давления, как правило, представляет собой один или более, выбранных из группы, состоящей из простых нетоксичных солей, таких как хлорид натрия, хлорид калия и т.п., или сахаридов, таких как один или более из глюкозы, маннита или ксилита и т.п. В некоторых вариантах реализации изобретения регулятор осмотического давления представляет собой хлорид натрия.
Концентрация регулятора осмотического давления зависит от количества, необходимого для достижения целевой изотоничности, например, изотоничности по отношению к плазме или легочной жидкости. Концентрация регулятора осмотического давления, как правило, составляет от около 0,01 мг/мл до около 10 мг/мл, и более типично от около 5 мг/мл до 9 мг/мл.
В некоторых вариантах реализации изобретения хелатирующий металл агент представляет собой один или более, выбранных из группы, состоящей из эдетовой кислоты и эдетата, такого как эдетат динатрия, эдетат динатрия-кальция и т.п. Предпочтительными являются эдетаты (например, соли кальция, соли натрия), и особенно предпочтительным является эдетат динатрия (ЭДТК-2Na).
Концентрация хелатирующего металл агента зависит от количества ионов металла, которое может быть введено во время получения фармацевтической композиции, и, как правило, составляет от около 0,01 мг/мл до около 40 мг/мл, предпочтительно от около 0,01 мг/мл до около 20 мг/мл, более предпочтительно от около 0,01 мг/мл до около 5 мг/мл, и еще более предпочтительно от около 0,01 мг/мл до около 2 мг/мл.
В некоторых вариантах реализации изобретения в фармацевтической композиции разбавитель может представлять собой любой фармацевтически приемлемый разбавитель. Разбавитель является пригодным для ингаляционного введения. Как правило, разбавитель представляет собой один или более, выбранных из группы, состоящей из воды, этанола и глицерина. Предпочтительным разбавителем является вода, а более предпочтительным разбавителем является стерильная вода.
В некоторых вариантах реализации изобретения в фармацевтической композиции количество используемого разбавителя может представлять собой подходящее количество, так что концентрация соединения Формулы (I) или его фармацевтически приемлемой соли или вспомогательного вещества в фармацевтической композиции находится в пределах определенного диапазона.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I) и Tween или Span; кроме того, Tween или Span представляет собой один или более, выбранных из группы, состоящей из Tween 20, Tween 80 и Span 20.
В некоторых вариантах реализации изобретения фармацевтическая композиция дополнительно содержит фосфат; кроме того, фосфат может быть выбран из группы, состоящей из дигидрофосфата натрия или его моногидрата и гидрофосфата динатрия; в некоторых вариантах реализации изобретения фармацевтическая композиция дополнительно содержит цитрат натрия или тартрат натрия.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), смесь Span и Tween (такую как Tween 80, Tween 20 или Span 20) и воду.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), смесь Span и Tween (такую как Tween 80, Tween 20 или Span 20), цитрат натрия или тартрат натрия и воду.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), Tween (такой как Tween 80 или Tween 20), дигидрофосфат натрия или его моногидрат, гидрофосфат динатрия, эдетат динатрия и воду.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), Tween (такой как Tween 80 или Tween 20), дигидрофосфат натрия или его моногидрат, гидрофосфат динатрия, хлорид натрия, эдетат динатрия и воду.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), Tween (такую как Tween 80 или Tween 20), цитрат натрия или тартрат натрия, хлорид натрия и воду.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), смесь Span и Tween (такую как Tween 80, Tween 20 или Span 20), цитрат натрия или тартрат натрия, хлорид натрия и воду.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), поверхностно-активное вещество, буферный агент, регулятор осмотического давления, хелатирующий металл агент и разбавитель, причем соединение Формулы (I) имеет концентрацию от 0,002 мг/мл до 50 мг/мл, поверхностно-активное вещество имеет концентрацию от 0,02 мг/мл до 3 мг/мл, буферный агент имеет концентрацию от 0,1 мг/мл до около 25 мг/мл, регулятор осмотического давления имеет концентрацию от 5 мг/мл до 9 мг/мл, а хелатирующий металл агент имеет концентрацию от 0,01 мг/мл до около 5 мг/мл.
В некоторых вариантах реализации изобретения в фармацевтической композиции поверхностно-активное вещество, буферный агент, регулятор осмотического давления, хелатирующий металл агент и разбавитель являются такими, как определено выше.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), поверхностно-активное вещество, буферный агент, регулятор осмотического давления, хелатирующий металл агент и разбавитель, причем соединение Формулы (I) имеет концентрацию от 0,002 мг/мл до 50 мг/мл, поверхностно-активное вещество имеет концентрацию от 0,02 мг/мл до 3 мг/мл, буферный агент имеет концентрацию от 0,1 мг/мл до около 25 мг/мл, регулятор осмотического давления имеет концентрацию от 5 мг/мл до 9 мг/мл, и хелатирующий металл агент имеет концентрацию от 0,01 мг/мл до около 5 мг/мл; поверхностно-активное вещество представляет собой одно или более из Tween 20, Tween 80 и Span 20, буферный агент представляет собой один или более из дигидрофосфата натрия или его моногидрата, гидрофосфата динатрия, винной кислоты и лимонной кислоты, регулятор осмотического давления представляет собой хлорид натрия, хелатирующий металл агент представляет собой один или более из эдетата динатрия и эдетата динатрия-кальция, и разбавитель представляет собой воду.
В некоторых вариантах реализации изобретения фармацевтическая композиция содержит соединение Формулы (I), Tween 80, дигидрофосфат натрия или его моногидрат, гидрофосфат динатрия, хлорид натрия, эдетат динатрия и воду, причем соединение Формулы (I) имеет концентрацию от 0,002 мг/мл до 50 мг/мл, Tween 80 имеет концентрацию от 0,02 мг/мл до 3 мг/мл, дигидрофосфат натрия и гидрофосфат динатрия имеют концентрацию от 0,1 мг/мл до 25 мг/мл, хлорид натрия имеет концентрацию от 5 мг/мл до 9 мг/мл, и эдетат динатрия имеет концентрацию от 0,01 мг/мл до около 5 мг/мл.
В некоторых вариантах реализации изобретения в фармацевтической композиции соединения Формулы (I) или его фармацевтически приемлемой соли соединение Формулы (I) находится в твердой форме; в некоторых вариантах реализации изобретения соединение Формулы (I) представляет собой кристаллическую форму соединения Формулы (I).
В некоторых вариантах реализации изобретения в фармацевтической композиции соединения Формулы (I) или его фармацевтически приемлемой соли соединение Формулы (I) представляет собой продукт, полученный путем контроля размера частиц кристаллической формы соединения Формулы (I).
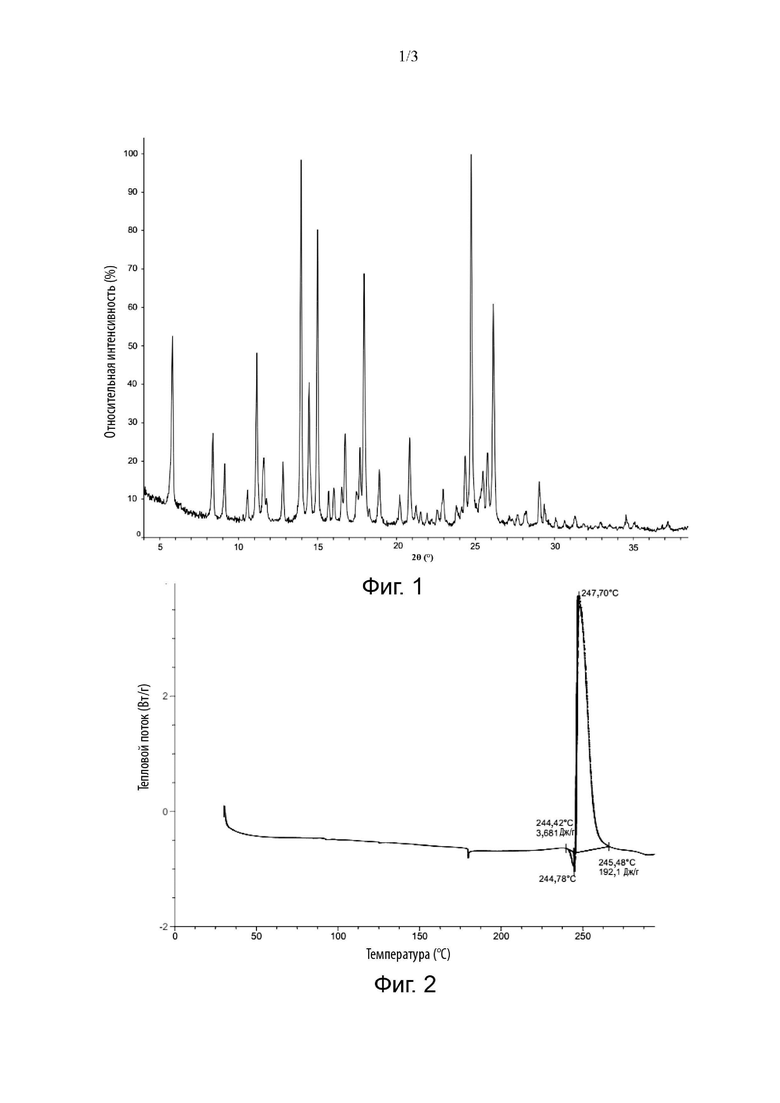
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет дифракционные пики на порошковой рентгенограмме с использованием Cu Kα-излучения при следующих значениях 2θ: 5,81±0,2°, 13,96±0,2°, 15,01±0,2°, 17,95±0,2° и 24,73±0,2°.
В некоторых вариантах реализации в настоящей заявке кристаллическая форма соединения Формулы (I) имеет дифракционные пики на порошковой рентгенограмме с использованием Cu Kα-излучения при следующих значениях 2θ: 5,81±9,2°, 8,38±9,2°, 11,16±9,2°, 13,96±9,2°, 14,47±9,2°, 15,91±9,2°, 17,95±9,2°, 24,73±9,2° и 26,13±9,2°.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет дифракционные пики на порошковой рентгенограмме с использованием Cu Kα-излучения при следующих значениях 2θ: 5,81±9,2°, 8,38±9,2°, 11,16±0,2°, 13,96±9,2°, 14,47±9,2°, 15,91±9,2°, 16,76±0,2°, 17,95±0,2°, 20,83±9,2°, 24,73±0,2° и26,13±0,2°.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет дифракционные пики на порошковой рентгенограмме с использованием Cu Kα-излучения при следующих значениях 2θ: 5,81±9,2°, 8,38±9,2°, 9,13±0,2°, 11,16±9,2°, 11,60±9,2°, 12,82±9,2°, 13,96±0,2°, 14,47±9,2°, 15,91±9,2°, 16,76±9,2°, 17,95±0,2°, 18,91±9,2°, 20,83±0,2°, 24,36±9,2°, 24,73±0,2°, 25,78±9,2° и 26,13±0,2°.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет 5, 6, 7, 8, 9, 10, 11, 12 или более дифракционных пиков на порошковой рентгенограмме с использованием Cu Kα-излучения при следующих значениях 2θ: 5,81±0,2°, 8,38±0,2°, 9,13±0,2°, 11,16±0,2°, 11,60±0,2°, 12,82±0,2°, 13,96±0,2°, 14,47±0,2°, 15,01±0,2°, 16,76±0,2°, 17,95±0,2°, 18,91±0,2°, 20,83±0,2°, 24,36±0,2°, 24,73±0,2°, 25,78±0,2° и26,13±0,2°.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет 5, 6, 7, 8, 9, 10 или 11 дифракционных пиков на порошковой рентгенограмме с использованием Cu Kα-излучения при следующих значениях 2θ: 5,81±0,2°, 8,38±0,2°, 11,16±0,2°, 13,96±0,2°, 14,47±0,2°, 15,01±0,2°, 16,76±0,2°, 17,95±0,2°, 20,83±0,2°, 24,73±0,2° и 26,13±0,2°.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет 5, 6, 7, 8 или 9 дифракционных пиков на порошковой рентгенограмме с использованием Cu Kα-излучения при следующих значениях 2θ: 5,81±0,2°, 8,38±0,2°, 11,16±0,2°, 13,96±0,2°, 14,47±0,2°, 15,01±0,2°, 17,95±0,2°, 24,73±0,2° и 26,13±0,2°.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет дифракционные пики на рентгенограмме РПД с использованием Cu Kα-излучения с положениями пиков и относительной интенсивностью, приведенными в Таблице 1 ниже:
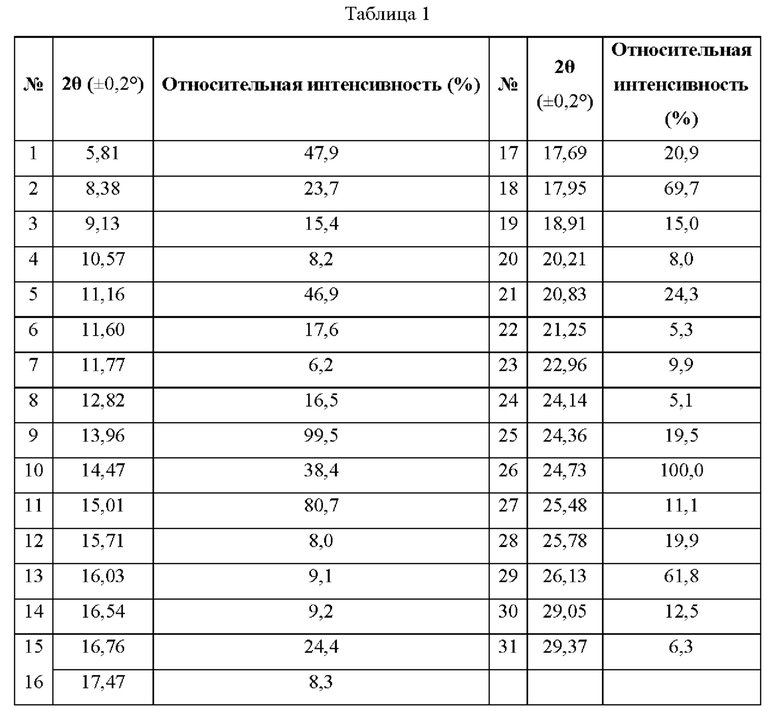
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет дифракционные пики на рентгенограмме РПД с использованием Cu Kα-излучения, как изображенные на Фиг. 1.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет экзотермический пик на кривой дифференциальной сканирующей калориметрии при 247,70°С±2°С.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет график дифференциальной сканирующей калориметрии, изображенный на Фиг. 2.
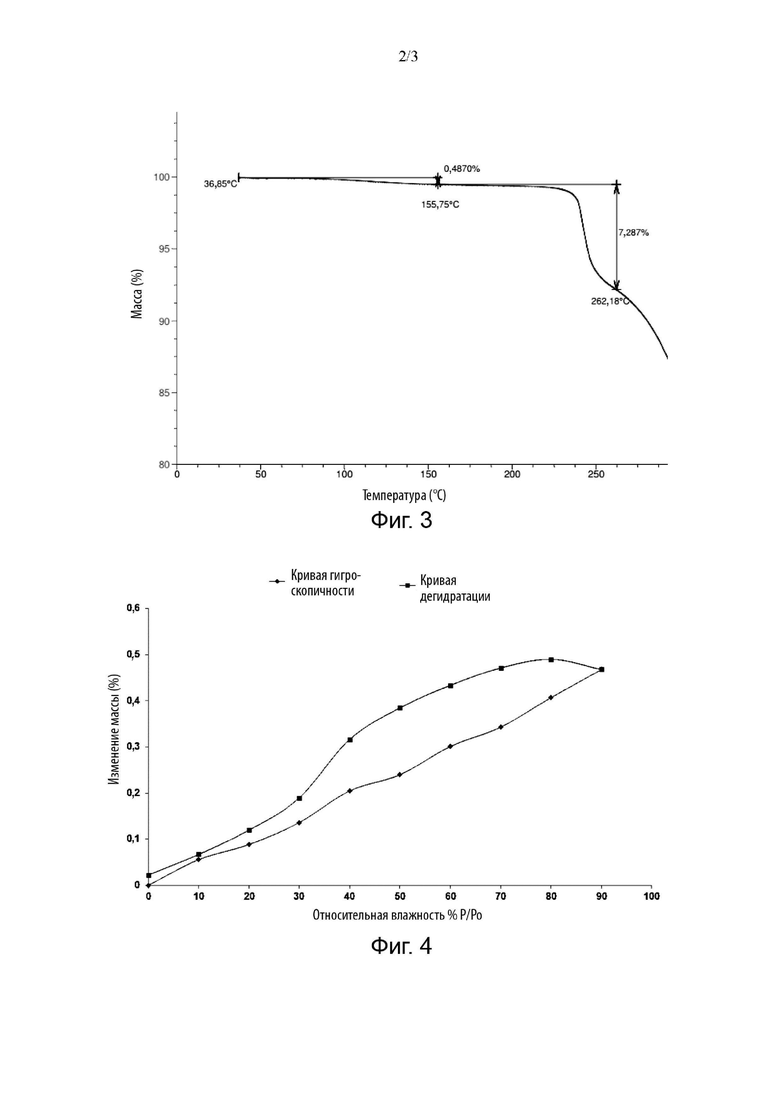
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет потерю массы 0,4870% при 155,75±2°С и потерю массы 7,287% при от 155,75±2°С до 262,18±2°С на кривой термогравиметрического анализа.
В некоторых вариантах реализации изобретения кристаллическая форма соединения Формулы (I) имеет график термогравиметрического анализа, изображенный на Фиг. 3.
В некоторых вариантах реализации изобретения в фармацевтической композиции соединение Формулы (I) или его фармацевтически приемлемая соль имеет размер частиц Х50≤10 мкм, предпочтительно от 0,1 мкм до 8 мкм.
В некоторых вариантах реализации изобретения в фармацевтической композиции соединение Формулы (I) или его фармацевтически приемлемая соль имеет размер частиц Х50≤5 мкм и Х90≤10 мкм.
Фармацевтическое соединение, раскрытое в настоящем документе, может находиться в различных дозированных формах, подходящих для перорального или ингаляционного введения человеку, например, в виде раствора.
В некоторых вариантах реализации изобретения фармацевтическую композицию, раскрытую в настоящем документе, вводят ингаляционно.
В некоторых вариантах реализации изобретения фармацевтическую композицию, раскрытую в настоящем документе, вводят перорально или назальной ингаляцией.
В некоторых вариантах реализации изобретения фармацевтическая композиция, раскрытая в настоящем документе, представляет собой раствор для ингаляции.
В некоторых вариантах реализации изобретения фармацевтическая композиция, раскрытая в настоящем документе, находится в форме суспензии.
В некоторых вариантах реализации изобретения фармацевтическая композиция, раскрытая в настоящем документе, находится в форме суспензии для ингаляции.
В другом аспекте в настоящей заявке предложен способ получения фармацевтической композиции, включающий: смешивание поверхностно-активного вещества и соединения Формулы (I) или его фармацевтически приемлемой соли. Предпочтительно способ включает смешивание поверхностно-активного вещества, соединения Формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного, выбранного из группы, состоящей из: хелатирующего металл агента, буферного агента, разбавителя и регулятора осмотического давления. Более предпочтительно, в настоящей заявке предложен способ получения фармацевтической композиции, включающий: смешивание смачивающего агента, буферного агента, регулятора осмотического давления, хелатирующего металл агента, соединения Формулы (I) или его фармацевтически приемлемой соли и разбавителя.
В некоторых вариантах реализации изобретения способ получения фармацевтической композиции включает:
1) смешивание смачивающего агента, буферного агента, регулятора осмотического давления, хелатирующего металл агента и разбавителя с получением раствора,
2) и смешивание соединения Формулы (I) или его фармацевтически приемлемой соли с раствором из этапа 1).
В некоторых вариантах реализации изобретения способ дополнительно включает этап 3): гомогенизацию продукта, полученного на этапе 2).
В некоторых вариантах реализации изобретения после процедуры гомогенизации соединение Формулы (I) или его фармацевтически приемлемая соль имеет размер частиц Х50≤5 мкм и Х90≤10 мкм.
В некоторых вариантах реализации изобретения способ включает этап наполнения.
В еще одном аспекте в настоящей заявке дополнительно предложен способ предотвращения или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4, у млекопитающего, включающий введение млекопитающему, предпочтительно человеку, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции.
В еще одном аспекте в настоящей заявке дополнительно предложено применение фармацевтической композиции для получения лекарственного средства для профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4.
В еще одном аспекте в настоящей заявке дополнительно предложено применение фармацевтической композиции для профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4.
В еще одном аспекте в настоящей заявке дополнительно предложена фармацевтическая композиция для применения в профилактике или лечении состояния, связанного с ФДЭ3 и/или ФДЭ4.
В некоторых вариантах реализации настоящей заявки состояние, связанное с ФДЭ3 и/или ФДЭ4, выбрано из группы, состоящей из астмы и хронической обструктивной болезни легких (ХОБЛ).
Технические эффекты
Соединение Формулы (I) и его фармацевтическая композиция, раскрытые в настоящем документе, обладают значительным двойным ингибирующим действием на ФДЭ3 и ФДЭ4, имеют значительное ингибирующее действие на ФНО-α в мононуклеарных клетках периферической крови человека (чМКПК), а также проявляют превосходный противовоспалительный эффект на модели острого повреждения легких у крыс, индуцированного липополисахаридом (ЛПС). Соединение имеет высокий плазменный клиренс in vivo, низкое распределение в плазме при пероральном введении и низкую пероральную биодоступность, а также хорошую безопасность при местном введении. Его ингибирующее действие на 5 изоферментов (CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4) микросомального цитохрома Р450 печени человека является низким, что позволяет избежать риска лекарственного взаимодействия. Кроме того, соединение снижает общее количество лейкоцитов в БАЛ, обладает выраженным противовоспалительным действием, действует в малых дозах и снижает показатель бронхоконстрикции Penh.
Кристаллическая форма соединения Формулы (I) и его фармацевтически приемлемая соль по настоящей заявке имеют преимущества с точки зрения фармацевтической активности, фармакокинетики, биодоступности, гигроскопичности, температуры плавления, стабильности, растворимости, чистоты, простоты получения и т.д., для удовлетворения требований фармацевтики в отношении производства, хранения, транспортировки, составления рецептуры и т.д. Кристаллическая форма соединения Формулы (I) имеет низкую гигроскопичность и благоприятна для абсорбции вдыхаемого соединения.
Фармацевтическая композиция соединения Формулы (I) имеет хорошую стабильность без увеличения примесей и показывает фармацевтически приемлемый уровень примесей. Фармацевтическая композиция имеет хорошую диспергируемость и стабильную динамику, не демонстрирует значительного осаждения частиц и увеличения размера частиц и находится в диапазоне, необходимом для эффективной доставки одобренных ингаляционных продуктов. Кроме того, композиция имеет однородный и средний размер частиц и высокую скорость абсорбции. Композиция имеет хорошую скорость доставки, точную дозировку, высокую долю вдыхаемых аэрозольных частиц и большое количество вдыхаемых мелких частиц.
Определения
Если иное не требуется в настоящей заявке, во всем описании и последующей формуле изобретения слово «содержать» и его варианты, такие как «содержит» и «содержащий» или эквиваленты, должны толковаться в открытом, всеобъемлющем смысле, т.е. «включает, но не ограничивается этим», указывая на то, что в дополнение к перечисленным элементам, компонентам и процедурам также могут быть включены другие неуказанные элементы, компоненты и процедуры.
«Один вариант реализации изобретения», «вариант реализации изобретения», «в другом варианте реализации изобретения» или «в некоторых вариантах реализации изобретения», используемые в описании, означают, что конкретный ссылочный элемент, структура или признак, описанные в связи с вариантом реализации изобретения, включены по меньшей мере в один вариант реализации изобретения. Таким образом, фразы «в одном варианте реализации изобретения», «в варианте реализации изобретения», «в другом варианте реализации изобретения» и «в некоторых вариантах реализации изобретения» в различных местах в описании не обязательно относятся к одному и тому же варианту (вариантам) реализации изобретения. Кроме того, конкретные элементы, структуры или признаки могут быть объединены любым подходящим образом в одном или более вариантах реализации изобретения.
Следует понимать, что, если явно не указано иное, формы единственного числа, используемые в описании и прилагаемой формуле настоящей заявки, включают ссылки во множественном числе; другими словами, термины в единственном числе в настоящем документе охватывают термины во множественном числе, и наоборот. Так, например, упомянутая реакция, включающая «катализатор», включает один катализатор или два или более катализаторов. Следует понимать, что, если конкретно не указано иное, термин «или», как правило, используется в своем значении, включая «и/или».
Термин «лечить» или «лечение» означает введение соединения или состава, описанного в настоящей заявке, для облегчения или устранения заболевания или одного или более симптомов, связанных с заболеванием, и включает:
(1) подавление заболевания или болезненного состояния, т.е. остановку его развития; и
(2) облегчение заболевания или болезненного состояния, т.е. вызывание его регрессии.
Термин «предотвращать» или «профилактика» означает введение соединения или состава по настоящей заявке для предотвращения заболевания или одного или более симптомов, связанных с заболеванием, и включает: предотвращение возникновения заболевания или болезненного состояния у млекопитающего, в частности когда такое млекопитающее предрасположено к болезненному состоянию, но еще не диагностировано.
Термин «терапевтически эффективное количество» относится к количеству соединения по настоящей заявке для (1) лечения или профилактики конкретного заболевания, состояния или расстройства; (2) облегчения, ослабления или устранения одного или более симптомов конкретного заболевания, состояния или расстройства, или (3) предотвращения или задержки появления одного или более симптомов конкретного заболевания, состояния или нарушения, описанных в настоящем документе. Количество соединения по настоящей заявке, составляющее «терапевтически эффективное количество», варьируется в зависимости от соединения, состояния заболевания и его тяжести, способа введения и возраста млекопитающего, подлежащего лечению, но может быть определено обычным путем специалистами в данной области техники в соответствии с их знаниями и настоящим раскрытием.
Как правило, размер частиц определяют количественно путем измерения характеристического эквивалентного сферического диаметра (именуемого объемным диаметром) с помощью лазерной дифракции, например, с помощью лазерного анализатора размера частиц.
В настоящей заявке распределение частиц по размерам выражается в терминах объемного диаметра (ОД).
Термин «Х10» относится к размеру частиц, соответствующему совокупному объемному распределению в процентах в 10%, что физически означает, что частицы с размерами меньше, чем данный, составляют 10% от общего объема.
Термин «Х50» относится к размеру частиц, соответствующему совокупному объемному распределению в процентах в 50%, что именуется объемным медианным диаметром и физически означает, что частицы с размерами меньше, чем данный, составляют 50% от общего объема.
Термин «Х90» относится к размеру частиц, соответствующему совокупному объемному распределению в процентах в 90%, что физически означает, что частицы с размерами меньше, чем данный, составляют 90% от общего объема.
Если в настоящем документе не указано иное, значения параметров (включая значения 2θ, условия реакции) следует толковать как модифицированные термином «около», чтобы отразить погрешность измерения и т.п., существующие в значениях, например, существует погрешность ±5% относительно заданного значения.
Все патенты, патентные заявки и другие указанные публикации явно целиком включены в настоящий документ посредством ссылки для целей описания и раскрытия. Эти публикации предоставлены исключительно потому, что они были раскрыты до даты подачи настоящей заявки. Все заявления относительно дат этих документов или описание содержания этих документов основаны на информации, доступной заявителю, и не являются признанием правильности дат или содержания этих документов. Кроме того, в любой стране или регионе любая ссылка на эти публикации в настоящем документе не должна рассматриваться как признание того, что публикации являются частью общепризнанных знаний в данной области техники.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 изображена рентгенограмма РПД кристаллической формы соединения Формулы (I);
На Фиг. 2 изображен график ДСК кристаллической формы соединения Формулы (I); На Фиг. 3 изображен график ТГА кристаллической формы соединения Формулы (I); На Фиг. 4 изображен график ДСП кристаллической формы соединения Формулы (I); На Фиг. 5 изображено общее количество лейкоцитов в БАЛ;
На Фиг. 6 изображен тест функции легких с метахолином (Мх) (показатель бронхоконстрикции Penh).
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Следующие конкретные примеры предназначены для того, чтобы позволить специалистам в данной области техники ясно понять и реализовать настоящую заявку. Эти конкретные примеры не следует рассматривать как ограничение объема настоящей заявки, а просто как иллюстративное описание и представление настоящей заявки.
Примеры
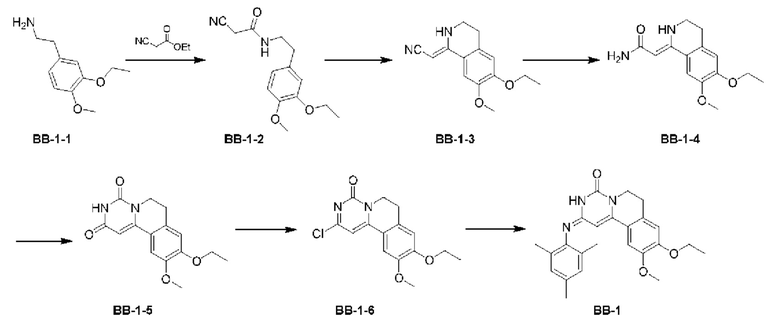
Этап 1: синтез соединения ВВ-1-2
Смесь соединения ВВ-1-1 (21,10 г) и этилцианоацетата (11,00 г, 10,38 мл) перемешивали при 100°С в течение 16 ч в атмосфере азота. После завершения реакции смесь охлаждали до 70°С, медленно по каплям добавляли этанол (30 мл), и большое количество твердого вещества выпадало в осадок. Полученную смесь фильтровали, и осадок на фильтре сушили при пониженном давлении с получением продукта ВВ-1-2.
1Н ЯМР (400 МГц, ДМСО-d6) δ=8,26 (t, J=5,2 Гц, 1Н), 6,86 (d, J=8,0 Гц, 1H), 6,79 (br s, 1H), 6,71 (d, 8,0 Гц, 1Н), 4,00 (q, J=6,8 Гц, 2Н), 3,72 (s, 3Н), 3,59 (s, 2Н), 3,31-3,23 (m, 2Н), 2,64 (t, J=7,2 Гц, 2Н), 1,32 (t, J=6,8 Гц, 3Н). МС-ИЭР m/z: 263,1 [М+Н]+.
Этап 2: синтез соединения ВВ-1-3
Оксихлорид фосфора (379,50 г, 230,00 мл) нагревали до 85°С в атмосфере азота и порциями добавляли соединение ВВ-1-2 (26,00 г). Реакционную смесь перемешивали при 85°С в течение 2 ч. После завершения реакции большую часть оксихлорида фосфора удаляли перегонкой при пониженном давлении. К остатку добавляли дихлорметан (200 мл), и смесь промывали водой (100 мл × 2). Затем органическую фазу сушили над безводным сульфатом натрия, фильтровали для удаления осушителя, и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали путем суспендирования с этилацетатом (20 мл) с получением соединения ВВ-1-3.
1Н ЯМР (400 МГц, CD3OD) δ=7,16 (s, 1H), 6,83 (s, 1H), 4,62 (s, 1H), 4,12 (q, J=6,8 Гц, 2Н), 3,86 (s, 3Н), 3,35 (d, J=6,4 Гц, 2Н), 2,84 (t, J=6,4 Гц, 2Н), 1,44 (t, J=6,8 Гц, 3Н). МС-ИЭР m/z: 245,1 [М+Н]+.
Этап 3: синтез соединения ВВ-1-4
Соединение ВВ-1-3 (1,00 г) порциями добавляли к 98% концентрированной серной кислоте (12,88 г, 128,69 ммоль, 7,00 мл) при 0°С. Реакционную смесь перемешивали при 27°С в течение 3 ч. После завершения реакции смесь добавляли к холодной воде (15 мл), а затем по каплям добавляли водный раствор гидроксида натрия (4 моль/л, 32 мл) для доведения рН до нейтрального с последующей экстракцией этилацетатом (100 мл × 3). Затем органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали для удаления осушителя и концентрировали при пониженном давлении с получением соединения ВВ-1-4.
МС-ИЭР m/z: 263,1 [М+Н]+.
Этап 4: синтез соединения ВВ-1-5
Натрий (2,42 г) порциями добавляли к этанолу (80 мл) при 0°С. После перемешивания смеси при 28°С в течение 0,5 ч к раствору порциями добавляли соединение ВВ-1-4 (6,90 г), и смесь перемешивали при 80°С в течение 0,5 ч. Затем одной порцией добавляли диэтилкарбонат (9,32 г, 9,51 мл), и смесь перемешивали в течение 5 ч при 80°С. После завершения реакции смесь охлаждали до комнатной температуры, медленно добавляли воду, охлажденную до температуры замерзания (30 мл), и затем добавляли разбавленную соляную кислоту (2 моль/л, 53 мл) для доведения смеси до нейтрального рН. Большое количество твердого вещества выпадало в осадок. Смесь фильтровали, и полученный осадок на фильтре очищали путем суспендирования с этанолом (10 мл) с получением соединения ВВ-1-5.
1Н ЯМР (400 МГц, ДМСО-d6) δ=11,22 (широкий s, 1Н), 7,35 (s, 1H), 6,95 (s, 1H), 6,22 (s, 1H), 4,09 (q, J=6,8 Гц, 2Н), 3,90 (широкий s, 2Н), 3,83 (s, 3Н), 2,89 (широкий s, 2Н), 1,35 (t, J=6,8 Гц, 3Н). МС-ИЭР m/z: 289,1 [М+Н]+.
Этап 5: синтез соединения ВВ-1-6
Соединение ВВ-1-5 (5,00 г) растворяли в оксихлориде фосфора (30 мл) при комнатной температуре. Смесь перемешивали при 100°С в течение 16 ч в атмосфере азота. После завершения реакции большую часть растворителя удаляли перегонкой при пониженном давлении. Добавляли воду (100 мл), и полученную смесь экстрагировали дихлорметаном (150 мл × 2). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали для удаления осушителя и концентрировали при пониженном давлении с получением соединения ВВ-1-6. МС-ИЭР m/z: 306,9 [М+Н]+.
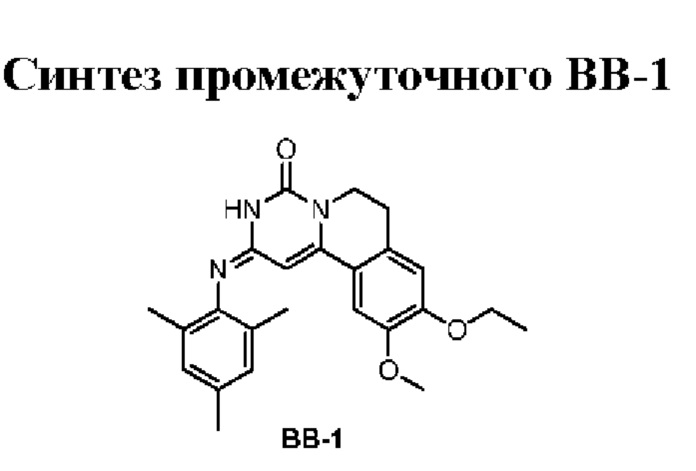
Этап 6: синтез соединения ВВ-1
Соединение ВВ-1-6 (925,67 мг) растворяли в изопропаноле (8 мл) при комнатной температуре и затем добавляли 2,4,6-триметиланилин (2,10 г). Смесь перемешивали при 90°С в течение 15 ч в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении, и полученный остаток очищали путем суспендирования с этанолом (6 мл) с получением соединения ВВ-1.
1Н ЯМР (400 МГц, ДМСО-d6) δ=8,85 (широкий s, 1Н), 7,27 (s, 1Н), 6,97 (s, 1H), 6,90 (s, 2Н), 6,45 (s, 1H), 4,10 (q, J=6,8 Гц, 2Н), 3,90 (t, J=6,0 Гц, 2Н), 3,86 (s, 3Н), 2,87 (t, J=6,0 Гц, 2Н), 2,45 (s, 3Н), 2,11 (s, 6Н), 1,37 (t, J=6,8 Гц, 3Н). МС-ИЭР m/z: 406,2 [М+Н]+.
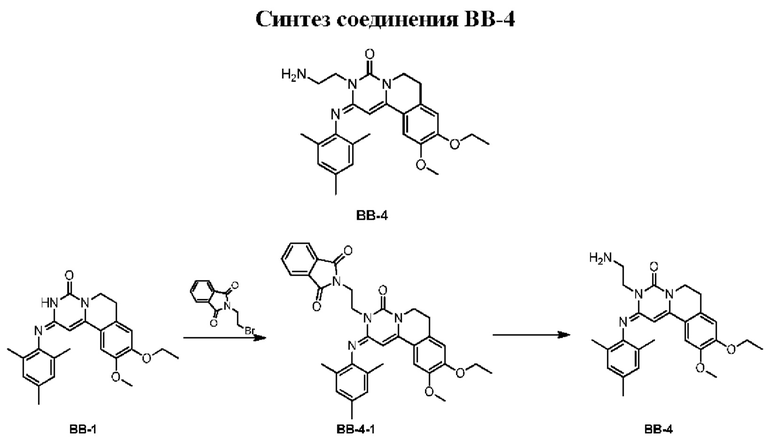
Этап 1: синтез соединения ВВ-4-1
Соединение ВВ-1 (1,00 г) растворяли в 2-бутаноне (35 мл) при комнатной температуре и последовательно добавляли 2-(2-бромэтил)изоиндолин-1,3-дион (3,76 г), карбонат калия (3,07 г) и йодит натрия (2,22 г). Смесь перемешивали при 85°С в течение 72 ч в атмосфере азота. После завершения реакции смесь концентрировали для удаления большей части органического растворителя, после чего для экстракции добавляли воду (30 мл) и этилацетат (25 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали для удаления осушителя и концентрировали при пониженном давлении. Полученный остаток очищали колоночной флэш-хроматографией на силикагеле (элюент: петролейный эфир/этилацетат =15/1-3/1) с получением соединения ВВ-4-1.
МС-ИЭР m/z: 579,3 [М+Н]+.
Этап 2: синтез соединения ВВ-4
Соединение ВВ-4-1 (500,00 мг) растворяли в трихлорметане (3 мл) и этаноле (3 мл) при комнатной температуре и добавляли гидразингидрат (152,67 мг, чистота 85%). Смесь перемешивали при 28°С в течение 16 ч в атмосфере азота. После завершения реакции смесь концентрировали для удаления большей части органического растворителя, после чего для экстракции добавляли воду (15 мл) и дихлорметан (15 мл × 3). Затем органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали для удаления осушителя и концентрировали при пониженном давлении с получением соединения ВВ-4.
1Н ЯМР (400 МГц, ДМСО-d6) δ=6,95 (s, 1Н), 6,85 (широкий s, 2Н), 6,66 (s, 1H), 5,31 (s, 1H), 4,14 (t, J=6,8 Гц, 2Н), 4,05 (q, J=6,8 Гц, 2Н), 3,91 (t, J=6,4 Гц, 2Н), 3,62 (s, 3Н), 2,90-2,86 (m, 4Н), 2,22 (s, 3Н), 1,95 (широкий s, 6Н), 1,33 (t, J=6,8 Гц, 3Н). МС-ИЭР m/z: 449,2 [М+Н]+.
Пример 1: Получение соединения Формулы (I)
5-гидрокси-3-метил-1,2,3-триазол-4-карбоновую кислоту (18,50 мг) растворяли в ДХМ (1 мл) при 20°С. Добавляли HATU (8,80 мг) и триэтиламин (57,40 мкл), и смесь перемешивали в течение 2 ч с последующим добавлением соединения ВВ-4 (50 мг) и перемешиванием при температуре в течение 16 ч. Смесь разбавляли до 10 мл с помощью ДХМ, промывали водой (30 мл × 3), сушили над безводным сульфатом натрия и фильтровали для удаления осушителя, а фильтрат концентрировали при пониженном давлении для удаления растворителя с получением неочищенного продукта. Неочищенный продукт отделяли и очищали препаративной ВЭЖХ с получением целевого соединения Формулы (I) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CD3OD) δ=6,94 (s, 2Н), 6,87 (s, 1H), 6,77 (s, 1H), 5,52 (s, 1Н), 4,48 (t, J=6,0 Гц, 2Н), 4,15 (s, 3Н), 4,12-4,08 (m, 2Н), 4,01 (t, J=6,0 Гц, 2Н), 3,87 (t, J=6,0 Гц, 2Н), 3,69 (s, 3Н), 2,94 (t, J=6,0 Гц, 2Н), 2,29 (s, 3Н), 2,06 (s, 6Н), 1,41 (t, J=6,8 Гц, 3Н). MCm/z[M+H]+ 574,1.
Пример 2: Получение кристаллической формы соединения Формулы (I)
50 мг соединения Формулы (I) добавляли в стеклянную бутыль емкостью 4 мл, добавляли 1 мл безводного этанола и 0,2 мл воды, смесь нагревали до 40°С и перемешивали в течение 48 ч. Смесь охлаждали естественным путем до комнатной температуры, центрифугировали для отделения твердого вещества и сушили в вакууме с получением 46 мг твердой кристаллической формы. Рентгенограмма РПД изображена на Фиг. 1, график ДСК изображен на Фиг. 2, а график ТГА изображен на Фиг. 3.
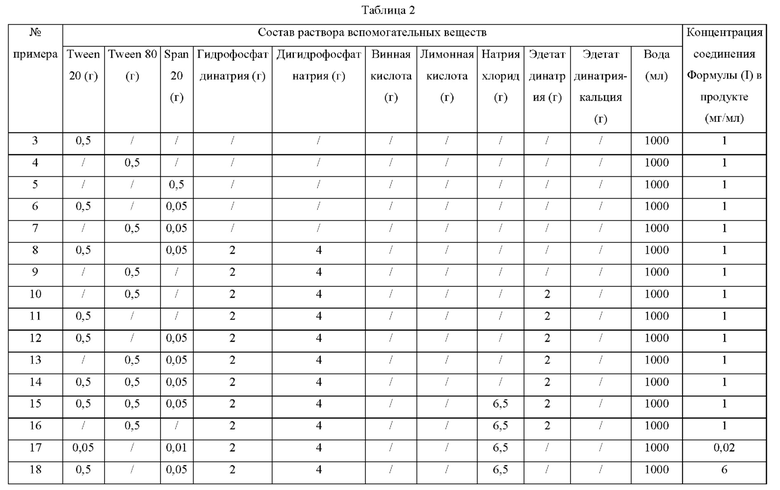
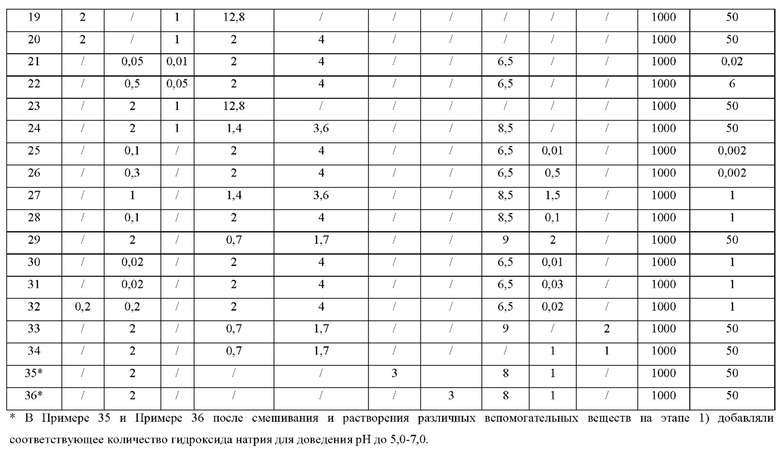
Примеры 3-36
Процедуры:
1) в резервуар для приготовления добавляли различные вспомогательные вещества, и смесь перемешивали для растворения с получением раствора вспомогательных веществ;
2) к приготовленному раствору вспомогательных веществ добавляли соединение Формулы (I), и смесь перемешивали до образования однородной суспензии;
3) суспензию дополнительно подавали в гомогенизатор высокого давления, микроструйную или песочную мельницу и т.п., и размер частиц соединения Формулы (I) в препарате контролировали на уровне Х50≤5 мкм и Х90≤10 мкм; и
4) получали продукт.
Количества конкретных вспомогательных веществ и продуктов приведены в следующей Таблице 2.
Экспериментальный пример 1: Исследование стабильности твердой кристаллической формы соединения Формулы (I)
Высокоэффективная жидкостная хроматография (ВЭЖХ)
Хроматографические условия метода ВЭЖХ приведены в Таблице ниже:
Хроматографическая колонка: Zorbax SB С-18, 4,6 мм × 150 мм, 5 мкм (PDS-HPLC-007)
Мобильная фаза А: 0,1% ТФК в воде
Мобильная фаза В: 100% АЦН
Подготовка образца: образец растворяли в смешанном растворителе из ацетонитрила и воды (ацетонитрил:вода = 50:50 (об./об.))
Метод разбивки стабильности твердого вещества
Изучали стабильность соединения в следующих условиях, и образцы отбирали в разные моменты времени для определения содержания. Около 5 мг кристаллической формы соединения Формулы (I), полученной в Примере 2, дважды точно взвешивали, переносили в сухую и чистую стеклянную бутыль, распределяли тонким слоем в качестве испытуемых образцов и помещали в экспериментальные условия факторов воздействия ((60°С), (относительная влажность 92,5%), освещенность (общая освещенность 1,2×106 лк⋅ч/практически УФ энергия 200 Вт⋅ч/м2), (40°С, относительная влажность 75%), или (60°С, относительная влажность 75%)). Образцы были покрыты алюминиевой фольгой, имеющей отверстия, и, таким образом, полностью подвергались воздействию условий. Анализ образцов проводили через 5 дней, 10 дней, 1 месяц, 2 месяца и 3 месяца. Образцы полностью подвергались освещению (видимый свет 1200000 лк, УФ 200 Вт) при комнатной температуре. Результаты экспериментов приведены в Таблице 3.
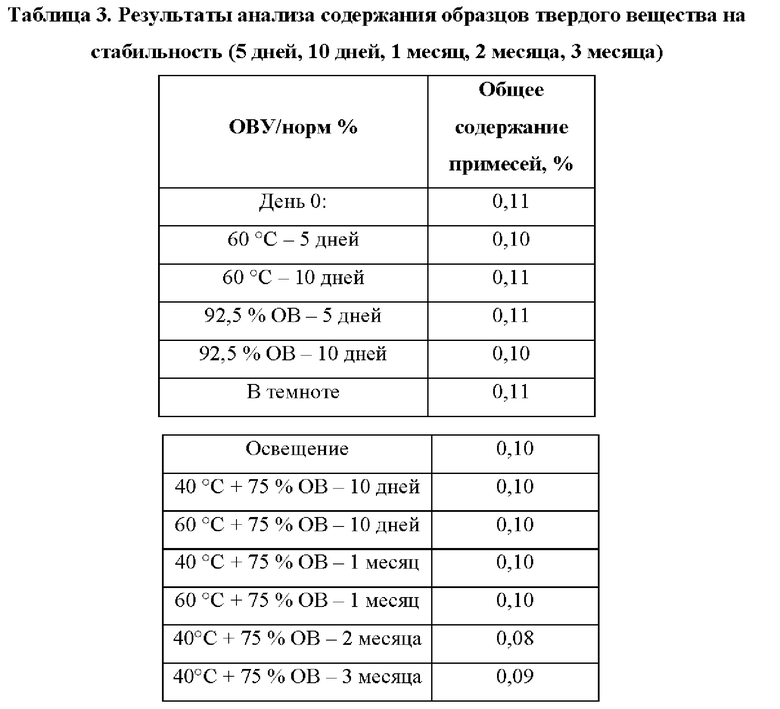
Как видно, кристаллическая форма соединения Формулы (I) по настоящей заявке обладает хорошей стабильностью в условиях высокой температуры, высокой влажности или освещения без увеличения количества примесей во время испытания.
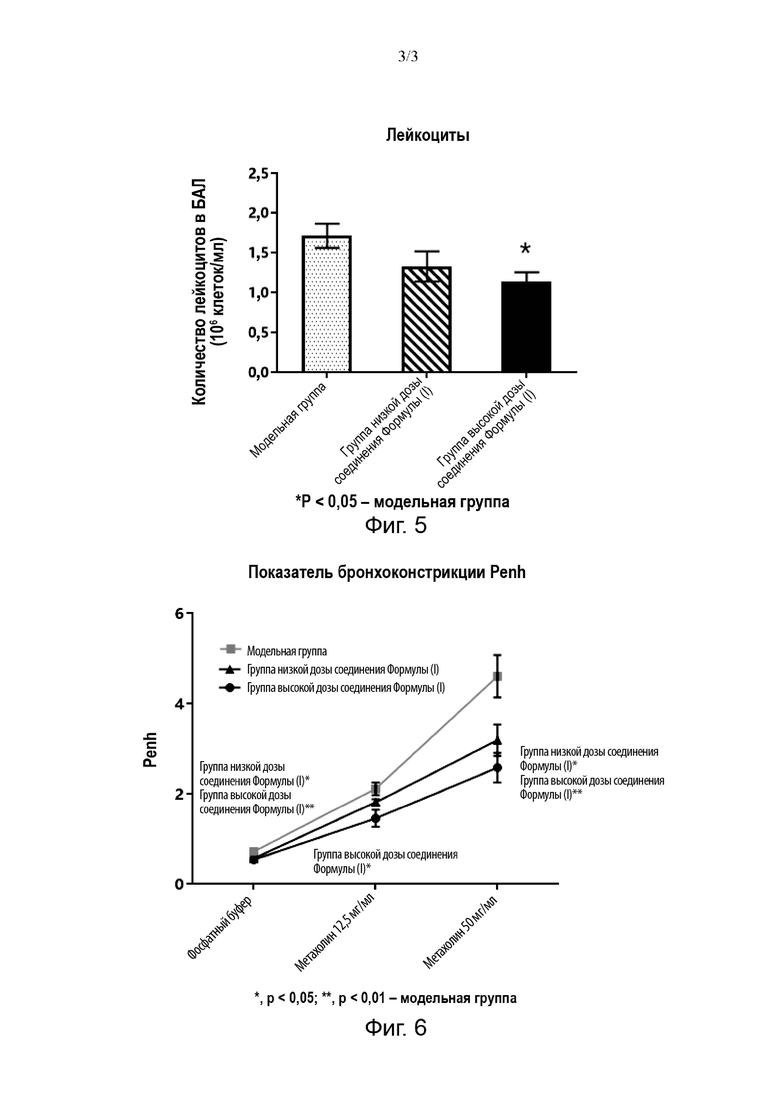
Экспериментальный пример 2: Изучение гигроскопичности кристаллической формы соединения Формулы (I)
Модель прибора: SMS DVS Advantage
Условия испытаний: образец (10-20 мг, кристаллическая форма, приготовленная в Примере 3) помещали в лоток для образцов ДСП для испытаний.
Подробные параметры ДСП следующие:
Температура: 25°С
Балансировка: dm/dt=0,01%/мин (самый короткий: 10 мин, самый длинный: 180 мин)
Сушка: сушка при 0% ОВ в течение 120 мин.
Градиент ОВ (%) при испытании: 10%
Диапазон градиента ОВ (%) при испытании: 0%-90%-0%. Полученный график динамической сорбции паров (ДСП) изображен на Фиг. 4.
Как видно из Фиг. 4, кристаллическая форма соединения Формулы (I) по настоящей заявке имеет небольшую гигроскопичность.
Экспериментальный пример 3: Обнаружение ингибирующей активности соединения в отношении фермента ФДЭ 3А in vitro
Задача: определить экспрессию АМФ/ЦМФ на основе флуоресцентной поляризации, т.е. отслеживать связывание АМФ/ЦМФ с антителом, чтобы показать активность фермента.
Реагенты:
Буферный раствор: 10 мМ Трис-HCl (рН 7,5), 5 мМ MgCh2, 0,01% Brij 35, 1 мМ дитиотреитола (ДТТ) и 1% ДМСО.
Фермент: рекомбинантный ФДЭ3А человека (инвентарный номер гена: NM_000921; аминокислота 669-конец) экспрессировался бакуловирусом в клетках насекомых Sf9 с использованием N-концевой GST-метки с молекулярной массой 84 кДа.
Ферментный субстрат: 1 мкМ цАМФ
Обнаружение: антитело АМФ2/ЦМФ2 Transcreener® и трейсер АМФ2/ЦМФ2 AlexaFluor633.
Процедуры:
1. Каждый из рекомбинантного фермента ФДЭ3А человека и ферментного субстрата (1 мкМ цАМФ) растворяли в свежеприготовленном экспериментальном буферном растворе;
2. Буферный раствор фермента ФДЭ3А переносили в реакционные лунки;
3. Соединение, растворенное в 100% ДМСО, добавляли в реакционные лунки, содержащие буферный раствор фермента ФДЭ3А, акустическим методом (эхо 550; диапазон миллилямбда), и смесь инкубировали в течение 10 мин при комнатной температуре;
4. Буферный раствор ферментного субстрата добавляли в вышеуказанные реакционные лунки, чтобы инициировать реакцию;
5. Полученную смесь инкубировали при комнатной температуре в течение 1 ч;
6. Смесь для обнаружения (антитело АМФ2/ЦМФ2 Transcreener® и трейсер АМФ2/ЦМФ2 AlexaFluor633) добавляли для остановки реакции, и полученную смесь инкубировали в течение 90 мин при медленном перемешивании. Диапазон измерения флуоресцентной поляризации составил Ex/Em=620/688.
Анализ данных: сигнал флуоресцентной поляризации был преобразован в нМ на основе стандартной кривой АМФ/ЦМФ и процент активности фермента относительно контрольного ДМСО был рассчитан с помощью Excel. GraphPad Prism использовался для подгонки кривой (рисование медицинской иконки). Результаты приведены в Таблице 4.
Экспериментальный пример 4: Обнаружение ингибирующей активности соединения в отношении фермента ФДЭ 4В in vitro
Задача: определить экспрессию АМФ/ЦМФ на основе флуоресцентной поляризации, т.е. отслеживать связывание АМФ/ЦМФ с антителом, чтобы показать активность фермента.
Реагенты:
Буфферный раствор: 10 мМ Трис-HCl (рН 7,5), 5 мМ MgCl2, 0,01% Brij 35, 1 мМ ДТТ и 1% ДМСО.
Фермент: рекомбинантный ФДЭ4 В человека (инвентарный номер гена: NM_002600; аминокислота 305-конец) экспрессировался бакуловирусом в клетках насекомых Sf9 с использованием N-концевой GST-метки с молекулярной массой 78 кДа.
Ферментный субстрат: 1 мкМ цАМФ
Обнаружение: антитело АМФ2/ЦМФ2 Transcreener® и трейсер АМФ2/ЦМФ2 AlexaFluor633.
Процедуры:
1. Каждый из рекомбинантного фермента ФДЭ4В человека и ферментного субстрата (1 мкМ цАМФ) растворяли в свежеприготовленном буферном растворе.
2. Буферный раствор фермента ФДЭ4В переносили в реакционные лунки.
3. Соединение, растворенное в 100% ДМСО, добавляли в реакционные лунки, содержащие буферный раствор фермента ФДЭ4В, акустическим методом (эхо 550; диапазон миллилямбда), и смесь инкубировали в течение 10 мин при комнатной температуре;
4. Буферный раствор ферментного субстрата добавляли в вышеуказанные реакционные лунки, чтобы инициировать реакцию.
5. Полученную смесь инкубировали при комнатной температуре в течение 1 ч.
6. Смесь для обнаружения (антитело АМФ2/ЦМФ2 Transcreener® и трейсер АМФ2/ЦМФ2 AlexaFluor633) добавляли для остановки реакции, и полученную смесь инкубировали в течение 90 мин при медленном перемешивании. Диапазон измерения флуоресцентной поляризации составил Ex/Em=620/688.
Анализ данных: сигнал флуоресцентной поляризации был преобразован в нМ на основе стандартной кривой АМФ/ЦМФ, и процент активности фермента относительно контрольного ДМСО был рассчитан с помощью Excel. GraphPad Prism использовался для подгонки кривой (рисование медицинской иконки).
Результаты представлены в Таблице 4:
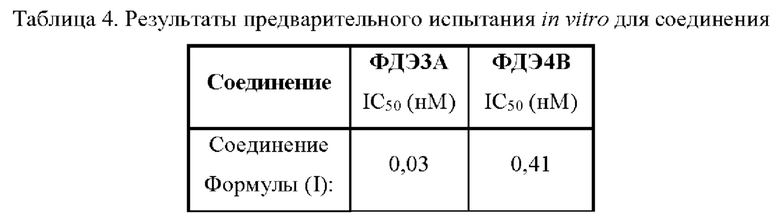
Активный ингредиент в фармацевтической композиции по настоящей заявке оказывает значительное двойное ингибирующее действие на ФДЭ3 и ФДЭ4.
Экспериментальный пример 5: Фармакокинетическое исследование на собаках породы бигль
В этом исследовании в качестве подопытных животных были выбраны самцы собак породы бигль. ЖХ-МС/МС использовали для количественного измерения концентрации лекарственного средства в плазме собак породы бигль в различные моменты времени после внутривенной инъекции или внутрижелудочного введения соединения Формулы (I) с целью оценки фармакокинетики соединения Формулы (I) у собак породы бигль.
Прозрачный раствор соединения Формулы (I) вводили инъекционно двум собакам породы бигль массой 10-12 кг через головную вену или подкожную вену, и прозрачный раствор соединения Формулы (I) вводили внутрижелудочно двум собакам породы бигль массой 10-12 кг (ночное голодание). У всех животных каждый раз брали около 500 мкл крови из периферических вен через 0,0333, 0,0833, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 ч после введения дозы, и кровь переносили в промышленные пробирки для центрифуги, содержащие 0,85-1,15 мг антикоагулянта К2 ЭДТА⋅2H2O, и плазму отделяли центрифугированием при 3000 g в течение 10 мин при 4°С. Концентрацию в плазме измеряли с помощью ЖХ-МС/МС, и соответствующие фармакокинетические параметры рассчитывали с использованием WinNonlin™ Version 6.3 (Pharsight, Маунтейн Вью, Калифорния), программного обеспечения для фармакокинетики, методом линейно-логарифмической трапеции без разделения на модели.
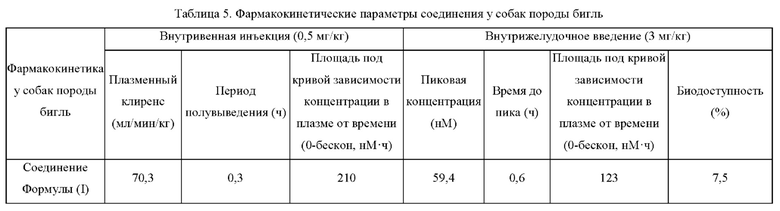
Активный ингредиент в фармацевтической композиции по настоящей заявке имеет высокий плазменный клиренс in vivo, низкое системное распределение в плазме при пероральном введении и низкую биодоступность при пероральном введении.
Экспериментальный пример 6: Ингибирующее влияние на активность изоферментов (CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4) микросомального цитохрома Р450 печени человека
В общей сложности 5 специфических маркерных субстратов 5 изоферментов CYP, а именно фенацетина (CYP1A2), диклофенака (CYP2C9), (S)-мефенитоина (CYP2C19), декстрометорфана (CYP2D6) и мидазолама (CYP3A4) инкубировали, совместно с микросомами печени человека и соединением Формулы (I), а затем для инициирования реакции добавляли восстановленный никотинамидадениндинуклеотидфосфат (НАДФ). После завершения реакции образцы обрабатывали и количественно определяли с помощью ЖХ-МС/МС концентрации 5 метаболитов (ацетаминофена, 4'-гидроксидиклофенака, 4'-гидроксимефенитоина, декстрорфана и 1'-гидроксимидазолама), полученных из конкретных субстратов для расчета соответствующих концентраций полумаксимального ингибирования (IC50).
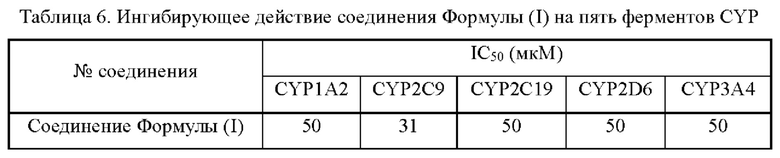
Активный ингредиент в фармацевтической композиции по настоящей заявке оказывает слабое ингибирующее действие на 5 изоферментов (CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4) микросомального цитохрома Р450 печени человека.
Экспериментальный пример 7: Фармакодинамическое исследование на модели острого повреждения легких у крыс, вызванного сигаретным дымом
Животные
Самцы крыс Sprague-Dawley (предоставленные Shanghai SLAC Laboratory Animal Co., Ltd.), класс SPF, около 200 г.
Процедуры:
1. Животные были случайным образом разделены на 6 групп в зависимости от массы тела после прибытия и недельной акклиматизации;
2. На 1-3-й день эксперимента соответствующее соединение каждой группы распыляли в течение 30 мин. Затем животных в модельной группе и экспериментальных группах подвергали воздействию сигаретного дыма в течение 1 ч, а после 4-часового перерыва животных снова подвергали воздействию сигаретного дыма в течение 1 ч. Сигаретный дым давали два раза в день в течение 3 дней подряд. Животные контрольной группы находились на комнатном воздухе;
3. На 4-й день эксперимента соответствующее соединение каждой группы распыляли в течение 30 мин, а затем животным в модельной группе и экспериментальных группах давали распыленный ЛПС 150 мг/мл путем ингаляции в течение 15 мин. Через 3 ч (с момента начала распыления) животных подвергали воздействию сигаретного дыма в течение 1 ч, после чего исследовали функцию легких (Penh и F) животных; жидкость бронхоальвеолярного лаважа (БАЛ) собирали для подсчета клеток после эвтаназии животных с помощью CO2.
4. Введение
Режим введения: испытуемое соединение и эталонное соединение вводили путем распыления с максимальной скоростью распыления (около 12 мл) с помощью устройства распыления, воздействующего на все тело, в течение 30 мин.
Частота введения: лекарственное средство или растворитель вводили путем распыления в течение 30 минут каждое утро перед воздействием сигаретного дыма и перед ингаляцией распыленного ЛПС на 4-й день.
5. Измерения фармакодинамических конечных точек
(1) Общее количество лейкоцитов в БАЛ (жидкости бронхоальвеолярного лаважа);
(2) проба функции легких по Мх (показатель бронхоконстрикции Penh);
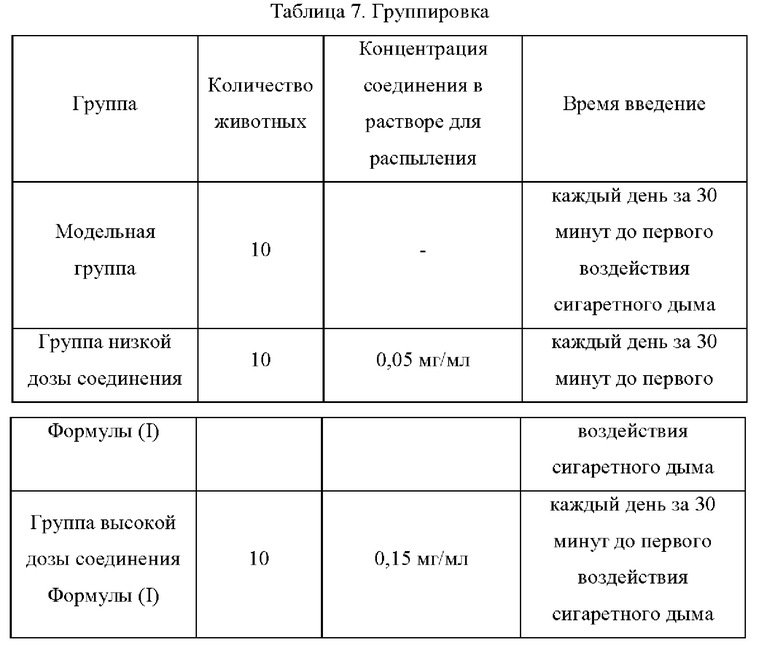
Экспериментальные результаты изображены на Фиг. 5 и Фиг. 6.
Как видно из Фиг. 5 и Фиг. 6, активный ингредиент в фармацевтической композиции по настоящей заявке может эффективно снижать количество лейкоцитов в жидкости бронхоальвеолярного лаважа и показатель бронхоконстрикции Penh.
Экспериментальный пример 8: Выявление ингибирующей активности соединения in vitro в отношении ФНО-α в мононуклеарных клетках периферической крови человека
Задача: измерить противовоспалительную активность испытуемого соединения на клеточном уровне на основе уровня ФНО-α в мононуклеарных клетках периферической крови человека (чМНПК).
Процедуры:
1. Нормальная человеческая цельная кровь была собрана в антикоагулянтную пробирку с ЭДТА;
2. МНПК отделяли центрифугированием в градиенте плотности фиколла и подсчитывали, а концентрацию клеток доводили до 2 × 106 клеток/мл;
3. В каждую лунку 96-луночного планшета с U-образным дном добавляли 2 × 105 клеток, 1 мкг/мл ЛПС и растворы соединения Формулы (I) в ДМСО в концентрациях 100 мкМ, 10 мкМ, 1 мкМ, 100 нМ, 10 нМ, 1 нМ, 100 пМ и 10 пМ, при объеме системы 200 мкл на лунку;
4. Смесь инкубировали в течение 24 ч, затем собирали надосадочную жидкость;
5. Уровень ФНО-α в надосадочной жидкости определяли с помощью ELISA, строили кривую ингибирования с использованием программного обеспечения Graphpad Prism и рассчитывали IC50.
Результаты приведены в Таблице 8:
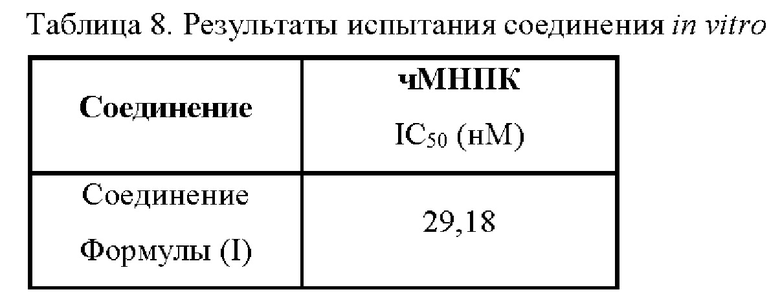
Следовательно, активный ингредиент в фармацевтической композиции по настоящей заявке проявляет мощную противовоспалительную активность и оказывает значительное ингибирующее действие на ФНО-α в мононуклеарных клетках периферической крови человека (чМНПК).
Экспериментальный пример 9: Испытание на стабильность
Продукт, полученный в Примере 28, помещали на 6 месяцев в условия ускоренного испытания (40°С±2°С/ОВ 25%±5%) и условия длительного испытания (30°С±2°С/ОВ 65%±5%), и результаты приведены в Таблице 9.
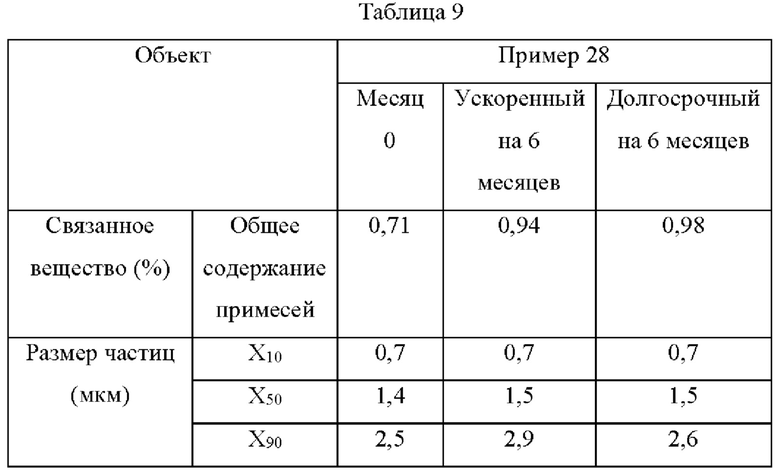
Как видно, фармацевтическая композиция по настоящей заявке показывает хорошую стабильность в условиях ускоренного испытания и в условиях длительного испытания без значительного увеличения примесей и размера частиц.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКИЙ АГЕНТ ДЛЯ ИНГИБИРОВАНИЯ ФОСФОДИЭСТЕРАЗЫ И СВЯЗАННЫХ С НЕЙ ПАТОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2018 |
|
RU2815010C2 |
| ТРАНСВАГИНАЛЬНЫЕ ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗЫ ДЛЯ ЛЕЧЕНИЯ БЕСПЛОДИЯ | 2012 |
|
RU2601913C2 |
| СТАБИЛЬНЫЕ ПЕПТИДНЫЕ КОМПОЗИЦИИ | 2018 |
|
RU2832964C2 |
| КОМПОЗИЦИЯ АНТИТЕЛ ПРОТИВ CTLA-4 | 2006 |
|
RU2356579C1 |
| КОНФОРМАЦИОННО ОГРАНИЧЕННЫЕ ПЕПТИДЫ | 2020 |
|
RU2838107C2 |
| СОСТАВЫ ДЛЯ ПАРЕНТЕРАЛЬНОЙ ДОСТАВКИ СОЕДИНЕНИЙ И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2539387C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЦИКЛОСПОРИН | 2006 |
|
RU2421209C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПЛАЗМИНОГЕН, И ЕЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2711989C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ПЕМБРОЛИЗУМАБА И ЕЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2791857C2 |
| Водная фармацевтическая композиция рекомбинантного моноклонального антитела к ФНОа | 2017 |
|
RU2764521C2 |
Группа изобретений относится к области фармацевтики, а именно к фармацевтической композиции трициклического соединения двойного ингибитора ФДЭ3/ФДЭ4, способу его получения и его применения, и более конкретно к фармацевтической композиции соединения формулы (I) 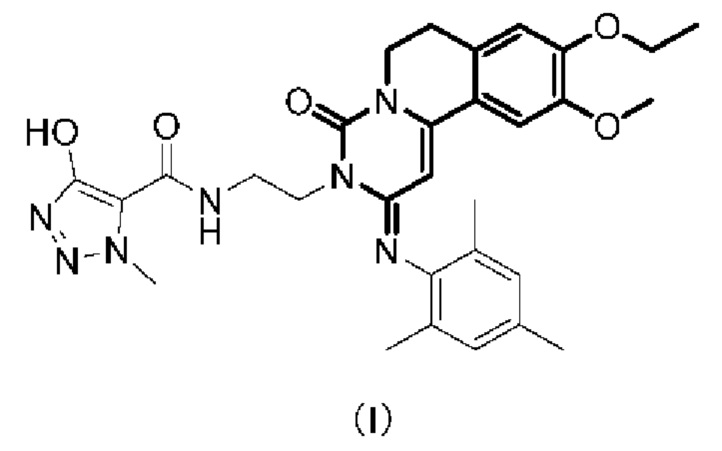 или его фармацевтически приемлемой соли, способу ее получения и ее применения. Фармацевтическая композиция для предотвращения или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4, содержащая: терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемую соль и поверхностно-активное вещество, где указанное поверхностно-активное вещество представляет собой сложный эфир полиоксиэтиленсорбитана и жирной кислоты или сложный эфир сорбитана и жирной кислоты, и где массовое отношение соединения формулы (I) или его фармацевтически приемлемой соли к поверхностно-активному веществу составляет от 1:200 до 100:1, и масса соединения формулы (I) или его фармацевтически приемлемой соли приведена на основании соединения формулы (I). Способ получения вышеуказанной композиции, включающий смешивание поверхностно-активного вещества, соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного, выбранного из группы, состоящей из: хелатирующего металл агента, буферного агента, разбавителя и регулятора осмотического давления. Способ профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4, у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества вышеуказанной композиции. Применение вышеуказанной композиции для получения лекарственного средства для профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4. Указанная группа изобретений позволяет получить и использовать стабильную фармацевтическую композицию с приемлемым уровнем примесей, хорошей диспергируемостью, пригодную для эффективной доставки ингаляционных продуктов, применяемых для профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4. 4 н. и 31 з.п. ф-лы, 6 ил., 9 табл., 9 пр.
или его фармацевтически приемлемой соли, способу ее получения и ее применения. Фармацевтическая композиция для предотвращения или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4, содержащая: терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемую соль и поверхностно-активное вещество, где указанное поверхностно-активное вещество представляет собой сложный эфир полиоксиэтиленсорбитана и жирной кислоты или сложный эфир сорбитана и жирной кислоты, и где массовое отношение соединения формулы (I) или его фармацевтически приемлемой соли к поверхностно-активному веществу составляет от 1:200 до 100:1, и масса соединения формулы (I) или его фармацевтически приемлемой соли приведена на основании соединения формулы (I). Способ получения вышеуказанной композиции, включающий смешивание поверхностно-активного вещества, соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного, выбранного из группы, состоящей из: хелатирующего металл агента, буферного агента, разбавителя и регулятора осмотического давления. Способ профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4, у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества вышеуказанной композиции. Применение вышеуказанной композиции для получения лекарственного средства для профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4. Указанная группа изобретений позволяет получить и использовать стабильную фармацевтическую композицию с приемлемым уровнем примесей, хорошей диспергируемостью, пригодную для эффективной доставки ингаляционных продуктов, применяемых для профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4. 4 н. и 31 з.п. ф-лы, 6 ил., 9 табл., 9 пр.
1. Фармацевтическая композиция для предотвращения или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4, содержащая: терапевтически эффективное количество соединения Формулы (I) или его фармацевтически приемлемой соли и поверхностно-активное вещество,
где указанное поверхностно-активное вещество представляет собой сложный эфир полиоксиэтиленсорбитана и жирной кислоты или сложный эфир сорбитана и жирной кислоты, и
где массовое отношение соединения Формулы (I) или его фармацевтически приемлемой соли к поверхностно-активному веществу составляет от 1:200 до 100:1, и масса соединения Формулы (I) или его фармацевтически приемлемой соли приведена на основании соединения Формулы (I).
2. Фармацевтическая композиция по п. 1, дополнительно содержащая хелатирующий металл агент.
3. Фармацевтическая композиция по п. 1 или 2, дополнительно содержащая буферный агент.
4. Фармацевтическая композиция по любому из пп. 1-3, дополнительно содержащая регулятор осмотического давления.
5. Фармацевтическая композиция по любому из пп. 1-4, дополнительно содержащая разбавитель.
6. Фармацевтическая композиция по любому из пп. 1-5, содержащая соединение Формулы (I) или его фармацевтически приемлемую соль, поверхностно-активное вещество и по меньшей мере один из буферного агента, регулятора осмотического давления, хелатирующего металл агента и разбавителя.
7. Фармацевтическая композиция по любому из пп. 1-6, отличающаяся тем, что поверхностно-активное вещество представляет собой одно или более веществ, выбранных из группы, состоящей из Tween и Span.
8. Фармацевтическая композиция по п. 7, отличающаяся тем, что поверхностно-активное вещество представляет собой одно или более веществ, выбранных из группы, состоящей из Tween 20, Tween 80 и Span 20.
9. Фармацевтическая композиция по любому из пп. 1-8, отличающаяся тем, что поверхностно-активное вещество имеет концентрацию от 0,01 мг/мл до 8 мг/мл.
10. Фармацевтическая композиция по п. 3, отличающаяся тем, что буферный агент представляет собой один или более, выбранных из группы, состоящей из серной кислоты, соляной кислоты, гидроксида натрия, лимонной кислоты, цитрата натрия, молочной кислоты, лактата натрия, уксусной кислоты, ацетата натрия, фосфата тринатрия, дигидрофосфата натрия, гидрофосфата динатрия, дигидрофосфата калия, винной кислоты, тартрата натрия, глицина, борной кислоты и фталевой кислоты.
11. Фармацевтическая композиция по п. 10, отличающаяся тем, что буферный агент выбран из группы, состоящей из лимонной кислоты, цитрата, винной кислоты, тартрата, фосфорной кислоты и фосфата.
12. Фармацевтическая композиция по п. 11, отличающаяся тем, что буферный агент выбран из группы, состоящей из цитрата, тартрата и фосфата.
13. Фармацевтическая композиция по любому из пп. 3 и 10-12, отличающаяся тем, что буферный агент имеет концентрацию от 0,01 мг/мл до 50 мг/мл.
14. Фармацевтическая композиция по любому из пп. 3 и 10-13, отличающаяся тем, что буферный агент используют для регулирования рН фармацевтической композиции в диапазоне от 3,0 до 8,5.
15. Фармацевтическая композиция по п. 4, отличающаяся тем, что регулятор осмотического давления представляет собой один или более, выбранных из группы, состоящей из хлорида натрия, хлорида калия, глюкозы, маннита и ксилита.
16. Фармацевтическая композиция по п. 2, отличающаяся тем, что хелатирующий металл агент представляет собой один или более, выбранных из группы, состоящей из эдетовой кислоты, эдетата динатрия и эдетата динатрия-кальция.
17. Фармацевтическая композиция по п. 5, отличающаяся тем, что разбавитель представляет собой один или более, выбранных из группы, состоящей из воды, этанола и глицерина.
18. Фармацевтическая композиция по любому из пп. 1-17, содержащая соединение Формулы (I), поверхностно-активное вещество, буферный агент, регулятор осмотического давления, хелатирующий металл агент и разбавитель, отличающаяся тем, что соединение Формулы (I) имеет концентрацию от 0,002 мг/мл до 50 мг/мл, поверхностно-активное вещество имеет концентрацию от 0,02 мг/мл до 3 мг/мл, буферный агент имеет концентрацию от 0,1 мг/мл до 25 мг/мл, регулятор осмотического давления имеет концентрацию от 5 мг/мл до 9 мг/мл, а хелатирующий металл агент имеет концентрацию от 0,01 мг/мл до 5 мг/мл.
19. Фармацевтическая композиция по любому из пп. 1-18, содержащая соединение Формулы (I), Tween 80, дигидрофосфат натрия или его моногидрат, гидрофосфат динатрия, хлорид натрия, эдетат динатрия и воду.
20. Фармацевтическая композиция по п. 19, отличающаяся тем, что соединение формулы (I) имеет концентрацию от 0,002 мг/мл до 50 мг/мл, Tween 80 имеет концентрацию от 0,02 мг/мл до 3 мг/мл, дигидрофосфат натрия и гидрофосфат динатрия имеют концентрацию от 0,1 мг/мл до 25 мг/мл, хлорид натрия имеет концентрацию от 5 мг/мл до 9 мг/мл, а динатрия эдетат имеет концентрацию от 0,01 мг/мл до 5 мг/мл.
21. Фармацевтическая композиция по любому из пп. 1-20, отличающаяся тем, что соединение Формулы (I) представляет собой кристаллическую форму соединения Формулы (I), которая содержит 5, 6, 7, 8, 9, 10 или 11 дифракционных пиков на порошковой рентгенограмме с использованием излучения Cu Kα, выбранных из следующих углов 2θ: 5,81±0,2°, 8,38±0,2°, 11,16±0,2°, 13,96±0,2°, 14,47±0,2°, 15,01±0,2°, 16,76±0,2°, 17,95±0,2°, 20,83±0,2°, 24,73±0,2°, 26,13±0,2°.
22. Фармацевтическая композиция по любому из пп. 1-20, отличающаяся тем, что соединение Формулы (I) представляет собой кристаллическую форму соединения Формулы (I), имеющую дифракционные пики на порошковой рентгенограмме с использованием Cu Kα-излучения при следующих значениях 2θ: 5,81±0,2°, 13,96±0,2°, 15,01±0,2°, 17,95±0,2° и 24,73±0,2°.
23. Фармацевтическая композиция по любому из пп. 1-20, отличающаяся тем, что соединение Формулы (I) представляет собой кристаллическую форму соединения Формулы (I), имеющую дифракционные пики при следующих 2θ: 5,81±0,2°, 8,38±0,2°, 11,16±0,2°, 13,96±0,2°, 14,47±0,2°, 15,01±0,2°, 16,76±0,2°, 17,95±0,2°, 20,83±0,2°, 24,73±0,2° и 26,13±0,2°.
24. Фармацевтическая композиция по любому из пп. 1-23, отличающаяся тем, что соединение Формулы (I) или его фармацевтически приемлемая соль имеет размер частиц Х50 менее или равный 10 мкм.
25. Фармацевтическая композиция по п. 24, отличающаяся тем, что соединение Формулы (I) или его фармацевтически приемлемая соль имеет размер частиц Х50 менее или равный 5 мкм и Х90 менее или равный 10 мкм.
26. Фармацевтическая композиция по п. 1, отличающаяся тем, что массовое отношение соединения Формулы (I) или его фармацевтически приемлемой соли к поверхностно-активному веществу составляет от 1:150 до 50:1.
27. Фармацевтическая композиция по п. 26, отличающаяся тем, что массовое отношение соединения Формулы (I) или его фармацевтически приемлемой соли к поверхностно-активному веществу составляет от 1:50 до 25:1.
28. Фармацевтическая композиция по п. 27, отличающаяся тем, что массовое отношение соединения Формулы (I) или его фармацевтически приемлемой соли к поверхностно-активному веществу составляет от 1:1 до 15:1.
29. Фармацевтическая композиция по любому из пп. 1-28, отличающаяся тем, что фармацевтическая композиция находится в форме суспензии.
30. Способ получения фармацевтической композиции по п. 6, включающий: смешивание поверхностно-активного вещества, соединения Формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного, выбранного из группы, состоящей из: хелатирующего металл агента, буферного агента, разбавителя и регулятора осмотического давления.
31. Способ профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4, у млекопитающего, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции по любому из пп. 1-29.
32. Способ по п. 31, отличающийся тем, что млекопитающее представляет собой человека.
33. Способ по п. 32, отличающийся тем, что состояние, связанное с ФДЭ3 и/или ФДЭ4, выбрано из группы, состоящей из астмы или хронической обструктивной болезни легких.
34. Применение фармацевтической композиции по любому из пп. 1-29 для получения лекарственного средства для профилактики или лечения состояния, связанного с ФДЭ3 и/или ФДЭ4.
35. Применение по п. 34, отличающееся тем, что состояние, связанное с ФДЭ3 и/или ФДЭ4, выбрано из группы, состоящей из астмы или хронической обструктивной болезни легких.
| CN 110403935 A, 05.11.2019 | |||
| CN 106794157 B, 31.05.2017 | |||
| Насадка на сваю | 1985 |
|
SU1348453A1 |

