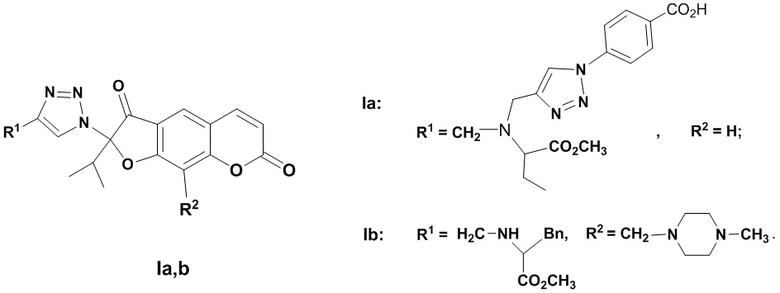
Изобретение относится к новым химическим соединениям, конкретно к 2-(1,2,3-триазолил)-замещенным линейным фурокумаринам общей формулы (I)
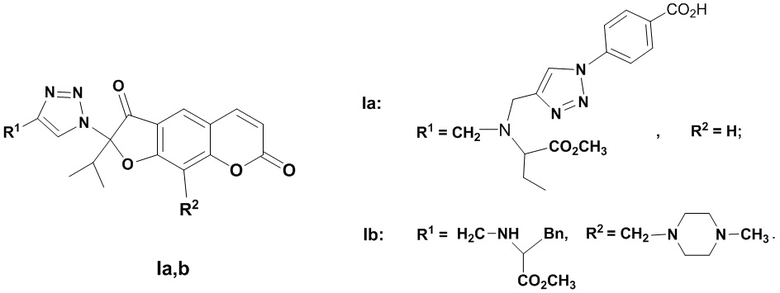 ,
,
I
обладающим антивирусным действием в отношении вируса гриппа А/H1N1. Указанные свойства позволяют предполагать возможность использования соединения в медицине.
Грипп - это респираторная инфекция, характеризующаяся выраженной общей интоксикацией, поражением эпителия верхних дыхательных путей и геморрагическим синдромом. Из-за высокой заразности сезонный грипп быстро приобретает характер эпидемии, иногда пандемии, охватывающей весь земной шар [Neumann G., Noda T., Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus // Nature. - 2009. - V. 459. - P. 931-939].
На основании различий в структуре внутренних антигенов выделяют три подтипа вируса гриппа, имеющих значение для человеческой популяции: А, В и С. Наиболее опасным является вирус гриппа А. Печально известная «испанка» была пандемией вируса гриппа А, свиной грипп и птичий грипп также являются вариантами вируса гриппа А. Вирусы гриппа В и С могут вызывать локальные вспышки заболевания, но причиной массовой эпидемии не являются. Общепризнанным методом профилактики гриппа и снижения ущерба от его эпидемий является вакцинация. Но вакцинация не решает всех проблем по обеспечению защиты от инфекции в связи с быстрой эволюцией вируса.
Применяемые в медицинской практике производные адамантана амантадин и ремантадин (ингибиторы оболочечного белка М2), эффективны против инфекции, вызываемой различными штаммами вируса гриппа типа А [Машковский М.Д. Лекарственные средства // Москва, Новая волна. - 2010. - С. 884]. Отечественный препарат арбидол (умифеновир) по механизму противовирусного действия относится к ингибиторам слияния (фузии), взаимодействует с гемагглютинином вируса и препятствует слиянию липидной оболочки вируса и клеточных мембран, одобрен для использования в профилактике инфекции вируса гриппа у взрослых и детей и проявляет хорошую эффективность до развития симптоматики заболевания [Киселев О.И., Малеев В.В., Деева Э.Г., Ленева И.А., Селькова Е.П., Осипова Е.А. Клиническая эффективность препарата Арбидол (умифеновир) в терапии гриппа у взрослых: промежуточные результаты многоцентрового двойного слепого рандомизированного плацебоконтролируемого исследования. Терапевтический архив. - 2015. - Т. 87(1). - С. 88-96].
При гриппе эффективен препарат ингавирин (имидазолилэтанамид пентандиовой кислоты), механизм действия которого реализуется на уровне инфицированных клеток за счет активации факторов врожденного иммунитета, подавляемых вирусными белками [Геппе Н.А., Малахов А.Б., Кондюрина Е.Г. Обоснование выбора противовирусной терапии ОРВИ в педиатрии (мета-анализ клинических исследований эффективности имидазолилэтанамида пентандиовой кислоты у детей разных возрастных групп // Вопросы практической педиатрии. - 2020. - Т. 15(3). - С. 106-114.]. Необходимо отметить, что перечисленные препараты эффективны для профилактики гриппа или в первые 3 дня болезни, затем их эффективность резко снижается. Дополнительно, необходимо применение патогенетических и симптоматических средств: жаропонижающие, противовоспалительные, противокашлевые.
Препараты занамивир, осельтамивир и перамивир (ингибиторы нейраминидазы), направлены специфично против вирусов гриппа типа А и В, подавляют активность нейраминидазы вирусов и, соответственно, препятствуют высвобождению вирионов и дальнейшему распространению инфекции. Ряд недавних работ, однако, продемонстрировал, что использование осельтамивира обусловливает селекцию вирусов, резистентных к воздействию препарата [Nicholson K.G., Wood J.M., Zambon M. Influenza // Lancet. - 2003. - V. 362. - P. 1733-1745; Samson M, Pizzorno A, Abed Y, Boivin G. Influenza virus resistance to neuraminidase inhibitors // Antiviral Res. - 2013. - V. 98(2). - P.174-185].
Кроме того, применение ингибиторов нейраминдазы лимитируется их токсичностью [Alame, M. M.; Massaad, E.; Zaraket, H. Peramivir: A novel intravenous neuraminidase inhibitor for treatment of acute influenza infections // Front. Microbiol. - 2016. - V. 7. - P. 450].
В последнее время внимание привлекают препараты - ингибиторы вирусной полимеразы - фавипиравир, пимодивир и балоксавир, которые ингибируют в доклинических моделях вирусы гриппа А, включая вирусы, представляющие пандемическую угрозу, и вирусы, устойчивые к одобренным в настоящее время противовирусным препаратам, а два (фавипиравир и балоксавир) также ингибируют вирусы гриппа В [Shiraki K., Daikoku T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections // Pharmacology and Therapeutics - 2020. - V. 209. - Article N 107512]. Комбинации новых ингибиторов полимеразы с ингибиторами нейраминидазы демонстрируют синергизм на доклинических моделях и в настоящее время проходят клинические испытания на госпитализированных пациентах [Valenzuela-Sánchez F, Valenzuela-Méndez B, Rodríguez-Gutiérrez JF, Estella Á. Latest developments in early diagnosis and specific treatment of severe influenza infection // J. Intensive Med. - 2023. - V. 4(2). - P. 160-174].
Несмотря на прогресс в разработке противовирусных средств, появление новых и возвращающихся инфекций, большая распространенность сочетанных форм, а также лекарственная устойчивость обуславливают важность и актуальность синтеза и изучения новых химических соединений, обладающих противовирусной активностью, в частности, антивирусным действием в отношении вируса гриппа А/H1N1.
Известны природные и синтетические фурокумарины, обладающие противовирусной активностью вируса гриппа A/H1N1. Так, линейный фурокумарин псорален (II) проявляет антивирусную активность, однако обладает высокой фототоксичностью и способностью сшивать дезоксирибонуклеиновую кислоту (ДНК). Ангулярный фуранокумарин ангелицин (III) явился стартовой молекулой-хитом для поиска агентов, эффективных против штамма вируса гриппа A/H1N1. Получены ключевые представления о молекулярном механизме производных ангелицина, которые указывают на вирусные рибонуклеопротеины в качестве вероятной молекулярной мишени для этих агентов [Yeh J.Y., Coumar M.S., Horng J.T., Shiao H.Y., Kuo F.M., Lee H.L., Chen I.C., Chang C.W., Tang W.F., Tseng S.N., Chen C.J., Shih S.R., Hsu J.T., Liao C.C., Chao Y.S., Hsieh H.P. Anti-influenza drug discovery: structure-activity relationship and mechanistic insight into novel angelicin derivatives // J. Med. Chem. - 2010. - V. 53. - P. 1519-1533].
Линейный фурокумарин изоимператорин (IV) проявляет активность в отношении вирусов гриппа (H1N1, H9N2) in vitro (значения EC50 7.67 ± 0.93 μM и 6.72 ± 0.51 μM, соответственно) [Lee B.W., Ha T.K.Q., Cho H.M., An J.-P., Kim S.K., Kim C.S., Kim E., Oh W.K. Antiviral activity of furanocoumarins isolated from Angelica dahurica against influenza a viruses H1N1 and H9N2. J. Ethnopharmacol. - 2020. - V. 259. - Article N 112945]. Механизм действия изоимператорина (IV) связан с ингибированием репликации вируса гриппа путем блокирования белка нейраминизазы [Lai Y., Han T., Zhan S., Jiang Y., Liu X., Li G. Antiviral activity of isoimperatorin against influenza A virus in vitro and its inhibition of neuraminidase // Front. Pharmacology. - 2021. - V. 12. - Article N 657826].

Для создания новых ингибиторов вирусной нейраминидазы выполнены модификации противовирусных препаратов осельтамивира и занамивира с введением 1,2,3-триазольных заместителей (соединения типов Va, Vb, VI) [Wang P., Oladejo B.O., Li C., Fu L., Zhang S., Qi J., Lv X., Li X. Structure-based design of 5′-substituted 1,2,3-triazolylated oseltamivir derivatives as potent influenza neuraminidase inhibitors // RSC Adv. - 2021. - V. 11. - P. 9528-9541; Das A., Adak A.K., Ponnapalli K., Lin C.-H., Hsu K.-C., Yang J.-M., Hsu T.-A., Lin C.-C. Design and synthesis of 1,2,3-triazole-containing N-acyl zanamivir analogs as potent neuraminidase inhibitors // Eur. J. Med. Chem. - 2016. - V. 123. - P. 397-406]. Предложенные модификации с введением триазольных фрагментов оказались весьма успешными в плане повышения противовирусной активности. Наибольшую ингибирующую активность в отношении нейроминидазы в ряду соединений (Va,b) проявило соединение, содержащее 3-аминофенильный заместитель в положении С-4 триазольного цикла. Это соединение продемонстрировало приемлемую эффективность (IС50 = 11μM) и низкую цитотоксичность. Соединения типа (VI) проявили ингибирующую активность в наномолярном диапазоне и, в основном, показали большую ингибирующую активность по сравнению с N-алкилированными или модифицированными карбоксамидом производными (IС50 = 2.9 - 33 μM). При этом ингибирующая активность снижалась с увеличением длины углеродной цепи от N-ацилгуанидиногруппы (увеличением значения n).
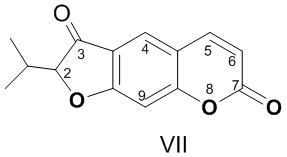
Задачей предлагаемого изобретения является создание новых противовирусных агентов химической модификацией доступного линейного фурокумарина ореозелона (VII). Соединение (VII) выделяли из корней растений рода горичник (Peucedanum morisonii Bess., Peucedanum ruthenicum M. Bieb., Р. tauricum M. Bieb.) по ранее описанным методикам [Шульц Э.Э., Петрова Т.Н., Шакиров М.М., Черняк, Е.И., Покровский Л.М., Нехорошев С.А., Толстиков Г.А. Кумарины корней горичника Морисона (Peucedanum morisonii Bess.) // Химия в интересах устойчивого развития. - 2003. - Т. 11. - С. 683-688; Лазуревский, Г.В.; Терентьева, И.В.; Шамшурин А.А. Практические работы по химии природных соединений. - Изд-во Высшая школа, Москва, 1966, 334 с.].
Поставленная задача решается новыми химическими соединениями - производными линейного фурокумарина ореозелона (VII), общей формулы (I), содержащими бис-гетероциклический заместитель в положении С-2 бис-(1,2,3-триазолильный) или гетероциклические заместители в положениях С-2 (1,2,3-триазолильный) и С-9 (N-метилпиперазино-метильный).
I
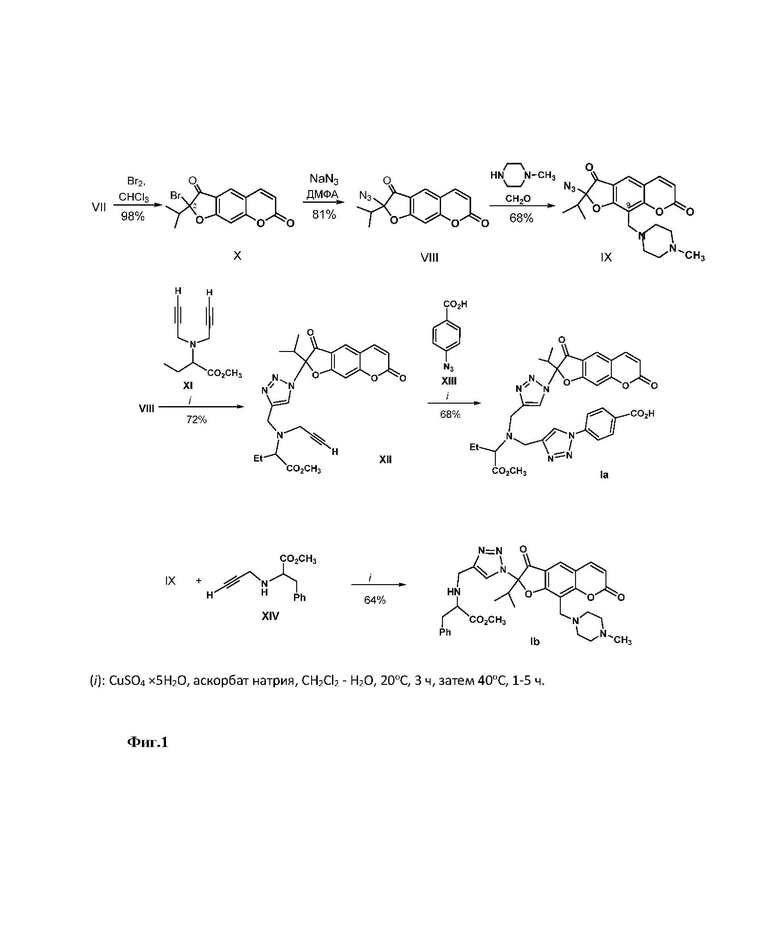
Способ получения 2-(1,2,3-триазолил)замещенных линейных фурокумаринов (Ia,b) приведен на Фиг. 1. В качестве исходных соединений использовали 2-азидоореозелон (VIII) или 2-азидо-9-(4-метилпиперазинометил)ореозелон (IX), которые синтезировали из ореозелона (VII). Бромирование ореозелона (VII) эквивалентным количеством брома в хлороформе гладко приводило к образованию 2-бромореозелона (Х) (выход 98%) [Осадчий C.А., Шульц Э.Э., Шакиров М.М., Толстиков Г.А. Исследование растительных кумаринов. Некоторые превращения пеуцеданина // Известия PАН, Сер. хим. - 2006. - С. 362-366]. Реакцией 2-бромореозелона (X) с азидом натрия при нагревании в ДМФА синтезировали 2-азидоореозелон (VIII) (выход 81%). Реакцией Манниха соединения (VIII) с формальдегидом и N-метилпиперазином при кипячении в растворе метанола получали 2-азидо-9-(4-метилпиперазинометил)ореозелон (IX) (выход 78%). Взаимодействие азида (VIII) с метил 2-(дипроп-2-иниламино)бутаноатом (XI) (получали реакцией метилового эфира аминомасляной кислоты с пропаргилом бромистым) (0.5 экв.) в присутствии медного купороса и аскорбата натрия в водн. хлористом метилене (0.01М раствор азида VIII в СH2Сl2) приводило к образованию 2-[(4-пропиниламинометил)-1,2,3-триазол-1-ил]ореозелона (XII) (выход 72%). Реакцией алкинилзамещенного фурокумарина (XII) с 4-азидобензойной кислотой (XIII) в присутствии медного купороса и аскорбата натрия синтезировали бис-триазолилзамещенный фурокумарин (Ia) (выход 68%).
Взаимодействие азидa (IX) с метил 3-фенил-2-(проп-2-иниламино)бутаноатом (XIV) в условиях CuAAC-реакции (присутствие медного купороса и аскорбата натрия в водн. хлористом метилене) приводило к триазолилзамещенному фурокумарину (Ib) (выход 64%).
Достоинством изобретения являются новые химические соединения (Iа,b), обладающие противовирусной активностью. Физико-химические данные впервые полученных соединений (Iа,b) приведены в примерах 1, 2.
На первой стадии биологических экспериментов определяли 50% цитотоксическую дозу (ЦТД50) агентов (Ia,b), исходного фурокумарина ореозелона (VII) и препаратов сравнения в культуре клеток MDCK. Далее оценивали противовирусную активность каждого агента и определяли 50% ингибирующую концентрацию (ЭД50), а также вычисляли химиотерапевтический индекс (ХТИ) - отношение ЦТД50 к ЭД50. Это соотношение отражает эффективность исследуемого соединения как противовирусного агента. Полученные данные сравнивали с соответствующими показателями препаратов сравнения - ремантадин и осельтамивир карбоксилат.
Исследования биологической активности соединений (Ia,b) показали их высокую эффективность как ингибиторов репродукции вируса гриппа A/ Puerto Rico/8/34 (Н1N1) (Таблица). Соединения (Iа,b) ингибировали репродукцию вируса с ЭД50 в микромолярном диапазоне, на порядок и более превосходили по активности (ингибирующей концентрации) препарат сравнения ремантадин, хотя и уступали препарату сравнения осельтамивир карбоксилату. Кроме того, значение ЭД50 1,2,3-триазолилзамещенных кумаринов (Ia,b) более чем на порядок ниже, чем у исходного соединения (VII), что свидетельствует об усилении противовирусных свойств в результате модификации.
Таблица. Результаты тестирования соединений (Ia,b) и (VII) в отношении вируса гриппа A/ Puerto Rico/8/34 (Н1N1) в клетках MDCK
ЦТД50 (MDCK) - концентрации, вызывающие 50% гибель клеточной линии MDCK; ЭД50(H1N1) представляет собой концентрацию, приводящую к 50% ингибированию цитопатогенного действия вируса гриппа H1N1. ХТИ - химиотерапевтический индекс, отношение ЦТД50/ЭД50. Представленные данные являются средними из трех независимых экспериментов.
Технический результат: повышение эффективности подавления репродукции вируса гриппа и расширение ассортимента ингибиторов репродукции вируса гриппа для преодоления лекарственной устойчивости современных вирусных штаммов.
Изобретение иллюстрируется следующими примерами.
Пример 1
Синтез 4-(4-{[({1-(2-изопропил-3,7-диоксо-2,3-дигидро-7H-фуро[3,2-g]хромен-2-ил)-1H-1,2,3-триазол-4-ил}метил) -(1-метокси-1-оксобутан-2-ил)амино]метил}-1H-1,2,3-триазол-1-ил)бензойной кислоты (Iа). Получение 2-азидоореозелона (VIII) описано в работе [Lipeeva A.V., Shults E.E., Makhneva E.A., Shakirov M.M., Tolstikov G.A. Study of plant coumarins. 12. Synthesis of 2-(1,2,3-triazolyl)modified furocoumarins // Chem. Heterocycl. Compd. - 2013. - V. 49. - P. 551-560]. Стадия1. Получение метил 2-[({1-(2-изопропил-3,7-диоксо-3,7-дигидро-2H-фуро[3,2-g]хромен-2-ил)-1H-1,2,3-триазол-4-ил}метил)(проп-2-инил)амино]бутаноата (XII). К раствору азида ореозелона (VIII) (0.86 г, 3.0 ммоль) в 300 мл CH2Cl2 при перемешивании одной порцией прибавили раствор аскорбата натрия (45 мг, 10 мол. %, 0.3 ммоль) и медного купороса (18 мг, 5 мол. %, 0.15 ммоль) в 10 мл Н2О, а затем прикапывали раствор метил 2-(дипроп-2-иниламино)бутаноата (XI) (0.59 г, 3.05 ммоль) в 20 мл СH2Сl2. Реакционную смесь перемешивали в течение 5 ч при комнатной температуре и 1 ч при нагревании до 40°С (контроль ТСХ). По охлаждении смесь разбавили водой (30 мл), органический слой отделили, водный слой обработали хлористым метиленом (5×20 мл), объединенные органические экстракты промыли насыщенным раствором соли, сушили над MgSO4, отфильтровали осушитель и растворитель упарили в вакууме. Остаток хроматографировали на колонке с силикагелем, собрали фракцию соединения (XII). Выход 72% (1.04 г). Желтое масло. ИК спектр, ν, см-1: 3180, 3062, 2248, 1735, 1670, 1625, 1581, 1500, 1390, 1351, 1284, 1222, 1203, 1126, 1093, 1035, 948, 863, 827, 752, 742, 700. УФ спектр (EtOH), λмакс., нм (lgε): 255 (4.43), 294 (3.92), 306 (3.84), 351(3.97). Спектр ЯМР 1H (400 MГц, CDCl3, δ, м.д., J, Гц): 0.88 (3Н, д, J = 7.0, CH3), 0.92 (3Н, д, J = 7.0, CH3), 1.25 (3H, т, J = 6.8, CH3), 2.18 (2H, ддд, J = 8.5, 6.8, 4.2, H-9'), 2.51 (1H, с, ≡CH), 2.81 (1Н, м, J 7.0, CH-iPr), 3.35 (2H, c, H-1''), 3.67 (3H, с, OCH3), 3.75 (2H, с, H-6'), 3.85 (1H, дд, J = 8.5, 4.2, H-8'), 6.36 (1H, д, J = 9.6, H-6), 7.06 (1H, с, H-9), 7.57 (1H, с, H-5'), 7.70 (1Н, д, J = 9.6, H-5), 7.82 (1H, с, H-4). Спектр ЯМР 13С (150 МГц, СDCl3, δ, м.д.): 11.6 (CH3), 15.4 (CH3), 15.8 (CH3), 29.7 (C-9'), 33.8 (CH-iPr), 45.7 (C-1''), 47.8 (C-6'), 51.4 (OCH3), 69.6 (C-8'), 73.9 (C-3''), 79.6 (C-2''), 96.1 (С-2), 101.3 (C-9), 115.6 (C-6), 115.8 (C-4a), 116.7 (C-5a), 125.9 (C-5'), 127.3 (C-4), 143.3 (C-4'), 145.8 (C-5), 158.6 (C-8a), 161.3 (C-7), 161.8 (C-9а), 171.8 (C=O), 191.6 (C-3). Найдено, %: C 62.38; H 5.14; N 11.59. C25H26N4O6. Вычислено, %: C 62.75; H 5.48; N 11.71. Стадия 2. Получение соединения (Iа). К раствору 4-азидобензойной кислоты (XIII) (0.36 г, 2.2 ммоль) в хлористом метилене (50 мл) при перемешивании последовательно добавили раствор аскорбата натрия (37 мг, 10 мол. %, 0.2 ммоль) и медного купороса (16 мг, 5 мол. %, 0.1 ммоль) в 10 мл Н2О, а затем раствор терминального алкина (XII) (0.96 г, 2 ммоль) в хлористом метилене (5 мл). Смесь перемешивали 3 ч при комнатной температуре и 5 ч при 40°С (контроль ТСХ). Охлажденную реакционную массу разбавили водой (10 мл), проэкстрагировали хлористым метиленом (5×50 мл). Объединенные органические слои сушили над MgSO4, осушитель отфильтровали и удалили растворитель в вакууме. Остаток хроматографировали на колонке с силикагелем, после перекристаллизации фракции продукта из этанола получали соединение (Iа). Выход 68% (0.86 г). Бесцветный кристаллический порошок. Т.пл. 201-202°C (этанол). ИК спектр, ν, см-1: 3440, 3417, 1730, 1708, 1652, 1610, 1505, 1459, 1402, 1378, 1280, 1186, 1145, 1124, 1087, 998, 977, 850, 823, 765, 722. УФ спектр, λмакс., нм (lg ε): 255(4.29), 292 (3.92), 305 (3.71), 350 (3.81). Спектр ЯМР 1H (400 МГц, CDCl3, δ, м. д., J, Гц): 0.90 (3Н, д, J = 7.0, CH3), 1.13 (3Н, д, J = 7.0, CH3), 1.19 (3H, т, J = 6.8, CH3), 2.31 (2H, ддд, J = 8.5, 6.8, 4.2, H-9'), 2.81 (1Н, м, J 7.0, CH-iPr), 3.58 (2H, с, H-6'), 3.63 (3H, с, OCH3), 3.76 (2H, c, H-1''), 3.85 (1H, дд, J = 8.5, 4.2, H-8'), 6.38 (1H, д, J = 9.6, H-6), 7.03 (1H, с, H-9), 7.40 (2Н, д, J = 8.6, H-2''',6'''), 7.57 (1H, с, H-5'), 7.68 (1Н, д, J = 9.6, H-5), 7.62 (1H, с, H-4), 7.85 (1H, с, H-5''), 8.21 (2Н, д, J = 8.6, H-3''',5'''), 10.55 (1H, с, OH). Спектр ЯМР 13С (150 МГц, СDCl3, δ, м.д.): 12.6 (CH3), 15.0 (CH3), 15.5 (CH3), 29.7 (C-9'), 31.6 (CH-iPr), 46.5 (C-6'), 51.2 (OCH3), 54.8 (C-1''), 67.2 (C-8'), 100.5 (С-2), 107.9 (C-9), 115.2 (C-6), 115.5 (C-4a), 116.2 (C-5a), 122.23 (C-5''), 123.8 (C-2''',6'''), 124.6 (C-5'), 126.2 (C-4), 128.5 (C-4'''), 135.3 (C-3''',5'''), 138.9 (C-1'''), 141.1 (C-4''), 142.8 (C-4'), 144.5 (C-5), 158.1 (C-8a), 161.3 (C-7), 162.2 (C-9а), 171.1 (C=O), 171.8 (C=O), 191.6 (C-3). Найдено, %: C 60.19; H 4.53; N 15.01. C32H31N7O8. Вычислено, %: C 59.90; H 4.87; N 15.28.
Пример 2
Синтез метил 2-[(1-{2-изопропил-9-[(4-метилпиперазин-1-ил)метил]-3,7- диоксо-3,7-дигидро-2H-фуро[3,2-g]хромен-2-ил}-1H-1,2,3-триазол-4-ил)метиламино]-3-фенилпропаноат (Ib). Получение 2-азидо-9-(4-метилпиперазинометил)ореозелона (IX) описано в работе [Lipeeva A.V., Pokrovsky M.A., Baev D.S., Shakirov M.M., Bagryanskaya I.Y., Tolstikova T.G., Pokrovsky A.G., Shults E.E. Synthesis of 1H-1,2,3-triazole linked aryl(arylamidomethyl) - dihydrofurocoumarin hybrids and analysis of their cytotoxicity // Eur. J. Med. Chem. - 2015. - V. 100. - P.119-128]. К раствору азида (IX) (0.79 г, 2.0 ммоль) в 10 мл CH2Cl2 при перемешивании прибавили раствор аскорбата натрия (37 мг, 10 мол. %, 0.2 ммоль) и медного купороса (16 мг, 5 мол. %, 0.1 ммоль) в 10 мл Н2О, а затем метил 3-фенил-2-(проп-2-иниламино)пропаноат (XIV) (0.48 г, 2.2 ммоль). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре и 3 ч при нагревании до 40°С (контроль ТСХ). По охлаждении смесь разбавили водой (10 мл), продукт экстрагировали хлористым метиленом (5×20 мл), объединенные органические экстракты промыли насыщенным раствором соли, сушили над MgSO4, осушитель отфильтровали, растворитель упарили в вакууме, остаток хроматографировали на колонке с силикагелем. Фракцию, содержавшую продукт, обработали эфиром, отфильтровали 2-(1,2,3-триазолил)-9-(4-метилпиперазинометил)ореозелон (Ib). Выход 64% (0.78 г), желтый аморфный порошок. ИК спектр, ν, см-1 : 3440, 3149, 3062, 1735, 1681,1625, 1580, 1498, 1454, 1390, 1351, 1284, 1222, 1203, 1126, 1093, 1035, 995, 948, 910, 863, 827, 752, 742, 700. УФ спектр, λмакс., нм (lg ε): 255 (4.12), 291(4.03), 308(3.92), 348(3.65). Спектр ЯМР 1H (400 МГц, CDCl3, δ, м. д., J, Гц): 0.97 (3Н, д, J = 7.0, CH3), 0.98 (3Н, д, J = 7.0, CH3), 2.22 (3H, с, Me), 2.28-2.47 (8H, м, 4×CH2), 2.88 (1Н, м, J 7.0, CH-iPr), 3.06 (1H, дд, J = 12.3, 4.0, H-9'), 3.51 (1H, дд, J = 12.3, 7.8, H-9'), 3.61 (3H, с, OCH3), 3.65 (2H, с, СH2 при H-9), 3.78 (2H, с, H-6'), 3.86 (1H, дд, J = 7.8, 4.0, H-8'), 6.36 (1H, д, J = 9.6, H-3), 7.09-7.22 (3H, м, H-2'',4'',6''), 7.50 (2H, м, H-3'',5''), 7.55 (1H, с, H-5'), 7.71 (1Н, д, J = 9.6, H-5), 7.88 (1H, с, H-4), 9.28 (1H, c, NH). Спектр ЯМР 13С (150 МГц, CDCl3, δ, м. д.): 15.2 (CH3), 15.6 (CH3), 31.1 (CH-iPr), 40.1 (C-9'), 42.3 (C-6'), 45.24 (CН2 при С-9), 46.1 (СН3), 51.7 (OCH3), 52.1, 52.8 (4×CН2-пиперазино), 66.2 (C-8'), 96.2 (С-2), 102.9 (C-9), 115.4 (C-6), 116.4 (C-4a), 116.6 (C-5a), 125.7 (C-4''), 126.2 (C-4), 126.5 (C-5'), 128.1 (C-2'',6''), 129.1 (C-3'',5''), 136.9 (C-1''), 142.8 (C-5), 144.5 (C-4'), 158.6 (C-8a), 161.3 (C-7), 163.6 (C-8а), 173.1 (C=O), 190.7 (C-3). Hайдено, %: C 64.24; H 6.19; N, 13.42. C33H38N6O6. Вычислено, %: C 64.48; H, 6.23; N, 13.67.
Пример 3
Изучение цитотоксичности новых 2-(1,2,3-триазолил)- или бис(триазолил)-замещенных производных фурокумаринов (Ia,b). Клетки Madin-Darby Canine Kidney (MDCK) (ATCC # CCL-34) культивировали в среде MEM с добавлением 5% эмбриональной бычьей сыворотки (FBS) и ципрофлоксацина (2 мг/мл). Соединения растворяли в ДМСО до концентрации 5 мг/мл и готовили серийные двукратные разведения (10-4 мг/мл). Клетки MDCK высевали в 96-луночные планшеты и культивировали в МЕМ с добавлением 5% фетальной телячьей сыворотки. После формирования монослоя клетки промывали бессывороточной МЕМ, наносили на клетки растворы изучаемых соединений и инкубировали 48 ч при 37°С. После инкубации клетки дважды промывали фосфатно-солевым буфером (PBS) и количество выживших клеток оценивали с помощью микротетразолиевого теста (МТТ). Вкратце, добавляли 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия бромид (ICN Biochemicals Inc., США, 0,5 мг/мл, 0,1 мл на лунку) с последующей инкубацией планшетов при 37°С в 5% CO2 в течение 60 мин. Окрашенный осадок растворяли в 100 мл ДМСО. Планшеты осторожно встряхивали и оставляли в темноте при комнатной температуре на 30 мин. Оптическую плотность измеряли с помощью спектрофотометра (Victor 1420, Perkin Elmer, Финляндия) при длине волны 535 нм. На основании этих данных для каждого соединения рассчитывали значение ЦТД50 (концентрация соединения, при которой погибает 50% клеток). Полученные результаты приведены в таблице.
Пример 4
Изучение противовирусной активности соединений (Ia,b) в отношении вируса гриппа A/PR/8/34 (H1N1). Вирусы гриппа A/Puerto Rico/8/34 (H1N1) были получены из коллекции вирусов ФГБУ Научно-исследовательского института гриппа им. А.А. Смородинцева МЗ РФ (Россия). Перед экспериментом вирусы размножали в аллантоисной полости 10-12-дневных куриных эмбрионов в течение 48 ч при 36°С (вирусы гриппа А). Инфекционный титр вируса определяли в клетках MDCK в 96-луночных планшетах.
Противовирусная активность в отношении вируса гриппа. Анализ снижения выхода вируса. Серийные двукратные разведения соединений готовили в среде МЕМ («Биолот», РФ), содержащей 2 мМ аргинина (Sigma-Aldrich, США), 2 мМ глутамина (Sigma-Aldrich, США) и 2 мг/мл трипсина (Sigma-Aldrich, США). ) и инкубировали с клетками MDCK в течение 1 ч при 36°С в присутствии 5% СО2 (объем 100 мл на лунку). Каждую концентрацию тестировали трижды; необработанные клетки использовали в качестве контроля. Затем культуру клеток дважды промывали PBS и инокулировали соответствующим вирусом при множественности заражения 0.1 (объем 100 мл на лунку) и соответствующем разведении соединения (объем 100 мл на лунку; общий объем каждой лунки составлял 200 мл) и инкубировали в течение 24 ч. После инкубации к клеткам MDCK добавляли десятикратные разведения супернатанта и затем инкубировали в течение 48 ч при 36°C в присутствии 5% CO2. Титры вируса в супернатанте определяли методом гемагглютинации. Реакцию гемагглютинации проводили следующим образом. 100 мкл супернатанта переносили в круглодонные лунки, смешивали со 100 мл 1% взвеси куриных эритроцитов и затем инкубировали 1 ч при комнатной температуре. Титр вируса рассматривали как обратную величину конечного разведения инокулята, при котором вирус был способен вызывать положительную гемагглютинацию в 50% лунок и выражали в 50% дозах, заражающих культуру ткани (TCID50). Противовирусную активность соединений оценивали по их способности снижать титр вируса. На основании полученных результатов рассчитывали ЭД50, 50% эффективную дозу (концентрация соединения, снижающую продукцию вируса в два раза по сравнению с контролем) и химиотерапевтический индекс (ХТИ, отношение ЦТД50 к ЭД50). Критерием оценки противовирусной активности является статистически значимое снижение титра вируса при применении препарата по сравнению с контролем.
Статистическая обработка данных. Статистический анализ выполнен с использованием пакета программ GraphPad Prism и Microsoft Office Excel. Экспериментальные результаты представлены как средние значения данных, полученные из трех независимо проведенных экспериментов. Полученные результаты приведены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3-ЗАМЕЩЕННЫЕ 6-(МЕТОКСИКАРБОНИЛ)УМБЕЛЛИФЕРОНЫ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ГРИППА A/Н1N1 | 2024 |
|
RU2838462C1 |
| Производные 7-гидроксикумарина, соединенные с остатками монотерпенов через триазольный линкер, как ингибиторы репродукции респираторно-синцитиального вируса (РСВ) | 2024 |
|
RU2826560C1 |
| 6,13,13-ТРИМЕТИЛ-6,8,9,12-ТЕТРАГИДРО-6,9-МЕТАНОАЗЕПИНО[2,1-b]ХИНАЗОЛИН-10(7Н)-ОН В КАЧЕСТВЕ ИНГИБИТОРА ВИРУСОВ ГРИППА А | 2017 |
|
RU2664331C1 |
| ИМИНОПРОИЗВОДНЫЕ КАМФОРЫ - ЭФФЕКТИВНЫЕ ИНГИБИТОРЫ РЕПРОДУКЦИИ ВИРУСА ГРИППА (штамм A/California/07/09 (H1N1)pdm09) | 2014 |
|
RU2554934C1 |
| O-Ациламидоксимы и 1,2,4-оксадиазолы, содержащие фрагмент бицикло[2.2.1]гептанона-2, в качестве ингибиторов репродукции филогенетически различных вирусов гриппа А: штаммы A/Puerto Rico/8/34 (H1N1), A/Anhui/1/2013 (H7N9) | 2022 |
|
RU2798171C1 |
| ИМИНОПРОИЗВОДНЫЕ КАМФОРЫ, СОДЕРЖАЩИЕ АРОМАТИЧЕСКИЙ ИЛИ ГЕТЕРОАРОМАТИЧЕСКИЙ ФРАГМЕНТ, - ИНГИБИТОРЫ РЕПРОДУКЦИИ ВИРУСА ГРИППА (штамм A/California/07/09 (H1N1)pdm09) | 2015 |
|
RU2607451C1 |
| Водорастворимые сульфосодержащие полимеры с собственной противовирусной активностью и способ их получения | 2023 |
|
RU2814298C1 |
| Производное декагидро-клозо-декарборатного аниона с метиловым эфиром L-триптофана и его противовирусная активность вируса гриппа А | 2024 |
|
RU2834695C1 |
| Композиция на основе пептида, подавляющего репликацию вируса гриппа А | 2018 |
|
RU2695336C1 |
| СИММЕТРИЧНЫЕ ДИИМИНЫ НА ОСНОВЕ КАМФОРЫ - ИНГИБИТОРЫ РЕПРОДУКЦИИ ВИРУСА ГРИППА (ШТАММ A/California/07/09 (H1N1)pdm09) | 2013 |
|
RU2520967C1 |
Изобретение относится к применению 2-(1,2,3-Триазол-1-ил)замещенных производных линейного фурокумарина ореозелона общей формулы (I), где соединение формулы (I) представляет собой Ia, где R1 представляет собой фрагмент, указанный в формуле изобретения, и R2 представляет собой Н; и Ib, где R1 представляет собой фрагмент (a) и R2 представляет собой фрагмент (b) в качестве средства, обладающего ингибирующей активностью в отношении вируса гриппа A/Н1N1. Технический результат - повышение эффективности подавления репродукции вируса гриппа и расширение ассортимента ингибиторов репродукции вируса гриппа для преодоления лекарственной устойчивости современных вирусных штаммов. 1 ил., 1 табл., 4 пр.

Применение 2-(1,2,3-Триазол-1-ил)замещенных производных линейного фурокумарина ореозелона общей формулы (I)

(I)
в качестве средства, обладающего ингибирующей активностью в отношении вируса гриппа A/Н1N1.
| Липеева А.В | |||
| Полифункциональные гетероциклические соединения на основе фурокумарина пеуцеданина | |||
| Синтез, свойства, биологическая активность | |||
| Диссертация на соискание ученой степени доктора химических наук, 2021, 392 с. | |||
| Липеева А.В | |||
| Триазолилсодержащие производные природных фурокумаринов как перспективные антимикробные агенты | |||
| Лаборатория и |

