1
(21)А0287 П/23-04
(22)05.01.87
(31)816475
(32)06.01.86
(33)US
(46) 23,10.89. Люл. № 39
(71)Е.Р.СквиОб энд Санз, Р1нк. (US)
(72)Джозеф Эдвард Сандин (US)
(53)547.718789.07Г088.8)
(56)Патент США № 4144333, кл. 424-244, опублик. 1979.
(54)СПОСОБ ПОЛУЧЕНИЯ МОНОСУЛЬсЬОКТАМА
(57)Изобретение касается гетероциклических веществ и, в частности, получения соединений общей формулы (I) :
N C(NHj) - S - СН (; - СК N - -О - С(СН7.) - С(0)рн, где К -С(0)- -im-CH-CR Rj- NY - с 0; Y -05(0) - ОН; R, и R - низший ал- кил или R,+ R - (СН,,)-, которые как антибактериальные средства могут быть использованы в медицине. Цель изобретения - создание новых более активных веществ указанного класса. Синтез ведут удалением из карбоксил- защищенного (дифенилметилим, фенил- метилом или трет-бутилом) соединения формулы (I) указанной эфирной группы. Новые вещества малотоксичны и активны в отношении инфекций мочеполового тракта, респираторных инфекций, т.е. против грамположительных и гран- отрицательных бактерий. 1 табл.
(О







| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения азетидиновых соединений | 1983 |
|
SU1329617A3 |
| Способ получения @ -лактамов (его варианты) | 1981 |
|
SU1272981A3 |
| РЕТИНОИДОПОДОБНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2163590C2 |
| ПРИМЕНЕНИЕ ПЕРФТОРАЛКИЛСОДЕРЖАЩИХ КОМПЛЕКСОВ МЕТАЛЛОВ В КАЧЕСТВЕ КОНТРАСТНЫХ ВЕЩЕСТВ В МАГНИТНО-РЕЗОНАНСНОЙ ТОМОГРАФИИ ДЛЯ ВИЗУАЛИЗАЦИИ БЛЯШЕК, ОПУХОЛЕЙ И НЕКРОЗОВ | 2001 |
|
RU2290206C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОБЕНЗО[b][1,4]ДИАЗЕПИН-2-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ I mGluR2 | 2002 |
|
RU2270197C2 |
| ФЕНИЛСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ ИЗОКСАЗОЛА И СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2133742C1 |
| ФЕНИЛОКСАЗОЛИДИНОНЫ, ИМЕЮЩИЕ С-С-СВЯЗЬ С 4-8-ЧЛЕННЫМИ ГЕТЕРОЦИКЛИЧЕСКИМИ КОЛЬЦАМИ | 1996 |
|
RU2175324C2 |
| Способ получения производных пиримидона-4 или их кислотно-аддитивных солей | 1979 |
|
SU999971A3 |
| КАРБАМАТЫ И КАРБАМИДЫ, ИНДУЦИРУЮЩИЕ ПРОИЗВОДСТВО ЦИТОКИНОВ | 1994 |
|
RU2135515C1 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 2006 |
|
RU2446171C2 |
Изобретение касается гетероциклических веществ и, в частности, получения соединений общей формулы I : N=C(NH2)-S-CH=C - CK=N -O-C (=CH2)- C(O)OH, где K= - C( O)-NH-CH-CR1R2-NY -C = O
Y = -OS(O2)-OH, R1 и R2- низший алкил, или R1+R2 = -(CH2)3-, которые, как антибактериальные средства, могут быть использованы в медицине. Цель изобретения - создание новых более активных веществ указанного класса. Синтез ведут удалением из карбоксилзащищенного (дифенилметилом, фенилметилом или трет-бутилом) соединения формулы I указанной эфирной группы. Новые вещества малотоксичны и активны в отношении инфекций мочеполового тракта, распираторных инфекций, т.е. против грамположительных и грамотрицательных бактерий. 1 табл.
Изобретение относится к снособу получения новых химических биологически активных соединений, конкретно производных моносульфоктама, проявляющих антибактериальную активность. Указанное свойство предполагает возможность применения этих соединений в медицине.
Цель изобретения - получение новых производных моносульфоктяча, обладающих улучшенной антибактериальной активностью в ряду моноциклических R -лактамовых антибиотиков.
Пример 1. (±)-И-2- 1- -(2-Амино-4-тиазолил)-2- 4,4-диме- ТИ.П-2-ОКСО-1 -(сул1 факси)-3-азетиди- нил амино -2-оксоэтил11деиЗпм1-шо - окси -2-акриловая кислота.
А. 2- 1,3-Диоксо-2Н-изоиндол-2- -ил)окси -2-акриловая кислота.
Раствор сложного трет-бутилового эфира 2-{( ,3-диоксо-2Н-изоиндол- -2-ил)окси |-2-акриловой кислоты (1,75 г, 5,7 ммоль) в метиленхлори- ;Де (10 мл) и анизоле (10 мл) обра- ,батьшают трифторуксусной кислотой (5 мл). После перемешивания в течение ночи при комнатной температуре прибавляют толуол и реакционную смесь концентрируют в вакууме. Остаток растирают в порошок дважды, используя гексан, с получением 1,54 г указанного в заголовке соединения,
В, Сложный дифенилметиловый эфир- -2 ( ,3-диоксо-2Н-изоиндол-2-ил)ок- си -2-акриловой кислоты,
2- (1 ,3-Диоксо-2Н-изои1ЩОЛ-2-ил) окси1-2-акриловую кислоту (1,54 г, 4,8 ммоль) растворяют в 25 мп ацето- нитрила и по каплям прибавляют растСП
to
О4
10
31517762
вор дифенилдиазометана 1,17 г, 5,9А NtMonb/50 мл ацетонитрила). После прибавления приблизительно 1,1 г- экв дифенилдиазометана тонкослойная хроматография не показывает никакого исходного материала, который бы оставался в смеси. Избыточный дифенил- диазометан расщепляют прибавлением небольшого количества уксусной кислоты. Реакционный раствор концентрируют до остатка, растворяют в этил- ацетате, промывают последовательно 1н. раствором бикарбоната натрия и соляным раствором, сушат в присут- |г ствии безводного сульфата магния и упаривают до получения твердого тела. После порошкования гексаном получают 1,9 г указанного в заголовке соединения.2п
С. 2-Амино-(С- -(дифенилметок- си)карбонил винил окси -имино -4- -тиазолуксусная кислота.
К раствору сложного дифенилметило- вого эфира 2-( 1 , 3-ДИОКСО-2Н-ИЗОИН- 25 дол-2-ил)окси -2-акриловой кислоты (0,8 г, 2 ммоль) в 50 мл метиленхло- рида под аргоном при О С прибавляют гидразингидрат (100 мг, 2 ммоль в 1 мл абсолютного этанола). Реакционную смесь медленно нагревают от О С до комнатной температуры в течение 1 ч и затем перемешивают при комнатной температуре в течение 2 ч. Белый осадок отфильтровьшают, раствор разбавляют диэтиловым эфиром и фильтруют вновь. Летучие компоненты затем удаляют из фильтрата с получением 2-аминоокси-2-акриловой кислоты в качестве остатка. 2-Аминоокси-2-ак- риловую кислоту затем растворяют в этаноле (6 мл) и воде (4 мл), после чего к раствору прибавляют 2-аминоти азол-4-глиоксиловую кислоту (0,31 г, 1,8 ммоль). После перемешивания при комнатной температуре в течение 17 ч тонкослойная хроматография показывает, что реакция является неполной. Прибавляют дополнительное количество этанола и воды, а для солюбилизации реагентов прибавляют не- -50
значительное количество диметилформ- амида. После перемешивания в течение 72 ч тонкослойная хроматография не показывает оставшейся 2-аминоокси- -2-акриловой кислоты. Упаривание при- 55 водит к получению неочищенного соединения, указанного в заголовке, которое хроматографируют на колонке НР20,
30
35
40
45
элю во пр ац вод за ос ни в
-( ам
Nлиги су -1 ди ре пр 13 си фу к ра по Не ют пе то ют су до 13 с го
с$к Ьпост аз (1 ка ма оч го ва тр по пи
ни 20 9,
0
г п
5 0
55
0
5
0
5
элюируемой градиентом ацетонитрил/ вода (0-80 %). Фракции, содержащие продукт, концентрируют для удаления ацетонитрила. Фильтрация полученной водной суспензии дает указанное в заголовке соединение в качестве осадка. После сушки в вакууме в течение ночи получают 167 мг указанного в заголовке соединения.
D.Ы-(т-Бутилоксикарбонил)-Н - -(фенилметокси)-В, L-3-оксивалин- амид.
Раствор 24,84 г (106,6 ммоль) Nт-бyтилoкcикapбoнил-D, L-3-оксива- лина и 16,33 г (106,6 ммоль) моногидрата оксибензотиазола в 500 мл сухого тетрагидрофурана охлаждают до -10 С и прибавляют 22 г (106,6 ммоль) дициклогексилкарбодиимида. Смесь перемешивают под азотом в течение 1 ч при О (. Вслед за этим раствор 13,13 г (106,6 ммоль) 0-бензилгидрок- силамина в 250 мл сухого тетрагийро- фурана прибавляют в течение 15 мин к активированной смеси сложного эфира, и полученную смесь перемешивают, под азотом в течение 1 ч при О С. Нерастворимый материал отфильтровывают и фильтрат упаривают в вакууме до пены. Пену экстрагируют этилацета- том и нерастворимый материал удаляют фильтрацией. Органическую фазу сушат (сульфат натрия) и упаривают до сиропа, который кристаллизуют из 130 мл простого изопропилового эфира с получением 24,7 г указанного в заголовке соединения; т. пл. 76-78°С.
E.Пиридиниевая соль Н-(т-бутил- с$ксикарбонш1)(фенилметокси)-0, ЬЗ-(сульфоокси)валинамида.
Сухой пиридин (8,08 мл, 0,10 моль) помещают в круглодонную колбу емкостью 500 мл и охлаждают до -10 С под азотом. Триметилсилилхлорсульфонат (15,6 мл, 0,10 моль) прибавляют по каплям (энергичное перемешивание с магнитной мешалкой), после чего очень густую реакционную смесь (благодаря осаждению продукта) перемешивают в течение 0,5 ч при 0°С. Хлор- триметилсилан удаляют в вакууме, получая 15 г комплексного соединения пиридин - трехокись серы..
N-(т-бyтилoкcикapбoнил)-N -(фе- нилметокси)-0,Ь-3-оксивалинамид (16,92 г, 50 ммоль) растворяют в 200 мл сухого пиридина и прибавляют 9,87 г (62,5 ммоль) комплексного
51
поединения пиридни - трехокись се- рЫг, Смесь перемешивают при под л:ютог. в течение 2 ч. Прибавляют другую порцию (790 мг, 5 ммоль) комплекса пиридин - трехокись серы и пе- ремепшвапие продолжают еще 1 ч. Реак циопнук смесь десорбируют в вакууме Е масло. Масло отгоняют 3 раза в вакууме из ацетонитрила с получепием псочишонпого продукта, указапиого и заголоткс, п нпде пеиы 1)Ьгход принимают как количестпеинын.
Г. (i-)-3- (т-бутилоксикарбонил) амипо -4 ,4-диметил-1 - (|})епилметоксм)- -2-азетидипоНо
Колбу, содержащую неочищенную пи- ридиние)-ю соль N-(т-бyтилoкcикnpбo- Hi-ui)-N - (ф ,нилмптокси)-0,Ь-3- (суль- боксм) - гзг;лин, 1мида (приблиз птельип 50 ммоль), noMGitiaioT в панну со льдом и при энергичном перемешивании прибавляют 400 Л этилапетата, затем раствор 42,8 г (0,3) моль) карбоната калия в 90 M:I воды. Полученную смесь -чиергичио пспеметивают с обратным ХОЛПДИЛ1-НПКОМ н течение 2 ч под язо- Т(1М. Реакл1;онную смесь охллж;т,ают до комнатноГу температуры и фазы разде- пя от. Водь.ую фазу пкС грагирутот 2 X 200 мл ,етата и все органические фазы об7:(;диняют, сушат (с;ульфат натрия) и упаривают в вакууме. Масло берут в 40%-иой смеси зтилацетат/гек сан (125 мл) и быстро фильтру| 1Т че- роз 350 мл подушки (10 см) Mallin- krodt Silic AR CC-7, используй 3 - 4 л 40%-iroii смеси этилацетат/гексан о Фильтрат упаривают в вакууме до твердого остатка (12,2 г). Кристаллизапи из 50 мл И зопропилового эфира дает 7,15 г указанного в заголовке соединения ; т. пх; „ 110 С.
G. (±)-3-- (т-бутилоксикарбонип( ампно -1 -окси-4,4-диметил-2-азети- динон,
(i)-3 (т-бутилоксикарбонил)ами- ,4-димотш1-1-(фенилметокси)-2- -азетидинон (8,07 г, 25 ммоль) гидрируют при атмосферном давлении и температуре окр 7каю цей среды в 40 мл метанола с 0,5 г- 10 %-ного палладиро- ианного угля в качестве катализатора Б течение 2 ч. Реакционную смесь фильтруют через подушку Целита и фильтрат концентрируют с получением 5,78 г указанного в заголовке соединения в виде твердого о статка„
77626
Н. Тетрабутиламмониевая соль (±)-3-С(т-бутилоксикарбонил)-амино -4,4-диметил-2-оксо-1-азетидипил- сульфата.
Раствор хлоруксусной кислоты (12,27 г, 0,105 моль) в 210 мл ди- хлорметана при -40 С под азо- обрабатывают по каплям 20,5 г
Q (0,26 моль) пиридина в течение
10 мин. Смесь перемешивают 10 мин при О Г, и 10 мин при 25°Г.. Суспечзию (t)-3-(т-бутилоксикарбонил)ампно - -1-окси-4,4-диметил-2-азетидинона в
5 20 мл дихлорметана прибавляют к смеси, которую затем перемешивают при в течение 3,5 ч. Почти гомогенный раствор обрабатывают затем 250 .л воды и 17 г (0,05 моль) .кислого суль0 фата тетрабутиламмония. Смеет, хорсччс неремешивают и органический слой отделяют и сушат в присутстнии сутыр.а- та натрия. Упаривание в вакууме даст пену, которую очищают растжтре тием i;
5 этилацетате, удаляя нераствор1П тые щества, и упаривают до указанного в заголовке соединения в виде пены, 30 г.
I. ()-3-Лмино-4,4-диметил-2-окQ со-1-азетидинилсульфат.
Раствор тетрабутиламмониевой соли -, (1:)-3-(т-бутилоксикарбо}Шл) пмпно |- -4 ,4-диметил-2-оксо- 1 -азетидин.и;- сульфата (30 г, 0,05 моль) Б 125 мл дихлорметана и 10 мл анизола при -5 С под аргоном обрабатывают 50 мл трифторуксусной кислоты по кайля,. Р течение 10 мин. После перемешивания в течение 2,5 ч при температуре от
-5 до О С смесь разбавляют 50 мл этилацетата и фильтруют. Твердое тело промывают дихлорметаном и затем этилацетатом и сушат в с получением 9,4 г указанного в заголовке соединения в виде белого зернистого твердого остатка.
J. Дифенилметиловый сложный эфир (i)- (7.)-2- 1 - (2-амино-4-тиазолил)- -2- 4 ,4-диметил-2-оксо- 1 - (сульфок- си)-3-азетидинил1-амино1 -2-оксоэтиГ -ами
лиден амино -окси -2-акриловой кислоты.
к раствору 2-амино-с1б- (дифе- нилметокси)карбонил винш -окси ими- но -4-тиаэолуксусной кислоты (167 г, 0,4 ммоль) и триэтиламина (56 мкл, 1,0 г-экв .) под аргоном при -30° С прибавляют дифенилхлорфосфат (107 мг.
0,4 ммоль). Реакционную смесь перемешивают при -ЗО С в течение 1 ч с образованием смешанного ангидрида. (4)-3-Амино-А,Д-димeтил-2-oкco-l- -aзeтидинилcyльфaт (126мг, 0,6 ммоль) растворяют в диметилформамиде при О С, этот раствор и триэтиламин (71 мкл, 0,85 г-экв.) одновременно прибавляют к раствору смешанного ангидрида при -30 С. Реакционную смесь перемешивают в течение 0,5 ч при -30°С и затем медленно нагревают до О С за 1 ч. Реакционную смесь концентрируют в вакууме, сохраняя в смеси ацетон/вода, и регулируют до рН 6,5 посредством 1 и. раствора бикарбоната калия. Затем смесь пропускают через колонку Dowex AG50 (К), элюируемую 30 %-ной смесью ацетон/ вода. Подходящие фракции объединяют и концентрируют, а остаточный водный раствор подвергают воздействию колонки НР20. Колонку элюируют водой и затем градиентом ацетон/вода (0-100 %). Подходящие фракции объединяют и лио- филизуют с получением указанного в заголовке соединения, которое полностью используют в следуюшей стадии.
К. (ч-)- (Z)-2- 1 - (2-Амино-4-тиа- золил)-2- 4 ,4-диметил-2-оксо-1 - -(cyльфoкcи)-3-aзeтидинилJ aMHHoj-2- оксоэтилиден -амино окси -2-акрило- вая кислота.
В колбу, содержащую сложный ди- фенилметиловый эфир (+)-(Z) -(2-амино-4-тиазолил),4-диме- ТИЛ-2-ОКСО-1-(сульфокси)-З-азетиди- нил амино -2-oкcoэтилидeн aминoJoк- си -2-акриловой кислоты, прибавляют метиленхлорид (10 мл) и анизол (10 мл). После охлаждения до-5 С прибавляют трифторуксусную кислоту (8 мл) и реакционную смесь переметиО
вают при температуре от -5 до О С под аргоном в течение 45 мин. Прибавляют толуол и реакционную смесь упаривают до остатка. Прибавляют к нему воду и гексан и слои разделяют. Водный слой регулируют до рН 2,5 10%-ны раствором бикарбоната калия. Используют колонку НР20, элюируемую вначал водой, затем градиентом ацетон/вода. Соответствующие фракции объединяют и лиофилизуют с получением указанного в заголовке соединения в виде белого твердого остатка.
(1:1) Dj.O/ CD,CN,: J : 1,57 (с, ЗН), 1,75 (с, ЗН), 5,09 (с, 1Н),
0
5
0
5
0
5
0
5
5
5,75 (д, I 2,3 Гц, 1Н), 5,86 (д, I 2,3 Гц, 1Н), 7,42 (с, 1Н).
Пример 2. 3S(Z) -(2-Амино-4-тиазолил)-2- (4,4-диме- тил-2-оксо-1-(сульфокси)-3-азетиди- нилЗамино -2-оксоэтилиден -амино 1- -окси -2-акриловая кислота.
А. 2- (1,3-Диоксо-2Н-изоиндол- -2-ил)-окси -2-акриловая кислота.
Раствор сложного т-бутилового эфира-2- f(l,3-диоксо-2Н-изоиндол- -2-ил)окси -2-акриловой кислоты (49,9 г, 0,173 моль) в метиленхлори- де (300 мл) и анизоле (150 мл) обрабатывают трифторуксусной кислотой (300 мл). После перемешивания в течение ночи при комнатной температуре прибавляют 800 мл сухого толуола и реакционную смесь концентрируют в вакууме. Остаток дважды порошкуют гексаном с получением 39,6 г указанного в заголовке соединения„
Во Сложный дифенилметиловый эфир 2- ( 1 ,3-ДИОКСО-2Н-ИЗОИНДОЛ-2-ИЛ) ок- си -2-акриловой кислоты.
2-(1,3-Диоксо-2Н-изоиндол-2-ил) окси -2-акриловую кислоту (39,6 г, 0,17 моль) растворяют в 800 мл ацето- нитрила и при О С в течение 3 ч по каплям прибавляют раствор дифенил- диазометана (43,4 г, 0,224 моль/ 1000 мл ацетонитрила). Реакционный раствор упаривают до твердого остатка, который растирают в порошок гексаном. Полученное твердое вещество растворяют в дихлорметане и фильтруют через подушку силикагеля (Kiesel-, gel 60). Прибавление гексана приводит к получению 47,7 г указанного в заголовке соединения.
С. 2-Aминo-oi-LLLL(дифeиилмeтoк- си)карбонил винил оксй -имино -4- -тиазолуксусная кислота.
К раствору сложного дифенилмети- лового эфира-2- |(1,3-диоксо-2Н-изо- индол-2-ил)окси -2-акриловой кислоты (6,07 г, 15,2 ммоль) в 375 мл метиленхлорида под аргоном при О С прибавляют гидразингидрат (0,76 г, 15,2 ммоль) в 4 МП абсолютного этанола. После перемешивания в течение 1 ч при 0°С смесь упаривают до сухости при и порошкуют этиловым эфиром. Фильтрация и концентрация фильтратов приводит к получению сложного дифенилметилового эфира 2-амино- окси-2-акриловой кислоты в качестве остатка. Это соединение обраблтывают при 20°С раствором 2-ами- и.о-4-тиазолглиоксиловой кислоты (2,61 г, 15,2 ммоль) в диметилформ- амиде (50 мл) с последующей обработкой 5 мл воды. Реакционную смесь перемешивают при 20°С в течение 20 ч и затем охлаждают и разбавляют 250 м воды. Перемешивание полученной камед дает зернистое твердое вещество, которое фильтруют, промьгеают водой и затем подвергают азеотропии ацето- нитрилом до сухости. Сухое твердое вещество суспендируют 100 мл ацето- нитрила, фильтруют и наконец промывают последовательно ацетонитрилом, этиловым эфиром и гексаном. Сушка на BO3j:yxe позволяет получить 1,97 г указанного в заголовке соединения.
D.«..-Мс-тилбензиламиновая соль
N- (т-j% тилоксикарбонил)-,-3-оксива- лн ла.
Раг гпор N-т-бутилоксикарбонил- -В,Ь-3 о ссивалина (7,02 г, 30 ммоль) п 250 мл этилового эфира обрабатываю 3,63 г (30 ммоль) S-(-)-« -метилбен- зиламипа. Через 8 ч полученное твер- до1 пещпство фильтруют„ При перекрис талпизации из ацетонитрила дают 5,ВО г указанного п заголовке соединения; т. пл. 1А6 - , - -,j° (С 2,0, метанол).
E.N-(т-бутилоксикарбонил)-Ь-З- -оксивги1ии.
Смесь 204,6 г (0,577 моль) тилбензиламиновой соли Ы-(т-бутил- оксикароонил)-Ь-3-оксивалина, этил- .нетата (3л) раствора 100 г бисульфата калия и 150 г хлористого натрия в 1 л воды встряхивают, слои разделяют и органическую фазу промывают водой и сушат в присутствии сульфата магния. Концентрация органического раствора и порошкование с помощью 800 мл гексана приводят к получению 136 г указанного в заголгвке соединения; т. пл. 120 - 121°C,oi,.p - + 7,81°.
F.К-(т-бутилоксикарбонил)-Н - -(фенилметокси)-Ь-З-оксивалинамидо
Следуя методике примера 1 (часть I), но замещая Ы-(т-бутилоксикарбо- нил)-В,Ь-3-оксивалин N-(т-бутилокси- карбонил)-Ь-3-окоивалином, получают указанное в заголовке соединение.
G.(3S)-3-(т-бутилоксикарбонил) амино -4,4-диметил-1-(фенилметокси)- -2-азетидинон.
Раствор 2-метилпиридина (296 мл, 3,0 моль) в метилизобутилкетоне (2700 мл) под аргоном при -78 с об- с рабатывают по каплям хлорсульфоно- вой кислотой (80 мл, 1,2 моль) в течение 30 мин. После завершения прибавления температуру смеси доводят до 25 С в течение 30 мин и поддержи- Q вают на этом уровне еще 30 мин. К этой суспензии прибавляют Ы-(т-бу- тилоксикарбонил)-Ы -(фенилметокси)- -L-3-оксивалинамид (338,4 г, 1,0 моль) и перемешивание продолжают 2ч. К 5 этой смеси прибавляют метилизобутил- кетон (800 мл), , 4 (1222 г, 4,0 моль) и воду (2700 мл) и смесь нагревают до 70°С, При нагревании к смеси прибавляют по кап- 0 лям в течение 15 мин. 2н„ раствор гидроокиси калия (1000 мл, 2 моль) и смесь нагревают еще 55 мин. Слои разделяют и водную фазу экстрагируют 500 мл метилизобутилкетона. Объеди- 5 ненные органические слои охлаждают до 0°С, промывают 3 л холодного 20 %-ного раствора бисульфата калия, 1 л ледяной воды, а также раствором 50 г бикарбоната натрия и 100 г хло- 0 ристого натрия в 1 л воды. Органический слой сушат в присутствии сульфата натрия и концентрируют в вакууме. Остаток кристаллизуют из 1,75 л изопропилового эфира с получением 161 г указанного в заголовке соединения; т. пл. 121 - 122 С, СобЗ- -f 21,06° (С 2,55, ,,).
Н. (35)-3-(т-бутилоксикарбонш1) аминоЗ-1-окси-4,4-диметил-2-азети- 0 динон.
Раствор 14,77 г (0,0461 моль) (ЗЯ)-3-(т-бутилоксикарбонил)-ами- но -4,4-диметил-1-(фенилметокси)- -2-азетидинона в 15 мл этанола и 5 85 мл этилацетата гидрируют в присутствии 0,75 г 5 %-ного палладиро- ванного угля в качестве катализатора в течение 1,5 ч при давлении 1 атм. Реакционную смесь фильтруют и кон- Q центрируют с получением белого твердого вещества. Перекристаллизация из этилацетата позволяет получить 8,82 г указанного в заголовке соединения; т. пло 148 - 149°С, «ill, + 31 (, этилацетат).
Следуя методике примера 1, но замещая (±)-3- (т-бутилоксикарбонил) амино -1-окси-А,Д-димeт ш-2-aзeтиди- нoн соединением (3S)-3-(т-бутилокси карбонил)амино -1-окси-А,4-диметил- -2-азетидинон, получают указанное в заголовке соединение в виде пены„
J. (35)-3-Амино-А,4 диметил-2-ок- со-1-азетидинилсульфат„ .
Следуя методике примера 1 (часть I), но замещая тетрабутилам- мониевую соль ()-3-(т-бутилокси- карбонил)амино -4,4-диметил-2-оксо- -1-азети,Цинилсульфата тетрабутилам- мониевой солью (35)-3-(т-бутилокси- карбонил)амино -4,4-диметил-2-оксо- -1-азетидинилсульфата, получают указанное в заголовке соединение„Перекристаллизация малой пробы из смеси этанол/вода приводит к получению указанного в заголовке соединения в ви- .де кристаллического твердого вещества; т, пл. 140 - 142 С (d),W.p 74,8° (С 1, Н,()).
К. Тетрабутиламмониевая соль сложного дифенилметилового эфира 38 (ZД 1 - (2-амино-4-тиазолил)-2- Lt ,4-диметил-2-оксо-1 (сульфокси)- -3-азетидинил амино -2-оксоэтилиден амино -окси -2-акрилояой кислоты.
К раствору 2-амино-о - С 1-(ди- фенилметокси)карбонил винил -окси аминоЗ-4-тиазолуксусной кислоты (1,423 г, 3,36 ммоль) и триэтиламина (0,404 г, 3,88 ммоль) под аргоном при - 30°С прибавляют дифенилхлор- фосфат (0,902 г, 3,36 ммоль). Реакционную смесь перемешивают при -30 С в течение 1 ч с образованием смешан- ноге ангидрида (35)-2-амино-4,4-ди- метил-22-оксо-1-азетидинилсульфат (0,706 г, 3,36 ммоль), растворяют в 4 мл диметилформамида при и этот раствор и триэтиламин (0,404 г, 3,88 ммоль) одновременно прибавляют к раствору смешанного ангидрида при - 30°С „ Реакционную смесь медленно нагревают до О С за 1 ч. Реакционную смесь обрабатывают триэтиламином (0,338 г, 3,36 ммоль) и затем концентрируют в вакууме. Остаток обрабатывают водой с получением камеди, которую отделяют от водного слоя и промьшают дополнительным количеством воды. Камедь растворяют в 100 мл метиленхлорида и встряхивают с раствором кислого сульфата тетрабутилам- мония (1,1 г, 3,36 ммоль) в 30 мл
0 5
о
, д 5
0
5
воды„ Органическую фазу отделяют и промывают трижды водой, сушат в присутствии сульфата натрия и упарива- . ют до пены. Эту пену растворяют в 30 мл метиленхлорида и разбавляют до 120 мл этилацетатом. Указанное в заголовке соединение кристаллизуют с получением 1,73 г белого твердого вещества; т. пл. 170 - 172 С.
L. ЗЯ((2-Амино-4-тиа- золил),4-диметил-2-оксо-1 - -(cyльфoкcи)-3-aзeтидинидJaминoJ-2- -оксоэтилиден амино -окси -2-акри- ловая кислота
Раствор тетрабутиламмониевой соли сложного дифенилметилового эфира 35(7.)(2-амино-4-тиазолил - -2-(4 ,4-диметил-2-оксо-1 -(сульфок- си)-3-азетидинил амино1-2-оксоэтили- денЗамино -оксиЗ-2-акриловой кислоты (2,17 г, 2,54 ммоль) в метиленхло- риде (24 мл) и анизоле (1 мл) при - 12°С обрабатьшают трифторуксусной кислотой (8 мл) и реакционную смесь перемешивают при - под аргоном в течение 1 ч. Смесь обрабатьшают по каплям 60 мл этипацетата и оставшуюся суспензию перемешивают в течение 20 мин и затем фильтруют и промывают этилацетатом и гексаном. После сушки на воздухе твердое вещество суспендируют в 50 мл воды при 20°С. Кристаллы образуются в течение нескольких минут После кристаллизации раствор фильтруют и твердое вещество промьтают водой и сушат в вакууме с получением 0,9 г указанного в заголовке соединения; т. пл. 140 - 170 С, разложение
Пример 3. (±)-(Z)-rCCl-(2- -Амино-4-тиазолил)-2-оксо-2- 2-ок- со-1-(сульфокси)-1-азаспиро(3.3) гепт-3-ил амино этилиден амино ок- си||-2-акриловая кислота.
А. Сложньй бензиловый эфир N- -(т-бутилоксикарбонил)(1-оксицик- ,лобутил)глицина.
Раствор диизопропиламина (9,7 мл, 70 ммоль) в 150 мл сухого тетрагидро- фурана при - 40 С под аргоном обрабатывают 39 мл (64,5 ммоль) 1,71 н. раствора н-бyтилfIития в гексане, и бледно-желтый раствор перемешивают при -40°С в течение 20 мин. Раствор охлаждают до -78 С, после чего в него вливают по каплям в течение 5 мин раствор 7,95 г (30 ммоль) сложного бензилового эфира К-(т-бутоксикарбонил)глицина в 30 мл сухого тетра- гидрофурана, что приводит к получению темно-желтого раствора и через 20 мин - к незначительному помутнению. Через 0,5 ч прибавляют раствор 2,42 г (2,0 мл, 34,5 ммоль) циклобу- танона в 30 мл тетрагидрофурана. Полученную желтую мутную смесь перемешивают, при -78°С в течение 15 мин, затем помещают в ванну с ледяной водой () на 2 ч. При внутренней температуре - (1 ч) раствор становится светлым, а при температуре - 15°С становится темно-пурпурным. Его перемешивают при 0°С в течение 0,5 ч, затем обрабатывают 3,96 г (66 ммоль) ледяной уксусной кислоты в 15 мл тетрагидрофурана с обпазова- нием мутной, светло-желтой смесио Эту смесь выливают в 500 мл холодной воды и дважды экстрагируют этиладе- татом. Экстракты промывают 2 %-ным раствором бисульфата калия, 5 %-ным раствором бикарбоната натрия и соляным раствором, высушивают (сульфат натрия) и упаривают до вязкого масла. Хроматография на 800 мл L PS-1 в смеси гексан/этилацетат (2: ) и соединение фракции продукта (Rj 0,29) приводит к получению 7,8 г продукта в виде масла.
B.N-(т-Бутоксикарбонил)-о.-(1 -ок- сициклобутил)глицин.
Сложный бензиловый эфир N-(T-Cy- токсикарбонил)(1-оксициклобутил) глицина (7,8 г, 23,3 ммоль) гидрируют при 1 атм в присутствии паллади- рованного угля в качестве катализатора в 150 мл абсолютного этанола в течение 4 ч при 25°С. Катализатор фильтруют и растворитель упаривают в вакууме. Прибавляют бензол и смесь упаривают дважды с получением 5,0 г продукта в виде жесткой пены
C.N-(Бензипокси)(т-бутокси- карбонил) (1 -оксициклобутил) глицин амид.
N- (т-бутоксикарбонил) (1 -оксициклобутил) глицин (5,0 г, 20,4 ммоль) растворяют в 150 мл сухого тетрагидрофурана под аргоном. Гидрат оксибен зотриазола (3,12 г, 20,4 ммоль) прибавляют к раствору, который затем охлаждают до 0°С и обрабатывают 4,20 г (20,4 ммоль) дициклогексил- карбодиимида. Через 1,75 ч при О С прибавляют раствор 0-бензилгидроксил амина в 15 мл тетрагидрофурана и
смесь перемешивают при О - 25 С в течение 17 ч. Смесь тетрагидрофурана затем охлаждают до - 10 С за 20 мин и полученные твердые тела фильтруют и промывают сухим тетрагидрофураном. Фильтрат упаривают и остаток, взятый в этилацетате, промывают быстро 2 %-ным бисульфатом калия, соляным
Q раствором, 5 %-ным бикарбонатом натрия и соляным раствором, затем сушат (сульфат натрия) и упаривают до пены. Порошкование изопропиловым эфиром приводит к получению 4,69 г продукта
5 в виде белого твердого вещества; т. пл. 95 - 97 с.
D.1- (Бензилокси)-З- |(т-бутилок- сикарбонил)аминoj-2-оксо-1-азаспи- ро(3.3)гептан.
0 Ы-(Бензилокси)-К -(т-бутоксикар- бонил).- (2-оксициклобутил)-глицин- амид (3,50 г, 10 ммоль) в 200 мл сухого тетрагидрофурана при под аргоном обрабатывают 2,4 мл (15 ммоль)
5 диэтилазодикарбоксилата, затем раствором трифенилфосфина (5,2 г, 20 ммоль) в 50 мл тетрагидрофурана в течение 10 мин и смесь перемешивают при 0°С в течение 1 ч. Остается
Q смесь желтого цвета, так что прибавляют дополнительное количество (0,52 г, 2 ммоль) трифенилфосфина. Через 15 мин упаривание в вакууме позволяет получить масло. Порошкование 100 мл смеси гексан/этилацетат (2:1) дает белое твердое тело, которое фильтруют. Хроматография фильтрата на 800 мл LPS-1 приводит к получению фракций продукта (R 0,8 в смеси гексан/этилацетат, 1:1) загрязненного плотной примесью, которую удаляют путем порошкования изопропи:; ловым эфиром, с получением продукта в виде белого твердого вещества
,. (1,07 г), т. пл. 156 - 157 С.
E.Мононатриевая соль 3-(т-бу- токсикарбонил)амино |-2-оксо-1-(суль- фокси)- -азаспиро(3.3)гептана.
1-(Бензилокси)-3-(т-бутоксикарбо- нил)амино -2-оксо-1-азаспиро(3. 3)гептан (1,07 г, 3,22 ммоль) гидрируют при 1 атм в 30 мл абсолютного этанола в присутствии 0,4 г 10 %-ного пал0
0
лядиевого угля F качестве катализатора в течение 3 ч при 25 С. Катализатор фильтруют и растворитель удаляют в вакууме при 10 С с получением твердого остатка. Этот твердый остаток, взятый в 19 мл сухого пиридийа.
15
обрабатывают 1,44 г (9 ммоль) комплексного соединения пиридин-трех- окись серы при 25°С под аргоном. Через 4 ч летучие компоненты удаляют в вакууме, остаток обрабатывают водой и рН регулируют от 5,40 до 6,45 раэ бавленнь1М раствором бикарбоната натрия. Пропускание через колонку 40 мл Dowex АС50 (К ) в воде элюирует про- в пределах 300 мл, Лиофилизация приводит к получению белого твердого вещества, которое хроматографируют на колонке НР--20 вначале в воде, затем градиентным увеличением ацетона (20 %). Фракции продукта лиофилизи- руют с получениям 0,75 г продутста в шлп бслп;-;; гпттгп и:р .
F. З-Лмиио-2-OKCo-l(сульфокси) азаспиро(3,3)гептан.
Г 1ононатрир;1у1 ) со :ь 3- (т-бутокси- к;1 рбонил) амиткГ -2 - оксо-1 - (сульфок- си)-1-лзаспиро(3.3)гептана (0,3 г, 0,87 п-юль) суспендируют в 2,5 мл су хс го дихлорметипа и 1,0 мл анизола - 0°С под аргоном, а затем обра бат.гопюг Д.О мл трифторуксусной кислоты. Через 0,5 ч образуется твердое вещест}ю. Через 1,5 ч прибавляют 4 м сухого толуола и смесь упаривают в вакууме с получением твердого вещества - 3-л;(п.ю--3-оксо-1 - (сульфокси)- -1-лзаспиро(3.3)геШ ана, которое растирают в пороиютч дважды гексаном и
п гО
сушат в натгууме при z3 С в течение
1 ч.
G. Тетрабутиламмониевая солт сложного дифенилметилового эфира (1)- -(Z) (амино-4-тиазолил)-2-ок- со-2- 2-оксо-1- (сульфок си)- -аза- спиро (3. 3) гeнт-3-илJ aминoJ этшчиден амино oKciiJ-2-акриловой кислоты.
Следуя методике npnj iepa 2 (часть К), но замещая (35)-3-амино- -4,4 диметил-2-оксо-1-азетидинил- сульфат соединением З-амино-2-оксо- -1 -(сульфокси)- 1-азаспиро(3.3)гептан, получают указанное в заголовке соединение.
Н. (±)-(Z) -(2-Амино-4-тиазо- лил)-2 оксо-2- 2-оксо-1-(сульфок- си)- -аз спиро(3.3)гепт-3-шЛамино этилиден aминo -oкcиJ-2-акриловая кислота,
Следуя методике примера 2 (часть I), но заметая тетрабутилам- мониевуто coj;), гложног(5 дифенилмети- лового эфира 3S(X)(2-ами- но-4-тназолил) ,4-диметил 2-ок
1776216
со- 1 - (сульфокси)-3-азеТ1щинил71аминр - -2-оксоэтилиден амино окси -2-акри- ЛОБОЙ кислоты соединением тетрабу- тиламмониевой соли сложного дифенил- метилового эфира (i:)-(Z) 1-(2- -амино-4-тиазолил)-2-оксо-2- П2-оксо- -1 -(сульфокси)-1-азаспиро(3.3)гепт- -З-ил -амино этилиден амино окси - Q -2-акриловор1 кислоты, а также хрома- тографируя осадок от растворения этиладетатом на колонке НР-20 (смола) вместо нерекристаллизации из воды,
0
5
0
5
юлS чaют указанное в заголовке соединение; т. пл. 170-200°С, разложение .
Соединения настоящего изобретения проявляют активность против грампо- ложительньгх и грамотрицательных организмов и могут использоватьсп в качестве средства борьбы с бактериальными инфекциями (включающими инфекции мочеполового тракта и респираторные инфекщти) в млеконитаюш,их видах, как, например, одомашненные жипотныс (собаки, котки, коровы, лошади и тому подобное) и люди.
Для борьбы с бактериальньми ин- фекпиями в млекопитающих предлагаемые соединения могут вводиться в млекопитающее в количестве от 1,4 до 350 мг/кг/день, предпочтительно от 14 до 100 мг/кг/день„ Все пути введения, которые использовались в нроишом для достгшки пенициллинов и дефалоспоринов к месту заражения, также рассматриваются для использования с и-лактамами изобретения. Такие способы введения включают пе- роральное, внутривенное, внутримышечное и в виде суппозиториев с
Соединение настоящего изобретения (обозначено SQ 31299) при испытании на антибактериальную активность сравнивали с тигемонамом формулы
СН-,
OSO,
где при R - тигемонам,
при R -с - СООН - соединение SO 31299.
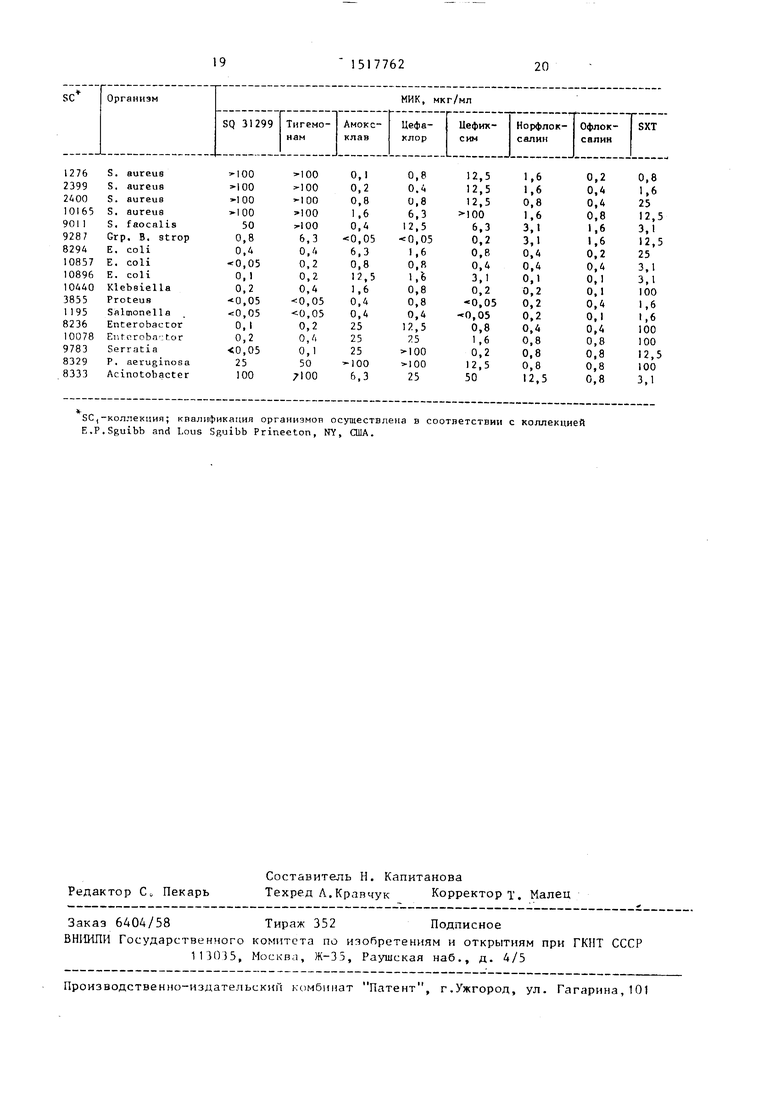
Кроме того, в таблице, где представлены результаты испытаний новых соединений на антибактериальную активность, приведены сравнительные данные соединения по изобретению с известными антибиотиками: амоксилин- клавулановой кислоты (обозначенной амокс клав), цефаклором, цефиксимом, нерфлоксалином, офлоксалином и три- метоприм-сульфаметоксазолом (в таблице - SXT при соответственном отношении 1:19 по весу).
Минимальную ингибирующую концент- рацию (МИК) в микрограммах на 1 мл определяли разбавлением агаром„ Антибиотики получены и разбавлены в среде агара двукратным разбавлением. Бактериальные инокулюмы затем разме- щены на поверхности агара при помощи мультикапельного инокулятора, подающего известный объем (Denley Instr. or Mast SystemLtD). Чашки инкубированы при 37 С в течение 18 - 24 ч в соответствующей атмосфере
В качестве агарной среды использовали (Yeast- Beet) агар говяжей закваски (Difco).
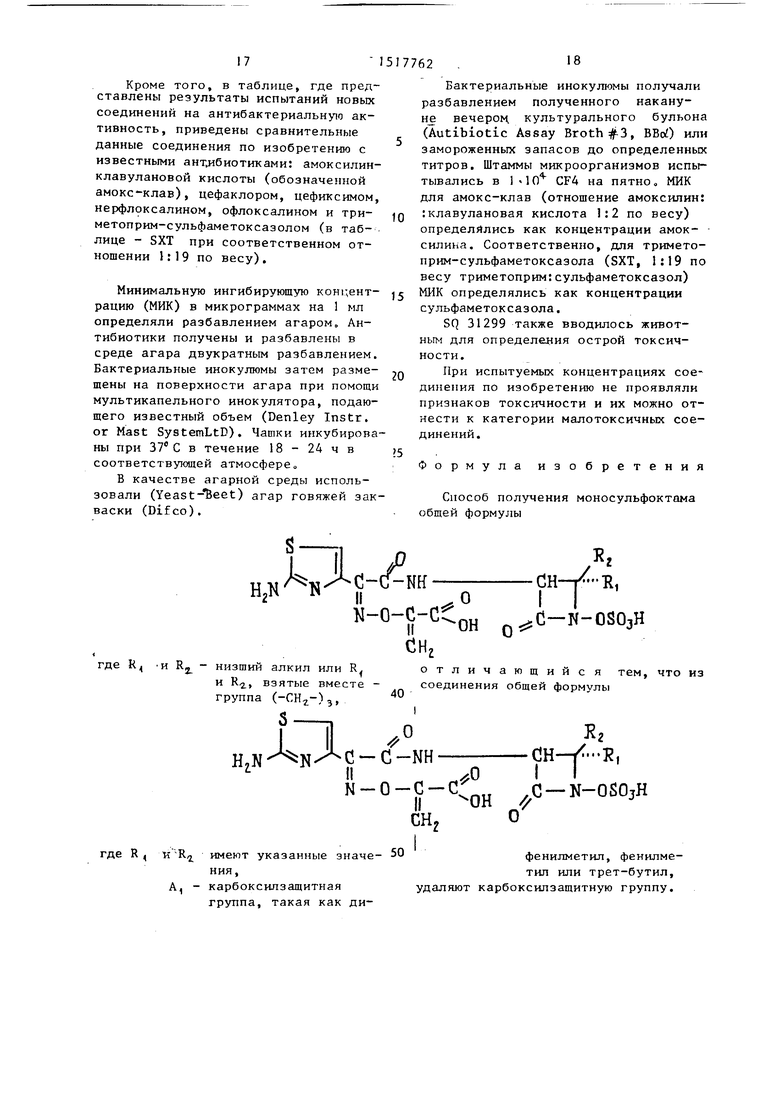
н
g
Nгде H И Rj - низший алкил или R
и R, взятые вместе - группа ()з,
где R, к R;ji. имеют указанные значе- 50фенилметил, фенилмения,тил или трет-бутил,
А, - карбоксилзащитнаяудаляют карбоксилзащитную группу, группа, такая как диБактериальные инокулюмы получали разбавлением полученного накану- Hje вечерон культурального бульона (Autibiotic Assay Broth :), ВВо) или замороженных запасов до определенных титров. Штаммы микроорганизмов испытывались в I lO CF4 на пятно, МИК для амокс-клав (отношение амоксилин: :клавулановая кислота 1:2 по весу) определялись как концентрации амок- - силина. Соответственно, для тримето- прим-сульфаметоксазола (SXT, 1:19 по весу триметоприм:сульфаметсксазол) МИК определялись как концентрации сульфаметоксазола.
SQ 31299 также вводилось животным для определения острой токсичности.
При испытуемых концентрациях соединения по изобретению не проявляли признаков токсичности и их можно отнести к категории малотоксичных соединений.
Формула изобретения
Способ получения моносульфоктама общей формулы
к,
Лн
. о 0-C-CCoH
-/Н,
сн,
о
сн
I
;С-N-OSOjH
отличающийся тем, что из соединения общей формулы
SC,-коллекция; квалификация организмов осуществле)а в соответствии с коллекцией E.P.Sguibb and Lous Sguibb Prinecton, NY, США.
