1
Изобретение предназначено для разделения многокомпонентных смесей органических соединений по температуре кипения и может быть использовано для анализа или выделения индивидуальных соединений в промышленном масштабе.
Для разделения многокомпонентных смесей по температуре кипения используют четкую ректификацию или препаративную газовую хроматографию с программированием температуры колонки.
Разделительная способность ректификационных колонок не превышает 100 теоретических тарелок, одна операция лабораторного фракционирования продолжается от 1 до 3 дней, вследствие чего изменяется структура нестабильных веш,еств, кроме того, используемые для этих целей установки не пригодны для разделения количеств вещества менее 50 г.
Разделительная способность препаративных хроматографических колонок обычно выше 1000 теоретических тарелок, одна операция разделения продолжается 0,5-2 час, но количество разделяемого вешества не превышает нескольких граммов. Разделяемое вещество подают в колонку в один прием. Для предотвращения перегрузки колонки соотношение вещества и неподвижной фазы в колонке не должно превышать определенной величины,
что ограничивает пропускную способность колонок. Количественное выделение разделенных фракций из газа-носителя затруднено.
Для повышения степени разделения предлагается способ, по которому пары смеси в потоке газа-носителя непрерывно подают в хроматографическую колонку с сорбентом, где их подвергают хроматографическому разделению при такой же программе повышения температуры, как и при испарении смеси. Кроме того, для повышения степени улавливания фракций в качестве газа-носителя используют пары легкоконденсирующихся жидкостей, например воды.
Разделение проводится сочетанием методов ректификации и газовой хроматографии. Предварительное грубое разделение проводят методом ректификации, а точное разделение методом газовой хроматографии. Вещество, разделенное в ректификационной колонке, поступает в хроматографическую колонку непрерывно, вследствие чего исключается перегрузка хроматографической колонки. Температуру ректификационной и хроматографической колонок поднимают синхронно.
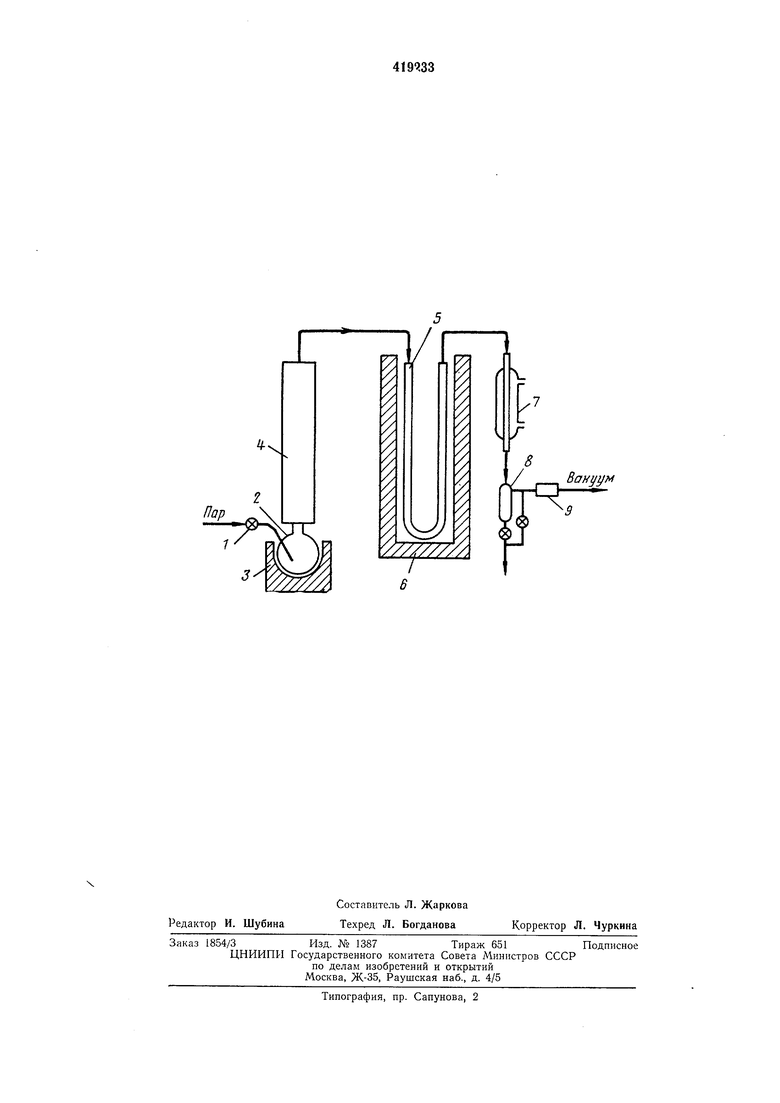
Для упрощения схемы разделения, уменьшения эксплуатационных расходов и обеспечения количественных выходов в качестве газа-носителя используют пары воды или других жидкостей, нерастворяющих разделяемые вещества, а ток пара через колонки осуществляется применением вакуума в конце системы. Хроматографирование в вакууме уменьшает также расход пара и понижает температуру колонок. На чертеже показана схема нотока разделяемого вещества н узлы установки, применяемой для реализации предлагаемого способа. Установка включает в себя дроссельный вентиль I, куб 2, нагреватель 3 куба, ректификационную колонку 4, хроматографическую колонку 5, термостат 6 хроматографической колонки, конденсатор 7, вакуумный нриемник 8, маностат 9. Пар жидкости, применяемый в качестве газа-носителя, может иметь низкое, а также атмосферное давление. Количество пара и вакуум в системе регулирует дроссельный вентиль 1. Пар проходит через куб 2, в котором паходится разделяемое вещество, и ректификационную колонку 4, которая имеет разделительную снособность 5-10 теоретических тарелок. Грубо фракционированное вещество попадает в хроматографическую колонку 5, температуру которой поднимают линеарно с помощью термостата 6 со скоростью 30-60 град/час. В отличие от обычной ректификации температуру ректификационного куба поднимают также линеарно, синхронно с температурой хроматографической колонки, обе температуры являются близкими. Для разделения компонентов по температуре кипения в хроматографической колонке пригодны только ненолярные неподвижные фазы (апиезоны, силиконовые масла и т. п.), а сопротивление насадки не должно превышать 500-600 мм рт. ст. В остальном колонки не отличаются от обычных препаративных колонок. Вещество, попадающее в хроматографическую колонку, растворяется в неподвижной фазе в начале колонки, и каждый компонент передвигается дальше только тогда, когда температура колонки достигает онтимальной температуры выхода этого комнонента. Разделенные компоненты, выходящие из колонки, конденсируются вместе с паром (газом-носителем) в конденсаторе 7, это исключает потери с уходящим газом-носителем. Фракции отбирают через каждые 5-15 мин через вакуумный приемник 8. Вакуум регулируется маностатом 9, при применении водяного шара остаточное давление поддержнвается не ниже 50 мм рт. ст. Для установления корреляции: температура кипения отбираемых фракций - температура газохроматографической колонки, необходимо калибровать применяемый прибор эталонами, например, в случае фракционирования углеводородов - н-парафинами. Пропускная способность установки зависит, в основном, от количества неподвижной фазы в газохроматографической колонке, это должно в 3--6 раз нревышать количество фракций, отбираемых в 1 час. Расход водяного пара следует регулировать таким образом, чтобы его количество превышало среднее количество отбираемых фракций в 2,5-8 раз. Предлагаемым способом можно фракционировать соединения, кинящие до 400°С. Так как температуру установки поднимают медленно, можно применять газохроматографические колонки с большим диаметром, чем при обычной препаративной хроматографии, и соответственно достигать высокой производительности установки. Пример лабораторного разделения. Вещество: 25 г сланцевой смолы полукоксования, кипящей в пределах 80-300°С. Хроматографическая колонка: длина 2,5 м, диаметр 12 мм, насадка 23 г аниезона L, 22% от носителя (рисорб С, 0,3-0,4 мм). Расход водяного пара 16 г/час. Температура колонок в начале программирования 90°С, в конце - 230°С, скорость подъема температуры 40 град/час. Продолжительность разделения 4 час. Фракции отбирали через каждые 10 мин, их количество составляло 0,2-1,2 г. Н-парафины, присутствующие в смоле, были разделены друг от друга количественно. Частично были разделены и н-парафины и соответствующие I-олефины, температуры кипения которых отличаются на 2-3°. Предмет изобретения 1. Способ разделения многокомпонентных жидких смесей по температуре кипения, заключающийся в испарении смесей при постепенном повышении температуры в поток газа-посителя, регистрации состава пара и улавливании его путем конденсации, отличающ и и с я тем, что, с целью новышения степени разделения, пары смеси в потоке газа-носителя непрерывно подают в хроматографическую колонку с сорбентом, где их подвергают хроматографическому разделению при такой же программе повышения температуры, как и при испарении смеси. 2. Способ по п. 1, отличающийся тем, что, с целью повышения степени улавливания фракций, в качестве газа-носителя используют пары легкоконденсирующихся жидкостей, например воды.
Вануу

| название | год | авторы | номер документа |
|---|---|---|---|
| Способ выделения меркаптанов из высокосернистого газоконденсата | 1986 |
|
SU1395628A1 |
| СПОСОБ ХРОМАТОГРАФИЧЕСКОГО РАЗДЕЛЕНИЯ ПАРОФАЗНОЙ СМЕСИ | 1994 |
|
RU2065606C1 |
| СПОСОБ ХРОМАТОГРАФИЧЕСКОГО РАЗДЕЛЕНИЯ ПАРОФАЗНОЙ СМЕСИ | 1991 |
|
RU2008667C1 |
| Способ выделения этилбензола | 1980 |
|
SU929620A1 |
| СПОСОБЫ ОЧИСТКИ ВЫСОКОМОЛЕКУЛЯРНЫХ АЛМАЗОИДОВ И СОСТАВЫ, СОДЕРЖАЩИЕ ТАКИЕ АЛМАЗОИДЫ | 2002 |
|
RU2307822C2 |
| СПОСОБ ГАЗОХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА ОПТИЧЕСКИХ И СТРУКТУРНЫХ ИЗОМЕРОВ | 2007 |
|
RU2356047C2 |
| СПОСОБ ГАЗОЖИДКОСТНОЙ ПРЕПАРАТИВНОЙ ХРОМАТОГРАФИИ | 1970 |
|
SU269569A1 |
| ПАТСНТНО- ГЬХЙИЧЕСКА5 БИБЛИОТЕКАч А llAILninU- 4 Л'" | 1969 |
|
SU250121A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКООКТАНОВЫХ БЕНЗИНОВЫХ ФРАКЦИЙ И АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2019 |
|
RU2708620C1 |
| Способ анализа ароматических углеводородов | 1981 |
|
SU1112272A1 |