(54),СПОСОБ ПОЛУЧЕНИЯ 2,б-ДИНИТРОПРОИЗВОДНЫХ N-АЛКИЛ ИЛИ N,N-ДИАЛКИЛАНИЛИНОВ
1
Изобретение относится к органическому синтезу, в частности к получению 2,6-динитропроизводных N-алкил или N,N-диалкиланилинов, которые могут найти применение в качестве гербицидов .
Обычно нитрование N-алкилированных вторичных анилинов проводят с предварительной i защитой атома водорода JQ в аминогруппе например ацетилированием 13
Одйако такой метод многостадиен и не позволяет получить целевые про- 5 дукты с достаточно высоким выходом.
Наиболее близким к предлагаемому по технической сущности и достигаемому результату является способ получения 2,6-динитро-третичных анилинов, 20 в которых оба N-заместителя являются галоидалкилом, нитрованием, по крайней мере, пятикратным избытком в начале реакции 50-90%-ной и в конце реакции 50%-ной азотной-кислотойв при-25 сутствии каталитического количества, азотистой кислоты 2.
Однако известный способ не .позволяет получить N-алкилированныв производные вторичных анилинов с высо- 30
ким выходом в достаточно простых условиях.
Цель изобретения-увеличение выхода и упрощение процесса.
Поставленная цель достигается тем, что согласно способу получения 2,6-динитропроизводных N-алкил или N,N-диалкиланилинов обгпвй формулы
-алкил, с числом углеродгде Y ных атомов 1-4, галоген;
-водород, галоген, алкил с
Z числом углеродных атомов 1-4, алкокси или монозамещенный алкил, в котором заместителем является галоген или алкокси;
-алкил, моногалоидалкил или алкоксиалкил с числом углеродных атомов 1-4, циклоал кил с числом углеродных атомов 4-6;
Rj - водород или R ; W и V - водород или нитро при условии , 4to W и V не оба нит ро;
путем нитрования соответствующего N-алкилированного анилина нитрирующей смесью, нитрующая смесь имеет следующий состав: 15-78% воды, 8-68% 95-98%-ной серной кислоты и 2-60% 70-75%-ной ,азотной кислоты.
Нитрующие агенты, используёлйе, согласно предлагаемому способу,представлены в виде трапеции, т,е. композиции площади, определяемой линия|ми,соединяющими точки, соответствующие, вес. HNO, 8 HnS04 , 50 HNO, 35 , 15 , 2HN04, 6 HjSO , 30 0 и 2 HNOj, 20. , 78 H ( ,
Три предпочтительных компонента, нитрующих состав, находятся внутри трапеции. Это площадь, заключена между линиями, соединяющими точки, со ответствующие, вес.%: 45 HNOa , 19 Hi2SOi , 36 , 45 HNCU, 19 HnO, 20 HNO, 52 ,, 28 Hijt) и 20 HNOj, 27 ... 53 .
Оптимальное число молей азотной кислоты на моль соединения I будет зависить от нитруемого соединения и состава используемого нитрующего агента. Обычно соединения формулы предпочтительно нитровать, используя от 2,2 до 5,0 моль азотной кислоты на моль М-алкилированного анилина. Ьнутри этой широкой области ндибоЬее предпочтительным является уровень от 2,5 до 3,5 моль азотной кислоты. В тех случаях, когда W или V являются нитро, предпочтительно использовать от 1,2 до 4,0 моль азотной кислоты на моль N-алкилированного анилина. Втури этой области обьачно более предпочтительной является узкая область от 1,5 до 2,5 мол азотной кислоты.
Используемое в нитрованиях предлагаемого способа мольное отношение ,серной кислоты к N-алкилированному 1анилину находится от 1,5:1 до 15:1, ио предпочтительно составляет от 12,0:1 до 10,0:1. Для серной кислоты эти области соответствуют количествам от 30 до 70 вес.% с предпочтительной областью, примерно, от 35 до 65 вес.%.
Присутствующее в нитрующей смеси количество воды является важным фактором, связанным с оптимальной температурой. Обычно реакционные смеси, содержащие большие количества воды, требуют более высоких реакционных температур. Количество воды в исходной нитругадей смеси должно находиться, примерно, от 15 до 78% от веса нитрующей смеси. Для превращения любого N-алкилированного мононитроанилина в N-алкилированный-2,6-динитроанилин должна быть использована достаточно высокая температура.
Соединения 1)Ормулы I могут нитроваться при температурах, изменяющихся от О до 70С, однако температуры ниже 15с стремятся помешать завершению динитрации и поэтому не являются особенно желательными. Температуры выше являются нежелательными потому, что реакцией становится трудно упр влять. Так как реакция является экзотермической, то , обычно требуется охлаждение для поддержания температуры ниже верхнего предела и желательно в оптимальной области. Оптимальная температура будет меняться в зависимости от использованного исходного М-алкилированного анилина и от состава нитрующего агента. Наиболее предпочтительной температурной областью является область 35 - ,
При работе в области 0-70 С (предпочтительно 35-60с) нитрование со смешанной кислотой легко регулируется. Во избежание протекания неконтролируемой реакции нитрование с концентрированной азотной кислотой должно проводиться при температуре ниже 10°С, Вследствие относительно простого контроля, требуемого для смешанных кислот, для этой системы требуется значительно меньшая мощность холодильных устройств. При перерывах или нехватках электроэдзергии смешанная кислота может быть легко обработана, а с концентрированной кислотой может произойти взрыв.
Другой особенностью нитрования смешанной кислотой является маленький избыток (О,50-1,50 моль) азотной кислоты, необходимый для завершения реакции. В случае концентрированной азотной кислоты требуется, по крайней мере, 5-10 моль. В тех случаях, когда извлечение азотной кислоты не,существенно для экономи ки процесса СТОИМОСТЬ и потенциальный риск при работе с концентрированной азотной кислотой будут значительно больше, чем со смешанной кислотой.
N-алкилированный анилин может реагировать с нитрующим раствором в виде жидкости, твердого вещества или, будучи растворенным в таком инертном растворителе, как этилендихлорид хлороформ, четыр ххлористый углерод или нитрометан. Предпочтительным является использование N-элкилированного анилина в этилендихлориде, в котором отношение миллилитров этилендихлорида на грамм исходного соединения составляет примерно 3,О : 1- - 0,5: 1, предпочтительно 0,75:1.
Способ добавки не является существенным фактором. В зависимости от конкретной ситуации может добавляться либо нитрующий агент к исходному
соединению, либо исходное соединение к нитрующему агенту.
Целевой продукт может быть отделен после завершения реакции нитрования. Точка завершения определяется такими обычными методами, как тонкослойная хроматограЛия или ядерно-магнитно-резонансная (ЯМР) спектроскопия. Однако в случае соединений формулы I, в которой R есть водород высокоэффективной является дополнительная обраГютка реакционной смеси от ступени нитрования ленитрозирующим агентом, предпочтительно ком.бинацией соляной и сульАаминовой кислот.
Мольное отношение сульЛаминовой кислоты к N-( нитрозосоединению составляет 2:1. Температура на ступени денитрозации может изменяться от 20 до 100 С и должна находиться между 80 и . Хотя ступень денитрозации может проводиться при атмосферЙом давлении, ее предпочитают проводить под давлением для сохранения соляной кислоты.«
Такие денитрозирующие агенты, как соляная кислота и хлорид железа также могут быть использованы в данном процессе, однако использование соля ной и сульфаминовой кислот является предпочтительным.
Предлагаемый способ проводят обычно как периодический процесс, однако разрабатывается также и непрерывный процесс.
Пример 1. Нитрование N-fl-этилпропил)-3,4-диметиланилина.
К раствору смешанной кислоты, приготовленной добавлением 145,2 г
Нитрование N-(1-этйлпропил)-3,4-диметиланилина
70,5%-ной азотной кислоты (1,625 моль) и 116,6 г 94,5%-ной серной кислоты (1,125 моль)к 58,8 г воды, добавляют в течение 2 ч при 35°С раствор из . 101,0 г 94,6%-ного М-(1-этилпропил)5 -3,4-динитрометиланилина (0,5 моль ) 143,5 мл этилендихлорида (ЭДХ) Реакционную смесь выдерживают при 35°С в течение 1 ч и затем отделяют |ВОдную фазу. Органическую фазу последоQ вательно промывают мл 5%-нЬго раствора каустика и 300 мл воды. Органический раствор высушивают над безводнЕлм сульфатом магния, фильтруют и концентрируют при 70°С под вакуумом до получения 141,5 г сухого продукта,
5 содержащего 117 г (82,62)М-(1-этилпропил)-3,4-диметил-2,6-динитроанилина и 14,2 г (10,0%) N-нитpoзo-N-(1-этилпропил)-3,4-диметил-2,6-динитроанилина. Общий выход 2,6-динитроанилина сос0тавил 72,6%.
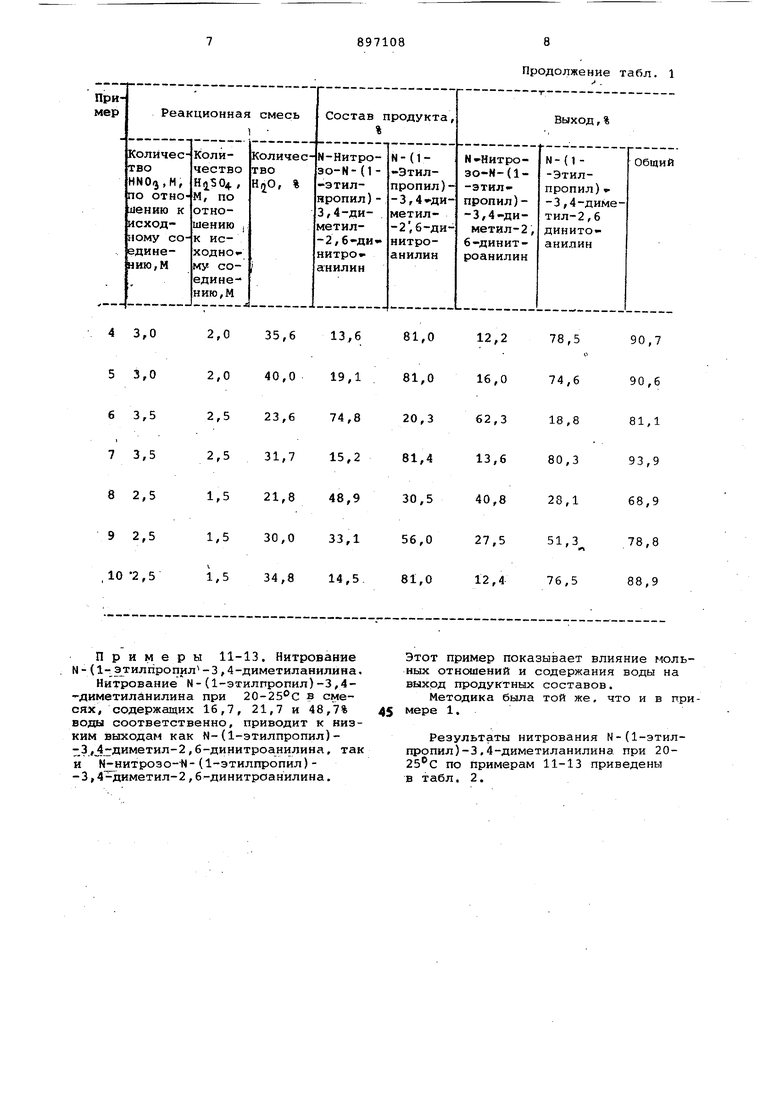
Примеры 2-10. Результаты их получены нитрованием при N-(1-этилпропил)-3,4-диметиланилина в реакционных смесях, содержащих
5 одинаковые мольные отнсяаения азотной и серной кислоты по отношению к исходному материалу, но разное количество воды.
Во всех случаях основные реакционные условия были такими же, как
0 и в примере 1. В некоторых случаях исходный материал добавляли без растворителя и реакционные смеси впос ледствни экстрагировали толуолом или ксилолсмл и обрабатывали обычным спо5собом .
Результаты нитрования М-(1-этил-, пропил)-3,4-димететиланилина приведены в табл. 1.
Таблица при 35°С






| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ N-АЛКИЛДИНИТРОАНИЛИНОВ ОТ N-НИТРОЗОАНИЛИНОВ | 1989 |
|
RU1617889C |
| Способ получения 4,6-динитро-5,7-дихлорбензофуроксана | 2020 |
|
RU2752080C1 |
| Способ получения композиции 5-нитро-4,6-дихлорбензофуроксана и 4,6-динитро-5,7-дихлорбензофуроксана | 2021 |
|
RU2771002C1 |
| Способ очистки динитроанилинов от нитрозаминов | 1978 |
|
SU833156A3 |
| СПОСОБ ПОЛУЧЕНИЯ 4-НИТРО-N-МЕТИЛФТАЛИМИДА | 1992 |
|
RU2044726C1 |
| Способ получения 4-окси-3-нитрокарбостирилов | 1974 |
|
SU578869A3 |
| СУЛЬФИРОВАННЫЕ НИТРОФЕНОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПОЛИМЕРИЗАЦИИ | 2005 |
|
RU2378242C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИНИТРОДИАЗААЛКАНОВ И ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ | 2001 |
|
RU2274637C2 |
| Способ получения 3-третбутил6-метил-5-нитросалициланилида | 1972 |
|
SU508179A3 |
| СРЕДСТВА УПРАВЛЕНИЯ ЭКЗОТЕРМИЧЕСКОЙ РЕАКЦИЕЙ СТИРОЛЬНЫХ МОНОМЕРОВ С СУЛЬФОНОВЫМИ КИСЛОТАМИ | 2006 |
|
RU2393143C2 |
2,0 27,0 48,6 43,1 2 3,0
2,0 31,1 31,9 61,0 29,5 48,0
3 3,0
77,5 41,2 40,3- 81,5
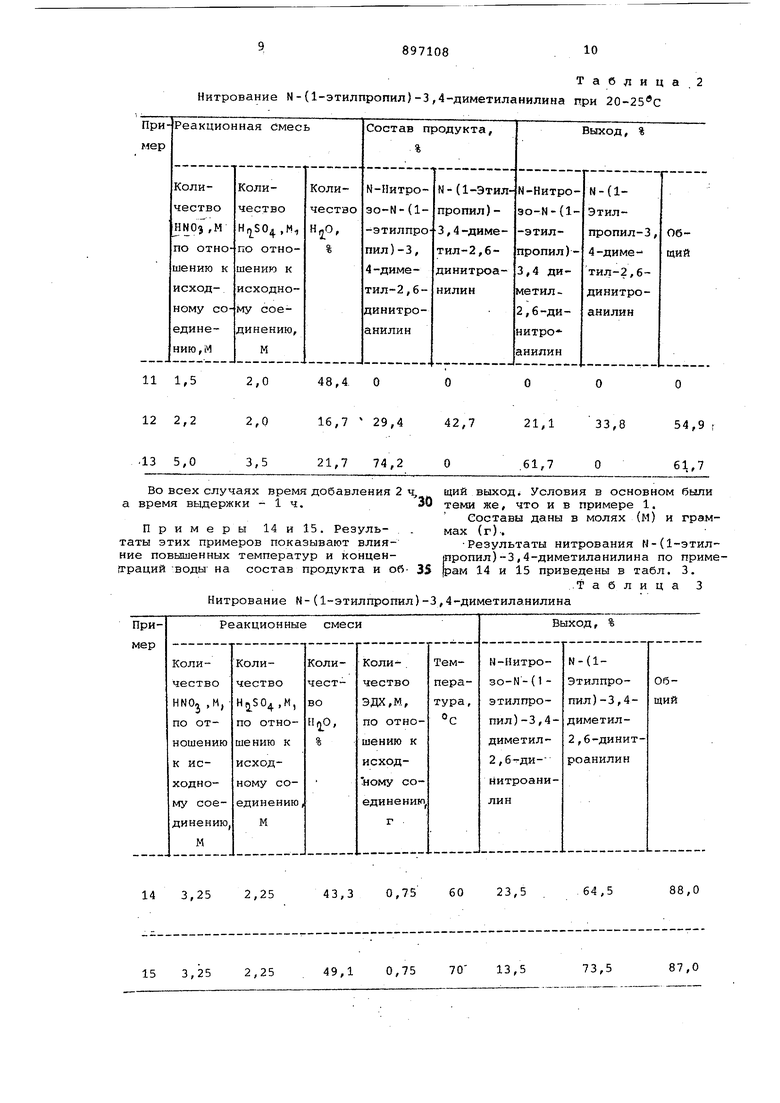
Примеры 11-13. Нитрование N- (1- этилпропил -3,4-диметиланилина.
Нитрование N -(1-этилпропил)-3,4-диметил анилин а при 20-25 0 в смесях, содержащих 16,7, 21,7 и 48,7% воды соответственно, приводит к низким выходам как N-(1-этилпропил)-3 ,-диметил-2,б-динитроанилина, так
к М;; НИТРОЗО-М- ( 1-ЭТИЛПрОПИЛ ) -3,4-диметил-2,6-динитроанилина.
Продолжение табл. 1
.
Этот пример показывает влияние мольных отношений и содержания воды на выход продуктных составов.
Методика была той же. что и в примере 1.
Результаты нитрования N-(1-этилпропил )-3 ,4-диметиланилина при 2025 С по примерам 11-13 приведены в табл. 2. 9 Нитрование N Во всех случаях время добавления 2 ч, щий выход Условия в основном бьши а время вьщержки - 1 ч.30 теми же, что и в примере 1. Примеры 14 и 15. Резуль- . мах (г)-.
таты этих примеров показывают влия- -Результаты нитрования Н-(1-этилние повышенных температур и кончен- тропил)3,4-диметиланилина по примеГграций -воды на состав продукта и об- 35 рам 14 и 15 приведены в табл. 3. Нитрование N-(1-этилпропил)-3,4-диметиланилина
43,3 0,75
2,25
14 3,25
49,1
2,25
3,25
15
Составы даны в молях (м) и грам.Таблица 3
88,0
64,5
23,5
60
87,0
73,5
13,5
70 {1-этилпропил)-3,4-диметиланилина при 20-25 с 89710810 Та б лица. 2
Пример 16. Денитрозация сырого нитрованного продукта от N- (1-этялпропил)-3,4 диметиланилина.
К сырому нитрующему раствору, содержащему 1550 г 2 ,6-динитpo-N.-(l-этилпpoпил -3,4-диметилайилина и 340 г 2,б-динитро--М-нитрозо-М-(1-этилпропил -3,4-диметиланилина в 3700 МП этилендихлорида, добавили 212 г сульфаминовой кислоты и 2850 мл 37,7%-ной соляной кислоты. Смесь дефлегмирозали при 80°С в течение б ч, добавили 2 л воды и водную фазу отделили. Водную фазу экстрагировали 500 мл этилендихлорида и органические фаДенитрозация сырых продуктов нитрования из -3,4-диметиланилина
17 17,2
22
3,1
2,5
23
24
2,5
Реакцию проводили в автоклаве. Во всех случаях процент Ni-нитрозо- (1-этилпропил) -3,4-диметил-2,6-динитроанилинп в конечном продукте был менее 0,1 вес.%.
зы объединили. Объединенную органическую фазу промыли 3 л воды, водную фазу отделили и этилендихлорид удалили под вакуумом при с извлечением 1915 г продукта, содержащего 1820 г 2,б-динитpo-N-(l-этилпpoпил)3, 4-диметиланилина и менее 0,1% 2 , б-динитро--М-нитрозо-М- (1-этилпропил)-3,4-диметилпнилина.
Примеры 17-24. Денитрозация сырых продуктов нитрования N-(1этилпропил -3,4-диметиланилина проведена при условиях примера 16.
Результаты денитрозации приведены в табл. 4.
90
97,0 94,3 97,0 97,2 95,5 96,4 96,5 Таблица 4 N -(1-этилпропил)Приведенные в табл. 4 мольные отно шения выражают число солей реагента на моль N-нитрозо-побочного продукта. В большинстве случаев реакцию Проводили в автоклаве. Пример 25. Нитрование N-(2-: -бутил)- 3,4-диметиланилина. К раствору смешанной кислоты, приготовленной добавлением 134,1 г 70,5%-ной азотной кислоты (,50молъ) и 101,1 г 96,9%-ной серной кислоты (1,0 моль)к 87,2 г воды при SO-SS C в течение 2 ч добавляли раствор из 92,3 95,9%-ного N-(2-бутил)-3,4-диметилани лина в 66,5 мл этилендихлорида. Реакцию проводили при 35 С в течение дополнительного часа и затем отделяли водную фазу.
Нитрование N-(2-бутил)-3,4-диметиланилина
20,6 70,8
4,0
26
2,0
27,6 19,8
2,5
27
3,5
31,7 10,5
2,5
3,5
28
Использование смешанной кислоты, содержащей большее количество воды и меньшее мольное отношение азотной кислоты, приводит к более низкому содержанию побочного продукта N-нитрозо соединения.
Во всех случаях использовалась основная методика примера 1 при 35°С, Добавляли раствор из 4,53 г (2-бутил )-3 ,4-диметиланилина в 15 мл этилен
Таблица 5
18,2
54,3
72,5
21,4
16,7
83,3
70,6
66,6
80,9
72,0
8,9
76,3
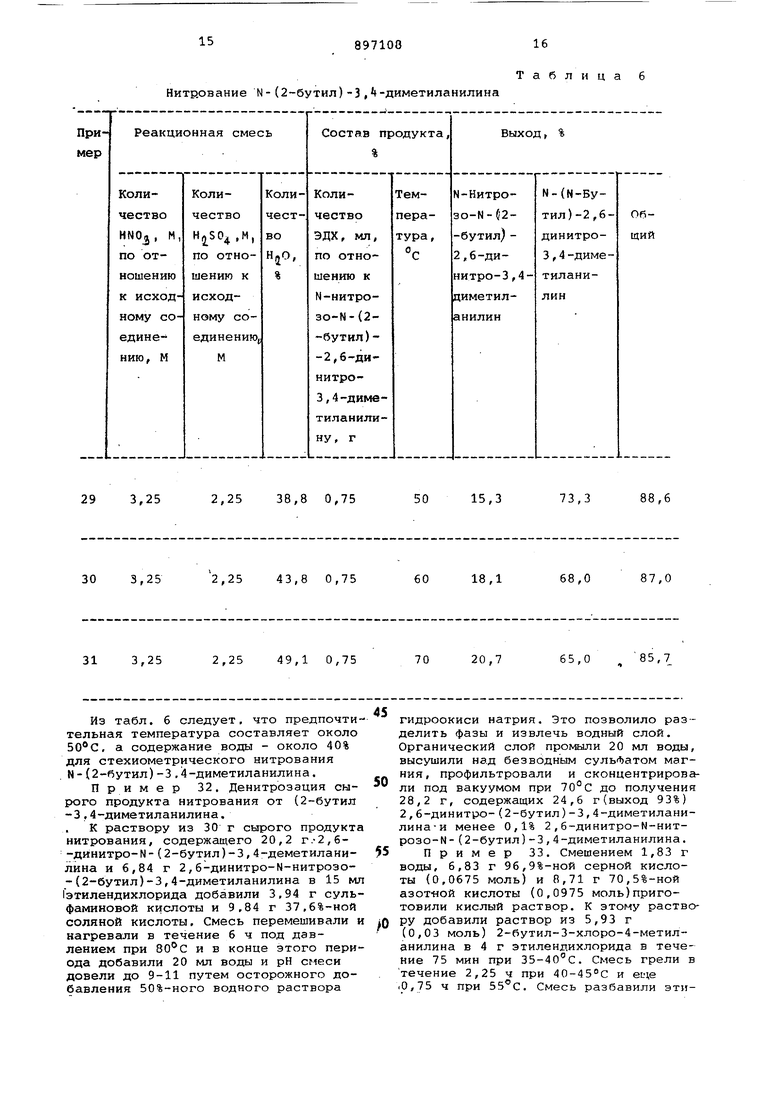
дихлорида в качестве исходного материала на протяжении 2 ч и затем поддерживали реакционную смесь при 35°С. Примера 29-31. Примеры, приведенные в табл. 6, показывают изменение общего выхода .и состава продукта в зависимости от изменения содержания воды и температуры реакционной меси при нитровании N-(2-6yтил)-37 4-циметиланилина. Органическую Лазу последовательно Промывали 500 мл 5%-ного водного раствора каустика и 500 мл воды; Органическую фазу затем высушивали над безводным сульфатом магния, фильтровали и концентрировали при 70с под вакуумом до получения 130,0 г сырого продукта, содержащего 100 г (выход 75,5%) 2,б-динитро-N-(2-бутил)-3,4-диметиланилина и 22,0 г (выход 14,9%) 2,6-динитро-Н-нитрозо-М-(2-бутил)-3,4-диметиланилина. Общий выход целевого 2,6 -динитроанилинового продукта 90,4%. Примеры 26-28. Нитрование N-(2-бутил)-3,4 диметиланилина показывает влияние содержания воды в нитруюадей смеси на состав продукта. Результаии нитрования N- (2-бутил)-3,4-диметиланилина приведены в табл.5.
Нитр.ование N- (2-бутил) -3 . -диметиланилина
3,25
2,25
38,8 0,75
29
2,25
3,25
30
43,8 0,75
49,1 0,75
2,25
31
3,25
Из табл. 6 следует, что предпочтительная температура составляет около , а содержание воды - около 40% для стехиометрического нитрования N-(2-бутил)-3,4-диметиланилина.
Пример 32. Денитроэация сырого продукта нитрования от (2-бутил -3.4-диметиланилина,
К раствору из 30 г сырого продукта нитрования, содержащего 20,2 г.-2,6-динитро-N-(2-бутил)-3,4-деметиланилина и 6,84 г 2,6-динитро-М-нитрозо-(2-бутил)-3,4-диметиланилина в 15 мл 1этилендихлорида добавили 3.94 г сульфаминовой кислоты и 9.84 г 37.6%-ной соляной кислоты. Смесь перемешивали и нагревали в течение 6 ч под давлением при и в конце этого периода добавили 20 мл воды и рН смеси довели до 9-11 путем осторожного добавления 50%-ного водного раствора
Таблица 6
15,3
88,6
73,3
50
87,0
18,1
68,0
60
85,7
65,0
20,7
70
гидроокиси натрия. Это позволило разделить фазы и извлечь водный слой. Органический слой промыли 20 мл воды, высушили над безводным суль|1)атом магния, профильтровали и сконцентрировали под вакуумом при 70°С до получения 28,2 г, содержащих 24,6 г(выход 93%) 2,6-динитро-(2-бутил)-3,4-диметиланилина -и менее 0,1% 2,6-динитро-Ы-нитрозо-N-(2-бутил)-3,4-диметиланилина. Пример 33. Смешением 1,83 г воды, 6,83 г 96,9%-ной серной кислоты (0,0675 моль) и 8,71 г 70,5%-ной азот-ной кислоты (0,0975 моль)приготовили кислый раствор. К этому раствору добавили раствор из 5,93 г (0,03 моль) 2-бутил-3-хлоро-4-метиланилина в 4 г этилендихлорида в течение 75 мин при 35-40°с. Смесь грели в течение 2,25 ч при 40-45°С и eL,e .0,75 ч при . Смесь разбавили этилендихлоридом и промыли разбавленным водным раствором каустика (до рН 1012), промыли свежей водой, высушили над сульфатом магния и профильтровал После концентрирования получили 7,8 масла. При анализе этого масла методом тонкослойной хроматографии было обнаружено, что оно содержит много одного компонента и незначительное к личество трех других компонентов. Это масло растворили в 20 мл этилендихлорида, к раствору добавили 13 мл концентрированной соляной кислотыи 1 г сульфаминовой кислоты. Смесь нагревали при дефлегмировании 7 ч. Анализ аликвоты тонкослойной хр матографией показал, что один из трех присутствующих в небольшом количестве компонентов исчез. Смесь охладили и водный компонент отбросили. Органический слой промыли водой высушили над сульфатом магния, профильтровали и сконцентрировали до получения 5,7 г масла. Главный компонент этого масла отделили в чистом виде жидкостной хромотогра ией и протонной магнитной резонансной спектроскопией. Было показано, что он является целевым N-(2-бутил)-2,6 динитро-3-хлоро-р-толуидином. Пример 34. Смешением 2,55 г воды, 6,91 г 95,7%-ной серной кислоты и 8,71 г 70,5%-ной азотной кислоты приготовили кислый раствор. К это му раствору на протяжении около 2 ч добавляли раствор из 6,3 г N-2-бутил-4-М-бутиланилинав 19 мл этилендихлорида при 33-37 С. Реакционную смесь перемешивали 1,5 ч при 40-45 С и 1 ч при . Дополнительно добавили 1,93 г 95,3%-ной серной кислоты и смесь перемешивали более 0,5 ч при 50°С. Испытания показа ли, что в реакционной смеси остался непрореагировавший N-2-бут.ил-4-Мбутиланилин или его моно-нитропроизводные. Смесь охладили, органическую фазу отделили и промыли разбавленным водным раствором каустика, а затем водой. Затем к органической фазе добавили 13 мл концентрированной соляной кислоты и 1,0 г сульфаминовой ки лоты. Эту смесь грели при дефлегмиро вании в течение 4 ч, затем ее охладили и кислый слой отбросили. Органическую фазу промыли водой и сконцентрировали до получения 14,4 г мас ла. Это масло поместили в колонну с силикагелем и элюировали толуолом. Наиболее прочно удерживаемый компонент экстрагировали из кремния с использованием метиленхлорида. Экстракт сконцентрировали до получения 3,4 г (более 38% от теоретического количества) желтого твердого вещества, которое было идентифицировано как К-2-бутил-2,б-динитро-4-М-бутиланилин. Пример 35. Получение N-фтор-бутил-2-нитро-3,4-диметиланилина и N-фтор-бутил-б-нитро-З,4-диметиланилина. N-Фтор-бутил-3,4-диметиланилин (5,3 г, 0,03 моль)растворили в 10 мл дихлорэтана и осторожно обработали смешанной кислотой (6 мл концентрированной серной кислоты и 2,7 г концентрированной азотной кислоты) при 15-25 С. После завершения растворения смесь вылили в воду. Органический слой отделили и очистили колонной хроматографией на силикагеле с использованием в качестве элгоента гексана. Первым элюированным соединением был М-фтор-бутил-2-нитро-3,4-диметиланилин, идентифицированный по ЯМР- и ИК-спектру. Вторым элюированным соединением был М-Фтор-бутил6-нитро-3,4-диметиланилин, который также характеризовался своим ЯМРспектром. Пример 36. Получение N-(1 этилпропил)-2,б-динитро-3,4-диметиланилина. N-(1-Этилпропил)-2-нитро-З, 4- . -диметиЛанилин приготовили по методике примера 35 с использованием вза мен N-фтор-бутил-3,4-диметиланр{лина N-(1-этилпропил)-3,4-диметиланилина. Целевое 2,6-динитро соединение получили с хорошим выходом и чистотой путем нитрования 2-нитро соединения с помощью методики примера 1. Пример 37. Способом,аналогичным примеру 36, с использованием методики примера 35 с заменой N-фтор-бутил-3 4-диметиланилина на N-(1-этилпропил)-3,4-диметаланилин приготовили N -(1-этилпропил)-6-нитро-3,4диметиланилин. Используя методику примера 1, 6-нитро-соединение превратили в N-(1-этилпропил)-2,6-динитро3,4-диметиланилин. Пример 38. Добавлением 95,3%-ной серной кислоты (5,79 г; 0,056 моль) и 70,5%-ной азотной кислоты(7,27 г; 0,081 моль) к воде (1,27 г) приготовили смешанный кислотный раствор. К смешанным кислотам, перемешивая, добавили раствор из N,Н-бис-(2-хлорэтил)3,4-диметиланилина (6,15 г; 0,025 моль) в 15 мл этилендихлорида. Добавление проводили на протяжении 90 мин при 35-45 С. После завершения добавления смесь перемешивали дополнительно в течение 2 ч при 30-40 с. Нижний слой затем отделили и сбросили. Верхний слой промыли 10 мл порциями разбавленного раствора каустика и воды поочередно. Органический раствор высушили над безводным сульфатом магния и затем подвергли хроматографическому разделению на колонне с силикагелем. Элюиройание бензолом позволило выделить 3,73 г (44,5%) желтого твердого
вещества, которое ЯМР-спектросКопией было определено как М,М-бис-(2-ХЛОрэтил)-2,б-дйнитро-3,4-диметиланилин.
Формула изобретения
Способ получения 2,6-динитропроизводных М-ЕШКИЛ или N,N-диалкиланилинов общейформулы
КгТ-«г
Y
алкил, с числом углеродных атомов 1-4, галоген
Z водород, галоген, алкил .с числом углеродных атомов 1-4, алкокси или монозамещенный алкйл, в котором заместителем является галоген или елкокси; R - алкил, моногалоидалкил или алкоксиалкил с числом углвродных атомов 1-4, циклоалкил с числом углеродных атомов 4-6
Pj - водород или R путем нитрования соответствующего -алкилированного анилина нитрующей месью, содержащей азотную кислоту,, серную кислоту и воду, о т л и ч ащ и и с я тем, что, с целью увеличения выхода целевого продукта и упрощения процесса, нитрующая смесь имеет следующий состав: 15-78% воды, 8-68% серной кислоты с концентрацией 95-98% и 2-60% азотной кислоты с конентрацией 70-75%.; Источники информации, принятые во внимание при экспертизе
кл. С 07 С 87/60, 1967 (прототип).
