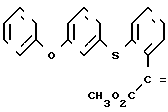
Изобретение относится к области кислот, в частности к способу получения производных пропеновой кислоты общей формулы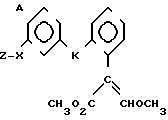
 и их стереоизомеров, где A - водород, галоген, С1-С4-алкил, С1-С4-алкокси, гидрокси, фенокси или С1-С4-алкилкарбонил; К представляет собой кислород или серу; Х - O, S(O)n, NH, NR1: CH2, CHR2, CO, CH2CH2, CH = CH, OCH2, (CH2)mO, CHR1O, OCH2O, S(O)nCH2, S(O)CH2O, NR1CH2, COO, OOC, SO2O, COCH2O, COCHR1O, CONH, NHCO, NHSO2, COS, SCO, N = N, CH2OCO, CH2SCO, CH2NHCO, CH2ON = CH2, OCH2CH2O, NR1N = CH, CH2OCON, CH = CHCH2O, (R2)2P+CH2Q-, N(COR1), N = CH, CH(OH), CO2CH2, SCH2O, NR1CO, S(O)2NH или CONR1; R1 - C1-C4-алкил; R2 - фенил; n - 0,1 или 2; m - 1,2,3,4 или 5; Q - галоидный анион; Z - фенил (возможно монозамещенный С1-С6-алкилом, С1-С4-алкокси, С1-С4-галоидалкилом, фенокси,
и их стереоизомеров, где A - водород, галоген, С1-С4-алкил, С1-С4-алкокси, гидрокси, фенокси или С1-С4-алкилкарбонил; К представляет собой кислород или серу; Х - O, S(O)n, NH, NR1: CH2, CHR2, CO, CH2CH2, CH = CH, OCH2, (CH2)mO, CHR1O, OCH2O, S(O)nCH2, S(O)CH2O, NR1CH2, COO, OOC, SO2O, COCH2O, COCHR1O, CONH, NHCO, NHSO2, COS, SCO, N = N, CH2OCO, CH2SCO, CH2NHCO, CH2ON = CH2, OCH2CH2O, NR1N = CH, CH2OCON, CH = CHCH2O, (R2)2P+CH2Q-, N(COR1), N = CH, CH(OH), CO2CH2, SCH2O, NR1CO, S(O)2NH или CONR1; R1 - C1-C4-алкил; R2 - фенил; n - 0,1 или 2; m - 1,2,3,4 или 5; Q - галоидный анион; Z - фенил (возможно монозамещенный С1-С6-алкилом, С1-С4-алкокси, С1-С4-галоидалкилом, фенокси,
фенилом, амино, гидрокси, 1-(С1-С4-алкоксикарбонил)-2-(С1-С4-алкокси)-винил, С1-С4-галоидалкокси или С1-С4-алкоксикарбонил, или моно- или дизамещенный галогеном, нитро, С1-С4-алкилом или цианогруппой); нафтил, хинолинил, пиридинил (возможно монозамещенный С1-С4-алкилом, С1-С4-алкоксикарбонилом, амино, галогеном, нитро, С1-С4-алкилкарбониламино, ди-(С1-С4-алкилсульфонил)амино или CH(O)NH, или моно- или дизамещенный С1-С4-галоидалкилом или цианогруппой, или дизамещенный аминогруппой и одной из циано, галоген или С1-С4-алкоксигруппы, или дизамещенный нитрогруппой и одной из циано, галоген, ди-(С1-С4-алкил)амино или С1-С4-алкоксигруппы, или замещенный цианогруппой и двумя С1-С4-алкильными группами); пиримидинил (возможно: монозамещенный С1-С4-алкилом, С1-С4-галоидалкилом, С1-С4-алкилтио, циано, нитро, фенилом, HO2C, С1-С4-алкоксикарбонилом или С1-С4-алкилсульфонилом, или моно- или дизамещенный С1-С4-алкокси, или моно-, ди- или тризамещенный галогеном, или дизамещенный галогеном и одной из С1-4 алкил или С1-4 алкилтио, или дизамещенный С1-4 алкил и С1-4 галоалкил), пиразинил (возможно монозамещенный галогеном или циано, или дизамещенный С1-С4-алкилом), пиридазинил (возможно монозамещенный С1-С4-алкокси, фенилом или аминокарбонилом, или моно- или дизамещенный галогеном, или дизамещенный галогеном и С1-С4-алкилом), бензотиазолил, тиенил (возможно монозамещенный пиразолилом), который сам по себе дизамещен С1-С4-алкилом и С1-С4-галоидалкилом (или пиридинилом), который сам по себе, возможно, монозамещен нитро (или дизамещенный галогеном), 1,2,4-триазолил, хиноксалинил (монозамещенный галогеном), 1,3,5-триазинил (дизамещенный галогеном, или дизамещенный галогеном и С1-С4-алкокси), тиазолил (возможно монозамещенный нитро или моно-, или дизамещенный С1-С4-алкилом), бензоксазолил, пиридинил-N-оксид, тиено[2,3-d] пиримидинил, пирролил (возможно монозамещенный С1-С4-алкилом), изоксазолил (монозамещенный С1-С4-алкилом), 1,3,4,-тиадиазолил, пиразолил (замещенный галогеном и двумя С1-С4-алкильными группами) или 1,2,4-триазинил (монозамещенный фенилом); при условии, что, когда Z представляет собой незамещенный фенил, а Х и К оба представляют собой кислород, тогда A не является водородом; которые обладают фунгицидной активностью.
Известны фунгициды - манкозеб, карбоксин, пироксифур. Однако их активность недостаточно высока.
Целью изобретения является разработка способа получения новых соединений, обладающих фунгицидной активностью.
Поставленная цель достигается предложенным способом получения производных пропеновой кислоты общей формулы I и их стереоизомеров, отличительной особенностью которого является то, что осуществляют взаимодействия соединений формулы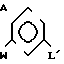
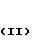 с соединением формулы
с соединением формулы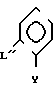
 в присутствии основания, где одна из L' и L" представляет собой отщепляемую группу, тогда как другая представляет собой КН; W представляет собой ZX или группу, которую превращают в ZX путем:
в присутствии основания, где одна из L' и L" представляет собой отщепляемую группу, тогда как другая представляет собой КН; W представляет собой ZX или группу, которую превращают в ZX путем:
(а) взаимодействия в присутствии основания соединения формулы
H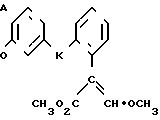

(i), когда Х представляет собой O, с соединением ZL; или
(ii), когда Х представляет собой SO2O, с соединением ZSO2O; или
(iii), когда Х представляет собой CH2O, с соединением ZCH2L; или
(iv), когда Х представляет собой CO.O, с соединением ZCO.Hal;
или (b) взаимодействия в присутствии основания соединения формулы
LCH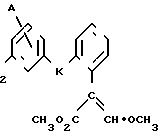

(i), когда Х представляет собой OCH2, с соединением ZOH; или
(ii), когда Х представляет собой (C6H5)2P + CH2Q- и L представляет собой галоид, с фосфином формулы Z(C6H5)2P; или
(iii), когда Х представляет собой CO.OCH2, с соединением ZCO.OH;
(с) когда Х представляет собой CH = CH, взаимодействие в присутствии основания соединения формулы
(R′O)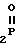 CH
CH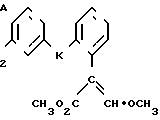
 где R1 представляет собой С1-С6-алкил, с соединением формулы ZCHO; или
где R1 представляет собой С1-С6-алкил, с соединением формулы ZCHO; или
(d), когда Х представляет собой CH = CH, взаимодействие соединения формулы (I), где Х представляет собой (C6H5)2P + CHR2Q- последовательно с основанием и карбонильным соединением формулы ZCHO; или
(е), когда Х представляет собой CONH, взаимодействие соединения формулы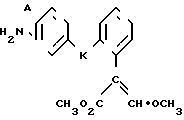
 в присутствии основания с галоидом кислоты формулы ZCO.Q; или
в присутствии основания с галоидом кислоты формулы ZCO.Q; или
(f), когда Х представляет собой СН(OH), взаимодействие альдегида формулы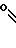
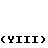 с реактивом Гриньяра формулы ZMgHAI; или
с реактивом Гриньяра формулы ZMgHAI; или
(g), когда Х представляет собой O.CO, взаимодействие хлорида кислоты формулы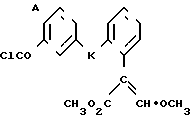
 с соединением формулы ZOH в подходящем растворителе в присутствии основания; или
с соединением формулы ZOH в подходящем растворителе в присутствии основания; или
(h), когда Х представляет собой OCH2, взаимодействие соединения формулы
HOCH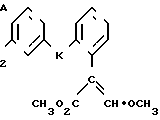
 с соединением формулы ZL; или
с соединением формулы ZL; или
(j), когда Х представляет собой N = N, взаимодействие формулы
H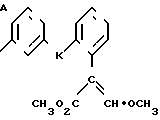
 с соединением формулы ZN+ = N Hal-; или
с соединением формулы ZN+ = N Hal-; или
(k), когда Х представляет собой NHSO2, взаимодействие соединения формулы
ClSO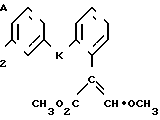
 с соединением формулы ZNH2; где: A, B, E, X, K и Z имеют значения, приведенные выше, L представляет собой отщепляющуюся группу, предпочтительно атом галогена, и Q представляет собой Hal; а Y представляет собой группу CH3O.CH = C-(CO2CH3) или половину CH2CO2CH3, которую можно превратить в подобную группу посредством взаимодействия с метилформиатом в присутствии основания и последующего метилирования в щелочных условиях.
с соединением формулы ZNH2; где: A, B, E, X, K и Z имеют значения, приведенные выше, L представляет собой отщепляющуюся группу, предпочтительно атом галогена, и Q представляет собой Hal; а Y представляет собой группу CH3O.CH = C-(CO2CH3) или половину CH2CO2CH3, которую можно превратить в подобную группу посредством взаимодействия с метилформиатом в присутствии основания и последующего метилирования в щелочных условиях.
Желательно соединения формулы III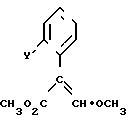 где Y' представляет собой галоген, подвергают взаимодействию с фенолом общей формулы II
где Y' представляет собой галоген, подвергают взаимодействию с фенолом общей формулы II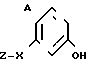 (II B) где A, X и Z имеют вышеуказанные значения в присутствии основания, или с солью фенола (IIb) в присутствии катализатора - соли меди или смесь соли меди и металлической меди.
(II B) где A, X и Z имеют вышеуказанные значения в присутствии основания, или с солью фенола (IIb) в присутствии катализатора - соли меди или смесь соли меди и металлической меди.
Изобретение иллюстрируется соединениями, перечисленными в нижеследующих таблицах I, II, III и IV. Везде в табл.I, II, III и IV метил-3-метоксипропеноатная группа имеет (Е)-конфигурацию.
Табл. 3 включает 446 соединений вышеприведенной формулы. Для первых 445 соединений все значения X, D, G, A, B и Е указаны в табл.1, т.е. соединения NN 1-445 те же, что и в табл.1, за исключением значения К, которому в табл. 1 соответствует кислород, а в табл.3 - сера. Соединение N 446 отвечает вышеприведенной формуле, где Х означает кислород и A, B, D, E и G все означают водород. Описание получения соединения N 446 приведено в примере 11.
Табл. 4 включает 320 соединений вышеприведенной общей формулы, где все значения Z, X, A, B и Е указаны в табл.2, т.е. соединения NN 1-320 те же, что и в табл.2, за исключением значения К, которому в табл.2 соответствует кислород, а в табл.4 - сера.
В табл.5 приведены данные протонного ЯМР для некоторых соединений табл. 1,2,3 и 4. Химические сдвиги обозначают в млн-1 от тетраметилсилана, во всех случаях в качестве растворителя используют дейтерированный хлороформ. Столбец, озаглавленный "частота", относится к рабочим частотам спектрометров ЯМР. В табл.5 используют следующие сокращения:
уш - уширенная полоса
с - синглет
д - дублет
т - триплет
к - квартет
м - мультиплет.
Соединения формулы I являются активными фунгицидами и могут быть использованы для борьбы со следующими патогенами: Pyricularia oryzae на рисе, Puccinia recondita, puscinis herdei и другие виды ржавчины на ячмене и также ржавчина на других культурных растениях, например: кофе, груша, яблоки, арахис, овощи и декоративные растения. Erisiphe graminis (настоящая мучнистая роса) на ячмене и пшенице и другие виды настоящей мучнистой росы на различных культурных растениях, такие как Sphaerotheca macularis на хмеле, Sphaerotheca fuliginea на тыквенных (например, огурцах), podosphaera feucolricha на яблоках и Uncinula necator на винограде Helminthosporium, Phynchosporium, Septoria, pseudocercosprella herepotrichoides и Caeumannomyces graminis на зерновых. Cercospora arachidieda и cerasporidium personata на арахисе и другие виды Cercospora на других культурных растениях, например сахарной свекле, бананах, сое и рисе.
Botrytis cinerea (серная мучнистая роса) на помидорах, клубнике, овощах и других культурных растениях.
Alternaria на овощах (например, огурцах), рапсе, яблоках, помидорах и других культурных растениях.
Vehtura inaegualis (парша) на яблоках.
Plasmopara vitocola на винограде.
Другие виды ложной мучнистой росы, такие, как Bremia lactucae на салате, peronospora - на сое, табаке, луке и других культурных растениях, pseudoperounospora - на хмеле и pseudoperonuspora cubeusis - на тыквенных. Phytophtora infestous - на картофеле и помидорах, и другие виды phytophtora - на овощах, землянике, авокадо, перце, декоративных растениях, табаке, какао и других культурных растениях. Thanatephorus cucumeris - на рисе и другие виды Rhizoctonia на различных культурных растениях, таких, как пшеница и ячмень, овощи, хлопок и трава для газонов.
Некоторые соединения проявляют широкий спектр активности против грибков in vitro. Соединения могут также проявлять активность к заболеваниям фруктов, наступающих после их уборки (например, penicillium digitatum и italicum и Trichoderma viride на апельсинах, Gloesporium musarum на бананах и Botrytis cinerea на винограде).
Кроме того, отдельные соединения могут проявлять активность при протравлении семян против грибков вида Fusarium, Septeria Tilletia (твердая головная в семенах пшеницы). Ustilago, Helminthosporium на зерновых, Rhizochtonia solani на хлопке и Pyriculare oryzae на рисе.
Соединения могут подвергаться систематическому продвижению в растениях. Кроме того, соединения могут оказаться достаточно летучими, чтобы проявлять активность в паровой фазе против грибков на растениях.
Многие соединения формулы I, в том числе и те соединения, в которых Х = 0, более безопасны по отношению к культурным растениям (например, винограду), по сравнению с известными близкими по строению соединениями.
Таким образом, предлагается способ борьбы с грибками, заключающийся в нанесении на растение, семена растений или место, где расположено растение или семена, эффективного количества соединения согласно вышеприведенному определению или содержащей его композиции.
Соединения могут также оказаться полезными в качестве промышленных (в противоположность сельскохозяйственным) фунгицидов, например, для профилактики от грибкового заражения древесины, выделанной кожи, кожных изделий и особенно пленок краски.
Соединения могут быть использованы в качестве фунгицидов, но обычно использованием носителя или разбавителя на основе соединений получают композиции. Таким образом, предлагаются фунгицидные композиции, содержащие соединение общей формулы I и фунгицидно приемлемый носитель или разбавитель.
При использовании в качестве фунгицидов соединения могут быть нанесены самыми различными путями. Например, они могут быть нанесены в виде композиции и как таковые непосредственно на листву растений, семена или среду обитания растений или выращивания растений или же могут быть нанесены путем опрыскивания, в виде дуста, крема или пасты, в виде паров или гранул замедленного действия. Обработке может быть подвергнута любая часть растения, в том числе: листва, ствол, ветви или корни или земля вокруг корней, или семена перед высевом, или почва в целом, а также вода для орошения или системы гидропонического выращивания растений. Соединения изобретения могут быть в виде инъекций введены в растения или нанесены опрыскиванием с использованием техники для электродинамического опрыскивания или других малоемких способов.
Понятие "растения" в используемом здесь значении включает рассаду, кустарники и деревья. Фунгицидный способ изобретения включает превентивную, защитную, профилактическую и истребительную обработку.
Соединения рекомендуют применять в виде композиции для сельскохозяйственных и садовых целей, причем тип используемой в каждом случае композиции зависит от конкретной цели применения.
Композиции могут иметь вид дустируемых порошков или гранул, содержащих активный компонент (соединение изобретения) и твердый носитель или разбавитель, например такие наполнители, как каолин, бентонит, кизельгур, доломит, карбонат кальция, тальк, порошковый оксид магния, фуллерова земля, гипс, диатомовая земля и китайская глина. Такие гранулы могут иметь вид предварительно полученных гранул, пригодных для использования без дополнительной обработки. Такие гранулы могут быть приготовлены либо пропиткой таблетированного носителя активным компонентом, либо таблетированием смеси активного компонента и порошкового носителя. Композиции для протравления семян могут включать средство (например, минеральное масло), способствующее наливанию композиции на семена. Или же активный компонент может быть введен в состав для протравливания семян использованием органического растворителя (например, N-метилпирролидона, пропиленгликоля или диметилформамида). Композиции также могут иметь вид смачиваемых порошков из диспергируемых в воде гранул, содержащих смачивающее или диспергирующее средство, способствующее диспергированию в жидкости. Порошки и гранулы могут также содержать наполнители и суспендирующие средства.
Эмульгируемые концентраты или эмульсии могут быть приготовлены растворением активного компонента в органическом растворителе, возможно содержащем смачивающее или эмульгирующее средство, с последующим добавлением смеси к воде, которая также может содержать смачивающее или эмульгирующее средство. Приемлемые органические растворители включают ароматические растворители, такие, как алкилбензолы и алкилнафталины, кетоны, такие, как изофорон, циклогексанон, метилциклогексанон, хлорированные углеводороды, такие, как хлорбензол и трихлорэтан, спирты, такие, как бензиловый спирт, фурфуриловый спирт, бутанол и простые гликолевые эфиры.
Концентрированные суспензии почти нерастворимых твердых продуктов могут быть приготовлены размолом в шаровой мельнице в присутствии диспергирующего средства с включением суспендирующего средства, препятствующего осаждению твердого продукта.
Композиции, предназначенные для использования в виде опрыскивания, могут иметь вид аэрозолей, в этом случае состав содержится в контейнере под давлением в присутствии пропелланта, например фтортрихлорметана или дифтордихлорметана.
Соединения формулы I могут быть смешаны в сухом состоянии с пиротехнической смесью с образованием композиции, пригодной для образования в замкнутом пространстве дыма, содержащего указанные соединения.
Или же соединения могут быть использованы в виде микрокапсул или могут быть введены в состав с биодеградируемыми полимерами с достижением медленного регулируемого выделения активного вещества.
Включением соответствующих добавок, например добавок, способствующих распределению частиц, увеличивающих липучесть и устойчивость к дождю на обработанных поверхностях, т.е. различные композиции могут быть лучше приспособлены для различных применений.
Соединения формулы I могут быть использованы в смеси с удобрениями (например, азотными, калиевыми и фосфорными удобрениями). Рекомендуются композиции, содержащие только гранулы удобрения, включающие, например, в виде покрытия соединение изобретения. Такие гранулы предпочтительно содержат до 25 мас. % соединения изобретения. Таким образом, изобретением дается композиция, состоящая из удобрения и соединения общей формулы I или его соли, или его комплекса с металлом.
Смачиваемые порошки: эмульгируемые концентраты и концентрированные суспензии обычно содержат поверхностно-активное вещество, например смачивающее средство, диспергирующее средство, эмульгатор или суспендирующее средство. Эти вещества могут быть катионного, анионного или неионного типа.
Приемлемыми катионными поверхностно-активными веществами являются четвертичные аммониевые соединения, например цетилтриметиламмонийбромид. Приемлемые катионные поверхностно-активные вещества включают: мыла, соли алифатических моноэфиров серной кислоты (например, натрийлаурилсульфат) и соли сульфированных ароматических соединений (например, натрийдодецилбензолсульфонат, натрий-, кальций- или аммонийлигносульфонат, смесь натрийдиизопропил- и триизопропилнафталинсульфоната).
Приемлемыми неионными поверхностно-активными веществами являются продукты конденсации окиси этилена с жирными спиртами, такими, как олеиловый или цетиловый спирт, или с алкилфенолами, такими, как октил- или нонилфенол и октилкрезол. Среди других неионных поверхностно-активных веществ можно назвать неполные эфиры, образованные длинноцепными жирными кислотами и ангидридами гексита, продукты конденсации указанных неполных эфиров с оксидом этилена и лецитинами. К приемлемым суспендирующим средствам относятся гидрофильные коллоиды (например, поливинилпирролидон и натрийкарбоксиметилцеллюлоза), а также набухающие глины, такие, как бентонит и аттапульгит.
Композиции, предназначенные для использования в виде водных дисперсий или эмульсий, как правило, поставляются в форме концентрата с высоким содержанием активного компонента, такие концентраты перед употреблением разбавляют водой. Эти концентраты предпочтительно должны выдерживать длительные сроки хранения и после такого хранения должны сохранять способность образовать водные препараты, остающиеся однородными в течение достаточного времени, необходимого для их распыления с помощью оборудования для опрыскивания. Концентраты, как правило, содержат до 95%, предпочтительно 10-85%, например, 25-60 мас. %, активного компонента. После разбавления с образованием водных препаратов полученные препараты могут содержать различное количество активного компонента, зависящее от намечаемого применения препарата, могут быть использованы водные препараты, содержащие 0,00055 или 0,01-10 мас.% активного компонента.
Композиции могут содержать другие биологически активные соединения, например соединения с аналогичной или дополняющей фунгицидной активностью или с регулирующей рост растений, гербицидной или инсектицидной активностью.
Фунгицидным соединением, которое может присутствовать в композиции изобретения, может быть соединение, способное противостоять заболеваниям колосьев зерновых культур (например, пшеницы), вызываемых такими возбудителями, как Septoria, Gibberella и Helminthosporium, заражению семян и заражению почвы, ложной, и настоящей мучнистой росе на винограде, настоящей мучнистой росе и парше на яблоках и т.д. Включением другого фунгицида композиция приобретает более широкий спектр активности, по сравнению с использованием одного только соединения формулы I. Кроме того, другой гербицид может оказывать синергическое действие на фунгицидную активность соединения общей формулы I. Примеры фунгицидных соединений, которые могут быть включены в композицию изобретения, включают: карбендазим, беномил, тиофанат-метил, тиабендазол, фуберидазол, этридазол, дихлофуанид, цимоксанил, оксадиксил, офурас, металаксил, фуралаксил, беналаксил, фозетил-алюминий, фенаримол, ипродион, протиокарб, процимидон, винхлозолин, пенконазол, михлобутанил, пропамокарб, диниконазол, пиразофос, этиримол, диталимфос, тридеморф, трифорин, нуаримол, триазбутил, гуазатия, триацетатную соль 1,1'-иминоди(октаметилен)дигуанидина, бутиобат, пропиконазол, прохлораз, флутриафол, гексаконазол, (2RS, 5RS)-5-(2,4-дихлорфенил)тетрагидро-5-(1Н-1,2,4-триазол-1-илметил)- 2-фурил-2,2,2-трифторэтиловый эфир, ципроконазол, тербуконазол, 1-[(2P, 4P; 2RS; 4RS)-4-бром-2-(2,4-дихлорфенил)тетрагидрофурфурил] -1Н-1,2,4-триазол, пирролнитрин, 5-этил-5,8-дигидро-8-оксо(1,3)-диоксоло(4,5-о)хинолин-7- карбоновая кислота, 3-(2,4-дихлорфенил)-2-(1Н, 1,2,4-триазол-1-ил)хиназолин-4-(3Н)-он, (RS)-1-аминопропилфосфоновая кислота, флузилазол, триадимефон, триадеменол, дихлорбутразол, фенпропиморф, пирифенокс, фенпропидин, хлорозолинат, имазалил, фенфурам, карбоксин, оксикарбоксин, метфуроксам, додеморф, BAS 454, бластицидин S, казугамицин, эдифенфос, катазин Р, циклогексимид, фталид, пробеназол, изопротиолан, трициклазол, 4-хлор-П-(циано(этокси)метил)бензамид, пирохилон, хлоробензтиазон, неоазозин, полиоксин D, валидамицин A, мепронил, флутоланил, пенцикурон, дихломезин, феназин-оксид, никельдиметилдитиокарбамид, техлофталам, битертанол, бупиримат, этаконазол, гидроксиизоксазол, стрептомицин, ципрофурам, билоксазол, хинометионат, диметиримол, 1-(2-циано-2-метоксииминоацетил)-3-этилмочевину, фенапанил, толхлофосметил, пироксифур, полирам, манеб, манкозеб, каптафол, хлороталонил, анилазин, тирам, каптан, фолпет, зинеб, пропинеб, серу, динокап, дихлон, хлоронеб, банакприл, нитротализопропил, додин, дитианон, фентигидроксид, фентинацетат, текназем, хинтозен, дихлоран, медьсодержащие соединения, такие, как оксихлорид меди, сульфат меди и бордоская смесь, а также ртутьорганические соединения.
Соединения общей формулы I могут быть смешаны с почвой, торфом или другой средой для корней с целью защиты растений от заражающих семена, заражающих почву или листву грибковых инфекций.
Приемлемые инсектициды, которые могут быть включены в композицию изобретения, включают: пиримикарб, диметоат, деметонсимм.-метил, формотион, карбарил, изопрокарб, ХМС, БРМС, карбофуран, карбосульфан, диазинон, фентион, фентротион, фентоат, хлорпирифос, изоксатион, пропафос, монокротофас, бупрофезин, этропроксифен, циклопротрин.
К регулирующим рост растений соединениям относятся соединения, регулирующие образование сорняков или всходов, или селективно регулирующие рост менее желательной растительности (например, травы).
Примеры приемлемых для совместного использования с соединениями формулы I регуляторов роста растений включают: гиберилины (например, ГА3, ГА4 или ГА7), ауксины (например, индолуксусную кислоту, индолмасляную кислоту, нафтоксиуксусную кислоту или нафтилуксусную кислоту), цитокинины (например, кинети, дифенилмочевину, бензимидазол, бензиладенин или бензиламинопурин), феноксиуксусные кислоты (например, 2,4-D или МХФК), замещенные бензойные кислоты (например, трийодбензойную кислоту), морфактины (например, хлорфлуорокол), гидразид малеиновой кислоты, глифозат, глифозин, длинноцепные жирные спирты и кислоты, дикегулак, паклобутразол, флуоридамид, мефлуидин, замещенные четвертичные аммониевые или фосфониевые производные (например, хлормекват хлорофония или меркватхлорид), этефон, карбетамид, метил-3,6-дихлоранизат, даминозид, азултам, абсцизовую кислоту, изопиримол, 1-(4-хлорфенил)-4,6-диметил-2-оксо-1,2-дигидропири-дин-3-карбоновую кислоту, гидроксибензонитрилы (например, бромксинил), дифензокват, бензоилпропэтил-3,6-дихлорпиколиновую кислоту, фенпентезол, инабенфид, триапентенол и текназен.
Нижеследующие примеры приведены для иллюстрации изобретения. Во всех примерах термин "эфир" относится к диэтиловому эфиру, для осушки растворов применяют сульфат магния, растворы концентрируют при пониженном давлении. Реакции с участием чувствительных к влаге промежуточных соединений проводят в атмосфере азота и растворители перед использованием высушивают там, где это необходимо. Если нет особых указаний, хроматографии проводят на заполненной силикагелем колонке. Там, где указано, данные инфракрасной и ЯМР-спектроскопии селективны без указания во всех случаях всех полос поглощения. 1Н-ЯМР-спектры записывают с использованием в качестве растворителя CDCl3, если нет особых указаний. Везде используют следующие сокращения:
ДМЭ - диметоксиэтан, ТГФ - тетрагидрофуран, ДМФА - N,N-диметилформамид, ЯМР - ядерный магнитный резонанс, ИК - инфракрасный, с - синглет, д - дуплет, т - триплет, м - мультиплет.
m.p. - температура плавления
GC = газовая хроматография
TLC = тонкослойная хроматография
HPLC = высокопроизводительная жидкостная хроматография
Br = широкая
ppm = миллионные доли.
Сущность изобретения иллюстрируется следующими примерами.
П р и м е р 1. Этим примером иллюстрируется получение (Е)-метил-2-[2-(3-бензилоксифенокси)фенил]-3-метоксипропеноата (соединение N 23, табл.1).
Смесь 100 г (0,54 моль) 2-бромбензальдегида, 67,03 г (1,08 моль) этиленгликоля, 0,5 г толуолсульфоновой кислоты и толуола нагревали до температуры кипения и выдерживали при этой температуре с обратным холодильником в течение 6 ч. В течение этого времени отгонялось 23 мл азеотропной смеси воды и этиленгликоля. После этого смесь охлаждали и добавляли к ней 1 л эфира. Эфирный раствор промывали насыщенным раствором бикарбоната натрия (200 мл), затем трижды водой, порциями по 150 мл и, наконец, 150 мл насыщенного раствора хлористого натрия. После высушивания и фильтрации, а затем упаривания эфирного раствора получали 121,96 г (выход 98,6%) 2-(2-бромфенил)-1,3-диоксолана в виде маслянистой жидкости.
1Н ЯМР: (60 МГц), δ : 3,4 (4Н, м), 6,0 (1Н, с), 6,9-7,6 (4Н, м), м.д.
Полученный продукт без дополнительной очистки использовали на следующей стадии.
35,2 г (0,63 моль) гранулированного гидроксида калия растворяли в 50 мл воды и к полученному раствору добавляли 78 г (0,63 моль) 3-метоксифенола с 250 мл толуола. Реакционную смесь нагревали до температуры кипения и выдерживали при этой температуре до прекращения отгонки воды (всего собиралось 65 мл воды). После этого смесь охлаждали до 80оС и добавляли к ней 120 г (0,524 моль) 2-(2-бромфенил-1,3-диоксана, 200 мл ТМФ и 0,2 г хлорида меди. Смесь медленно нагревали до 150-155оС и отгоняли толуол. После этого ее выдерживали в течение 6 ч при 150-155оС, а затем охлаждали до 25оС и добавляли 500 мл воды. Смесь фильтровали, остаток промывали 200 мл эфира, а фильтр подвергали экстракции эфиром (трижды, порциями по 150 мл). Объединенные эфирные фазы дважды промывали 2 н раствором гидроксида натрия, (порциями по 150 мл), четырежды водой (порциями по 150 мл) и затем 150 мл рассола. После высушивания и фильтрации эфирный раствор упаривали, получая в результате 124,1 г (выход 87,1%) 2-[2-(3-метоксифенокси)фенил]-1,3-диоксолана в виде маслянистой жидкости.
1Н ЯМР: (60 МГц), δ : 3,65 (3Н, с), 3,95 (4Н, д), 6,12 (1Н, с), 6,6-7,6 (8Н, м) м.д.
Полученный продукт без дополнительной очистки использовали на следующей стадии.
32,7 г (0,12 моля) 2-[2-(3-метоксифенокси)фенил]-1,3-диоксолана перемешивали в смеси 95 мл воды и 5 мл концентрированной соляной кислоты при температуре окружающей среды в течение 19 ч. Затем смесь дважды подвергали экстракции эфиром, порциями по 60 мл, объединенные эфирные фазы промывали 30 мл насыщенного водного раствора бикарбоната натрия, трижды водой (порциями по 30 мл) и, наконец, 30 мл рассола. Полученный раствор высушивали, фильтровали и концентрировали, получая в результате 26,17 г (выход 95,4%) довольно чистого 2-(3-метоксифенокси)-бензальдегида (A) в виде маслянистой жидкости. Полученный продукт без дополнительной очистки использовали на следующей стадии. Пробу для анализа, однако, готовили с помощью хроматографии, используя в качестве элюента смесь эфира и гексана. В результате получали маслянистую жидкость янтарного цвета.
1Н ЯМР: (90 МГц), δ : 3,79 (3Н, с), 6,58-7,97 (8Н, м), 10,49 (1Н, д) м. д.
ИКмакс. (пленка): 1691, 1599 см-1.
Смесь 25,0 г (0,109 моль) 2-(3-метоксифенокси)-бензальдегида, 13,64 г (0,11 моль) метил-метилтиометилсульфоксида, 8,0 мл 30%-ного раствора гидроксида бензилтриметиламмония в метаноле и 150 мл ТГФ перемешивали в течение 45 мин при температуре кипения с обратным холодильником. Образующийся в результате раствор упаривали досуха и остаток подвергали хроматографии, используя в качестве элюента смесь эфира и гексана. В результате получали 27,67 г (75,3%-ный выход) сульфоксида (B) в виде смолы янтарного цвета.
1Н ЯМР: (60 МГц), δ : 2,2 (3Н, с), 2,55 (3Н, с), 3,65 (3Н, с), 6,35-8,15 (9Н, м) м.д.
20 мл ацетилхлорида добавляли по каплям в течение 15 мин к 200 мл абсолютного метанола, охлаждая смесь на водяной бане и поддерживая температуру 20-25оС. Затем к смеси добавляли в один прием раствор 27,67 г (0,83 моль сульфоксида (B) и 40 мл метанола и образующийся раствор перемешивали при температуре окружающей среды в течение 18 ч. Метанольный раствор упаривали досуха при пониженном давлении и остаток (22,78 г смолы коричневого цвета) растворяли в 200 мл эфира. Эфирный раствор промывали насыщенным водным раствором бикарбоната натрия, отфильтровывали небольшое количество нерастворившегося материала и упаривали досуха. Остаток подвергали хроматографии, используя в качестве элюента смесь эфира и гексана. В результате получали 15,62 г (выход 69,3%) 2-(3-метоксифенокси)-фенилацетата (С) в виде вязкой маслянистой жидкости.
12,89 г (0,051 моль) трибромида бора растворяли в 50 мл дихлорметана и полученный раствор охлаждали до 0-5оС, после чего добавляли к нему по каплям в течение часа, при перемешивании, раствор 7,0 г (0,026 моль) 2-(3-метоксифенокси)-фенилацетата в 80 мл дихлорметана. После перемешивания в течение 20 мин при 0-5оС смесь добавляли по каплям, при перемешивании, к 100 мл абсолютного метанола, поддерживая температуру 0-5оС. Образующийся раствор выливали в 250 мл воды с растворенными в ней 12 г бикарбоната натрия и смесь подвергали экстракции 500 мл эфира. Органическую фазу трижды промывали водой (порциями по 200 мл), а затем 150 мл насыщенного раствора хлористого натрия. После высушивания, фильтрации и упаривания получали 6,12 г (выход 92,3%) метил-2-(3-оксифенокси)-фенилацетата (D) в виде смолы коричневого цвета. Полученный продукт без дополнительной очистки можно было использовать на следующей стадии. С помощью хроматографии с использованием в качестве элюента смеси эфира и гексана, однако получали высокочистый продукт в виде вязкой золотистой маслянистой жидкости, которая быстро темнела при выдержке ее на воздухе.
Кроме вышеописанного, метил-2-(3-оксифенокси)-фенилацетат (D) получали следующим способом.
Смесь 30 г (0,18 моль) 2-хлорфенилуксусной кислоты, 48,6 г (0,34 моль) карбоната калия и 43,5 (0,35 моль) 3-метоксифенола нагревали при перемешивании при 140оС в присутствии каталитического количества хлорида меди (I). Через 3 ч по данным газовой и тонкослойной хроматографии исходная кислота полностью прореагировала. Реакционной смеси давали остыть (добавляя к ней 5 мл сухого ДМФ) до 70оС, чтобы она не стала слишком вязкой, после чего выливали ее в воду и подкисляли соляной кислотой. Образующуюся смесь подвергали экстракции эфиром, объединенные эфирные фазы промывали водой до нейтральной реакции, высушивали и упаривали, получая в результате смесь 3-метоксифенола (49% ) и 2-(3-метоксифенокси)-фенилуксусной кислоты (41%) в виде коричневой подвижной маслянистой жидкости, которую без дополнительной очистки использовали на следующей стадии.
Коричневую маслянистую жидкость кипятили в течение 2,5 ч с обратным холодильником в 70 мл метанола с добавкой с 2 мл концентрированной серной кислоты. Реакционную смесь охлаждали затем до комнатной температуры и выливали в воду. Образующуюся смесь дважды подвергали экстракции эфиром, эфирные фазы объединяли, промывали вначале разбавленным водным раствором гидроксида натрия, а затем водой до нейтральной реакции и высушивали. После упаривания получали 34,9 г сырого метил-2-(3-метоксифенокси)-фенилацетата в виде оранжево-коричневой маслянистой жидкости (содержание целевого продукта, определенное с помощью газовой хроматографии, равнялось 86%). Полученный сырой продукт объединяли с другой порцией (8,2 г) такого же продукта, полученного тем же самым методом. После повторной молекулярной перегонки (50-150оС при давлении 4 х 10-2 Мбар) получали 37,1 г очищенного (степень чистоты 95%, выход около 60% в расчете на 2-хлорфенилуксусную кислоту) метил-2-(3-метоксифенокси)-фенилацетата. Повторяя вышеописанные операции, получали нужное количество продукта.
97 г (0,36 моль) метил-2-(3-метоксифенокси)-фенилацетата нагревали в течение 8 ч при 110оС с 194 мл концентрированной бромистоводородной кислоты в 150 мл уксусной кислоты. Смесь оставляли стоять на ночь при комнатной температуре, после чего добавляли к ней 100 мл концентрированной бромистоводородной кислоты и снова нагревали до 110оС. После выдержки реакционной смеси при этой температуре в течение 7 ч весь исходный материал вступал в реакцию. Реакционную смесь охлаждали до комнатной температуры, выливали в рассол и дважды экстрагировали дихлорметаном. После отгонки дихлорметана получали маслянистую жидкость, которую нагревали при 70оС с 400 мл метанола и 2 мл концентрированной серной кислоты в течение 2 ч. Реакционную смесь затем охлаждали до комнатной температуры, выливали в рассол и дважды экстрагировали дихлорметаном. Объединенные экстракты промывали водой до нейтральной реакции, высушивали, фильтровали и упаривали, получая 92,8 г коричневой маслянистой жидкости. После молекулярной перегонки (при 150оС и давлении 1 х 10-3 Мбар) части продукта (72,8 г) получали 41,4 г (выход 57% в расчете на метил-2-(3-метоксифенокси)-фенилацетат) метил-2-(3-оксифенокси)-фенилацетата (D) в виде золотистого сиропа.
1Н ЯМР: (60 МГц), δ : 3,57 (3Н, с), 3,63 (2Н, с), 5,82 (1Н, с), 6,4-7,35 (8Н, м) м.д.
ИКмакс. (пленка): 3408, 1713 см-1.
К суспензии 0,558 г (0,023 моль) гидрида натрия в 20 мл ДМФ добавляли по каплям раствор 2,0 г (0,0077 моль) метил-2-(3-оксифенокси)-фенилацетата (D) в 10 мл ДМФ и 10 г (0,167 моль) метилформиата. После перемешивания в течение 45 мин к смеси добавляли 100 мл воды и проводили экстракцию 50 мл эфира. Водный слой подкисляли соляной кислотой до рН 3-4 и смесь дважды подвергали экстракции эфиром (порциями по 40 мл). Объединенные эфирные экстракты трижды промывали водой (порциями по 30 мл), а затем 30 мл насыщенного раствора хлористого натрия и высушивали. Эфир отгоняли, а остаток растворяли в 20 мл ДМФ и добавляли к раствору 0,64 г (0,0046 моль) безводного карбоната калия и 0,55 г (0,0044 моль) диметилсульфата. Смесь перемешивали при температуре окружающей среды в течение часа, после чего добавляли к ней 100 мл воды и дважды подвергали экстракции эфиром (порциями по 40 мл). Объединенные эфирные экстракты трижды промывали водой (порциями по 20 мл), а затем 20 мл насыщенного раствора хлористого натрия, высушивали, фильтровали, выпаривали досуха и затем подвергали хроматографии с использованием в качестве элюента смеси эфира и гексана. В результате получали (Е)-метил-2-/2-(3-оксифенокси)фенил/-3-метоксипропеноат (Е) в виде смолы янтарного цвета. После растирания ее со смесью гексана и дихлорметана получали 0,7 г (выход 30% в расчете на метил-2-(3-оксифенокси)-фенилацетат (D)) белого твердого вещества. Температура плавления 115-116оС.
Кроме того, (Е)-метил-2-[2-(3-оксифенокси)фенил]-3-метоксипропеноат (Е) получали следующим образом.
Раствор 12 г (0,0465 моль) метил-2-(3-оксифенокси)фенилацетата (D) и 55,8 г (0,93 моль) метилформиата в 35 мл ДМФ добавляли по каплям в течение 45 мин к перемешиваемой суспензии гидрида натрия (6,696 г 50%-ной дисперсии в масле, 0,1395 моль, предварительно промыта петролейным эфиром 40-60) в 65 мл ДМФ. Реакционную смесь перемешивали при комнатной температуре в течение 2,5 ч, выливали в 200 мл воды, подкисляли до рН 3 концентрированной соляной кислотой и затем дважды подвергали экстракции эфиром (порциями по 200 мл).
Объединенные органические экстракты дважды промывали рассолом (порциями по 200 мл), высушивали, фильтровали и упаривали, получая в результате 12,5 (0,0433 моль) желтой маслянистой жидкости.
12,5 г (0,0433 моль) полученной маслянистой жидкости растворяли в 100 мл ДМФ и добавляли к раствору 5,98 г (0,0433 моль) карбоната натрия. После перемешивания в течение 10 мин к смеси в один прием добавляли 5,19 г (0,042 моль) диметилсульфата в 10 мл ДМФ. Полученную смесь перемешивали в течение ночи при комнатной температуре, выливали в 200 мл воды и подвергали экстракции эфиром (дважды порциями по 200 мл). Объединенные эфирные экстракты трижды промывали рассолом (порциями по 200 мл), высушивали, фильтровали и упаривали, получая клейкую смолу. После перекристаллизации из смеси дихлорметана и гексана получали 9,54 г (73%) (Е)-метил-2-[2-(3-оксифенокси)фенил] -2-ме-токсипропеноата (Е). Т.пл. 117-118оС.
1Н ЯМР (90 МГц), δ : 3,58 (3Н, с), 3,75 (3Н, с), 5,38 (1Н, с), 6,39-7,33 (8Н, м), 7,4 (1Н, с) м.д.
ИКмакс. (нуйол): 3295, 1672, 1630 см-1.
Смесь 1,0 г (0,0033 моль) (Е)-метил-2-[2-(3-оксифенокси)фенил]-3-метоксипропеноата, 0,57 г (0,0033 моль) бензилбромида, 0,8 г (0,0053 моль) карбоната калия и 15 мл сухого ДМФ перемешивали в течение 3 ч при температуре окружающей среды. После этого к смеси добавляли 50 мл воды и дважды подвергали ее экстракции эфиром (порциями по 30 мл). Объединенные органические экстракты дважды промывали водой (порциями по 20 мл) и после высушивания и фильтрации эфирный раствор упаривали досуха и подвергали хроматографии, используя в качестве элюента смесь эфира и гексана. В результате получали 1,11 г (выход 85%) целевого соединения (F) в виде бесцветной смолы.
1Н ЯМР: (90 МГц), δ : 3,55 (3Н, с), 3,7 (3Н, с), 4,97 (2Н, с), 6,5-7,32 (13Н, м), 7,44 (1Н, с) м.д.
ИКмакс. (пленка): 1710, 1638 см-1.
П р и м е р 2. Настоящим примером иллюстрируется получение (Е)-метил-3-метокси-2-[2-(3-фенилсульфонилоксифенокси) -фенил]- пропеноата (соединение N 51 из табл.1).
Смесь 0,5 г (0,00166 моль) (Е)-метил-2-[2-(3-оксифенокси)фенил]-3-метоксипропеноата, полученного с помощью способа, описанного в примере 1, 0,36 г (0,002 моль) бензолсульфонилхлорида и 10 мл пиридина перемешивали в течение 3 ч при 60-70оС. После этого смесь охлаждали до 25оС, добавляли к ней 60 мл воды и дважды подвергали экстракции эфиром (порциями по 30 мл). Объединенные эфирные экстракты промывали 20 мл воды, 20 мл разбавленной соляной кислоты, трижды водой (порциями по 200 мл) и 20 мл насыщенного раствора хлористого натрия. Эфирный раствор высушивали, фильтровали, концентрировали и подвергали хроматографии, используя в качестве элюента смесь хлороформа и гексана. В результате получали 0,21 г (выход 28,7%) целевого соединения в виде бесцветной смолы.
1Н ЯМР (90 мГц), δ : 3,56 (3Н, с), 3,75 (3Н, с), 6,52-7,96 (13Н, м), 7,40 (1Н, с) м.д.
П р и м е р 3. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-{ 2-[3-(4-нитрофенокси)фенокси]фенил}- пропеноата (соединение N 133 из табл.1).
Смесь 1,2 г (0,004 моль) Е-метил-2-[2-(3-оксифенокси)фенил]-3-метоксипропеноата, полученного описанным в примере 1 способом, 0,68 г (0,008 моль) карбоната калия и 15 мл ДМФ перемешивали в течение 16 ч при температуре окружающей среды, после чего выливали смесь в воду (80 мл) и дважды подвергали ее экстракции эфиром (30 мл). Объединенные органические экстракты трижды промывали водой (25 мл), а затем 25 мл насыщенного раствора хлористого натрия. После этого их высушивали, фильтровали, концентрировали и подвергали хроматографии, используя в качестве элюента смесь хлороформа и гексана. В результате получали 0,93 г (выход 55,2%) целевого соединения.
1Н ЯМР: (90 мГц), δ : 3,55 (3Н, с), 3,72 (3Н, с), 6,67-8,41 (12Н, м), 7,44 (1Н, с) м.д.
П р и м е р 4. Этим примером иллюстрируется получение (Е)-метил-2-{2-[3-(4-фторфенокси)фенокси] фенил}-3-метокси-пропеноата (соединение N 124 из табл.1).
Смесь 1,0 (0,0033 моль) (Е)-метил-2-/2-(3-оксифенокси)фенил/-3-метоксипропеноата, полученного описанным в примере 1 способом, 2,63 г (0,0069 моль) бис-4-(фторфенил)иодонийбромида, 0,5 мл триэтиламина, 0,5 г порошка меди и 15 мл абсолютного метанола кипятили в течение 6 ч с обратным холодильником. После этого добавляли к смеси еще 1 г (4-фторфенил)йодонийбромида и кипятили ее с обратным холодильником в течение еще 3 ч. После охлаждения и фильтрации к фильтрату добавляли 80 мл воды и смесь дважды подвергали экстракции эфиром (порциями по 30 мл). Объединенные эфирные экстракты трижды промывали водой (порциями по 15 мл), а затем 15 мл насыщенного раствора хлористого натрия.
После высушивания и фильтрации эфирный раствор концентрировали, получая в результате 0,16 г (выход 12,3%) целевого соединения в виде смолы янтарного цвета.
1Н ЯМР: (60 мГц), δ : 3,42 (3Н, с), 3,51 (3Н, с), 6,35-7,30 (12Н, м), 7,35 (1Н, с) м.д.
ИКмакс. (пленка): 1710, 1641 см-1.
П р и м е р 5. Этим примером иллюстрируется получение (Е)-метил-2-[2-(3-бензоилоксифенокси)фенил] -3-метоксипропеноата (соединение N 49 из табл. 1).
Смесь 0,5 г (0,00166 моль) (Е)-метил-2-[2-(3-оксифенокси)-фенил]-3-метоксипропеноата, полученного описанным в примере 1 способом, 0,26 г (0,00185 моль) бензоилхлорида, 0,23 г (0,00166 моль) карбоната калия и 10 мл ДМФ перемешивали в течение 1,5 ч при температуре окружающей среды. После этого к смеси добавляли еще 0,26 г (0,00166 моль) бензоилхлорида и 0,23 г (0,00166 моль) карбоната калия и продолжали перемешивание в этих же условиях в течение еще 16 ч. Затем к ней добавляли 80 мл воды и дважды подвергали экстракции эфиром (порциями по 40 мл). Объединенные эфирные экстракты трижды промывали водой (порциями по 20 мл воды) и 20 мл насыщенного раствора хлористого натрия, затем высушивали, фильтровали, концентрировали и подвергали хроматографии, используя в качестве элюента смесь эфира и гексана. В результате получали более твердое вещество. После перекристаллизации еще из водного раствора метанола получали 0,23 г (выход 47,7%) чистого целевого соединения в виде белого твердого вещества. Т.пл. 94-95оС.
1Н ЯМР: (90 мГц), δ : 3,62 (3Н, с), 3,74 (3Н, с), 6,76-8,38 (13Н, м), 7,46 (1Н, с) м.д.
ИКмакс. (нуйол): 1741, 1627 см-1.
П р и м е р 6. Этим примером иллюстрируется получение (Е,Е)-метил-2-{ 2-[3-(4-хлорфенилазо)-4-оксифенокси]фенил}-4- метоксипропеноата (соединение N 282 из табл.1).
2,5 мл 1М соляной кислоты добавляли к 6,64 мл 0,25М водного раствора 3-хлоранилингидрохлорида и смесь охлаждали до температуры ниже 10оС. Затем к ней добавляли по каплям 3,32 мл 0,5М водного раствора нитрита натрия и перемешивали в течение 10 мин при температуре ниже 10оС. Образующийся раствор хлористого 3-хлордибензолдиазония добавляли по каплям при перемешивании к смеси 0,5 г (0,00166 моль) (Е)-метил-2-[2-(4-оксифенокси)фенил]-3-метоксипропеноата, полученного таким же образом, как это описано в примере 1 для соответствующего 3-окси-производного, в 16 мл 0,1М водного раствора гидроксида натрия и 30 мл ацетона. Одновременно продолжали добавлять раствор гидроксида натрия для поддержания рН в пределах 8-10, поддерживая температуру ниже 10оС. После перемешивания в течение 20 мин смесь дважды подвергали экстракции эфиром (порциями по 40 мл). Объединенные эфирные экстракты трижды промывали водой (порциями по 15 мл), а затем 15 мл насыщенного раствора хлористого натрия, после чего их высушивали, фильтровали, концентрировали и подвергали хроматографии, используя в качестве элюента смесь эфира и гексана. В результате получали твердое вещество оранжевого цвета. После перекристаллизации его из смеси гексана и дихлорметана получали 99,3 мг (выход 13,6%) чистого целевого соединения; т.пл. 143-144оС.
П р и м е р 7. Этим примером иллюстрируется получение (Е)-метил-2-{2-[3-(3-метоксифенокси)фенокси] фенил}-3-метокси-пропеноата (соединение N 129 из табл.1).
К перемешиваемому раствору 0,61 г натрия в 10 мл метанола добавляли в один прием 4,34 г резорцинола. После перемешивания образующейся смеси в течение получаса при комнатной температуре избыток метанола отгоняли при пониженном давлении. К остатку в виде маслянистой жидкости оранжевого цвета добавляли 6,6 мл пиридина, 14,74 г 3-броманизола и 192 мг хлористой меди. Смесь перемешивали в течение 66 ч при 125оС, после чего охлаждали и выливали в разбавленную соляную кислоту, а затем подвергали экстракции эфиром. Эфирные экстракты подвергали реэкстракции разбавленным водным раствором гидроксида натрия. Водные экстракты подкисляли разбавленной соляной кислотой и подвергали экстракции эфиром. Полученные эфирные экстракты промывали последовательно водой и рассолом, а затем высушивали и концентрировали, получая в результате 3,72 г маслянистой жидкости красного цвета. После перегонки этой жидкости (температура печи 170оС, давление 0,05 мм рт.ст.) получали 1,71 г 3-(3-метоксифенокси)-фенола в виде маслянистой жидкости густого бледно-желтого цвета.
1Н ЯМР, δ : 3,78 (3Н, с), 4,93 (1Н, с) м.д.
К перемешиваемому раствору 0,18 г натрия в 4 мл метанола добавляли в один прием 1,70 г 3-(3-метоксифенокси)фенила. После перемешивания образующейся смеси в течение получаса при комнатной температуре избыток метанола отгоняли при пониженном давлении. К остатку в виде маслянистой жидкости оранжевого цвета добавляли 0,85 г о-бромфенилуксусной кислоты и 40 мг хлористой меди и реакционную смесь перемешивали в течение часа при 130оС. После этого к ней добавляли еще 0,4 г о-бромфенилуксусной кислоты и 0,13 г этоксида натрия и продолжали перемешивание при 130оС в течение еще 3 ч.
Затем смесь охлаждали, подкисляли разбавленной соляной кислотой и подвергали экстракции эфиром. Эфирные экстракты промывали последовательно водой и рассолом, затем высушивали и концентрировали, получая в результате 3,12 г маслянистой жидкости красного цвета, содержащей 2-[3-(3-метоксифенокси)фенокси]-фенилуксус-ную кислоту. К 3,12 г этой кислоты добавляли 40 г метанола и 3 капли концентрированной серной кислоты. Реакционную смесь перемешивали в течение часа при 90оС, затем медленно охлаждали, выливали в воду и подвергали экстракции эфиром. Эфирные экстракты промывали последовательно разбавленным водным раствором гидроксида натрия, водой и рассолом, высушивали и концентрировали, получая в результате 1,33 г маслянистой жидкости желтого цвета. После перегонки ее (температура печи 160оС, давление 0,07 мм рт.ст.) получили 1,03 г (выход 36%) в расчете на 3-(3-метоксифенокси)фенол 2-[3-(3-метоксифенокси)-фенокси]-фенилацетата.
1Н ЯМР, δ : 3,62 (3Н, с), 3,68 (2Н, с), 3,78 (3Н, с) м.д.
Смесь 1,00 г метил-[3-(3-метоксифеноксил)фенокси]-фенилацетата и 3,34 мл метилформиата в 1 мл ДМФ добавляли по каплям в течение 10 мин и к перемешиваемой суспензии 0,13 г гидрида натрия в 10 мл ДМФ, охлажденной льдом до температуры ниже 10оС (при этом происходило выделение пузырьков газа). По окончании добавления реакционную смесь перемешивали в течение 2 ч при комнатной температуре, выливали затем в воду, подкисляли разбавленной соляной кислотой и подвергали экстракции эфиром. Экстракты промывали водой, высушивали и концентрировали, получая в результате 1,09 г желтой маслянистой жидкости. К перемешиваемому раствору этой жидкости в 20 мл ДМФ добавляли 0,76 г карбоната калия и 0,33 г диметилсульфата и полученную смесь перемешивали при комнатной температуре в течение 2,5 ч, затем выливали ее в воду и подвергали экстракции эфиром. Экстракты промывали водой, высушивали, концентрировали и подвергали хроматографии, используя в качестве элюента смесь эфира и бензина (1:1). В результате получали 0,61 г (выход 55% в расчете на метил-2-[3-(3-метоксифенокси)фенокси]-фенилацетат целевого соединения в виде бесцветной вязкой маслянистой жидкости.
1Н ЯМР, δ : 3,60 (3Н, с), 3,75 (3Н, с), 3,78 (3Н, с), 6,55-6,72 (5Н, м), 6,97 (1Н, д), 7,10-7,30 (6Н, м), 7,48 (1Н, с) м.д.
ИКмакс. (нуйол): 1713, 1638 см-1.
П р и м е р 8. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-{ 2-[3-(феноксиметил)фенокси]фенил}-про-пеноата (соединение N 21 из табл.1).
0,50 г (Е)-метил-3-метокси-2-[2-(3-метилфенокси)фенил] -пропеноата, полученного из 3-метилфенола и 2-бромбензальдегида описанным в примере 1 способом, и 0,30 г N-бромсукцинимида кипятили с обратным холодильником в 25 мл четыреххлористого углерода со следами азобисизобутиронитрила (АИБН) в течение 4,5 ч, добавляя через каждые 1,5 ч следы АИБН. Ход реакции контролировали с помощью газовой хроматографии. После этого реакционную смесь оставляли стоять на ночь при комнатной температуре, добавляли еще следовые количества АИБН и продолжали кипятить ее до тех пор, пока газовая хроматография показывала почти полное отсутствие исходного материала (1 ч). После этого реакционную смесь фильтровали через целит, промывали водой и упаривали, получая 0,69 г бледно-желтой смолы. По данным газовой хроматографии и ЯМР эта смола состояла из (Е)-метил-2-[2-(3-бромметилфенокси)фенил]- 3-метоксипропеноата (80% ), соответствующего дибромметильного производного (11%) и непрореагировавшего исходного материала (пропеноата, 8%).
1Н ЯМР, данные для основного компонента, δ , 3,61 (3Н, с), 3,77 (3Н, с), 4,42 (2Н, с), 6,90-7,40 (3Н, м), 7,48 (1Н, с), м.д.
Полученный материал использовался далее без дополнительной очистки.
Часть этого сырого материала (0,42 г, концентрация 80%) перемешивали с 0,105 г фенола и 0,077 г карбоната калия в 20 мл ДМФ и нагревали в течение часа при 60оС. Затем смесь оставляли на ночь при комнатной температуре, после чего нагревали при 60оС в течение еще часа, охлаждали, выливали в воду и подвергали экстракции этилацетатом. Органическую фракцию промывали водой, высушивали и упаривали, получая в результате 0,42 г бледно-желтой маслянистой жидкости. После очистки с помощью высокопроизводительной жидкостной хроматографии с использованием в качестве элюента смесь бензина и этилацетата, взятых в соотношении 3:1, получали 0,13 г целевого соединения в виде бесцветной смолы, содержащей в качестве примеси 20% (Е)-метил-2-[2-(3-дибромметилфенокси)фенил]-3-метоксипропеноата.
1Н ЯМР, данные для целевого соединения: δ : 3,58 (3Н, с), 3,70 (3Н, с), 4,98 (2Н, с), 6,88-7,36 (с), (13Н, м), 7,46 (1Н, с). м.д.
П р и м е р 9. Этим примером иллюстрируется получение (Е) метил-2-[2-(2-ацетил-5-феноксифенокси)фенил] -3-метоксипропеноата и (Е)-метил-2-/2-(4-ацетил-3-феноксифенокси)фенил/-3 -метоксипропеноата (соединение N 366 и 365 соответственно из табл.1).
Метил-2-(3-феноксифенокси)фенилаце- тат получали из 3-феноксифенола и 2-бромбензальдегида таким же образом, как это описано в примере 1 для получения метил-2-(3-метоксифенокси)фенилацетата. Полученный продукт переводили в (Е)-метил-3-метокси-2-[2-(3-феноксифенок-си)фенил]-пропеноат (1Н ЯМР (250 мГц) 3,61 (3Н, с), 3,78 (3Н, с), 6,68-7,35 (13Н, м), 7,48 (1Н, с) м,д.), используя гидрид натрия и метилформиат, а затем карбонат калия и диметилсульфат, по способу, описанному в примере 1, для получения (Е)-метил-2-[2-(3-оксифенокси)фенил] -3-метоксипропеноата, с той разницей, что в данном случае использовали два эквивалента гидрида натрия.
К перемешиваемому раствору 0,722 г (1,92 ммоль) (Е)-метил-3-метокси-2-/2-(3-феноксифенокси)фенил/пропеноата в 20 мл сухого дихлорметана добавляли при 0-5оС 0,512 г (3,84 ммоль) порошкообразного хлорида алюминия, а затем по каплям, в течение 10 мин, раствор 0,151 г (1,92 ммоль) ацетилхлорида в 3 мл сухого дихлорметана. Полученную смесь перемешивали в течение ночи и давали нагреться до температуры окружающей среды. Реакционную смесь затем разбавляли 125 мл эфира, дважды промывали 2н. соляной кислоты, 10%-ным водным раствором карбоната натрия и наконец водой. Остаток, полученный после отгонки растворителя, подвергали очистке с помощью флеш-хроматографии, используя в качестве элюента смесь эфира и бензина. В результате получали 0,424 г смеси двух целевых соединений в примерном соотношении 3:1. (разделение на отдельные компоненты не проводили) в виде бесцветной смолы. Часть этой смолы (0,400 г) разделяли с помощью высокопроизводительной жидкости хроматографии на силикагеле, используя в качестве элюента смесь гексана, дихлорметана и метил-трет-бутилового эфира с соотношением компонентов 70: 25:5. В результате получали (I) региоизомер A (0,179 г), выходивший первым и являющийся основным компонентом смеси, в виде белого кристаллического вещества. Т.пл. 90-92оС.
1Н ЯМР, (250 мГц), δ : 2,52 (3Н, с), 3,56 (3Н, с), 3,72 (3Н, с), 6,48 (1Н, д), 6,64 (1Н, кв), 6,9-7,4 (9Н, м), 7,43 (1Н, с), 7,84 (1Н, д) м.д. и (II) региоизомер B (0,061 г, с примесью около 5% региоизомера A), выходившего вторым и содержавшегося в смеси в меньшем количестве, в виде белого кристаллического вещества; т.пл. 82-85оС.
1Н ЯМР (250 мГц), δ : 2,51 (3Н, с), 3,60 (3Н, с), 3,75 (3Н, с), 6,45 (1Н, д), 6,59 (1Н, кв), 6,9-7,4 (9Н, м), 7,48 (1Н, с), 7,82 (1Н, д) м.д.
П р и м е р 10. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-[2-(3-пиримидин-2-ил-оксифенокси)фенил] пропеноата (соединение N 22 из табл.2).
Смесь 0,5 г (Е)-метил-2-[2-(3-оксифенокси)фенил]-3-метоксипропеноата, полученного описанным в примере 1 способом, 0,46 г карбоната калия, 0,23 г 2-хлорпиримидина и 0,01 г хлорида меди в 15 мл ДМФ кипятили в течение 4 ч с обратным холодильником. После охлаждения смесь выливали в воду и фильтровали. Фильтрат подвергали экстракции эфиром. Объединенные эфирные экстракты промывали последовательно водой и рассолом, высушивали, концентрировали и подвергали хроматографии, используя в качестве элюента смесь эфира и гексана. В результате получали 0,26 (выход 41% ) целевого соединения, ИК (пленка): 1707, 1633 см-1.
1Н ЯМР (90 мГц), δ : 3,54 (3Н, с), 3,68 (3Н, с), 6,74-7,34 (9Н, м), 7,38 (1Н, с), 8,28 (2Н, д) м.д.
П р и м е р 11. В этом примере описано получение (Е)-метил-3-метокси-2-[2-(3-феноксифенилтио)фенил]пропеноата (соединение N 446 из табл.3).
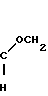
2-Меркаптофенилуксусную кислоту получали описанным в литературе способом (см.D.Papa и др., J.Org.Chem. 1949, 24.723, R.H.Glauert, F.G.Mann, J. Chem. Soc. 1952, 2127 и ссылки в этой статье). 1,68 г 2-меркаптофенилуксусной кислоты добавляли к перемешиваемому раствору 0,8 г гидроксида натрия в 10 мл метанола (сравни D, C.Atkinson и др. J.Med.Chem., 1983, 26, 1361). Образующийся оранжевый раствор перемешивали при комнатной температуре в течение 90 мин, затем концентрировали при пониженном давлении, удаляя остатки метанола путем азеотропной перегонки с толуолом. В результате получали желтое твердое вещество. К перемешиваемому раствору этого желтого вещества в 20 мл ДМФ добавляли 0,2 г хлорида меди и раствор 2,49 г 3-феноксибромбензола, полученного из 3-феноксифенола и трифенилфосфиндибромида по способу, описанному J.P.Schaefer и др. Org.Synth., Coll, т.5, 142) в 10 мл ДМФ. Образующуюся смесь нагревали при 95оС в течение часа, при 125оС в течение 2 ч и затем при температуре кипения в течение еще 2 ч. После охлаждения реакционную смесь выливали в водный раствор гидроксида натрия и трижды промывали эфиром. Водный раствор подкисляли концентрированной соляной кислотой и трижды подвергали экстракции эфиром.
Экстракты промывали водой, высушивали и концентрировали, получая в результате 2,2 г маслянистой жидкости пурпурного цвета, состоящей в основном из 2-(3-феноксифенилтио)-фенилуксусной кислоты. Раствор этой маслянистой жидкости в 20 мл метанола добавляли к кислому метанолу, полученному путем осторожной обработки 30 мл метанола 3,5 мл ацетилхлорида, и образующуюся смесь перемешивали в течение 90 мин при комнатной температуре. Реакционную смесь затем концентрировали и остаток распределяли между эфиром и водным раствором бикарбоната натрия. Органический слой отделяли и промывали последовательно водным раствором гидроксида натрия (дважды) и трижды водой, после чего высушивали и концентрировали, получая в результате 2,06 г сырого 2-(3-феноксифенилтио)фенилацетата в виде маслянистой жидкости пурпурного цвета.
ИКмакс. (пленка): 1740 см-1, степень чистоты 94% (определена с помощью газовой хроматографии). Сырой метил-2-(3-феноксифенилтио)-фенилацетат переводили в целевое соединение с выходом 53% в две стадии, таким же образом, как это описано в примере 7 для перевода метил-2-[3-(3-метоксифенокси)фенокси] фенилацетата в (Е)-метил-2-[2-(3-метоксифенокси)фенокси) фенил] -3-метоксипропеноат, а именно путем формирования метилформиатом и гидридом натрия и последующего O-метилирования диметилсульфатом и карбонатом калия. Полученный продукт представлял собой смолу оранжевого цвета (степень чистоты, определенная с помощью газовой хроматографии, 98%), которая кристаллизовалась при стоянии. Т.пл. 48-51,5оС.
ИКмакс. (пленка): 1710 и 1632 см-1.
1Н ЯМР (270 мГц), δ: 3,62 (3Н, с), 3,73 (3Н, с), 6,78 (1Н, дд), 6,88-7,00 (4Н, м), 7,05-7,36 (7Н, м), 7,42 (1Н, д), 7,48 (1Н, с) м.д.
П р и м е р 12. Этим примером иллюстрируется получение (Е)-метил-2-[2-(3-пиримидин-2-ил-оксифенилтио)фенил]-3 -метоксипропеноата (соединение N 22 из табл.4).
Смесь натриевой соли 3-метокситиофенола, полученной путем обработки 2,8 г 3-метокситиофенола 0,8 г гидроксида натрия в 20 мл метанола с последующим выпариванием раствора досуха, 4,3 г 2-бромфенилуксусной кислоты и 0,4 г хлорида меди (I) в 25 мл сухого ДМФ кипятили в течение ночи с обратным холодильником. Реакционную смесь затем охлаждали, выливали в воду и подкисляли разбавленной соляной кислотой. Водную смесь трижды экстрагировали эфиром, а объединенные эфирные экстракты в свою очередь дважды подвергали экстракции разбавленным раствором гидроксида натрия. Объединенные водные гидроксидные экстракты подкисляли разбавленной соляной кислотой и трижды реэкстрагировали эфиром.
Объединенные эфирные экстракты трижды промывали водой, высушивали и упаривали, получая в результате 3,5 г (содержание 96,8%, определенного с помощью газовой хроматографии) маслянистой жидкости оранжевого цвета. Полученную маслянистую жидкость выдерживали в течение ночи при комнатной температуре с кислым метанолом. После обычной переработки получали 2,9 г (содержание 91% , определенное с помощью газовой хроматографии) метил-2-(3-метоксифенилтио)фенилацетата в виде жидкости желтого цвета, которую без дополнительной очистки использовали на следующей стадии.
1Н ЯМР, δ : 3,64 (3Н, с), 3,74 (3Н, с), 3,86 (2Н, с) м.д.
ИКмакс. (пленка): 1739 см-1.
0,86 г метил-2-(3-метоксифенилтио)фенилацетата и 2,08 г (избыток) пиридинийгидрохлорида нагревали при 200оС в атмосфере азота. Через 3 ч реакционную смесь охлаждали и распределили между разбавленной соляной кислотой и этилацетатом. Кислый водный слой дважды подвергали экстракции этилацетатом и объединенные органические фазы трижды экстрагировали разбавленным раствором гидроксида натрия. Объединенные щелочные фазы подкисляли концентрированной соляной кислотой и трижды подвергали экстракции этилацетатом. Эти органические экстракты объединяли, трижды промывали водой, высушивали и упаривали, получая в результате 0,64 г грязно-белого твердого вещества. Полученное грязно-белое твердое вещество обрабатывали метанольным раствором хлористого водорода и после стандартной обработки получали 0,44 г метил-2-(3-оксифенилтио)фенилацетата в виде красной маслянистой жидкости (степень чистоты, определенная с помощью газовой хроматографии, 90,5%), которую без дополнительной очистки использовали на следующей стадии.
ИКмакс.: 3384, 1738 см-1.
Раствор 0,44 г сырого метал-2-(3-оксифенилтио)фенилацетата и 1,92 мл метилформиата в 2 мл сухого ДМФ добавляли по каплям к перемешиваемой суспензии 0,21 г (55%-ная дисперсия в масле, предварительно промытая петролейным эфиром) гидрида натрия в 3 мл сухого ДМФ при 0-5оС. Через 15 мин смеси давали нагреваться до комнатной температуры. Через 2,5 ч реакционную смесь выливали в воду, подкисляли концентрированной соляной кислотой и трижды подвергали экстракции эфиром. Объединенные эфирные экстракты трижды промывали водой, высушивали и упаривали, получая в результате 0,49 г смолы красного цвета. Полученную красную смолу растворили в 5 мл ДМФ и охлаждали раствор до 0оС. К приготовленному раствору добавляли 0,132 г карбоната калия, а затем по каплям раствор 0,111 г диметилсульфата в ДМФ. После перемешивания в течение 4,5 часа реакционную смесь выливали в воду и трижды подвергали экстракции эфиром. Объединенные эфирные экстракты трижды промывали водой, высушивали и упаривали, получая в результате 0,45 г метил-2-[2-(3-оксифенилтио)фенил]-3-метоксипропеноата в виде смолы красного цвета.
ИКмакс. 3240, 1709, 1665 см-1, М+ 316.
1Н ЯМР, δ : 3,65 (3Н, с), 3,76 (3Н, с), 7,47 (1Н, с) м.д.
0,4 г сырого (Е)-метил-2-[2-(3-оксифенилтио)фенил]-3-метоксипропеноата обрабатывали 0,45 г 2-хлорпиримидина и 0,17 г карбоната калия в 10 мл сухого ДМФ при 80-90оС в атмосфере азота. Как показали результаты газовохроматографического анализа, через 4,5 ч в реакционной смеси присутствовал один продукт реакции, реакционную смесь охлаждали, выливали в воду и четырежды подвергали экстракции эфиром. Объединенные эфирные экстракты желтого цвета дважды промывали водой, высушивали и упаривали, получая в результате 0,39 г смолы оранжевого цвета. После хроматографии с использованием в качестве элюента эфира получали 0,34 г целевого соединения в виде вязкой смолы оранжевого цвета.
ИКмакс. 1706, 1632 см-1.
1Н ЯМР, δ : 3,64 (3Н, с), 3,75 (3Н, с), 6,97-7,06 (3Н, м), 7,08-7,12 (1Н, д), 7,25-7,35 (4Н, м), 7,46-7,48 (1Н, д), 7,49 (1Н, с), 8,53-8,56 (2Н, д) м.д.
П р и м е р 13. Этим примером иллюстрируется получение (Е)-метил-2-/2-(3-фенилтиофенокси)фенил]-3-метоксипропено-ата (соединение N 1 из таблицы 1).
2,02 г (0,01 моль) 3-оксидифенилсульфида, 1,35 г (0,005 моль) (Е)-метил-2-(2-бромфенил)-3-метоксипропеноата, полученного из о-бромфенилацетата, метилформиата и гидрида натрия с последующей добавкой карбоната калия и диметилсульфата в две стадии по способу, описанному в примере 7 для аналогичного превращения, 0,69 г (0,005 моль) безводного карбоната калия и каталитического количества хлорида меди смешивали и нагревали при перемешивании до 175оС. Через 10 ч реакционную смесь охлаждали до температуры окружающей среды и растворяли в 50 мл ДМФ. Полученный раствор выливали в 100 мл воды и образующуюся эмульсию дважды подвергали экстракции эфиром (порциями по 100 мл). Объединенные эфирные экстракты промывали последовательно водой (дважды, порциями по 100 мл), дважды 2М раствором гидроксида натрия (порциями по 100 мл) и дважды водой (порциями по 100 мл). Полученный после промывки эфирный раствор высушивали, фильтровали и упаривали досуха при пониженном давлении. После хроматографии с использованием в качестве элюента хлороформа получали 0,83 г целевого соединения в виде вязкой маслянистой жидкости.
1Н ЯМР (60 мГц), δ : 3,52 (3Н, с), 3,64 (3Н, с), 6,5-7,3 (13Н, м), 7,42 (1Н, с) м.д.
П р и м е р 14. Этим примером иллюстрируется получение (Е)-метил-2-[2-(3-фенилтиофенокси)фенил]-3-метоксипропеноат-S,S- диоксида (соединение N 3 из табл.1).
3,66 г (0,0156 моль) 3-оксидифенилсульфонатa, 1,5 г (0,0055 моль) (Е)-метил-2-(2-бромфенил)-3-метоксипропеноата, полученного описанным в примере 13 способом, и 1,1 г (0,0079 моль) безводного карбоната калия смешивали с каталитическим количеством хлорида меди и медной бронзы. Смесь нагревали в течение 10 ч при 170оС в атмосфере азота. После этого расплав охлаждали до температуры окружающей среды и остаток растворяли в 50 мл ДМФ. Полученный раствор разбавляли 100 мл эфира и фильтровали для удаления неорганических солей. Раствор последовательно промывали 100 мл воды, дважды 2М раствором гидроксида натрия (порциями по 100 мл), снова 100 мл воды и 100 мл насыщенного раствора хлорида натрия. Эфирный раствор высушивали, фильтровали и упаривали досуха при пониженном давлении. После хроматографии остатка с использованием в качестве элюента гексана и хлороформа получали 0,66 г целевого соединения.
1Н ЯМР (60 мГц), δ : 3,46 (3Н, с), 3,57 (3Н, с), 6,6-8,0 (14Н, м) м.д.
П р и м е р 15. Этим примером иллюстрируется получение (Е)-метил-2-/2-(3-анилинфенокси)фенил/-3-метоксипропеноата (соединение N 4 из табл.1).
1,365 г (0,0074 моль) 3-оксидифениламина, 1 г (0,0037 моль) 2-(2-бромфенил)-3-метоксипропеноата, полученного описанным в примере 13 способом, и 0,517 г (0,0037 моль) безводного карбоната калия смешивали с каталитическими количествами хлорида меди и медной бронзы. Смесь нагревали в течение 9 ч при 170оС, затем охлаждали и растворяли в 20 мл ДМФ. Полученный раствор распределяли между эфиром и водой. Эфирный слой промывали водой (дважды порциями по 100 мл) и дважды 1М раствором гидроксида натрия (порциями по 100 мл), после чего высушивали, фильтровали и упаривали досуха при пониженном давлении. Остаток в виде смолы очищали с помощью хроматографии, используя в качестве элюентов гексан и дихлорметан. В результате получали 0,40 г целевого соединения.
1Н ЯМР (60 мГц), δ : 3,57 (3Н, с), 3,67 (3Н, с), 5,75 (1Н, широкая), 6,3-7,4 (13Н, м), 7,44 (1Н, с) м.д.
П р и м е р 16. Этим примером иллюстрируется получение (Е)-метил-2-[2-(3-N-метиланилинфенокси)фенил] -3-метоксипропе- ноата (соединение N 5 из табл.1).
300 мг (0,01 моль) 80% -ной дисперсии гидрида натрия в масле дважды промывали гексаном (порциями по 50 мл) для удаления масла, после чего суспендировали в 10 мл сухого ДМФ. К полученной суспензии добавляли раствор (290 мг) (Е)-метил-2-[2-(3-анилинфенокси)фенил] -3-метоксипропеноа- та, полученного описанным в примере 15 способом, в 10 мл ДМФ с такой скоростью, чтобы обеспечить равномерное выделение пузырьков газа. После прекращения выделения газа смесь перемешивали в течение еще 15 мин и добавляли к ней в течение 5 мин 2 мл (больший избыток) иодметана. Перемешивание продолжали в течение еще 30 мин, после чего суспензию осторожно разбавляли 50 мл воды. Водную эмульсию дважды подвергали экстракции эфиром (порциями по 50 мл), высушивали, фильтровали и упаривали досуха при пониженном давлении, получая в результате 211 мг целевого соединения в виде вязкой маслянистой жидкости.
1Н ЯМР (60 мГц), δ : 3,20 (3Н, с), 3,54 (3Н, и с), 3,65 (3Н, с), 6,3-7,4 (12, м), 7,44 (1Н, с) м.д.
П р и м е р 17. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-{ 2-[3- (α-оксибензил)фенокси]фенил}-пропеноа-та (соединение N 380 из табл.1).
Мелко измельчали 31,0 г 3-оксибензилового спирта и смешивали его с 34,6 г карбоната калия, 26,9 г 2-бромфенилуксусной кислоты и хлоридом меди (большой шпатель) в атмосфере азота. Смесь нагревали до 140оС и интенсивно перемешивали при этой температуре в течение 3,5 ч. Затем к перемешиваемому расплаву добавляли 60 мл ДМФ, раствор охлаждали, выливали в воду и подкисляли разбавленной соляной кислотой. Водный слой подвергали экстракции эфиром, эфирные экстракты промывали водой, высушивали и упаривали, получая в результате 42,03 г 2-(3-оксиметилфенокси)фенилуксусной кислоты и в виде коричневой маслянистой жидкости, которую без дополнительной очистки использовали на следующей стадии.
41,0 г полученной сырой кислоты кипятили в течение 3,5 ч с обратным холодильником в 600 мл метанола с добавкой 2,5 мл концентрированной серной кислоты. После отгонки метанола остаток растворяли в этилацетате, промывали разбавленным водным раствором гидроксида натрия, а затем водой, высушивали и упаривали, получая в результате 26,31 г коричневой маслянистой жидкости. 1,31 г ее очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь этилацетата и гексана, взятых в соотношении 1: 1. В результате получали чистый 2-(3-оксиметилфенокси)фенилацетат в виде бледно-желтой маслянистой жидкости.
1Н ЯМР (400 мГц), δ : 2,12 (1Н, с), 3,60 (3Н, с), 3,69 (3Н, с), 4,62 (2Н, с), 6,95 (1Н, с), 6,85-6,90 (2Н, т), 7,04-7,14 (2Н, м), 7,21-7,32 (3Н, м) м.д.
ИК (пленка): 3450, 1742 см-1.
Смесь 25,0 г сырого метил-2-/3-оксиметилфенокси/фенилацетата и 56 мл метилформиата в 50 мл сухого ДМФ добавляли по каплям в течение 30 минут при 5оС к гидриду натрия (7,35 г 60%-ной дисперсии в масле, промытой гексаном). После перемешивания в течение еще 30 мин при 5оС смеси давали нагреваться до комнатной температуры в течение нескольких часов и затем оставляли стоять на ночь. После этого реакционную смесь выливали в воду и подвергали экстракции эфиром. Водный слой подкисляли разбавленной соляной кислотой и подвергали экстракции эфиром. Эфирные экстракты высушивали и упаривали, получая в результате 32,19 г сырого метил-2-[2-(3-оксиметилфенокси)]-фенилпропеноата в виде оранжевой маслянистой жидкости 32,10 г сырого метилового эфира перемешивали в 80 мл ДМФ при 5-10оС с 25,4 г карбоната калия и добавляли к смеси по каплям раствор 11,6 г диметилсульфата в 20 мл ДMФ в течение 10 мин. Смеси давали нагреться до комнатной температуры в течение нескольких часов и оставляли стоять на ночь. После этого ее выливали в воду, подкисляли разбавленной соляной кислотой и подвергали экстракции эфиром. Эфирные экстракты промывали водой, высушивали и упаривали, получая в результате 14,38 г оранжево-коричневой маслянистой жидкости. После очистки с помощью высокопроизводительной жидкостной хроматографии получали 7,8 г (Е)-метил-3-метокси-2-[2-(3-оксиметилфенокси)фенил] пропеноата в виде розоватого кристаллического вещества.
1Н ЯМР (270 мГц), δ : 2,55 (1Н, с), 3,58 (3Н, с), 3,74 (3Н, с), 4,55 (2Н, с), 6,8-7,28 (8Н, м), 7,44 (1Н, с) м.д.
ИКмакс., (нуйол): 3515, 1705, 1625 см-1.
Часть полученного спирта (0,314 г) перемешивали в 5 мл сухого метиленхлорида, добавляли 0,564 г дихромата пиридиния и смесь перемешивали в течение 4 ч при комнатной температуре. Затем ее фильтровали и осадок промывали эфиром. Метиленхлорид и эфир после промывки объединяли и упаривали, получая в результате 0,309 г (Е)-метил-3-метокси-2-[2-(3-формилфенокси) фенил]пропеноата в виде коричневой маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,59 (3Н, с), 3,65 (3Н, с), 6,98 (1Н, д), 7,17-7,36 (4Н, м), 7,40-7,47 (3Н, м), 7,47 (1Н, с), 7,55 (1Н, д) м.д.
ИКмакс. (пленка): 1710, 1640 см-1.
0,50 г (Е)-метил-3-метокси-2-[2-(3-формилфенокси)фенил] -пропеноата перемешивали в 20 мл сухого ТГФ при -20оС в атмосфере азота. К приготовленной смеси медленно добавляли по каплям фенилмагнийбромид (0,53 мл 3М эфирного раствора) в виде разбавленного раствора в сухом ТГФ (5 мл). По окончании добавления реакционную смесь перемешивали при -20оС в течение 30 мин и затем медленно, в течение часа, нагревали до комнатной температуры, после чего оставляли стоять на ночь. Смесь затем охлаждали до 5оС, добавляли к ней воду и подвергали экстракции этилацетатом. После промывки рассолом и высушивания этилацетатный раствор упаривали, получая в результате желтую маслянистую жидкость, которую очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь гексана и эфира, взятых в соотношении 2:1. В результате получали 0,340 г целевого соединения в виде бесцветной маслянистой жидкости.
1Н ЯМР (400 мГц), δ : 2,30 (1Н, д), 3,57 (3Н, с), 3,72 (3Н, с), 5,78 (1Н, д), 6,82 (1Н, д), 6,91 (1Н, д), 7,02-7,08 (2Н, м), 7,10-7,16 (1Н, м), 7,20-7,38 (8Н, м), 7,45 (1Н, с) м.д.
ИКмакс. (пленка): 3460, 1715, 1635 см-1.
П р и м е р 18. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-{2-[3-(2-пиридилоксиметил)фенокси]фенил} пропеноата (соединение N 6 из табл.2).
0,28 г карбоната серебра добавляли к 0,75 г (степень чистоты 70%) (Е)-метил-3-метокси-2-[2-(3-бромметилфенокси)фенил] - пропеноата, полученного описанным в примере 8 способом, и 0,19 г 2-пиридона в гексане. Смесь кипятили в течение 3 ч с обратным холодильником в темноте, для чего заворачивали реакционную колбу в фольгу, и затем оставляли стоять на ночь. После этого гексан отгоняли, а остаток растворяли водным раствором бикарбоната натрия, а затем водой, высушивали и упаривали, получая в результате 0,72 г оранжевой смолы. Полученную смолу очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь эфира и гексана, взятых в соотношении 1:1. В результате получали 0,188 г целевого соединения в виде бесцветной смолы.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,76 (3Н, с), 5,32 (2Н, с), 6,78 (1Н, д), 6,84-6,96 (3Н, м), 7,04-7,16 (1Н, м), 7,21-7,31 (3Н, м), 7,48 (1Н, с), 7,52-7,60 (1Н, м), 8,15 (1Н, д), м.д.
ИКмакс. (пленка): 1715, 1670, 1645, 1600 см-1.
П р и м е р 19. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-[2-(3-пиримидин-2-ил-оксиметилфенокси) фенил] пропеноата (соединение N 85 из табл.2).
0,5 г (Е)-метил-3-метокси-[2-(3-оксиметилфенокси)фенил]пропеноата, полученного описанным в примере 17 способом, в нескольких мл ДМФ добавляли к гидриду натрия (0,072 г 60%-ной дисперсии в масле, промытой гексаном) и перемешивали в 10 мл сухого ДМФ при комнатной температуре. По окончании добавления смесь перемешивали в течение 5 мин, добавляли к ней 0,92 г 2-хлорпиримидина и оставляли стоять на ночь. Затем ее выливали в воду, подкисляли и подвергали экстракции эфиром. Эфирные экстракты высушивали и упаривали, получая в результате 0,95 г желтой маслянистой жидкости. После очистки ее с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь этилацетат и гексана, взятых в соотношении 1:1, получали 0,104 г чистого целевого соединения в виде маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,75 (3Н, с), 5,39 (2Н, с), 6,86-6,96 (3Н, м), 7,03-7,31 (6Н, м), 7,49 (1Н, с), 8,50 (2Н, д) м.д.
ИКмакс. (пленка): 1713, 1640 см-1.
П р и м е р 20. Этим примером иллюстрируется получение (Е,Е)- и (Е, Z)-метил-3-метокси-2-{2[3-(4-нитростирил)фенокси] фенокси}фенил- пропеноата (смесь соединений N 403 из табл.1).
1,39 г диметилфосфита в 5 мл сухого ДМФ добавляли по каплям к перемешиваемой суспензии гидрида натрия (0,61 г 50%-ной дисперсии в масле, промытой гексаном) в 10 мл сухого ДМФ при 20оС. По окончании добавления и перемешивания смеси в течение еще 20 мин к ней добавляли по каплям 7,0 г (степень чистоты 70%) (Е)-метил-3-метокси-2-[2-(3-бромметилфенокси)фенил] пропеноата, полученного описанным в примере 8 способом. Реакционную смесь оставляли стоять в течение 60 часов, после чего нагревали при 55оС в течение 10 ч, выливали в воду и подвергали экстракции этилацетатом. Экстракт высушивали и упаривали, получая в результате вязкую смолу желтого цвета, которую очищали с помощью флеш-хроматографии, используя в качестве элюента 5% -ный метанол в этилацетате. В результате получали 1,50 г фосфоната (Е)-метил-3-метокси-2-(3-/диметилфосфономе- тил/фенокси)- фенилпропеноата в виде почти бесцветной масляной жидкости.
1Н ЯМР (400 мГц), δ : 3,13 (2Н, д), 3,62 (3Н, с), 3,66 (3Н, с), 3,68 (3Н, с), 3,78 (3Н, с), 6,85 (1Н, д), 6,92 (2Н, д), 7,00 (1Н, д), 7,13 (1Н, т), 7,20-7,31 (4Н, м), 7,48 (1Н, с) м.д.
ИКмакс. (пленка): 1715, 1645 см-1.
0,61 г полученного фосфоната в 5 мл сухого ДМФ добавляли по каплям к гидриду натрия (0,072 г 50% -ной дисперсии в масле, промытой гексаном), перемешиваемой в 10 мл сухого ДМФ при 5оС в атмосфере азота. По окончании добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение 15 мин. После этого к ней добавляли медленно по каплям 0,227 г N-нитробензальдегида в 5 мл сухого ДМФ и перемешивали в течение ночи при комнатной температуре, добавляя затем воду, и подвергали экстракции эфиром. Эфирный слой высушивали и упаривали, получая в результате вязкую желтую жидкость, которую очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь гексана и этилацетата, взятых в соотношении 3:1. В результате получали 0,20 г целевого соединения в виде желтой смолы, представляющего собой смесь (в соотношении 5:1) /Z/- и /E/-стильбеновых изомеров.
1Н ЯМР (270 мГц), δ : (данные для /Z/-изомера) 3,57 (3Н, с), 3,74 (3Н, с), 6,58 (1Н, д), 6,72 (1Н, д), 6,72-6,98 (3Н, м), 7,05-7,36 (7Н, м), 7,45 (1Н, с), 8,06 (2Н, д) м.д.
Кипячением смеси с обратным холодильником в толуоле со следами йода получали смесь с соотношением (Е)- и (Z)-изомеров 85:15.
1Н ЯМР (400 мГц), δ (данные для (Е)-изомера): 3,62 (3Н, с), 3,78 (3Н, с), 6,92-7,35 (10Н, м), 7,49 (1Н, с), 7,61 (2Н, д), 8,22 (2Н, д) м.д.
П р и м е р 21. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-[2-(3-бензоилоксиметилфеноксил)фенил] пропеноата (соединение 398 из табл.1).
0,5 г (степень чистоты 75%) (Е)-метил-3-метокси-2-(3-бромметилфенокси)фенилпро- пеноата, полученного описанным в примере 8 способом, 0,13 г бензойной кислоты и 0,076 г карбоната калия перемешивали в течение ночи при комнатной температуре в сухом ДМФ. После этого к смеси добавляли воду и подвергали ее экстракции разбавленным водным раствором бикарбоната, высушивали и упаривали, получая в результате 0,49 г желтой вязкой маслянистой жидкости, которую очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь гексана и этилацетата, взятых в соотношении 5:2. В результате получали 0,120 г целевого соединения.
1Н ЯМР (400 мГц), δ : 3,60 (3Н, с), 3,75 (3Н, с), 5,31 (2Н, с), 6,93 (1Н, д), 6,96 (1Н, д), 7,06 (1Н, с), 7,12 (1Н, д), 7,16 (1Н, д), 7,44 (2Н, т), 7,25-7,32 (2Н, м), 7,47 (1Н, с), 7,55 (1Н, д), 8,05 (2Н, д).
П р и м е р 22. Этим примером иллюстрируется получение бромистой соли (Е)-метил-3-метокси-2-{ 2-[3-(трифенилфосфорфо- нийметил)фенокси] фенил}-пропеноата (соединение N 404 из табл.1).
Перемешивали в течение 4 часов при комнатной температуре 4,58 г (степень чистоты 70% ) (Е)-метил-3-метокси-2[2-(3-бромметилфенокси)фенил] пропеноата, полученного описанным в примере 8 способом, и 2,33 г трифенилфосфина в 40 мл сухого ДМФ и полученную смесь оставляли стоять на ночь. После этого растворитель отгоняли и получали густой остаток, который растирали в смеси эфира и этилацетата, получая в результате 4,38 г целевого соединения в виде желто-белого кристаллического вещества; т.пл. 176-177оС.
1Н ЯМР (270 мГц), δ : 3,56 (3Н, с), 3,74 (3Н, с), 5,28 (2Н, д), 6,48 (1Н, с), 6,62 (1Н, д), 6,77 (1Н, д), 6,97 (1Н, д), 7,04 (1Н, т), 7,10-7,28 (3Н, м), 7,40 (1Н, с), 7,54-7,80 (15Н, м) м.д.
П р и м е р 23. Этим примером иллюстрируется получение (Е,Е)-метил-3-метокси-2-[2-(3-стирилфенокси)фенил]пропеноата (соединение N 18 из табл.1).
1,0 г бромистой соли (Е)-метил-3-метокси-2-[2-(3-трифенилфосфонийметил)фенокси] фенилпропеноата, полученной описанным в примере 22 способом, в 5 мл сухого ДМФ добавляли по каплям к гидриду натрия (0,075 г 50%-ной дисперсии в масле, промытой гексаном) в 5 мл сухого ДМФ. В результате получали оранжевый раствор. По окончании выделения водорода (2 ч) к нему добавляли 0,66 г бензальдегида в 5 мл сухого ДМФ и реакционную смесь перемешивали при комнатной температуре в течение 20 ч, а затем нагревали в течение 2 ч при 60оС. После этого к ней добавляли воду и подвергали экстракции этилацетатом. Органический слой высушивали и выпаривали, получая в результате 1,3 г желтой маслянистой жидкости, которую очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь ТГФ и гексана, взятых в соотношении 1:4. В результате получали 0,362 г смеси целевого соединения и соответствующего (Z)-стирильного изомера в соотношении 1:1.
Кипячением этой смеси и с обратным холодильником в течение нескольких часов в толуоле с добавкой кристаллического иода получали (Е)-изомер в виде бесцветной смолы.
1Н ЯМР (270 мГц), δ : 3,64 (3Н, с), 3,77 (3Н, с), 6,88 (1Н, д), 6,96-7,40 (10Н, м), 7,49 (1Н, с), 7,48 (2Н, м), м.д.
ИКмакс. (пленка): 1710, 1640 см-1.
П р и м е р 24. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-[2-(3-феноксикарбонилфенокси)фенил] пропеноата (соединение N 50 из табл.1).
К 2,43 г (Е)-метил-3-метокси-2-[2-(3-оксиметилфенокси)фенил]-пропеноата, полученного в примере 17 способом, перемешиваемого в 100 мл ацетона при 5-10оС, добавляли хромовую кислоту, полученную растворением 6,5 г триоксида хрома в 18,5 мл воды с добавкой 5,5 мл концентрированной серной кислоты, до получения стойкой красноватой окраски и вступления (по данным газовой хроматографии) всего количества исходного спирта в реакцию. После этого смесь выливали в воду и подвергали экстракции эфиром. Эфирные экстракты промывали водой, высушивали и упаривали, получая в результате 2,495 г (Е)-метил-3-метокси-2-[2-(3-карбоксифенокси) -фенилпропеноата] в виде бледно-желтой маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,76 (3Н, с), 6,95 (1Н, д), 7,14-7,40 (5Н, м), 7,50 (1Н, с), 7,66 (1Н, с), 7,78 (1Н, д), 9,35 (1Н, широкая с) м.д.
ИКмакс. (пленка): 3500-2500, 1725, 1640 см-1.
0,33 г карбоновой кислоты, полученной на предыдущей стадии, перемешивали в 10 мл сухого ДМФ и обрабатывали 0,11 мл оксалилхлорида и одной каплей сухого ДМФ. Реакционную смесь перемешивали в течение 45 мин, оставляли стоять на ночь и упаривали, получая в результате сырой (Е)-метил-3-метокси-2-[2-(3-хлоркарбонилфенокси) фенил] пропеноат в виде оранжево-желтой маслянистой жидкости.
ИКмакс. (пленка): 1760, 1715, 1640 см-1.
К хлорангидриду кислоты, полученному на предыдущей стадии, в 15 мл сухого ТГФ добавляли 0,090 г фенола и 0,096 г триэтиламина в 5 мл сухого ДМФ. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч, выливали в воду и подвергали экстракции эфиром. Эфирные экстракты промывали разбавленным раствором гидроксида натрия, а затем водой, высушивали и упаривали, получая в результате 136 мг целевого соединения в виде оранжевой маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,77 (3Н, с), 6,94 (1Н, д), 7,14 (2Н, т), 7,23-7,38 (3Н, м), 7,47 (1Н, с), 7,61 (1Н, т), 7,72 (1Н, д) м.д.
ИКмакс. (пленка): 1755, 1710, 1640 см-1.
П р и м е р 25. Этим примером иллюстрируется получение (Е)-метил-2-{ 2-[3-(6-хлорпиримидин-4-илокси)фенокси] фенил} -3- метоксипропеноата (соединение N 89 из табл.2).
К перемешиваемому раствору 1,0 г (Е)-метил-2-[2-(3-оксифенокси)фенил] -3-меток-сипропеноата, полученного описанным в примере 1 способом, в 10 мл добавляли последовательно 0,46 г карбоната калия, 0,027 г хлорида меди и 0,41 г 4,6-дихлорпиримидина и полученную смесь перемешивали в течение 10 ч при комнатной температуре. Затем ее разбавляли водой и подвергали экстракции эфиром. Экстракты промывали последовательно водным раствором бикарбоната натрия и водой, высушивали, концентрировали и хроматографировали, используя в качестве элюента смесь эфира и гексана, взятых в соотношении 1: 1. В результате получали 0,39 г (выход 28%) целевого соединения в виде бесцветной маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,60 (2Н, с), 3,76 (3Н, с), 6,74 (1Н, т), 6,81 (1Н, дд), 6,90 (2Н, м), 7,03 (1Н, м), 7,17 (1Н, т), 7,26-6,36 (3Н, м), 7,49 (1Н, с), 8,59 (1Н, с) м.д.
П р и м е р 26. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-[2-(3-пиримидин-4-илоксифенокси)фенил] пропеноата (соединение N 92 из табл.2).
Раствор 0,27 г гипофосфита натрия в 5 мл воды добавляли по каплям при перемешивании к смеси 0,4 г (Е)-метил-2-{2-[3-(6-хлорпиримидин-4-илокси)фенокси] фенил}-3- метоксипропеноата, полученного описанным в примере 25 способом, 0,2 г карбоната калия и 0,08 г 5%-ного палладия на угле в 4 мл ТГФ. Образующуюся смесь перемешивали в течение 2 ч при комнатной температуре, фильтровали через "Hyflo" и промывали этилацетатом и водой. Объединенные фильтрат и промывочные жидкости разделяли на водный и органический слои. Последний высушивали, концентрировали и хроматографировали, используя в качестве элюента смесь эфира и гексана, взятых в соотношении 1:1. В результате получали 0,19 г (выход 52%) целевого соединения в виде бесцветной маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,61 (3Н, с), 3,75 (3Н, с), 7,75 (1Н, т), 7,48 (1Н, с), 8,56 (1Н, д), 8,76 (1Н, с) м.д.
П р и м е р 27. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-{ 2-[3-(3-нитрофенокси)фенокси]фенил} -пропеноата (соединение N 132 из табл.1).
Смесь 1,7 г 3-(3-нитрофенокси)фенола, 2,09 г (Е)-метил-2-(2-бромфенил)-3-метоксипропеноата, полученного описанным в примере 13 способом, 1,0 г карбоната калия и 1,0 г хлорида меди перемешивали в течение 5 ч при 170-180оС, затем охлаждали, разбавляли водой и подвергали экстракции эфиром. Экстракты промывали последовательно водным раствором гидроксида натрия и рассолом, высушивали и концентрировали, получая в результате 3,12 г коричневой маслянистой жидкости. Путем хроматографирования с использованием в качестве элюента смесей с различным соотношением эфира (до 20%) и гексана получали 1,06 г (выход 34%) целевого соединения в виде желтой маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,76 (3Н, с), 6,66-6,83 (3Н, м), 7,02 (1Н, д), 7,18 (1Н, д), 7,22-7,38 (3Н, м), 7,45-7,52 (1Н, м), 6,49 (1Н, с), 7,78 (1Н, м), 7,92-7,97 (1Н, м) м.д.
П р и м е р 28. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-{ 2-[3-(3-метоксифеноксиметил)фенокси] - фенил} - пропеноата (соединение N 372 из табл.1).
0,50 г (Е)-метил-3-метокси-2-(2-[3-метилфенокси]фенил)пропеноата, полученного описанным в примере 8 способом, 0,32 г 1,3-дибром-5,5-диметилгидантоина и 0,033 г азобутиронитрила кипятили с обратным холодильником в 40 мл четыреххлористого углерода, облучая вольфрамовой лампой мощностью 400 Вт. Через час смесь охлаждали и выливали в воду. Органический слой отделяли, промывали водой, высушивали и упаривали, получая в результате 0,825 г желтой вязкой маслянистой жидкости, содержащей около 65% (Е)-метил-2-метокси-2-[2-(3-бромметилфенокси)фенил] - пропеноата, который использовали далее без дополнительной очистки (см. пример 8, данные по 1Н ЯМР).
Раствор 0,41 г сырого бромида в 4 мл сухого ДМФ добавляли к раствору 3-метоксифеноксида натрия (полученного из 3-метоксифенола и гидрида натрия) в 6 мл сухого ДМФ и смесь перемешивали в течение 4 ч, после чего оставляли стоять на ночь. Реакционную смесь выливали затем в разбавленный водный раствор соляной кислоты и подвергали экстракции этилацетатом. Органические фракции высушивали и упаривали, получая в результате коричневую маслянистую жидкость, которую очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь петролейного эфира 40/60 и этилацетата, взятых в соотношении 7:3. В результате получили 0,20 г целевого соединения в виде бесцветной смолы.
ИКмакс. (пленка): 1715, 1640 см-1.
1Н ЯМР (400 мГц), δ : 3,60 (3Н, с), 3,73 (3Н, с), 3,77 (3Н, с), 4,99 (3Н, с), 6,50-6,55 (3Н, м), 6,88-6,95 (2Н, м), 7,05 (1Н, с), 7,09-7,20 (3Н, м), 7,24-7,31 (3Н, м), 7,47 (1Н, с) м.д.
П р и м е р 29. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-[2-(3-бензоилфенокси)фенил]-пропеноата (соединение N 12 из табл.1).
10,0 г метил-2-(3-оксиметилфенокси)фенилацетата, полученного описанным в примере 17 способом, и 10 г целита перемешивали в 100 мл метиленхлорида и добавляли к смеси в один прием 15,85 г хлорхромата пиридиния. После перемешивания смеси при комнатной температуре в течение 2,5 ч ее фильтровали и фильтрат упаривали, получая в результате 8,48 г метил-2-(3-формилфенокси)фенилацетата в виде оранжевой маслянистой жидкости, которая была достаточно чистой, чтобы ее можно было использовать без дополнительной очистки.
ИКмакс. (пленка): 1740, 1700 см-1.
1Н ЯМР (270 мГц), δ : 3,59 (3Н, с), 3,69 (2Н, с), 6,92 (1Н, д), 7,14-7,20 (1Н, т), 7,23-7,37 (3Н, м), 7,43 (1Н, м), 7,50 (1Н, т), 7,60 (1Н, дд), 9,95 (1Н, с) м.д.
К 2,30 г охлажденного перемешиваемого раствора полученного на предыдущей стадии альдегида в ТГФ добавляли по каплям фенилмагнийбромид (2,84 мл 3М раствора в эфире) с такой скоростью, чтобы температура не поднималась выше - 30оС. После окончания добавления (35 мин) реакционную смесь медленно нагревали до комнатной температуры, перемешивали в течение ночи и затем охлаждали на ледяной бане, осторожно добавляя воду. После этого к ней добавляли разбавленную соляную кислоту и подвергали экстракции этилацетатом. Экстракты высушивали и упаривали, получая в результате желтую маслянистую жидкость, которую очищали с помощью флеш-хроматографии, используя в качестве элюента смесь гексана и этилацетата, взятых в соотношении 2:1. В результате получали 1,69 г 2-[(3- α-окси)бензилфенокси]фенилацетата в виде бледно-желтой маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,57 (3Н, с), 8,68 (2Н, с), 5,79 (1Н, с), 6,79-6,90 (2Н, м), 7,05-7,13 (3Н, м), 7,18-7,40 (9Н, м) м.д.
0,91 г оксиэфира, полученного на предыдущей стадии, перемешивали при комнатной температуре в 25 мл метиленхлорида с добавкой двух шпателей целита. Затем к смеси добавляли 0,65 г хлорхромата пиридиния и перемешивали ее в течение 3 ч, после чего фильтровали, фильтрат упаривали и очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь гексана и этилацетата, взятых в соотношении 3:1. В результате получали 0,56 г метил-2-(3-бензоилфенокси)фенилацетат в виде бледно-желтой смолы.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,70 (2Н, с), 6,93 (1Н, д), 7,10-7,63 (10Н, м), 7,81 (2Н, д) м.д.
ИКмакс. (пленка): 1740, 1660 см-1.
Полученный продукт переводили в целевое соединение путем обработки гидридом натрия и метилформиатом с последующим добавлением карбоната калия и диметилсульфата, обработку промывали в две стадии, как это описано в примере 7 для аналогичного превращения.
ИКмакс. (пленка): 1710, 1660, 1635 см-1.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,75 (3Н, с), 6,98 (1Н, д), 7,12-7,20 (2Н, м), 7,26-7,52 (8Н, м), 7,47 (1Н, с), 7,55-7,63 (1Н, м), 7,80 (2Н, дд) м.д.
П р и м е р 30. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-[2-(3-бензилфенокси)фенил]пропеноата (соединение N 9 из табл.1).
К 1,68 г метил-2-{[3-( -окси)бензил]фенокси}фенилацетата, полученного описанным в примере 29 способом, добавляли по каплям при 5оС и перемешивали 3,28 г трифторуксусной кислоты. По окончании добавления к полученной смеси добавляли по каплям медленно 2,24 г триэтилсилана. Образующийся прозрачный раствор перемешивали в течение ночи, разбавляли водным раствором бикарбоната натрия, высушивали, концентрировали и очищали с помощью высокопроизводительной жидкостной хроматографии, используя в качестве элюента смесь гексана и эфира, взятых в соотношении 4:1. В результате получали 1,03 г метил-2-(3-бензилфенокси)фенилацетата в виде бесцветной маслянистой жидкости.
ИКмакс. (пленка): 1742 см-1.
Полученный продукт переводили в целевое соединение путем обработки гидрида натрия и метилформиатом и последующим добавлением карбоната калия и диметилсульфата. Обработку проводили в две стадии, как это описано для аналогичного превращения в примере 7.
ИК макс. (пленка): 1708, 1635 см-1.
1Н ЯМР (270 мГц), δ : 3,56 (3Н, с), 3,72 (3Н, с), 3,93 (2Н, с), 6,76-6,93 (4Н, м), 7,08-7,31 (10Н, м), 7,47 (1Н, с) м.д.
П р и м е р 31. Этим примером иллюстрируется получение (Е)-метил-3-метокси-2-{2-[3-(N-фенилсульфонамидо)фенокси]фе- нил}- пропеноата (соединение N 78 из табл.1).
Смесь 21,5 г 2-бромфенилуксусной кислоты, 29,2 г 3-нитрофенола, 27,6 г карбоната калия и 0,5 г хлорида меди нагревали в течение 6 ч при перемешивании при 130оС.
После охлаждения смесь выливали в 500 мл воды, подкисляли концентрированной соляной кислотой и трижды проводили экстракцию этилацетатом (порциями по 200 мл). Экстракты высушивали, фильтровали и концентрировали, получая темную маслянистую жидкость, которую растворяли в 400 мл метанола с добавкой 4 мл концентрированной серной кислоты и полученный раствор кипятили в течение 3 ч. После этого реакционную смесь концентрировали и остаток растворяли в 300 мл этилацетата. Полученный раствор последовательно промывали 1М водным раствором гидроксида натрия (дважды, порциями по 100 мл) и рассолом, высушивали, фильтровали и концентрировали, получая темную маслянистую жидкость. После перегонки ее (температура печи 220оС, давление 0,2 мм рт. ст.) получали 16,74 г (выход 58% в расчете на 2-бромфенилуксусную кислоту) метил-2-(3-нитрофенокси)-фенилуксусной кислоты в виде прозрачной бледной маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,70 (2Н, с), 6,9-8,0 (8Н, м) м.д.
ИКмакс. (пленка): 1739 см-1.
Смесь 15 г метил-2-(3-нитрофенокси)фенилацетата, 100 мл метанола, 100 мл ледяной уксусной кислоты и 15,0 г железного порошка мягко нагревали при перемешивании до температуры кипения. Через 30 мин смесь охлаждали и отфильтровывали избыток железного порошка. Фильтрат выливали в 700 мл воды и дважды подвергали экстракции эфиром. Эфирные экстракты нейтрализовали путем перемешивания с водным раствором бикарбоната натрия, затем высушивали, фильтровали и концентрировали, получая в результате 13,0 г (выход 97%) 2-(3-аминофенокси)фенилацетата в виде бледно-желтой маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,63 (3Н, с), 3,68 (2Н, с), 3,9 (1Н, широкий с), 6,2-7,3 (8Н, м) м.д.
ИКмакс. (пленка): 3400, 3373, 1733 см-1.
Смесь 11,54 г метил-2-(3-аминофенокси)фенилацетата и 27,7 мл метилформиата в 25 мл ДМФ добавляли по каплям при перемешивании к охлаждаемой с помощью льда до температуры ниже 10оС суспензии 3,25 г гидрида натрия в 50 мл ДМФ (выделение пузырьков газа). По окончании добавления реакционную смесь перемешивали при комнатной температуре в течение 3 ч, выливали в воду, подкисляли концентрированной соляной кислотой и подвергали экстракции этилацетатом. Экстракты промывали рассолом, высушивали и концентрировали, получая вязкую маслянистую жидкость желтого цвета. К перемешиваемому раствору этой жидкости в 50 мл ДМФ добавляли последовательно 12,4 г карбоната калия и 4 мл диметилсульфата. Полученную смесь перемешивали в течение 3 ч при комнатной температуре, выливали затем в воду и подвергали экстракции этилацетатом. Экстракты промывали водой, высушивали и концентрировали, получая в результате 13,67 г (выход 93%) (Е)-метил-2-[2-(3-формамидофенокси)фенил]-3-метоксипропеноата в виде прозрачной смолы зеленого цвета.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,78 (3Н, с), 7,47 (1Н, с) м.д.
ИКмакс. (пленка): 3309, 1702, 1606 см-1.
К перемешиваемому раствору (Е)-метил-2-[2-(3-формамидофенокси)фенил]-3- метоксипропеноата (13,67 г) в 100 мл метанола добавляли по каплям 7,8 мл фосфорилхлорида, поддерживая температуру в процессе добавления с помощью охлаждающей бани ниже 50оС. После перемешивания в течение 20 мин реакционную смесь выливали в 500 мл воды, нейтрализовывали с помощью бикарбоната натрия и подвергали экстракции эфиром. Экстракты высушивали и концентрировали, получая желтую маслянистую жидкость, которую подвергали хроматографии, используя в качестве элюента эфир. В результате получали 8,57 г (выход 68%) (Е)-метил-2-[2-(3-аминофенокси)фенил]-3-метоксипропеноата в виде твердого вещества желтого цвета. Т.пл. 83-85оС.
1Н ЯМР (270 мГц), δ : 3,6 (2Н, широкий с), 3,62 (3Н, с), 3,77 (3Н, с), 6,2-6,4 (3Н, м), 6,9-7,3 (5Н, м), 7,48 (1Н, с), м.д.
ИКмакс. (пленка): 3450, 3370, 1703, 1632 см-1.
Раствор (Е)-метил-2-[2-(3-аминофенокси)фенил]-3-метоксипропеноата (0,4 г) в 2 мл ледяной уксусной кислоты обрабатывали при - 10оС 1 мл 5,8М соляной кислотой. После этого перемешиваемый раствор обрабатывали при - 10оС раствора 0,1 н. нитрита натрия в 2 мл воды. Через 20 мин образующийся раствор (содержащий соль диазония) добавляли к перемешиваемой смеси 0,5 мл ледяной уксусной кислоты, насыщенной диоксидом серы, с добавкой 0,1 г хлорида меди (выделение пузырьков газа). Через 30 мин реакционную смесь выливали в воду и подвергали экстракции эфиром. Эфирные экстракты нейтрализовывали насыщенным водным раствором бикарбоната натрия, высушивали, концентрировали, получая в результате 0,14 г (Е)-метил-2-[2-(3-хлорсульфонилфенокси) фенил] -3-метоксипропеноата в виде желтой маслянистой жидкости.
ИКмакс. (пленка): 1710, 1636 см-1.
К раствору 0,14 г метил-2-[2-(3-хлорсульфонилфенокси)фенил]-3-метоксипропено-ата в 0,5 мл пиридина добавляли по каплям при комнатной температуре и перемешивании 0,05 мл анилина.
Через 3 ч реакционную смесь выливали в воду, добавляли к ней 2М соляную кислоту до слабокислой реакции и подвергали экстракции эфиром. Эфирные экстракты промывали рассолом, высушивали, концентрировали и подвергали хроматографии, используя в качестве элюента эфир. В результате получали 0,145 г целевого соединения в виде прозрачной маслянистой жидкости.
ИКмакс. (пленка): 3240, 1693, 1935, 1600 см-1.
1Н ЯМР (270 мГц), δ : 3,55 (3Н, с), 3,69 (3Н, с), 6,53 (1Н, широкий с), 6,8 (1Н, м), 7,0-7,4 (12Н, м), 7,43 (1Н, с) м.д.
П р и м е р 32. Этим примером иллюстрировали получение (Е)-метил-2-{ 2-[3-(3-бромбензоиламино)фенокси] фенил}-3 -метоксипропеноата (соединение N 421 из табл.1).
К перемешиваемому раствору 0,5 г (Е)-метил-2-[2-(3-аминофенокси)фенил] -3-мето- ксипропеноата, полученного описанным в примере 31 способом, в 20 мл дихлорметана с добавкой 0,17 г триэтиламина добавляли 0,37 г 3-бромбензоилхлорида. Через 3 ч реакционную смесь выливали в воду и дважды подвергали экстракции дихлорметаном (порциями по 50 мл). Экстракты высушивали, концентрировали и подвергали хроматографии, используя в качестве элюента эфир. В результате получали 0,61 г целевого соединения в виде бледно-желтой пены.
ИКмакс. (нуйол): 1710, 1680, 1640, 1605 см-1.
1Н ЯМР (270 мГц), δ : 3,62 (3Н, с), 3,78 (3Н, с), 6,73-8,0 (13Н, м), 7,47 (1Н, с) м.д.
П р и м е р 33. Этим примером иллюстрируется получение (Е)-метил-2-[2-(3-пиридин-2-илоксифенокси)фенил]-3-метоксипропеноата (соединение N 1 из табл.2).
К перемешиваемому раствору 2,0 г (Е)-метил-2-[2-(3-оксифенокси)фенил]-3-мето- ксипропеноата, полученного описанным в примере 1 способом, в 15 мл ДМФ добавляли последовательно 0,2 г карбоната калия, каталитическое количество хлорида меди и 1,94 г 2-фторпиридина. Полученную смесь перемешивали в течение 3 ч при 130оС. После охлаждения ее разбавляли водой и дважды экстрагировали эфиром. Объединенные экстракты промывали последовательно водным раствором гидроксида натрия, водой и рассолом, затем высушивали и концентрировали. После хроматографии с использованием в качестве элюента смеси эфира и гексана получали 1,68 г (выход 67%) целевого соединения в виде оранжево-желтой маслянистой жидкости.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,73 (3Н, с), 6,72-7,32 (10Н, м), 7,48 (1Н, с), 7,67 (1Н, м), 8,19 (1Н, м) м.д.
П р и м е р 34. Этим примером иллюстрируется получение (Е)-метил-2-{ 2-[3-(6-хлорпиридиазин-3-илокси)фенокси] фенил} -3- метоксипропеноата (соединение N 130 из табл.2).
К перемешиваемому раствору 2,01 г (Е)-метил-2-[2-(3-оксифенокси)фенил] -3-мето- ксипропеноата, полученного описанным в примере 1 способом, в 30 мл ДМФ добавляли последовательно 0,93 г карбоната калия, каталитическое количество хлорида меди и 1,0 г 3,6-дихлорпиридазина. Приготовленную смесь перемешивали в течение 1 ч при 95оС. После охлаждения ее разбавляли водой и подвергали экстракции (дважды) эфиром. Объединенные экстракты промывали последовательно водным раствором гидроксида натрия, водой и рассолом, затем высушивали и концентрировали. После хроматографии с использованием в качестве элюента смеси эфира и гексана получали 1,71 г (выход 62%) целевого соединения в виде желтой смолы.
1Н ЯМР (270 мГц), δ : 3,60 (3Н, с), 3,73 (3Н, с), 6,73-7,36 (9Н, м), 7,46 (1Н, м), 7,50 (1Н, с) м.д.
Ниже приведены примеры композиций, приготовленных на основе соединений в соответствии с настоящим изобретением, которые могут использоваться для обработки сельскохозяйственных и садовых культур. Такие композиции также являются предметом настоящего изобретения. Приведенные проценты являются массовыми.
П р и м е р 35. Концентрат эмульсии готовили путем смешения и последующего перемешивания компонентов до их растворения (%).
Соединение N 212 из табл.1 10
Бензоловый спирт 30
Додецилбензолсульфонат
кальция 5
Нонилфенолэтоксилат
(13 моль этиленоксида) 10
Алкилбензолы 45
П р и м е р 36. Активный компонент растворяли в метиленхлориде и полученный раствор напыляли на гранулы аттаппульгитовой глины (%). После испарения растворителя получали гранулированный препарат.
Соединение N 212 из табл.1 5
Аттапульгитный гранулят 95
П р и м е р 37. Композицию для протравливания семян получали путем измельчения и смешения трех компонентов (%). Соединение N 212 из табл.1 50 Минеральное масло 2 Каолин 48
П р и м е р 38. Порошок для распыления получали путем измельчения и смешения активного компонента с тальком (%). Соединение N 212 из табл.1 5 Тальк 95
П р и м е р 39. Концентрат суспензии готовили путем измельчения в шаровой мельнице компонентов с образованием водной суспензии измельченной смеси (% ) Соединение N 212 из табл.1 40 Лигносульфонат натрия 10 Бентонитовая глина 1 Вода 49
Полученная композиция может использоваться непосредственно для опрыскивания семенного материала или для опрыскивания после предварительного разбавления водой.
П р и м е р 40. Смачивающийся порошок получали путем смешения и последующего измельчения компонентов до образования однородной смеси (%).
Соединение N 212 из табл.1 25
Лаурилсульфат натрия 2
Лигносульфонат натрия 5
Диоксид кремния 25
Каолин 43
П р и м е р 41. Соединения формулы I испытывали по отношению к различным грибковым заболеваниям листьев растений. Испытания проводили следующим образом.
Растения выращивали в John Innes Potting Compost (N 1 или 2) в горшочках диаметром 4 см. Препараты испытуемых соединений готовили или путем измельчения в шаровой мельнице с водным дисперсолом Т, или путем растворения в ацетоне, или смеси ацетона и этанола и последующего разбавления до нужной концентрации непосредственно перед использованием. В случае болезней листьев композициями (концентрация активного компонента, если это не оговорено, 100 м.д.) опрыскивали листья или их вносили в почву. При опрыскивании композиции наносили в максимальном удерживающемся на листьях количестве. При введении же их в почву к корневой системе конечная концентрация соответствовала примерно 40 мл активного компонента в расчете на сухую почву. При обработке злаковых к композициям добавляли Tween 20 в таком количестве, чтобы конечная концентрация его равнялась 0,05%.
В большинстве случаев испытуемое соединение вносили в почву или им опрыскивали листья за один или два дня до заражения растения возбудителем болезни. Исключение составляли опыты с Erysi phegraminis. В этом случае растения заражались за 24 ч до обработки. Заражение возбудителями заболеваний листьев осуществляли путем опрыскивания листьев испытуемых растений суспензиями спор. После заражения растения выдерживали в условиях, благоприятных для развития болезни, до тех пор, пока появлялись явные ее признаки, по которым можно было сделать оценку степени ее развития. Время между моментами заражения и оценки степени развития болезни варьировалось от 4 до 14 дней в зависимости от болезни и окружающих условий.
Степень развития болезни оценивали в баллах по следующей системе: 4 = отсутствие болезни; 3 = заболевание от следов до 5% от необработанных растений; 2 = заболевание 6-25% от необработанных растений; 1 = заболевание 26-59% от необработанных растений; 0 = заболевание 60-100% от необработанных растений. Полученные результаты приведены в табл.6, эталоны - в табл.7.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПРОПЕНОВОЙ КИСЛОТЫ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ | 1989 |
|
RU2024496C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПЕНОВОЙ КИСЛОТЫ И ИХ СТЕРЕОИЗОМЕРОВ | 1989 |
|
RU2026282C1 |
| ПРОИЗВОДНЫЕ АКРИЛОВОЙ КИСЛОТЫ И ИХ СТЕРЕОИЗОМЕРЫ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2037487C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА | 1992 |
|
RU2043990C1 |
| Способ получения производных пиридина | 1988 |
|
SU1811528A3 |
| СПОСОБ ПОЛУЧЕНИЯ Е-ИЗОМЕРОВ ПРОИЗВОДНЫХ АКРИЛОВОЙ КИСЛОТЫ | 1988 |
|
RU2024511C1 |
| ПРОИЗВОДНЫЕ АКРИЛОВОЙ КИСЛОТЫ | 1991 |
|
RU2044723C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРИМИДИНА | 1990 |
|
RU2019543C1 |
| ЗАМЕЩЕННЫЕ АЦИЛАМИНОБЕНЗАМИДЫ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2034829C1 |
| ПРОИЗВОДНЫЕ АКРИЛОВОЙ КИСЛОТЫ | 1991 |
|
RU2039044C1 |
Способ получения производных пропеновой кислоты и их стереоизомеров. Сущность изобретения: производные пропеновой кислоты формулы: (Z - X, А) - C6H2-K-C6H3-C(COOCH3)=CHOCH3 , где К - кислород или сера, А - водород, галоген, C1-C4 -алкил, C1-C4 -алкокси, гидрокси, фенокси или C1-C4 -алкилкарбонил. Реагент 1: соединение формулы (A, W, L′)C6H3 . Реагент 2: соединение формулы (L″, Y)C6H4 , где L, L′ - общепляемая группа. 1 с.п., 1 з.п. ф - лы, 5 табл.
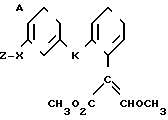
где A - водород, галоген, C1-C4-алкил, C1-C4-алкокси, гидрокси, фенокси или C1-C4-алкилкарбонил;
К - кислород или сера;
X - кислород, S(O)n, NH, NR1, CH2, CHR2, CO, CH2CH2, CH=CH, OCH2, (CH2)mO, CHR1O, OCH2O, S(O)nCH2, S(O)CH2O, NR1CH2, COO, OOC, SO2O, COCH2O,
COCHR1O, CONH, NHCO, NHSO2, COS, SCO, N=N, CH2OCO, CH2SCO, CH2NHCO, CH2ON= CH, OCH2CH2O, NR1N= CH, CH2OCONH, CH=CHCH2O, (R2)2P+CH2Q-, N(COR1), N=CH, CH(OH), CO2CH2, SCH2O, NR1CO, S(O)2NH или CONR1;
R1 - C1-C4-алкил;
R2 - фенил;
n=0,1 или 2;
m=1 - 5;
Q - галоидный анион;
Z - фенил (возможно, монозамещенный C1-C6-алкилом, C1-C4-алкокси, C1-C4-галоидалкилом, фенокси, фенилом, амино, гидрокси, 1-(C1-C4-алкоксикарбонил)-2-(C1-C4-алкокси)-винил, C1-C4-галоид-алкокси,
или C1-C4-алкоксикарбонил, или моно- или дизамещенный галогеном, нитро, C1-C4-алкилом или цианогруппой), нафтил, хинолинил, пиридинил (возможно, монозамещенный C1-C4-алкилом, C1-C4-алкоксикарбонилом, амино, галогеном, нитро, C1-C4-алкилкарбониламино, ди-(C1-C4-алкилсульфонил)амино или CH(O)NH, или моно- или дизамещенный C1-C4-галоидалкилом или цианогруппой, или дизамещенный аминогруппой и одной из циано, галоген- или C1-C4-алкоксигрупп, или дизамещенный нитрогруппой и одной из циано, галоген, ди-(C1-C4-алкил)амино или C1-C4-алкоксигрупп, или замещенный цианогруппой и двумя C1-C4-алкильными группами), пиримидинил (возможно, монозамещенный C1-C4-алкилом, C1-C4-галоидалкилом, C1-C4-алкилтио, циано, нитро, фенилом, HO2C, C1-C4-алкоксикарбонилом или C1-C4-алкилсульфонилом, или моно- или дизамещенный C1-C4-алкокси, или моно-, ди- или тризамещенный галогеном, или дизамещенный галогеном и одной из C1-C4-алкил или C1-C4-алкилтио, или дизамещенный C1-C4-алкил и C1-C4 галоалкил), пиразинил (возможно, монозамещенный галогеном или циано или дизамещенный C1-C4-алкилом), пиридазинил (возможно, монозамещенный C1-C4-алкокси, фенилом или аминокарбонилом, или моно- или дизамещенный галогеном, или дизамещенный галогеном и C1-C4-алкилом), бензотиазолил, тиенил (возможно, монозамещенный пиразолилом, который сам по себе дизамещен C1-C4-алкилом и C 1-C4-галоидалкилом, или пиридинилом, который сам по себе, возможно, монозамещен нитро, или дизамещенный галогеном), 1,2,4-триазолил, хиноксалинил (монозамещенный галогеном), 1,3,5-триазинил (дизамещенный галогеном или дизамещенный галогеном и C1-C4-алкокси), тиазолил (возможно, монозамещенный нитро или моно- или дизамещенный C1-C4-алкилом), бензоксазолил, пиридинил-N-оксид, тиено [2,3-d]пиримидинил, пирролил (возможно, монозамещенный C1-C4-алкилом), изоксазолил (монозамещенный C1-C4-алкилом), 1,3,4-тиадиазолил, пиразолил (замещенный галогеном и двумя C1-C4-алкильными группами) или 1,2,4-триазинил (монозамещенный фенилом) при условии, что когда Z - незамещенный фенил, а X и K-оба кислород, тогда A не является водородом;
и их стереоизомеров, отличающийся тем, что осуществляют взаимодействие в присутствии основания соединений общей формулы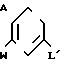
с соединением общей формулы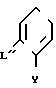
где одна из L' и L'' - отщепляемая группа, а другая - КН;
W - ZX или группа, которую превращают в ZX путем
а) взаимодействия в присутствии основания соединения общей формулы
H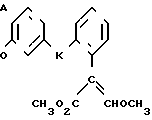
когда X - кислород, с соединением формулы ZL;
или, когда X - SO2O, с соединением формулы ZS O2O;
или,
когда X - CH2O, с соединением формулы ZCH2L;
или,
когда X - CO · O, с соединением формулы ZCO · Hal;
или
б) взаимодействия в присутствии основания соединения формулы
LCH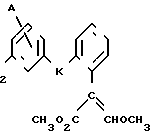
когда X - OCH2, с соединением формулы ZOH;
или
когда X - (C6H5)2P+CH2O- и L - галоид, с фосфином формулы Z (C6H5)2P;
или
когда X - CO · OCH2, с соединением формулы Z CO ·OH;
в) когда X - CH=CH, взаимодействие в присутствии основания соединения общей формулы
(R1O)2-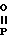 -CH
-CH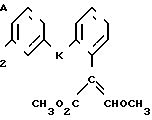
где R1 - C1-C6-алкил,
с соединением формулы Z CHO;
или
г) когда X - CH= CH, взаимодействие соединения формулы I, где X - (C6H5)2P+CHR2Q-, последовательно с основанием и карбонильным соединением формулы Z CHO;
или
д) когда X - CONH, взаимодействия соединения общей формулы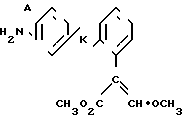
в присутствии основания с галоидом кислоты формулы Z CO·Q;
или
е) когда X - CH(OH), взаимодействия альдегида общей формулы
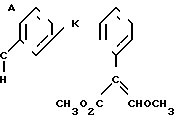
с реактивом Гриньяра формулы Z MgHal;
или
ж) когда X - O·CO, взаимодействия хлорида кислоты общей формулы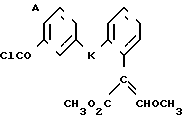
с соединением формулы Z OH в подходящем растворителе в присутствии основания;
или
з) когда X - OCH2, взаимодействия соединения общей формулы
HOCH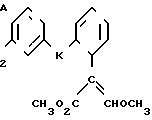
с соединением формулы ZL;
или
и) когда X - N=N, взаимодействия соединения общей формулы
H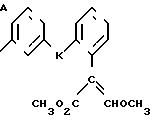
с соединением формулы ZN+ = NHal-;
или
к) когда X - NHSO2, взаимодействия соединения общей формулы
ClSO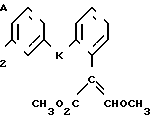
с соединением формулы
Z NH2;
где A, K и Z имеют указанные значения;
L - отщепляющая группа, предпочтительно, атом галогена и кислорода, представляет собой HaI;
л) - группа CH3O. CH=C-(CO2CH3) или половина CH2CO2CH3, которую можно превратить в подобную группу посредством взаимодействия с метилформиатом в присутствии основания и последующего метилирования в щелочных условиях.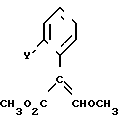
где Y' - галоген,
подвергают взаимодействию с фенолом общей формулы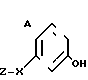
где A, X и Z имеют указанные значения,
в присутствии основания, или с солью фенола указанной формулы в присутствии катализатора - соли меди или смеси соли меди и металлической меди.
Приоритеты по признакам:
22.01.88 при A - гидрокси, фенокси, C1-C4-алкилкарбонил; X - CH(C6H5), C(= O)CH2O, CH(OH), CO2CH2, COCH (C1-C4-алкил)O-, SCH2O; K - кислород; Z - незамещенный фенил или замещенный галогеном;
17.03.88 при K - сера; X - кислород, A - водород, K -фенил, Z - пиримидинил;
21.06.88 при A - водород, X - Ph2 P+ CH2Br-, CH2OCO; CH2NHCO, CH2SCO; K - кислород; Z - фенил;
08.09.88 при A - водород, X - OCH2O, S(O)CH2O, CH2ON=CH, (CH2)3O, (CH2)5O, OCH2CH2O, CH2OCONH; K - кислород, Z - фенил.
