Изобретение относится к способу получения новых гетероциклических производных акриловой кислоты, обладающих ценными фунгицидными свойствами и которые могут найти применение в сельском хозяйстве.
Известно применение стробилурина в качестве фунгицидного средства.
Недостатком этого известного средства является его сравнительно невысокая эффективность.
Цель изобретения - способ получения новых производных акриловой кислоты, обладающих более высокой фунгицидной активностью.
Цель достигается способом получения Е-изомеров производных акриловой кислоты общей формулы I.
Нижеследующие примеры иллюстрируют способ по изобретению и фунгицидные свойства получаемых соединений.
П р и м е р 1. /E/-Метил-2-/2'-(5"-нитропиридин-2"-илокси)фенил/-3-метоксиакри- лат (соединение 133, табл.1).
2-/Гидроксифенил/уксусную кислоту /50 г/ добавляют к раствору хлористого водорода в метаноле (приготовленного из ацетилхлорида /25 мл/ и метанола /250 мл/). Раствор перемешивают при комнатной температуре в течение 3 ч, а затем выдерживают в течение 15 ч. Полученную смесь концентрируют при пониженном давлении, остаток берут в простой эфир /250 мл/ и промывают водным раствором бикарбоната натрия до тех пор, пока не прекратится бурное выделение газа. Эфирный раствор сушат, а затем концентрируют при пониженном давлении. Полученное твердое вещество перекристаллизовывают из смеси простой эфир/бензин; получают метил-(2-гидроксифенил)ацетат (50 г, 92% выход) в виде белых кристаллов, т.пл. 70-72оС; ИК (нуйол): 3420, 1715 см-1; 1Н ЯМР (90 МГц); δ, м.д.: 3,70 (2Н, с), 3,75 (3Н, с), 6,80-6,95 (2Н, м), 7,05-7,10 (1Н, м), 7,15-7,25 (1Н, м), 7,40 (1Н, с).
Метил (2-гидроксифенил)ацетат (21,0 г) растворяют в диметилформамиде (200 мл), добавляют карбонат калия (19,35 г) в виде одной порции. К смеси по каплям добавляют бензилбромид (23,94 г) в диметилформамиде (50 мл) при перемешивании при комнатной температуре. Спустя 18 ч смесь выливают в воду (500 мл) и экстрагируют диэтиловым эфиром (2х400 мл). Экстракты промывают водой (3х150 мл) и солевым раствором (100 мл), сушат и фильтруют через силикагель (50 г, Мерк 60), затем концентрируют при пониженном давлении, получают желтое масло, перегонка которого (160о/0,5 мм рт.ст.) дает метил-2-бензилоксифенилацетат в виде бесцветного масла (26,99 г, 83% выход), ИК (пленка): 1730 см-1; 1Н ЯМР (90 МГц), δ, м.д.: 3,60 (3Н, с), 3,75 (2Н, с), 4,10 (2Н, с), 6,80-7,40 (9Н, м).
Смесь метил 2-бензилоксифенилацетата (26,99 г) и метилформата (126,62 г) в сухом диметилформамиде (300 мл) добавляют по каплям к перемешанной суспензии гидрида натрия (50% дисперсия в масле, 10,13 г) в ДМФ (300 мл) при 0оС. После перемешивания при 0оС в течение 2 ч смесь выливают в воду (1000 мл) и промывают диэтиловым эфиром (2х150 мл). Водный слой подкисляют до рН 4 с помощью 6 М соляной кислоты, затем экстрагируют диэтиловым эфиром (2х350 мл). Экстракт сушат и концентрируют при пониженном давлении, получают неочищенный метил-2/2'-бензилоксифенил/-3-гидроксиакрилат в виде желтого масла, ИК (пленка) 1720, 1660 см-1.
Неочищенный метил 2-/2'-бензилоксифенил/-3-гидроксиакрилат растворяют в сухом диметилформамиде (100 мл) и в виде одной порции добавляют карбонат калия (29,0 г). Затем по каплям добавляют диметилсульфат (16,00 г) в сухом ДМФ (100 мл) при перемешивании. Спустя 90 мин добавляют воду и раствор экстрагируют диэтиловым эфиром (2х300 мл). После промывания водой (3х150 мл) и солевым раствором экстракты сушат и концентрируют при пониженном давлении, полученное желтое масло затвердевает после растирания со смесью диэтилового эфира и бензина. Перекристаллизация из сухого метанола дает (Е)-метил-2(2'-бензилоксифенил)-3-метоксиак- рилат в виде белого кристаллического твердого вещества (5,44 г, 17% выход из расчета на метил-2-бензилоксифенилацетат), т.пл. 76-77оС; ИК (нуйол): 1710, 1640 см-1; 1Н ЯМР (90 МГц), δ, м. д. : 3,63 (3Н, с), 3,75 (3Н, с), 5,06 (2Н, с), 6,80-7,40 (9Н, м), 7,50 (1Н, с).
(Е)-Метил-2-(2'-бензилоксифенил)-3-ме- токсиакрилат (5,44 г) растворяют в этилацетате (50 мл) и добавляют 5% палладий на угле (0,25 г). Перемешиваемую смесь гидрируют при давлении 3 атм при перемешивании до прекращения поглощения водорода, затем фильтруют через целит силикагель (50 г, Мерк 60). Концентрирование фильтрата при пониженном давлении дает (Е)-метил-2-(2'-гидроксифенил)-3-метоксиакрилат в виде белого кристаллического твердого вещества (3,76 г, 99% выход), т.пл. 125-126оС; ИК (нуйол): 3400, 1670 см-1; 1Н ЯМР (270 МГц), δ, м.д.:3,80 (3Н, с), 3,90 (3Н, с), 6,20 (1Н, с), 6,80-7,00 (2Н, м), 7,10-7,30 (2Н, м), 7,60 (1Н, с).
(Е)-Метил-2-(2'-гидроксифенил)-3-мет-оксиакрилат (0,30 г, 1,44 ммоля), 2-хлор-5-нитропиридин (0,48 г, 2,88 ммоля) и карбонат калия (0,40 г, 2,88 ммоля) перемешивают в ДМФ (20 мл) при комнатной температуре в атмосфере азота. Через 18 ч реакционную смесь выливают в воду, а затем экстрагируют дважды диэтиловым эфиром. Объединенные эфирные слои промывают два раза водой и солевым раствором, а затем сушат. Полученный раствор фильтруют через слой силикагеля, а затем концентрируют, получают розовое твердое вещество. Хроматография (элюент - диэтиловый эфир) дает (Е)-метил 2-[2'-(5"-нитропиридин-2"-илокси)фенил] -3-метоксиакрилат (240 мг) в виде желтой смолы, которая кристаллизуется при стоянии, т.пл. 107-109оС.
П р и м е р 2. (Е)-Метил 2-[2'-(4"-хлорпиримидин-2"-илокси)фенил]-3-метоксиакри- лат (соединение N 61 таблицы 1).
Е-Метил 2-(2'-гидроксифенил)-3-метоксиакрилат (0,63 г), 2,4-дихлорпиримидин (0,75 г) и карбонат калия (0,69 г) перемешивают в ДМФ при комнатной температуре. Спустя 2 ч реакционную смесь выливают в воду (50 мл) и экстрагируют два раза диэтиловым эфиром. Объединенные эфирные слои промывают водой (х3) и солевым раствором (х1), а затем сушат. Фильтруют и упаривают растворитель при пониженном давлении, получают светлое масло. Хроматография с использованием в качестве элюента диэтилового эфира дает (Е)-метил 2-[2'-(4"-хлорпиримидин-2"-илокси/фенил] -3-метоксиакрилат (0,35 г) в виде масла, которое кристаллизуется после растирания с эфиром; т.пл. 120-121,5оС; 1Н ЯМР, δ, м.д.: 3,60 (3Н, с), 3,80 (3Н, с), 6,60 (1Н, д, I = 4 Гц), 7,40 (1Н, с), 8,40 (1Н, д, I = 4 Гц).
П р и м е р 3. (Е)-метил-2-[2'-(6"-метилпиридин-3"-илокси)фенил]-3-метоксиакрилат (соединение 45, табл.1).
6-Метил-3-гидроксипиридин (9,5 г) суспендируют в толуоле (30 мл) и обрабатывают водной гидроокисью калия (4,9 г в воде, 3 мл). Смесь энергично перемешивают в течение 15 мин, затем упаривают при пониженном давлении. Последние следы воды удаляют с помощью повторного упаривания в присутствии толуола. Образовавшееся коричневое полутвердое вещество обрабатывают смесью 2-(2-бромфенил)-1,3-диоксолана (10,0 г), хлористой меди (60 мг) и трис [2-(2-метоксиэтоксиэтил] амина (0,194 г) для солюбилизации медной соли в сухом ДМФ (25 мл), и смесь нагревают при 155оС при перемешивании в атмосфере азота в течение 30 часов. Добавляют дополнительно хлористую медь (60 мг) и нагревание продолжают в течение 14 часов.
Смесь охлаждают, выливают в воду и экстрагируют этилацетатом. Экстракт промывают 2 норм. водным раствором гидроокиси натрия и воды с последующим экстрагированием 2 норм. соляной кислотой. Кислый водный экстракт обрабатывают твердым карбонатом калия до рН 8, а затем экстрагируют этилацетатом. Органический экстракт сушат, а затем упаривают при пониженном давлении, получают 2-/6'-метилпиридин-3'-илокси бензальдегид (2,2 г) в виде масла; ИК (пленка) 1697, 1606, 1480 см-1; 1Н ЯМР, δ,м.д.: 2,58 (3Н, с), 6,86 (1Н, с), 7,28 (3Н, м), 7,55 (1Н, т), 7,95 (2Н, м), 8,36 (1Н, м), 10,53 (1Н, с).
2-(6'-Метилпиридин-3'-илокси)бензаль- дегид (2,08 г) и метилсульфинилметилсульфид (1,21 г) растворяют в сухом ТГФ (15 мл) и Тритон В (1,5 мл) добавляют медленно по каплям при перемешивании при комнатной температуре. Смесь выдерживают в течение ночи, разбавляют водой и экстрагируют этилацетатом. Экстракт сушат, а затем упаривают при пониженном давлении, получают оранжево-коричневое масло (3,2 г). Масло обрабатывают метанольным раствором хлористого водорода (25 мл, 2,6 норм.) и выдерживают в течение ночи при комнатной температуре. Раствор затем разбавляют водой и доводят до рН 8 путем добавления карбоната натрия. Смесь экстрагируют этилацетатом, экстракт сушат и упаривают, получают коричневое масло (2,23 г), которое очищают с помощью жидкостной хроматографии высокой разрешающей способности (элюент 1: 1, смесь этилацетат: гексан), получают метил[2-(6'-метилпиридин-3'-илокси)фенил] ацетат в виде желтого масла (1,53 г), ИК (пленка) 1747, 1488, 1237 см-1; 1Н ЯМР, δ, м.д.: 2,54 (3Н, с), 3,63 (3Н, с), 3,64 (2Н, с), 6,84 (1Н, д), 7,24 (5Н, м), 8,3 (1Н, д).
Смесь метил [2-(6'-метилпиридин-3'-илокси)фенил] ацетата (1,3 г) и метилформиата (1,52 г) в ДМФ (5 мл) добавляют по каплям к суспензии гидрида натрия (316 мг 50% дисперсии в масле) в ДМФ (5 мл) при перемешивании при 5оС. После перемешивания в течение 4 ч смесь разбавляют водой, делают слабо кислой путем добавления ледяной уксусной кислоты (рН 4-5) и экстрагируют этилацетатом. Экстракт после сушки и упаривания при пониженном давлении дает метил 2-[(2'-(6"-метилпиридин-3"-илокси)-фенил]-3-гидроксиакрилат в виде желтого масла (1,15 г), 1Н ЯМР, δ, м.д.: 2,53 (3Н, с), 3,63 (3Н, с), 6,89 (1Н, с), 7,2 (5Н, м), 8,21 (1Н, д).
Масло (1,14 г) растворяют в ДМФ (15 мл) добавляют карбонат калия (1,1 г) и смесь перемешивают в течение 15 мин. Диметилсульфат (0,53 г) растворяют в ДМФ (5 мл) и этот раствор добавляют к смеси. Получающуюся смесь перемешивают в течение 80 мин, затем разбавляют водой и получающуюся эмульсию экстрагируют этилацетатом. Экстракт сушат и упаривают при пониженном давлении, получают желтое масло (2,06 г), которое очищают с помощью хроматографии высокого давления (элюент:этилацетат), получают (Е)-метил-2-[2'-(6"-ме- тилпиридин-3"-илокси)фенил] -3-метоксиак- рилат в виде бледно-желтого масла (0,73 г), ИК (пленка) 1705, 1642, 1488 см-1; 1Н ЯМР, δ, м.д.: 2,52 (3Н, с), 3,63 (3Н, с), 3,81 (3Н, с), 6,88 (1Н, д), 7,04-7,32 (5Н, м), 7,51 (1Н, с), 8,26 (1Н, д).
П р и м е р 4. Используя методику примера 1, получают следующие соединения:
1. (Е)-метил 2-[2'-(5"-хлорпиридин-2"-илокси)фенил]-3-метоксиакрилат, т. пл. 77-78оС; ИК 1700, 1625, 1260, 1200 см-1; 1Н ЯМР (CДCl3): δ, м.д.: 3,57 (3Н, с), 3,74 (3Н, с), 6,75 (1Н, д), 7,41 (1Н, с), 8,10 (1Н, шир.с.), 7,1-7,6 (м).
2. (Е)-метил-2-[2'-(5"-цианопиридин-2"-илокси)фенил] -3-метоксиакрилат (соединение N127, табл.1), в виде белых кристаллов т.пл. 108,5-109,5оС. 1Н ЯМР, δ, м.д.: 3,58 (3Н, с), 3,75 (3Н, с). 6,9 (1Н, д), 7,1 (1Н, д), 7,28-7,4 (4Н, м), 7,45 (1Н, с), 7,85 (1Н, кв), 8,45 (1Н, д).
3. (Е)-метил-2-2'-(5"-хлорпиридин-2"-илтио/фенил-3-метоксиакрилат (соединение 14, табл.1) в виде густой смолы, ИК 1700, 1630 см-1;
4. (Е)-метил 2-[2'-(5"-бромпирид-2"-илсульфонил)фенил-3-метоксиакрилат в виде смолы.
5. (Е)-метил 2-[2'-(5"-бромпирид-2"-илсульфонил)фенил]-3-метоксиакрилат в виде аморфного твердого вещества.
6. (Е)-метил 2-[2'-(5"-метоксикарбонилпиридин-2"-илокси)фенил] 3-метоксиакрилат (соединение 141, табл. 1). 1Н ЯМР, δ, м.д.: 3,47 (3Н, с), 3,62 (3Н, с), 3,82 (3Н, с), 6,75-7,3 (5Н, м), 7,32 (1Н, с), 8,15 (1Н, кв), 8,72 (1Н, д).
7. (Е)-метил 2-[2'-(5"-бензилоксикарбонилпиридин-2"-илокси)фенил]-3-метоксиак- рилат (соединение 184, табл.1), в виде смолы; 1Н ЯМР, δ, м.д.: 3,55 (3Н, с), 3,60 (3Н, с), 5,35 (2Н, с), 6,82 (1Н, д), 7,18-7,48 (м), включая синглет одного протона при 7,39, 8,25 (1Н, кв), 8,25 (1Н, д).
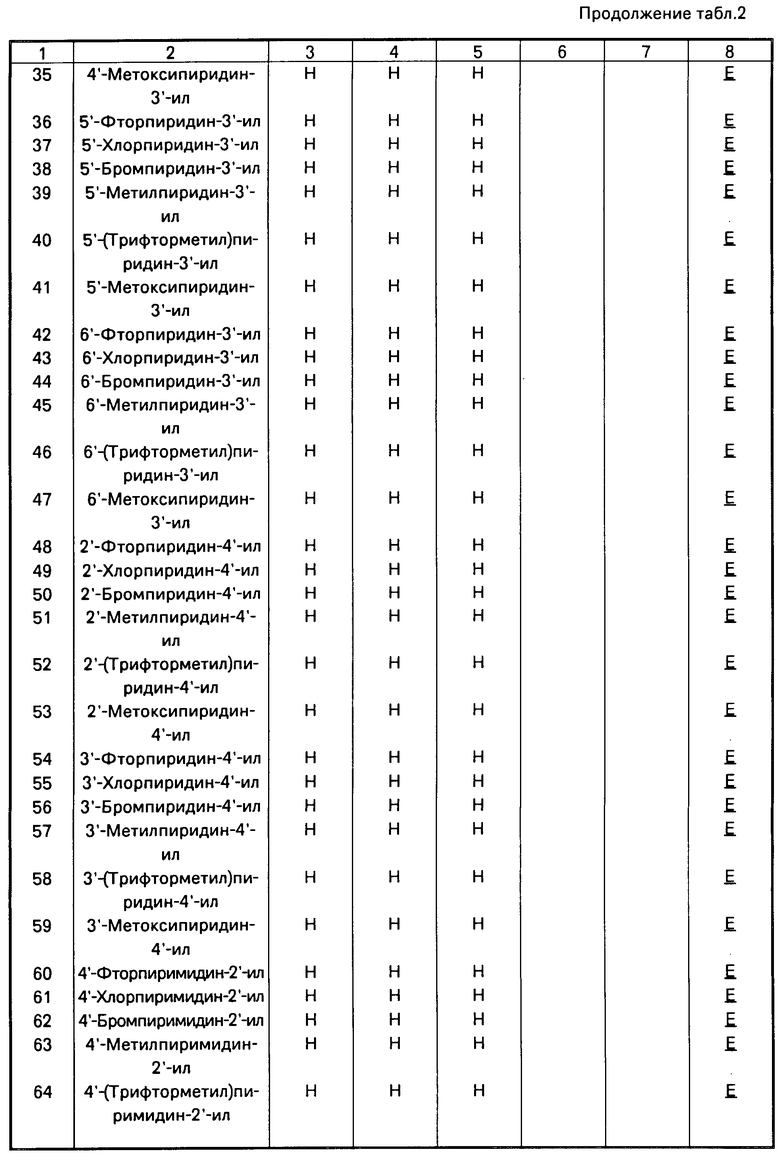
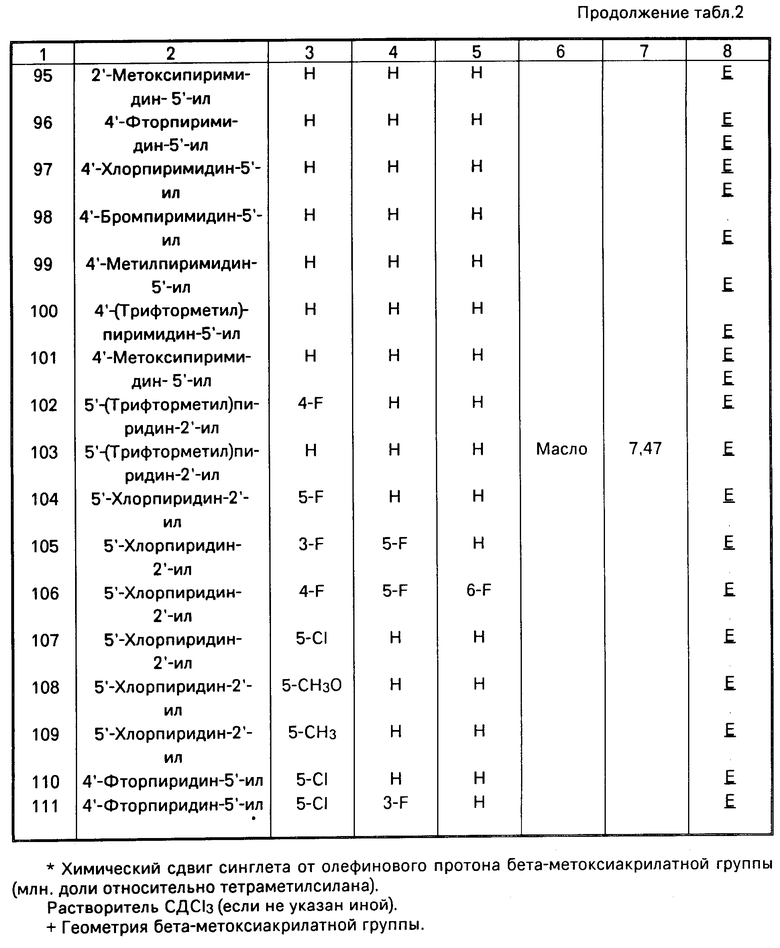
Аналогично получают другие соединения табл.I, II и III, строение которых также подтверждено спектрами ПМР.
Ниже следуют примеры композиций, пригодные для сельскохозяйственного и садового применения, которые могут быть преобразованы в готовые препаративные формы из соединений данного изобретения.
П р и м е р 5. Эмульгирующий концентрат готовят путем смешения и перемешивания ингредиентов до полного растворения,%: Соединение 61, табл.1 10 Бензиловый спирт 30
Додецилбензолсульфонат кальция 5
Нопилфенолэтоксилат (13 молей окиси этилена) 10 Алкилбензолы 45
П р и м е р 6. Активный ингредиент растворяют в метилендихлориде, полученную жидкость разбрызгивают на гранулы аттапульгитной глины. Растворителю затем дают возможность испариться, получают композицию в виде гранул,%: Соединение 14, табл.1 5 Гранулы аттапульгита 95
П р и м е р 7. Композицию, пригодную для протравливания семян, готовят с помощью измельчения и смешения трех ингредиентов,%: Соединение 61, табл.1 50 Минеральное масло 2 Китайская глина 48
П р и м е р 8. Дустовый порошок готовят с помощью измельчения и смешения активного ингредиента с тальком, %: Соединение 61, табл.1 5 Тальк 95
П р и м е р 9. Суспензионный концентрат готовят измельчением в шаровой мельнице ингредиентов с образованием водной суспензии измельченной смеси в воде,%: Соединение 61 40 Лигносульфонат натрия 10 Бентонитная глина 1 Вода 49
Данная препаративная форма может использоваться в виде спрея путем разбавления водой или применяться непосредственно для обработки семян.
П р и м е р 10. Форму смачиваемого порошка готовят путем смешения и измельчения ингредиентов до тех пор, пока они все не смешиваются тщательно,%: Соединение 61, табл.1 25 Лаурилсульфат натрия 2 Лигносульфонат натрия 5 Кремнезем 25 Китайская глина 43
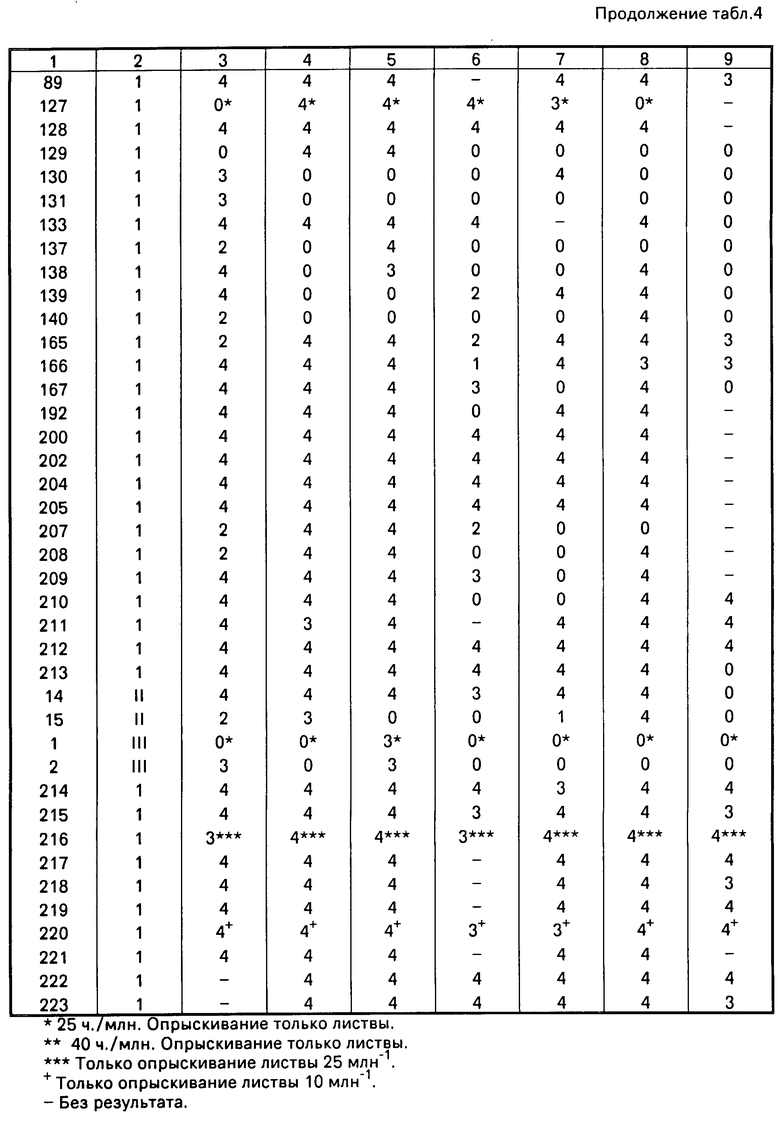
П р и м е р 11. Соединения испытывались против ряда грибковых заболеваний листвы растений. Технология, применяемая при испытания, заключалась в следующем.
Растения выращивались в компосте для горшков John Innes (1 или 2) в минигоршках диаметром 4 см. Испытуемые соединения преобразовывались в готовую форму препарата или измельчением шариками с водным Дисперзолем Т, или в виде раствора в ацетоне, или смеси ацетона и этанола, который разбавлялся до требуемой концентрации непосредственно перед использованием. В случае заболеваний листвы препарата (100 ч. на 1 млн. активного ингредиента) распылялись на листья и вносились к корням растений в почве. Спреи применялись до максимального удержания на поверхности, а корни увлажнялись до конечной концентрации, эквивалентной приблизительно 40 ч на 1 млн. активного ингредиента в сухой почве. Когда спреи применялись по отношению с злаковым, добавлялся Твин 20 для получения конечной концентрации 0,05%.
В случае большинства испытаний соединение применялось по отношению к почве (к корням) и к листве (опрыскиванием) за один или два дня до заражения растения болезнью. Исключением было испытание против Erysiphe graminis, при котором растения инокулировались за 24 ч до обработки. Лиственные патогенные организмы применялись путем разбрызгивания суспензий спор на листья, используемых растений. После инокуляции растения помещались в соответствующую окружающую среду для обеспечения возможности развития заболевания, а затем инкубировались до тех пор, пока инокуляция не была готовой для оценки. Период между инокуляцией и оценкой варьировал от 4 до 14 дней, в зависимости от болезни и окружающих условий.
Борьба с заболеванием регистрировалась по следующей шкале:
4 - отсутствие болезни;
3 - следы - 5% заболевания на необработанных растениях;
2 - 6-25% болезни на необработанных растениях;
1 - 26-59% болезни на необработанных растениях;
0 - 60-100% болезни на необработанных растениях.
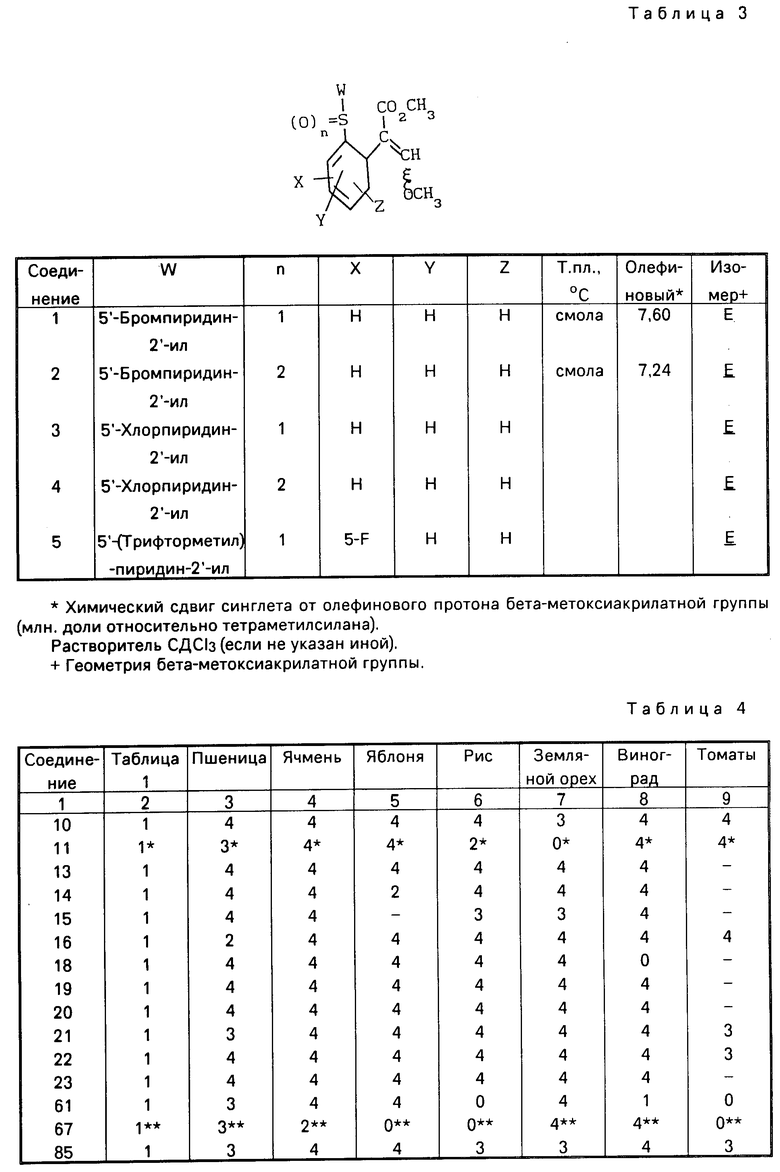
Результаты приведены в табл.4.
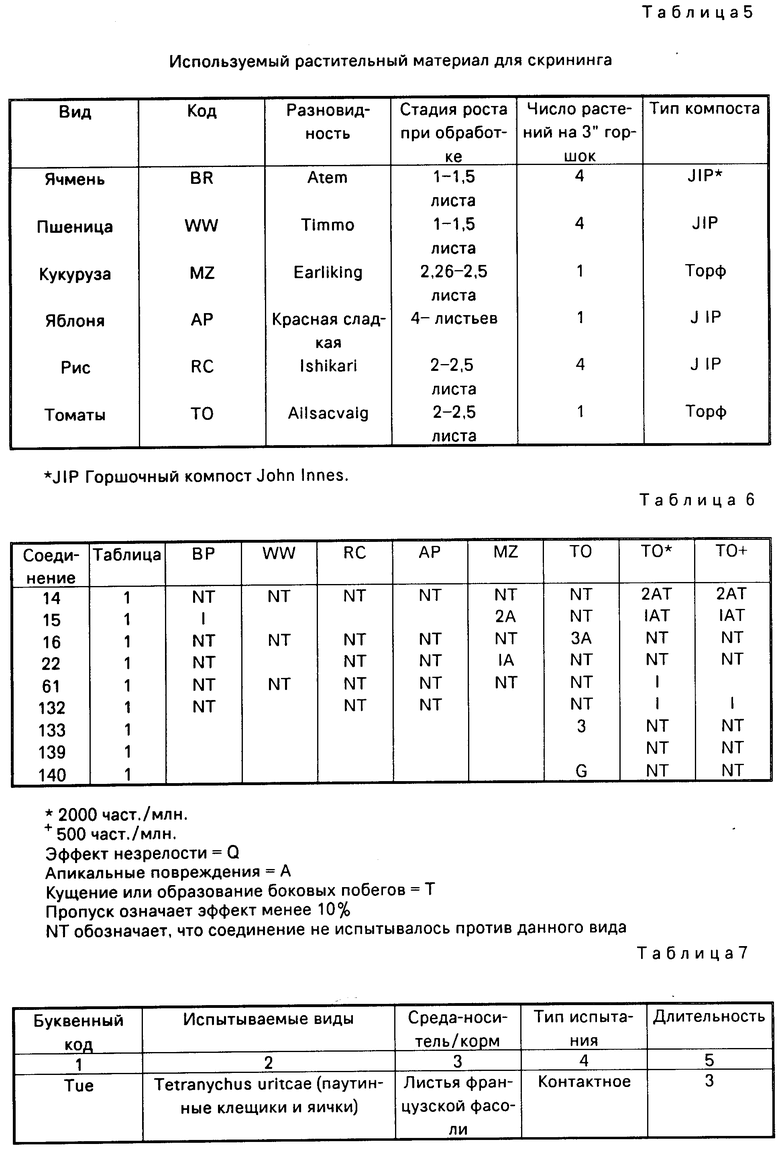
П р и м е р 12. Данный пример иллюстрирует свойства соединений 14-16, 22, 61, 132 и 138-140 (табл.1) регулировать рост растений.
Эти соединения испытывались при проведении скрининга на активность регулирования роста растений на шести видах растений. Виды растений, используемые в данном скрининге, представлены в табл.5 с указанием листовой стадии, на которой растения опрыскивались.
Препарат каждого химиката применялся в количестве 4000 ч./млн, (4 кг/га при полевом объеме 1000 л/га) с использованием гусеничного опрыскивателя и сопла SS8004E (Teejet). Дополнительные испытания проводились на томатах с использованием 2000 и 500 ч./млн.
После опрыскивания растения выращивались в теплице с температурой днем 25оС и ночью - 22оС. Исключением в данном испытании были злаковые умеренного климата, пшеница и ячмень, которые выращивались при дневных температурах 13-16оС и температурах ночью - 11-13оС. При необходимости для обеспечения средней продолжительности фотопериода 16 ч (минимум 14 ч) применялось дополнительное освещение.
После 2-6 недель нахождения в теплице в зависимости от вида и времени года растения оценивались визуально в отношении морфологических характеристик по сравнению с контрольными растениями, опрыскиваемыми бланк-препаратами без активного вещества. Результаты представлены в табл.6.
П р и м е р 13. Данный пример иллюстрирует инсектицидные свойства некоторых соединений формулы 1.
Активность каждого соединения определялась при использовании различных насекомых, клещей и нематодных вредителей. Соединение использовалось в виде жидких препаратов, содержащих 100-500 ч. на 1 млн. (по весу). Препараты приготавливались с помощью растворения соединения в ацетоне и разбавления растворов водой, содержащей 0,1% по массе смачивающего агента, продаваемого под торговым наименованием "СИНПЕРОНИК" NX, до тех пор, пока жидкие препараты не будут содержать требуемую концентрацию продукта.
Процедура испытания, применяемая в отношении каждого из вредителей, была в основном одной и той же и предусматривала помещение ряда вредителей на среду, которой было обычно растение-хозяин или корм, которым питались вредители, и обработку или среды, или вредителей или и того и другого препаратами. Смертность вредителей оценивалось через периоды, обычно варьирующиеся от одного до семи дней после обработки.
Результаты испытаний даны в табл.5 для каждого из продуктов, взятых в количестве в частях на миллион, данном во второй колонке, при этом шкала оценки смертности обозначалась как 9,5 или 0, где 9 обозначает 80-100% смертность (70-100% снижение отрастания корней по сравнению с необработанными растениями в случае Meloidogyne incognita, 5 означает 50-79% смертность (50-69% уменьшение отрастания корней в случае Meloidogyne incognita и 0 означает менее, чем 50% смертность (уменьшение отрастания корней для Meloidogyne incognita).
В табл.8 используемый организм вредитель обозначается буквенным кодом, а виды вредителей, среда или корм, и тип и длительность испытания даются в табл.7.
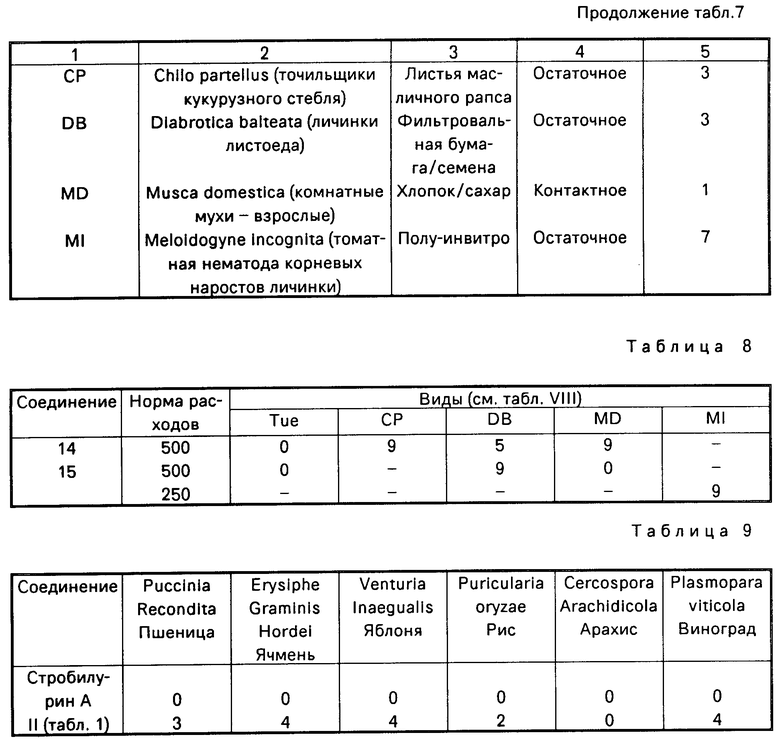
Фунгицидное действие соединений по изобретению (соединение II табл.1) сравнивалось со стробилурином А формулы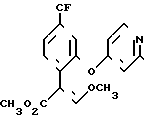
Испытание проводилось в соответствии с процедурой, описанной в примере 11, но соединения испытывались в концентрации 25 ч/млн. Соединение 11 наносили путем опрыскивания листы, а стробилурин А наносили на листву и в почву под корни. Получены результаты (табл.9) с применением оценок, которые определены в примере 16.
Эти результаты показывают, что соединение 11 табл.1 является более активным, чем стробилурин А в качестве фунгицида для растений даже в том случае, когда стробилурин А использовали путем сочетания опрыскивания листвы и пропитки корней, в то время как соединение 11 использовали только путем опрыскивания листвы.
Соединения I относятся к группе малотоксичных соединений.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных акриловой кислоты или их стереоизомеров | 1987 |
|
SU1598872A3 |
| ПРОИЗВОДНЫЕ АКРИЛОВОЙ КИСЛОТЫ | 1991 |
|
RU2039044C1 |
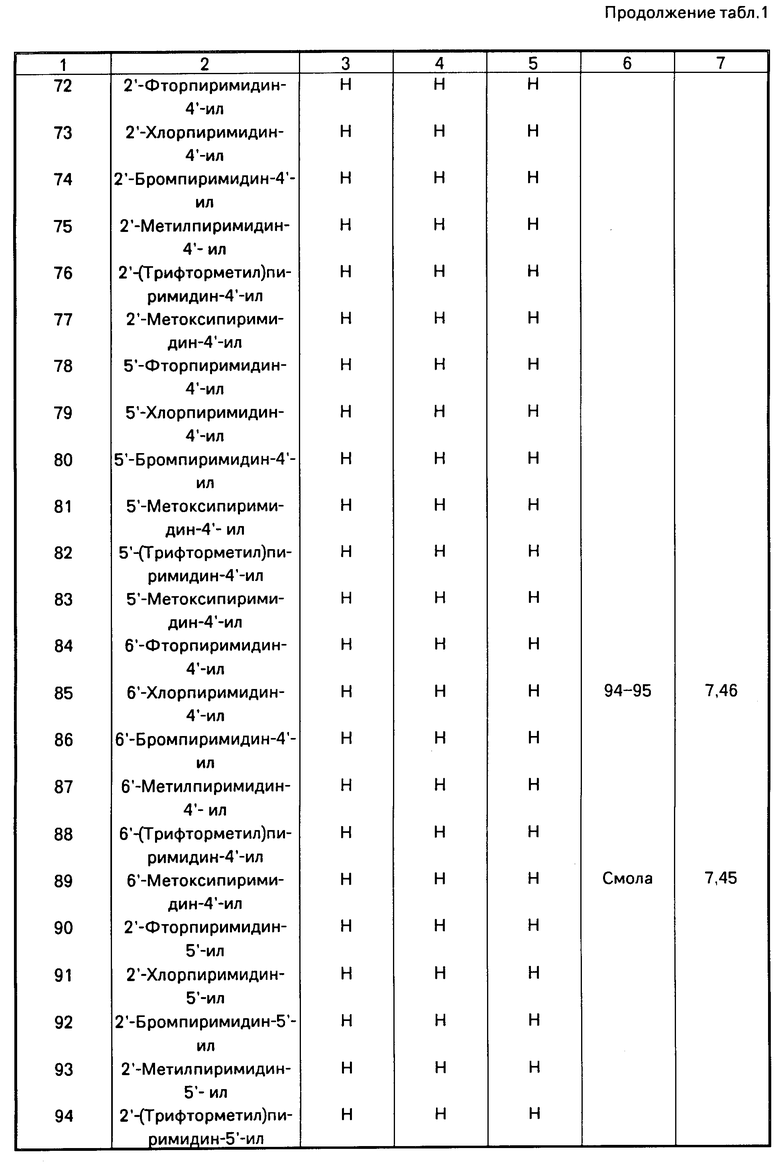
| Способ получения Е-изомеров производных акриловой кислоты | 1988 |
|
SU1665875A3 |
| ПРОИЗВОДНЫЕ ПРОПЕНОВОЙ КИСЛОТЫ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ | 1989 |
|
RU2024496C1 |
| ЗАМЕЩЕННЫЕ АЦИЛАМИНОБЕНЗАМИДЫ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2034829C1 |
| ПРОИЗВОДНЫЕ АКРИЛОВОЙ КИСЛОТЫ И ИХ СТЕРЕОИЗОМЕРЫ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2037487C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АЦИЛАМИНОБЕНЗАМИДОВ | 1991 |
|
RU2032662C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРИМИДИНА | 1990 |
|
RU2019543C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА | 1992 |
|
RU2043990C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПЕНОВОЙ КИСЛОТЫ И ИХ СТЕРЕОИЗОМЕРОВ | 1988 |
|
RU2014320C1 |
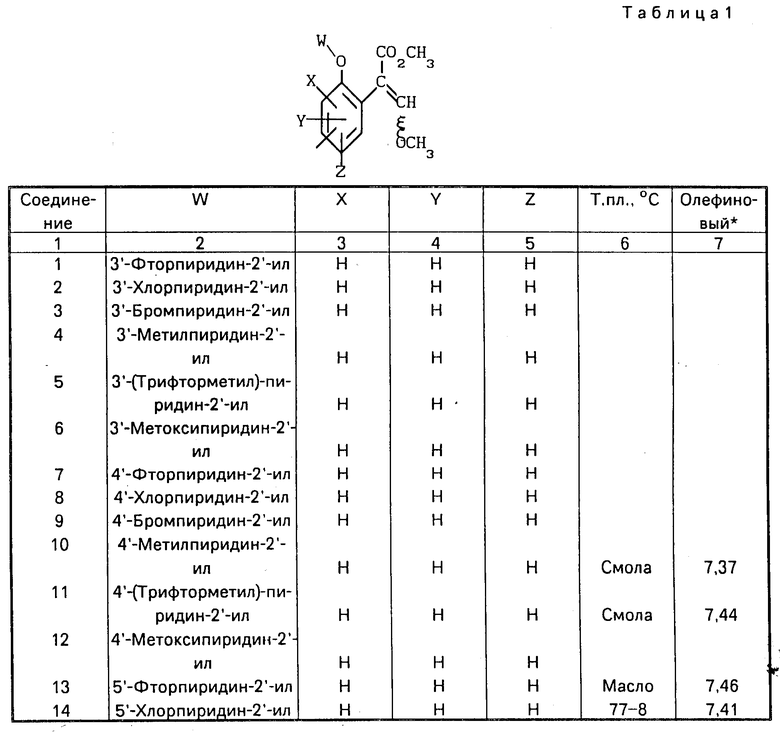
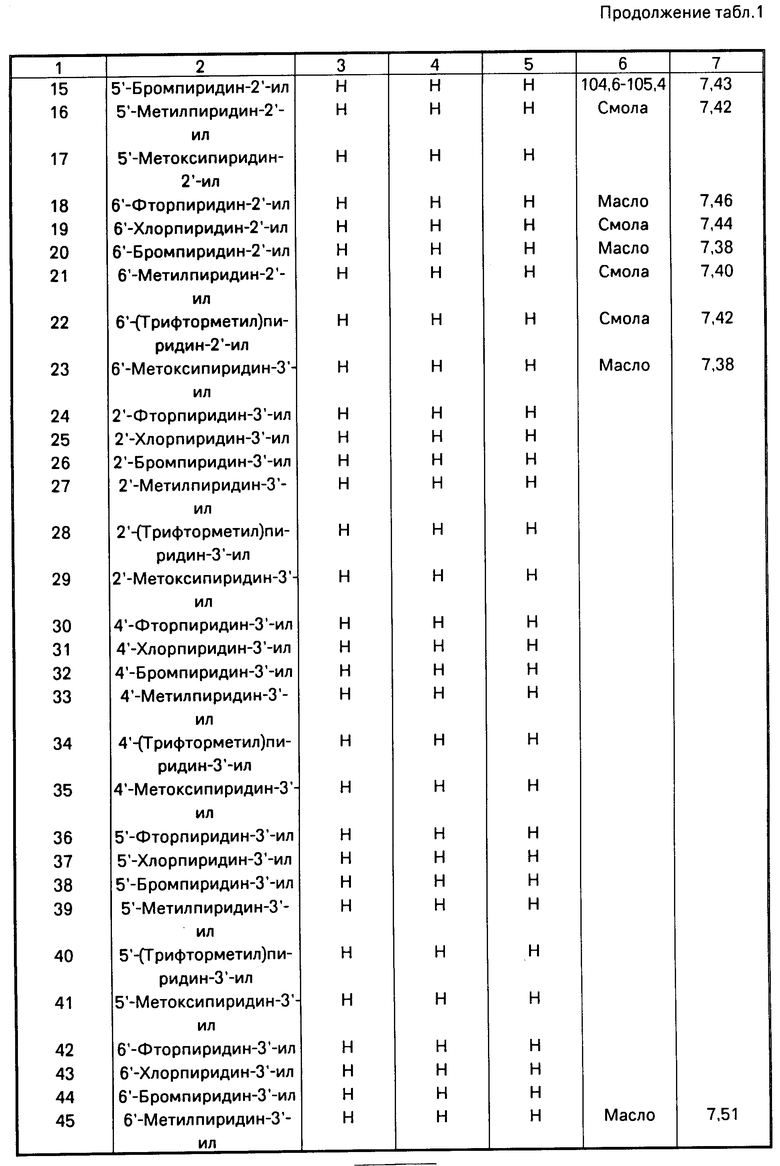
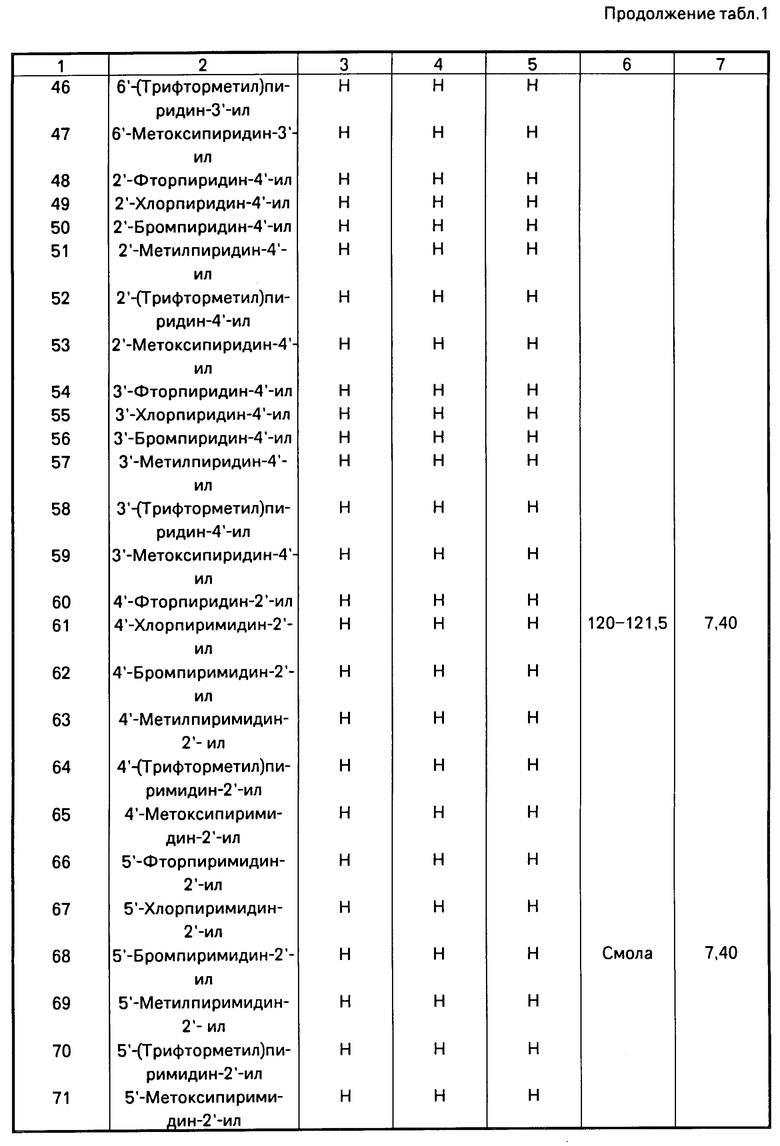
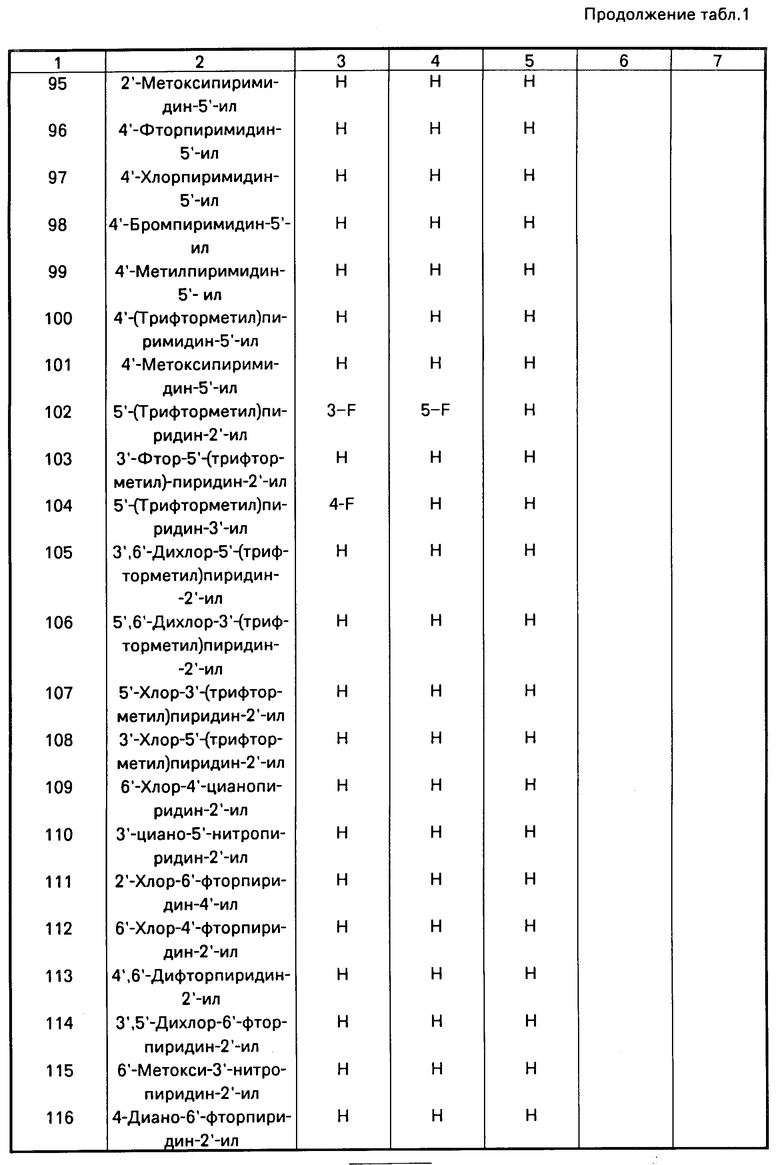
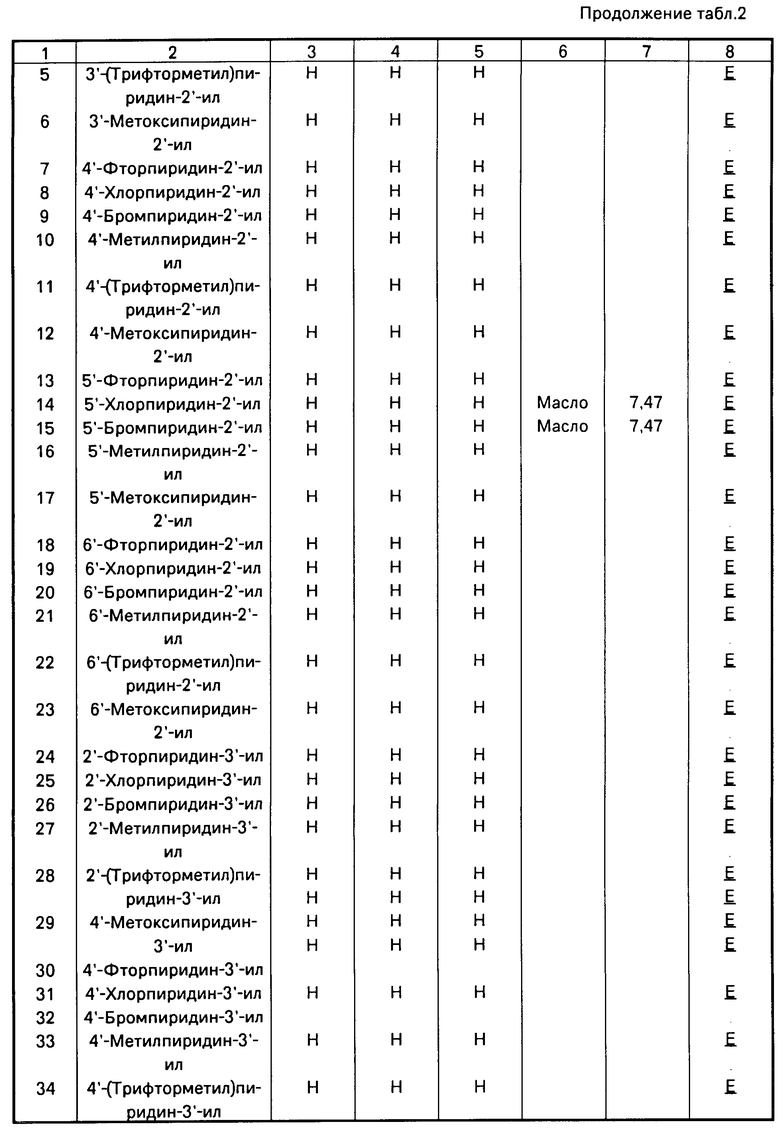
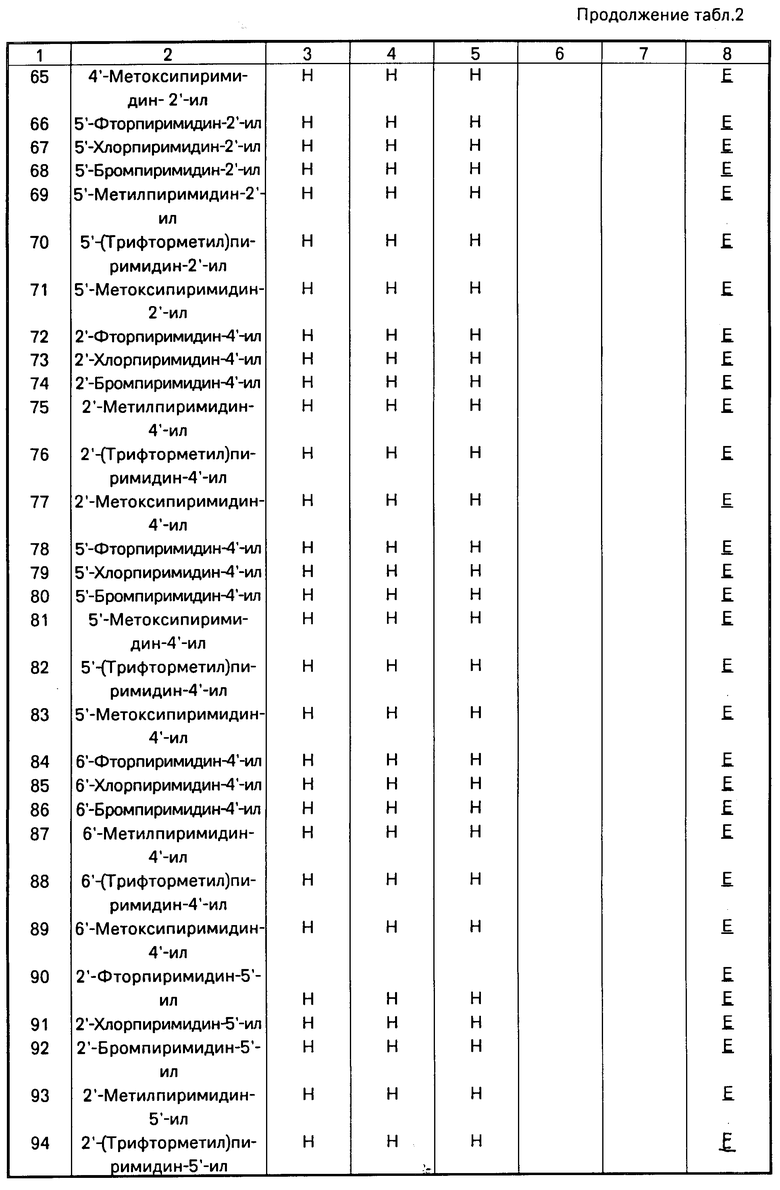
Использование: в качестве фунгицидов в сельском хозяйстве. Сущность изобретения: продукт: Е - изомеры производных акриловой кислоты общей формулы 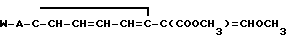 , где W - пиридинил- или пиримидинилгруппа, замещенная галогеном, C1-C4 -алкилом, который, в свою очередь, может быть замещен гомогеном, фенилом; C1-C4 -алкоксилом, феноксигруппой, которая может быть замещена 1-метоксикарбонил-2-метоксиэтенилом, галогеном, циано- или нитрогруппой, амино-, формамидо-, нитро-, циано- или N-оксидной группой, или W-хинолинил- или хиназолинилгруппа, возможно замещенная галогеном, и связанные с А одним из атомов углерода цинка, А - кислород или группа S(O)n , где n=0, или 1, или 2 при условии, что когда W - 5-трифторметилпиридинил-2, то А не является кислородом. Реагент 1: соединение общей формулы
, где W - пиридинил- или пиримидинилгруппа, замещенная галогеном, C1-C4 -алкилом, который, в свою очередь, может быть замещен гомогеном, фенилом; C1-C4 -алкоксилом, феноксигруппой, которая может быть замещена 1-метоксикарбонил-2-метоксиэтенилом, галогеном, циано- или нитрогруппой, амино-, формамидо-, нитро-, циано- или N-оксидной группой, или W-хинолинил- или хиназолинилгруппа, возможно замещенная галогеном, и связанные с А одним из атомов углерода цинка, А - кислород или группа S(O)n , где n=0, или 1, или 2 при условии, что когда W - 5-трифторметилпиридинил-2, то А не является кислородом. Реагент 1: соединение общей формулы 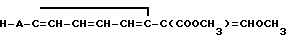 . Реагент 2: W - L, где L - галоген. Условия процесса: в присутствии основания в органическом растворителе, возможно в присутствии катализатора из числа переходного металла или его соли. 9 табл.
. Реагент 2: W - L, где L - галоген. Условия процесса: в присутствии основания в органическом растворителе, возможно в присутствии катализатора из числа переходного металла или его соли. 9 табл.
СПОСОБ ПОЛУЧЕНИЯ Е-ИЗОМЕРОВ ПРОИЗВОДНЫХ АКРИЛОВОЙ КИСЛОТЫ общей формулы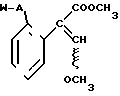
где W - пиридинил- или пиримидинилгруппа, замещенная галогеном, C1- C4-алкилом, который, в свою очередь, может быть замещен галогеном, фенилом, C1 - C4-алкоксилом, феноксигруппой, которая может быть замещена 1-метоксикарбонил-2-метоксиэтенилом, галогеном, циано- или нитрогруппой, амино-, формамидо-, нитро-, циано- или N-оксидной группой, или W - хинолинил- или хиназолинилгруппа, возможно замещенная галогеном, и связанные с А одним из атомов углерода цикла;
А - кислород или группа S(O)n, где n=0, или 1, или 2 при условии, что, когда - W-5-трифторметилпиридинил-2-, то А не является кислородом,
отличающийся тем, что соединение общей формулы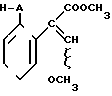
где А имеет указанные значения,
подвергают взаимодействию с соединением общей формулы
W - L,
где W имеет указанные значения;
L - галоген,
в присутствии основания в органическом растворителе, возможно в присутствии катализатора из числа переходного металла или его соли.
| Вейганд-Хильгетаг Методы эксперимента в органической химии, М.:Химия, 1968, с.340. |
