Изобретение относится к новым 7-замещенным фтор-2 β-ацетил-2 α,4 α, 5,12-тетраокси-1,2,3,4-тетрагидро-6,11-нафтацендио- нам формулы I, производным антрациклина формулы II, нафтацендионам формулы III, нафтацендионам формулы IV.
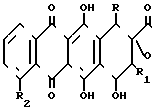 H
H (I)
(I)
(I) где X является атомом водорода или гидроксильной группой; R и R1различны и могут быть равными, являются атомом водорода или атомом фтора; R2 может быть атомом водорода или гидроксильной группой или низшей алкоксигруппой.
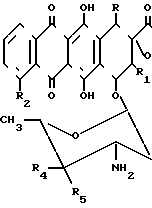 H
H (II) где X,R,R1,R2 являются такими, как это определено выше, R4 и R5 - атом водорода или гидроксил, но не могут быть одновременно гидроксилом. Антрациклины формулы II обладают противоопухолевой и противовирусной активностью.
(II) где X,R,R1,R2 являются такими, как это определено выше, R4 и R5 - атом водорода или гидроксил, но не могут быть одновременно гидроксилом. Антрациклины формулы II обладают противоопухолевой и противовирусной активностью.
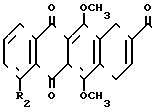 (III) где R2 является таким, как это указано выше.
(III) где R2 является таким, как это указано выше.
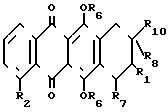 (IV) где R2 имеет указанные значения, когда R6 - метил, R7 - водород, R10 - группа
(IV) где R2 имеет указанные значения, когда R6 - метил, R7 - водород, R10 - группа  CH3 , то R8 гидроксил, R1 - фтор или R8 и R1 вместе с атомом углерода образуют оксирановый цикл; когда R6 - водород, R7 - водород, R8 - гидроксил, R1 - фтор, то R10 - группа
CH3 , то R8 гидроксил, R1 - фтор или R8 и R1 вместе с атомом углерода образуют оксирановый цикл; когда R6 - водород, R7 - водород, R8 - гидроксил, R1 - фтор, то R10 - группа 
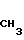 или
или 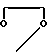 , когда R6 - водород, R7 - гидроксил, R8 - гидроксил, R1 - фтор, то R10 - группа
, когда R6 - водород, R7 - гидроксил, R8 - гидроксил, R1 - фтор, то R10 - группа  .
.
Целью изобретения является изыскание новых производных нафтацеидионов, позволяющих получить антрациклины с улучшенными свойствами, изыскание новых производных антрациклинов с улучшенными свойствами.
Поставленная цель достигается предложенными соединениями формул I, II, III, IV.
Следующие примеры иллюстрируют способ получения заявленных соединений.
П р и м е р 1. Va (2-ацетил-5,12-диметокси-1,4-дигидро-6,11-нафтацендион).
В смеси 50 мл диметилацетамида и 10 мл диоксана растворяются в атмосфере азота 29 г (193 ммоля) NaI. Растворение экзотермично: когда температура спадает до 40оС в один прием добавляется 6,6 г (97 ммолей) бутиона IV. Температура смеси доводится до 65оС и в течение периода времени в один час, по каплям добавляется раствор, состоящий из 5 г (11 ммолей) IIIа и 100 мл диметилацетамида, сохраняя температуру вблизи первоначального значения. По окончании добавления перемешивание продолжается при 65-70оС в течение еще 1 ч и смесь затем охлаждается и выливается в 600 мл воды. Полученное твердое вещество фильтруется, промывается хорошо с помощью воды и высушивается под вакуумом над P2O5, чтобы получить 3,85 г Ya. Т.пл. 209-213оС.
1Н-ЯМР, δ: 2,40 (синглет, 3Н); 3,60-3,80 (мультиплет, 4Н); 3,91 (синглет, 3Н); 3,92 (синглет, 3Н); 7,00-7,10 (мультиплет, 1Н); 7,64-7,76 (мультиплет, 2Н); 8,10-8,22 (мультиплет, 2Н). Масс-спектр m/Z (%): 362 (М+, 100); 360 (91); 347 (27); 344 (14); 331 (49); 320 (30); 319 (60).
ТСХ: н-гексан/ТГФ 2:1 Rf = 0,33.
П р и м е р 2. Vc (2-ацетил-5,7,12-триметокси-1,4-дигидро-6,11-нафтацендион).
В смеси 50 мл диметилацетамида и 10 мл диоксана растворяются в атмосфере азота 29 г (193 ммолей) NaI. Растворение является экзотермичным: когда температура спадает до 40оС в один прием добавляются 6,6 г (97 ммолей) бутинона IV. Температура смеси доводится до 65оС и, в течение 1 ч, по каплям добавляется раствор, состоящий из 5,3 г (11 молей) III и 100 мл диметилацетамида, поддерживая температуру близко к исходному значению. По окончании добавления перемешивание продолжается при 65-70оС в течение еще 1 ч, и смесь затем охлаждается и выливается в 600 мл воды. Полученное твердое вещество отфильтровывается, промывается тщательно водой и высушивается под вакуумом над P2O5, чтобы получить 3,9 г сырого вещества. Продукт реакции Vc отделяется с помощью хроматографии на силикагелевой колонке, элюируя с помощью н-гексана/ТГФ 2:1.
Получают 1,9 г чистого продукта реакции.
Масс-спектр, m/Z (%) 392 (М+, 100).
ТСХ: н-гексан/ТГФ 2:1 Rf = 0,3.
П р и м е р 3. VIa (2-ацетил-2,3-эпокси-5,12-диметокси-1,2,3,4-тетрагидро-6,11-наф- тацендион).
В 400 мл хлороформа в течение 7 ч кипятят с обратным холодильником в атмосфере азота 3,0 г (8,3 ммолей) Va и 18 г 55%-ной мета-хлорпербензойной кислоты (57 ммолей). Реакционная смесь, охлажденная до комнатной температуры, обрабатывается 10%-ным раствором бисульфита натрия (2 х 100 мл), затем 5%-ным раствором бикарбоната натрия (3 х 100 мл) и наконец водой. Органическая фаза высушивается над сульфатом натрия и растворитель выпаривается. Сырой продукт реакции очищается путем суспендирования его в кипящем четыреххлористом углероде, охлаждается и отфильтровывается и твердое вещество промывается тщательно с помощью того же растворителя, чтобы получить 850 мг VIа с 90%-ной чистотой (выход 30%). Т.пл. 241-245оС (разл.).
1Н ЯМР, δ: 2,16 (синглет, 3Н); 3,00-3,09, 3,55-4,10 (мультиплет, 5Н); 3,88 (синглет, 3Н); 3,89 (синглет, 3Н); 7,65-7,80 (мультиплет, 2Н); 8,10-8,20 (мультиплет, 2Н).
Масс-спектр, m/Z (%): 378 (М+, 70); 347 (26); 346 (41); 335 (61); 291 (50); 189 (59); 176 (68); 165 (100).
П р и м е р 4. VIc (2-ацетил-2,3-эпокси-5,7,12-триметокси-1,2,3,4-тетрагидро-6,11- -нафтацендион).
В 250 мл хлороформа кипятят с обратным холодильником в атмосфере азота в течение 7 ч 1,9 г (4 ммолей) Vс и 8,7 г 55%-ной мета-хлорпербензойной кислоты (27,5 ммолей). Реакционная смесь, охлажденная до комнатной температуры, обрабатывается 10%-ным раствором бисульфита натрия (2 х 100 мл), затем 5% -ным раствором бикарбоната натрия (3 x100 мл) и наконец водой. Органическая фаза высушивается над сульфатом натрия и растворитель выпаривается. Сырой продукт реакции очищается путем суспендирования его в кипящем четыреххлористом углероде, охлаждается и отфильтровывается и твердое вещество промывается тщательно с тем же самым растворителем, чтобы получить 600 мг VIс с 90%-ной чистотой. Масс-спектр, m/Z (%): 408 (М+, 80).
П р и м е р 5. VIIa (2 β-ацетил-2α -окси- 3 β-фтор-5,12-диметокси-1,2,3,4-тетрагидро-6,11-нафтацендион).
В полиэтиленовую колбу вводятся 1,4 г (3,7 ммолей) VIa и 50 мл 70%-ной HF в пиридине и подвергаются реакции в течение 24 ч при комнатной температуре в атмосфере азота. Реакционная смесь выливается на 600 г льда, с энергичным перемешиванием затем полученное твердое вещество отфильтровывается и промывается повторно водой. Это сырое вещество высушивается под вакуумом над P2O5 и суспендируется в 100 мл кипящего ацетона; суспензия оставляется охлаждаться, а затем фильтруется, причем твердое вещество промывается малым количеством ацетона для того, чтобы получить 880 мг VIIa. Т. пл. 241-245оС (разл.) (выход: 60%).
1Н ЯМР, δ : 2,49 (дублет, 5IHF = 2,6 Гц, 3Н); 2,90-3,45 (мультиплет, 4Н); 3,90 (синглет, 3Н); 3,92 (синглет, 3Н); 4,25 (синглет, 1Н, OH); 4,76 (двойной триплет, 2IHF = 49,6 Гц, 3IHH = 2,6 Гц); 7,65-7,80 (мультиплет, 2Н); 8,10-8,25 (мультиплет, 2Н). Масс-спектр, m/Z (%): 398 (М+, 100); 380 (12); 255 (36); 335 (30); 291 (47).
П р и м е р 6. VIIc (2 β-ацетил-2 α-окси-3β -фтор-5,7,12-триметокси-1,2,3,4-тетрагидро--6,11-нафтацендион).
В полиэтиленовую колбу вносится 1 г (2,5 ммолей) VIс и 30 мл 70%-ного HF в пиридине и подвергаются реакции в течение 24 часов при комнатной температуре в атмосфере азота. Реакционная смесь выливается на 300 г льда с энергичным перемешиванием, затем полученное твердое вещество отфильтровывается и промывается повторно водой. Это сырое вещество высушивается под вакуумом над P2O5 и суспендируется в 50 мл кипящего ацетона; суспензию оставляют охлаждаться и затем фильтруют, причем твердое вещество промывается малым количеством ацетона для того, чтобы получить 600 мг УПС.
Масс-спектр, m/Z (%): 428 (М+, 100).
П р и м е р 7. VIIIa (2 β-ацетил-2 α,5,12-триокси-3β-фтор-1,2,3,4-тетрагидро-6,11- -нафтацендион).
В 15 мл безводного хлористого метилена суспендируется в атмосфере азота 82 мг (0,21 ммолей) VIIа и, после доведения температуры до -65оС с помощью бани ацетона с сухим льдом, добавляются в течение 5 мин. периода времени 3 мл 1 М раствора треххлористого бора (3 ммоля) в хлористом метилене. Смесь перемешивается в течение дополнительных 30 мин при -65оС прежде, чем медленно добавлять 4 мл метанола и позволять реакционной смеси возвратиться до комнатной температуры. Путем выпаривания растворителя в конце концов получаются 79 мг (100%) VIIIa. Т.пл. 247-253оС.
1Н-ЯМР, (ДМСО) δ: 2,35 (синглет, 3Н); 2,85-3,25 (мультиплет, 4Н); 5,26 (дублет, широкий, 2IHF = 47,6 Гц); 6,34 (синглет, широкий, 1Н, OH); 7,85-8,00 (мультиплет, 2Н); 8,15-8,30 (мультиплет, 2Н); 13,24 (синглет, 1Н); 13,28 (синглет, 1Н).
Масс-спектр, m/Z (%): 370 (М+, 24); 352 (55); 332 (63); 327 (33); 307 (100); 187 (80).
П р и м е р 8. VIIIc (2 β-ацетил-2 α,5,12-триокси-3-фтор-7-метокси-1,2,3,4-тетрагид- ро-6,11-нафтацендион).
В 15 мл безводного хлористого метилена суспендируются в атмосфере азота 90 мг (0,21 ммоля) VIIс и после доведения температуры до -65оС с помощью бани ацетона и сухого льда добавляются в течение 5 мин периода времени 3 мл 1 М раствора треххлористого бора (3 ммоля) в хлористом метилене. Смесь перемешивается в течение еще 30 мин при -65оС прежде, чем медленно добавляется 4 мл метанола и оставить реакционную смесь возвратиться к комнатной температуре. Выпариванием растворителя в конце концов получают 84 мг (100%) VIIIс.
Масс-спектр, m/Z (%): 400 (М+, 35).
П р и м е р 9. IXa (2 β-[2-метил-1,3-диоксоланил-2-ил]-2 α,5,12-триокси-3 β-фтор-1,2,3,4-тетрагидро-6,11-нафтацендион).
В 250-миллилитровую колбу с ловушкой Дина-Старка для реакции с нагреванием с обратным холодильником в атмосфере азота вводятся следующие соединения: 320 мг (0,86 ммоля) VIIIa и 7,77 г (7 мл, 124 ммолей) этиленгликоля в 100 мл бензола, в присутствии 70 мг (0,4 ммоля) пара-толуолсульфоновой кислоты, удаляя азеотроп из реакционной среды по мере того, как он отгоняется. По окончании растворитель концентрируется до малого объема и полученное твердое вещество фильтруется, промывая его последовательно малым количеством бензола, этанола, воды, 5%-ным водным раствором бикарбоната натрия и снова воды. Наконец его высушивают под вакуумом при 80оС, чтобы получить 283 мг (0,68 ммоля) IXa: Т.пл. 237-240оС. (Выход 79,1%). Т.пл. 239-241оС (толуол).
ТСХ: CCl4/этилацетат 2:1 Rf = 0,54.
П р и м е р 10. IXc (2 β-[2-метил-1,3-диоксоланил-2-ил]-2 α ,5,12-триокси-3 β-фтор-7-метокси-1,2,3,4-тетрагидро-6,11-нафтацен- дион).
В колбу объемом 250 мл с ловушкой Дина-Старка для реакции с нагреванием с обратным холодильником в атмосфере азота вводятся следующие соединения: 350 мг (0,88 ммоля) VIIIс и 7,77 г (7 мл, 124 ммолей), этиленгликоля в 100 мл бензола, в присутствии 70 мг (0,4 ммоля) пара-толуолсульфоновой кислоты, удаляя азеотроп из реакционной среды по мере того, как он отгоняется. По окончании растворитель концентрируется до малого объема и полученное твердое вещество отфильтровывается, промывая его последовательно малым количеством бензола, этанола, воды, 5%-ного водного раствора бикарбоната натрия и снова воды. Наконец его высушивают под вакуумом при 80оС, чтобы получить 310 мг (0,68 ммоля) IXc.
ТСХ: ССl4/этилацетата 2:1 Rf = 0,5.
П р и м е р 11. Xa (2 β-[2-метил-1,3-диоксоланил-2-ил]2 α, 4 α, 5,12-тетрокси-3 β-фтор-1,2,3,4-тетрагидро-6,11-нафтацендион).
В 200 мл четыреххлористого углерода, в атмосфере азота суспендируются 880 мгл VIIIa вместе с 880 мг карбоната калия. Добавляется раствор 500 мг брома в 20 мл четыреххлористого углерода. Облучением с помощью 500-ваттной лампы смесь доводится до слабого кипения с обратным холодильником. После примерно 5 мин продукт растворяется. Реакция останавливается после 1 ч. Смесь разбавляется с помощью 200 мл хлороформа и промывается сначала бикарбонатом, а затем водой. Она делается безводной (обезвоживается) над сульфатом натрия и растворитель выпаривается. Остаток хроматографируется на Si, элюируя сначала смесью 3:1 четыреххлористого углерода и этилацетата, чтобы удалить остаточный исходный продукт и побочные продукты. Затем он (остаток) элюируется этилацетатом до тех пор, пока не получится 300 мг Xa.
Н-ЯМР, δ: 1,53 (дублет, 5JHF = 2,2 Гц, 3Н); 2,95 (дублет 4JHF = 2,2, 1H, OH2); 3,02 (Н1акс, часть В ABMX-спектра, 2JHH = =18,8 Гц, 4JHF= 2,6 Гц); 3,26 (Н1 экв, часть А ABMX-спектра, 2JHH = 18,8 Гц, 4JHH = 0,6 Гц, 4JHF = 2,7 Гц); 4,02-4,18 (мультиплет, 4Н); 3-70 (дублет, 3JHH = 10,3 Гц, 1Н, OH4); 5,17 (тройной дублет, 3JHH = 10,3 Гц, 3IHH = 2,3 Гц, 3JHF = 14,1 Гц, 1Н); 5,18 (тройной дублет, 2JHF = 45,6 Гц, 3JHH = 2,3 Гц, 4JHH = 0,6 Гц, 1Н); 7,80-7,90 (мультиплет, 2Н); 8,30-8,43 (мультиплет, 2Н); 13,41 (синглет, 1Н); 13,63 (синглет, 1Н).
П р и м е р 12. Xc (2 β-[2-метил-1,3-диоксоланил-2-ил]-2 α , 4α , 5,12-тетраокси-3 β-фтор-7-метокси-1,2,3,4-тетрагидро-6,11- нафтацендион).
В 200 мл четыреххлористого углерода суспендируются в атмосфере азота 900 мг VIIIc вместе с 880 мг карбоната калия. Добавляется раствор 500 мг брома в 20 мл четыреххлористого углерода. Облучением с помощью 500-ваттной лампы смесь доводится до слабого кипения с обратным холодильником. После примерно 5 мин продукт растворяется. Реакция останавливается после 1 ч. Смесь разбавляется с помощью 200 мл хлороформа и промывается сначала бикарбонатом, а затем водой. Она обезвоживается над сульфатом натрия и растворитель выпаривается. Остаток хроматографируется на силикагеле, элюируя сначала 3: 1 смесью четыреххлористого углерода и этилацетата, чтобы удалить остаточный исходный продукт и побочные продукты. Затем он элюируется этилацетатом до тех пор, пока не получится 330 мг Хс.
П р и м е р 13. XIa (2 β-ацетил-2 α,4 α, 5,12-тетраокси-3 β-фтор-1,2,3,4-тетрагидро-6,11-нафтацендион).
В 5 мл безводного хлористого метилена растворяются в атмосфере азота 82 мг (0,19 ммоля) Xa и полученный раствор охлаждается до 0оС. Затем добавляются 5 мл (5 ммоля) 1М раствора BCl3 в CH2Cl2 и ледяная баня удаляется. Смесь перемешивается при комнатной температуре в течение еще 2 дней, затем осторожно добавляются 15 мл воды и, после разбавления с помощью 20 мл CH2Cl2, смесь промывается водой (3 x 20 мл). Органическая фаза высушивается над сульфатом натрия, растворитель выпаривается и сырое вещество очищается с помощью силикагелевой колоночной хроматографии (элюент: 4:1 смесь хлороформа и ацетона). Получают 40 мг XIa. (Выход 50%).
1Н-ЯМР, δ: 2,54 (дублет, 5JHF = 2,2 Гц, 3Н); 3,18 (Н1акс, 2JHH = 18,5 Гц, 4JHF = 2,4 Гц); 3,3 (Н1экв, 2JHH = 18,5 Гц, 4JHF = 3,1 Гц); 3,65 (уширенный дублет, 4JHF = 2,9 Гц, 1Н, OH2); 4,6 (уширенный синглет, 1Н, OH4); 4,98 (двойной дублет, 2JHF = 46,2 Гц, JHHapp = =2,2 Гц, 1Н); 5,18 (уширенный дублет, 3JHF = =13 Гц, 1Н); 7,80-7,90 (мультиплет, 2Н); 8,20-8,43 (мультиплет, 2Н); 13,29 (синглет, 1Н); 13,53 (синглет, 1Н).
П р и м е р 14. XIс (2 β-ацетил-2 α,4α,- 5,12-тетраокси-3β-фтор-7-метокси- 1,2,3,4 -тетрагидро-6,11-нафтацендион).
В 5 мл безводного СН2Сl2 растворяются в атмосфере азота 88 мг (0,19 ммоля) Xc и полученный раствор охлаждается до 0оС. Затем добавляются 5 мл (5 ммоля) 1М раствора BCl3 в CH2Cl2 и ледяная баня удаляется. Смесь перемешивается при комнатной температуре в течение еще 2 дней, затем осторожно добавляются 15 мл воды и после разбавления с 20 мл CH2Cl2смесь промывается водой (3 x 20 мл). Органическая фаза высушивается над сульфатом натрия, растворитель выпаривается и сырое вещество очищается с помощью колоночной хроматографии на силикагеле (элюент: 4:1 смесь хлороформа и ацетона). Получают 50 мг XIc.
Масс-спектр, m/Z (%): 462 (М+, 100).
П р и м е р 15. (4-Деметокси-8-фтор-дауномицин).
В растворе 45 мг XIа в 8,3 мл безводного хлористого метилена и 7 мл безводного эфира суспендируется 142 мг (-)-3-N-трифторацетил-1,4-бис-(О-пара-нитробензоил)-1-дау- нозамина, в атмосфере азота, в присутствии 600 мг гранулированных молекулярных сит 4  . Смесь охлаждается до 0оС и добавляется 0,08 мл триметилсилилтрифлата. После 3 ч реакционная смесь обрабатывается с 50 мл этилацетата и 100 мл насыщенного раствора бикарбоната натрия и затем органическая фаза промывается раствором хлористого натрия. После обезвоживания растворитель выпаривается и остаток очищается с помощью хроматографии на силикагеле, элюируя смесь 4:1 хлорофоpма и ацетона. Получается 80 мг продукта реакции, который растворяется в 0,6 мл хлористого метилена и 37 мл метанола. Раствор охлаждается до 0оС и в атмосфере азота добавляется 1,2 мл 0,1М раствора гидроокиси натрия. Раствор перемешивается в течение 30 мин, а затем добавляется ледяная уксусная кислота до тех пор, пока окраска раствора не станет бледнооранжевой. Раствор обрабатывается с 60 мл этилацетата и 60 мл раствора хлористого натрия. Органическая фаза промывается дважды раствором хлористого натрия, затем обезвоживается и выпаривается. Остаток растворяется в 15 мл 0,1М раствора гидроокиси натрия при 0оС и перемешивается в течение 20 мин в атмосфере азота, pH раствора доводится до 8 с помощью 5М HCl, а затем раствор экстрагируется повторно хлороформом. Органическая фаза промывается водой, обезвоживается и концентрируется под вакуумом. Остаток растворяется в малом количестве хлороформа и метанола (9:1), раствор HCl в 0,25 М метанола добавляется до тех пор, пока pH не станет равным 3,5, а затем добавляется эфир до тех пор, пока не выпадет хлоргидрат требуемого продукта реакции.
. Смесь охлаждается до 0оС и добавляется 0,08 мл триметилсилилтрифлата. После 3 ч реакционная смесь обрабатывается с 50 мл этилацетата и 100 мл насыщенного раствора бикарбоната натрия и затем органическая фаза промывается раствором хлористого натрия. После обезвоживания растворитель выпаривается и остаток очищается с помощью хроматографии на силикагеле, элюируя смесь 4:1 хлорофоpма и ацетона. Получается 80 мг продукта реакции, который растворяется в 0,6 мл хлористого метилена и 37 мл метанола. Раствор охлаждается до 0оС и в атмосфере азота добавляется 1,2 мл 0,1М раствора гидроокиси натрия. Раствор перемешивается в течение 30 мин, а затем добавляется ледяная уксусная кислота до тех пор, пока окраска раствора не станет бледнооранжевой. Раствор обрабатывается с 60 мл этилацетата и 60 мл раствора хлористого натрия. Органическая фаза промывается дважды раствором хлористого натрия, затем обезвоживается и выпаривается. Остаток растворяется в 15 мл 0,1М раствора гидроокиси натрия при 0оС и перемешивается в течение 20 мин в атмосфере азота, pH раствора доводится до 8 с помощью 5М HCl, а затем раствор экстрагируется повторно хлороформом. Органическая фаза промывается водой, обезвоживается и концентрируется под вакуумом. Остаток растворяется в малом количестве хлороформа и метанола (9:1), раствор HCl в 0,25 М метанола добавляется до тех пор, пока pH не станет равным 3,5, а затем добавляется эфир до тех пор, пока не выпадет хлоргидрат требуемого продукта реакции.
ТСХ: (свободное основание) Rf = 0,52 (CHCl3/MeOH/H2O = 13:6:1).
П р и м е р 16. 8-Фтор-дауномицин.
В растворе 48 мг XIс в 8,3 мл безводного хлористого метилена и 7 мл безводного эфира суспендируется 142 мг (-)-3-N-трифторацетил-1,4-бис-(О-пара-нитробензоил)-1-да-унозамина, в атмосфере азота, в присутствии 600 мг гранулированных молекулярных сит размером пор 4  . Смесь охлаждается до 0
. Смесь охлаждается до 0 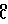 и добавляется 0,08 мл триметилсилил-трифлата. После 3 ч реакционная смесь обрабатывается с 50 мл этилацетата и 100 мл насыщенного раствора бикарбоната натрия, а затем органическая фаза промывается раствором хлористого натрия. После обезвоживания растворитель выпаривается и остаток очищается с помощью хроматографии на силикагеле, элюируя 4: 1 - смесью хлороформа и ацетона. Получают 85 мг продукта реакции, который растворяется в 0,6 мл хлористого метилена и 37 мл метанола. Раствор охлаждается до 0оС и, в атмосфере азота добавляется 1,2 мл 0,1М раствора гидроокиси натрия. Раствор перемешивается в течение 30 мин, а затем ледяная уксусная кислота добавляется до тех пор, пока окраска раствора не станет бледно-оранжевой. Раствор обрабатывается с 60 мл этилацетата и 60 мл раствора хлористого натрия. Органическая фаза промывается дважды раствором хлористого натрия, затем обезвоживается и выпаривается. Остаток растворяет в 15 мл 0,1М раствора гидроокиси натрия при 0оС и перемешивается в течение 20 мин в атмосфере азота. pH раствора доводится до 8 с помощью 5 М HCl, а затем раствор экстрагируется повторно хлороформом. Органическая фаза промывается водой, обезвоживается и концентрируется под вакуумом. Остаток растворяется в малом количестве 9:1 смеси хлороформа и метанола, добавляется HCl в 0,25М метаноле до тех пор, пока pH не станет 3,5, а затем эфир добавляется до тех пор, пока не выпадет в осадок хлоргидрат целевого продукта реакции.
и добавляется 0,08 мл триметилсилил-трифлата. После 3 ч реакционная смесь обрабатывается с 50 мл этилацетата и 100 мл насыщенного раствора бикарбоната натрия, а затем органическая фаза промывается раствором хлористого натрия. После обезвоживания растворитель выпаривается и остаток очищается с помощью хроматографии на силикагеле, элюируя 4: 1 - смесью хлороформа и ацетона. Получают 85 мг продукта реакции, который растворяется в 0,6 мл хлористого метилена и 37 мл метанола. Раствор охлаждается до 0оС и, в атмосфере азота добавляется 1,2 мл 0,1М раствора гидроокиси натрия. Раствор перемешивается в течение 30 мин, а затем ледяная уксусная кислота добавляется до тех пор, пока окраска раствора не станет бледно-оранжевой. Раствор обрабатывается с 60 мл этилацетата и 60 мл раствора хлористого натрия. Органическая фаза промывается дважды раствором хлористого натрия, затем обезвоживается и выпаривается. Остаток растворяет в 15 мл 0,1М раствора гидроокиси натрия при 0оС и перемешивается в течение 20 мин в атмосфере азота. pH раствора доводится до 8 с помощью 5 М HCl, а затем раствор экстрагируется повторно хлороформом. Органическая фаза промывается водой, обезвоживается и концентрируется под вакуумом. Остаток растворяется в малом количестве 9:1 смеси хлороформа и метанола, добавляется HCl в 0,25М метаноле до тех пор, пока pH не станет 3,5, а затем эфир добавляется до тех пор, пока не выпадет в осадок хлоргидрат целевого продукта реакции.
ТСХ: (свободное основание) Rf = 0,49 (СНСl3/MeOH/H2O = 13:6:1).
П р и м е р 17. XIIIc (1-фтор-2 β-ацетил-2γ, 4γ,5,12-тетраокси-1,2,3,4-тетрагидро-6,11- нафтацендион).
При комнатной температуре с перемешиванием обрабатываются 1 г XIIс с 15 мл 70% HF в пиридине (реагент Олаха). После 1 ч смесь выливается на лед, продукт реакции экстрагируется хлористым метиленом и органическая фаза тщательно промывается водой. Органическая фаза обезвоживается, растворитель выпаривается и продукт реакции очищается с помощью хроматографии на силикагеле, элюируя 4:1 смесью хлороформа и ацетона.
ТСХ (Хлороформ-ацетон 4:1):Rf = 0,47.
1Н-ЯМР, δ: 1,53 (дублет, 5JHF = 2,2 Гц, 3Н); 2,95 (дублет, 4JHF = 2,2 Гц, 1Н, OH2); 3,02 (Н1акс, часть В ABMX-спектра, 2JHH = =18,8 Гц, 4JHF = 2,6 Гц); 3,26 (Н1экв, часть А ABMX-спектра, 2JHH = 18,8 Гц, 4JHH= 0,6 Гц, 4JHF = 2,7 Гц); 4,02-4,18 (мультиплет, 4Н); 3,70 (дублет, 3JHH= 10,3 Гц, 1Н, OH4); 5,17 (тройной дублет, 3JHH = 10,3 Гц, 3JHH = =2,3 Гц, 3JHF = 14,1 Гц, 1Н); 5,18 (тройной дублет, 2JHF = 45,6 Гц, 3JHH = 2,3 Гц, 4JHH = =0,6 Гц, 1Н); 7,80-7,90 (мультиплет, 2Н); 8,30-8,43 (мультиплет, 2Н); 13,41 (синглет, 1Н); 13,63 (синглет, 1Н).
П р и м е р 18. (10-Фтор-4'-эпи-дауномицин).
В 24 мл безводного хлористого метилена и 20 мл безводного эфира растворяются 280 мг (0,60 ммоля) (-)-3-N-(аллилокси)-карбонил-1-акозамина и 110 мг XIIIc, в атмосфере азота, в присутствии гранулированных молекулярных сит размером пор 4 А. Смесь охлаждается до 0оС и при перемешивании добавляется 0,22 мл О-триметилсилил трифторметансульфоната. После 2 ч реакционная смесь разбавляется хлористым метиленом и органическая фаза промывается насыщенным раствором бикарбоната натрия, а затем водой. Она обезвоживается и растворитель выпаривается.
Остаток растворяется в 30 мл этилацетата, а затем добавляются 40 мг тетракис-(трифенилфосфин) палладия, 40 мг трифенилфосфина и 350 мг 2-этилгексановой кислоты, в атмосфере азота и при комнатной температуре. Раствор перемешивается в течение 24 ч, затем разбавляется хлористым метиленом и вслед за этим промывается насыщенным раствором бикарбоната натрия, а затем водой. Раствор обезвоживается и растворитель выпаривается. Остаток растворяется в малом количестве 9: 1 смеси хлороформа и метанола, 0,25М HCl в метаноле добавляется до тех пор, пока pH не станет 3,5, а затем эфир добавляется до тех пор, пока не выпадет в осадок хлоргидрат целевого продукта реакции.
ТСХ (хлороформ/ацетон 6:1): Rf = 0,2.
П р и м е р 19. (10-Фтор-4'-эпи-доксорубицин).
Соединение, полученное по примеру 18, превращают в 10-фтор-4'-эпидоксорубицин, выделенный в виде хлоргидрата с выходом примерно 60%.
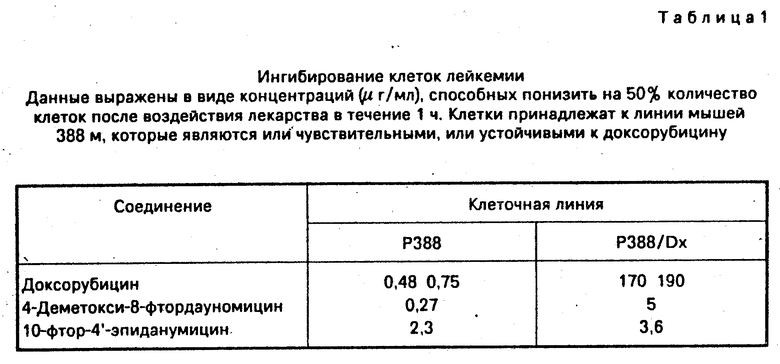
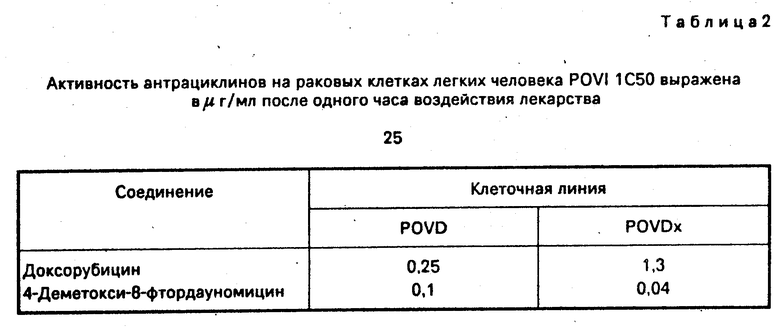
Соединения, которые составляют цель изобретения, были оценены "ин витро" на культурах опухолевых клеток человека и "ин виво" против трансплантируемых опухолей мышей. Данные приведены в табл.1-4.
Соединения показывают большую активность, чем даунорубицин и доксорубицин, используемые в качестве контрольных веществ.
П р и м е р 19бис. 100 мг высушенного 8-фтор-дауномицин-гидрохлорида, полученного в примере 16, растворяют в смеси метанола и диоксана и обрабатывают раствором брома в хлороформе при 0-5оС. После 5 ч продукт осаждают с помощью добавления смеси диэтилового эфира и н-гексана. Полученное красное твердое вещество растворяют в смеси ацетона и воды, и обрабатывают натриевой солью муравьиной кислоты, до тех пор, пока анализ с помощью тонкослойной хроматографии не покажет конец реакции. Продукт экстрагируют хлороформом и выделяют как гидрохлорид.
Масс-спектр m/Z: 561 (М+, свободное основание).
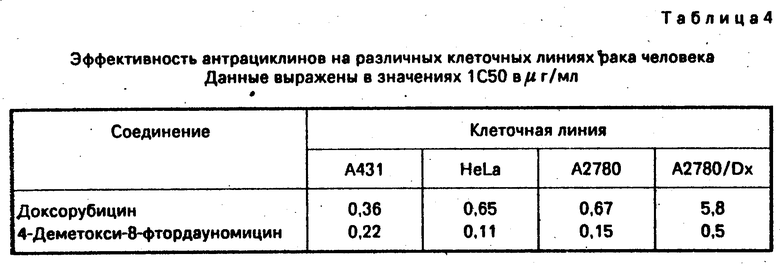
Оценку 8-фтор-замещения в молекулярной структуре типа дауномицин-доксорубицин продолжили на других клеточных линиях опухоли человека, включая, среди других, спиноклеточную (spinocellular) карциному (линия А431), опухоль шейки матки (линия Hela) и яичниковый рак (А2780). Результаты представлены в табл.IV.
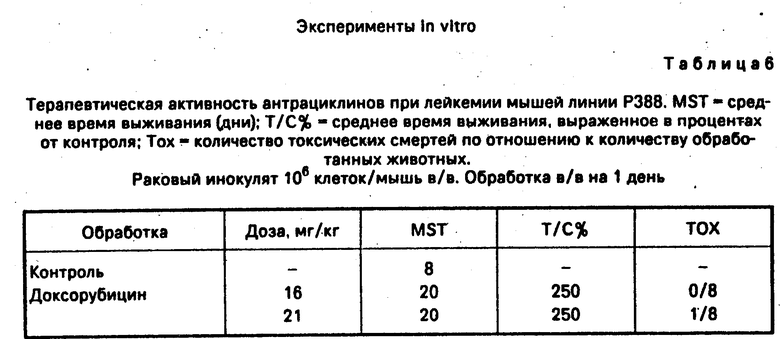
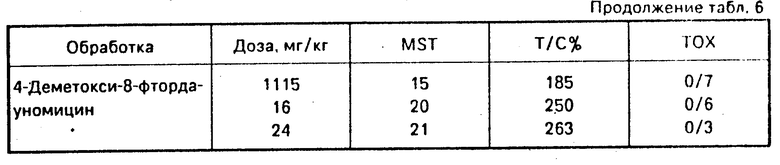
Другие эксперименты in vitro приведены в табл.5 и 6.

Использование: в медицине в качестве противоопухолевых препаратов. Сущность изобретения: продукт-производные нафтацендионов ф-л 1, 2, 3, производные антрациклина ф-лы 4. Реагент 1: 1,4-диметокси-2,3-бис(бромметил)-антрахинон. Реагент 2: 3-бутин-2-он. Реагент 3: пероксидное соединение. Реагент 4: нуклеофильный фтор. Реагент 5: фенил, содержащий гидроксил. Реагент 6: нафтацендион. Реагент 7: бром. Условия реакции: циклизация, эпоксидирование, деметилирование, бромирование. Структура соединений ф-л 1, 2, 3, 4. 4 с.п. ф-лы, 6 табл.
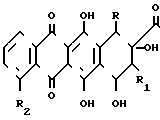

где R и R1 - различные, водород или фтор;
R2 - водород или низшая алкоксигруппа;
Х - водород, гидроксильная группа,
в качестве промежуточных соединений для получения производных антрациклина, обладающих противоопухолевой и противовирусной активностью.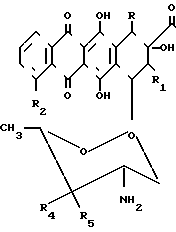

где R и R1 - различные, водород или фтор;
R2 - водород или низшая алкоксигруппа;
R4 и R5 - водород или гидроксильная группа, но они не могут быть одновременно гидроксилом,
или их фармацевтически пригодные соли, обладающие противоопухолевой и противовирусной активностью.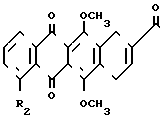
где R2 - водород или низшая алкоксигруппа,
в качестве промежуточных соединений для получения 7-замещенных фтор- 2β -ацетил- 2α , 4α , 5 , 12 -тетраокси-1,2,3,4-тетрагидро-6,11-нафтацендиона.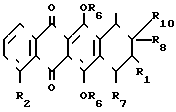
где R2 - водород или низшая алкоксигруппа;
когда R6 - метил, R7 - водород, R10 - группа
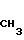
то R8 - гидроксил, R1 - фтор или R8 и R1 вместе с атомом углерода образуют оксирановый цикл; когда R6 - водород, R7 - водород, R8 - гидроксил, R1 - фтор, то R10 - группа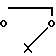 или
или 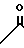
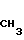
когда R6 - водород, R7 - гидроксил, R8 - гидроксил, R1 - фтор, то R10 группа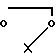
в качестве промежуточных соединений для получения 7-замещенных фтор- 2β-ацетил-2α , 4α , 5 , 12 -тетраокси-1,2,3,4-тетрагидро-6,11-нафтацендиона.
Приоритет по признакам:
13.11.89 при R - H, R1 - F, R2 - H, X - H; R4 и R5 - все значения, R6, R7, R8, R10 - все значения;
20.02.90 при R - F, R1 - H, R2 - H, OH, OCH3, X - H, OH;
30.03.90 при R - H, R1 - F, R2 - OCH3, X - H.
| ТОПЛИВНЫЙ НАСОС | 1997 |
|
RU2169284C2 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
