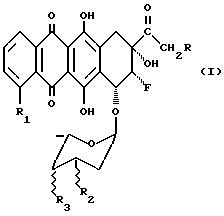
Описаны гликозидные производные 8-фрорантрациклинона общей формулы (1):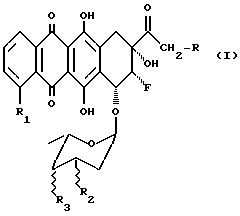
где R выбран из группы, состоящей из H, ОН, OR4, где R4 выбран из группы, состоящей из CHO, COCH3, ацильного производного, содержащего до 6 углеродных атомов;
R1 выбран из группы, состоящей из H, OH, OCH3;
R2 выбран из группы, состоящей из H, OH, NH2;
R3 выбран из группы, состоящей из H, OH, NH2, остатка формулы (А)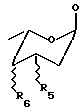
где R5 и R6, одинаковые или разные, выбраны из группы, состоящей из H, OH, NH2, и символ  означает, что заместители R2, R3, R5 и R6 могут находиться в осевой или экваториальной конфигурации;
означает, что заместители R2, R3, R5 и R6 могут находиться в осевой или экваториальной конфигурации;
и где группы 8-F и 9-OH находятся в положении цис;
их фармацевтически приемлемые соли, способы их получения и содержащие их фармацевтические композиции.
Уровень техники
Даунорубицин (дауномицин) и 4-деметоксидаунорубицин (идарубицин) и их производные, содержащие гидроксилированную боковую цепь (доксорубицин), являются гликозидами, обладающими хорошо известными противоопухолевыми свойствами; их получение и применение уже описаны (F. Arcamone "Doxorubicin: Anticancer Аntibiotics" Medinal Chemistry Series Vol. 17, Academic press, 1981).
8-фторантрациклины - это класс антрациклинов, уже известных (см., например, ЕР-А-0436474; ЕР-А-0457215; WO 95/09173) их более высокими активностью и избирательностью по сравнению с соответствующими нефторированными соединениями.
Теперь неожиданно обнаружено (и это является объектом настоящей заявки), что когда два заместителя 8-F и 9-ОН имеют цис-стереохимию, то соответствующие производные необычайно более активны, в частности в отношении опухолевых клеток, стойких к уже известным соединениям.
Подробное описание изобретения
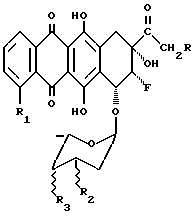
В соответствии с настоящим изобретением предлагаются гликозидные производные 8-фторантрациклинона общей формулы
где R выбран из группы, состоящей из H, OH, OR4, где R4 выбран из группы, состоящей из CHO, COCH3, ацильного производного, содержащего до 6 углеродных атомов;
R1 выбран из группы, состоящей из H, OH, OCH3;
R2 выбран из группы, состоящей из H, OH, NH2;
R3 выбран из группы, состоящей из H, OH, NH2, остатка формулы (А)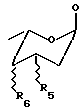
где R5 и R6, одинаковые или разные, выбраны из группы, состоящей из H, OH, NH2 и символ  означает, что заместители R2, R3, R5 и R6 могут находиться в осевой или экваториальной конфигурации;
означает, что заместители R2, R3, R5 и R6 могут находиться в осевой или экваториальной конфигурации;
и где группы 8-F и 9-ОН находятся в положении цис;
их фармацевтически приемлемые соли, способы их получения и содержащие их фармацевтические композиции.
Более конкретно, в соответствии с настоящим изобретением предлагаются следующие соединения:
1) 4-деметокси-8-(R)-фтордаунорубицин
(1:R = R1 = H; R2 =NH2; R3 = OH),
2) 4-диметокси-8-(R)-фтор-3'-деамино-4'-деокси-4'- аминодаунорубицин (1: R = R1 = R2 = H; R3 = NH2),
3) З-деметокси-8-(R)-фтор-3'-деамино-4'-деокси-4'- эпиаминодаунорубицин (1:R= R1 = R2 = H; R3 = NH2),
4) 4-деметокси-8-(R)-фтор-4'-эпидаунорубицин (1:R = R1 = H; R2 = NH2; R3 = OH),
5) 4-деметокси-8-(R)-фтор-7-(дауносаминилфукозил) даунорубицинон (1:R = R1 = H; R3 = A; R2 = R6 = OH; R5 = NH2),
6) 4-деметокси-8-(R)-фтор-7-(дауносаминилрамнозил) даунорубицинон (1:R = R1 = H; R3 = А; R2 = R6 = OH; R5 = NH2),
7) 4-деметокси-8-(R)-фтор-7-(2'', 3'', 4''-тридеокси-4''- аминоэзапиранозилфукозил)даунорубицинон (1:R = R1 = R5 = H; R2 = A; R2 = OH; R6 = NH2),
8) 4-деметокси-8-(R)-фтор-7-(2'', 3'', 4''-тридеокси-4''- аминоэзапиранозилрамнозил)даунорубицинон (1:R = R1 = R5 = H; R3 = a; R2 = OH; R6 = NH2),
9) 4-деметокси-8-(R)-фтор-7-(фукозил-4'-O-фукозил) даунорубицинон (1:R = R1 = H; R3 = А; R2 = R5 = R6 = OH),
10) 4-деметокси-8-(R)-фтор-7-(фукозил-4'-O-рамнозил) даунорубицинон (1:R = R1 = H; R3 = A; R2 = R5 = R6 = OH),
11) 8-(R)-фтордаунорубицин (1:R = H; R1 = OCH3; R2 = H2; R3 = OH),
12) 8-(R)-фтор-3'-деамино-4'-деокси-4'-аминодаунорубицин (1:R = R2 = H; R1 = OCH3; R3 = NH2),
13) 8-(R)-фтор-3'-деамино-4'-деокси-4'-эпиаминодаунорубицин (1:R = R2 = H; R1 = OCH3; R3 = NH2),
14) 8-(R)-фтор-4'-эпидаунорубицин (1: R = H; R1 = OCH3; R2 = NH2; R3 = OH),
15) 8 -(R)-фтор-7-(дауносаминилфукозил)даунорубицинон (1: R = H; R1 = OCH3; R3 = A; R2 = R6 = OH; R5 = NH2),
16) 8-(R)-фтор-7-(дауносаминилрамнозил)даунорубицинон (1: R = H; R1 = OCH3; R3 = A; R2 = R6 = OH; R5 = NH2),
17) 8-(R)-фтор-7-(2'', 3'', 4''-тридеокси-4''- аминоэзапиранозилфукозил)даунорубицинон (1:R = R5 = H; R1 = OCH3; R3 = A; R2 = OH; R6 = NH2),
18) 8-(R)-фтор-7-(2'', 3'', 4''-тридеокси-4''- аминоэзапиранозилрамнозил)даунорубицинон (1:R = R5 = H; R1 = OCH3; R3 = A; R2 = OH; R6 = NH2),
19) 8-(R)-фтор-7-(фукозил-4'-O-фукозил)даунорубицинон (1: R = H; R1 = OCH3; R3 = A; R2 = R5 = R6 = OH);
20) 8-(R)-фтор-7-(фукозил-4'-О-рамнозил)даунорубицинон (1: R = H; R1 = OCH3; R3 = А; R2 = R5 = R6 = OH),
21) 4-деметокси-8-(R)-фтордоксорубицин и его сложные эфиры по C-14 (1:R = R3 = OH; R1 = H; R2 = NH2),
22) 4-деметокси-8-(R)-фтор-3'-деамино-4'-деокси-4'- аминодоксорубицин и его сложные эфиры по C-14 (1:R = PH; R1 = R2 = H; R3 = NH2),
23) 4-деметокси-8-(R)-фтор-3'-деамино-4'-деокси-4'- эпиаминодоксорубицин и его сложные эфиры по C-14 (1:R = OH; R1 = R2 = H; R3 = NH2),
24) 4-деметокси-8-(R)-фтор-4'-эпидоксорубицин и его сложные эфиры по C-14 (1:R = R3 = OH; R1 = H; R2 = NH2),
25) 4-деметокси-8-(R)-фтор-7-(дауносаминилфукозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = R6 = OH; R3 = A; R1 = H; R5 = NH2),
26) 4-деметокси-8-(R)-фтор-7-(дауносаминилрамнозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = R6 = OH; R3 = А; R1 = H; R5 = NH2),
27) 4-деметокси-8-(R)-фтор-7-(2'',3'',4''-тридеокси-4''- аминоэзапиранозилфукозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = OH; R3 = А; R1 = R5 = H; R6 = NH2),
28) 4-деметокси-8-(R)-фтор-7-(2'',3'',4''-тридеокси-4''- аминоэзапиранозилрамнозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = OH; R3 = А; R1 = R5 = H; R6 = NH2),
29) 4-деметокси-8-(R)-фтор-7-(фукозил-4'-O-фукозил) доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = R5 = R6 = OH; R3 = А; R1 = H),
30) 4-деметокси-8-(R)-фтор-7-(фукозил-4'-O-рамнозил) доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = R5 = R6 = OH; R3 = А; R1 = H),
31) 8-(R)-фтордоксорубицин и его сложные эфиры по C-14 (1:R = R3 = OH; R1 = OCH3; R2 = NH2),
32) 8-(R)-фтор-3'-деамино-4'-деокси-4'-аминодоксорубицин и его сложные эфиры по C-14 (1:R = PH; R1 = OCH3; R2 = H; R3 = NH2),
33) 8-(R)-фтор-3'-деамино-4'-деокси-4'-эпиаминодоксорубицин и его сложные эфиры по C-14 (1:R = OH; R1 = OCH3; R2 = H; R3 = NH2),
34) 8-(R)-фтор-4'-эпидоксорубицин и его сложные эфиры по C-14 (1:R = R3 = OH; R1 = OCH3; R2 =NH),
35) 8-(R)-фтор-7-(дауносаминилфукозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = R6 = OH; R1 = OCH3; R3 = A; R5 = NH2),
36) 8-(R)-фтор-7-(дауносаминилрамнозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = R6 = OH; R3 = А; R1 = OCH3; R5 = NH2),
37) 8-(R)-фтор-7-(2'', 3'', 4''- тридеокси-4''- аминоэзапиранозилфукозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = OH; R1 = OCH3; R3 = А; R5 = H; R6 = NH2),
38) 8-(R)-фтор-7-(2'', 3'', 4''-тридеокси-4''- аминоэзапиранозилрамнозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = OH; R1 = OCH3; R3 = А; R5 = H; R6 = NH2),
39) 8-(R)-фтор-7-(фукозил-4'-O-фукозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = R5 = R6 = OH; R3 = A; R1 = OCH3),
40) 8-(R)-фтор-7-(фукозил-4'-O-рамнозил)доксорубицинон и его сложные эфиры по C-14 (1:R = R2 = R5 = R6 = OH; R1 = OCH3; R3 = A).
Соединения формулы (I) и их фармацевтически приемлемые соли получают способом, включающим следующие стадии.
Осуществляют конденсацию между 8-фторантрациклиноном формулы (II):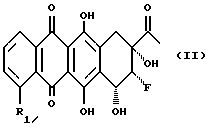
где R1 - такой, как указан выше, и соединением формулы (III)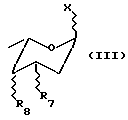
где X представляет уходящую группу, способную создавать (в условиях конденсации) устойчивый карбокатион, который может взаимодействовать с гидроксилом в положении С-7 соединения (II). Такую уходящую группу целесообразно выбирать из тех групп, которые используют в реакциях гликозидирования (например, галоген, пара-нитробензоилоксигруппа); R7 представляет H или группу OH или NH2, подходящим образом защищенную;
R8 представляет H или группу OH или NH, подходящим образом защищенную, или остаток формулы (III'):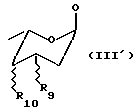
где R9 и R10, одинаковые или разные, выбраны из H, OH и NH2, подходящим образом защищенных.
Группы OH защищают пара-нитробензоатом или аллилоксикарбонилом.
Группы NH2 защищают аллилкарбоксиамидом или трифторацетамидом.
Символ  означает, что заместители R7, R8, R9, R10 могут находиться в осевой и/или экваториальной конфигурации. Конденсация между соединениями (II) и (III) дает гликозид формулы (IV)
означает, что заместители R7, R8, R9, R10 могут находиться в осевой и/или экваториальной конфигурации. Конденсация между соединениями (II) и (III) дает гликозид формулы (IV)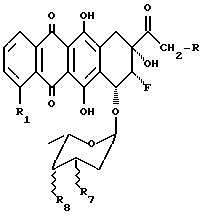
Реакцию гликозидирования осуществляют в органическом растворителе в присутствии конденсирующего агента. Конденсирующими агентами являются, например, трифторметансульфат серебра, перхлорат серебра, смеси оксида или бромида ртути, галогенида бора, ионообменной смолы, такой как Амберлит.
Предпочтительно реакцию гликозидирования осуществляют с использованием инертного органического растворителя, такого как бензол, толуол, диэтиловый эфир, тетрагидрофуран, диоксан, хлороформ, метиленхлорид или дихлорэтан и их смеси.
Температуру реакции можно изменять в пределах между -40oC и 40oC, предпочтительно между -20oC и 20oC, а необходимое для реакции время может колебаться между 5 минутами и 2 часами.
В реакционной смеси может присутствовать обезвоживающее средство, такое как активированные молекулярные сита.
Во время реакции или в ее конце к реакционной смеси может быть добавлено органическое основание, такое как пиридин, коллидин, триэтиламин.
Удаление защитных групп с групп OH и/или NH2 в соединениях формулы (IV), чтобы получить целевые соединения формулы (I), можно изменять в соответствии с использованной защитной группой.
Когда R7 и/или R8, и/или R9, и/или R10 (одинаковые или разные) представляют группу NH2, защищенную трифторацетамидом, и/или группу OH, защищенную пара-нитробензоатом, то реакцию удаления защиты осуществляют в полярном растворителе, таком как вода, метанол, этанол, пиридин, диметилформамид или их смеси, в присутствии стехиометрического или избыточного количества неорганического основания, такого как NaOH, KOH, LiOH, Ba(OH)2 и их карбонаты. Температура реакции может колебаться в пределах от 0oC до 50oC, а время реакции - от 3 часов до 3 дней.
Когда R7 и/или R8, и/или R9, и/или R10 (одинаковые или разные) представляют группу NH2, защищенную аллилкарбоксиамидом, и/или группу OH, защищенную аллилоксикарбонатом, то реакцию удаления защиты осуществляют в инертном растворителе и в присутствии металлического комплекса, такого как (тетракистрифенилфосфин) палладий, как описано, например, в Tetrahedron Letters, 30 (1989), 3773, или (тетракарбонил)никель, как описано, например, в J. Org. Chem. 38 (1973) 3233.
При желании соединения формулы (I), где R1, R2 и R3 - такие, как указаны выше, и R представляет группу OH, могут быть получены из гликозидов формулы (I) или их фармацевтически приемлемых солей, где R1, R2 и R3 и символ  - такие, как указаны выше, и R представляет H путем бромирования атома углерода в положении 14 бромом в хлороформе с последующим гидролизом полученных описанным образом 14-бромпроизводных с формиатом натрия при комнатной температуре в течение 48 часов.
- такие, как указаны выше, и R представляет H путем бромирования атома углерода в положении 14 бромом в хлороформе с последующим гидролизом полученных описанным образом 14-бромпроизводных с формиатом натрия при комнатной температуре в течение 48 часов.
Тот же самый продукт может быть получен также путем бромирования атома углерода в положении 14 (в соответствии с описанной выше процедурой) агликона формулы (II), где R1 - такой, как указан выше, и R представляет H. Это соединение после гидролиза, которое преобразует R в группе OH, гликозилируют соединением формулы (III), определенным выше, путем осуществления такой, как описана выше, реакции гликозидирования для получения целевого продукта.
Если это является предпочтительным, гликозиды формулы (I) могут быть преобразованы в соответствующие фармацевтически приемлемые соли, например гидрохлориды, путем обработки хлороводородной кислотой в метаноле.
Другими объектами данной заявки являются 8-фторантрациклиноны формулы (II), где группы 8-F и 9-OH находятся взаимно в положении цис и где R и R1 - такие, как указаны выше, и способ их получения.
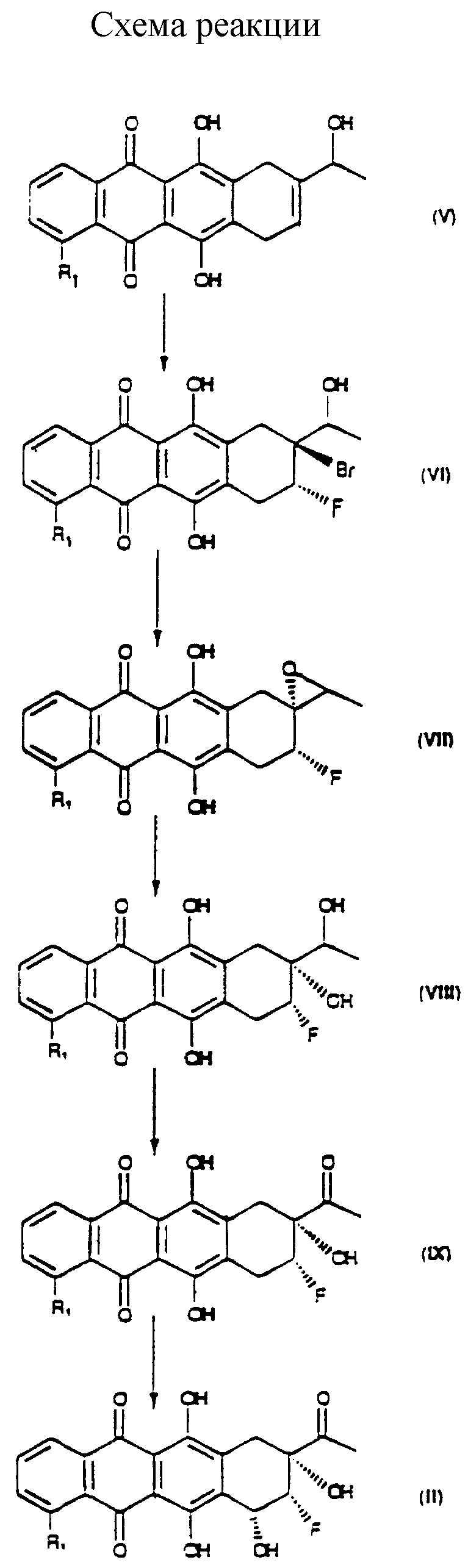
Способ проиллюстрирован в схеме, приведенной в конце описания.
Первой стадией является бромфторирование аллилового спирта формулы (V), где R1 - такой, как указан выше, для получения соединения формулы (VI).
Бромфторирование соединения (V) может быть осуществлено разными способами, описанными в литературе (например, Cazz. Chim. It. 121 (1991) 537-545; J.O.C. 58 (1993) 2791-2796), для бромфторирования алкенов.
Второй стадией является образование эпоксида из бромидрина (VI) для получения соединения формулы (VII), где R1 - такой, как указан выше. Реакция может быть осуществлена с использованием сильного основания в соответствии с известными способами, описанными в литературе в связи с этим типом реакции.
Третьей стадией является раскрытие эпоксидного цикла для получения продукта формулы (VII), где R1 - такой, как указан выше. Способы осуществления реакции в этом случае тоже описаны в литературе, относящейся к раскрытию эпоксидного цикла с получением диола путем кислотного катализа, предпочтительно в смесях воды, минеральных кислот и инертных органических растворителей.
Четвертой стадией является реакция окисления диола (VIII) в гидроксикетон формулы (IX). Реакция окисления может быть осуществлена в соответствии с известными способами. Предпочтительными являются способы, включающие использование диметилсульфоксида, как, например, реакция Моффата и тому подобное, или комплексов пиридин-хром, как, например, хлорхромат пиридиния.
Последней стадией является преобразование фторгидроксикетона формулы (IX) в соединение формулы (II). Это может быть осуществлено известными методами, как, например, бромирование и сольволиз, при необходимости с возможной защитой кетогруппы (Can. J. Chem. 49 (1973) 2712; J.A.C.S. 98 (1976) 1969; J.A.C.S. 98 (1976) 1967).
Настоящее изобретение касается также фармацевтических композиций, содержащих в качестве активного начала антрациклингликозид формулы (I) или его фармацевтически приемлемую соль.
Терапевтически активное количество соединения по настоящему изобретению (2-200 мг/м2 поверхности тела, или 0,05-0,5 мг/кг массы тела, если продукт вводят внутривенно, или 10-200 мг/м2 поверхности тела или 0,25-5 мг/кг массы тела в случае перорального введения) смешивают с инертным носителем. Могут быть использованы традиционные носители и композицию изготавливают известными методами.
Предпочтительными являются композиции, пригодные для внутривенного или перорального введения.
Соединения по настоящему изобретению полезны в лечении людей и животных. В частности, соединения по настоящему изобретению полезны в качестве противоопухолевых средств для введения терапевтически активных количеств соединения пациенту, проходящему лечение.
В частности, соединения проявили необычайную активность против плотных опухолей (например, опухоль яичника, молочной железы, легкого, матки), а также против тех, которые проявили стойкость к известным, обычно используемым противоопухолевым средствам.
Для лучшей иллюстрации (но без цели ограничения) изобретения даны следующие примеры.
Пример I.
9-(1'-Гидроксиэтил)-7,10-дигидро-6,11-дигидрокси-8- фтор-9-бром-5,1-нафтацендион (VI), R1 = H)
К суспензии 9-(1'-гидроксиэтил)-7,10-дигидро-6,11- дигидрокси-5,12-нафтацендиона ((V), R1 = H) (9,4 г, 28 ммоль) в метиленхлориде (900 мл)э добавляли с перемешиванием при -5oC раствор дигидротрифторида тетрабутиламмония (18,4 г при 55%, 33,6 ммоль) в метиленхлориде, после чего добавляли также при -5oC (при этом реактор держали в темноте) N-бромсукцинимид (6,0 г, 33,6 ммоль). Реакция длилась 5 часов. Большее время реакции не дает увеличения выхода. Добавляли 10%-ный раствор NaHCO3 до получения нейтрального pH. Органическую фазу сушили на Na2SO4, фильтровали и выпаривали. Остаток очищали путем флэш-хроматографии с использованием CHCl3 в качестве элюента.
Было получено 3,6 г (выход 29%) соединения (VI) (R1 = H).
ЯМР (CDCl3, δ ): 1,45 (3H, д); 3,0-3,5 (2H, кв); 3,2-3,4 (2H, кв); 3,93 (1H, м); 5,4-5,6 (1H, д); 7,90 (2H, м); 8,30 (2H, м); 13,36 (1H, с); 13,46 (1H, с).
Аналогичным образом был получен 4-метокси-9-(1'-гидроксиэтил)-7,10-дигидро-6,11-дигидрокси-8-фтор-9- бром-5,12-нафтацендион ((VI), R1 = OCH3).
Пример II
9-(9-1'-Эпоксиэтил)-7,10-дигидро-6,11-дигидрокси-8-фтор-5,12- нафтацендион ((VII), R1 = H)
Раствор 9-(1'-гидроксиэтил)-7,10-дигидро-6,11-дигидрокси-8- фтор-9-бром-5,12-нафтацендиона (3,6 г, 8,3 ммоль), полученный в соответствии с примером I, в 5%-ной NaOH (16 мл) оставляли перемешиваться на 2 часа при комнатной температуре. Реакционную смесь обрабатывали 1 н. раствором HCl и продукт экстрагировали CHCl3; органическую фазу сушили на Na2SO4, фильтровали и выпаривали. Полученный остаток очищали путем флэш-хроматографии с использованием CHCl3 в качестве элюента. Получили 1,6 г (выход 57%) указанного в заголовке соединения.
ЯМР (CDCl3, δ ): 1,4 (3H, д); 2,62-3,48 (2H, кв); 2,9-3,1 (2H, кв); 3,26-3,68 (2H, м); 3,10 (1H, м); 4,80-4,96 (1H, д); 7,80 (2H, м); 8,40 (2H, м); 13,35 (1H, с); 14,42 (1H, с).
Аналогичным образом был получен 4-метокси-9-(9-1'-эпоксиэтил) -7,10-дигидро-6,11-дигидрокси-8-фтор-5,12-нафтацендион ((VII), R1 = OCH3).
Пример III
9-(1'-Гидроксиэтил)-7,10-дигидро-6,9,11-тригидрокси-8-фтор- 5,12-нафтацендион ((VIII), R1 = H)
К раствору 9-(9-1'-эпоксиэтил)-7,10-дигидро-8,11-дигидрокси-8- фтор-5,12-нафтацендиона (1,8 г, 4,7 ммоль), полученному в соответствии с примером II, в диоксане (700 мл) добавляли H2O (700 мл) и затем, держа реактор в бане при 0oC, медленно капали в полученную смесь олеум. Закончив добавление олеума, давали температуре подняться до комнатной и перемешивали раствор в течение 6 часов. Добавляли к смеси NaHCO3 до получения нейтрального pH и продукт экстрагировали CH2Cl2. Органическую фазу, высушенную на Na2SO3, выпаривали. В результате получили указанный в заголовке продукт (1,7 г, выход 100%).
ЯМР (CDCl3, δ ): 1,40 (3H, д); 2,8-3,1 (2H, кв); 3,0-3,3 (2H, кв); 4,05 (2H, кв); 5,0-5,2 (1H, кв); 7,80 (2H, м); 8,30 (2H, м); 13,30 (1H, с); 13,22 (1H, с).
Аналогичным образом был получен 4-метокси-9-(1'-гидроксиэтил) -7,10-дигидро-6,9,11-тригидрокси-8-фтор-5,12-нафтацендион ((VIII), R1 = OCH3).
Пример IV
9-Ацетил-8(8H)-фтор-7,10-дигидро-6,9, 11-тригидрокси-5,12- нафтацендион ((IX), R1 = H).
9-(1'-Гидроксиэтил)-7,10-дигидро-6,9,11-тригидрокси-8-фтор- 5,12-нафтацендион (1,7 г, 4,7 ммоль), полученный в соответствии с примером III, в безводных условиях растворяли в ДМСО (90 мл), после чего к раствору добавляли смесь пиридина (1,14 мл, 14,1 ммоль) и трифторуксусной кислоты 0,83 мл, 10,8 ммоль) в ДМСО (16 мл). К смеси добавляли уксусный ангидрид (4,44 мл, 47 ммоль) и раствор оставляли перемешиваться при комнатной температуре в течение 4 часов. Затем к раствору добавляли H2O, в результате чего в осадок выпадал целевой продукт.
Путем экстрагирования посредством CHCl3 извлекали все еще, возможно, присутствующий в растворе дополнительный продукт. Полученный указанным образом продукт очищали путем флэш-хроматографии с использованием CHCl3 в качестве элюента. В результате получили указанное в заголовке соединение (1,2 г, выход 70%).
ЯМР (CDCl3, δ ): 2,48 (3H, с); 3,18 (2H, с); 3,1-3,6 (2H, м); 3,90 (1H, с); 5,2-5,4 (1H, с); 7,82 (2H, м); 8,38 (2H, м); 13,34 (1H, с); 13,36 (1H, с).
Аналогичным образом был получен 4-метокси-9-ацетил-8(8H), фтор-7,10-дигидро-6,9,11-тригидрокси-5,12-нафтацендион ((IX), R1 = OCH3).
Пример V
9-Ацетил-8(8H)-фтор-10-гидро-6,7(7H), 9,11-тетрагидрокси- 5,12-нафтацендион ((II), R1 = H).
К находящейся в колбе, снабженной устройством Дина-Старка, суспензии 9-ацетил-8(8H)-фтор-7,10-дигидро-6,9,11-тригидрокси- 5,12-нафтацендиона (1,2 г, 3,3 ммоль), полученного в соответствии с примером IV, в безводном бензоле (200 мл) добавляли этиленгликоль (5 мл) и каталитическое количество паратолуолсульфоновой кислоты (60 мг, 0,3 ммоль). Смесь нагревали с обратным холодильником при 120oC в течение 15 часов. Карбонильную функциональную группу в положении 13 защищали путем кетализации (ацетализации). По окончании реакции повышали температуру до комнатной с выпадением в осадок продукта, который собирали и промывали водой.
Полученный продукт бромировали в положении 7 с PHPP и AlB в CCl4 путем нагревания с обратным холодильником при 120oC в потоке NH2 в течение 8 часов. Температуру реакционной смеси доводили до комнатной, отфильтровывали осадок и раствор выпаривали. Остаток гидролизовали с трифторуксусной кислотой (240 мл) и водой (60 мл) путем нагревания с обратным холодильником в течение 1 часа. Температуру доводили до комнатной, а затем снижали до 7oC. Выпавший в осадок продукт промывали водой и высушивали. В результате получили смесь эпимеров в положении 7 (а = 70%, b = 30%). Целевой эпимер выделяли путем ВЭЖХ с получением указанного в заголовке продукта (312 мг, выход 24%).
ЯМР (CDCl3, δ ): 2,50 (3H, с); 3,1-3,4 (2H, м); 3,85 (1H, д); 4,82 (1H, с); 5,04-5,22 (1H, кв); 5,50 (1H, м); 7,85 (2H, м); 8,40 (2H, м); 13,28 (1H, с); 14,54 (1H, с).
Аналогичным образом был получен 4-метокси-9-ацетил-8(8H)- фтор-10-гидро-6,7(7H),9,11-тетрагидрокси-5,12-нафтацендион ((II), R1 = OCH3).
Пример VI
Защищенный 4-деметокси-8(R)-фтордаунорубицин (IV, R = R1 = H, R7 = NHCO2CH2CH=CH2, R8 = p-CO-C6-H4-NO2)
К раствору 9-ацетил-8(8H)-фтор-10-гидро-6,7(7H), 9,11- тетрагидрокси-5,12-нафтацендиона (77 мг, 0,2 ммоль), полученного в соответствии с примером V, и защищенного дауносамина ((III), X = R8 = OCOC6H4NO2; R7 = NHCO2CH2CH= CH2) (127 мг, 0,24 ммоль) в безводном метиленхлориде (40 мл) и безводном диэтиловом эфире (10 мл) добавляли молекулярные сита 4 ангстрем (1,2 г). К этому раствору, который держали при -10oC под струей безводного азота, добавляли триметилсилилтрифлат (107 мг, 0,48 ммоль). Реакцию проводили при -10oC в течение 30 минут. Раствор обрабатывали раствором NaHCO3, органическую фазу экстрагировали, сушили на Na2SO4 и выпаривали. Полученный при этом неочищенный продукт очищали путем флэш-хроматографии (CHCl3 + 1% iPrOH). Получили 61 мг (выход 40%) указанного в заголовке соединения.
ЯМР (CDCl3, δ ): 1,2 (3H, д); 2,0 (2H, м); 2,5 (3H, д); 3,1-3,5 (2H, кв); 4,2-4,7 (5H, м); 5,0-5,3 (3H, м); 5,4-5,6 (2H, м); 5,8 (1H, с); 5,9 (1H, м); 7,8 (2H, м); 8,3 (6H, м); 13,7 (1H, с); 13,2 (1H, с).
Аналогичным образом был получен защищенный 8-(R)-фтордаунорубицин (IV, R = H, R1 = OCH3, R7 = NHCO2CH2CH=CH2, R8 = p-CO-C6H4-NO2).
Пример VII
4-Деметокси-8-(R)-фтордаунорубицингидрохлорид ((I), R = R1 = H; R2 = NH2 • HCl, R3 = OH)
Продукт, описанный в примере VI, необходимо подвергнуть двум реакциям удаления защиты для преобразования в соответствующее соединение 1.
Полученный в примере VI продукт (61 мг, 0,08 ммоль) суспендировали в смеси MeOH/H2O и обрабатывали 0,5 М раствором K2CO3 для снятия защиты с гидроксигруппы в положении 41.
Полученный продукт обрабатывали Ph3P, Тетракисом, 2-метилмасляной кислотой в CH2Cl2 в безводных условиях, в темноте и при комнатной температуре и после снятия защиты с аминогруппы в положении 3' получили целевое соединение (32 мг, 75%).
Солеобразование путем обработки этого соединения 0,01 н. раствором HCl дало указанное в заголовке соединение.
ЯМР (CDCl3, δ ): 1,3 (3H, д); 2,0-2,2 (2H, м); 2,5 (3H, с); 3,2-3,4 (2H, м); 3,6-3,8 (2H, м); 4,4-4,5 (1H, м); 5,2-5,4 (1H, м); 5,5 (1H, м); 5,5 (1H, м); 5,7 (1H, м); 8,0 (2H, с); 8,4 (2H, с).
Аналогичным образом был получен 8-(R)-фтордаунорубицингидрохлорид (1, R = H; R1 = OCH3; R2 = NH2 • HCl; R3 = OH).
Приложение 1
Сравнительные испытания
Противоопухолевую активность in vitro (см. табл.1) следующих соединений:
Ia: 4-деметокси-8-(R)-фтордаунорубицина гидрохлорид (соединение формулы I, где: R = R1 = H, R2 = NH2, R3 = OH)
Ib: 4-деметокси-8-(R)-фтор-3'-дезамино-4'-дезокси-4'- эпиаминодаунорубицина гидрохлорид (соединение формулы I, где: R = R1 = R2 = H, R3 = NH2),
испытывали в отношении нескольких линий клеток в сравнении с соответствующими 8-(S)-фтор-аналогами:
Ic: 4-деметокси-8-(S)-фтордаунорубицина гидрохлоридом,
Id: 4-деметокси-8-(S)-фтор-3'-дезамино-4'-дезокси-4'- эпиаминодаунорубицина гидрохлоридом.
Использовали следующие линии опухолевых клеток человека, как DXR-чувствительные, так и DXR-устойчивые:
A431 (плоскоклеточная карцинома шейки матки),
H460 (крупноклеточная карцинома легкого), A2780 (карцинома яичников) и A2780/DX,
MCF7 (карцинома молочной железы) и MCF7/DX,
LoVo (аденокарцинома толстой кишки) и LoVo/DX
Обратите внимание, что DXR обозначает доксорубицин, а клеточные линии DXR-устойчивые обозначены с помощью .../DX.
8(R)-фтор-производные Ia и Ib более цитотоксичны в отношении испытываемых клеточных линий, чем 8(S)-соединения, Ic и Id. Кроме того эта повышенная цитотоксичность еще более значительно выражена, если мы рассматриваем активность этих соединений в отношении соответствующих DXR-устойчивых опухолей.
Подобные результаты показывают прямую корреляцию между стереохимией фтора в положении C-8 агликона и цитотоксической активностью.
Приложение 2
Анализ и определение транс- и цис-8,9-фторгидриновой системы
Относительную ориентацию атома фтора в отношении гидроксильных групп у C(7) и C(9) определяли, используя эксперименты с ядерным эффектом Оверхаузера (Nuclear Overhouser Effect (NOE)).
В случае агликона II, имеющего цис-8,9-фторгидриновую систему, облучение H-8 не приводило к усилению сигналов для гидроксильных водородов (OH-7 и OH-9), указывая на то, что в этом соединении протон H-8 был на противоположной стороне кольца А по отношению к двум гидроксильным группам.
С другой стороны, для агликона, имеющего транс-8,9-фторгидриновую систему, облучение H-8 давало усиление сигналов для OH-7 и OH-9, показывая, что все три из этих протонов находятся на той же самой стороне кольца А.
Все эти аргументы изложены также в статье одного из авторов изобретения (см. Lombardi P. et al., Acta Biochemica Polonica, 42, 433, 1995).
Приложение 3
Фармацевтические композиции
Лиофилизованные лекарственные формы соединения Ia, 4-деметокси-8-(R)-фтордаунорубицина гидрохлорида [(I) R = R1 = H; R2 = NH2; R3 = OH; в виде соли с HCl] получали по методике, изложенной ниже.
Относительные количества различных компонентов (на флакон), взятые для приготовления формы:
Соединение Ia - 10,00 мг
Лактоза - 50,00 мг
Вода для инъекций, достаточное количество - (q.s.) до 2,50 мл.
Соединение Ia и лактозу последовательно растворяли при перемешивании в воде для инъекций и деаэрировали пропусканием азота (примерно 90% от необходимого конечного объема воды). Затем добавляли деаэрированную воду для инъекций до получения конечного объема. В каждый флакон вставляли стерильные пробки для лиофильной сушки и флаконы загружали в аппарат для лиофильной сушки при комнатной температуре.
Раствор в сосудах замораживали при температуре -40oC в течение 4-5 часов.
Проводили лиофилизацию, высушивая продукт на конечной стадии при температуре 43-45oC в течение 6 - 7 часов. В завершение цикла сосуды закупоривали пневматически при пониженном давлении и закрывали алюминиевыми колпачками.
По аналогичной методике получали лиофилизованные лекарственные формы соединения Ia, содержащие 50 мг активного вещества:
Соединение Ia - 50,00 мг
Лактоза - 250,00 мг
Вода для инъекций - q.s. до 5,0 мл.
Лиофильную сушку проводили в аппарате (тип 1, Aluglas В.V., Uithoorn, Нидерланды), имеющем номинальную производительность 50/57 мл.
Приложение 4
Биологическая активность
Активность in vivo соединения Ia, 4-деметокси-8-(R)-фтордаунорубицина гидрохлорида [(I) R = R1 = H; R2 = NH2; R3 = OH; в виде соли с HCl].
Противоопухолевая активность in vivo
Противоопухолевую активность соединения Ia изучали в отношении человеческих опухолей А-2780 (яичников) и А- 431 (шейка матки) пересаженных голым мышам.
Исследования по эффективности:
для оценки реакции опухоли на соединение Ia его активность in vivo определяли при дозе 1,5 мг/кг при режиме введения 7дх3, чтобы оптимизировать противоопухолевую активность, принимая во внимание зависимость цитотоксического эффекта в экспериментах in vitro в клеточной культуре от времени экспозиции. В качестве лекарственного препарата для сравнения использовали доксорубицин (ДКР), применяя его в дозе (7 мг/кг) и при схеме введения (7дх3), который, как было установлено, является оптимальным режимом дозирования.
Материалы и методы
Для исследований in vivo линии человеческих опухолевых клеток поддерживали с помощью серийных подкожных (п/к) пассажей фрагментов опухолей с двух сторон у взрослых бестимусных голых мышей. Регулярно производили проверку фрагментов опухоли человеческого происхождения с помощью гистологического исследования после окрашивания гематоксилином и эозином и с помощью картины электрофореза лактатдегидрогеназных изоферментов.
Бестимусным мышам (Charles River, Caico, Italy) п/к вводили фрагменты опухоли с обеих сторон (исследования эффективности) или с одной стороны (сравнительные контрольные исследования). За ростом опухолей следили путем измерений с помощью микрометра длины и ширины в определенные сроки (еженедельно или дважды в неделю). Вес опухоли (ВО) рассчитывали, используя формулу: мг = объем в мм3 = ширина2 • длина/2 (принимая плотность = 1), через 7/8 дней после последнего введения лекарства. Время увеличения опухоли вдвое (ВДУ) рассчитывали для каждой линии опухоли по кривой с наилучшей аппроксимацией на полулогарифмической сетке для каждой контрольной опухоли, построенной по отношению к уровню выравнивания (экспоненциальная фаза роста). В исследованиях использовали средние значения ВДУ.
Мышам, имеющим опухоли, вводили внутривенно (в/в) лекарственные средства, начиная в разные сроки при разных опухолях. Дни первой и последней инъекции были одинаковыми для двух лекарственных препаратов во всех экспериментах. Каждая экспериментальная группа состояла из, по меньшей мере, пяти опухолей.
Оценивали следующие эффекты, достигнутые с помощью лечения лекарственными препаратами:
Снижение веса опухоли, % (% СВО): у мышей, получавших лечение, по сравнению с контрольными мышами, определенное через 7-10 дней после последнего введения лекарства.
Log гибели клеток (LГK) у получающих лечение мышей по формуле: (Л-К/ВДУ • 3,32, где Л и К представляют количество дней, потребовавшееся у лечившихся (Л) и контрольных (К) животных для того, чтобы опухоли достигли среднего веса, точно определенного в каждом эксперименте.
Гибель от интоксикации: число умерших мышей, у которых выявлена не измеряемая масса опухоли, или мыши умерли до первой смерти в контрольной группе.
Результаты
Модели карцином А-2780 (яичников) и А-431 (шейки матки), взятых от не получавшего лечения больного использовали в испытании из-за их устойчивости к доксорубицину (ДКР) и идарубицину (ИДА). В отношении этих опухолей % СВО и LГK, достигнутые с помощью использования соединения Ia, были всегда значительно превосходившими значения, полученные после лечения ДКР и ИДА (табл.2).

Описаны новые 8-фторантрациклины формулы I, где R = Н; R1=Н; OCH3; R2= Н, NH2; R3= ОН, NН2; символ  означает, что заместители R2, R3 могут находиться в осевой или экваториальной конфигурации, и группы 8-F и 9-ОН находятся в положении цис; или их фармацевтически приемлемые соли. Они проявляют необычайно высокую противоопухолевую активность. Описаны также способ получения заявленных соединений и содержащие их фармацевтические композиции. 5 с. и 1 з.п.ф-лы, 2 табл.
означает, что заместители R2, R3 могут находиться в осевой или экваториальной конфигурации, и группы 8-F и 9-ОН находятся в положении цис; или их фармацевтически приемлемые соли. Они проявляют необычайно высокую противоопухолевую активность. Описаны также способ получения заявленных соединений и содержащие их фармацевтические композиции. 5 с. и 1 з.п.ф-лы, 2 табл.
где R = H, R1 = H, OCH3, R2 = H, NH2, R3 = OH, NH2;
символ  означает, что заместители R2, R3 могут находиться в осевой или экваториальной конфигурации;
означает, что заместители R2, R3 могут находиться в осевой или экваториальной конфигурации;
и группы 8-F и 9-ОН находятся в положении цис,
или их фармацевтически приемлемые соли.
4-деметокси-8-(R)-фтордаунорубицин,
4-деметокси-8-(R)-фтор-3'-дезамино - 4' - дезокси- 4' - эпиаминодаунорубицин,
8- (R) -фтордаунорубицин.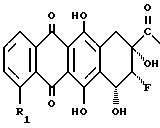
где R1 = H, OCH3 и группы 8-F и 9-ОН имеют относительную стереохимию цис.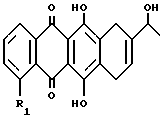
с получением соединения формулы VI
которое путем обработки основанием преобразуют в эпоксид формулы VII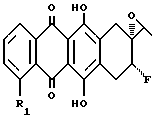
который путем обработки кислотами в водном растворе преобразуют в соответствующий диол формулы VIII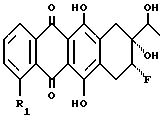
который окисляют во фторгидроксикетон формулы IX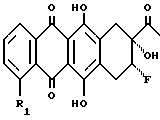
который, наконец, бромируют и подвергают сольволизу в положении 7 традиционными агнетами с получением агликона II.
