Изобретение относится к способу получения новых соединений, которые обладают биологической активностью, сходной с активностью ретеновой кислоты, более конкретно к способам и промежуточным продуктам, используемым при синтезе двухзамещенных ацетиленовых соединений, обладающих сходной с ретеновой кислотой активностью.
Известны тетрагидронафталины, имеющие этинилбензойные кислоты, а также соединения, в которых три олефиновых звена в кислотной части ретеновой кислоты замещены этинилфенильной группой. Эти соединения обладают биологической активностью, подобной активности ретеновой кислоты.
Известны также такие дизамещенные ацетиленовые соединения, в которых один из заместителей ацетилиновой (этиловой) группы представляют собой замещенную фенильную группу, и вторым заместителем является замещенная или незамещенная хроманил, тиохроманил или тетрагидрохинолинил-группа. Эти соединения обладают биологической активностью, подобной активности ретеновой кислоты.
Известны типы дизамещенных ацетиленовых соединений, в которых один заместитель ацетиленовой (этиловой) группы представляет собой замещенную фенильную или замещенную гетероарильную группу, а другой заместитель - замещенную или незамещенную хроманил, тиохроманил или тетрагидрохинолинил-группу. Эти дизамещенные ацетиленовые соединения обладают значительной ретенокислотно-подобной активностью.
Активность, подобно ретеновой кислоте, специалисты обычно связывают с полезной биологической активностью, конкретно соединения, обладающие ретенокислотно-подобной активностью, используются в качестве регуляторов клеточного разрастания и особенно в качестве препаратов для лечения дерматозов, таких как прыщи, болезнь Дарье, псориаз, ихтиоз, экзема, атопический дерматит и эпителиальные злокачественные заболевания, для лечения артритов и других иммунологических нарушений, например лупус эриматозус, для ускорения заживления ран, для лечения синдрома Съегрена и для устранения эффектов солнечного поражения кожи.
Что касается процессов предлагаемого синтеза, которые включают либо формирование ацетиленовой (этинильной) функции в соединениях по данному изобретению, либо сочетание этих соединений, которые уже обладают этинильной функцией, с галоидозамещенным фенилом или гетероарильной группой.
Изобретение относится к новому способу получения дизамещенных ацетиленовых соединений, обладающих биологической активностью, сходной с активностью ретеновой кислоты, и имеющих в качестве одного заместителя замещенную или незамещенную хроманил, тиохроманил или тетрагидрохинолинил-группу. В изобретении также раскрываются промежуточные соединения, которые могут использоваться при синтезе указанных биологически активных дизамещенных ацетиленов.
В соответствии с изобретением осуществляют реакцию 6-тиохроманил-этинового, 6-хроманил-этинового и 6-тетрагидрохинолинил-этинового соединения формулы II или соответствующих солей металла с соответствующим галоидозамещенным фенильным или гетероарильным соединением формулы III с получением соединений формулы I. Соединения формулы I либо обладают ценной билогической активностью, подобной ретеновой кислоте, либо легко стандартными операциями, например этерификацией, диэтерификацией, гомологизацией, окислением, восстановлением, амидированием и тому подобными способами могут быть превращены в соединения, обладающие активностью, аналогичной активности ретеновой кислоты.
Реакционная схема I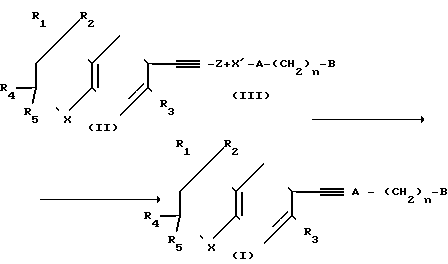 где R1, R2, R4 и R5 - могут быть одинаковыми или различными и представляют собой С1-С6-алкил;
где R1, R2, R4 и R5 - могут быть одинаковыми или различными и представляют собой С1-С6-алкил;
R3 - атом водорода или алкил с числом атомов углерода 1-6;
Х - кислород или сера;
Z - водород, ион металла или ион металла, связанный с анионом, причем этот ион металла образует соль с этинильной группой соединения;
А - фенил или пиридил;
n - целое число, равное 0;
В - группа СООR6, где R6 - C1-C6-алкил.
Реакцию, приведенную в реакционной схеме I, т.е. сочетание между соединениями формулы II и соединениями формулы III осуществляют в соответствии с предлагаемым способом в присутствии йодида меди (I), a также в присутствии Pd(PQ3)2Cl2, где Q - фенил, когда Z - водород, и в присутствии Pd(PQ3)4, где Q - фенил, или аналогичного комплекса, когда Z - ион металла, такой как ZnCl+.
Соединения, соответствующие формуле II, представляют собой другой аспект изобретения. Эти соединения используются в качестве промежуточных соединений для синтеза биологически активных и ценных ретенокислотноподобных соединений формулы I.
Фармацевтически приемлемая соль может быть получена для любого соединения, изготовленного в соответствии с настоящим изобретением, при условии, что соединение обладает функциональной способностью образовывать такую соль, например кислотную или аминную функцию. Фармацевтически приемлемой солью может быть любая соль, которая сохраняет активность исходного соединения и не оказывает отрицательного или неблагоприятного эффекта на объект, на который они направлены, в условиях применения.
Такая соль может быть получена из любой органической или неорганической кислоты или основания. Солью может быть моно- или поливалентный ион. Особый интерес в случае кислотной функции представляют неорганические ионы, натрий, калий, кальций, магний.
Соли органических аминов особенно аммониевые соли могут быть приготовлены из аминов таких, как моно-, ди-, триалкиламины или этаноламины. Соли также могут быть получены из кофеина, тометамина и аналогичных молекул. В том случае, если существует азот, достаточно щелочной, чтобы образовывать молекулярные соли с кислотами, такие соли могут получаться с любыми неорганическими или органическими кислотами или алкилирующим агентом, таким как метилйодид. Предпочтительными солями являются такие, которые получают из неорганических кислот, таких как соляная кислота, серная кислота или фосфорная кислота. Может также применяться любое число простых органических кислот таких, как моно-, ди- или трех-основных кислот.
Соединениям, которые предпочтительно используются в процессе предлагаемого синтеза для введения замещенной хроманил, замещенной тиохроманил и замещенной тетрагидрохинолинил-группы в качестве одного из заместителей этиновой группы биологически активных соединений, и которые следовательно являются предпочтительными промежуточными соединениями в изобретении, отвечает формула IV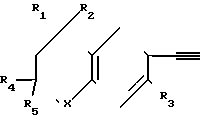


Такими предпочтительными соединениями и промежуточными веществами являются следующие:
Соединение 1: X=S; R1=R2=CH3, R3=R4=R5=H.
Соединение 2: X=S; R1=R2=R3=CH3, R4=R5=H.
Соединение 3: X=S; R1=R2=R4=R5=CH3, R3=H.
Соединение 4: X=O; R1=R2=CH3, R3=R4=R5=H.
Соединение 5: X=O; R1=R2=R4=R5=CH3, R3=H.
Cоединение 6: X=S; R1=R2=R3=R4=R5=CH3.
Соединение 7: X=O; R1=R2=R3=R4=R5=CH3.
4,4-диметил-6-этинилтиохроман (соединение 1)
4,4,7-триметил-6-этинилтиохроман (соединение 2)
2,2,4,4-тетраметил-6-этинилтиохроман (соединение 3)
4,4-диметил-6-этинилхроман (соединение 4)
2,2,4,4-тетраметил-6-этинилхроман (соединение 5)
2,2,4,4,7-пентаметил-6-этинилтиохром- ан (соединение 6)
2,2,4,4,7-пентаметил-6-этинилхроман (соединение 7).
В предлагаемом способе также предпочтительны соли металла, преимущественно цинка, в качестве промежуточных предпочтительных соединений.
Соединения формулы II и их соли металлов, таких как цинк, предпочтительно соединения формулы IV (или их соли), взаимодействуют с соединениями формулы III с получением соединений формулы I.
Когда предпочтительные соединения формулы IV взаимодействуют с соединениями формулы III, тогда в получаемых соединениях формулы I заместители Х, R1, R2, R3, R4 и R5 соответствуют определениям, данным для соединений 1-7.
Реакционные условия для осуществления реакции сочетания соединений формулы II с соединениями формулы III в соответствии с предлагаемым способом так же, как и процессы синтеза соединений формулы II, подробно раскрываются в последующем.
В отношении биологической активности соединений формулы I, полученных в соответствии с предлагаемым способом (используя промежуточные соединения по изобретению), необходимы следующие пояснения. Активность этих соединений, подобная активности ретеновой кислоты, подтверждается стандартным измерением активности ретеновой кислоты, включая действие ретеновой кислоты на орнитиндекарбоксилазу. Известна работа, устанавливающая связь между ретеновой кислотой и снижением размножения клеток. В этой работе раскрывается, что активность орнитиндекарбоксилазы (ОДС) усиливает склонность к биосинтезу полиамина. Было установлено, что увеличение синтеза полиаминов может коррелироваться с клеточным размножением. Так при подавлении ОДС-активности может меняться гиперразмножение клеток. Хотя неизвестны все причины, вызывающие увеличение ОДС-активности. Известно однако, что 12-О-тетрадеканоилфорбол-13-ацетат (ТФА) стимулирует ОДС-активность. Ретеновая кислота подавляет это стимулирование ОДС-активности ТФА. Соединения по изобретению также подавляют ТФА-стимулирование ОДС, что иллюстрируется опытами известной методики.
В качестве примера активности, подобной активности ретеновой кислоты, следует указать, что в эксперименте, осуществленном в соответствии с методикой Бермана и Ботвелла, следующие примеры соединений (соединения 10, 11 и 12), полученные в соответствии с предлагаемым способом, вызывают 80%-ное подавление ТФА-вызванной ОДС-активности при следующих концентрациях (IC80): Cоединение IC80 конц.ммоль 10 0,69 11 0,13 12 0,12
Соединения 10, 11 и 12 характеризуются со ссылкой на формулу I:
Соединение 10: X=S, R1=R2=R4=R5=CH3
R3=H, A-(CH2)n-B=этил 6-никотинат
Соединение 11: X=O, R1=R2=R4=R5=CH3
R3=H A-(СH2)n-B=этил 6-никотинат
Соединение 12 Х=О, R1=R2=R3=R4=R5=CH3 A-(CH2)n=B=этил-6-никотинат
В силу ретеноподобной активности соединения формулы III могут использоваться длительно или местно при лечении различных заболеваний.
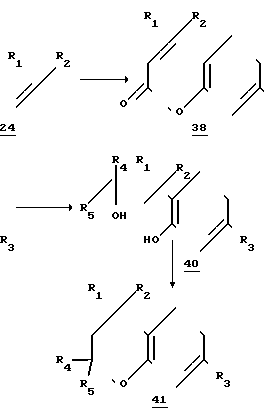
Соединения по изобретению, которые соответствуют формуле II и являются ключевыми промежуточными соединениями в стадиях предлагаемого синтеза, могут быть получены разными путями. Для иллюстрации процесса этого синтеза приведен ряд стадий, которые гарантируют получение соединений формул II и I при строгом следовании описанной методике. Указанные условия являются конкретными примерами осуществления, которые применимы к любому и всем соединениям по формулам II и I. Очевидно, что описываемые стадии могут меняться и/или регулироваться без отклонения от объема и смысла предлагаемого способа. Также следует иметь ввиду, что некоторые из стадий и способов, используемых в приведенных примерах соединений формулы II, являются новыми и новаторскими.
Со ссылкой на соединение формулы II схема 2 реакции иллюстрирует пример их синтеза, когда Х=сера; R4 и R5 - водородные атомы.
Реакционна схема 2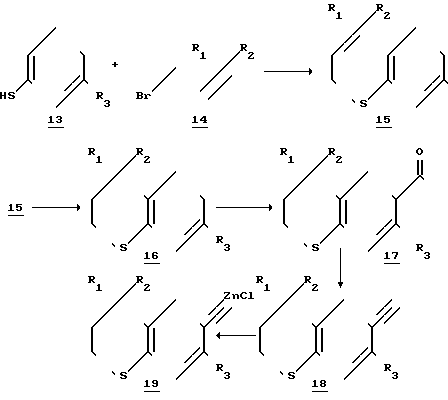

Учитывая реакционную схему 2, тиофенол 13, который может иметь R3-заместитель (определенный выше как атом водорода или низший алкил), алкилируют, предпочтительно в жестких щелочных условиях, например в едком натре, в полярном растворителе (ацетон при комнатной температуре) соединением 14, которым является либо 1-бром-3-метил-2-бутен (R1 и R2 - метил фирмы Олдрич), либо его производное, где либо R1 либо R2, либо оба являются алкилами, отличными от метила. Полученный алкилированный тиофенол (сульфид, соединение 15) затем подвергают реакции циклизации по Фриделю-Крафтсу или в подобных условиях, обычно при кипячении с обратным холодильником в инертном растворителе, таком как бензол или толуол, в присутствии фосфорного ангидрида и фосфорной кислоты. Получаемый в соответствии со схемой 2 тиохроман (соединение 16) не содержит заместителей в положении 2 и предпочтительно в соответствии со схемой 2 R1 и R2 - метилы и R3 - водород.
Соединение 16 ацетилируют по Фриделю-Крафтсу или при аналогичных условиях, предпочтительно ацетилхлоридом (AlCl3, CH2Cl2, кипячение с обратным холодильником) с получением 6-ацетил-тиохромана (соединение 17). Ацетильную группу соединения 17 превращают в ацетиленовую (этинильную) функцию с помощью диизопропиламида лития, или аналогичного основания при пониженной температуре. Промежуточное соединение, полученное из соединения 17, предположительно литиевая соль соответствующего енола (на схеме 2 не показана), этерифицируют обработкой диэтилхлорфосфатом (или подобным соединением) и вновь осуществляют реакцию при пониженной температуре, например -78оС, с диизопропиламидом лития, приводящую к образованию тройной связи, предположительно в ходе реакции отщепления, с получением 6-этинилтиохроманового производного (соединение 18).
В связи с этим следует отметить, что предлагаемый синтез не ограничен и не зависит от конкретного механизма реакции.
6-этинил-тиохроман (соединение 18) либо используется непосредственно в реакции сочетания, указанной в схеме I, либо предварительно его превращают в металлическую (цинковую) соль (соединение 19).
Хлоридцинковые соли (соединение 19) готовят в отсутствии воды и кислорода. В качестве растворителя может использоваться безводный эфирного типа растворитель, такой как серный эфир или циклический простой эфир, такой как фуран или пиран, особенно тетрагидрофуран. Раствор соединения 18 сначала получают в инертной атмосфере (аргона или азота) и затем добавляют сильное основание, такое как н-бутиллитий (примерно при 10%-ном молярном избытке). Реакцию начинают при пониженной температуре в интервале -10 - +10оС, предпочтительно примерно при 0оС. Реакционную смесь перемешивают в течение короткого промежутка времени (в интервале 0,5-2 ч и затем обрабатывают примерно при 10% -ном молярном избытке плавленным хлоридом цинка, растворенным в реакционном растворителе. Смесь перемешивали дополнительно 1-3 ч при примерно начальной температуре, затем температуру повышали примерно до температуры окружающей среды в течение 10-40 мин.
Такое получение хлоридцинковых солей, соответствующих соединению 19, также применимо с такими модификациями, которые обеспечивают получение всех цинкохлоридных солей, соответствующих формуле II.
Реакционная схема 3 показывает другой способ получения соединения формулы II, когда Х - сера и R4 и R5 - водород. Приведенная на схеме 3 последовательность реакций является предпочтительной (но не обязательной)/ когда R3 - не является водородом/ тогда как схема 2 предпочтительно осуществляетя (но не обязательно)/ когда R3 - водород.
Таким образом/ следуя схеме 3/ 4-бромтио фенол (соединение 20)/ предпочтительно алкил-замещенное в положении 3/ алкилируют соединением 14. Получаемые 4-бромфенилсульфиды замыкают в кольцо при условиях/ аналогичных замыканию кольца соединения 15/ описанному в реакционной схеме 2.
Реакционная схема 3.
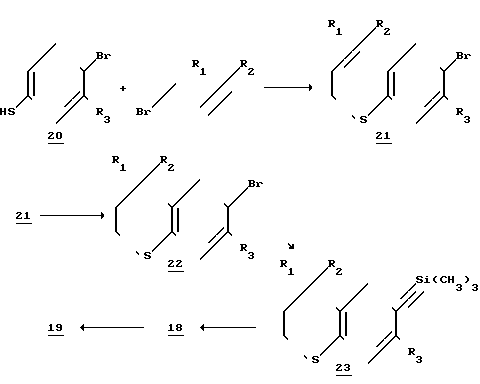
Для введения ацетиленовой группы в молекулу осуществляют реакцию замещенного 6-бромтиохромана (соединение 22) с триметилсилилацетиленом и в присутствии йодида меди (I) и соответствующего катализатора, которому обычно отвечает формула Pd(PQ3)2Cl2 (Q - фенил). Реакцию обычно осуществляют в присутствии катализатора - хлористого бис(трифенилфосфин)палладия (II), кислотного акцептора, (такого как триэтиламин) в атмосфере инертного газа (аргона) путем нагрева в герметично укупоренной трубке. Получаемый 6-триметилсилилэтинилтиохроман показан в виде соединения 23 на схеме 3.
Как далее следует из схемы 3, триметилсилил удаляют из 6-триметилсилилэтинил-ти- охромана (23) в следующей стадии синтеза, получая 6-этинил-тиохромановое производное с кольцевым заместителем (соединение 18). Последнюю реакцию осуществляют при щелочных условиях, предпочтительно в атмосфере инертного газа.
6-этинил-тиохроман 18 может использоваться непосредственно в реакции сочетания, как указано в реакционной схеме I, или перед сочетанием может быть превращено в соответствующую цинкхлоридную соль, как было описано.
Реакционная схема 4 описывает получение соединений, соответствующих формуле II, где Х - сера и по меньшей мере один из радикалов R4 и R5представляют собой низший алкил, предпочтительно, когда оба R4 и R5представляет собой низшие алкилы, и еще более предпочтительно, когда R4и R5 идентичны друг другу.
Схема 4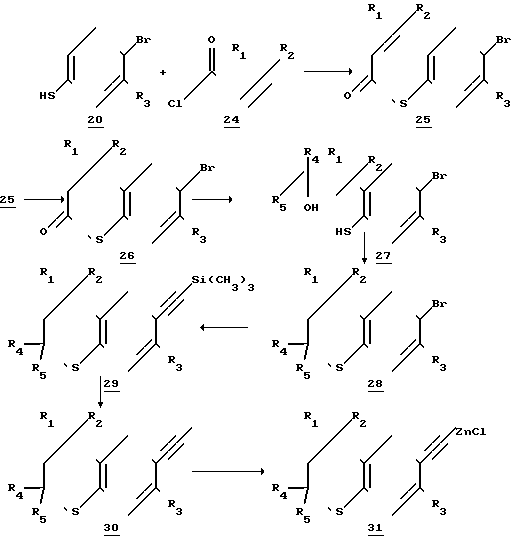
Следуя схеме 4, 2-замещенные, предпочтительно 2,2-дизамещенные, 6-этинил-тиохроманы могут быть получаться следующим образом: 4-бром-тиофенол (соединение 20) ацилируют с помощью ацилирующего агента, такого как хлорангидрид кислоты (соединение 24), получаемой из соответствующим образом замещенной акриловой кислоты. Ацилирование осуществляют в инертном растворителе (таком как тетрагидрофуран) в присутствии сильного основания (например, гидрида натрия). Получаемый тиоэфир (соединение 25), который содержит олефиновую связь акриловокислой группы, замыкают в кольцо в присутствии катализатора Фриделя-Крафтса (такого как алюминийхлорид) путем перемешивания в соответствующем растворителе, такой как дихлорметан. Получаемый 2-оксо-6-бром-тиохроман (соединение 26) обычно выделяют в кристаллической форме.
R4 и/или R5 заместители (оба из которых не могут быть одновременно атомами водорода в случае осуществления процесса по схеме 4) и которые предпочтительно идентичны друг другу, например оба метилы, вводят путем обработки 2-оксо-6-бромтиохромана (соединение 26) реактивом Гриньяра, несущим алкил-заместители R4 и R5 (таким как метилмагнийбромид, когда R4и R5 - метил). Очевидно, что в зависимости от молекулярного соотношения реактива Гриньяра и оксо-тиохромана (соединения 26), а также в зависимости от реакционных условий первичные продукты реакции могут представлять собой такие соединения, в которых либо одна, либо обе алкильные группы вводятся в ходе реакции Гриньяра. Когда реактив Гриньяра, такой как метилмагнийбромид, используется в избытке, тиохромановое кольцо раскрывается и получается третичный спирт - производное 4-бром-тиофенол (соединение 27).
Раскрытие кольца тиофенольного производного (соединение 27), содержащего требуемые R1, R2, R3, R4 и R5 заместители, осуществляется путем нагрева в кислых условиях, предпочтительно путем нагрева соединений 27 в водном растворе кислоты. Получаемый 6-бромтиохроман, который содержит необходимые алкильные (или водородные) заместители R1, R2, R3, R4 и R5 показан на реакционной схеме как соединение 28.
6-бромтиохроманы 28 (колторве отличаются от6-бромтиохроманов 22 только тем/ что соединение 28 в положении 2 тиохроманового кольца содержит заместитель) превращают в 6-(2-триметилсилил)- энитильное производное 29/ и затем в 6-этинильное производное 30 и далее/ если необходимо/ в ZnCl-соли (31) в стадиях реакции/ которые аналогичны соответствующим стадиям/ описанным в связи с реакционной схемой 3.
Реакционная схема 5 в общих чертах раскрывает получение соединений формулы II, в которых X - кислород. Другими словами/ схема 6 раскрывает получение 2-замещенных и предпочтительно 2/2-дизамещенных 6-этинилхроманов.
Реакционная схема 5.
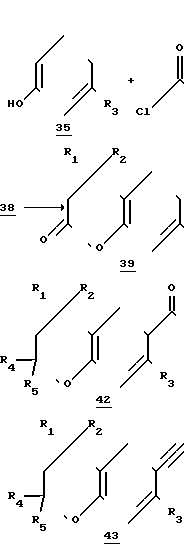

Так, в соответствии со схемой 5 фенол или замещенный в мета-положении фенол алкилзаместителем (R3) (соединение 35) ацилируют ацилирующим реагентом, таким как хлорангидрид (соединение 24), получаемым из соответственно замещенной акриловой кислоты. В схеме 5, также как и в схеме 4, R1 и R4 - заместители целевых соединений вводят с помощью этого акриловокислого производного 24. Ацилирование хлорангидридом 24 предпочтительно осуществляют в присутствии сильного основания (например, гидрида натрия) в инертном растворителе (таком как тетрагидрофуран). В реакционной схеме 2 в качестве соединения 38 показан получаемый замещенный фенилакрилат.
Осуществляя реакцию по Фриделю-Крафтсу (катализатор AlCl3, инертный растворитель, такой как дихлорметан) из замещенного фенилакрилата 38 получают кольцевое соединение, такое как 2-оксо-хроман (соединение 39), у которого в положении 4 имеется два R1 и R2 заместителя и в положении 6 R3 - заместитель. Аналогично 2-оксо-тиохроману 26 в схеме 4, 2-оксохроман 39 по схеме 6 обрабатывают реактивом Гриньяра для введения R4 и R5 заместителей. Как ранее уже указывалось, в этой схеме R4 и R5оба не могут быть атомами водорода, и в предпочтительных примерах осуществления R4 и R5 являются идентичными, например оба являются метил- или этилгруппами. Когда R4 и R5 являются метилами, в качестве реактива Гриньяра предпочтительно используется метилмагнийхлорид, растворенный в тетрагидрофуране (ТГФ). Раствор соединения 39 в подходящем растворителе, например в безводном серном эфире, добавляют к указанному реактиву Гриньяра. На схеме 6 в качестве примера соединения 40 показан получаемый фенол, содержащий третичную спиртовую боковую цепь (это есть молекула, в которой раскрыто хромановое кольцо).
Соединение 40, в котором уже имеются необходимые R1, R2, R3, R4 и R5 заместители, образует в кислых условиях (например, путем нагрева водной серной кислоты) ядро, давая хромановое производное (соединение 41). Следует отметить, что вплоть до этого момента последовательность реакций синтеза включает сходное или аналогичные стадии получения как 2,2-дизамещенных производных тиохромана (реакционная схема 4), так и 2,2-дизамещенных производных тиохромана (реакционная схема 4), так и 2,2-дизамещенных производных хромана (реакционная схема 5), с единственным отличием, что в схеме 5 исходное фенольное производное не содержит галоидного заместителя (такого как бром).
Как дополнительно раскрывается в схеме 5, этинильную группу вводят в замещенный в положении 2 (предпочтительно 2,2-дизамещенный) хроман, осуществляя последовательность реакций, аналогичную стадиям схемы 2 для введения этильной функции в 4-замещенные тиохромана. В соответствии с описанной методикой 6-этинилхроман (43) также может использоваться непосредственно в реакции сочетания с соединениями формулы II или может быть превращен в подходящую соль металла, предпочтительно ZnCl-соль.
Соединения формулы III сочетаются в соответствии с предлагаемым способом с соединениями формулы II, обеспечивая получение биологически активных соединений формулы I. Соединение формулы III сами по себе не являются новыми и могут быть получены известными способами.
В соответствии с одним предпочтительным примером осуществления предлагаемого синтеза группы А в формуле III представляет собой фенильную группу, Х1 - галоид, предпочтительно бром или йод. Этиловый эфир 4-йодобензойной кислоты является одним из предпочтительных примеров реагента формулы III, используемого при сочетании с соединениями формулы II. Другими примерами, в которых А - фенил, являются: этиловый эфир 4-йодофенилуксусной кислоты, этиловый эфир 4-йодофенилпропионовой кислоты, этиловый эфир 4-йодо-фенилбутановой кислоты, этиловый эфир 4-йодо-фенилпентановой кислоты. Также могут использоваться в предлагаемом способе реагентами в соответствии с формулой III, в которой Х1 и (СН2)n-B - заместители соответственно находятся в мета- или орто-положении фенильного ядра.
Получение таких соединений формулы III, пригодных для сочетания с соединениями формулы II, должно использоваться в свете предлагаемого синтеза.
Когда А-группа формулы III представляет собой гетероарил, примерами предпочтительных реагентов являются следующие соединения:
этиловый эфир 6-хлорникотиновой кислоты;
этиловый эфир 2-(2-хлорпирид-5-ил)уксусной кислоты;
этиловый эфир 5-(2-хлорпирид-5-ил)-пентановой кислоты;
этиловый эфир 2-(2-йодофур-5-ил)уксусной кислоты;
этиловый эфир 5-(2-йодофур-5-ил)пентановой кислоты;
этиловый эфир 2-(2-йодотиен-5-ил)уксусной кислоты;
этиловый эфир 5-(2-йодотиен-5-ил)пентановой кислоты;
этиловый эфир 5-(3-хлорпиридазин-6-ил)пентановой кислоты, и соответствующие хлор или иные галогено-замещенные пиримидинил или пиразинил-аналоги указанных сложных эфиров. В этом ряду также В-группа соединения формулы 2 может быть защищена или незащищена спиртовой альдегидной, кето-, амидной или другой группой, указанной в связи с формулой II.
В объем изобретения также входит осуществление сочетания соединений формулы II с соединениями формулы III с последующим осуществлением обычных операций, используемых при синтезе, таких как блокировка и деблокировка, гомологация, восстановление и окисление, получение сложного эфира, омыление и тому подобные операции, над получаемой молекулой (особенно той ее частью, которая получается от реагента формулы III), в результате чего получают дополнительные аналоги в пределах объема формулы I и обладающие рентгеноподобной биологической активностью.
Что касается новой реакции сочетания в предлагаемом способе, то следует сделать необходимые пояснения. Сочетание обычно осуществляется в присутствии йодида меди (I), подходящего катализатора, обычно формулы Pd(PQ3)2Cl2, и кислотного акцептора, такого как триэтиламин, путем нагрева в герметически укупоренной трубке в атмосфере инертного газа.
Альтернативно, соединение формулы II сначала превращают в соль (препочтительно ZnCl соль) для сочетания. Пример осуществления способа получения солей ZnCl и соединений формулы II (где Z превращают из водорода в ZnCl), описан ранее для соединения 19.
Сочетание ZnCl солей 6-тиохроманил, 6-хроманил и 6-(1,2,3,4-тетрагидрохинолинил)этиновых соединений с соединениями формулы III осуществляют в присутствии Pd(PQ3)4 катализатора (Q - фенил). Более подробно условия протекания реакций раскрываются в примерах получения конкретных соединений.
Фенил-3-метилбут-2-анилсульфид (соединение 60).
В течение 2,5 ч кипятили с обратным холодильником смесь 14,91 г (135,324 ммоль) тиофенола и 5,5 г (137,5 ммоль) едкого натра в 100 мл ацетона, и затем по каплям добавляли раствора 20 г (134,19 ммоль) 1-бром-3-метил-2-бутена в 20 мл ацетона. Полученный раствор кипятили с обратным холодильником в течение 40 ч и затем перемешивали при комнатной температуре 24 ч. Растворитель затем отгоняли в вакууме и остаток растворяли в воде и экстрагировали трижды серным эфиром порциями по 50 мл. Эфирные экстракты объединяли и промывали трижды порциями по 30 мл 5%-ного раствора едкого натра, затем водой, насыщенным раствором хлорида натрия и сушили над сульфатом магния. Растворитель затем отгоняли в вакууме, и остаток дополнительно очищали перегонкой с шариковым дефлегматором (80оС, 0,75 мл) с получением целевого соединения в виде бледно-желтого масла.
ЯМР-спектр (СDСl3) δ1,57 (3H, синглет), 1,68 (3Н, синглет), 3,52 (2Н, дуплет, J 7,7 Гц), 5,29 (1Н, триплет, J 7,7 Гц), 7,14 (1Н, триплет, J 7,0 Гц), 7,24 (2Н, триплет, J 7,0 Гц), 7,32 (2Н, дуплет, J 7,0 Гц).
Следуя описанной методике и используя вместо тиофенола соответствующие 3-алкилтиофенолы, были получены следующие соединения:
3-метилфенил-3-метилбут-2-енилсуль-фид;
3-этилфенил-3-метилбут-2-енилсульфид;
3-пропилфенил-3-метилбут-2-енилсульфид;
3-бутилфенил-3-метилбут-2-енилсульфид;
3-пентилфенил-3-метилбут-2-енилсульфид;
3-гексилфенил-3-метилбут-2-енилсульфид.
4,4-Диметилтиохроман (соединение 61).
К раствору 15,48 г (86,824 ммоль) фенил-3-метилбут-2-енилсульфида (соединение 60) в 160 мл бензола последовательно добавляли 12,6 г (88,767 ммоль) фосфорного ангидрида и 11 мл 85%-ной фосфорной кислоты. Этот раствор кипятили с обратным холодильником при энергичном перемешивании в атмосфере аргона в течение 20 ч, затем охлаждали до комнатной температуры. Всплывший верхний органический слой декантировали, и сиропообразный остаток экстрагировали трижды эфирными порциями по 30 мл. Органические фракции объединяли и промывали водой, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток очищали перегонкой с шариковым дефлектором (при 80оС и 0,5 мм рт.ст.), получая целевое соединение в виде бледножелтого масла.
ЯМР-спектр (СDСl3): δ1,30 (6Н, синглет), 1,90-1,95 (2Н, мультиплет), 2,95-3,00 (2Н, мультиплет), 6,96-7,00 (2Н, мультиплет), 7,04-7,07 (1Н, мультиплет), 7,30-7,33 (1Н, мультиплет).
Такой способ может использоваться для получения аналогов с алкилами в положении 7, что иллюстрируется следующими соединениями:
4,4,7-триметилтиохроман (соединение 2)
4,4-диметил-7-этилтиохроман
4,4-диметил-7-пропилтиохроман
4,4-диметил-7-бутилтиохроман;
4,4-диметил-7-гексилтиохроман.
4,4-Диметил-6-ацетилтиохроман (соединение 62).
Раствор 14,3 г (80,21 ммоль) 4,4-диметилтиохромана (соединение 61) и 6,76 г (86,12 ммоль) ацетилхлорида в 65 мл бензола охлаждали на ледяной бане и по каплям добавляли 26,712 г (102,54 ммоль) хлористого олова (IV). Смесь перемешивали при комнатной температуре в течение 12 ч, затем промывали 65 мл воды и 33 мл концентрированной соляной кислоты и нагревали с обратным холодильником в течение получаса. После охлаждения до комнатной температуры органический слой отделяли, и водный слой экстрагировали пятью порциями по 50 мл бензола. Органические фракции объединяли и промывали 5% -ным карбонатом натрия, водой, насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакуме, и остаток очищали флешхроматографическим методом (на силикагеле, 5% этилацетата в гексанах) с последующей дистилляцией с шариковым дефлегматором (150оС, 0,7 мм рт.ст.), получая целевой продукт в виде бледно-желтого масла.
ЯМР-спектр (СDCl3): δ1,35 (6Н, синглет), 1,92-1,98 (2Н, мультиплет), 2,54 (3Н, синглет), 3,02-3,08 (2Н, мультиплет), 7,13 (1Н, дуплет, J 8,6 Гц), 7,58 (1Н, дуплет, дуплет, J 8,6 Гц, 2 Гц), 7,99 (1Н, дуплет, J 2 Гц).
Описанная процедура предназначена для ацетилирования всех соединений, которые могут получаться способом, описанным для синтеза соединения 61.
4,4-Диметил-6-этинилтиохроман (соединение 1)
К раствору 1,441 г (14,2405 ммоль) диизопропиламина в 30 мл сухого тетрагидрофурана в атмосфере аргона при -78оС по каплям добавляли 9 мл (14,4 ммоль) н-бутиллития в гексане. После перемешивания этого раствора при -78оС в течение часа по каплям добавляли раствор 2,95 г (13,389 ммоль) 4,4-диметил-6-ацетилтиохромана (соединение 62) в 5 мл сухого тетрагидрофурана. После дополнительного перемешивания в течение часа при -78оС раствор обрабатывали 2,507 г (14,53 ммоль) диэтилхлорфосфата и доводили до комнатной температуры, после чего перемешивали в течение 3,75 ч. Раствор переводили в раствор литийдиизопропиламида (приготовленного из 2,882 г (28,481 ммоль) диизопропиламина и 18 мл 1,6М (28,8 ммоль) н-бутиллития в гексане)) в 60 мл сухого тетрагидрофурана при -78оС, используя двухконцевую иглу. Охлаждающую баню удаляли, и раствор перемешивали при комнатной температуре в течение 15 ч, затем гасили водой и подкисляли до рН 1 трехнормальной соляной кислотой. Смесь экстрагировали пятью порциями по 50 мл пентана, и объединенные органические фракции промывали трехнормальной соляной кислотой, водой, насыщенным раствором бикарбоната натрия и насыщенным раствором поваренной соли, затем сушили над сульфатом магния. Растворитель затем удаляли в вакууме, остаток очищали с шариковым дефлектором (100оС, 0,7 мм рт.ст.), получая целевой продукт в виде бледножелтого твердого вещества.
ЯМР-спектр (СDCl3): δ1,34 (6Н, синглет), 1,94-1,99 (2Н, мультиплет), 3,04-3,08 (3Н, мультиплет), 7,06 (1Н, дуплет, J 8,4 Гц), 7,17 (1Н, дуплет-дуплет, J 8,4 Гц, 2,1 Гц), 7,51 (1Н, дуплет, J 2,1 Гц).
Аналогично, у всех соединений, приготовленных так же, как и соединение 62, ацетильная группа может быть превращена в этинильную группу.
4,4-Диметил-6-этинилтиохроман ZnCl (соединение 63) и этиловый эфир 4-(4,4-диметилтиохроман-6-ил-этинил)бензойной кислоты (соединение 64)
Использовали реакторы, высушенные на горелке под вакуумом, и все операции осуществляли в отсутствии кислорода в атмосфере аргона или азота. К раствору 533,9 мг (2,6389 ммоль) 4,4-диметил-6-этинил-тиохромана (соединение I) в 4 мл безводного тетрагидрофурана при 0оС по каплям добавляли 1,7 мл 1,6 М (2,72 ммоль) н-бутиллития в гексане. Реакционную массу перемешивали при 0оС в течение 10 мин и при комнатной температуре в течение 15 мин, вновь охлаждали до 0оС и затем обрабатывали раствором 410 мг (3,005 ммоль) расплавленного хлорида цинка в 4 мл безводного тетрагидрофурана, используя иглу с двумя концами. Затем раствор перемешивали при 0оС в течение 45 мин, при комнатной температуре в течение 20 мин и получали соединение 63. Продукт без выделения использовали в последующей реакции. Раствор 724,4 мг (2,6243 ммоль) этилового эфира 4-йодобензойной кислоты в 4 мл безводного тетрагидрофурана переводили двухконцевой иглой в суспензию 520 мг (0,45 ммоль) тетракисфенилфосфинпалладия в 5 мл безводного тетрагидрофурана и перемешивали при комнатной температуре в течение 20 мин, затем обрабатывали раствором алкинилцинкхлорида, приготовленного выше, используя при введении двухконцевую иглу. Смесь перемешивали при комнатной температуре 18 ч, затем гасили льдом и 30 мл трехнормальной соляной кислоты. Продукт выделяли экстракцией тремя порциями по 75 мл серного эфира. Эфирные экстракты объединяли и последователно промывали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали хроматографически (на силикагеле, 5% -ным этилацетатом в гексане), а затем методом жидкостной хроматографии при высоком давлении (Вотмен Партисил М-9 10/50; 4% этилацетата в гексане), получая этиловый эфир 4-(4,4-диметилтиохроман-6-ил-этинил)бензойной кислоты (соединение 64) в виде бесцветного масла.
ЯМР-спектр (СDCl3): δ 1,36 (6H), 1,42 (3Н, триплет, J 7 Гц), 1,93-1,99 (2Н, мультиплет), 3,03-3,08 (2Н, мультиплет), 4,40 (2Н, квартет, J 7 Гц), 7,09 (1Н, дуплет, J 8,4 Гц), 7,22 (1Н, дуплет, дуплет, J 7,8 Гц), 8,04 (2Н, дуплет, J 7,8 Гц).
(3-Метил-4-бром-фенил)-3-метилбут-2-енилсульфид (соединение 65)
К перемешиваемому раствору 9,52 г (68 ммоль) 3-метил-4-бромтиофенола в 80 мл ацетона добавляли 2,86 г (68 ммоль) порошкованной гидроокиси натрия, и смесь перемешивали до завершения растворения. Затем реакционную смесь нагревали с обратным холодильником, и затем обрабатывали раствором 11,26 г (68 ммоль) 4-бром-2-метил-2-бутена в 20 мл ацетона. Смесь нагревали с обратным холодильником в течение 0,5 ч, охлаждали до комнатной температуры, и растворитель отгоняли в вакууме. Остаток растворяли в 35 мл воды и экстрагировали серным эфиром. Эфирные экстракты объединяли и последовательно промывали водой и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния.
Растворитель отгоняли в вакууме, и остаток подвергали дистилляции с шариковым дефлегматором (140-145оС, 0,2 мм рт.ст.), получая целевой продукт в виде бесцветного масла.
ЯМР-спектр (СDCl3): δ1,58 (3Н, синглет), 1,70 (3Н, синглет), 2,33 (3Н, синглет), 3,49 (2Н, дуплет, J 7,8 Гц), 5,26 (1Н, триплет, J 7,8 Гц), 6,98 (1Н, дуплет, дуплет, J 8,3 Гц, 2,3 Гц), 7,17 (1Н, дуплет, J 2,3 Гц), 7,38 (1Н, дуплет, J 8,3 Гц).
4,4,7-Триметил-6-бромтиохроман (соединение 66).
К 40 г энергично перемешиваемой смеси 10%-ного фосфорного ангидрида в метансульфокислоте медленно добавляли 6,0 г (28,8 ммоль) (3-метил-4-бромфенил)-3-метилбут-2-енилсульфида (соединение 65). Смесь перемешивали при комнатной температуре в течение 2 ч и затем выливали в лед. Смесь экстрагировали дважды с поршнями серным эфиром по 40 мл каждая и объединенные эфирные экстракты последовательно промывали водой и насыщенным раствором хлорида натрия и затем сушили. Растворитель отгоняли в вакууме, и остаток подвергали дистилляции с шариковым дефлегматором при 130оС и 0,07 мм рт.ст. , получая целевое соединение в виде вязкого масла.
ЯМР-спектр (CDCl3): δ1,28 (6Н, синглет), 1,84-1,93 (2Н, мультиплет), 2,26 (3Н, синглет), 2,95-3,03 (2Н, мультиплет), 6,94 (1Н, синглет), 7,46 (1Н, синглет).
4,4,7-Триметил-6-триметилсилилэти- нилтиохроман (соединение 67).
Смесь 624 мг (3,0 ммоль) 4,4,7-триметил-6-бромтиохроман (соединение 66), 314 мг (3,2 ммоль) триметилсилилацетилена, 40 мг (0,21 ммоль) иодида меди (I), 80 мг (0,11 ммоль) хлорида бис (трифенилфосфин) палладия (II) и 1 мл триэтиламина дегазировали в атмосфере азота и нагревали в запаянной трубке при 85оС в течение 15 ч. Смесь затем дополнительно обрабатывали 20 мг (0,11 ммоль) иодида меди (I) и 40 мг (0,06 ммоль) палладиевого (II) катализатора. Затем смесь нагревали в атмосфере азота в запаянной трубке при 100оС в течение 64 ч. Триэтиламин отгоняли в вакууме, и остаток очищали хроматографически на силикагеле, используя для элюирования гексана, получая целевой продукт в виде желтого масла.
ЯМР-спектр (CDCl3): δ0,28 (9Н, синглет), 1,30 (6Н, синглет), 1,88-1,97 (2Н, мультиплет), 2,33 (3Н, синглет), 2,97-3,05 (2Н, мультиплет), 6,92 (1Н, синглет), 7,43 (1Н, синглет).
4,4,7-Триметил-6-этинилтиохроман (соединение 2)
Смесь 380 мг (1,69 ммоль) триметилсилил-(4,4,7-триметилтиохроман-6-ил)ацети-лена. Соединение 67, 4 мл изопропанола и 2,5 мл водного однонормального едкого калия дегазировали в атмосфере азота и перемешивали при комнатной температуре в течение 16 ч. Смесь концентрировали под вакуумом и экстрагировали двумя порциями серного эфира (по 10 мл каждая). Эфирные экстракты объединяли и последовательно промывали водой и насыщенным раствором хлорида натрия, и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, получая целевое соединение в виде желтого масла.
ЯМР-спектр (СDCl3): δ1,31 (6H, синглет), 1,88-1,96 (2Н, мультиплет), 2,35 (3Н, синглет), 3,00-3,08 (2Н, мультиплет), 3,25 (1Н, синглет), 6,94 (1Н, синглет), 7,47 (1Н, синглет).
S-(4-бромфенил)овый эфир 3,3-диметилтиоакриловой кислоты (соединение 69)
К охлаждаемому в ледяной бане раствору 1,92 г (80 ммоль) гидрида натрия, приготовленного из 60% -ной суспензии в минеральном масле промывкой 3х15 мл гексана, в 30 мл безводного тетрагидрофурана медленно добавляли в атмосфере аргона раствор 15,1 г (80 ммоль) 4-бромтиофенола в 60 мл безводного тетрагидрофурана в течение 1 ч. Смесь перемешивали при 0оС в течение последующих 30 мин и затем обрабатывали раствором 10,1 г (85 ммоль) диметилакрилоилхлорида в 30 мл безводного тетрагидрофурана. Охлаждающую баню затем удаляли, и смесь перемешивали при комнатной температуре в течение 40 ч. Реакционную смесь выливали в 200 мл воды, содержащей 2 мл ледяной уксусной кислоты, и органический слой отделяли. Этот органический слой промывали дважды водой (порциями по 75 мл) и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, получая целевое соединение в виде желтого масла.
ЯМР-спектр (CDCl3): δ1,91 (3H, синглет), 2,14 (3Н, синглет), 6,03-6,06 (1Н, мультиплет), 7,28 (2Н, дуплет, J 8,6 Гц), 7,53 (2Н, дуплет, J 8,6 Гц).
4,4-Диметил-6-бром-2-оксо-тиохроман (соединение 70).
К перемешиваемой охлаждаемой льдом суспензии 15,9 г (119 ммоль) алюминийхлорида в 140 мл дихлорметана добавляли в атмосфере азота раствор 21,64 г (79,9 ммоль) S-(4-бромфенил)ового эфира 3,3-диметилтиоакриловой кислоты (соединение 69) в 100 мл дихлорметана. Смесь затем перемешивали при комнатной температуре в течение 72 ч и затем выливали в 250 г льда и рассола. Смесь экстрагировали дихлорметаном, и объединенные органические экстракты промывали насыщенным раствором поваренной соли и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток подвергали перекристаллизации из гексанов, получая целевое соединение в виде белых кристаллов.
ЯМР-спектр (СDCl3): 1,40 (6Н, синглет), 2,67 (2Н, синглет), 7,31-7,40 (3Н, мультиплет);
Масс-спектр m/e 269,9714 (расчетная величина для С11Н11SOBr равна 269,9714).
4-Бром-2-(1,1,3-триметил-3-оксибутил)-тиофенол (соединение 71).
К 3,49 г (32,8 ммоль) перхлората лития в атмосфере аргона добавляли 35 мл 3,0 М (105 ммоль) метилмагний бромида в серном эфире. Полученную смесь по каплям при перемешивании обрабатывали раствором 2,961 г (10,926 ммоль) 4,4-диметил-6-бром-2-оксо-тиохромана (соединение 70), и реакционную смесь затем кипятили с обратным холодильником в течение 70 ч. Реакционной смеси затем давали возможность охладиться и вливали ее в смесь 100 г льда и 8 мл концентрированной серной кислоты. Отделяли органический слой, и водный слой дважды экстрагировали серным эфиром (порциями по 25 мл каждая). Органические слои объединяли и последовательно промывали дважды по 25 мл насыщенным раствором бикарбоната натрия, 25 мл воды и 25 мл насыщенного раствора поваренной соли и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали хроматографически, получая целевой продукт в виде желтого масла.
ЯМР-спектр (CDCl3): δ1,05 (6Н, синглет), 1,52 (6Н, синглет), 2,30 (2Н, синглет), 3,71 (1Н, синглет), 7,22 (1Н, дуплет-дуплет, J 8,5 Гц, 2,1 Гц), 7,28 (1Н, дуплет, J 8,5 Гц), 7,35 (1Н, дуплет, J 2,1 Гц).
Используя этилмагнийбромид вместо метилмагнийбромида, получали соответствующий 4-бром-2-(1,1-диметил-3-этил-3-оксипентил)-тиофенол.
2,2,4,4-Тетраметил-6-бромтиохроман (соединение 72).
Смесь 500 мг (1,49 ммоль) 4-бром-2-(1,1,3-триметил-3-оксибутил)тиофенола (соединение 71) и 8 мл 20%-ной водной серной кислоты кипятили с обратным холодильником 24 ч. Смесь экстрагировали гексаном, органические экстракты объединяли и последовательно промывали водой, насыщенным раствором бикарбоната натрия, вновь водой, насыщенным раствором поваренной соли и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали методом флеш-хроматографии на силикагеле гексанами, получая целевое соединение в виде бесцветного масла.
ЯМР-спектр (CDCl3): δ1,35 (6Н, синглет), 1,40 (6Н, синглет), 1,93 (2Н, синглет), 7,17 (1Н, двойной дуплет, J 8,4 Гц, 2,1 Гц), 7,23 (1Н, дуплет, J 8,4 Гц), 7,26 (1Н, дуплет, J 2,1 Гц).
Масс-спектр m/e 284,0221 (расчетная величина для С13Н17SBr 284,0234).
2,2,4,4-Тетраметил-6-триметилсилил- этинил-тиохроман (соединение 73).
Раствор 600 мг (2,11 ммоль) 2,2,4,4-тетраметил-6-бромтиохромана (соединение 72) в 1,5 мл триэтиламина помещали в толстостенную трубку и дегазировали и затем обрабатывали в атмосфере аргона 1,4 г (14,3 ммоль) триметилсилилацетилена и порошкованной смесью 75 мг (0,39 ммоль) иодида меди (I) и 150 мг (0,21 ммоль) хлористого бис(трифенилфосфин)палладия (II). Реакционную смесь вновь дегазировали, затем помещали в атмосферу аргона, и трубку запаивали. Смесь нагревали при 100оС в течение 24 ч, давали ей охладиться до комнатной температуры и затем обрабатывали дополнительно 1,4 г (14,3 ммоль) триметилсилилацетилена и порошкованной смесью 75 мг (0,39 ммоль) йодида меди (I) и 150 мг (0,21 ммоль) хлористого бис-(трифенилфосфин)палладия (II). Смесь дегазировали, помещали в атмосферу аргона и затем выдерживали в запаянной трубке при 100оС в течение 96 ч. Смесь охлаждали до комнатной температуры и экстрагировали трижды серным эфиром порциями по 10 мл каждая. Органические экстракты объединяли, последовательно промывали 25 мл воды и 25 мл насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток подвергали очистке методом флеш-хроматографии на силикагеле, используя в качестве элюирующего растворителя гексаны, а затем 3%-ный раствор этилацетата в гексанах. В результате получали целевое соединение в виде желтого кристаллического вещества.
ЯМР-спектр (СDCl3): δ0,23 (9Н, синглет), 1,36 (6Н, синглет), 1,39 (6Н, синглет), 1,94 (2Н, синглет), 7,17 (1Н, двойной дуплет, J 8,2 Гц, 1,8 Гц), 7,2 (1Н, дуплет, J 1,8 Гц), 7,30 (1Н, дуплет, J 8,2 Гц).
Масс-спектр m/e 302,1519 (расчетное для С18Н26SSi 382,1524).
2,2,4,4-Тетраметил-6-этинилтиохроман (соединение 3).
К раствору 527,6 мг (1,75 ммоль) 2,2,4,4-тетраметил-6-триметилсилил-этинилтиохро- мана (соединение 73) в 4 мл изопропанола в атмосфере аргона добавляли 4 мл однонормального раствора едкого калия. Реакционную смесь перемешивали при комнатной температуре в течение 20 ч, и изопропанол затем отгоняли в вакууме. Остаток экстрагировали серным эфиром и объединенные эфирные экстракты последовательно промывали водой и насыщенным раствором хлорида натрия и сушили над сульфатом магния. Растворитель отгоняли в вакууме, получая целевое соединение в виде желтого масла.
ЯМР-спектр (СDCl3): δ1,34 (6Н, синглет), 1,37 (6Н, синглет), 1,91 (2Н, синглет), 2,99 (1Н, синглет), 7,17 (1Н, двойной дуплет, J 8,1 Гц, 1,8 Гц), 7,26 (1Н, дуплет, J 1,8 Гц), 7,30 (1Н, дуплет, J 8,1 Гц).
Масс-спектр m/e 230,1122 (расчетное для С15Н18S 230,1129).
Дифенил-3-метил-3-бутен-1-илфосфат (соединение 75).
К охлаждаемому льдом раствору 12,2 г (141,65 ммоль) 3-метил-3-бутен-1-ола (Олдрич) и 11,9 г (150,44 ммоль) пиридина в 100 мл тетрагидрофурана по каплям добавляли в атмосфере аргона раствор 38,5 г (143,21 ммоль) дифенилхлорфосфата (соединение 32) в 100 мл тетрагидрофурана. Смесь кипятили с обратным холодильником в течение трех часов, затем охлаждали и фильтровали. Фильтрат концентрировали в вакууме, остаток растворяли в 400 мл смеси серного эфира и гексана (1:1) и затем промывали водой 2х200 мл, 75 мл насыщенного раствора хлорида натрия и сушили над сульфатом магния. Растворитель отгоняли в вакууме, получая целевое соединение в виде бледно-желтого масла.
ЯМР-спектр (СDCl3): δ1,69 (3Н, синглет), 2,37 (2Н, триплет, J 7 Гц), 4,32 (2Н, квартет, J 7 Гц), 4,72 (1Н, синглет), 4,80 (1Н, 7,10-7,35 (10Н, мультиплет).
4,4-Диметилхроман (соединение 76).
В сухую, охлаждаемую льдом колбу, содержащую 34,95 г (0,134 моль) хлористого олова (IV), быстро добавляли в атмосфере аргона 63,0 г (0,669 моль) фенола. Смесь перемешивали при 0оС в течение получаса и затем обрабатывали 43,0 г (0,135 моль) дифенил-3-метил-3-бутен-1-илфосфата (соединение 75) с последующим добавлением 5 мл сероуглерода. Смесь перемешивали при комнатной температуре в течение 21 ч и затем гасили вливанием в 700 г льда и 1 л 1,5н. едкого натра. Смесь экстрагировали 1х600 мл и 2х300 мл серного эфира. Соединенные эфирные фракции промывали двухнормальным едким натром, насыщенным раствором хлорида натрия и сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали методом флеш-хроматографии на силикагеле, элюируя 2%-ным серным эфиром в гексане и получая целевое соединение в виде бесцветного масла.
ЯМР-спектр (СDCl3): δ1,34 (6Н), 1,80-1,85 (2Н, мультиплет), 4,15-4,20 (2Н, мультиплет), 6,80 (1Н, двойной дуплет, J 8,1 Гц, 1,5 Гц), 6,87 (1Н, триплет-дуплет, J 8,1 Гц, 1,5 Гц), 7,07 (1Н, триплет-дуплет, J 8,1 Гц, 1,5 Гц), 7,26 (1Н, двойной дуплет, J 8,1 Гц, 1,5 Гц).
Аналогичным образом, но используя соответствующий 3-алкилфенол вместо фенола, получали следующие соединения: 4,4,7-триметилхроман; 4,4-диметил-7-этилхроман; 4,4-диметил-7-пропилхроман; 4,4-диметил-7-пентилхроман.
4,4-Диметил-6-ацетилхроман (соединение 77).
К перемешиваемому раствору 7,94 г (48,9425 ммоль) 4,4-диметилхромана (соединение 76) в 70 мл нитрометана добавляли в атмосфере аргона 4,0 г (50,96 ммоль) ацетилхлорида, а затем 6,8 г (51 ммоль) хлорида алюминия. Эту смесь перемешивали при комнатной температуре в течение 5,5 ч и затем охлаждали в ледяной бане и обрабатывали медленно 70 мл 6н. соляной кислоты. Полученную смесь перемешивали при комнатной температуре в течение 10 мин и затем обрабатывали 100 мл серного эфира. Органический слой отделяли, промывали водой, насыщенными растворами бикарбоната натрия и хлорида натрия и сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток подвергали очистке методом флеш-хроматографии на силикагеле, элюируя 10%-ным этилацетатом в гексанах. Продукт подвергали дистилляции на шариковом дефлегматоре (95-100оС, 0,15 мм рт.ст.), получая целевое соединение в виде бесцветного масла.
ЯМР-спектр (CDCl3): δ1,40 (6Н), 1,95-2,00 (2Н, мультиплет), 2,58 (3Н), 4,25-4,30 (2Н, мультиплет), 6,83 (1Н, дуплет, J 8,0 Гц), 7,62 (1Н, двойной дуплет, J 8,0 Гц, 1,5 Гц), 8,00 (1Н, дуплет, J 1,5 Гц).
Аналогичным образом превращали другие хромановые соединения, полученные аналогично соединению 76, в их соответствующие ацетильные аналоги.
4,4-Диметил-6-этинилхроман (соединение 4).
К раствору 2,47 г (24,41 ммоль) диизопропиламина в 40 мл безводного тетрагидрофурана в атмосфере аргона при -78оС по каплям добавляли 15,2 мл 1,6М (24,32 ммоль) н-бутиллития в гексане. Смесь перемешивали при -78оС в течение часа и затем по каплям добавляли раствор 4,98 г (24,38 ммоль) 4,4-диметил-ацетилхромана (соединение 77) в 1 мл безводного тетрагидрофурана. После перемешивания при -78оС в течение часа раствор обрабатывали 4,2 г (24,36 ммоль) диэтилхлорфосфата. Охлаждающую баню затем удаляли и реакционную смесь перемешивали при комнатной температуре в течение 2,75 ч. Раствор затем переводили с помощью двухконцевой иглы в раствор литийдиизопропиламида, приготовленного из 4,95 г (48,92 ммоль) диизопропиламина и 30,5 мл 1,6 М (48,8 ммоль) н-бутиллития в гексане, в 80 мл безводного тетрагидрофурана при -78оС. Охлаждающую баню удаляли и смесь перемешивали при комнатной температуре в течение 18 ч и затем гасили 50 мл и 25 мл трехнормальной соляной кислоты. Смесь экстрагировали по 100 мл и трижды по 50 мл пентаном и объединенные органические экстракты промывали трехнормальной соляной кислотой, водой, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель затем отгоняли в вакууме и остаток очищали методом флеш-хроматографии на силикагеле, используя для элюирования 10%-ный этилацетат в гексане и дистилляцией с шариковым дефлегматором (70оС, 0,35 мм рт.ст.). Получали целевое соединение в виде бесцветного кристаллического твердого вещества.
ЯМР-спектр (CDCl3): δ1,33 (6Н), 1,81-1,86 (2Н, мультиплет), 3,00 (1Н, синглет), 4,19-4,24 (2Н, мультиплет), 6,75 (1Н, дуплет, J 8,5 Гц), 7,22 (1Н, двойной дуплет, J 8,5 Гц, 2,3 Гц), 7,44 (1Н, дуплет, J 2,3 Гц).
Следуя тому же методу, ацетилпроизводные, приготовленные аналогично соединению 77, превращали в этинильную форму.
4,4-Диметил-6-этинилхроман ZnCl (соединение 79) и этиловый эфир 4-(4,4-диметилхроман-6-ил-этинил)бензойной кислоты (соединение 80).
Используемые реакторы сушили над горелкой в вакууме и все последующие процедуры осуществляли без доступа кислорода в атмосфере азота или аргона. К раствору 509,4 мг (2,74 ммоль) 4,4-диметил-6-этинилхромана (соединение 78) в 4 мл безводного тетрагидрофурана при 0оС по каплям добавляли 1,72 мл 1,6 М (2,75 ммоля) н-бутиллития в гексане. Перемешивание начинали при 0оС в течение 30 мин и продолжали при комнатной температуре 15 мин, после чего раствор вновь охлаждали до 0оС и затем обрабатывали раствором 380 мг (2,79 ммоль) плавленного хлорида цинка в 5 мл сухого тетрагидрофурана, используя двухконцевую иглу. Полученный раствор перемешивали при 0оС в течение часа и затем в течение 15 мин при комнатной температуре, получая соединение 79.
Получаемый продукт без выделения использовали следующим образом. Раствор 628,6 мг (2,74 ммоль) этилового эфира 4-бромбензойной кислоты в 4 мл безводного тетрагидрофурана переводили с помощью двухконцевой иглы в суспензию 380 мг (0,33 ммоль) тетракистрифенилфосфинпалладия в 5 мл безводного тетрагидрофурана и перемешивали при комнатной температуре в течение 15 мин, затем обрабатывали с помощью двухконцевой иглы раствором алкинилцинкхлорида, приготовленного выше. Смесь перемешивали при комнатной температуре в течение 20 ч и затем гасили льдом и 30 мл трехнормальной соляной кислоты. Смесь затем экстрагировали трижды по 75 мл серным эфиром, и эфирные экстракты объединяли и последовательно промывали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток дополнительно очищали методом флеш-хроматографии на силикагеле с элюированием 10%-ным этилацетатом в гексане. Получали белый твердый продукт (соединение 80).
ЯМР-спектр (CDCl3):δ1,36 (6Н), 1,42 (3Н, триплет, J 7,3 Гц), 1,82-1,86 (2Н, мультиплет), 4,21-4,25 (2Н, мультиплет), 4,40 (2Н, квартет, J 7,3 Гц), 6,79 (1Н, дуплет, J 8,1 Гц), 7,28 (1Н, 7,58 (2Н, дуплет, J 8,7 Гц), 8,03 (2Н, дуплет, J 8,7 Гц).
Фениловый эфир 3,3-диметилакриловой кислоты (соединение 81).
К охлаждаемому в ледяной бане раствору 1,29 г (54 ммоль) гидрида натрия, приготовленного из 60%-ной суспензии в минеральном масле трехкратной промывкой гексаном (трижды по 10 мл), в 20 мл безводного тетрагидрофурана медленно добавляли в присутствии кислорода раствор 5 г (53 ммоль) фенола в 50 мл безводного тетрагидрофурана. Смесь обрабатывали раствором 7 г (59 мммоль) диметилакрилсилхлорида в 30 мл безводного тетрагидрофурана. Охлаждающую баню затем удаляли и смесь перемешивали в течение последующих 2,5 ч. Реакционную смесь вливали в 150 мл воды, содержащей 1 мл ледяной уксусной кислоты. Смесь экстрагировали 150 мл серного эфира и эфирный экстракт промывали насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель удаляли в вакууме, и остаток очищали флеш-хроматографически на силикагеле, используя для элюирования 5%-ный серный эфир в гексанах. Получали целевое соединение в виде желтого масла.
ЯМР-спектр (СDCl3): δ1,99 (3Н, синглет), 2,24 (3Н, синглет), 5,93 (1Н, широкий синглет), 7,10 (2Н, триплет, J 7,8 Гц).
4,4-Диметил-2-оксо-хроман (соединение 82).
К перемешиваемой охлаждаемой на ледяной бане суспензии 10,4 г (78 ммоль) алюминийхлорида в 160 мл дихлорметана медленно добавляли в атмосфере аргона раствор 7 г (39,8 ммоль) фенилового эфира 3,3-диметилакриловой кислоты (соединение 81) в 40 мл дихлорметана. Охлаждающую баню убирали и смесь перемешивали в течение последующих 42 ч. Смесь вливали в смесь льда и рассола, и отделяли органический слой. Водный слой экстрагировали дихлорметаном, органические экстракты объединяли и промывали насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали методом флеш-хроматографии на силикагеле с помощью 10%-ного серного эфира в гексане. Получали целевой продукт в виде бесцветного масла.
ЯМР-спектр (СDCl3): δ1,30 (6Н, синглет), 2,56 (2Н, синглет, 7,06 (1Н, дв. дуплет, J 8,0 Гц, 1,4 Гц), 7,16 (1Н, триплет-дуплет, J 8,0 Гц, 1,4 Гц), 7,26 (1Н, триплет-дуплет, J 8,0 Гц, 1,7 Гц), 7,33 (1Н, дв.дуплет, J 8,0 Гц, 1,7 Гц).
Масс-спектр m/e 176,0852 (расчетное для С11Н12О2 176,0837).
2-(1,1,1-Триметил-3-оксибутил)фенол (соединение 83).
К 11 мл 3,0 М (33 ммоль) метилмагнийхлорида в тетрагидрофуране, охлажденным в ледяной бане, добавляли в атмосфере азота раствор 1,96 г 4,4-диметил-2-оксо-хромана (соединение 82) в 35 мл безводного серного эфира. Затем охлаждающую баню убирали и смесь перемешивали при комнатной температуре 72 ч. Реакционную смесь вливали в смесь 100 г льда в 3 мл концентрированной серной кислоты и перемешивали вплоть до растворения магниевых солей. Отделяли органический слой, и водный слой экстрагировали дважды по 50 мл серным эфиром. Органические слои объединяли и последовательно промывали водой, насыщенным раствором бикарбоната натрия и насыщенным раствором поваренной соли и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали методом флеш-хроматографии на силикагеле, используя для элюирования 20%-ный этилацетат в гексане. Получали целевое соединение в виде бледно-желтого твердого продукта.
ЯМР-спектр (СDCl3):δ1,13 (6Н, с), 1,48 (6Н, с), 1,89 (1Н, с), 2,23 (2Н, с), 6,60 (1Н, дв.д, J 7,9 Гц, 1,4 Гц), 6,83 (1Н, с), 6,84 (1Н, тр.д. J 7,9 Гц, 1,4 Гц), 7,07 (1Н, тр.д. J 7,9 Гц, 1,6 Гц), 7,31 (1Н, дв.д. J 7,9 Гц, 1,6 Гц).
Масс-спектр m/e 208,1458 (расчетный для С13Н20О2 208,1464).
2,2,4,4-Тетраметил-хроман (соединение 84).
С обратным холодильником в атмосфере азота в течение 4 ч кипятили смесь 2,98 г (14,3 ммоль) 2-(1,1,3-триметил-3-оксибутил)фенола (соединение 83) и 40 мл 20% водной серной кислоты. Смесь затем перемешивали при комнатной температуре в течение 72 ч и разбавляли 50 мл воды. Смесь экстрагировали трижды 20 мл порциями гексанов. Органические экстракты объединяли и последовательно промывали водой и насыщенным раствором хлорида натрия и сушили над сульфатом магния. Растворитель затем отгоняли в вакууме, получая целевое соединение в виде бесцветного масла.
ЯМР-спектр (СDCl3):δ1,36 (6Н, с), 1,37 (6Н, с), 1,83 (2Н, с), 6,71 (1Н, дв.д. J 8,2 Гц, 1,5 Гц), 6,92 (1Н, тр.д. J 8,2 Гц, 1,5 Гц), 7,09 (1Н, тр.д. J 8,2 Гц, 1,5 Гц), 7,29 (1Н, дв.д. J 8,2 Гц, 1,5 Гц).
2,2,4,4-Тетраметил-6-ацетилхроман (соединение 85).
К охлажденному на ледяной бане раствору 2 г (10,53 ммоль) 2,2,4,4-тетраметилхромана (соединение 84) в 25 мл нитрометана добавляли в атмосфере азота 941 мг (11,99 ммоль) ацетилхлорида и затем 1,59 г (11,92 ммоль) алюминийхлорида. Охлаждающую баню убирали, и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь вновь охлаждали на ледяной бане и обрабатывали 25 мл концентрированной соляной кислоты. Затем смесь фильтровали, и остаток промывали дихлорметаном. Фильтрат концентрировали в вакууме и получаемый остаток очищали методом флеш-хроматографии на силикагеле, используя для элюирования 10%-ный этилацетат в гексанах. Получали целевой продукт в виде желтого масла.
ЯМР-спектр (СDCl3):δ1,38 (6Н, с), 1,39 (6Н, с), 1,87 (2Н, с), 2,56 (3Н, с), 6,83 (1Н, д,J 8,7 Гц), 7,71 (1Н, дв.д, J 8,7 Гц, 2,1 Гц), 7,98 (1Н, д, 2,1 Гц).
Масс-спектр m/e 232,1468 (расчетный для С13Н20О2 232,1464).
2,2,4,4-Тетраметил-6-этинилхроман (соединение 5).
К охлаждаемому (при -78оС) раствору 522 мг (5,17 ммоль) диизопропиламина в 8 мл безводного тетрагидрофурана медленно в атмосфере азота добавляли 3,23 мл 1,6 М (5,17 ммоль) н-бутиллития в гексане. Смесь перемешивали при -78оС в течение 40 мин и затем обрабатывали раствором 1,24 г (5,17 ммоль) 2,2,4,4-тетраметил-6-ацетилхромана (соединение 85) в 2 мл безводного тетрагидрофурана. Смесь перемешивали при -78оС в течение последующего часа и затем добавляли 895 мг (5,19 ммоль) диэтилхлорфосфата. Реакционной смеси позволяли принять комнатную температуру и переводили иглой с двумя концами в раствор диизопропиламида лития в тетрагидрофуране при -78оС, приготовленного как описано выше из 1,04 г (10,34 ммоль) диизопропиламина и 6,46 мл 1,6 М (10,34 ммоль) н-бутиллития в гексане. Охлаждающую баню убирали, и смесь перемешивали при комнатной температуре в течение 16 ч. Затем смесь обрабатывали 10 мл ледяной воды и подкисляли до рН 2 10%-ной соляной кислотой. Отделяли органический слой, водный слой экстрагировали трижды по 30 мл пентаном. Органические экстракты объединяли и последовательно промывали 2,30 мл разбавленной соляной кислоты, водой, трижды по 30 мл насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток очищали флеш-хроматографически на силикагеле, используя в качестве элюирующего растворителя 2%-ный этилацетат в гексане. Получали целевое соединение в виде бледно-желтого масла.
ЯМР-спектр (СDCl3): δ1,31 (6Н, синглет), 1,32 (6Н, синглет), 1,50 (2Н, синглет), 3,00 (1Н, синглет), 6,72 (1Н, дуплет, J 8,4 Гц), 7,20 (1Н, дв. дуплет, J 8,4 Гц, 2,1 Гц), 7,42 (1Н, дуплет, J 2,1 Гц).
Масс-спектр m/e 214,1251 (расчетное для С15Н18О 214,1357).
3-Метил-фенил-3,3-диметилакрилат (соединение 87).
60% суспензию гидрида натрия 3,22 г (81 ммоль) в минеральном масле промывали гексаном трижды по 10 мл и затем обрабатывали 30 мл безводного тетрагидрофурана. Смесь охлаждали в ледяной бане и затем обрабатывали раствором 8,6 г (79,5 ммоль) м-крезола в 80 мл безводного тетрагидрофурана. Реакционную смесь перемешивали в течение 10 мин и затем обрабатывали раствором 10,5 г (88,5 ммоль) диметилакрилоилхлорида в 40 мл безводного тетрагидрофурана. Реакционную смесь перемешивали при комнатной температуре 96 ч и затем вливали в смесь 150 мл воды и 1 мл ледяной уксусной кислоты. Смесь перемешивали 10 мин и отделяли органический слой. Водный слой экстрагировали серным эфиром дважды порциями по 50 мл. Органические слои объединяли и последовательно промывали водой и насыщенным раствором поваренной соли и сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали флеш-хроматографически на силикагеле с 10%-ным этилацетатом в гексане. Получали целевое соединение в виде бледно-желтого масла.
ЯМР-спектр (CDCl3): δ1,95 (3Н, д, J 1,3 Гц), 2,21 (3Н, д, J 1,2 Гц), 2,34 (3Н, с), 5,90 (1Н, шир. 6,86-6,93 (2Н, м), 7,01 (1Н, д, J 8,2 Гц), 7,24 (1Н, триплет, J 7,2 Гц).
2-(1,1,3-Триметил-3-оксибутил)-5-метил-фенол (соединение 88).
К суспензии 13 г (97,5 ммоль) алюминийхлорида в 200 мл метиленхлорида в 200 мл метиленхлорида, охлаждаемой в ледяной бане, добавляли по каплям в атмосфере аргона раствор 9,0 г (47,4 ммоль) 3-метил-фенил-3,3-диметилакрилата (соединение 87) в 100 мл дихлорметана. Реакционную смесь перемешивали при 0оС в течение последующих 30 мин и затем при комнатной температуре 15 ч. Реакционную смесь вливали в 200 мл смеси ледяной воды и соли, органический слой отделяли, и водный слой экстрагировали серным эфиром (50 мл). Органические слои объединяли и последовательно промывали водой и насыщенным раствором поваренной соли и затем сушили. Растворитель отгоняли в вакууме, и остаток подвергали очистке методом флеш-хроматографии на силикагеле, используя 5% этилацетата с гексаном. Получали смесь изомерных продуктов при соотношении 4,4,7-триметил-2-оксо-хромана и 4,4,5-триметил-2-оксо-хромана примерно 2,5:1 в виде бледно-желтого масла. К раствору 3,8 г (20 ммоль) этой смеси изомерных 2-оксо-хромана в 60 мл серного эфира при 0оС добавляли в атмосфере аргона 20 мл 3,0 М (60 ммоль) метилмагнийбромида в серном эфире. Реакционную смесь перемешивали при комнатной температуре 48 ч и затем вливали в смесь льда и 1 мл концентрированной серной кислоты. Органический слой отделяли и водный слой экстрагировали серным эфиром дважды порциями по 50 мл. Органические слои объединяли и последовательно промывали водой, насыщенным раствором бикарбоната натрия, вновь водой и затем насыщенным раствором поваренной соли в вакууме, и остаток очищали методом флеш-хроматографии на силикагеле, используя смесь 15%-ного этилацетата с гексанами. Получали целевое соединение в виде бесцветного масла.
ЯМР-спектр (СDCl3):δ1,14 (6Н, с), 1,45 (6Н, с), 2,19 (3Н, с), 2,21 (2Н, с), 6,39 (1Н, д, J 1,8 Гц), 6,67 (1Н, дв.д. J 7,9 Гц, 1,8 Гц), 7,16 (1Н, д. J 7,9 Гц), 7,44 (1Н, с).
2,2,4,4,7-Пентаметилхроман (соединение 89).
К 2,16 г (11,7 ммоль) 2-(1,1,3-триметил-3-оксибутил)-5-метилфенола (соединение 88) добавляли в атмосфере азота 50 мл 20%-ной водной серной кислоты. Реакционную смесь кипятили с обратным холодильником в течение 13 ч и затем охлаждали. Органический слой отделяли, и водный слой экстрагировали серным эфиром. Органические экстракты объединяли и промывали последовательно водой, насыщенным раствором бикарбоната натрия, вновь водой и насыщенным раствором поваренной соли и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, получая целевое соединение в виде желтого масла.
ЯМП-спектр (CDCl3): δ1,32 (6Н, синглет), 1,34 (6Н, синглет), 1,81 (2Н, синглет), 2,26 (3Н, синглет), 6,63 (1Н, синглет), 6,72 (1Н, дуплет, J 7,9 Гц), 7,15 (1Н, дуплет, J 7,9 Гц).
К охлаждаемому в ледяной бане раствору 1,96 г (9,6 моль) 2,2,4,4,7-пентаметил-хромана (соединение 89) в 30 мл нитрометана добавляли в атмосфере аргона 1,059 г (13,5 ммоль) ацетилхлорида, а затем 1,9 г (14,3 ммоль) алюминийхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч и затем охлаждали в ледяной бане и обрабатывали 25 мл концентрированной соляной кимслоты. Смесь нагревали при комнатной температуре и разбавляли серным эфиром и водой. Органический слой отделяли, и водный слой экстрагировали серным эфиром. Органические экстракты объединяли и последовательно промывали водой, насыщенным раствором бикарбоната натрия, вновь водой и насыщенным раствором поваренной соли и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток очищали методом флеш-хроматографии на силикагеле, используя для элюирования смесь 5%-ного этилацетата и гексанов. Получали целевое соединение бледно-желтого масла.
ЯМР-спектр (CDCl3): δ1,36 (6Н, синглет), 1,37 (6Н, синглет), 1,86 (2Н, синглет), 2,49 (3Н, синглет), 2,56 (3Н, синглет), 6,65 (1Н, синглет), 7,74 (1Н, синглет).
2,2,4,4,7-Пентаметил-6-тинил-хроман (соединение 9).
К раствору 455 г (4,5 ммоль) диизопропиламина в 5 мл безводного тетрагидрофурана при -78оС в атмосфере аргона добавлядли 3 мл 1,5 М н-бутиллития в гексане. Смесь перемешивали при -78оС в течение последующих 45 мин и затем обрабатывали раствором 1,07 г (4,3 ммоль) 2,2,4,4,7-пентаметил-6-ацетил-хромана (соединение 90) в 4 мл безводного тетрагидрофурана. Реакционную смесь перемешивали при -78оС в течение часа и затем обрабатывали 776 мг (4,5 ммоль) диэтилового эфира хлорфосфата. Смеси предоставляли возможность самопроизвольно принять комнатную температуру и затем переводили двухконцевой иглой в раствор литийдиизопропиламида в 10 мл безводного тетрагидрофурана при -78оС, который получали в соответствии с методикой выше, используя 910 мг (9,0 ммоль) диизопропиламина и 6 мл 1,5 М (9,0 ммоль) н-бутиллития в гексане. Смесь перемешивали при комнатной температуре в течение 15 ч и затем вливали в 10 мл ледяной воды. Смесь подкисляли до рН 2 10% -ной водной соляной кислотой. Отделяли органический слой, и водный слой экстрагировали пентаном. Органические экстракты объединяли и последовательно промывали водой, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток очищали дистилляцией с шариковым дефлегматором (82оС, 0,3 мм рт. ст.), получая целевое соединение в виде бледно-желтого масла.
ЯМР-спектр (CDCl3): δ1,32 (6Н, синглет), 1,34 (6Н, синглет), 1,81 (2Н, синглет), 2,36 (3Н, синглет), 3,18 (1Н, синглет), 6,64 (1Н, синглет), 7,40 (1Н, синглет).
Масс-спектр m/e 228,1520 (расчетное для С16Н20О 228,1514).
N-(4-бромфенил)-3,3-диметилакрила- мид (соединение 92).
К раствору 9,48 г (80 ммоль) 3,3-диметилакрилоилхлорида в 200 мл безводного тетрагидрофурана при энергичном встряхивании добавляли раствор 13,76 г (80 ммоль) 4-броманилина в 300 мл безводного тетрагидрофурана. Смесь оставляли при комнатной температуре на два часа и затем обрабатывали 80 г льда, а затем 200 мл гексана. Отделяли органический слой, и водный слой экстрагировали гексанами дважды порциями по 50 мл. Органические слои объединяли и последовательно промывали 30 мл воды и насыщенным раствором поваренной соли, дважды порциями по 30 мл и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали путем перекристаллизации из смеси этилацетата и гексанов, получая бесцветные кристаллы целевого соединения.
ЯМР-спектр (CDCl3): δ1,91 (3Н, синглет), 2,23 (3Н, синглет), 5,73 (1Н, широкий синглет), 7,38-7,55 (5Н, мультиплет).
4,4-Диметил-6-бром-2-оксо-1,2,3,4-тет- рагидрохинолин (соединение 93).
В течение 25 мин к 6,7 г (26,02 ммоль) расплавленного N-(4-бромфенил)-3,3-диметилакриламида (соединение 92), нагретого до 135оС, добавляли 4,15 г (31,09 ммоль) алюминийхлорида. Реакционную смесь перемешивали при 130оС в течение 16 ч и затем гасили обрабатывали дополнительным 1 г (7,5 ммоль) алюминийхлорида. Реакционную смесь выдерживали при 130оС в течение последующих 9 ч и затем охлаждали до комнатной температуры. Реакционную смесь затем гасили медленным добавлением 100 мл ледяной воды при слабом подогреве колбы для усиления перемешивания. Смесь экстрагировали серным эфиром однократно 100 мл и четырежды порциями по 50 мл. Органические экстракты объединяли и промывали 25 мл насыщенного раствора поваренной соли и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток очищали методом флеш-хроматографии на силикагеле, используя 30% этилацетата с гексанами. Получали бледно-желтый твердый целевой продукт.
ЯМР-спектр (CDCl3): δ1,37 (6Н, синглет), 2,53 (2Н, синглет), 6,85 (1Н, дуплет, I 8,4 Гц), 7,32 (1Н, дуплет-дуплет, J 8,4 Гц, 2,1 Гц), 7,43 (1Н, дуплет, J 2,1 Гц), 10,12 (1Н, широкий синглет).
4,4-Диметил-6-бром-1,2,3,4-тетрагидрохинолин (соединение 94).
С помощью двухконцевой иглы к 23,5 мл 1,0 М (23,5 ммоль) алюмогидрата лития в тетрагидрофуране, нагретом до кипения с обратным холодильником, в атмосфере азота добавляли раствор 4,95 г (19,48 ммоль) 4,4-диметил-6-бром-2-оксо-1,2,3,4-тетраги- дрохинолина (соединение 93) в 50 мл безводного тетрагидрофурана и 100 мл безводного серного эфира. Смесь кипятили с обратным холодильником в течение двух часов и затем охлаждали до комнатной температуры. Реакционную смесь затем гасили медленным добавлением 25 мл воды и затем 50 мл 5%-ного раствора едкого натра. Смесь экстрагировали серным эфиром дважды порциями по 25 мл, органические экстракты объединяли и последовательно промывали 25 мл воды и 25 мл насыщенного раствора поваренной соли и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток очищали методом флеш-хроматографии на силикагеле, используя 15% этилацетата в гексанах. Получали коричневое маслянистое целевое соединение.
ЯМР-спектр (CDCl3): δ1,27 (6H, синглет), 1,67-1,74 (2Н, мультиплет), 3,23-3,32 (2Н, мультплет), 3,90 (1Н, широкий синглет), 6,33 (1Н, дуплет, J 8,4 Гц), 7,10 (1Н, дуплет-дуплет, J 8,4 Гц, 2,3 Гц), 7,25 (1Н, дуплет J 2,3 Гц).
4,4-Диметил-6-триметилсилилэтинил-1, 2,3,4-тетрагидрохинолин (соединение 95).
В толстостенной трубке дегазировали под аргоном раствор 1,608 г (6,7 ммоль) 4,4-диметил-6-бром-1,2,3,4-тетрагидрохиноли- на (соединение 94) в 1,5 мл триэтиламина и затем обрабатывали 75 мг (0,39 ммоль) иодида меди (I) и 150 мг (0,21 ммоль) хлорида бис- (трифенилфосфин) палладия (II). Смесь вновь дегазировали под аргоном, обрабатывали 2,09 г (21,2 ммоль) триметилсилилацетилена и трубку герметизировали. Смесь нагревали при 50оС в течение 48 ч. После охлаждения до комнатной температуры к реакционной смеси добавляли метиленхлорид и смесь фильтровали. Фильтрат концентрировали в вакууме и остаток очищали методом флеш-хроматографии на силикагеле, используя 10% этилацетата в гексанах. Получали желтый маслянистый целевой продукт.
ЯМР-спектр (CDCl3): δ0,20 (9Н, синглет), 1,20 (6Н, синглет), 1,57-1,63 (2Н, мультиплет), 3,16-3,25 (2Н, мультиплет), 4,02 (1Н, широкий синглет), 6,24 (1Н, дуплет, J 8,2 Гц), 7,00 (1Н, дуплет-дуплет), J 8,2 Гц и 1,8 Гц), 7,26 (1Н, дуплет, J 1,8 Гц).
4,4-Диметил-6-этинил-1,2,3,4-тетрагид- рохинолин (соединение 6).
К раствору 569 мг (2,21 ммоль) 4,4-диметил-6-триметилсилилэтинил-1,2,3,4-тетра- гидрохинолина (соединение 95) в 3 мл изопропанола добавляли в атмосфере аргона 1 мл однонормального водного раствора едкого калия. Реакционную смесь перемешивали при комнатной температуре в течение 36 ч и изопропанол отгоняли в вакууме. Остаток экстрагировали серным эфиром, эфирные экстракты последовательно промывали водой и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток очищали методом флеш-хроматографии на силикагеле, используя смесь 10% этилацетата с гексанами. Получали коричневое маслянистое целевое соединение.
Этиловй эфир 6-хлорникотиновой кислоты (соединение 98).
Смесь 15,75 г (0,1 ммоль) 6-хлорникотиновой кислоты, 6,9 г (0,15 ммоль) этанола, 22,7 г (0,1 ммоль) дициклогексилкарбодиимида и 3,7 г диметиламинопиридина в 200 мл дихлорметана кипятили с обратным холодильником два часа. Смеcи позволяли охлаждаться, растворитель отгоняли в вакууме и остаток подвергали флеш-хроматографической очистке, получая целевое соединение в виде белого твердого вещества с низкой температурой плавления.
ЯМР-спектр (CDCl3):δ1,44 (3Н, триплет, J 6,2 Гц), 4,44 (2Н, квартет, J 4,4 Гц), 7,44 (1Н, дуплет, J 8,1 Гц), 8,27 (1Н, дуплет-дуплет, J 8,1 Гц, 3 Гц), 9,02 (1Н, дуплет, J 3 Гц).
Описанная процедура может использоваться для этерификации любых других галогено-замещенных кислот, используемых при получении этих соединений, таких как этиловй эфир 2-(2-хлорпирид-5-ил) уксусной кислоты, этиловый эфир 5-(2-хлорпирид-5-ил) пентановой кислоты, этиловый эфир 2-(2-йодофур-5-ил) уксусной кислоты, этиловый эфир 5-(2-йодофур-5-ил)пентановой кислоты, этиловый эфир 2-(2-йодотиен-5-ил) уксусной кислоты, этиловый эфир 5-(2-йодотиен-5-ил)пентановой кислоты, этиловый 2-(3-хлорпиридазин-6-ил)уксусной кислоты, этиловый эфир 5-(3-хлорпиридазин-6-ил)пентановой кислоты, и соответствующие хлор- или другими галоидами замещенные пиримидинил- или пиразинил-аналоги таких сложных эфиров.
ЯМР-спектр (CDCl3): δ1,26 (6Н, синглет), 1,65-1,72 (2Н, мультиплет), 2,96 (1Н, синглет), 3,27-3,34 (2Н, мультиплет), 6,34 (1Н, дуплет, J 8,3 Гц), 7,08 (1Н, дуплет-дуплет, J 8,3 Гц, 1,6 Гц), 7,33 (1Н, дуплет, J 1,6 Гц).
Этиловый эфир 4-йодобензойной кислоты (соединение 97).
К суспензии 10 г (40,32 ммоль) 4-йодобензойной кислоты в 100 мл абсолютного этилового спирта добавляли 2 мл тионилхлорида и смесь кипятили с обратным холодильником 3 ч. Растворитель отгоняли в вакууме и остаток растворяли в 100 мл серного эфира. Эфирный раствор промывали насыщенным раствором бикарбоната натрия и насыщенным раствором поваренной соли и сушили над сульфатом магния. Затем растворитель отгоняли в вакууме, и остаток подвергали дистилляции с шариковым дефлегматором (100оС, 0,55 мм рт.ст.), получая бесцветный маслянистый целевой продукт.
ЯМР-спектр (CDCl3): δ1,42 (3Н, триплет, J 7 Гц), 4,4 (2Н, квартет, J 7 Гц), 7,8 (4Н).
Аналогичным образом, используя вместо 4-йодобензойной кислоты, соответствующую кислоту, получали следующие соединения: этиловый эфир 4-йодофенилуксусной кислоты, этиловый эфир 3-(4-йодофенил)пропионовой кислоты, этиловый эфир 4-(4-йодофенил)-бутановой кислоты и этиловый эфир 5-(4-йодофенил)пентановой кислоты.
Этиловый эфир 6-4(4,4-диметилтиохроман-6-ил)этинил)никотиновой кислоты (соединение 99).
Реакционные сосуды, используемые в настоящей процедуре, сушили на горелке под вакуумом, и все операции осуществляли в бескислородной атмосфере аргона или азота. К раствору 465,7 мг (2,3019 ммоль) 4,4-диметил-6-этинил-тиохромана (соединение 1) в 4 мл безводного тетрагидрофурана при 0оС по каплям добавляли 1,5 мл 1,6 М (2,4 ммоль) н-бутиллития в гексане. Реакционную смесь перемешивали при 0оС в течение 10 мин и при комнатной температуре в течение 10 мин, вновь охлаждали до 0оС и затем обрабатывали раствором 330 мг (2,4215 ммоль) плавленного хлорида цинка в 4 мл безводного тетрагидрофурана, используя двухконцевую иглу. Раствор перемешивали при 0оС в течение получаса, затем при комнатной температуре в течение 10 мин. Раствор 426,3 мг (2,2967 ммоль) этилового эфира 6-хлорникотиновой кислоты (соединение 98) в 4 мл безводного тетрагидрофурана переводили с помощью двухконцевой иглы в суспензию 430 мг (0,37 ммоль) тетракистрифенилфосфинпалладия в 4 мл безводного тетрагидрофурана и перемешивали при комнатной температуре в течение 10 мин, затем обрабатывали с помощью двухконцевой иглой раствором алкинилцинка, приготовленного выше. Эту смесь перемешивали при комнатной температуре в течение 18 ч, затем гасили добавлением 100 мл воды. Продукт трижды экстрагировали серным эфиром порциями по 75 мл. Эфирные реакции объединяли и промывали насыщенным раствором хлорида натрия и сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток очищали флеш-хроматографически на силикагеле, используя 5% этилацетата в гексане, а затем методом жидкостной хроматографии при высоком давлении (Вотмен Партисил М-9 10/50; 4% этилацетата в гексане). Получали белое твердое целевое соединение.
ЯМР-спектр (CDCl3):δ1,36 (6Н, с), 1,45 (3Н, т, J 7 Гц), 1,96-2,00 (2Н, м), 3,05-3,09 (2Н, м), 4,45 (2Н, кв, J 7 Гц), 7,11 (1Н, д, 8,4 Гц), 7,29 (1Н, дд, J 8,4 Гц, 2,2 Гц), 7,59 (1Н, д, J 7,8 Гц), 7,66 (1Н, д, J 2,2 Гц), 8,30 (1Н, дд, J 7,8 Гц, 2,3 Гц), 8,22 (1Н, д. J 2,3 Гц).
Альтернативный синтез.
Этиловый эфир 6-[(4,4-диметилтиохроман-6-ил]этинил)никотиновой кислоты (соединение 99).
Соединение 99 получали следующим образом. Раствор 15,4 г (76,2 ммоль) 4,4-диметил-6-этинил-тиохроман (соединение 1) и 14,0 г (75,5 ммоль) этилового эфира 6-хлорникотиновой кислоты (соединение 98) в 35 мл свежеперегнанного триэтиламина дегазировали и затем обрабатывали в атмосфере азота тонко порошкованной смесью 1 г (5,25 ммоль) йодида меди (I) высокой чистоты и 2 г (2,85 ммоль) хлорида бис (трифенилфосфин) палладия (II). Смесь выдерживали в атмосфере азота при 55оС 20 ч и затем охлаждали до комнатной температуры. Триэтиламин отгоняли в вакууме и остаток перегоняли с 200 мл смеси 1:4 этилацетата и гексанов. Смесь фильтровали через силикагель, и фильтрат концентрировали в вакууме. Полученный остаток очищали флеш-хроматографически на силикагеле, используя 15% этилацетата с гексанами. Перекристаллизацией из смеси этилацетата и гексанов получали целевое соединение в виде бледно-желтого твердого вещества.
Этиловый эфир 6-[(4,4,7-триметилтиохроман-6-ил)этинил]никотиновой кислоты (соединение 100).
Смесь 86 мг (0,4 ммоль) 4,4,7-триметил-6-этинил-тиохромана (соединение 2), 85 мг (0,46 ммоль) этилового эфира 6-хлор-никотиновой кислоты (соединение 98) и 0,8 мл триэтиламина дегазировали в атмосфере азота и затем обрабатывали смесью 10 г (0,05 ммоль) йодида меди (I) и 20 мг (0,03 ммоль) хлорида бис-(трифенилфосфин) палладия (II). Реакционную смесь нагревали при 55оС в атмосфере азота в течение 18 ч, затем экстрагировали 1,5 мл смеси 40% этилацетата, остальное - гоексаны и очищали методом флеш-хроматографии на силикагеле, используя для элюирования 10% этилацетата и гексаны. Получали желтое твердое целевое соединение.
ЯМР-спектр (CDCl3): δ1,32 (6Н, с), 1,43 (33Н, т. J 7,2 Гц), 2,44 (3Н, с), 3,01-3,05 (2Н, м), 4,42 (2Н, ква. J 7,2 Гц), 6,98 (1Н, с), 7,54-7,63 (2Н, м), 8,27 (1Н, дд, J 8,3 Гц, 2,3 Гц), 9,21 (1Н, д. J 2,3 Гц).
Этиловый эфир 6-[(4,4-диметилхроман-6-ил)этинил] никотиновой кислоты (соединение 101).
Используемые в этом примере реакторы сушили над горелкой под вакуумом, и все операции проводили в отсутствии кислорода в атмосфере аргона или азота. К раствору 509,4 мг (2,74 ммоль) 4,4-диметил-6-этинилхромана (соединение 4) в 4 мл безводного тетрагидрофурана при 0оС по каплям добавляли 1,72 мл 1,6 М (2,75 ммоль) н-бутиллития в гоексане. Перемешивание начинали при 0оС, которое продолжалось в течение получаса, затем при комнатной температуре в течение 15 мин, а затем раствор вновь охлаждали до 0оС и обрабатывали раствором 380 мг (2,79 ммоль) плавленного хлорида цинка в 5 мл безводного тетрагидрофурана, используя двухконцевую иглу. Полученный раствор перемешивали при 0оС в течение часа и затем при комнатной температуре в течение 15 мин. Раствор 628,6 мг (2,74 ммоль) этилового эфира 6-хлорникотиновой кислоты (соединение 98) в 4 мл безводного тетрагидрофурана переводили с помощью двухконцевой иглы в суспензию 380 мг (0,33 ммоль) тетракистрифенилфосфинпалладия в 5 мл безводного тетрагидрофурана, смесь перемешивали при комнатной температуре в течение 15 мин и затем с помощью двухконцевой иглы вводили раствор алкинилхлорида, приготовленного ранее. Смесь перемешивали при комнатной температуре в течение 20 ч и затем реакцию гасили льдом и 30 мл трехнормальной соляной кислоты. Смесь экстрагировали серным эфиром трижды по 75 мл и эфирные экстракты объединяли и последовательно промывали насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме, и остаток дополнительно очищали методом флеш-хроматографии на силикагеле с 10% этилацетата в гексане, получая твердое желтое целевое соединение.
ЯМР-спектр (CDCl3): δ1,36 (6Н, с), 1,44 (3Н, тр. J 7,1 Гц), 1,83-1,87 (2Н, м), 4,22-4,26 (2Н, м), 4,44 (2Н, кв. J 7,1 Гц), 6,80 (1Н, д, J 7,6 Гц), 7,35 (1Н, д, J 8,9 Гц), 7,58 (1Н, д. J 7,6 Гц), 7,60 (1Н, м), 8,28 (1Н, д. J 8,9 Гц), 9,21 (1Н, с).
Этиловый эфир 6-[(2,2,4,4-тетраметил-тиохроман-6-ил)этинил]никотиновой кислоты (соединение 102).
Раствор 232 мг (1,01 ммоль) 2,2,4,4-тетраметил-6-этинилтиохромана (соединение 3) и 190 мг (1,03 ммоль) этилового эфира 6-хлорникотиновой кислоты (соединение 98) в 2 мл триэтиламина помещали в толстостенную стеклянную трубку, дегазировали, создавали аргоновую подушку и затем обрабатывали порошкованной смесью 53 мг (0,28 ммоль) йодида меди (I) и 84 мг (0,12 ммоль) хлорида бис (трифенилфосфин)палладия (II). Смесь вновь дегазировали, помещали в атмосферу аргона и трубку укупоривали. Реакционную смесь выдерживали при 55оС в течение 60 ч и затем охлаждали до комнатной температуры. Смесь обрабатывали водой и серным эфиром, и органический слой отделяли. Водные слои экстрагировали серным эфиром. Органические слои объединяли и промывали насыщенным раствором поваренной соли и затем сушили. Растворитель отгоняли в вакууме и полученный остаток очищали методом флеш-хроматографии на силикагеле, используя для элюирования 10% этилацетата в гексанах. В результате получали целевое соединение в виде темно-желтого масла.
ЯМР-спектр (CDCl3): δ1,32-1,43 (15Н, м), 1,92 (2Н, с), 4,38 (2Н, кв, J 7,1 Гц), 7,28 (1Н, дд. J 8,3 Гц, 1,8 Гц), 7,32-7,38 (2Н, м), 7,53 (1Н, д, J 8,3 Гц), 8,24 (1Н, д-д, J 8,2 Гц, 2,2 Гц), 9,16 (1Н, д, J 2,2 Гц).
Масс-спектр m/e 379/1594 (расчетное для С23Н25NO2S 379,1606).
Этиловый эфир 6-[(2,2,4,4-тетраметилхроман-6-ил)этинил]никотиновой кислоты (соединение 103).
Раствор 233 мг (1,09 ммоль) 2,2,4,4-тетраметил-6-этинилхроман (соединение 5) и 209 мг (1,09 ммоль) этилового эфира 6-хлорникотиновой кислоты (соединение 98) в 1 мл триэтиламина дегазировали и затем обрабатывали в атмосфере аргона порошкованной смесью 50 мг (0,26 ммоль) йодида меди (l) и 100 мг (0,14 ммоль) хлорида бис (трифенилфосфин) палладия (II). Реакционную смесь выдерживали в атмосфере аргона при 55оС в течение 80 ч и затем охлаждали до комнатной температуры. Удаляли в вакууме триэтиламин, и остаток очищали методом флеш-хроматографии на силикагеле, используя 5% этилацетата в гексанах. В результате получали целевое соединение в виде желтого масла.
ЯМР-спектр (CDCl3): δ1,36 (12Н, с), 1,42 (3Н, т, J 7,2 Гц), 1,85 (2Н, с), 4,37 (2Н, J 7,2 Гц), 6,79 (1Н, д, J 4 Гц), 7,34 (1Н, дд J 8,4 Гц, 2,1 Гц), 7,56 (1Н, д, J 8,7 Гц), 7,60 (1Н, д, 2,1 Гц), 8,27 (1Н, дд, J 8,7 Гц, 2,4 Гц), 9,19 (1Н, д, J 2,4 Гц).
Масс-спектр m/e 363, 1837 (расчетн. для C23Н25О3N, 363,1834).
Этиловый эфир 6-[(2,2,4,4,7-пентаметил-6-хроманил)этинил] никотиновой кислоты (соединение 104).
Раствор 300 мг (1,316 ммоль) 2,2,4,4,7-пентаметил-6-этинилхромана (соединение 9) и 245,6 мг (1,3276 ммоль) этилового эфира 6-хлорникотиновой кислоты (соединение 98) в 2 мл триэтиламина помещали в толстостенную трубку и через раствор в течение 15 мин пробулькивали азот. Трубку затем заполняли аргон и к раствору добавляли тонко измельченную смесь 100 мг (0,1425 ммоль) хлорида бис (трифенилфосфин)палладия (II) и 50 мг (0,2625 ммоль) йодида меди (I). Трубку герметизировали, и реакционную смесь нагревали до 60оС в течение 72 ч. Смесь охлаждали до комнатной температуры, и отгоняли в вакууме триэтиламин. Остаток очищали методом флеш-хроматографии на силикагеле, используя для элюирования 10% этилацетата в гексане. Получали желтое твердое целевое соединение.
ЯМР-спектр (CDCl3): δ1,37 (6Н, с), 1,38 (6Н, с), 1,44 (3Н, тр. J 7,2 Гц), 1,85 (2Н, с), 2,49 (3Н, с), 4,43 (2Н, кв. J 7,2 Гц), 6,70 (1Н, с), 7,55-7,61 (2Н, м), 8,28 (1Н, д-д. J 8,2 Гц, J 2,1 Гц), 9,22 (1Н, д. J 2,1 Гц).
Масс-спектр m/e 377,1982 (расчетное для C24Н27О3N 377,1991).
Этиловый эфир 4-[(2,2,4,4,7-пентаметилхроман-6-ил)этинил] бензойной кислоты (соединение 105).
Через раствор 200 мг (0,877 ммоль) 2,2,4,4,7-пентаметил-6-этинилхромана (соединение 9) и 245,3 мг (0,888 ммоль) этилового эфира 4-йодобензойной кислоты (соединение 97) в 2 мл триэтиламина пробулькивали в течение 15 мин азот. Затем смесь помещали в атмосферу аргона и обрабатывали тонко измельченной смесью 50 мг (0,2625 ммоль) йодида меди (I) и 100 мг (0,1425 ммоль) хлорида бис(трифенилфосфин) палладия (II). Затем реакционный сосуд соединяли с обратным холодильником и смесь выдерживали в течение 72 ч при 55оС в атмосфере аргона. После этого отгоняли в вакууме триэтиламин и остаток очищали флеш-хроматографически на силикагеле, используя этилацетата в гексане. В результате получали целевое соединение в виде желтого масла.
ЯМР-спектр (CDCl3):δ1,32 (12Н, синглет), 1,37 (3Н, триплет, J 7,0 Гц), 1,80 (2Н, синглет), 2,40 (3Н, синглет), 4,36 (2Н, квартет, J 7,0 Гц), 6,66 (1Н, синглет), 7,42 (1Н, синглет), 7,54 (2Н, дуплет, J 8,6 Гц), 7,99 (2Н, дуплет, J 8,6 Гц).
Масс-спектр m/e 376,2038 (расчетный для С25Н28О3 376,2038).
Этиловый эфир 4-[(2,2,4,4-тетраметилхроман-6-ил-этинил]бензойной кислоты (соединение 106).
Раствор 233 мг (1,088 ммоль) 2,2,4,4-тетраметил-6-этинилхромана (соединение 9) и 308 мг (1,087 ммоль) этилового эфира 4-йодобензойной кислоты (соединение 97) в 1 мл триэтиламина помещали в толстостенную трубку и дегазировали в атмосфере аргона. Смесь обрабатывали тонко измельченной смесью 50 мг (0,263 ммоль) йодида меди (I) и 100 мг (0,142 ммоль) хлорида бис-(трифенилфосфин) палладия (II) и затем трубку герметизировали. Реакционную смесь нагревали при 55оС в течение 48 ч. Отгоняли затем в вакууме триэтиламин и остаток очищали методом флеш-хроматографии на силикагеле с 5% -ным этилацетатом в гексанах. Получали целевое соединение в виде желтого масла.
ЯМР-спектр (CDCl3): δ1,33 (6Н, синглет), 1,34 (6Н, синглет), 1,34 (6Н, с), 1,37 (3Н, тр. I 7.2 Гц), 1,83 (2Н, с), 4,35 (2Н, кв, J 7,2 Гц), 6,75 (1Н, д, J 8,4 Гц), 7,24 (1Н, д-д, J 8,4 Гц, 2,1 Гц), 7,46 (1Н, д, J 2,1 Гц), 7,54 (2Н, д, 8,1 Гц), 7,99 (2Н, д, J 8,1 Гц).
Масс-спектр m/e 362,1880 (расчетный для С24Н26О3 362,1881).
Этиловый эфир 4-[(4,2,2,4,4-тетраметил-тиохроман-6-ил)этинил]бензойной кислоты (соединение 107).
В толстостенную стеклянную трубку помещали раствор 110,7 мг (0,481 ммоль) 2,2,4,4-тетраметил-6-этинилтиохромана (соединение 3) и 142,3 мг (0,516 ммоль) этилового эфира 4-йодобензойной кислоты. Смесь обрабатывали тонко измельченной смесью 42 мг (0,221 ммоль) йодида меди (I) и 63 мг (0,09 ммоль) хлорида бис-(трифенилфосфин) палладия. Реакционную смесь дегазировали в атмосфере аргона и трубку укупоривали. Смесь перемешивали при комнатной температуре в течение 40 ч. Триэтиламин отгоняли в вакууме и остаток очищали методом флеш-хроматографии на силикагеле с 3%-ным этилацетатом в гексанах. Получали бледно-желтое твердое целевое соединение.
ЯМР-спектр (CDCl3): δ1,37-1,42 (15Н, мультиплет), 1,96 (2Н, синглет), 4,38 (2Н, квартет, J 7,0 Гц), 7,25 (1Н, дуплет-дуплет, J 8,2 Гц, 1,87 Гц), 7,33 (1Н, дуплет, J 1,8 Гц), 7,37 (1Н, дуплет, J 8,2 Гц), 7,65 (2Н, дуплет, 8,6 Гц), 8,01 (2Н, дуплет, J 8,6 Гц).
Масс-спектр m/e 378,1638 (расчетное для С24Н26О2, 378,1653).
Этиловый эфир 4-(4,4-диметил-1,2,3,4-тетрагидрохинолин-6-ил)этинил[бензойной кислоты (соединение 108).
В толстостенной трубке через раствор 145 мг (0,7838 ммоль) 4,4-диметил-6-этинил-1,2,3,4-тетрагидрохинолина (соединение 6) и 220 мг (0,797 ммоль) этилового эфира 4-йодобензойной кислоты (соединение 97) в 2 мл тетрагидрофурана пробулькивали газообразный азот. Затем раствор обрабатывали тонко измельченной смесью 31 мг (0,163 ммоль) йодида меди (I) и 62 мг (0,088 ммоль) хлорида бис(трифенилфосфин) палладия (II). Трубку затем заполняли аргоном и укупоривали. Реакционную смесь выдерживали при 55оС в течение 72 ч. Смесь позволяли охладиться и затем обрабатывали 1 мл триэтиламина. Смесь дополнительно обрабатывали измельченной смесью 15 мг (0,079 ммоль) йодида меди (I) и 30 мг (0,003 ммоль) хлорида бис (трифенилфосфин) палладия (II). Трубку заполняли аргоном, укупоривали и смесь выдерживали при 55оС в течение последующих 24 ч. Смесь охлаждали и обрабатывали серным эфиром и водой и органический слой отделяли. Водный слой экстрагировали серным эфиром и объединенные экстракты последовательно промывали водой и насыщенным раствором хлорида натрия и затем сушили над сульфатом магния. Растворитель отгоняли в вакууме и остаток очищали методом флеш-хроматографии на силикагеле с 10% этилацетата в гексане. Получали целевое соединение в виде желтого масла.
ЯМР-спектр (CDCl3): δ1,31 (6Н, с), 1,40 (3Н, тр. J 7,2 Гц), 1,68-1,78г (2Н, м), 3,32-3,40 (2Н, м), 4,17 (1Н, шир. с.), 4,38 (2Н, кв. J 7,2 Гц), 6,41 (1Н, J 8,2 Гц), 7,15 (1Н, д-д, J 8,2 Гц), 1,9 Гц), 7,39 (1Н, д, J 1,9 Гц), 7,54 (2Н, д, J 8,4 Гц), 8,00 (2Н, д, J 8,4 Гц).
Масс-спектр m/e 333,1729 (расчетное для С22Н23О2N 333,1728).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗОКСАЗОЛОНА, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2024512C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОКСАЗОЛОНА | 1990 |
|
RU2036913C1 |
| ПРОИЗВОДНЫЕ ГЛИЦЕРИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1989 |
|
RU2040521C1 |
| N-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ (3R, 4R)-3-ЭТИЛ-4-[(1-МЕТИЛ-1H-ИМИДАЗОЛ-5-ИЛ)-МЕТИЛ]-2-ПИРРОЛИДОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ОБЛАДАЮЩИЕ АНТИГЛАУКОМНЫМ ДЕЙСТВИЕМ | 1990 |
|
RU2015978C1 |
| Способ получения производных 2-оксо-1-азетидинсульфокислоты или их солей с щелочными металлами | 1983 |
|
SU1195908A3 |
| ЗАМЕЩЕННЫЕ ЦИКЛОПЕНТАНЫ, ОБЛАДАЮЩИЕ ПРОСТАГЛАНДИНОВОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2505530C2 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2047602C1 |
| ПРОИЗВОДНЫЕ ИНДОЛОНА В ВИДЕ РАЦЕМАТА ИЛИ ОПТИЧЕСКИ АКТИВНОГО ИЗОМЕРА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ АДДИТИВНЫЕ СОЛИ КИСЛОТЫ И КЕТОПРОИЗВОДНЫЕ ИНДОЛОНА | 1991 |
|
RU2068414C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАНИЛ-ЦИАНОГУАНИДИНА | 1990 |
|
RU2057129C1 |
| Способ получения производных арилтиазолов или их хлористоводородных или бромистоводородных солей | 1985 |
|
SU1380614A3 |
Использование: в медицине, в частности в качестве цитостатического средства на основе ацетиленовых производных хромана. Сущность изобретения: продукт - ацетиленовые производные хромана ф-лы 1, приведенной в описании, где R1, R2, R4, R5=C1-C6 -алкил; R3 = H или C1-C6 -алкил; X = о или S; A - фенил или пиридил; n = о; B = C(O)OR6; R6=C1-C6 -алкил; а также производные хромана или тиохромана ф-лы II, приведенной в описании, где R1, R2, R4, R5=C1-C6 -алкил; R3 = Н или C1-C6 -алкил; X = о или S; Z = Н. Реагент 1: соединения ф-лы 1, где R1-R5 см. выше; Z = H или ион металла, или связанный с анионом ион металла, причем последний образует с этинильной группой соль соединения ф-лы 1. Реагент 2: соединение ф-лы III, приведенной в описании: X′-A-(CH2)n-B , где X′ = Hal, A и B, n- см. выше. Условия реакции: в присутствии одного или двух катализаторов, т.е. в случае, когда Z = H, то используют катализатор - иодид меди (1) и Pd(PQ3)2Cl2 , где Q - фенил, а в случае, когда Z = ZnCL(+), то катализатор - Pd(PQ3)4 , где Q - фенил. Соединения ф-лы 1 вызывают 80%-ное подавление ТФА, вызванной ОДС-активности при JC80 = 0,12 - 0,69. 2 с.п. ф-лы.
СПОСОБ ПОЛУЧЕНИЯ АЦЕТИЛЕНОВЫХ ПРОИЗВОДНЫХ ХРОМАНА ИЛИ ТИОХРОМАНА общей формулы I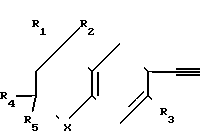


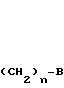
где R1, R2, R4 и R5 - C1 - C6- алкил;
R3 - водород или C1 - C6-алкил;
Х - кислород или сера;
А - фенил или пиридил;
n - целое число, равное 0;
В - СООR6-группа, где R6 - С1 - С6-алкил,
отличающийся тем, что соединение общей формулы II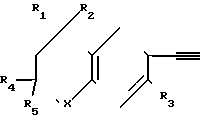

где R1 - R5 имеют указанные значения;
Z - водород, ион металла или ион металла, связанный с анионом, причем этот ион металла образует с этинильной группой соль соединения указанной формулы,
подвергают взаимодействию с соединением формулы III
X' - A - (CH2)n - B,
где Х1 - галогена;
А, В и n имеют указанные значения,
в присутствии одного или двух катализаторов, при условии, что, если Z - водород, то в качестве катализатора используют иодид меди (I) и Pd(PQ3)2Cl2, где фенил - Q, и если Z - ZnCl+, то в качестве катализатора используют Pd(PQ3)4, где Q - фенил.
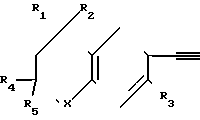

| Берма и ботвели | |||
| Cancer Reseaz, 1977, 37, с.21996. |

