Ы
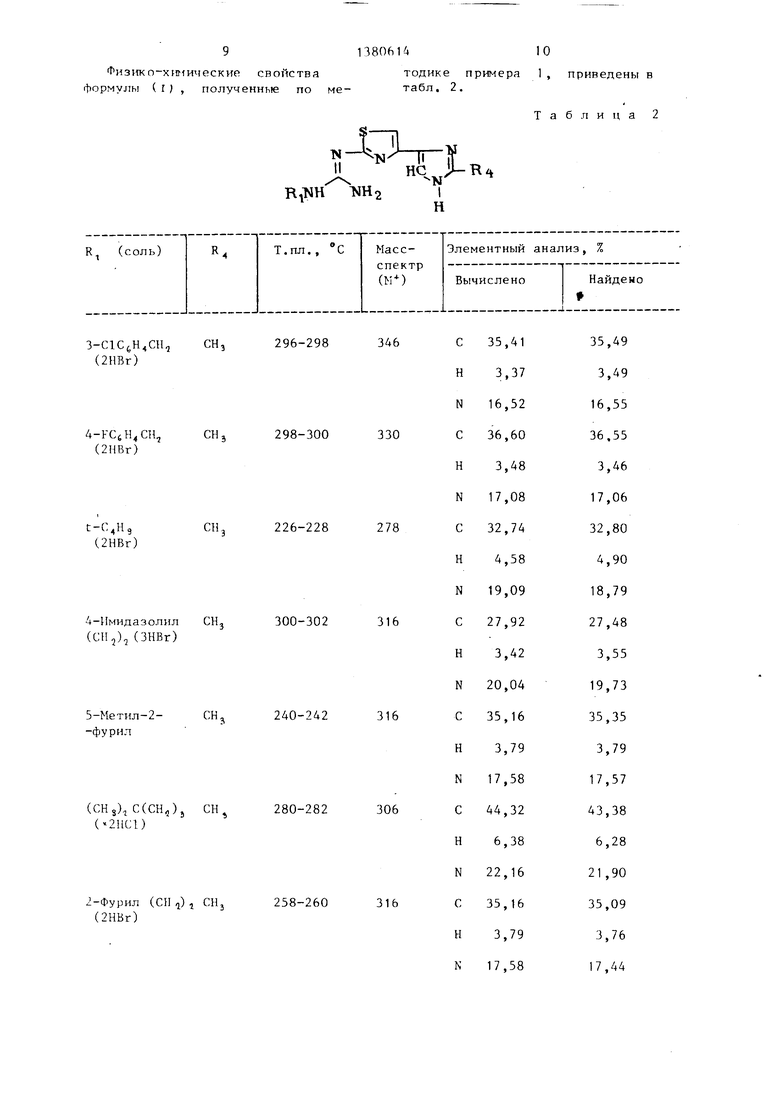
Изобретение относится к способу получения новых производных арилтиа- золов общей формулы
N II
.,
или их хлористоводородных или бро- мистоводородных солей, где при X, равном NH, R, - прямой или разветвленный (С4-Сз)алкил, фенил или группа (R,)tАг(СН,)„,где п - целое число от 1 до 4; RS - одинаковые или различные и означают Н, F, С1, Аг - остаток фенила, нафтила, фури- ла, тиенила, пиридила, тиазолила, имидазолила; F 4 Н, (С ,-0 4)-алкил, , при X, равном S, R, - н-гек- сил; Р.-метил, R,-водород.
Эти новые соединения обладают активностью как средства, препятствую- шие секреции, антагонисты гистамин- -Н рецептора и/или как ингибиторы образования язвы желудка, вызываемой алкоголем. Соединения могут быть использованы для ингибирования (т.е. предотвращения и лечения) пепсиновых язв у млекопитающих, включая человека.
Цель изобретения - синтез новых производных арилтиазолов, обладающих антисекреторной и цитозащитной активностью.
Спектры ядерного магнитного резонанса (ЯМР) измерены для растворов в дейтерированном хлороформе (CDClj), дейтерированном метаноле (CDjO) или дейтерированном диметил- сульфоксиде (DMCO-d), и пиковые положения сообщаются в долях на миллион в нисходящей области (downfidd) от тетраметилсилана.
Пример 1. 2-(Ы-Гексил-ы -гуанидино)-4-(2-метилимидазол-4-)ил -тиазол дигидрохлорид.
А. К раствору 1 3,08 г (46,05 ммоль) 2-бром-1 -(2-метилимидазол-4-ил)этанол гидробромида в 150 мл ацетона добавляют раствор 10,25 г (50,66 ммоль) 1-(н-гексилгуанил)ти- окарбамида в 50 мл ацетона, смесь нагревают с обратным холодильником 6 ч и оставляют стоять при комнатной температуре 16 ч. Смесь нагревают с обратным холодильником дополнительно 1 ч, охлаждают и продукт
собирают, что дает 19,44 г (98%) желтого твердого вещества (двубро- мистоводородная соль). Растворяют ее
5 в 300 мл воды и добавляют к раствору 20,59 г (166 ммоль) натрия карбоната моногидрата в 200 мл воды. После 15 мин стояния твердое вещество отфильтровывают и промывают во0 дои. Влажное вещество растворяют в 400 мл ацетона, фильтруют для удаления нерастворимого материала и фильтрат обрабатывают 8 мл 37%-ной (по весу на объем) концентрированной
5 хлористоводородной кислоты. Подкисленную смесь перемешивают 1,5 с, фильтруют и собранное твердое вещество высушивают, что дает 16,31 г светло-желтого твердого вещества. Раст0 воряют его в 50 мл метанола, обрабатывают активированным углем и фильтруют через диатомитовую землю. Фильтрат разбавляют изопропиловым эфиром и твердый осадок собирают фильтрова - нием и высушивают, что дает 11,5 г (66%) целевого продукта в виде светло-желтого порошка, т.пл. 303-305°С. Масс-спектр (т/е): 306 (М); Н-ЯМР (DMCO-d), доли на млн. (дельта):
0 0,7-1,8 (мультиплет, 11Н); 2,70 (син- глет, ЗН); 3,25 (мультиплет, 2Н); 7,83 (синглет, 1Н); 8,03 (синглет, 1Н); 8,6 /мультиплет, ЗН).
5 Вычислено, % С 44,32; Н 6,38; N 22,15.
С,4 2НС1 Найдено,%: С 43,83; Н 6,29; N 21,89.
0 В. Повторяют указанную выше процедуру,имея 114 мг (0,71 ммоль) 4- -хлорацетил-2- метилимидазола, 114 мг (0,71 ммоль) 1-(н-гексилгуанил) ти- окарбамида и 8 ммоль хлористого во- 5 дорода в 7 мл ацетона, 65 ч нагревают с обратным холодильником и получают целевой продукт в виде порошка рыжевато-коричневого цвета, выход 36%. Пример 2. Применяя соотQ ветствующий п-замещенный гуанилтиог- карбамид взамен 1-(н-гексилгуанил) тиокарбамида по методике примера 1 получают соединения формулы (1а) в виде солей, получаемых присоединениг ем кислоты SKjNH
СН,
31380614
Значения R и характеристики соед
единений приведены в табл.1.
Таблица 1
















Изобретение касается производных арилтиазолов (AT), в частности соединений общей формулы 1 R,R2,N - C(NHj) где К - -С СН CH-S, X - CR N, при а) X - Н; RI - прямой или изо-С.j - -алкил; фенил; группа RjRjAr-(CH)„; п 1,2, 3, 4;R } одинаковые или разные, Н, F, С1, CHjO; Ar - фенил, нафтил, фурил, тиенил, пиридил, ти- азолил, имидазолил; С -алкил, R,-H или б) X-S г; R,-H- гексил; , NHj; , которые, как противоязвенные средства,могут быть использованы в медицине.Цель - создание новых активных веществ указанного класса с широким спектром дей- ствия. Их синтез ведут из соединений формулы II и. Ill - С (NH) -NH - C(S)NHj (II); Hal - СИ, -С(0)К (III), где К, R,, R, - указаны выше; Hal - хлор или бром, в среде инертного растворителя при 50-60 С с последующим выделением целевого продукта в свободном виде или в виде НС1 или НВг-солей, преимущественно последней. В качестве растворителя целесообразно использовать ацетон или диметилформамид. Новые вещества обладают дополнительным цито- защитным действием при дозе 30 мг/кг без токсического эффекта, доза инги- бирования пептических язв составляет 0,1-2 мг/кг (орально). 2 з.п. ф-лы, 8 табл. СО 00 00 о о: 4
Э L /HirL tl
CH,(CHj)j-CHсн.
CHj(CH,)4
CHjCCH,),
CH,(CH,)j
снесен,).
g СН,(СН,)-,
CH,(CH)j
iCHj(CH,),-CHсн.
2HBr208-209
СНз(СН, )5-СН/
сн.
2HBr X 174-176334
X H,0
kC.HS
2HBr X X 1,5 X X H,0
1CtEgCHj
2HBr273-275
mC HjCCH,)
2HBr 290-292 326
(CH,),
2HBr X 264-266 X H,0
C HsCCH.,)
2HBr259-261
pA-CIC H CH,
2HBr297-298
q4-CiqH4(CH), 2HBr268-270
Продолжение табл.1
320
70
298
312
340
354
346
360
Ьромистоводородные соли получают, применяя 48%-ную бромистоводород- ную кислоту взамен, хлористоводородной кислоты.
Продолжение табл.1
Фнз1жо-хииическуте свойства формулы ( I ) , полученные по ме3-С1С(,Н4С1ЦСН,
(2НВг)
296-298
,СН.
(2НВг)
298-300
t-C,H9 (2НВг)
СИ,
226-228
4-Имидазолил CHj (СИ,,), (ЗНВг)
300-302
5-Метнл-2- СН, -фурил
240-242
(СНз)г C(CH)j СН,, («21КЛ)
280-282
2-Фурил (СИ ) г CHj (2НВг)
258-260
тодике примера 1 , приведены в табл. 2.
Таблица 2
Пример 3, 2-(Ы-н-Гексил-Ы - -гуанидино)-4-(имидазол-4-ил)тиазол дигидробромид (R, H-CjH , R, Н).
Раствор 511 мг (4,64 ммоль) 4-аце тилимидазола в 6 мл уксусной кислоты обрабатывают 1 мл (9 ммоль) 48%-ной бромистоводородной кислоты и затем 741 мг (4,64 ммоль) брома в 4 мл уксусной кислоты. Смесь перемешивают при 50 С 4 ч, добавляют 939 г (4,64 ммоль) 1-(н-гексилгуанил) ти- окарбамида и перемешивание продолжают 20 ч при 50°С. Охлажденную реакционную смесь разбавляют ацетоном и фильтруют, что дает 735 мг (35%) указанного в заголовке соединения в виде белого порошка, т.пл. 254 - 255°С. Масс-спектр (т/е): 292 (); Н-ЯМР (DMCO-d), доли на млн. (дель
1380614
12 Продолжение табл. 2
та): 0,7-1,9 (мультиплет, 11Н), 3,5 (мультиплет, 2Н); 7,87 (синглет, 1Н); 8,25 (синглет, 1Н); 8,6 (синглет, широкий, ЗН); 9,28 (синглет, 1Н).
Вычислено,% С 34,37; Н 4,88; N 18,50.
С,зН,,Н,5 2НВг
Найдено,%: С 34,24; Н 5,01; N 18,48.
Пример 4, 2-(Н-н-гексил-Н - -гуанидино)-4-(2-этилимидазол-4-ил) тиазол дигидрохлорид (R , ; R, Н; R С,Н,.).
Повторяют методику предыдущего примера, однако исходят из 1,45 г (10,5 ммоль) 4-ацетил-2-этилимида- зола, до добавления эквимолекулярного количества 1-(1 -гексилгуанил) ти- окарбамида перемешивают 6 ч при 50°С, и после добавления 20 ч при . Затем реакционную смесь концентрируют в вакууме до снропа, растворяют ацетоном и охлаждают, что дает 1,58 г (31%) рыжевато-коричневого твердого вещества. Обрабатывают его насыщенным водным раствором бикарбоната натрия, экстрагируют этилацетатом, экстракты высушивают, концентрируют и остаток очищают колоночной.хро- матографиер на силикагеле. Фракции продукта соединяют, концентрируют до масла, растворяют в 100 мл этанола и обрабатывают 2 мл 37%-ной хлористоводородной кислоты. После выпари- вания в вакууме получают желтое твердое вещество, которое растирают в порошок с ацетоном, фильтруют. Получают 776 мг (19%) рыжевато-коричневого порошка. Масс-спектр (т/е): 320 (M-); MI-ЯМР (DMCO-d,), доли на млн. (дельта): 0,7-2,0 (мультиплет, 14Н); 3,05 (квартет, 7 Гц, 211); 3,5 (мультиплет, 2Н), 7,88 (синглет, 1Н) 8,02 (синглет, 1Н); 8,7 (мульти- плет, ЗН).
Вычислено, С 45,80; Н 6,66; N 21 ,37.
С 2НС1
11айдено,%: С 45,42; Н 6,61; N 21 ,10.
При мер 5. 4-(2-Аминотиа- зол-4-ил)-2-(1-н-гексил-З-гуаниди- но)тиазол (R, ,; R Н;
R 4 Н,).
А. 2-Амино-4-бромацетилти&зол
гидробромид.
К суспензии 1,11 г (5,0 ммоль) 2-амино-4-ацетилтиазол гидробромида, приготовленного из 1-бром-2,3-бутан- диона и тиокарбамида в 50 мл уксусной кислоты, добавляют 5 капель А8%- ной бромистонодородной кислоты и 799 мг (5,0 ммоль) брома и смесь нагревают при 60 С 4 ч. Продукт, выпавший в осадок, собирают фильтрованием, промывают уксусной кислотой и ацетоном, что дает 1,32 г (88%) светлого, рьшевато-коричневого порошка, т.пл, 198 С (разложение без плавления) .
Масс-спектр (т/е): 220 (М ), 222 (h- + 2); Н-ЯМР (DMSO-d ) , доли на млн. (дельта): 4,83 (синглет,2Н); 9,27 (синглет, 1Н).
Вычислено,%: С 19,88; Н 2,00; N 9,28.
CjHsN-jOSBrHBr
п n 5
с
Q с
0
0
Найдено,%: С 20,33; Н 2,04; N 9,28.
В, Смесь 1,316 г (4,35 ммоль) 2-амино-4-бромацетилтиазола гидробромида и 880 мг (4,35 ммоль) 1-(н- -гексилгуанил) тиокарбамида в 44 м.п диметилформамида нагревают при 60 С 4 ч. Охлажденную реакционную смесь концентрируют в вакууме до масла, которое подщелачивают добавлением 10%-ного раствора карбоната натрия и экстрагируют этилацетатом. Высушенный экстракт фильтруют, концентрируют в вакууме и остаток дважды хро- матографируют на колонках с силика- гелем, отмывают из адсорбента 9:1 хлороформ с метанолом. Очищенный продукт извлекают выпариванием растворителя, экстрагируют этилацетатом и обрабатывают избытком газообразного хлористого водорода. Полученное твердое вещество собирают и промывают этилацетатом, что дает 640 мг (37%) целевого продукта в виде бесцветно- го порошка. Т.пл. 205-207°С (разложение). Масс-спектр (т/е): 324 (М); Н-ЯМР (DMCO-dj), доли на млн, (дельта): 0,89 (мультиплет, ЗН); 1,32 (мультиплет, 6Fi); 1,60 (мультиплет, 2Н); 3,44 (мультиплет, 2Н); 7,36 (синглет, 1Н); 7,80 (синглет, 1Н); 8,63 (синглет, широкий, 2Н); 8,92 (синглет,широкий, 1Н),
Вычислено,%: С 39,29; Н 5,58; N 21,15.
С,зН,„Г1,5,-2НС1
Найдено,. %: С 38,39; Н 5,56; N 21,09,
Пример 6, 2-(1-н-Гeкcил- гyaнидинo)-4-(2-мeтил-тиaзoл-4-ил) тиазол (R, ,, ; R ,j - П; R CHj),
А. 2-Ацетил-З-метилтиазол гидробромид.
Раствор 1,13 г (15,0 ммоль) ти- ацетамида и 2,47 г (15,0 ммоль) 1- -бром-2,3-бутандиона в изопропаноле перемешивают при комнатной температуре 10 дней. Продукт, выпавший в осадок, собирают фильтрованием и промывают изопропанолом, что дает 0,619 г (18%) бесцветного порошка. Выпаривание фильтрата и растирание в порошок полученного твердого вещества с ацетоном приводит к 2,329 г (70%) желтоватого порошка. Т.пл, 200 С (возгоняется). Масс-спектр (т/е): 1,41 (К); Н-ЯМР (DMCO-d«),
15
доли на млн. (дельта); 2,60 (син- глет, ЗН); 2,78 (синглет, ЗН); 8,42 (синглет, 1Н).
Вычислено, %: С 32,44; Н 3,63; В 6,31.
HBr
Найдено, %: С 32,41; Н 3,71; N 6,29.
В. 4-Бромацетил-2-метилтиазол гидробромид.
Суспензию 828 г (3,73 ммоль) 4-ацетил-2-метилтиазола гидробромида в 37 мл уксусной кислоты обрабатывают 4 каплями 48%-ной бромистово дородной кислоты и 596 мг (3,73 ммоль брома и смесь подогревают до бО С. После 3 ч выдерживания при этой температуре смесь охлаждают до комнатной температуры, добавляют затравочный кристалл и оставляют стоять на всю ночь. Выпавший в осадок продукт собирают, промывают уксусной кислотой и высушивают, что дает 1,01 г (90%) коричневых кристаллов, т.пл. 187-189°С. Масс-спектр (т/е): 219 (1-Г); 221 (М + 2); Н-ЯМР (DMCO-dj), доли на млн. (дельта): 2,77 (синглет, ЗН); 4,82 (синглет, 2Н); 8,30 (синглет, 1Н).
Вычислено, %: С 23,94; Н 2,34; N 4,65.
CjHjNOS Br
HBr С 23,/1; H 2,30;
Найдено, % N 4,51.
С. Смесь 960 мг (3,19 ммоль) 4- -бромацетйл-2-метилтиазол гидробромида, 645 мг (3,19 ммоль) 1-(н-гек- силгуанил)тиокарбамида и 32 мл ди- метилформамида нагревают при 60°С 4 ч. Охлажденную смесь разбавляют этиловым эфиром, осадок собирают, что дает 801 мг (52%) не вполне белго порошка. Перекристаллизация дает 332 мг (21%), т.пл. 160-162 С. Масс спектр (т/е): 323 (М), Н-ЯМР (DMCO-dj), доли на млн. (дельта): 0,7-2,0 (мультиплет, 11Н); 2,75 (сиглет, ЗН); 3,43 (мультиплет, 2Н); 7,60 (синглет, 1Н); 7,98 (синглет, 1Н); 8,55 (синглет, широкий, 2Н); 8,90 (синглет, широкий 1Н).
Вычислено, %: С 34,64; Н 4,78; N 14,43.
C,«H,, ,. 2НВг
Найдено, %: С 34,26; Н 4,68; N 14,28.
В приведенных ниже примерах приводятся методики получения промежу380614
16
точных продуктов, используемых при получении исходных реагентов и целевых продуктов.
Пример 7. Общий способ получения N-замещеиных-3-цианогуани- динов (препарат А).
Ы-замещенные З-пианогуанидины получают по схеме:
R,R,NH
+ HN(CN), R,R NC NCN
/ NH.
(i) 1-н-Гексил-З-цианогуанидин (R, „; R, H).
Смесь 13,8 г (0,10 ммоль) н-гек- силамина гидрохлорида ,8,9 г (0,125 ммоль) дицианимида и 75 мл н-бутанола перемешивают при нагревании с обратным холодильником 3 ч. Затем смесь охлаждают,фильтруют для удаления остаточной соли и фильтрат выпаривают до сиропа и кристаллизуют из диоксана. Масс-спектр (т/е): 169 молекулярного иона.
(ii) Следующие N-замещенные 3-ци- аногуанидины указанной далее формулы, приведенные в табл.3, получаются из соответствующего амина R,R2NH по указанному вьпие способу.
R.P.C NCN NH
35
Таблица J
40
45
50
н-Гептил н-Октил
Н Н Н
183
197 160
Продолжение табл.3,
(iii) Применяя подходящий амин и дицианимид, NH(CN), указанным способом получают соответствующие соединения R. NCN, указанNH.
ные в табл.4.
Таблица 4
R,
ПА
снесен,)э
(СН,),СН(СИг)-,
н
Пример 8. Получение N-заме щенных гуанилтиокарбамидов (препарат В) ,
Общий способ получения N-замещен ных гуанилтиокарбамидов заключается в следующей реакции:
R, - С NCN 1Ц8 - NH,
SS
R, N CN11,; v R,RjN - С NHC NH NH -;NH
10
(i) 1-(и-гексилгуанил) тиокарба- мид (R, ,5; R, Н).
В смесь 4,5 г (0,027 ммоль) 1-н- 5 -гексил-3-цианогуанидина , 75 мл метанола и 0,5 мл диэтиламина вводят путем барботирования газообразный сероводород в течение 8 ч. Смесь перемешивают всю ночь при комнатной температуре, затем снова пропускают сероводород 6 ч и опять перемешивают всю ночь. На данном этапе присутствие исходного материала в реакционной смеси регистрируют по- 15 средством тонкослойной хроматографии. Смесь нагревают с обратным холодильником при одновременном барботиро- вании сероводорода дополнительно
6ч и нагревание с обратным холодиль- 20 НИКОМ продолжают всю ночь. Растворитель вьшаривают в вакууме и остаток очищают по способу мгновенной (flash) хроматографии. Для вымыва-- ния из адсорбента используют смесь
25 9:1 хлороформ: метанол, что дает 4,49 г продукта. асс-спектр (т/е) : 202 ().
(ii) Иначе препарат В можно получить по способу Кутлера и Калита,как
30 пояснено далее для 1-(бензилгуанил) тиокарбамида.
К раствору 6,73 г (38,6 ммоль) 1-бензил-3-цианогуанидина в 100 мл метанола добавляют 2 мл диэтиламина, смесь охлаждают до 0°С и насыщают газообразным сероводородом. Охлажденный раствор переносят в реактор высокого давления из нержавеющей стали, герметично закрывают его и нагревают при 80°С 48 ч. Затем смесь переносят в колбу, прадувают азотом для удаления избытка сероводорода и растворитель выпаривают в вакууме. Получающееся остаточное масло очидс щают мгновенной хроматографией (силикагель), отмывают из адсорбента 20:1 смесью хлороформа с метано- . лом и получают 3,06 г продукта в виде светло-желтого масла. Масс- спектр (т/е): 209 (И).
(iii) Другие 1-замещенные 3-ци- аногуанидины, указанные в примере
7в части (ii), превращают в N-замещенные гуанилтиокарбамиды
S 55«
формулы NCNH, по указанN14ным выше способам (табл.5):
35
40
50
Таблица 5
Пример 9. Получение 2-окси- метил-4-бромацетилимидазол гидробромида (препарат С).
(i) З-Бром-А-этокси-З-бутен-2-он.
Смесь 400 мл абсолютного этанола и 60 мл толуола нагревают с обратным холодильником и 20 мл азеотропа удаляют посредством отделителя по Дину - Старку. К этанол-толуоловому раствору добавляют 33,0 г (0,2 ммоль) 3- -бром-4-окси-3-бутен-2-она и продолжают нагревание с обратным холодильником 2 ч. За это время три равные доли по 20 мл этанол-толуола удаляют через отделитель. Раствор сокращают в объеме в вакууме, что дает 38,6 г (100%) 3-бром-4-этокси-3-бутеи-2-она в виде подвижного масла.
14
20
0
5
0
5
0
5
0
5
0
5
Н-ЯМР (DMCO-d), доли на млн. (дельта): 8,21 (синглет, 1 fl) ; 4,23 (квартет, 2Н) ; 2,39 (синглет, 311); 1,31 (синглет, ЗИ) .
(ii) 2-Оксиметил-4-ацетилимидазол
9,7 г (0,05 ммоль) З-бром-4-это- кси-З-бутен-2-она объединяют с 5,35 г (0,05 ммоль) оксиацетамидина гидрохлорида в 100 мл ацетона так, чтобы получилась суспензия. К суспензии при 25°С добавляют 11,5 г (0,1 ммоль) 1,1,3,3-тетраметилгуани- дина за 5 мин. После 48 ч перемешивания суспензию фильтруют и маточные растворы концентрируют в вакууме. Полученное масло хроматографиру- ют «а Силикагеле-60, используя хлороформ для отмывки с адсорбента, что дает 1,48 г (21%) 2-оксиметил-4- -ацетилимидазола.Т.пл. 147-148 С.
Н-ЯМР (DMCO-d), доли на млн. (дельта): 7 (синглет, 1Н); 5,46 (синглет, очень широкий, 1Н); 4,5 (синглет широкий, 2Н); 2,4 (синглет, ЗН).
(iii) 1,826 г (0,013 ммоль) 2-ок- симетил-4-ацетилимидазола растворяют в 40 мл 48%-ной бромистоводород- ной кислоты и добавляют 2,1 г (0,013 ммоль) брома. Реакционную смесь нагревают при 80°С 2 ч, затем концентрируют в вакууме до твердого вещества. Этот материал растирают в порошок с изопропиловым эфиром, полученное твердое вещество собирают фильтрованием, промывают эфиром и высушивают, что дает 2,2 г (56%) 2-ок- симетил-4-бром-ацетилимидазола гидробромида, т.пл. 183°С с разложением.
Н-ЯМР (DMCO-d j), части на млн. (дельта): 8,8 (синглет, 1Н); 4,8 (синглет, 2х2В).
Пример 10. Получение 4-аце- тил-2-метилимидазола (препарат D).
(L) 1,2-Дихлор-1-бутен-3-он
(CHjCOC СНС1) /
. С1
Смесь 392 г (5,0 ммоль) ацетилхло рида и 1817 г (18,75 ммоль) цис, транс-1,2-дихлорэтилена в условиях безводной среды охлаждают до 0°С (ванна из ацетона с твердой углекислотой) . К этой смеси добавляют порциями 734 г (5,6 ммоль) безводного хлорида алюминия. Смесь выдерживают ниже 25 С. Хлорид алюминия промывают добавочными 606 г (6,25 ммоль)
1, -дихлорэтана . По окончании добав- охлаждающую ванну убирают и смесь иагренают с обратным холодильником (30-60°С) всю ночь. Охлажден- ную реакционную смесь выливают на лед, органический слой отделяют, а водный слой экстрагируют 3x500 мл метиленхлорида. Соединенные органические слои энергично перемешивают, добавляют 450 г хлорида натрия и удаляют небольшое количество выделившейся воды. Органический слой фильтруют через диатомитовую землю (целит) для удаления неорганических со- единений, затем добавляют к раствору 748 г (6 ммоль) натрийкарбоната моногидрата в количестве воды, достаточном для получения 2,5 л раствора. Образующуюся смесь перемешивают 1,5 ч, выпавшее в осадок твердое вещество удаляют фильтрованием и промывают метиленхлоридом. Органический слой отделяют, водную часть экстрагируют 2x200 мл метиленхлорида и со- единенные органические слои высушивают сульфатом натрия. Растворитель удаляют выпариванием в вакууме и остаточное масло перегоняют, что дает 517,5 Г (74,5%) продукта в виде свет ло-желтой жидкости, т.кип. 40-52 С при В мм. Н-ЯМР (CDClj), доли на млн. (дельта): 2,50 (синглет, ЗН); 7,55 /синглет, 1Н).
(ii) 2-хлор-1,1-диметокси-3-бу- танон
CHjCOCHCH / OCHj/, J
Cl
К раствору 297 г (5,5 ммоль) ме- тилата натрия в 5 мл метанола при 0°С добавляют слабой струей 695 г (5,0 ммоль) 1,2-дихлор-1-бутен-3-она, По окончании добавления смесь пере
мешивают при 0°С 1 ч, добавляют дополнительно 54 г (1,0 ммоль) метила- та натрия и перемешивание при 0 С продолжают 1 ч. Смесь оставляют при перемешивании при комнатной температуре на всю ночь, добавляют еще 1 г-моль метилата натрия и продолжают перемешивание 1 ч. Смесь фильтруют (с добавлением вспомогательного средетва,способствующего фильтрованию) для удаления солей и промывают свежим метанолом. Фильтрат концентрируют в вакууме до образования суспензии, которую поглощают, эк0 5 0 5 о
5
0
5
0
страгируют изопропиловым эфиром и промывают поочередно водой, насыщенным раствором бикарбоната натрия и рассолом, после чего высушивают над безводным сульфатом магния. Экстракт концентрируют в вакумме, что дает остаточное масло, которое перегоняют в вакууме. Получают основную фракцию 628 г (75%) продукта, т.кип. 66 - 75 С при 8 мм. Н-ЯМР (CDC1 j), доли на млн. (дельта): 2,33 (синглет, ЗН); 3,43 (синглет, ЗН); 3,47 (синглет, ЗН); 4,23 (дуплет, 1Н); 4,63 (дуплет, 1.Н).
(iii) а) К 500.мл диоксана добавляют 83,5 г (0,50 ммоль) 2-хлор-1,1- -диметокси-2-бутанона, 94,5 г (1,0 моль) ацетамидина гидрохлорида с 123 г (1,5 моль) ацетата натрия, затем смесь нагревают с обратным холодильником всю ночь. Охлажденную реакционную смесь фильтруют через си- ликагелевую подкладку на воронке для фильтрования с пластинкой из спеченных стеклянных зерен, промывая 3500 мл диоксана. Фильтрат и промывные жидкости соединяют и выпаривают в вакууме, что дает остаточное масло. Этот продукт очищают хроматографией на колонке с силикагелем (600 г), отмывают из адсорбента этилацетатом. Собирают фракции по 200 нл каждая. После 16 фракций производят отмывания из адсорбента 95:5 этилацетатом с метанолом. Фракции 18-35 соединяют и растворитель выпаривают в вакууме, что дает 28,82 г (46,4%) целевого продукта. Перекристаллизация из 1:1 этилацетат изо- пропиловый эфир дает 19,27 г (31%) кристаллов, т.пл. 132-133 С. Дополнительно 4,24 г (6,8%) получают за счет переработки маточного раствора. Н-ЯМР (CD-CD), доли на млн. (дельта): 2,40 (синглет, ЗН); 2,43 (синглет, ЗН); 7,68 (синглет, 1Н).
(iii) b) Смесь 1,66 г (10 ммоль) 2-хлор-1,1-диметокси-З-бутанона, 1,43 г (15 ммоль) ацетамидина гидрохлорида и 2,05 г (25 ммоль) ацетата, натрия в 50 мл диоксана нагревают с обратным холодильником 24 ч. Диок- сан выпаривают в вакууме и остаточное масло обрабатывают посредством мгновенного хроматографирования на силикагеле (40:60 этилацетат гек- сан, 40 мм), что дает три фракции. Третью фракцию -белое твердое вещест23Г
во (1,121 I ) lorrropiio хроматографн- руют (40 мм, ацетон), что дает 933 мг (75,1%) продукта в виде бкло- го твердого вещества, которое является чистым в результате его оценки по в CDC , также посредстном тонкослойной хроматографии на силикагеле (1 пятно, 1:9 метанол/хлороформ).
Пример 11. Получение 4-аце- тил-2-мети.гшмидазол (препарат F.) .
(I) 1-Бензил-2-метилимидазол.
К суспензии 2,4 г (0,1 моль) гидрида натрия в 50 мл диметилформа- мида в атмосфере азота добавляют при перемешивании 8,2 г (0,1 ммоль) 2-метилимидазола.Происходит реакция с постепенным выделением тепла, температура достигает A3 С. Если вьще- ление тепла убывает, реакционную смесь подогревают на паровой бане до 70-75 С 0,5 ч, затем при 95°С 15 мин для завершения реакции (прекращения выделения газа). Затем реакционную смесь охлаждают до 68°С и добавляют каплями 12,7 г (0,1 моль) бензилхлорида. Происходит экзотермическая реакция, температура достигает 95°С. После 30 мин перемешивания, следующего за окончанием пере- мешивания, реакционную смесь вливают в 600 мл воды и продукт экстрагируют этилацетатом (2x200 мл). Соединенные экстракты промыв, ют последовательно водой (1x400 мл), насьпденным водным раствором натрия хлорида (1x100 мл, затем 6 н. НС1 (1x50 мл) Промывную жидкость после обработк, НС1 экстрагируют эфиром (1x25 мл), после чего подщелачивают добавлением натрия гидроокиси. Отделившееся желтое масло экстрагируют эфиром, экстракт высушивают (сульфат магния) и упаривают при пониженном давлении, что дает светло-желтое масло. Выход 11,5 г (60,5%). ЯМР показывает, что соединение получено как моногидрат, который используется для реакции ок- симетилирования.
(И) 1-Бензил-4-оксиметш1-2-ме- тилимидазол.
Смесь 8,5 г (0,05 моль) 1-бензил- -2-метилимидазола моногидрата, 50 мл 36%-ного формальдегида, 6 мл уксусной кислоты и 8,0 г (0,098 моль) аце
тата натрия перемешивают и нагрева ют с обратным холодильником 26 ч. Затем перемешивание продолжают в течение около 65 ч при комнатной тем
58П6
Q
15 20 25ЗО . Q дс
55
1424
пературе, после чего нейтра.чияуют твердым карбонатом натрия.
Не14трал11ный раствор экстрагируют ттилацетатом, экстракт высутивя от (сульфат магния) и выпаривают при пониженном давлении до масла. К маслу добавляют воду (10 №i) и итопро- нанол (50 мл), раствор перемешивают всю ночь, затем в 1паривают при пониженном давлении. Полученньй маслообразный остаток поглотают водой и раствор делают силр.но щелочным путем добавления твердой гидроокиси натрия. Раствор подвергают резкому охлаждению, покрывают слоем.диэтило- вого эфира и полученное белое твердое вещество удаляют фильтро ванием и высушивают на воздухе. Выход 1,8 г (18%), т.пл. иО-иб С. Вещество . очищают растворением в 30 мл горячего (50 С) этилацета.та и фильтрованием. Концентрирование фильтрата примерно до 2/3 первоначального объема и резкое охлаждение дают 1,3 г белот о твердого вещества, т.пл. 147-151 С. Тонкослойная хроматография в системе этилацетат ; метанол : диэтила- мин (80 : 10 : 10) показала единичное пятно.
(III)1-Бензил-2-метилимидазол-З- -карбоксальдегид.
Суспензию, содержащую 9,0 г (0,466 моль) 1-бензил-4-оксиметил- -2-метилимидазола; 750 мл метиленхло- рида и 50,0 г (0,575 моль) марганца двуокиси перемешивают при комнатной температуре 2 ч. Затем фильтруют, оставшуюся на фильтре массу промывают метиленхлоридом и соединенные фильтраты, также растворы после про- мьшки упаривают при пониженном давлении, что дает масло. Масло поглощают 100 мл диэтилового эфира, добавляют 100 мл гексана и в раствор вносят затравку для кристаллизации в виде нескольких кристаллов предлагаемого соединения. Концентрирование раствора путем продувки азотом с периодическим возмещением гексана дает кристаллический продукт, которьо отделяют фи.пьтрованием: 7,2 г, выход 81%, т.пл. 57-60°С.
Вторую фракцию (0,75 г) получают концентрированием фипьтрата, т.пл. 57-59,5°С. Общий выход 89,4%.
(IV)1-Бензил-4-(1-оксизтил)-2- ме тилимида з ол.
К растнору 7,2 г(О,306 моль) 1- -бен: 1{.ч-2-импдазол-4-карбоксальде- 1 идя п 100 мл тетрагидрофурана добавляют 15 мл 2,9 М метилмагнийхло- рнда (0,14Л моль) в тетрагидрофура- не. Тотчас же получается белый осадок. Смесь перемешивают при комнатной температуре 30 мин, затем нагревают с 50 мл 25%-ного водного раствора хлорида аммония. Осадок отфильтровывают, промывают тетрагидро фураном и высушивают на воздухе, Соединенные фильтраты и промывные растворы высушивают (сульфат натрия и концентрируют в вакууме до твердого остатка. Остаток растворяют в 300 r-t:r кипящего этилацетата, высушивают (сульфат натрия) и концентрируют до половины объема при пони- женном )влении. Твердое вещество: Bbinatjniee в осадок при охлаждении, отфи:11)Тронызают и высушивают на воздухе, Общий выход: 7,1 г (90%), т.пл. 1fi2,5-l67,5°C.
(V)4-(1-Оксиэтил)-2-метилимидаЗО.ТГ .
Аппарат Парра для перемешивания встряхиванием загружают 10,0 г (А,6,23 ммоль) 1-бензил-4-( 1-окси- отил)-2-метилимидазола, 60 мл метанола и 2,0 г 5%-ного палладия на угле (50% воды). Нагнетают газообразный водород до давления 2,04 ат (30 фунтов /кв. дюйм), смесь нагре- вают до и встряхивают 16 ч. Ох лаждарлт до 30°С, фильтруют через диатомовую землю и отфильтрованный осадок промывают 10 мл метанола. Выиаривапие соединенных фильтрата и промывной жидкости при пониженном давлении дает 6,44 г (97%-ньсй выход предлагаемого продукта в виде масла
Продукт можно кристаллизовать добавлением тетрагидрофурана в количе стве, достаточном для растворения масла и перемешивания раствора при температуре окружающей среды 2ч. Белое кристаллическое твердое вещесво собирают фильтрованием и высушивают на воздухе, т.пл. 107-111 С.
(VI)К )1агреваемой при действии обратного холодильника смеси 1240 г (9,989 моль) 4, 5-(1-оксиэтил)-2-ме тилимидазола в 10л тетрагидрофурана добавляют 2200 г (25,293 моль) марганца двуокиси в течение 10 мин. Смесь нагревают с обратным холодильником (18 ч), затем фильтруют горячей через диатомовую землю. Отфильтрованный осадок промывают 4 л тетрагидрофурана.
Соединенные фильтраты и промывные жидкости после проведения двух таких реакций перемешивают и концентрируют при атмосферном давлении до объема около 6 л. В этот момент смесь затвердевает. Добавляют этил- ацетат (2 л), смесь нагревают до образования раствора, что позволяет дополнительно удалять тетрагидрофу- ран. Если смесь затвердевает, добавляют еще 2 л этилацетата и снова нагревают. Если смесь становится твердой, нагревание и перемешивание прекращают и смеси дают остыть в течение ночи. Добавляют этилацетат (3,8 л) и разбивают твердую массу, пользуясь шпателем. Если смесь уже можно будет перемешивать, суспензию |нагревают при 50 с 3 ч, затем охлаждают при 5°С 1 ч и фильтруют при отсасывании. Желтый остаток после филв трования промывают 1,5 л этилацетата при 5°С, затем высушивают на воздухе Выход 1887 г (76,08%), т.пл. 128 - 130°С.
Пример 12. Получение 4-хлор ацетил-2-метилимидазола и его хлористоводородной соли (препарата Г).
Через раствор 248 мг (2,0 ммоль) 4-ацетил-2-метилимидазола в 20 мл метиленхлорида пропускают сухой газообразный хлористый водород 5 мин, затем добавляют 192 мг (6 молей) сухого метанола. К полученному раствору добавляют 297 мг (2,2 ммоль) хлористого сульфурила и смесь перемешивают 1 ч при комнатной температуре. Для гарантирования завершения реакции затем добавляют две дополнительных порции хлористого сульфурила по 155 мг каждая с 10 мин интервалами. Добавляют несколько миллилитров метанола и смесь выпаривают в вакууме досуха. Полученное бесцветное масло подщелачивают твердым бикарбонатом натрия. Выпавшее в осадок твердое вещество собирают, промывают водой и высущивают в высоком вакууме, что дает 167 мг (33%) твердого вещества в виде мелкого белого порошка.Н-ЯМР (DMCO-d), доли на млн. (дельта): 2,25 (синглет, ЗН); 4, 75, (синглет, 2Н); 7,8 (синглет, 1Н). Масс-спектр (т/е): 158 (М), 109 (M-CHjCl).
27
Если указанную процедуру повторять в том же масштабе, но без мета нопа при последующих добавлениях хлористого сульфурила и 2 ч перемешивания при KOMHaTHOti температуре, то получают 395 мг (100%) хлористоводородной соли, т.пл. 159-166 С, разложение .
Пример 13. Получение 2-ами нометилнафталина (препарата G).
Смесь 10,0 г (65,3 ммоль) 2-циан нафталина, 2,0 г скелетного никелевого катализатора по Ренею, 100 мл этанола и 9 мл концентрированной гидроокиси аммония гидрируют при 2,53 кг/см 4,5 дня. Смесь фильтруют и фильтрат концентрируют в вакууме до масла. Масло перегоняют з вакууме, что приводит к целевому ам ну в виде бесцветной жидкости, затвердевающей при стоянии. Выход 2,02 г. Тонкослойная хроматография на силикагелевьсх пластинах показана одно пятно при Rf 0,1 после проявления 19:1 хлороформ/метанолом.
Пример 14. Общий способ получения 3-арилпропиламинов формулы
(Rj) Ar (CH,)jNH,
3-(4-н-пропилфенил)пропиламин.
(i) Этил-2-циано-3-(4-н-пропил- феиил)акрилат.
Смесь 20,0 г (90 ммоль) 4-н-про- пилбензальдегид диэтилацеталя, 20,4 г (180 ммоль) этилцианацетата, 7,2 г (93,4 ммоль) ацетата аммонил и 60 мл толуола нагревают с обратным холодильником 6 ч, охлаждают и выливают в воду. Полученную смесь экстрагируют этиловым эфиром, высушивают (сульфат магния) и летучие упаривают в вакууме, что дает 23,0 г сырого желтого масла, которое очищают колоночной хроматографией на силикагеле. Отмывка из адсорбента смесью 2:1 метиленхлорид/гексан дает 20,58 г (94%) продукта, Н-ЯМР (CDCI.J), доли на млн. (дель- та): 0,85-1,95 (мультиплет, 8Н); 2,45-2,70 (триплет, 2Н); 4,15-4,60 (квартет, 2Н); 7,15-8,05 (квартет,
:4Н); 8,25 (синглет, 1Н).
(ii) 3-(4-н-Пропилфенил)пропио- нитрил.
Смесь 20,50 г (84,3 ммоль) продута по разделу (i); 8,75 г магния в
28
15
стружках и 200 м.п метанола перемеши- нают в атмфере азота 6 ч при периодическом охлаждении для поддержания температуры на уровне около 30 С. Смесь подкисляют хлористоводородной кислотой, экстрагируют этиловым эфиром, экстракты промывают раствором бикарбоната натрия, водой, рассолом
)0 и высушивают над сульфатом магния. Выпаривание растворителя дает 23,8 г сырого продукта, которьш очищают колоночной хроматографией на силикагеле, отмывают из адсорбет1та метил с нхлоридом, что обеспечивает 11,55 г (59%) очищенного метил-2- -циано-3-(4-н-пропил-фенил) пропио- ната. Этот продукт соединяют с 4,17 г хлористого натрия, 175 мл диметил- 20 сульфоксида и 5 мл воды в атмосфере азота и смесь нагревают при 150 С 5 ч. Реакционную смесь охлаждают, выливают в 700 воды и экстрагируют 2x500 мл этилацетата.
25 Объединенные экстракты промывают рассолом (300 мл) высушивают над безводным сульфатом натрия и концентрируют в вакууме, что дает 12,5 г целевого нитрила, который очищают перегонкой, т.пл, 124-125 С (1,0 мм). (CDCl,), доли на млн. (дельта): 0,75-1,15 (триплет, ЗН); 1,30 - 2,00 (мультиплет, 2Н); 2,40-3,10 (мультиплет, 6Н) ; 7,15 (синт-лет,4Н) .
(iii) Смесь 14,13 г (81,6 ммоль) указанного выше нитрила (перегнанного), 1,5 г скелетного никелевого катализатора по Ренею, 60 мл этанола и 8 мл концентрированной гидроокиси аммония гидрируют при 3,5 кг/см 18 ч. Смесь продувают азотом, катализатор удаляют фильтрованием и фильтрат концентрируют в вакууме, что дает 12,3 г (84,8%) прозрачного масла. Масло перегоняют, что обеспечивает 8,60 г (59%) чистого амина в виде бесцветного масла. Н-ЯМР (CD( lj), доли на млн, (дельта): 0,75-1,05 (триплет, ЗН); 1,05 (синглет, ЗН) ; 1,15-1,75 (мультиплет, 4Н); 2,30 - 2,85 (мультиплет, 6Н); 6,95-7,10 (мультиплет, 4Н).
Пример 15. Получение Д-( Л- -хлорфенил)-бутиламина (препарата V), Общий способ получения (-арил55 бутиламинов формулы
(R),j Аг (СН,) NH, поясняется далее.
30
35
40
45
50
(1) Д-(4-Хлорфенил)-3-бутеновая кислота.
Смесь 4-хлорбензальдегида (10,0 68,2 ммоль), 34,0 г (81,9 ммоль) бромида З-(трифеннлфосфоний) пропи- оновой кислоты (приготовленного реакций трифенилфосфина и 3-бромпро- пионовой кислоты в ксилоле), 12,5 г гидрида натрия (50% в минеральном масле) и 200 мл диметилсульфоксида нагревают лри 120°С 5 ч, охлаждают и выливают в ледяную воду. Смесь пощелачивают карбонатом натрия, экстрагируют этиловым эфиром и экстраты отбрасывают. Водную фазу подкислют, экстрагируют снова этилацетатом высушивают (сульфат магния) и эфир испаряют в вакууме, что дает 6,9 г (51%) целевой кислоты.
Н-ЯМР (CDClj), доли на млн. , (дельта): 3,10-3,00 (дублет, 2Н); 6,10-6,35 (мультиплет, 2Н); 7,20 (синглет, 4Н); 11,55-11,75 (широкий синглет, 1Н).
(IT) 4-(4-хлорфенил) масляная кислота.
Смесь 19,5 г (98,2 ммоль) нена- сьпценной кислоты, полученной по разделу (I), 1,95 г катализатора палладий на углероде и 200 мл этилацет та гидрируют при 3,5 кг/см и обрабатывают обычным образом, что дает целевую насьш1енную кислоту с 91%- ным выходом. Н-ЯМР (CDClj), доли на млн. (дельта): 1,75-2,80 (мультиплет, 6Н) ; 6,95-7,40 (квартет, 4Н); 9,15-10,25 (широкий синглет, 1Н).
(III) амид-А-(4-хлорфенил)масляной кислоты.
Смесь 8,8 г (44,3 ммоля) насыщенной кислоты по разделу (II) и 45 мл хлористого тиснила нагревают с обратным холодильником 3 ч. Смесь охлаждают и избыток хлористого тио- нила удаляют выпариванием в вакууме Неочищенный хлорид кислоты растворяют в 20 мл этилового эфира и к раствору добавляют каплями 67 мл коцентрированной гидроокиси аммония при 0°С в течение 20 мин. Сразу же образуется рыжевато-коричневое твердое вещество. Смесь перемешивают 1 при 0°С, добавляют 80 мл и воды и смесь экстрагируют 3x100 мл этилового эфира. Объединенные эфирные слои промывают рассолом, высущивают (сульфат масиия) и концентрируют в
вакууме, что дает 8,70 г (97%) амида. Н-ЯМР (CDC1,), доли на мл. (дельта): 1,60-2,40 (мультиплет,4Н); 5 2,45-2,85 (триплет, 2Н); 5,25-6,10 (широкий синглет, 2Н); 6,90-7,30 (квартет, 4Н).
(IV) Смесь 8,70 г (44 ммоль) амида по разделу (III) и 71 мл 1,0 М
0 бороводорода (тетрагидрофуран) в
60 мл тетрагидрофурана перемешивают 4 ч и реакцию останавливают смешением с 6 н. хлористоводородной кислотой (36 мл), Смесь экстрагируют эти- 5 ловым эфиром, экстракты высушивают (сульфат натрия) и концентрируют в вакууме. Остаточное масло перемешивают с изоцропиловым эфиром, фильтруют и фильтрат выпаривают в вакууме,
0 что дает 2,08 г, дополнительно 2,4 г получают экстрагированием жидкости этилацетатом. Н-ЯМР (CDClj), доли на млн. (дельта): 1,15 (синглет, 2Н); 1,30-1,90 (мультиплет, 4Н);
5 2,40-2,90 (квартет, 4Н); 6,90-7,35 (квартет, 4Н).
Пример 16. Активность в отношении воспрепятствования секреции |.
Q кислоты в желудке.
Активность предлагаемых соединений, препятствующую секреции кислоты в желудке, определяют на привязанных на всю ночь здоровых санитарных собаках породы хайденхайн. Для стимулиро вания выделения кислоты применяют Пентагастрин (Петаволон-Ауэрст) посредством непрерывного внутривенного вливания в поверхностную вену ноги на участке от колена до ступни. Вводимые дозировки соответствуют ранее определенным для стимулирования практически наибольшего выделения кислоты из желудочного кармана. Желудочный сок собирают через 30-минутные интервалы, начиная от момента внутривенного вливания Пен- тагастрина и замеряют с точностью, близкой к 0,1 мл. Во время опыта сборы производят у каждой собаки.
0 Концентрацию кислоты определяют путем титрования 1,0 мл желудочного сока с применением автобюретки и стеклянного электрода в составе аппарата для измерения рН (радиометр).
5 Лекарственный препарат или растворитель, также индифферентную среду препарата дают внутривенно или через рот через 90 мин после начала внут0
5
ривеиного вливания пентагастрина при дозировки 2 мг/кг или менее. Действия, препятствующие выделению кислоты в желудке, вычисляют по сравнению самого низкого выделения кислоты после введения лекарства с средним выделением кислоты непосредственно до введения лекарства.
Продукты f, g, n и о примера 2 при приеме через рот с дозировкой 2 мг/кг ингибируют желудочную секрецию на 24%, Предпочтительные соединения Ь, с, J, 1 и т, испытанные в виде гидробромида примера 2, ингибируют секрецию в желудке минимум на 64% при такой же дозе. При дозе 0,1 мг/кг /внутривенно) соединение примера 1 (испытано в виде гидробромида) дает 58%-ное ингибирование.
Пример 17. Активность в отношении противодействия к гистамину
Активность предлагаемых соединений, противодействующую гистамину -Н, определяют по следующей методике .
Морских свинок быстро умерщвляют ударом по голове, удаляют сердце и правую артерию препарируют так, чтобы она была свободной. Артерию под- вещивают изометрически в тканевой ванне с регулируемой температурой (32i2°C). Эта ванна (10 м.п), содержит
NRNH ТШ.
А. Соединения формулы(I), где X C HjCHj 7,31,0
C.Hj7,21,04
СП,(СН,)7,80,72
0
5
0
5
0
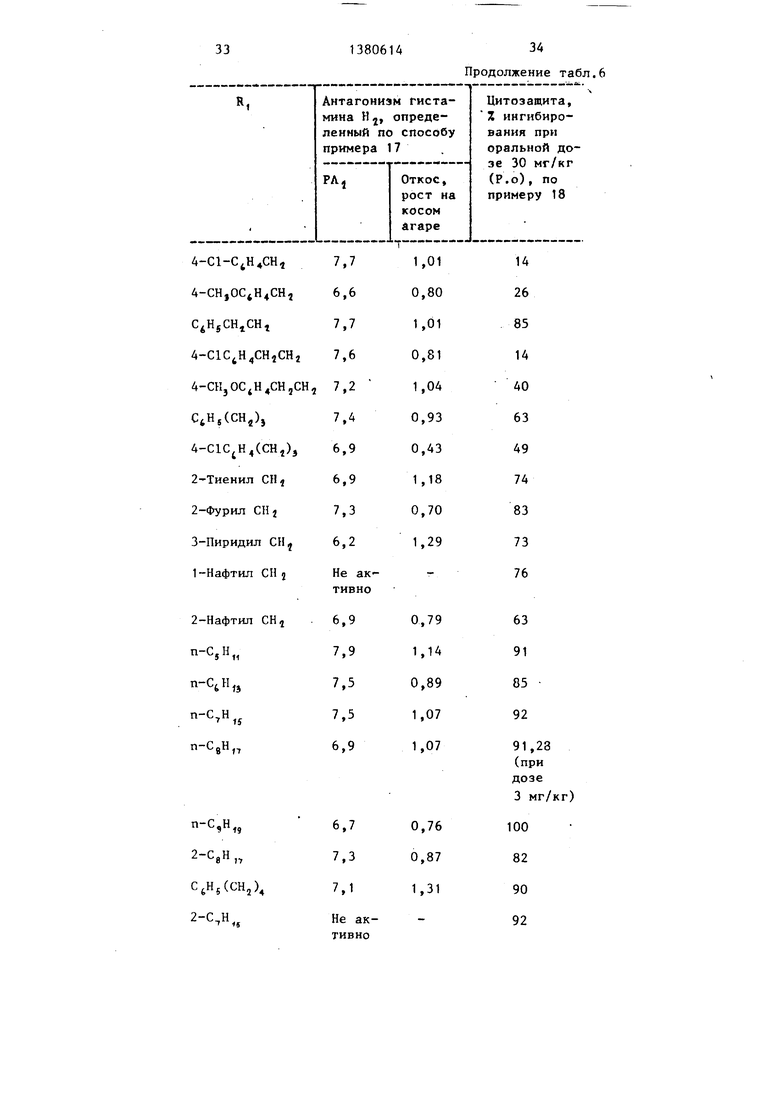
оксиг емированньп fQS% О-,; С0,(у- фер (рН 7,4) Крсогп-Хентелейга. Оставляют для стабилизации примерно на 1 ч. За это время чканевую яан- ну промывают несколько раз ciL ibiun i струей жидкости. Отдельные coKjiame- ния, относящиеся к предсердию,прослеживают с помощью приспособления, передающего смещение от усилия, соединенного с кардиотахометром и регистрирующим полиграфом Грасса. После получения кривой, выражающей зависимость между дозой и ответной реакцией со стороны гистамина, ванну, содержащую каждое предсердие, промывают несколько раз сильной струей свежего буфера и предсердие приводят в равновесие с основными частотами. После возвращения к основной частоте добавляют испытуемые соединения при избранных окончательных концентрациях и снова определяют кривую доза гистамина - ответная реакция. Определяют отношение концентрации гистамина, требуемое для получения половины максимального стимулирования в присутствии или отсутствии антагониста, и вычисляют константу кажущейся диссоциатши Н, - рецептора антагониста рА. Результаты антисекреторной и цитоза- щитной активности соединений форьгу- лы (I) представлены в таГхп.Ь и 7.
Таблица 6
N
СН
-Tjl
л
Nn, R СИ,
7,0 61 63
33
13806U
34
Продолжение табл.6
35
S-ClCjH CH A-FCflUCH 1-Бутил А-Пиридил СН J 2-11иридил СН,
4-Имидазолил (CHjOj
5-Метил-1-2- -фурил СН,
4-Метилпен- тил
2-Фурил-СН,СН 2-Тиазолил-СН 3-CHjCjH CH
ЕД ЕД
6,9 при подкожном введение (sc),
(P.O.) / ЕД,. (sc) 1.
138061436
Продолжение табл.6
36
70
70
78
О
83
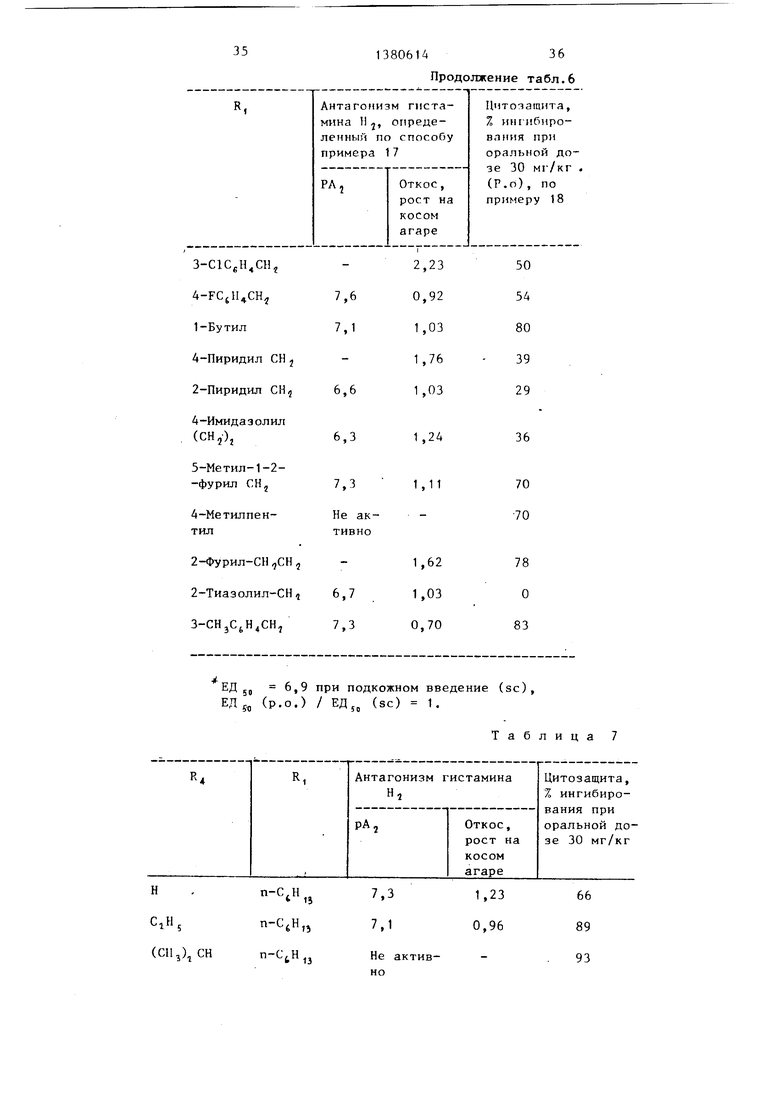
Таблица 7
трет-Бутил n-CjHu
CHjOH PnCHj
В. Соединение формулы (I) где Y СН, X NH, R, и R как определено
С. Соединения формулы (1),где X определено ниже
СН.
NH.
n-CtH,j n-CjH,5
Предпочтительное известное соединение.
Циметидин Н-циано-Н -метш1-Ы -2-(5-метил-1Ы-имидазол- -4-ил)-метил(тио)этилгуанидин7 - коммерческий стандартный препарат для лечения язвы желудка и двенадцатиперстной кишки.
Пример 18. Ингибирование образования язв, вызываемых этанолом у крыс.
Противоязвенную активность продуктов по настоящему изобретению также определяют у крысы посредством проверки язвы, вызванной этанолом. При этом испытании самцам крыс, зафиксированным всю ночь, давали лекарство. (при 30 или 3 мг/кг или воду путем приема через рот за 15 мин до приема через рот дозы абсолютного Зта- нола (1,0 мл). Через 1 ч после приема этанола животных (по 8 в группе) убивают и исследуют желудок на налиПродолжение табл.7
39
38
X
SjR как
1,05 0,77
13 76
0
5
чие повреждений. Путем оперативного вмешательства вскрывают желудок и у привратника желудка устанавливают замыкающий гемостат. В желудок вводят инъекцией 6 мл 4%-ного раствора формальдегида через желудочный зонд и для запирания пищевода устанавливают второй замыкающий челюстат. Желудок извлекают, вскрывают вдоль наибольшей кривизны и исследуют на образование язв.
Система подсчета для желудка, применяемая для определения количества повреждений, вызванных этанолом, приводится в табл.8.
Метка подсчета
Определение
Желудок обычного вида
Незначительные по величине повреждения
Повреждений, два или меньше, могут присутст- 15 вовать точечные повреждения
Число повреждений более двух,могут присутствовать 20 точечные повреждения
Повреждения с кровотечением
Для каждой группы животных вычисляют индекс язвы, как показано далее ,
Индекс язвы равен сумме повреждений для каждой группы, умноженной на сумму числа язв в каждой группе и умноженной на долю в группе, имеющую язвы в любой степени развития.
Ингибирование образования язв, выраженное в %, вычисляют так: % ин- гибирования 100 х (индекс язвы у контрольной) - (индекс язвы у получивших лекарство) : (.индекс язвы к контрольных).
Результаты испытаний приведены в табл.6 и 7. При испытаниях на цито защиту, как это описано в примере 18 (ингибирование образования язв, вызываемых этанолом у крыс), соединения, полученные предлагаемым способом, применялись в дозах 30 мг/кг без токсического эффекта.
Эта информация совместно с дозой применения соединений формулы (I) (оральное введение в дозе 0,1 - 20 мг/кг веса тела) показывает, что имеется обоснованный коэффициент безопасности для этих соединений. Таким образом, при предпочтительной дозе 0,2-2,5 мг/кг имеется по крайней мере коэффициент безопасности от 12 до 150 раз.
t
R
где при X, равном NH,R , - прямой
или разветвленный - алкил, фенил или группа (ЕЗ), Аг (СН,)„ где п 1, 2, 3, 4; R, - одинаковые или различные, Н, F, С1, CHjO;
0
5
0
5
0
Аг R. R,
остаток фенила, нафтила, фурила, тиенила, пириди- ла, тиазолила, имида- золила;
водород, С -С -алкил, СНуОН; - водород ,
или при X, равном S,R -н-гексил;
R
R,
- метил, NH
г
.,; - водород, или их хлористоводородных или бро- мистоводородных солей, отличающийся тем, что соединение общей формулы
7У1Н S II II
T lR2 C--NH-C-7 H2,
где R, и R указаны подвергают взаимодействию с эквимолекулярным количеством соединения формулы
HalCHgC -г-N .
ц о
R
где X и R - указаны;
Hal - С1 или Вг,
в присутствии инертного органическо- го растворителя при 50-60 С с последующим выделением целевого продукта в свободном виде или в виде хлористоводородной или бромистоводородной соли.
| Эльдерфильд Р | |||
| Гетероциклические соединения, т.5, с.536 | |||
| Патент США № 4374843, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
