Изобретение относится к способу получения производных бензоксазолонам, а именно к способу получения производных определенных бензоксазолона, замещенным не только в положении 6 аминовой боковой цепью, но также в положении 4, 5 или 7 бензольным кольцом. Такие соединения оказывают ингибирующее действие на ферменты липоксигеназу и/или циклоксигеназу и могут использоваться в качестве ингибиторов указанных ферментов, как таковых. Соединения по изобретению также могут использоваться для лечения различных аллергических и воспалительных заболеваний у млекопитающих.
Известны бензоксазолоновые соединения с алкиламиновой группой в положении 6, которые являются ингибиторами липоксигеназы и/или циклоксигеназы.
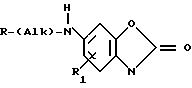
Изобретение раскрывает способ получения новых бензоксазолоновых соединений формулы
R-(Alk)-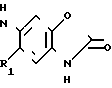
(I) или их фармацевтически приемлемые соли аддукты кислот, где Alк неразветвленная или разветвленная двухкалентная алкильная группа Сn, где n равно 0, 1, 2, 3 или 4,
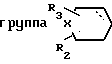
R выбирают из группы, включающей 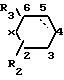 где R2 и R3 каждый независимо друг от друга водород, или (С1-С4) алкил;
где R2 и R3 каждый независимо друг от друга водород, или (С1-С4) алкил;
Х метилен, кислород, сера или сульфоксид;
и пунктирная линия между положениями 3 и 4 представляет собой необязательную связь или R группа формулы
 и ломаная линия указывает, что группа, содержащая такую ломаную линию, может представлять собой эндо или экзо-7-оксабицикло [2,2,1]гептан-1-ил; или R представляет группу
и ломаная линия указывает, что группа, содержащая такую ломаную линию, может представлять собой эндо или экзо-7-оксабицикло [2,2,1]гептан-1-ил; или R представляет группу
СН3 (СН2)m Y где m равно 1, 2 или 3 и
Y кислород, сера или сульфоксид.
Соединения формулы (1) могут содержать ассиметрический центр и, следовательно, могут существовать в виде пары энантиомеров. Изобретение включает каждый чистый энантиомер, их рацематы, смеси энантиомеров, частично или полностью оптически расщепление.
Фармацевтические применимые соли соединений формулы (1) продукты кислот включают те из них, которые получают из кислот, образующих нетоксичные соли, например солянокислые, бромистоводородные, сульфаты или бисульфаты, фосфаты или кислотные фосфаты, ацетаты, цитраты, фумараты, глюконаты, лактаты, малеаты, сукцинаты, тартраты, метансульфонаты, бензолсульфонаты, толуолсульфонаты и формиаты.
Новые соединения формулы (I) могут быть получены в соответствии со следующей схемой реакции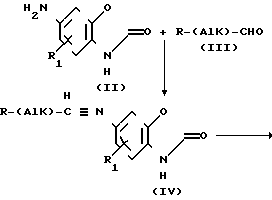

В приведенных формулах Alk, R и R1 соответствуют данным выше определениям. На первой стадии примерно эквимолярные количества реагентов, таких как амин (II) и альдегид (III), смешивают в подходящем органическом растворителе, хотя реакцию предпочтительно вести при температуре окружающей среды, возможно применение и более высоких температур, например температуры кипения с обратным холодильником, и это не будет приводить к каким-либо значительным нежелательным осложнениям. К числу подходящих органических растворителей относятся (С1-С4) алканолы (например метанол или этанол), бензол, толуол и тетрагидрофуран. Возможно, что применение дегидратирующего вещества будет давать определенные преимущества. В качестве дегидратирующего вещества предпочтительны молекулярные сита. Для ускорения реакции произвольно добавляют малые количества низких алкановых кислот, например уксусной кислоты. Реакция существенно завершается в течение 24 ч. Продукты реакции (IV) может выделяться и подвергаться очистке стандартными способами, например перекристаллизации или хроматографии, когда получаемый имин сопрягается с ненасыщенной группой. Однако более предпочтительно продукт не выделять, а подвергать его (т.е. на месте) реакции во второй стадии.
На второй стадии реакции осуществляют восстановление С N двойной связи под действием приемлемого источника водорода. Хотя восстановление может осуществляться с помощью широкого спектра восстановителей, которые известны как восстановители углеродазотной двойной связи, в соответствии с предпочтительным способом по изобретению применяют гидрид металла или осуществляют реакцию каталитического гидрирования. К числу приемлемых для этой реакции гидридов относятся борогидрид натрия, цианоборогидрид натрия и цианоборогидрид лития. Обычно восстановление осуществляют при температуре олкружающей среды в присутствии избытка гидрида в (С1-С4) алканоле, таком как метанол или этанол. Каталитическое гидрирование осуществляют в присутствии каталитического количества катализатора благородного металла, такого как палладий на угле или PtO2 в атмосфере водорода. После завершения восстановления целевой продукт формулы (I) выделяют стандартными способами. Очистку осуществляют стандартными методами, например, перекристаллизацией или хроматографированием. Или, альтернативно, соединение формулы II подвергают взаимодействию с кетоном формулы
R где Х метилен, незамещенный или замещенный одной метильной группой, азот, незамещенный или замещенный защитной группой, кислород, сера или SO и R2 и R3 определены выше. Например, в реакции сочетания соединения (II) с кетоном (V) получают имин формулы (VI)
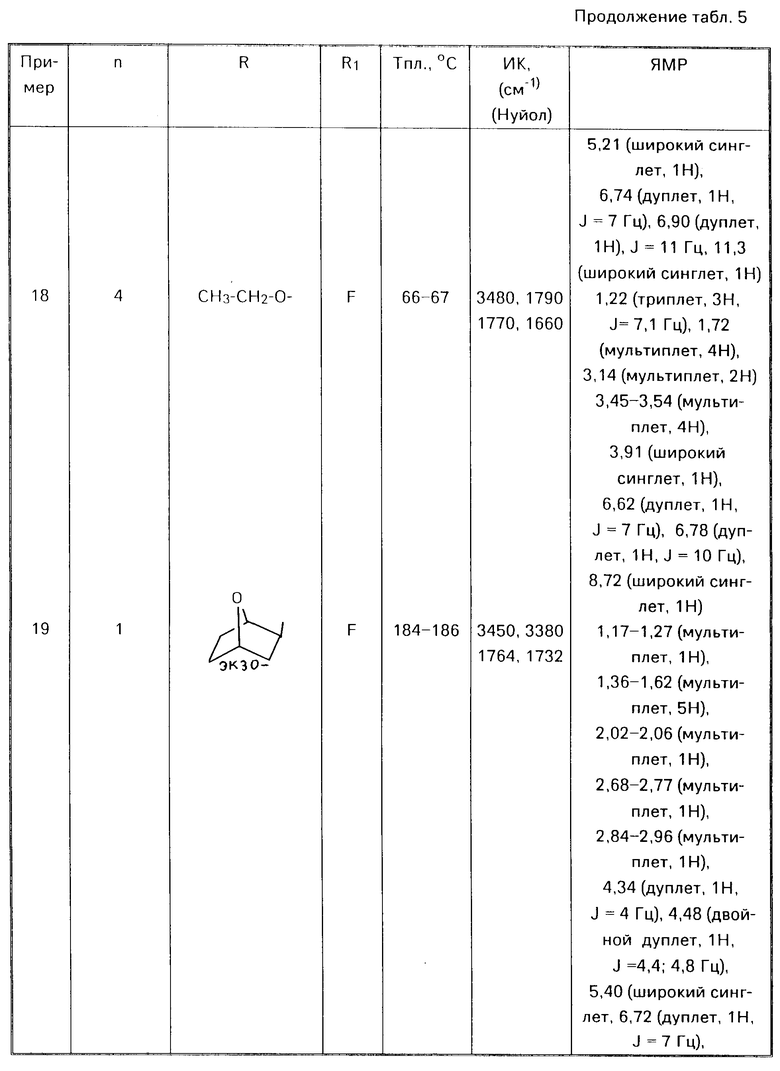
где Х метилен, незамещенный или замещенный одной метильной группой, азот, незамещенный или замещенный защитной группой, кислород, сера или SO и R2 и R3 определены выше. Например, в реакции сочетания соединения (II) с кетоном (V) получают имин формулы (VI)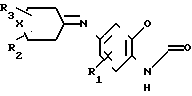 где R1, R2, R3 и X имеют указанные значения.
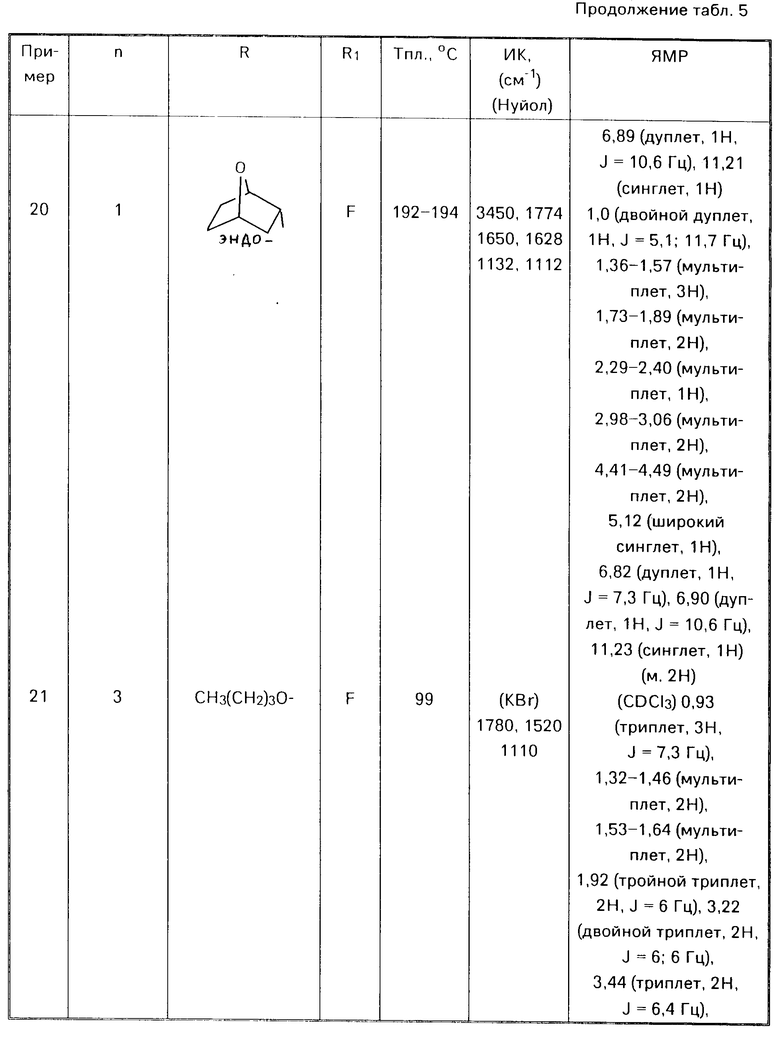
где R1, R2, R3 и X имеют указанные значения.
Последнее соединение легко восстанавливается до целевого соединения. Условия реакции для осуществления указанной двухступенчатой конверсии незначительно отличаются от тех, которые применяют при синтезе соединений (I), у которых n не равно 0.
Соединение формулы (I), в которых Х или Y атом серы переводят окислением в соединение формулы I, где или Y сульфоксид. Приемлемые условия окисления включают, но не ограничиваются ими, реакцию таких соединений с метапериодатом натрия на окиси алюминия, проводимую в приемлемом растворителе, таком как (С1-С3) алканол и/или тетрагидрофуран.
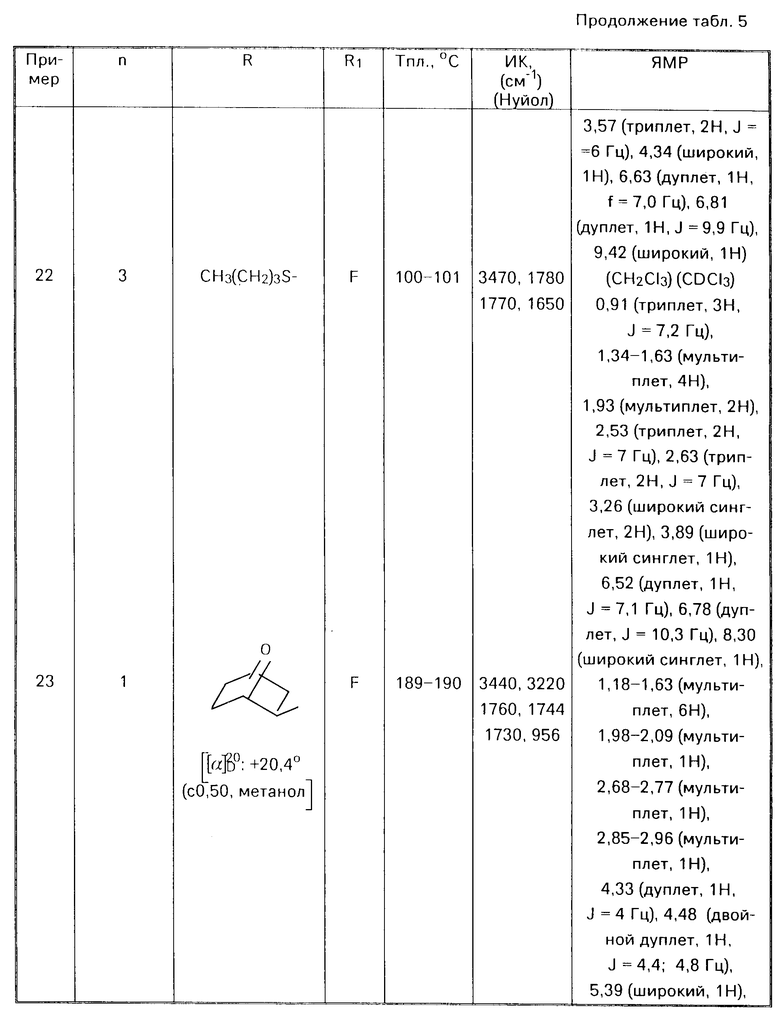
Или, в случае необходимости, восстанавливают соединения формулы I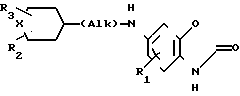
(1) -
-
Особо предпочтительный способ такого восстановления включает гидрирование соединений формулы (I), которые восстанавливаются водородом в присутствии катализатора благородного металла в соответствующем растворителе. Приемлемыми растворителями при таком гидрировании являются, например, серный эфир, тетрагидрофуран, диоксан, этилацетат и (С1-С3) алканол, такой как метанол или этанол. Катализаторами на основе благородных металлов являются, например, никель, палладий, платина и родий. Особо предпочтительными катализаторами являются оксид платины и палладий на угле. Платиновый катализатор иногда более предпочтителен, так как он труднее отравляется серой. Для такого гидрирования не требуется давления водорода (1-4 атм.), и оно протекает при температуре окружающей среды. После завершения гидрирования (в течение примерно 2-24 ч) катализатор отфильтровывают и затем выделяют продукт формулы (IX) и при необходимости очищают стандартным способом.
6-аминобензоксазолиноны-2 (II) получают различными известными в технике способами и иллюстрируются в примерах. Требующиеся для синтеза альдегиды и кетоны выпускаются промышленностью либо они могут быть получены известными способами.
Фармацевтически приемлемые соли новых соединений по изобретению легко могут быть синтезированы путем контакта указанных соединений с соответствующими минеральными или органическими кислотами, взятыми с стехиометрическом количестве, и такой контакт осуществляют либо в водном растворе, либо в приемлемом органическом растворителе. Соль в дальнейшем либо отделяют осаждением, либо выпариванием растворителя.
Соединения по изобретению подавляют активность ферментов липоксигеназа и/или циклоксигеназа. Это подавление демонтируется в экспериментах на крысах на резидентных клетках брюшной полости, в ходе которых определяется эффект указанных соединений на метаболизм арахидоновой кислоты.
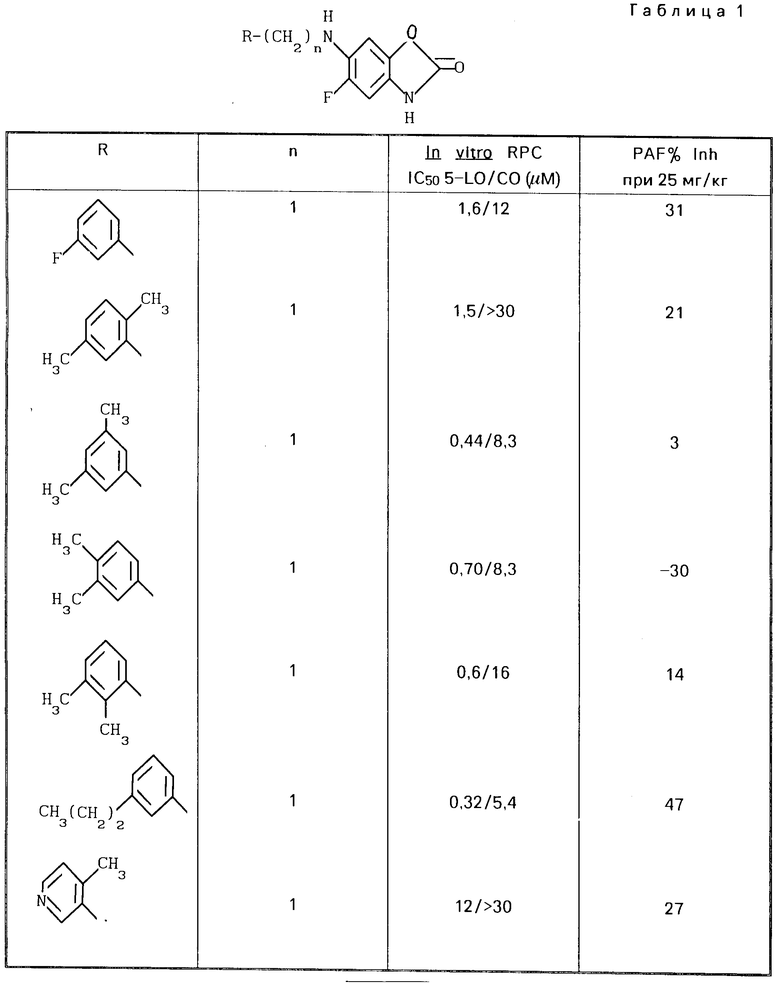
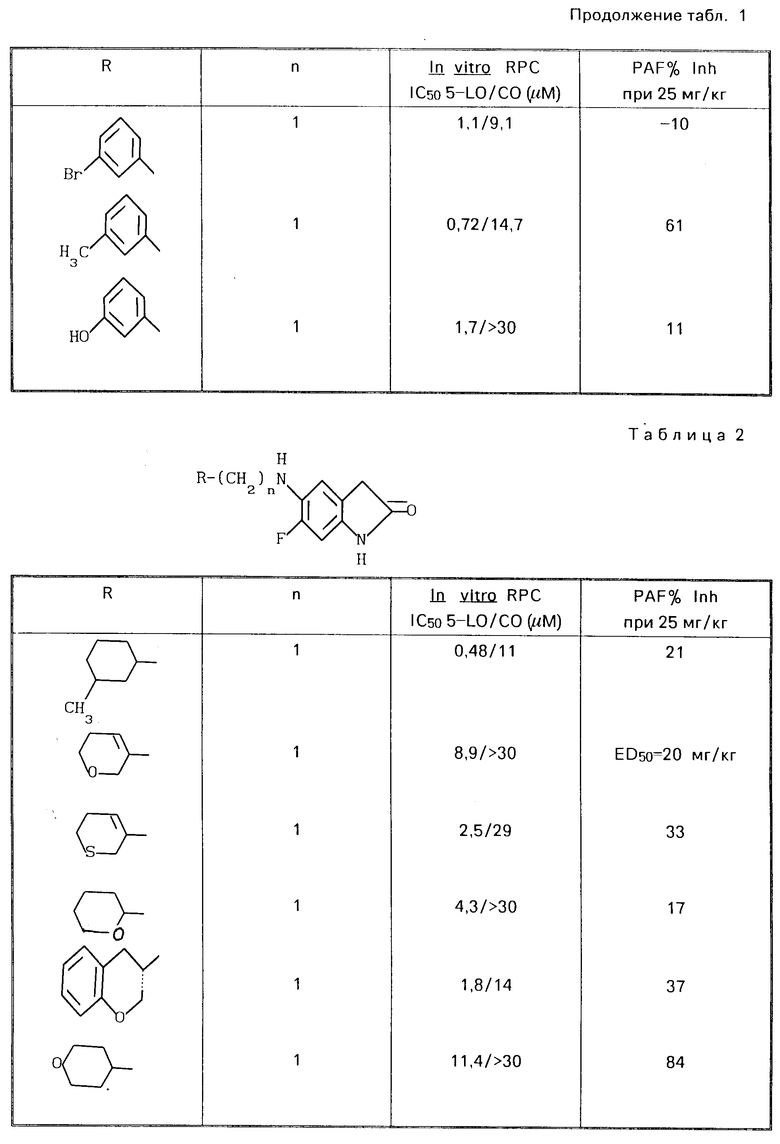
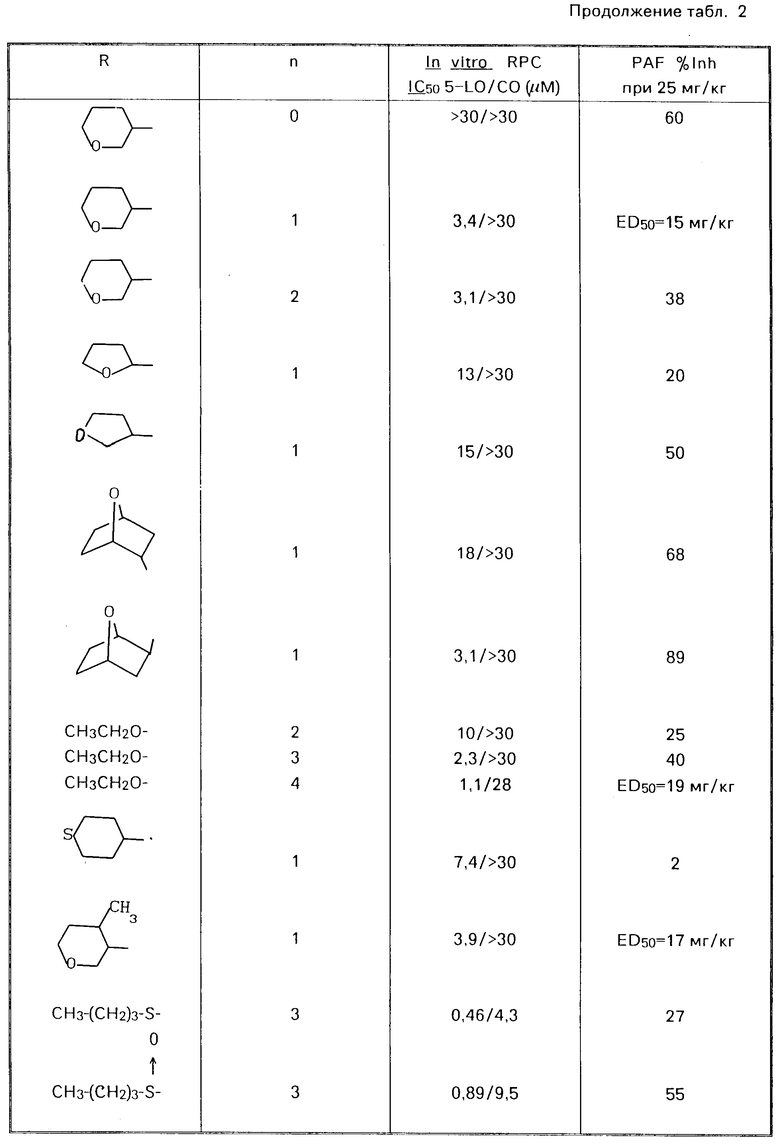
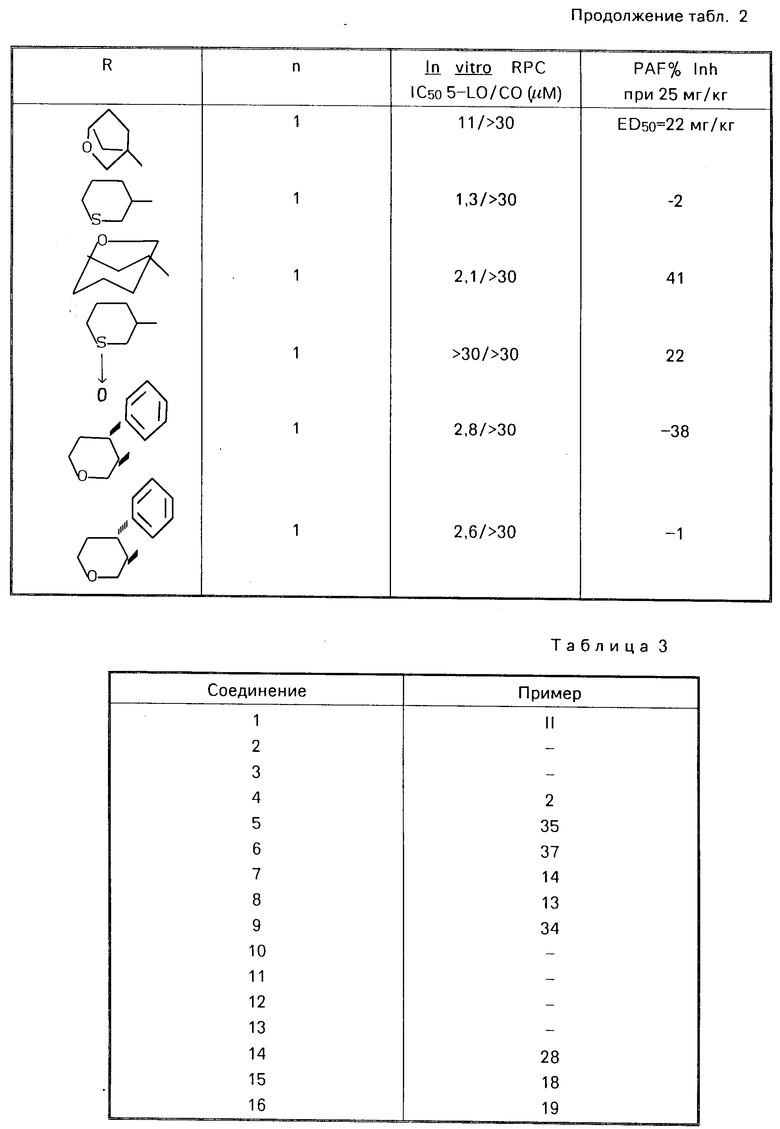
В этом эксперименте некоторые предпочтительные соединения показали низкие IС50 величины, в диапазоне 0,5-30 мкМ, в отношении подавления как липоксигеназы, так и циклоксигеназы.
Способность соединений по изобретению подавлять ферменты липоксигеназа и/или циклоксигеназа позволяет применять их для регулирования симптомов, вызываемых эндогенными метаболитами, являющихся результатом распада арахидоновой кислоты у млекопитающих. Соединения, следовательно, являются ценными для предотвращения и лечения таких заболевания, причиной которых является накопление метаболитов арахидоновой кислоты, например аллергическая бронхиальная астма, ревматоидный артрит, остеоартрит, тромбоз и кожные заболевания.
Активность предлагаемых соединений также может быть проиллюстрирована в стандартных опытах на крысах и в случае каррагенином индуцированной эдемы (С.А.Винтер с сотр. Proc.Soc.Exp.Biol. III, р. 544, 1962).
Так, соединения формулы (I) и их фармацевтически приемлемые соли особенно полезны при лечении или облегчении аллергических или воспалительных заболеваний у людей, а также при подавлении ферментов циклоксипеназа и липоксигеназа.
При лечении различных описанных заболеваний соединения формулы (I) и их фармацевтически приемлемые соли могут вводиться пациентами либо одни, либо в сочетании с фармацевтически приемлемыми носителями или разбавителями в фармацевтической композиции, что предпочтительнее в соответствии с обычной практикой.
Соединение может вводиться разными путями, в том числе перорально, парентерально и путем ингаляции. При пероральном введении суточная доза пациентам колеблется в пределах примерно от 0,1 до 20 мо/кг массы больного, предпочтительно примерно 0,1-10 мг/кг, и используется как розовая или делится на несколько дозировок. При парентеральном введении суточная эффективная доза колеблется в пределах 0,1-1,0 мг/кг массы пациента. В некоторых случаях может оказаться необходимым применять дозировки, выходящие за указанные выше пределы, так как дозировки необходимо менять в зависимости от возраста, массы и реакции конкретных пациентов, равно как и сложности симптомов и активности конкретного используемого соединения.
При пероральном введении соединения формулы (I) и их фармацевтически приемлемые соли могут входить в состав таких форм, как таблетки, порошки, сиропы, капсулы, пилюли. В случае использования при пероральном приеме таблеток в качестве носителя обычно применяют лактозу и зерновой крахмал. Кроме того, обычно добавляют смазочное вещество, такое как стеарат магния. При применении капсулированных форм разбавителями служат лактоза и сухой крахмал. Когда при пероральном применении используются водные суспензии, активный компонент объединяют с эмульгатором и суспензирующим веществом. При необходимости возможно добавление определенных сладких и/или ароматизирующих веществ. При внутримышечном, внутрибрюшинным, подкожным и внутривенном введении обычно применяют стерильные растворы активного компонента и требуется соответствующее регулирование рН растворов и введение в них буферных компонентов. При внутривенном применении суммарная концентрация раствора должна регулироваться таким образом, чтобы он был изотоническим.
Привoдятся табл. 1 и 2. Табл. 1 перечисляет данные in vitro RPC I C50 для 5-LO/CO и данные о летальности PAF для некоторых соединений ЕР 249, 407. Табл. 2 перечисляет эти данные для соединений изобретения. Тест in vitro RPC был проведен согласно опубликованной методике Cheng Jet al Eur J.Phannacol, 107 215 (1985). Тест о летальности PAF был проведен согласно опубликованной методике Joung I.M. et al. Prostaglanding, 30, 545 (1985).
Далее на основе доступной информации соединения изобретения не имеют высокой токсичности.
Табл. 3 устанавливает соответствие между соединениями из табл. 2 и номерами примеров.
Изобретение иллюстрируется следующими примерами. Однако следует иметь ввиду, что изобретение не ограничивается конкретными деталями этих примеров. Спектр протонного ЯМР измеряли при 270 МГц, если особо не оговорено, в растворах полностью дейтерированного диметилсульфоксида (ДМСО-d6). Положение пиков выражали в частях на миллион, отсчитывая от тетраметилсилана. Форма пиков выражалась следующим образом:
S синглет (с), d дуплет (д), t триплет (т), m мультиплет (м), Br широкий (ш).
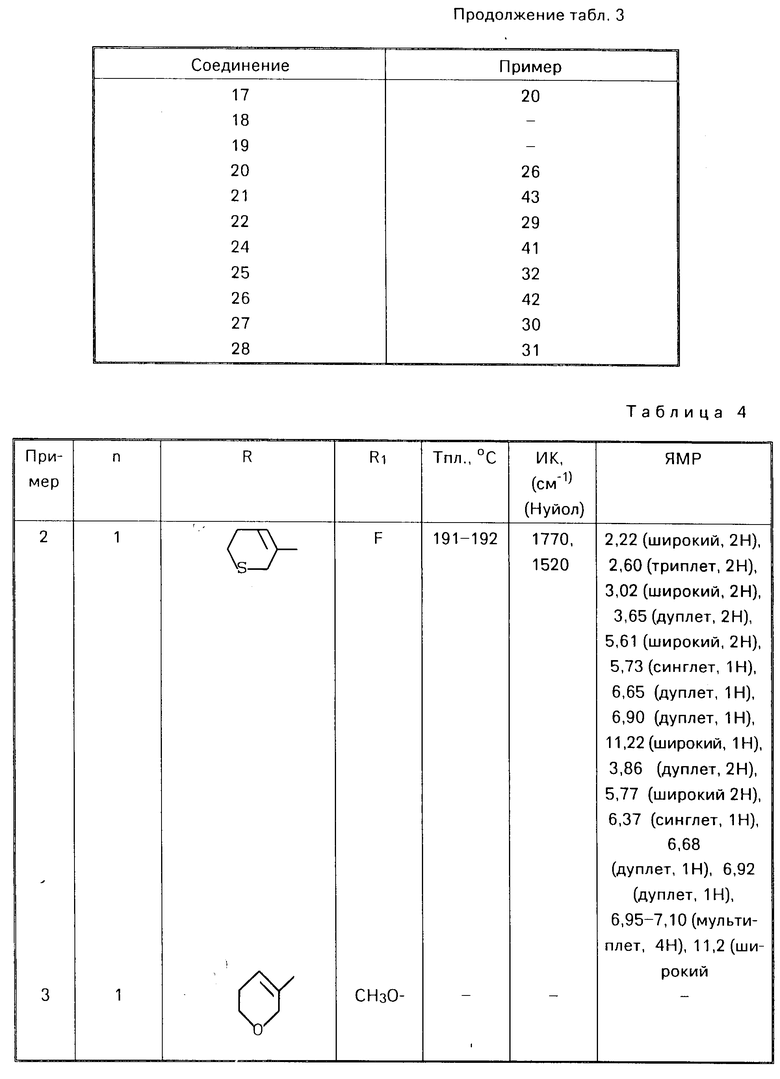
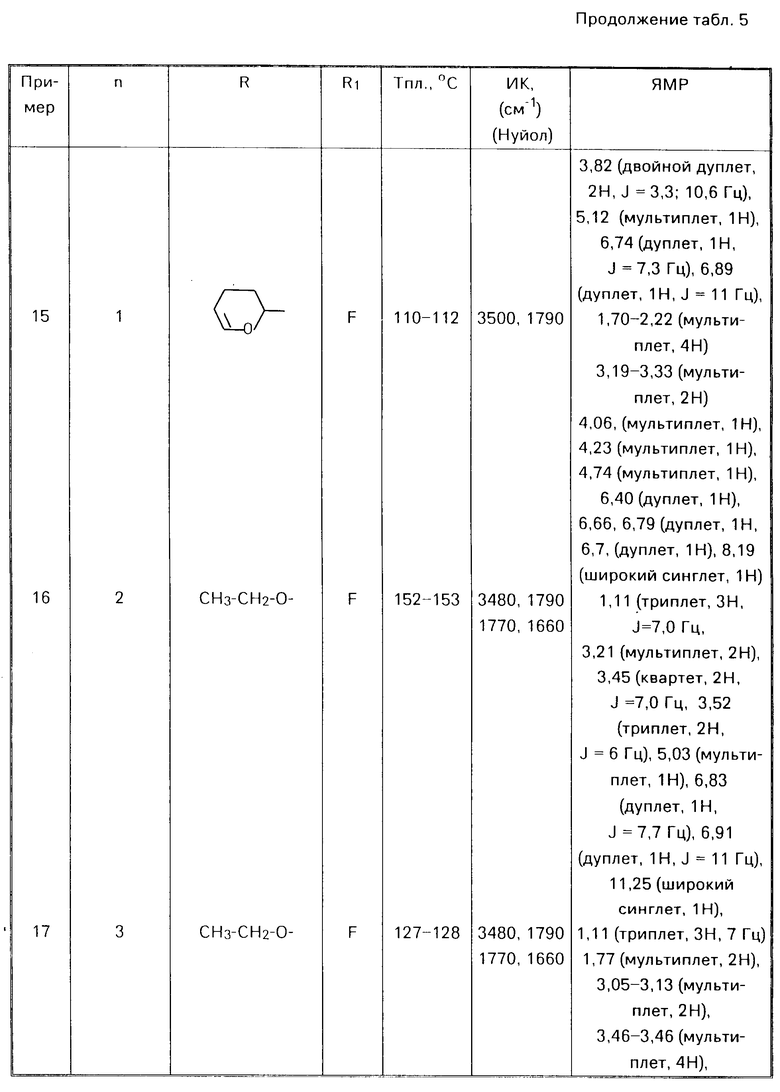
П р и м е р 1. 5-фтор-6-(5,6-дигидро-2Н-пиран-3-ил)метиламино бензоксазолин-2-он.
К раствору 0,97 г (5,7 ммоля) 6-амино-5-фтор-бензоксазолин-2-она и 0,71 г (6,3 ммоля) 3-формил-5,6-дигидро-2Н-пирана в 40 мл этанола добавляли 1 г молекулярных сит 4Ао. Смесь кипятили с обратным холодильником в течение 3 ч. После охлаждения смесь фильтровали, и фильтрат концентрировали о получения твердого продукта, который промывали этанолом. Продукт растворяли в метаноле (200 мл) и затем по частям добавляли при комнатной температуре борогидрид натрия. Реакционную смесь перемешивали в течение нескольких часов. Реакционную смесь концентрировали и добавляли воду. Органический материал экстрагировали этилацетатом. Объединенные экстракты промывали рассолом, сушили над сульфатом магния и концентрировали. Полученный остаток перекристаллизовывали из метанола, получая 420 мг целевого продукта (28%) с точкой кипения 175-176оС.
ИК спектр (КВr): 1790, 1529, 1100, 960 см-1
ЯМР спектр 2,03 (ш, 2Н), 3,61 (м, 4Н), 3,97 (ш, 2Н)
5,53 (ш, 1Н), 5,74 (ш, 1Н), 6,68 (д, 1Н),
6,90 (д, 1Н), 11,23 (с, 1Н).
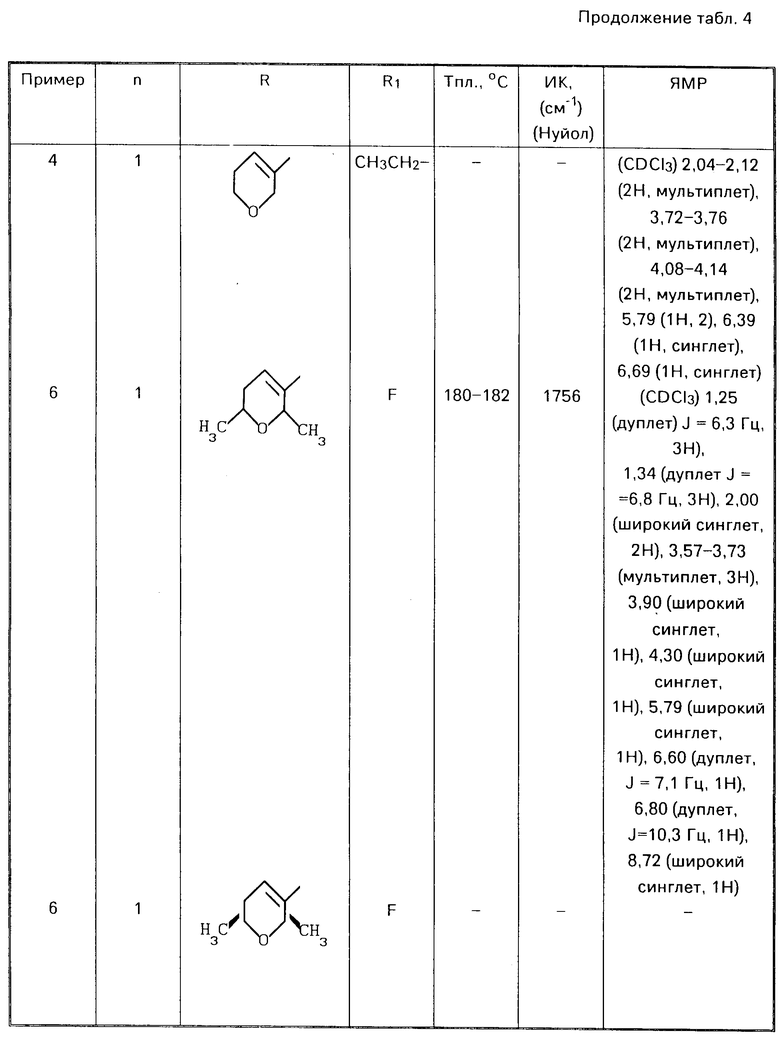
П р и м е р ы 2-6. Аналогичным образом, применяя соответствующие альдегиды (III) в процедуре, описанной в примере 1, получали соответствующие соединения формулы (I), представленные в табл. 4.
П р и м е р 7. 5-фтор-6-(тетрагидро-4Н-пиран-3-ил)пропиламино бензоксазолин-2-он.
При комнатной температуре к раствору 2,1 г (12,5 ммоля) 6-амино-5-фтор-бензоксазолин-2-она в 80 мл метанола добавляли 1,95 г (13,7 ммоля) 3-(тетрагидро-2Н-пиран-3-ил) пропионового альдегида и 1 мл уксусной кислоты, и смесь перемешивали в течение 1 ч. Добавляли 0,867 г (13,7 ммоля) цианоборогидрида натрия и продолжали перемешивание реакционной смеси в течение 17 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме, и остаток обрабатывали водным раствором хлористого аммония. Органическое вещество экстрагировали этилацетатом-тетрагидрофураном. Экстракты промывали рассолом, сушили над сульфатом магния и концентрировали, получая сырой продукт. Последний перекристаллизовывали из метанола и получали 1,40 г целевого соединения с точкой плавления 144-145оС.
ИК спектр (КВr) 1770, 1090 см-1
ЯМР спектр: 1.00-1,30 (м, 3Н), 1,38-1,63 (м, 5Н),
1,75-1,85 (м, 1Н), 2,91-3,03 (м, 3Н), 3,20-3,28.
(м, 1Н), 3,73 (ш.д. 2Н), 5,13 (ш, 1Н).
6,73 (д, 1Н), 6,89 (д, 1Н), 11,21 (ш, 1Н).
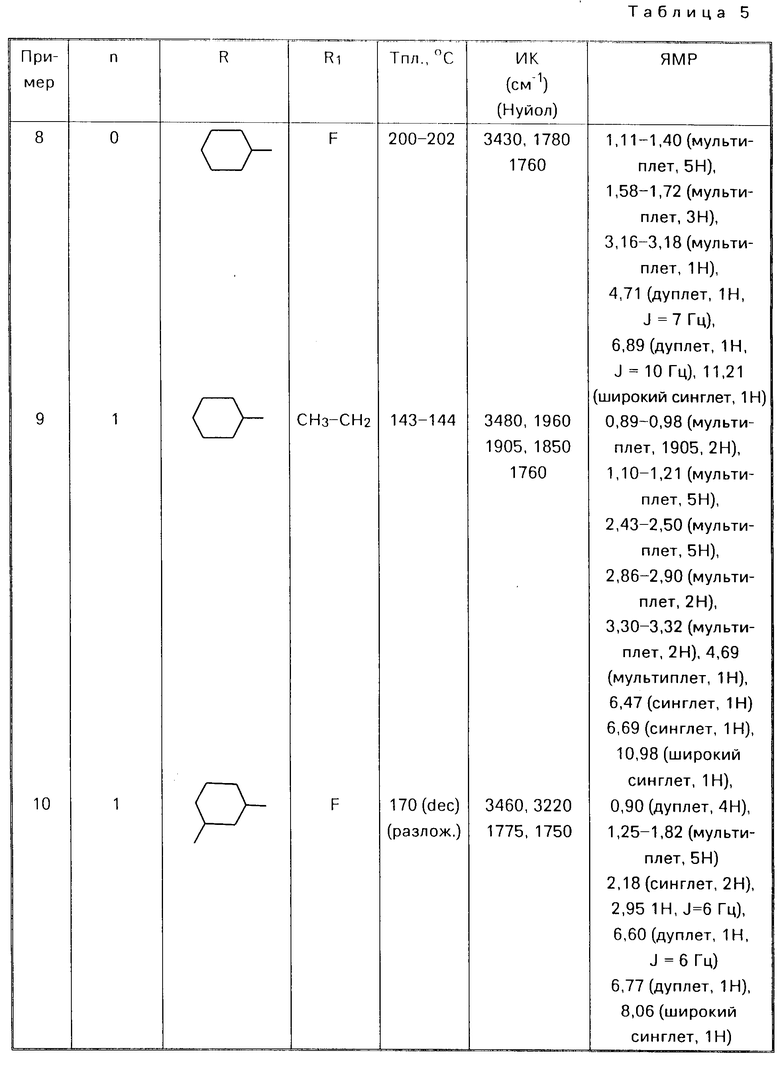
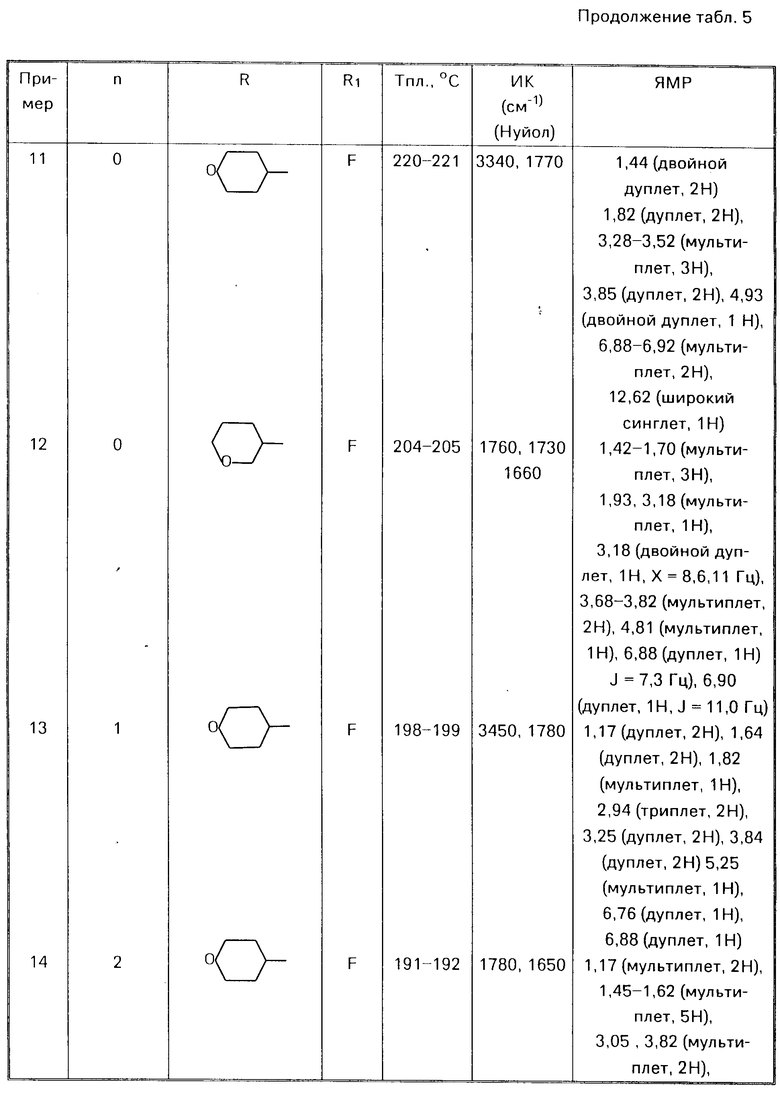
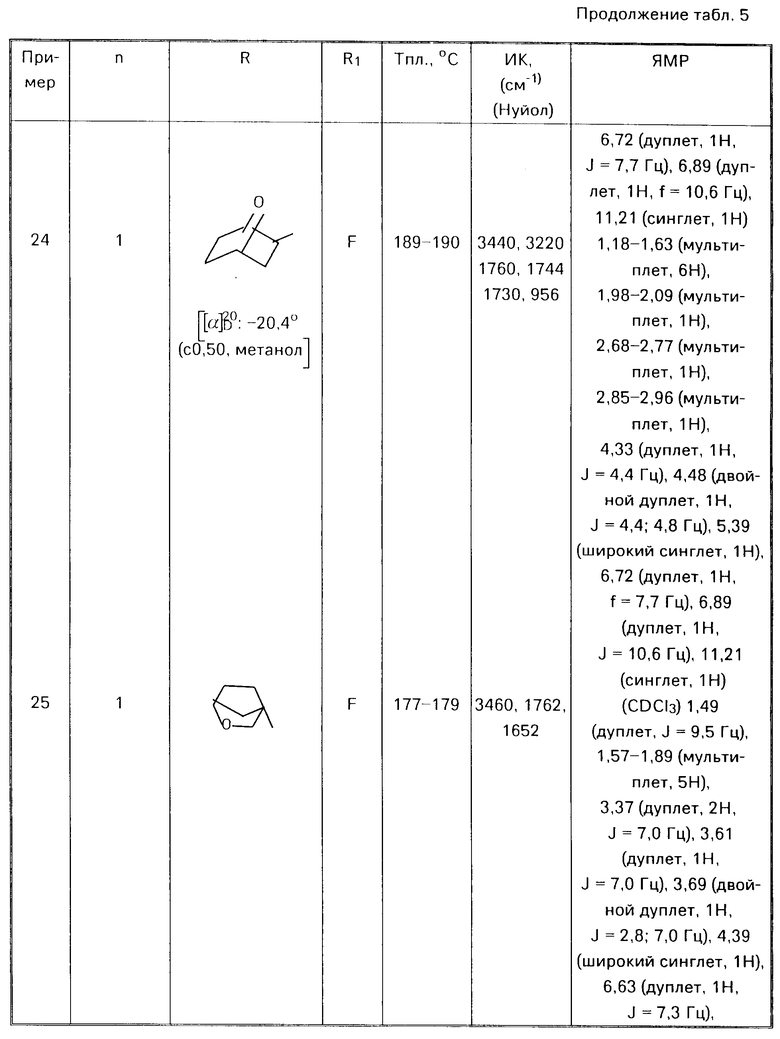
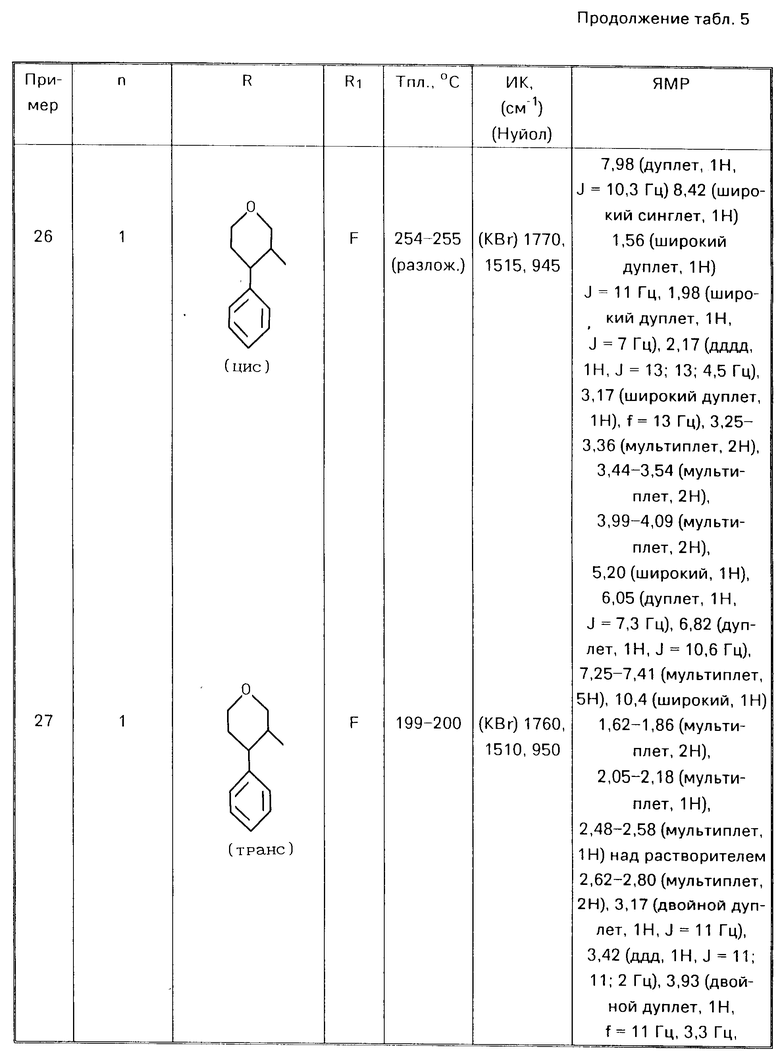
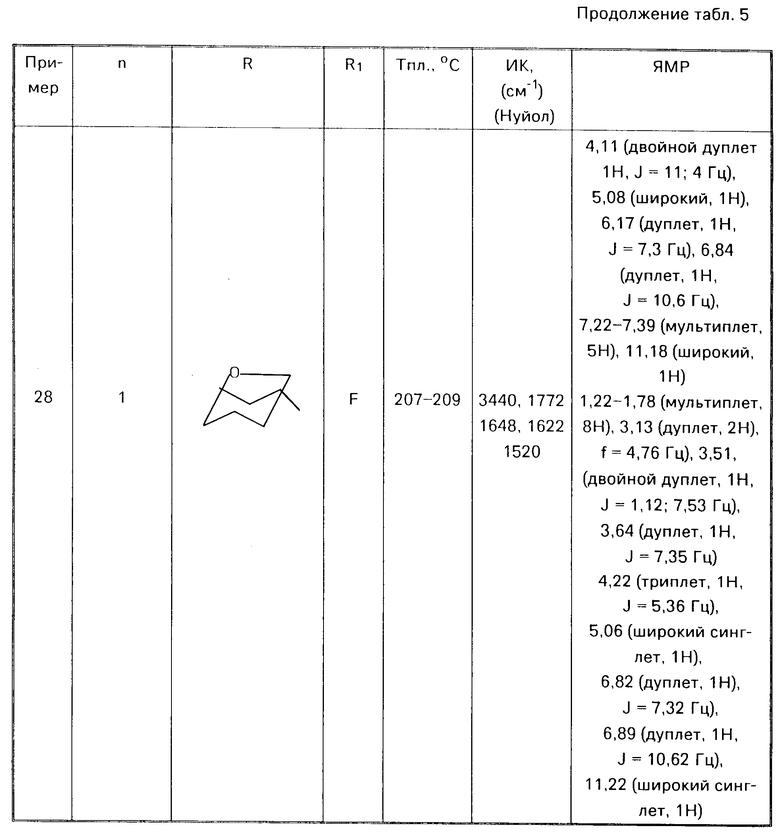
П р и м е р ы 8-28.
Аналогичным способом используя соответствующие альдегиды (III) или кетон (V) в соответствии с процедурой примера 7 получали соответствующие соединения формулы (I)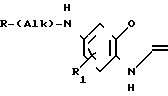 O где Alk есть (СН2)n, представлены в табл. 5
O где Alk есть (СН2)n, представлены в табл. 5
П р и м е р 29. 5-фтор-6-(тетрагидро-4Н-пиран-3-ил)метиламино-2-бензоксазолон.
К раствору 1,0 г (3,88 ммоля) 6-(5,6-дигидро-2Н-пиран-3-ил)метиламино-5-фтор-2- бензоксазолона в 100 мл метанола добавляли 50 мг оксида платины, смесь гидрировали при давлении 1 атм и при комнатной температуре в течение 1 ч. Затем смесь фильтровали, фильтрат концентрировали в вакууме и полученный твердый остаток промывали этанолом. Перекристаллизацией из этанола получали 0,48 г целевого продукта (47%) с точкой плавления 177-178оС.
ИК-спектр (КВr): 1760, 1510, 1090, 950 см-1
ЯМР-спектр: 1,15-1,30 (М, 1Н), 13,35-1,65 (м, 2Н),
1,75-1,90 (м, 2Н), 2,90-2,95 (м, 2Н),
3,05-3,15 (м, 1Н), 370-3,85 (м, 2Н),
5,25 (ш, 1Н), 6,75 (д, 1Н), 6,89 (д, 1Н),
11,30 (ш, 1Н).
П р и м е р 30. 5-фтор-6-(тетрагидропиран-2-ил)-метиламино-бензоксазолин-2-он.
Способом, аналогичным описанному в примере 29, из 5-фтор-6-(3,4-дигидро-2н-пиран-2-ил)метиламино) бензоксазолин-2-она получали целевое соединение с точкой плавления 185-186оС.
ИК-спектр (СН2Сl2): 3500, 1790, 1780 см-1
ЯМР-спектр (CDCl3): 1,36-1,67 (м), 1,89 (м, 1Н), 3,04 (дд, 1Н, J 8, 12 Гц), 3,16, (дд, 1Н, J 3,5, 12 Гц) 3,42-3,61 (м, 2Н), 4,03 (м, 1Н), 4,27 (м, 1Н), 6,62 (д, 1Н, J 7,1 Гц), 6,78 (д, 1Н, J 10,3 Гц), 8,52 (ш.с. 1Н).
П р и м е р 31. 5-фтор-6-(1,2,3,4-тетрагидро-2-нафтил)метиламино) бензоксазолин-2-он
Следуя процедуре примера 29 и используя в качестве исходного реагента 5-фтор-6-(3,4-дигидро-2-нафтил)метиламино)бензо-ксазолин-2-он, а вместо оксида платины 5%-ный палладий на угле, получали целевой продукт с т.пл. 177-178оС.
ИК-спектр (КВr): 1770, 1660, 1520 см-1.
ЯМР-спектр: 1,30-1,50 (м, 1Н), 1,90-2,10 (м. 2Н), 2,49 (дд, 1Н).
2,65-2,95 (м, 3Н), 3,06 (дд, 2Н), 5,36 (м, 2Н), 6,79 (д. 1Н), 6,93 (д, 1Н), 11,21 (с, 1Н).
П р и м е р 32. 5-фтор-(6)-(хроман-3-ил)метиламино)бензоксазолин-2-он.
Следуя процедуре примера 29, используя в качестве исходного реагента 5-фтор-6-(4-хлор-2Н-хромен-3-ил-метиламино)бензо- ксазолин 2-он и гидрируя в присутствии триэтиламина, получали целевое соединение с точкой плавления 223-234оС.
ИК-спектр (КВr): 1750, 1510, 1490 см-1.
ЯМР-спектр: 2,25-2,38 (м, 1Н), 2,53-2,60 (м, 1Н), 2,87 (д, 1Н),
3,06 (д, 1Н), 3,09 (д, 1Н), 3,87 (дд, 1Н), 4,22 4,27 (м, 1Н), 6,71-6,75 (м, 1Н), 6,78-6,84 (м, 2Н)
6,91 (д, 1Н), 7,01-7,08 (д, 2Н), 11,20 (ш, 1Н).
П р и м е р 33. 5-метокси-6-(тетрагидропиран-3-ил)метила-мино-бензоксазолин-2- он.
Следуя процедуре примера 29 из 5-метокси-6-5,5-дигидро-Н)-2Н-пиран-3-ил)ме-тиламино)бензоксазолин-2-она получали целевое соединение с точкой плавления 155оС (с разложением).
ИК-спектр (Нуйол): 3200, 1780, 1680, 1640 см-1
ЯМР-спектр: 1,20-1,25 (м, 1Н), 1,42-1,60 (м, 2Н), 1,72-1,91 (М, 2Н), 2,87-2,94 (м, 2Н), 3,08-3,15 (м, 1Н), 3,69-3,81 (м, 6Н), 6,58 (с, 1Н), 6,62 (с, 1Н), 11,09 (ш.с. 1Н).
П р и м е р 34. 5-этил-6-[(тетрагидропиран-3-ил)метиламино]-бензоксазолин-2-он.
Следуя процедуре примера 29, из 5-этил-6-[(5,6-дигидро-2Н-пиран-3-ил)метил- амино](бензоксазолин-2-она получали целевой продукт с точкой плавления 148-150оС.
ИК-спектр (Нуйол): 3200, 2750, 1775, 1630 см-1.
ЯМР-спектр: 1,25-1,30 (м, 1Н), 1,86-2,00 (м, 4Н), 2,04 (с, 3Н), 2,91-2,95 (м, 2Н), 3,13-3,17 (м, 2Н), 3,72 (ш. с, 1Н), 3,84 (ш.с, 1Н), 5,01-5,03 (м, 1Н), 6,40 (с, 1Н), 6,43 (с, 1Н).
П р и м е р 35. 5-фтор-6-(2,6-диметилтетрагидропиран-3-ил)метил-амино(бензокса- золин-2-он.
Следуя процедуре примера 29, из 5-фтор-6-(2,6-диметилдигидро-2Н-пиран-3-ил) метиламино)бензоксазолин-2-она и получали целевой продукт с точкой появления 123-139оС (метанол)
ИК-спектр (Нуйол): 1757, 1788 см-1.
ЯМР-спектр (СDCl3): 1,22 (д, 3Н, J 6,1 Гц, 1,27 (д, 3Н, J 6,6 Гц), 1,44 (м, 2Н), 1,77 (м, 2Н), 1,90-2,06 (м, 1Н), 3,17-3,39 (м, 2Н), 3,55 (м, 1Н), 3,77 (м, 1Н), 4,03 (ш.с. 1Н), 6,61 (д, 1Н, 7,1 Гц), 6,79 (д, 1Н, J 10,3 Гц), 8,70 ш.с. 1Н).
П р и м е р 36. 5-фтор-6-(тетрагидротиопиран-3-ил)метиламино-бензоксазолин-2-он.
Следуя процедуре примера 29, из 5-фтор-6-(дигидро-2Н-тиопиран-3-ил)метил-амино(бензоксазолин-2-она получали целевое соединение.
П р и м е р 37. 5-фтор-6-(тетрагидротиопиран-1-оксидо-3-ил)метил-амино)бензо- ксазолин-2-он.
К раствору 1,2 г (4,25 ммоля) 5-фтор-6-(тетрагидро-тиопиран-3-ил)метиламино)бен- зоксазолин-2-она в 200 мл этанола и 50 мл тетрагидрофурана добавляли 5,7 г метапериодата натрия на окиси алюминия (К.Т.Лиу и др.д О. С7 43: 2717 (1989). Смесь перемешивали 20 ч при комнатной температуре. Окись алюминия отфильтровывали и растворитель удаляли в вакууме. Сырой продукт очищали из хроматографической колонке с силикагелем, осуществляя элюирование этилацетатом (тетрагидрофураном) метанолом (80:20:5), и перекристаллизацию из метанола) серного эфира. В результате получали целевой продукт в количестве 0,28 г (22% выход).
ИК-спектр (КВr): 1765, 1520, 940 см-1.
ЯМР-спектр: 1,1-1,25 (m, 1Н), 1,4-1,55 (m, 1Н, одного изомера, Е или Z в положении 1,3 тетрагидротиопиран-1-оксидного ядра), 1,65-2,15 (m, 3Н), 2,3-2,6 (m, 2Н+1Н одного изомера) 2,75-2,85) (m, 1Н одного изомера), 2,9-3,15 (м, 2Н одного изомера), 5,4-5,55 (m, 1Н), 6,77-6,85 (m, 1Н), 6,91 (d, 1Н, J 10,6 Гц), 11,2 (вг, 1Н).
П р и м е р 38. 5-фтор-6-[3-(бутилтионил)пропиламино]бензоксазолин-2-он.
Следуя методике примера 37, из 5-фтор-6-[3-бутилтио)пропиламино]бензоксазолин-2-она получали целевой продукт с точкой плавления 116-117оС (метанол).
ИК-спектр (СН2Сl2):3480, 1780, 1660 см-1.
ЯМР-спектр: 0,90 (т. 3Н, J 7,3 Гц), 1,40 (м, 2Н), 1,60 (м, 2Н), 1,91 (м, 2Н), 2,57-2,88 (м, 4Н), 3,18 (м, 2Н), 5,38 (м, 1Н), 6,79 (д, 1Н, J 7,3 Гц), 6,90 (д, 1Н, J 11,0 Гц), 11,23 (ш.с. 1Н).
Пример подготовительный А. 6-амино-5-фторбензоксазолин-2-он.
А.1-4-фтор-2-нитрофенол.
К механически перемешиваемому раствору 400 мл концентрированной азотной кислоты при 0оС по каплям добавляли раствор 204 г (1,8 моля) 4-фторфенола в 200 мл уксусной кислоты в течение двух часов. Перемешивание продолжали в течение последующих двух часов при 5оС. Реакционную смесь вливали в лед, и полученное твердое желтое вещество собирали и промывали водой. Вещество перекристаллизовывали из водного метанола (5 ч. метанола, 1 ч воды). В результате получали 198 г целевого продукта. ЯМР спектроскопия показала наличие поглощения при 7,17 (двойной дуплет, 1Н, J 9,5 Гц), 7,44-7,52 (мультиплет, 1Н) и 7,80 (двойной дуплет, 1Н, J 8,3 Гц).
А.2. 2-амино-4-фторфенол.
К раствору 48,3 г (0,30 моля) 4-фтор-2-нитрофенола в 300 мл этанола добавляли 0,24 г оксида платины в атмосфере азота. Смесь гидрировали в трясучке Нарра в течение 8 ч при давлении 3,15 атм (45 фунтов/кв.дюйм). Катализатор отфильтровывали, и фильтрат концентрировали, получая 40,5 г целевого продукта в виде коричневого порошка. ЯМР-спектроскопия показала наличие поглощения при 4,79 (широкий синглет, 2Н), 6,11 (мульфиплет, 1Н), 6,36 (двойной дуплет, 1Н, J 11,3 Гц), 6,53 (двойной дуплет, 1Н, J 5,9 Гц) и 8,89 (синглет, 1Н).
А.3. 5-фторбензоксазолин-2-он.
К раствору 40,5 г (0,32 моля) 2-амино-4-фторфенола в 400 мл тетрагидрофурана при 0оС по каплям добавляли 44,8 мл (0,32 моля) трихлорметилхлорформиата. Реакционной смеси давали возможность самопроизвольно принять комнатную температуру. Перемешивание продолжали в течение двух часов. Затем реакционную смесь вливали в лед и органическое вещество экстрагировали этилацетатом (трижды по 500 мл). Объединенные экстракты промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом магния и концентрировали, получая 44,3 г целевого продукта в виде коричневой твердой массы.
ЯМР-спектроскопия показала наличие поглощения при 6,86-6,90 (мультиплет, 1Н), 7,01 (двойной дуплет, 1Н, J 8,3 Гц), 7,30 (двойной дуплет, 1Н, J 9,5 Гц) и 11,82 (широкий синглет, 1Н).
А.4. 5-фтор-6-нитробензоксазолин-2-он.
При комнатной температуре к перемешиваемому раствору 300 мл концентрированной азотной кислоты добавляли порцией 73,2 г (0,48 моля) 5-фторбензоксазолин-2-она. Реакционную массу нагревали до 50оС и перемешивали в течение 4 ч. После охлаждения реакционную смесь выливали в лед. Полученный осадок промывали водой и сушили, получая 72,8 г целевого продукта в виде коричневого порошка.
Точка плавления продукта 207-209оС.
ИК-спектр (Нуйол): 3300, 1810, 1780, 1630 см-1.
ЯМР-спектр, 7,35 (дуплет, 1Н, J 11,0 Гц), 8,16 (дуплет, 1Н, J 6,6 Гц), 12,6 (широкий синглет, 1Н).
А.5.6-амино-5-фторбензосалин-2-ОН.
К раствору 20 г (0,1 моля) 5-фтор-6-нитро-бензоксазолин-2-она в 300 мл тетрагидрофурана в атмосфере азота добавляли 2 г палладия на угле (5%). Смесь гидрировали при непрерывном встряхивании в течение 10 ч при 3,15 атм (45 фунтов/кв. дюйм). Получаемый осадок растворяли добавлением тетрагидрофурана. Катализатор отфильтровывали и фильтрат концентрировали, получая 18,1 г целевого соединения в виде коричневого твердого вещества.
Точка плавления продукта 180-182оС (с разложением).
ИК-спектр (Нуйол): 3400, 3280, 1750, 1630 см-1.
ЯМР-спектр: 4,93 (широкий синглет, 2Н), 6,71 (дуплет, 1Н, J 7,3 Гц), 6,84 (дуплет, 1Н, J 10 Гц), 11,2 (широкий синглет, 1H).
Подготовительный пример. 6-амино-5-этилбензоксазолин-2-он.
5-этил-2-бензоксазолин синтезировали конденсацией 2-амино-4-этилфенола с мочевиной, следуя при этом методике, описанной У.Дж.Клоссом с сотр. S.Am. Chеm. 71, 1265 (1949). Аналогично подготовительному примеру А из 5-этилбензоксазолин-2-она получали 6-амино-5-этилбензоксазол с точкой плавления 146-147оС.
ИК-спектр (Нуйол): 3430, 3340, 3130, 1710, 1640 см-1.
ЯМР-спектр: 1,10 (т, 3Н, J 7,3 Гц), 2,43 (кв. 2Н, J 7,3 Гц).
4,73 (ш.с. 2Н), 6,56 (с. 1Н), 6,64 (с, 1Н), 10,99 (ш.с. 1Н).
Подготовительный пример В. Использовали процедуру подготовительного примера А для синтеза 6-амино-4-метилбензоксазолин-2-она, 6-амино-5-метил- бензоксазолин-2-она, 6-амино-5-трифторметилбензоксазолин-2-она, 6-амино-5-метоксибензоксазолин-2-она, 6-амино-5-метилтиобензоксазолин-2-она, 6-амино- 5-феноксибензоксазолин-2-она, 6-амино- 5-фенилтиобензоксазолин-2-она, 6-амино-7-хлорбензоксазолин-2-она и 6-амино-7- фторбензоксазолин-2-она.
Подготовительный пример Г. 3-(тетрагидропиран-3-ил)-пропионовый альдегид.
Г.1. Этил-3-(5,6-дигидро-2Н-пиран-3-ил)акрилат.
К перемешиваемой суспензии гидрида натрия (60% в минеральном масле, 1,43 г, 35,7 ммоля) и в 50 мл тетрагидрофурана при комнатной температуре по каплям добавляли 8,35 г (37,2 ммоля) триэтилфосфонацетата в атмосфере азота. Реакционную смесь перемешивали в течение 15 мин. К ней добавляли по каплям раствор 3,34 г (29,8 ммоля) 3-формил-5,6-дигидро-2Н-пирана (Японская публикация 59-167584, ВА SF) в 20 мл тетрагидрофурана. Полученную смесь перемешивали 1 ч. Реакцию прекращали добавлением уксусной кислоты. Затем реакционную смесь концентрировали, и добавляли водный раствор бикарбоната натрия. Органическое вещество экстрагировали этилацетатом. Экстракт промывали рассолом, сушили над сульфатом магния и упаривали до получения масла. Сырое масло очищали на хроматографической колонке с силикагелем, элюируя 25%-ным этилацетатгексаном. Получали 3,1 г целевого соединения.
ЯМР-спектр показал наличие поглощения при 1,26-1,38 (м, 3Н), 2,34 (ш. 2Н), 3,8 (тр. 2Н, J 5 Гц), 4,15 4,30 (м, 4Н), 5,63 (д, 1Н, J 17 Гц, 6,28 (ш. 1Н) и 7,21 (д. 1Н, J 17 Гц).
Г. 2. Этил-3-(тетрагидро-2Н-пиран-3-ил)пропионат. В атмосфере водорода при комнатной температуре над катализатором 0,15 г палладия на угле (5%) гидрировали раствор 3,1 г этил-3-(5,6-дигидро-2Н-пиран-3-ил) акрилата в 50 мл метанола. Катализатор отфильтровывали и фильтрат концентрировали, получая сырое масло. Сырой продукт очищали на хроматографической колонце с силикагелем, элюируя этилацетат гексановой смесью (1:1). Получали 3,0 г целевого продукта.
ЯМР-спектроскопия показала наличие поглощения при 1,10-1,20 (м, 1Н), 1,26 (тр. 3Н, J 7 Гц), 1,45-1,63 (м, 5Н), 1,82-1,91 (м, 1Н), 2,27-2,34 (м. 2Н); 3,06 (дд. 1Н, J 9,5, 11 Гц) 3,30-3,40 (м, 1Н), 3,83-3,89 (м, 2Н) и 4,13 (кв. 2Н, J 7 Гц).
Г.3. 3-(тетрагидро-2Н-пиран-3-ил)пропиональдегид.
В атмосфере азота при температуре 78оС к раствору 3,0 г этил-3-(тетрагидро-2H-пиран-3-ил)пропионата по каплям добавляли 16 мл полуторамольного толуольного раствора ДИВАЛ. Перемешивание осуществляли в течение одного часа. Реакцию прекращали добавлением смеси метанол-вода. Полученному раствору позволяли принять комнатную температуру. Удаляли образующийся твердый осадок. Фильтрат сушили над сульфатом магния и концентрировали, получая сырое масло. Сырой продукт очищали перегонкой, и получали 2,0 г целевого продукта.
ЯМР-спектр показал наличие поглощения при 1,09-1,28. (м, 1Н), 1,42-1,65 (м, 5Н), 1,80-1,91 (м, 1Н), 2,42-2,49 (м, 2Н), 3,07 (дд. 1Н, J 9, 11 Гц), 3,31-3,40 (м, 1Н), 3,84-3,89 (м, 2Н) и 9,78 (с, 1Н).
Подготовительный пример Д. 3-метилциклогексанкарбоксальдегид.
Д.1. 3-метилциклогексанкарбоновая кислота.
В атмосфере азота к раствору 13,6 г (0,1 моля) метафенилуксусная кислота в уксусной кислоте добавляли 0,1 г оксида платины. Смесь гидрировали в мешалке Парра при 2,45 атм (35 фунтов/кв.дюйм). После завершения катализатор отфильтровывали, и фильтрат концентрировали досуха, получая 12 г целевого соединения. ЯМР-спектр показал наличие поглощения при 0,84 (дуплет, 3Н), 0,90 (дуплет, 3Н), 0,99-1,13 (мультиплет, 1Н), 2,21-1,46 (мультиплет, 3Н), 1,54-1,65 (мультиплет, 3Н), 1,70-1,98 (мультиплет, 2Н) и 1,23-2,41 (мультиплет, 1Н).
Д. 2. 1-оксиметил-3-метилциклогексан. К боран-сернистому метиловому комплексу (1,7 мл, 0,028 ммоля) в 7 мл тетрагидрофурана при 0оС по каплям добавляли 2 г 3-метилциклогексанкарбоновой кислоты (0,014 моля) в 7 мл тетрагидрофурана. Перемешивали в течение 1 ч. Реакционную смесь разбавляли эфиром и промывали одновременно однонормальным водным раствором едкого натра и затем рассолом. Концентрирование и перегонка позволили получить 1,34 г целевого продукта. ЯМР-спектр показал наличие поглощения при 0,54-0,74 (мультиплет, 1Н) 0,90-0,93 (синглет), 1,17-1,53 (мультиплет, 3Н), 1,65-1,77 (мультиплет, 3Н) и 3,39-3,52 (мультиплет, 2Н).
Д.3. 3-метилциклогексанкарбоксальдегид. В атмосфере азота к раствору 6,8 г (0,053 моля) 1-оксиметилциклогексана в 150 мл дихлорметана добавляли 22,9 г (0,106 моля) рсс. Смесь перемешивали 1 ч при комнатной температуре. Твердый осадок отфильтровывали на Флоризиле и фильтрат концентрировали, получая 8 г целевого соединения.
ЯМР-спектр показал наличие поглощения при 0,90, 0,95 (дуплет, 3Н), J 8 Гц), 0,86-2,32 (мультиплет, 10Н) и 9,68, 9,70 (дуплет, 1Н) J 2 Гц.
Подготовительный пример Е. Энд-7-оксабицикло (2,2,1)гептан-2-карбоксальдегид.
Следуя методике примера Г. З эндо-2-карбометокси-7-оксабицикло (2,2,1)гептан (м. п. Кунстман с сотр. S.Am.Chem. Soc. 84, 4115 (1962) (2,13 г. 12,5 ммоля) восстанавливали до целевого соединения (1,51 г.) ЯМР-спектр выявил наличие поглощения при 1,46-1,95 (мультиплет), 3,07 (мультиплет, 1Н), 4,68 (мультиплет, 1Н), 4,86 (двойной дуплет, 1Н, J 5,6, 5,6 Гц) и 9,73 (дуплет, 1Н, J 1,5 Гц).
Аналогичным образом экзо-2-карбометокси-7-оксабицикло (2,2,1) гептан восстанавливали до соответствующего экзо-7-оксабицикло (2,2,1)-гептан-2-карбоксальдегида.
Подготовительный пример Ж. 5-фтор-6-[(4-хлор-2Н-хромен-3-ил)-метиламино] -бен- зоксазолин-2-он.
Ж.1.4-хлор-3-формил-2Н-хромен.
Следуя методике, описанной Дж. А.Виджилио с сотр. Органический синтез и методы 14,9 (1982), получали целевое соединение.
Ж.2. 5-фтор-6-[(4-хлор-2Н-хромен-3-ил)метиламино]бензо-ксазолин-2-он.
К раствору 2,02 г (12 ммоля) 6-амино-5-фторбензоксазолин-2-она в 100 мл этанола добавляли продукт их Ж.1 (2,53 г, 13 ммолей). Смесь перемешивали 6 ч при комнатной температуре. Затем реакционную смесь концентрировали в вакууме, получая твердый осадок. Твердый продукт промывали этанолом. Продукт растворяли в 150 мл метанола и порциями добавляли борогидрид натрия при комнатной температуре. Перемешивание продолжали в течение нескольких часов. Реакционную смесь концентрировали и добавляли водный хлористый аммоний. Органическое вещество экстрагировали этилацетатом/тетрагидрофураном. Объединенные экстракты промывали рассолом, сушили над сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле, элюируя этилацетатом/гексаном (1:3). Получали сырой продукт, после перекристаллизации которого из этанола получали 0,90 г целевого соединения (22% выход) с точкой плавления 197оС (при разложении).
Подготовительный пример 3. 6-оксабицикло[3.2.1]окт.-1'-илметанол.
3.1. 6-оксабицикло[3.2.1]окт.-1'-илметанол.
Смесь 10,0 г (70 ммолей) 3-циклогексен-1,1-диметанола (Олдрич Кемикл. Компани) и 13,7 г (77 ммолей) NBS 200 мл дихлорметана перемешивали при комнатной температуре 13 ч. Затем реакционную смесь промывали дважды водой (по 100 мл) и рассолом и сушили над сульфатом натрия. Растворитель отгоняли и получали бледно-желтое масло (17,0 г). К смеси этого масла и 20 мл толуола добавляли 0,2 г АZBN и затем 21,5 г (84 ммоля) и н-трибутилоловогидрида при перемешивании. Смесь нагревали до 110оС и перемешивали в течение 1,5 ч. Пропуская продукт (150 г 50% этилацетат/гексан, дважды) через колонку с силикагелем и получали целевое соединение (7,75 г, 77% выход).
ЯМР-спектр (СDCl3) показал поглощение при 1,28-1,52 (м, 3Н), 1,66-1,84 (м, 6Н), 3,57 (дд, 2Н, J 1,84) Гц, 5,50 Гц) 3,65 (дд, J 1,84 Гц, 7,69 Гц) 3,84 (д, 1Н, J 7,69 Гц), 4,40 (тр. 1Н, J 5,31 Гц).
3.2. 6-оксабицикло[3,2,1.окт-1'-илкарбоксальдегид.
В течение 1 ч при комнатной температуре перемешивали смесь 3,55 г (25 ммолей) 6-оксабицикло[3,2,1]окт-1'-илметанола, рсс (8,08 г, 37,5 моля) и 100 мл дихлорметана. Полученную смесь разбавляли 100 мл серного эфира и фильтровали через силикагель. Силикагель семь раз промывали серным эфиром по 100 мл каждый раз. Фильтрат и промывки объединяли, растворитель выпаривали и полупоследовательно промывали 200 мл воды и 200 мл рассола, сушили над безводным сульфатом магния и концентрировали в вакууме, получая 18,0 г бледно-желтого масла.
Разделение диастереизомерных имидов осуществляли на препаративном жидкофазном хроматографе (система 500  , используя два силикагельных Ргер-РАК-500 брикета (57 мм х 30 см, эфир/н-гексан (1:5), скорость потока 250 мл/мин при трехкратном пропуске. Время удерживания менее полярного и более полярного имидов соответственно равны 16 и 22 мин. Менее полярный имид (6,47 г), который содержал неизвестную примесь, очищали перекристаллизацией из эфирагексана и получали 4,47 г (выход 29%) чистого, менее полярного имида, (4S-3[(1S, 2R, 4R)-7-оксабицикло-[2,2,1]-гепт-2-илкарбонил]-4-изопропилок-сазолидин-2-он, (более 99% de). Структура менее полярного имида определена рентгеноструктурным анализом, используя кристалл, получаемый при другой медленной перекристаллизации в эфире-гексане. Более полярный имид (4S)-3-[(1R, 2S, 4S)-7-окса-бицикло [2.2.1] гепт-2-илкарбонил] -4-изопропилоксазолидин-2-он (6,36 г, 42% выхода, 98,5%-ое) использовали без дополнительной очистки.
, используя два силикагельных Ргер-РАК-500 брикета (57 мм х 30 см, эфир/н-гексан (1:5), скорость потока 250 мл/мин при трехкратном пропуске. Время удерживания менее полярного и более полярного имидов соответственно равны 16 и 22 мин. Менее полярный имид (6,47 г), который содержал неизвестную примесь, очищали перекристаллизацией из эфирагексана и получали 4,47 г (выход 29%) чистого, менее полярного имида, (4S-3[(1S, 2R, 4R)-7-оксабицикло-[2,2,1]-гепт-2-илкарбонил]-4-изопропилок-сазолидин-2-он, (более 99% de). Структура менее полярного имида определена рентгеноструктурным анализом, используя кристалл, получаемый при другой медленной перекристаллизации в эфире-гексане. Более полярный имид (4S)-3-[(1R, 2S, 4S)-7-окса-бицикло [2.2.1] гепт-2-илкарбонил] -4-изопропилоксазолидин-2-он (6,36 г, 42% выхода, 98,5%-ое) использовали без дополнительной очистки.
И.2. Метиловый эфир (1S, 2R, 4R)-7-оксабицикло[2.2.1]гептан-2-карбоновой кислоты.
К охлажденному до 0оС раствору 4,35 г (17 ммолей) (4S)-3-[(1S, 2R, 4R)-7-оксабицикло [2.2.1] гепт-2-илкарбонил]-4-изопропилоксазолидин-2-она в 350 мл тетрагидро- фурана медленно по каплям добавляли водный раствор гидроперексида лития (приготовленный из 15 мл 30%-ой перекиси водорода, 1,28 г (30 ммолей) гидроксида лития и 120 мл воды), при перемешивании. После перемешивания в течение 1 ч при 0оС реакцию останавливали добавлением по каплям 300 мл 2 и сульфита натрия. После перемешивания полученного шлама в течение 15 мин при 0оС смесь подщелачивали насыщенным бисульфитом натрия и органический растворитель отгоняли в вакууме. Остающуюся водную смесь промывали 200 мл дихлорметана. После подкисления концентрированной соляной кислоты, хиральную кислоту экстрагировали десятикратно 300 мл дихлометана. Объединенные органические фазы сушили над сульфатом магния и концентрировали в вакууме, получая неочищенную кислоту в виде бледно-желтого масла. Неочищенную кислоту разбавляли 100 мл эфира и обрабатывали избытком диазометана в эфире. 15 мин избыток диазометана удаляли пробулькиванием азота через раствор. Полученный раствор концентрировали при пониженном давлении и очищали хроматографически (100 г силикагеля, эфир/гексан 1:1), получая 2,03 г (76% выход) целевого соединения в виде прозрачного летучего масла. Аналитический образец очищали перегонкой: точка кипения 106-109оС при 0,9 мм рт.ст.
ИК-спектр (Нуйол): 3000, 2970, 2880, 1736, 1064, 1002, 938 см-1.
ЯМР-спектр (СDCl3): 1,42-1,55 (м, 2Н), 1,67-1,80 (м, 3Н), 2,09-2,17 (м, 1Н), 2,61 (дд, 1Н, J 4,9 Гц, 9,1 Гц), 3,70 (с, 3Н), 4,66 (дд, 1Н, J 4,9 Гц, 5,1 Гц), 4,84 (д, 1Н, J 4,9 Гц).
[α]D20 + 31,3о (С 1,00, метанол).
И.З. (1S, 2R, 4R)-7-оксабицикло[2.2.1]гептан-2-карбоксальдегид.
Cледуя процедуре примера Г.3. в целевое соединение переводили метиловый эфир (1S, 2R, 4R)-7-оксабицикло 2.2.1. гептане-2-карбоновой кислоты. ЯМР-спектр (СDCl3) показал поглощение при 1,46-1,95 (мультиплет, 6Н), 3,07 (мультиплет, 1Н), 4,68 (мультиплет, 1Н), 4,86 (двойной дуплет, 1Н, J 5,6 Гц, 5,6 Гц), 9,73 (дуплет, 1Н, J 1,5 Гц).
Подготовительный пример К.
(1R, 2S, 4S)-7-оксабицикло-[2.2.1]гептан-2-карбоксальдегид.
К.1. Метиловый эфир (1R, 2S, 4S)-7-оксабицикло 2.2.1 гептан-2-карбоновой кислоты.
Следуя методике примера И.2 и используя (4S-3[(1R, 2S, 4S)-7-оксабицикло [2.2.1] гепт-2-илкаробонил] -4-изопропилок- сазолидин-2-он, получали целевое соединение с выходом 96% Точка кипения 94-98оС при 0,5 мм рт.ст.
ИК-спектр (Нуйол): 300, 2970, 2880, 1736, 1064, 1002, 938 см-1.
ЯМР-спектр (CDCl3): 1,42-1,55 (м, 2Н), 1,67-1,80 (м, 3Н), 2,09-2,17 (м, 1Н), 2,61 (дд, 1Н), J 4,9 Гц, 9,1 Гц), 3,70 (с, СН) 4,66 (дд, 1Н, J 4,9 Гц, 5,1 Гц) 4,84 (д, 1Н, J 4,9 Гц).
[ α]D20: 29,8о (с 1,00, метанол).
К 2. (1R, 2S, 4S)-7-оксабицикло[2.2.1]гептан-2-карбоксальдегид.
Следуя процедуре примера Г.3 метиловый эфир (1R, 2S, 4S)-7-оксабицикло[2.2.1] гептан-2-карбоновой кислоты превращали в целевое соединение. ЯМР-спектр (CDCl3) показал наличие поглощения при 1,46-1,95 (мультиплет, 6Н), 3,07 (мультиплет, 1Н), 4,68 (мультиплет, 1Н), 4,86 (двойной дуплет, 1Н, J 5,6 Гц, 5,6 Гц), 9,73 (дуплет, 1Н, J 1,5 Гц).
Подготовительный пример Л. 2-оксабицикло[2.2.1]гепт-4-илметанол.
При 0оС и перемешивании к смеси 3,74 г (29 ммолей) 3-циклопентен-1,1-диметанола, приготовлено в соответствии с Дж.П.Депре с сотр. S.Org.Chem 49:928 (1984) и Х.Паулзен с сотр. Chem. Ber. 144:346 (1981), 100 мл дихлорметана и 100 мл тетрагидрофурана добавляли NBC (5,71 г, 32 ммоля). После завершения добавления NBC ледяную баню убирали и реакционную смесь перемешивали при комнатной температуре. Спустя 2,5 ч добавляли другую порцию NBC (5,71 г, 32 ммоля) и смесь дополнительно перемешивали 1 ч при комнатной температуре. Реакционную смесь разделяли между 200 мл СНСl3 и 200 мл воды. Водную фазу экстрагировали 100 мл хлороформа. Объединенные органические фазы промывали 0,5 н сульфитом натрия и затем рассолом, сушили над сульфатом магния и упаривали. Остаточное масло пропускали через колонку с силикагелем 150 г) и элюировали 33% этилацетатом/гексаном до примерно 50% этилацетата/гексана. Объединяли фракции, содержащие в качестве основного компонента целевой продукт. Испарением растворителя получали 3,47 г бледно-желтого масла. Смесь масла, 5,44 г (18,7 ммоля) три-н-бутилоловогидрида, 0,05 г AZBN и 4 мл толуола кипятили с обратным холодильником в течение 80 мин. Очисткой на силикагеле (150 г, смесью этилацетата гексана от 50% до 67%) получали целевое соединение в количестве 1,10 г.
ЯМР-спектр (CDCl3) показал наличие поглощения при 1,41 (дуплет, 1Н, J 9,5 Гц), 1,54-1,79 (мультиплит, 5Н), 3,67 (двойной дуплет, 1Н, J 2,8 Гц, 6,8 Гц), 3,83 (синглет, 2Н), 4,36 (синглет, 1Н).
Использование: в медицине, в частности в средствах подавления липоксиназы и/или циклоксигеназы и при лечении аллергических и воспалительных заболеваний. Сущность изобретения: продукт - производные бензоксазолона (BL) ф-лы 1, R-Alk-NH-[R1]Bl, где R, R1, Alk - соответсвующие значения. Реагент 1: незамещенный амин ф-лы 2: NH-[R1] Bl. Реагент 2: альдегид ф-лы 3: R-(Alk)-C(O)H, в среде органического соединения. Реагент 3: соединение ф-лы 4: Alk-CHN -[R1] BL с последующим выделением соединения ф-лы 1 или соединение ф-лы 2 подвергают взаимодействию с соединением ф-лы 5, где R2,R3, X- соответствующие значения. Реагент 4: соединение ф-лы 6 подвергают восстановлению и в случае необходимости соединение ф-лы 1, где X - атом серы, окислением переводят в соединение ф-лы 1, где R - группа ф-лы 7, в которой пунктирная линия обозначает дополнительную связь, восстанавливают и переводят в соединение ф-лы 1, где в группе ф-лы 7 отсутствует двойная связь. Структура соединений ф-л 5, 6, 7 (см. рис.). 1 з. п. ф-лы.
где Alk Cn неразветвленный двухвалентный алкильный радикал, где n 0 4;
R1 галоген;
R выбирают из группы, включающей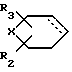
где R2 и R3 каждый независимо друг от друга водород или С1-С4-алкил;
Х метилен, кислород, сера или SO,
пунктирная линия между положениями 3 и 4 указывает на наличие необязательной связи,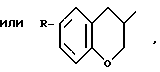
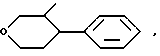
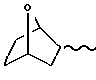


OP линия указывает, что группа, содержащая такую волнистую линию, может быть эндо- или экзо-7-оксабицикло (2,2,1)-гептан-1-ил, или -CH3 - (CH2)m-У-группа, где m 1 3 целевое число, У кислород, сера или SO-группа,
отличающийся тем, что соединение общей формулы II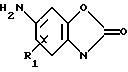
где R1 имеет указанное значение,
подвергают взаимодействию с эквимолярным количеством соединения общей форму III
R (Alk) CHO,
где R и Alk имеют указанные значения,
в среде органического растворителя с последующим восстановлением полученного соединения общей формулы IY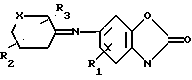
где R, R1 и Alk имеют указанные значения,
и выделяют соединения формулы I или формулы II, подвергают взаимодействию с соединением общей формулы V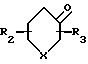
где X, R2 и R3 имеют указанные значения,
с последующим восстановлением полученного соединения общей формулы YI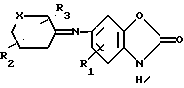
где R1, R2, R3 и Х имеют указанные значения,
и выделяют целевой продукт, или в случае необходимости, соединение формулы I, где Х или У сера, окислением переводят в соединение формулы I, где Х или У -SO-группа, или в случае необходимости соединение формулы I, где R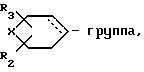
где R2, R3 и Х имеют указанные значения, и пунктирная линия - дополнительная связь, восстанавливают и выделяют соединение формулы I, где R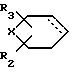
где Х, R2 и R3 имеют указанные значения.
| БАК ДЛЯ ЗАКАЛКИ ПРОТЯЖНЫХ ИЗДЕЛИЙ | 0 |
|
SU249407A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
