Изобретение относится к органической химии, в частности, к способу получения 2,6-диметил-10-метилен-4-С1-С4-алкоксика- рбонил-2,6,11-додекатриена.
Целью изобретения является разработка способа получения 2,6-диметил-10-метилен-4-С1-С4-алкоксикарбонил-2,6,11-додекатрие- на, позволяющего при использовании его в качестве промежуточного соединения при синтезе витамина Е получать последний с более высоким выходом.
Сущность изобретения иллюстрируется следующими примерами.
Исходный 3-хлормирцен получают из мирцена с выходом 84,6% по известной методике.
П р и м е р 1. В колбу на 100 мл вводят в атмосфере аргона 10 мл сухого пентана, 2,07 диизопропиламина (20 ммолей). После охлаждения до 0оС добавляют 12,5 мл раствора н-бутиллития в гексане (1,6 М/л), что соответствует 20 ммолям. Реакционную смесь оставляют на 20 мин при 0оС, после чего ее охлаждают до -78оС. К реакционной смеси медленно добавляют 2,78 г этилового эфира 4-метил-3-пентеновой кислоты (20 ммолей), после чего ее оставляют на 20 мин при -78оС, а затем в течение 1 часа - при температуре около 20оС.
Во вторую колбу на 100 мл вводят в атмосфере аргона 20 мг (С3Н5PdCl)2 (0,1 ммоля), 2,30 мг P(C6H5)3 (0,87 ммоля), 5 мл сухого толуола и 3,5 г 6-хлор-3-метилен-7-метил-1,7-октадиена (20 ммолей).
С помощью иглы для перевода реагентов во вторую колбу вводят карбанион, полученный в первой колбе, эта операция занимает 20 мин, после чего реакционную смесь оставляют на 2 ч при температуре около 20оС. Цвет раствора меняется от светло-желтого до оранжевого. Реакционную смесь выливают в 30 мл 10%-ного раствора соляной кислоты. Водную фазу отделяют декантированием и извлекают эфиром. Органические фазы собирают и сушат над сульфатом магния. После фильтрования и отгонки растворителя получают 5,08 г масла желтого цвета. В результате разгонки при пониженном давлении (температура 111-118оС при 1,4 мм рт.ст.; 0,18 кПа) получают 4,5 г смеси продуктов 1 и 2: 1) (CH3)2C= CH-CH(CO2C2H5)-CH2-C(CH3)= CH-CH2-C(= CH2)-CH= CH2 2) CH2= C(CH3)-CH-/-CH(CO2C2H5)-CH= = C(CH3)2/-CH2-CH2-C(= CH2)-CH= CH2 Выход составляет 81,5%. Анализ методом хроматографии в газовой фазе дает соотношение количеств 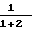 = 98%.
= 98%.
Структура полученных продуктов подтверждается ИК-спектрами, масс-спектрами и спектрами протонного ядерного магнитного резонанса (ПМР).
Соединение 1: масс-спектр: m/e = =276 (М+)
ИК-спектр (пленка), см-1 3090 (=С-Н), 1735 (С=О), 1640-1600 (С=С), 990 (С=СН2) и 900 (СН=СН2; С=СН2)
спектр ПМР (360 Мгц, CDCl3, смещение в частях на миллион)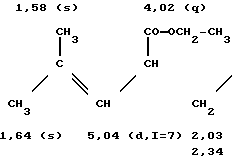
 m - комплексный массив; S = синглет; d = дублет; t = триплет; q = четырехплет; I = константа связи в гц.
m - комплексный массив; S = синглет; d = дублет; t = триплет; q = четырехплет; I = константа связи в гц.
Соединение 2: масс-спектр: m/e = 276 (М+)
спектр ПМР (360 Мгц, CDCl3, смещение в частях на миллион)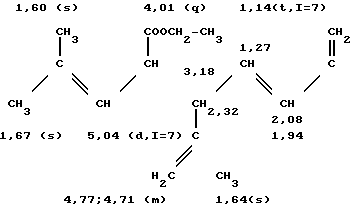
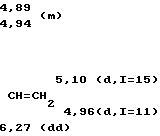
П р и м е р 2 иллюстрирует возможность использования продукта 1 в для синтеза витамина Е.
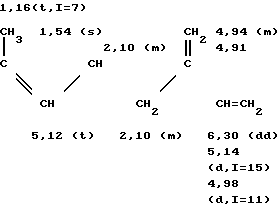
а) В трехгорлую колбу на 100 мл, оборудованную обратным холодильником и термометром, вводят при температуре около 20оС 22,9 г продукта сочетания, полученного согласно примеру 1, что соответствует 87,4 ммолям, 0,0458 г /Ph Cl (1,5-циклооктадиен)/(0,186 мграмматома Ph), 0,0847 г Na2CO3 (0,8 ммоля), 1,110 г трехнатриевой соли три(метасульфофенил)фосфина (TPPTSNa), 20 мл смеси этанола с водой (75-25 по объему) и 22,36 г метилацетилацетата (192 ммоля). Реакционную смесь оставляют при 75оС на 24 ч. После охлаждения раствора к нему добавляют 100 мл эфира. После декантирования органическую фазу промывают тремя порциями по 30 мл воды. Растворитель отгоняют и извлекают оставшуюся массу 100 мл пентана. Затем вновь промывают органическую фазу тремя порциями по 30 мл воды, объединяют органические фазы и сушат их над сульфатом магния. После фильтрования и отгонки растворителя получают 33,03 г желтого масла, которое содержит 30,97 г смеси продуктов формулы: (CH3)2C= CH-CH(CO2CH3)-CH2-C(CH3)= = CH-CH2CH2-C(CH3)= CH-CH2-CH(CO2 CH3)- -CO-CH3 и (CH3)2C=CH-CH(CO2CH3)-CH2-C(CH3)= =CH-CH2-CH2-C(= CH2)-CH2-CH2- -СH(CO2CH3)-CO-CH3 Выход составляет 93,7%.
Масс-спектр: m/e = 378 (М+)
ИК-спектр (пленка), см-1: 1740 [C=0 (сложный эфир)], 1720 [CO (кетон)], 1645 (С=С), 1160 (С-О) и 895 (С=СН2; С = СН)
спектр ПМР (360 Мгц, CDCl3 смещение в частях на миллион).

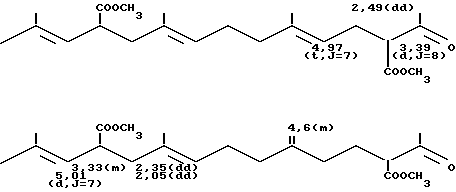 б) В колбу на 100 мл вводят 29,95 г продукта, полученного согласно примеру 2а (79,2 ммоля), 31 г раствора 30,75% гидрата окиси натрия, что соответствует 0,238 моля гидрата окиси натрия, и 60 мл воды. Реакционную смесь оставляют на 1 час при температуре 40оС до появления гомогенного желтого раствора, после чего к ней добавляют 7 мл 95%-ной серной кислоты и 18 мл воды. Реакционную смесь вновь оставляют в течение 1 часа при температуре 25оС. При этом наблюдается обильное выделение углекислого газа. Полученный раствор экстрагируют тремя порциями по 30 мл эфира, объединяют органические фракции и сушат их над сульфатом магния. После фильтрования и отгонки растворителя получают с выходом 95% 23,19 г смеси продуктов формул: (CH3)2C= CH-CH(CO2H)-CH2-C(CH3)= =CH-CH2-CH2-C(CH3)=CH-CH2-CH2- -CO-CH3 (CH3)2C=CH-CH(CO2H)-CH2-C(CH3)= =CH-CH2CH2-C(=CH2)-CH2-CH2-CH2-CO-CH3
б) В колбу на 100 мл вводят 29,95 г продукта, полученного согласно примеру 2а (79,2 ммоля), 31 г раствора 30,75% гидрата окиси натрия, что соответствует 0,238 моля гидрата окиси натрия, и 60 мл воды. Реакционную смесь оставляют на 1 час при температуре 40оС до появления гомогенного желтого раствора, после чего к ней добавляют 7 мл 95%-ной серной кислоты и 18 мл воды. Реакционную смесь вновь оставляют в течение 1 часа при температуре 25оС. При этом наблюдается обильное выделение углекислого газа. Полученный раствор экстрагируют тремя порциями по 30 мл эфира, объединяют органические фракции и сушат их над сульфатом магния. После фильтрования и отгонки растворителя получают с выходом 95% 23,19 г смеси продуктов формул: (CH3)2C= CH-CH(CO2H)-CH2-C(CH3)= =CH-CH2-CH2-C(CH3)=CH-CH2-CH2- -CO-CH3 (CH3)2C=CH-CH(CO2H)-CH2-C(CH3)= =CH-CH2CH2-C(=CH2)-CH2-CH2-CH2-CO-CH3
Масс-спектр: m/e = 306 (М1)
ИК-спектр (пленка), см-1: 1710 (С=О), 1600 (С=С), 895 (С=СН2) и 840 см-1 (С=СН)
спектр ПМР (360 Мгц, CDCl3, смещение в частях на миллион).
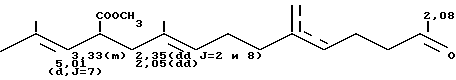
в) В автоклав на 125 мл из нержавеющей стали вводят в атмосфере аргона 5 г продукта, полученного согласно примеру 2б (16,3 ммоля), 30 мл пентана и 0,2 г 10%-ного палладия на активированном угле, после чего вводят водород до создания давления в 100 бар. Реакционную смесь оставляют в течение 12 ч при температуре около 20оС. После фильтрования и отгонки растворителя получают бесцветное масло, содержащее 98% продукта формулы: (CH3)2CH-CH2-CH(CO2H)-CH2-CH(CH3)- -CH2-CH2-CH2-CH(CH3)-CH2-CH2-CH2-
-CO-CH3
Структура полученного продукта подтверждается масс-спектрами, ИК-спектрами и спектрами протонного ядерного магнитного резонанса и углеродного (С13) ядерного магнитного резонанса.
В результате термической обработки полученного продукта при 150оС получают фитон формулы:(CH3)2CH-CH2-CH2-CH2-CH(CH3)-CH2- -CH2-CH2-CH(CH3)-CH2-CH2-CH2-CO- -CH3. Выход 90%.
Продукт гидрогенизации перед термообработкой:
масс-спектр: m/e = 312 (М+)
ИК-спектр (пленка), см-1: 1710 (C=О).
ИК-спектр фитона идентичен ИК-спектру, описанному в литературе.
г) Перевод фитона в витамин Е известным методом.
Таким образом предложенный способ позволяет получить 2,6-диметил-10-метилен-4-С1-С4-алкокси-карбонил-2,6,11-доде- натрием, использование которого в качестве промежуточного продукта, в синтезе витамина Е позволяет получать последний с более высоким выходом (54,1%).
СПОСОБ ПОЛУЧЕНИЯ 2,6-ДИМЕТИЛ-10-МЕТИЛЕН-4- C1-C4-АЛКОКСИКАРБОНИЛ-2,6,11-ДОДЕКАТРИЕНА, отличающийся тем, что C1-C4-алкиловый эфир 4-метил-3-пентеновой кислоты подвергают анионизации бутиллитием в присутствии диизопропиламина с получением аниона формулы
CH3-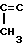 H-CHCOOR ,
H-CHCOOR ,
где R - C1 - C4-алкил,
который подвергают взаимодействию с 7-метил-3-метилен-6-хлор-1,7-октадиеном при 20 - 25oС в присутствии палладиевого катализатора, модифицированного трифенилфосфином в среде апротонного растворителя.
| Болторезная головка | 1935 |
|
SU44771A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
