Изобретение касается новых замещенных бензанилидов и родственных им бензиламидов, гербицидных композиций, содержащих их, и их использования при подавлении нежелательного роста растений.
Согласно данному изобретению предлагаются соединения общей формулы: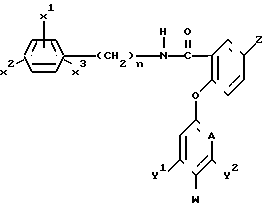
(I)
где Х1, Х2 и Х3 - каждый независимо водород или атом галогена или С1-С4алкильную группу;
n - 0 или 1;
Z - атом водорода или галогена, или амино- С1-С4-алкильную, С1-С4-галоидалкильную, С1-С4-алкилтио- или С1-С4-алкокси-группу или фенокси-группу, необязательно замещенную С1-С4 галоидалкилом;
А - СН или N;
Y1 и Y2 - каждый независимо атом водорода или галогена, или С1-С4-алкильную, С1-С4-галоидалкильную, С1-С4-алкокси- или С1-С4-галоидалкокси группу;
и, или W - атом водорода, или, когда А представляет СН и, по крайней мере, один из Y1 и Y2 является иным, чем водород, или, когда А представляет N, W - атом водорода или галогена;
при условии, что когда А представляет N, тогда каждый из Y1 и Y2должен представлять атом водорода.
Соединения общей формулы I, как было найдено, имеют полезную гербицидную активность. Соответственно, изобретением является гербицидная композиция, которая включает носитель, и в качестве активного ингредиента, соединение формулы I. Также предлагается способ изготовления такой композиции, который включает сочетание соединения формулы I, определенной выше, с по крайней мере одним носителем.
Предлагаемая композиция содержит 0,164%-95% предпочтительно в пределах 0,5-95% по массе активного ингредиента.
Носителем в предлагаемой композиции является любой материал, с которым активный ингредиент формируется с образованием препаративной формы для облегчения применения к обрабатываемому локусу или участку, которым может быть, например, растение, семена или почва, или для облегчения хранения, транспортировки или обращения с ними. Носитель может быть твердым веществом или жидкостью, включая материал, который обычно является газообразным, но который при сжатии образует жидкость, или любой из носителей, обычно используемых при изготовлении готовых препаративных форм гербицидных композиций.
Композиция может также содержать другие активные ингредиенты, например соединения, обладающие инсектицидными или фунгицидными свойствами, или другие гербициды.
Предлагается применение в качестве гербицидов соединения общей формулы I или композиции как способ борьбы с нежелательным ростом растений на локусе с помощью такого соединения или композиции. Локусом, например, может быть почва или растения в посевах сельскохозяйственных культур. Доза активного ингредиента может быть, например до 10 кг/га, предпочтительно до 5 кг/га.
П р и м е р 1. Получение 2-(3-трифторметил-фенокси)-5-фторбензойной кислоты - 2' ,4' -дифторанилида. (Х1 = 2-F; X2 = 4-F; X3 = H; n = 0; Z = F; A = CH; Y1 = CF3; Y2 = H; W = H).
Получение 2-(3-трифторметилфенокси)-5-фторбензонитрила.
К раствору 3-трифторметилфенола (29 г, 0,18 моль), растворенного в диметилформамиде (100 мл) добавляли измельченную в тонкий порошок гидроокись калия (15 г, 0,19 моль). Реакционную смесь нагревали приблизительно до 60оС при перемешивании в течение одного часа, после чего добавляли 2,5-дифторбензонитрил (25 г, 0,19 моль), и температура увеличивалась до 110оС. Спустя приблизительно полчаса, реакционной смеси дали возможность охладиться и избыток диметилформамида удаляли в вакууме перед тем как добавляли 500 мл 50: 50 смеси воды и трихлорметана. Органический слой отделяли, промывали и сушили, а остаток очищали с помощью хроматографии с использованием колонки с окисью кремния, с трихлорметаном в качестве элюента с последующей перегонкой и перекристаллизацией. Получали целевое соединение в виде твердого вещества.
Точка плавления 51оС.
Вычислено,%: С 59,8; Н 2,5; N 5,0.
Найдено,%: С 59,8; Н 2,7; N 5,2.
Получение 2-(3-трифторметилфенокси)-5-бензойной кислоты.
2-(3-Трифторметилфенокси)-5-фторбен- зонитрил, полученный на предыдущей стадии, (12 г, 0,04 моль) добавляли к этиленгликолю (15 мл) и к данному раствору добавляли гидроокись калия (10 г, 0,125 моль), растворенную в воде (10 мл). Реакционную смесь нагревали с обратным холодильником в течение приблизительно 4 ч, охлаждали и добавляли воду (150 мл). Смесь затем экстрагировали диэтиловым эфиром (100 мл) и водный слой подкисляли перед дальнейшей экстракцией трихлорметаном (3 х 150 мл). Органический слой отделяли, промывали, сушили и хроматографировали на колонке с SiO2 с использованием 3: 2 (объем/объем) смеси трихлорметана и этилацетата в качестве элюента, получая целевое соединение в виде бесцветного твердого вещества (10 г-моль, выход 74%).
Точка плавления: 106оС.
Вычислено,%: С 56,0; Н 2,7.
Найдено,%: С 55,9; Н 2,7.
Получение 2-(3-трифторметилфенокси)-5-фторбензойной кислоты - 2' ,4' -дифторанилида.
К раствору 2-(3-трифторметилфенокси)-5-бензойной кислоты, полученной на пре- дыдущей стадии (6 г, 0,02 моль), в толуоле (60 мл) добавляли тионилхлорид (6 мл). Реакционную смесь нагревали с обратным холодильником в течение одного часа и растворитель впоследствии удаляли, по- лучая остаток. Остаток затем растворяли в толуоле (25 мл) и получающийся раствор добавляли к смеси 2,4-дифторанилина (2,6 г, 0,02 моль) и триэтиламина (3 г, 0,03 моль) в толуоле (25 мл) и реакции дали возможность протекать при комнатной температуре (примерно 20оС). После того, как реакция закончилась, реакционную смесь фильтровали, и фильтрат хроматографировали на колонке с SiO2 с использованием трихлорметана в качестве элюента, получая 2',4'-дифторанилид-2-(3-трифторметилфенокси)-5-фторбен- зойной кислоты в виде бесцветного твердого вещества (6,2 г, 0,015 моль, выход 76%).
Точка плавления 98оС.
Вычислено,%: С 58,4; Н 2,7; N 3,4.
Найдено,%: С 58,9; Н 2,8; N 3,7.
П р и м е р 2. Получение 2',4'-дифторанилида 2-(3-трифторметилфенокси)-5-трифторметил-бензойной кислоты (Х1 = 2-F; X2 =4-F; X3 = H; n=0; Z = CF3; A=CH; Y1 = CF3; Y2 = H; W=H)
Получение 3-карбокси-4-фторбензотрифторада.
К раствору 4-фторбензотрифторида (75 мл) в тетрагидрофуране (570 мл) в атмосфере азота и при температуре приблизительно -60оС добавляли бутиллитий (25 N, 200 мл) в течение одного часа. Реакционную смесь перемешивали при температуре от -70 до -60оС в течение 4 ч, а затем выливали на избыток сухого льда. Спустя 15 мин избыток сухого льда выпаривали, и растворитель удаляли, получая остаток. Остаток раствора выливали в воду (500 мл) и гидроокись натрия (1 N, 30 мл) перед промывкой этилацетатом (2 х 300 мл). Водную фазу подкисляли концентрированной соляной кислотой и экстрагировали этилацетатом (2 х 300 мл). В конце объединенные органические экстракты промывались водой (500 мл), сушились, а затем выпаривали, получая сырой остаток, который после перекристаллизации из смеси толуола и гексана дал целевое соединение в виде твердого вещества (77 г, 0,37 моль, выход 81%).
Точка плавления: 100оС.
Вычислено,%: С 46,2; Н 1,9.
Найдено,%: С 46,1; Н 1,7.
Получение 2',4'-дифторанилида 2-фтор-5-трифторметилбензойной кислоты.
3-Карбокси-, 4-фторбензотрифторид, полученный на предыдущей стадии (10 г, 0,05 моль), растворяли в толуоле (100 мл) и по каплям добавляли избыток тионилхлорида (10 мл) при перемешивании в атмосфере азота. Спустя один час, растворитель удаляли, получая остаток, который впоследствии повторно растворяли в толуоле (50 мл) и добавляли к смеси 2,4-дифторанилина (6,2 г, 0,05 моль) и триэтиламина (7 г, 0,07 моль) в толуоле (50 мл). Реакционную смесь фильтровали с получением свободного от осадка фильтрата, из которого растворитель впоследствии удаляли, получая остаток. Очистка остатка на кpемнеземной колонке с использованием трихлорметана в качестве элюента дала целевое соединение в виде бесцветного твердого вещества (10,6 г, 0,03 моль, выход 69%).
Точка плавления 126оС.
Вычислено,%: С 52,7; Н 2,2; N 4,4.
Найдено,%: С 53,3; Н 2,2; N 4,7.
Получение 2', 4'-дифторанилида 2-(3-трифторметилфенокси)-5-трифторметилбен- зойной кислоты.
К суспензии свободного от масла гидрида натрия (0,7 г, 0,03 моль) в сухом тетрагидрофуране (50 мл) добавляли 3-трифторметилфенол (3,6 г, 0,022 моль) небольшими порциями и 2',4'-дифторанилид 2-фтор-5-трифторметилбензойной кислоты (7 г, 0,22 моль) в виде единственной порции. Нагревание реакционной смеси с обратным холодильником в течение одного часа сопровождалось удалением растворителя и распределением остающегося остатка между трихлорметаном и водой (500 мл, 50:50 об/об). Органический слой отделяли, промывали, затем сушили и наконец очищали на кремнеземной колонке с использованием трихлорметана в качестве элюента, получая целевое соединение в виде бесцветного твердого вещества (6,8 г, выход 67%, 0,015 моль).
Точка плавления 105оС.
Вычислено,%: С 54,7; Н 2,4; N 3,0.
Найдено,%: С 55,6; Н 2,6; N 3,0.
П р и м е р 3. Получение 2',4'-дифторанилида 2-(3-трифторметилфенокси)-5-метоксибензойной кислоты
(Х1 = 2-F; X2 = 4-F; X3 = H; n = 0; Z = OCH3; A=CH; Y1 = CF; Y2 = H; W = H)
Получение метил-2-(3-трифторметилфенокси)-5-метоксибензоата.
Натрий (5,4 г, 0,23 моль) в метаноле (90 мл) добавляли к раствору 3-трифторметилфенола (36 г, 0,22 моль) в ксилоле (200 мл) и ксилол затем удаляли. Добавляли свежий ксилол (200 мл) и впоследствии выпаривали в вакууме. Добавляли еще свежий ксилол (200 мл) с последующим добавлением хлористой меди (6 г, 0,06 моль) и пиридина (100 мл). Затем добавляли метиловый эфир 2-бром-5-метоксибензойной кислоты (50 г, 0,20 моль) в ксилоле (50 мл) и получившуюся смесь нагревали с обратным холодильником на протяжении ночи перед тем, как ее вылили в воду (1000 мл), подкисляли разбавленной соляной кислотой и экстрагировали диэтиловым эфиром (2х500 мл). Органический слой отделяли и хроматографировали на колонке с SiO2использованием смеси 50:50 (об/об) трихлорметана и гексана в качестве элюента, получая целевое соединение в виде бесцветного масла (55,2 г, 0,17 моль, выход 83%).
Точка кипения 140оС приблизительно при 1 мм рт.ст.
Вычислено,%: С 58,9; Н 4,0.
Найдено,%: С 58,6; Н 4,1.
Получение 2-(3-трифторметилфенокси)-5-метоксибензойной кислоты.
Метил-2-(3-трифторметилфенокси)-5- метоксибензоат, полученный на предыдущей стадии (10,0 г, 0,03 моль) растворяли в метаноле (20 мл) и добавляли 10% -ный водный раствор гидроокиси калия (50 мм). Реакционную смесь нагревали с обратным холодильником до гомогенного состояния (30 мин), подкисляли 2N соляной кислотой, и добавляли воду (300 мл). Получившуюся смесь экстрагировали трихлорметаном (2х150 мл) и объединенные экстракты хроматографировались на колонке с SiO2 с использованием 80:20 (об/об) смеси трихлорметана и этилацетата в качестве элюента, получая бесцветное твердое вещество. Перекристаллизация твердого вещества из смеси толуола и гексана дала целевое соединение в виде бесцветных кристаллов (7,5 г, 0,024 моль, выход 78%).
Точка плавления 124оС.
Вычислено,%: С 57,7; Н 3,5.
Найдено,%: С 57,6; Н 3,9.
Получение 2',4'-дифторанилида-2-(3-трифторметилфенокси)-5-метоксибензой- ной кислоты.
К раствору 2-(3-трифторметилфенокси)-5-метоксибензойной кислоты, полученному на предыдущей стадии (6 г, 0,02 моль), в толуоле (60 мл), добавляли тионилхлорид (6 мл). Реакционную смесь нагревали в течение одного часа и растворитель впоследствии удалили, получая остаток. Данный остаток затем повторно растворяли в толуоле (25 мл) и получившийся раствор добавляли к смеси 2,4-дифторанилина (2,6 г, 0,02 моль) и триэтиламина (3 г, 0,03 моль) и реакции давали возможность протекать при комнатной температуре (приблизительно 20оС). После того, как реакция завершилась, реакционную смесь фильтровали и фильтрат хроматографировали на колонке с SiO2 с использованием трихлорметана в качестве элюента, получив бесцветное твердое вещество. Перекристаллизация твердого вещества из смеси гексана и этилацетата дала целевое соединение в виде бесцветных кристаллов (6,5 г, 0,015 моль, выход 80%).
Точка плавления 112оС.
Вычислено,%: С 59,6; Н 3,3; N 3,3.
Найдено,%: С 59,9; Н 3,5; N 3,7.
П р и м е р 4. Получение 2',4'-дифторанилида 2-(5-хлор-2-пиридилокси)бензойной кислоты.
(X1 = 2-F; X2 = 4-F; X3 = H; n = 0; Z = H; A = N; Y1 = H; Y2 = H; W = Cl)
Получение этил-2-(5-хлор-2-пиридилокси)бензоата
Этилсалицилат (30 г, 0,18 моль) добавляли по каплям при перемешивании к суспензии свободного от масла гидрида натрия (5 г, 0,21 моль) в сухом диметилформамиде (100 мл) в атмосфере азота. Спустя один час, добавляли 2,5-дихлорпиридин (26 г, 0,17 моль) и реакционную смесь нагревали с обратным холодильником в течение 48 ч. Диметилформамид затем удаляли в вакууме и к получающемуся остатку добавляли 11 мл 50:50 (об/об) смеси воды и трихлорметана. Органический слой отделяли, промывали, сушили и, наконец, хроматографировали на колонке с SiO2 с использованием смеси трихлорметана и гексана (3:1, об/об) в качестве элюента, получая целевое соединение в виде бесцветного масла (32,1 г, 0,115 моль, выход 64%).
Точка кипения 150оС при приблизительно 1 мм рт.ст.
Вычислено,%: С 60,5; Н 4,3; N 5,1.
Найдено,%: С 60,5; Н 4,4; N 5,1.
Получение 2-(5-хлор-2-пиридилокси)бензойной кислоты.
К раствору этил-2-(5-хлор-2-пиридилокси)бензоата, полученного на предыдущей стадии (30 г, 0,11 моль), растворенного в этаноле (50 мл), добавляли 10% -ный водный раствор гидроокиси калия (120 мл). Реакционную смесь нагревали с обратным холодильником при перемешивании в течение 30 мин до тех пор, пока смесь не становилась гомогенной. Реакционную смесь затем подкисляли (до рН 2-3) водной соляной кислотой и экстрагировали трихлорметаном (500 мл). Оpганический слой отделяли, сушили и впоследствии хроматографировали на колонке с SiO2 с использованием 50:50 (об/об) смеси трихлорметана и этилацетата в качестве элюента, получая целевое соединение в виде бесцветного твердого вещества (19,7 г, 0,08 моль, выход 73%).
Точка плавления 159оС.
Вычислено,%: С 57,7; Н 3,2; N 5,6.
Найдено,%: С 57,7; Н 3,2; N 5,7.
Получение 2', 4'-дифторанилида 2-(5-хлор-2-пиридилокси)бензойной кислоты.
2-(5-хлор-2-пиридилокси)бензойную кислоту, полученную на предыдущей стадии (1,5 г, 0,006 моль) растворяли в сухом тетрагидрофуране (20 мл), охлаждали до -15оС, а затем обрабатывали последовательно N-метилморфолином (1 мл) и изобутилхлорформиатом (1 мл). Получающуюся смесь перемешивали приблизительно при -15оС в течение одной минуты перед тем, как добавлялся 2,4-дифторанилин (0,8 г, 0,006 моль). Смесь перемешивали в течение дополнительных 30 мин при той же температуре, после чего добавляли 10%-ный (об/об) водный раствор лимонной кислоты (50 мл). После экстрагирования этилацетатом (3 х 50 мл) органические экстракты объединяли, сушили, а затем хроматографировали с SiO2, получая целевое соединение в виде бесцветного твердого вещества (1,2 г, 0,0033 моль, выход 54%).
Точка плавления 138оС.
Вычислено,%: С 59,9; Н 3,1; N 7,8.
Найдено,%: С 60,2; Н 3,1; N 7,5.
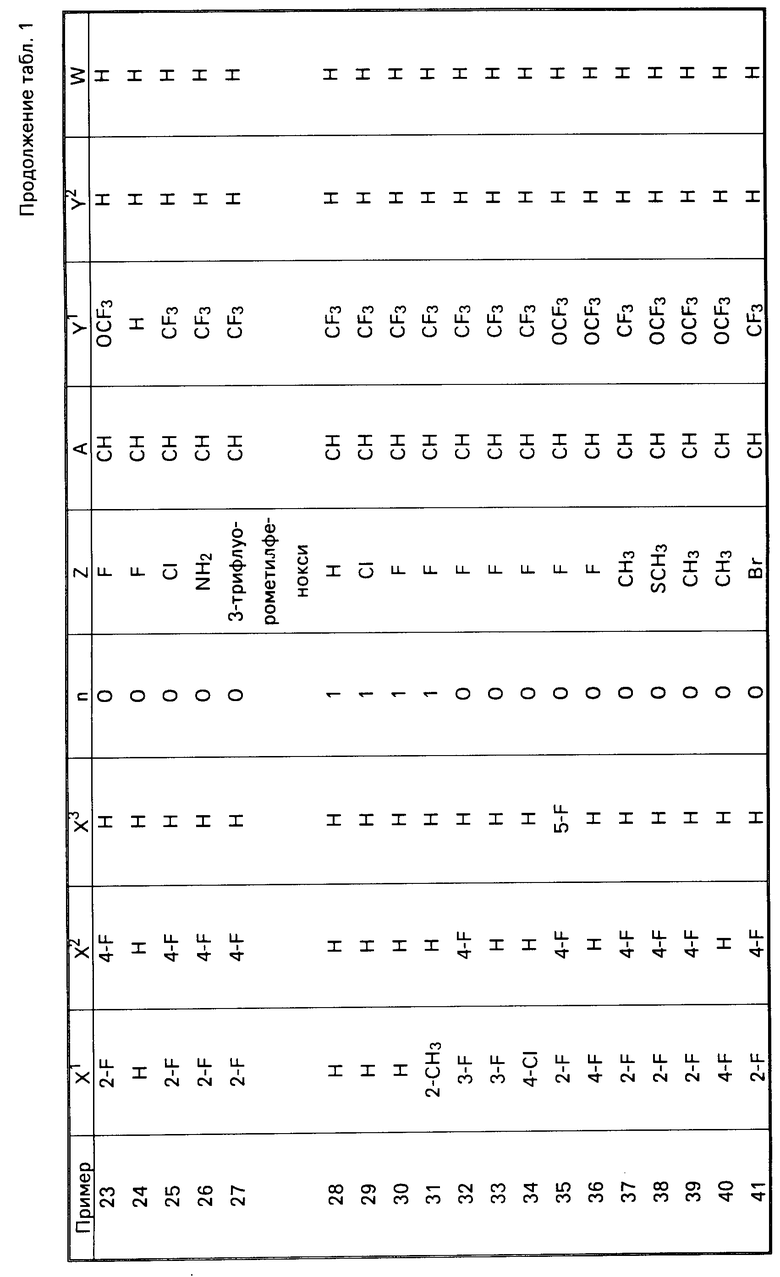
П р и м е р ы 5-41. С помощью методик, аналогичных описанным в примерах 1-4, получали предлагаемые соединения, подробно описанные в табл.1. В табл. 1 соединения идентифицируюся ссылками на формулу I.
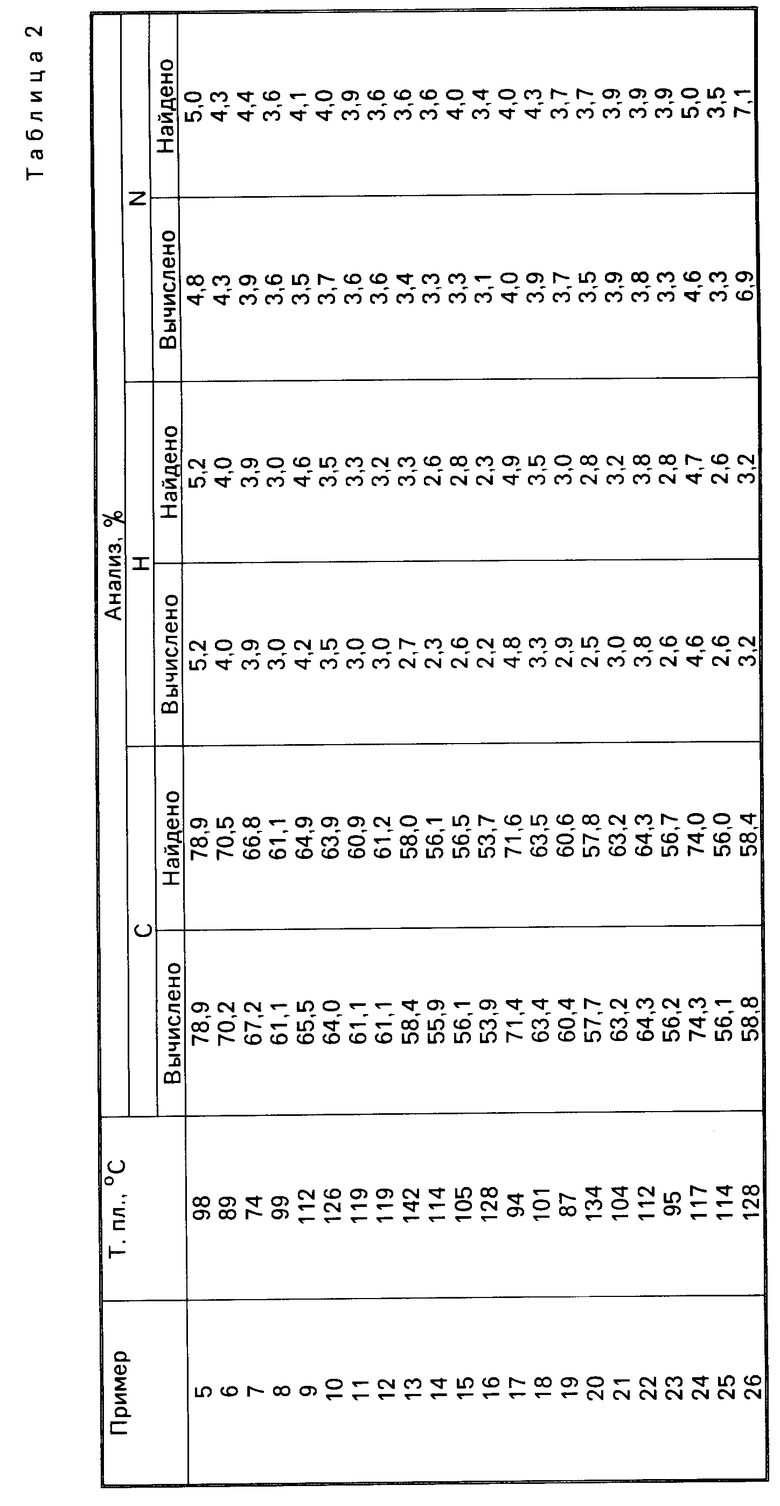
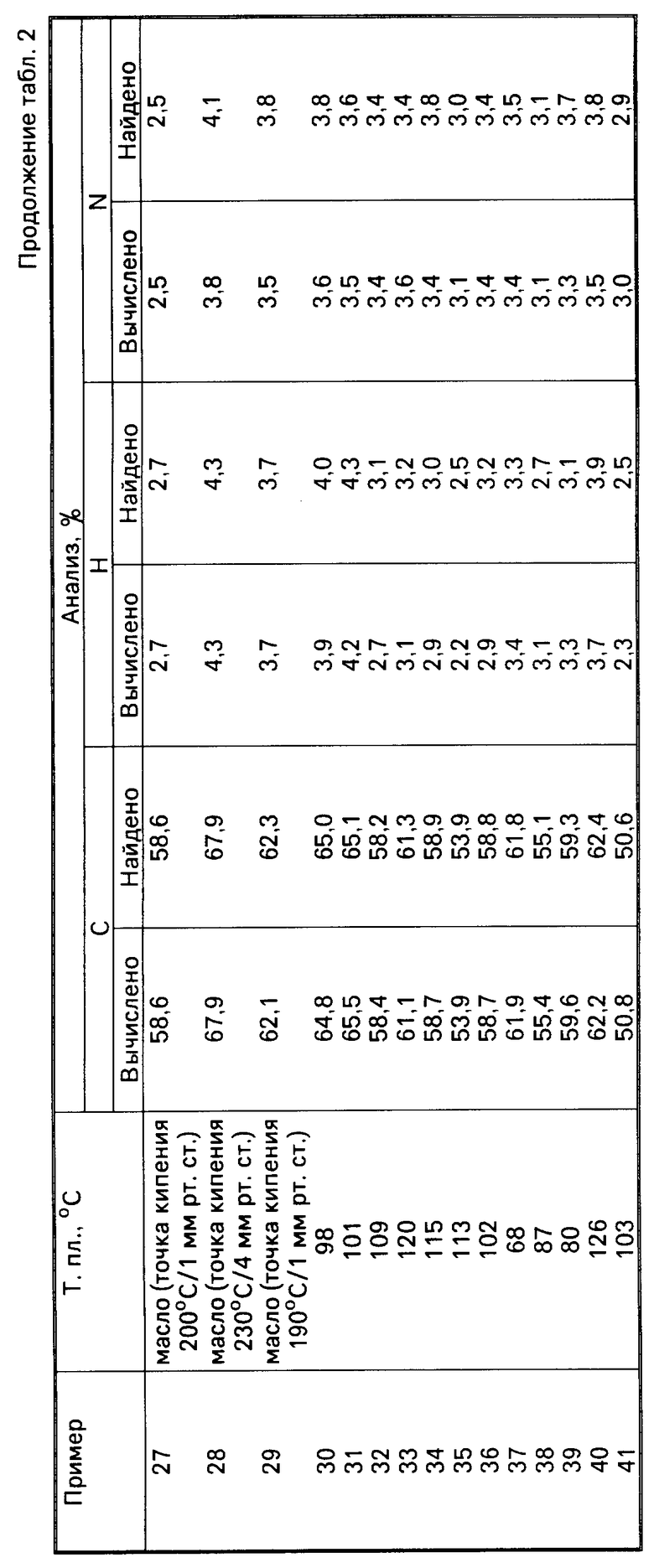
Данные точек плавления/кипения и данные элементного анализа для соединения примеров 5-41 приводятся в табл.2.
П р и м е р 42. Гербицидные композиции.
А) Смачиваемый порошок:
95 г активного ингредиента (соединение I)
3 г полиметакрилата натрия (диспергирующий агент)
2 г октил сульфосукцинат натрия (смачивающий агент)
В) Смачиваемая композиция:
50 г активного ингредиента
3 г полиметакрилата натрия
2 г октил сульфосукцината
45 г сухой чистой глины
С) Эмульгируемый концентрат:
25 г активного ингредиента
45 г ксилола
20 г циклогексанона
10 г эмульгатора (смесь додецилбензолсульфалата Са и этоксилата ионил фенола)
Д) Раствор:
1 г активного ингредиента
10 г ацетона
0,04 г Тритона Х-155 (торговая марка конденсата алкилфенола и этиленоксида)
600 г воды
П р и м е р 43. Гербицидная активность.
Для оценки гербицидной активности соединения согласно изобретению испытывались с использованием в качестве представителей ряда растений: кукурузы, Zea mays (Mz); риса (Oryza sativa (R)); ежовника Echinochloa crusgalli (BG), овса Avena sativa (O), льна Linum usitatissimum (L), горчицы (Sinapsis alba (M), сахарной свеклы Beta vulgaris (SB) и сои Glycine max (S).
Испытания подразделялись на две категории, довсходовые и после появления всходов. Предвсходовые испытания включали опрыскивание жидкой препаративной формой соединения почвы, в которую незадолго были высеяны семена указанных выше растений. Послевсходовые испытания включали два типа испытания, а именно орошение почвы и испытание с опрыскиванием листвы. В испытаниях по орошению почвы почва, в которой росли сеянцы растений указанных видов, орошались жидкой препаративной формой, содержащей соединение изобретения, а в испытаниях по опрыскиванию листвы сеянцы растений опрыскивались такой препаративной формой.
Почвой, используемой в опытах, был садовый суглинок.
Препаративные формы, используемые в испытаниях, приготавливали из растворов испытываемых соединений в ацетоне, содержащем 0,4 мас.% конденсата алкилфенола и окиси этилена, доступного под торговой маркой ТРИТОН Х-155. Эти ацетоновые растворы разбавляли водой и получившиеся препаративные формы применили в дозах, соответствующих: 5 кг или 1 кг активного ингредиента на гектар в объеме эквивалентном 600 л на гектар при опрыскивании почвы и опрыскивании листвы в дозе, эквивалентной 10 кг активного ингредиента на гектар в объеме, эквивалентном приблизительно 3000 л/га в испытании по орошению почвы.
В качестве контроля в довсходовых испытаниях применялась необработанная засеянная почва, а в послевсходовых испытаниях применялась необработанная почва с сеянцами растений.
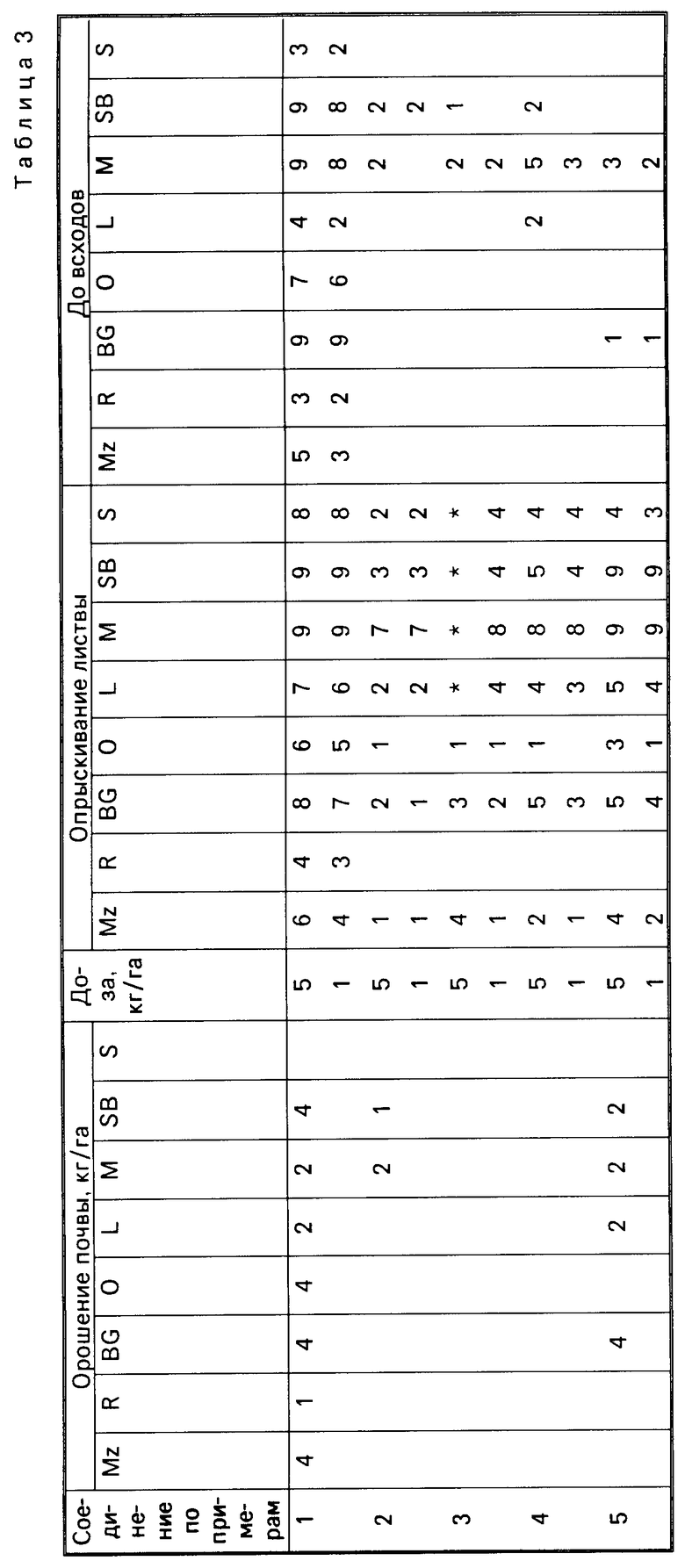
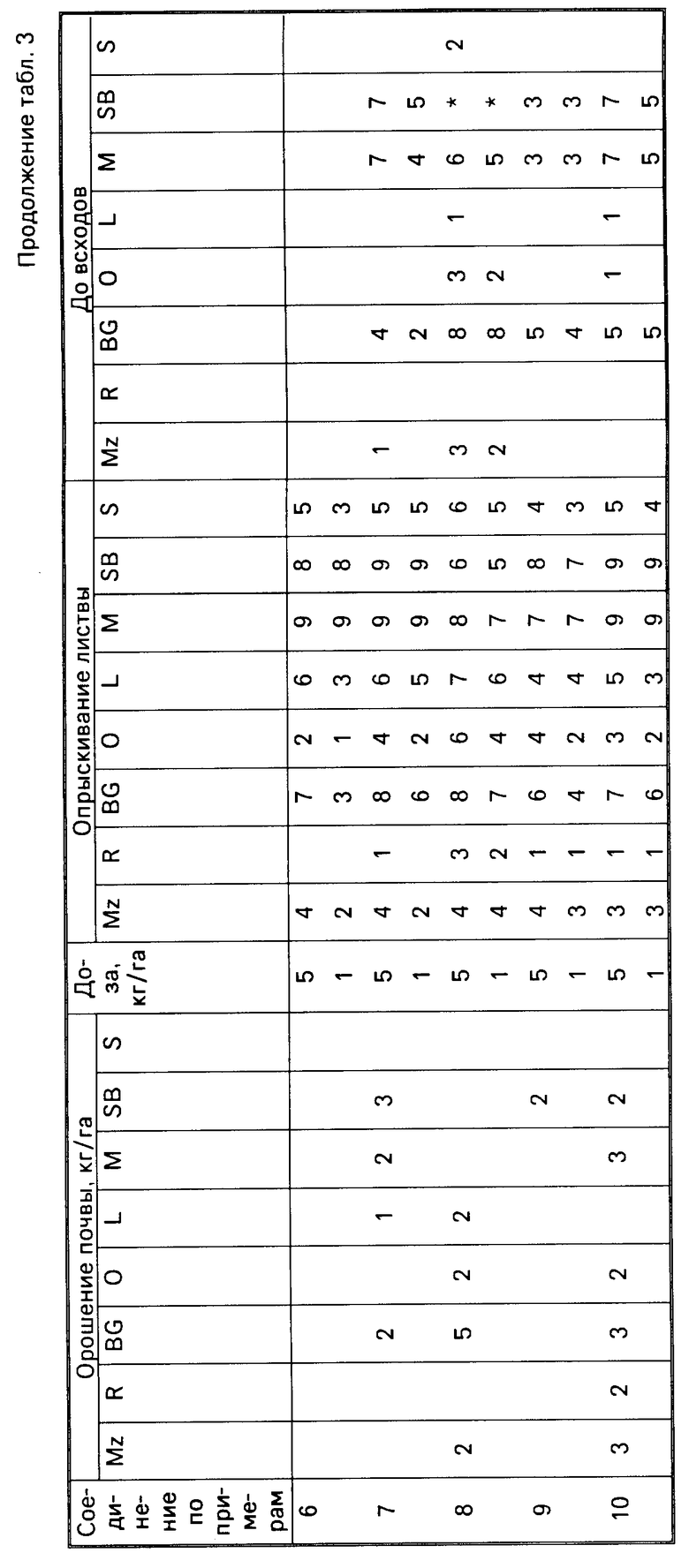
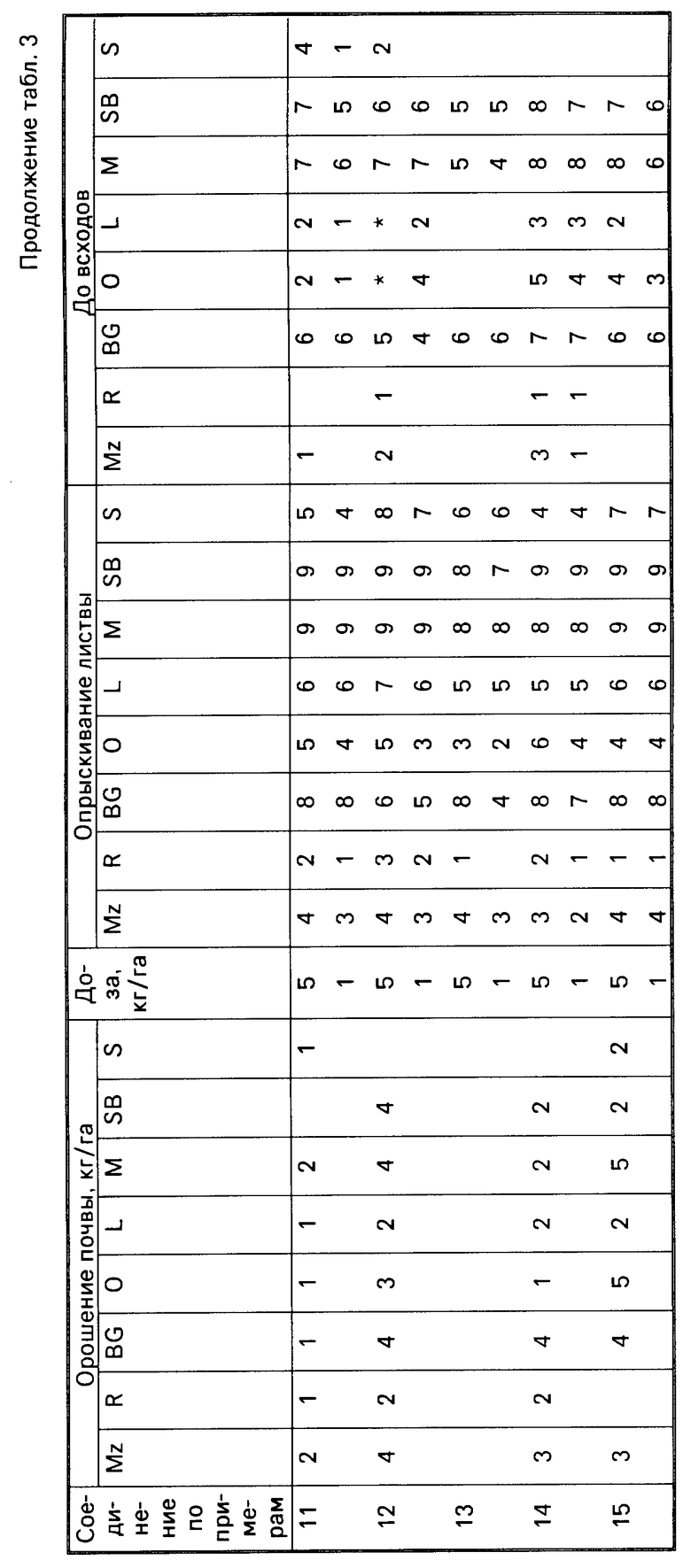
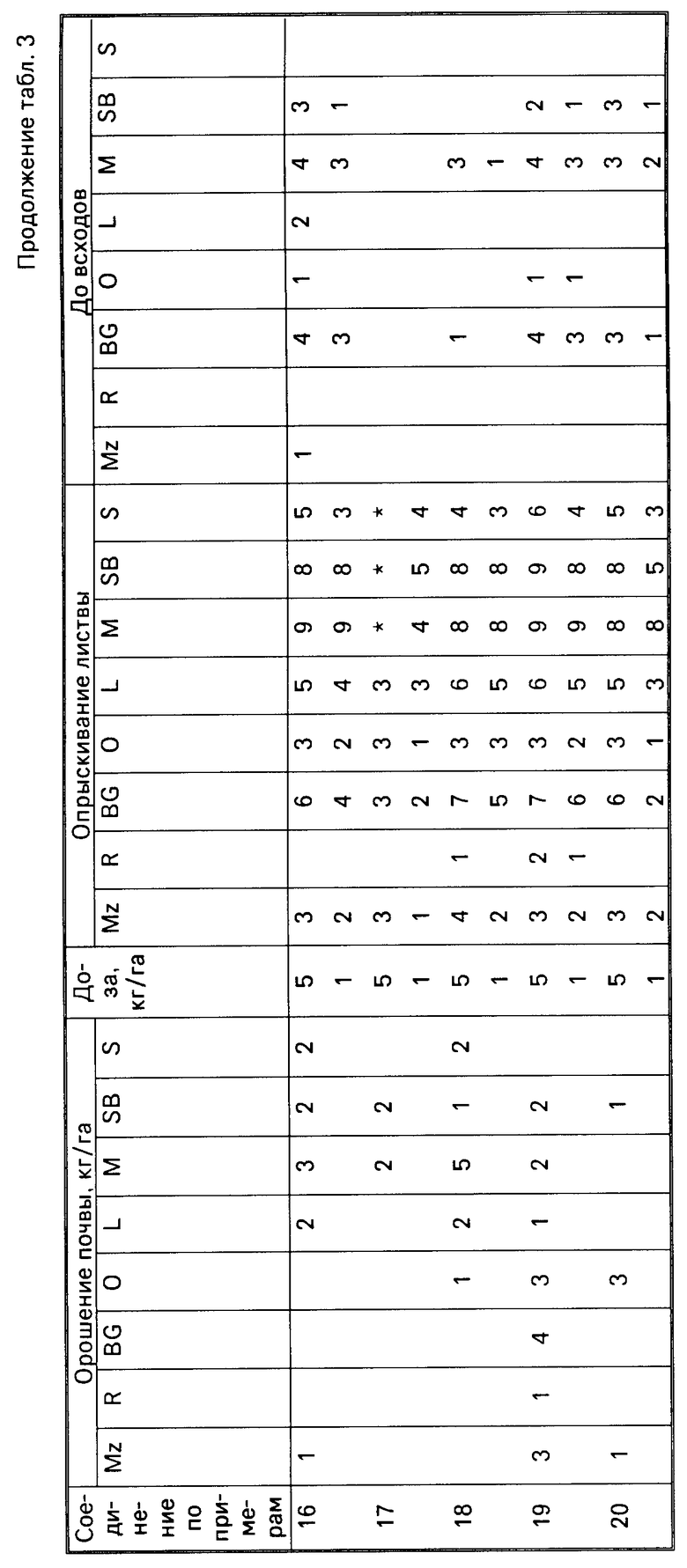
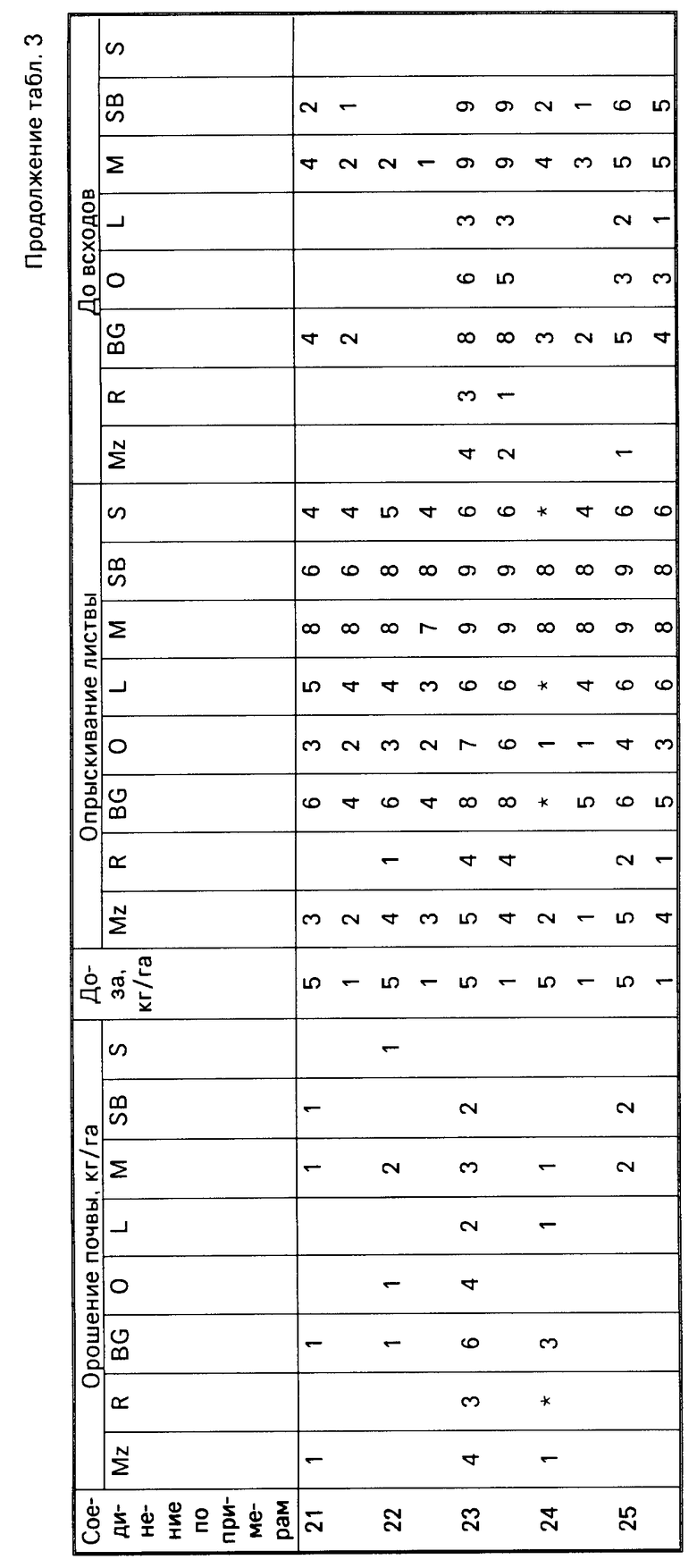
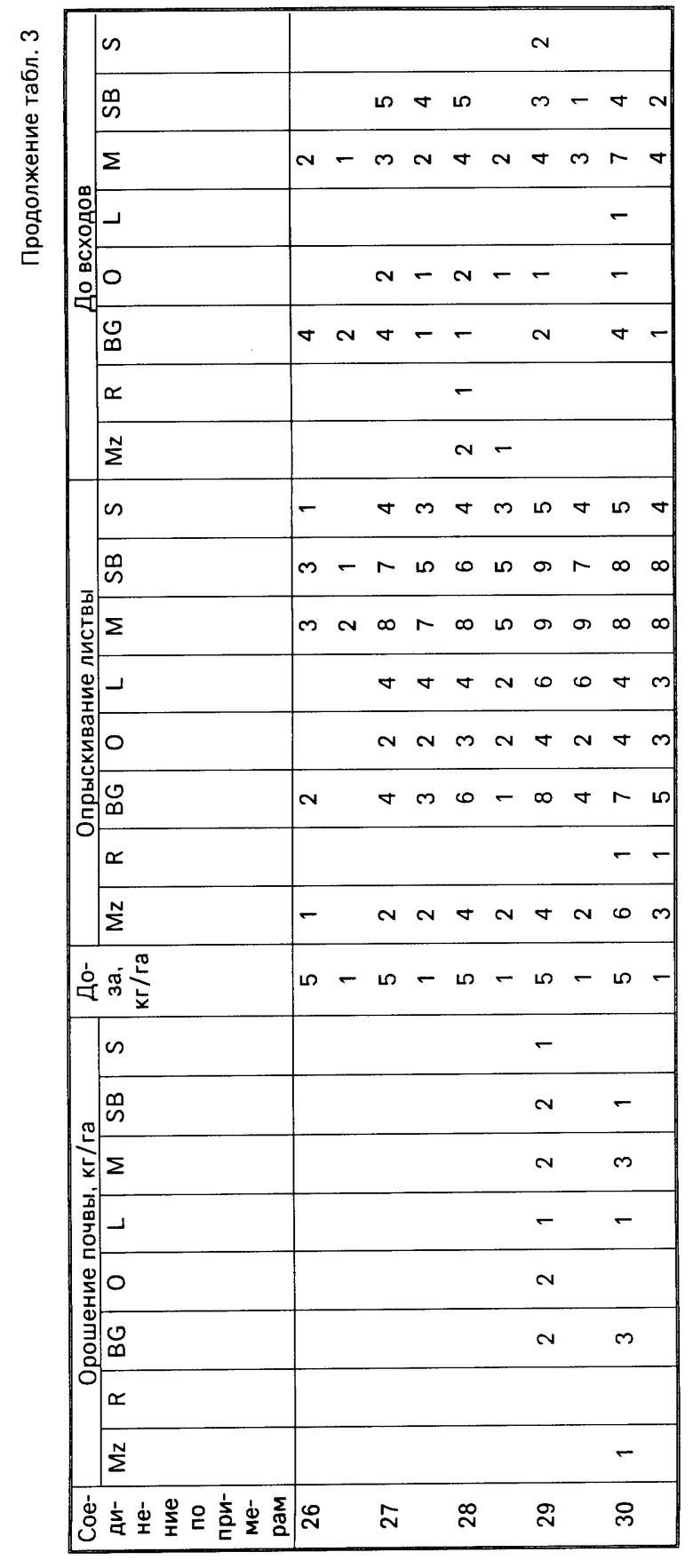
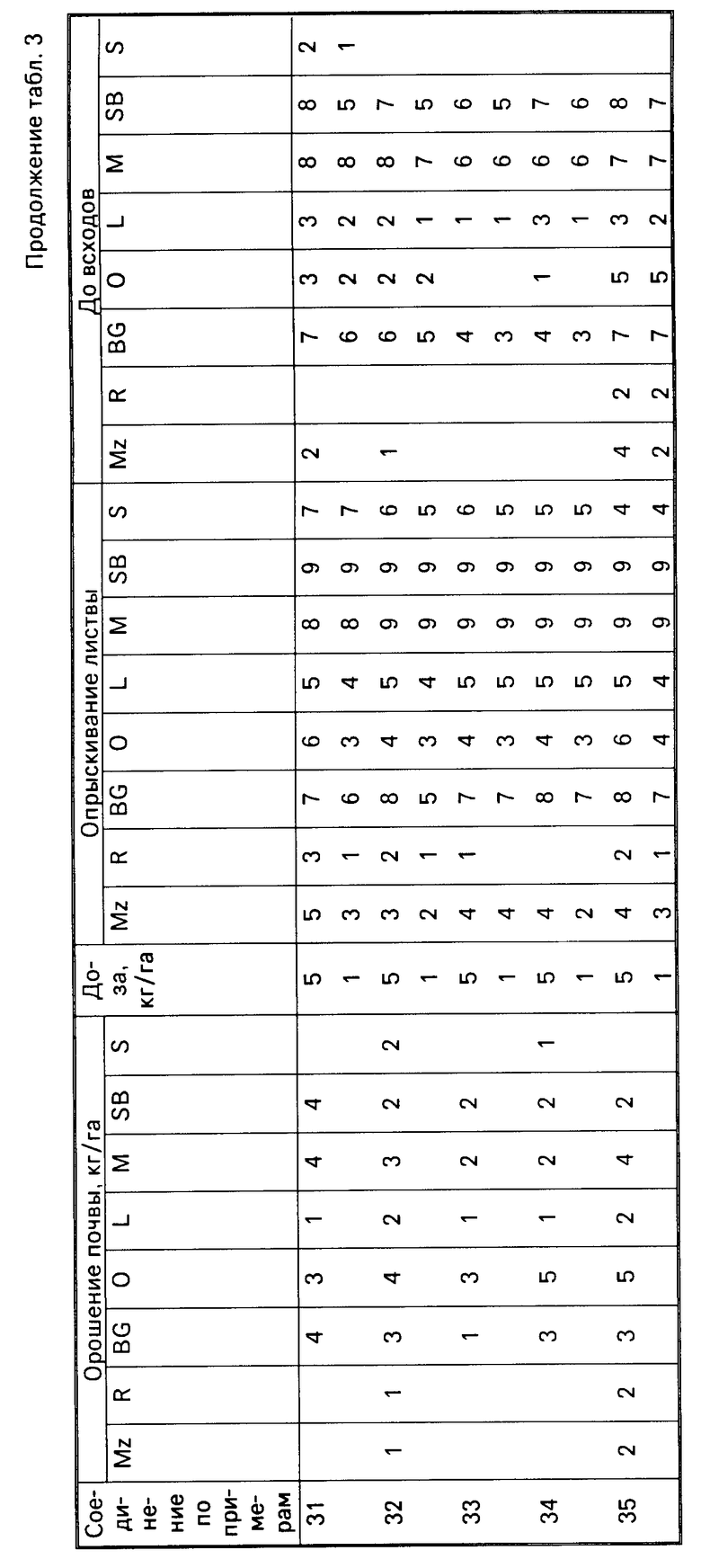
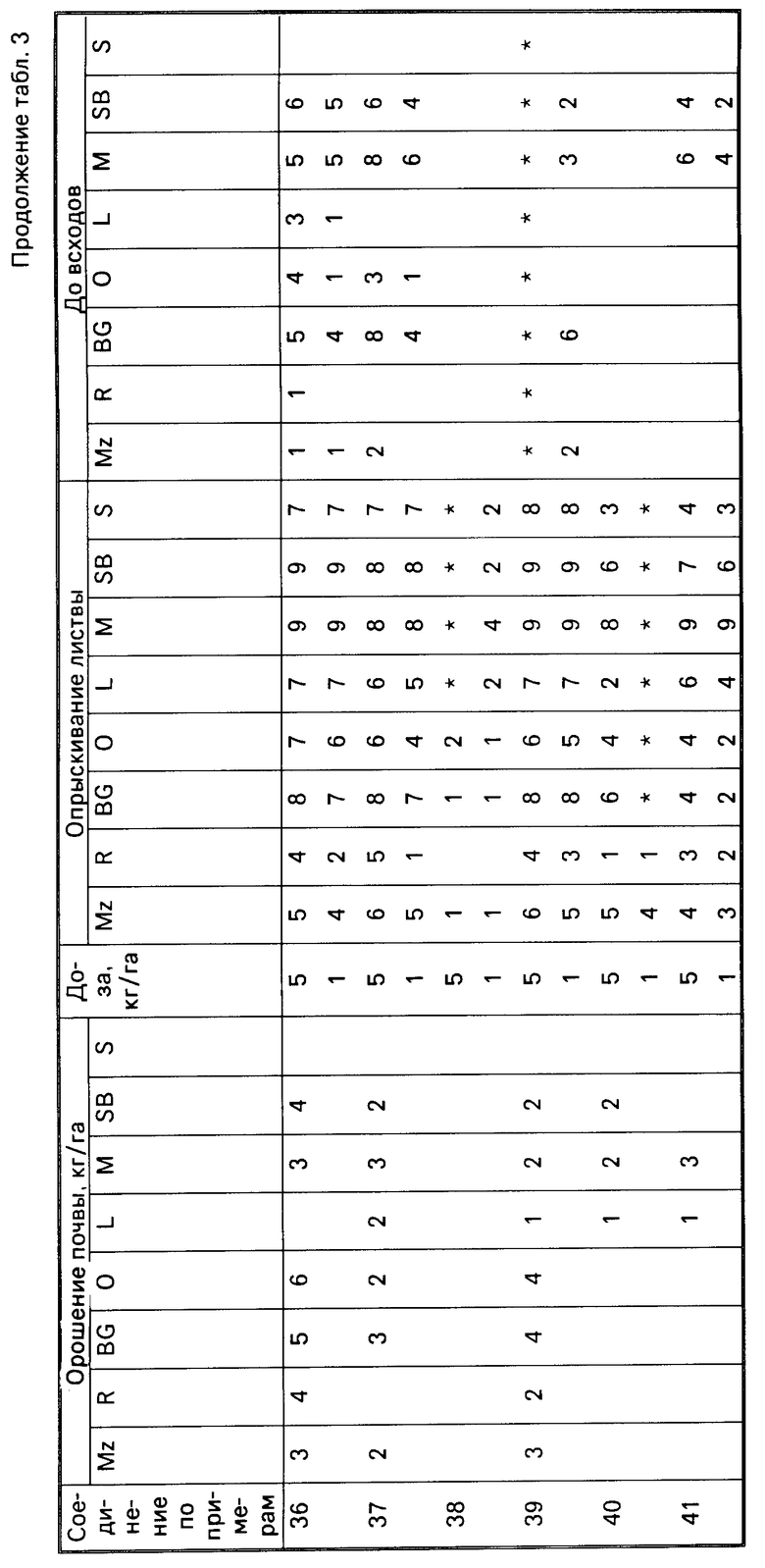
Гербицидные эффекты испытываемых соединений оценивались визуально через 12 дней после опрыскивания листвы и почвы и через 13 дней после орошения почвы, и оценивались по шкале 0-9. Оценка 0 указывает на рост, в случае необработанного контроля, оценка 9 означает гибель. Увеличение на единицу по линейной шкале приблизительно соответствует 10% увеличению по уровню эффективности.
Результаты испытаний представлены в табл.3, в которой соединения идентифицируются ссылками на предшествующие примеры. Отсутствие числовой величины в таблице указывает на нулевую оценку, звездочка указывает на то, что результат не получен.

Использование: в сельском хозяйстве, так как обладает гербицидной активностью и может быть использоваться для борьбы с ростом нежелательной растительности, а также в гербицидной композиции. Сущность изобретения: продукт - производные бензанилидов и бензамидов ф-лы I, указанной в описании, где x1, x2, x3 каждый независимо представляет H или Gal, или C1-C4 -алкил; n-0 или 1; Z-H, Gal, амино, C1-C4 -алкил, C1-C4 -галоидалкил, C1-C4 -алкилтио, C1-C4 -алкокси или фенокси-, необязательно замещенный C1-C4 -галоидалкилом; A-CH или N; Y1 и Y2 каждый независимо H, Gal, C1-C4 -алкил, C1-C4 -галоидалкил, C1-C4 -алкокси или C1-C4 -галоидалкокси; W-H или когда A означает CH и по крайней мере, один из Y1 и Y2 отличен от H или, когда A -N, W-H или Gal; при условии, что когда А-N, тогда каждый из Y1 и Y2 -H. Реагент 1: соединение ф-лы II, где М - удаляемая группа. Реагент 2: фенол или пиридинол или их реакционноспособные производные. Выход 70 - 80 проц. 4 з.п. ф-лы, 3 табл.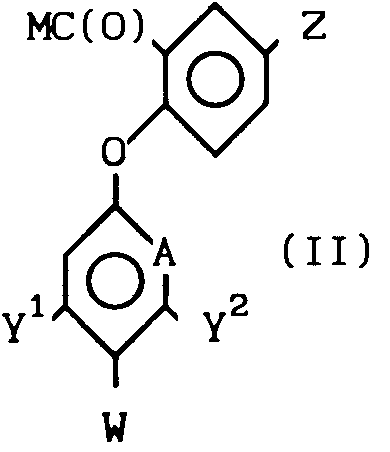
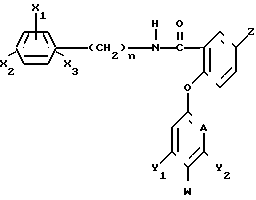
где X1, X2 и X3 - каждый независимо водород, или галоген, или C1-C4-алкил;
n = 0 или 1;
Z - водород, галоген, амино, C1-C4-алкил, C1-C4-галоидалкил, C1-C4-алкилтио, C1-C4-алкокси- или феноксигруппа, не обязательно замещенная C1-C4-галоидалкилом;
А - CH или N;
Y1 и Y2 - каждый независимо водород, галоген, C1-C4-алкил, C1-C4-галоидалкил, C1-C4-алкокси или C1-C4-галоидалкокси;
W - водород, или, когда А - CH и по крайней мере один из Y1 и Y2отличен от водорода или когда А - азот, то W - водород или галоген при условии, что когда А - азот, то Y1 и Y2 - водород.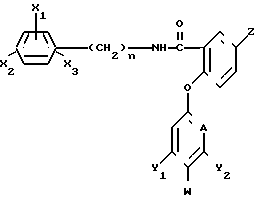
где X1, X2, X3 - каждый независимо водород, или галоген или C1-C4-алкил;
n = 0 или 1;
Z - водород или галоген, амино, C1-C4-алкил, C1-C4-галоидалкил, C1-C4-алкилтио или C1-C4-алкоксигруппа или феноксигруппа, не обязательно замещенная C1-C4-галоидалкилом;
А - CH или N;
Y1 и Y2 - водород, или галоген, или C1-C4-алкил, C1-C4-галоидалкил, C1-C4-алкокси- или C1-C4-галоидалкоксигруппа;
W - водород или, когда А - CH и по крайней мере один из Y1 и Y2отличен от водорода, или, когда А - N, то W - водород или галоген при условии, что когда А - N, то Y1 и Y2 - водород,
в эффективном количестве, целевые добавки - остальное.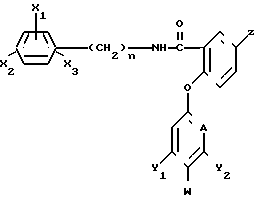
где X1, X2 и X3 - каждый независимо водород, или галоген, или C1-C4-алкил;
n = 0 или 1;
Z - водород или галоген, амино, C1-C4-алкил, C1-C4-галоидалкил, C1-C4-алкилтио, или C1-C4-алкокси- или феноксигруппа, не обязательно замещенная C1-C4-галоидалкилом;
А - CH или N;
Y1 и Y2 - каждый независимо водород, или галоген или C1-C4-алкил, C1-C4-галоидалкил, C1-C4-алкокси- или C1-C4-галоидалкоксигруппа;
W - водород или, когда А - CH и по крайней мере один из Y1 и Y2отличен от водорода или когда А - N, то W - водород или галоген, при условии, что, когда А - N, то Y1 и Y2 - водород,
в количестве 1 - 10 кг/га.
| Патент США N 3719707, C 07C103/32, 1973 | |||
| Chem | |||
| Abstr, 1958, 54, 3404. |
