Изобретение относится к аналитической химии, а именно к способам определения новокаинамида в объектах биологического происхождения: печени, крови и моче при проведении химико-токсикологических, судебно-химических и клинических исследований.
Известен способ определения новокаинамида в объектах биологического происхождения путем обработки анализируемой пробы водными растворами кислот и органическими растворителями с последующим фотоколориметрическим способом окрашенных растворов [1].
Недостатком способа является малая точность, обусловленная мешающим влиянием балластных веществ.
Наиболее близким к описываемому по технической сущности и достигаемым результатам является способ определения новокаинамида в объектах биологического происхождения путем обработки анализируемой пробы уксусной и трихлоруксусной кислотами и органическими растворителями, очисткой от балластных веществ электрофорезом при рН 9 с последующей десорбцией соляной кислотой и фотоколориметрированием окрашенных растворов [2ъ.
Недостатком способа является малая точность и большая продолжительность определения - около 25 ч.
Целью изобретения является повышение точности способа и сокращение времени определения.
Поставленная цель достигается описываемым способом определения новокаинамида в объектах биологического происхождения путем обработки анализируемой пробы 1 н.хлорной кислотой, очистке от балластных веществ на хроматографической колонке с силикагелем при рН 6-11 при соотношении массы силикагеля в граммах и объему крови в мл (0,4-0,5):(5-6) при содержании новокаинамида от 10 до 100 мкг с последующим фотоколориметрированием окрашенного раствора.
Отличительным признаком способа является последовательное осуществление всех приведенных выше стадий.
Примеры осуществления способа
Построение калибровочного графика.
В мерные колбы на 25 мл с помощью микропипетки вносят по 0,01; 0,02; 0,03...0,40 мл стандартного раствора новокаинамида с исходной концентрацией 100 мкг/мл, а затем во все колбы прибавляют 1 н. соляную кислоту до 1 мл. К растворам в мерных колбах прибавляют по 1 мл 0,1%-ного раствора нитрита натрия, по 1 мл 0,1%-ного раствора 3- α,γ -дикарбоксипропилроданина в 95%-ном этиловом спирте. Жидкость во всех колбах взбалтывают и объемы растворов в них доводят до метки боратной буферной смесью. Содержимое колб тщательно перемешивают и измеряют оптическую плотность окрашенных в красный цвет растворов с помощью фотоколориметра КФЕ-2 светофильтр λэфф = 490±10 нм в кювете с толщиной рабочего слоя 50 мм. В качестве растворов сравнения используют смесь всех реактивов взятых в указанных количествах. рН растворов аэроданина должен находиться в интервале значений 10,7-12,1.
Боратную буферную смесь получают смешиванием 0,1 н. раствора гидроксила натрия с боратным буферным раствором в соотношении 1:0,975. Боратный буферный раствор готовят путем смешивания 0,05 М раствора буры с 0,1 н. раствором гидроксида натрия в соотношении 1:1 (рН 11,02).
По полученным данным строят калибровочный график, откладывая по оси абсцисс введенные в реакцию количества первичных ароматических аминов, а по оси ординат - соответствующие им величины оптической плотности. Светопоглощение окрашенных растворов подчиняется закону Бугера-Ламберта-Бера в пределах концентраций 1-40 мкг.
Описание хроматографической колонки.
Хроматографическая колонка представляет собой трубку длиной 70 мм с внутренним диаметром 13 мм, закрытую снизу перфорированной перегородкой (дно корпуса шприца одноразового использования с 5-6 отверстиями). Сверху на перфорированную перегоpодку кладут диск из фильтровальной бумаги "красная лента", затем вносят 0,5 г силикагеля марки 15/40, ЧССР, уплотняя его поршнем от шприца до упора. Сверху снова кладут диск фильтровальной бумаги и ставят прижимное кольцо из тефлона или полиэтилена.
Клейкой лентой размером 20 х 45 мм хроматографическая колонка соединяется снизу с приемником, представляющим собой стеклянную пробирку длиной 50 мм с внутренним диаметром 13 мм. В колонку вносят 6 мл буферного раствора рН которого соответствует рН нейтрализованной биопробы. Помещают колонку в центрифужный приемник и центрифугируют при 500 g в течение 7 мин. Приготовленная колонка рассчитана на проведение одного анализа.
П р и м е р 1. Определение содержания свободного новокаинамида в ткани печени.
В центрифужные стаканы емкостью 250 мл помещают по 100 г мелкоизмельченной печени трупа человека, погибшего в результате травмы. В каждый стакан вносят по 5 мл раствора новокаинамида в буферном растворе с рН 7,4, исходная концентрация которого 100 мкг/мл.
Содержимое стаканов оставляют стоять при периодическом перемешивании в течение суток, затем к содержимому каждого стакана прибавляют по 100 мл 1 н. хлорной кислоты, настаивают в течение 2 ч и центрифугиpуют 5 мин при 500 g. Надосадочные жидкости сливают с твердых частиц биоматериала и операцию изолирования осуществляют новыми порциями 1 н. хлорной кислоты еще 3 раза. Вытяжки объединяют и измеряют объем полученного извлечения.
40 мл безбелковой вытяжки, исследуемой на содержание новокаинамида, вносят в мерную колбу на 50 мл, доводят рН кислотного извлечения до рН 6-8-40%-ным раствором гидроксида натрия и прибавляют дистиллированную воду до метки, после чего в четыре предварительно приготовленные как указано выше хроматографические колонки с оптимальным содержанием силикагеля (0,4-0,5 г для 5-6 мл пробы) вносят определенные, точно отмеренные объемы биопробы с рН 6-8.
Колонки с приемниками помещают в центрифугу и центрифугируют при 500 g в течение 7 мин. Приемники освобождают от центрифугатов, вносят в каждую колонку по 5 мл фосфатного буферного раствора с рН 6-8 и центрифугируют как указано выше. Промывание колонки новыми порциями буферного раствора 6-8 проводят еще два раза, центрифугаты отбрасывают, после чего препарат элюируют из колонок. С этой целью в колонки вносят по 3 мл 2 н. соляной кислоты и приступают к центрифугированию в режиме, описанном выше. Промывание колонок новыми порциями 2 н. соляной кислоты (объемом по 3 мл) повторяют еще два раза. Элюаты, полученные с четырех колонок, объединяют, собирая их в мерную колбу емкостью 50 мл, объем жидкости содержимого в колбе доводят до метки дистиллированной водой и после перемешивания определяют содержание новокаинамида.
Для этого в четыре мерные колбы емкостью 25 мл вносят точно отмеренные объемы полученных элюатов. В первые три колбы прибавляют по 1 мл 0,1%-ного раствора нитрата натрия, затем по 1 мл 0,1%-ного раствора 3- α,γ -дикаpбоксипропилроданина. После перемешивания к содержимому колбы прибавляют 10% -ный раствор гидроксида натрия до появления розово-красного окрашивания. После перемешивания объем раствора в колбе доводят до метки дистиллированной водой. Для получения раствора сравнения к содержимому в 4-й колбе прибавляют 10%-ный раствор гидроксида натрия в таком объеме, какой был добавлен в первые три колбы для получения розово-красного окрашивания, затем прибавляют 1 мл 0,1%-ного раствора 3- α,γ-дикарбоксипропилроданина, после чего 1 мл 0,1% -ного раствора нитрита натрия. После перемешивания объемы жидкостей во всех колбах доводят дистиллированной водой до 25 мл.
При определении суммарного содержания новокаинамида (включая и ацетилированную форму новокаинамида) технология сорбционной очистки не меняется. В этом случае солянокислые центрифугаты переносят в колбу на 50 мл и подвергают гидролизу при нагревании на кипящей водяной бане в течение 20 мин. После этого содержимое колб охлаждают, добавляют объем в колбе до метки дистиллированной водой и проводят фотометрический анализ как указано.
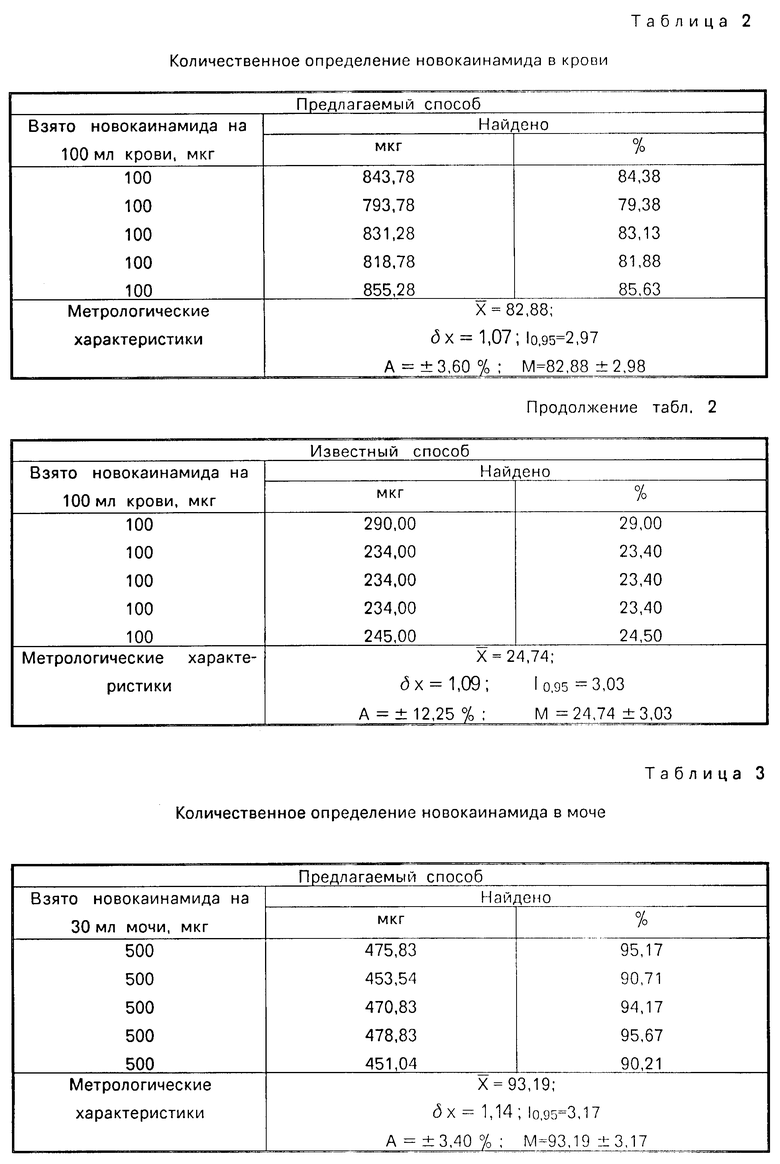
Результаты определения новокаинамида в ткани печени приведены в табл.1. Ошибка способа определения новокаинамида составляет ±3,76%.
Содержание новокаинамида в ткани печени рассчитывают по формуле
y = 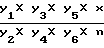 , где Y - содержание новокаинамида в 100 г биоматериала
, где Y - содержание новокаинамида в 100 г биоматериала
Y1 - суммарный объем кислотного извлечения (мл)
Y2 - объем кислотного извлечения, взятый на нейтрализацию (мл);
Y3 - объем полученной нейтрализованной биовытяжки (мл);
Y4 - объем нейтрализованной биовытяжки, внесенный на колонку (мл);
Y5 - суммарный объем солянокислых элюатов (мл);
Y6 - объем солянокислого элюата, взятый на проведение реакции (мл);
Х - количество (мкг) новокаинамида в 25 мл фотометрируемого раствора, найденное по калибровочному графику;
n - число колонок.
Для конкретного примера Y = 0,26 х Y1.
П р и м е р 2. Определение свободного новокаинамида в крови.
В мерную колбу емкостью 100 мл прибавляют 10 мл раствора новокаинамида в буферном растворе с рН 7,4 и исходной концентрацией 100 мкг/мл. После этого доводят донорской кровью человека объем жидкости в колбе до метки. Содержимое колбы перемешивают и инкубируют в течение 24 ч. По истечении указанного времени из колбы отбирают пробу объемом 10 мл, внося ее в центрифужную пробирку и прибавляют туда же 10 мл дистиллированной воды.
Смесь перемешивают и через 5 мин в пробирку вносят 5 мл раствора 5,0 н. хлорной кислоты. Содержимое пробирки центрифугируют при 500 g в течение 5 мин. Центрифугат переносят в мерную колбу на 50 мл. К осадку в центрифужной пробирке прибавляют еще 5,0 мл 5 н. хлорной кислоты и центрифугируют 5 мин. Центрифугаты объединяют и доводят рН раствора до рН 6-8 40%-ным раствором гидроксида натрия, объем содержимого в колбе доводят до метки дистиллированной водой. После тщательного перемешивания нейтрализованных извлечений проводят сорбционную очистку пробы как указано в примере 1, затем фотометрический анализ элюатов.
Результаты определения новокаинамида в крови представлены в табл.2. Ошибка способа определения новокаинамида в крови составляет ±3,64%.
Расчет содержания новокаинамида в крови проводят по формуле:
y = 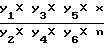 , где Y1 - объем крови, затравленной новокаинамидом (мл);
, где Y1 - объем крови, затравленной новокаинамидом (мл);
Y2 - объем крови, содержащей новокаинамид, взятой для проведения анализа (мл);
Y3 - объем нейтрализованного кислотного извлечения (мл);
Y4 - объем нейтрализованного кислотного извлечения, внесенный на колонку (мл);
Y5 - суммарный объем солянокислых элюатов (мл);
Y6 - объем солянокислого элюата, взятый на проведение реакции (мл);
Х - количество (мкг) новокаинамида в 25 мл фотометрируемого раствора, найденное по калибровочному графику;
n - число колонок.
Для конкретного примера Y - 104,17 х, Y6 = 10 мл.
П р и м е р 3. Определение свободного новокаинамида в моче.
В мерную колбу на 50 мл вносят 5 мл раствора новокаинамида в буферном растворе с рН 7,4 и исходной концентрацией 100 мкг/мл, а затем в колбу добавляют 30 мл донорской мочи человека. После этого рН раствора доводят до рН 6-8 10%-ным раствором гидроксида натрия, объем содержимого в колбе доводят до метки дистиллированной водой. После тщательного перемешивания содержимого колбы, проводят сорбционную очистку пробы как указано в примере 1, а затем проводят фотометрический анализ элюатов.
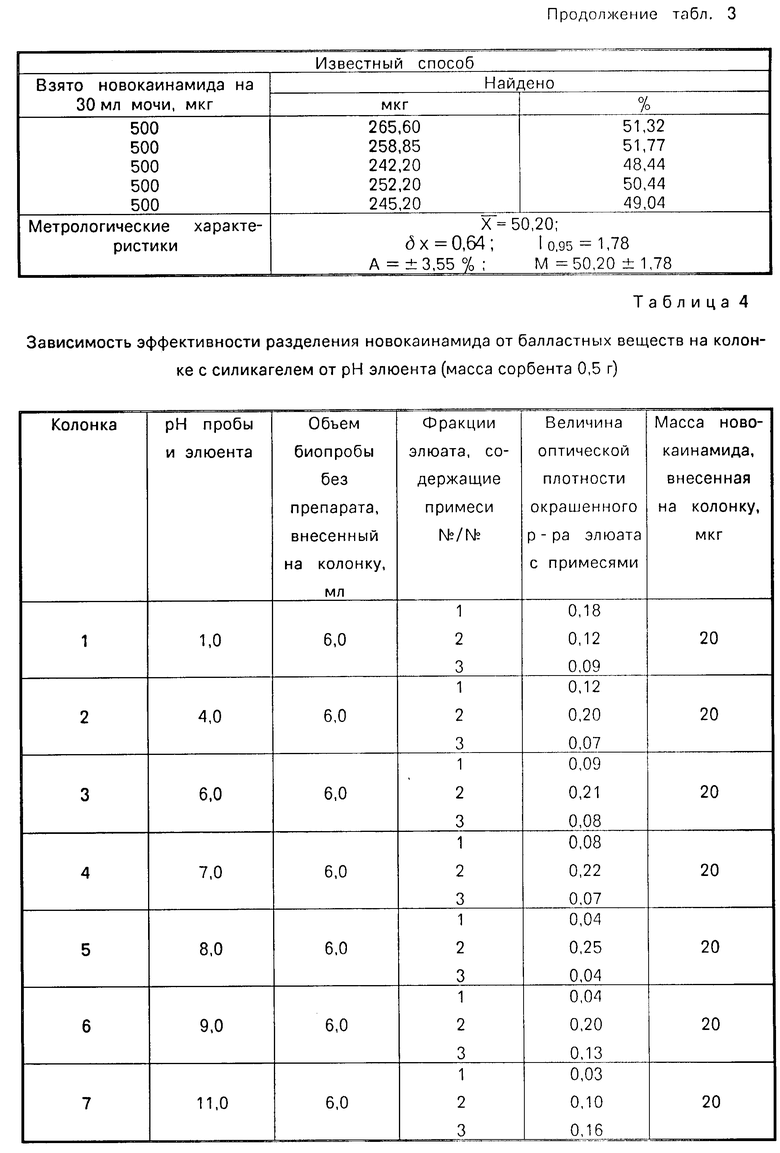
Результаты определения новокаинамида в моче представлены в табл.3. Ошибка способа определения новокаинамида в моче составляет ±3,40%.
Расчет содержания новокаинамида в моче проводят по формуле:
y = 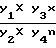 , где Y1 - объем нейтрализованной мочи, взятой на исследование содержания новокаинамида (мл);
, где Y1 - объем нейтрализованной мочи, взятой на исследование содержания новокаинамида (мл);
Y2 - объем нейтрализованной пробы, внесенный на колонку (мл);
Y3 - объем солянокислых элюатов суммарный (мл)
Y4 - объем солянокислого элюата, взятый на проведение реакции (мл)
х - количество (мкг) новокаинамида в 25 мл фотометрируемого раствора, найденное по калибровочному графику.
n - число колонок.
Для конкретного примера Y = 20,833х, Y4 = 5 мл.
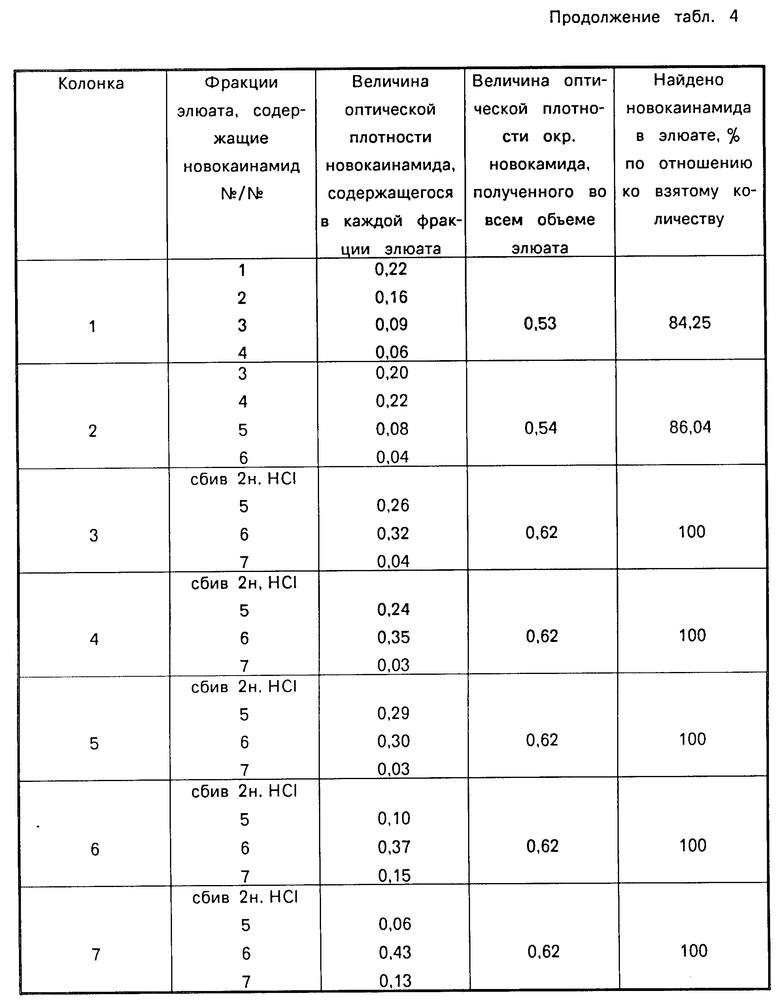
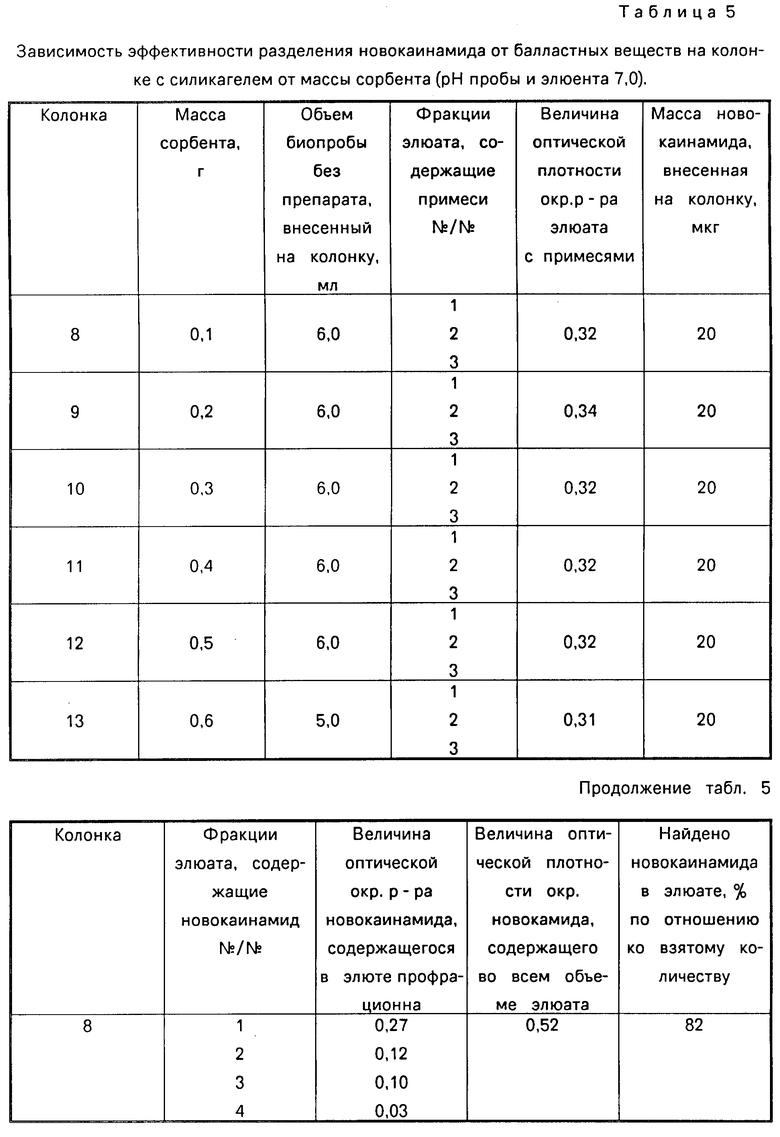
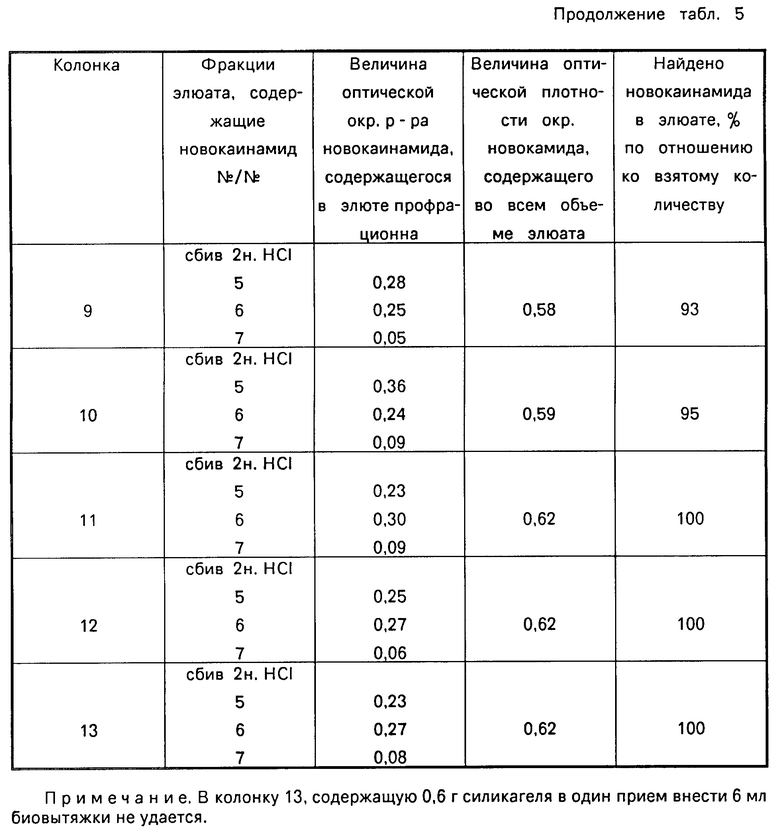
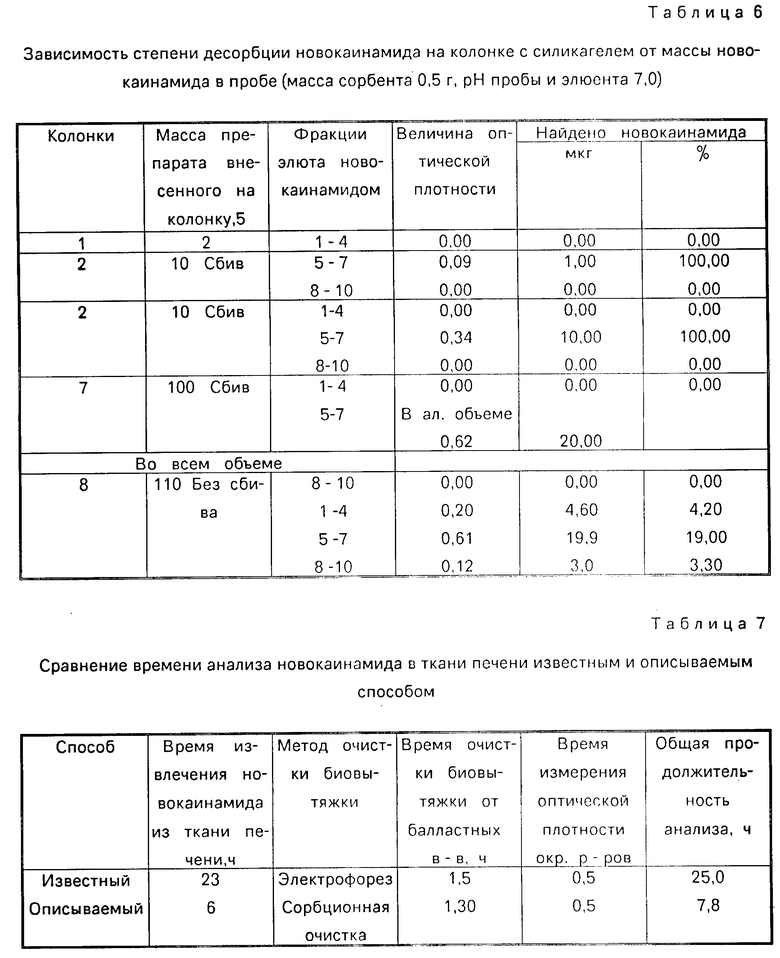
В табл. 4 приведена зависимость эффективности разделения новокаинамида на колонке с сорбентом от рН элюента, в табл.5 - от массы сорбента, в табл. 6 - от массы новокаинамида в анализируемой пробе,
Как видно из таблиц оптимальными условиями проведения определения являются следующие: рН 6-11, соотношение массы силикагеля (г) к объему крови (мл) (0,4-0,5): (5-6) при массе новокаинамида в анализируемой пробе 10-100 мкг.
Сравнение времени анализа новокаинамида в объектах биологического происхождения известным и описываемым способами представлено в табл.7.
Как видно из табл.1-3, 7 описываемый способ позволяет повысить точность определения новокаинамида в объектах биологического происхождения:
в ткани печени - на 49%
в крови - на 52%
в моче - на 43% и сократить время анализа на 17,2 ч.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения сульфаниламидных препаратов в объектах биологического происхождения | 1989 |
|
SU1663516A1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЦИКДОМЕТИАЗИДА | 1994 |
|
RU2090866C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ТЕТРАЭТИЛТИУРАМДИСУЛЬФИДА В КРОВИ | 2010 |
|
RU2417372C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 2,4-ДИХЛОРФЕНОКСИУКСУСНОЙ КИСЛОТЫ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2011 |
|
RU2453848C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 4-НИТРОФЕНОЛОВ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 1994 |
|
RU2121681C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 2,6-БИС-[БИС-(БЕТА-ОКСИЭТИЛ)-АМИНО]-4,8-ДИ-N-ПИПЕРИДИНО-ПИРИМИДО(5,4-D)ПИРИМИДИНА В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2006 |
|
RU2322674C1 |
| Способ количественного определения фурагина | 1989 |
|
SU1698716A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ Н-БУТИЛОВОГО ЭФИРА 2-[4-(5-ТРИФТОРМЕТИЛПИРИДИЛ-2-ОКСИ)ФЕНОКСИ]ПРОПИОНОВОЙ КИСЛОТЫ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2005 |
|
RU2287812C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 2-МЕТОКСИ-4-АЛЛИЛГИДРОКСИБЕНЗОЛА В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2008 |
|
RU2395081C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СЛОЖНОГО НИТРОФЕНОЛЬНОГО ПРЕПАРАТА "НИТРАФЕН" В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 1999 |
|
RU2153169C1 |
Использование: в аналитической химии при проведении химико-токсилогических, судебно - химических и клинических исследований. Сущность изобретения: анализируемую пробу обрабатывают хлорной кислотой, доводят pH до 6 - 11, пропускают через хроматографическую колонку с силикагелем при соотношении массы силикагеля в граммах к объему пробы в мл (0,4 - 0,5) : (5 - 6) при содержании нокаинамида 10 - 100 мкг в пробе с последующим фотоколориметрированием окрашенного раствора. Относительная ошибка ± 3.8 % , время анализа 8 ч. 7 табл.
СПОСОБ ОПРЕДЕЛЕНИЯ НОВОКАИНАМИДА В ОБЪЕКТАХ БИОЛОГИЧЕСКОГО ПРОИСХОЖДЕНИЯ путем обработки анализируемой пробы раствором кислоты, очистки от балластных веществ и фотокалориметрирования окрашенного раствора, отличающийся тем, что, с целью повышения точности способа и сокращения времени определения, обработку анализируемой пробы ведут 1 н хлорной кислотой, очистку от балластных веществ ведут на хроматографической колонке с силикагелем при pH 6 - 11 при соотношении массы силикагеля (г) и объема пробы (мл) 0,4 - 0,5 : 5 - 6 при содержании новокаинамида 10 - 100 мкг.
| Песахович Л.В | |||
| Химико-токсикологическое доказательство новокаинамида, в книге "Современные методы исследования судебно-медицинских объектов, Рига", РМИ, 1977, с.114-117. |
