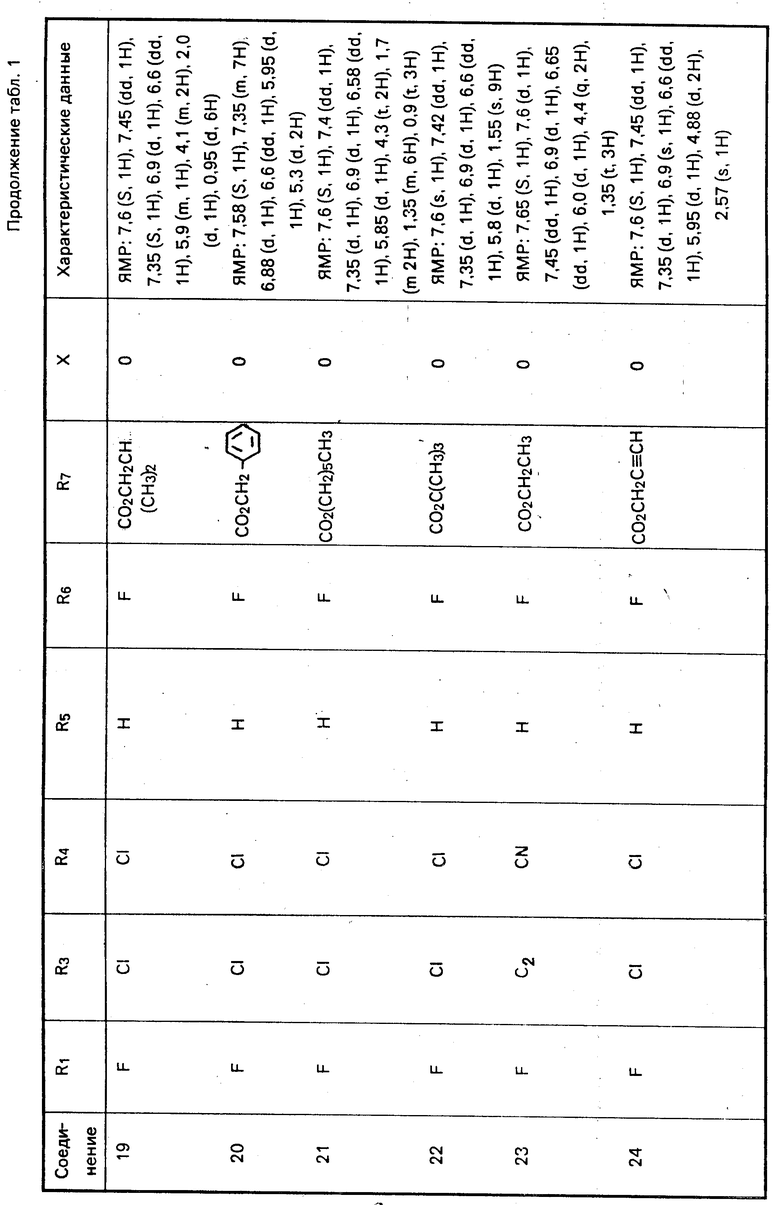
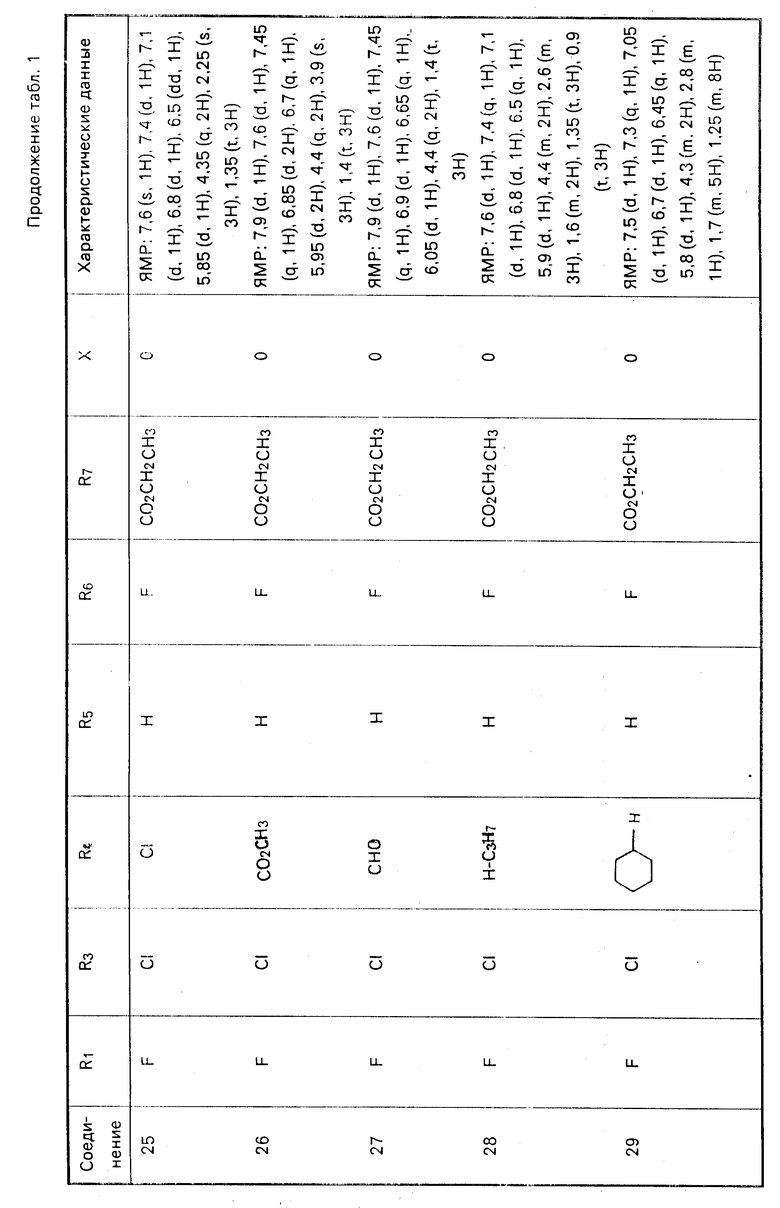
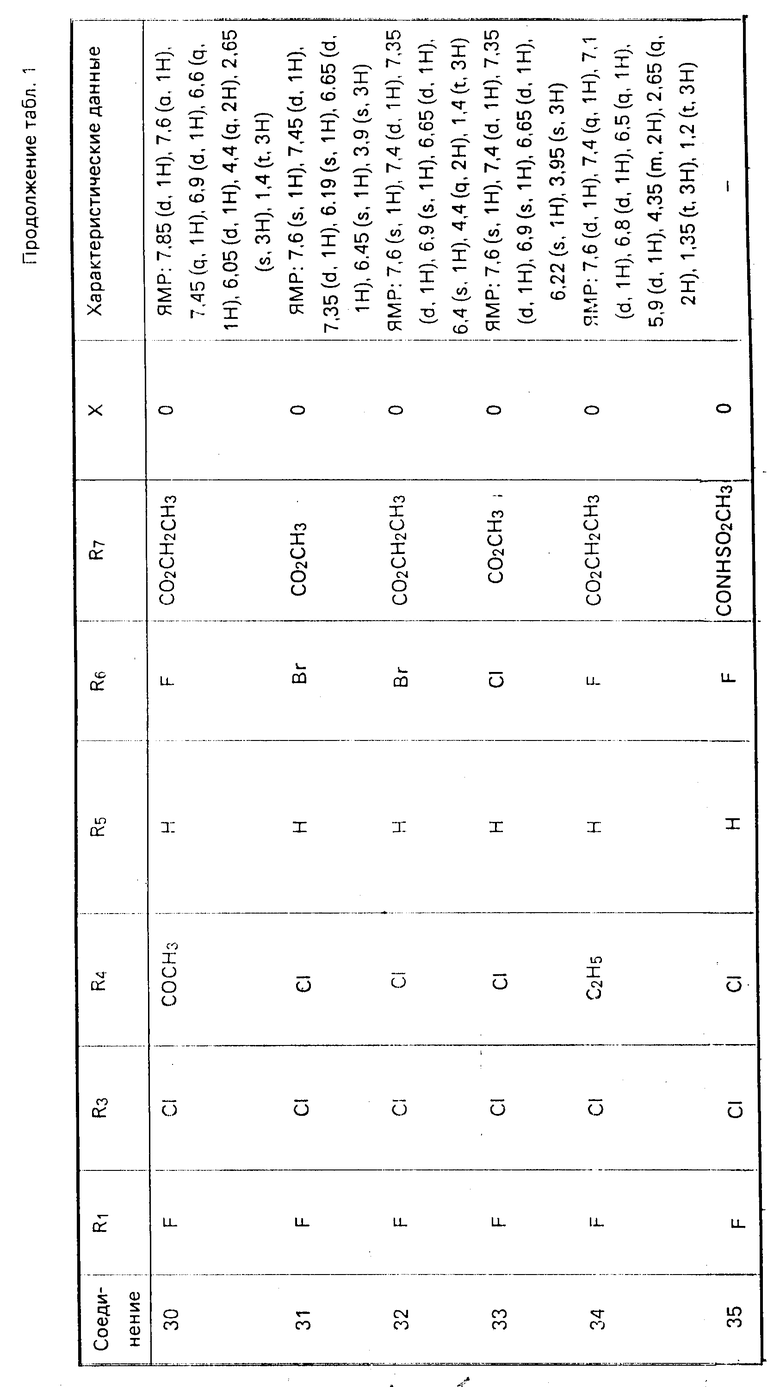
Изобретение относится к производным дифенилового эфира общей формулы I
F3C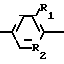 O
O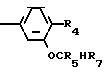 где R1 - Н или галоид;
где R1 - Н или галоид;
R2 - N или СR3, где R3 - галоид или NO2;
R4 - Н, галоид, NO2, CN, CO2CH3;
R5 - галоид;
R7 - COOR8, CHO, CONR8R9;
R8 - Н, С1-С6-алкил, необязательно замещенный фенилом, алкенил, С2-С6;
R8 и R9 - независимо представляют Н или С1-С6-алкил, обладающие гербицидной активностью.
Структурная формула I, представленная выше, включает таутомерные формы представленной структуры, а также физически различные модификации соединений, возникающие различными путями, в которых молекулы упорядочиваются в кристаллической решетке, или возникающие благодаря неспособности части молекул свободно вращается относительно других частей молекул, или возникающие благодаря геометрической изомерии, или при образовании внутримолекулярной или межмолекулярной водородной связи, или иным образом.
Некоторые из соединений изобретения могут существовать в энантиомерных формах. Изобретение включает и энантиомеры, и смеси двух энантиомеров во всех соотношениях.
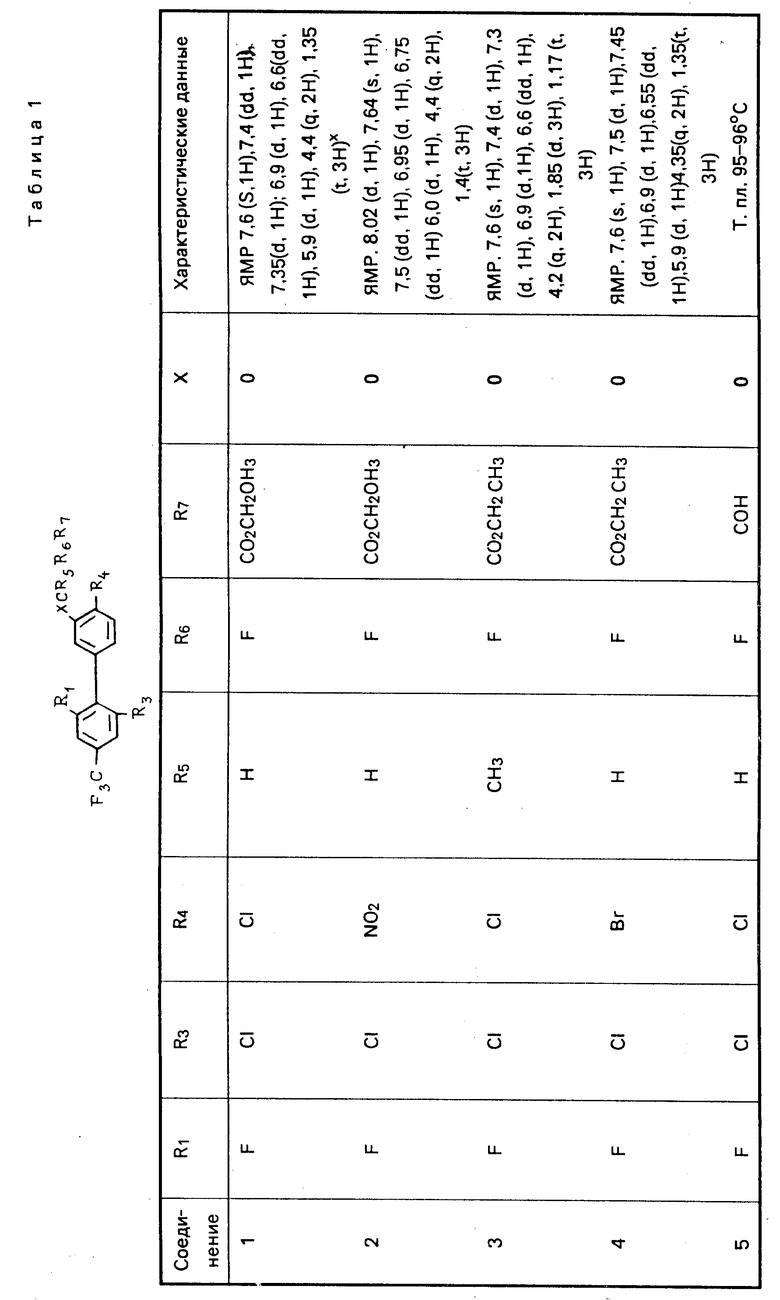
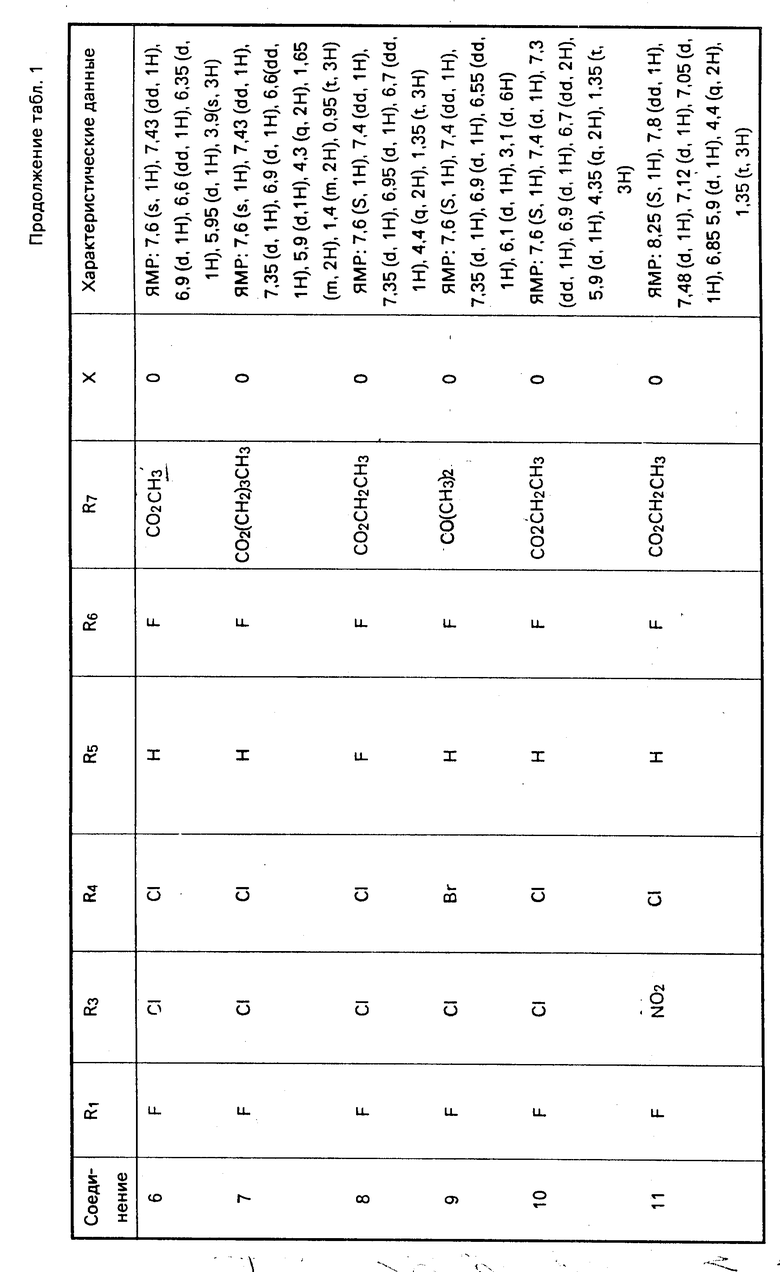
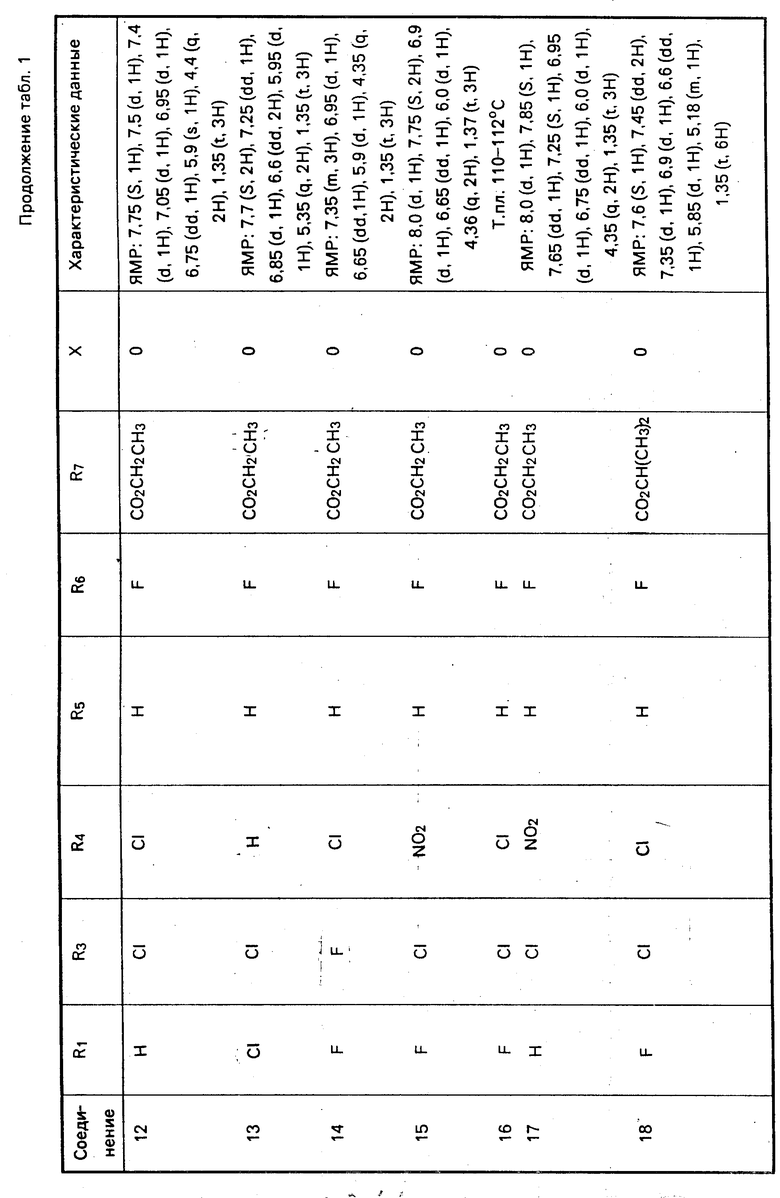
Характерные примеры соединений изобретения приводятся в табл. 1, как и другие соли и их цвиттер-ионы.
Дополнительным характерным примером является соединение формулы
F3C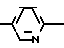 O
O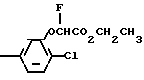 с характеризующими его данными ЯМР: 8,25 (s, 1H), 8,0 (d, 1H), 7,5 (d, 1H), 7,1 (d, 1H), 7,0 (dd, 1H), 5,8-6,05 (d, 1H), 4,4 (q, 2H), 1,4 (t, 3H).
с характеризующими его данными ЯМР: 8,25 (s, 1H), 8,0 (d, 1H), 7,5 (d, 1H), 7,1 (d, 1H), 7,0 (dd, 1H), 5,8-6,05 (d, 1H), 4,4 (q, 2H), 1,4 (t, 3H).
Соединения формулы I могут быть получены взаимодействием соединения формулы II в которой X, R1, R2 и R4 являются такими, как это определено для формулы I, с соединением формулы III
в которой X, R1, R2 и R4 являются такими, как это определено для формулы I, с соединением формулы III
Y- CR5R6R7 в которой R5, R6 и R7 являются такими же, как в формуле I, а Y является уходящей группой.
Предпочтительно реакция проводится в присутствии основания, такого как гидроокись натрия или карбонат калия.
Примеры уходящих групп для Y включают галоген, такой как хлор, бром или иод, мезилатную или тозилатную группу.
Реакция предпочтительно проводится в инертном органическом растворителе, таком как диметилсульфоксид, при температуре, например, 40-120оС. Затем, если это необходимо, могут быть проведены одна или более следующих стадий реакций:
а) когда R7 является алкоксикарбонильной группой, осуществляют гидролиз до соответствующей кислоты,
б) когда R7 является СООН, этерифицируют или переводят в соль, амидное, сульфонамидное, гидразидное или гидразинийпроизводное.
Соединения формулы II могут иметь гербицидную активность сами по себе и использование этих соединений в гербицибных формах является еще одним аспектом изобретения. Кроме того, ряд соединений формулы II являются новыми и это дополнительный аспект изобретения.
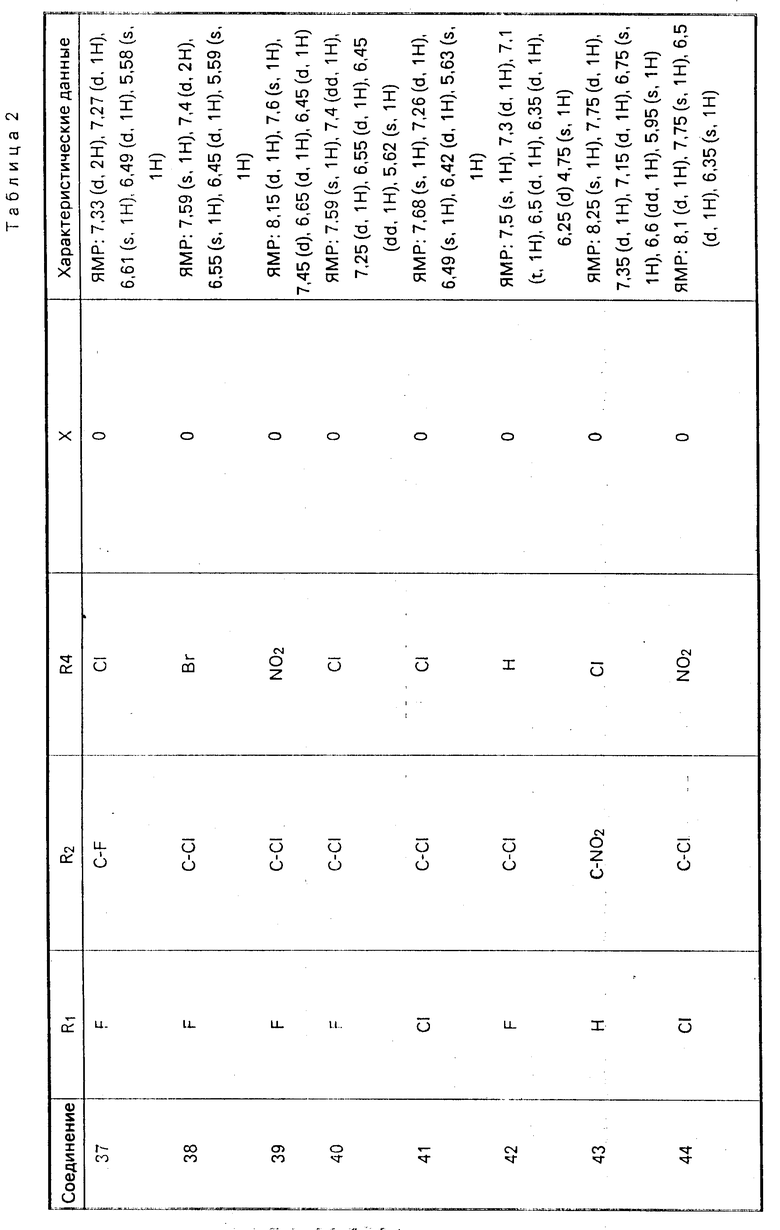
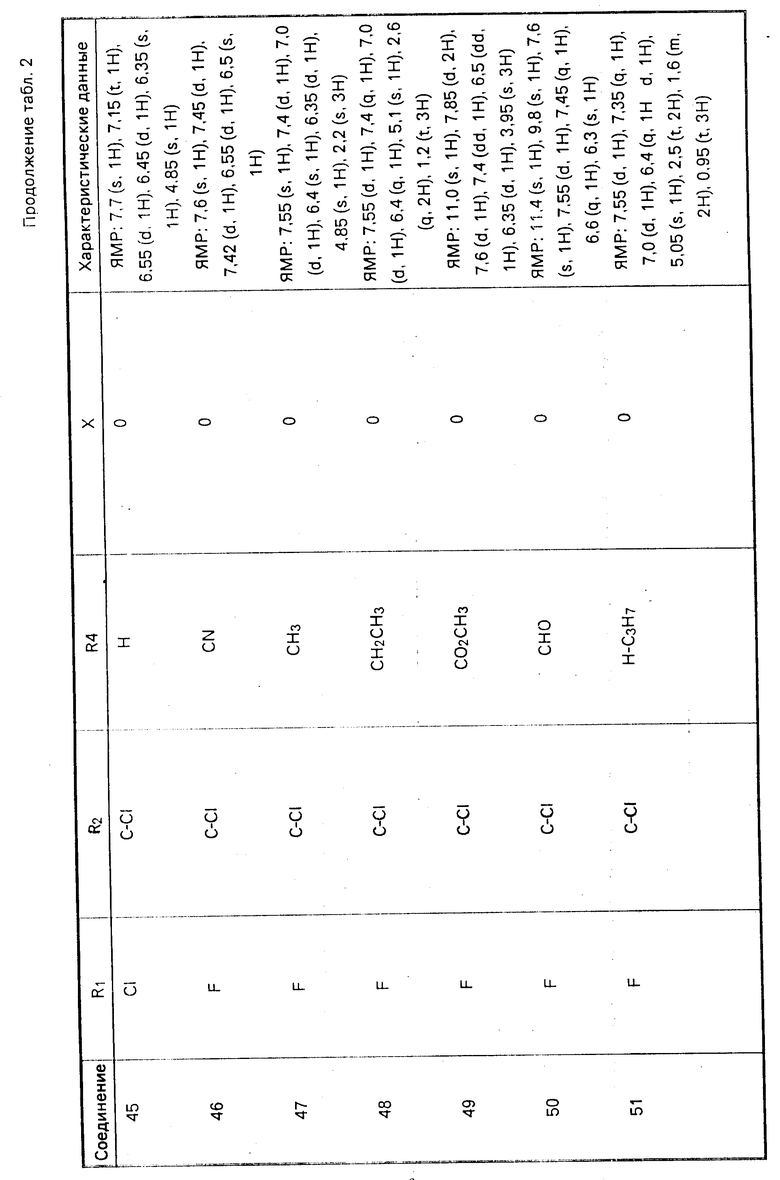
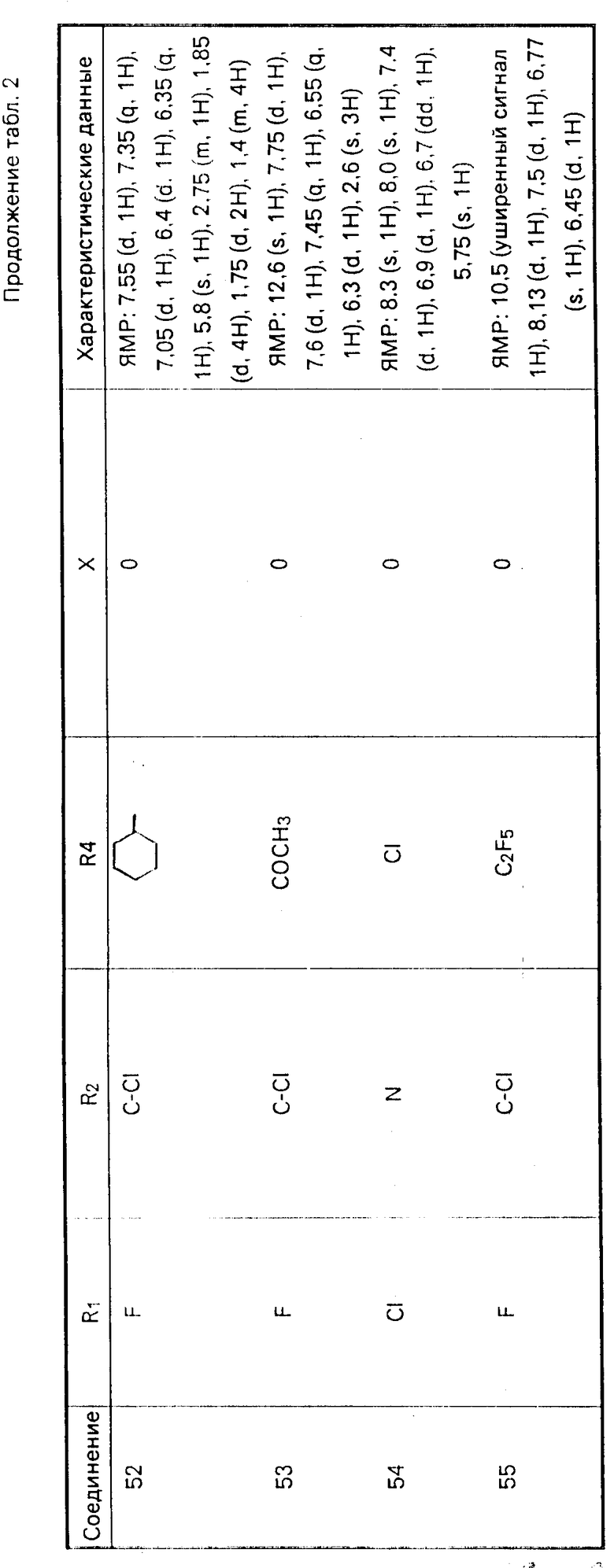
Примеры соединений формулы II представлены в табл. 2.
Соединения формулы II могут быть получены путем взамодействия соединения формулы IV
F3C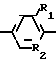 R16 в которой R1 и R2 являются такими, как определено для формулы I, а R16 является галогеном, предпочтительно фтором или хлором, с соединением формулы V
R16 в которой R1 и R2 являются такими, как определено для формулы I, а R16 является галогеном, предпочтительно фтором или хлором, с соединением формулы V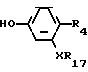 где Х и R4 являются такими, как определено для формулы I, а R17 является Н или защитной группой, предпочтительно алкилом, и после этого, если необходимо, удаляют любые защитные группы R17.
где Х и R4 являются такими, как определено для формулы I, а R17 является Н или защитной группой, предпочтительно алкилом, и после этого, если необходимо, удаляют любые защитные группы R17.
Реакция предпочтительно проводится в присутствии основания. Подходящие основания включают карбонат калия.
Реакция предпочтительно проводится в инертном органическом растворителе, таком как диметилсульфоксид, при температурах, например, 20-120оС, необязательно в инертной атмосфере.
Дополнительно соединения формулы I могут быть получены непосредственно путем взаимодействия соединения формулы IV с соединением формулы VI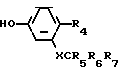 в которой Х, R4, R5, R6 и R7 являются такими, как определено для формулы I.
в которой Х, R4, R5, R6 и R7 являются такими, как определено для формулы I.
Условия реакции, применяемые при этом, должны быть одинаковыми с условиями, описанными для реакции соединения формулы IV с соединением формулы V.
Соединения формулы I или II, где R4 является нитрогруппой или галогеном, могут быть получены путем нитрования или галогенирования соответственно соединения формулы VII
F3C O
O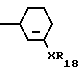 в которой R1, R2 и Х являются такими, как определено для формулы I, а R18 является водородом или защитной группой, такой, как алкил, или группой CR5R6R7, где R5, R6 и R7 являются такими, как определено для формулы I, и после этого, если необходимо, удаляют любые защитные группы R18.
в которой R1, R2 и Х являются такими, как определено для формулы I, а R18 является водородом или защитной группой, такой, как алкил, или группой CR5R6R7, где R5, R6 и R7 являются такими, как определено для формулы I, и после этого, если необходимо, удаляют любые защитные группы R18.
Реакция нитрования может быть проведена при обычно используемых условиях, например путем взаимодействия с нитрирующим агентом, таким как азотная кислота в серной кислоте или нитрат меди и уксусный ангидрид в уксусной кислоте.
Аналогичным путем может быть осуществлено галогенирование путем взаимодействия с галоидирующим агентом, таким как хлор или бром, например, в уксусной кислоте. Соединения формулы VII могут быть получены путем взаимодействия соединения формулы IV с соединением формулы VIII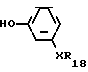 где Х является таким, как определено для формулы I, а R18 является такими, как опрделено для формулы VII. Условия реакции должны быть одинаковыми с условиями, описанными для реакции соединения формулы IV и соединения формулы V.
где Х является таким, как определено для формулы I, а R18 является такими, как опрделено для формулы VII. Условия реакции должны быть одинаковыми с условиями, описанными для реакции соединения формулы IV и соединения формулы V.
Удаление защитных групп R17 и R18 может быть осуществлено путем взаимодействия с кислотой, такой как бромистоводородная кислота в уксусной кислоте, или с хлоргидратом пиридина, или с трехбромистым бором.
Соединения формулы IV, V, VI и VIII являются известными соединениями или могут быть получены из известных соединения по обычным методикам.
Соединения формулы II, в которой R4 является галоидалкилом, могут быть получены путем взаимодействия соединения формулы II, в которой R4 является бромом или иодом, с соответствующим иодистым галоидалкилом при повышенных температурах в присутствии медного порошка.
Соединения формулы I, в которой R7 является СООН-группой, может быть получено гидролизом соответствующего сложного эфира, производные кислоты, например сложные эфиры, амиды, альдегиды, могут быть получены с помощью стандартных методик.
Соединения формулы I, где R5 или R6 являются хлором или бромом, а R7 является СООR8, могут быть получены галоидированием соответствующего соединения, у которого R5 и R6 - H. Подходящие реагенты включают N-бромсукцинимид или газообразный хлор в присутствии света или радикальных ингибиторов. Полученные сложные галоидэфиры могут быть превращены в другие производные кислоты по стандартным методикам.
Соединения изобретения способны регулировать рост широкого ряда растений, в частности некоторые показывают полезную селективность для таких культур, как рис, хлебные злаки, кукуруза, соя и сахарная свекла, тогда как другие показывают активность более широкого спектра. Они могут быть внесены в почву до появления растений (предвсходовая обработка) или могут быть нанесены на надпочвенные части растущих растений (послевсходовая обработка). Обычно в большинстве случаев соединения являются более активными при послевсходовой обработке. Изобретение также предлагает способ ингибирования роста неделательных растений путем нанесения на растения или на места расположения их соединения формулы I, как описано выше. Норма обработки, требуемая для ингибирования роста нежелательных растений, должна зависеть, например, от конкретно выбираемого для использования соединения формулы I и от конкретного вида растений, рост которого необходимо контролировать. Обычное количество 0,01-5,0 кг/га, предпочтительно 0,025-2 кг/га.
Следующие примеры иллюстрируют способ получения соединений формулы I.
П р и м е р 1. Иллюстрирует получение соединения 40 в табл. 2.
3-Хлор-4,5-дифторбензотрифторид (1,08 г) растворяют в сухом диметилсульфоксиде (15 мл) и по частям добавляют 4-хлоррезорцин (0,72 г), после чего прибавляют безводный карбонат калия (1,38 г). Реакционную смесь перемешивают и нагревают приблизительно до 60оС в течение 4 ч. Затем содержимое оставляют стоять при комнатной температуре в течение двух дней, впоследствии выливают в избыток воды и подкисляют с помощью разбавленной соляной кислоты.
После двух экстракций в этилацетата объединенный экстракт промывают водой и солевым раствором и высушивают над сульфатом магния. Концентрирование дает соединение 40 в виде коричнево-оранжевого масла (0,42 г).
Соединения 37, 38, 41, 43 и 54 получают аналогичными методами, используя соответствующие реагенты.
П р и м е р 2. Иллюстрируют получение соединения I в табл. 1.
Соединение 40, полученное по описанному методу примера 1 (5 г) растворяют в сухом диметилсульфоксиде (50 мл) и добавляют карбонат калия (2,1 г). Затем добавляют этиловый эфир хлорфторуксусной кислоты (2,1 г) в диметилсульфоксиде (10 мл).
Смесь перемешивают и нагревают при 90оС в течение 5 ч, охлаждают и оставляют стоять при комнатной температуре в течение ночи, выливают в избыток воды и подкисляют с помощью 2М раствора соляной кислоты. После экстракции в диэтиловом эфире экстракт промывают водой, высушивают и концентрируют до получения соединения I в виде масла (5,73 г).
Соединения 3, 11, 12, 13, 14, 16, 25, 26, 27, 28, 29, 30, 34 и 36 получают аналогичными методами, используя соответствующие реагенты.
П р и м е р 3. Иллюстрируют получение соединения 2 в табл. 1.
Стадия а.
3-Метоксифенол (2,48 г) растворяют в диметилфорамиде (20 мл) и медленно добавляют гидрид натрия (0,48 г). Реакционную смесь перемешивают при комнатной температуре в течение 0,5 ч. Раствор 3-хлор-4,5-дифторбензотрифторида (4,32 г) в диметилформамиде (10 мл) прибавляют затем по каплям. Реакционную смесь перемешивают при комнатной температуре в течение 3 ч и в дальнейшем выливают в смесь льда с водой и подкисляют с помощью разбавленной соляной кислоты.
После экстракции диэтиловым эфиром экстракт промывают водой, высушивают и концентрируют, чтобы получить соединение формулы
F3C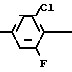 O
O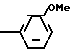 в виде масла (5,49 г).
в виде масла (5,49 г).
Стадия б.
Образец масла, полученного в стадии а (10,01 г) растворяют в уксусном ангидриде (60 мл), перемешивают и по каплям с перемешиванием прибавляют раствор нитрата меди (II) (7,45 г) в ледяной уксусной кислоте. После дополнительного перемешивания при комнатной температуре в течение 5 ч реакционную смесь выливают в избыток воды и экстрагируют диэтиловым эфиром. Экстракт промывают, сушят и концентрируют, чтобы получить масло, которое при растирании с пентаном дает желтое твердое вещество (8,05 г) формулы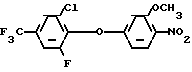
т.пл. 100-103оС
Стадия в.
Описывают получение соединения 39 в табл. 2.
Соединение, полученное в стадии б (3,66 г), растворяют в сухом дихлорметане (20 мл), охлаждают в бане с твердым углекислым газом и изопропиловым спиртом до -60оС и помещают в атмосферу азота. Раствор трехбромистого бора (2,51 г) в дихлорметане прибавляют медленно по каплям, обеспечивая температуру не выше -50оС. Как только добавление завершается, реакционной смеси позволяют достигнуть комнатной температуры, а затем выливают в избыток смеси льда с водой и экстрагируют дихлорметаном. Экстракт сушат и концентрируют, чтобы получить темное вязкое масло, которое при перегонке под высоким вакуумом дает оранжевое масло, которое превращается в твердую массу при стоянии, что дает соединение 39 (2,91 г) в виде желтого твердого вещества.
Стадия г.
Соединение номер 39 (0,5 г), полученное по описанной в стадии в методике, добавляют к гидриду натрия (0,04 г), суспендированному в ДМФА (1 мл). Раствор этилового эфира хлорфторуксусной кислоты (0,2 г) в ДМФА (1 мл) добавляют по каплям.
Смесь перемешивают и нагревают при 100оС в течение 6 ч, выливают в ледяную воду, подкисляют разбавленной соляной кислотой и экстрагируют диэтиловым эфиром. После экстракции экстракт промывают, сушат и концентрируют, чтобы получить масло, которое затем разделяют, используя тонкослойную хроматографию (ТСХ) с помощью элюента из диэтилового эфира 60; гексана 40; уксусной кислоты 5. Средний слой извлекают и соединение 2 получают в виде вязкого масла (0,157 г).
Соединения 4,15 и 17 получают аналогичными методами, используя соответствующие реагенты.
П р и м е р 4. Иллюстрирует получение соединения 5 в табл. 1.
Образец соединения 1 (4,17 г), полученного, как описано в примере 2, растворяют в изопропиловом спирте (60 мл) и по каплям добавляют к смеси 1,1 М раствор гидроокиси натрия в воде (10 мл). Реакционную смесь нагревают с обратным холодильником при 100оС в течение 4 ч. После охлаждения и стояния в течение ночи смесь выливают в избыток воды и подкисляют затем разбавленной соляной кислотой. После двух экстракций в диэтиловом эфире объединенный экстракт промывают, сушат и концентрируют до получения масла. Растирание в порошок с помощью пентана, фильтрация и высушивание дают соединение 5 (2,115 г) в виде не совсем белого твердого вещества.
П р и м е р 5. Иллюстрирует получение соединения 6 в табл. 1.
Образец соединения 5, полученного, как описано в примере 4 (0,63 г), растворяют в чистом для анализа метаноле (15 мл) и добавляют несколько капель концентрированной серной кислоты. Реакционную смесь перемешивают и нагревают с обратным холодильником в течение 5 ч. Затем смесь оставляют стоять при комнатной температуре в течение ночи, концентрируют на роторном выпаривателе, разбавляют водой и экстрагируют в диэтиловом эфире. Экстракт промывают, сушат и концентрируют, чтобы получить масло, которое затем разделяют, используя ТСХ с разбавителем из гексана 50, диэтилового эфира 30, уксусной кислоты 5. Соединение 6 выделяют в виде вязкого желтого масла (0,471 г).
Соединения 7, 18 и 19 получают по аналогичным методам, используя соответствующие реагенты.
П р и м е р 6. Иллюстрирует получение соединение 8 в табл. 1.
Соединение 40 (0,51 г), полученное, как описано в примере 1, растворяют в сухом ДМФА (10 мл) и добавляют затем гидрид натрия (0,04 г), за которым следует прибавление раствора этилового эфира бромдифторуксусной кислоты (0,31 г) в ДМФА (2 мл). Реакционную смесь оставляют стоять при комнатной температуре в течение ночи, а затем выливают в воду, подкисляют 2М раствором соляной кислоты и экстрагируют диэтиловым эфиром. Экстракт промывают, сушат и концентрируют, чтобы получить вязкое оранжево-коричневое масло, которое разделяют, используя ТСХ спомощью элюента из диэтилового эфира 30, гексана 60, уксусной кислоты 5. Соответствующий слой отделяют и соединение 8 получают в виде масла (0,082 г).
П р и м е р 7. Иллюстрирует получение соединения 9 в табл. 1.
Образец соединения 5 (0,5 г), полученного как описано в примере 4, растворяют в хлористом тиониле (10 мл) и смесь нагревают с обратным холодильником в течение 1,5 ч. Затем смесь концентрируют и подвергают азеотропной перегонке с толуолом. Полученное масло растворяют в сухом толуоле (15 мл) и через раствор пропускают диметиламин в течение 5 мин. Реакционную смесь перемешивают в течение 2 ч и оставляют при комнатной температуре в течение ночи. Смесь концентрируют, добавляют воду и экстрагируют этилацетатом. Экстракт промывают, сушат и концентрируют, чтобы получить масло, которое разделяют, используя ТСХ с помощью разбавителя из диэтилового эфира 30, гексана 60, уксусной кислоты 5. Соединение 9 выделяют в виде масла (0,385 г).
Соединения 20, 21 и 24 получают аналогичными методами, используя соответствующие реагенты.
П р и м е р 8. Иллюстрирует получение соединения 42 в табл. 2.
Образец масла (2,21 г), полученного, как описано в стадии а примера 3, растворяют в дихлорметане (15 мл) и охлаждают до -70оС. По каплям прибавляют 1 М раствор трехбромистого бора в дихлорметане (10 мл). Затем, когда добавление завершится, реакционную смесь оставляют до достижения комнатной температуры и после этого перемешивают в течение 4 ч. После оставления на ночь реакционную смесь добавляют ко льду и органический раствор разбавляют большим количеством дихлорметана. Органический раствор промывают водой, сушат над сульфатом магния и концентрируют, чтобы получить соединение 42 в виде масла (3,195 г).
П р и м е р 9. Иллюстрирует получение соединения 10 в табл. 1.
Образец соединения 42, полученного, как описано в примере 8 (0,92 г), растворяют в сухом диметилсульфоксиде (15 мл), добавляют безводный карбонат калия (0,83 г) и реакционную смесь перемешивают. По каплям добавляют раствор этилового эфира хлорфторуксусной кислоты (0,42 г) в сухом диметилсульфоксиде и реакционную смесь перемешивают и нагревают до 100оС в течение 3 ч, затем выдерживают в течение ночи. Реакционную смесь разбавляют водой и подкисляют разбавленной соляной кислотой. После экстракции диэтиловым эфиром экстракт промывают, сушат и концентрируют, чтобы получить соединение 10, которое очищают препаративной ТСХ, используя диэтиловый эфир 30, гексан 60, уксусную кислоту 5, в качестве элюента, чтобы получить продукт реакции в виде вязкого зелено-желтого масла (0,232 г).
П р и м е р 10. Иллюстрирует получения соединения 46 в табл. 2.
Стадия а.
2,4-Диметоксибензонитрил (2 г) смешивают с хлористым пиридинием (5,67 г) и реакционную смесь расплавляют при 210оС в течение 2 ч. После этого смесь оставляют стоять при комнатной температуре в течение ночи, а затем ее разбавляют водой и экстрагируют диэтиловым эфиром. Эфирные экстракты промывают водой, сушат над сульфатом магния и концентрируют, чтобы получить розовое твердое вещество, которое промывают бензином и сушат воздухом, чтобы получить 2,4-диоксибензонитрил (1,17 ч), т.пл. 176-178оС.
Стадия б.
Продукт реакции из стадии а растворяют в сухом диметилсульфоксиде (15 мл) и добавляют карбонат калия (3,59 г). Смесь перемешивают в течение 0,5 ч и затем добавляют раствор 3-хлор-4,5-дифторбензофрифторида (1,88 г) в диметилсульфоксиде (5 мл). Реакционную смесь нагревают приблизительно до 100оС и перемешивают в течение 3 ч. После этого смесь оставляют стоять в течение ночи, а затем выливают ее в избыток воды, подкисляют 2М раствором соляной кислоты и экстрагируют диэтиловым эфиром. Эфирные экстракты промывают, сушат и концентрируют, чтобы получить масло. Разделяют, используя препаративную ТСХ с помощью элюента диэтиловый эфир : гексан : уксусная кислота в соотношении 30 : 60 : 5, получают соединение 46, т.пл. 142-144оС.
П р и м е р 11. Иллюстрирует получение соединения 23 в табл. 1.
Соединение 46, полученное, как описано в примере 10 (0,663 г), растворяют в сухом диметилсульфоксиде (10 мл) и добавляют безводный карбонат калия (0,41 г). Смесь перемешивают в течение 0,5 ч и добавляют раствор этилового эфира хлорфторуксусной кислоты (0,281 г) в диметилсульфоксиде (3 мл). Добавляют каталитическое количество иодистого калия и реакционную смесь перемешивают и нагревают в течение 4 ч приблизительно при 80оС. Смесь оставляют на 48 ч, а затем выливают в избыток воды и подкисляют 2М раствором соляной кислоты. Смесь экстрагируют диэтиловым эфиром и экстракты промывают, высушивают и концентрируют, чтобы получить масло. Разделяют с помощью препаративной ТСХ, используя элюент диэтиловый эфир : гексан : уксусная кислота в соотношении 30 : 60 : 5, получают соединение 23 (0,437 г) в виде масла. Структура подтверждается ЯМР- и ИК-спектроскопией.
П р и м е р 12. Иллюстрирует получение соединения 22 в табл. 1.
Образец соединения 5, полученного, как описано в примере 4 (0,63 г), растворяют в сухом дихлорметане (10 мл) и добавляют трет-бутанол (0,22 г), за которым следует прибавление 4-диметиламинопиридина (0,04 г). Смесь охлаждают до 0оС и добавляют раствор N,N-дициклогексилкарбодиимида (0,31 г) в дихлорметане (5 мл). Реакционную смесь перемешивают в течение 30 мин, позволяют достигнуть комнатной температуры и перемешивают в течение дополнительных 4 ч. После стояния в течение ночи реакционную смесь фильтруют и фильтрат концентрируют до получения маслянистого остатка. Остаток разделяют, используя ТСХ с помощью разбавителя из гексана 60, диэтилового эфира 30, уксусной кислоты 5. Передний (верхний) слой разделяют и соединение 22 получают в виде масла (0,244 г).
П р и м е р 13. Иллюстрирует получение соединения 47 в табл. 2.
Гидрид натрия (0,24 г 55%-ного) промывают бензином, затем перемешивают в ДМФА (10 мл). Суспензию охлаждают в бане со льдом и водой. Раствор 2-метилрезорцина (0,6 г) и 3-хлор-4,5-дифторбензотрифторида (0,8 г) в ДМФА (10 мл) добавляют по каплям в атмосфере азота в течение 30-минутного периода времени. Реакционную смесь выливают затем на смесь льда и 2М раствора соляной кислоты. Смесь быстро экстрагируют эфиром, промывают и сушат. После фильтрации и концентрирования получают светло-желтое масло. Масло разделяют, используя ТСХ с помощью элюента из эфира 60, гексана 40, уксусной кислоты 5. Слой при РГ 6,5 удаляют и экстрагируют, чтобы получить соединение 47 в виде светло-желтого масла (0,2 г).
Соединения 48, 49, 50, 51, 52 и 53 получают аналогичными методами, используя соответствующие реагенты.
П р и м е р 14. Иллюстрирует получение соединения 31 в табл. 1.
Стадия а.
Соединение 40, полученое, как описано в примере 1 (3,4 г), растворяют в сухом диметилсульфоксиде (ДМСО) (30 мл) и добавляют метиловый эфир бромуксусной кислоты (1,6 г), а затем прибавляют карбонат калия (3,5 г). Смесь нагревают до 75оС и перемешивают в течение 4 ч. После охлаждения до комнатной температуры реакционную смесь выливают на лед и разбавленную соляную кислоту. Полученную смесь экстрагируют диэтиловым эфиром. Экстракт промывают, высушивают, фильтруют и концентрируют, чтобы получить соединение формулы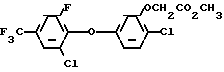 в виде оранжевого масла (3,5 г).
в виде оранжевого масла (3,5 г).
ЯМР: 7,6 (s, 1H), 7,4 (d, 1H), 7,3 (d, 1H), 6,55 (s, 1H), 6,35 (d, 1H), 4,7 (s, 2H), 3,8 (s, 3H).
Продукт реакции из стадии а (1 г) растворяют в четыреххлористом углероде. Добавляют N-бромсукцинимид (0,8 г) и смесь перемешивают и нагревают с обратным холодильником в течение 6 ч. Смесь фильтруют, чтобы удалить светло-желтое твердое вещество. Фильтрат выпаривают, чтобы получить соединение 31 в виде желтого масла (0,35 г).
Соединения 32 и 33 получают аналогичными методами, используя соответствующие реагенты.
Биологические данные.
Эти данные иллюстрируют гербицидные свойства соединений табл. 1 и 2. Соединения были подвергнуты гербицидным испытаниям, как описано ниже.
Каждое соединение при соответствующей концентрации вводят в 4%-ную эмульсию метилциклогексанона и 4%-ную смесь из 3,6 ч. Твина 20 и 1 ч. Спана 80. Твин 20 является торговой маркой для поверхностно-активного вещества, включающего конденсат 20 мол. ч., этиленоксида с сорбитан-лауратом. Спан 80 является торговой маркой для поверхностно-активного вещества, включающего сорбитан-монолаурат. Составление состава осуществляют растворением соединения в требуемом количестве смеси растворителя и поверхностно-активнго вещества и разбавлением водой до конечного разбрызгиваемого объема 45 мл. Если соединение не растворяется, добавляют стеклянные бусы, общий объем жидкости достигает 5 мл, вместе с водой и смесь встряхивают, чтобы осуществить диспергирование соединения. Состав, полученный таким образом, после удаления бусинок разбавляют затем водой до конечного разбрызгиваемого объема (45 мл).
Разбрызгиваемыми композициями, полученными таким образом, опрыскивают на горшочную рассаду (послевсходовая обработка) при норме, эквивалентной 1000 л/га. Повреждение растений оценивают спустя 13 дней после опрыскивания по сравнению с необработанными растениями по шкале от 0 до 5, где 0 означает 0-10% -ное повреждение; 1 - 11 - 25%-ное повреждения; 2 - 26-50%-ное повреждение; 3 - 51-80%-ное повреждение; 4 - 81-95%-ное повреждение; 5 - 96-100%-ное повреждение.
При испытании, проводимом для определения предвсходовой гербицидной активности, семена испытуемого вида помещают на поверхность пластмассовых лотков с компостом и опрыскивают композициями при норме 1000 л/га. Семена покрывают затем дополнительным компостом. Спустя 20 дней после опрыскивания всходы в опрыскиваемых пластмассовых лотках сравнивают со всходами в необработанных контрольных лотках, причем повреждение оценивается по той же самой шкале от 0 до 5.
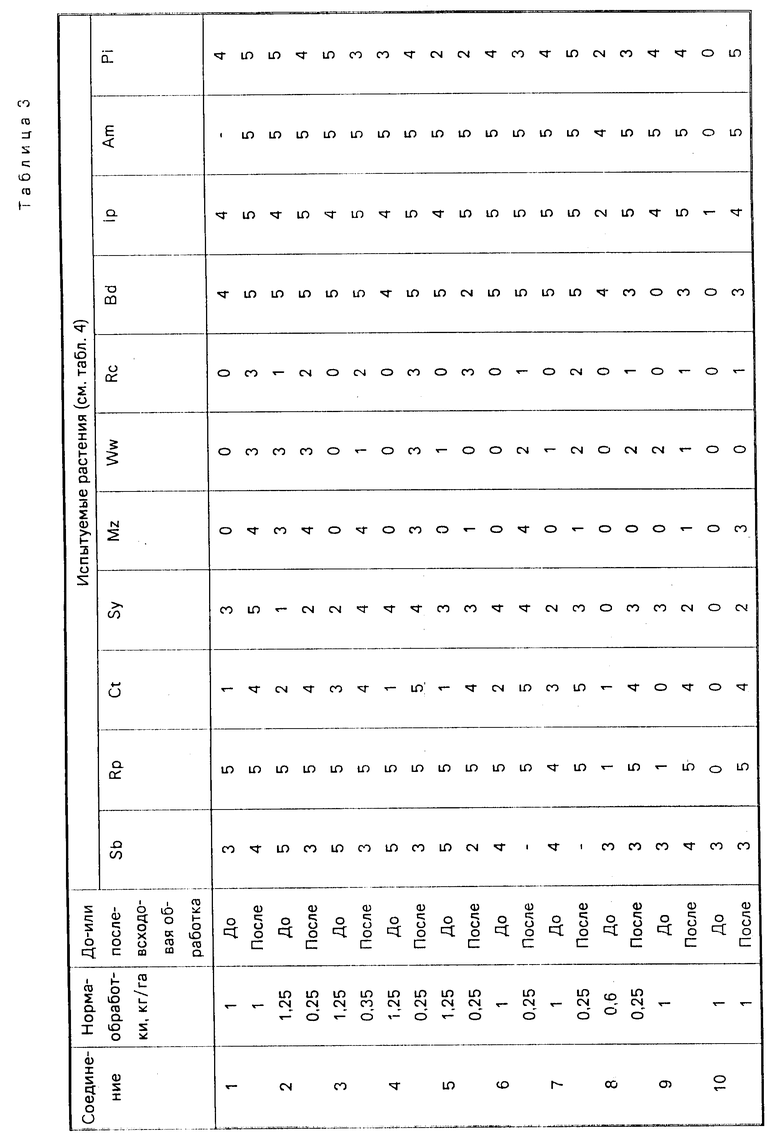
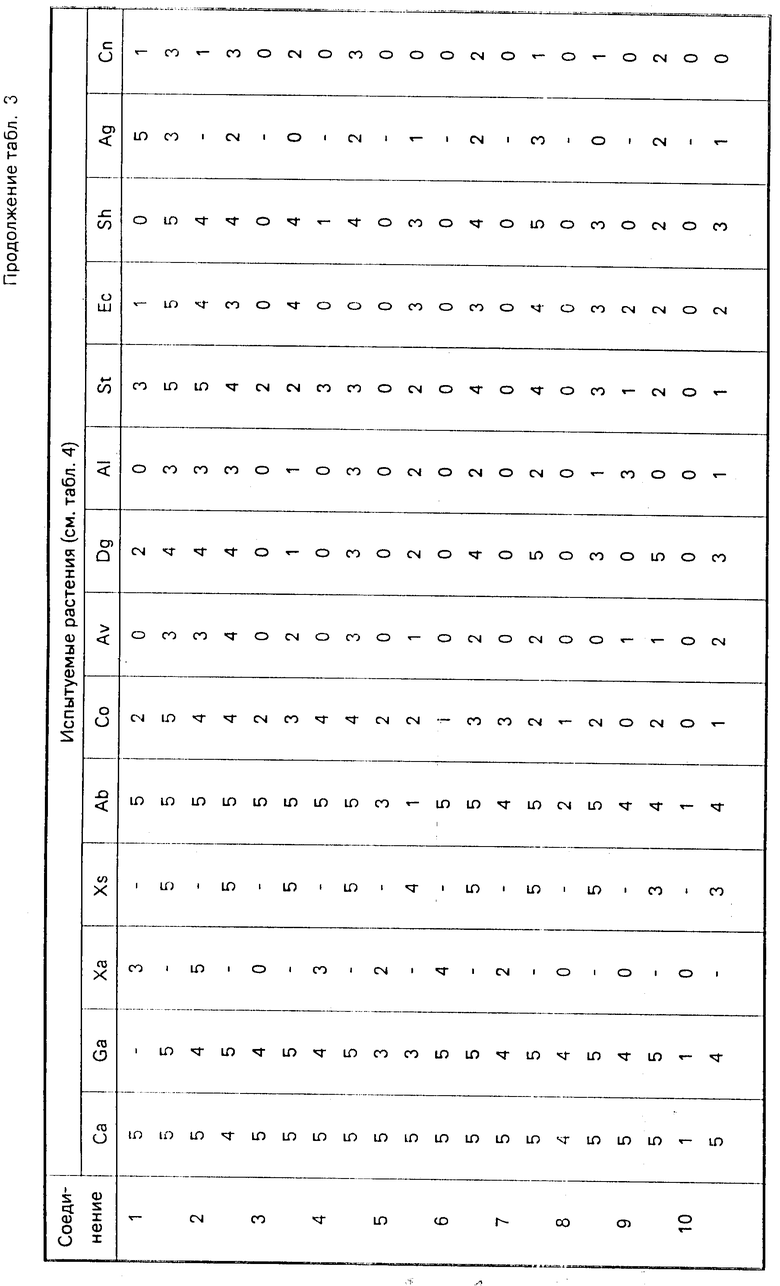
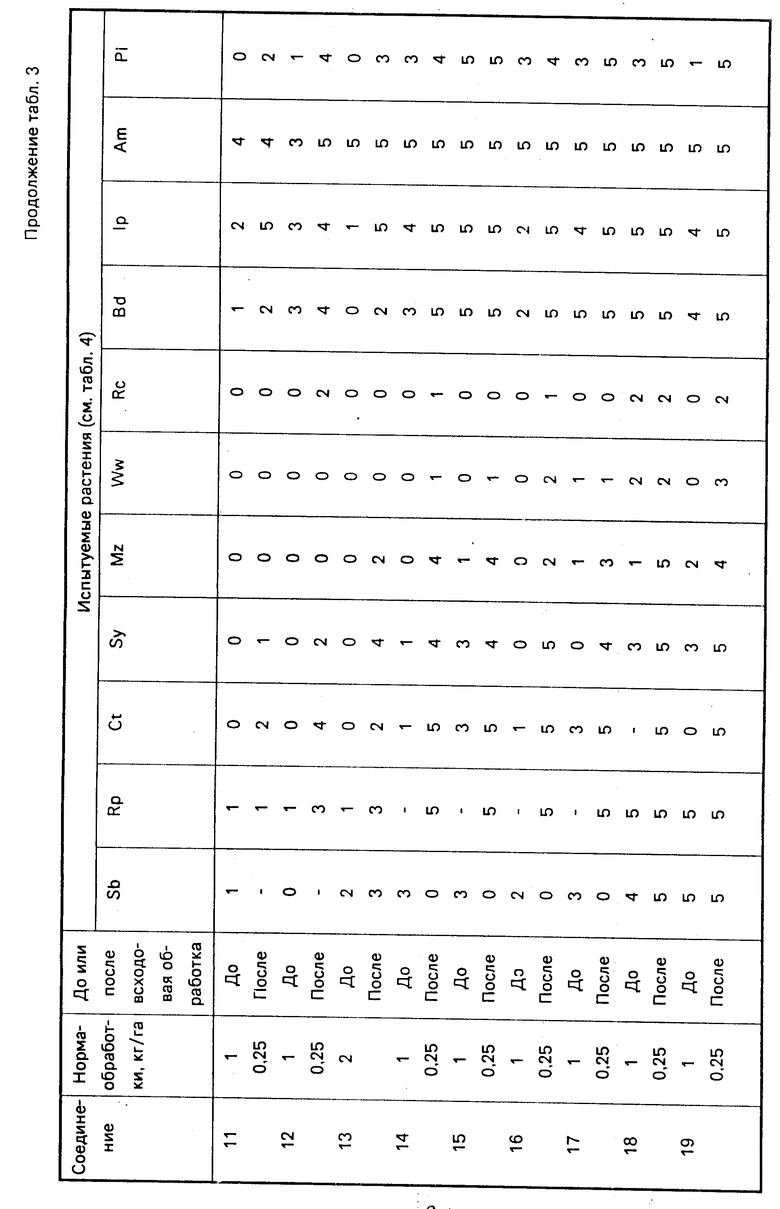
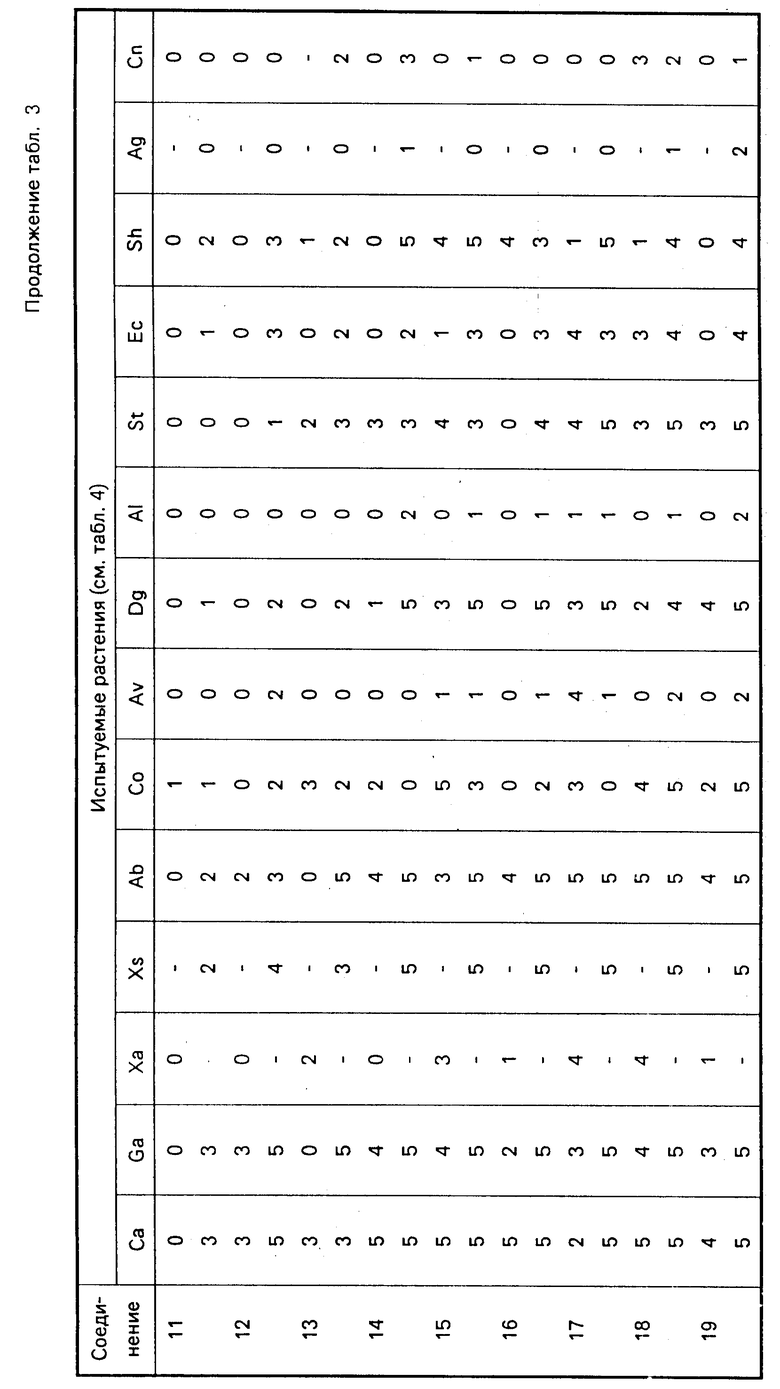
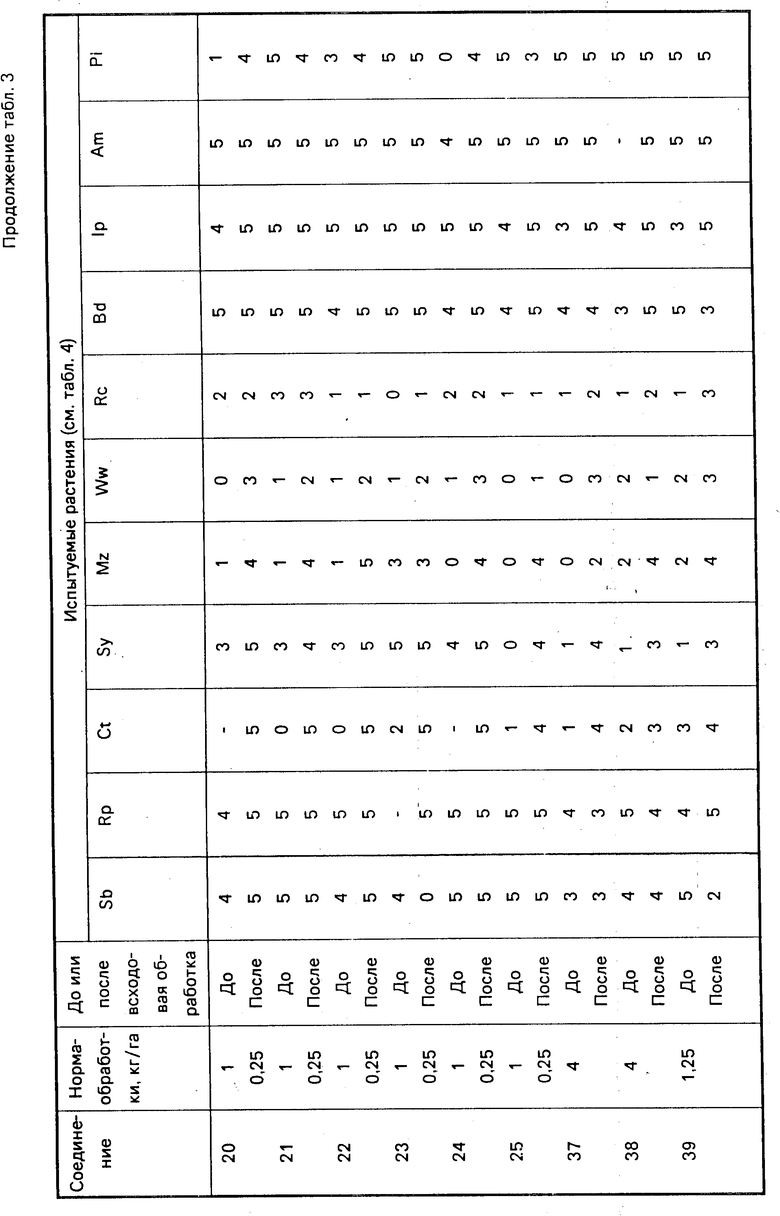
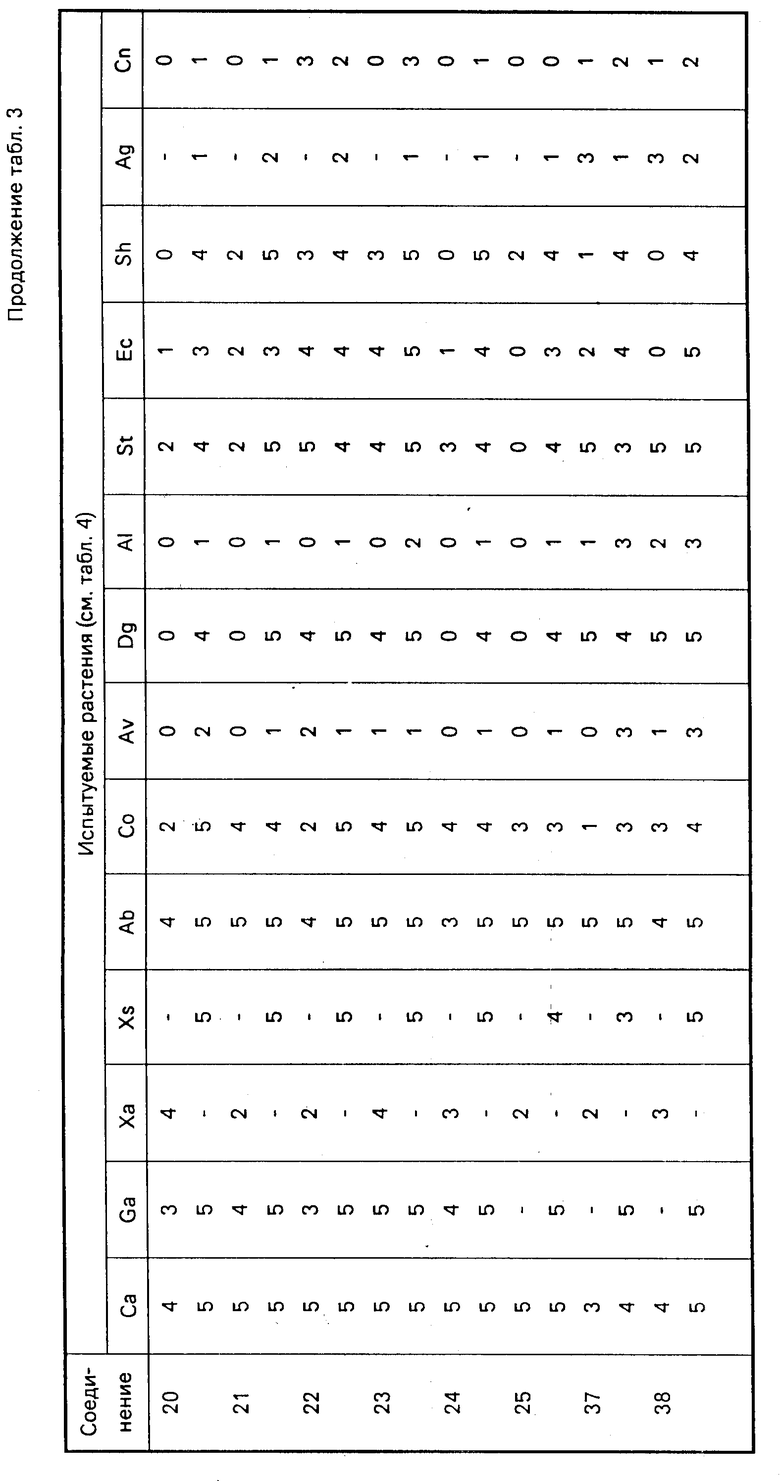
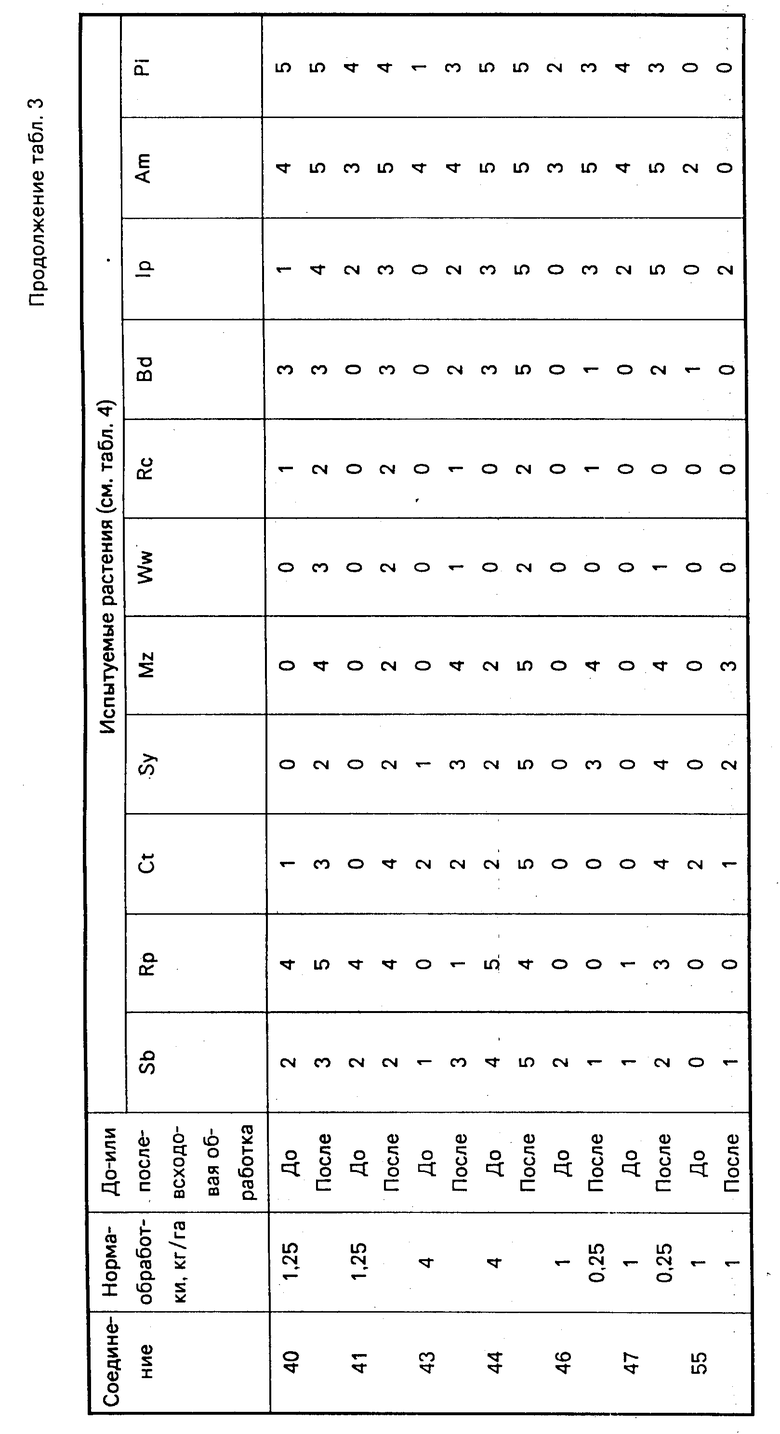
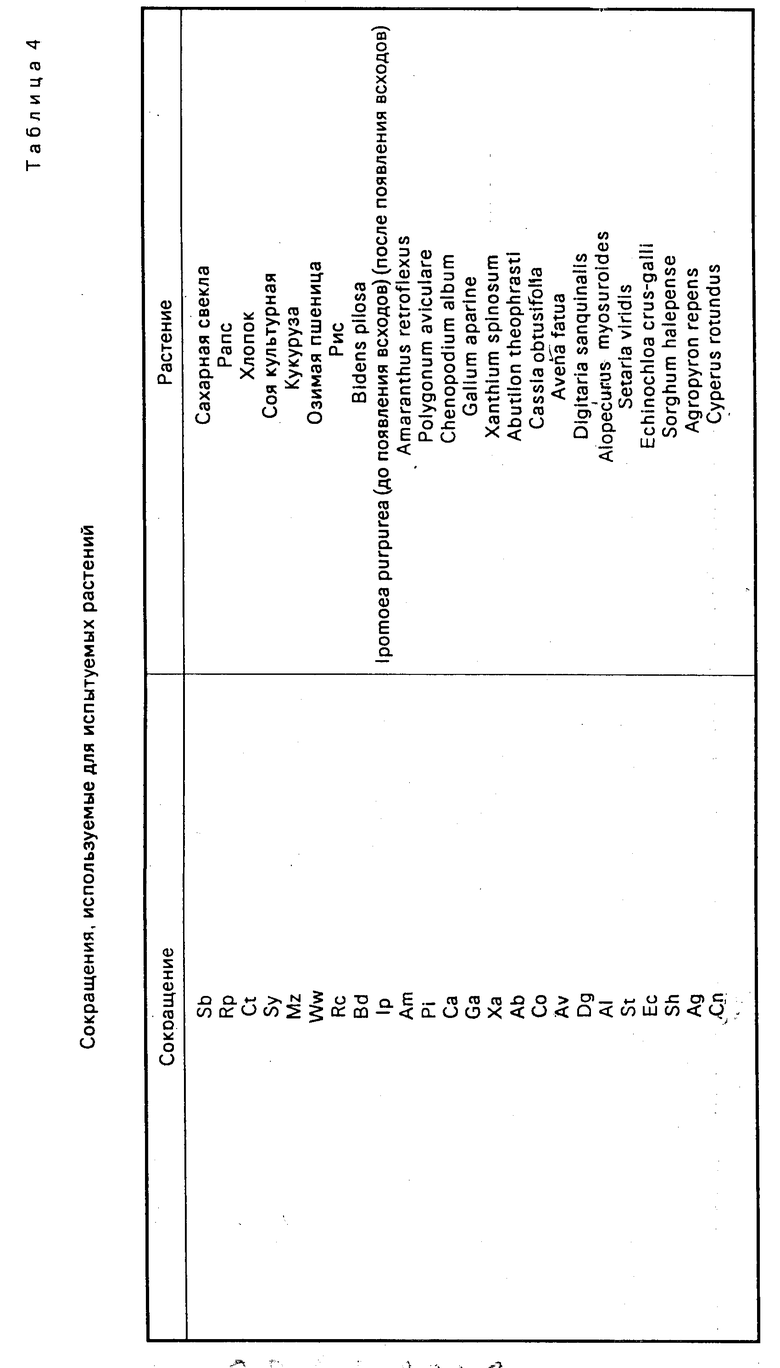
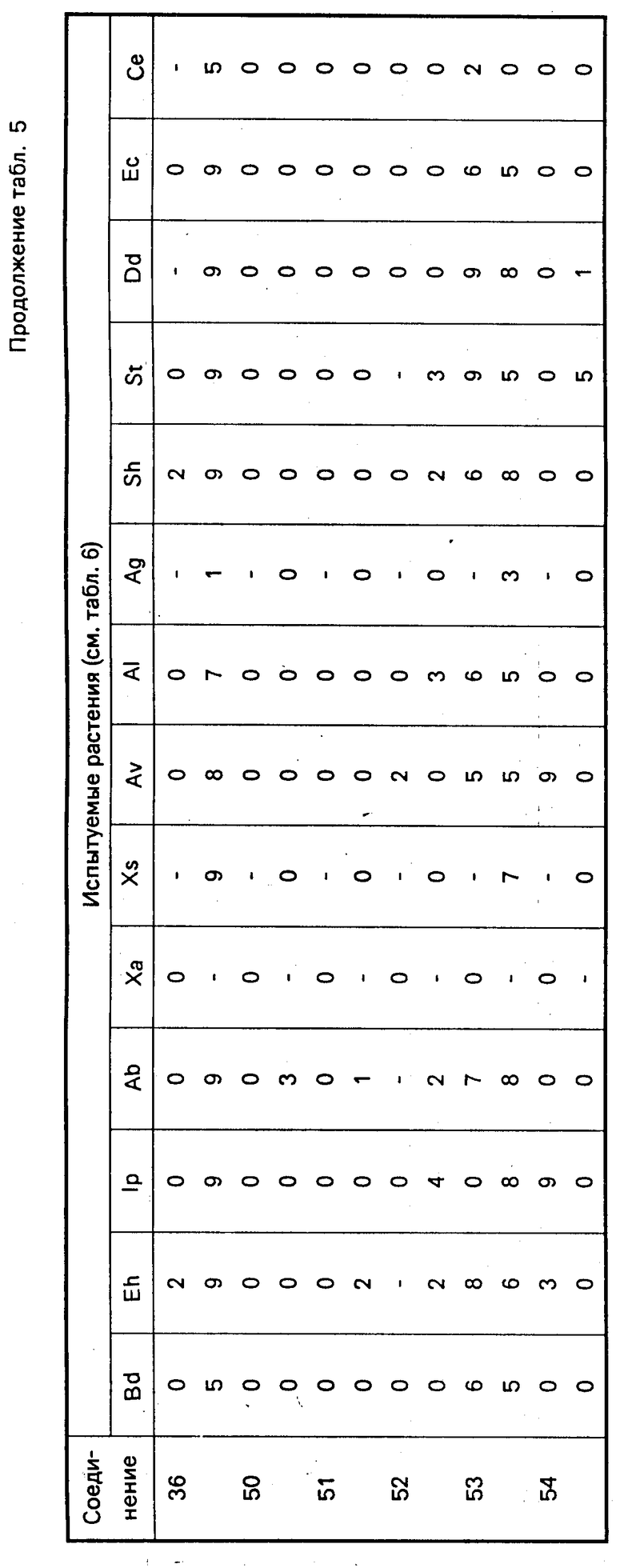
Результаты испытаний представлены ниже в табл. 3, расшифровка названий испытуемых растений см. в табл. 4.
Гербицидная активность некоторых соединений испытана альтернативным методом следующим образом.
Каждое соединение при соответствующей концентрации вводят в 4%-ную эмульсию метилциклогексанона и 0,4% -ную смесь и 3,6 ч. Твина 20 и 1 ч. Спана 80. Составление состава осуществляют путем растворения соединения в необходимом количестве смеси растворителя и поверхностно-активного вещества и разбавления водой до конечного объема разбрызгивания 45 мл. Если соединение нерастворимо, добавляют стеклянные бусинки, общий объем жидкости достигает 5 мл вместе с водой и смесь встряхивают до образования дисперсии соединения. Полученный таким образом состав после удаления бусинок разбавляют затем водой до конечного объема разбрызгивания (45 мл).
Композициями для разбрызгивания, полученными таким образом, опрыскивают горшочную рассаду (послевсходовая обработка) при норме, эквивалентной 1000 л/г. Повреждение у растений оценивают спустя 13 дней после опрыскивания по сравнению с необработанными растениями по шкале от 0 до 9, где 0 означает 0% -ное повреждение; 1 - 1-5%-ное повреждение; 2 - 6-15%-ное повреждение; 3 - 16-25% -ное повреждение; 4 - 26-35%-ное повреждение; 5 - 36-59%-ное повреждение; 6 - 60-69%-ное повреждение; 7 - 70-79%-ное повреждение; 8 - 80-89%-ное повреждение; 9 - 90-100%-ное повреждение.
При испытании, проводимом для определения предвсходовой гербицидной активности, семенная культура засевается на глубину 2 см (то есть Sb, Ct, Rp, Ww, Rc, Sy), а семена сорных растений на глубину 1 см под компост и опрыскивают композициями при норме 1000 л/га. Спустя 20 дней после опрыскивания всходы в опрысканных пластмассовых лотках сравнивают с всходами в необработанных контрольных лотках, причем повреждение оценивается по той же самой шкале от 0 до 9.
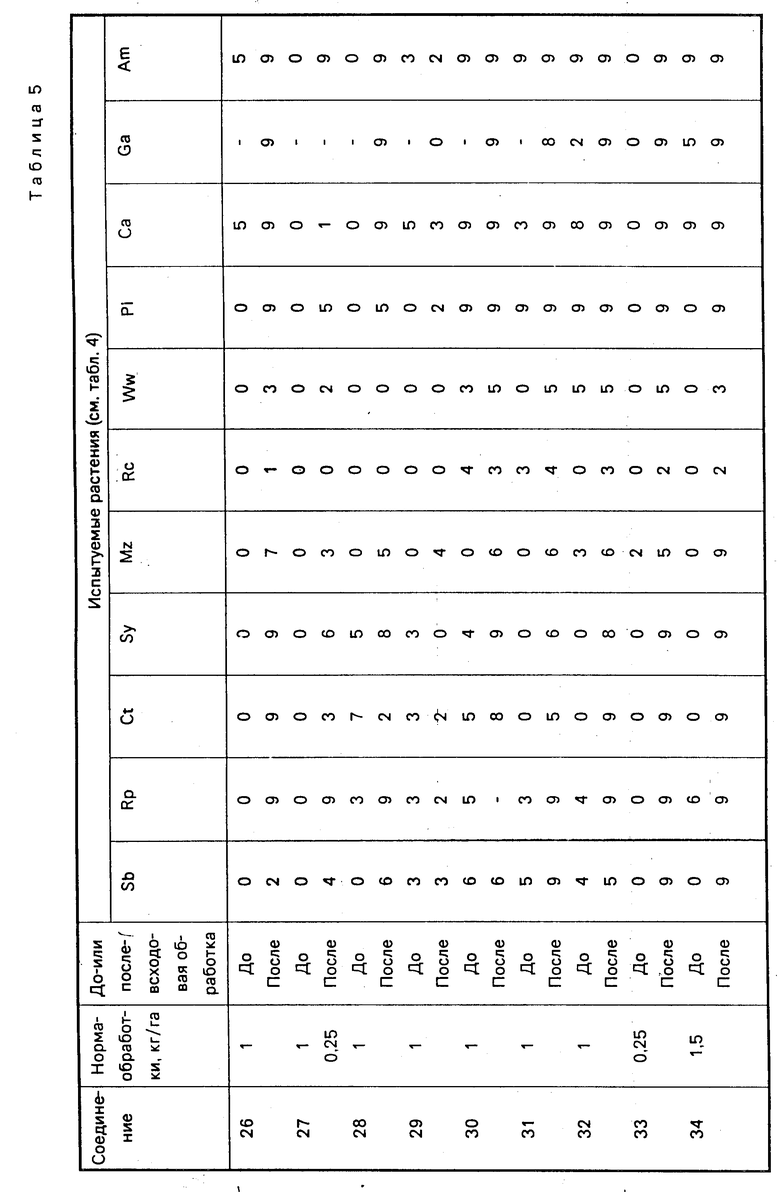
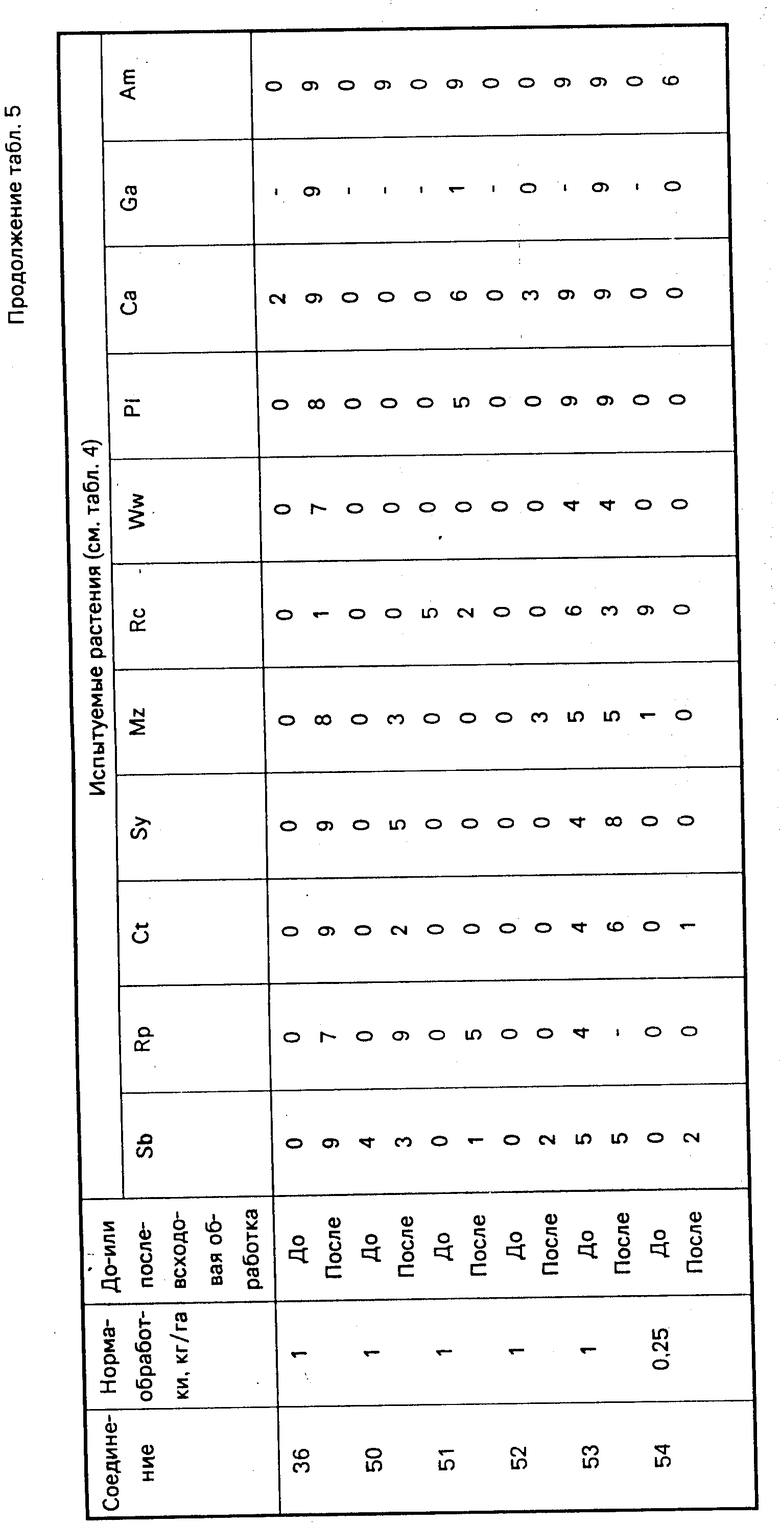
Результаты испытаний представлены ниже в табл. 5, расшифровка названий испытуемых растений дана в табл. 6.
Следующие соединения испытывали на сорняках, перечисленных ниже:
Соединение А - известное соединение
соединение В - соединение 17 настоящей патентной заявки
Am - Amaranthus retroflexus
Pi - Polygonym aviculare
Ca - Chenopodium album
Ga - Galium aparine
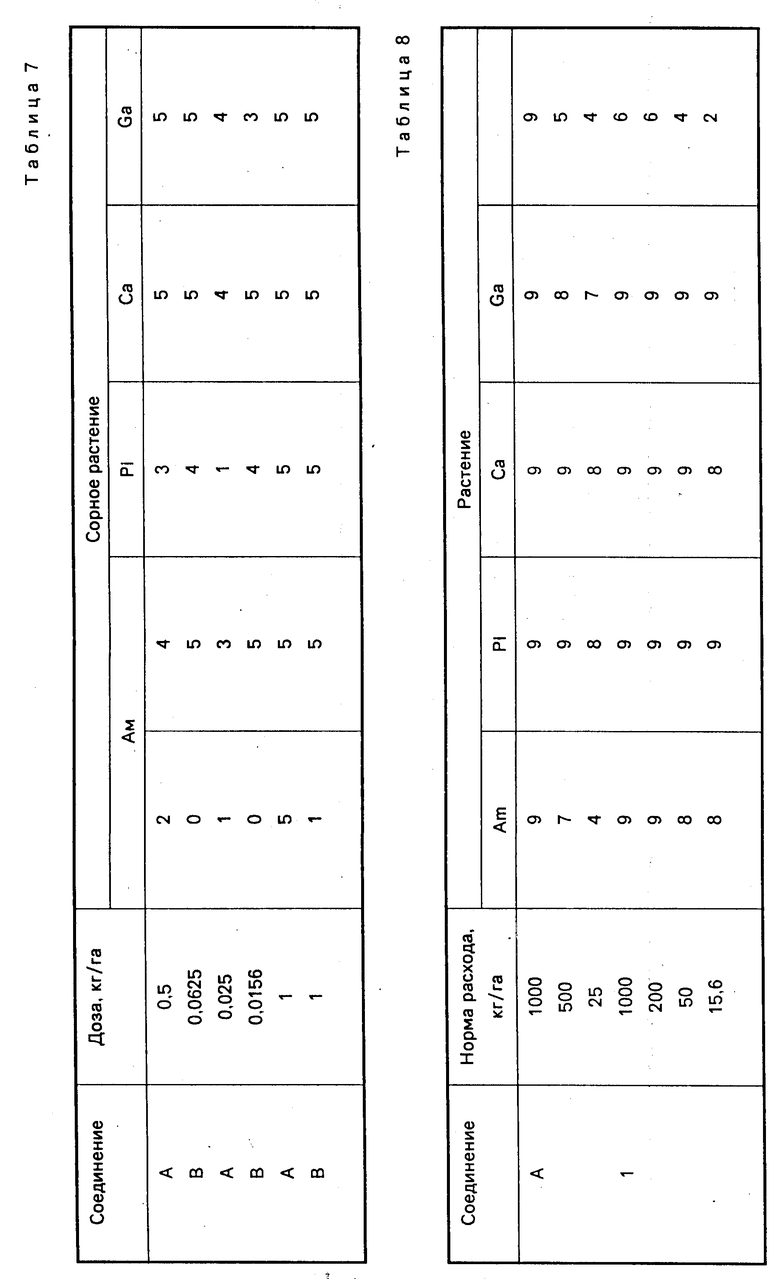
Испытания проводили после появления всходов в различное время для каждого соединения, используя методику и систему оценок, описанную в патентной заявке на с. 47. Система оценок, использованная для соединения А, была той же, что представлена в патентном описании. В испытаниях, в которых было включено соединение В, использовали весьма похожую систему оценок, а именно Балл % уничтожения 0 0-10 1 11-25 2 26-50 3 51-80 4 81-95 5 96-100
Полученные результаты приведены в табл. 7.
Как можно видеть из приведенных данных, соединение 17 изобретения является более активным при меньших дозах при применении на сорняках, которые растут в злаках, по сравнению с соединением, предложенным для сравнения. Известное соединение не только дает более слабый контроль над сорняками при более низких дозах, но наносит существенно большой ущерб пшенице по сравнению с соединением 17 настоящей патентной заявки, когда эти соединения применяли в одинаковых дозах. Такие тенденции очевидны, даже если принять во внимание ожидаемое несоответствие из-за того факта, что испытания не проводили по принципу "бок о бок" и имелись небольшие различия в системе оценок испытаний.
Таким образом, соединения, являющиеся предметом изобретения, обладают лучшими свойствами по сравнению с известными соединениями.
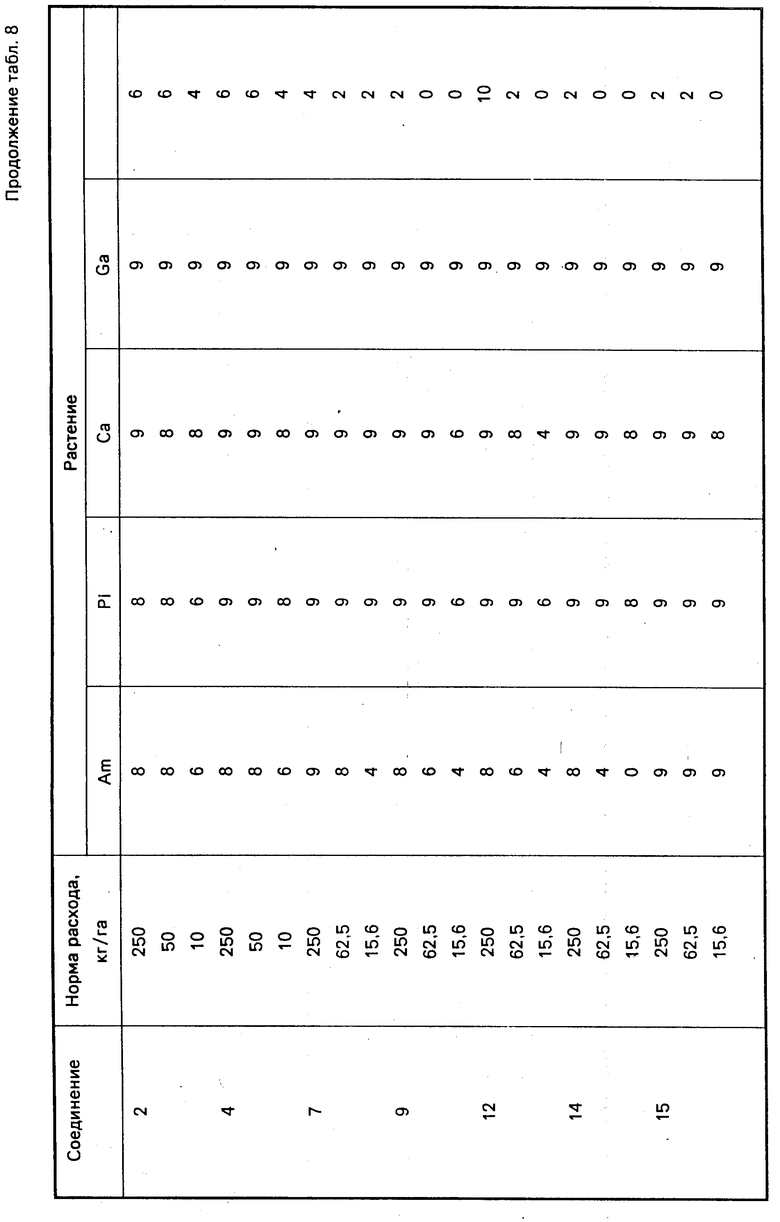
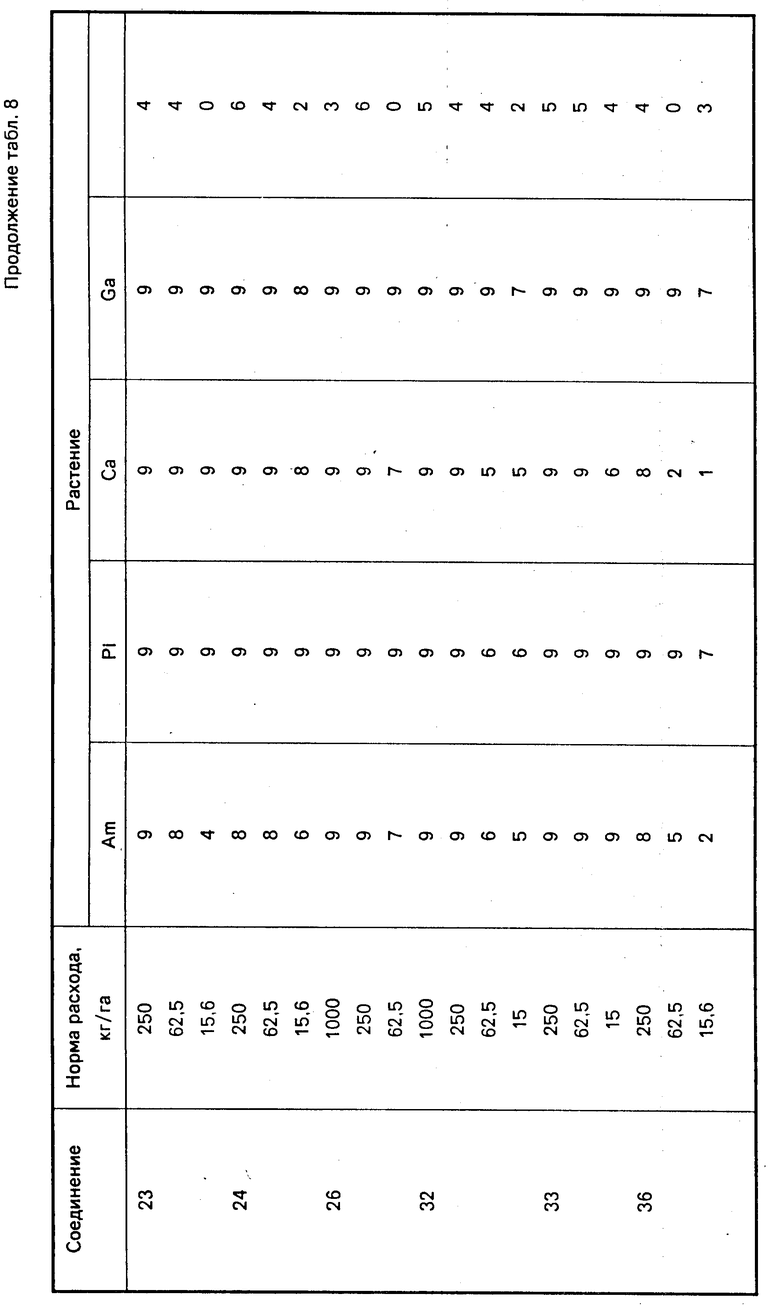
Данные приведены в табл. 8.


Использование: в качестве гербицида в сельском хозяйстве. Сущность изобретения: производные диметилового эфира ф-лы I, где R1 -H или Hal; R2 -N или CR3, где R3 -Nal или NO2; R4 -H, Hal, NO2, CN или COOCH3; R5 -Hal; R7 - COOR8 или CONR8 <195> R9, R8 -H, C1-C6 -алкил, необязательно замещенный фенилом C2-C6 - алкенил или C2-C6; R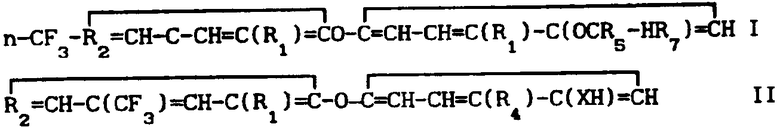
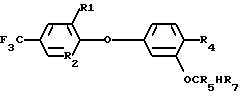
R1 или галоид;
R2 или CR3, где R3 - галоид или NO2;
R4 - галоид, NO2, CN или COOCH3;
R5 - галоид;
R7 - COOR8 или CONR8R9;
R8 - водород, C1-C6 - алкил, необязательно замещенный фенилом, C2-C6 - алкенил или C2-C6 - алкинил;
R8 и R9 - независимо водород или C1-C6 - алкил,
обладающие гербицидной активностью
2. Соединения по п.1, отличающиеся тем, что R1 - фтор, а R2-C-Cl.
| Способ борьбы с сорняками | 1982 |
|
SU1181517A3 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
