Изобретение относится к способу получения трансгенных растений, клетки которых способны экспрессировать химерный ген, кодирующий кристаллический σ -эндотоксин Bacillus thuringiensis. Такой кристаллический протеин представляет собой протоксин, который превращается в токсин в ходе заглатывания личинками насекомых разновидностей Lepidoptero, Coleoptera и Diptera.
Кристаллический протеин из Вt представляет собой потенциально важный инсектицид, не оказывающий вредного воздействия на людей, других млекопитающих, птиц, рыб или других насекомых помимо личинок насекомых разновидностей Lepidoptera, Coleoptera и Diptera. Bt токсин обладает чрезвычайно высокой активностью и лишь нанограммовые его количества требуются для уничтожения личинок восприимчивых насекомых. Другие преимущества использования кристаллического протеина из Bt в качестве инсектицида включают его широкий спектр активности против личинок насекомых разновидностей Lepidoptera, Coleоptera и Diptera, а также явные затруднения в развитии такими личинками устойчивости к действию кристаллического протеина даже в случае его использования в крупном масштабе. Указанные личинки представляют собой большую проблему для сельского хозяйства и лесного хозяйства и особенно для культивации хлопка.
Кристаллический протеин является эффективным инсектицидом в том случае, когда он применяется на растениях, подверженных заражению личинками насекомых разновидностей Lepidoptera, Coleoptera или diptera. Такие растения включают спаржевую капусту, латун и хлопок. Заражение личинками Lepidoptera представляет собой особенно серьезную проблему для хлопковых растений.
До сих пор Вt кристаллический протеин (протоксин) выделяли из Bacillus и применяли на растениях такими стандартными методами, как опыление и опрыскивание. Препараты, содержащие Bt кристаллический протеин, находят коммерческое использование в качестве биологических инсектицидов. Например, Бактоспеин, выпускаемый компаниями Биокем Продактс Лтд. Дипел, Аббот Лабораториз и Турцид, Сандоз АГ.
Тот факт, что Вt продуцирует кристаллический протеин только в ходе споруляции, представляет собой значительный недостаток, связанный с производством и использованием такого биологического инсектицида. Ограничение такой фазой роста, особенно при реализации промышленного процесса, может приводить к неудобствам и чрезмерной длительности производства. Кроме этого, затраты, связанные с указанным производством, могут затруднять конкуренцию такого биологического инсектицида с другими выпускаемыми промышленностью продуктами на основе таких химических веществ, как, например, производные пиретроида.
Другим недостатком, связанным с использованием Вt токсина, является, например, тот факт, что протеин обычно остается на поверхности растения, подвергаемого обработке, где он эффективен лишь по отношению к личинкам, питающимся на поверхности, и где он дезактивируется в результате длительного воздействия ультрафиолетового излучения. Такая дезактивация может представлять собой, по крайней мере, одну из причин потери стойкости кристаллического протеина под воздействием окружающей среды. В результате приходится осуществлять многократное и дорогостоящее применение кристаллического протеина.
Указанные и другие недостатки могут быть преодолены путем внедрения и экспрессии гена, кодирующего Bt кристаллический протеин или протеин, обладающий такими же инсектицидными токсическими свойствами, что и Вt кристаллический протеин, в растения. В настоящем изобретении описывается путь преодоления указанных недостатков в результате внедрения и экспрессии гена, кодирующего Вt кристаллический протеин или протеин, обладающий инсектицидно токсическими свойствами Bt кристаллического протеина, в протопласты зерновой культуры и регенерации продуктивных трансгенных зерновых растений из трансформированных протопластов и культивации таких зерновых растений, устойчивых к действию насекомых.
Методы генной инженерии описаны в качестве усовершенствованных путей продуцирования кристаллического протеина. Так, например, в патентах США 4 448 885 и 4 467 036 описываются плазмиды для продуцирования кристаллического протеина в бактериальных штаммах, отличных от Bt. Такие методы позволяют получать кристаллический протеин, но не снимают недостатки, связанные с использованием кристаллического протеина в качестве коммерческого инсектицида.
Предложено непосредственно клонировать Bt токсичные гены в растения с целью реализации такой ситуации, когда растения защищают сами себя (Клауэнер, 1984). В заявке на Европейский патент ЕР-0142924 (Аграгенатикс) предлагается способ клонирования токсичных генов из Bt в табак (с.59) и этот же способ предлагается для защиты хлопка (с.77).
Способы трансформации клеток хлопка и развития из таких клеток растений описаны в заявке на патент США с серийным номером 122 200 - "Регенерация и трансформация хлопка", фирмы Фитоген, а также в заявке на патент США с серийным N 122 162 под названием "эффективный способ регенерации хлопка из культивированных клеток" фирмы Циба-Гейге. Способ трансформации клеток хлопка в заявке фирмы Фитоген на патент с серийным номером 122 200 и способ регенерации хлопковых растений в заявках на патенты фирм Фитоген и Циба-Гейги с серийными NN 122 200 и 122 162 соответственно включены в объем настоящей заявки.
Цель изобретения - создать способ защиты хлопковых растений от повреждения насекомыми, заключающийся в продуцировании инсектицидно эффективного количества Bt кристаллического протеина или протеина, обладающего инсектицидно токсическими свойствами Bt кристаллического протеина в клетке растения, причем такое количество должно быть достаточно эффективным для уничтожения или борьбы с насекомыми, питающимися указанными растениями.
Цель изобретения достигнута в результате разработки химерных генов, способных экспрессировать в хлопковых клетках полипептид, обладающий в значительной степени инсектицидно-токсическими свойствами Bt кристаллического протеина (далее в тексте, Bt токсичный ген).
На фиг.1 показана конструкция mp/19bt плазмиды, содержащей 5' окончание Bt протоксинового гена; на фиг.2 - конструкция mp/19bt ca/del плазмиды, содержащей VI промотор СаМV гена, слитый с 5' окончанием Вt протоксиновой кодирующей последовательности; на фиг.3 - конструкция p702/bt плазмиды, содержащей 3' кодирующий участок протоксина, слитый с СаМV сигналами обрыва транскрипции; на фиг.4 - конструкция рВR 322)bt14, содержащая полную протоксиновую кодирующую последовательность, от которой ответвляются последовательности СаМV промотора и терминатора; на фиг.5 - конструкция pRK252/Tn903/Bg III; на фиг.6 - конструкция pС1B5; на фиг.7-8 - конструкция рС1B4; на фиг. 9 - конструкция pС1B2; на фиг.10 - конструкция pС1B10, плазмиды с широким набором возможных хозяев, содержащей Т-ДНК границы и ген для селекции растения; на фиг.11 - конструкция pС1B10/19 sbt; на фиг.12 - конструкция pС1B710; на фиг. 13 - конструкция pС1B10/710; на фиг.14 - конструкция pС1B10/35 sbt; на фиг.15 - конструкция pС1B10/35 sbt (КрпI); на фиг. 16 - конструкция pС1B10/35 sbt (ВсII); на фиг. 17 - конструкция pС1B10/35 sbt (607); на фиг.18 - конструкция pС1B1300, плазмиды, имеющей химерный ген, содержащий СаМV 35S промотор/АМV лидер (Bt (BcI) делецию/35 терминатор; на фиг. 19, 20 и 21 - конструкция pС1B 1301, имеющей химерный ген, содержащий хлопковую rbs-gx промотор/Bt (607 делеция) кодирующую последовательность; на фиг.22 - конструкция pС1B1302, имеющей химерный ген, содержащий хлопковую rbs-gv промотор/Вt (607 делеция) кодирующую последовательность; на фиг. 23 - рестрикционная карта хлопковых геномных клонов, несущих rbc-gx и rbc-gv; на фиг.24 - нуклеотидная и аминокислотная последовательность rbc-gv (первый АТG и метионин переходного пептида показаны в виде блока); на фиг.25 - нуклеотидная и аминокислотная последовательности rbc-X. Первый АТG и метионин переходного пептида показаны в виде блока.
В соответствии с настоящим изобретением перечисленные ниже плазмиды и/или микроорганизмы были депонированы Международным депозидарием "Американская коллекция типовых культур, Роквилл, Мэриленд" в соответствии с требованиями Будапештского договора.
1) Escherichia coli MC1061, pCIB10/35S BT...АTCC 67329 (дата депонирования: 27 февраля 1987 г.)
2) Escherichia coli HB101, pCIB10/19sBT...АTCC 67330 (дата депонирования 27 февраля 1987 г.)
3) Плазмида р LVIII АТСС 40235 (дата депонирования: 14 мая 1986 г.)
4) Фаг λ /rbc-gv АТСС 40486 (дата депонирования, 25 августа 1988 г.)
5) Фаг λ /rbc-X АТСС 40487 (дата депонирования, 25 августа 1988 г.)
Рассматриваемые хлопковые клетки включают клетки любых и всех хлопковых растений, в которые может быть введена, реплицирована и экспрессирована гетерологичная. В качестве примеров подходящих разновидностей хлопковых растений можно отметить Gossypium hirsutum, Gossypium arborcum и Gossypium barbadeuse. Разновидность Gossypium hirsutum является предпочтительной и она может быть обдирного или сборного типов. Обдирной или сборный хлопок отличаются способом сбора урожая, причем коробочки обдирного хлопка очень прочно присоединены к растению и они не опадают даже при послесезонных штормах. При сборе обрывного хлопка растение фактически разрушается. Сборный хлопок присоединен к стеблю менее прочно и его собирают менее разрушительными методами. Некоторые выпускаемые промышленностью разновидности G. hirsutum, которые можно регенерировать методом настоящего изобретения включают: AcaIa 1515-75, AcaIa SI-2, AcaIa-SI-4, AcaIa SI-5, AcaIa-SIC-1, AcaIa-SIC-22, AcaIa-SIC-28, AcaIa-SIC-30, AcaIa B-1644. AcaIa-B-1810, AcaIa B-2724, AcaIa-GC-510.
Coker 304, Coker 315, Coker 201, Coker 310, Coker 312, DP 41, DP 90.
DPL 50, DPL 20, DPL 120, DPL 775.
Hankert 611, lankert 57,
Paymaster 145, Paymaster HS 26,
Stoneville 306, Stoneville 825,
Funk 519-2, Funk FC 3008, Funk FC 3024, Funk C 1568R, Funk FC 2005,
Funk C 0947B, Funk C 2028, Funk FC 2017, Funk C 1379,
McNair 235, Tomcot SR 21-Siokra Tx-CAB-CS.
Предпочтительными разновидностями являются Acala SI-2, AcaIa SIC-1, AcaIa GC 510, AcaIa SIC-28, AcaIa SIC-30, AcaIa B-1644 и Siokra.
AcaIa SI-2, AcaIa GC 510, AcaIa B-1644 и Siokra особенно предпочтительны. Термин "растительные клетки" относится к любой клетке хлопкового растения. Некоторые примеры клеток, входящих в объем настоящего изобретения, включают дифференцированные клетки, представляющие собой часть живого растения; недифференцированные клетки в культуре; клетки недифференцированной ткани, такой как каллюс или опухоли; семена; амбрионы; побеги и пыльцу.
Химерный ген настоящего изобретения содержит последовательность регуляции транскрипции, включающую промотор и 5' и 3' нетранслированные последовательности, являющиеся функциональными в хлопковых растениях. Такие последовательности, независимо друг от друга, могут быть получены из любого источника, например вирусного, растительного или бактериального гена.
Подходящие для использования вирусные промоторы и 5' и 3' нетранслированные последовательности являются функциональными для хлопковых растений и они могут быть получены, например, из таких растительных вирусов, как вирус мозаики цветной капусты (СаМУ). Для использования предпочтительными промоторами САМV являются 19S и 35S промотор.
Для выделения СаМV 19S промотора и, необязательно, соседнего 5' нетранслированного участка, рестрикционный фрагмент СаМУ генома, содержащего желаемую последовательность, подвергают селекции. Подходящим рестрикционым фрагментом, содержащим 19S промотор и 5' нетранслированный участок, является фрагмент между Pst1 сайтом, начинающимся в положении 5386, и Hind III сайтом, начинающимся в положении 5850 (Хон и др., 1982, 194-220). Как описывается ниже, аналогичными методами может быть получен 35S промотор СаМV.
Нежелательные нуклеотиды в рестрикционном фрагменте могут быть необязательно удалены стандартными методами. Некоторые подходящие методы делеции нежелательных нуклеотидов включают использование экзонуклеаз (Маниатис с сотр., 1982) и регулируемого нуклеотидом мутагенеза (Золлер и Смит, 1982).
Аналогичная методика может использоваться для получения желаемого 3' нетранслированного участка. Так, например, подходящая СаМV 19S генная 3' нетранслированная последовательность может быть получена выделением участка между ЕсоРУ сайтом в положении 7342 и Bg III сайтом в положении 7643 СаМV генома (Хон с сотр.).
Примеры растительных генных промоторов и 5' и 3' нетранслированных участков, подходящих для использования в настоящем изобретении, также включают те гены, которые кодируют мелкие субзвенья рибулозо-1,5-бифосфат карбоксилазы и хлорофил а/b связующий протеин. Такие участки растительного гена могут быть выделены из растительных клеток методами, аналогичными тем, что описаны выше для выделения соответствующих участков из СаМV (см. статью Морелли с сотр., 1985).
Подходящие промоторы и 5' и 3' нетранслированные участки бактериальных генов включают фрагменты, присутствующие в Т-ДНК участке плазмид Аgrobacterium. Примерами подходящих плазмид Agrobacterium могут служить Тi плазмида A. tumefaciens и Ri плазмида A.rhizogeus. Agrobacterium промоторы, а также 5' и 3' нетранслированныe участки, используемые в настоящем изобретении, представляют собой, главным образом, те фрагменты, которые присутствуют в генах, кодирующих октофин синтазу и нопалин синтазу. Такие последовательности могут быть получены методами, аналогичными тем, что были описаны выше для выделения СаМV и растительных промоторов, а также нетранслированных последовательностей (см. статью Бивана с сотр. 1983).
Кодирующий участок химерного гена содержит нуклеотидную последовательность, которая кодирует полипептид, в значительной мере обладающий токсическими свойствами Bt σ -эндотоксинового кристаллического протеина, если он проявляет инсектицидную активность к тому же набору личинок насекомых, что и кристаллический протеин подразновидности Bt. Некоторые подходящие для такой цели подразновидности включают, например, Bt var. Kurstaki, Bt var. berliner, Bt alesti, Bt var. tolworthi, Bt var. sotto, Bt var. dendrolimus; Bt var. tenebrionis; Bt var. san. diego; и Bt var. aizanai. Предпочтительными разновидностями являются Bt var. Kurstaki и особенно Bt var. Kurstaki НDI.
Кодирующий участок может существовать в Bt в естественном состоянии. С другой стороны, кодирующий участок может содержать последовательность, которая отличается от последовательности существующей в Bt, но является эквивалентной вследствие вырожденности генетического кода.
Кодирующая последовательность химерного гена может также кодировать полипептид, отличающийся от встречающегося в природе кристаллического протеинового δ -эндотоксина, но, в значительной степени, обладающий инсектицидно-токсическими свойствами кристаллического протеина.
Предпочтительно, чтобы нуклеотидная последовательность была в значительной мере гомологичной по крайней мере той части или тем частям природной последовательности, которые ответственны за инсектицидную активность.
Полипептид, экспрессированный химерным геном настоящего изобретения, будет обычно обладать по крайней мере некоторыми иммунологическими свойствами природного Bt кристаллического протеина, поскольку он имеет по крайней мере некоторые общие антигенные детерминанты.
Полипептид, кодированный химерным геном настоящего изобретения, предпочтительно должен быть структурно родственным кристаллическому δ-эндотоксиновому протеину, продуцируемому Bt. Bt продуцирует кристаллический протеин в присутствии субзвена, представляющего собой протоксин со значением Мr в интервале 130000-140000. Такой протеин может расщепляться под действием протеаз или щелочей с образованием инсектицидных фрагментов, имеющих Mr порядка 80 000, предпочтительно 70 000, более предпочтительно около 60 000 и возможно даже ниже. Такие фрагменты предпочтительно имеют максимальное значение Mr порядка 120 000, более предпочтительно порядка 110 000 и наиболее предпочтительно 100 000. Химерные гены, кодирующие такие фрагменты протоксина или даже более мелкие его части, согласно настоящему изобретению могут быть сконструированы в том случае, если фрагменты или части таких фрагментов обладают достаточной инсектицидной активностью. Протоксин, инсектицидные фрагменты протоксина и инсектицидные части таких фрагментов могут быть слиты с другими молекулами, например, с полипептидами и протеинами.
Кодирующие области, подходящие для использования в настоящем изобретении, могут быть получены из генов кристаллического протеина, выделенных из Bt (см. например, заявку РСТWO 86/01536 и патенты США 4448 885 и 4 467 036). Предпочтительной последовательностью нуклеотидов, которая кодирует кристаллический протеин, является последовательность, указанная как нуклеотиды 156-3623 в последовательности формулы 1 или более короткая последовательность, кодирующая инсектицидный фрагмент такого кристаллического протеина. Описание такой последовательности данно в статье Гейзера с сотр. (1986). Формула I дана ниже.
Кодирующая область, определенная нуклеотидами 156-3623 последовательности (I), кодирует полипептид, последовательность которого отвечает формуле (II).
Токсины некоторых Bt штаммов являются токсичными в отношении других насекомых, помимо разновидности lepidoptera. Так, например, токсин Bt var tenebrienis является токсичным по отношению к насекомым разновидности coleoptera. Токсичность Bt штамма san diego в отношении насекомых разновидности coleoptera и последовательность соответствующего токсинового гена раскрыты в ЕР-0.202.739 и в ЕР-0.213.818. Для введения химерного гена настоящего изобретения в растительные клетки такой ген вначале внедряют в вектор. Если не имеется достаточного количества гена для трансформации, то вектор может быть усилен репликацией в клетке-хозяине. Наиболее удобными клетками хозяевами для такого усиления являются бактериальные или дрожжевые клетки. В том случае, когда имеется достаточное количество химерного гена, его вводят в хлопковые клетки или ткани. Введение гена в хлопковые растительные клетки или ткани может осуществляться с помощью того же вектора, что использовался для репликации или с помощью другого вектора.
Некоторые примеры бактериальных клеток-хозяев, подходящих для репликации химерного гена, включают клетки, выбранные из группы, состоящей из такого вида Escherichia как Е.coli и такого вида Agrobacterium как A.tumefaciens или A.rhizogenes. Методы клонирования гетерологичных генов в бактериях описаны в патентах США 4 237 224 и 4 468 464.
Репликация генов, кодирующих кристаллический протеин Bt в Е.coli описаны Вонгом с сотр. (1983).
Предпочтительной бактериальной клеткой-хозяином для амплификации химерных В генов настоящего изобретения является Agrobacterium. Преимуществом амплификации гена в Agrobacterium является то, что Agrobacterium может далее использоваться для внедрения амплифицированного гена в растительные клетки без дополнительных генетических манипуляций.
Некоторые примеры дрожжевых клеток-хозяев, подходящих для репликации генов настоящего изобретения, включают клетки вида Saccharomyces.
Любой вектор, в который может быть внедрен химерный ген и который реплицирует в подходящей клетке-хозяине, например в бактерии или дрожжах, может использоваться для амплификации генов настоящего изобретения. Примерами векторов - производных фагов, которые используются в настоящем изобретении, являются векторы, полученные из М13 и λ . Подходящие векторы, являющиеся производными М13, включают М13mр18 и M13mр19. Подходящие векторы, являющиеся производными λ , включают λ gt II, λ gt 7 и λ chazon 4.
Векторы, являющиеся производными плазмид, особенно подходящие для репликации в бактериях, включают pВR 322 (Боливар с сотр., 1977), pYС18 и pYС19 (Норрандер с сотр., 1983); а также Тi плазмиды (Бенан с сотр., 1983). Предпочтительными векторами для амплификации генов в бактериях являются pВR322, pYС18 и pYС19.
Внедрение или сборка гена в вектор осуществляется стандартными методами, такими как использование техники рекомбинантных ДНК (Маннатис с сотр., 1982) и гомологичная рекомбинация (Хиннен с сотр., 1978).
Подходящие рестрикционные эндонуклеазы включают такие, которые образуют тупые концы, например, SmaI, HpaI и ЕcoRY, а также такие, которые образуют липкие концы например, EcoRI, SaCI и BamHI.
Bt токсиновые гены настоящего изобретения могут непосредственно вводиться в растительные клетки с помощью некоторых Agrobacterium, примеры которых включают Ti плазмиду вида A. tumefaciens и Ri плазмиду вида A. rhizogenes.
Такие плазмиды содержат участки (Т-ДНК), способные встраиваться в геном клеток растений.
Т-ДНК области, которые существуют в природе, являются онкогенными. Онкогенные части таких Т-ДНК областей могут быть частично или полностью удалены до или совместно с внедрением желаемой ДНК-последовательности. Такие плазмиды, содержащие модифицированные Т-ДНК области, являются безопасными.
Гены, пригодные для использования в настоящем изобретении, встраиваются в Т-ДНК, векторную систему с помощью методов, известных в данной области (Бартон и Чилтон, 1983; Чилтон, 1985). Т-ДНК векторы могут быть онкогенными (Хернальстингс с сотр. , 1980), частично безопасными (Бертон и Чилтон, 1983), полностью безопасными (Замбриски с сотр., 1983) или могут основываться а искусственных Т-ДНК векторах, имеющих синтетические Т-ДНК фланкирующие последовательности (Ванг с сотр., 1984). Подходящие, лишенные опасных признаков векторы, содержащие Т-ДНК флокирующие последовательность, включают pGA436, pGA437 и pGA438 в соответствии с описанным в работе Эна с сотр. (1985); pMON 120 (см. статью Фрэйли с сотр., 1983) и pC1B10 (Розштейн с сотр., 1987). Перенос Т-ДНК обычно осуществляют инкубированием Agrobacterium в присутствии протопластов растительных клеток или тканей поврежденных растений (см. статью Каплана с сотр., 1983).
Помимо химерного гена, кодирующего Bt или Bt-подобный токсин, векторы предпочтительно включают также ДНК-последовательность, позволяющую осуществлять селекцию или скрининг клеток хлопкового растения, содержащих вектор, в присутствии клеток, не содержащих вектор.
Предпочтительный селективный маркер представляет собой ген, кодирующий антибиотическую устойчивость. Гены, которые сообщают устойчивость к хлорамфениколу, канамицину, гигромицину, С418 или, в принципе, к любому другому антибиотику, могут использоваться в качестве способного к селекции маркера.
Примерами генов, сообщающих устойчивость к антибиотику, могут служить гены, кодирующие неомицин фосфотрансферазу (канамицин и С418 устойчивость, Велтен с сотр., 1984); гигромицин фосфотрансферазу (гигромициновая устойчивость, ван-ден Эльзен с сотр., 1985) и хлорамфеникол ацетилтрансферазу.
Примером гена, используемого главным образом в качестве селективного маркера в тканевой культуре, для идентификации растительных клеток, содержащих сконструированные методами генной инженерии векторы, является ген, кодирующий энзим, имеющий хромогенный субстрат. Так, например, если ген кодирует энзим β -галактеозидазу, то растительные клетки высевают в среду тканевой культуры, содержащую хромогенный субстрат Xgаl(5-хлор-4-бром-3-индолил-β -Д-галактозид), и в соответствующих условиях растительные клетки, содержащие копии такого гена, окрашиваются в голубой цвет под действием красителя индиго, который выделяется при расщеплении Xgal β -галактозидазой.
Введение химерных генов в растения согласно настоящему изобретению может осуществляться с помощью любой Т-ДНК векторной системы, способной к введению генов в хлопковые растительные клетки. Такая векторная система может быть, например, Ко-интегральной системой (Коман с сотр., 1983; Замбриски с сотр. , 1983), векторной системой с расщепленными концами (Фрэйли с сотр., 1985), как это описано Чилтоном (1985). С другой стороны, векторная система может представлять собой бинарную систему (де Фрамон с сотр., 1983); Гоекема с сотр., 1983), или Тi плазмиду, содержащую ген в Т-ДНК (Матске и Чилтон, 1981). Другим возможным вариантом является система, в которой Т-ДНК находится на плазмиде, а вирулетные гены - на хромосональной ДНК.
Предпочтительная Т-ДНК векторная система представляет собой бинарную векторную систему и особенно систему, в которой используется pC1B10 (Ротштейн с сотр., 1987) (см. фиг.10).
Введение гетерологичных генов с использованием техники рекомбинантных ДНК в бинарную векторную систему описано Кли с сотр., 1935 г. Внедрение генов в Т-ДНК вектор может представлять собой гомологическую рекомбинацию с использованием стратегии двойной рекомбинации (Матске и Чилтон, 1981); стратегии одинарной рекомбинации (Коман и сотр., 1093); (Замбриски с сотр., 1983); стратегии одинарной рекомбинации без повторений в Т-ДНК (Фрэйли с сотр., 1985), в соответствии с описанным Чилтоном (1985).
Если векторы, содержащие химерный ген, не собираются в Agrobacterium, то они могут вводиться в Agrobacterium методами, известными в данной области. Такие методы включают трансформацию и конъюгацию. Трансформация Agrobacterium описана Холстерсом с сотр., 1978.
Конъюгация описана Коман с сотр. (1983) и Чилтон с сотр. (1976).
Примеры включают использование A. tumefaciens, A.rhizogenes и A.radiobacter.
Среда, способная поддерживать конкретную растительную клетку в культуре, зависит от конкретного вида хлопковой растительной клетки. Так, например, некоторые подходящие среды включают примерно 10 мг/л 2,4-дихлорфеноксиуксусной кислоты и неорганические соли Мурашиги и Скуга (Мурашиги и Скуг, 1962) или неорганические соли В-5 Гaмборга (Гамборг с сотр., 1968).
Эмбрион хлопка (Gossypium spp.), способный к прорастанию и возобновлению, может эффективно продуцироваться в результате соматического эмбриогенеза путем развития про-эмбрионных клеточных масс и из них эмбрионов в клеточной суспензионной культурной системе.
Способ изобретения позволяет продуцировать, например, в стандартной колбе Де-Лонга емкостью 250 мл около 10.000 сферических эмбрионов, из которых может быть получено примерно 1000 зрелых эмбрионов и примерно 50 растений.
Полученные в соответствии с таким способом хлопковые растения могут быть культивированы или оставлены в некультивированном состоянии.
Особенно предпочтительным является способ трансформации хлопковых клеток, подвергаемых суспензионной культивации на каллюсовой растительной среде, который после цикла роста суспензионной субкультуры включает стадии:
а) выделения клеток и любого эмбриогенного каллюса из каллюсовой растительной среды;
b) ресуспендирования клеток и эмбриогенного каллюса в каллюсовой растительной среде, содержащей Agrobacterium вектор, имеющий ген, который сообщает устойчивость антиобиотическому гигромицину хлопковых клеток при сохранении условий суспензионного роста в течение времени, достаточного для трансформации суспендированных клеток;
с) выделения суспендированных клеток из каллюсовой растительной среды, содержащей Agrobacterium;
d) обработки трансформированных клеток и эмбриогенного каллюса антибиотиком, применяемым в концентрации, достаточной для уничтожения;
е) контактирования клеток и эмбриогенного каллюса с антиобиотическим гигромицином с целью селекции трансформированных клеток и эмбриогенного каллюса;
f) фильтрации суспензии с целью удаления эмбриогенного каллюса размером более 600 мкм.
Стадия а.
Эмбриогенный хлопковый каллюс. Первая стадия заключается в индуцировании образования хлопкового каллюса из хлопковой эксплантатной ткани. Примеры подходящей хлопковой эксплантатной ткани включают соматические эмбрионы, зрелые и незрелые зиготные эмбрионы, котиледоны или гипокотили из сеянцев, а также молодую ткань зрелого растения. Предпочтительными являются соматические эмбрионы и котиледоны или гипокотили сеянцев.
Зиготные эмбрионы, например, могут быть получены вырезанием из семяпочек. Такие семяпочки срезаются, предпочтительно, через 7-30 дней после опыления, предпочтительно через 10-21 день после опыления и наиболее предпочтительно через 12-16 дней опыления.
Котиледоны и гипокотили могут быть получены из молодых сеянцев. Такие сеянцы имеют возраст 3-21 день, более предпочтительно 4-9 дней, и наиболее предпочтительно около 7 дней. Гипокотили срезают в продольном направлении и разрезают на удобные части размером, например, 1-20 мм, предпочтительно около 2 мм. Котиледоновую ткань разрезают на участки площадью 1-400 мм2, предпочтительно 5-100 мм2, и наиболее предпочтительно около 10 мм2.
Соматические эмбрионы, полученные по такой методике, представляют собой наиболее предпочтительный источник получения эмбриогенного каллюса согласно настоящему способу.
Соматические эмбрионы могут быть получены, например, путем использования описанного выше способа в отношении гипокотильной и котиледонной ткани в качестве источника эксплантата. Подходящим является любой соматический эмбрион, снятый до первичного развертывания листа. Размер соматического эмбриона не имеет решающего значения. Предпочтительно, чтобы соматический эмбрион имел длину менее 5 мм.
Молодая ткань зрелого хлопкового растения может быть легко получена путем отрезания верхушек побегов длиной 10 см, предпочтительно 5 см. Стеблевую и черешковую ткань разрезают в продольном направлении и далее разрезают на части того же размера, что и в случае гипокотилей (см. выше). Ткань листьев разрезают на части того же размера, что и котиледоновую ткань (см. выше).
Ткань хлопкового растения помещают в среду, подходящую для индуцирования каллюса при 20-40оС, предпочтительно 23-34, более предпочтительно при 31оС. В таком способе регенерации может использоваться среда, способная индуцировать каллюс из ткани. Такая среда может быть жидкой или твердой, хотя твердая среда предпочтительна в связи с тем, что она более удобна.
Среда, способная индуцировать каллюс в условиях настоящего изобретения, содержит неорганические соли, витамины, источник углерода, ауксин и цитокинин. pH такой среды устанавливают в интервале 3,5-7,5, предпочтительно 4,5-6,5 и наиболее предпочтительно 5,7.
Для этой цели подходят любые неорганические соли и витамины, способные вносить свой вклад в индуцирование каллюса. Примерами подходящих неорганических солей и витаминов могут служить вещества, описанные Мурашиги и Скугом (1962) (MS) и Гамборгом с сотр., (1968) (В-5). Другим примером может служить модификация MS или В-5 Гамборга, описанная Ченгом с сотр. (1980). Предпочтительные неорганические соли представляют собой MS неорганические соли. Предпочтительные витамины представляют собой витамины В-5 Гамборга.
В качестве источника углерода может применяться любой его источник, в котором может расти каллюс. Предпочтительными источниками углерода являются сахара и их производные. Предпочтительными сахарами являются глюкоза и сахароза. Особенно желательно инициировать каллюс в среде для его индуцирования, содержащей глюкозу с тем, чтобы уменьшить степень потемнения ткани и затем перенести каллюс в среду для его индуцирования, содержащую сахарозу.
Концентрация источника углерода составляет 5-60 г/л, предпочтительно 30 г/л.
Ауксин, присутствующий в среде для индуцирования каллюса, может представлять собой любой ауксин, способный индуцировать каллюс. Подходящие для этой цели ауксины включают нафталин, уксусную кислоту, пиклорам, 2,4,5-трихлорфеноксиуксусную кислоту, 2,4-дихлорфеноксиуксусную кислоту, индол-3-масляную кислоту, индол-3-молочную кислоту, индол-3-пировиноградную кислоту, индол-3-уксусную кислоту и п-хлорфеноксиуксусную кислоту. Предпочтительный ауксин представляет собой а-нафталинуксусную кислоту.
В способе настоящего изобретения может использоваться любая концентрация ауксина, способная индуцировать образование каллюса. Подходящая концентрация составляет 0,1-10 мг/л. Предпочтительная концентрация составляет 2 мг/л, особенно в том случае, когда ауксин представляет собой а-нафталинуксусную кислоту.
Цитокинин, присутствующий в среде для индуцирования каллюса, может представлять собой любой цитокинин, способный индуцировать каллюс. Подходящие для этой цели цитокинины включают кинетин, 6-бензиладенин, 2-изопентениладенин и зоатин. Предпочтительным цитокинином является кинетин.
В способе настоящего изобретения может применяться любая концентрация цитокинина, способная индуцировать образование каллюса. Подходящие для этой цели концентрации составляют 0,1-10 мг/л. Предпочтительная концентрация составляет 1 мг/л, особенно в том случае, когда цитокинин представляет собой кинетин.
В случае твердой среды она содержит компонент, вызывающий затвердевание, например, около 0,8% такого агара, как агар Noble (Difco) или около 0,8% агарозы. (Все процентные соотношения в описании даны в массовом выражении).
Ткань культивируют на среде, индуцирующей каллюс в течение времени, достаточного для образования каллюса. Так, например, ткань может культивироваться на среде, индуцирующей каллюс и содержащей глюкозу в качестве источника углерода. В этом случае типичным является пятинедельный период индуцирования. Если необходимо, для предотвращения потемнения предварительно образуют субкультуры. Предпочтительными являются недельные субкультуры.
Образующий каллюс может быть неорганизованным или может содержать про-эмбрионные клеточные массы, эмбриогенный каллюс и/или эмбрионы. Обычно в том случае, когда в качестве источника эксплантата используются гипокотили или котиледоны, каллюс, по-видимому, является неорганизованным. При использовании в качестве источника трансплантата соматических эмбрионов по крайней мере часть каллюса включает эмбриогенный каллюс, который характеризуется светло-желтым цветом и образованием узелков.
Полученный в результате каллюс далее может быть перенесен в среду субкультивирования каллюса, которая похожа на среду индуцирования каллюса за исключением того, что она содержит в качестве источника углерода сахарозу, на период времени до 5 мес. Предпочтительным является субкультивирование в течение одного или двух месяцев с переносом через месяц в свежую среду, на сахарозосодержащей среде для индуцирования каллюса.
Каллюс может индуцироваться в темноте, но предпочтительно индуцирование проводят на свету. Свет может иметь интенсивность, например, 0,5-150 Еm-2с-1 (=41,75-12525 лк).
Стадия b.
Групповые агрегаты про-эмбрионных клеточных масс. Каллюс со стадии (а) суспендируют в жидкой среде, промотирующей развитие про-эмбрионных или пролиферирующих эмбрионных клеточных масс. Важно, чтобы плотность клеток была низкой. Поэтому суспендируют не более 40 мг каллюса/мл культурной среды, предпочтительно не более 15 мг каллюса/мл культурной среды и более предпочтительно не более 5 мг каллюса/мл культурной среды.
В качестве среды, используемой стадии (b), может применяться любая среда, способная индуцировать про-эмбрионные клеточные массы. Такая среда содержит неорганические соли, витамины, источник углерода, а также ауксин. Такая среда может также содержать источники органического азота, цитокинины, аминокислоты и другие адденды, такие как казеиновый гидрализат или кокосовую воду.
Неорганические соли и витамины могут быть теми же, что на стадии (а) (см. выше). Предпочтительными веществами являются MS неорганические соли и витамины В-5.
Источник углерода может быть тем же, что на стадии (а) (см. выше). Предпочтительным источником является сахароза. Концентрация источника углерода составляет 0,1-100 г/л. Предпочтительная концентрация составляет 20 г/л, особенно в том случае, когда источником углерода является сахароза.
Ауксин может выбираться из ауксинов, используемых на стадии (а). Предпочтительными ауксинами являются 2,4-дихлорфеноксиуксусная кислота и пиклорам. Наиболее предпочтительным веществом является циклорам.
Концентрация ауксина на стадии (b) относительно низка. Точная концентрация зависит от природы используемого ауксина. Такая относительно низкая концентрация ауксина обычно аналогична концентрации, используемой в суспензионной культурной среде, и она значительно ниже концентрации ауксина на стадии (с). При использовании пиклорама в качестве ауксина стадии (b) его концентрация составляет 0,01-5 мг/л, предпочтительно 0,1-1 мг/л, и наиболее предпочтительно около 0,5 мг/л. При использовании 2,4-дихлорфеноксиуксусной кислоты в качестве ауксина на стадии (b) его концентрация составляет 0,01-0,5 мг/л, предпочтительно 0,05-0,25 мг/л, и наиболее предпочтительно около 0,1 мг/л.
Индуцирование про-эмбрионных клеточных масс, предпочтительно, осуществляют в аэрированной среде при 20-35оС, предпочтительно при 22-33оС, и наиболее предпочтительно при 25-31оС. Среду можно аэрировать любым известным способом, например встряхиванием. Стадию (b) можно проводить в темноте или при освещении мощностью 75 Em-2 c-1 (=6262,5 лк), предпочтительно при 5-10 Em-2 с-1 (=417,5-835).
Каллюс находится в среде, предпочтительно, в отсутствии субкультуры до тех пор, пока не образуются групповые агрегаты про-эмбрионных клеточных масс и не начнется быстрая пролиферация. Начало быстрой пролиферации обычно наблюдается в промежутке 3-8 недель, более типично, в промежутке между 5 и 7 неделями. В ходе индукционного периода среда может быть заменена свежей порцией, хотя предпочтительно в ходе этого периода не нарушать среду.
Изменение в системе при переходе от каллюса к агрегатам про-эмбриогенных клеточных масс может быть легко зарегистрировано специалистом в области культивации растительных тканей. Его можно различить по светло-желтому цвету и групповой природе про-эмбрионных клеточных масс.
Как только групповые агрегаты про-эмбриогенных клеточных масс начинают быстро пролиферировать, они могут непосредственно вводиться в среду, описанную на стадии (с), или они могут субкультивироваться с целью предотвращения потемнения. Лучше всего проводить субкультивацию каждые 3-7 дней, предпочтительно каждые 5-7 дней. Клеточные массы выживают без субкультивации в течение примерно четырнадцати дней.
Стадия с.
Тонко диспергированные про-эмбрионные клеточные массы. Групповые агрегаты про-эмбрионных клеточных масс со стадии (b) переносили в жидкую среду, способную тонко диспергировать групповые агрегаты про-эмбрионных клеточных масс. Такая среда может быть аналогична среде, описанной для стадии (b), за исключением того, что среда со стадии (с) содержит относительно высокую концентрацию ауксина. В качестве ауксина может использоваться ауксин со стадии (а). Предпочтительными ауксинами являются 2,4,5-трихлорфеноксиуксусная кислота и 2,4-дихлорфеноксиуксусная кислота. Наиболее предпочтительный ауксин - 2,4-дихлорфеноксиуксусная кислота.
Концентрация зависит от природы используемого ауксина. Концентрация ауксина в среде стадии (с) обычно выше или по крайней мере находится на уровне наибольшего значения из указанного интервала концентраций, обычно используемых в суспензионной культурной среде, и в любом случае такая концентрация значительно выше, чем концентрация ауксина на стадии (b).
Так, например, при использовании 2,4-дихлорфеноксиуксусной кислоты в качестве ауксина стадии (с) его концентрация может составлять 0,5-200 мг/л, предпочтительно 1-10 мг/л, и наиболее предпочтительно 2,5-7,5 мг/л.
За исключением концентрации и, возможно, природы ауксина на стадии (c) может использоваться также среда, температура, освещенность, что и на стадии (b).
Условия стадии (с) поддерживаются до тех пор, пока групповые агрегаты про-эмбрионных клеточных масс превратятся в более мелкие, более тонко диспергированные про-эмбрионные клеточные массы. Внешний вид более мелких, более тонко диспергированных про-эмбрионных клеточных масс легко определяется специалистами в данной области. Такие клеточные массы характеризуются их желтым цветом, гладкой поверхностью, промежуточным значением плотности и мелким размером. Изменение в системе, связанное с образованием более мелких, более тонко диспергированных клеточных масс обычно происходит в течение 6 недель, более типично, в течение 2 недель. Культура мелких тонко диспергированных про-эмбрионных клеточных масс может сохраняться в течение неопределенного длительного времени и она может подвергаться субкультивации с целью поддержания активного роста. Удобно проводить субкультивацию в течение, например, каждых 3-28 дней, предпочтительно 5-10 дней.
Стадия d.
Зрелые эмбрионы. Мелкие, более тонко диспергированные про-эмбрионные клеточные массы добавляют к среде, которая индуцирует развитие зрелых эмбрионов. Такая среда предпочтительно является жидкой.
Эмбрионы проходят через ряд стадий развития перед тем, как они становятся зрелыми и способными к проpастанию. Такие стадии включают сферическую сердцевинную, торпедную и зрелую стадии. Названия стадий базируются на примерных формах эмбрионов.
В качестве среды для стадии (d) может использоваться любая среда, индуцирующая развитие зрелых эмбрионов. Подходящая для такой цели среда содержит неорганические соли, витамины, источник углерода и органическое соединение, содержащее восстановленный азот.
Соли и витамины, а также их концентрации могут быть теми же, что указаны для стадии (а). В качестве источника углерода может использоваться один из источников, описанных для стадии (а). Концентрация источника углерода составляет 1-10 г/л, предпочтительно 2-6 г/л. Предпочтительным источником углерода является сахароза.
В качестве источника органического азота может использоваться любое соединение, которое при добавлении в среду стадии (d) индуцирует развитие зрелого эмбриона. Предпочтительными соединениями являются аминокислоты. Предпочтительной аминокислотой является глютамин.
Концентрация источника органического азота зависит от природы используемого соединения. Эффективная концентрация глютамина в качестве источника органического азота составляет 2-260 мМ, предпочтительно 5-100 мМ, и наиболее предпочтительно 10-50 мМ.
Среда для стадии (d) может содержать ауксин. Ауксины желательны в ходе ранних стадий развития эмбрионов, но не на более поздних стадиях. Поэтому, если ауксины присутствуют в системе, они должны находиться в ней лишь до сердцевидной стадии развития. После этого эмбрионы переносят в среду, не содержащую ауксина.
В случае присутствия ауксина его концентрация составляет 0,01-0,1 мг/г.
В качестве ауксина может использоваться один из ауксинов, описанных для стадии (a). Предпочтительным ауксином является пиклорам и 2,4-дихлорфеноксиуксусная кислота.
Эмбрионы могут культивироваться в среде со стадии (d) при 20-35оС в темноте или при освещении. Интенсивность освещения может составлять, например, 5-75 Em-2с-1 (=6262,5 лк).
Эмбрионы находятся в среде стадии (d) до тех пор, пока они не созревают до торпедообразного или зрелого состояния. Специалисты в области культивации растительных тканей могут легко распознать сферические, сердцевидные, торпедообразные и зрелые эмбрионы по мере их созревания. Эмбрионы созревают обычно за 2-5 недель, как правило за 3-4 недели. Обычно нет необходимости в субкультивации эмбрионов или их переносе в свежую среду, за исключением возможной замены ауксинсодержащей среды на среду, не содержащую ауксин на первой сердцевидной стадии.
Стадия е.
Прорастание. Зрелые эмбрионы помещают на твердую среду, способную индуцировать прорастание. Такая среда содержит неорганические соли, витамины и источник углерода. Среда затвердевает в присутствии такого подходящего отверждающего агента, как Гелрит (Калко, Сан-Диего, Калифорния), агароза или агар.
В качестве неорганических солей могут использоваться соли, описанные для стадии (а), модифицированные таким образом, что нитрат присутствует в высокой концентрации, тогда как аммоний либо отсутствует, либо присутствует в очень низкой концентрации. Концентрация нитрата может составлять 20-60 мМ, предпочтительно 30-60 мМ, более предпочтительно, 35-46 мМ. Концентрация ионов аммония не должна быть выше 5 мМ.
Источником углерода, предпочтительно, служит сахар. Предпочтительным сахаром является сахароза. Концентрация источника углерода зависит от природы используемого источника. Так, например, при использовании сахарозы в качестве источника углерода его концентрация составляет 0,1-6 мас.%, предпочтительно 0,5-4 мас.%, более предпочтительно 1-3 мас.%.
В среде стадии (е) необязательно присутствует источник органического азота. Такое органическое соединение предпочтительно представляет собой аминокислоту или смесь аминокислот, способную поддерживать прорастание. Предпочтительными аминокислотами или их смесями является глютамин или гидролизат казеина.
Концентрация источника органического азота зависит от природы используемого соединения. Так, например, при использовании глютамина его концентрация может составлять 2-50 мМ, предпочтительно 5-30 мМ, более предпочтительно 10-20 мМ. При использовании казеинового гидролизата или модифицированного казеинового гидролизата его концентрация составляет 100-3000 мг/л, предпочтительно 1000-2800 мг/л, более предпочтительно 1500-2500 мг/л.
Предпочтительно, чтобы прорастание происходило в среде, содержащей источник органического азота, до появления побегов. Затем эмбрионы переносят в среду, не содержащую источника органического азота, для удлинения корней.
Плотность эмбрионов в среде ограничена значением ниже того, при котором развитие становится самоингибированным. Подходящие значения плотности составляют 1-100 эмбрионов в чашке Петри диаметром 9 см, содержащей 10-75 мл среды, предпочтительно 25-50 мл среды и наиболее предпочтительно около 35 мл среды.
Среда или среды со стадии (а) поддерживаются при 20-30оС. Предпочтительная температура составляет 25оС. Для стадии (е) требуется некоторое освещение. Интенсивность освещения обычно составляет 5-150 Em-2с-1 (=417,5-12525 лк), предпочтительно 10-75 Em-2с-1 (=835-6266,5 лк).
Эмбрионы выдерживают в среде или средах стадии (е) до стадии их прорастания, обычно в течение 1-20 дней, как правило, в течение 2-4 дней. Специалисту в данной области известен срок прорастания эмбрионов.
Стадия f.
Растения. После прорастания ростки переносят в почву для выращивания растений. Вначале перенесенные растения накрывают стеклянным колпаком для поддержания высокой влажности. Через неделю пребывания под таким колпаком такие ростки или растения не требуют специальной обработки.
Применение-размножение.
Зрелые эмбрионы могут использоваться для массового размножения и клонирования. Под этим подразумевается любое прорастание эмбрионов и трансплантация ростков в почву, другие растущие субстраты или в среду, окружающую другие растущие растения. Зрелые эмбрионы могут также упаковываться в искусственную семенную оболочку и высеваться в виде "соматических семян". Массовое размножение и клонирование будет выгодным в том случае, если гибридные родители или сам гибрид нуждается в массовом продуцировании.
Растительные клетки настоящего изобретения содержат химерный ген и могут использоваться для продуцирования полипептида, обладающего, в значительной мере, инсектицидно-токсическими свойствами Bt кристаллического протеина. Растительные клетки могут perse образовывать инсектицид. Растительные клетки, используемые непосредственно в качестве инсектицидов, могут представлять собой культивированные растительные клетки или могут являться компонентами живых растений.
Токсин может быть также выделен из растительных клеток с помощью таких известных способов, как экстракция или хроматография. Такой экстракт может представлять собой весь экстракт растительных клеток, частично очищенный экстракт, или чистый препарат полипептида. Любой такой экстракт или хроматографический изолят может использоваться так же, как кристаллический протеин из Bt (см. , например, статью Дикона, 1983, и Миллера с сотр., 1983).
Общие методы, основанные на использовании ДНК техники рекомбинантных.
А. Переваривание с помощью рестрикционной эндонуклеазы.
Обычно ДНК присутствует в реакционной смеси в количестве 50-500 мкг/мл буферного раствора, рекомендованного производителем, Нью Ингланд Биолабс, Беверли, МА. 2-5 единиц рестрикционных эндонуклеаз добавляют на каждый 1 мкг ДНК и реакционную смесь инкубируют при температуре, рекомендованной производителем, в течение 1-3 ч. Реакцию обрывают нагреванием до 65оС в течение 10 мин или путем экстракции фенолом с последующим осаждением ДНК этанолом. Такая методика описана также на стр. 104-106 работы Манниатиса с сотр.
В. Обработка ДНК полимеразой с целью создания полных концов.
Фрагменты ДНК добавляли в реакционную смесь с концентрацией 50-500 мкг/мл в буфере, рекомендованном производителем, Нью Инглэнд Биолабс. Реакционная смесь содержит все четыре деоксинуклеотидных трифосфата с концентрацией 0,2 мМ. Реакционную смесь инкубируют при 15оС в течение 30 мин и затем реакцию прерывают нагреванием при 65оС в течение 10 мин. Для фрагментов, полученных перевариванием с рестрикционными эндонуклеазами, которые обеспечивают получение 5'-выступающих концов, таких как EcoRI и BamIII, используется крупный фрагмент или фрагмент Кленова ДНК-полимеразы. Для фрагментов, продуцируемых эндонуклеазами, которые обеспечивают получение 3'-выступающих концов, таких как PstI и SacI, используют Т4 ДНК-полимеразу. Использование двух таких энзимов описано на стр.113-121 работы Манкатиса с сотр.
С. Электрофорез на геле агарозы и очистка фрагментов ДНК от гелей.
Электрофорез на геле агарозы осуществляется в горизонтальном устройстве в соответствии с описанным на стр.150-163 работы Маниатиса с сотр. Используемый буфер представляет собой описанный в этой работе Трис-боратный буфер. ДНК-фрагменты могут наблюдаться визуально в результате окрашивания 0,5 мкг/мл этилбромида, который присутствует в геле и буфере в ходе электрофореза, или добавляется после электрофореза. ДНК можно визуально наблюдать при освещении коротковолновым или длинноволновым ультрафиолетовым светом. В том случае, когда фрагменты выделяют из геля, используют агарозу с низкой температурой стеклования, выпускаемую компанией Сигма Кэмикл, Сант-Льюис, Миммури. После электрофореза желаемый фрагмент вырезают, помещают в пластиковую трубку, нагревают до 65оС в течение примерно 15 мин, затем трижды экстрагируют фенолом и дважды осаждают из этанола. Такая методика имеет некоторые отличия от описанной на стр.170 статьи Маниатиса с сотр.
Д. Присоединение фрагментов синтетического линкера к концам дНК.
В том случае, когда к концу молекулы ДНК желательно присоединить сайт новой рестрикционной эндонуклеазы, такую молекулу, если это необходимо, вначале обрабатывают ДНК-полимеразой с целью создания боковых концов в соответствии с описанным в предыдущем разделе.
Примерно 0,1-1,0 мкг такого фрагмента добавляют к 100 нг фосфорилированной линкерной ДНК, выпускаемой компанией Нью Инглэнд Биолабс в объеме 20-30 мкл, содержащем 2 мкл Т4 ДНК-лигазы, выпускаемой Нью Инглэнд Биолабс и 1 мМ АТР в среде буфера, рекомендованного производителем. После инкубирования в течение ночи при 15оС реакцию обрабатывают путем нагревания смеси до 65оС в течение 10 мин. Затем реакционную смесь разбавляют примерно 100 мкл буфера, пригодного для рестрикционной эндонуклеазы, которая расщепляет по синтетической линкерной последовательности, и добавляют примерно 50-200 единиц такой эндонуклеазы. Полученную смесь инкубируют при соответствующей температуре в течение 2-6 ч, затем фрагмент подвергают электрофорезу на геле агарозы и очищают в соответствии с описанным выше. Полученный в результате фрагмент имеет концевые группы, полученные перевариванием с рестрикционной эндонуклеазой. Такие окончания обычно оказываются когезивными, в результате чего полученный фрагмент легко лигатируется с другими фрагментами, имеющими аналогичные когвезивные окончания.
Е. Удаление 5'-терминальных фосфатов из фрагментов ДНК.
В ходе стадий клонирования плазмиды обработка векторной плазмиды фосфатазой уменьшает рециркуляризацию вектора (что обсуждается на стр.13 работы Маниатиса с сотр.). После переваривания ДНК с соответствующей рестрикционной эндонуклеазой добавляют одну единицу телячьей кишечной щелочной фосфатазы, выпускаемой компанией Борингер-Маннхейм, Индианополис, 1N ДНК инкубируют в течение 1 ч при 37оС, затем дважды экстрагируют фенилом и осаждают из этанола.
F. Легирование ДНК-фрагментов.
При соединении фрагментов с комплементарными когезивными окончаниями примерно 100 нг каждого фрагмента инкубируют в реакционной смеси объемом 20-40 мкл, содержащей примерно 0,2 единицы Т4 ДНК лигазы, полученной от Нью Инглэнд Биолабс в буфере, рекомендованном производителем. Инкубирование проводят в течение 1-20 ч при 15оС. В том случае, когда соединяют ДНК-фрагменты с боковыми концами, их инкубируют в соответствии с описанным выше, за исключением того, что количество Т4 ДНК-лигазы увеличивают до 2-4 единиц.
С. Трансформация ДНК в E.coli.
В большинстве экспериментов использовали штамм E.coli НВ101. ДНК вводили в E.coli с использованием кальций хлоридной методики, описанной в работе Маниатиса с сотр. на с. 250-251. Трансформированные бактерии способны к селективному росту на среде, содержащей соответствующие антибиотики. Такая селективная способность позволяет отличать желательные бактерии от бактерии-хозяина, не получающей трансформированной ДНК. Установление желательного антибиотика является общепринятой операцией, которая дает сведения о генах, устойчивых к лекарству и присутствующих во входящей трансформирующей ДНК, и о чувствительности бактерии-хозяина к действию лекарства. Так, например, когда известно, что бактерия-хозяин чувствительна к ампициллину и на входящей трансформирующей ДНК имеется функциональный ген, устойчивый к действию ампициллина, то такой антибиотик является подходящим для селекции трансформантов.
Н. Скрининг E.coli на плазмиды.
После трансформации полученные в результате E.coli подвергают скринингу на присутствие желаемой плазмиды, используя для этой цели метод быстрого выделения плазмиды. Две удобные методики описаны на с. 366-369 работы Маниатиса с сотр.
I. Выделение плазмидной ДНК в крупном масштабе.
Методы выделения больших количеств плазмид в E.coli описаны на с. 88-94 работы Маниатиса с сотр.
J. Клонирование в М13 фаговые векторы.
В следующем ниже описании следует иметь в виду, что двутяжная репликационная форма производных фага М13 используется для таких общепринятых операций, как рестрикционное эндонуклеазное переваривание, лигатирование и т. п.
П р и м е р 1. Конструирование химерного гена в плазмиде рВР322.
Для слияния СаМV генного VI промотора с протоксиновой кодирующей последовательностью конструируют производное фагового вектора mp19 (Яниш-Перрон с сотр., 1985).
Вначале ДНК-фрагмент, содержащий примерно 155 нуклеотидов в положении 5' к протоксиновой кодирующей области и соседние 1346 нуклеотидов кодирующей последовательности, внедряют в mp19. Фаговую mp19dsrf (двутяжная репликативная форма) ДНК переваривают с рестрикционными эндонуклеазами SaCI и SmaI и векторный фрагмент размером примерно 7,2 kbр (килопар оснований) очищают после электрофореза через агарозу с низкой температурой стеклования согласно стандартным методикам. Плазмида рКУ25/4, содержащая примерно 10 kbp BtДНК, включающей протоксиновый ген, была получена от д-ра Дж. Коэха, Циба-Гейги Лтд., Базель, Швейцария. Нуклеотидная последовательность протоксинового гена, присутствующего в плазмиде рКУ25/4, соответствует формуле 1. Плазмиду КУ25/4 переваривали с эндонуклеазами HpaI и SacI и фрагмент размером 1503 bp (содержащий нуклеотиды 2-1505 в формуле (1)) очищали в соответствии с описанным выше. (Такой фрагмент содержит примерно 155 bp бактериальной промоторной последовательности и примерно 1346 bp начала протоксиновой кодирующей последовательности). Смешивали примерно по 100 нг каждого фрагмента, добавляли Т4 ДНК-лигазу и в течение ночи проводили инкубирование при 15оС. Полученную в результате смесь трансформировали в штамм E.coli НВ101, смешивали с индикаторной бактерией E.coli IM 101 и высевали в соответствии с описанным Мессингом (1983). Для последующего конструирования описанного ниже (фиг.1) использовали названный mp19/bt.
После этого фрагмент ДНК, содержащий САМV генный VI промотор и некоторые из кодирующих последовательностей гена VI, вставляли в mp18/bt фаговую mp19/bt ds rg ДНК переваривали с BamH1, обрабатывали крупным фрагментом ДНК-полимеразы для создания боковых концов и повторно расщепляли в присутствии эндонуклеазы PSt1. Крупный векторный фрагмент очищали электрофорезом в соответствии с описанным выше. Плазмида рАВД1 описана Разковски с сотр., 1984 г. Плазмидную рАВД1 ДНК переваривали с pst I и Hind III. Очищали фрагмент длиной примерно 465bp, содержащий СаМV генный VI промотор и примерно 75bp кодирующей последовательности гена VI. Два этих фрагмента лигетировали и высевали в соответствии с описанным выше. В следующих экспериментах использовали один из полученных в результате фагов, названный mp19/btca.
Фаг bt19/btca содержит СаМV генные VI промоторные последовательности, часть генной VI кодирующей последовательности, примерно 155bpBt ДНК расположенных вверх от протоксиновой кодирующей последовательности и, примерно, 1346bp протоксиновой кодирующей последовательности. С целью точного слияния СаМV промоторных последовательностей с протоксиновыми кодирующими последовательностями, производили делецию промежуточной ДНК с использованием олигонуклеотид-контролируемого мутагенеза mp19/btca ДНК. ДНК олигонуклеотид с последовательностью (5') TTGGCATTGTTATGGATCCTTCCACCTCTCA (3') синтезировали общепринятым методом с использованием ДНК-синтезатора фирмы Эплайд Биосистемс. Такой олигонуклеотид является комплементарным к тем последовательностям фаговой mp19/btca ДНК, которые находятся 3' окончании СаМV промотора (нуклеотиды 5762-5778 из статьи Хона с сотр., 1982) и к началу протоксиновой кодирующей последовательности (нуклеотиды 156-172 в формуле 1). Общая методика мутагенеза описана Золлером и Смитом (1983). Примерно 5 мкг однотяжной фаговой mp19/btca ДНК смешивали с 0,3 мкг фосфорилированного олигонуклеотида в объеме 40 мкл. Полученную смесь нагревали до 65оС в течение 5 мин, охлаждали до 50оС и медленно охлаждали до 4оС. После этого добавляли буфер, нуклеотид трифосфаты, АТР, Т4 ДНК-лигазу и крупный фрагмент ДНК-полимеразы и проводили в течение ночи инкубирование при 15оС в соответствии с известной методикой (Золлер и Смит, 1983). После электрофореза на геле агарозы сферическую двутяжную ДНК очищали и переносили в E.coli штамм IМ101. Полученные в результате пятна подвергали скринингу на последовательности, которые гибридизируются с оликонуклеотидом, меченным изотоном 32Р, и фаг анализировали методом с использованием ДНК ограничительной эндонуклеазы. Среди полученных в результате фаговых клонов будут присутствовать такие, в которых правильно вычеркнуты нежелательные последовательности между САМV генным VI промотором и протоксиновой кодирующей последовательностью. Такой фаг обозначают как mp/19btca/del (см. фиг.2).
Далее конструировали плазмиду, в которой 3' кодирующую область протоксинового гена сливали с сигналами обрыва СаМV транскрипции. Вначале плазмидную рАВД1 ДН переваривали с эндонуклеазами ВamHI и Вg III и выделяли фрагмент размером 0,5 kbp, содержащий СаМV транскрипционные терминаторные последовательности. Далее плазмиду pYС19 (Яниш-Перрон, 1985) переваривали с BamHI, смешивали с 0,5 kbp фрагментом и инкубировали в присутствии Т4 ДНК-лигазы. После трансформации ДНК в E.coli штамм НВ101 получали один из клонов, обозначенный как плазмида p702, который имел структуру, показанную на фиг.3.
После этого плазмидную p702 ДНК расщепляли эндонуклеазами SacI и SmaI и более крупный фрагмент размером примерно 3,2 kbp выделяли гельэлектрофорезом. Плазмидную pКY25/4 ДНК переваривали с эндонуклеазами Aha III и SaCI и путем электрофореза выделяли 2,3 kbp фрагмент (нуклеотиды 1502-3773 в приведенной выше формуле 1), содержащий 3' часть протоксиновой кодирующей последовательности (нуклеотиды 1504-3773 последовательности, показанной в формуле I). Эти два фрагмента ДНК смешивали, инкубировали в присутствии Т4 ДНК-лигазы и трансформировали в E.coli штамм НВ101. Полученная в результате плазмида представляла собой р702/bt (см. фиг.3).
Наконец части фаговой mp19/btca/del ds rf ДНК и плазмиды р702bt соединяли друг с другом с получением плазмиды, содержащей полную протоксиновую кодирующую последовательность с боковыми СаМV промоторной и терминаторной последовательностями. Фаговую mp/19btca/del ДНК переваривали с эндонуклеазами SacI и SphI и фрагмент размером примерно 1,75 kbp очищали методом электрофореза на геле агарозы. Аналогичным образом плазмидную р702/bt ДНК переваривали с эндонуклеазами SacI и CalI и выделяли фрагмент, размером примерно 3,5 kbp. Наконец, плазмидную pBR322 ДНК (Боливер с сотр., 1977) переваривали с SalI и SphI и выделяли более крупный фрагмент размером 4,2 kbp. Все три ДНК фрагмента смешивали и инкубировали в присутствии Т4 ДНК-лигазы и трансформировали в E.coli штамм НВ101. Полученная в результате плазмида pBR322bt14 представляет собой производное pBR322, содержащее СаМV генный VI протоморный сигнал и сигнал начала трансляции, слитые с Bt кристаллической протеиновой, кодирующей последовательностью, после которой следуют сигналы обрыва СаМV транскрипции (показано на фиг.4).
П р и м е р 2. Конструирование вектора на основе Тi плазмиды.
Вектор рС1B10 (Ротштейн с сотр. , 1987) представляет собой вектор - производное Ti плазмиды, используемый для переноса химерного гена к растениям через Agrobacterium tumefaciens. Такой вектор является производным плазмиды pРК252 с широким набором хозяев, которая может быть получена от д-ра В.Бариса, Вашингтонский Университет. Сэнт Льюис, Монтана. Такой вектор содержит также ген, ответственный за устойчивость к нанамицину в Agrobacterium из Tn 903 (Ока с сотр., 1981), а также левую правую Т-ДНК бордюрные последовательности из Ti плазмиды pTiТ37. Между бордюрной последовательностью находится полилинкерная область из плазмиды pYС18 и химерный ген, придающий растениям устойчивость к действию канамицина.
Вначале плизмиду pRK252 модифицируют таким образом, чтобы заменить ген, придающей устойчивость к действию тетрациклина на ген, обеспечивающий канамициновую устойчивость, из транспосона Tn903, а также модифицируют путем замены E.coli сайта в pRK252 на BgIII сайт (см.фиг.5, на которой изображено резюме таких модификаций). Плазмиду pRK252 вначале переваривают с эндонуклеазами SalI и SmAI, затем обрабатывают крупным фрагментом ДНК-полимеразы I с целью создания боковых концов и крупный векторный фрагмент очищают агарозагелевым электрофорезом. Далее плазмиду p368, которая содержит Tn903 на BamНI фрагменте размером 1050 bp, переваривают с эндонуклеазой BamHI, обрабатывают крупным фрагментом ДНК-полимеразы и после электрофореза на геле агарозы выделяют фрагмент размером 1050 bp; этот фрагмент содержит ген транспосона Тn903, который придает устойчивость к действию антибиотика - канамицина (Ока с сотр., 1981). Затем оба фрагмента обрабатывают крупным фрагментом ДНК-полимеразы с целью получения боковых концов. Оба фрагмента смешивают и инкубируют в присутствии Т4 ДНК-лигазы в течение ночи при 15оС. После трансформации в E.coli штамм НВ101 и селекции на канамицин-устойчивые колонии, получают плизмиду pRK252/Tn903 (см. фиг.5).
Плазмиду pRK252/Tn 903 переваривают по ее уникальному EcoRI сайту, после чего обрабатывают крупным фрагментом E.coli ДНК-полимеразы с целью получения боковых концов. Этот фрагмент добавляют к линкерам синтетического Bg III рестрикционного сайта и проводят инкубирование в течение ночи в присутствии Т4 ДНК-лигазы. Полученную в результате ДНК переваривают с избытком Bg III рестрикционной эндонуклеазы и более крупный векторный фрагмент очищают методом электрофореза на агарозогеле. Полученный в результате фрагмент снова инкубируют в присутствии Тк ДНК-лигазы с целью рециркуляризации фрагмента через его вновь присоединенные BgIII когезивные концы. После трансформации в E.coli штамме НВ101 получают плазмиду pRK252/Tn903/Bg III (см.фиг.5).
Конструируют производное плазмиды pBR322, которое содержит Ti плазмидные Т-ДНК границы, полилинкерную область плазмиды pYC19 и способный к селекции ген, ответственный за устойчивость растений к действию канамицина (см. фиг. 6). Плазмида pBR325/Eco29 содержит EcoRI фрагмент размером 1,5 kbp из нопалиновой Ti плазмиды pTi T37. Такой фрагмент содержит Т-ДНК левую граничную последовательность (Ядав с сотр., 1982). Для замены EcoR1 концов такого фрагмента на Hind III концы, плазмидную pBR325/Eco29 ДНК переваривают с EcoRI, затем инкубируют с нуклеазой SI, после чего проводят инкубирование с крупным фрагментом ДНК-полимеразы с целью получения боковых концов, после чего смешивают с синтетическими Hind III линкерами и инкубируют в присутствии Т4 ДНК-лигазы. Полученную в результате ДНК переваривают с эндонуклеазами ClaI и избытком Hind III и полученный в результате фрагмент размером 1,1 kbp, содержащий левую границу Т-ДНК, очищают гельэлектрофорезом. Далее полилинкерную область плазмиды pYC19 выделяют перевариванием плазмидной ДНК с эндонуклеазами EcoRI и Hind III и меньший фрагмент (размером примерно 53 bp) выделяют методом электрофореза на агарозогеле. После этого плазмиду pBR322 переваривают с эндонуклеазами EcoRI и ClaI, смешивают с двумя другими выделенными фрагментами, инкубируют в присутствии Т4 ДНК-лигазы и трансформируют в E.coli штамм НВ101. Полученная в результате плазмида pC1B5 содержит полилинкер и Т-ДНК левую границу в производном плазмиды pBR322 (см. фиг. 6).
Конструировали плазмиду, содержащую ген, экспрессирующий канамициновую устойчивость в растения (см. фиг.7 и 8). Плазмиду pBIN6 получали от д-ра М. Бивена, Институт разведения растений, Кэмбридж, Англия. Такая плазмида описана в статье Бивана, 1984 г. Плазмидную pBIN6 ДНК переваривали с EcоRI и Hind III и фрагмент размером 1,5 kbp, содержащий химерный неомицин фосфотрансферазный (NPT) ген выделяли из системы и очищали методом электрофореза на геле агарозы. Затем такой фрагмент смешивали с плазмидой pYC18 ДНК, которую расщепляли эндонуклеазами EcoRI Hind III. После инкубирования в присутствии Т4 ДНК-лигазы полученную в результате ДНК трансформировали в E. coli штамм НВ101. Полученной в результате плазмиде давали наименование pYC18/n ео. Такая плазмида содержит нежелательную BamHI распознающую последовательность между неомицин фосфотрансферазным геном и терминаторной последовательностью нопалин синтазного гена (см. статью Бивана, 1984). С целью удаления такой распознавательной последовательности плазмиду pYC18) n ео переваривали с эндонуклеазой BamHI, после чего обрабатывали крупным фрагментом ДНК-полимеразы с целью получения боковых концов. Затем такой фрагмент инкубировали в присутствии Т4 ДНК-легазы для рециркуляризации фрагмента и трансформировали в E. coli штамм НВ101. Полученная в результате плазмида pYC18/n ео (Bam) теряет свою BamHI распознающую последовательность.
Затем Т-ДНК правую граничную последовательность присоединяли к участку после химерного NРТ гена (см. фиг.8). Плазмида pBR325/Hind 23 содержит 3,2 kbp Hind III фрагмент плазмиды pTi T37. Такой фрагмент содержит правую Т-ДНК граничную последовательность (Биван с сотр. , 1983). Плазмидную pBR325/Hind 23 ДНК расщепляли эндонуклеазами Sac II и Hind III и фрагмент размером 1,0 kbp, содержащий правую границу выделяли и очищали методом электрофореза на агарозoгеле. Плазмидную pYC18/n ео (Bam) ДНК переваривали в присутствии эндонуклеаз Sac II и Hind III и фрагмент вектора размером 4,0 kbp выделяли электрофорезом на геле агарозы. Эти два фрагмента смешивали, инкубировали в присутствии Т4 ДНК-лигазы и транcформировали в E.coli штамм НВ101. Полученная в результате плазмида, pC1В4 (показанная на фиг.8) содержит Т-ДНК правую границу и маркер способного к селекции растения на канамициновую устойчивость в производном плазмиды pYC18.
После этого конструировали плазмиду, которая содержит Т-ДНК правую и левую границы, между которыми находится ген, способный к селекции растений, обладающий устойчивостью к канамицину и полилинкер pYC18 (см. рис.9). Плазмидную pC1B4 ДНК переваривают с эндонуклеазой Hind III, после чего обрабатывают крупным фрагментом ДНК-полимеразы с целью получения боковых концов и переваривают с эндонуклеазой EcoRI. Фрагмент размером 2,56 kbp, содержащий химерный устойчивый к канамицину ген, и правую границу Т-ДНК выделяют электрофорезом на агарозовом геле. Плазмидную pC1B5 ДНК переваривали с эндонуклеазой AatII, обрабатывали Т4 ДНК-лигазой с целью получения боковых концов, затем расщепляли эндонуклеазой EcoRI. Больший векторный фрагмент очищали электрофорезом на геле агарозы, смешивали с pC1B4 фрагментом, инкубировали в присутствии Т4 ДНК-лигазы и трансформировали в E.coli штамм НВ101.
Полученная в результате плазмида pCIB2 (показанная на фиг.9), представляет собой производное плазмиды pBR322, содержащее желаемую последовательность между этими двумя Т-ДНК-границами.
Следующие стадии завершают конструирование вектора pC1B10 (см.фиг.10). Плазмидную pCIB2 ДНК переваривали с эндонуклеазой EcoRY и присоединяли синтетические линкеры, содержащие Bg III распознающие сайты. После переваривания с избытком Bg III эндонуклеазы методом электрофореза на агарозовом геле выделяли фрагмент размером примерно 2,6 kbp. Плазмиду pRK252/Tn903/Bg III, описанную выше (см. фиг.5), переваривали с эндонуклеазой Bg III и затем обрабатывали фосфатазой для предотвращения рециркуляризации. Два фрагмента ДНК смешивали, инкубировали с Т4 ДНК-лигазой и трансформировали в E.coli штамм НВ101. Полученная в результате плазмида представляет собой завершенный вектор, pC1B10.
П р и м е р 3. Внедрение химерного протоксинового гена в вектор pC1B10.
Следующие ниже стадии показаны на фиг.11. Плазмидную pBR322/bt14 ДНК переваривали с эндонуклеазами pVVI и SalI и затем частично переваривали с эндонуклеазой BamHI BamHI-SalI фрагмент, размером 4,2 kbp, содержащий химерный ген, выделяли электрофорезом на агарозовом геле и смешивали с плазмидой pC1B10 ДНК, которую переваривали с эндонуклеазами BamHI и SalI. После инкубации с Т4 ДНК-лигазой и трансформации в E.coli штамм НВ101 получали плазмиду pC1B10/19sbt (см. фиг.11).
Такая плазмида содержит химерный протоксиновый ген в плазмидном векторе pC1B10.
Для переноса плазмиды pC1B10/19sbt из E.coli НВ101 в Agrobacterium использовали промежуточный штамм S17-I хозяина E. coli (Симон с сотр., 1983). Такой штамм, полученный из агрогенетической исследовательской корпорации, Бсулдер, Ко., содержит мобилизационные функции, которые могут переносить плазмиду pC1B10/19sbt непосредственно к Agrobacterium посредством конъюгации, в результате чего отпадает необходимость в трансформации голой плазмидной ДНК непосредственно в Agrobacterium. Вначале плазмидную pC1B10/19sbt ДНК вводят в обработанные хлористым кальцием клетки S17-I. Далее культуры трансформированных S17-I клеток и Agrobacterium tumefaciens штамм IBA 4404 (Оoмс с сотр., 1982) смешивают друг с другом и проводят спаривание на пластине с агаром N(Difco) в течение ночи при комнатной температуре. Полную петлю полученных в результате бактерий наносят штрихами на АВ минимальную среду (Чилтон с сотр., 1974), засеянную 50 мкг/мл канамицина, и проводят инкубирование при 28оС. Колонии повторно наносят штрихами на эту же среду, затем наштриховывают на пластины с N агаром. Медленно растущие колонии отбирают, повторно наносят штрихами на АВ минимальную среду с канамицином и выделяют единичные колонии. В результате такой методики осуществляют селекцию Agrobacterium, содержащей pC1B10/19sbt плазмиду.
П р и м е р 4. Конструирование Bt протоксинового химерного гена с СаМV 35 S промотором.
4.1. Конструирование СаМV 35S промоторной кассеты.
Плизмиду pC1B710 конструировали, как показано на фиг.12. Такая плазмида содержит СаМV промотор и последовательности обрыва транскрипции для 35 РНК матрицы (Ковэй с сотр., 1981), 1149 bp BgIII рестрикционный фрагмент СаМV ДНК (bp 6494-7643 в работе Хона с сотр., 1982) выделяли из плазмиды pIVIII (полученной от д-ра Хауэлла, Калифорнийский университет, Сан-Диего). С другой стороны, такой фрагмент может быть выделен непосредственно из СаМV ДНК препаративным электрофорезом на геле агарозы в соответствии с описанным ранее и смешан с расщепленной BamHI плазмидной pYC19 ДНК, обработанной Т4 ДНК-лигазой, после чего трансформирован в E.coli (следует отметить, что BamHI рестрикционный сайт в полученной в результате плазмиде разрушен лигированием Bg III когезивных концов с BamHI когезивными концами). Полученную в результате плазмиду, (pYC19)35S, затем использовали в олигонуклеотид-управляемом in vitro мутагенезе с целью внедрения BamHI распознающей последовательности GGATCC сразу после СаМV нуклеотида 7483 согласно работе Хона. Полученная в результате плазмида, pC1B710, содержит СаМV 35S промоторную область и участок обрыва транскрипции, отделенный BamHI ограничительным сайтом. ДНК-последовательности, внедренные в такой BamHI сайт, будут экспрессировать в растениях с такими СаМV транскрипционно-регулирующими последовательностями. (Следует также отметить, что pC1B710 не содержит каких-либо АТС-кодонов инициирования трансляции между началом транскрипции и BamHI сайтом).
4.2. Внедрение СаМV 35S промотор/терминаторной кассеты в pC1B10.
Следующие ниже стадии показаны на фиг.13. Плазмидные pC1B10 и pC1B710 ДНК переваривали с EcoRI и SalI, смешивали и лигатировали. Полученная в результате плазмида, pC1B10/710, имеет СаМV 35S промотор/терминаторную кассету, внедренную в вектор трансформации растения pC1B10. СаМV 35S последовательности находятся между Т-ДНК границами в pC1B10 и, таким образом, они будут вставляться в растительный геном в экспериментах по трансформации растений.
4.3. Внедрение Bt протоксинового гена в pC1B10/710.
Следующие ниже стадии показаны на фиг.14. Источник протоксинового гена, плазмиду pC1B10/19sbt переваривали с BamHI и NcoI и фрагмент размером 3,6 kbp, содержащий протоксиновый ген, выделяли препаративным гельэлектрофорезом. Затем этот фрагмент смешивали с синтетическим NcoI-BamHI адаптером с последовательностью 5'CATGGCCGGATCCGGC-3' и после этого переваривали с BamHI. Такая стадия приводит к образованию когезивных концов на обоих окончаниях протоксинового фрагмента. Затем этот фрагмент вставляли в BamHI-расщепленную pC1B10/710. Полученная в результате плазмида, pC1B10/35sbt, показанная на фиг.14, содержит протоксиновый ген между СаМV 35sbt промоторной и транскрипционной терминаторной последовательности.
4.4. Перенос плазмиды pC1B10/35sbt Agrobacterium tumefaciens с целью трансформации растения.
Плазмиду pC1B10/35sbt переносили в A.tumefaciens штамм LBA4404 в соответствии с описанным в примере 4.
П р и м е р 5. Конструирование рТОХ, содержащего химерных ген, кодирующий инсектицидно-токсический ген Bt var. tenebrionis.
Ген, кодирующий инсектицидный ген кристаллического протеина Bt var. tenebrionis, был охарактеризован и была установлена его аминокислотная последовательность (Секар с сотр., 1987). Такую кодирующую последовательность выделяли на удобном рестрикционном фрагменте, таким как Hind III - фрагмент размером, примерно, 3 kbp и внедряли ее в соответствующий растительный экспрессионный вектор, например в плазмиду pC1B770 (Ротштейн с сотр., 1987). Плазмида pC1B770 содержит химерный канамициновый ген для экспрессирования в растения, а также промотор и терминатор 35 РНК матрицы СаМV, разделенные особым BamHI сайтом. Рестрикционный фрагмент, несущий последовательность, кодирующую токсин, становится совместимым с особым BamHI сайтом pC1B770 в результате использования соответствующего молекулярного адаптера и совместного лигатирования.
П р и м е р 6. Конструирование pSAN, содержащего химерный ген, кодирующий инсектицидно-токсический ген Bt штамма san diego.
Ген, кодирующий инсектицидный протеин Bt штамма san diego, был охарактеризован и его последовательность была установлена Хaрнштадтом с сотр., ЕР-020-739 и ЕР-0-213-818. Такую кодирующую последовательность выделяли на удобном рестрикционном фрагменте и внедряли в соответствующий растительный экспрессионный вектор, например, такой как pC1B770. Плазмида pC1B770 содержит химерный канамициновый ген для экспрессирования в растения, а также промотор и терминатор 35S RHK матрицы СаМV, разделенные особым BamH сайтом. Рестрикционный фрагмент, несущий последовательность, кодирующую токсин, становится совместимым с особым BamHI сайтом pC1B770 в результате использования соответствующего молекулярного адаптера и совместного лигатирования.
П р и м е р 7. Конструирование подвергнутого делеции Bt протоксинового гена, кодирующего полипептид из ок. 725 аминокислот, и конструирование химерного гена, содержащего такой делетированный ген с СаМV 35S промотора.
Делетированный протоксиновый ген, кодирующий полипептид из, примерно, 725 аминокислот, получали путем удаления СООН-терминальной части гена путем расщепления по месту КpnI сайта рестрикционной эндонуклеазы в положении 2325 в последовательности, показанной формулой I.
Плазмиду pC1B10/35 sbt (фиг.14) переваривали с BamHI и КpnI и 2.2 kbp BamHI/KpnI фрагмент, содержащий подвергнутый делеции протоксиновый ген, выделяли препаративным электрофорезом на геле агарозы. Для превращения KpnI сайта на 3' окончании в BamHI сайт этот фрагмент смешивали с KpnI/BamHI адаптерным олигонуклеотидом и легировали. Затем этот фрагмент смешивали с BamHI-расщепленной pC1B10/710 (фиг.13). Полученные в результате трансформанты, обозначенные как pC1B10/35sbt (KpnI) и показанные на фиг. 15, содержат подвергнутый делеции протоксиновый ген, кодирующий полипептид из, примерно, 725 аминокислот. Эти трансформанты подвергали селекции на канамицине.
П р и м е р 8. Конструирование подвергнутого делеции Bt протоксинового гена, кодирующего полипептид из, примерно, 645 аминокислот и конструирование химерного гена, содержащего такой делетированный ген с помощью СаМV 35S промотора.
Делетированный протоксиновый ген, кодирующий полипептид из, примерно, 645 аминокислот получали путем удаления СООН-терминальной части гена в результате расщепления по месту ВсII сайта рестрикционной эндонуклеазы в положении 2090 последовательности, показанной формулой I.
Плазмиду pC1В10/35sbt (фиг.14) переваривали с BamHI и BcII и фрагмент BamHI/BcII размером, примерно, 1,9 kbp, содержащий делецированный протоксиновый ген, выделяли препаративным электрофорезом на агарозовом геле. Поскольку ВсII создает когезивный конец, совместимый с BamHI, не требуется дополнительных манипуляций перед легированием такого фрагмента в BamHI-расщепленную pC1B10/710 (фиг.13). Полученную в результате плазмиду pC1B10/35sbt (ВсII), имеющую структуру, показанную на фиг.16, подвергали селекции на канамицин.
П р и м е р 9. Конструирование подвергнутого делеции Bt протоксинового гена, кодирующего полипептид размером, примерно 607 аминокислот, и конструирование химерного гена, содержащего такой делетированный ген с СаМV 35S промотором.
Делетированный протоксиновый ген получали путем введения BamHI расщепляющего сайта (CGATCC) после нуклеотида 1976 в последовательности, показанной формулой I.
Такую операцию проводили путем клонирования BamHI фрагмента, содержащего протоксиновую последовательность из pC1B10/35sbt в mp18 и используя стандартные методики мутагенеза олигонуклеотидов. После мутагенеза двухтяжную репликационную форму ДНК готовили из М13 клона, который затем переваривали с BamHI. Фрагмент размером 1,9 kbp, содержащий делетированный протоксиновый ген, внедряли в BamHI-расщепленную pC1B10/710. Полученную в результате плазмиду pC1B10/35sbt (607) со структурой, показанной на фиг.17, подвергали селекции на канамицин.
В оставшихся примерах приведены конкретные протоколы трансформации хлопковых клеток и регенерации хлопковых растений из хлопковых клеток и каллюса. Следует иметь в виду, что в данной области можно изменять некоторые детали таких протоколов. Так, например, известны многочисленные растительные тканевые культурные среды, некоторые из которых подробно описаны ниже. Известно, как нужно изменять такие растворы для того, чтобы достичь таких же или аналогичных результатов. Так, например, в примере 1 описан модифицированный раствор Вайта, используемый в качестве среды для всхода семян и развития каллюса, используемый в качестве среды для роста/сохранения каллюса; в примере 13 описан раствор Бизли и Тинга, используемый в качестве среды для прорастания растений. В области культивации тканей должно быть известно, как нужно изменять такие растворы для достижения результатов, аналогичных полученным в примерах. Так, например, в качестве сахара для среды роста каллюса может использоваться глюкоза, которая минимизирует фенольные секреции, или сахароза, которая промотирует образование эмбриогенного каллюса.
Эксплантаты, используемые в способе трансформации, могут быть взяты из любого подходящего источника, например из сеянцев, особенно гипокотиль или котиледон, или из незрелых эмбрионов развивающегося плода.
Любой антибиотик, обладающий токсичностью в отношении Agrobacterium может использоваться для уничтожения остаточного Agrobacterium после стадии трансформации. Предпочтительным антибиотиком является цефотаксим.
П р и м е р 10. Регенерация хлопковых растений.
10.1 Среда. Все среды настоящего примера содержат неорганические соли Мурашига и Скуга и витамины В-5 Гамборга, имеют значение pH 5,7 и следующий состав, мг/л:
Макропитательные вещества: MgSO4 x 7H2O 370 KH2PO4 170 KNO3 1900 NH4NO3 1650 CaCl2 x 2H2O 440
Микропитательные вещества H3BO3 6,2 MnSO4 x H2O 15,6 ZnSO4 x 7H2O 8,6 NaMoO4 x 2H2O 0,25 CuSO4 x 5H2O 0,025 CaCl2 x 6H2O 0,025 KI 0,83 FeSO4 x 7H2O 27,8 Na2ЕДТА 37,3
Витамины Тиамин х HCl 10 Пиридоксин х HCl 1 Никотиновая кислота 1 Мио-Инозит 100
Кроме этого, различные среды содержат следующие компоненты:
Среды с индексами 25, 28 и 31оС, помимо указания температуры, относятся к фотопериоду 16 ч освещения: 8 ч темноты при интенсивности освещения 20 мкEm-2.с-1 (=1670 лк).
10.2. Стерилизация и высевание семян.
Семена хлопка (Gossypium hirsutum Кокер 310) очищали от хлопкового пуха, помещая их в концентрированную H2SO4 на 2 мин. Затем семена 4 раза промывали стерильной, дистиллированной водой, погружали в 95% этанол, стерилизовали на пламени и высевали на среде 1 при 31оС.
10.3. Индуцирование каллюса. Через семь дней после высевания гипокотили сеянцев отрезали, надрезали в продольном направлении, разрезали на 2 мм части и помещали на среду #2 при 31оС. В недельный срок частички гипокотиля (2 мм) переносили на свежую Среду #2 и эти культуры также выдерживали при 31оС. После 4-недельных переносов на среду #2 пролиферацию каллюсовой ткани на частицах гипокотиля удаляли с исходного трансплантата и помещали на среду # 3 при 31оС. Каллюс переносили на свежую среду #3 через месяц и выдерживали еще в течение 1-2 мес.
10.4. Индуцирование суспензионной культивации, Для инициирования суспензионных культур 100 мг каллюсовой ткани помещали на 35 мл среды #4 в колбе Делонга емкостью 125 мл. Суспензии перемешивали в течение 6 недель со скоростью мешалки 140 об/мин при 28оС и в течение этого времени они начинали быстро пролифирировать.
10.5. Развитие эмбрионов и регенерация растений. Эмбрионы, образовавшиеся в среде #4, пролифирируют даже быстрее при замене среды #4 на среду #5. Такую эмбриогенную суспензию разделяли и субкультивировали каждые 3-7 дней на свежей среде #5. Для развития эмбрионной пролиферации в среде #5 эмбрионы промывали средой #6 и затем помещали в эту среду. Через 3-4 недели после переноса в среду # 6 зрелые эмбрионы помещали на твердую среду при 25оС. Твердая среда состоит из модифицированной среды MS, содержащей MS соли, в которых 40 мМ KNO3используют вместо KNO3 и NH4NO3, В-5 витамины, 2% сахарозы, 15 мМ глютамина и затвердевает в присутствии 0-2% Герлита (pH 5.7). Эмбрионы помещали в чашки Петри при 25оС. Развитие побегов на такой среде протекает спорадически, а удлинение корней усиливается при переносе эмбрионов на указанную выше среду MS, не содержащую глютамина. Проросшие эмбрионы затем высевали в чашки с вермикулятом и накрывали стаканом (25оС). После появления на вермикулите побегов стакан снимали. Через неделю пребывания при 28оС побеги помещали в теплицу для дальнейшего развития в растения.
П р и м е р 11. Среда для прорастания семян и развития каллюса. (Композиция на основе модифицированного основного раствора Вайта (1961), на который ссылаются в настоящем описании (табл. 1).
П р и м е р 12. Среда для роста/сохранения каллюса. (Композиция на основе основных растворов Мурашига и Скуга (MS) (табл. 2).
П р и м е р 13. Среда для всхода растений. (Композиция на основе основных растворов Бизли и Тинга (1973)) (табл. 3).
П р и м е р 14. Регенерация растений исходя из эксплантатов котиледона.
Семена AcaIa хлопковой разновидности SI2 Gossypium hirsutum стерилизовали путем контактирования с 95% спиртом в течение 3 мин, затем дважды промывали стерильной водой и погружали в 15%-ный раствор гипохлорита на 15 мин, после чего промывали стерильной водой. Стерилизованные семена проращивали на среде базального агара в темноте, в течение 14 дней с целью появления сеянцев. Котиладоны сеянцев разрезали на сегменты размером 2-4 мм2, которые переносили асептически в среду, индуцирующую каллюс (см. выше), состоящую из основных и вспомогательных солей Мурашига и Скуга (МS) и дополненных 0,4 мг/л тиамин-HCl, 30 г/л глюкозы, 2,0 мг/л нафталинуксусной кислоты (NAA), 1 мг/л кинетина, 100 мг/л м-инозита и агаром (0,8%). Полученные культуры инкубировали при 30оС в условиях 16-часового освещения и 8-часового периода темноты в инкубаторе Персиваля, снабженного флуоресцентными лампами (холодный дневной свет) с интенсивностью света 2000-4000 лк. За 3-4 недели на культивированных тканевых сегментах образовывался каллюс, имеющий цвет от белого до зеленовато-серого. Образовавшиеся наплывы субкультивировали каждый три-четыре недели в среде для роста каллюса, содержащей MS среду, включающую 100 мг/л м-инозита, 20 г/л сахарозы, 2 мг/л нафталинуксусной кислоты (NAA) и агар. Через 4-6 мес после первого помещения в среду соматические эмбрионы образуют тканевые эксплантаты на среде для индуцирования каллюса. Каллюс и эмбрионы выдерживают на среде для роста каллюса путем субкультивации в свежей среде для роста каллюса каждые три-четырех недели.
Соматические эмбрионы, которые образуются на кусочках ткани, эксплантируют в свежую среду для роста каллюса или в среду Бизли и Тинга (среда для прорастания эмбрионов).
Соматические побеги, которые образуются из соматических эмбрионов, переносили в среду Бизли и Тинга, которая содержала 1200 мг/л нитрата аммония и 500 мг/л казеинового гидролизата в качестве источника органического азота. Такая среда отвердевала под воздействием отвердителя (Герлит) и побеги помещали в ящики Magenta.
Cоматические эмбрионы развиваются в побеги примерно за три месяца. Побеги давали корневую систему на стадии 6-8 листьев (длиной 7,5-10 см) и их переносили в почву, выдерживали в инкубаторе в условиях высокой влажности в течение 3-4 недель, после чего переносили в теплицу. После того, как растения окрепли, их переносили в открытую вспаханную почву.
П р и м е р 15. Регенерация растений исходя из котиледоновых эксплантатов. Вариант I.
Повторяли методику, описанную в примере 15, используя в качестве среды систему MS, концентрация компонентов которой была уменьшена в половину. Получены практически аналогичные результаты.
П р и м е р 16. Регенерация различных разновидностей хлопка из котиледоновых трансплантатов.
Методику примеров 15 и 16 повторяли на разновидностях хлопка AcaIa SI4, SI2C-1, GC510, B1644, B2724, B1810, сборной разновидности Siokra и обрывной разновидности F02017. Во всех случаях была проведена успешная регенерация.
П р и м е р 17. Регенерация хлопковых растений из котиледоновых трансплантатов при использовании в качестве промежуточной стадии суспензионной культивации клеток.
Методику примера 15 повторяли до стадии получения каллюса, способного к образованию соматических эмбрионов.
Кусочки активно растущего эмбриогенного каллюса массой 750-1000 мг суспендировали в 8 мл единицах жидкой суспензионной культурной среды, включающей MS основные и побочные соли, дополненные 0,4 мг/л тиамина HCl, 20 г/л сахарозы, 100 мг/л м-инозита и нафталинуксусной кислотой (2 мг/л) в Т-образных пробирках и полученную смесь помещали в барабан, вращающийся со скоростью 1,5 об/мин в режиме освещение: темнота 16:8. Интенсивность освещения 2000-4500 лк обеспечивалась флуоресцентными лампами (холодный дневной свет).
Через четыре недели суспензию отфильтровывали через нейлоновую сетку с отверстиями в 840 мкм с целью удаления крупных сгустков клеток. Фракцию с размером частиц менее 840 мкм осаждали, промывали 20-25 мл свежей суспензии культурной среды. Такую клеточную суспензию переносили в Т-пробирки (по 2 мл на пробирку) и каждую пробирку разбавляли 6 мл свежей суспензионной культурной среды. Культуры выдерживали, повторяя указанные выше операции с интервалами 10-12 дней. Для каждой субкультуры суспензию отфильтровывали и лишь фракцию, содержащую агрегаты клеток мельче 840 мкм переносили в свежую суспензионную культурную среду. Во всех случаях фракцию, содержащую сгустки клеток размером более 840 мкм, помещали в среду для роста каллюса с целью получения зрелых соматических эмбрионов.
Соматические эмбрионы, образующиеся на среде для роста каллюса, удаляли и переносили в среду для прорастания эмбрионов. В соответствии с описанным в примере 15 происходило их прорастание, развитие в побеги и далее в выращенные в полевых условиях растения.
П р и м е р 18. Регенерация хлопковых растений из котиледоновых трансплантатов при использовании в качестве промежуточной стадии суспензионной культивации клеток. Вариант I.
Повторяли методику примера 18, за исключением того, что суспензионные культуры образовывались в результате переноса 750-1000 мг эмбриогенного каллюса в колбу Де-Лонга, содержащую 15-20 мл жидкой среды MS, включающей 2 мг/л NAA. Колбу, содержащую культуру, помещали на роторный встряхиватель, работающий с частотой 100-110 встряхиваний в 1 мин. Через три недели суспензию отфильтровывали через нейлоновую сетку с размером отверстий 840 мкм с целью удаления крупных клеточных сгустков для роста растений в соответствии с методикой примера 18. Суспензии с частицами меньше 840 мкм давали оседать, промывали жидкой средой MS и повторно суспендировали в 2-5 мл жидкой среды MS. Такую суспензию подвергали субкультивации путем переноса в свежую среду, находящуюся в колбе Де-Лонга, содержащую 1-2 мл суспензии и 15 мл свежей жидкой среды MS. Культуры выдерживали, повторяя описанную выше методику с интервалам 7-10 дней. Субкультивации подвергали только суспензии с размером частиц менее 840 мкм, а большие сгустки (840 мкм и более) использовали для роста растений.
П р и м е р 19. Продуцирование растений из крупных колоний суспензионных культивированных клеток.
После трех или четырех субкультиваций согласно методу роста в суспензии, описанному в примерах 18 и 19, 1,5-2,0 мл клеточной суспензии из Т-пробирок и колбы Де-Лонга высевали на отвержденную агаром среду MS, содержащую 2 мг/л NAA и среду Бизли и Тинга, содержащую 500 мг/л казеинового гидролизата. Через три-четыре недели эмбриогенный каллюс с развивающимися эмбрионами становился видимым. Клеточные сгустки с размером частиц более 840 мкм высевали на среду для роста каллюса, в результате чего образовывались эмбриогенные колонии с развивающимися эмбрионами, которые вырастали в растения.
П р и м е р 20. Трансформация клеток хлопковой суспензионной культуры в опухолевый фенотип под действием Agrobacterium LBA 4434.
20.1. Пост суспензионной культуры растений.
Суспензионную культуру хлопка AcaIa (описанную в примере 18) подвергали субкультивации в "Т" пробирках в присутствии среды (среда MS, содержащая 2 мг/л NAA), которую заменяли каждые семь-десять дней. После замены "Т" пробирку вращали при 90оС и клеткам давали оседать. Верхний слой удаляли пипеткой перед трансформацией и полученные в результате клетки обрабатывали в соответствии с описанным ниже.
20.2. Описание вектора Agrobacterium.
Agrobacterium штамм LBA 4434 (Хоекема с сотр., 1983) содержит бинарную растительную трансформационную систему на основе Ti плазмиды. В такой бинарной системе одна плазмида содержит Т-ДНК Ti-плазмиды, вторая плазмида содержит vir область Ti плазмиды и совместное действие двух этих плазмид заключается в трансформации растения. В Agrobacterium штамме LBA 4434. Т-ДНК плазмида pAL1050 cодержит TLpTiAch5 октопиновую Ti-плазмиду. vir-плазмида в штамме LBA 4434, pA14404, содержит интактные вирулентные области pTiAch5 (ОсмС с сотр., 1982). Штамм LBA 4434 получен от д-ра Роберта Шилперурта, Биохимическое отделение Лейденского Университета, Голландия.
20.3. Рост Agrobacteria. Трансформирующий Agrobacterium штамм отбирали из глицеринового основного раствора, инокулированного в малой культуре после выдержки в течение ночи, 50 мл этой культуры инокулировали на следующий день Agrobacteria, выращивали на среде УЕВ (УЕВ в расчете на 1 л воды содержат 5 г говяжьего экстракта, 1 г дрожжевого экстракта, 5 г пептона, 5 г сахарозы, при pH 7,2 установленным с помощью NaOH). После выдерживания в автоклаве добавляли 1 мл 2М раствора MgCl2, к которому, если нужно, добавляли антибиотики. Регистрировали оптическую плотность 50 мл культуры ночной выдержки при длине волны 600 нм (ОД600), культуру центрифугировали и гранулы повторно суспендировали в растительной среде для роста клеток (среда MS плюс NAA в количестве 2 мг/л) до конечной оптической плотности 0,5 при длине волны 600 нм. 8 мл такой бактериальной суспензии добавляли в каждую "Т" пробирку, содержащую растительные клетки из раздела 21.1.
20.4. Инфецирование. Содержимое "Т" пробирки, состоящее из растительных и бактериальных клеток, перемешивали для повторного суспендирования всех клеток и возвращали во вращающийся барабан на три часа с тем, чтобы Agrobacteria присоединились к растительным клеткам. Затем клеткам давали отстаиваться и верхний слой удаляли. В каждую "Т" пробирку добавляли свежую аликвоту растительной среды и содержимое пробирок инкубировали во вращающемся барабане в течение 18-20 ч в присутствии остаточного количества Agrobacteria. После этого клеткам снова давали оседать, верхний слой удаляли и клетки дважды промывали раствором среды для роста, содержащей цефтотаксим (200 мкг/мл). После промывaния клетки из каждой Т-пробирки повторно суспендировали в 10 мл растительной среды, содержащей цефотаксим (200 мкг/мл), и 1 мл аликвоты таких суспензий высевали на чаши Петри.
20.5. Рост трансформированных клеток. Клетки инфецированные Agrobacterium растут в растительной среде в отсутствиe фитогормонов, что указывает на тот факт, что ткань получила гены фитогормонов в Т-ДНК. Такие клетки развиваются в опухоли, что является дополнительным указанием на трансформацию культур.
П р и м е р 21. Трансформация клеток хлопковой суспензионной культуры в канамицин-устойчивый не опухолевый фенотип.
Следовали методике примера 20, но использовали различные трансформирующие Agrobacterium и селекционную среду, которая содержала антибиотик для селекции трансформированных растительных тканей.
21.1. Рост растительной ткани. Повторяли методику, описанную в части 21.1 примера 21.
21.2. Описание Agrobacterium вектора. Трансформирующие Agrobacteria содержат Т-ДНК, включающую бинарный вектор pC1B10 (Ротштейн с сотр., 1987), а также pA14404 vir-плазмиду, Т-ДНК pC1B10 содержит химерный ген, включающий промотор нопалин-синтазы, кодирующую область Тn5 (кодирующую энзим неомицин фосфотрансферазу и терминатор нопалин синтазы. Agrobacterium штамм IBA4404, содержащий vir-плазмиду-помощника pA14404 (описанную выше), получали также от д-да Шлиперурта.
21.3. Рост Agrobacteria.
Agrobacteria, содержащие pC1B10, выращивали на УКВ, содержащей канамицин (50 мкг/мл). Другие условия были теми же, что в части 21,3 примера 21.
21.4. Инфецирование. Трансформацию проводили в соответствии с методикой, описанной в примере 21, по 1 мл аликвоты, полученные в части 21,3, сразу же высевали на среду, содержащую селективные антибиотики. Среда для селекции содержала канамицин (50 мкг/мл) или С418 (25 мкг/мл). Экспрессирование nos/ned nos химерного гена в ткани трансформированных растений позволяет осуществлять селекцию такой ткани на любой из указанных антибиотиков.
21.5. Рост трансформированной ткани. Среда для роста растений в этом и следующих примерах содержит фитогормоны, указанные в примере 15. Через 2-4 недели трансформированная ткань становится видимой на селекционных пластинах. Неинфецированная ткань или контрольная ткань не проявляет признаков роста, становится темной и погибает. Трансформированная ткань очень хорошо растет в присутствии канамицина или С418. Кусочки хорошо растущей ткани субкультивируют в свежую селекционную среду.
21.6. Рост соматических эмбрионов. На таких кусочках ткани образуются соматические эмбрионы. Соматические эмбрионы эксплантировали в свежую среду (не селективная среда).
21.7. Прорастание. Когда эмбрионы начинают размножаться и прорастать, т. е. к моменту образования корней и появления 2 или трех листьев, их переносят в боксы Magenta, содержащие растительную среду. Осуществляют рост до стадии образования на всходах 6-8 листьев и в этот момент их извлекают из агаровой среды.
21.8. Рост всходов. Всходы помещают в почву, находящуюся в стаканчиках, накрывают химическим стаканом для поддержания влажности и помещают в инкубатор Персиваля на 4-8 недель. После этого стакан снимают и растение переносят в теплицу.
21.9. Рост растения в теплице. Растения в теплице растут, цветут и сбрасывают семена.
П р и м е р 22. Трансформация клеток хлопковой суспензионной культуры в глифосат-толерантный фенотип.
Повторяли методику, описанную в примере 21, за исключением изменений, отмеченных ниже. Использовали различные трансформирующие Agrobacteria. После селекции ткани растений на действие антибиотика с целью селекции трансформированных материалов их также дополнительно подвергали селекции на устойчивость к действию гербицида.
22.1. Рост растительной ткани. Повторяли методику, описанную в части 21.1 примера 21.
22.2. Описание вектора Agrobacterium. Трансформирующие Agronacteria содержат Т-ДНК вектор pMC85/587 (Филлати с сотр., 1987), а также pAI4404 vir-плазмиду. Плазмида pMC85/587 несет три химерных гена, способных к экспрессированию в растения. Два их этих генов кодируют неомицин фосфотрансферазу (NPT), которая придает устойчивость к действию канамицина или С418. Третий химерный ген, содержащий кодирующую последовательность мутантного aroA гена Salmоnella typhimurium, придает толерантность к гербициду глифосату (Коман с сотр., 1983).
22.3. Рост Agrobacteria. Agrobacteria, cодержащие pPMC85/587, выращивали на среде, содержащей канамицин (100 мкг/мл).
22.4. Инфецирование. Трансформацию осуществляли в соответствии с методикой, подробно описанной в примере 21, за тем исключением, что 1 мл аликвоты, полученной в части 21,3, сразу высевали на среду, содержащую селективные антибиотики. Такая селекционная среда содержит канамицин (50 мкг/л) или С418 (25 мкг/л). Экспрессирование NPT химерного гена в трансформированной растительной ткани позволяет проводить селекцию ткани на любой из указанных антибиотиков.
22.5. Рост трансформированной ткани. За 2-4 недели трансформированная ткань становится видимой на селекционных пластинах. Растительный материал вначале подвергали селекции на канамицин.
Затем растительную ткань (индивидуальные эмбрионы или каллюс) помещали на среду, содержащую гербицидный глифосат. Трансформированная ткань продолжала хорошо расти.
П р и м е р 23. Трансформация клеток хлопковой суспензионной культуры в гигромицин-устойчивый не опухолевый фенотип.
Повторяли методику, описанную в примере 22, за исключением специально указанных случаев. Использовали различные трансформирующие Agrobacteria и среда для селекции растений содержала антибиотик, подходящий для селекции трансформированной растительной ткани.
23.1. Рост растительной ткани. Проводили по методике, описанной в части 21.1 примера 21.
23.2. Описание Agrobacterium. Трансформирующие Agrobacteria содержат Т-ДНК, включающую бинарный вектор pC1B2115 (Ротштейн с сотр., 1987), а также vir-плазмиду. Т-ДНК pC1B2115 содержит химерный ген, состоящий из промотома и терминатора СаМV 35S матрицы (Оделл с сотр., 1985), и последовательность, кодирующую гигромицин В фосфотрансферазу (Гретц и Дэвис, 1983).
23.3. Рост Agrobacteria. Agrobacteria, содержащие рС1B2115, выращивали на УЕВ, содержащей канамицин (5 мкг/мл).
23.4. Инфецирование. Трансформацию проводили по методике примера 21, за тем исключением, что 1 мл аликвоты, полученной в части 21,3, сразу высевали на среду, содержащую селективные гербициды. Селекционная среда содержала 50 мкг/мл гигромицина. Экспрессирование химерного гигромицинового гена в трансформированную растительную ткань позволяет осуществлять селекцию такой ткани на среде, содержащей гигромицин.
23.5. Рост трансформированной ткани. Повторяли методику, описанную в части 22,5 примера 22, но гигромицин использовали в растительной среде для роста растений.
П р и м е р 24. Методика экстракции растения.
Растительную ткань гомогенизировали в экстракционном буфере (примерно 100 мг в 0,1 мл экстракционного буфера).
Буфер для экстракции листьев Na2CO3(pH 9,5) 50 мМ ЕДТА 10 мМ Тритон Х-100 0,05% Твин 0,05% NaCl 1000 мМ PMSF (добавляли перед применением) лейпептин (добавляли пе- ред использованием) 1мМ
После экстракции добавляли 2М Трис, pH 7,0, с целью установления pH экстракта на значении 8,0-8,5. Затем такой экстракт центрифугировали в течение 10 мин на микрофуге Бекмана и верхний слой использовали для анализа ELISA.
П р и м е р 25. Анализ растительной ткани методом ELISA.
Метод ELISA (иммуносорбентный анализ связанным энзимом) представляет собой очень чувствительный, специфический анализ на антигенный материал. Метод ELISA очень полезен для изучения экспрессирования полипептидных генных продуктов. Развитие методов ELISA как общего инструмента анализа описано Кларком с сотр., (1986); на эту работу ссылаются в настоящем документе.
Метод ELISA был разработан для Bt токсина с использованием стандартных методик и он используется для анализа трансгенного растительного материала на способность экспрессирования Bt последовательностей. Стадии, применяемые в таком методе, описаны ниже.
Среда и буферы.
EPBS (ELISA фосфатный буферный раствор)
10 мМ фосфата Nа: Na2HPO4 4,68 г/4 л.
Na2HPO4·H2O 0,976 г/4 л.
140 мМ NaCl NaCl 32,7 г/4 л.
pH следует поддерживать равным 7,4.
Боратный буферный рассол
100 мМ борной кислоты
25 мМ бората Na
75 мМ BaCl
Если необходимо, то pH поддерживают равным 8,4-8,5 с помощью HCl или NaOH.
ELISA блокирующий буфер в EPBS
1% BSA
0,02% азида Na
ELISA промывной буфер
10 мМ Трис-НCl, pH 8,0
0,05% Твина 20
0,02% азида Na
2,5М Трис
ELISA разбавитель
В EPBS:
0,05% Твина 20
1% BSA
0,02% азида Na
ELISA субстратный буфер
В 500 мл:
48 мл диэтаноламина
24,5 мг MgCl2
pH устанавливают равным 9,8 с помощью HCl.
ELISA cубстрат
15 мг п-нитрофенил фосфата в 25 мл субстратного буфера.
Методика.
1. Пластины для анализа ELISA предварительно обрабатывают этанолом.
2. Очищенную по сродству кроличью анти-Bt токсиновую антисыворотку (50 мкл) c концентрацией 3 мкг/мл в среде боратного буферного рассола добавляли на пластину и полученную систему инкубировали в течение ночи при 4оС. Антисыворотку получали в результате реакции на иммунизацию кроликов градиентно-очищенными Bt токсиновыми кристаллами (Энг и Никерсон, 1978), солюбилизированными натрий додецил сульфатом.
3. Проводят промывку ELISA промывным буфером.
4. Проводят обработку в течение 1 ч при комнатной температуре блокирующим буфером.
5. Промывают ELISA промывным буфером.
6. Добавляют растительный экстракт в количестве, обеспечивающем 50 мкг протеина (обычно примерно 5 мкл экстракта). Лиственный экстракционный буфер описан в примере 25, протеин определяли по методу Брэдфорда (Брэдфорд, 1976) с использованием выпускаемого промышленностью набора (Бис-Рад, Ричмонд, Калифорния). Если необходимо разбавление лиственного экстракта, то используют ELISA разбавитель. Полученную систему инкубируют в течение ночи при 4оС.
7. Проводят промывку ELISA промывным буфером.
8. Добавляют 5 мкл очищенной по сродству козьей анти-Bt токсиновой антисыворотки с концентрацией 3 мкг протеина/мл в ELISA разбавителе. Полученную систему инкубируют в течение 1 ч при 37оС.
9. Проводят промывку ELISA промывным буфером.
10. Добавляют 50 мкл кроличьего анти-козьего антитела, связанного с щелочной фосфатазой (выпускается фирмой Сигма Кэмикл Сэнт-Льюис-Мо.). Полученную смесь разбавляют разбавителем в соотношении 1:500. Систему инкубируют в течение 1 ч при 37оС.
11. Проводят промывание ELISA промывным буфером.
12. Добавляют 50 мкл субстрата (0,6 мг/мл п-нитрофенил фосфата в ELISA субстратном буфере). Систему инкубируют в течение 30 мин при комнатной температуре.
13. Обрывают реакцию добавлением 50 мкл 3М NaOH.
14. Регистрируют оптическую плотность при длине волны 405 нм в модифицированном оптическом устройстве ELISA (Хьюлетт Пакард, Стэнфорд, Кв). Растительная ткань, трансформированная с помощью pC1B10/35sbt (BcII) (см. фиг. 16) конструкции, дает положительную реакцию при анализе, выполненном методом ELISA, что указывает на экспрессирование Bt гена.
П р и м е р 26. Биоанализ трансформированного хлопка.
Кладку яиц Heliothis virescens на кусочках марли получали по контролю табачных насекомых при университете штата Северная Каролина, Ралей, Северная Каролина. Кусочки марли переносили в большой закрытый стеклянный стакан и инкубировали при 29оС в присутствии влажных бумажных салфеток для поддержания влажности. Вылупливание из яиц происходит за три дня. Сразу после вылупливания личинки переносят в маленькие закрытые пластиковые стаканы (по одной личинке на стакан). Каждый стакан содержит кружки хлопковых листьев. Личинки переносят с использованием кисточки для рисования.
Кружки листьев диаметром 1 см выштамповывают из листьев хлопковых растений и помещают на круг влажной фильтровальной бумаги, который кладут в чашку с личинками. От каждого растения испытывают по 6-10 листьев, представляющих как молодые, так и старые экземпляры. Кружки листьев заменяют с интервалами в два дня, или, если необходимо, кормят ими личинок. Скорость роста (размер или общий вес всех личинок) и смертность личинок, поедающих листья трансформированных растений, сравнивают с аналогичными показателями для личинок, питающихся листьями неинфецированного хлопка.
Личинки, питающиеся на дисках хлопка трансформированного pC1B 10/35 sbt (BcII), демонстрируют понижение скорости роста и увеличение смертности по сравнению с контрольными опытами.
П р и м е р 27. Конструирование pC1B1300 для высокого уровня экспрессии в растении.
Методами генной инженерии получали pC1B1300 для высокой степени экспрессирования Bt токсинового гена, она содержала нетранслированную лидерную последовательность 5' и Bt токсиновому гену для усиления степени экспрессирования Bt токсинового гена в растения. Нетранслированный лидер представляет собой последовательность 5' величиной 50 bp к кодону инициирования Bt токсинового гена и 3' к СаМV 35S нетранслированному лидеру. Конечную pC1B 1300 конструкцию получали путем внедрения лидера размером 40 bp и делеции Bt токсинового гена в BamHI сайт pC1B10/710 (фиг.19). Фрагмент NcoI-BamHI размером 1,9 kbp из рC1B10/35sbt (BcI) делеции очищали на агарозе с низкой температурой стеклования. Лидер размером 40 bp химически синтезировали как двутяжный олигонуклеотид с 5' выступающим BamHI сайтом и 3' выступающим NcoI сайтом с использованием ДНК-синтезатора (плайд Биосистемс). Последовательность нетранслированного лидера, показанная в центре, фиг.19, получена из покровного протеина нетранслированного лидера альфа-мозаичного вируса (АМУ), описанного Копер-Швартхофф с сотр., (1977). Лидер размером 40 bp, Bt фрагмент размером 1,9 kbp и BamHI линеаризованный pC1B710 вектор соединяли тройным легированием с использованием Т4 ДНК лигазы с целью конструирования pC1B1300.
П р и м е р 28. Выделение кДНК клонов, кодирующих небольшую субъединицу RuВРС-азы в хлопке.
Растения Gossypium hirsutum (FunK линия PF522) выращивали из семян в теплице при 14-часовом дневном освещении. Общую РНК выделяли из молодых зеленых листьев по методике Ньюбари и Поссингхама (1977). ПолиА+RНК очищали согласно Миниатису с сотр. (1982), с.197. Двухнитевую сДНК (комплементарная ДНК( синтезировали согласно методике Окаяма и Берга (1982) при следующих измерениях:
А. Первый тяж сДНК затравливали олиго-dT;
B. После разветвления двухтяжной сДНК с олиго-dG с использованием нуклеотидил-трансферазы ее клонировали в олиго-dC, разветвленный pYC9 (сайт PStI от фирмы Фармация), и отжигали; и С.ДНК трансформировали в E.coli штамм НВ101.
Поскольку в присутствии хлорофил а) b связующего протеина (Cab)RuBP-аза представляет собой наиболее широкораспространенный протеин в зеленых листьях, сДНК-библиотеку затем подвергали скринингу на сДНК клоны наиболее распространенных мРНК. Нитроцеллюлозные (Глейхер и Шуэлл) фильторные копии сДНК клонов подвергали скринингу в присутствии первого сДНК тяжа, меченного радиоактивным изотопом а-dCT32p и реверстранскриптазой, причем матрицей была та же полиA+RНК, что использовалась для конструирования сДНК-библиотеки. Отбирали шесть сДНК клонов, начиная с 275, и подвергали их дальнейшему анализу.
Анализ по Нортерну, выполненный и описанный в статье Маниатиса с сотр., 1982, с. 202), показал, что два из таких кДНК клонов гибридизированы с классом RНК длиной 1100 нт. Они перекрестно гибридизируются с Cab генным зондом из табака. Четыре других клона гибридизируются с классом мРНК длиной 900-1000 нт, размер которых соответствует размеру rbcs (мелкая субъединица Rubisco) мРНК хлопковых листьев, после гибридной селекции с использованием одного из этих четырех сДНК клонов, выделяется и транслируется in vitro (как описано в статье Маниатиса 1982, с.329) с использованием in vitro трансляционного набора кроличьих ретикулоцитов (Промега Биотек.). В результате электрофореза продуктов трансляции на полиакриламидном геле установлено наличие одного главного полипептида с мол.м. 20 кД, который соответствует мол. м. предшественника RuВРС-азы. Другие три кДНК клона перекрестно гибридизируются с клоном, используемым в экспериментах по секреции гибрида.
В больших порциях таких кДНК клонов устанавливали последовательность с использованием дидеокси-цепного терминаторного метода (Сангер с сотр., 1977) после субклонирования в М13. Сравнение их последовательностей с ранее опубликованными rbcs последовательностями других разновидностей показало, что они действительно являются rbcs кДНК клонами.
П р и м е р 29. Выделение геномных клонов мелкой субъединичной RuВР-азы хлопка.
29.1. Геномный анализ хлопка по Саузерну.
Геномные пятна по Саузерну готовили стандартными методами с использованием нитроцеллюлозных фильтров. Условия предгибридизации, гибридизации и промывки описаны Классигом с сотр. (1983). Геномный анализ по Саузерну с использованием в качестве зонда rbcs кДНК клона, обнаружил наличие 4-5 геномных фрагментов в зависимости от рестрикционного энзима, применяемого для переваривания ДНК. Установлено, что rbcs мультигенное семейство хлопка содержит по крайней мере 5 членов.
29.2. Выделение rbcs геномных клонов.
Для конструирования геномной библиотеки хлопка, частичные sau3A перевары хлопковой геномной ДНК фракционировали по размеру на 10-40% сахарозном градиенте и легировали в λ ЕСВ13 цепи (Стратаген), переваренные в BamHI. После уплотнения λ рекомбинантов, выполненного с использованием набора Пакаген (Стратаген), проводили трансфекцию E.coli K802. Дупликатные реплики на нитроцеллюлозном фильтре подвергали скринингу по методике, описанной на с. 320 книги Маниатиса с сотр., (1982), с использованием в качестве зонда rbcs с ДНК клона, описанного выше. Очищали двенадцать положительных клонов из 450 000 пятен. ДНК выделяли из посевных лизатов таких рекомбинантных фагов в соответствии с описанным на с.80 работы Маниатиса с сотр. (1982).
После сравнения таких геномных клонов по их образцам ограничительного перевара с различными энзимами идентифицировали пять различных rbcs генов, каждый из них субклонировали в плазмидный вектор pB M13+ (Статаген). Такие субклоны затем картировали и частично устанавливали последовательность с тем, чтобы локализовать 5' окончание гена и первый АТG (сайт начала трансляции). Карта двух из таких геномных субклонов, rbc-gx и rbc-gv показана на фиг. 23. λ ЕМВ13 фаги, содержащие геномную ДНК субклонов rbc-gx и rbc-gv, депонированы в Международной депонентной коллекции американских типовых культур. Роквил, Мэрилэнд.
29.3. Исследование уровня экспрессии rbcs генных фрагментов в листья хлопка.
Сорок один дополнительный rbcs кДНК клон выделяли из библиотеки кДНК листьев хлопка. В результате анализа методом рестрикционного картирования, установления последовательностей и гибридизации таких кДНК клонов с ген-специфическими зондами был сделан вывод, что ген, принадлежащий геномному клону rbc-gx, ответственен за, примерно, 17% rbcs матриц хлопковых листьев.
П р и м е р 30. Конструирование химерных генов с использованием хлопкового rbcs промотора.
30.1. Внедрение NcoI сайта по месту первого АТG rbcs генов, кодирующих переходные пептиды.
Последовательность переходных пептидов rbc-gx и rbc-gv показаны на фиг. 25 и 24 соответственно, сайт NcoI расщепления (CCATGG) вводили по месту первого АТG таких двух генов, кодирующих переходный пептид. Эту операцию осуществляли клонированием pstI-EcoRI фрагмента гена rbc-gx и xbaI-SphI фрагмента гена rbc-gv (заштрихованные фрагменты на фиг.21 и 22 соответственно) в mp18 и mp19, соответственно и с использованием описанной выше стандартной методики управляемого олигонуклеотидным сайтом мутагенезa с целью введения NcoI сайта.
30.2. Конструирование pC1B 1301, плазмиды, несущей химерный ген, содержащий делетированный Bt протоксиновый ген (607 делеция) с rbc-gx генным промотором.
После сайт-специфического мутагенеза репликативную форму (dsrf) двухтяжной ДНК выделяли из М13 клона и затем переваривали с Hind III и EcoRI. Hind III EcoRI фрагмент, содержащий rbc-gx промотор, легировали совместно с Hind III и EcoRI, переваренной плазмидой pYC19, и затем легирующую смесь трансформировали в E.coli штамма НВ101. Плазмидную ДНК выделяли из трансформантов, подвергнутых селекции по ампициллину, и переваривали с Hind III. Концы молекулы делали тупыми в результате обработки субъединицей Кленова ДНК полимеразы I и с этими концами легировали Sal I линкеры. Полученную в результате линейную молекулу переваривали с Sal I и Nco I и очищали на геле. В результате тройного легирования очищенный на геле Sal I-Nco I фрагмент присоединяли к очищенному на геле BamHI-Sal I фрагменту из pC1B770, репликону с широким набором хозяев, используемому в качестве Agrobacterium Ti плазмидного клонирующего вектора (Ротштейн с сотр., 1987) и очищенному на геле NcoI-BamHI фрагменту, содержащему усеченный Bt ген на 607 аминокислот, легирующую смесь трансформировали в E.coli штамм НВ101. Полученную в результате плазмиду, pC1B1301, которая изображена графически на фиг.19, 20 и 21, подвергали селекции на канамицин.
30.3. Конструирование pC1B1302, плазмиды несущей химерный ген, содержащий делетированный Bt протоксиновый ген (607 делеция) с rbc-gv генным промотором.
После мутагенеза, двухнитевую реплекативную форму (dsrf) ДНК выделяли из М13 клона, который затем переваривали с Хba- I-NcoI. Фрагмент NcoI-BamHI размером, примерно, 1,97 kbp, содержащий делетированный протоксиновый ген, затем легировали совместно с XbaI-NcoI rbc-gv промоторным фрагментом, по трехмаршрутной реакции, в ХbaI-BamHI расщепленный pC1B10/710. Полученную в результате плазмиду, pC1B1302, структура которой показана на фиг.22, подвергали селекции на канамицин.

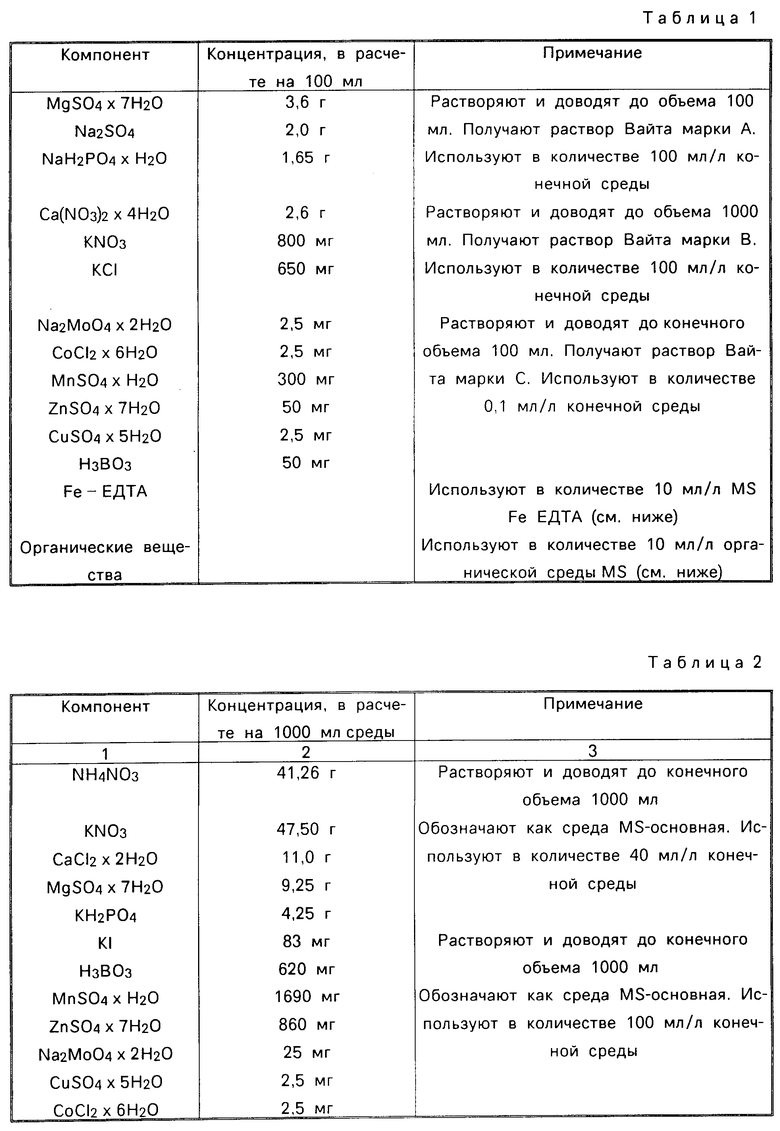

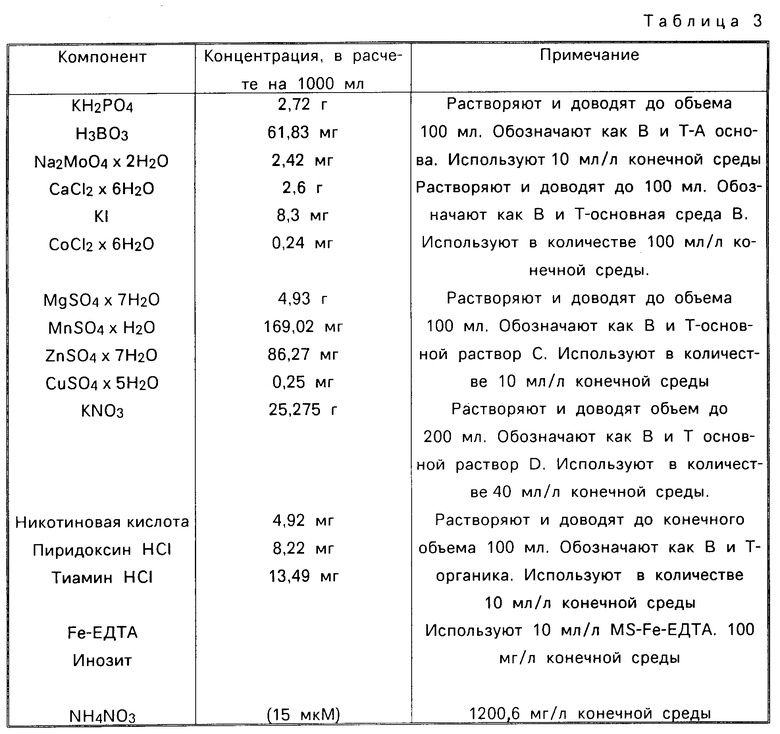




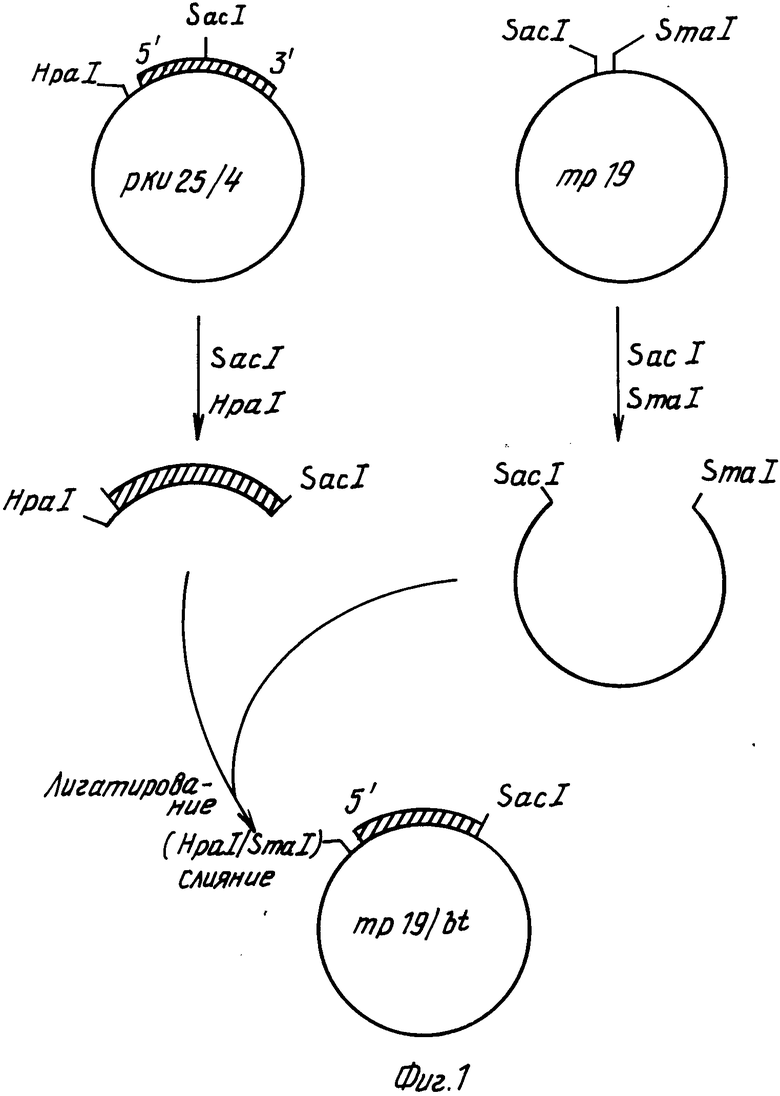
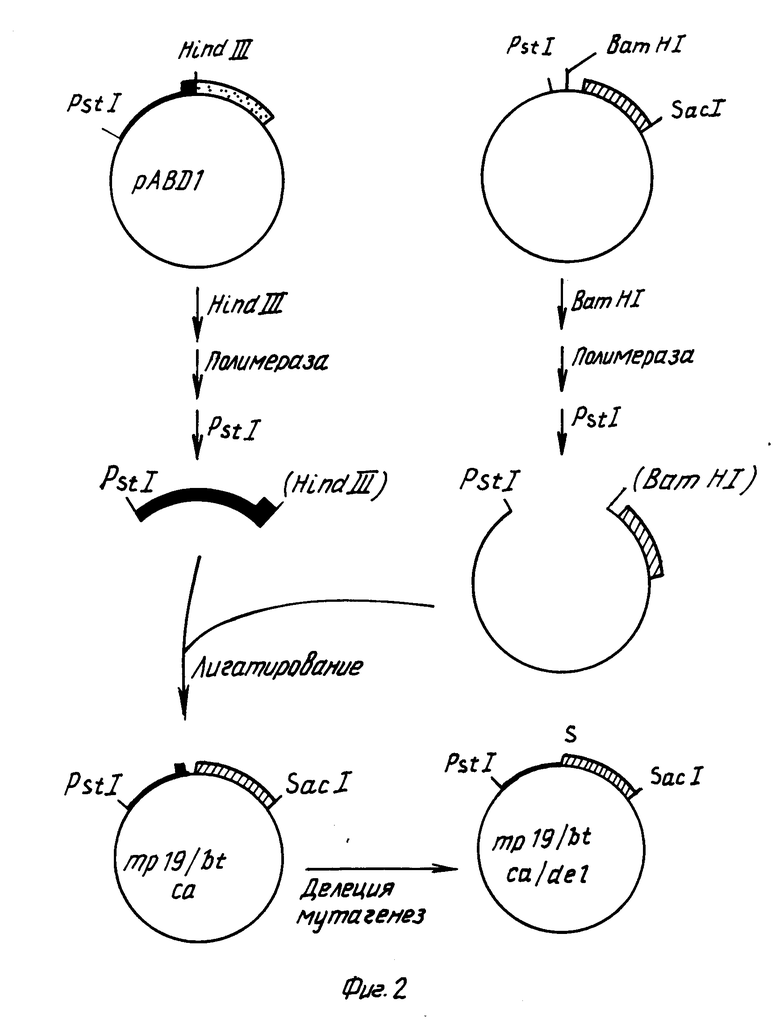
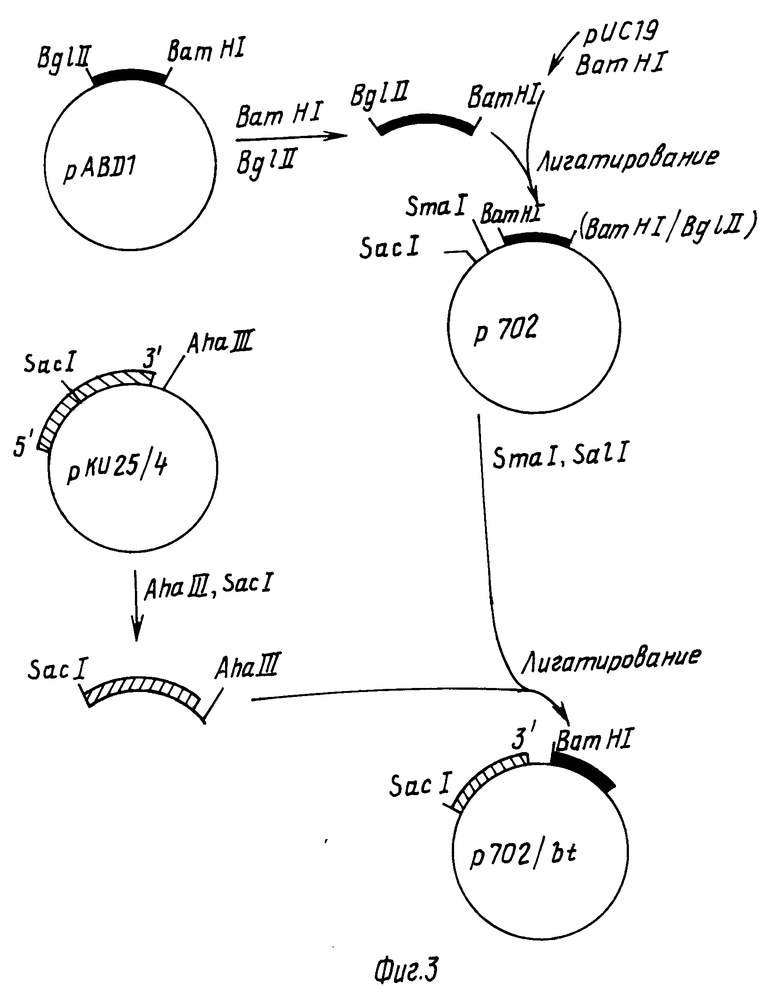
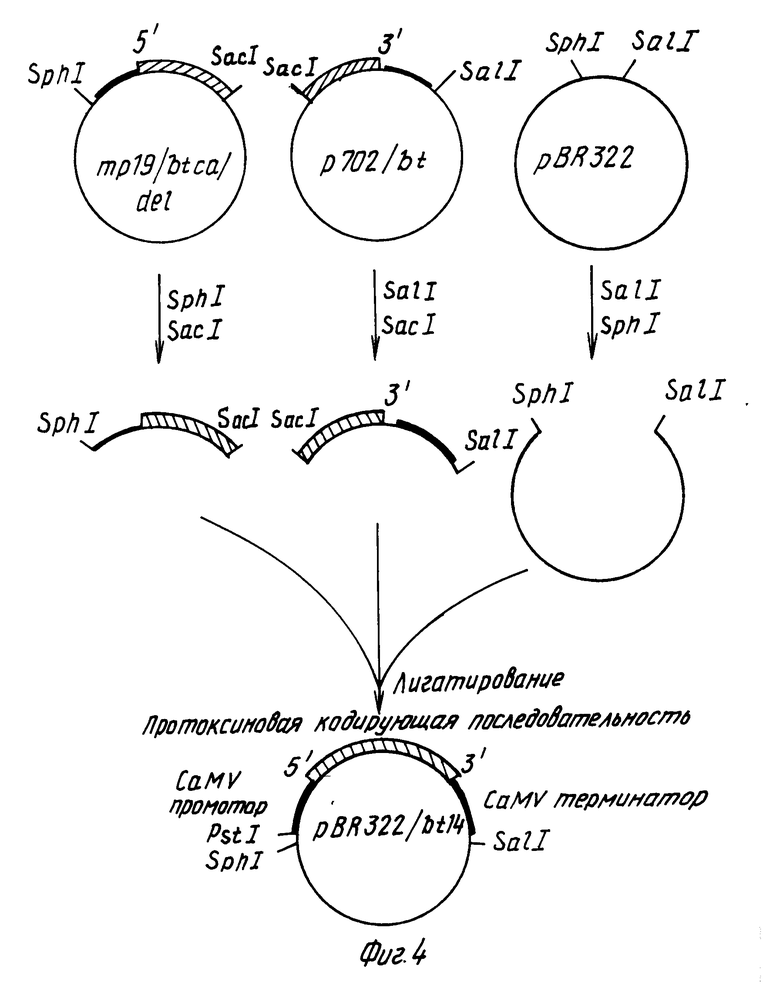
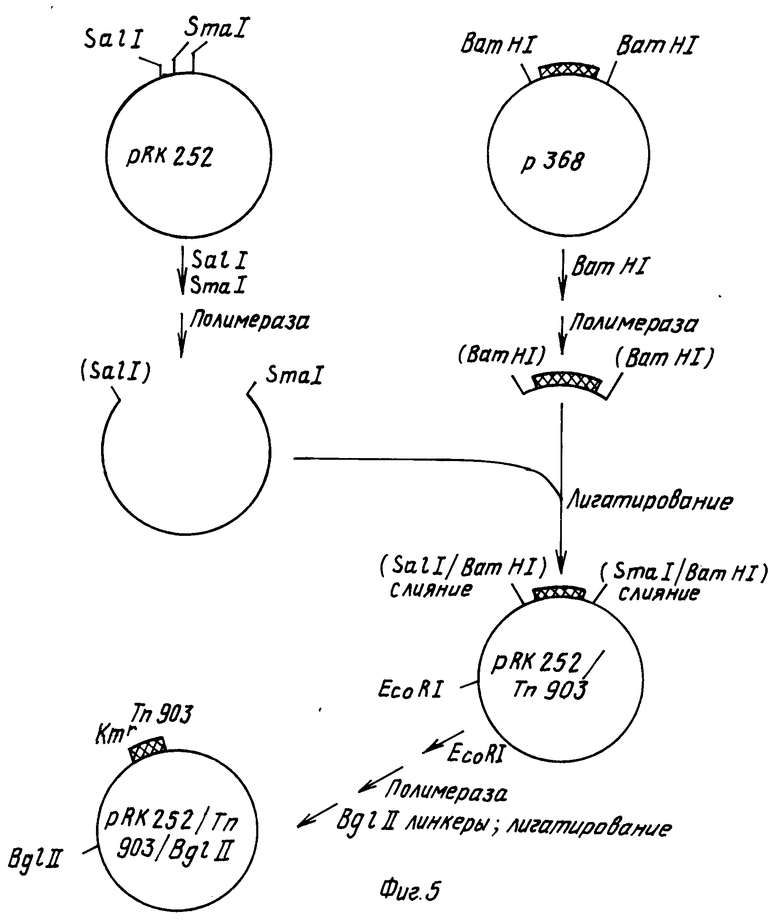
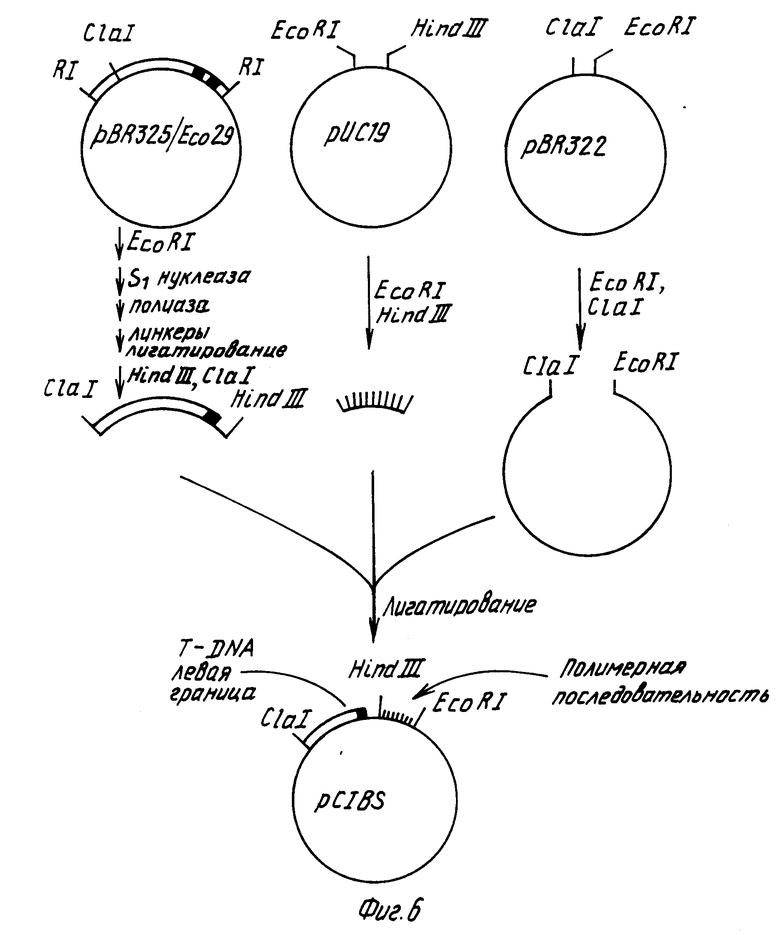
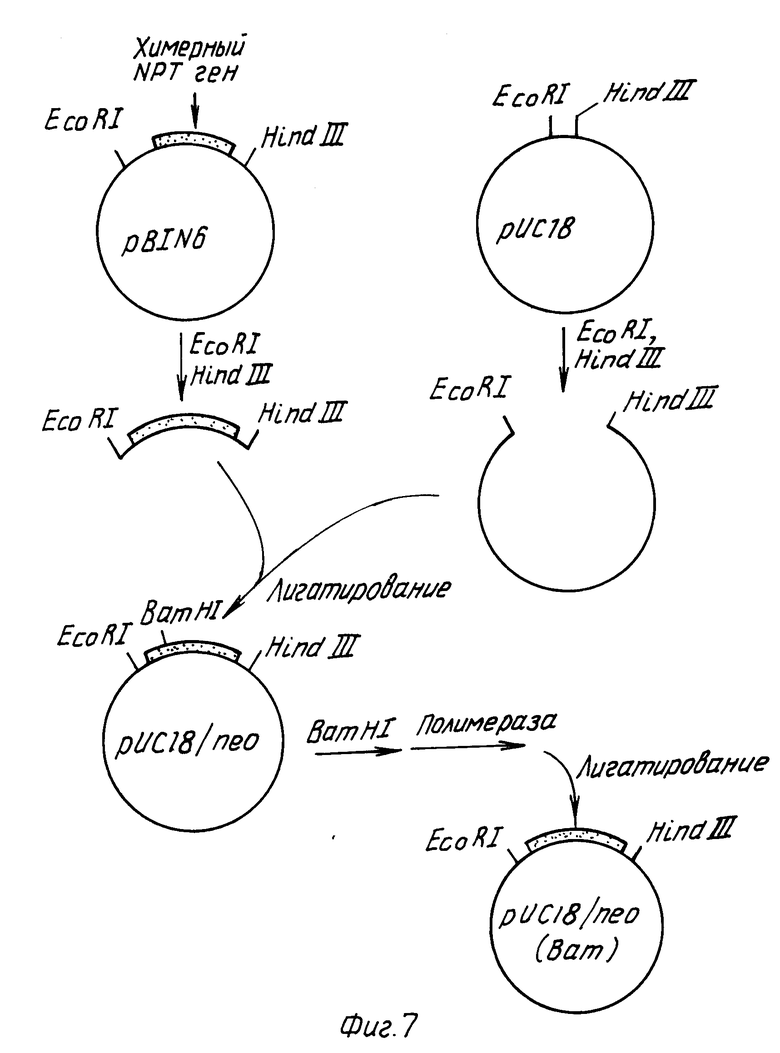
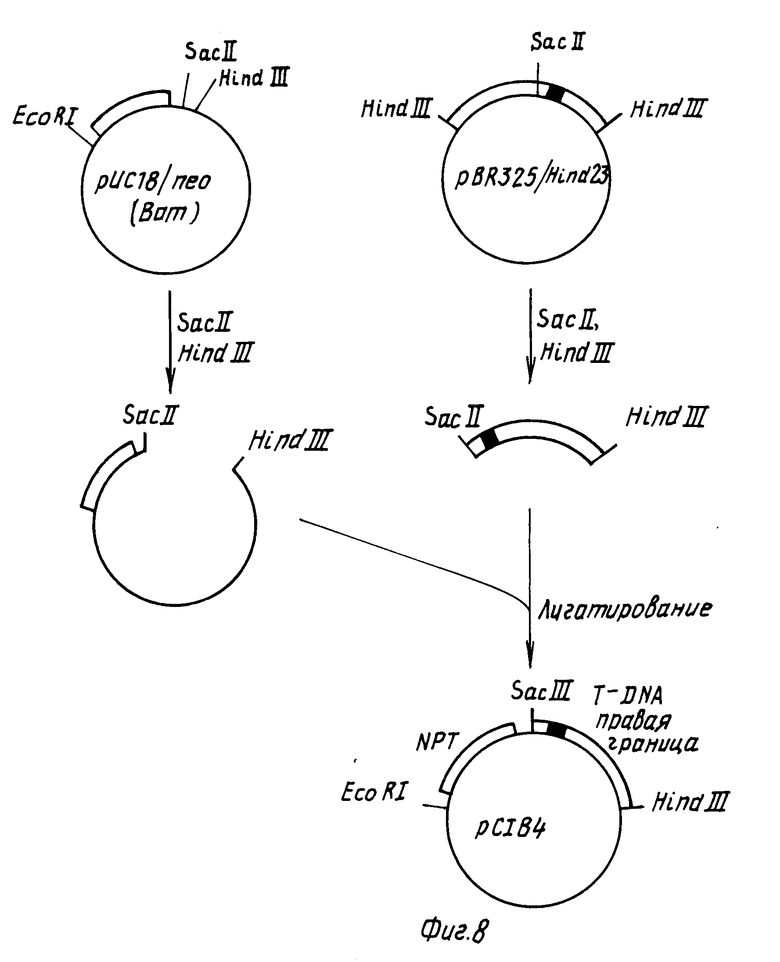
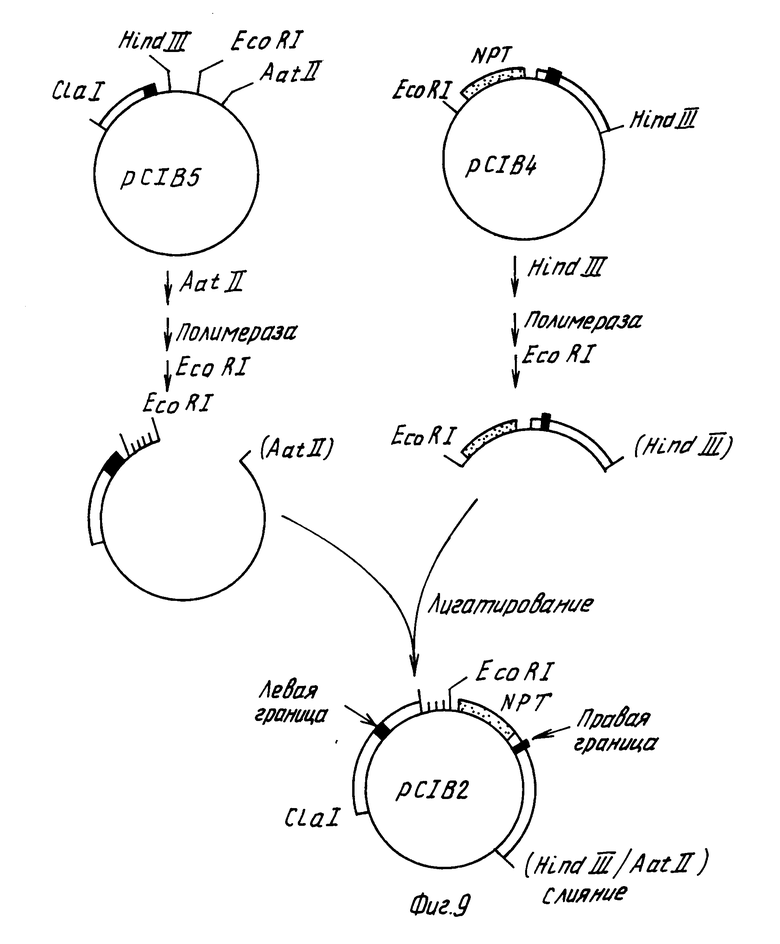
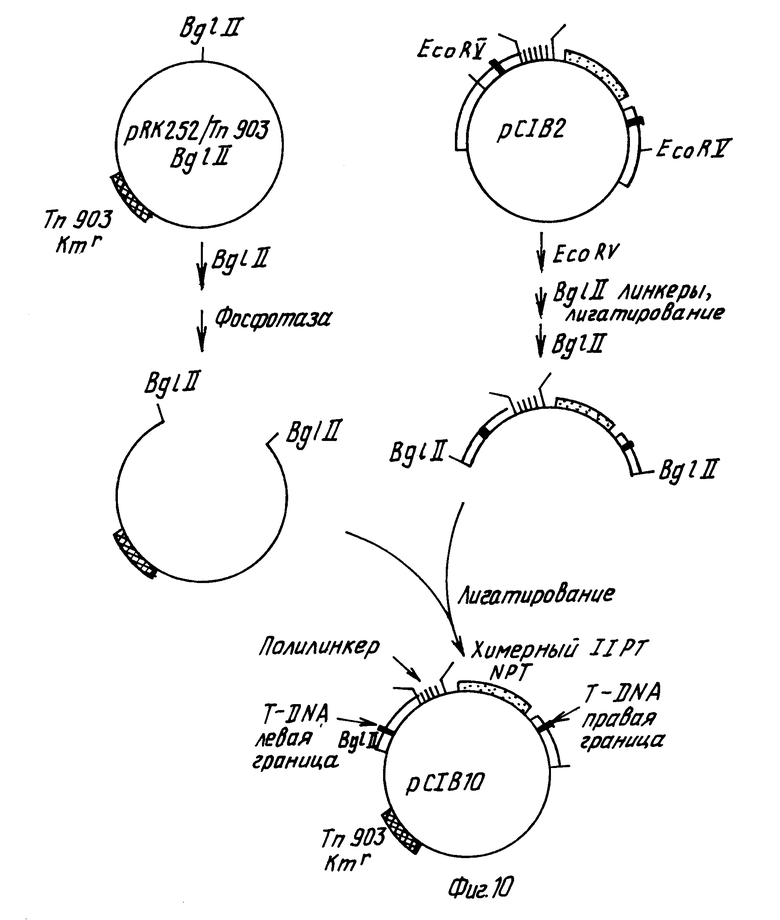
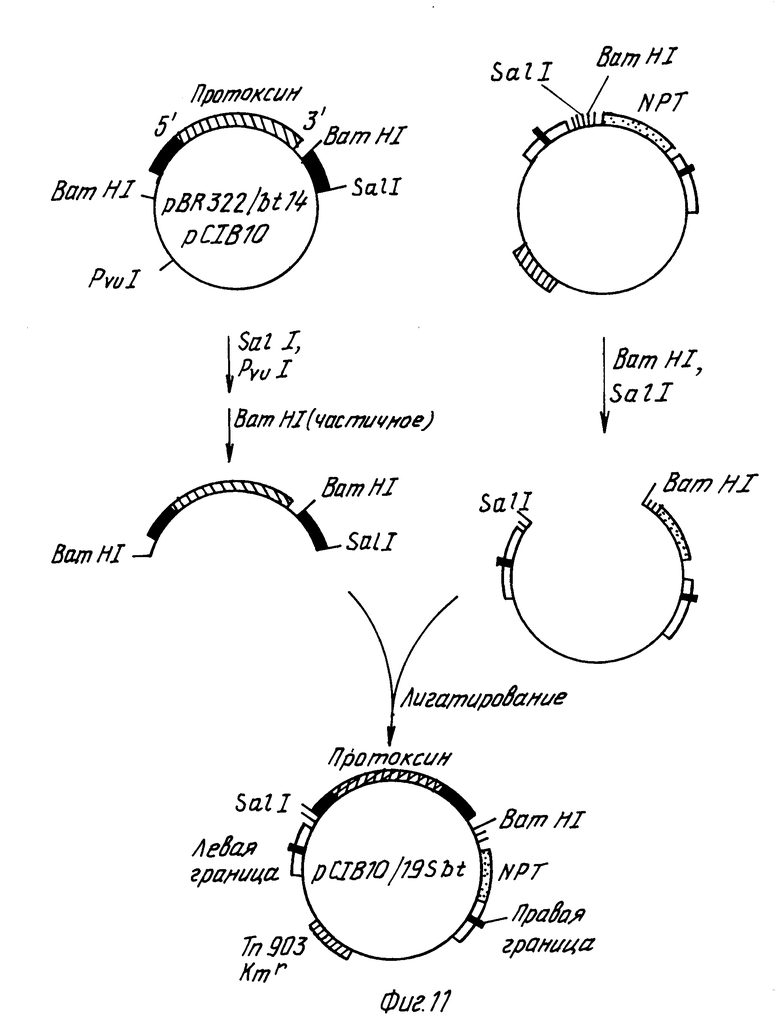
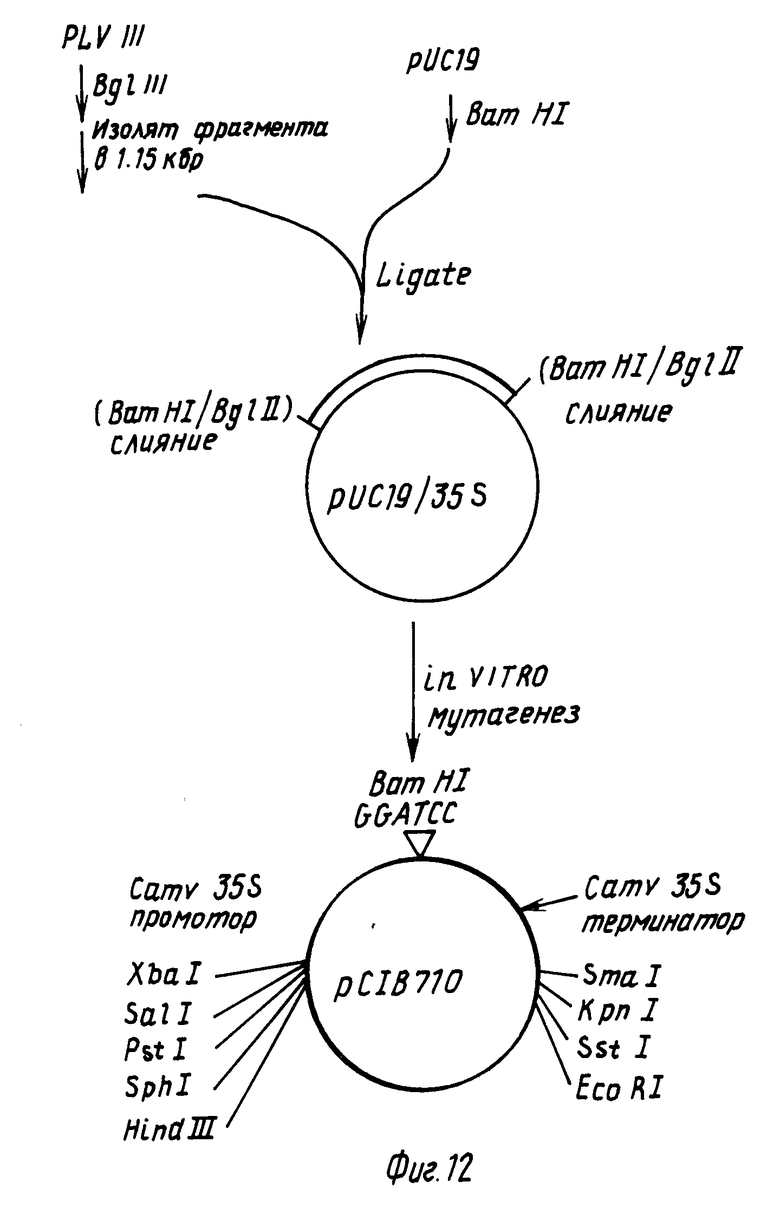
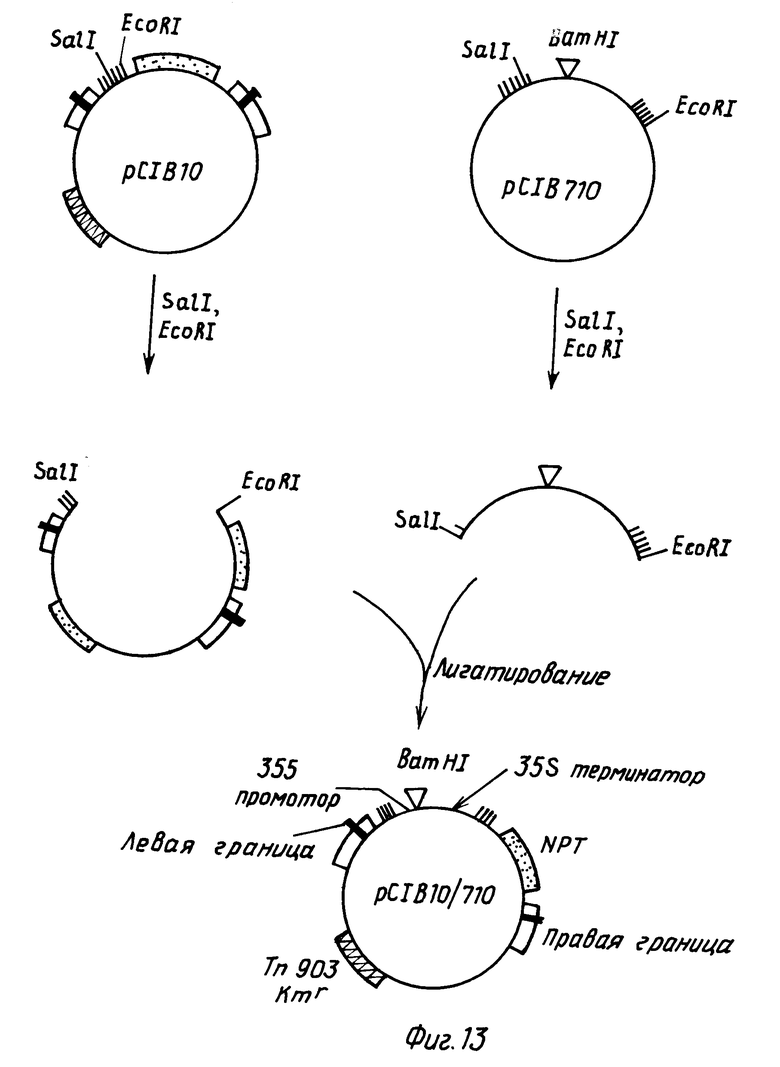
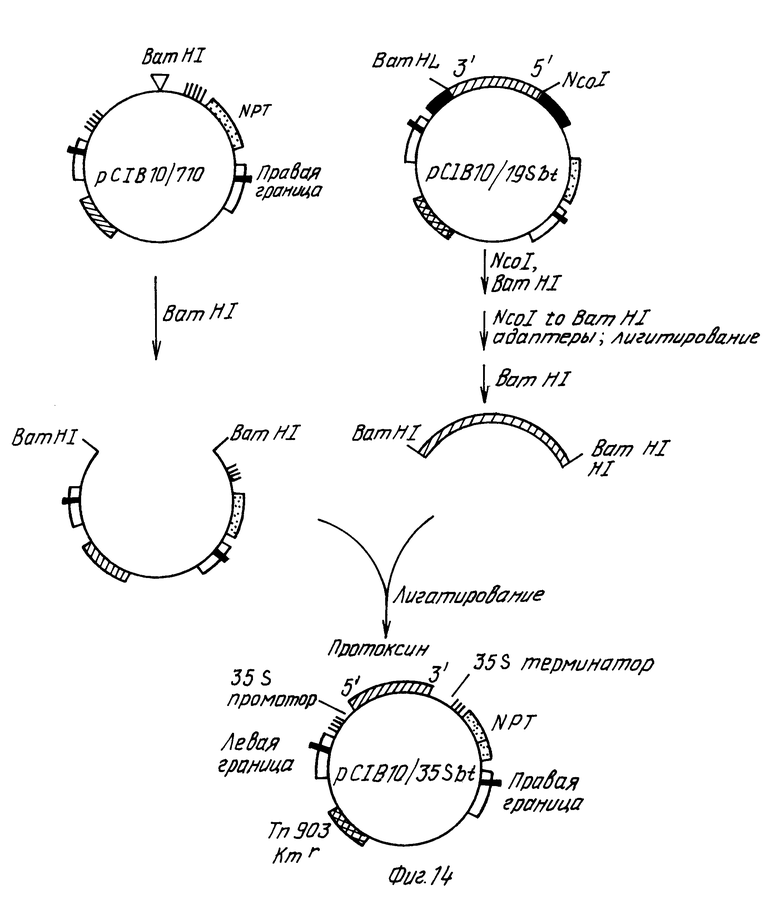
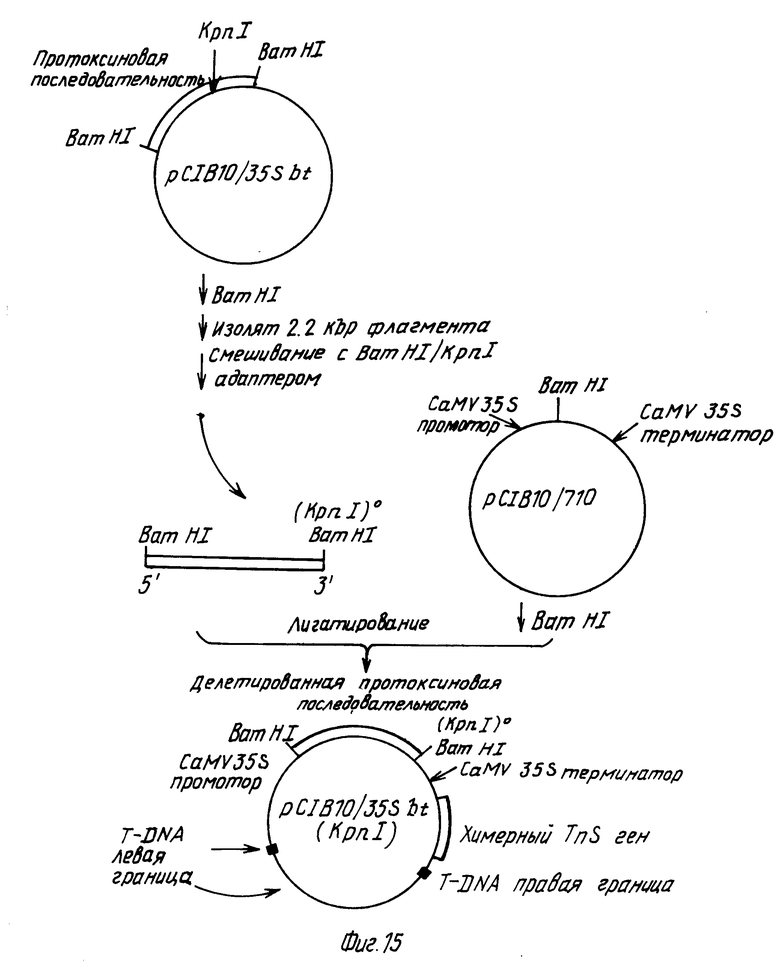
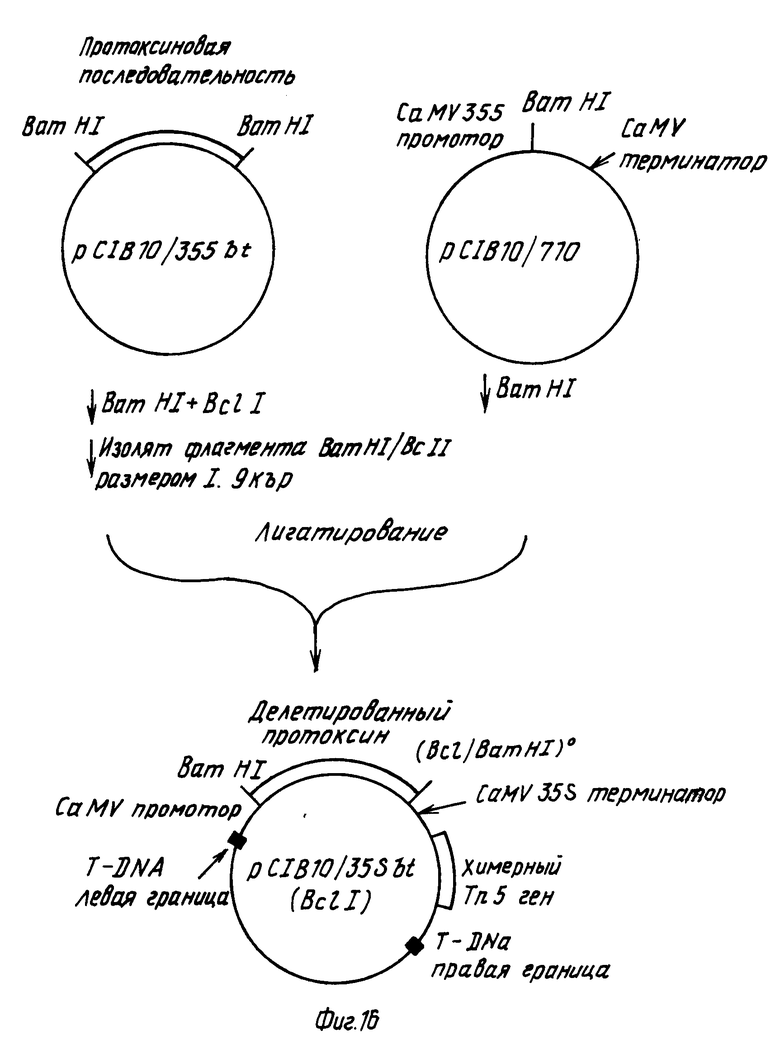
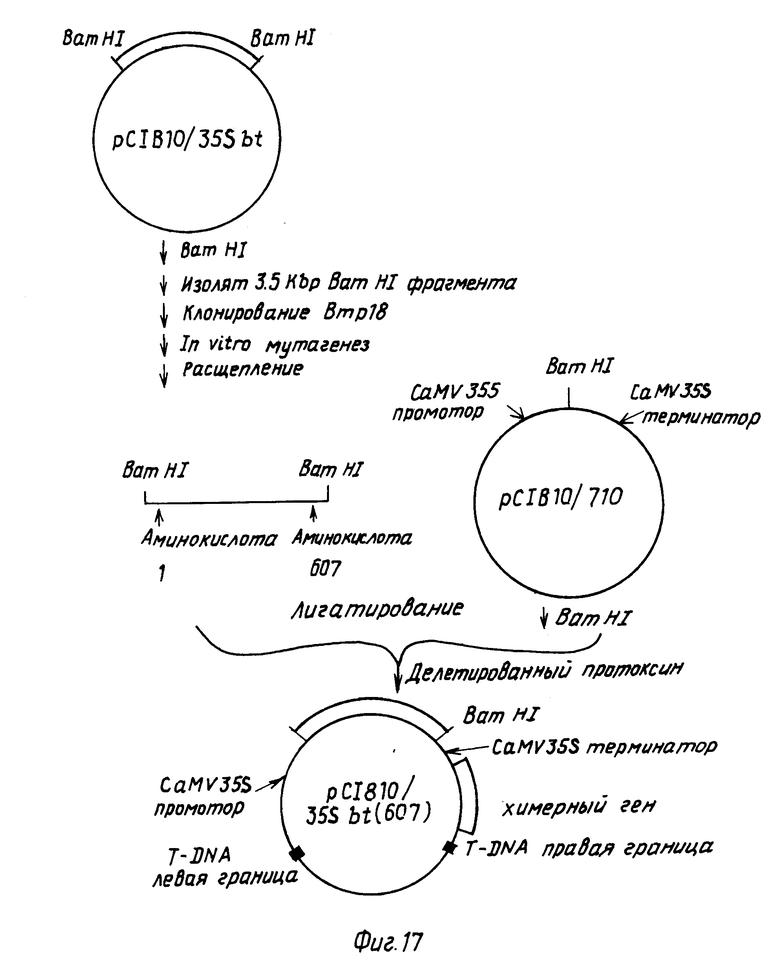
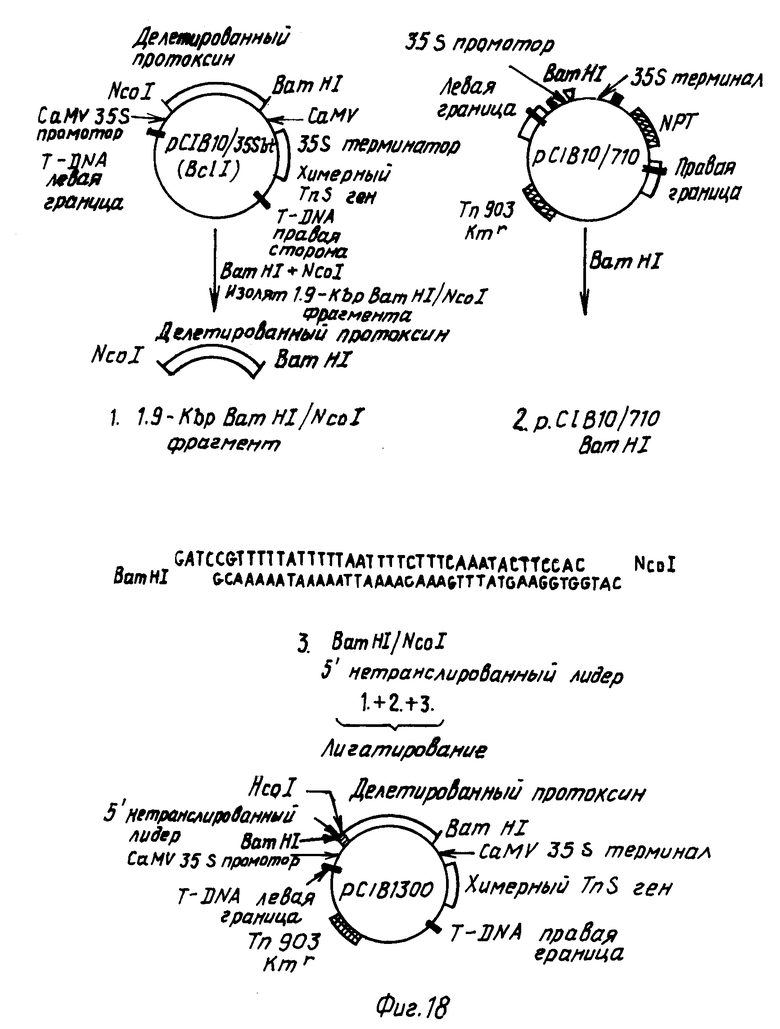
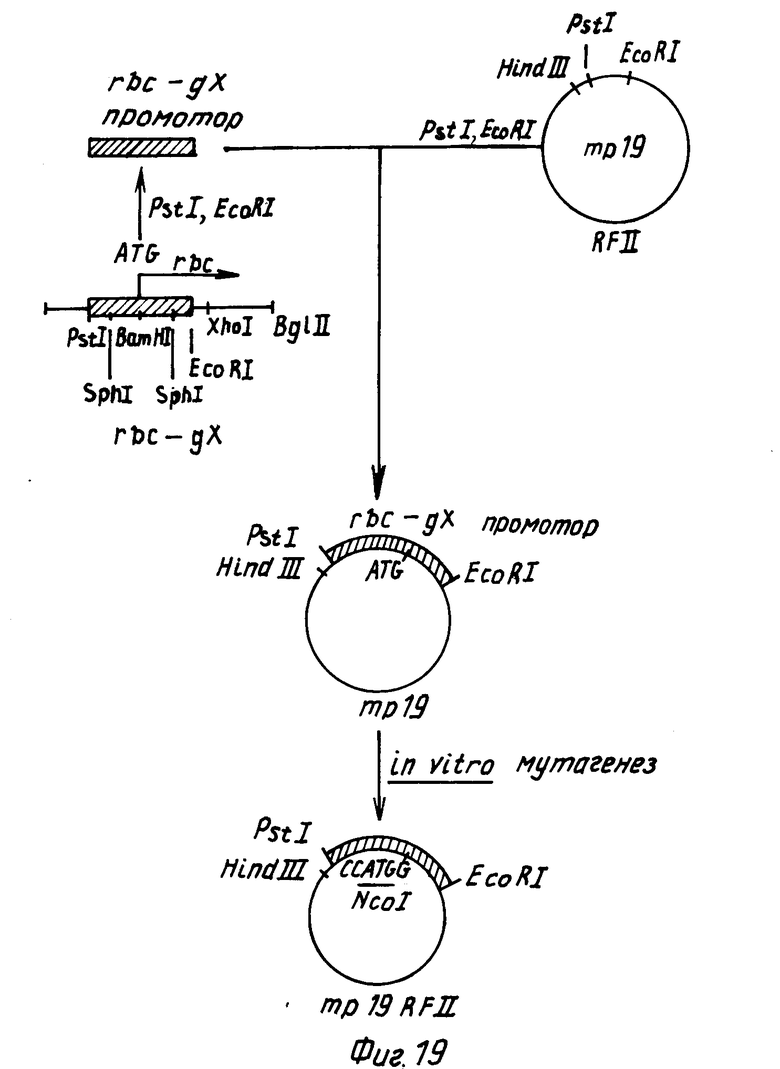
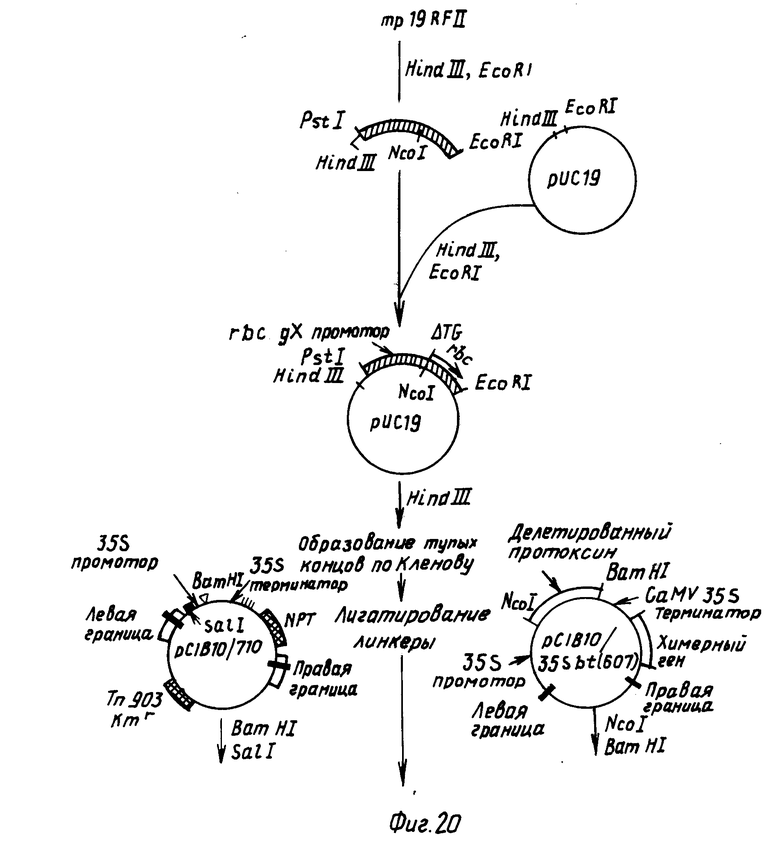
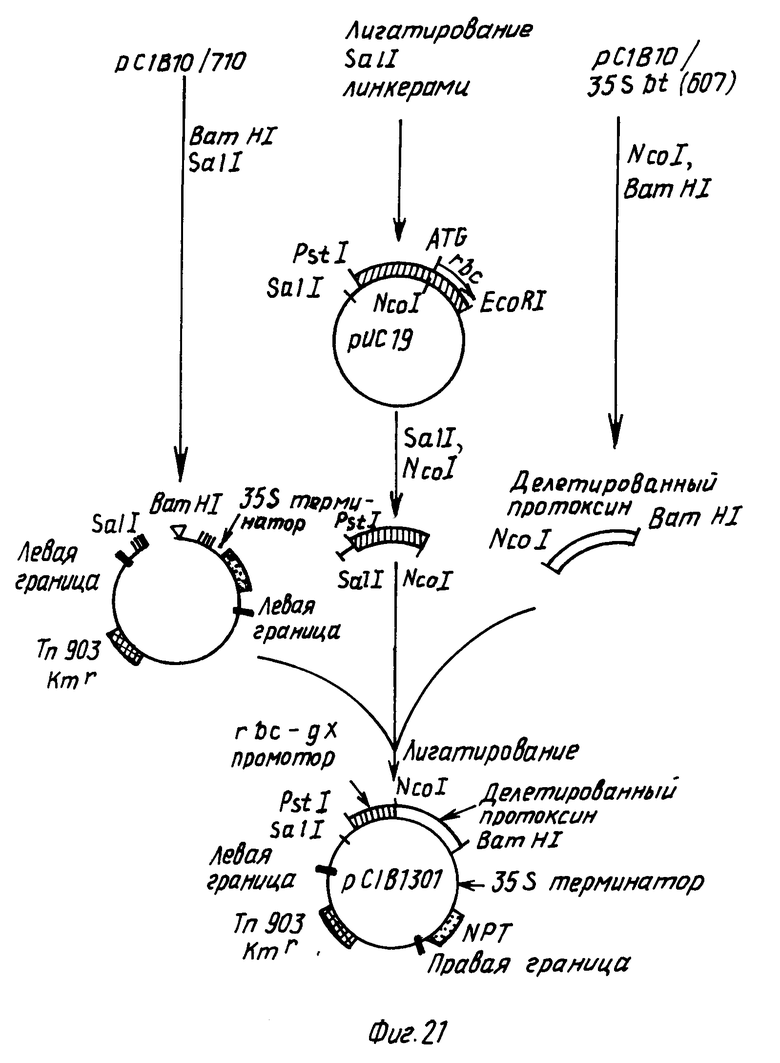
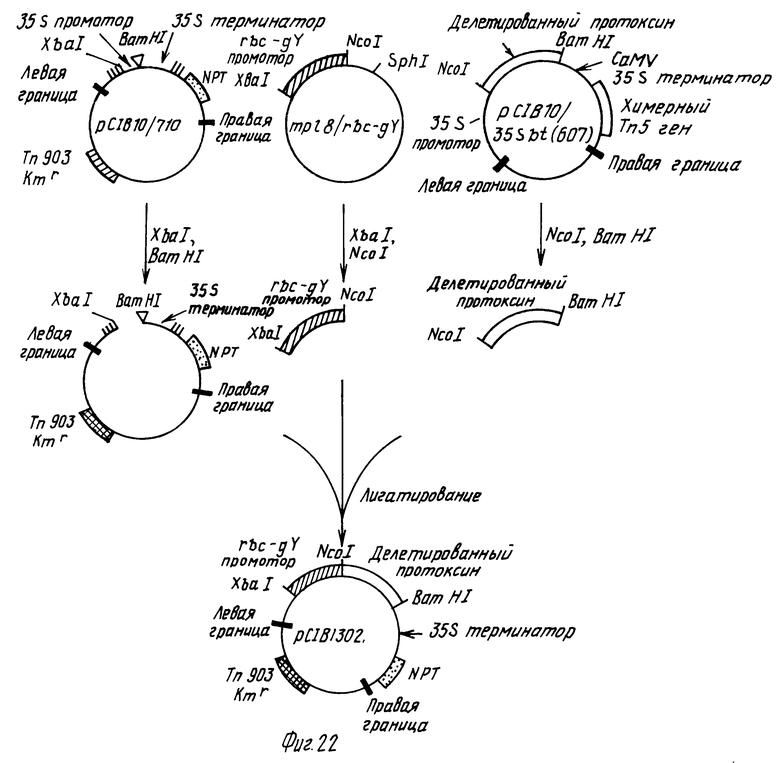
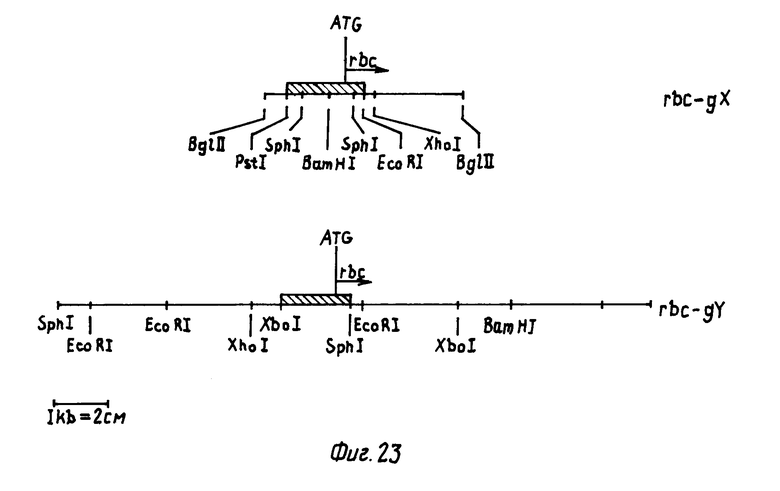


Использование: биотехнология, генетическая инженерия растений. Сущность изобретения: трансгенный хлопок получают в результате взаимодействия культивируемых клеток хлопка со штаммом бактерий Agrobacterium tumefaciens, содержащим рекомбинантную плазмидную ДНК с химерным геном, кодирующим кристаллический протеин δ -эндотоксина Bacillus thuringiensis, промотором, 5' и 3' нетранслируемыми областями и вторым геном, обеспечивающим устойчивость к антибиотику, инкубируют трансформированные клетки, переносят его в среду для выращивания, культивируют каллюс на среде для выращивания каллюсов, отбирают трансформированный эмбриогенный каллюс, затем фильтруют суспензию для отделения эмбриогенного каллюса размером 600 мкм с последующим получением регенераторов. 25 ил.
СПОСОБ ПОЛУЧЕНИЯ ТРАНСГЕННЫХ РАСТЕНИЙ GOSSYPIUM, УСТОЙЧИВЫХ К НАСЕКОМЫМ, заключающийся в том, что осуществляют взаимодействие культивируемых клеток хлопка со штаммом бактерий Agrobacterium tumefaciens, содержащим плазмиду pC1B 10/19 Sbt, или pC1B10/35Sbt, или pTOX, или pSAN, или pSIB10/35Sbt(KpnI), или pC1B10/35Sbt (Bc1 1) или pC1B10/35Sbt(607), или pC1B1300, или pC1B1301, или pC1B1302 с химерным геном, кодирующим кристаллический протеин δ -эндотоксина Bacillus thuringiensis, промотором, 5' и 3' нетранслируемыми областями и вторым геном, обеспечивающим устойчивость к антибиотику, причем просмотр и 3' нетранслируемые области получают из генов растений, или генов вирусов растений или фрагментов ДНК из плазмид Agrobacterium tumefaciens, затем инкубируют трансформированные клетки в среде роста каллюса, которая является модифицированной или немодифицированной средой Мурашиге и Скуга, или модифицированной или немодифицированной средой Гамборга В-5, содержащей 1 - 10 мг/л ауксина и парахлорфеноксиуксусную кислоту или цитокинин, в течение 15 - 200 ч при температуре 25 - 35oС на свету и 8 ч в темноте, затем переносят инкубированный каллюс в среду для выращивания, содержащую антибиотик, токсичный для Agrobacterium, культивируют каллюс на среде для выращивания каллюса с получением эмбриогенного каллюса, отбирают трансформированный эмбриогенный каллюс путем помещения его на среду с вторым антибиотиком и фильтруют суспензию для отделения эмбриогенного каллюса размером 600 мкм с последующим получением регенерантов.
