Настоящее изобретение относится к способу получения зерновых растений (Zea mays), резистентных к повреждениям, вызываемым насекомыми, которые регенерируют из протопластов или из клеток, происходящих из протопластов
Предпосылки изобретения
(Часть А) Регенерация растений Zea mays из протопластов
Описание регенерации Zea mays из каллюса датируется в основном серединой 70-х годов (например, Green and Phillips, Crop. Se. 15 (1975) 417-421; Harms et al., Z. Pflanzenzuechtg; 77 (1976) 437-352; европейские патентные заявки EP-0160390, Zowe and Smith (2985), EP-0176162, Cheng (1985), EP-0177738, Close (1985)). Были сделаны попытки получить деление и последующую регенерацию растений из протопластов Zea mays в течение более 12 лет (Potrykus et al., Jn. Cell Genetics in Higher Plants, Dudits et al., (eds), Akademiai Kiado, Budapest (1976) 129-140, и приведенные там ссылки, Harm "Maize and Cereal Protoplasts - Facte and Perspectives" Maize for Biological Research, W. F. Sheridan, ed. (1982); Dale in: Protoplasts (1983); Potrykus et al. (eds) Zecture Proceedings, Experientia Supplementum 46, Potrykys et al., eds Birkhauser, Basel (1983) 31-41 и ссылки там). Однако протопласты Zea mays всегда давали рост только в культурах, которые не были способны регенерировать фертильные растения (Potrykus et al., Mol. Gen. Genet 156 (1977) 347-350; Potrykus et al., Theor. Appl. Genet., 54 (1979) 209-214; Choury and Zurawski, Yheor. Appl. Genet. 59 (1981) 341-344; Vasil, in: Proc 5th Int. Cong, Plant Tissue and Cell Culture, Fujiwara Tokyo (1982) 101-104; Imbric-Milligan and Hodges, Planta, 168 (1986) 395-401; Vasil and Vasil, Theor. Appl. Genet. , 73 (1987) 793-798). Однако ни в одной из этих ссылок не приводится описания процедуры получения протопластов, способных к регенерации фертильных растений.
Способность продуцировать такие протопласты даст возможность генетических манипуляций с этой важной злаковой культурой, с помощью, например, трансформации или слияния протопластов или при использовании протопластов в качестве исходного материала для селекции ценных новых фенотипов.
Хотя был проявлен большой интерес к генетической трансформации Zea mays не имеется описания успешного метода, ин витро, который может привести к регенерированному трансформированному растению, хотя имелись описания трансформации протопласта Zea mays, которая привела к получению нерегенерируемого каллюса (Fromm et al., Nature, 319 (1986) 791-793). Один метод получения трансгенных растений Zea mays может быть найден путем стабильной трансформации протопластов, способных к регенерации целых фертильных растений. Однако не описано способа получения протопластов Zea mays, способных подвергаться дифференциации с возникновением целых фертильных растений.
Эта и другие цели были достигнуты в соответствии с настоящим изобретением при раскрытии способа получения протопластов Zea mays, которые могут делиться с образованием каллюсных колоний. Протопласты могут быть трансформированы, а каллюсы способны к регенерации фертильных растений Zea mays. Способ получения протопластов Zea mays, способных к делению и образованию каллюса, который затем индуцируют для регенерации в целые фертильные растения, требует специального типа каллюса. Такие каллюсы и способы их получения и идентификации описаны здесь далее.
Для начальной клеточной суспензии используют специальный тип каллюса, при субкультивировании, приводят к суспензиям, которые могут быть использованы для изоляции протопластов. Эти протопласты способны к делению и образованию каллюса, который может быть индуцирован на регенерацию растений. Клеточное деление или образование каллюса может быть промотировано некоторыми агентами, такими как O-низший алканоилсалициловая кислота (O-ацетилсалициловая кислота и ее гомологи), регуляторы роста растений или ДМСО, или сочетания этих агентов.
Регенерация растений из клеток, происходящих из протопластов Zea mays в культуре является существенной для применения соматической гибридизации, для получения улучшенных разновидностей Zea mays через сомаклональную вариабельность и для использования генетической инженерии при получении новых растений Zea mays сортов растений, культивированных или инбредных, с новыми фенотипическими признаками.
(Часть B) Инсектицидные растения кукурузы Zea mays
Bacillus thuringiensis (здесь далее  ) является штаммом бактерий, который продуцирует кристаллический белок, также упоминаемый как дельта-эндотоксин. Технически, этот кристаллический белок является протоксином, который превращается в токсин при проглатывании личинками чешуекрылых жесткокрылых и двукрылых насекомых.
) является штаммом бактерий, который продуцирует кристаллический белок, также упоминаемый как дельта-эндотоксин. Технически, этот кристаллический белок является протоксином, который превращается в токсин при проглатывании личинками чешуекрылых жесткокрылых и двукрылых насекомых.
Кристаллический белок  является потенциально важным инсектицидом, не обладающим известными вредными воздействиями на людей, других теплокровных, птиц, рыб или насекомых, других чем личинки чешуекрылых, жесткокрылых и двукрылых насекомых. Активность
является потенциально важным инсектицидом, не обладающим известными вредными воздействиями на людей, других теплокровных, птиц, рыб или насекомых, других чем личинки чешуекрылых, жесткокрылых и двукрылых насекомых. Активность  токсина является крайней высокой, так что требуются только нанограммовые количества для успешного уничтожения личинок насекомых. Другие преимущества использования кристаллического белка
токсина является крайней высокой, так что требуются только нанограммовые количества для успешного уничтожения личинок насекомых. Другие преимущества использования кристаллического белка  в качестве инсектицида включает его широкий спектр действия против личинок чешуекрылых, жесткокрылых и двукрылых насекомых, и для таких личинок возникают затруднения развития устойчивости к кристаллическому белку, даже когда кристаллический белок используют в широких масштабах. Указанные личинки являются основной проблемой в сельском и лесном хозяйствах и особенно при выращивании кукурузы.
в качестве инсектицида включает его широкий спектр действия против личинок чешуекрылых, жесткокрылых и двукрылых насекомых, и для таких личинок возникают затруднения развития устойчивости к кристаллическому белку, даже когда кристаллический белок используют в широких масштабах. Указанные личинки являются основной проблемой в сельском и лесном хозяйствах и особенно при выращивании кукурузы.
Кристаллический белок является эффективным в качестве инсектицида, когда его наносят на растения, зараженные личинками чешуекрылых, жесткокрылых или двукрылых насекомых.
До сих пор  кристаллический белок (протоксин) выделяют из Bacillus и наносят на растения с помощью стандартных методов, таких как опудривание дустом или распыление. Препараты, содержащие
кристаллический белок (протоксин) выделяют из Bacillus и наносят на растения с помощью стандартных методов, таких как опудривание дустом или распыление. Препараты, содержащие  , кристаллический белок, коммерчески используют в качестве биологических инсектицидов. Например, Бастоспейн, выпускаемый Биохем Продактс Лтд., Дипель, выпускаемый Эббот Лабораториз, и Турцид, выпускаемый Сандоз АГ. Тот факт, что
, кристаллический белок, коммерчески используют в качестве биологических инсектицидов. Например, Бастоспейн, выпускаемый Биохем Продактс Лтд., Дипель, выпускаемый Эббот Лабораториз, и Турцид, выпускаемый Сандоз АГ. Тот факт, что  продуцирует кристаллический белок только во время споруляции, является значительным недостатком для производства и применения этого биологического инсектицида. Такое ограничение фазы роста, особенно в промышленном процессе, может привести в результате к неудобствам и излишнему времени, требующемуся для производства.
продуцирует кристаллический белок только во время споруляции, является значительным недостатком для производства и применения этого биологического инсектицида. Такое ограничение фазы роста, особенно в промышленном процессе, может привести в результате к неудобствам и излишнему времени, требующемуся для производства.
Кроме того, затраты, связанные с указанным производством делает трудным для такого биологического инсектицида эффективную конкуренцию с другими коммерчески доступными продуктами на химической основе, такими как, например, пиретроидные производные.
Другим недостатком в отношении применения  токсина является, например, тот факт, что белок обычно остается на поверхности обработанных растений, где он является эффективным только против поверхностью питающихся личинок и где он инактивируется при продолжительном воздействии ультрафиолетового излучения. Такая инактивация может быть по крайней мере одной из причин общей потери персистентности кристаллического белка в окружающей среде. Следовательно, необходимо частое и дорогостоящее нанесение кристаллического белка.
токсина является, например, тот факт, что белок обычно остается на поверхности обработанных растений, где он является эффективным только против поверхностью питающихся личинок и где он инактивируется при продолжительном воздействии ультрафиолетового излучения. Такая инактивация может быть по крайней мере одной из причин общей потери персистентности кристаллического белка в окружающей среде. Следовательно, необходимо частое и дорогостоящее нанесение кристаллического белка.
Эти и другие недостатки могут быть устранены за счет введения и экспрессии гена, кодирующего  кристаллический белок или белок, обладающий по существу токсичностью по отношению к насекомым
кристаллический белок или белок, обладающий по существу токсичностью по отношению к насекомым  кристаллического белка, в растениях.
кристаллического белка, в растениях.
Настоящее изобретение описывает, как эти недостатки могут быть устранены путем введения и экспрессии гена, кодирующего  кристаллический белок или белок, обладающий по существу токсичностью по отношению к насекомым
кристаллический белок или белок, обладающий по существу токсичностью по отношению к насекомым  кристаллического белка, в протопластах кукурузы, и регенерации фертильных трансгенных растений кукурузы из трансформированных протопластов и выращивания этих устойчивых к насекомым растений кукурузы.
кристаллического белка, в протопластах кукурузы, и регенерации фертильных трансгенных растений кукурузы из трансформированных протопластов и выращивания этих устойчивых к насекомым растений кукурузы.
Используя преимущества генетической инженерии, ген, ответственный за продуцирование полезного полипептида, может быть трансформирован и в донорскую клетку, в которой ген обычно встречается, в клетку хозяина, в которой ген в природе не встречается; Cohen and Boyer, патенты США 4237224 и 4468464. Действительно, имеются немного ограничений для таких переносов. Гены могут быть перенесены между вирусами, бактериями, растениями и животными. В некоторых случаях перенесенный ген является функциональным или может быть сделан функциональным, в клетке хозяина. Когда клетка хозяин является растительной клеткой, целое растение может быть сделано функциональным, в клетке хозяина. Когда клетка хозяин является растительной клеткой, целое растение может быть иногда регенерировано из клетки.
Гены обычно содержат области последовательностей ДНК, включающие промотор и область транскрипции. Область транскрипции обычно содержит 5'-нетранслируемую область, кодирующую последовательность и 3'-нетранслируемую область.
Промотор содержит ДНК последовательность, необходимую для инициирования транскрипции, во время которой транскрибирующая область превращается в мРНК. В эукариотных клетках, полагают, что промотор включает область, распознаваемую РНК полимеразой, и область, которая помещает РНК полимеразу на ДНК для инициирования транскрипции. Эта последняя область упоминается как ТАТА-бокс, который обычно встречается примерно на 30 нуклеотидов выше от сайта инициирования транскрипции.
Область после промотора представляет собой последовательность, которая транскрибирована в мРНК, но не транслируется в полипептид. Эта последовательность состоит из так называемой 5'-нетранслируемой области и, как полагают, содержит последовательности, которые являются ответственными за инициирование транскрипции, такие как сайт связывания рибосом.
Кодирующая область представляет собой последовательность, которая находится сразу ниже 5'-нетранслируемой области в ДНК или соответствующей РНК. Она является кодирующей областью, которая транслируется в полипептиды в соответствии с генетическим кодом. Например,  имеет ген с кодирующей последовательностью, которая транслируется в аминокислотную последовательность инсектицидного протоксина кристаллического белка.
имеет ген с кодирующей последовательностью, которая транслируется в аминокислотную последовательность инсектицидного протоксина кристаллического белка.
Кодирующая область находится ниже последовательности, которая транскрибируется в мРНК, но не транслируется в полипептид. Эта последовательность называется 3'-нетранслируемой областью и как полагают, содержит сигнал, который приводит к окончанию транскрипции, и в эукариотной мРНК, сигнал, который вызывает полиаденилирование нити транскрибированной. Полагают, что полиаденилирование мРНК обладает функциями процессинга и транспортирующей функцией.
Природные гены могут быть перенесены целиком из донорской крови в клетку хозяина. Однако часто является предпочтительным конструировать ген, содержащий желаемую кодирующую последовательность с промотором и, в некоторых случаях, 5'- и 3'-нетранслируемые области, которые не существуют в природе в том же самом гене как кодирующая область. Такие конструкции известны как химерические гены.
Уже описаны методы генетической инженерии для усовершенствованных путей получения кристаллического белка. Например, Schnepf et al., патенты США 4448885 и 4467036, описали плазмиды для продуцирования кристаллического белка в бактериальных штаммах, других чем  . Эти методы позволяют продуцировать кристаллический белок, но не устраняют недостатков применения кристаллического белка в качестве коммерческого инсектицида.
. Эти методы позволяют продуцировать кристаллический белок, но не устраняют недостатков применения кристаллического белка в качестве коммерческого инсектицида.
Были сделаны предложения клонировать гены  токсина непосредственно в растения, чтобы позволить растениям защищать самих себя:, (Klausner A., Biotechnology, 2 (1984) 409 - 413). Adang et al. Европейская заявка на патент EP-0142924 (Агригенетикс) и De Greve H.M.J. et al. EP-0193269 (Плант Генетик Системс Н.В.) заявили способ клонирования генов токсина из Bt в табак. Ни в одной из этих публикаций не приводится примеров трансформации однодольных растений.
токсина непосредственно в растения, чтобы позволить растениям защищать самих себя:, (Klausner A., Biotechnology, 2 (1984) 409 - 413). Adang et al. Европейская заявка на патент EP-0142924 (Агригенетикс) и De Greve H.M.J. et al. EP-0193269 (Плант Генетик Системс Н.В.) заявили способ клонирования генов токсина из Bt в табак. Ни в одной из этих публикаций не приводится примеров трансформации однодольных растений.
Существует необходимость разработки новых способов получения кристаллического белка  в клетках растений кукурузы и новых способов контроля личинок насекомых, которые питаются на растениях кукурузы, содержащих
в клетках растений кукурузы и новых способов контроля личинок насекомых, которые питаются на растениях кукурузы, содержащих  кристаллический белок или подобный полипептид, обладающий по существу токсичностью по отношению к насекомым (инсектицидной активностью)
кристаллический белок или подобный полипептид, обладающий по существу токсичностью по отношению к насекомым (инсектицидной активностью)  кристаллического белка. Под "контролем" как здесь упоминается, понимают или умерщвление личинок или по крайней мере снижение их питания. Кроме того, существует необходимость создания способа защиты растений кукурузы против повреждений, вызываемых паразитами или патогенами, заключающегося в продуцировании количества пестицидного или антипатогенного белка в клетке растения и растении соответственно, это количество является достаточно эффективным для уничтожения или контроля паразита или патогена. Кроме того, существует необходимость в предпочтительном создании способа защиты растений кукурузы против повреждений насекомыми, заключающегося в продуцировании инсектицидно эффективного количества
кристаллического белка. Под "контролем" как здесь упоминается, понимают или умерщвление личинок или по крайней мере снижение их питания. Кроме того, существует необходимость создания способа защиты растений кукурузы против повреждений, вызываемых паразитами или патогенами, заключающегося в продуцировании количества пестицидного или антипатогенного белка в клетке растения и растении соответственно, это количество является достаточно эффективным для уничтожения или контроля паразита или патогена. Кроме того, существует необходимость в предпочтительном создании способа защиты растений кукурузы против повреждений насекомыми, заключающегося в продуцировании инсектицидно эффективного количества  кристаллического белка или белка, обладающего по существу токсичностью против насекомых
кристаллического белка или белка, обладающего по существу токсичностью против насекомых  кристаллического белка, в клетках растений. Далее, существует необходимость в создании способа защиты растений кукурузы от повреждений химическими агентами, такими как, например, гербициды, чтобы такие химикаты могли быть безопасно нанесены на растения кукурузы.
кристаллического белка, в клетках растений. Далее, существует необходимость в создании способа защиты растений кукурузы от повреждений химическими агентами, такими как, например, гербициды, чтобы такие химикаты могли быть безопасно нанесены на растения кукурузы.
Из уровня техники ко времени создания настоящего изобретения нельзя было предсказать, что зерновые растения (Zea mays), в частности, фертильные растения, могут быть регенерированы из протопластов, из клеток, происходящих из протопластов или каллуса, происходящего из протопластов. Еще менее можно было предположить, что протопласты Zea mays, содержащие стабильную включенную экзогенную ДНК можно регенерировать трансгенные растения, в частности в фертильные трансгенные растения Zea mays.
Объекты настоящего изобретения детально описаны ниже.
Способ получения растений Zea mays L., устойчивых к повреждениям, вызываемым насекомыми, включающий выделение ткани из незрелых зародышей, культивирование указанной ткани в среде для инициирования каллуса и/или в среде для поддержания пролиферации каллуса, до образования особого типа каллуса, характеризующегося нелипким, гранулярным и рыхлыми структурами и содержащего плотные цитоплазматически, делящиеся клетки, отбор вышеуказанного типа каллуса и последующую обработку его ферментным препаратом для удаления клеточных стенок, сбор, очистку и трансформацию полученных тотипотентных протопластов вектором, содержащим структурный ген, кодирующий полипептид кристаллического белка δ- эндотоксина Bacillus thuringiensis токсичного по отношению к насекомым, и фланкированного контролирующими транскрипцию последовательностями, содержащими промотор и 5'- и 3'-нетранслируемые последовательности, которые функциональны в зерновых, дальнейший посев трансформированных тотипотентных протопластов на питательную среду для деления и образования клеток, субкультивирование их на среде для регенерации и последующее получение фертильных растений Zea mays L., устойчивых к повреждениям, вызываемым насекомыми.
Изобретение далее направлено на способ по п. 1, отличающийся тем, что после отбора особого типа каллуса, осуществляют перенос его в жидкую питательную среду для формирования суспензии клеток или клеточных агрегатов, субкультивирование полученной суспензии в течение времени и с частотой, достаточной для поддержания клеток и клеточных агрегатов в жизнеспособном состоянии с последующим отбором и сохранением культур, содержащих агрегаты делящихся клеток с плотной цитоплазмой, которые характеризуются более гладкой поверхностью, чем другие клеточные агрегаты.
На фиг. 1 показан каллюс, который пригоден для использования в начальных эмбриогенных суспензиях Zea mays (увеличение примерно 7X). (Стадия a).
На фиг. 2 показаны плотные цитоплазматические, делящиеся клетки, которые удерживаются в культурах стадий b - d (увеличение примерно 20X).
На фиг. 3 представлена нуклеотидная последовательность гена эндотоксина Bacillus thuringiensis var. Kurstaki: HD1
На фиг. 4 представлена последовательность полипептида кристаллического белка δ- эндотоксина B. thuringiensis, кодируемая структурным геном.
Предпочтительная нуклеотидная последовательность, которая колирует кристаллический белок, является такой последовательностью, что показана нуклеотидами 156-3623 в последовательности, или более короткой последовательностью, которая кодирует инсектицидный фрагмент такого кристаллического белка; Geiser et al., Gene, 48 (1986) 109-118.
Определения
Zea mays /кукуруза: В настоящем описании или в формуле изобретения эти термины относятся к любому растению или части растения (протопластам, клеткам, тканям, органам, органеллам, эмбрионам, каллюсам, побегам и т.п.), принадлежащего к виду Zea mays. Термины, как использованы здесь, включают сорт, рассу, разновидность, инбредные линии и гибриды Zea mays. Настоящее изобретение включает, но не ограничивается подобными инбредными линиями кукурузы, например Funr 5N984, Funk 5N986, Funk 2717, Funk 211Д, Funk 2N217A, B73, A632, CM105, B37, B84, B14, Vc17, R 186, MS 71, A188, F A91, A641 и W 117. Предпочтительными являются все элитные генотипы (сорта, разновидности, инбредные линии, гибриды и т.д.), включая элитные инбредные линии Funk 5 N 984, Funk 5N 986, Funk 2717, Funk 211Д и Funk 2N217A, более предпочтительная элитная линия инбредная Funk 2717.
BMS: "Черная мексиканская сладкая кукуруза", старый мелкопочаточный сорт кукурузы с темно-голубой окраской околоплодника зерна, придающей зерну "черный" оттенок.
Клетка растения
Структурная и физиологическая единица растений, состоящая из протопласта и клеточной стенки.
Термин "клетка растения" относится к любой клетке, которая или является частью или отделена от растения.
Некоторые примеры клеток, охватываемых настоящим изобретением, включают дифференцированные клетки, которые являются частью живого растения; дифференцированные клетки в культуре; недифференцированные клетки в культуре; клетки недифференцированной ткани, такой как каллюс или опухоли, семена, зародыши (эмбрионы); побеги и пыльца.
Ткань растения
Группа растительных клеток, организованная в структурную и функциональную единицу. Любая ткань растения в растении или в культуре. Этот термин включает, но не ограничивается целым растением, клетками растений, органами растений, семенами растений, протопластами, культурой клеток и любой группой растительных клеток, организованных в структурные и/или функциональные единицы. Использование этого термина в сочетании с или в отсутствии какого-либо специфического типа растительной ткани, как перечислено выше, или другими словами, охватываемыми настоящим определением, не предназначены для исключения любого другого типа растительной ткани.
Орган растения
Отдельная и видимо структурированная и дифференциированная часть растений, такая как корень, ствол, лист, бутон цветка или зародыш. Орган растения может состоять из различных типов клеток растения или тканей растения.
Пропласт
Изолированная клетка растения без клеточной стенки.
Трансгенное растение. Растение, имеющее стабильно введенную экзогенную ДНК в своем генетическом материале. Термин включает экзогенную ДНК, которая может быть введена в протопласты в различной форме, включая, например, голую ДНК в кольцевой, линейной или суперспирализованной форме, ДНК, содержащуюся в нуклеосомах, или ядрах или в их частях, ДНК, комплексно связанную или ассоциированную с другими молекулами, ДНК, заключенную в липосомах, сферопластах, клетках или пропластах. На данном уровне техники известны различные способы введения экзогенной ДНК в пропласты и они могут быть использованы для получения трансгенных растений настоящего изобретения.
Культура
Пролиферирующая масса клеток в недифференциированном или частично дифференциированном состоянии. Термин "клеточная культура" включает каллюсные культуры, состоящий из массы клеток, растущих на отвержденной культуральной среде, и суспензионные клеточные культуры, растущие в дисперсном состоянии в перемешиваемой жидкой культуральной среде.
Эмбрион (зародыш)
Наиболее ранняя стадия развития растения, или происходящая из зиготы (затем называемой половым зародышем), или из эмбриогенной соматической клетки (затем называемой соматическим эмбрионом), со стадиями распознаваемой морфологии, структуры и клеточной организации, включающих клеточную до глобулярной стадии, до стадии семядоли. (Развитие зародыша кукурузы описано, например, у Randolph, L.F., J.Agril. Research, 53 (1936) 881-916; развитие злакового зародыша в общем описано, например, у Brown, W.V., Phytomorphology, 10 (1960) 215-223).
Побег
Многоклеточная структура, состоящая из ростка и корня в форме маленького растения.
Химерическая последовательность или химерический ген.
Последовательность ДНК, содержащая по крайней мере две гетерологических части, например, части, происходящую от или имеющую по существу последовательность, гомологичную ранее существующим последовательностям ДНК, которые не ассоциированы на ранее прошедших стадиях. Ранее существовавшие последовательности ДНК могут быть природного или синтетического происхождения.
Кодирующая последовательность ДНК.
Последовательность ДНК, которая при транскрипции и трансляции приводит в результате к образованию клеточного полипептида.
Ген
Дискретная хромосомная область, состоящая из регуляторных последовательностей ДНК, ответственных за контроль экспрессии, а именно, трансляцию и транскрипцию, и из колирующей последовательности, которая транскрибируется и транслируется, давая определенный полипептид.
Происходящий из:
в контексте этого описания гены, части генов или другие последовательности ДНК, "происходящие из" других источников ДНК, охватывают гены, части генов или другие последовательности ДНК, идентичные или по существу гомологичные материалу источника ДНК.
Фенотипический признак
Наблюдаемое свойство, являющееся результатом экспрессии одного или более генов.
Ранее существующая последовательность ДНК.
Последовательность ДНК, которая существует до ее применения, полностью или частично, в продукте или способе согласно настоящему изобретению. Хотя такое предсуществование обычно отражает природное происхождение. Ранее существующие последовательности могут быть природного или синтетического происхождения.
Существенная гомология последовательности
Существенная функциональная и/или структурная эквивалентность между последовательностями нуклеотидов или аминокислот. Функциональные и/или структурные различия между последовательностями, имеющими существенную гомологию последовательностей, будут минимальными.
Дикамба
3,6-Дихлор-2-метоксибензойная кислота;
MES 2-(N- морфолино) этансульфоновая кислота;
2,4-Д: 2,4-дихлорфеноксиуксусная кислота.
Пиклорам: 4-амино-3,6,6-трихлорпиколиновая кислота.
Трис-HCl: альфа, альфа, альфа-трис-(оксиметил)метиламин гидрохлорид;
EDTA: 1-этилендиамин-N,N,N',N'-тетрауксусная кислота;
PEG : полиэтиленгликоль.
Агароза: Приготовление и очистка агарозы описаны, например, Guiseley and Renn, ("The Agarose Monograph"; Marine Colloids Division FMC Corp. (1975)). Агароза является одним из компонентов агара. Коммерчески доступный агар обычно состоит из смеси нейтральной агарозы и ионного агаропектина с большим количеством боковых групп. Коммерческую агарозу обычно получают из агара традиционными способами. Обычно некоторое количество боковых цепей остается интактной и определяет физикохимические свойства агарозы, такие как гелеобразование и температура плавления. Низкоплавкая агароза, особенно Sea PlagueR агароза является предпочтительным отверждающим агентом в описанном здесь ниже процессе.
SH среда: Среда Шенка Р.Ю., с сотр.Can. H. Bot., 50(1972) 199-204; без регуляторов роста (SH среда может быть жидкой или отвержденной 0,8% (мас. /объем)агара или 0,5% (мас./объем) GelRiteR) Среду обычно стерилизуют с помощью методик, известных на данном уровне техники, например, путем нагревания или стерилизацией автоклавированием при 121oC и 15 фунт/дюйм2 (1,2 кг/см2) давления в течение примерно 15-20 мин.
MS-среда: Murashige T.and Shoog F., Physiol Plant., 15 (1962) 473
B5-среда: Gambord, O.et al., Exp. Cell Res., 50(1968) 151 N6 среда: Chu et al. Scientia sinica,18 (1975)659.
Состав приведен в табл. 1.
КМ-среда: KaO K. N.et.al., Planta, 126 (1975) 105 - 110. Эта среда может быть жидкой или отвержденной агаром, агарозой или Gel RiteR и также может быть приготовлена и использована без аскорбиновой кислоты, витамина D и витамина A. Компоненты среды за исключением отверждающего агента обычно стерилизуют фильтрацией через фильтр 0,2 мкм.
Состав использованной среды приведен в табл. 1.
(Макроэлементы и микроэлементы среды соответствуют приведенным в литературе).
Примечания к табл. 1:
a: макроэлементы обычно готовят в виде 10х концентрированного раствора для хранения, а макроэлементы в виде 1000х концентрированных растворов для хранения.
b: лимонную, фумаровую и малеиновую кислоты (каждая в конечной концентрации 40 мг/л) и пируват натрия (конечная концентрация 20 мг/л) готовят в виде 100х концентрированных растворов для хранения, устанавливают pH 6,5 с помощью NH4OH и прибавляют к этой среде.
c: Аденин (0,1 мг/л конечная концентрация) и гуанин, тимин, урацил, гипоксантин и цитозин (конечная концентрация каждого 0,03 мг/л) готовят в виде 1000х концентрированных растворов для хранения, устанавливают pH 6,5, как описано выше, и прибавляют к этой среде.
d: Следующие аминокислоты прибавляют к этой среде, используя 10х растворы для хранения (pH 6,5 с помощью NH4OH), чтобы получить заданные конечные концентрации: глютамин (5,6 мг/л), аланин, глютаминовую кислоту (каждая 0,6 мг/л), цистеин (0,2 мг/л), аспаргин, аспаргиновую кислоту, цистин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин и валин (каждый 0,1 мг/л).
e: Раствор для хранения витамина обычно готовят 100х концентрированным.
Целлюлоза RS: Целлюлоза RS Якулт Хонша Ко., Лтд., 1.1.19 Хигаши-Шинбаши, Минато-ку, Токио 105, Япония
Пектолиаза Y-23: Сейшин фармацетикал Ко, Лтд. 4-13 Коамихо, Нихонбаши, Токио, Япония.
 фильтр: Нальге Кол., Отделение Сирбон Корп. Рочестер, Нью-Йорк, 14602, США.
фильтр: Нальге Кол., Отделение Сирбон Корп. Рочестер, Нью-Йорк, 14602, США.
Bgl I[ Рестрикционный фермент Bgl I]; Нью Инглэнд Биолабс, 32 Тозер Роад, Беверли, МА, 01915, США, или любой другой коммерческий поставщик.
Bam HI Рестрикционный фермент Bam HI; Нью Ингленд Биолабс, 32 Тозер Роад, Беверли, МА, 01915, США, или любой другой коммерческий поставщик.
Гидромицин B: цитотоксин, гидромицин B, очищенный; кат. N 400050 Калбиохем Беринг Диагностика, Ла Йолла, СА 92037, США, Группа N 702296.
Голь  : ГельРайтГеллан Гум, Скотт Лабораториз Инк., Gel Rite Фискерсвилл, РИ 02823.
: ГельРайтГеллан Гум, Скотт Лабораториз Инк., Gel Rite Фискерсвилл, РИ 02823.
Prime Rit randon primer Rit of IBI Random primer Rit:
Интернейшнл Биотехнолоджиз Инк., НО Бокс 1565, Нью Хевен, СТ 07606, США (Каталожный N 77800; группа N F 630-01).
 контейнер. Маленький этерильный контейнер
контейнер. Маленький этерильный контейнер
(примерно 7 х 7 х 10 см) изготовлен из полупрозрачного пластика с полупрозрачной крышкой: используют для выращивания побегов, культур побегов и т. д. Поставщик: Магента Корп., 3800 Н.Милуокси Авеню, Чикаго, ИЛ, 60641, США. Может быть заменен любым подобным стерильным полупрозрачным контейнером (например, стеклянным или пластиковым).
Перевод микроЭнштейнов в люксы
мкЕ - люкс
0,1 - 200 - 8 - 16700
0,1 - 100 - 8 - 8350
30 - 90 - 2500 - 7500
10 - 40 - 830 - 3330 \\\ 30 - 80 - 2500 - 6670
Депонирование плазмид в микроорганизмах или в клетках растений
В соответствии с настоящим изобретением следующие перечисленные плазмиды (в микроорганизмах) и клетки растений были депонированы в соответствии с требованиями Будапештского договора:
(1) Zea mays суспензионная клеточная культура 6-2717 CG Funk 2717 депонирована в Коллекции культур американского типа (АТСС), Рокквилл, США, и имеет АТСС номер хранения 40326 (дата депонирования: 20 мая 1987).
(2) Плазмида pCIB712 (в E. coli штамм MC1061) депонирована в АТСС и имеет АТСС номер хранения 67407 (дата депонирования: 18 мая 1987 г.).
(3) Плазмида pSCHI (в E. coli штамм K12) депонирована в немецкой коллекции микроорганизмов (DMS) в Геттингене, ФРГ, и имеет DMS номер хранения 4129 (дата депонирования: 29 мая 1987 г.).
(4) Плазмида pCIB10 (35 SBt (в E. coli штамм MC1061) депонирована в АТСС и имеет АТСС номер хранения 67329 (дата депонирования: 27 февраля 1987 г.).
(5) Плазмида pE16 (8-C4) в E. coli штамм HB101) депонирована в АТСС и имеет АТСС номер хранения 67420 (дата депонирования: 29 мая 1987 г.).
(6) Плазмида pCIB10 (19 SBt (в E. coli штамм HB 101) депонирована в АТСС и имеет АТСС номер хранения 67330 (дата депонирования: 27 мая 1987 г.).
Детальное описание настоящего изобретения (Часть A) регенерация растений кукурузы Zea mays из протопластов
Каллюсы, использованные в настоящем изобретении, могут происходить из любой ткани растений Zea mays. Предпочтительно эта ткань является незрелой тканью, такой как незрелая метелка, незрелый зародыш, базальная часть молодого листа и в принципе любая другая ткань Zea mays, способная образовать каллюс. Особенно полезной тканью является щиток незрелого зародыша Zea mays.
В принципе все генотипы Zea mays (сорта, разновидности, инбредные линии, гибриды и т.п.) являются полезными в настоящем изобретении. Наиболее ценными являются коммерчески интересные генотипы.
Предпочтительными являются все элитные генотипы, особенно все инбредные линии, наиболее предпочтительны элитные инбредные линии. Подходящими инбредными линиями являются, например, Funk 2717, Funk 211D, Funk 2N 217A, B 73, A632, CM105, B37, B84, B14, Mo 17, R 168, MS71, A188, FA91, A641, и W117. Дополнительными примерами полезных генотипов Zea mays являются Funk 5 N 984 и Funk 5N986. Особенно предпочтительными являются элитные инбредные линии Funk 2717, Funk 211D, Funk 5N984 или Funk 2N217A, а наиболее предпочтительным является элитный генотип Funk 2717.
В приведенном ниже описании будет раскрыт способ получения эмбриогенных суспензий Funk 2717, из которых могут быть изолированы протопласты, которые способны регенерироваться в фертильные растения. Должно быть понятно, что могут быть использованы и другие генотипы кукурузы в описанном способе с применением в большинстве случаев рутинных методик, известных специалистам в данной области.
Стадия (a). Образование каллюса.
Выращивают растения Zea mays генотипа Funk 2717 до цветения и образования пыльцы. Незрелые зародыши удаляют из початков и помешают на среду для инициирования. Зародыши обычно имеют длину между 1 и 4 мм, предпочтительно между 1,5 и 3 мм, а более предпочтительно между 2 и 2,5 мм. Среда для инициирования может быть жидкой или отвержденной. Может быть использована любая среда для инициирования, способная индуцировать каллюс. Некоторые подходящие примеры включают, но не ограничиваются теми средами, которые основаны на MS-среде (Murashige T. and SRoog F., Physiol. Plant. 15 (1962) 473), среде N 6 (Chu et al., Scientia Sinica 18, (1975) 659), B5-среде (Gamborg o et al., Exp. Cell. Res. 50 (1968) 151), SH-среде или КМ-среде с соответствующими концентрациями сахаров и регуляторов роста растений. Другие подходящие сочетания описаны в Plant Culture Media "George E.E. et al/ /eds/, Exegetics Ltd, Edington, Westbury, Wiltshire, England /1987/.
Каллюс удаляют из культивированных незрелых зародышей и помещают на поддерживающую среду. Может быть использована любая поддерживающая среда, способная обеспечить пролиферацию каллюса. Некоторые подходящие примеры включают, но не ограничиваются средами, основанным на MS-cреде, N 6 среде, B5-среде или КМ-среде, с соответствующими концентрациями сахаров и регуляторов роста растений. Материал субкультивируют на свежей поддерживающей среде через соответствующий интервал времени, например, каждые 1-120 дней, предпочтительно 3-21 дня, более предпочтительно 5-14 дней. Инициирование и поддерживание может быть осуществлено на свету или в темноте, предпочтительно в темноте. Температура может быть между 0 и 50oC, предпочтительно между 20 и 32oC, более предпочтительно между 25 и 28oC.
Субкультивирование продолжают в тех же самых условиях до тех пор, пока каллюс не будет представлять собой каллюс специального типа. Специальный тип каллюса, пригодного для использования при инициировании суспензий в соответствии с настоящим изобретением, является относительно нелипким, гранулярным и рыхлым. На фиг. 1 представлен каллюс, который пригоден для использования при инициировании эмбриогенных суспензий в соответствии с настоящим изобретением. Пригодный каллюс субкультивируют с соответствующими интервалами времени, например, каждые 1-120 дней, предпочтительно 3-21 дня, более предпочтительно 5-14 дней. Указанный специальный тип каллюса, который способен регенерировать фертильные растения Zea mays и протопласты которого могут быть регенерированы в фертильные растения, может быть изолирован и представляет собой один из предпочтительных вариантов настоящего изобретения.
Стадия (b) - (d) суспензионного культивирования
Специальный тип каллюса со стадии (a) помещают на жидкую среду для формирования суспензии клеток и клеточных агрегатов. Некоторые подходящие примеры жидких сред включают, но не ограничиваются средами на основе M-среды, N 6 среды, B5-среды и KM-среды с соответствующими концентрациями сахаров и регуляторов роста растений. Предпочтительной средой является N6 среда.
Культуры субкультивируют с частотой, достаточной для поддержания клеток и клеточных агрегатов в жизнеспособном и пролиферирующем состоянии, например, каждые 1-120 дней, предпочтительно 3-21 дня, более предпочтительно 5-10 дней. Культуры, которые содержат агрегаты из плотных цитоплазматических, делящихся клеток, сохраняют. Культуры с высоким содержанием расширенных клеток, отбрасывают. Количество клеток, которые являются плотно цитоплазматическими, может увеличиваться со временем.
Желательным типом суспензионной культуры является культура, содержащая предпочтительно по крайней мере 50%, а более предпочтительно по крайней мере 70% клеток в агрегатах плотных, цитоплазматических делящихся клеток. Желаемый тип суспензионной культуры получают после подходящего периода времени, обычно после примерно 7 дней, предпочтительно после примерно 30 дней. Температура и световые условия являются такими же, как и в стадии (a). Суспензионная культура депонирована в АТСС, Рокквилл, Мэриленд, США, под номером хранения 40326 (дата депонирования: 20 мая 19687 г.).
Следовательно, в настоящем изобретении описан способ получения суспензионных культур клеток Zea mays и клеточных агрегатов, из которых могут быть изолированы протопласты, причем протопласты способны регенерировать фертильные растения, состоящие из стадий:
(a) получение каллюса, который является относительно нелипким, гранулярным и рыхлым, на индукционной и поддерживающей средах.
(b) переноса каллюса на жидкую среду для образования суспензии клеток или клеточных агрегатов.
(c) субкультивирования суспензии (i) в течение периода времени (предпочтительно в течение примерно 7 дней), достаточного для получения протопластов в жизнеспособной, деляющейся стадии, способных к регенерации в фертильные растения, и (ii) с частотой, достаточной для поддержания любых клеток и клеточных агрегатов в жизнеспособном состоянии, и
(d) отбора и сохранения тех культур из продукта стадии (c), которые содержат агрегаты из плотных, цитоплазматических, делящихся клеток.
Стадия (e1) - получение протопластов
Изоляцию протопластов проводят путем инкубации каллюсов или суспензионной культуры клеток или клеточных агрегатов (полученных в соответствии со стадиями (a) - (d) выше) с ферментным препаратом, который удаляет клеточные стенки. Подходящие ферментные препараты известны на данном уровне техники. Особенно подходящим ферментным препаратом является, например, препарат, содержащий 4% мас./объем RS целлюлозы (Якулт Фармацеутикал Инд., Ко, Лтд, Шингикан Шо, Нишиномия, Япония) с 1% мас./объем Розима (Ром энд Хаас, Филадельфия, ПА, США) в КМС солевом растворе (8,65 г/л KCl, 12,5 г/л CaCl2 • 2H2O, 16,47 г/л MgSO4 • 7H2O, 5 г/л MES pH 5,6). Переваривание осуществляют при температуре между 0 и 50oC, предпочтительно между 10 и 35oC, более предпочтительно между 26 и 32oC. Переваривание продолжают до тех пор, пока не освободятся протопласты. Время, требующееся для переваривания, обычно составляет от 30 минут до 5 дней, предпочтительно между 1 часом и 1 днем более предпочтительно между 3 и 5 часами.
Следовательно, кроме того, настоящее изобретение обеспечивает способ получения протопластов Zea mays, способных к регенерации фертильных растений, указанный способ состоит из стадий:
(a) получение каллюса, который является относительно нелипким гранулярным и рыхлым, на индуцирующей каллюс и поддерживающей каллюс средах,
(b) перенос каллюса на жидкую среду для получения суспензии клеток или клеточных агрегатов,
(c) субкультивирование суспензии (i) в течение периода времени (предпочтительно в течение примерно 7 дней), достаточного для получения протопластов в жизнеспособной, делящейся стадии, способных к регенерации в фертильные растения, и (ii) с частотой, достаточной для поддержания любых клеток и клеточных агрегатов в жизнеспособном состоянии,
(d) отбор и поддержание тех культур из продута стадии (c), которые содержат агрегаты плотных, цитоплазматических, делящихся клеток,
(e1) удаление клеточных стенок с помощью подходящих ферментов и изолирование полученных в результате протопластов.
Высвобожденные протопласты собирают и очищают с помощью стандартных методик, таких как фильтрация и центрифугирование.
Стадия (e2). Обработка протопластов экзогенной ДНК.
Протопласты могут быть обработаны экзогенной ДНК таким образом, чтобы получить клетки, которые содержат всю или часть экзогенной ДНК, стабильно введенную в их генетический материал. Экзогенная ДНК представляет собой любую ДНК, добавленную к протопласту. Экзогенная ДНК может содержать промотор, активный в Zea mays или может использовать промотор, уже имеющийся в геноме Zea mays.
Экзогенная ДНК может быть химерическим геном или его частью. Обработка протопластов экзогенной ДНК может быть проведена в соответствии с методиками, описанными в следующих публикациях:
(Paszkowski J. et al. The EMBO Journal 3, 12 (1984) 2717-2732; европейская заявка на патент EP-0 164 575 (Paszkowski, J. et al);
Shillito R.D. et al. Bio (Techonology, 3 (1985) 1099-1103; Potrykus I et al., Mol. Gen. Genet 199 (1985) 183-188; Zoers H., et al., Mol. Gen. Genet., 199 (1985) 178-182; Fromm M.E. et al., Nature, 319 (1986) 79)1-793; британская заявка на патент GB-2 140 822 (Mettler I.J.): и Negrutiu I. et al., Plant Mol. Biology 8 (1987) 363-373). Эти публикации приведены здесь в качестве уровня техники.
Экзогенная ДНК может быть добавлена в любой форме, такой как, например, незащищенная линейная или кольцевая ДНК, ДНК, капсулированная в липосомы, ДНК в шаропластах, ДНК и других растительных протопластах, ДНК, комплексно связанная с солями. ДНК в форме нуклеосом, хромосом или ядер или их частей и т.п. Внедрение инородной ДНК может быть стимулировано с помощью любого известного подходящего способа на данном уровне техники, включая методики, описанные в приведенных выше ссылках.
В первую очередь, химерические гены, рассматриваемые в настоящем изобретении, являются такими, которые обеспечивают трансформированным протопластам растений, происходящим из протопластов тканям и, наконец, происходящим из протопластов растениям, появление новых фенотипических свойств (ценных свойств), таких как повышенная устойчивость к паразитам (например, Arthropods, включая насекомых и клещей, нематод и т.п.); повышенная устойчивость к патогенам (например, к фитопатогенным грибам, бактериям, вирусам и т. п.); повышенная устойчивость к химикатам (например, гербицидам (таким как триазины, карбаматы, мочевины, динитроанилиды, сульфонилмочевины, имидазолиноны, триазоло-пиримидины, биалафос, глифосат и т.п.), инсектициды или другие биоциды), повышенная устойчивость к цитотоксинам (например, к гидромицину, канамицину, хлорамфениколу и т.п.); повышенная устойчивость к вредным воздействиям окружающей среды (почвенным или атмосферным) (например, к теплу, холоду, ветру, почвенным условиям, влаге, засухе и т.п.); с повышенным образованием желательных веществ (например, в листьях, семенах, клубнях, корнях, стеблях и т.п.). Желательные вещества, продуцируемые генетически измененным растением, включают белки, крахмалы, сахара, аминокислоты, алкалоиды, ароматические вещества, окрашивающие вещества, жиры и т.п.); пониженное образование нежелательных веществ (например, токсинов, ингибиторов протеаз, нежелательных ароматических веществ, нежелательных белков, жиров, углеводов или вторичных метаболитов и т.п.); или модифицированные морфологические или эволюционные фенотипические свойства (например, изменение высоты, характер произрастания, формы, цвета, крепкости, времени цветения и т.п.).
Устойчивость к цитотоксинам может быть придана геном, экспрессирующим в клетках растения фермент, который детоксифицирует цитотоксин, например, неомицин фосфотрансферазу типа II или аминогликозид фосфотрансферазу типа IV для детоксификации канамицина, гидромицина и других аминогликозидных антибиотиков, или глютатион S-трансферазу или цитохром P-450 или другой катаболический фермент, известный для детоксификации триазиновых, сульфонилмочевинных или других гербицидов. Устойчивость к цитотоксинам может быть также придана с помощью гена, который экспрессирует в растении форму "целевого фермента" (сайт действия цитотоксина), который является устойчивым к цитотоксину, например, форму ацетогидрокси кислой синтазы, которая является нечувствительной и ингибированию сульфонилмочевинами или имидазолинонами или другими гербицидами, действующими на этой стадии метаболизма, или форму 5-енолпирувил шикимат-3-фосфат (EPSP) синтазы, которая является нечувствительной к ингибированию глифосатом. Может быть целесообразным экспрессировать эти измененные целевые ферменты в форме, которая позволяет их транспорт в клетке растения в нужный клеточный отдел, а именно в хлоропласт, в приведенных выше примерах.
В некоторых случаях является целесообразным нацелить генный продукт в митохрондрии, вакуоли, в эндопламатические везикулы или другие частиц клеток или даже в межклеточное (апопластичное) пространство.
Устойчивость к некоторым классам грибов может быть придана при введении гена, который экспрессирует хитиназу в тканях растений. Многие патогенные для растений грибы содержат хитин как интегральную часть гифовой или споровой структуры, включая базидиомицеты (головни и ржавчины) и аскомицеты и несовершенные грибы (включая Alternaria и Bipolaris, Exerohilum turcicum, Colletotricum, Gleocercospora и Cercospora). Хитиназа может ингибировать рост мицелия некоторых патогенов ин винтро. Листья и корни растения, экспрессирующие хитиназу, защищены против многих типов грибных нападений.
Устойчивость к паразитам может быть придана геном, кодирующим пестицидный полипептид.
Устойчивость к Arthropods может быть придана, например, геном кодирующим полипептид, который является токсичным для стадий личинок и имаго, или придает растению непривлекательность для личинок, так что они меньше его едят.
Подходящим геном в этом контексте является кристаллический белок  Вторым классом белков, которые будут придавать устойчивость растению, являются ингибиторы протеаз. Ингибиторы протеаз являются обычными составляющими запасных структур растения (Ryan C., Ann. Rev. Plant Physiol; 24 (1973) 173 - 196). Ингибирование развития насекомых было приписано наличию ингибитра трипсина в муке (Lipke H. , Fraenkel G. S. and Liener I.E., J. Agg. Foos chem. 2 (1954) 410 - 414). Очищенный ингибитор протеазы Баумана-Бирка, выделенный из соевой муки, как было показано, ингибирует протеазы кишечника личинок Tenebrio (Berk V, Gertler A. and Khalef S., Biochem. Biophys. Acta, 67 (1963) 326 - 328). Ген, кодирующий ингибитор трипсина вигны китайской, описан Hilder et al (Nature, 330 (1987) 160 - 163).
Вторым классом белков, которые будут придавать устойчивость растению, являются ингибиторы протеаз. Ингибиторы протеаз являются обычными составляющими запасных структур растения (Ryan C., Ann. Rev. Plant Physiol; 24 (1973) 173 - 196). Ингибирование развития насекомых было приписано наличию ингибитра трипсина в муке (Lipke H. , Fraenkel G. S. and Liener I.E., J. Agg. Foos chem. 2 (1954) 410 - 414). Очищенный ингибитор протеазы Баумана-Бирка, выделенный из соевой муки, как было показано, ингибирует протеазы кишечника личинок Tenebrio (Berk V, Gertler A. and Khalef S., Biochem. Biophys. Acta, 67 (1963) 326 - 328). Ген, кодирующий ингибитор трипсина вигны китайской, описан Hilder et al (Nature, 330 (1987) 160 - 163).
Ген, кодирующий ингибитор протеазы, может быть помещен под контролем промотора растения, предпочтительно конститутивного промотора, такого как 35S прмотор вируса мозаики подсолнечника (CaM V), в подходящем векторе, таком как описанный ниже вектор pCIB 710.
Например, ген, кодирующий последовательность ингибитора протеазы Баумана-Бирка сои, может быть получен с использованием методов клонирования кДНК, описанных Хаммондом с сотр. (J. Biol. Chem. 259 (1984) 9883 - 9890). Соответствующий клон может быть идентифицирован или иммуноосаждением гибрида, выбранного мРНК, как описано Хаммондом с сотр., или зондированием при низкой волокнистости с синтетическим олигонуклеотидом, содержащим 50 или более оснований с 5'-конца последовательности, кодирующей заранее определенную аминокислотную последовательность (аминокислотные последовательности приведены в справочнике Г. Фасмана "Handbook of Biochemistry and Molecular Biology, 2 (1976) 611 - 620). Например, для клонирования гена для ингибитора трипсина Куница из сои в качестве зонда использован следующий нуклеотид:
AA Asp Phe Val Leu Asp Asn Glu Gly Asn Pro Leu Glu Asn Gly зонд 5'-GAC TTC GTT GTG GAC AAC GAA GGT AAC CCG GTG GAA AAC GGT Gly Thr Tyr Tyr Ile Leu Ser AGP Ile Thr Ala Phe Gly Gly GGT ACC TAC TAC ATC CTG TCC GAC ATC ACC GCT TTC GGT GGT-3'
Клонированный фрагмент ДНК может быть секвенсирован с использованием, например, методики Максима и Гилберта (Methods Enzymol., 65 (1980) 499 - 560) и идентифицирован начальный сайт трансляции. Если клонированный фрагмент ДНК длиннее, чем кодирующая последовательность, дополнительные 5'- и 3'-последовательности резектируются Bal 31. Если клонированный фрагмент ДНК короче, чем кодирующая последовательность, используют клонированную ДНК в качестве праймера для растяжения праймера мРНК (Maniatis et al., Molecular Cloning: A Laboratory Manual (1982), Cold Spring Harbor, N. Y.) и реклонируют расширенную ДНК.
Прибавляют соответствующие линкеры к ДНК, кодирующей белок и вставляют фрагмент в выбранный вектор (кассета), например pCIB 710, так что ген находится под контролем желаемого промотора (CaMV 35S и pCIB 710).
Альтернативным способом получения гена для ингибиторов протеаз с меньше, чем 100 аминокислотами, таких как ингибитор трипсина лимской фасоли, является их синтез. Кодирующая последовательность является заранее определенной соответствующими сайтами обратной трансляции и рестрикции для целевого вектора, включенными с каждого конца. Синтетический ген получают путем синтеза перекрывающихся олигонуклеотидов из 30 - 60 оснований. Эти фрагменты киназируют, лигируют (Maniatis et al., выше) и клонируют в соответствующий вектор.
Клон, вставка которого находится в нужной ориентации, может быть идентифицирован секвенсированием. Плазмиду ДНК изолируют и используют для введения в протопласты кукурузы.
Следовательно, в настоящем изобретении описан способ, в котором способ, описанный в стадиях (a) - (d), кроме того содержит стадию:
(e2) обработки протопластов со стадии (e1), способных к регенерации в фертильные растения, экзогенной ДНК, чтобы получить клетки, которые содержат всю или часть экзогенной ДНК, стабильно введенной в их генетический материал.
Способ получения Zea mays, имеющих стабильно введенную экзогенную ДНК, предпочтительно экзогенную ДНК, экспрессируемую в Zea mays, является еще одним предпочтительным объектом настоящего изобретения
Особенно предпочтительным способом получения трансгенных Zea mays является тот, где стабильно введенная экзогенная ДНК является химерным геном, который кодирует полипептид, обладающий по существу токсичностью по отношению к насекомым (инсектицидной активностью) кристаллического белка 
Стадия (f). Посев протопластов.
Протопласты высевают для культивирования или с или без обработки ДНК в жидкой или отвержденной культуральной среде. Некоторые примеры подходящих культуральных сред включают, но не ограничиваются средами, основанными на КМ-среде, N6-среде, B5-среде и MS-среде, с соответствующими концентрациями сахаров и регуляторов роста растений. Предпочтительной средой является KM-среда, содержащая отверждающий агент. Предпочтительным отверждающим агентом является агароза, особенно Sea PlagueR агароза (европейская заявка на патент EP-0 129 668, Shillito R.D. et al., (1984). При ее использовании концентрации Sea PlagueR агарозы может быть между 0,1% и 2,5% мас./объем, предпочтительно между 0,6% и 1,5% мас./объем. Оптимальная концентрация других отверждающих агентов может меняться, но может быть легко определена.
Среда, в которой обычно культивируют протопласты в присутствии подходящих регуляторов роста растений (таких как ауксины и/или цитокинины и т.п.), способствует делению протопластов и образованию колоний. Подходящие регуляторы роста растений включают, но не ограничиваются, 2,4-дихлорфеноксиуксусной кислотой (2,4-Д), 3,6-дихлор-2-метоксибензойной кислотой (дикамба), 4-амино-3,6,6-трихлорпиколиновой кислотой (пиклорам) и пара-хлорфеноксиуксусной кислотой (pCPA). Количество регулятора роста растений является эффективным для регулирования роста растений, нетоксичным количеством. Концентрация таких регуляторов роста растений обычно находится в интервале от 0,01 до 100 мг/л, предпочтительно 0,1 - 100 мг/л, более предпочтительно 0,1 - 10 мг/л, еще более предпочтительно 0,3 - 3 мг/л.
Неожиданно было обнаружено, что О-низший алканоилсалициловые кислоты (о-ацетилсалициловая кислота и ее гомологи/ одни или в сочетании с регуляторами роста растений и/или ДМСО промотрируют деление или образование колоний из протопластов растений, предпочтительно протопластов Zea mays Термин "низший алканоил" означает алканоильные группы, имеющие 1 - 7, предпочтительно 1 - 4, более предпочтительно 2 - 3 атома углерода.
Предпочтительными О-низший алканоилсалициловыми кислотами являются О-ацетилсалициловая кислота и О-пропионилсалициловая кислота, предпочтительно О-ацетилсалициловая кислота.
Количество О-низший алканоилсалициловой кислоты должно быть достаточным для промотирования деления клеток и образования колоний. Предпочтительным является эффективное рострегулирующее, нетоксичное количество. Подходящими концентрациями О-низший алкеноил-салициловых кислот является интервал от 0,1 до 3000 мг/л, предпочтительно в интервале от 10 до 300 мг/л, более предпочтительно от 10 до 100 мг/л, а еще более предпочтительно примерно 100 мг/л.
Количество ДМСО является достаточным для промотирования деления клеток или образования колоний. Подходящими концентрациями ДМСО являются концентрации в интервале от 0 до 3%, предпочтительно, 0,01 - 2%, более предпочтительно 0,1 - 1%.
Следовательно, в настоящем изобретении описан способ повышения деления клеток или образования колоний из протопластов растений, предпочтительно протопластов Zea mays, заключающийся в культивировании протопластов в присутствии эффективно регулирующего рост растений нетоксичного количества О-низший алканоилсалициловой кислоты, достаточного для промотирования деления клеток или образования колоний. Используют 0,01 - 100 мг/д, предпочтительно 0,1 - 100 мг/л, более предпочтительно 0,1 - 10 мг/л, еще более предпочтительно 0,3 - 3 мг/л регулятора роста растений.
Предпочтительно присутствие ДМСО в количестве, промотирующем клеток или образование колоний. Предпочтительными количествами ДМСО являются 0 - 3%, предпочтительно 0,01 - 2%, более предпочтительно 0,1 - 1%.
Этот способ повышения деления или образования колоний из протопластов является пригодным для всех растительных протопластов и не ограничивается Zea mays. Особенно пригодным он является для Angiospermae и Gymnospermae, например, растений следующих семейств: Solanaceae, Crueiferae, Liliaceae, Yitaceae, Chenopodiaceae, Rutaceae, Bromoliaceae, Rubidiaceae, Theaceae, Musaceae или Graminaceae и из порядка Leguminoseae, в особенности семейства Papilionaceae. Предпочтительные растения являются представителями Solanaceae, Cruciferae и Gramineae.
Среда, в которой культивируют протопласты, может содержать среду, которая ранее была сбалансирована для роста подходящих клеток, например, Zea mays или Dactylis glomerafa.
Протопласты могут быть культивированы на твердой или жидкой среде без субкультивирования в течение периода времени до 12 недель, предпочтительно до 6 недель, более предпочтительно в интервале от 3 до 4 недель. Альтернативно, твердая среда может быть помещена в жидкую среду, как описано в европейской заявке на патент EP-0 129 668, Shillito R.D. et al., (1984), или обработана некоторым другим способом, чтобы помочь делению и/или образованию колоний из протопластов. Одним из подходящих способов является культивирование вместе с выращиваемыми клетками, как описано Shneyour et al. / Plant Scince Zett. 33 (1984) 293) и Smitn et al. (Plant Science Zett 36 (1984) 67) и в примере 8S.
Протопласты культивируют на свету или, предпочтительно, в темноте при температуре между 0 и 50oC, предпочтительно между 20 и 32oC, более предпочтительно между 25 и 28oC. Интенсивность света обычно составляет 0,1 - 200 мкЕ/(м2•с) (микроЭнштйен) (м2•с), предпочтительно 0,1 - 100 мкЕ/(м2•с), более предпочтительно 30 - 90 мкЕ/(м2•с), еще более предпочтительно 10 - 40 мкЕ/(м2•с). Каллюс, развивается из протопластов, культивированных на твердой среде, а каллюс, происходящий из протопластов согласно стадии (f), может быть субкультивирован на поддерживающей среде, как описано для каллюса на стадии (a), а затем субкультивирован на регенерирующей среде согласно стадии (g), или может быть непосредственно субкультивирован на регенерирующей среде согласно стадии (g).
Стадия (g). Регенерация ростков.
Каллюс, который образовался из протопластов согласно стадии (f), субкультивируют один или более раз на подходящих средах, чтобы регенерировать фертильное растение. Некоторые примеры подходящих культуральных сред включают, но не ограничиваются теми средами, которые основаны на N6-среде, B5-среде, MS-среде или KM-среде, с соответствующими концентрациями сахаров, регуляторов роста растений и в некоторых случаях ДМСО. Предпочтительной средой является N6-среда без регуляторов роста, или содержащая цитоксин и/или другие регуляторы роста растений, стимулирующие регенерацию ростков и/или содержащие О-низший алканоилсалициловую кислоту, необязательно в дополнительном сочетании с ДМСО, как описано выше в стадии (1). Появляется каллюс, содержащий дифференциированные клетки и клеточные агрегаты (фиг. 6) после соответствующего периода времени, обычно 1 недели до 6 месяцев, более типично 1 - 3 месяца. Затем каллюс дифференциируется с образованием ростков после соответствующего дополнительного времени, обычно от 1 недели до 6 месяцев, более типично 1-3 месяцев.
Каллюс культивируют на этой стадии на свету. Интенсивность света обычно находится между 0,1 и 100 мкЕ/(м2•с), предпочтительно между 30 и 80 мкЕ/(м2•с).
Стадия (h). Получение фертильного растения из ростка.
Ростки переносят на подходящую среду, например, N6 среду без регуляторов роста растений. Альтернативно могут быть добавлены регуляторы роста, стимулирующие рост корней или побегов. Ростки культивируют на этой среде до тех пор, пока они не образуют корней. Время, требующееся для образования корней, обычно составляет 1 - 6 недель, более типично примерно 2 недели. Когда корни и ростки достаточно вырастут, растения переносят в почву и осторожно прижимают. Достаточной длиной корней на этой стадии является интервал от 1 до 10 см, более типично 2 - 5 см, а для ростков она находится в интервале от 1 до 15 см, более типично 2 - 10 см.
Растения выращивают до цветения и полового размножения или проводят вегетативное размножение обычным путем.
Трансгенные растения кукурузы, содержащие экзогенную ДНК, могут быть поддержаны в культуре или могут быть регенерированы в растения. Клетки растений кукурузы, в которые может быть введена экзогенная ДНК, включают любые клетки растений кукурузы, способные к восприятию, репликации и экспрессии чужеродных генов на подходящем уровне. Некоторыми пригодными клетками растений кукурузы являются, например, клетки, выделенные из инбредных линий Funk 57984, Funk 5H986, Funk 2717, 211D, Funk 2N217A, B73, A632, CM105, B37, B84, B14, Mo 17, R168, MS71, A188, FA91, A641, и W117. Предпочтительными являются все элитные генотипы. Более предпочтительными являются все элитные генотипы. Более предпочтительными являются все инбредные линии. Еще более предпочтительными являются все элитные инбредные линии, особенно элитные инбредные линии Funk 5N984, Funk 5N986, Funk 2717, Funk 211D или Funk 2N217A, а еще более предпочтительная Funk 2717.
Культуральная среда, способная поддерживать конкретную клетку растения в культуре, может зависеть от использованного генотипа растения кукурузы. Например, некоторые подходящие среды включают, но не ограничиваются средами, основанными на MS (Murashige and Skoog (1962)), N6 (Chu et al., (1975)), B5 (Gamborg et al. (1968)) и KM (Kao and Michayluk (1975)) средах с соответствующими концентрациями сахаров и регуляторов роста растений.
Следовательно, в настоящем изобретении описан способ регенерации растений Zea mays из протопластов, заключающийся в стадиях:
(e1) продуцирования протопластов, способных быть регенерированными в фертильные растений, из клеток и клеточных агрегатов стадий (d),
(f) посева протопластов на подходящую среду, в которой протопласты делятся и образуют клетки,
(g) субкультивирования клеток, полученных на стадии (f), чтобы регенерировать ростки, и (h) культивирование ростков на среде роста для получения фертильных растений.
Изобретение далее обеспечивает способ, в котором протопласты Zea mays содержат стабильно введенную экзогенную ДНК, предпочтительно экзогенную ДНК, экспрессируемую в растениях Zea mays. Более предпочтительным является культивирование протопластов Zea mays в присутствии 0,01 - 100 мг/л регулятора роста растений (предпочтительно 2,4-Д, дикамба, пиклорама или pCPA, наиболее предпочтительно 2,4-Д) и в присутствии О-низший алканоилсалициловой кислоты (предпочтительно 2,4-Д) и в присутствии О-низший алканоилсалициловой кислоты (предпочтительно О-ацетилсалициловой кислоты или О-пропионилсалициловой кислоты, наиболее предпочтительно О-ацетилсалициловой кислоты) в количестве, достаточном для промотирования деления клеток или образования колоний. Наиболее предпочтительным является способ, включающий дополнительно присутствие ДМСО в количестве, достаточном для промотирования деления клеток или образования колоний.
Дополнительно описан способ получения трансгенных протопластов Zea mays, способных регенерироваться в фертильные трансгенные растений, включающий обработку протопластов Zea mays, способных регенерироваться в фертильные растения, экзогенной ДНК, чтобы получить клетки, которые содержат всю или часть экзогенной ДНК, включенной в их генетический материал.
Подходящими экзогенными ДНК являются, например, химерические гены, описанные выше или впоследствии. Термин "ростки" включает любой растительный материал, способный к половому или неполовому размножению, или размножаемый ин виво или ин витро. Такие ростки предпочтительно состоят из протопластов, клеток, каллюсов, тканей, зародышей или семян регенерированных растений. Потомство регенерированных растений и регенерированных трансгенный растений включает мутанты и варианты.
Детальное описание.
(Часть B) инсектицидные растения кукурузы (Zea mays)
Настоящее изобретение обеспечивает способ получения химерного гена  токсина. Рассматриваемые клетки растений кукурузы включают все генотипы (сорта, разновидности, инбредные линии, гибриды и т.п.) растений кукурузы, в которых может быть введена чужеродная ДНК, реплицирована и экспрессирована. Некоторые примеры подходящих генотипов растений кукурузы включают, но не ограничиваются такими инбредными линиями, как Funk 2717, Funk 211D, Funk 2N217A, B73, A632, CM105, B37, B84, В14, Mo17, 168, M71, A188, FA91, A641 и W117. Funk 5N984 и 5N986 являются дополнительными примерами полезных генотипов Zea mays. Предпочтительно генотипом является элитный генотип, более предпочтительно элитная инбредная линия, выбранная в группе, состоящей из Funk 5N984, Funk 5N986, Funk 2717, Funk 211D или Funk 2N217A, а еще более предпочтительно им является инбредная линия Funk 2717.
токсина. Рассматриваемые клетки растений кукурузы включают все генотипы (сорта, разновидности, инбредные линии, гибриды и т.п.) растений кукурузы, в которых может быть введена чужеродная ДНК, реплицирована и экспрессирована. Некоторые примеры подходящих генотипов растений кукурузы включают, но не ограничиваются такими инбредными линиями, как Funk 2717, Funk 211D, Funk 2N217A, B73, A632, CM105, B37, B84, В14, Mo17, 168, M71, A188, FA91, A641 и W117. Funk 5N984 и 5N986 являются дополнительными примерами полезных генотипов Zea mays. Предпочтительно генотипом является элитный генотип, более предпочтительно элитная инбредная линия, выбранная в группе, состоящей из Funk 5N984, Funk 5N986, Funk 2717, Funk 211D или Funk 2N217A, а еще более предпочтительно им является инбредная линия Funk 2717.
Химерный ген, описанный в настоящем изобретении, содержит промоторную область, которая функционирует эффективно в растениях кукурузы, и кодирующую область, которая кодирует кристаллический белок  или полипептид, обладающий по существу инсектицидными свойствами кристаллического белка
или полипептид, обладающий по существу инсектицидными свойствами кристаллического белка  Кодирующая последовательность химерического гена не известна в ассоциации с указанным промотором в природных генах.
Кодирующая последовательность химерического гена не известна в ассоциации с указанным промотором в природных генах.
5'- и/или 3'-нетранслируемые области независимо могут быть ассоциированы в природе или с промотором или с кодирующей областью, или ни с промотором и ни с кодирующей областью. Предпочтительно или 5'- или 3'-нетранслиреумая область ассоциирована с промотором в природных генах, а еще более предпочтительно обе 5'- и 3'-области ассоциированы с промотором в природных генах.
На основании состояния уровня техники в настоящее время нельзя было предсказать, что клетки кукурузы будут экспрессировать инсектицидный полипептид на любом уровне, и особенно в достаточных количествах, чтобы придать инсектицидные свойства клеткам. В частности, считалось, что полипептид как большой и нерастворимый и как полипептид, обладающий инсектицидными свойствами кристаллического белка  будет особенно трудно экспрессироваться в клетках растений.
будет особенно трудно экспрессироваться в клетках растений.
Для того, чтобы считаться инсектицидным, клетки растений должны содержать инсектицидное количество токсина, обладающего инсектицидной активностью кристаллического белка  Инсектицидным количеством является такое количество, которое при наличии в клетках растений, убивает личинок насекомых или по крайней мере снижает существенно их кормление.
Инсектицидным количеством является такое количество, которое при наличии в клетках растений, убивает личинок насекомых или по крайней мере снижает существенно их кормление.
Ген.
Транскрипция контрольных последовательностей.
Химерный ген, описанный в настоящем изобретении, содержит последовательности контроля транскрипции, включающие промотор и 5'- 3'-нетранслируемые последовательности, которые являются функциональными в растениях кукурузы. Эти последовательности могут независимо происходить из любого источника, такого как гены вируса, растения или бактерий.
Вирусные промоторы и 5'- и 3'-нетранслируемые последовательности, пригодные для использования, являются функциональными в растениях кукурузы и их получают, например, из вирусов растений, таких как вирус мозаики подсолнечника (CaMV). Вирус подсолнечника был охарактеризован и описан Хоном в сорт. в Current Topics in Microbiology and Immunology, 96 (1982) 194-220 и в приложениях A - G. Это описание приведено здесь в качестве уровня техники.
CaMV является атипичным вирусом растений, в котором содержится двуниточная ДНК. По крайней мере два промотора CaMV являются функциональными в растениях, а именно 19S промотор, который является результатом транскрипции гена VI CaMV и промотор 35S транскрипта, 19S промотор и 356C промотор являются предпочтительными вирусными растительными промоторами для использования в настоящем изобретении.
CaMV 19S промоторы и 5'-нетранслируемые области могут быть получены с помощью рестрикционной карты, такой как карта, описанная на фиг.4 на странице 199 статьи Хона с сотр., упомянутой выше, или из последовательности, которая приведена в приложении C в статье Хона с сотрудниками.
Для того, чтобы изолировать CaMV 19S промотор и, в некоторых случаях, соседнюю 5'-нетранслируемую область, отбирают рестрикционный фрагмент CaMV генома, содержащий желаемый последовательности. Подходящим рестрикционным фрагментом, который содержит 195 промотор и 5'-нетранслируемую область, является фрагмент между Pst I сайтом, начинающимся в положении 5386, и Hind III сайтом, начинающимся в положении 5850 фиг. 4 и приложения C статья Хона с сотрудниками.
По аналогичным методикам может быть получен 35S промотор CaMV как описано в примере 6 ниже.
Нежелательные последовательности в рестрикционном фрагменте в некоторых случаях могут быть удалены с помощью стандартных методик. Некоторые подходящие методики для делеции нежелательных нуклеотидов включают использование экзонуклеаз (Maniatis et al., Molecular Cloning, Cold Spring Harbor Laboratory. (1982) 135-139) и олигонуклеотидно-направленный мутагенез (Loller and Smit, Meth. Enzymol., 100 (1983) 468-500).
Подробная процедура может быть использована для получения желаемой 3'-нетранслируемой области. Например, подходящая 3'-нетранслируемая последовательность CaMV 19S гена может быть получена путем изоляции области между EcoRV сайтом в положении 7342 и Bgl II сайтом в положении 7643 генома CaMV как описано на фиг.4 Приложении C статьи Хона с сотрудниками.
Примеры промоторов генов растений и 5'- и 3'-нетранслируемых областей, пригодные для использования в настоящем изобретении, также включают промоторы гена, кодирующего маленькую субъединицу риболоза бифосфат карбоксилазы и хлорофилл а/в - связующего белка. Эти области растительного гена могут быть изолированы из клеток растения с помощью способов, сравнимых со способами, описанными выше для изоляции соответствующих областей из CaMV смотри Morelli et al., Nanure, 315 (1985) 200-204.
Подходящие промоторы и 5'- и 3'-нетраслируемые области из бактериальных генов включают те, что присутствуют в Т-ДНК области Agrobacterium плазмид. Некоторые примеры подходящих Agrobacterium плазмид включают Ti плазмиду A. turefaciens плазмиду и Ri плазмиду
A. thizogenes. Agrobacterium промоторы и 5' и 3'- нетранслируемые области, полезные в настоящем изобретении, являются в частности теми, что присутствуют в генах, кодирующих октопин синтазу и нопалин синтезу. Эти последовательности могут быть получены по методикам, подобным описанным выше для изоляции CsMV и растительных промоторов и нетранслируемых последовательностей; смотри Bevan et al., Nabure, 304 (1983) 184-187.
Кодирующая область
Кодирующая область химерического гена содержит нуклеотидную последовательность, которая кодирует полипептид, обладающий по существу токсичностью  дельта-эндотоксина кристаллического белка. Полипептид для целей настоящего изобретения обладает по существу токсичностью
дельта-эндотоксина кристаллического белка. Полипептид для целей настоящего изобретения обладает по существу токсичностью  дельта-эндотоксина кристаллического белка, если он является инсектицидным для подобного ряда личинок насекомых, как и кристаллический белок от штаммов
дельта-эндотоксина кристаллического белка, если он является инсектицидным для подобного ряда личинок насекомых, как и кристаллический белок от штаммов  Некоторые подходящие штаммы включают, например,
Некоторые подходящие штаммы включают, например,  var. Rurstaki,
var. Rurstaki,  var. berliner,
var. berliner,  var. alesti;
var. alesti;  var. tolworthi;
var. tolworthi;  var. sotto;
var. sotto;  var. dandrolimus;
var. dandrolimus;  var. tenebrionis;
var. tenebrionis;  var. san diego; и
var. san diego; и  var. aizawai.
var. aizawai.
Предпочтительным штаммом являются  var. Rurstaki, а особенно
var. Rurstaki, а особенно  var. Rurstaki HDI.
var. Rurstaki HDI.
Кодирующая область может существовать в природе в  Альтернативно кодирующая область может содержать последовательность, которая отличается от последовательности, которая существует в
Альтернативно кодирующая область может содержать последовательность, которая отличается от последовательности, которая существует в  , но является эквивалентной из-за вырождения генетического кода.
, но является эквивалентной из-за вырождения генетического кода.
Кодирующая последовательность химерического гена может также кодировать полипептид, который отличается от пригодно встречающегося кристаллического белка дельта-эндотоксина, но который еще обладает по существу токсичностью по отношению к насекомым кристаллического белка. Такая кодирующая последовательность обычно будет вариантом природной кодирующей области. "Вариант" природной последовательности ДНК является модифицированной формой природной последовательности, которая осуществляет ту же функцию. Вариант может быть мутацией или может быть синтетической последовательностью ДНК и является по существу гомологичной соответствующей природной последовательности. "Существенная гомология последовательности" должна быть здесь понята как фрагмент ДНК, имеющей нуклеотидную последовательность достаточно подобную другому фрагменту ДНК, чтобы получить белок, обладающий подобными свойствами; или полипептид, имеющий аминокислотную последовательность, в достаточной мере подобную другому полипептиду, чтобы проявлять подобные свойства. Обычно последовательность ДНК является в значительной мере гомологичной второй последовательности ДНК, если по крайней мере 70%, предпочтительно по крайней мере 80%, а еще более предпочтительно по крайней мере 90% активных участков последовательности ДНК являются гомологичными. Два различных нуклеотида считаются гомологичными в кодирующей области последовательности ДНК для целей определения существенной гомологии, если замещение одного на другой составляет немую мутацию.
Предпочтительно, чтобы нуклеотидная последовательность являлась по существу гомологичной по крайней мере той части или тем частям природной последовательности, которые ответственны за инсектицидную активность.
Полипептид, экспрессированный химерическим геном, использованным в настоящем изобретении, обычно также будет обладать по крайней мере некоторыми иммунологическими свойствами природного  кристаллического белка, так как он имеет по крайней мере некоторые из одинаковых антигенных детерминат.
кристаллического белка, так как он имеет по крайней мере некоторые из одинаковых антигенных детерминат.
Следовательно, в настоящем изобретении полипептид кодированный химерическим геном предпочтительно структурно относится к дельта-эндоксину кристаллического белка, продуцируемому  .
.  продуцирует кристаллический белок с субъединицей, которая является протоксином, имеющим Мг примерно 130000-140000. Эта субъединица может быть расщеплена протеазами или щелочами с образованием инсектицидных фрагментов, имеющих Мг 50000 и возможно даже ниже. Химерические гены, которые кодируют такие фрагменты протоксина или даже его меньшие части, согласно настоящему изобретению могут быть сконструированы, поскольку фрагмент или части фрагментов имеют требуемую инсектицидную активность. Протоксин, инсектицидные фрагменты протоксина и инсектицидные части этих фрагментов могут быть слиты с другими молекулами, такими как полипептиды.
продуцирует кристаллический белок с субъединицей, которая является протоксином, имеющим Мг примерно 130000-140000. Эта субъединица может быть расщеплена протеазами или щелочами с образованием инсектицидных фрагментов, имеющих Мг 50000 и возможно даже ниже. Химерические гены, которые кодируют такие фрагменты протоксина или даже его меньшие части, согласно настоящему изобретению могут быть сконструированы, поскольку фрагмент или части фрагментов имеют требуемую инсектицидную активность. Протоксин, инсектицидные фрагменты протоксина и инсектицидные части этих фрагментов могут быть слиты с другими молекулами, такими как полипептиды.
Кодирующие области, пригодные для использования в настоящем изобретении, могут быть получены из генов кристаллического белкового токсина, изолированных из Bt например, смотри Whiteley et al., заявка PCT WO 86, 01536 и патенты США 4 448 885 и 4 467 036. Предпочтительная нуклеотидная последовательность, которая кодирует кристаллический белок, является такой, как показано от нуклеотида 156 до 3623 и последовательности формулы (1) или более короткой последовательности, которая кодирует инсектицидный фрагмент и такого кристаллического белка; Geiser et al., Gene 48 (1986) 109-118.
Следовательно, пригодным для использования в настоящем изобретении также является ген, имеющий нуклеотидную последовательность формулы I, гены, имеющие последовательность, по существу гомологичную гену формулы I, и гены, кодирующие полипептид, имеющий последовательность формулы II, или имеющий последовательность, по существу гомологичную последовательности формулы II, и предпочтительно такие из этих генов, которые экспрессируются в растениях, более предпочтительно в Zea mays.
Кодирующая область, определенная нуклеотидами 156 до 3623 последовательности I, кодирует полипептид последовательности формулы II.
Последовательность формулы II см. в конце описания.
Следовательно, в настоящем изобретении кроме того описан полипептид, имеющий последовательность формулы II, или инсектицидные части этого полипептида.
Кроме того, было показано, что некоторые штаммы  являются токсичными к другим, чем чешуекрылые насекомые. Токсичность
являются токсичными к другим, чем чешуекрылые насекомые. Токсичность  штамме tenebrionis по отношению к жесткокрылым насекомым и последовательность ассоциированного гена токсина обсуждается Herrustadt and Soares иEP-0 202 739 и E - 0 213 817.
штамме tenebrionis по отношению к жесткокрылым насекомым и последовательность ассоциированного гена токсина обсуждается Herrustadt and Soares иEP-0 202 739 и E - 0 213 817.
Векторы.
Для того, чтобы ввести химерический ген в клетки растений, ген сначала вставляют в вектор. Если ген не является доступным в количестве, достаточным для трансформации вектор может быть амплифицирован путем репликации в клетке хозяина. Наиболее удобными клетками хозяевами для амплификации являются бактериальные или дрожжевые клетки. Когда будет доступно достаточное количество химерического гена, его вводят в протопласты кукурузы согласно настоящему изобретению. Введение гена в растения кукурузы может быть осуществлено с помощью вектора, использованного для репликации или с помощью другого вектора.
Некоторые примеры бактериальных клеток хозяев, пригодных для репликации химерического гена, включают бактерии рода Escherihia, такие как E.coli, и Agrobacterium такие как A. tumefaciens или  rhizogenes. Методы клонирования гетерологических генов в бактерии описаны Coher et al., патенты США 4237224 и 468464.
rhizogenes. Методы клонирования гетерологических генов в бактерии описаны Coher et al., патенты США 4237224 и 468464.
Репликация генов, кодирующих кристаллический белок  в E.coli описана Wong et al., J. Biol. Chem. 258 (1983), 1960-1967.
в E.coli описана Wong et al., J. Biol. Chem. 258 (1983), 1960-1967.
Некоторые примеры дрожжевых клеток хозяев, пригодных для репликации генов настоящего изобретения, включают дрожжи вида Saccharomyces.
Любой вектор, в который химерический ген может быть вставлен и который реплицируется в подходящей клетке хозяина, такой как бактерия или дрожжи, может быть использован в настоящем изобретении для амплификации генов. Вектор может быть, например, происходящим из фага или плазмиды. Некоторые примеры векторов, происходящих из фагов, полезных в изобретении, включают векторы, происходящие из M13 или из лямбда. Некоторые подходящие векторы, происходящие из M13, включают M13 mp18 и M13 mp19. Некоторые подходящие векторы, происходящие из лямбда, включают лямбда-gt11, лямбда-gt7 и лямбда Шарон 4.
Некоторые векторы, происходящие из плазмид, особенно пригодные для репликации в бактериях, включают pBR322 (Bolivar et al., Gene 2 (1977) 95-113); pUC18 и pUC19 (Harrander et al., Gene, 26 (1983) 101-104); и Ti-плазмиды (Bevan et al., Nature, 304 (1983) 184-187). Предпочтительными векторами для амплификации генов в бактериях являются pBR322, pUC18 и pUC19.
Конструирование векторов для репликации.
Для конструирования химерического гена, пригодного для репликации в бактериях последовательность промотора, 5'-нетранслируемую последовательность, кодирующую последовательность и 3'-нетранслируемую последовательность вставляют в или объединяют в нужном порядке в подходящий вектор, такой как описанный выше вектор. Для того, чтобы он был пригоден, вектор должен быть способен к репликации в клетке хозяина.
Промотор, 5'-нетранслируемая область, кодирующая область и 3'-нетранслируемая область, которые составляют химерический ген, сначала могут быть объединены в единое целое вне вектора, а затем вставлены в вектор. Альтернативно участки химерического гена могут быть вставлены в вектор отдельно. Предпочтительно вектор также содержит ген, который придает свойство клетке хозяина, позволяющее селекцию клеток, содержащих вектор. Предпочтительным свойством является устойчивость к антибиотику. Некоторые примеры полезных антибиотиков включают ампициллин, тетрациклин, гигромицин, G418 и неомицин.
Вставка или объединение гена с вектором осуществляется по стандартным методикам, таким как использование рекомбинантной ДНК (Maniatis et al., (1982)) и рекомбинантная гомология (Hinnen et al., Proc. Natl. Acad. Sci 75 (1978) 1929-1933).
При использовании методов рекомбинантной ДНК вектор разрезают, вставляют желаемую последовательность ДНК между разрезанными участками вектора и концы желаемой последовательности ДНК лигируют к соответствующим концам вектора.
Наиболее удобно разрезать вектор с помощью подходящих рестрикционных эндонуклеаз. Некоторые подходящие эндонуклеазы включают те, которые образуют тупые концы, такие как Smal, Hpal и EcoRV и те, которые образуют липкие концы, такие как EcoR1, Sac1 и BamH1.
Желаемая последовательность ДНК обычно существует как часть большой молекулы ДНК, такой как хромосома, плазмида, транспозон, или фаг. Желаемую последовательность ДНК вырезают из ее источника и в некоторых случаях модифицируют таким образом, что ее концы могут быть соединены с концами разрезанного вектора. Если концы желаемой последовательности ДНК и разрезанного вектора являются тупыми концами, их соединяют лигазами для тупых концов, такими как Т4 ДНК лигаза.
Концы желаемой последовательности ДНК также могут быть соединены с концами разрезанного вектора в виде липких концов, в этом случае с помощью лигазы для липких концов, которой также может быть Т4 ДНК лигаза. Другие подходящие лигазы для липких концов включают, например, E.coli ДНК лигазу.
Липкие концы наиболее удобно образуются при разрезании желаемой последовательности ДНК и вектора одной и той же рестрикционной эндонуклеазой. В этом случае желаемая последовательность ДНК и разрезанный вектор имеют липкие концы, которые являются комплементарными друг другу.
Липкие концы также могут быть сконструированы при добавлении комплементарно гомополимерных хвостов к концам желаемой последовательности ДНК и к разрезанному вектору, используя концевую деоксинуклеотидилтрансферазу или при добавлении синтетической олигонуклеотидной последовательности, распознаваемой конкретной рестрикционной эндонуклеазой, известной как линкер, и расщепления последовательности эндонуклеазой: смотри, например Maniatis et al., (1962).
Конструирование векторов для трансформации растений.
В дополнение к химерическому гену, кодирующему  токсин или
токсин или  -подобный токсин, вектор предпочтительно также содержит последовательность ДНК, которая обеспечивает отбор или скрининг клеток растений кукурузы, содержащих вектор, в присутствии клеток, которые не содержат вектор. Такие отбираемые или скринируемые маркеры могут быть в векторе природно, в этот вектор введен химерический ген, или же маркер может быть введен в вектор до или после введения химерического гена. Альтернативно, отбираемый или скринируемый маркерный ген или его часть могут быть сначала соединены с желаемым химерическим геном или любой его частью, а рекомбинантные гены или сегменты генов могут быть введены в вектор как единое целое. Отбираемый или скринируемый маркер сам может быть химерическим.
-подобный токсин, вектор предпочтительно также содержит последовательность ДНК, которая обеспечивает отбор или скрининг клеток растений кукурузы, содержащих вектор, в присутствии клеток, которые не содержат вектор. Такие отбираемые или скринируемые маркеры могут быть в векторе природно, в этот вектор введен химерический ген, или же маркер может быть введен в вектор до или после введения химерического гена. Альтернативно, отбираемый или скринируемый маркерный ген или его часть могут быть сначала соединены с желаемым химерическим геном или любой его частью, а рекомбинантные гены или сегменты генов могут быть введены в вектор как единое целое. Отбираемый или скринируемый маркер сам может быть химерическим.
Предпочтительным отбираемым маркером является ген, кодирующий устойчивость к антибиотику. Ген должен быть способен к экспрессии в клетках, которые должны быть трансформированы. Клетки могут быть культивированы в среде, содержащей антибиотик, и отбирают те клетки, содержащие вектор, которые обладают повышенной способностью выживать в среде. Гены, которые придают устойчивость к токсинам кукурузы, таким как гигромицин или G413, могут быть полезными в качестве отбираемого маркера.
Некоторые примеры генов, которые придают устойчивость к антибиотикам, включают, например, неомицин фосфотрансферазу (устойчивость к канамицину и G418, Velten et al., EMBO J. 3 (1984) 2723-2730), гигромицин фосфотрансферазу (устойчивость к гигромицину, van den Elzen et al., Plant Molec. Biol., 5 (1985) 299-300).
Предпочтительные векторы для трансформации растений кукурузы представляют собой caMV 35S промотор, функционально соединенный с кодирующей областью  -токсина, и CaMV 35S промотор, функционально соединенный с отбираемым маркерным геном, таким как G418 устойчивость или устойчивость к гигромицину. Примерами таких векторов являются pCIB710 (355 (G418 устойчивость) и pIRV (устойчивость к гигромицину), как описано ниже.
-токсина, и CaMV 35S промотор, функционально соединенный с отбираемым маркерным геном, таким как G418 устойчивость или устойчивость к гигромицину. Примерами таких векторов являются pCIB710 (355 (G418 устойчивость) и pIRV (устойчивость к гигромицину), как описано ниже.
В изобретении также описаны фертильные растения кукурузы, клетки которых содержат химерический ген, который экспрессирует  кристаллический токсин или полипептид, обладающий по существу инсектицидной активностью
кристаллический токсин или полипептид, обладающий по существу инсектицидной активностью  кристаллического токсина.
кристаллического токсина.
Инсектициды.
Клетки растений, использованные в настоящем изобретении, содержат химерический ген и могут быть использованы для получения полипептида, обладающего по существу инсектицидной активностью  кристаллического белка. Клетки растений сами могут содержать инсектицид. Клетки растений, используемых непосредственно в качестве инсектицидов, могут быть культивируемыми клетками растений или могут быть составляющими живого растения.
кристаллического белка. Клетки растений сами могут содержать инсектицид. Клетки растений, используемых непосредственно в качестве инсектицидов, могут быть культивируемыми клетками растений или могут быть составляющими живого растения.
Токсин также может быть выделен из клеток растений с помощью известных методик, например, экстракцией или хроматографией. Экстракт может быть экстрактом всех клеток растения, частично очищенным экстрактом или чистым препаратом полипептида. Любой такой экстракт или хроматографический изолят может быть использован таким же путем, как кристаллический белок  смотри, например, Deacon, Aspeots of Microbiology, 7 (1983) Cole et al., ed, American Society for Microbiology and Miller et al., Seience 219 (1983) 715.
смотри, например, Deacon, Aspeots of Microbiology, 7 (1983) Cole et al., ed, American Society for Microbiology and Miller et al., Seience 219 (1983) 715.
Инсектицидные клетки являются токсичными для чешуекрылых насекомых, которые нападают на клетки и растения кукурузы, таких как совка хлопковая (Heliothis zea) кукурузный мотылек (Ostrinia nubilalis). Кроме того, клетки кукурузы, несущие ген кристаллического токсина  var. tenebrionis являются токсичными для жесткокрылых насекомых-паразитов. Важными насекомыми паразитами, принадлежащими к порядку Coleoptera, являются, например, колорадский картофельный жук, долгоносик хлопковый и кукурузный корневой червь.
var. tenebrionis являются токсичными для жесткокрылых насекомых-паразитов. Важными насекомыми паразитами, принадлежащими к порядку Coleoptera, являются, например, колорадский картофельный жук, долгоносик хлопковый и кукурузный корневой червь.
Следовательно, в настоящем изобретении описан способ получения в Zea mays полипептида, обладающего по существу инсектицидной активностью кристаллического белка Bacillus thuringiens, указанный способ включает:
(a) введение в клетки Zea mays гена, кодирующего полипептид, обладающий по существу инсектицидной активностью  кристаллического белка, в котором промотор, 5'-нетранслируемая область, и необязательно 3'-нетранслируемая область гена происходят из гена растения или гена вируса растения, и
кристаллического белка, в котором промотор, 5'-нетранслируемая область, и необязательно 3'-нетранслируемая область гена происходят из гена растения или гена вируса растения, и
(b) экспрессию полипептида
В настоящем изобретении также описан способ уничтожения личинок насекомых, заключающийся в кормлении личинок инсектицидным количеством трансгенных клеток Zea mays, содержащих ген, который кодирует кристаллический токсин Bacillus thuringiensis или полипептид, обладающий по существу инсектицидной активностью кристаллического белка Bacillus thuringiensis.
Кроме того, описан способ уничтожения личинок чешуекрылых (Lepidopteran) заключающийся в кормлении личинок инсектицидным количеством трансгенных клеток растений кукурузы, которые содержат химерический ген. Клетки растений могут быть культивируемыми клетками растения или могут быть составляющими живого растения. Кроме того, способ включает уничтожение личинок жесткокрылых (Coleopteran) путем кормления их инсектицидным количеством клеток, содержащих химерический ген, имеющий кодирующую последовательность кристаллического токсина  var tenebrionis или ее инсектицидные участки.
var tenebrionis или ее инсектицидные участки.
Полезность.
Способ, описанный в настоящем изобретении, обеспечивает протопласты Zea mays, которые могут быть регенерированы в фертильные растения. Эта возможность возникает за счет введения экзогенной ДНК (например, гена, кодирующего  кристаллический белок) стабильно в геном таких растений, и изменения их фенотипа. Кроме того, протопласты могут быть слиты с протопластами того же или другого сорта, чтобы получить новые сочетания ядерной ДНК или новые сочетания ядерной и органелль ДНК. Эта последняя возможность обеспечивает, например, конструирование новых растений с мужской стерильностью для целей селекции. Кроме того, протопласты могут быть использованы в качестве источника материала для клонирования, на котором может быть проведен мутагенез и/или селекция на желаемый фенотип. Примерами желаемых фенотипов могут быть, например, повышенная устойчивость к фитотоксичным химикатам, к патогенам, к паразитам, к вредным воздействиям окружающей среды, модифицированные морфологические или эволюционные свойства и измененное содержание метаболитов.
кристаллический белок) стабильно в геном таких растений, и изменения их фенотипа. Кроме того, протопласты могут быть слиты с протопластами того же или другого сорта, чтобы получить новые сочетания ядерной ДНК или новые сочетания ядерной и органелль ДНК. Эта последняя возможность обеспечивает, например, конструирование новых растений с мужской стерильностью для целей селекции. Кроме того, протопласты могут быть использованы в качестве источника материала для клонирования, на котором может быть проведен мутагенез и/или селекция на желаемый фенотип. Примерами желаемых фенотипов могут быть, например, повышенная устойчивость к фитотоксичным химикатам, к патогенам, к паразитам, к вредным воздействиям окружающей среды, модифицированные морфологические или эволюционные свойства и измененное содержание метаболитов.
Пример 1a. Получение специального типа каллюса Zea mays генотип Funk 2717.
Растения Zea mays генотипа Funk 2717 выращивают до цветения в теплице и опыляют пыльцой с того же генотипа. Незрелые кусочки кукурузы початков, содержащие зародыши, длиной 1,5 - 2,5 мм удаляют с растений и стерилизуют в 10% раствора КлороксR в течение 20 минут. Зародыши отделяют от зерен и помещают зародышевым стеблем вниз на среду Муросиги-Скуга (Physiol. Plant), 15(1962) 473 - 497), содержащую 0,1 мг/л 2,4-Д 6% мас./объем сахарозы и 25 мМ Z-пролина, отвержденную 0,24% мас./объем GelriteR (инициирующая среда). После двух недель культивирования в темноте при 27oC каллюс, развившийся на щитке, удаляют с зародыша и помещают на B5 среду (0,5 мг/л 2,4-Д), отверженную GelriteR. Материал субкультивируют (пассируют) каждые 2 недели на свежую среду. Примерно через 8 недель после помещения зародыша на инициирующую среду идентифицируют специальный тип каллюса по его характеристической морфологии. Этот каллюс далее субкультивируют на той же среде. После дополнительного периода в течение 2 месяцев каллюс переносят и стерильно субкультивируют на N6 среде, содержащей 2 мг/л 2,4-Д и отвержденной GelriteR.
Пример 1b. Получение специального типа каллюса Zea mays генотип Funk 5N984.
Растения Zea mays генотипа Funk 5N984 выращивают до цветения в теплице и опыляют пыльцой с того же генотипа. Незрелые кусочки початков, содержащие зародыши длиной 1,5 - 2,5 мм, удаляют с растений и стерилизуют в 15%-ном растворе CloroxR в течение 15 мин. Зародыши удаляют с зерновок и помещают зародышевым стеблем вниз на среду Шенка и Гильдебрандта (Can. J. Bot 50 (1972) 199 - 204) содержащую 1,0 мг/л 2,4-Д, 2% мас./объем сахарозы, 100 мг/л казеинового гидролизата, 100 мг/л мио-инозитола и 25 мМ Z-пролина, отвержденную 0,8% мас./объем ФитагараR (инициирующая среда).
После двух недель культивирования в темноте при 27oC предпочтительный каллюс отбирают по наличию хорошо сформировавшегося глобулярного, соматического зародыша, развившегося на щитке некоторых экплантатов. Эти каллюсы удаляют и помещают или на среду B5 (0,5 мг/л 2,4-Д плюс 2% мас./объм сахарозы, 100 мг/л казеинового гидролизата, 100 мг/л мио-инозитола и 25 мМ L-пролина/ или на среду Шенка или Гильдебрандта (описанную выше). Материал субкультивируют каждые 2 недели на свежую среду. После по крайней мере 8 недель от помещения зародыша на инициирующую среду идентифицируют специальный тип каллюса по его характеристической морфологии. Специальный тип каллюса может быть субкультивирован затем на той же среде, но предпочтительно его переносят и субкультивируют стериально на N6 - среды, содержащей 2 мг/л 2,4-Д и отвержденной 0,24% мас./объем GelriteR. Эмбриогенные суспензионные культуры могут быть получены из этих каллюсных культур по методике примера 2.
Пример 2. Приготовление суспензии Zea mays генотипа Funk 2717.
Каллюс, описанный в примере 1a, оставляют в культуре в течение по крайней мере 6 месяцев. Тип каллюса, выбранный для субкультивирования, представляет собой относительно не липкий гранулярный и очень рыхлый, так что он разделяется на индивидуальные клеточные агрегаты при помещении в жидкую среду. Культуры, содержащие агрегаты с большими, расширенными клетками не оставляют.
Аликвоты примерно 500 мг специального каллюса Zea mays описанного на стадии (a) выше, элитного генотипа Funk 2717 помещают в 30 мл N 6-среды, содержащей 2 мг/л 2,4-Д в недлинные колбы емкостью 125 мл. После 1 недели культивирования при 26oC в темноте на вращающейся качалке (130 об/мин, ход 2,5 см) среду заменяет свежей средой. Суспензии субкультивируют таким же путем снова каждую неделю.
За это время культуры осматривают и оставляют те культуры, в которых не имеется большого количества расширенных клеток. Суспензионные культуры, содержащие агрегаты с большими расширенными клетками, отбрасывают. Предпочтительная ткань состоит из плотных цитоплазматических делящихся клеточных агрегатов, которые имеют характерную более гладкую поверхность, чем большинство клеточных агрегатов. Эти оставленные культуры содержат по крайней мере 50% клеток, находящихся в этих мелких агрегатах. Это является желаемой морфологией.
Эти суспензии также обладают высокой скоростью роста со временем удвоения менее 1 недели. Суспензионные культуры еженедельно субкультивируют, перенося 0,5 мл уплотненного клеточного объема (РСУ: осажденный клеточный объем в пипетке) на 25 мл свежей среды. После 4 - 6 недель субкультивирования таким образом культуры увеличиваются в 2 - 3 раза за неделю субкультивирования. Для дальнейшего субкультивирования оставляют культуры, у которых более 75% клеток обладают желаемой морфологией. Поддерживают культуры, выбирая всегда для субкультивирования колбы, содержимое которых имеет лучшую морфологию. В некоторых случаях используют периодическое фильтрование через стальную сетку из нержавеющей стали с размером пор 630 мкм (каждые 2 недели) для повышения дисперсности культур, но это не является необходимым.
Сусиенжильная культура Zea mays 6-277-CG(Funk 2717) депонирована в АТСС под номером хранения 40326. Это депонирование было осуществлено в соответствии с Будапештским договором (дата депонирования: 20 мая 1987 г).
Пример 3. Приготовление протопластов из суспензионных культур Zea mays.
Инкубируют 1 - 1,5 мл PCV суспензионной культуры клеток, приготовленных, как описано в примере 2, в 10 - 16 мл смеси, состоящей из 4% мас./объем RS-целлюлазы с 1% мас./объем Розима в КМС солевом растворе (8,65 г/л KCl, 16,47 г/л MgCl2 6H2O и 12,5 г/л CaCl2, 2H2O, 5 г/л MES pH 5,6).
Переваривание осуществляют при 30oC на качающемся столе в течение 3 - 4 часов. Получение контролируют в обратном микроскопе для выделившихся протопластов.
Выделившиеся протопласты собирают следующим образом. Препарат фильтруют через сито 100 мкм меш, затем через сито 50 мкм. Протопласты промывают через сито объемом КМС солевого раствора, равным исходному объему ферментного раствора. Помещают 10 мл препарата протопластов в каждую из нескольких пластиковых свободных центрифужных трубок и 1,5 - 2 мл 0,6 М раствора сахарозы, забуферированного до pH 5,6 с помощью 0,1% мас./объем морфолиноэтансульфоновой кислоты (MES) и KOH наслаивают вниз. Трубки центрифугируют при 60 - 100 g в течение 10 минут и собирают протопласты с поверхности раздела и помещают в свежую трубку.
Препарат протопластов повторно суспендируют в 10 мл свежего КМС солевого раствора и центрифугируют в течение 5 минут при 60 - 100 g. Супернатант удаляют и отбрасывают, а протопласты осторожно ресуспендируют в оставшейся капле и затем постепенно прибавляют 10 мл КМС раствора крепостью 13/14. После 5 минут центрифугирования снова удаляют супернатант и протопласты повторно суспендируют в КМС раствора крепостью 6/7. Отбирают аликвот для подсчета и протопласты снова осаждают центрифугированием. Протопласты повторно суспендируют при концентрации 107 (мл в КМ среде (таблица 1) или в растворе маннита, имеющем такие же осмотические свойства, как и среда КМ, и содержащем 6 мМ MgCl2 или в любой другой подходящей среде, используемой для трансформации, как описано в следующих примерах. Эту суспензию протопластов используют для культивирования, как описано в примере 8a, или где выполняют, для трансформации протопластов, как описано в примерах 8 и 9.
Пример 4. Общие методики рекомбинантной ДНК.
Поскольку многие методики рекомбинантной ДНК, использованные в настоящем изобретении, являются рутинными для специалистов в данной области, краткое описание этих обычно используемых методик приведено здесь, а не в каждом случае, где они возникают ниже. За исключением отмеченного здесь все эти рутинные процедуры описаны в работе Maniatis et al. (1982).
А. Переваривание рестрикционными эндонуклеазами.
Обычно ДНК находится в реакционной смеси в количестве примерно 50 - 500 мкг/мл в буферном растворе, рекомендованном производителем, Нью Инглэд Биолабс, Беверли, МА, прибавляют 2 - 5 единиц рестрикционных эндонуклеаза на каждый 1 мкг ДРК и инкубируют реакционную смесь при температуре, рекомендованной производителем, в течение 1 - 3 часов. Реакцию заканчивают при нагревании при 65o в течение 10 минут или экстракцией фенолом с последующим осаждением ДНК этанолом. Эта методика также описана на страницах 104 - 106 в работе Маниатиса с сотр.
В. Обработке ДНК полимеразой для создания flush концов.
Фрагменты ДНК прибавляют к реакционной смеси при 50 - 500 мг/мл в буфере, рекомендованном производителем, Нью Инглэнд Биолабо. Реакционная смесь содержит все четыре деоксинуклеотидтрифосфата в концентрации 0,2 мМ. Реакционную смесь инкубируют при 15oC в течение 30 минут, а затем заканчивают при нагревании до 65oC в течение 10 минут. Для фрагментов, полученных перевариванием рестрикционными эндонуклеазами, которые создают 5'-выступающие концы, такие как EcoRI и Bam HI используют большой фрагмент или фрагмент Кленова ДНК полимеразы.
Для фрагментов, продуцированных эндонуклеазами, которые продуцируют 3'-выступающие концы, такие как PstI и SacI используют T4 ДНЕ полимеразу. Использование этих двух ферментов описано на страницах 113 -121 работы Маниатиса с сотр.
С. Электрофорез на агарозном геле и очистке фрагментов ДНК от гелей.
Электрофорез на агарозном геле осуществляют в горизонтальном устройстве, как описано на страницах 150 - 163 работы Маниатиса с сотрудниками. Использованный буфер представляют собой Трисборатный буфер, описанный здесь. Фрагменты ДНК визуализуют окрашиванием 0,5 мкг/мл этидинийбромида, который или присутствует в геле и tank буфере или прибавляется после электрофореза. ДНК визуализуют при освещении коротковолновым или длинноволновым ультрафиолетовым светом. Когда фрагменты должны быть выделены из геля, используют агарозу с низкотемпературным гелеобразованием, полученную от Сигма Кемикал, Сент-Луис, Миссури. После электрофореза вырезают желаемый фрагмент, помещают в пластиковую трубку, нагревают до 65oC в течение примерно 15 минут, затем экстрагируют три раза фенолом и дважды осаждают этанолом. Эта процедура является несколько модифицированной по сравнению с той, что описана в работе Маниатиса с сотрудниками на странице 170.
D. Присоединение синтетических линкерных фрагментов к концам ДНК.
Когда желательно добавить новый сайт рестрикции эндонуклеазы к концу молекулы ДНК, такую молекулу сначала обрабатывают ДНК полимеразой для создания flush концов, если необходимо, как описано в приведенном выше разделе. Приблизительно 0,1 - 1,0 мкг этого фрагмента прибавляют к примерно 10 нг фосфорилированного линкера ДНК, полученного от Нью Инглэд, в объеме от 20 до 30 мкл, содержащем 2 мкл T4 ДНК лигазы, от Нью Инглэд Биолабс и 1 мМ АТФ в буфере, рекомендованном производителем. После инкубации в течение ночи при 15oC реакции заканчивают при нагревании смеси при 65oC в течение 10 минут. Затем реакционную смесь разбавляют до примерно 100 мкл в буфере, пригодном для рестрикционных эндонуклеаз, которые расщепляют синтетическую линкерную последовательность, и прибавляют примерно 50 - 200 единиц этой эндонуклеазы. Смесь инкубируют при соответствующей температуре в течение 2 - 6 часов, затем фрагмент подвергают электрофорезу на агарозном геле и очищают фрагмент, как описано выше. Полученный в результате фрагмент теперь будет иметь концы с окончаниями, полученными при переваривании рестрикционной эндонуклеазой.
Эти концы обычно являются липкими, так что полученный в результате фрагмент теперь легко лигируется с другими фрагментами, имеющими такие же липкие концы.
E. Удаление 5'-концевых фосфатов из фрагментов ДНК.
Во время стадии клонирования плазмиды обработка плазмидного вектора фосфатазой уменьшает рециркулизацию вектора (обсуждается на странице 13 работы Маниатиса с сотрудниками). После переваривания ДНК соответствующей рестрикционной эндонуклеазой, прибавляют одну единицу желудочной щелочной фосфатазы коровы, полученной от Боерингер-Маннхейм, Индианаполис, ИН, ДНК инкубируют при 37oC в течение часа, затем экстрагируют дважды фенолом и осаждают этанолом.
F. Лигация фрагментов ДНК.
При соединении фрагментов, имеющих комплементарные липкие концы, инкубируют приблизительно 100 нг каждого фрагмента в реакционной смеси емкостью 20 - 40 мкл, содержащей приблизительно 0,2 единицы Т4 ДНК лигазы из Нью Биолабс в буфере, рекомендуемом производителем Инкубацию продолжают в течение 1 часа 20 минут при 15oC. При соединении фрагментов ДНК, имеющих тупые концы, их инкубируют, как описано выше, за исключением того, что количество Т4 ДНК лигазы увеличивают в 2-4 раза.
G. Трансформация ДНК в E. coli
Для большинства экспериментов используют E. coli штамм HB101. ДНК вводят в E. coli используя процедуру с хлористым кальцием, описанную Маниатисом с сотрудниками на страницах 250-251.
H. Скринирование E. coli для плазмид.
После трансформации полученные в результате колонии E. coli скринируют на присутствие целевой плазмиды с помощью процедуры быстрой изоляции плазмиды. Две удобные процедуры описаны на страницах 366-369 в работе Маниатиса с сотрудниками (1982).
1. Широкомасштабное выделение плазмиды ДНК.
Процедуры для выделения больших количеств плазмид в E. coli описаны на страницах 86-94 работы Маниатиса с сотрудниками (1982).
f. Клонирование в векторе фага N13.
В последующем описании должно быть понятно, что двуниточная репликативная форма производных фага M13 используется для рутинных процедур, таких как переваривание рестрикционной эндонуклеазой, лигирование и т.п.
Пример 5. Конструирование химерического гена в плазмиде pBR322.
Для слияния промотора гена VI CaMV и последовательностей, кодирующих протоксин, конструируют производное фага вектора mp 19 (Janiscn - Perron et al., (1985)).
Сначала вставляют в mp 19 фрагмент ДНК, содержащий приблизительно 155 нуклеотидов 5' к кодирующей протоксин области и соседние приблизительно 1346 нуклеотидов кодирующей последовательности. Переваривают фаг mp 19 ds rf двуниточная репликативная форма) ДНК рестрикционными эндонуклеазами Sac I и Sma I и очищают фрагмент вектор приблизительно 7,2 кпо после электрофореза через агарозу с низкой температурой гелеобразования с помощью стандартных процедур. Плазмида pK U 25/4, содержащая приблизительно 10 кпо (килопар оснований) Baccillus thuringiensis ДНК, включая ген протоксина, была получена от др. Дж. Нуеша, Циба-Гейги Лтд., Базель, Швейцария. Нуклеотидная последовательность гена протоксина, присутствующего в плазмиде pK U 25/43, показана в последовательности (1). Плазмиду pK U 25/4 ДНК переваривают эндонуклазами HpaI и SacI и очищают фрагмент 1503 по (содержащий нуклеотиды 2 по 1505) в последовательности (1)), как описано выше). Этот фрагмент содержит последовательности приблизительно 155 по бактериального промотора и приблизительно 1346 по старта последовательности, кодирующей протоксин). Приблизительно 100 нг каждого фрагмента затем смешивают, прибавляют Т4 ДНК лигазу и инкубируют всю ночь при 15oC. Полученную смесь трансформируют в E. coli штамм HB 101, смешивают с бактериальным индикатором E. coli JM101 и высевают, как описано (Messing J. Meth. Enzymol 101 (1983) 20). Ниже для дальнейшего конструирования используют один фаг, называемый mp 19 (b)
Затем фрагмент ДНК, содержащий промотор гена VI CaMV и некоторые кодирующие последовательности гена VI, вставляют в mp 19/bt. Фаг mp 19 /bt ds rf ДНК переваривают Ba HI, обрабатывают большой фрагмент ДНК полимеразой для создания тупых концов и повторно расщепляют эндонуклеазой Pst I. Большой векторный фрагмент очищают электрофорезом, как описано выше. Плазмида pABDI описана Пашковским с сотрудниками, EMBO J. 3 (1984) 2717-2722. Плазмиду pABDI ДНК переваривают Pst I и Hind III. Очищают фрагмент приблизительно 465 по длинной, содержащий промотор гена VI CaMV и приблизительно 75 по кодирующей последовательности гена VI.
Лигируют два фрагмента и высевают, как описано выше. Один из полученных в результате рекомбинантных фагов, называемый mp 19/ bt ca, используют в следующем эксперименте:
Фаг mp 19/ bt ca содержит последовательности промотора гена VI CaMV часть кодирующей последовательности гена VI, приблизительно 155 по Baccillus thuringiensis ДНК выше последовательности, кодирующей протоксин, и приблизительно 1346 по последовательности, кодирующей протоксин. Для слияния последовательностей промотора CaMV точно к последовательностям, кодирующим протоксин, делецируют внедрившуюся ДНК, используя направленный мутагенез олигонуклеотидов mp 19/ bt ca ДНК. ДНК олигонуклеотид с последовательностью
(5') TTCGGATTGTTATCCATGGTTGGAGGTCTGA
(3') синтезируют с помощью рутинных процедур с использованием синтезатора ДНК Апплайд Биосистемс. Этот олигонуклеотид является комплеметарным к этим последовательностям в фаге mp 19/ bt ca ДНК на 3'-конце промотора CaMV нуклеотиды 5762 по 5778 у Хону (1982)) и началу последовательности, кодирующей протоксин (нуклеотиды 156 по 172) в формуле 1 выше. Общая процедура для мутагенеза является такой, как описана у Цоллера и Смита (1983). Приблизительно 5 мкг однониточной ДНК фага mp 19/bt ca смешиваются 0,3 мкг фосфорилированного олигонуклеотида в объеме 40 мкл. Смесь нагревают до 65oC в течение 5 минут, охлаждают до 50oC и медленно охлаждают до 4oC. Затем прибавляют буфер, нуклеотид трифосфатазу, АТФ, Т4 ДНК лигазу и большой фрагмент ДНК полимеразы и инкубируют всю ночью при 15oC, как описано (смотри Цоллера и Смита (1983)). После электрофореза на агарозном геле очищают циркулярную двуниточную ДНК и трансфецируют в E. coli штамм JM101. Полученные в результате плашки скринируют на последовательности, которые гибридизуют с 32P-меченым олигонуклеотидом и анализируют фаг с помощью ДНК рестрикционного анализа эндонуклеазами. Среди полученных в результате клонов фага будут клоны, которые имеют правильно делецированные неожиданные последовательности между промотором гена VI CaMV и кодирующей протоксин последовательностью. Этот фаг называют mp19 (bt ca) del.
Затем конструируют плазмиду, в которой 3'-кодирующую область гена протоксина сливают с сигналами окончания транскрипции CaMV. Сначала плазмиду pABDI ДНК переваривают эндонуклеазами Bam HI и BglII изолируют фрагмент 0,5 кпо, содержащий последовательности терминатора транскрипции CaMV. Затем плазмиду pU C19, Yanish - Perronn et al., Gene 33 (1985 ) 103-119, переваривают Bam HI, смешивают с фрагментом 0,5 кпо и инкубируют с T4 ДНК лигазой. После трансформации НК в E. coli штамм HB101, получают один из полученных в результате клонов, называемый плазмидой p702.
Затем расщепляют плазмиду p702 ДНК эндонуклеазами SacI и SmaI и изолируют с помощью электрофореза больший фрагмент примерно 3,2 кпо. Плазмиду pK U 25/4 ДНК переваривают эндонуклеазами Aha III и Sai I и изолируют после гель-электрофореза фрагмент 2,3 кпо (нуклеотиды 1502 по 3773 в формуле I выше), содержащий 3'-участок последовательности, кодирующей протоксин /nt 1504 по 3773 последовательности, показанной в формуле I. Эти два фрагмента ДНК смешивают, инкубируют с T4 ДНК лигазой и трансформируют в E. coli штамм HB101. Полученной плазмидой является p702/bt.
Наконец соединяют частиц фага mp 19/bt ca/del ds rf ДНК и плазмиду p702/в t чтобы создать плазмиду, содержащую последовательность, кодирующую протоксин, фланкированную промотором CaMV и терминаторными последовательностями. Фаг mp 19/bt del ДНК переваривают эндонуклеазами Sea I и Spn I и очищают фрагмент примерно 1,75 кпо электрофорезом на агарозном геле подобным образом переваривают плазмиду p702/bt
ДНК эндонуклеазами ScaI и SalI изолируют фрагмент приблизительно 2,5 кпо. Наконец переваривают плазмиду pBk 322 ДНК (Bolivar et al., (19677) эндонуклеазами SalI и SphI и изолируют больший фрагмент 4,2 кпо. Смешивают все три фрагмента ДНК и инкубируют с Т4 ДНК лигазой и трансформируют в E. coli, штамм HB 101. Полученная в результате плазмида, pBK 322/bt 14 является производной из pBK 322, содержащей промотор гена VI CaMV, и сигналы начала трансляции, слитые с последовательностью, кодирующей  кристаллический белок, затем с терминатором транскрипции CaMV.
кристаллический белок, затем с терминатором транскрипции CaMV.
Пример 6a. Конструирование pTOX, содержащего химерический ген, кодирующий ген инсектицидного токсина Bacillus thuringiensis var. tenebrionis.
Ген, кодирующий ген инсектицидного кристаллического белка Bacillus thuringiensis var. benebrionia был охарактеризован и секвенсирован (Sekar V et al. , Proc. Natl. Acad. Sci. США 84 (1987) 7036-7040). Эту кодирующую последовательность изолируют на походящем рестрикционном фрагменте, таком как Hind III. Фрагмент размером приблизительно 3 ко, и вставляют в подходящий растительный вектор эксперсии, такой как плазмида pCIB 770 (Rothstein 5 et al., Gene, 63 (1987) 153-161). Плазмида pCIB 770 содержит химический канамициновый ген для экспрессии в растениях, а также промотор и терминатор 35S РНК транскрипта CaMV (вируса мозаики подсолнечника), разделенных единичным Bam HI сайтов. Рестрикционный фрагмент, несущий последовательность, кодирующую токсин, делают совместимым с единичным Bam HI сайтом pCIB 770 при использовании подходящего молекулярного адаптора и лигируют вместе.
Пример 6b. Конструирование pSAH, содержащей химерический ген, кодирующий ген инсектицидного токсина
Baccillus thuringiensis штамм San diego.
Ген, кодирующий инсектицидный белок Baccillus thuringiensis штамм san diego был охарактеризован и секвенсирован Herrnstadt et al., EP-0 202 739 и EP-0 213 818. Эту кодирующую последовательность изолируют на подходящем рестрикционном фрагменте и вставляют в подходящий растительный вектор экспрессии, такой как pCIB 770. Плазмида pCIB770 содержит химерический канамициновый ген для экспрессии в растениях, а также промотор и терминатор 35 S РНК транскрипта CaMV (вируса мозаики подсолнечника), разделенные единичным Bam HI сайтом. Рестрикционный фрагмент, несущий последовательностью, кодирующую токсин, делают совместимым с единичным Bam HI сайтом pCIB 770 при использовании подходящего молекулярного адаптера и лигируют вместе.
Пример 7А. Конструирование плазмиды pCIB 710.
Плазмида pZW III (доступная от S. Howell Калифорнийский университет, Сан Диего) состоит из трех маленьких EcoRI фрагментов BYI штамма вируса мозаики подсолнечника (CaMV) (Franck et al. (1980) Cell 21: 285 - 294), клонированных в pMB9. p ZW III переваривают и изолируют BgelII фрагмент 1150 по/ пары оснований номера 6494-7643, Hohn et al., Current Topics in Microbiology and Immunology 96 (1982) 193-216). Этот фрагмент лигируют по Bam HI сайту p UC 19. Этот рестрикционный фрагмент колирует как промотор для 35 S pHK CaMV так и поли А сигнал присоединения (т.е. терминатор) для этого транскрипта.
Мутагенез ин витро
Единичный Bam HI сайт вставляют между этим промотором и терминатором с помощью мутагенеза ин витро. Синтезируют олигонуклеотид 36-оснований, который является идентичный CaMV последовательности, в которую вставлена область с исключением такого сайта рестрикции Bam HI у пары оснований N 7463 в последовательности.
Клонируют BglII фрагмент 1150 по из приведенного выше, в М13 mp 19 и изолируют однониточную ДНК фага.
Эту однониточную ДНК отжигают с синтетическим олигонуклеотидом синтезируют новую нить, используя олигонуклеотид в качестве праймера и трансфецируют ДНК в штаммы JM101 (Zoller and Smit, DHK, 3(1984) 479-488), затем изолируют как описано у Цоллера и Смита (выше).
Селекция целевого мутантного фага
Олигонуклеотид 36 оснований метят путем Kinasing c 32-Р-АТФ.
Группу плашек трансфецированного М13 фага локализуют на нитроцеллюлозном фильтре. Этот фильтр гибридизуют с меченой 36-mer, фильтр промывают при повышенной температуре. Мутированные фаги являются стабильными при промывке при повышенных температурах.
Изолируют в секвенсируют один из этих фагов, стабильных при повышенных температурах, чтобы подтвердить наличие Bam HI сайта.
Окончательная сборка pCIB 710
Двуниточную ДНК, изолированную из этого фага, переваривают Hind III и EcoRI pUC19 расцепляют Hind III и EcoRI. Лигируют эти две переваренные ДНК. Трансформанты отирают по устойчивости к ампициллину. Один трансформант из этой лигации является pCIB710.
Плазмида pCIB710 не имеет АТC стартовых кодонов между этим Bam HI сайтом, который вставлен, и начальной точкой транскрипции.
Пример 76. Конструирование pCIB712
Ген устойчивости к гигромицину (Gritz, Z. and Davies J. Gene 25 (1983) 179-188) находится примерно на 1150 парах оснований BamHI фрагмента от плазмиды p LG90. Плазмида pLG90 была получена от др. Лиды Гриц. Прикладная Биотехнология, 80 Роджерс Ст., Кембридж, Массечусеттс 02142. Этот фрагмент линкируют с BglII линкерами, а затем вставляют в единичный BamHI сайт pCIB 710, разрушая BamHI сайт и конструируя плазмиду pCIB 712. Плазмида pCIB 712 была депонирована в АТСС, Рокквилл, Мэриленд, США, под номером хранения 67407. Это депонирование было осуществлено в соответствии с Будапештским договором (дата депонирована: 18 мая 1987 г).
Плазмиду pCIB712 линеаризуют, используя соответствующий фермент, например, SmaI.
Пример 7с. Конструирование pCIB10/34 Sbt
Плазмида pCIB710 описана выше.
Конструирование pCIB10
1. Т-ДНК фрагмент, содержащий левый край от pTi 737, изолируют из pB R 325 (EcoR 129) Yadav et al., Proc. Natl. Acad Sci. США, 79 (1982) 6322-6326, pBR 325 (EcoR 129 разрезают EcoRI и линкируют фрагмент 1,1 ко с Hind III линкерами (Нью Ингланд Биолабс).
2. Плазмиду pBR322 (Bolivar et al., Gene, 2(1977) 95-103) разрезают Hind III
3. Фрагмент, содержащий левый край, описанный в стадии 1%, лигируют в Hind перевар pBR 322.
4. Плазмиду, содержащий левый край, сконструированную на стадии 3, переваривают ClaI и Hind III и изолируют фрагмент Hind III/ClaI 1,1 ko (Hepburn et al., J.Mol. Appl. Genet., 2, (1983) 315-329).
5. Плазмиду p UC18 (Horrander et al., Genl 26(1983) 101-106) разрезают Hind III и Eco RI; полилинкер 60 пометят по концу, используя Т4 полинуклеотид киназу и гамма-32р-АТФ, и изолируют из акриламидного гена.
6. Плазмиду pBR 322 разрезают EcoRI и ClaI и изолируют большой фрагмент.
7. Полилинкер Hind III/EcoRI 60 по и фрагмент Hind III/ClaI 1,1 ко EcoR 129 лигируют в pBR 322, разрезанную ClaI и EcoRI, конструируя pCIB5.
8. Химерический ген, придающий устойчивость к канамицину (nos-neo) берут из Binb(Bevan, Nuc. Acid Res. 12(1984) 8711-8721) в виде фрагмента SalI/EcoRI
9. Плазмиду pUC18 разрезают EcoRI и SalI
10. Фрагмент SalI/EcoRI содержащий химерический ген стадии 8, лигируют в pUC18, разрезанную EcoRI и SalI
11. BanHI сайт распознавания в терминальной последовательности этого химерического гена разрушают, разрезая Bam HI, заполняют при использовании Т4 ДНК полимеразы и лигируют.
12. Полученную в результате плазмиду разрезают Sst II (Бетесда Рисерч Лабораториз) и Hind III.
13. Фрагмент, содержащий 5'-часть nos-промотора и правый край pTi T37, изолируют при разрезании pBR 325 (Hind 23) с помощью HindIII и Sst II и изолируют фрагмент 1,0 ко.
14. Этот Hind III/SstII фрагмент 1,0 ко лигируют в pUC18 вектор стадии 12, конструируют pCIB4.
15. pCIB5, содержащую левый край Т-ДНК, разрезают Aat II, создавая тупой конец с помощью обработки Т4 ДНК полимеразой, а затем разрезают EcoRI.
16. pCIB4 разрезают Hind III создавая тупой конец путем обработки с использованием фрагмента Кленова E. coli ДНК полимеразы 1, и разрезают EcoRI.
17. Вектор стадии 29 лигируют с большим фрагментом стадии 16, конструируя pCIB2, CoIEI репликон, содержащий левый и правый края T-ДНК, фланкирующие химерический Kmr ген и полилинкер.
18. Плазмиду pR 102 (Jorgensen et al Mol. Gen. Gemet 177 (1979) 65-72) переваривают Bam HI и заполняют при использовании фрагмента Кленова.
19. Готовят частичный Alul перевар плазмиды pAO3 (Оkа et al, Nature 276, (1978) 845-847 и Оkа et al, J. Mal. Biol., 147 (1981) 217-226).
20. AluI перевар лигируют в рестриктированную pRZ 102 со стадии 18, отбирая целевые трансформанты по устойчивости к канамицину.
21. Полученная в результате плазмида имеет кодирующую последовательность Tn 903, присутствующую на Bam HI фрагменте 1,05 ко; этот фрагмент изолируют после BamHI переваривания и заполнения фрагментом Кленовым.
22. Плазмида pRK 252, производная от больного ряда хозяев плазмиды pRK 2 получена от др. Дон Хелински из Калифорнийского университета, Сан Диего. Эта плазмида потеряла BglII сайт, присутствующий в родственной плазмиде pRK 290 (Ditta et al. , Proc. Nat. Acad. Sci., США, 77 (1980) 262-269). p RK 252 переваривают SmaI и SalI заполняют, используя фрагмент Кленова и изолируют большой фрагмент, полученный из этого перевара.
23. Фрагмент, содержащий TN903, изолированный на стадии 21, лигируют в большой фрагмент из pRK 252, конструируя pRK252 Км.
24. Плазмиду pPK252 Km разрезают EcoRI получают тупые концы с использованием фрагмента Кленова и линкируют BglII линкерами (Нью Инглэнд Биолабс).
25. Плазмиду pCI82 разрезают EcoRV и линкируют BglI линкерами (Нью Инглэнд Биолбс).
26. Bgl II линкированную pCIB2 со стадии 25 лигируют в вектор стадии 24, конструируя pC1810.
Пример 7. Конструирование pCIBIOa
Пример 7c может быть повторен с заменой pRK252 на pRK290.
1. Плазмиду pRZ102 (Jorgensen et al., (1979), выше) переваривают BamHI и заполняют, используя Кленова.
2. Готовят частичный перевар AluI плазмиды pA03 (Oka et al., (1978) выше).
3. AluI перевар лигируют в рестриктированную со стадии 18, пример 7c, отбирают целевые трансформанты с помощью устойчивости к канамицину.
4. Полученная в результате плазмида имеет кодирующую последовательность Tn903, присутствующую на фрагменте BamHI размером 1,05 kb; этот фрагмент выделяли после BamHI переваривания и заполняли фрагментом Кленова.
5. Плазмида pRK290, представляющая собой производное распространенной у большого числа хозяев плазмиды RK2, была получена от д-ра Дон Хелински из Калифорнийского университета, Сан-диего, pRK290 переваривали с SmaI и SaII заполняли с использованием фрагмента Кленова и выделяли крупный фрагмент, полученный в результате такого переваривания.
6. Tn908 содержащий фрагмент, выделенный на стадии 35, лигировали на крупный фрагмент из pRK290. Конструирующий pRKK290Km.
7. Плазмиду со стадии 6 переваривали с BgIII, заполняли с использованием фрагмента Кленова и лигировали, разрушая ее BgIII сайт, с целью конструирования pRK290Km.
8. Плазмиду pRK290Km разрезали с помощью EcoRI, соединяли тупыми концами с использованием фрагмента Кленова и соединяли с помощью BglII линкеров (Нью Инглэнд Биолабс).
9. Плазмиду pCIB2 разрезали с помощью EcoRV и соединяли с BglII линкерами (Нью Иншлэнд Биолабс).
10. Линкированную BglII pCIB2 со стадии 9 лигировали на вектор со стадии 7, конструируя pCIB10a.
Пример 7e. Конструирование химерного гена протоксина Bacillus thuringiensis с CaMV35S промотором.
A. Конструирование CaMV35S промоторной кассеты
Плазмиду pCIB710 конструировали в соответствии с изображенным на фиг. 107. Такая плазмида содержит CaMY промотор и транскрипционные терминаторные последовательности 35S РНК матрицы (Кови С.Н. Ломонософф Г.П., Хулл Р., Nucleic Acids Research (1981) 6735-6747), 1149 bp BgIII рестрикционный фрагмент CaMV ДНК (bp 7643-6494 в Хон с сотр. (1982)) выделяли из плазмиды pLVIII (полученной от д-ра С.Хоувелла, Калифорнийский университет, Сан-Диего; либо такой фрагмент может быть выделен непосредственно из CaMV ДНК) методом препаративного агарозагелевого электрофореза, описанного выше, и смешивали с BamHI-расщепленной плазмидой pUC19 ДНК, обрабатывали T4 ДНК-лигазой и и трансформировали в E. coli (Примечание: BamHI рестрикционный сайт в полученной в результате плазмиде был разрушен лигированием BglII липких концов с BamHi липкими концами).
Полученную в результате плазмиду, названную pUC19/35S затем использовали в олигонуклеотид-направленном in vitro мутагенезе с целью внедрения BamHI распозновательной последовательности GGATCC непосредственно после CaMV нуклеотида 7483 в ссылке Хона. Полученная в результате плазмида, pCIB710, содержит CaMV35S промоторный участок и участок окончания транскрипции, разделенные BamHI рестрикционным сайтом. ДНК последовательности, вставленные в такой BamHI сайт, могут быть экспрессированы в растения с помощью таких CaMV транскрипционных регуляторных последовательностей. (Следует также отметить, что pCIB710 не содержит каких-либо ATG трансляционных инициирующих кодонов между началом транскрипции и сайтом).
B. Внедрение CaMV35S промоторной/терминаторной кассеты в pCIB10
Следующие стадии отражены на фиг. 11
Плазмиды pCIB10 и pCIB70 переваривали с EcoRI и SalI, смешивали и лигатировали. Полученная в результате плазмида, pCIB10 710 имеет CaMV35S промотор/терминаторную кассету, вставленную в трансформационный вектор растений pCIB10. CaMV35S последовательности находятся между границами T-ДНК в PCIB10 и таким образом, они могут быть внедрены в геном растения в ходе экспериментов по трансформации растений.
C. Внедрение Bacillus thuringiensis протоксинового гена в pCB10/710.
Следующие ниже стадии изображены на фиг. 12. В качестве источника протоксинового гена плазмиду pCIB10/19B переваривали с BamHI и NcoI и методом препаративного гель-электрофореза выделяли фрагмент размером 3,6 kb, содержащий протоксиновый ген. Затем этот фрагмент смешивали с синтетическим NcoI-BamHI адаптором, имеющим последовательность 5'-CATGGCCGGATCCGGC-3', после чего переваривали с BamHI. На этой стадии образуются BamHI липкие концы с обоих концов протоксинового фрагмента. Затем полученный фрагмент вставляли в BamHI-расщепленную pCIB10/710. Полученная в результате плазмида, pCIB10/35S  показанная на фиг. 12, содержит протоксиновый ген между CaMV 35S промоторной и транскрипционной терминаторной последовательностями.
показанная на фиг. 12, содержит протоксиновый ген между CaMV 35S промоторной и транскрипционной терминаторной последовательностями.
Плазмиду pCIB10 (19SBt) [в штамме E.coli HB101] депонировали в ATCC и ей присвоили каталожный номер ATCC 67330 (дата депонирования: 27 февраля 1987 г).
pCIB10/35SBt в штамме MC1061 депонировали в ATCC, Роквилл, Мэриленд, США, под каталожным номером 67329. Такое депонирование осуществляли в соответствии с требованиями Будапештского договора (дата депонирования: 27 февраля 1987 г).
Пример 8. Трансформация протопластов Zea mays методом электропорации
(a) Все стадии за исключением теплового шока осуществляли при комнатной температуре. Протопласты повторно суспендировали на последней стадии примера 3 в 0,5М растворе маннита, содержащем 0,1% мас./об. MES и 6 мМ MgCl2 сопротивление такой суспензии измеряли в касере диалогового электропоратораR и поддерживали равным 1-1,2 Ком используя для этой цели 300 мМ раствор MgCl2. Протопласты подвергали действию теплового шока путем погружения трубки, содержащей образец на 5 минут в водяную баню с температурой 45oC, после чего проводили охлаждение льдом до комнатной температуры. К аликвотам такой суспензии объемом 0,25 мл добавляли 4 мкг линеаризованной pCIB712 плазмиды, содержащей гидромицин устойчивый ген и 20 мкг ДНК-носителя телячьего тимуса.
К протопластам добавляли 0,125 мл 24% мас./об. раствора полиэтиленгликоля (ПЭГ) (мол.м. ПЭГ 8000) в 0,5 М маннита, содержащего 30 мМ MgCl2. Полученную смесь хорошо, но осторожно перемешивали и инкубировали в течение 10 минут. Образец переносили в камеру электропоратора и через 10 секундные интервалы трижды воздействовали импульсами с начальными напряжениями 1000, 1200, 1500, 1800, 2300 и 2800 см-1, причем время экспоненциального спадания и пульса составляло 10 мкс).
Культивацию протопластов проводили следующим образом:
Образцы помещали в чашки Петри диаметром 6 см при комнатной температуре. В течение следующих 5-15 минут добавляли 3 мл среды КМ, содержащей 1,2% мас. /об. Sea PlagueR агарозы, 1 мг/л 2,4-Д и 100 мг/л O-ацил салициловой кислоты. Агарозу в протопласты хорошо перемешивали и среде давали желатинироваться.
(b) Повторяли методику стадии (a) за исключением того, что сопротивление протопластного препарата поддерживали равным 0,5-0,7 кОм.
(c) Повторяли методику стадии (a), за исключением того, что в качестве ПЭГ использовали ПЭГ с мол.м. 4000.
(d) Повторяли методику стадии (c), за исключением того, что сопротивление протопластного препарата поддерживали равным 0,5-0,7 кОм.
(e) Повторяли методику стадии (a), за исключением того, что ПЭГ не добавляли.
(f) Повторяли методику стадии (b), за исключением того, что ПЭГ не добавляли.
(g) Повторяли методику стадии (a), за исключением того, что половину объема 12% мас./об. ПЭГ.
(h) Повторяли методику (b), за исключением того, что добавляли половину объема 12% мас./об. ПЭГ.
(i) Повторяли методику стадии (c), за исключением того, что добавляли половину объема 12% мас./об. ПЭГ.
(j) Повторяли методику стадии (d), за исключением того, что добавляли половину объема 12% мас./об. ПЭГ.
(k) Повторяли методику, стадий (a)-(j) за исключением того, что импульсы применяли с интервалами в 3 с.
(l) Повторяли стадии (a)-(k) с тем отличием, что после электропорации протопласты помещали в чашки, находящиеся на пластине, охлажденной до 16oC.
(m) Повторяли стадии (a)-(l) с тем отличием, что после стадии электропорации протопласты помещали в трубки, промывали 10 мл раствора КМС крепостью 6/7, собирали центрифугированием при 60g в течение 10 мин, повторно суспендировали в 0,3 мл среды КМ и высевали согласно методике стадии (a).
(n) Повторяли методику стадии (m), за тем исключением, что среда, используемая для промывания протопластов, представляла собой раствор W5 (380 мг/л KCl; 18,375 г/д CaCl2•2HO; 9 г/л NaCl; 9 г/л глюкозы; pH 6,0).
(o) Повторяют стадии (a)-(n) за тем исключением, что среда, используемая для высевания протопластов представляла собой среду КМ, содержащую 30% об. среды Шенка и Хильдебрандта (1972), которая содержала 30 мкМ ДикамбаR, в котором предварительно выращивали клеточную суспензию Dactylis glomcrata.
(p) Повторяют стадии (a) - (n) с тем исключением, что в качестве среды, используемой для высевания протопластов применяли среду KM, содержащую 15% об. среды Шенка и Хильдебрандта (Can. J. Bot. 50 (1972), 199 - 204), содержащую 30 мкМ ДикамбаR в котором предварительно выращивали клеточные суспензии Dactylis glomerata.
(q) Повторяли стадии (a) - (n) с тем исключением, что в качестве среды для высевания протопластов использовали среду KM, содержащую 30% об. среды N6, в которой предварительно выращивали клеточные суспензии, описанные в примере 3.
(r) Повторяли стадии (a) - (n) с тем исключением, что в качестве среды для высевания протопластов использовали среду KM, содержащую 15% об. среды N6, в которой предварительно выращивали клеточные суспензии, описанные в примере 3.
(s) Повторяли стадии (a) - (r) с тем исключением, что протопласты переносили в культуральную среду (среда KM, содержащая 0,5 мг/л 2,4-Д и 100 мг/л O-ацетилсалициловой кислоты, а также 1,2% мас./об. Sea PlagulR агарозы) на фильтр для растительных культур, при плотности 0,5 миллионов протопластов/мл.
В этом варианте фильтры Durapore (каталожный номер CVW PO4700, размер пор 0,22 мкм, диаметр 47 мм) нумеровали с помощью свинцового карандаша по краю внешней поверхности по мере их извлечения из резервуара. Такую операцию проводили для того, чтобы быть уверенным, что верхняя сторона обращена к растительным культурам, и что для каждой культуры используется лишь один фильтр. Такие фильтры обрабатывали в автоклаве в дистиллированной, деионизированной воде в полупрозрачном контейнере (GA 7R или любой аналогичный контейнер) в течение 20 минут. После охлаждения воду заменяли на жидкую KM среду, описанную выше, но не содержащую гелеобразующих агентов, по крайней мере за 3 часа перед помещением фильтров на растительную культуру.
Посевные культуры готовили из суспензионной культуры черной мексиканской сладкой кукурузы (BMS) (Грин Hort. Sci 12 (1977) 131, Смит с сотр. Plant Sci Lett. 36 (1984) 67 / или из эмбриогенных суспензий, описанных в примере 3.
В том случае когда в качестве растительной среды использовали BMS стерильную среду KM, содержащую 0,5 мг/л 2,4-Д и 100 мг/л O-ацетилсалициловой кислоты (2 мг/мл в 2 об./об. DMCO, содержащем 0,1% мас./об. MES pH 6,0), добавляли к стерильной Sea PlagueR агарозе до получения конечной концентрации агарозы 1,2% мас./об. После нагревания для расплавления агарозы, среду охлаждали до 44oC на водяной бане, добавляли 1 мл PCV суспензии BMS в расчете на 10 мл среды и 5 аликвот распределяли в чашках Петри диаметром 60 мм. После отвердевания среды на поверхность помещали стерильные фильтры Durapore пронумерованные на верхней стороне.
В том случае, когда в качестве растительной культуры использовали эмбриогенные кукурузные суспензионные культуры, осмолярность среды N6 повышали с помощью глюкозы до значения 530 - 540 mOS /кг H2O, pH системы снова устанавливали равным 6,0 и среду подвергали стерилизации через фильтр 0,2 мкм. Добавляли 0,5 мг/л 2,4-Д и 100 мг/л O-ацетилсалициловой кислоты (2 мг/мл в 2% об./об. DMCO, содержащем 0,1% мас./ об. MES, pH 6,0). Эту среду добавляли к стерильной Sea PlagulR агарозе до конечной концентрации агарозы 1,2% вес. /об. После нагревания для расплавления агарозы, среду охлаждали до 44oC на бане с водой, добавляли 1 мл PCV эмбриогенной суспензионной культуры в расчете на 10 мл среды, а аликвоты объемом 5 мл распределяли в чашках Петри диаметром 60 мм. После затвердевания среды на ее поверхность помещали стерильные фильтры Durapore пронумерованной стороной вверх.
Протопластные препараты разбавляли охлажденной (44oC) KM средой с 1,2% мас./об. Sea PlagueR агарозой, содержащей 0,5 мг/л 2,4 - д и 100 мг - л O-ацетилсалициловой кислоты до концентрации 0,5 миллионов протопластов/мл. По 0,5 мл такого препарата медленно прикапывали пипеткой на поверхность каждого из фильтров.
(t) Повторяли стадии (a) - (s) с тем исключением, что ДНК-носитель телячьего тимуса не добавляли.
(u) Повторяли стадии (a) - (t) с тем исключением, что добавляли 50 мкг линеаризованной pC1B712 плазмиды.
(v) Повторяли стадии (a) - (u) с тем исключением, что плазмиду pCIB 712 не линеаризовали.
Пример 9. Трансформация протопластов Zea mays путем обработки полиэтиленгликолем
(a) Протопласты ресуспендировали на последней стадии примера 3 в 0,5M растворе маннита, содержащем 12 - 30 мМ MgCl2. В течение 5 мин при 45oC осуществляли тепловой шок, как описано в примере 8a. Протопласты распределяли аликвотами для трансформации в пробирках центрифуги, по 0,3 мл суспендированных протопластов на пробирку. В течение следующих 10 минут добавляли следующие вещества; ДНК (как указано в примерах 8a - 8V) в раствор ПЭГ (ПЭГ 6000 40% мас./об; Ca(NO3)2 0,1M; маннит 0,4M; pH 8 - 9 с помощью KOH) в результате чего обеспечивалась конечная концентрация 20% вес./об. Аликвоты инкубировали в течение 30 минут при периодическом мягком встряхивании, после чего протопласты помещали в чашки Петри (по 0,3 мл исходной суспензии протопластов на чашку диаметром, 6 см) и культивировали, как описано в примерах 8a или 8o - 8r.
(b) Протопласты повторно суспендировали на последней стадии примера 3 в 0,5M раствора маннита, содержащем 12 - 30 мМ MgCl2 при плотности 20 миллионов протопластов/мл. Тепловой шок осуществляли в течение 4 мин при 45oC, как описано в примере 8a. Протопласты распределяли аликвотами для трансформации в пробирках центрифуги, по 0,5 мл протопластов на пробирку. В течение последующих 10 минут добавляли следующие компоненты: ДНК (как описано в примере 5a) и раствор ПЭГ (ПЭГ 8000 (сигма или другой эквивалентный сорт), 36% мас./об., 0,1 M Ca(NO3)2 0,4 M маннита, 0,1% MES поддерживая в течение 3 часов pH 7 - 8 путем воздействия KOH, стерилизованной через фильтр 0,2 мкм до конечной концентрации 18% мас./об. Аликвоты инкубировали в течение 30 мин при мягком встряхивании и затем протопласты помещали в чашки Петри и культивировали, как описано в примерах 8a - 8s.
(c) Повторяли стадии (a) и (b) и протопласты промывали после 30 минут инкубирования в ПЭГ путем добавления 0,3 мл раствора W5 из примера 8n пять раз через 2/3-минутные интервалы, затем систему центрифугировали, надосадочный слой удаляли и протопласты культивировали, как описано в примерах 8a - 8s
(d) Согласно другой методике протопласты через 5-минутные интервалы промывали путем последовательного добавления 1 мл, 2 мл и 5 мл раствора W5 из примера 8n. Затем систему центрифугировали и протопласты культивировали, как описано в примерах 8a - 8s.
(c) Повторяли стадии (a) - (d) с тем исключением, что конечная концентрация ПЭГ составляла 15% мас./об.
(f) Стадии (a) - (e) повторяли с тем исключением, что конечная концентрация ПЭГ составляла 25% мас./об.
(g) Стадии (a) - (d) повторяли с тем исключением, что конечная концентрация ПЭГ составляла 12% мас./об.
(h) Повторяли стадии (b) - (f) с тем исключением, что протопласты промывали соленым раствором KMC согласно примеру 8n.
Пример 10a. Регенерация каллюса из протопластов
Пластины, одержащие протопласты в агарозе, помещали в темноту при 26oC. В течение 14 дней из протопластов развивались колонии клеток. Агарозу, содержащую колонии, переносили на поверхность чашек Петри диаметром 9 см, содержащих 30 мл среды N6 с 2 мг/л 2,4-Д, которая затвердевала при обработке 0,24% вес. /об. Гелрайта (2N6). Колонии подвергали культивированию с получением каллюса: Каллюс культивировали дополнительно в темноте при 26oC и кусочки каллюса субкультивировали каждые 2 недели на свежей среде Nb, содержащей 2 мг/л 2,4-Д (которая отвердевала при обработке 0,24% мас./об. ГелрайтаR).
Пример 10b. Регенерация каллюса их протопластов
Пластины, содержащие протопласты посевных культур, помещали в темноту при 26oC. В течение примерно 2 недель появлялись колонии. Полные фильтры затем переносили на поверхность чашек Петри диаметром 9 см, содержащих среду с 2 мг/л 2,4-Д, которая отвердевала при воздействии 0,24% вес./об., ГелрайтаR (2N 6), либо колонии снимали и по отдельности переносили в такую среду. Развившийся каллюс дополнительно культивировали в темноте при 26oC и кусочки каллюса субкультивировали каждые 2 недели на свежей среде N6, содержащей 2 мг/л 2,4-Д (которая отвердевала при обработке 0,24% мас./об. ГелрайтаR).
Пример 11a. Селекция трансформированного каллюса Zea mays.
Повторяли примеры 10a и 10b с тем исключением, что в среду 2N6 добавляли 50 мг/л гигромицина с тем, чтобы осуществить селекцию трансформированных клеток
Пример 11B. Селекция трансформированного каллюса Zea mays.
Примеры 10a и 10b повторяли с тем исключением, что в среду 2N6 добавляли 100 мг/л гигромицина с тем, чтобы осуществить селекцию трансформированных клеток.
Пример 11c. Селекция трансформированного каллюса Zea mays.
Примеры 10a и 10b повторяли с тем исключением, что в среду 2N6 добавляли 200 мг/л гигромицина с тем, чтобы осуществить селекцию трансформированного материала.
Пример 12a. Регенерация растений
Каллюс помещали на среду 2N6 для сохранения и на 0N6 (N6 среда без гормонов) и N61 (0,25 мг/л 2,4-Д + 10 мг/л кинетина) для инициирования регенерации. Выращивание материалов на среде 0N6 и N61 осуществляли на свету (16 ч дневного света с 10-30 мкЕ/м2• c от флуоресцентной лампы). Каллюс, выраженный на среде N61, переносили через 2 недели на среду 0N6 поскольку более продолжительное время пребывания на пластинах с N61 оказывается вредным. Каллюс субкультивировали каждые 2 недели даже в том случае, если его снова переносили на исходную среду.
Через 4-8 недель появлялись побеги
Как только побеги достигали высоты, по крайней мере, 2 см, их переносили на среду 0N6 в контейнерах G47 или других подходящих культуральных сосудах. В течение 2-4 недель появлялись корни. Если корни выглядели достаточно сформированными для поддержания роста, побеги переносили в торфяные горшочки, которые помещали в тень на первые 4-7 дней. Иногда оказывается полезным поставить на несколько дней над трансплантатами перевернутую пластмассовую чашку.
Сразу после адаптации растений их обрабатывали, как обычные Zea mays растений и выращивали в теплице до созревания с целью испытания на действие удобрений и наследование введенных генов.
Пример 12b. Регенерация растений.
Повторяли пример 12a с тем исключением, что в среду добавляли 50 мг/л гигромицина с целью поддержания каллюса.
Пример 12c. Регенерация растений
Повторяли методику примера 12a с тем исключением, что 100 мг/л гигромицина добавляли в среду, используемую для сохранения каллюса.
Пример 12d. Регенерация растений.
Повторяли методику примера 12a с тем исключением, что 200 мг/л гигромицина добавляли в среду, используемую для сохранения каллюса.
Пример 13. Трансформация и регенерация.
Повторяли примеры 8, 9, 10 и 11 за исключением того, что в качестве плазмиды ДНК использовали плазмиду pCIB10/35 SBt и на стадиях селекции гигромицин заменяли на антибиотик C418.
Пример 14. Конструирование pIRV содержащей как химерический ген устойчивости гигромицину, так и химерический ВТ ген
Плазмиду pCIB переваривали SmaI плазмиду pCIB10/35 Sb+ переваривали с SphI обрабатывали крупным фрагментом ДНК-полимеразы (фермент Кленова) с целью развития побеговых концов, затем переваривали SmaI. Фрагмент размером примерно 4,8 kO, содержащий химерический BT ген, очищали электрофорезом на агарозном геле и лигировали с SmaI расщепленной pCIB712ДНК. Полученная в результате плазмида pIRV содержит как химерический ген устойчивости к гигромицину, так и химерический  ген, pIRV линеаризовывали на соответствующем сайте, дистальном по отношению к химерическим генам, перед электропорацией.
ген, pIRV линеаризовывали на соответствующем сайте, дистальном по отношению к химерическим генам, перед электропорацией.
Пример 15. Трансформация и регенерация.
Примеры 8, 9, 10 и 11 повторяли с тем исключением, что в качестве плазмиды ДНК использовали плазмиду pIRV а для селекции применяли гигромицин.
Пример 16. Конструирование плазмиды, несущей химерический хитиназный и химерический ген устойчивости к гигромицину.
cДНК клон, PSCHI, содержащий кодирующий участок табачного хитиназного гена, идентифицировали в библиотеке табачных ДНК, клонированных в PstI сайт pBR322. Гибридселективная трансляция давала протеиновый продукт, который гибридно реагирует с антителом к хитиназе бобов (Шинши с сотр.: Proc.Nate. Acad. Sci USA 84 (1987) 89-93). Из лямбда библиотеки табачных ДНК соответствующий геномный клон идентифицировали с использованием в качестве зонда cДНК-клона (Маниатис цит. ссылка). Для идентификации Pst I сайта общего с сДНК-клоном и геномным клоном использовали методы гибридизации и рестрикционной картографии.
Установление последовательности геномной ДНК выше такого общего PstI сайта позволило обнаружить начало открытой рамки считывания для хитиназа-структурального гена, а также сайт SphI распознавания в положении 35, считая со стартового сайта. Описанные ниже стадии использованы для того, чтобы объединить этот верхний фрагмент геномной ДНК с идущим вниз фрагментом сДНК, формируя полную длину кодирующей области хитиназы:
а) Ген полный длины конструировали в плазмидном векторе pGVI, который содержит CaMV 35S промоторный фрагмент, сигнал терминации полиценилирования от CaMV и полилинкер между ними. Полярность полилинкерных сил ориентирует верхний фрагмент; точная ориентация нижнего фрагмента определяется анализом последовательности.
Верхний SphI/PstI фрагмент геномного клона выделяют и очищают; затем его лигируют в плазмиду pGYI, которая была переварена SphI и PstI.
b) Полученную в результате промежуточную плазмиду переваривали PstI. Нижний PstI фрагмент выделяли из клона pSCHI и лигировали в такой переваренной промежуточный вектор.
c) Клон, имеющий правильную ориентацию и слияние в рамке фрагмента cДНК с верхним геномным фрагментом ДНК, подтверждали анализом последовательности ДНК. Полученную в результате плазмиду называли плазмидой pSCHI. Плазмиду pSCHI (в штамме E.coli K12/депонировали в Deutsche Sammlung von Mikroorganismen. Геттинген, ФРГ, каталожный номер DSM 4129. Такое депонирование было произведено в соответствии с требованиями Будапештского договора (дата депонирования: 29 мая 1987 года).
Полный хитиназный ген, фланкированный CaMV 35S промоторный и терминаторной областями, вырезали из pSCHI путем переваривания EcoRI. Фрагменту создавали тупые концы путем использования крупного фрагмента ДНК полимеразы I (Маниатис, приведенная выше ссылки). Рецепторную плазмиду pCIB712, которая содержит химерический ген устойчивости к гигромицину, который экспрессируется в клетках растения, переваривали SmaI • SmaI - переваренную pCIB 712 и фрагмент с тупыми концами, содержащий химерический хитиназный ген, подвергали совместному лигированию с использованием T4 лигазы (Маниатис, указанная ссылка). Полученную в результате плазмиду назвали pCIBcht. Перед трансформацией протопласта pCIBcht линеаризовали по месту сайта, дистального в отношении химерических генов.
Пример 17. Конструирование вектора, несущего химерический AHAS ген, придающий устойчивость к сульфонилмочевине.
a. Выделение сульфонилмочевина устойчивых (SU-R) растений Arabidopsis.
Семена M2 Arabidopsis thaliana (Колумбийской расы), мутагенизированные действием этилметансульфоната (EMS) получали согласно методу Хауна и Сомервилла (Mol. Gen.Genet. 204 (1986) 430-434), а также методу Сомервилла и Огрина (Методы хлоропластной молекулярной биологии, изд. Эдельман и др. (1982) 129-138). EMS использовали в концентрации 0,3% в течение 14-16 часов и семена M1 высевали и выращивали с плотностью 2-3 растения на см2.
Сульфонилмочевина устойчивые (SU-R) растения Arabidopsis подвергали селекции и выращивали из семян M2 согласно методу Хауна и Сомервилла (см. ссылку, приведенную выше).
b. Конструирование рекомбинантной лямбда фаговой библиотеки SU-R Arabidopsis ДНК.
ДНК выделяли из SU-R растений вида Arabidorsis согласно методу Джена и Хилтона (J.Bacteriol. 166 (1986) 491-499). ДНК Arabidopsis полностью переваривали с XlaI (Нью Инглэнд Биолабс).
Из агарозного геля выделяли фрагменты ДНК размером 4-8 kho c использованием NA45 DEAE мембраны (Шлейцер и Шуэлл, Ким, Н.Г.Каталог N 03431) и клонировали в лямбда-ongC-X вектор (Стратеген, 3770, Танси стр. Сан-Диего, СА 92121 согласно методикам, рекомендованным производителями. Рекомбинантную фаговую ДНК инкапсулировали с использованием набора Гигипак-плюс, полученного от Стратегена.
с. Клонирование SU-R ацетоксигидрокси кислотного, синтазного (AHAS) гена растений разновидности SU-R Arabidopsis.
Плазмиду pE16 /8-c4, содержащую IZV2 локус Saccharomgeces cerevisiol (Жд. Полайна, Carlsberg Res. Comn. 49(1984 (577-584), полученную от Дж.Полайна, трансформировали в E.coli HB101 получали крупномасштабный плазмидный препарат. Плазмиду pE16/8-c4 депонировали в Американской коллекции типовых культур, под каталожным номером 67420. Такое депонирование осуществляли согласно требованиям Будапештского Договора (дата депонирования: 29 мая 1987 г. ). Плазмиду pE16/8-04 ДНК переваривали EcoRI и фрагмент размером 2,1 kho (Eco 2.1), содержащий AHAS кодирующую последовательность (Фалкс с сотр. Nucl. Acid. Res. 13 (1985) 4011-4027) выделяли из агарозного геля. Фрагмент Eco 2,1 подвергали радиационной метке изотопом фосфора-32 до удельной активности 5-8 • 108 cpm/мкг с помощью набора Прайм тайм (Интернейшнл Биотекнолоджиз, ИНК).
Рекомбинантные фаги, содержащие Arabidopsis Xba I фрагменты, высевали на E. coli VCS 257 согласно методу Маниатиса с сортр. (1982) (Колд Спринг Харбор, Нью-Йорк) и протоколам Стратегена. Лифты дупликатных лямда бляшек получали с каждой фаговой пластины с помощью Colony / Plague ScreenR мембраны (нью Инглэнд Ньюклер Резерч Продактс) в соответствии с предписаниями производителей. Лифты бляшек обрабатывали следующим образом:
i) в течение 6 ч промывали 5X SSC, 2% SDS при 65oC (все предписания даны Маниатисом с сотр. (см. ссылку, приведенную выше);
ii) прегибридизировали в гибридизационной смеси (5 x SSC, 1% SDS, 5 x раствор Денхардта, 200 мкг/мл денатурированной ДНК семги, 25% формамида, 10% декстансульфата; 10 мл смеси на 150 мм мембраны) в течение 16-20 ч при 42oC;
iii) подвергали гибридизации в гибридизационной смеси (10 мл на 150 мм мембраны), содержащей 2 нг/мл денатурированного радиомеченного Eco 2,1 фрагмента в течение 18-20 часов при 42oC;
iv) промывали 2 x SSC, 1% SDS при комнатной температуре в течение 30 мин;
v) промывали 5 x SSC, 1% SDS, 25% формамида в течение 6-8 часов при температуре 42oC при четырехкратной замене промывного раствора.
Мембраны экспонировали на рентгеновскую пленку (Кодак X-Омат AR). Фаговые бляшки, проявляющие соответствующую гибридизацию на дупликатных мембранах, очищали с помощью второй партии лифтов лямбда-бляшек и гибридизировали с помощью Eco 2.1 зонда.
Лизаты бляшек рекомбинантного фага, содержащего вставку, гомологичную дрожжевой AHAS последовательности, готовили согласно методу Маниатиса с сотр. (см. выше). Фаговую ДНК готовили из лизатов бляшек в присутствии Lambda Sorb (Promega. 2800. S. Fish Hatchery Rd., Madison 53711 в соответствии с методиками, рекомендованными производителями.
d. Конструирование и подтверждение трансформационного вектора pC1B808, содержащего SU-R Arabidopsis AHAS ген
Рекомбинантную фаговую ДНК переваривали Xbal и фрагмент вставку размером 5,8 kho клонировали b Xba-I-переваренный pC1B10 трансформационный вектор с получением pC1B803. Плазмиду pC1B803 переносили с хозяина E.coli HE101 на Agrobacterium tumefaciens C1B542 путем трипарентального скрещивания (Дитта с сотр. Proc. Acad. Sci USA 77 (1980 (262-269). A tumefaciens pC1B803/C1B542 использовали для трансформации эксплантатов листьев Arabidopsis thaliana в соответствии с методом Лойда с сотр. (Science 234 (1980 (464/-466) и Шейхолеслама и Викса (Plant. Mol. Biol, 8, (1986/291-298). Трансформированные ткани регенерировали в растениях согласно методу Ллойда с сотр. (см. ссылку, приведенную выше) и Фельдмана и Маркса (Plant Sei 47, (1987/63-69). Было показано, что рост трансформированных тканей в культуральной среде толерантен по отношению к 3-10 нг/мл хлорсульфурона. Всхожесть и рост трансгенных растений Arabidopsis на среде агара, как было показано, толерантны к действию 10-30 нг/мл хлорсульфурона.
e. Конструирование трансформационного вектора pC1B804, содержащего химерический SU-R AHAS ген.
Расположение AHAS кодирующей последовательности внутри Xba фрагмента размером 5,8 определяли методом нуклеазного-S1 картирования (Маниатис с сотр. см. приведенную выше ссылку), методом расширения праймера (Макнайт с сорт., Cell, 25 (1981)385-398) и подтверждали путем установления последовательности ДНК (Сангер с сотр. Proc. Nat. Acad. Sci, USA 74 (1977) 5463-5467/ и путем сравнения протеиновой последовательности с бактериальными и дрожжевыми AHAS последовательности с бактериальными и дрожжевыми AHAS последовательностями (Фалко с сотр. Nucl. Acid. Res 13 (1985) (4011-4027) Xbal фрагмент размером 5,8 кb клонировали в Блюскрипт (Стратеген) и последовательности выше AHAS кодирующей последовательности делецировали в пределах 10-20 bp находящегося выше AHAS инициирующего кодона, с использованием делеционного набора Exo/Mung (Стратеген). Соответствующий линкер присоединяли к концевой точке делеции. Резекцированный AHAS ген разрезали по месту соответствующего рестрикционного сайта, по крайней мере на 10bp выше AHAS терминационного кодона и тот же линкер, что использовали на 5'-окончании гена, присоединяли к 3'-окончанию гена. Фрагмент, содержащий AHAS кодирующую последовательность вырезали на вектора путем разрезания по линкерным сайтам и клонировали в Bam HI сайт pC1B710.
BamHI сайт pC1B710 адаптировали или превращали в сайт, совместимый с линкером, использованным выше, если это необходимо, с тем, чтобы получить линкерный SU-R AHAS ген. Ориентацию AHAS генной вставки в pC1B710 определяли с помощью соответствующих рестрикционных ферментов. Клон, содержащий AHAS кодирующую последовательность, правильно ориентированную относительно транскрипционального направления CaMV 35S промотора, обозначали как pC1B04.
Пример 18. Промотирование образования каллюса из протопластов Zea mays под воздействием 2,4-Д ацетилсалициловой кислоты
Протопласты ресуспендировали на последней стадии примера 3 в растворе КМС крепостью 6/7 в количестве 7,5 миллионов на мл. О-Ацетилсалициловую кислоту растворяли в количестве 2,5 мг/мл в среде КМ. Полученный раствор стерилизовали на фильтре. Затем, непосредственно перед использованием культуральной среде добавляли О-ацетилсалициловую кислоту. Аликвоты протопластовой суспензии помещали в чашки Петри диаметром 6 см при комнатной температуре. 3 мл среды КМ, содержащей 1,2% мас./об. Sea PlagueR агарозы, 2,4-Д и О-ацетилсалициловую кислоту добавляли таким образом, чтобы получить правильную концентрацию 2,4-Д и О-ацетилсалициловой кислоты и плотность протопластов 0,5 миллионов на мл. Агарозу и протопласты тщательно перемешивали и среде давали желатинироваться.
Чашки подвергали культивации, как описано в примере 7. Через 3 недели подсчитывали число макроскопических колоний, появившихся в чашках. В табл. 2 и 3 представлены результаты, полученные для протопластов из двух различных независимо полученных эмбриогенных суспензионных культур.
В табл. 2 представлено образование колоний протопластов Zea maus в присутствии О-ацетилсалициловой кислоты и 2,4-Д (цифрами обозначены номера образовавшихся колоний)
В табл 3 представлено образование колоний протопластов Zea mays в присутствии О-ацетилсалициловой кислоты и 2,4-Д. (Цифрами обозначено число образовавшихся колоний)
Пример 19. Усиленное образование колоний из протопластов в результате добавления O-ацетилсалициловой кислоты и 2,4-Д.
Протопласты готовили и высевали для культивации в соответствии с описанным в примере 18, используя для этой цели 2 мл среды и чашки Петри диаметром 6 см.
Чашки подвергали культивации в соответствии с описанным в примере 10. Через 3 недели подсчитывали число макроскопических колоний, появившихся в чашках. Полученные результаты представлены в табл. 4. Снова было установлено, что O-ацетилсалициловая кислота стимулирует образование колоний. Однако при концентрациях 300 мг/л и выше наблюдался токсический эффект. Было предложено, что такой эффект связан с проблемой установления pH и возможно, что эффект может быть исключен введением в среду соответствующего буфера.
И табл. 4 представлено образование колоний (в расчете на чашку) протопластов в присутствии O-ацетилсалициловой кислоты и 2,4-Д
(Цифрами обозначено число образовавшихся колоний)
Пример 20. Промотирование образования колоний из протопластов Zea mays под воздействием ДМСО и O-ацетилсалициловой кислоты и в присутствии 2,4-Д.
Протопласты готовили и высевали в количестве 0,75 миллионов на мл в соответствии с описанным в примере 18, с использованием 1 мл среды и чашек Петри диаметром 3,5 см. В среду вводили O-ацетилсалициловую кислоту. O-ацетилсалициловую кислоту растворяли в ДМСО и затем добавляли в среду перед высеванием протопластов. Для получения раствора в ДМСО, 1 г O-ацетилсалициловой кислоты растворяли в 10 мл ДМСО и добавляли в 90 мл дистиллированной воды. Затем полученный раствор перед использованием стерилизации на фильтре.
Чашки подвергали культивации, как описано в примере 10. Через 3 недели посчитывали число макроскопических колоний, образовавшихся в чашках. Полученные результаты представлены в таблице 5. Использование только ДМСО (1% об. /об. ) не обеспечивает значительного повышения или понижения эффективности образования колоний противопластов Zea mays и явно усиливает стимуляторную активность O-ацетилсалициловой кислоты.
В табл. 5 представлено влияние O-ацетилсалициловой кислоты на образование колоний протопластов Zea mays демонстрирующее стимуляторный эффект растворения O-ацетилсалициловой кислоты в ДМСО (число образовавшихся колоний)
Пример 21. Усиление образования колоний протопластов в результате добавления 2,4-Д и родственных регуляторов роста растений (ауксинов).
Протопласты ресуспендировали на последней стадии примера 3 в растворе КМС крепостью 6/7 в количестве 5 миллионов на мл.
Аликвоты протопластовой суспензии, помещали в чашки Петри диаметром 6 см при комнатной температуре. Добавляли 2 мл среды КМ, содержащей 1,2% вес./об. Sea PlagueR агарозы, а также 2,4-Д, Пиклорам, Дикамба или пара-хлорфеноксиуксусную кислоту (pCPA) с целью достижения правильных концентраций регулятора роста растений. Использовали конечную плотность протопластов 0,54 миллиона на мл. Агарозу и протопласты хорошо перемешивали и среде давали желатинироваться.
Чашки культивировали, как описано в примере 10. Через 3 недели подсчитывали число макроскопических колоний, появившихся в чашках. Полученные результаты представлены в табл. 6.
В табл. 6 представлено образование колоний из культивированных протопластов Zea mays в присутствии различных регуляторов роста растений (ауксины) / число образовавшихся колоний)
Пример 22. Конструирование делецированного Bt протоксинового гена, содержащего примерно 725 аминокислот, и конструирование химерного гена, содержащего такой делецированный ген с CaMV 35s промотором
Делецированный протоксиновый ген, содержащий примерно 725 аминокилсот, получали удалением COOH-терминальной части гена путем расщепления по сайту Kn pI рестрикционной эндонуклеазы в положении 2325 последовательности. Плазмиду pCIB10 /35 sBr (фиг. 12) переваривали с Bam HI и Kn pI и фрагмент Bam HI/Kn pI размером примерно 2,2 k b p, содержащий делеционный протоксиновый ген, выделяли методом препаративного электрофореза на агарозном геле. Для превращения KnpI сайта на окончании 3' в BAm HI сайт, такой фрагмент смешивали с KnpI/Bam HI адаптерным олигонуклеотидом и лигировали. Затем такой фрагмент смешивали с Bam HI-расщепленной pCIВ10/710. Полученные в результате трансформанты, обозначенные как pCIB10/35SBt (Kn p1 и показанные на фиг. 15, содержат делецированный протоксиновый ген размером в примерно в 725 аминокислот. Такие транформанты подвергали селекции на канамицине.
Пример 23. Конструирование делецированного  протоксинового гена, содержащего примерно 645 аминокислот и конструирование химерного гена, содержащего такой делецированный ген с CaMV 35S промотором.
протоксинового гена, содержащего примерно 645 аминокислот и конструирование химерного гена, содержащего такой делецированный ген с CaMV 35S промотором.
Делецированный протоксиновый тип, содержащий примерно 645 аминокислот, получали путем удаления COOH-терминальной части гена расщеплением на сайте Bcl I рестрикционной эндонуклеазы в положении 2090 последовательности. Плазмиду pCIB10 /35S  (фиг. 12) переваривали с Bam HI и BCl I и Bam HI/Bcl I, фрагмент размером 1,9 k b p, содержащий делецированный протоксиновый ген, выделяли методом препаративного электрофореза агарозном геле. Поскольку BCII создает липкий конец, совместимый с Bam HI, никакого дополнительного манипулирования не требуется перед лигированием такого фрагмента на BamHI - расщепленную pCIB10/710. Полученную в результате плазмиду, со структурой pCIB10/35 SBt (BcII), подвергали селекции не канамицине.
(фиг. 12) переваривали с Bam HI и BCl I и Bam HI/Bcl I, фрагмент размером 1,9 k b p, содержащий делецированный протоксиновый ген, выделяли методом препаративного электрофореза агарозном геле. Поскольку BCII создает липкий конец, совместимый с Bam HI, никакого дополнительного манипулирования не требуется перед лигированием такого фрагмента на BamHI - расщепленную pCIB10/710. Полученную в результате плазмиду, со структурой pCIB10/35 SBt (BcII), подвергали селекции не канамицине.
Пример 24. Конструирование делецированного  протоксинового гена, содержащего примерно 607 аминокислот и конструирование химерического гена, содержащего делецированный ген с CaMV 35S промотором.
протоксинового гена, содержащего примерно 607 аминокислот и конструирование химерического гена, содержащего делецированный ген с CaMV 35S промотором.
Делецированный протоксиновый ген получали введением Bam HI сайта расщепления (GGATCC) после нуклеотида 1976 в последовательности. Такую операцию осуществляли клонированием BamHI фрагмента, содержащего протоксиновую последовательность, из pCIB10/35 S  b mp18 и использованием стандартных методик олигонуклеотидного мутагенеза, описанных выше. После мутагенеза, двухнитевую репликационную ДНК готовили из М13 клона, который затем переваривали с Bam HI. Фрагмент размером около 1,9 k b p, содержащий делецированный протоксиновый ген, вставляли в Bam HI - расщепленную pCIB10/710. Полученную в результате плазмиду, имеющую структуру pCIB10/35S
b mp18 и использованием стандартных методик олигонуклеотидного мутагенеза, описанных выше. После мутагенеза, двухнитевую репликационную ДНК готовили из М13 клона, который затем переваривали с Bam HI. Фрагмент размером около 1,9 k b p, содержащий делецированный протоксиновый ген, вставляли в Bam HI - расщепленную pCIB10/710. Полученную в результате плазмиду, имеющую структуру pCIB10/35S  (607), подвергали селекции на канамицине.
(607), подвергали селекции на канамицине.

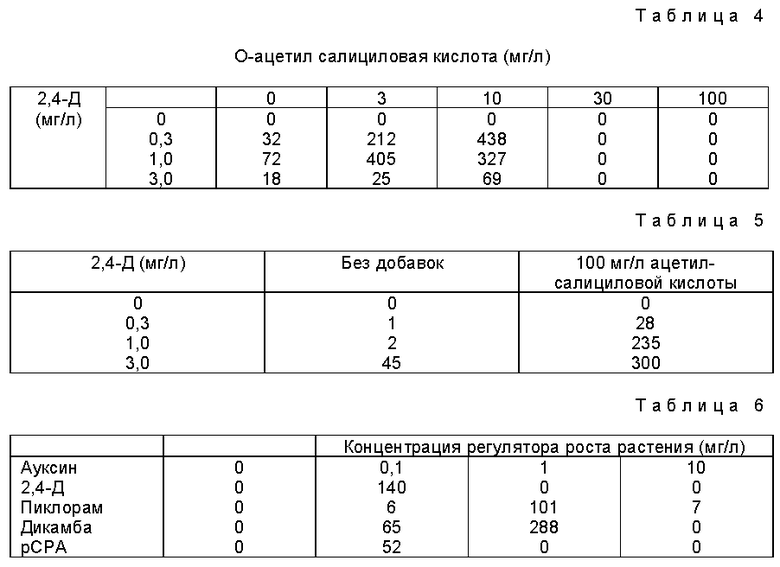







Способ предназначен для создания трансгенных растений, устойчивых к повреждениям, вызываемым насекомыми, и может быть использован при трансформации тотипотентных протопластов вектором, содержащим структурный ген, кодирующий полипептид кристаллического белка δ-эндотоксина Bacillus thuringiensis. Перед трансформацией выделяют ткань из незрелых зародышей, культивируют ее до образования особого типа каллуса. Каллус обрабатывают ферментным препаратом для получения тотипотентных протопластов, после трансформации которых из них генерируют фертильные растения. Регенерация растений из клеток протопластов позволяет применить соматическую гибридизацию для получения улучшенных разновидностей Zea mays L. 30 з.п. ф-лы, 4 ил., 6 табл.
или инсектицидную часть указанного полипептида.
| СПОСОБ ПРЕДОТВРАЩЕНИЯ ОСМОЛЕНИЯ САХАРНЫХРАСТВОРОВ | 0 |
|
SU198259A1 |
| Глубинный вибратор для уплотнения преимущественно бетонной смеси | 1961 |
|
SU142924A1 |
| US 4677247A, 1985 | |||
| Калинин Ф.Л | |||
| и др | |||
| Методы культуры тканей в физиологии и биохимии растений | |||
| - Киев: Наукова Думка, 1980, с.247 | |||
| Bot.Caz., 143(3) рр | |||
| Перепускной клапан для паровозов | 1922 |
|
SU327A1 |
| Gmicn., 2(1) рр | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Ребристый каток | 1922 |
|
SU121A1 |
| Способ получения камфоры | 1921 |
|
SU119A1 |

