Изобретение относится к замещенным пиридином диоксигептеновым кислотам, в частности к новой 3R,5S-(+)-7-[4-(4-фторфенил)-2,6-диизопропил-5-метоксиметил-пи- рид-3-ил]-3,5-диоксигептеновой кислоте в эритро-(Е)-конфигурации в виде физиологически переносимой соли металла, служащей в качестве ингибитора биосинтеза холестерина.
Известны замещенные пиридином диоксигептеновые кислоты, в частности рацемический сложный метиловый эфир 7-[4-(4-фторфенил)- 2,6-диизопропил-5-метоксиметил-пирид-3-ил] -3,5-диоксигептено- вой кислоты в эритро-(Е)-конфигурации (см. ЕР 325 130 А2, кл. С 07 D 213/65, 1989).
Целью изобретения является получение новой замещенной пиридином диоксигептеновой кислоты, являющейся лучшим ингибитором биосинтеза холестерина, чем указанное известное соединение.
Поставленная задача решается 3R,5S-(+)-7-[4-фторфенил)-2,6-диизопропил-5-ме-токсиметил-пирид-3-ил]-3,5-ге птенкислотой формулы I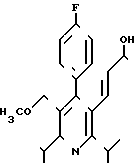

 в эритро-(Е)-конфигурации в виде физиологически переносимой соли металла.
в эритро-(Е)-конфигурации в виде физиологически переносимой соли металла.
Предпочтительными солями металла являются соли натрия, калия, магния или кальция.
Предлагаемое соединение указанной формулы I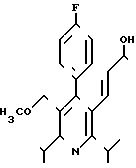

 получают путем взаимодействия рацемического сложного эфира формулы II
получают путем взаимодействия рацемического сложного эфира формулы II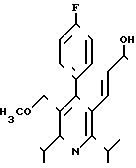

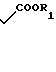 где R1 - низший алкил или бензил,
где R1 - низший алкил или бензил,
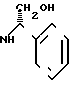
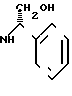
с(+) - энантиомерным амином формулы III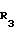 _
_ 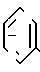
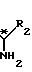 где R2 - низший алкил, незамещенный или замещенный гидроксильной группы, а
где R2 - низший алкил, незамещенный или замещенный гидроксильной группы, а
R3 - атом водорода, атом галогена, низший алкил или низший алкоксил, разделения получаемой при этом смеси соответствующих диастереомерных амидов формулы (IV)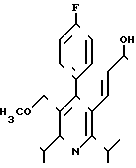


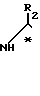
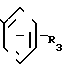 путем хроматографии или кристаллизации с получением желаемого диастереомера, который подвергают гидролизу до получения чистого энантиомера.
путем хроматографии или кристаллизации с получением желаемого диастереомера, который подвергают гидролизу до получения чистого энантиомера.
Способ поясняется следующей схемой.
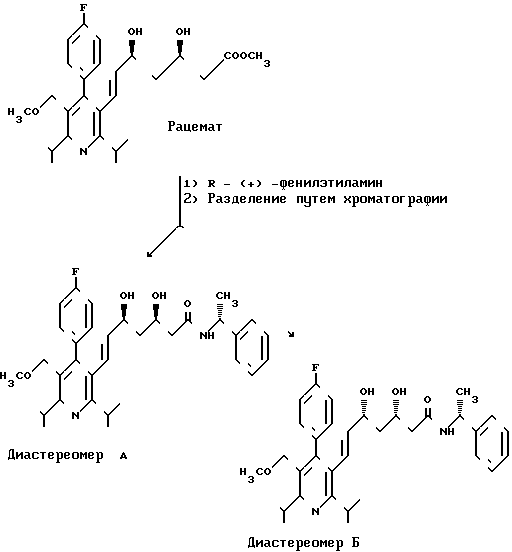

Взаимодействие сложных эфиров формулы II с (+) - аминами формулы III обычно осуществляют в среде инертного растворителя.
При этом в качестве растворителя можно использовать те органические растворители, которые обычно используют при амидировании. Предпочтительно используют простые эфиры, например, простой диэтиловый эфир, диоксан или тетрагидрофуран, или хлорированные углеводороды, например, метиленхлорид или хлороформ, или диметилформамид. Особенно предпочтительно, однако, соответствующий амин формулы III используют в избытке, в случае необходимости, в среде тетрагидрофурана или диоксана в качестве растворителя.
Взаимодействие обычно осуществляют при 0оС - 100оС, предпочтительно 20-80оС, и атмосферном давлении. Однако возможно также осуществление гидролиза при пониженном или повышенном давлении. При осуществлении взаимодействия оказалось выгодным использование амина или в качестве растворителя в особенно большом избытке, или, при использовании дальнейшего растворителя, в избытке до 10 раз.
Гидролиз диастереомерных амидов формулы IV осуществляют обычными методами, например, путем обработки амидов основаниями или кислотами в среде инертного растворителя.
При этом в качестве инертного растворителя можно использовать воду и/или органические растворители. В качестве органических растворителей предпочтительно используют спирты, например, метанол, этанол, пропанол или изопропанол, или простой эфир, например, простой диэтиловый эфир, диоксан или тетрагидрофуран. Особенно предпочтительно используют воду или смесь воды со спиртом.
Для осуществления гидролиза можно использовать неорганические или органические кислоты. Предпочтительно используют соляную кислоту, бромистоводородную кислоту, серную кислоту, метансульфокислоту или толуолсульфокислоту.
В качестве основания для гидролиза амидов можно использовать обычные неорганические основания, например гидроокись натрия или калия, метанолат или этанолат натрия или калия или карбонат натрия или калия.
В случае фенэтиламидов гидролиз предпочтительно осуществляют в этанольной соляной кислоте, а в случае фенилглициноамидов - натровым щелоком, в случае необходимости, в присутствии спирта.
Гидролиз диастереомерных амидов формулы IV осуществляют обычно при 0-150оС, предпочтительно 20-100оС, и атмосферном давлении. Однако возможно также осуществление гидролиза при пониженном или повышенном давлении.
Кроме того, целевой продукт можно также получать путем разделения соответствующего рацемата известными методами хроматографии.
Получение соединений формулы I иллюстрируется следующими примерами.
П р и м е р 1. Натриевая соль 3R,5S-(+)-эритро-(Е)-7-[4-(4-фторфенил)-2,6-диизопропил-6-метоксиметил-пирид -3-исигепт-6-еновой кислоты
H3CO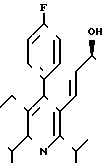
Вариант А.
4,7 г (10 ммоль) сложного метилового эфира эритро-(Е)-4-фторфенил)-2,6-диизопропил-5-метоксиметил-пирид-3-ил)- 3,5-диокси-гепт-6-еновой кислоты растворяют в 20 мл R мл R-(+)-фенилэтиламина и нагревают при 40оС в течение 72 ч. Реакционный раствор выливают в 150 мл воды и добавлением 1 н. соляной кислоты раствор доводят до значения рН 4. Затем несколько раз экстрагируют простым диэтиловым эфиром. Объединенные органические экстракты промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и сгущают. После предварительной очистки путем хроматографии на силикагеле (величина зерен 63-200 мкм, элюент: смесь сложного этилового эфира уксусной кислоты и петролейного эфира в объемном соотношении 4:6 до 6:4) разделяют путем хроматографии на силикагеле величины зерен 15 мкм (элюент: смесь сложного этилового эфира уксусной кислоты и петролейного эфира в объемном соотношении 1:1).
Выход: 2,1 г (37,4% теории) диастереомера АI формулы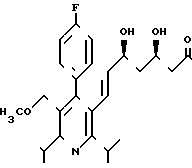
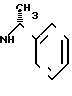 и 1,5 г (26,6% теории диастереомера БI формулы
и 1,5 г (26,6% теории диастереомера БI формулы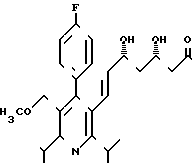
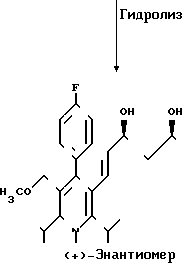
2,1 г (3,7 ммоль) диастереомера АI растворяют в 70 мл 15%-ного этанола и после добавления 13 мл 1 н.соляной кислоты нагревают с обратным холодильником в течение 48 ч. После охлаждения надосадочный раствор фильтруют и остаток несколько раз размешивают с этанолом. Объединенные этанольные растворы сгущают и остаток поглощают в 50 мл воды и 50 мл дихлорметана. Добавлением 1 н. соляной кислоты раствор доводят до значения рН 3,5 и затем раствор несколько раз экстрагируют дихлорметаном. Объединенные органические растворы сушат над сульфатом натрия и сгущают. Остаток поглощают в 50 мл смеси тетрагидрофурана и воды в соотношении 1:1, и добавлением 1н. натриевого щелока раствор доводят до значения рН 7,5. Тетрагидрофуран удаляют путем упаривания на ротационном испарителе, и остающийся водный раствор лиофилизуют. Сырой продукт лиофилизации очищают путем хроматографии на силикагеле (элюент: смесь ацетонитрила и воды в объемном соотношении 30:70). В результате сушки вымораживанием фракций получают 850 г (48% теории) целевого продукта.
1Н-ЯМР (DMCO-d6): δ (частей на миллион) = 1,0 (м, 1Н); 1,28 (д, 6Н); 1,3 (м, 1Н); 1,75 (ДД, 1Н); 3,07 (с, 3Н); 3,2-3,4 (м, 3Н); 3,52 (м, 1Н); 4,02 (м, 2Н); 5,28 (дд, 1Н); 6,17 (д, 1Н); 7,1-7,3 (м, 4Н).
Показатель вращения (этанол): [α]D20 = =24,1о (С = 1,0).
Вариант Б
418 г (0,88 моль) сложного метилового эфира эритро-(Е)-7-[4-(4- фторфенил)-2,6-диизопропил-5-метоксиметил-пирид-3-ил]-3,5-диокси- гепт-6-еновой кислоты и 360 г (2,6 моль) S-(+)-фенилглицинола растворяют в 1 л абсолютного тетрагидрофурана и нагревают при температуре 50оС в течение 96 ч.
После охлаждения до комнатной температуры добавляют 1 л воды, добавлением 5 н. соляной кислоты раствор доводят до значения рН 4, и трижды экстрагируют, каждый раз используя 400 мл простого диэтилового эфира. Объединенные органические фазы промывают 400 мл насыщенного раствора хлорида натрия, сушат над сульфатом натрия и сгущают на ротационном испарителе. Остаток (500 г сырого продукта) в двух порциях предварительно разделяют путем хроматографии на колонке, наполненной примерно 1,8 кг силикагеля (элюент: смесь сложного этилового эфира уксусной кислоты и петролейного эфира в объемном соотношении 8: 2). Получают 350 г предварительно очищенного сырого продукта, почти исключительно состоящего из двух диастереомерных амидов. Предварительно очищенный сырой продукт в 7 порциях по 50 г разделяют путем хроматографии на силикагеле величины зерен 20 мкм (колонка длиной 63 см и диаметром 7 см).
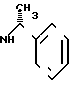
Выход 195 (38,2% теории) диастереомера А2 формулы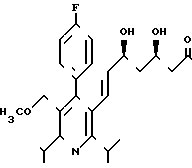
Диастереомер Б2 формулы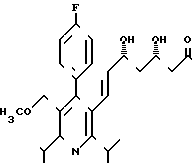
 не выделяют в чистом виде, однако, его получают в качестве сырого продукта при промывке колонок.
не выделяют в чистом виде, однако, его получают в качестве сырого продукта при промывке колонок.
195 г (0,34 моль) чистого диастереомера А2 растворяют в 1л этанола и после добавления 1,2 л 1 н. натрового щелока нагревают с обратным холодильником в течение ночи. После охлаждения до комнатной температуры надосадочный раствор удаляют декантированием, и масляный остаток трижды размешивают с этанолом, каждый раз используя 50 мл этанола. Растворы объединяют и сгущают, и остаток поглощают в 500 мл воды и 500 мл метиленхлорида. Добавлением 1 н. соляной кислоты раствор доводят до значения рН 3,5, и затем органическую фазу удаляют, и водную фазу трижды экстрагируют метиленхлоридом, каждый раз используя 400 мл метиленхлорида. Объединенные органические фазы сушат над сульфатом натрия и сгущают. Остаток поглощают в 100 мл тетрагидрофурана, и раствор разбавляют добавлением 500 мл воды. Добавлением 1 н. натрового щелока раствор доводят до значения рН 7,5, тетрагидрофуран удаляют путем упаривания на ротационном испарителе, и остающийся водный раствор лиофилизуют.
Получают 142 г сырого продукта лиофилизации, который с целью обессоливания в 27 порциях по 5 г и 2 порциях по 3,5 г дальше очищают и обессоливают путем хроматографии на силикагеле величиной зерен 30 мкм (колонка длиной 40 см и диаметром 3 см, элюент: смесь ацетонитрила и воды в объемном соотношении 30:70). Все фракции объединяют, ацетонитрил упаривают на ротационном испарителе и водный остаток подвергают лиофилизации.
Выход 102 г (62,5% теории) целевого продукта, имеющего ту же характеристику, что и целевой продукт, полученный по варианту А.
Как уже указывалось, предлагаемое соединение проявляет намного лучшую активность в качестве ингибитора биосинтеза холестерина, чем известное рацемическое соединение формулы 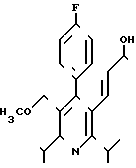

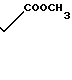 Для оценки данной активности определяют способность соединений к торможению редуктазы 3-окси-3-метил-глутарил-соэнзима А следующим образом.
Для оценки данной активности определяют способность соединений к торможению редуктазы 3-окси-3-метил-глутарил-соэнзима А следующим образом.
Самцов крыс массой 300-400 г в течение 11 дней кормили порошковым кормом типа альтромина, содержащим 40 г холестирамина на кг корма. Животным отрезали голову, затем удаляли почки, которые подали на лед. Почки измельчали и трижды гомогенизировали в 3 объемных частях буфера с рН 7,2, содержащего 0,1-молярную сахарозу, 0,05-молярный хлорид калия, 0,04-молярный бифосфат калия (смеси К2НРО4 и КН2РО4), 0,03-молярную этилендиаминтетрауксусную кислоту, 0,002-молярный дитиотреит. Затем подвергали центрифугированию в течение 15 мин и остаток удаляли. Надосадочную жидкость центрифугировали в течение 75 мин при 100000 g. Осадок поглощали в 1/4 части указанного буфера, повторно гомогенизировали и затем опять центрифугировали в течение 60 мин. Осадок поглощали в пятикратном объеме указанного буфера, гомогенизировали и для хранения замораживали при -78оС.
Для определения активности исследуемые соединения и стандартное вещество мевинолин растворяли в диметилформамиде при давлении 5 об.% 1 н.гидроокиси натрия и для осуществления опыта использовали по 10 мкл раствора, содержащего каждое вещество в разных концентрациях. Сперва соединения инкубировали вместе с энзимом при 37оС в течение 20 мин. Для осуществления опыта использовали смесь в количестве 0,380 мл, содержащую 4 мкмоль глюкоза-6-фосфата, 1,1 мг альбумина сыворотки крупного рогатого скота, 2,1 мкмоль дитиотреита, 0,35 мкмоль β-никотинамин-аденин-динуклеотид-фос-фата, 1 единицу глюкоза-6- фосфатдегидрогеназы, 35 мкмоль указанной смеси бифосфатов калия со значением рН 7,2, 20 мкл препарата энзима и 56 нмоль 3-окси-3-метилглутарил соэнзима А (глутарил-3-14С) с удельной активностью 100000 распадов в минуту.
Инкубировали в течение 60 мин при 37оС. Затем реакцию заканчивали добавлением 300 мкл 0,25-молярной соляной кислоты. По окончании дополнительной инкубации в течение 60 мин при 37оС центрифугировали, и 600 мкл надосадочной жидкости подают на колонку размером 0,7 х 4 см, наполненную 5-хлорид-анионитом величиной зерен 100-200 меш. Дополнительно промывали 2 мл дистиллированной воды, к продукту очистки и промывочной воде добавляли 3 мл сцинтилляционной жидкости и считывали в сцинтилляционном счетчике. Величины КТ50 (концентрация вещества, обеспечивающая 50%-ное торможение) определяли с помощью интраполяции на основе торможения в % и концентрации соединения. Для определения относительной ингибирующей активности величину КТ50 стандартного вещества мевинолина взяли за 100 и сравнивали с одновременно определяемой величиной КТ50 обоих исследуемых соединений. Таким образом определяли относительную активность обоих исследуемых соединений по сравнению со стандартным веществом, относительная активность которого равна 1. В результате данного опыта выявлено, что относительная активность предлагаемого соединения составляет 110, тогда как известное соединение проявляет относительную активность, равную 30.
С учетом результатов описанного опыта рекомендуется давать предлагаемое соединение в количестве 0,1 мкг/кг массы тела.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ HMG-COA РЕДУКТАЗЫ | 2003 |
|
RU2299196C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ МЕВАЛОНОВОЙ КИСЛОТЫ, ИНГИБИРУЮЩИХ ГМГ-СОА РЕДУКТАЗУ | 2003 |
|
RU2335500C2 |
| ПРОИЗВОДНЫЕ ПИРИДОБЕНЗОКСАДИАЗИНКАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ИСПОЛЬЗУЕМЫЕ ГИДРАТЫ И СОЛИ, А ТАКЖЕ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ НА ИХ ОСНОВЕ | 1995 |
|
RU2138504C1 |
| ПРОИЗВОДНЫЕ 7-ИЗОИНДОЛИНИЛХИНОЛОНА И 7-ИЗОИНДОЛИНИЛНАФТИРИДОНА | 1993 |
|
RU2111964C1 |
| ПРОИЗВОДНЫЕ ЦИКЛОАЛКАНО-ПИРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2194698C2 |
| ПРОИЗВОДНЫЕ ХИНОЛОН- И НАФТИРИДОНКАРБОНОВОЙ КИСЛОТЫ В ВИДЕ СМЕСИ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫХ ИЗОМЕРОВ, ИХ ГИДРАТЫ И СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2105770C1 |
| ПРОИЗВОДНЫЕ ПИРИДОБЕНЗОКСАДИАЗИНА, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ, ИХ ГИДРАТЫ И СОЛИ | 1994 |
|
RU2131430C1 |
| ПИПЕРИДИЛМЕТИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ХРОМАНА И ИХ СОЛИ С НЕОРГАНИЧЕСКИМИ КИСЛОТАМИ | 1992 |
|
RU2102392C1 |
| ПРОИЗВОДНЫЕ 1-АЛКИЛ-3,5-ДИАЦИЛ-1,4-ДИГИДРОПИРИДИНА В ФОРМЕ СМЕСИ ИХ ИЗОМЕРОВ | 1995 |
|
RU2158259C2 |
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ СОЕДИНЕНИЕ, ИМЕЮЩЕЕ АНГИОТЕНЗИН II АНТАГОНИСТИЧЕСКУЮ АКТИВНОСТЬ | 1997 |
|
RU2188013C2 |
Сущность изобретения: продукт - 3R,5S-(+)-7-/4-(4-фтофенил) -2,6-диизопропил-5-метоксиметил-пирид-3-ил/ -3,5-диоксигептеновая кислота. БФ C26H34NO5F . Выход 48% . Реагент 1: метиловый эфир эритро-(Е)-7-[4- (4-фторфенил)-2,6-диизопропил-5-5-метоксиметил -пиридид-3-ил] -3,5 -диоксигепт-6-еновой кислоты. Реагент 2: R-(+)-фенилэтиламин или S-(+)-фенилглицинол. Условия реакции: при нагревании в среде растворителя с последующим гидролизом в кислой среде.
3R,5S-(+)-7-[4-(4-ФТОРФЕНИЛ)-2,6- ДИИЗОПРОПИЛ-5-МЕТОКСИМЕТИЛ-ПИРИД-3-ИЛ] -3,5- ДИОКСИГЕПТЕНОВАЯ КИСЛОТА В ЭРИТРО-(E)-КОНФИГУРАЦИИ В ВИДЕ ФИЗИОЛОГИЧЕСКИ ПЕРЕНОСИМОЙ СОЛИ МЕТАЛЛА В КАЧЕСТВЕ ИНГИБИТОРА БИОСИНТЕЗА ХОЛЕСТЕРИНА.
3R, 5S-(+) -7-[4-(4-Фторфенил)-2,6-диизопропил-5-метоксиметил-пирид-3-ил] -3,5-диоксигептеновая кислота формулы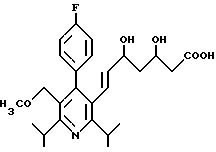
в эритро-(E)-конфигурации в виде физиологически переносимой соли металла в качестве ингибитора биосинтеза холестерина.
| 0 |
|
SU325130A1 | |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

