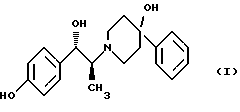
Изобретение относится к способу получения мезилат тригидрата соединения формулы (I), (1S,2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанола: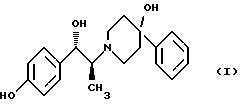
Данное изобретение относится также к способу получения (2S)-(+)-энантиомера формулы (II):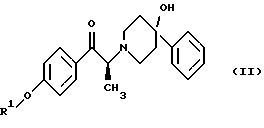
где R1 представляет собой защитную группу, выбранную из группы, включающей бензил, (C1-С6) алкилбензил, (C1-C6) алкоксибензил, три(C1-С6)алкилсилил, ацил (например, ацетил) и ароил (например, бензоат). Кроме того, данное изобретение относится к промежуточным продуктам, полезным в указанных способах.
Соединение формулы (I), (1S, 2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанол, проявляет значительную активность в качестве антагониста рецептора NMDA (N-метил-D-аспаргиновой кислоты) и пригодно для лечения эпилепсии, состояния страха, церебральной ишемии, мышечных спазм, деменции обширного инфаркта, травматического повреждения мозга, боли, слабоумия, вызванного СПИДом, гипогликемии, мигрени, амиотрофического латерального склероза, привыкания к чрезмерному применению лекарственных средств или хронического алкоголизма, синдрома отмены приема лекарственных средств или алкоголя, психотических состояний, недержания мочи и дегенеративных расстройств ЦНС (центральной нервной системы), таких как удар, болезнь Альцгеймера, болезнь Паркинсона и болезнь Хутингтона.
Мезилат тригидратная форма (1S, 2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанола по своим свойствам в качестве активного терапевтического средства превосходит безводный мезилат. Мезилат тригидрат имеет более стабильную кристаллическую форму, чем безводный мезилат, и, следовательно, существенно более длительный срок хранения. Тригидрат также менее подвержен разрушению кристаллической структуры благодаря включению воды в кристалл. В патенте США 6008233 описывается мезилат тригидрат, безводный мезилат и свободное основание - (1S, 2S)-1-(4-гидроксифенил)-2-(4-гидрок-си-4-фенилпиперидин-1-ил)-1-пропанол, а также способы их получения.
Кроме того, свободное основание формулы (I), его безводный мезилат и способы их получения описаны также в патенте США 5185343, выданном 9 февраля 1993 года. Их применение для лечения некоторых из указанных выше расстройств описано в патенте США 5272160, выданном 21 декабря 1993 года, и Международной заявке на патент PCT/IB 95/00380, которая разработана в США, заявлена 18 мая 1995 года и опубликована как WО 96/06081. Их применение в сочетании с соединением, способным повышать и таким образом восстанавливать баланс стимулирующей обратной связи от вентрального латерального ядра таламуса в кору для лечения болезни Паркинсона, описано в Международной заявке на патент PCT/IB 95/00398, которая разработана в США, подана 26 мая 1995 года и опубликована как WО 96/37226. Перечисленные выше патенты и заявки США включены в описание во всей полноте в качестве ссылок.
Разработанные ранее способы получения (1S, 2S)-1-(4- гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанола осуществлялись посредством рацемического синтеза с разделением оптически активных изомеров на стадиях, предшествующих получению терапевтической соли. Одна из проблем, связанная с разделением соединений на относительно более поздних стадиях схемы, состоит в потере и снижении эффективности, которая заключается в осаждении значительных количеств неактивных или менее активных энантиомеров и диастереомеров. Для достижения максимальной эффективности желательно разработать синтез, посредством которого вводятся центры оптической активности в предшественники целевых молекул в начале синтеза. Соответственно, способ превращения рацемического исходного вещества в оптически активный структурно-образующий блок для направления синтеза по хиральному пути с получением соединения формулы (I) был бы в значительной степени преимущественным.
Хотя способы асимметрической перегруппировки рацемических смесей в хиральные вещества был описан, возможность успешно получать оптически активные продукты зачастую строго ограничивается конкретными обстоятельствами и соединениями, которые участвуют в синтезе. Получение оптически активных α-аминопропиофенонов достигалось путем асимметрической перегруппировки. (Takamatsu, J. Pharm. Soc. Japan, 76(11), 1219-1222 (1956)). Кроме того, было описано превращение рацемического 3-(RS)-амино-1,3-дигидро-1-метил-5-фенил-2Н-1,4-бензодиазепин-2-она в его почти оптически чистый (S)-энантиомер посредством асимметрической перегруппировки, индуцируемой кристаллизацией (Reider et al., J. Org. Chem., 52, 955-957 (1987).
Данное изобретение относится к способу получения метансульфонатной соли, тригидрата (1S, 2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанола: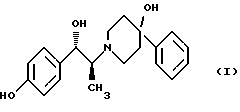
который включает следующие стадии:
(i) восстановление карбонильной группы соединения формулы (II):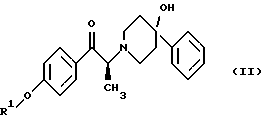
где R1 представляет собой защитную группу, выбранную из группы, включающей бензил, (C1-С6) алкилбензил, (C1-C6) алкоксибензил, три(C1-С6)алкилсилил, ацил (например, ацетил) и ароил (например, бензоат), посредством взаимодействия с боргидридом щелочного металла; и
(ii) удаление защитной группы R1 соединения формулы (III):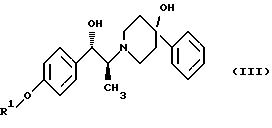
в присутствии метансульфоновой кислоты.
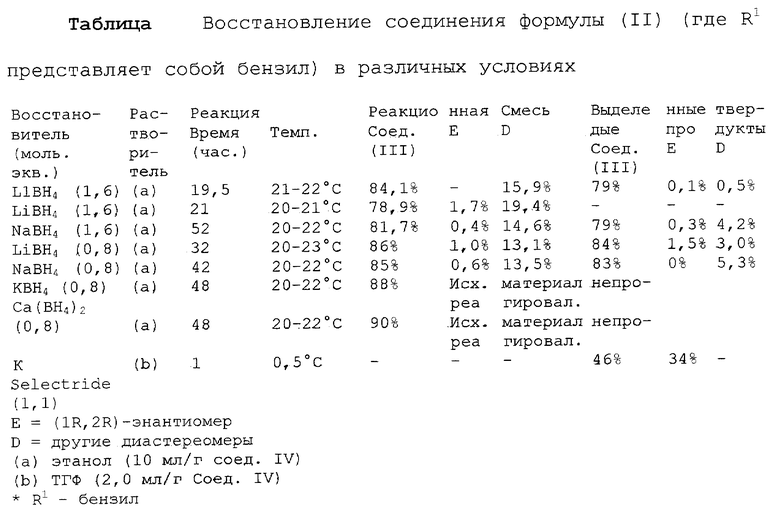
Предпочтительным воплощением данного изобретения является воплощение, в котором защитная группа R1 представляет собой бензил, (C1-С6) алкилбензил или (C1-C6) алкоксибензил. Другим предпочтительным воплощением данного изобретения является воплощение, в котором боргидрид щелочного металла представляет собой боргидрид лития или боргидрид натрия. Более предпочтительным воплощением данного изобретения является воплощение, в котором R1 представляет собой бензил и боргидрид щелочного металла представляет собой боргидрид лития.
Еще одним предпочтительным воплощением данного изобретения является воплощение, в котором защитная группа R1 представляет собой бензил и удаление защитной группы на стадии (ii) представляет собой гидрогенолиз, осуществляемый в присутствии газообразного водорода и 5-20% палладия на углероде. Более предпочтительным воплощением данного изобретения является воплощение, в котором группа R1 представляет собой бензил и гидрогенолиз проводится в присутствии газообразного водорода и 5% палладия на углероде. Предпочтительным является воплощение изобретения, в котором стадии (i) и (ii) проводятся в (C1-C6)алканольном растворителе, необязательно смешанном с водой. Более предпочтительным является воплощение изобретения, в котором растворитель, используемый на стадиях (i) и (ii), представляет собой этанол, смешанный с водой.
Изобретение также относится к способу получения соединения формулы (II):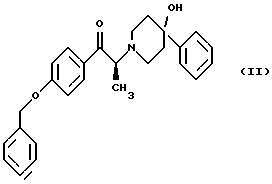
включающему следующие стадии
(i) обработку соединения формулы (IV)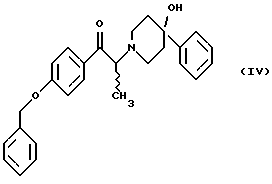
диароил-D-тapтpaтoм;
(ii) обработка продукта стадии (i) - D-тартратной соли слабым основанием.
Термин "слабое основание" в данном описании относится к основному соединению, основность которого недостаточна для удаления α-протона из соединения формулы (IV). Предпочтительным воплощением данного изобретения является воплощение, в котором стадии данного способа проводятся в низшем алкилкетоне в качестве растворителя, более предпочтительно - в ацетоне. Более предпочтительным воплощением изобретения является воплощение, в котором стадии данного способа проводятся в ацетоне при температуре в интервале от 25oС до температуры кипения, более предпочтительно в интервале от 48 до 52oС.
Предпочтительным воплощением данного изобретения является воплощение, в которым слабое основание представляет собой три (C1-C6)алкиламин или карбонат, бикарбонат или алкилкарбоксилат щелочного/щелочноземельного металла, например, NаНСО3, NaCO3, NаООССН3 и т.д. Более предпочтительно, когда слабым основанием является водный раствор NаНСО3, смешанный с органическим растворителем, таким как этилацетат или метиленхлорид, более предпочтителен, этилацетат.
Данное изобретение относится также к (2S)-(+)-энантиомеру формулы (II):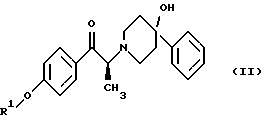
или его соли, где R1 представляет собой водород или защитную группу, выбранную из группы, включающей бензил, (C1-С6) алкилбензил, (C1-С6) алкоксибензил, три (C1-C6) алкилсилил, ацил (например, ацетил) и ароил (например, бензоат) и соль представляет собой диароил-D-тартрат. Предпочтительным воплощением данного изобретения является воплощение, в котором R1 представляет собой бензил. Другим предпочтительным воплощением данного изобретения является воплощение, в котором диароильная соль представляет собой дибензоил-D-тартрат или ди-п-толуоил-D-тартрат.
Подробное описание изобретения
Мезилат тригидрат (1S, 2S)-1-(4-гидроксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-1-пропанола представляет собой твердое белое кристаллическое вещество, которое имеет единственную кристаллическую форму и хорошо растворимо в воде (25 и 15 мг/мл в буферных растворах с рН 3 и 7 соответственно). Мезилат тригидрат, как известно, образуется из безводного мезилата как равновесная форма при относительной влажности окружающей среды 81%. Ранее при получении мезилат тригидрата перед его образованием, требовалось разделение рацемата threo-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанола. В указанной методике было необходимо удаление наименее активного/неактивного (1R, 2R)-изомера после разделения.
Данное изобретение позволяет получать мезилат тригидрат соединения формулы (I) введением хирального центра во 2 положение пропанольной цепи конечного продукта на более ранней стадии синтеза, чем в прежней методике синтеза мезилат тригидрата. Такое введение хирального центра на более ранней стадии приводит к большей эффективности и получению с более высоким выходом мезилат тригидрата без значительного образования энантиомерных и диастереомерных примесей.
Приведенная далее схема 1 синтеза (см. в конце описания) иллюстрирует способ данного изобретения. Если не указано других значений, R1 принимает значения, указанные выше.
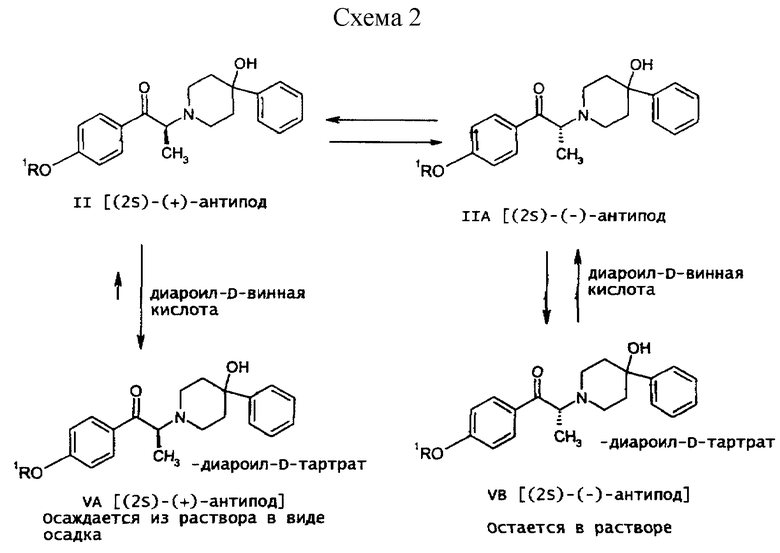
Как показано на схеме 1, рацемическое соединение формулы (IV), содержащее защитную группу, подвергается превращению посредством асимметрической перегруппировки, индуцируемой кристаллизацией, в диароил-D-тартратную соль (2S)-соединения формулы (VA), где ароил представляет собой бензоил или п-толуил. Кислотность α-протона позволяет хиральному центру рацемизироваться и устанавливает равновесие между (2S)-соединением и его (2R)-антиподом, как показано на схеме 2 (см. в конце описания). Как видно на схеме 2, представленной ниже, в присутствии диароил-D-винной кислоты кристаллическая диароил-D-тартратная соль (2S)-(+)-соединения формулы (VA) выводится из равновесного состояния вследствие ее относительной нерастворимости, смещая равновесие так, что (2R)-(-)-антипод в конечном счете трансформируется в желаемую (2S)-(+)-форму.
Такая асимметрическая перегруппировка, индуцируемая кристаллизацией, наилучшим образом достигается в растворителях, таких как низшие алкилкетоны растворители, например, ацетон. Оптимально данная стадия проводится при нагревании раствора соединения формулы (IV) и дибензоил-D-винной кислоты в ацетоне в инертной атмосфере в течение 6-7 часов при температуре от 48 до 52oС с охлаждением до комнатной температуры (20-25oС), гранулированием полученной суспензии, фильтрованием и последующей сушкой полученной соли.
Как показано на схеме 1, диароил-D-тартратная соль (2S)-энантиомера формулы (VA) затем подвергается обработке основанием, предпочтительно водным бикарбонатом натрия, в присутствии этилацетата или метиленхлорида, предпочтительно - этилацетата. После этого органическую фракцию отделяют, затем концентрируют, добавляют к охлажденным гексанам и затем гранулируют получая соединение формулы (II) в виде свободного основания.
После этого соединение формулы (II) подвергают обработке, в результате которой карбонильный фрагмент восстанавливается без сопутствующей рацемизации в α-положении. Это может достигаться при выдерживании соединения формулы (II) в условиях умеренного восстановления, например, при обработке боргидридом щелочного металла, таким как боргидрид лития или боргидрид натрия, предпочтительно боргидрид лития, в растворителе, таком как тетрагидрофуран или этанол, предпочтительно этанол. Сравнение различных условий восстановления показано в таблице 1.
Защитная группа R1 продукта - соединения формулы (III) затем наилучшим образом удаляется с помощью гидрогенолиза, если эта защитная группа представляет собой бензил, (С1-С6)алкилбензил или (C1-С6)алкоксибензил. Когда защитная группа R1 представляет собой три (C1-C6) алкилсилил, ацил (например, ацетил) или ароил (например, бензоат), она может удаляться стандартными методами, известными в области химии, то есть обработкой ионом фтора для удаления силильной группы или гидролизом для отщепления ацил/ароил-сложного эфира.
Когда R1 представляет собой бензил, то такая защитная группа эффективно удаляется при использовании газообразного водорода с 5-20% палладием на углероде в подходящем растворителе, таком как тетрагидрофуран, для получения свободного основания. Однако реакция гидрогенолиза может проводиться в присутствии или отсутствии метансульфоновой кислоты в зависимости от того, является ли целевой продукт свободным основанием или мезилатной солью. В присутствии метансульфоновой кислоты реакция гидрогенолиза проводится в (C1-С6)алканоле, необязательно смешанном с водой, предпочтительно в водном этаноле, и мезилатная соль образуется сразу. В ходе реакции в концентрированный фильтрат реакционной смеси гидрогенолиза также может добавляться вода с последующей фильтрацией с выделением мезилат тригидрата соединения формулы (I) в качестве конечного продукта. Если используются условия, обеспечивающие низкое содержание пирогена или его отсутствие, выделенная соль - мезилат тригидрат пригодна для применения при парентеральном введении.
Если удаление защитной группы гидрогенолизом проводится в отсутствие мезилат тригидрата, реакция может проводиться в менее полярном растворителе, например, в тетрагидрофуране, для получения свободного основания. При необходимости, получение мезилат тригидрата может осуществляться на отдельной стадии, исходя из свободного основания.
Соль мезилат тригидрат, так же как и безводный мезилат и свободное основание, обладает селективной нейрозащитной активностью, которая основана на ее антиишемической активности и способности блокировать возбуждающие аминокислотные рецепторы. Предпочтительная методика оценки нейрозащитной активности данного соединения описана в публикации Ismail A. Shalaby, et аl., J. Pharm. Exper. Ther., 260, 925 (1992). Данная статья приведена в описании в качестве ссылки, а методика описана ниже.
Культура клеток. Клетки гиппокампа 17-дневного плода крысы (CD, Charles River Breeding Laboratories, Inc., Wilmington, Mass.) выращивают на культуральных планшетах PRIMARIA (Falcon Co., Lincoln Park, N.J.) в течение 2-3 недель в сыворотке, содержащей культуральную среду (минимальная незаменимая среда (minimum essential medium) с заменимыми аминокислотами, содержащая 2 мМ глутамина, 21 мМ глюкозы, пенициллин/стрептомицин (5000 Ед. каждого), 10% фетальную телячью сыворотку (дни 1-7), и 10% сыворотку лошади (дни 1-21). Клетки помещают либо на 96-луночные титрационные микропланшеты с плотностью 80000 клеток на ячейку, либо на 24-луночные культуральные планшеты с плотностью 250000 клеток на ячейку. Культуры выращивают при 37oС в гумифицированном СО2 инкубаторе культур ткани, содержащем 5% СО2/95% воздуха. Пролиферацию ненейронных клеток контролируют добавлением 20 мкМ уридина и 20 мкМ 5-фтор-2-деоксиуридина (Sigma Chemical Co. , St. Louis Mo.) в 6-8 дневную культуру. Культуральную среду заменяют каждые 2-3 дня свежей биомассой.
Глутаматная токсичность. Культуры испытывают на активность в отношении глутамата через 2-3 недели после начального посева. Культуральную среду удаляют и культуры дважды промывают CSS (миллимоли): NaCl, 12; КС1, MgCl2, 0,8; ' CaCl2, 1,8; глюкоза, 15; 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота, 25 мМ (рН 7,4). Затем культуры выдерживают в течение 15 минут (37oС) в растворах глутамата различных концентраций. После инкубирования культуры 3 раза промывают не содержащим глутамата CSS и дважды свежей культуральной средой без сыворотки. Затем культуры инкубируют в течение 20-24 часов в не содержащей сыворотки культуральной среде. Испытываемые соединения добавляют за 2 минуты до и во время 15-минутной экспозиции в глутамате. В некоторых экспериментах соединение добавляют в разное время после экспозиции в глутамате и в течение следующих 20-24 часов.
Жизнеспособность клеток обычно оценивают через 20-24 часа после экспозиции, токсической для стимулирующего действия, измерением активности цитозольного фермента LDH. LDH активность определяют из культуральной среды каждой из 96 лунок титрационных микропланшетов. Образец 50 мкл среды добавляют к равному объему фосфатно-натриевого буфера (0,1 М, рН 7,4), содержащего 1,32 мМ пирувата натрия и 2,9 мМ NADH. Оптическую плотность всей реакционной массы при 340 нм для каждой из 96 лунок определяют каждые 5 секунд в течение 2 минут с помощью автоматического микротитровального планшета-ридера (Molecular Devices; Menio Park, Calif.). Скорость абсорбции определяют автоматически с помощью программы IBM SOFTmax (версия 1,01; Molecular Devices) и используют в качестве показателя LDH активности.
Морфологическое определение нейронной жизнеспособности осуществляют с использованием фазово-контрастной микроскопии. 96-луночные культуральные планшеты не позволяют получить хорошее фазово-контрастное изображение, поэтому клетки выращивают на 24-луночных планшетах. Количественно оба вида культуральных планшетов являются в равной степени чувствительными к глутаматной токсичности и показывают 2-3-кратное повышение LDH активности через 24 часа после экспозиции в 0,1-1,0 мМ глутамате.
Реагенгы. DTG может закупаться в Aldrich Chemical Company (Milwaukew, Wis.), а галоперидол - в Research Biochemicals Inc. (Natick, Mass.). Спермин может закупаться в Sigma Chemical Co. (St. Louis, Mo.). Сыворотка лошади и фетальная телячья сыворотка могут закупаться в Hylone (Logan, Utah). Культуральная среда, глутамин и пенициллин/стрептомицин могут закупаться в Gibco Co. (Grand Island, N.Y.).
Анализ данных. Нейротоксичность может количественно оцениваться измерением активности LDH, присутствующей в культуральной среде через 20-24 часа после экспозиции в глутамате. Повышенная активность LDH в культуральной среде коррелирует с деструкцией и дегенерацией нейронов (Koh and Choi. 1987). Поскольку фактические содержания LDH изменяются в зависимости от различных культур, данные обычно выражают относительно обработанных буфером соседних ячеек одного культурального планшета. Для получения показателя активности LDH глутаматной и обработанной лекарством культур значения LDH контрольных культур вычитают из значений обработанных культур. Данные обработок лекарственными средствами выражают в виде процента повышения LDH, индуцированной 1 мМ глутамата (или NMDA) для каждого эксперимента. Концентрации NVDA антагонистов, необходимые для возврата 50% повышения LDH индуцированного токсинами возбуждения (IC50), вычисляют с использованием log-пробит анализа из объединенных результатов трех независимых экспериментов.
Селективная нейрозащитная антиишемическая активность и активность блокирования возбуждающей аминокислоты мезилат тригидрата согласно данному изобретению делает ее полезной для лечения расстройств, выбранных из дегенеративных расстройств ЦНС, таких как удар, болезнь Альцгеймера, болезнь Паркинсона и болезнь Хутингтона; эпилепсии, состояния страха, церебральной ишемии, мышечных спазм, деменции обширного инфаркта, травматического повреждения мозга, боли, слабоумия, вызванного СПИДом, гипогликемии, мигрени, амиотрофического латерального склероза, привыкания к чрезмерному применению лекарственных средств или хронического алкоголизма, синдрома отмены приема лекарственных средств или алкоголя, психотических состояний и недержания мочи.
В системном лечении таких расстройств дозировка обычно составляет от примерно 0,02 до 250 мг на кг массы тела в день (0,001-12,5 г в день при обычной массе тела человека 50 кг) в одной или нескольких дозах, независимо от способа введения, Более предпочтительная дозировка составляет от примерно 0,15 мг на кг массы в день до примерно 250 мг на кг массы в день. Вместе с тем, в зависимости от точной природы заболевания и состояния пациента лечащим врачом могут назначаться дозы, выходящие за указанные пределы. Пероральный способ введения обычно предпочтителен. Однако в том случае, когда пациент не способен проглатывать или пероральная абсорбция ослаблена иным образом, предпочтительным способом введения будет парентеральное (внутримышечное, внутривенное) или местное.
Мезилат тригидрат может вводиться в форме фармацевтических композиций вместе с фармацевтически приемлемым носителем или разбавителем. Такие композиции обычно приготавливают стандартным образом с использованием твердых или жидких наполнителей или разбавителей в зависимости от способа желательного введения: для перорального введения - в форме таблеток, твердых или мягких желатиновых капсул, суспензий, гранул, порошков и т.п.; для парентерального введения - в форме растворов или суспензий для инъекций и т.п.; и для местного введения - в форме растворов, лосьонов, мазей, бальзамов и т.п.
Приведенные далее примеры иллюстрируют способы данного изобретения и получение соединений изобретения. Температуры плавления не корректировались. Данные ЯМР приводятся в миллионных долях (5) и относятся к дейтерий-запирающему сигналу (deuterium lock signal) растворителя образца (дейтерохлороформ, если не указано, другого растворителя). Коммерческие реагенты использовались без дополнительной очистки.
Пример 1
(23)-1-(4-Бензилоксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-1-пропанон, дибензоил-D-тартратная соль
Рацемический 1-(4-бензилоксифенил)-2-(4-гидрокси-4-фенил-пиперидин- 1-ил)-1-пропанон (100 г, 0,24 моль) и дибензоил-D-винную кислоту (86,3 г, 0,24 моль) добавляют в ацетон (1,5 л) под атмосферой азота с получением желтоватого раствора. После выдерживания раствора в течение 1 часа при температуре 48-52oС образуется густая белая суспензия. Суспензию выдерживаю при указанной температуре в течение дополнительных 6,5 часов и затем охлаждают до 20-25oС. Твердое вещество гранулируют в течение 1 часа при 20-25oС, фильтруют и после этого осадок на фильтре промывают свежеперегнанным ацетоном (0,2 л). Твердое белое вещество сушат в вакууме в течение 12-15 часов при 35-40oС, получают 155,6 г указанного в заглавии соединения (выход 84%), т.пл. 140, 1-141, 1oС; [α]D 25+65,4 (с 4,5; СН3ОН). Хиральная ВЭЖХ показывает, что соль содержит 0,9% (-)энантиомера, (2R)-1-(4-бензилоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанона.
Пример 2
(2S)-1-(4-Бензилоксифенил)-2-(4-гидрокси-4-фенилпипери-дин-1-ил)-1-пропанон
Под атмосферой азота дибензоил-D-тартратную соль (2S)-1-(4-бензилоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанона (150,0 г, 0,19 моль) суспендируют в этилацетате (0,45 л, 3,0 мл/г тартратной соли) и воде (0,75 л, 50 мл/г тартратной соли), содержащей NаНСО3 (51/0 г, 0,61 моль). Смесь перемешивают в течение 2 часов при 20-25oС с выделением CO2(рНf 8,1). Перемешивание прекращают и смесь оставляют для разделения на прозрачные слои. Нижний водный слой отделяют и затем этилацетатный слой концентрируют до 0,1 л при 25-30oС при пониженном давлении. Концентрат медленно, в течение 2 часов, добавляют в гексан (0,5 л), охлажденные до 15-20oС. Суспензию концентрируют до 0,4 л, твердый продукт гранулируют в течение 1 часа при 15-20oС, фильтруют и затем дополнительно промывают гексанами (80 мл). (2S)-1-(4-бензилоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанон сушат в вакууме в течение 12 часов при 40-45oС с получением 77,8 г свободного основания в виде белого вещества (выход 96,7%). Т.пл. 102,5-103,8oС; [α] D 25+18,9 (с 8,9, СН3ОН). 1H ЯМР (СDСl3) δ 8,13 (д, J=8,7 Гц, 2Н), 7,2-7,4 (м, 10Н), 7,00 (д, J=8,7 Гц, 2Н), 5,13 (с, 2Н), 4,11 (кв. J=6,8 Гц, 1Н), 2,6-2,9 (м, 2Н), 2,0-2,2 (м, 2Н), 1,7 - 1,8 (м, 2Н), 1,31 (д, J=6,8 Гц, 3Н). 13С ЯМР (CDCl3) δ 199,69, 162,75, 136,47, 131,49, 129,72, 128,96, 128,55, 128,50, 127,77, 127,23, 124,80, 114,58, 71,44, 70,34, 64,78, 47,83, 44,62, 39,14, 38,79 и 12,28. Хиральная ВЭЖХ показывает, что (-)-энантиомер, (2R)-1-(4-бензилоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанон, присутствует в количестве 1,2%.
Пример 3
(1S, 2S)-1-(4-бензилоксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-1-пропанол
В течение 20 минут (2S)-1-(4-бензилоксифенил)-2- (4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанон (75 г, 0,18 моль) добавляют к суспензии боргидрида лития (3,15 г, 0,15 моль) в этаноле (0,75 л) под атмосферой азота при 20-25oС. После перемешивания в течение примерно 5 минут выделяется небольшое количество теплоты, что приводит к повышению температуры до 27oС. Суспензию перемешивают в течение 42 часов при 20-25oС, когда ВЭЖХ показывает, что реакция завершена. Добавляют воду (37,5 мл) и суспензию гранулируют в течение 1 часа при 20-25oС. Твердый белый продукт фильтруют и затем промывают этанолом (75 мл), водой (150 мл) и наконец этанолом (75 мл). Продукт сушат в вакууме при 40-45oС в течение 20 часов, получают 65,3 г указанного в заглавии соединения. Выход аминоспиртового продукта составляет 78,3%, и продукт содержит только 2,3% диастереомеров. Т. пл. 158-161oС, [α]D 25+38,7 (с 6,1, СН3ОН).
Пример 4
Метансульфонат (1S, 2SR)-1-(4-гидроксифенил)-2-(4- гидрокси-4-фенилпиперидино)-1-пропанола, тригидрат
Катализатор - пятипроцентный палладий на углероде (0,75 г, 50% водн. вл. ), (1S, 2S)-l-(4-бензилоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанол (5,0 г, 12,0 ммоль), этанол (62,5 мл) и метансульфоновую кислоту (1,15 г, 12,0 ммоль) соединяют в реакторе Парра под атмосферой азота под давлением. Атмосферу азота замещают водородом (3•35 фунтов/кв.дюйм=3•241,32 кПа) и затем давление водорода повышают до 50-55 фунтов/кв.дюйм (344,74-379,21 кПа). Смесь нагревают и перемешивают при 50-55oС в течение 5 часов, когда ВЭЖХ показывает, что реакция завершена. Газообразный водород медленно стравливают, реактор продувают азотом и затем теплую (50oС) реакционную смесь фильтруют через целит. Осадок на целитном фильтре промывают этанолом (5 мл). Объединенные промывной раствор и фильтрат концентрируют в вакууме до 10 мл. Добавляют воду (17/5 мл) и раствор концентрируют при атмосферном давлении до достижения температуры дистиллята 76oС. Прозрачный раствор медленно охлаждают в течение часа до 15-20oС и затем охлаждают до 0-5oС. После гранулирования в течение 1 часа при 0-5oС густую суспензию фильтруют и осадок на фильтре промывают холодной водой (5oС, 2,5 мл). Твердый продукт сушат в течение 18 часов при 20-25oС, получают 4,71 г указанного в заглавии соединения с 83% выходом. Продукт идентичен подлинному образцу указанного в заглавии соединения. Если в описанной выше методике применяются вода с низким содержанием пирогенного препарата и условия, обеспечивающие отсутствие пирогенной воды, выделенное соединение, указанное в заголовке, подходит для парентеральных применений.

Изобретение относится к новому способу получения мезилат тригидрада соединения формулы (I), (1S, 2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-1-пропанола: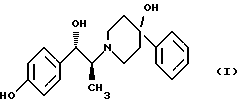
восстановлением карбонильной группы (2S)-соединения формулы (II)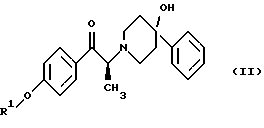
где R1 представляет собой защитную группу, с последующим удалением защитной группы R1 из восстановленного соединения. Изобретение также относится к новым соединениям формулы (II) и способу их получения. 4 с. и 15 з. п.ф-лы, 1 табл.
включающий следующие стадии:
(i) восстановление карбонильной группы (2S)-соединения формулы (II)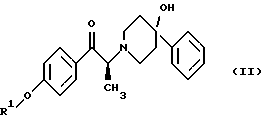
где R1 представляет собой защитную группу, выбранную из группы, включающей бензил, (С1-С6)алкилбензил, (C1-С6)алкокси-бензил, три(C1-С6)алкилсилил, ацил и ароил, посредством взаимодействия с боргидридом щелочного металла; и
(ii) удаление защитной группы R1 (2S)-соединения формулы (III)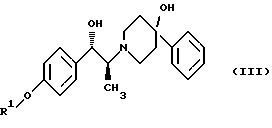
в присутствии метансульфоновой кислоты.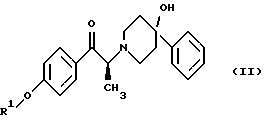
где R1 представляет собой защитную группу, выбранную из группы, включающей бензил, (C1-C6)алкилбензил, (C1-C6)алкоксибензил, три(C1-C6)алкилсилил, ацил и ароил; включающий следующие стадии:
(i) смешение соединения формулы (IV)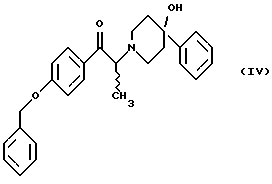
с диароил-D-тартратным соединением;
(ii) обработку продукта стадии (i) D-тартрата (2S)-энантиомера слабым основанием.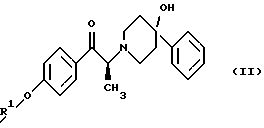
или его соль,
где R1 представляет собой защитную группу, выбранную из группы, включающей бензил, (C1-С6)алкилбензил, (C1-C6) алкоксибензил, три(C1-C6)алкилсилил, ацил и ароил, и соль представляет собой диароил-D-тартрат.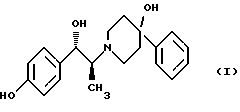
включающий следующие стадии:
(i) смешение соединения формулы (IV)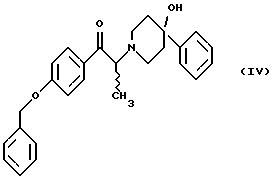
с диароил-D-тартратным соединением, выбранным из группы, включающей дибензоил-D-тартрат и ди-п-толуоил-D-тартрат;
(ii) обработку продукта стадии (i) D-тартратной соли слабым основанием для получения соединения формулы (II)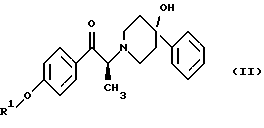
(iii) восстановление карбонильной группы соединения формулы (II) посредством взаимодействия с боргидридом щелочного металла; и
(iv) удаление защитной группы R1 соединения формулы (III)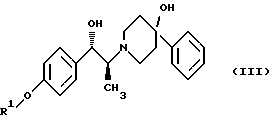
в присутствии метансульфоновой кислоты.
| US 6008233 A, 28.12.1999 | |||
| US 5272160 A, 21.12.1993 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Способ получения 4-фенил-4-пиперидинола | 1983 |
|
SU1109390A1 |

