Предпосылки к созданию изобретения
Изобретение относится к области лечения болезни Паркинсона (также называемой паркинсонизмом). Конкретно, это изобретение относится к лечению млекопитающих, страдающих паркинсонизмом, которое включает в себя введение указанному млекопитающему синергического количества селективного в отношении переднего мозга антагониста N-метил-D-аспартата (далее называемого NMDA) и соединения, способного усиливать возбуждающую обратную связь между вентральным латеральным ядром таламуса и корой таким образом, что баланс возбуждающей обратной связи от вентрального латерального ядра таламуса к коре у указанного млекопитающего, страдающего болезнью Паркинсона, восстанавливается.
Паркинсонизм - это постепенно развивающееся заболевание, которое характеризуется симптомами, включающими прогрессирующий тремор, брадикинезию и ригидность. Это заболевание может окончиться смертью больного в течение 5-10 лет от его начала (появлении симптомов). Основополагающей причиной болезни Паркинсона является дегенерация дофаминергических нейронов нигростриарного пути. Эти дофаминергические нейроны являются частью цепи нейрональной обратной связи, контролирующей двигательную функцию. Дегенерация этих нейронов приводит к токсическим изменениям активности нескольких нейрональных путей этой двигательной цепи. Следствием этих изменений является значительное ослабление обратной связи между вентролатеральным таламусом и премоторной зоной коры. Эта потеря обратной связи и порождает двигательные симптомы названного заболевания.
В настоящее время единственным успешным лечением паркинсонизма является терапия леводопой. Леводопа является предшественником дофамина. Лечение этим агентом частично восстанавливает дефицит дофамина в полосатом теле, который образуется вследствие дегенерации нигростриарных дофаминергических нейронов. Применение леводопы значительно улучшало качество жизни и предполагаемую продолжительность жизни пациентов, страдающих паркинсонизмом. Тем не менее применение леводопы для лечения паркинсонизма имеет ряд недостатков. В настоящее время наиболее часто леводопу назначают с ингибитором декарбоксилазы леводопы, таким как карбидопа или бензеразид. Это предотвращает превращение леводопы в дофамин вне центральной нервной системы и дополнительно улучшает соотношение между благоприятными и побочными эффектами этой терапии. Тем не менее терапия леводопой лечит только симптомы болезни, но не замедляет и не предотвращает прогрессирование заболевания. Помимо этого благоприятное влияние лечения леводопой на двигательные симптомы заболевания уменьшается в течение нескольких лет от начала лечения. Это снижение эффективности также часто сопровождается развитием тяжелых дискинезий и индуцированных леводопой спутанности сознания, галлюцинаций, паранойи и делирия. Очевидно, что существует потребность в новой терапевтической стратегии, которая обеспечит усиленное лечение симптомов паркинсонизма и в то же время уменьшит побочные эффекты, которые лимитируют лечение в настоящее время.
Рассматриваются и другие подходы к лечению болезни Паркинсона. Некоторые из этих подходов ставят своей целью замещение потери дофаминовой функции полосатого тела. Это замещение включает: агонисты дофаминовых рецепторов, включая агонисты дофаминовых рецепторов D1, агонисты дофаминовых рецепторов D2, агонисты дофаминовых рецепторов D5 и агонисты дофаминовых/опиатных рецепторов, ингибиторы поглощения дофамина, стимуляторы тирозин-гидроксилазы, ингибиторы моноаминоксидазы и ингибиторы моноаминооксидазы-B, а также ингибиторы КОМТ. Другие обсуждаемые способы лечения направлены на восстановление обратной связи между вентролатеральным таламусом и премоторной корой с помощью ряда механизмов. Это включает: антагонисты АМФК, агонисты ГАМК, агонисты аминергических рецепторов, антагонисты мускариновых рецепторов, аденозинрегулирующие агенты, антагонисты опиатных рецепторов, стимуляторы ЛДГ, агонисты рецепторов ССК, антагонисты рецепторов ССК, агонисты адренорецепторов, антагонисты ИЛ-1, факторы роста, противовоспалительные агенты, антиоксиданты, иммуностимуляторы, ингибиторы обратного захвата беротонина и ингибиторы обратного захвата аминов. Однако в то время как некоторые из этих подходов демонстрируют обнадеживающие результаты в экспериментах на животных, ни один из этих способов лечения не доказал свою эффективность для лечения болезни Паркинсона человека.
Chenard в патентах США NN 5185343, 5272160 и 5338754, включенных в настоящий документ в качестве ссылок, указывает, что соединения формул
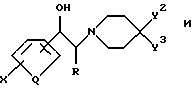
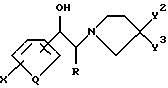
являются антагонистами NMDA и в качестве таковых находят применение для лечения болезни Паркинсона.
Butler в патенте США N 5356905, также включенном в настоящий документ в качестве ссылки, указывает, что соединения формулы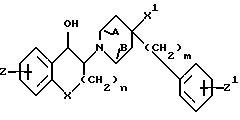
являются антагонистами NMDA и в качестве таковых находят применение для лечения болезни Паркинсона.
При скрининге, описанном Williams и др. Neuron 10, 267-278 (1993), антагонисты NMDA, описанные Butler и Chenard, оказались селективными для NR2B подтипа антагонистами NMDA. Специалисты хорошо осведомлены, что обозначения "NR2A", "NR2B", "NR2C" и "NR2D" используются для описания рецепторов NMDA у крыс и что подобные рецепторы существуют у других млекопитающих, включая человека, которые называются по другому.
Настоящее изобретение утверждает, что селективные в отношении переднего мозга антагонисты NMDA, которые являются селективными для рецепторов, содержащих субъединицу NR2B, также синергически взаимодействуют с леводопой в реверсировании двигательных дефицитов на экспериментальных животных с моделью болезни Паркинсона. Рецептор NMDA состоит из субъединицы NR1 в комбинации с одной или более субъединицей NR2, NR2A, NR2A, NR2B, NR2C или NR2D (Monyer и др., Science, 256, 1217-1221 (1992)). NR2B-селективный антагонист NMDA является агентом, ингибирующим функцию рецепторов NMDA, содержащих субъединицу NR2B, но менее эффективным в отношении рецепторов NMDA, не имеющих этой субъединицы. В настоящее время сообщается о том, что только неселективные антагонисты NMDA действуют синергически с леводопой на экспериментальных животных с моделью болезни Паркинсона. Примерами неселективных антагонистов NMDA являются MK801, CGS-19,755 и CNS-1102. Настоящее изобретение утверждает, что именно NR2B-содержащие рецепторы NMDA являются важным местом действия этих неселективных агентов.
Побочные эффекты, которые наблюдаются при лечении некоторыми антагонистами NMDA, не встречаются или значительно уменьшены при использовании селективных антагонистов NMDA настоящего изобретения. Таким образом, настоящее изобретение позволяет использовать полный терапевтический потенциал леводопы посредством применения в синергической комбинации с ней антагонистов NMDA.
Сущность изобретения
Настоящее изобретение относится к способу лечения болезни Паркинсона у млекопитающих, который включает в себя введение указанному млекопитающему эффективного для лечения болезни Паркинсона количества комбинации селективного в отношении переднего мозга антагониста N-метил-D-аспартата (NMDA) и агента, способного усиливать возбуждающую обратную связь между вентральным латеральным ядром таламуса и корой (агента, усиливающего возбуждающую обратную связь).
Предпочтительным способом в объеме настоящего изобретения является способ лечения болезни Паркинсона у млекопитающих, которые страдают болезнью Паркинсона, как описано в предыдущем параграфе, при котором указанный агент, способный усиливать возбуждающую обратную связь между вентральным латеральным ядром таламуса и корой, выбирают из группы, содержащей агонисты дофамина, агонисты дофамина D1, агонисты дофамина D2, агонисты дофамин/β-адренергических рецепторов, агонисты дофамин-ингибитор поглощения 5-HT/5-HT-1A, дофамин/опиатные агонисты, агонисты адренорецепторов, антагонисты α 2-адренорецепторов/дофамина, агонисты α 2-адренорецепторов/дофамина D2, ингибиторы поглощения дофамина, ингибиторы моноаминооксидазы-В, ингибиторы КОМТ и леводопы.
Более предпочтительным способом в объеме настоящего изобретения является способ, описанный в предыдущем параграфе, в котором названный селективный в отношении переднего мозга антагонист NMDA является селективным в отношении подтипа NR2B антагонистом NMDA.
Еще более предпочтительным способом в объеме настоящего изобретения является способ, описанный в предыдущем параграфе, в котором названный агент, усиливающий возбуждающую обратную связь, является леводопой.
Еще более предпочтительным способом в объеме настоящего изобретения является способ, описанный в предыдущем параграфе, который дополнительно включает лечение названного млекопитающего ингибитором декарбоксилазы леводопы.
Еще более предпочтительным способом в объеме настоящего изобретения является способ, описанный в предыдущем параграфе, в котором названный селективный в отношении подтипа NR2B антагонист NMDA является соединением формулы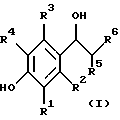
или его фармацевтически приемлемой солью присоединения кислоты, где:
(a) R2 и R5 берутся раздельно и R1, R2, R3 и R4, каждый независимо представляют водород, (C1-C6)алкил, галоген, CF3, OH или OR7, а R5 представляет метил или этил, или
(b) R2 и R5 берутся вместе и представляют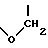
образуя хроман-4-оловое кольцо, а R1, R3 и R4, каждый независимо представляют водород, (C1-C6)алкил, галоген, CF3, OH или OR7,
R6 представляет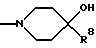
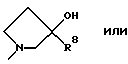
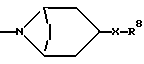
R7 представляет метил, этил, изопропил или н-пропил,
R8 представляет фенил, необязательно замещенный до трех заместителями, независимо выбранными из группы, состоящей из (C1-C6)алкила, галогена и CF3,
X представляет O, S или (CH2)n, и
n равно 0, 1, 2 или 3.
Особенно предпочтительными в объеме настоящего изобретения, как описано в предыдущем параграфе, являются три соединения. Этими соединениями являются: (+)-(1S,2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанол, (1S, 2S)-1-(4-гидрокси-3-метоксифенил)- 2-(4-гидрокси-4-фенилпиперидино)-1-пропанол и (3R, 4S)-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4,7-диол.
Особенно предпочтительный способ в объеме предыдущего параграфа включает в себя введение ингибитора декарбоксилазы леводопы, карбидопы, с одним из особенно предпочтительных соединений.
Также в объем настоящего изобретения входят фармацевтические композиции, включающие в себя эффективное для лечения болезни Паркинсона количество сочетания селективного в отношении переднего мозга антагониста NMDA и агента, усиливающего возбуждающую обратную связь, и фармацевтически приемлемый растворитель или носитель.
Особенно предпочтительными композициями в объеме настоящего изобретения, как описано в предыдущем параграфе, являются композиции, где:
(a) указанным селективным в отношении переднего мозга антагонистом NMDA является (+)-(1S,2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидино)-1-пропанол, а указанным агентом, усиливающим возбуждающую обратную связь, является леводопа,
(b) указанным селективным в отношении переднего мозга антагонистом NMDA является (1S, 2S)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанол, а указанным агентом, усиливающим возбуждающую обратную связь, является леводопа, и
(c) указанным селективным в отношении переднего мозга антагонистом NMDA является (3R, 4S)-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4,7-диол, а названным агентом, усиливающим возбуждающую обратную связь, является леводопа.
Более предпочтительной композицией в объеме настоящего изобретения является композиция, как описано в предыдущем параграфе, дополнительно включающая карбидопу.
Также в объеме настоящего изобретения находится первая фармацевтическая композиция для использования со второй фармацевтической композицией для получения антипаркинсонического эффекта у млекопитающих, страдающих болезнью Паркинсона, причем этот эффект больше суммы антипаркинсонических эффектов, получаемых при введении названных первой и второй фармацевтических композиций в отдельности, а вторая фармацевтическая композиция включает в себя количество агента, усиливающего возбуждающую обратную связь, и фармацевтически приемлемый носитель или растворитель, названная первая фармацевтическая композиция включает в себя количество селективного в отношении переднего мозга антагониста NMDA и фармацевтически приемлемый носитель или растворитель.
Особенно предпочтительными композициями в объеме настоящего изобретения, как описано в предыдущем параграфе, являются композиции, в которых:
(a) указанным селективным в отношении переднего мозга антагонистом NMDA является (+)-)(1S, 2S)-1-(4-гидроксифенил)-2-(4- гидрокси-4-фенилпиперидино)-1-пропанол,
(b) указанным селективным в отношении переднего мозга антагонистом NMDA является (1S, 2S)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанол, и
(c) указанным селективным в отношении переднего мозга антагонистом NMDA является (3R, 4S)-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4,7-диол.
Помимо этого в объем настоящего изобретения входит первая фармацевтическая композиция применения со второй фармацевтической композицией для получения антипаркинсонического эффекта у млекопитающих, страдающих болезнью Паркинсона, причем этот эффект больше, чем сумма антипаркинсонических эффектов, получаемых при введении названных первой и второй фармацевтических композиций по отдельности, а вторая фармацевтическая композиция включает в себя количество леводопы, количество ингибитора декарбоксилазы леводопы и фармацевтически приемлемый носитель или растворитель, указанная первая фармацевтическая композиция включает в себя количество селективного в отношении переднего мозга антагониста NMDA и фармацевтически приемлемый носитель или растворитель.
Особенно предпочтительными композициями в пределах объема настоящего изобретения, как описано в предыдущем параграфе, являются композиции, в которых:
(a) указанным селективным в отношении переднего мозга антагонистом NMDA является (+)-(1S, 2S))-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидино)-1-пропанол,
(b) указанным селективным в отношении переднего мозга антагонистом NMDA является (1S,2S)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанол, и
(c) указанным селективным в отношении переднего мозга антагонистом NMDA является (3R, 4S)-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4,7-диол.
Также в объем настоящего изобретения входит первая фармацевтическая композиция для получения антипаркинсонического эффекта у млекопитающих, страдающих болезнью Паркинсона, причем этот эффект больше, чем сумма антипаркинсонических эффектов, получаемых при введении названных первой и второй фармацевтических композиций по отдельности, а вторая фармацевтическая композиция включает в себя количество селективного в отношении переднего мозга антагониста NMDA и фармацевтически приемлемый носитель или растворитель, указанная первая фармацевтическая композиция включает в себя количество агента, усиливающего возбуждающую обратную связь, и фармацевтически приемлемый носитель или растворитель.
Особенно предпочтительными композициями в объеме настоящего изобретения, как описано в предыдущем параграфе, являются такие композиции, в которых названным агентом, усиливающим возбуждающую обратную связь, является леводопа.
Также в объем настоящего изобретения входит первая фармацевтическая композиция для получения антипаркинсонического эффекта у млекопитающих, страдающих болезнью Паркинсона, причем этот эффект больше, чем сумма антипаркинсонических эффектов, получаемых при введении названных первой и второй фармацевтических композиций по отдельности, а вторая фармацевтическая композиция включает в себя количество селективного в отношении переднего мозга антагониста NMDA и количество ингибитора декарбоксилазы леводопы, а также фармацевтически приемлемый носитель или растворитель, названная первая фармацевтическая композиция включает в себя количество леводопы.
Также в объем настоящего изобретения входит первая фармацевтическая композиция для применения со второй фармацевтической композицией и третьей фармацевтической композицией для получения антипаркинсонического эффекта у млекопитающих, страдающих болезнью Паркинсона, причем этот эффект больше, чем сумма антипаркинсонических эффектов, получаемых при введении названных первой и второй фармацевтических композиций по отдельности, а вторая фармацевтическая композиция включает в себя количество леводопы и фармацевтически приемлемый носитель или растворитель и названная третья фармацевтическая композиция включает в себя ингибитор декарбоксилазы леводопы и фармацевтически приемлемый носитель или растворитель, названная первая фармацевтическая композиция включает в себя селективный в отношении переднего мозга антагонист NMDA и фармацевтически приемлемый носитель или растворитель.
Особенно предпочтительными композициями в объеме настоящего изобретения, как описано в предыдущем параграфе, являются такие композиции, в которых:
(a) указанным селективным в отношении переднего мозга антагонистом NMDA является (+)-(1S,2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4-фенилпиперидино)-1-пропанол,
(b) указанным селективным в отношении переднего мозга антагонистом NMDA является (1S, 2S)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанол, и
(c) указанным селективным в отношении переднего мозга антагонистом NMDA является (3R, 4S)-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4,7-диол.
Помимо этого в объем настоящего изобретения входит первая фармацевтическая композиция для применения со второй фармацевтической композицией и третьей фармацевтической композицией для получения антипаркинсонического эффекта у млекопитающих, страдающих болезнью Паркинсона, причем этот эффект больше, чем сумма антипаркинсонических эффектов, получаемых при введении названных первой и второй фармацевтических композиций по отдельности, а вторая фармацевтическая композиция включает в себя количество селективного в отношении переднего мозга антагониста NMDA и фармацевтически приемлемый носитель или растворитель и названная третья композиция включает в себя ингибитор декарбоксилазы леводопы и фармацевтически приемлемый носитель или растворитель, названная первая фармацевтическая композиция включает в себя количество леводопы и фармацевтически приемлемый носитель или растворитель.
Также в объем настоящего изобретения входит способ получения синергического антипаркинсонического эффекта у млекопитающих, нуждающихся в таком эффекте, который включает в себя введение названному млекопитающему количеств двух терапевтических агентов, выбранных из группы, состоящей из: (a) селективного в отношении переднего мозга антагониста NMDA и (b) агента, усиливающего возбуждающую обратную связь, причем количество в отдельности (a) и в отдельности (b) недостаточно для получения терапевтического эффекта, и комбинированный эффект количеств этих терапевтических агентов больше, чем сумма терапевтических эффектов количеств индивидуальных терапевтических агентов, введенных по отдельности.
Предпочтительным способом в объеме настоящего изобретения, как описано в предыдущем параграфе, является способ, при котором количество селективного в отношении переднего мозга антагониста NMDA и количество агента, усиливающего возбуждающую обратную связь, вводят одновременно.
Еще одним предпочтительным способом в объеме настоящего изобретения является способ, как описано в параграфе, предшествующем непосредственно предыдущему параграфу, при котором количество селективного в отношении переднего мозга антагониста NMDA и количество агента, усиливающего возбуждающую обратную связь, вводят последовательно в любом порядке.
Особенно предпочтительными способами в объеме настоящего изобретения являются способы, как описано в двух параграфах, непосредственно предшествующих этому параграфу, при которых указанные способы дополнительно включают в себя введение количества карбидопы.
В особенно предпочтительном варианте осуществления настоящее изобретение касается способа получения синергического антипаркинсонического эффекта у млекопитающего, нуждающегося в таком эффекте, который включает в себя введение названному млекопитающему количеств двух терапевтических агентов, выбранных из группы, состоящей из: (a) селективного в отношении переднего мозга антагониста NMDA и (b) агента, усиливающего возбуждающую обратную связь, причем количество только (a) и количество только (b) недостаточно для получения терапевтического эффекта, а комбинированный эффект введенных количеств этих терапевтических агентов больше, чем сумма терапевтических эффектов количеств индивидуальных терапевтических агентов, введенных по отдельности, а названный селективный в отношении переднего мозга антагонист NMDA выбирают из группы, состоящей из (+)-(1S,2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанола, (1S,2S)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанола и (3R,4S)-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4,7-диола.
Выражение "фармацевтически приемлемые соли присоединения кислот" включают, но не ограничиваются, такие соли как гидрохлорид, гидробромид, сульфат, кислый сульфат, фосфат, кислый фосфат, двукислый фосфат, ацетат, сукцинат, цитрат, тартрат, лактат, манделат, метансульфонат (мезилат) и п-толуолсульфонат (тозилат).
Соли присоединения кислот соединений настоящего изобретения легко получить путем взаимодействия основных форм с соответствующей кислотой. Когда соль является солью одноосновной кислоты (например, гидрохлорид, гидробромид, п-толуолсульфонат, ацетат), кислой формой двуосновной кислоты (например, кислый сульфат, сукцинат) или двукислой формой трехосновной кислоты (например, двукислый фосфат, цитрат), применяют по меньшей мере одномолярный эквивален и обычно молярный избыток кислоты. Однако, когда желательно получить такие соли, как сульфат, гемисукцинат, кислый фосфат или фосфат, обычно используют определенные и точные химические эквиваленты кислоты. Свободное основание и кислоту обычно смешивают в сорастворителе, из которого выпадает в осадок желаемая соль, или же она может выделяться концентрацией и/или добавлением нерастворителя.
Подробное описание изобретения
Селективные в отношении переднего мозга антагонисты NMDA, которые применяют в синергической комбинации настоящего изобретения, получаются легко.
Соединение формулы (I) по настоящему изобретению, в которых R2 и R5 берутся вместе, образуя хроман-4-оловое кольцо, а R1, R3 и R4 представляют водород, приготавливаются способами, аналогичными тем, которые описаны в патенте США N 5356905, описание которых включено в настоящий документ в качестве ссылки.
Соединения формулы (I) по настоящему изобретению, в которых R2 и R5 берутся отдельно, а R1, R2, R3 и R4 представляют водород, получают способами, аналогичными тем, которые описаны в патентах США N 5185343, 5272160 и 5338754, описание которых включено в настоящий документ в качестве ссылок.
Соединения формулы (I) по настоящему изобретению, в которых R2 и R5 берутся вместе, образуя хроман-4-оловое кольцо, и по меньшей мере один из R1, R3 и R4 не является водородом, и соединения формулы (I) настоящего изобретения, в которых R2 и R5 берутся раздельно, и по меньшей мере один из R1, R2, R3 и R4 не является водородом, получают, как описано ниже.
Конкретно, соединения формулы (I) обычно получают путем снятия защиты у фенольного спиртового промежуточного соединения. Эту группу, защищающую фенол, удаляют обычными способами. Фенольную группу предпочтительно защищают в виде обычных силильных эфиров, таких как триизопропил-, трет-бутилдиметилсилил, трифенилсилил и т.п., или в форме бензиловых или замещенных бензиловых эфиров. По предпочтительному способу удаления указанных силиловых групп применяют 1-1,1 мольные эквиваленты фторида тетрабутиламмония или другого обычного источника фтора в реакционно инертном растворителе, таком как тетрагидрофуран. Эту реакцию обычно осуществляют при около 0-50oC и, наиболее удобно, при комнатной температуре, чтобы избежать затрат на нагревание или охлаждение реакционной смеси и свести к минимуму распад продукта в процессе нагревания. По одному способу удаления бензиловых или замещенных бензиловых эфиров применяют стандартное восстановление эфиров до спиртов действием водорода над катализатором из благородного металла, такого как палладий или никель, в реакционно инертном растворителе, например, с использованием 10% палладия на угле в качестве катализатора, предпочтительно, при низком давлении (например, 1-10 атмосфер) и температуре (например, 20-75oC) и обычно в реакционно инертном растворителе, таком как метанол или этанол. По другому способу гидрирования используют формиат аммония в качестве источника водорода в реакционно инертном растворителе при низкой температуре (например, от 20oC до кипячения с обратным холодильником). Подходящие реакционно инертные растворители для такой реакции гидрирования включают эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан, низшие спирты, такие как метанол или этанол, или их сочетания. Особенно предпочтительным соединением растворителей для этого гидрирования является смесь тетрагидрофурана и метанола.
Используемое в предыдущем параграфе и везде в настоящем описании выражение "реакционно инертный растворитель" относится к любому растворителю, который не взаимодействует с исходными продуктами, реагентами, промежуточными веществами или продуктами таким образом, что может неблагоприятно повлиять на выход желаемого продукта.
Соединения формулы (I), в которых гидроксильная группа фенола защищена, можно получить обычным гидридным восстановлением 3-пиперидино-хромен-4-она, 3-пирролидино-хромен-4-она, 3-(8-аза-бицикло(3,2,1)октанил)-хромен-4-она, 2-пиперидино-4'-гидроксипропиофенона, 2-пирролидино-4'-гидроксипропиофенона, 2-(8-аза-бицикло(3,2,1)октанил)-4'-гидроксипропиофенона, 3-пиперидино-хроман-4-она, 3-пирролидино-хроман-4-она, или 2-8-аза-бицикло(3,2,1)окстанил-хроманона, например,

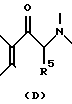
в результате которого обычно получается смесь цис- и транс-изомеров, например, соответственно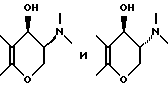
Разумеется, в отдельных случаях один из этих цис- или транс-изомеров часто может преобладать.
Такие гидридные восстановления проводят, используя обычные гидридные восстанавливающие агенты, такие как, например, NaBH4 или LiAlH4. Последний гидридный реагент обычно применяют в избытке (например, моль на моль) в реакционно инертном растворителе, таком как тетрагидрофуран, при пониженных температурах (например, от -15 до 75oC). Любые защищающие группы, которые все еще остаются на месте после восстановления кетона, затем удаляются обычными способами, описанными выше.
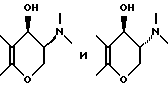
Промежуточные продукты типа (B), изображенные выше, где R2 и R5 берутся вместе, и промежуточные соединения типа (D), изображенные выше, в которых R2 и R5 берутся раздельно, обычно получают путем взаимодействия соответствующего монобромпроизводного хроманона с подходящим замещенным пиперидином, пирролидином или 8-азабицикло(3,2,1)октаном, например,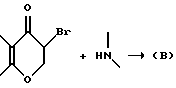
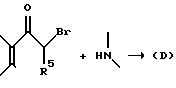
соответственно. Специалистам хорошо известно, что для целей этой реакции альфа-бром-группа может быть заменена на другую нуклеофильно замещаемую группу, такую как хлор, алкансульфонилокси или арилсульфонилокси. Эту реакцию осуществляют в условиях, обычно типичных для нуклеофильных замещений. В случае если оба реагента доступны примерно одинаково, можно использовать их приблизительно молярные эквиваленты, если же один из указанных реагентов более доступен, нежели другой, обычно предпочитают использовать более доступный реагент в избытке для стимулирования бимолекулярного нуклеофильного замещения к завершению в более короткий период времени. Указанное взаимодействие обычно проводят в присутствии по меньшей мере одномолярного эквивалента основания, самого аминопроизводного, если он доступен, но, более часто третичного амина, который по меньшей мере сравним по основной силе с нуклеофильным амином, и в реакционно инертном растворителе, таком как ацетонитрил, этанол, метанол и т. п. Если желательно, реакцию можно выполнять в присутствии катализатора путем добавления вплоть до одномолярного эквивалента или более иодида (например, NaI, KI). Температура не является важным параметром, но обычно ее слегка повышают для стимулирования завершения реакции за более короткий промежуток времени, но не настолько, чтобы вызвать разложение соединений. Температура в пределах 20-120oC обычно является удовлетворительной. Специалистам известно, что при использовании повышенной температуры удобнее следить за ходом реакции и провести ее за возможно более короткий промежуток времени при минимальном разложении соединений. Удобно, когда температура представляет температуру кипения с обратным холодильником реакционной смеси.
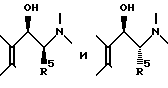
Промежуточные соединения типа (C), описанные выше, в которых R2 и R5 берутся вместе, обычно получают путем взаимодействия соответствующего альфа, альфа-дибромпроизводного хроманона с подходящим замещенным пиперидином, пирролидином или 8-азабицикло(3,2,1)октаном, например,
За исключением применения по меньшей мере одного добавочного молярного эквивалента основания (для нейтрализации HBr, образующегося при одновременной дегидрогалогенизации), условия аналогичны описанным для получения соединений типов (B) и (D) посредством нуклеофильного замещения.
Соединения формулы (I) содержат два асимметричных атома углерода - соответствующих двум рацематам и четырем оптически активным соединениям. Одним из этих рацематов является вышеупомянутый цис-изомер, а другим - транс-изомер. Каждый из этих рацематов способен разделяться на два энантиомера через образование диастереоизомерных солей прибавления кислот с оптически активной кислотой. Альтернативно, рацемический спирт превращается в соответствующие диастереоизомерные сложные эфиры или уретаны соответствующей оптически активной кислотой или изоцианатом. Такие ковалентно связанные производные подвергают разделению различными способами (например, хроматографией). Такие диастереоизомерные сложные эфиры образуются из спирта и оптически активной кислоты с помощью обычных способов, обычно таких, в которых участвует активное производное кислоты, например, такое как хлорангидрид этой кислоты, смесь ангидрид с алкилхлорформатом или с дегидратирующим связывающим агентом, таким как дициклогексилкарбодиимид. После разделения полученных дистереоизомерных сложных эфиров, например, посредством хроматографии, их гидролизуют обычными способами, например водной кислотой или водным основанием, с получением энантиомерных, оптически активных спиртовых соединений формулы (I). В намерения заявителя входит не ограничивать настоящее изобретение рацемическими цис- и транс-соединениями, приведенными в примерах ниже, а включить все оптически активные энантиомеры соединений формулы (I) настоящего изобретения.
Альфа-гало-кетоновые исходные продукты для синтеза соединений по настоящему изобретению обычно получают взаимодействием соответствующего ацилгалогенида с ароматическим галогенидом в условиях ацилирования Фриделя-Крафтца или в других условиях ароматического ацилирования, хорошо известных специалистам. Когда ацилгалогенид не содержит галоген-заместителя в положении альфа по отношению к карбонильной группе, продукт указанного ароматического ацилирования взаимодействует в условиях обычного бромирования, хорошо известных специалистам. Остальные исходные продукта и реагенты, требующиеся для синтеза соединений по настоящему изобретению, легко доступны либо коммерчески, согласно литературным способам, либо с помощью способов, приведенных в качестве примеров в разделе Приготовления ниже.
Следует учесть, что другие селективные в отношении переднего мозга антагонисты NMDA входят в объем способа по настоящему изобретению. Включенные в этот объем другие антагонисты NMDA являются соединениями формулы (II), называемые также ифенпродилом, и соединениями формулы (III), называемыми также элипродилом. Ифенпродил получают способами, аналогичными описанным в патенте США N 3509164, включенном в настоящий документ в качестве ссылки. Элипродил получают способами, аналогичными описанными в патенте США N 4690931, также включенном в настоящий документ в качестве ссылки.

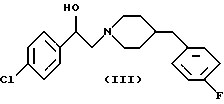
Настоящие соединения формулы (I) являются селективными в отношении переднего мозга антагонистами NMDA, как было показано в биологических экспериментах. В социты лягушки инъецировали с помощью методики, хорошо известной специалистам, РНК, экспрессирующую подтипы NR1 и NR2A, NR1 и NR2B, NR1 и NR2D рецепторов NMDA млекопитающих. Это гарантировало, что определенный социт будет содержать только NR2A, NR2B, NR2C или NR2D подтип рецептора NMDA. Подготовленные таким способом социты лягушки подвергали воздействию испытуемым соединением, согласно процедуре, описанной Williams и др., Neuron 10, 267-278 (1993). Соединения, которые показывали наибольшую активность на социтах, содержащих рецепторы NR2B, по сравнению с другими рецепторами NMDA, являлись селективными в отношении подтипа NR2B антагонистами NMDA и, следовательно, являлись селективными в отношении переднего мозга антагонистами NMDA.
Показано, что настоящие соединения формулы (I) связывают рецепторы в области переднего мозга в следующем ауторадиографическом эксперименте. Самцов крыс Sprague-Dawley обезглавливали и быстро удаляли мозг, который замораживали в порошковом сухом льду. Мозг помещали на столик Tissul-Tek и приготавливали 20 микронные срезы, получая гистологические микропрепараты с заливкой хромалюм/желатином.
Эти микропрепараты до использования хранили при -20oC. Эти микропрепараты необходимо использовать в течение 48 часов. В день эксперимента микропрепараты оттаивали до комнатной температуры и инкубировали с меченным тритием испытуемым соединением (обычно около 10 нМ) в течение 20 минут при 30oC в растворе 50 мМ Трис HCl буфера при pH 7,4. Неспецифическое связывание определяли добавлением 100 мкМ не меченного тритием испытуемого соединения. Микропрепараты повторно отмывали в ледяном буфере в течение 2-10 секунд, а затем высушивали струей теплого воздуха. Микропрепараты помещали в кассеты, где они воздействовали на чувствительную к тритию пленку в течение 14-18 дней при 4oC. Пленку проявляли, фиксировали и высушивали. Просмотр слайдов выявил участки мозга, в которых меченное тритием соединение соединялось с рецепторным участком.
Сообщалось о многих соединениях и классах соединений, используемых при лечении болезни Паркинсона. Среди этих соединений те из них, которые действуют, усиливая возбуждающую обратную связь между вентральным латеральным ядром таламуса и корой (агенты, усиливающие возбуждающую обратную связь), входят в объем настоящего изобретения. Классы соединений, которые содержат агенты, усиливающие возбуждающую обратную связь, включают в себя, но не ограничиваются ими, агонисты дофаминовых рецепторов, агонисты дофаминовых рецепторов D1, агонисты дофаминовых рецепторов D2, агонисты дофаминовых/β-адренергических рецепторов, агонисты дофамин/ингибитор поглощения 5-HT/5-HT-1A, дофамин/опиатные агонисты, агонисты адренорецепторов, антагонисты α 2-адренорецепторов/агонисты дофамина, агонисты α 2-адренорецепторов/дофамина D2, ингибиторы поглощения дофамина, ингибиторы моноаминооксидазы-B, ингибиторы КОМТ и леводопу.
Агонисты дофаминовых рецепторов, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, дигидроэргокриптин, этисулергин, AF-14, алаптид, перголид и пирибедил. Агонисты дофаминовых рецепторов D1, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, A-68939, A-77636, дигидрексин и SKF-38393. Агонисты дофаминовых рецепторов D2, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, карберголин, лизурид, N-0434, наксаголид, PD-118440, прамипексол, хинпирол и ропинирол. Агонисты дофаминовых/β-адренергических рецепторов, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, DPDMS и допексамин. Агонисты дофамин/ингибитор поглощения 5-HT/5-HT-1A, используемые по способу настоящего изобретения, включают, но не ограничиваются им, роксиндол. Дофамин/опиатные агонисты, полезные для способа настоящего изобретения, включают, но не ограничиваются им, N1H-10494. Агонисты адренорецепторов, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, дроксидопу, ибопамин и мазиндол. Антагонисты α 2-адренорецепторов/агонисты дофамина, используемые по способу настоящего изобретения, включают, но не ограничиваются им, тергурид. Агонисты α 2-адренорецепторов/дофамина D2, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, эрголины и талипексол. Ингибиторы поглощения дофамина, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, GBR-12909, GBR-13069, GYK1-52895 и NS-2141. Ингибиторы моноаминооксидазы-B, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, селегилин, N-(2-бутил)-N-метилпропаргиламин, N-метил-N-(2-пентил)-пропаргиламин, AGN-1133, производные спорыньи, лазабемид, LU-53439, MD-280040 и мофегилин. Ингибиторы КОМТ, используемые по способу настоящего изобретения, включают, но не ограничиваются ими, CGP-28014, энтакапон и толкапон. Эти соединения хорошо известны в литературе и легко получаются с помощью описанных там способов.
Следующие агенты, усиливающие возбуждающую обратную связь, особенно полезны по способу настоящего изобретения: леводопа, полученная, как описано в патенте США N 3405159, дигидроэргокриптин, полученный как описано в патенте США N 3755328, этилсулергин и AF-14, полученные, как описано в патенте США N 4348392, алаптид, полученный, как описано в патенте США N 4083985, перголид, полученный, как описано в патенте США N 4166182, пирибедил, полученный, как описано в патенте США N 3299067, лизурид, полученный, как описано в Coll. Czech. CC, 25, 1992 (1960), бромкриптин, полученный, как описано в патенте США N 3752814, амантадин, полученный, как описано в патенте США N 3162180, 3-PPP, полученный, как описано в Европейском патенте N 105243, A-68930, полученный, как описано в J. Med. Chem., 33, 2848 (1990), A-77636, полученный, как описано в J. Org. Chem., 57, 7115 (1992), SKF-38393, полученный, как описано в J. Med. Chem., 23, 973 (1980), N-0434, полученный, как описано в патенте США N 4465692, наксаголид, полученный, как описано в патенте США N 4420480, PD-118440, полученный, как описано в патенте США N 4650805, прамипексол, полученный, как описано в патенте США N 4886812, хинпирол, полученный, как описано в J. Med. Chem., 26, 1112 (1983), ропинирол, полученный, как описано в патенте США N 4452808, хинелоран, полученный, ка описано в патенте США N 4501890, DPDMS, полученный, как описано в Biochem. Pharm. 33, 2371, (1984), допексамин, полученный, как описано в патенте США N 5013760, роксиндол, полученный, как описано в патенте США N 4251538, N1H-10494, полученный, как описано в J. Med. Chem., 30, 1906 (1987), дроксидопа, полученная, как описано в патенте США N 3920728, ибопамин, полученный, как описано в патенте США N 4218470, мазиндол, полученный, как описано в патенте США N 3597445, тергурид, полученный, как описано в патенте США N 3953454, талипексол, полученный, как описано в патенте США N 3804849, GBR-12909, полученный, как описано в патенте США N 4202896, GBR-13069, полученный, как описано в Eг. J. Chem. Chim. Ther., 15, 363 (1980), GYK1-52895, полученный, как описано в патенте Германии DE N 3727226, селегилин, полученный, как описано в патенте Нидерландов NL N 6605956, N-(бутил)-N-метилпропаргиламин и N-метил-N-(2-пент)пропаргиламин, полученные, как описано в J. Med. Chem., 35, 3705 (1992), AGN-1133, полученный, как описано в патенте США N 3201470, лазабемид, полученный, как описано в патенте США N 4764522, MD-280040, полученный, как описано в патенте США N 4971995, мофегилин, полученный, как описано в патенте США N 4454158, CGP-28014, полученный, как описано в патенте США N 4863938, знтакапон, полученный, как описано в патенте США N 5135950, толкапон, полученный, как описано в патенте Австралии N 90/603788, и SDZHDC 912, полученный, как описано в патенте США N 4950672. Описания всех упомянутых патентов США включены в настоящий документ в качестве ссылок.
Селективный в отношении переднего мозга антагонист NMDA, как описано выше, применяется в комбинации с агентом, усиливающим возбуждающую обратную связь, описанным выше, для синергического лечения болезни Паркинсона. Для определения активности настоящей комбинации селективного в отношении переднего мозга антагониста NMDA и агента, усиливающего возбуждающую обратную связь, селективный в отношении переднего мозга антагонист NMDA объединяют в удобных соотношениях с агентом, усиливающим возбуждающую обратную связь, и эту комбинацию испытывают согласно способу, хорошо известному специалистам, как описано в Greenamyrl и др., Annals Neurology, 35, 655-61 (1994).
Используемый здесь термин "синергическое количество" селективного в отношении переднего мозга антагониста NMDA и соединения, способного восстанавливать баланс возбуждающей обратной связи между вентральным латеральным ядром таламуса и корой (агента, усиливающего возбуждающую обратную связь), означает такое количество, которое, будучи введено млекопитающему, страдающему болезнью Паркинсона, достаточно для проявления более сильного действия на названную болезнь Паркинсона, чем сумма действий, которая наблюдается при независимом введении селективного в отношении переднего мозга антагониста NMDA и агента, усиливающего возбуждающую обратную связь, в отдельности.
Ввиду природы болезни Паркинсона синергические эффекты селективного в отношении переднего мозга антагониста NMDA и агента, усиливающего возбуждающую обратную связь, будут
наблюдаться в широких границах доз. Болезнь Паркинсона развивается вследствие прогрессирующей гибели дофаминовых нейронов, исходящей из среднего мозга стриатума. Поскольку клинический эффект этой гибели нейронов накапливается по мере прогрессирования болезни, введение терапевтических агентов должно тщательно подбираться по мере протекания болезни. Это изменение доз без труда выполняется клиническим специалистом по лечению млекопитающих, страдающих болезнью Паркинсона, что облегчается настоящим описанием.
При введении леводопы в отдельности начальная доза обычно составляет 14 мг на килограмм веса тела пациента в день. Дозу повышают по мере течения заболевания до максимально переносимой дозы 114 мг на килограмм веса тела пациента в день. При совместном введении леводопы и карбидопы начальная доза обычно составляет 1,4 мг леводопы на килограмм веса тела пациента в день. Дозу повышают по мере течения заболевания до максимально переносимой дозы 11,4 мг леводопы на килограмм веса тела пациента в день. Карбидопу вводят одновременно в дозах по меньшей мере 1,4 мг на килограмм веса тела пациента в день. При лечении болезни Паркинсона (+)-(1S,2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанолом и леводопой, (+)-(1S,2S)-1-(4-гидроксифенил)-2-(4-гидрокси-4- фенилпиперидино)-1-пропанол вводят в дозах приблизительно от 15 до 100 μ г на килограмм веса тела пациента в день, а леводопу вводят совместно в начальной дозе от 1,4 до 7,1 мг на килограмм веса тела пациента в день. Когда синергическая комбинация включает по меньшей мере 1,4 мг карбидопы на килограмм веса тела пациента в день, доза леводопы составляет от 0,14 мг до 0,71 мг на килограмм веса тела пациента в день. Дозу леводопы можно повышать, как это требуется, вплоть до максимально переносимой дозы. Однако применение селективного в отношении переднего мозга антагониста NMDA в комбинации с леводопой будет предупреждать необходимость повышения дозировки леводопы на поздних стадиях болезни Паркинсона.
Подобным образом, дозы других агентов, усиливающих возбуждающую обратную связь, также будут составлять от 1/2 до 1/10 от дозы, в которой это соединение оказывает терапевтическое действие, будучи введено отдельно, или после развития болезни Паркинсона, в которой соединение максимально переносится. Такое изменение дозы легко определяется специалистом по лечению болезни Паркинсона, что облегчается настоящим описанием.
Синергическая комбинация по настоящему изобретению обычно вводится в форме фармацевтической композиции, включающей в себя один из селективных в отношении переднего мозга антагонистов NMDA и один из агентов, усиливающих возбуждающую обратную связь, совместно с фармацевтически приемлемым носителем или растворителем. Такие композиции обычно получают обычными способами с использованием твердых или жидких наполнителей или растворителей в соответствии с предполагаемым способом введения. Альтернативно, селективный в отношении переднего мозга антагонист NMDA и агент, усиливающий возбуждающую обратную связь, могут вводиться одновременно в раздельных формах, что приводит к возникновению синергической комбинации указанного селективного в отношении переднего мозга антагониста NMDA и названного агента, усиливающего возбуждающую обратную связь, после инкорпорации в организм пациента. При этих условиях селективный в отношении переднего мозга антагонист NMDA помещается в одну стандартную фармацевтическую композицию, а агент, усиливающий возбуждающую обратную связь, помещается в отдельную стандартную фармацевтическую композицию. Не обязательно, если агентом, усиливающим возбуждающую обратную связь, является леводопа, вводится ингибитор декарбоксилазы леводопы, такой как карбидопа. Обычно ингибитор декарбоксилазы леводопы вводится в виде отдельной фармацевтической композиции.
Индивидуальные элементы комбинации по настоящему изобретению, а именно селективный в отношении переднего мозга антагонист NMDA или его фармацевтическая композиция и агент, усиливающий возбуждающую обратную связь, или его фармацевтическая композиция могут вводиться также в виде индивидуальных композиций, которые, будучи введены совместно, как одновременно, так и последовательно, демонстрируют сигергические антипаркинсонические эффекты.
Для перорального введения могут применяться таблетки, содержащие наполнители, такие как цитрат натрия, карбонат кальция и кислый фосфат кальция, вместе с различными разрыхляющими агентами, такими как крахмал, предпочтительно картофельный крахмал или крахмал из тапиоки, альгиновая кислота и некоторые сложные силикаты, со связующими агентами, такими как поливинилпирролидон, сахароза, желатин и аравийская камедь. Помимо этого для целей таблетирования часто очень полезны смазывающие агенты, такие как (но не ограничивающиеся этим перечнем) стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции сходного типа могут использоваться также в качестве наполнителей для мягких эластичных и твердых желатиновых капсул, предпочтительные в этой связи материалы, включают, но не ограничиваются этим перечнем, например, лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Если для перорального введения желательны водные суспензии и/или эликсиры, необходимый активный ингредиент можно комбинировать с различными корригентами, красителями или окрашенными веществами, и, если это желательно, с эмульгирующими и/или суспендирующими агентами, совместно с растворителями, такими как вода, этанол, пропиленгликоль, глицерин и различные их сочетания.
Термин "антипаркинсонический", используемый в настоящем документе, означает эффект, облегчающий симптоматику болезни Паркинсона.
Настоящее изобретение иллюстрируется нижеприведенными примерами, но не ограничивается их деталями.
Все неводные реакции проводят в атмосфере азота для удобства и повышения выходов продуктов. Все растворители/разбавители высушивались обычными способами или приобретались в предварительно высушенном виде. Все реакционные смеси перемешивались с помощью магнитных мешалок или механически. Спектры ЯМР записывались при 300 МГц и представлены в миллионных долях. Растворителем при ЯМР был CDCl3, если не указывается иной. Спектры ИК представлены в см-1, обычно фиксируя только сильные сигналы.
ПРИМЕРЫ
ПРИМЕР 1
(1R*, 2R*)-1-(3-фтор-4-гидроксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин)-1-ил)-пропан-1-ол мезилат
Смесь 3-фтор-4-триизопропилсилилокси- α -бромпропиофенона (соединения по приготовлению 1, 1,19 г, 2,95 ммоль), 4-(4-фторфенил)-4-гидроксипиперидина (0,864 г, 4,43 ммоль) и триэтиламина (1,03 мл, 7,38 ммоль) в этаноле (25 мл) нагревали с обратным холодильником в течение 4 часов, а затем перемешивали при комнатной температуре в течение 64 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли и органический слой промывали насыщенным раствором соли, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1,5 х 3 дюйма) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (350 мл), ноль, 20% этилацетат/гексан (150 мл), ноль, 20% этилацетат/гексан (450 мл), 0,437 г (29%) 1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-(4-фторфенил)-4- гидроксипиперидин)-1-ил)-пропан-1-она в виде желтого масла, у которого: ЯМР δ 7,88 (дд, J = 2,11 Гц, 1H), 7,81 (д, J = 8,5 Гц, 1H), 7,43 (дд, J = 5,5, 9 Гц, 1H), 7,00 (м, J = 9 Гц, 1H), 6,95 (м, J = 8,5 Гц, 1H), 4,05 (кв, J = 6,5 Гц, 1H), 2,93-2,72 (м, 2H), 2,72-2,53 (м, 2H), 2,03 (сим.м, 2H), 1,82-1,58 (м, 3H), 1,35-1,22 (м, 5H), 1,10 (д, J = 7 Гц, 18H).
Смесь боргидрида натрия (0,027 г, 0,718 ммоль) и этанола (10 мл) перемешивали 10 минут, а затем добавляли 1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,371 г, 0,717 ммоль в 10 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали, а остаток распределяли между этилацетатом и водой. Фазы разделяли и органический слой промывали насыщенным раствором соли, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (0,75 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (200 мл), ноль, 10% этилацетат/гексан (100 мл), ноль, 20% этилацетат/гексан (200 мл), в результате чего получали 0,22 г (59%) (1R*,2R*)-1-(3-фтор-4- триизопропилсилилоксифенил)-2-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-пропан-1-ола. Образец, перекристаллизованный из эфира, имел т.пл. 159-160oC.
Продукт описанной выше реакции (0,192 г, 0,37 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли фторид тетрабутиламмония (0,407 мл, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 30 минут при комнатной температуре, а затем концентрировали. Остаток распределяли между этилацетатом и водой, а фазы разделяли. Органический слой промывали насыщенным раствором соли, высушивали над сульфатом кальция и концентрировали с получением 0,122 г (91%) твердого белого продукта. Это твердое вещество суспендировали в метаноле (6 мл) и прибавляли метансульфокислоту (0,022 мл, 0,34 ммоль). Смесь концентрировали при кипячении до 0,5 мл. При охлаждении получали белые кристаллы, которые собирали фильтрованием, в результате чего получали 0,062 г (36%) (1R*,2R*)-1-(3-фтор-4-гидроксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилата, с т. пл. 239-241oC. Анализ, (вычислено) рассчитано для C20H23F3NO3•CH4SO3: C 54,89, H 5,92, N 3,05. Найдено: C 55,17, H 6,08, N 3,11.
ПРИМЕР 2
(1R*, 2R*)-1-(4-гидрокси-3-метилфенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3-метил-4-триизопропилсилилокси- α -бромпропиофенона (соединение по приготовлению 6, 9,17 г, 22,97 ммоль), 4-(4-фторфенил)-4-гидроксипиперидина (6,73 г, 34,45 ммоль) и триэтиламина (8,0 мл, 57,43 ммоль) в этаноле (180 мл) нагревали с обратным холодильником в течение 6 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным раствором соли, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (3 х 3,5 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (1000 мл), ноль, 20% этилацетат/гексан (700 мл), ноль, 20% этилацетат/гексан (1300 мл) и 25% этилацетат/гексан (600 мл) 7,66 г (65%) 1-(3-метил-4-триизопропилсилилоксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-она в виде желтой пены, используемого далее без дополнительной очистки. Образец перекристаллизовывали из этилацетата/гексана в виде белых кристаллов с т.пл. 78-82oC.
Смесь боргидрида натрия (0,564 г, 14,92 ммоль) и этанола (60 мл) перемешивали 10 минут, а затем добавляли 1-(3-метил-4-триизопропилсилилоксифенил)-2-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (7,66 г, 14,92 ммоль в 10 мл этанола) с промывкой два раза по 30 мл этанола. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Выпавшее в осадок твердое белое вещество собирали фильтрованием и высушивали с получением 5,72 г (74%) (1R*,2R*)-1-(3-метил-4-триизопропилсилилоксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола, используемого далее без дополнительной очистки и имеющего т.пл. 188-189oC.
Продукт описанной выше реакции (5,72 г, 11,1 ммоль) растворяли в тетрагидрофуране (150 мл) и добавляли фторид тетрабутиламмония (12,21 мл, 12,21 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 1 час при комнатной температуре, а затем концентрировали. Остаток распределяли между этилацетатом и водой, а фазы разделяли. Органический слой концентрировали и суспендировали с метиленхлоридом. Выпавшее в осадок белое твердое вещество собирали фильтрованием и высушивали с получением 3,41 г (85%) (1R*, 2R*)-1-(4-гидрокси-3-метилфенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола. Образец (0,16 г, 0,447 ммоль) превращали в его мезилат. Последний суспендировали в метаноле (8 мл) и добавляли метансульфокислоту (0,029 мл, 0,45 ммоль). Смесь фильтровали и концентрировали, затем остаток перекристаллизовывали из этанола с получением 0,152 г (58%) мезилата с т.пл. 215-216oC. Анализ, рассчитано для C21H25FNO3•CH4SO3: C 58,01, H 6,64, N 3,07. Найдено: C 57,99, H 6,72, N 3,17.
ПРИМЕР 3
(1R*, 2R*)-1-(3,5-диметил-4-гидроксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3,5-диметил-4-триизопропилсилилокси- α -бромпропиофенона (соединение по приготовлению 18, 1,50 г, 3,63 ммоль), 4-(4-хлорфенил)-4-гидроксипиперидина (1,00 г, 4,03 ммоль) и триэтиламина (1,7 мл, 12,2 ммоль) в этаноле (30 мл) нагревали с обратным холодильником в течение 4,5 часов, а затем перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 5 дюймов в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (750 мл), ноль, 10% этилацетат/гексан (250 мл), и 20% этилацетат/гексан (500 мл), с получением 0,82 г (41%) 1-(3,5-диметил-4-триизопропилсилилоксифенил)-2-(4-(4-хлорфенил)-4- гидроксипиперидин-1-ил)-пропан-1-она в виде желтой пены, используемого далее без дополнительной очистки и у которого: ЯМР δ 7,37 (с, 2H), 7,36 (ABкв, ΔV1-3 = 30,5 Гц, J = 8,5 Гц, 4H), 4,15 (кв, J = 6,7 Гц, 1H), 2,85-2,75 (м, 2H), 2,67-2,53 (м, 1H), 2,31 (с, 6H), 2,25-1,97 (м, 1H), 1,74-1,60 (м, 2H), 1,60 (с, 1H), 1,40-1,18 (м, 6H), 1,13 (д, J = 7,2 Гц, 18H).
Смесь боргидрида натрия (0,054 г, 1,43 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-диметил-4-триизопропилсислилоксифенил)-2-(4-(4-хлорфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,77 г, 1,42 ммоль в 25 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Выпавшее в осадок твердое белое вещество собирали фильтрованием и высушивали с получением 0,44 г (56%) (1R*,2R*)-1-(3,5-диметил-4-триизопропилсилилоксифенил)- 2-(4-(4-хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола, удобного для использования без дополнительной очистки и имевшего т.пл. 211,5-212,5oC.
Продукт описанной выше реакции (0,40 г, 0,73 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли фторид тетрабутиламмония (0,81 мл, 0,81 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 30 минут при комнатной температуре, а затем концентрировали. Остаток распределяли между этилацетатом и водой и фазы разделяли. Органический слой промывали рассолом, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3 дюйма с гексаном) с элюированием, проводимым следующим образом: 50% этилацетат/гексан (300 мл), ноль, 50% этилацетат/гексан (100 мл) и этилацетат (200 мл), 0,247 г (88%) (1R*,2R*)-1-(3,5-диметил-4-гидроксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола. Образец (0,24 г, 0,616 ммоль) превращали в его мезилат. Последний суспендировали с метаноле (15 мл) и добавляли метансульфокислоту (0,040 мл, 0,616 ммоль). Смесь фильтровали и концентрировали, затем остаток перекристаллизовывали из 9: 1 этанола/воды с получением 0,228 г (58%) соли мезилата в виде пушистого твердого белого вещества с т.пл. 202,5-203oC. Анализ, рассчитано для C22H28ClNO3•CH4SO3: C 56,84, H 6,64, N 2,88. Найдено: C 57,01, H 6,83, N 2,94.
ПРИМЕР 4
(1R*, 2R*)-1-(3,5-диметил-4-гидроксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3,5-диметил-4-триизопропилсилилокси- α -бромпропиофенона (соединения по приготовлению 18, 1,50 г, 3,63 ммоль), 4-(4-фторфенил)-4-гидроксипиперидина (0,78 г, 4,00 ммоль) и триэтиламина (1,0 мл, 7,2 ммоль) в этаноле (30 мл) нагревали с обратным холодильником в течение 4,5 часов, а затем перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (500 мл), ноль, 20% этилацетат/гексан (500 мл), 0,96 г (50%) 1-(3,5-диметил-4-триизопропилсилилоксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-она в виде оранжевой пены, удобной для использования без дополнительной очистки и у которой: ЯМР δ 7,74 (с, 2H), 7,48-7,43 (м, 2H), 7,02 (т, J = 8,8 Гц, 2H), 4,15 (кв, J = 6,7 Гц, 1H), 2,90-2,77 (м, 3H), 2,68-2,57 (м, 1H), 2,31 (с, 6H), 2,28-2,03 (м, 2H), 1,77-1,66 (м, 2H), 1,56 (с, 1H), 1,41-1,19 (м, 5H), 1,13 (д, J = 7,2 Гц, 18H).
Смесь боргидрида натрия (0,065 г, 1,72 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-диметил-4-триизопропилсилилоксифенил)-2-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,90 г, 1,71 ммоль в 25 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение уикенда. Выпавшее в осадок твердое белое вещество собирали фильтрованием и высушивали с получением 0,365 г (40%) (1R*,2R*)-1-(3,5-диметил-4-триизопропилсилилоксифенил)- 2-(4-(4-фторфенил)-4-гидросипиперидин-1-ил)-пропан-1-ола, удобного для использования без дополнительной очистки и имевшего т.пл. 186,5-187oC. Анализ, рассчитано для C31H48FNO3Si•0,125H2O: C 69,69, H 9,15, N 2,62. Найдено: C 69,65, H 9,29, N 2,57.
Продукт описанной выше реакции (0,31 г, 0,59 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли фторид тетрабутиламмония (0,65 г, 0,65 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 30 минут при комнатной температуре, а затем концентрировали. Остаток распределяли между этилацетатом и водой и фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 50% этилацетат/гексан (150 мл), ноль, 50% этилацетат/гексан (50 мл) и этилацетат (200 мл), 0,200 г (91%) (1R*, 2R*)-1-(3,5-диметил-4-гидроксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола. Образец превращали (0,194 г, 0,519 ммоль) в его соль мезилат. Последнюю суспендировали в метаноле (15 мл) и добавляли метансульфокислоту (0,034 мл, 0,524 ммоль). Смесь фильтровали и концентрировали, затем остаток перекристаллизовывали из 9:1 этанола/воды с получением соли мезилата в виде пушистого твердого белого вещества (0,174 г) с т.пл. 179-180oC. Анализ, рассчитано для C22H28FNO3•CH4SO3•0,25H2O: C 58,27, H 6,91, N 2,95. Найдено: C 58,30, H 7,24, N 3,00.
ПРИМЕР 5
(1R*, 2R*)-1-(3,5-дифтор-4-гидроксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3,5-дифтор-4-триизопропилсилилокси- α -бромпропиофенона (соединения по приготовлению 20, 1,50 г, 3,56 ммоль), 4-(4-хлорфенил)-4-гидроксипиперидина (1,00 г, 4,03 ммоль) и триэтиламина (1,7 мл, 12,2 ммоль) в этаноле (30 мл) нагревали с обратным холодильником в течение 4,5 часов, а затем перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (250 мл), ноль, 10% этилацетат/гексан (250 мл) и 20% этилацетат/гексан (250 мл), 0,79 г (40%) 1-(3,5-дифтор-4-триизопропилсислилоксифенил)-2-(4-(4-хлорфенил)-4- гидроксипиперидин-1-ил)-пропан-1-она в виде оранжевой пены, удобной для использования без дополнительной очистки и у которой: ЯМР δ 7,73 (продолжительно сопряженные д, J = 9,0 Гц, 2H), 7,37 (ABкв, ΔV1-3 = 26,3 Гц, J = 8,7 Гц, 4H), 4,03 (кв, J = 6,8 Гц, 1H), 2,95-2,81 (м, 2H), 2,66-2,61 (м, 2H), 2,17-1,93 (м, 2H), 1,80-1,55 (м, 3H), 1,39-1,21 (м, 5H), 1,12 (д, J = 7,2 Гц, 18H).
Смесь боргидрида натрия (0,058 г, 1,40 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-дифтор-4-триизопропилсислилоксифенил)-2-(4-(4-хлорфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,76 г, 1,38 ммоль в 20 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение уикенда. Выпавшее в осадок твердое белое вещество собирали фильтрованием и высушивали с получением 0,43 г (57%) (1R*,2R*)-1-(3,5-дифтор-4-триизопропилсилилоксифенил)- 2-(4-(4-хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола, удобного для использования без дополнительной очистки и имевшего т.пл. 192-192,5oC. Анализ, рассчитано для C29H42ClF2NO3Si•0,25H2O: C 62,35, H 7,67, N 2,51. Найдено: C 62,37, H 7,81, N 2,73.
Продукт описанной выше реакции (0,39 г, 0,70 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли фторид тетрабутиламмония (0,80 мл, 0,80 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 30 минут при комнатной температуре, а затем концентрировали. Остаток распределяли между этилацетатом и водой и фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма) с элюированием, проводимым следующим образом: 50% этилацетат/гексан (200 мл), ноль, этилацетат (200 мл), ноль, 2% метанол/этилацетат (200 мл) и 5% метанол/этилацетат (200 мл) с получением 0,232 г (86%) (1R*,2R*)-1-(3,5-дифтор-4-гидроксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола. Образец (0,226 г, 0,589 ммоль) превращали в его соль мезилат. Последнюю суспендировали в метаноле (15 мл) и добавляли метансульфокислоту (0,038 г, 0,587 ммоль). Смесь фильтровали и концентрировали, затем остаток перекристаллизовывали из 9:1 этанола/воды с получением соли мезилата в виде белого твердого вещества (0,240 г) с т.пл. 239,5-240oC. Анализ, рассчитано для C20H22ClF2NO3•CH4SO3• H2O: C 50,65, H 5,67, N 2,81. Найдено: C 50,94, H 5,54, N 2,85.
ПРИМЕР 6
(1R*, 2R*)-1-(3,5-диметил-4-гидроксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3,5-диметил-4-триизопропилсилилокси- α -бромпропиофенона (соединения по приготовлению 18, 2,00 г, 4,84 ммоль), 4-(4-трифторметилфенил)-4-гидроксипиперидина (1,78 г, 7,26 ммоль) и триэтиламина (1,4 мл, 10,0 ммоль) в этаноле (30 мл) нагревали с обратным холодильником в течение 7,75 часов, а затем перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1,5 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (500 мл), ноль, 25% этилацетат/гексан (250 мл), 1,39 г (50%) 1-(3,5-диметил-4-триизопропилсилилоксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-она в виде оранжевой пены, удобной для использования без дополнительно очистки и у которой: ЯМР δ 7,74 (с, 2H), 7,60 (м, 4H), 4,17 (кв, J = 6,8 Гц, 1H), 2,92-2,79 (м, 2H), 2,71-2,58 (м, 1H), 2,31 (с, 6H), 2,25-2,00 (м, 2H), 1,76-1,65 (м, 2H), 1,41-1,18 (м, 6H), 1,13 (д, J = 7,2 Гц, 18H).
Смесь боргидрида натрия (0,090 г, 2,38 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-диметил)-4-триизопропилсислилоксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-он (1,30 г, 2,25 ммоль в 25 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Выпавшее в осадок твердое белое вещество собирали фильтрованием и высушивали с получением 0,408 г (31%) (1R*, 2R*)-1-(3,5-диметил-4- триизопропилсилилоксифенил)-2-(4-(4-трифторметилфенил)-4- гидроксипиперидин-1-ил)-пропан-1-ола, удобного для использования без дополнительной очистки и имевшего т.пл. 177-177,5oC. Анализ, рассчитано для C32H48F3NO3Si•0,25H2O: C 65,78, H 8,37, N 2,40. Найдено: C 65,65, H 8,51, N 2,57.
Продукт описанной выше реакции (0,348 г, 0,60 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли фторид тетрабутиламмония (0,60 мл, 0,60 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли водой и эфиром и интенсивно перемешивали. Выпавшее в осадок твердое вещество отфильтровывали и промывали эфиром и взвешивали 0,166 г (65% продукта). Фильтрат экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3 дюйма) с элюированием, проводимым следующим образом: 50% этилацетат/гексан (100 мл), ноль, 50% этилацетат/гексан (100 мл) и этилацетат (75 мл), 0,077 г продукта. Таким способом было получено 0,243 г (96%) (1R*, 2R*)-1-(3,5-диметил-4-гидроксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола. Этот продукт превращали в его соль мезилат. Последнюю суспендировали в 9:1 этаноле/воде (5 мл) и добавляли метансульфокислоту (0,038 г, 0,587 ммоль). Смесь фильтровали и концентрировали до приблизительно 0,5 мл, в продукт собирали с получением 0,184 г соли мезилата в виде твердого белого вещества с т.пл. 147-148oC. Анализ, рассчитано для C23H28F3NO3•CH4SO3• 1,25H2O: C 53,18, H 6,42, N 2,58. Найдено: C 53,18, H 6,63, N 2,58.
ПРИМЕР 7 (1R*,2R*)-1-(3,5-дихлор-4-гидроксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3,5-дихлор-4-триизопропилсилилокси- α -бромпропиофенона (соединения по приготовлению 14, 1,00 г, 2,20 ммоль), 4-(4-фторфенил)-4-гидроксипиперидина (0,64 г, 3,28 ммоль) и триэтиламина (0,62 мл, 4,45 ммоль) в этаноле (20 мл) нагревали с обратным холодильником в течение 6 часов, а затем перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (250 мл), ноль, 10% этилацетат/гексан (350 мл), 0,12 г (10%) 1-(3,5-дихлор-4-триизопропилсилилоксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-она в виде оранжевого масла, которое переносили непосредственно на следующий этап.
Смесь боргидрида натрия (0,010 г, 0,26 ммоль) и этанола (1 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-дихлор-4-триизопропилсилилоксифенил)-2-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,12 г, 0,211 ммоль в 4 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь охлаждали водой и концентрировали при 40oC. Остаток распределяли между этилацетатом и водой, а фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (0,75 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (200 мл), ноль, 20% этилацетат/гексан (150 мл), 0,003 г (27%) (1R*,2R*)-1-(3,5-дихлор-4-триизопропилсилилоксифенил)- 2-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола в виде желтого масла, удобного для использования без дальнейшей очистки.
Продукт описанной выше реакции (0,033 г, 0,058 ммоль) растворяли в тетрагидрофуране (5 мл) и добавляли фторид тетрабутиламмония (0,060 мл, 0,060 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 3 часа при комнатной температуре, а затем концентрировали. Остаток распределяли между этилацетатом и водой, а фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (0,75 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 25% этилацетат/гексан (200 мл), ноль, 50% этилацетат/гексан (150 мл), до получения 0,014 г (58%) (1R*, 2R*)-1-(3,5-дихлор-4-гидроксифенил)- 2-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества. Образец превращали в его соль мезилат. Последнюю суспендировали в метаноле и добавляли метансульфоновую кислоту (0,0022 мл, 0,0034 ммоль). Смесь концентрировали, затем остаток стирали с 20:1 эфир/этанолом с получением 0,013 г соли мезилата в виде белого твердого вещества, у которого: ЯМР (D2O/DMSO-d6) δ 7,70 (ABкв, ΔV1-3 = 23,8 Гц, J = 8,5 Гц, 4H), 7,42 (с, 2H), 4,70 (д, J = 10,2 Гц, 1H), 3,71-3,50 (м, 4H), 3,37-3,32 (м, 1H), 2,75 (с, 3H), 2,60-2,42 (м, 2H), 2,15-2,05 (м, 2H), 1,11 (д, J = 6,8 Гц, 3H).
ПРИМЕР 8
(1R*, 2R*)-1-(3,5-дихлор-4-гидроксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3,5-дихлор-4-триизопропилсилилокси- α -бромпропиофенона (соединение по приготовлению 14, 1,00 г, 2,20 ммоль), 4-(4-трифторметилфенил-4-гидроксипиперидина (0,80 г, 3,26 ммоль) и триэтиламина (0,62 г, 4,45 ммоль) в этаноле (20 мл) нагревали с обратным холодильником в течение 6 часов, а затем перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагале (1 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (250 мл), ноль, 10% этилацетат/гексан (250 мл), 0,18 (13%) 1-(3,5-дихлор-4-триизопропилсилилоксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-она в виде оранжевого масла, которое переносили непосредственно на следующий этап.
Смесь боргидрида натрия (0,012 г, 0,317 ммоль) и этанола (1 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-дихлор-4-триизопропилсилилоксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-он (0,18 г, 0,291 ммоль в 4 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь охлаждали водой и концентрировали при 40oC. Остаток распределяли между этилацетатом и водой, а фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагале (0,75 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (200 мл), ноль, 20% этилацетат/гексан (150 мл), 0,072 г (40%) (1R*,2R*)-1-(3,5-дихлор-4- триизопропилсилилоксифенил)-2-(4-(4-трифторметилфенил)-4- гидроксипиперидин-1-ил)-пропан-1-ола в виде желтого масла, удобного для использования без дополнительной очистки.
Продукт описанной выше реакции (0,072 г, 0,116 ммоль) растворяли в тетрагидрофуране (5 мл) и добавляли фторид тетрабутиламмония (0,120 мл, 0,120 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре, а затем концентрировали. Остаток подвергали флэш-хроматографии на силикагале (0,75 х 4 дюймах в гексане) с элюированием, проводимым следующим образом: 25% этилацетат/гексан (200 мл), ноль, 50% этилацетат/гексан (100 мл), 0,028 г (52%) (1R*, 2R*)-1-(3,5-дихлор-4- гидроксифенил)-2-(4-(4-трифторметилфенил)-4-гидрокси-пиперидин-1-ил)- пропан-1-ола в виде белого твердого вещества. Образец превращали в его соль мезилат. Последнюю суспендировали в метаноле и добавляли метансульфокислоту (0,003 г (0,0039 мл, 0,006 ммоль). Смесь концентрировали, затем остаток стирали с 20:1 эфир/этанолом с получением 0,022 г соли мезилата в виде белого твердого вещества с т.пл. 208-208,5oC. ЯМР (D2O/DMSO-d6) 7,49-7,42 (м, 6H), 4,70 (д, J = 10,2 Гц, 1H), 3,72-3,47 (м, 4H), 3,36-3,28 (м, 1H), 2,75 (с, 3H), 2,55-2,33 (м, 2H), 2,14-2,02 (м, 2H), 1,10 (д, J = 6,8 Гц, 3H).
ПРИМЕР 9
(1R*, 2R*)-1-(3,5-дихлор-4-гидроксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3,5-дихлор-4-триизопропилсилилокси- α -бромпропиофенона (соединения по приготовлению 14, 1,00 г, 2,20 ммоль), 4-(4-хлорфенил)-4-гидроксипиперидина (0,81 г, 3,26 ммоль) и триэтиламина (0,93 мл, 6,67 ммоль) в этаноле (20 мл) нагревали с обратным холодильником в течение 6 часов, а затем перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагале (1 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (250 мл), ноль, 10% этилацетат/гексан (250 мл), 0,08 г (6%) 1-(3,5-дихлор-4-триизопропилсилилоксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-она в виде оранжевого масла, которое переносили непосредственно на следующий этап.
Смесь боргидрида натрия (0,010 г, 0,26 ммоль) и этанола (1 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-дихлор-4-триизопропилсилилоксифенил)-2-(4-(4-хлорфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,08 г, 0,137 ммоль в 4 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь охлаждали водой и концентрировали при 40oC. Остаток распределяли между этилацетатом и водой и фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (0,75 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (200 мл), ноль, 20% этилацетат/гексан (150 мл), 0,03 г (40%) (1R*,2R*)-1-(3,5-дихлор-4-триизопропилсилилоксифенил)- 2-(4-(4-хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола в виде желтого масла, удобного для использования без дополнительной очистки.
Продукт описанной выше реакции (0,030 г, 0,051 ммоль) растворяли в тетрагидрофуране (5 мл) и добавляли фторид тетрабутиламмония (0,053 мл, 0,053 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре, а затем концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (0,75 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 25% этилацетат/гексан (200 мл), ноль, 50% этилацетат/гексан (150 мл), 0,009 г (41%) (1R*, 2R*)-1-(3,5-дихлор-4- гидроксифенил)-2-(4-(4-гидроксипиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества. Образец превращали в его соль мезилат. Последнюю суспендировали в метаноле и добавляли метансульфокислоту (0,0014 мл, 0,002 ммоль). Смесь концентрировали, затем остаток стирали с 10:1 эфир/этанолом до получения 0,0085 г соли мезилата в виде белого твердого вещества с т.пл. 223-223,5oC. ЯМР (D2O) δ 7,54-7,46 (м, 6H), 4,70 (д, 1H частично скрытый растворителем), 3,74-3,53 (м, 4H), 3,37 (бр,д, J = 13,2 Гц, 1H), 2,80 (с, 3H), 2,60-2,27 (м, 2H), 2,20-2,07 (м, 2H), 1,15 (д, J = 6,8 Гц, 3H).
ПРИМЕР 10
(1R*, 2R*)-1-(3,5-дифтор-4-гидроксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 4-бензилокси-3,5-дифтор-α-бромпропиофенона (соединения по приготовлению 22, 1,00 г, 2,82 ммоль) и 4-(4-фторфенил)-4-гидроксипиперидина (1,1 г, 5,63 ммоль) в этаноле (25 мл) нагревали с обратным холодильником в течение ночи. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 5% этилацетат/гексан (500 мл), ноль, 15% этилацетат/гексан (500 мл), ноль, 20% этилацетат/гексан (250 мл), 0,59 г (45%) 1-(4-бензилокси-3,5-дифторфенил)-2-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-пропан-1-она в виде светло-желтого масла, удобного для использования без дополнительной очистки, у которого: ЯМР δ 7,75 (длительно сопряженный, д, J = 9,2 Гц, 2H), 7,48-7,30 (м, 7H), 7,03 (длительно сопряженный, т, J = 8,7 Гц, 2H), 5,31 (с, 2H), 4,01 (кв, J = 6,7 Гц, 1H), 2,93 (дт, J = 2,6, 11,2 Гц, 1H), 2,80-2,75 (м, 1H), 2,70-2,60 (м, 2H), 2,18-1,92 (м, 2H), 1,81-1,62 (м, 2H), 1,30 (д, J = 6,7 Гц, 3H).
Смесь боргидрида натрия (0,050 г, 1,32 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(4-бензилокси-3,5-дифторфенил)-2-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,55 г, 1,17 ммоль в 20 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Выпавшее в осадок твердое белое вещество собирали фильтрованием и высушивали с получением, 0,34 г продукта. Фильтрат концентрировали при пониженном давлении и 40oC. Остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 30% этилацетат/гексан (300 мл) с получением 0,059 г продукта. Таким образом получали 0,399 г (73%) (1R*,2R*)-1-(4-бензилокси-3,5-дифторфенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола, который был удобен для использования без дополнительной очистки и имел т.пл. 169-171oC, ЯМР δ 7,53-7,44 (м, 4H), 7,41-7,30 (м, 3H), 7,06 (длительно сопряженный т, J = 8,7 Гц, 2H), 6,92 (длительно сопряженный д, J = 8,9 Гц, 2H), 5,27 (с, 1H), 5,15 (с, 2H), 4,18 (д, J = 9,7 Гц, 1H), 3,08 (дт, J = 2,3, 11,6 Гц, 1H), 2,71-2,68 (м, 2H), 2,59-2,48 (м, 2H), 2,26-2,01 (м, 2H), 1,83 (бр,д, J = 13,9 Гц, 2H), 1,57 (с, 1H), 0,86 (д, J = 6,7 Гц, 3H).
Продукт описанной выше реакции (0,34 г, 0,721 ммоль) растворяли в тетрагидрофуране (10 мл) и метаноле (10 мл) и и добавляли формиат аммония (0,45 г, 7,14 ммоль) и 10% палладий на угле (0,19 г). Реакционную смесь перемешивали 2 часа при комнатной температуре, а затем фильтровали через диатомовую землю. Прокладку фильтра промывали этанолом и водой. Фильтрат концентрировали, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали без потерь материала. Прокладку фильтра из сульфата магния растворяли в воде и серое твердое вещество отфильтровывали, промывали водой и высушивали воздухом. Серое твердое вещество весило 0,195 г и было очищено посредством флэш-хроматографии на силикагеле (1 х 4 дюйма). Элюирование проводили следующим образом: 50% этилацетат/гексан (100 мл), ноль, этилацетат (200 мл), ноль, 10% метанол/этилацетат (200 мл), ноль, 25% метанол/этилацетат (200 мл) и 50% метанол/этилацетат (200 мл), 0,097 г (36%) (1R*,2R*)-1-(3,5-дифтор-4-гидроксифенил)-2-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества. Продукт превращали в его соль мезилат.
Продукт суспендировали в метаноле (10 мл) и добавляли метансульфокислоту (0,017 мл, 0,262 ммоль). Смесь фильтровали и концентрировали, затем остаток перекристаллизовывали из 9:1 этанол/воды с получением соли мезилата в виде кристаллического белого твердого вещества (0,099 г) с т.пл. 239-239,5oC. Анализ, рассчитано для C20H22F3NO3•CH4SO3: C 52,82, H 5,49, N 2,93. Найдено: C 52,80, H 5,76, N 2,99.
ПРИМЕР 11
(1R*, 2R*)-1-(3,5-дифтор-4-гидроксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 4-бензилокси-3,5-дифтор- α -бромпропиофенона (соединения по приготовлению 22, 1,14 г, 3,21 ммоль), 4-(4-трифторметилфенил)-4-гидроксипиперидина (0,87 г, 3,55 ммоль) и триэтиламина (0,90 мл, 6,5 ммоль) в этаноле (25 мл) нагревали с обратным холодильником в течение 1,75 часа и перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 15% этилацетат/гексан (500 мл), ноль, 25% этилацетат/гексан (250 мл), 1,09 г (65%) 1-(4-бензилокси-3,5-дифторфенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-она в виде светло-оранжевого масла, удобного для использования без дополнительной очистки и у которого: ЯМР δ 7,74 (длительно сопряженный, д, J = 9,4 Гц, 2H), 7,61 (с, 4H), 7,48-7,34 (м, 5H), 5,32 (с, 2H), 4,03 (кв, J = 6,7 Гц, 1H), 2,95-2,83 (м, 2H), 2,67-2,62 (м, 2H), 2,16-1,98 (м, 2H), 1,81-1,67 (м, 2H), 1,57 (бр, с, 1H), 1,31 (д, J = 6,8 Гц, 3H).
Смесь боргидрида натрия (0,085 г, 2,25 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(4-бензилокси-3,5-дифторфенил)-2-(4-(4-трифторметилфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (1,02 г, 1,96 ммоль в 30 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Выпавшее в осадок белое твердое вещество собирали фильтрованием и высушивали с получением 0,66 г (65%) (1R*,2R*)-1-(4-бензилокси-3,5-дифторфенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола, удобного для использования без дополнительной очистки и имевшего т. пл. 201-202oC. Анализ, рассчитано для C28H28F5NO3•0,25H2O: C 63,93, H 5,46, N 2,66. Найдено: C 63,98, H 5,49, N 2,70.
Продукт описанной выше реакции (0,60 г, 1,15 ммоль) растворяли в тетрагидрофуране (15 мл) и метаноле (15 мл) и добавляли формиат аммония (0,73 г, 11,6 ммоль) и 10% палладий на угле (0,30 г). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, а затем фильтровали через диатомовую землю. Прокладку фильтра промывали этанолом и водой. Фильтрат концентрировали, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 0,517 г (1R*, 2R*)-1-(3, 5-дифтор-4-гидроксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества. Образец (0,50 г, 1,16 ммоль) превращали в его соль мезилат. Продукт суспендировали в метаноле (15 мл) и добавляли метансульфокислоту (0,075 г, 1,16 ммоль). Смесь фильтровали и концентрировали, затем остаток перекристаллизовывали из 9:1 этанол/воды с получением соли мезилата в виде пушистого твердого белого вещества (0,475 г) с т. пл. 218-219oC. Анализ, рассчитано для C21H22F5NO3• CH4SO3•0,75H2O: C 48,84, H 5,12, N 2,59. Найдено: C 48,88, H 5,37, N 2,59.
ПРИМЕР 12
(1R*, 2R*)-1-(3-фтор-4-гидроксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3-фтор-4-триизопропилсилилокси- α -бромпропиофенона (соединение по приготовлению 11, 1,25 г, 3,10 ммоль), 4-(4-хлорфенил)-4-гидроксипиперидин (1,0 г, 4,03 ммоль) и триэтиламина (1,51 мл, 10,85 ммоль) в этаноле (25 мл) нагревали с обратным холодильником в течение 4 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3,5 дюйма в 10% этилацетат/гексане) с элюированием, проводимым следующим образом: гексан (150 мл), ноль, 10% этилацетат/гексан (350 мл), ноль, 20% этилацетат/гексан (300 мл), 0,535 г (32%) 1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-(4-хлорфенил)-4- гидроксипиперидин-1-ил)-пропан-1-она в виде желтой маслянистой пены, у которой: ЯМР δ 7,87 (дд, J = 2,11 Гц, 1H), 7,80 (д, J = 8,5 Гц, 1H), 7,39 (д, J = 8,5 Гц, 2H), 7,28 (д, J = 8,5 Гц, 2H), 6,95 (т, J = 8,5 Гц, 1H), 4,07 (кв, J = 7 Гц, 1H), 2,95-2,78 (м, 2H), 2,78-2,57 (м, 2H), 2,04 (сим.м, 2H), 1,78-1,64 (м, 2H), 1,30 (д, J = 7 Гц, 3H), 1,10 (д, J = 7 Гц, 18H).
Смесь боргидрида натрия (0,032 г, 0,85 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-(4-хлорфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,454 г, 0,850 ммоль в 10 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Образовавшийся белый осадок собирали фильтрованием с получением 0,245 г (54%) (1R*,2R*)-1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидина-1-ил)-пропан-1-ола, у которого: ЯМР δ 7,39 (ABкв, ΔV1-3 = 35,2 Гц, J = 8,5 Гц, 4H), 7,06 (дд, J = 2, 11,5 Гц, 1H), 6,96-6,82 (м, 2H), 5,15 (с, 1H), 4,18 (д, J = 9,5 Гц, 1H), 3,04 (дт, J = 2,5, 11,5 Гц, 1H), 2,78-2,67 (м, 1H), 2,67-2,52 (м, 3H), 2,12 (сим.м, 2H), 1,80 (искаженный д, J = 14 Гц, 2H), 1,54 (с, 1H), 1,36-1,19 (м, 3H), 1,08 (д, J = 7 Гц, 18H), 0,80 (д, J = 6,5 Гц, 3H). Продукт содержал также около 8% эритро диастереоизомера, но был удобен для использования без дополнительной очистки.
Продукт описанной выше реакции (0,220 г, 0,41 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли фторид тетрабутиламмония (0,452 мл, 0,45 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 30 минут при комнатной температуре, а затем концентрировали. Остаток распределяли между этилацетатом и водой и фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагале (0,75 х 3,5 дюйма) с элюированием, проводимым следующим образом: 5% этилацетат/гексан (100 мл), ноль, 15% этилацетат/гексан (200 мл), ноль, 25% этилацетат/гексан (200 мл), ноль, 35% этилацетат/гексан (200 мл), ноль, 35% этилацетат/гексан (200 мл), 0,106 г (68%) белого твердого продукта. Этот твердый продукт суспендировали в метаноле (4 мл) и добавляли метансульфокислоту (0,018 мл, 0,28 ммоль). Смесь фильтровали, затем концентрировали при кипении до 0,5 мл с добавлением нескольких капель этанола. Охлаждение давало белые кристаллы, которые собирали фильтрованием с получением 0,084 г (43%) (1R*,2R*)-1-(3-фтор-4-гидроксифенил)-2-(4-(4- хлорфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилата с т.пл. 233-235oC. Анализ, рассчитано для C20H23ClFNO3•CH4SO3: C 52,99, H 5,72, N 2,94. Найдено: C 53,06, H 5,91, N 3,03.
ПРИМЕР 13
(1R*, 2R*)-1-(3-фтор-4-гидроксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3-фтор-4-триизопропилсилилокси- α -бромпропиофенона (соединения по приготолению 11, 1,35 г, 3,35 ммоль), 4-(4-трифторметилфенил)-4-гидроксипиперидина (1,15 г, 4,69 ммоль) и триэтиламина (1,20 мл, 8,38 ммоль) в этаноле (25 мл) нагревали с обратным холодильником в течение 4 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3,5 дюйма в 10% этилацетат/гексане) с элюированием, проводимым следующим образом: гексан (150 мл), ноль, 10% этилацетат/гексан (350 мл), ноль, 20% этилацетат/гексан (350 мл), 0,841 г (44%) 1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-(4-трифторметилфенил)-4- гидроксипиперидин-1-ил)-пропан-1-она в виде желтой маслянистой пены, у которой: ЯМР δ 7,88 (дд, J = 2, 11,5 Гц, 1H), 7,80 (сим.м, 1H), 7,60-7,57 (м, 4H), 6,96 (т, J = 8,5 Гц, 1H), 4,08 (кв, J = 7 Гц, 1H), 3,32 (бр. м, 1H), 2,95-2,78 (м, 2H), 2,78-2,56 (м, 2H), 2,08 (сим.м, 2H), 1,78-1,63 (м, 2H), 1,31 (д, J = 7 Гц, 3H), 1,10 (д, J = 7 Гц, 18H).
Смесь боргидрида натрия (0,049 г, 1,30 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-(4-трифторметилфенил)-4- гидроксипиперидин-1-ил)-пропан-1-он (0,738 г, 1,30 ммоль в 10 мл этанола) с промывкой 5 мл этанола. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Образовавшийся белый осадок собирали фильтрованием с получением 0,335 г (45%) (1R*,2R*)-1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола, у которого: ЯМР δ 7,63 (с, 4H), 7,07 (дд, J = 2, 11,5 Гц, 1H), 6,98-6,84 (м, 2H), 5,13 (с, 1H), 4,20 (д, J = 9,5 Гц, 1H), 3,06 (сим. м, 1H), 2,81-2,71 (м, 1H), 2,70-2,50 (м, 3H), 2,15 (сим.м, 2H), 1,81 (искаженный д, J = 14 Гц, 2H), 1,59 (с, 1H), 1,33-1,19 (м, 3H), 1,08 (д, J = 7 Гц, 18H), 0,81 (д, J = 6,5 Гц, 3H). Этот продукт также содержал около 7% эритро диастереоизомера, он был удобен для использования без дополнительной очистки.
Продукт описанной выше реакции (0,300 г, 0,527 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли фторид тетрабутиламмония (0,58 мл, 0,58 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 30 минут при комнатной температуре, а затем концентрировали. Остаток распределяли между этилацетатом и водой и фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (0,75 х 3,5 дюйма) с элюированием, проводимым следующим образом: 5% этилацетат/гексан (100 мл), ноль, 15% этилацетат/гексан (200 мл), ноль, 25% этилацетат/гексан (200 мл), ноль, 35% этилацетат/гексан (350 мл), 0,156 г (72%) белого твердого продукта. Этот твердый продукт суспендировали в метаноле (4 мл) и добавляли метансульфокислоту (0,025 мл, 0,38 ммоль). Смесь фильтровали, затем концентрировали. Остаток перекристаллизовывали из этанола с получением 0,085 г (32%) (1R*, 2R*)-1-(3-фтор-4-гидроксифенил)-2-(4-(4- трифторметилфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол мезилата с т.пл. 155-157oC. Анализ, рассчитано для C21H23F4NO3•CH4SO3: C 51,86, H 5,34, N 2,75. Найдено: C 51,94, H 5,58, N 2,76.
ПРИМЕР 14
(1R*, 2R*)-4-{ 2-(3-(4-хлорфенилсульфанил)-8- азабицикло(3.2.1)окт-8-ил)-1-гидроксипропил}-2-метилфенол
Смесь 3-метил-4-триизолпропилсилилокси- α -бромпропиофенона (соединения по приготовлению 6, 1,25 г, 3,14 ммоль), 3-(4-хлорфенилсульфанил)-8-азабицикло
(3.2.1)октана (соединения по приготовлению 41, 1,11 г, 4,40 ммоль) и триэтиламина (1,09 мл, 7,85 ммоль) в этаноле (17 мл) нагревали с обратным холодильником в течение 16 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между метиленхлоридом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: гексан (150 мл), ноль, 5% этилацетат/гексан (300 мл), сброшенный отгон, 10% этилацетат/гексан (200 мл) и 20% этилацетат/гексан (150 мл), 1,325 г (74%) 1-(3-метил-4-триизопропилсилилоксифенил)-2-{3-(4- хлорфенилсульфанил)-8-азабицикло(3.2.1)-октан-8-ил} -пропан-1-она в виде желтого масла, которое переносили непосредственно на следующий этап.
Смесь боргидрида натрия (0,082 г, 2,18 ммоль) и этанола (10 мл) перемешивали 10 минут, а затем добавляли 1-(3-метил-4-триизопропилсилилоксифенил)-2-{3-(4-хлорфенилсульфанил)-8- азабицикло(3.2.1)октан-8-ил}-пропан-1-он (1,247 г, 2,18 ммоль в 5 мл этанола) с двумя по 5 мл промывали этанолом. Реакционную смесь перемешивали при комнатной температуры в течение ночи, затем концентрировали. Остаток распределяли между метиленхлоридом и водой и фазы разделяли. Органическую фазу промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (200 мл), ноль, 20% этилацетат/гексан (500 мл), 0,475 г (38%) (1R*,2R*)-1-(3-метил-4-триизопропилсилилоксифенил)- 2-{3-(4-хлорфенилсульфонил)-8-азабицикло(3.2.1)октан-8-ил} -пропан-1-ола в виде масла, у которого: ЯМР δ 7,29 (ABкв, ΔV1-3 = 23 Гц, J = 8,5 Гц, 4H), 7,07 (д, J = 2 Гц, 1H), 6,94 (дд, J = 2,8 Гц, 1H), 6,70 (д, J = 8 Гц, 1H), 5,11 (бр,с, 1H), 4,00 (д, J = 8 Гц, 1H), 3,42 (бр,с, 1H), 3,27 (сим.м, 1H), 3,16 (бр,с, 1H), 2,59 (сим, м, 1H), 2,20 (с, 3H), 1,90-1,51 (м, 8H), 1,34-1,20 (м, 3H), 1,08 (д, J = 7 Гц, 18H), 0,79 (д, J = 6,5 Гц, 3H). Этот продукт также содержал около 10% эритро диастереоизомера, но был удобен для использования без дополнительной очистки. Необходимо отметить, что дальнейшее элюирование флэш-хроматографической колонки 25% этилацетат/гексаном (250 мл) и 30% этилацетат/гексаном (200 мл) дало 0,291 г эритро диастереоизомера в виде масла.
Продукт вышеописанной реакции (0,475 г, 0,828 ммоль) растворяли в тетрагидрофуране (14 мл) и добавляли фторид тетрабутиламмония (0,91 мл, 0,91 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 1 час при комнатной температуре, а затем концентрировали. Остаток распределяли между метиленхлоридом и водой и фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (0,75 х 3 дюйма) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (150 мл), ноль, 30% этилацетат/гексан (200 мл) и 40% этилацетат/гексан (300 мл), 0,183 г (52%) (1R*, 2R*)-4-{ 2-(3-(4-хлорфенилсульфанил)-8- азабицикло(3.2.1)окт-8-ил)-1-гидроксипропил}-2-метилфенола в виде белого твердого продукта. Образец, перекристаллизованный из этилацетата, имел т.пл. 168-169oC, ЯМР δ 7,31 (ABкв, ΔV1-3 = 19,5 Гц, J = 8,5 Гц, 4H), 7,09 (д, J = 2 Гц, 1H), 7,00 (дд, J = 2,8 Гц, 1H), 6,68 (д, J = 8 Гц, 1H), 5,10 (бр,с, 2H), 4,02 (д, J = 8 Гц, 1H), 3,45 (бр, с, 1H), 3,30 (сим.м, 1H), 3,22 (бр,с, 1H), 2,62 (сим.м, 1H), 2,23 (с, 3H), 1,92-1,68 (м, 5H), 1,68-1,55 (м, 3H), 0,82 (д, J = 6,5 Гц, 3H).
ПРИМЕР 15
(1R*, 2R*)-4-{ 2-(3-(4-хлорфенилсульфанил)-8- азабицикло(3.2.1)-окт-8-ил)-1-гидроксипропил}-2,6-диметилфенол
Смесь 3,5-диметил-4-триизопропилсилилокси- α -бромпропиофенона (соединения по приготовлению 41, 1,3 г, 3,14 ммоль), 3-(4-хлорфенилсульфанил)-8-азабицикло(3.2.1)
октана (1,11 г, 4,40 ммоль) и триэтиламина (1,09 г, 7,85 ммоль) в этаноле 17 мл) нагревали с обратным холодильником в течение 16 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между метиленхлоридом и водой. Фазы разделяли, а органичесий слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимы следующим образом: гексан (150 мл), ноль, 5% этилацетат/гексан (300 мл), сброшенный отгон, 10% этилацетат/гексан (200 мл) и 20% этилацетат/гексан (150 мл), 1,175 (64%) 1-(3,5-диметил-4-триизопропилсилилокси-фенил)-2-{ 3-(4- хлорфенилсульфанил)-8-азабицикло(3.2.1)октан-8-ил}-пропан-1-она в виде желтого масла, которое использовали непосредственно на следующем этапе).
Смесь боргидрида натрия (0,070 г, 1,86 ммоль) и этанола (10 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-диметил-4-триизопропилсилилоксифенил)-2-{ 3-(4- хлорфенилсульфанил)-8-азабицикло(3.2.1)октан-8-ил}-пропан-1-он (1,09 г, 2,86 ммоль в 5 мл этанола) с тремя промывками по 5 мл этанола. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Выпавший белый осадок собирали и высушивали с получением 0,22 г эритро продукта (1R*,2S*). Фильтрат концентрировали, а остаток распределяли между метиленхлоридом и водой. Фазы разделяли, и органическую фазу промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3,5 дюйма) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (200 мл), ноль, 20% этилацетат/гексан (500 мл), 0,208 г (19%) (1R*,2R*)-1-(3,5-диметил-4- триизопропилсилилоксифенил)-2-{ 3-(4-хлорфенилсульфанил)-8- азабицикло(3.2.1)октан-8-ил} -пропан-1-ола в виде масла, у которого: ЯМР δ 7,29 (ABкв ΔV1-3 = 22,5 Гц, J = 8,5 Гц, 4H), 6,88 (с, 2H), 5,08 (бр с, 1H), 3,98 (д, J = 7,5 Гц, 1H), 3,41 (бр с, 1H), 3,26 (сим.м, 1H), 3,14 (бр с, 1H), 2,60 (сим с, 1H), 2,22 (с, 6H), 1,90-1,50 (м, 8H), 1,34-1,20 (м, 3H), 1,08 (д, J = 7 Гц, 18H), 0,80 (д, J = 6,5 Гц, 3H). Этот продукт содержал более 10% эритро диастереоизомера и был удобен для использования без дополнительной очистки. Дальнейшее элюирование флэш-хроматографической колонки 20% этилацетат/гексаном (250 мл) дало 0,126 г эритро диастереоизомера в виде масла, в результате чего общее количество эритропродукта составило 0,346 г.
Продукт описанной выше реакции (0,196 г, 0,33 ммоль) растворяли в тетрагидрофуране (7 мл) и добавляли фторид тетрабутиламмония (0,37 мл, 0,37 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, а затем концентрировали. Остаток распределяли между метиленхлоридом и водой и фазы разделяли. Органический слой промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (0,75 х 2,5 дюйма) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (140 мл), ноль, 30% этилацетат/гексан (200 мл) и 40% этилацетат/гексан (75 мл), 0,144 г (100%) (1R*,2R*)-4-{2-(3-(4-хлорфенилсульфанил)-8- азабицикло(3.2.1)окт-8-ил)-1-гидроксипропил} -2,6-диметилфенола в виде светло-желтого масла. Образец, перекристаллизованный из этилацетата, имел т.пл. 143-144,5oC. ЯМР δ 7,31 (ABкв ΔV1-3 = 19,5 Гц, J = 8,5 Гц, 4H), 6,39 (с, 2H), 5,19 (бр с, 1H), 4,59 (брс, 1H), 3,98 (д, J = 8,5 Гц, 1H), 3,45 (брс, 1H), 3,29 (сим.м, 1H), 3,22 (брс, 1H), 2,62 (сим, м, 1H), 2,23 (с, 6H), 1,95-1,56 (м, 8H), 0,81 (д, J = 6,5 Гц, 3H).
ПРИМЕР 16
3R*, 4S* 6-фтор-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хроман-4,7-диол
Смесь 3,3-дибром-6-фтор-7-бензилоксихроман-4-она (соединения по приготовлению 31, 0,91 г, 2,12 ммоль), 4-(4-фторфенил)-4-гидроксипиперидина (0,83 г, 4,25 ммоль) и триэтиламина (0,60 мл, 4,30 ммоль) в ацетонитриле (30 мл) перемешивали в течение ночи при комнатной температуре. Выпавший желтый осадок собирали фильтрованием. Этот материал подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в метиленхлориде) с элюированием, проводимым следующим образом: 2% метанол-метиленхлорид (200 мл), ноль, 3% метанол/метиленхлорид (100 мл), 0,16 г (16%) 7-бензилокси-6-фтор-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)- хромен-4-она, который использовали без дополнительной очистки.
Смесь боргидрида натрия (0,13 г, 3,44 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 7-бензилокси-6-фтор-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)- хромен-4-он (0,16 г, 0,345 ммоль в 10 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь охлаждали водой и концентрировали. Остаток стирали с водой и фильтровали с получением 0,136 г белого твердого вещества, которое переносили непосредственно на следующий этап.
Продукт описанной выше реакции (0,13 г, 0,28 ммоль) растворяли в тетрагидрофуране (6 мл) и метаноле (6 мл) и добавляли формиат аммония (0,18 г, 2,85 ммоль) и 10% палладий на угле (0,09 г). Реакционную смесь перемешивали в течение ночи при комнатной температуре, а затем фильтровали через диатомовую землю. Прокладку фильтра промывали метанолом. Фильтрат концентрировали, а остаток интенсивно перемешивали с водным бикарбонатом. Твердые вещества (0,057 г) собирали и перекристаллизовывали из этанола с получением 0,022 г (20%) (3R*, 4S*)-6-фтор-3-(4-(4-фторфенил)-4- гидроксипиперидин-1- ил)-хроман-4,7-диола в виде белого твердого вещества с т. пл. 160-161oC, ЯМР (DMSO-d6) δ 9,84 (брс, 1H), 7,50 (дд, J = 5,6, 8,9 Гц, 2H), 7,11 (т, J = 8,9 Гц, 2H), 6,95 (д, J = 11,4 Гц, 1H), 6,31 (д, J = 7,7 Гц, 1H), 4,90 (брс, 1H), 4,86 (с, 1H), 4,62 (с, 1H), 4,20 (дд, J = 2,3, 10,3 Гц, 1H), 4,02 (т, J = 10,5 Гц, 1H), 2,95 (брд, J = 10,8 Гц, 1H), 2,85 (брд, J = 10,9 Гц, 1H), 2,73-2,60 (м, 2H), 2,57-2,52 (м, 1H частично затемнен растворителем ЯМР), 1,96-1,86 (м, 2H), 1,56 (брд, J = 13,4 Гц, 2H).
ПРИМЕР 17
(3R*, 4S*)-5-бром-6-метил-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хроман-4,7-диол
Смесь 3,3-дибром-6-метил-7-триизопропилсилилоксихроман-4-она и 6-метил-3,3,5-трибром-7-триизопропилсилилоксихроман-4-она (соединений по приготовлению 34, 1,0 г), 4-(4-фторфенил)-4-гидроксипиперидина (0,79 г, 4,05 ммоль) и триэтиламина (0,60 мг, 4,30 ммоль) в ацетонитриле (30 мл) перемешивали 30 минут при комнатной температуре. Выпавший осадок собирали фильтрованием с получением 0,188 г 5-бром-6-метил-7-триизопропилсилилокси-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хромен-4-она. Фильтрат подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (100 мл), 0,115 г 6-метил-7-триизопропилсилилокси-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4-она в виде светло-желтого твердого вещества с т.пл. 193-195oC, 20% этилацетат/гексан (100 мл) и 40% этилацетат/гексан (100 мл), 0,07 г смеси, 40% этилацетат/гексан (100 мл) и 60% этилацетат/гексан (400 мл), 0,30 г 6-метил-7-гидрокси-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)-хромен-4-она.
Смесь боргидрида натрия (0,11 г, 2,91 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 5-бром-6-метил-7- триизопропилсилилокси-3-(4-(4-фторфенил)-4-гидроксипиперидин-1- ил)-хромен-4-он (0,15 г, 0,285 ммоль в 10 мл этанола и 5 мл тетрагидрофурана). Реакционную смесь перемешивали при комнатной температуре в течение уикенда. Реакционную смесь охлаждали водой и концентрировали. Остаток стирали с водой и фильтровали с получением 0,14 г твердого вещества кремового цвета. Это твердое вещество подвергали флэш-хроматографии на силикагеле (1 х 3,5 дюйма в гексане) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (200 мл) и 30% этилацетат/гексан (100 мл), ноль, 30% этилацетат/гексан (100 мл) и 50% этилацетат/гексан (150 мл), 0,094 г (63%) (3R*,4S*)-5-бром-6-метил-7-триизопропилсилилокси-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)- хроман-4-ола в виде бледно-желтого твердого вещества с т.пл. 201-202,5oC. Анализ, рассчитано для C30H43BrFNO4Si: C 59,20, H 7,12, N 2,23. Найдено: C 59,30, H 7,41, N 2,25.
Продукт описанной выше реакции (0,09 г, 0,17 ммоль) растворяли в тетрагидрофуране (5 мл) и добавляли фторид тетрабутиламмония (0,175 мл, 0,175 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали в течение ночи при комнатной температуре, а затем концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (200 мл), ноль, 40% этилацетат-гексан (200 мл), ноль, 60% этилацетат/гексан (100 мл), ноль, 60% этилацетат/гексан (100 мл), 0,045 г (71%) (3R*, 4S*)-5-бром-6-метил-3-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4,7-диола в виде ярко-белого твердого вещества. Образец перекристаллизовывали из этанол/эфира с получением 0,035 г продукта с т.пл. 195,5-196oC. Анализ, рассчитано для C21H23BrFNO4: C 55,76, H 5,13, N 3,10. Найдено: C 55,70, H 5,23, N 3,07.
ПРИМЕР 18
(3R*, 4S*)-6-метил-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хроман-4,7-диол
Смесь 6-метил-7-гидрокси-3-(4-(4-фторфенил)-4-гидроксипиперидин-1- ил)-хромен-4-она (соединения по примеру 17, 0,30 г, 0,81 ммоль), карбоната калия (0,22 г, 1,59 ммоль) и бензилбромида (0,10 мл, 0,84 ммоль) в ацетоне нагревали с обратным холодильником в течение 6 часов. Реакционную смесь концентрировали, а остаток распределяли между 2:1 этилацетат/тетрагидрофураном и водой, с подогреванием для более эффективного растворения. Фазы разделяли, а органический слой промывали водой и насыщенным солевым раствором. Органическую фазу высушивали над сульфатом магния и концентрировали с образованием желтого твердого вещества. Это твердое вещество стирали с эфиром с получением 0,31 г (84%) 7-бензилокси-6-метил-3-(4-(4-фторфенил)-4-гидроксипиперидин)-1- ил)-хромен-4-она с т.пл. 245-245,5oC. Анализ, рассчитано для C28H26FNO4: C 73,19, H 5,70, N 3,05. Найдено: C 72,87, H 5,76, N 3,21.
Смесь боргидрида натрия (0,25 г, 6,61 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 7-бензилокси-6-метил-3-(4-(4-фторфенил)-4-гидроксипиперидин-1-ил)- хромен-4-он (0,30 г, 0,653 ммоль в 20 мл этанола и 15 мл тетрагидрофурана). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Дополнительно добавляли боргидрид натрия (0,12 г) и перемешивание продолжали в течение уикенда. Реакционную смесь охлаждали водой и концентрировали. Остаток стирали с водой и фильтровали с получением твердого вещества, которое представляло собой 2:1 смесь исходного материала и продукта. Этот материал перемешивали с горячим этанолом и фильтровали. Собранное твердое вещество весило 0,2 г и представляло собой чистый исходный материал, который можно было возвращать на этот этап восстановления. Этаноловый фильтрат концентрировали с получением 0,113 г (3R*, 4S*)-7-бензилокси-6-метил-3-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4-ола с т.пл. 201-202oC. Этот материал переносили непосредственно на следующий этап.
Продукт описанной выше реакции (0,080 г, 0,173 ммоль) растворяли в тетрагидрофуране (3 мл) и этаноле (3 мл) и добавляли формиат аммония (0,14 г, 2,22 ммоль) и 10% палладий на угле (0,06). Реакционную смесь перемешивали в течение уикенда при комнатной температуре, а затем фильтровали через диатомовую землю. Прокладку фильтра промывали тетрагидрофураном и этанолом. Фильтрат концентрировали, а остаток стирали с водой. Твердые вещества (0,045 г) собирали и перекристаллизовывали из этанол/эфира с получением 0,030 г (46%) (3R*, 4S*)-6-метил-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хроман-4,7-диола в виде белого твердого вещества с т.пл. 173,5-174oC, ЯМР (DMSO-d6) δ 9,10 (с, 1H), 7,37-7,32 (м, 2H), 6,94 (т, J = 8,9 Гц, 2H), 6,71 (с, 1H), 6,02 (с, 1H), 4,69 (с, 1H), 4,55 (д, J = 4,3 Гц, 1H), 4,43 (брс, 1H), 4,01 (д, J = 7,7 Гц, 1H), 3,83 (т, J = 10 Гц, 1H), 2,81 (брд, J = 11,2 Гц, 1H), 2,69 (брд, J = 10,8 Гц, 1H), 2,55-2,43 (м, 2H), 1,85 (с, 3H), 1,79-1,71 (м, 2H), 1,40 (брд, J = 13,3 Гц, 2H).
ПРИМЕР 19
(3R*, 4S*)-6,8-диметил-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хроман-4,7-диол
Смесь 3,3-дибром-6,8-диметил-7-триизопропилсилилоксихроман-4-она (соединения по приготовлению 28, 0,62 г, 1,22 ммоль), 4-(4-фторфенил)-4-гидроксипиперидина (0,48 г, 2,46 ммоль) и триэтиламина (0,68 г, 4,88 ммоль) в ацетонитриле (20 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь фильтровали, а фильтрат концентрировали. Остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Этот остаток подвергали флэш-хроматографии на силикагеле (1 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (250 мл), 0,23 г 3-бром-6,8-диметил-7-триизопропилсилилоксихромен-4-она, 20% этилацетат/гексан (250 мл), 0,14 г (21%) 6, 8-диметил-7-триизопропилсилилокси-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хромен-4-она, который использовали без дополнительной очистки.
Смесь боргидрида натрия (0,082 г, 2,17 ммоль) и этанола (3 мл) перемешивали 10 минут, а затем добавляли 6,8-диметил-7-триизопропилсилилокси-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хромен-4-он (0,117 г, 0,217 ммоль в 12 мл этанола и 3 мл тетрагидрофурана). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь охлаждали водой и концентрировали. Остаток стирали с водой и фильтровали с получением 0,110 г твердого вещества. Этот материал подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 25% этилацетат/гексан (300 мл), ноль, 25% этилацетат/гексан (300 мл), 0,064 г (54%) (3R*,4S*)-6,8-диметил-7-триизопропилсилилокси-3-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4-ола в виде белого твердого вещества с т.пл. 198-199oC. ЯМР δ 7,46 (дд, J = 5,5, 8,5 Гц, 2H), 7,04 (т, J = 8,7 Гц, 2H), 6,95 (с, 1H), 4,72 (д, J = 2,8 Гц, 1H), 4,38 (дд, J = 2,9, 10,4 Гц, 1H), 4,06 (т, J = 10,5 Гц, 1H), 3,09 (брд, J = 11,1 Гц, 1H), 2,80-2,68 (м, 4H), 2,19 (с, 3H), 2,11-2,02 (м, 4H), 1,90-1,76 (м, 3H), 1,38-1,21 (м, 3H), 1,12 (д, J = 7,1 Гц, 18H).
Продукт описанной выше реакции (0,060 г, 0,110 ммоль) растворяли в тетрагидрофуране (5 мл) и добавляли фторид тетрабутиламмония (0,115 мл, 0,115 ммоль, 1 М раствора тетрагидрофурана). Реакционную смесь перемешивали 1,5 часа при комнатной температуре, а затем концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (200 мл), ноль, 50% этилацетат/гексан (200 мл), ноль, 75% этилацетат/гексан (400 мл), бесцветное масло, которое отвердевало с получением 0,035 г (81%) (3R*, 4S*)-6,8-диметил-3-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-хроман-4,7-диола в виде белого твердого вещества. Образец перекристаллизовывали из этанол/эфира с получением 0,016 г продукта в двух сборах, с т.пл. 185,5-186oC, ЯМР (DMSO-d6) δ 8,17 (с, 1H), 7,52 (дд, J = 5,7, 8,7 Гц, 2H), 7,12 (т, J = 8,9 Гц, 2H), 6,76 (с, 1H), 4,86 (с, 1H), 4,69 (с, 1H), 4,61 (с, 1H), 4,29 (брд, J = 7,8 Гц, 1H), 4,02 (т, J = 10,5 Гц, 1H), 3,01 (брд, J = 10,7 Гц, 1H), 2,89 (брд, J = 12,4 Гц, 1H), 2,72-2,60 (м, 2H), 2,54-2,49 (м, 1H, частично затемненный растворителем ЯМР), 2,08 (с, 3H), 1,97-1,89 (с перекрыванием м, 5H), 1,58 (брд, J = 13 Гц, 2H).
ПРИМЕР 20
(3R*, 4S*)-6,8-диметил-3-(4-(4-фторфенил)-4- гидроксипиперидин-1-ил)-хроман-4,7-диол
Смесь 3-бром-6,8-диметил-7-триизопропилсилилоксихромен-4-она (соединения по примеру 19, 0,23 г, 0,54 ммоль), 4-(4-фторфенил)-4-гидроксипиперидина (0,22 г, 1,12 ммоль) и триэтиламина (0,3 мл, 2,15 ммоль) в ацетонитриле (15 мл) перемешивали в течение уикенда при комнатной температуре. Выпавший осадок собирали и промывали водой и эфиром. Это твердое вещество подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (100 мл), ноль, 25% этилацетат/гексан (200 мл), 0,065 г (22%) 6,8-диметил-7-триизопропилсилилокси-3-(4-(4- фторфенил)-4-гидроксипиперидин-1-ил)-хромен-4-она с т. пл. 226,5-227oC. Анализ, рассчитано для C31H42FNO4Si: C 68,98, H 7,84, N 2,59. Найдено: C 69,00, H 7,94, N 2,37. Этот продукт был идентичен продукту, выделенному на первом этапе примера 19, и превращался в указанный в заглавии продукт согласно процедуре примера 19.
ПРИМЕР 21
(1R*, 2R*)-1-(4-гидрокси-3-метоксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ол
Смесь 4-бензилокси- α -бром-3-метоксипропиофенона (соединения по приготовлению 46, 1,00 г, 2,86 ммоль), 4-гидрокси-4-фенилпиперидина (0,60 г, 3,39 ммоль) и триэтиламина (0,80 г, 5,74 ммоль) в этаноле (30 мл) нагревали с обратным холодильником в течение 3,5 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 1,25 г (98%) 1-(4-бензилокси-3-метоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1- ил)-пропан-1-она в виде светло-оранжевой пены, удобной для использования без дополнительной очистки, у которой: ЯМР δ 7,76 (дд, J = 2, 8,4 Гц, 2H), 7,71 (д, J = 2 Гц, 1H), 7,49-7,23 (м, 10H), 6,89 (д, J = 8,4 Гц, 1H), 5,22 (с, 2H), 4,16-4,11 (м, 1H), 3,93 (с, 3H), 2,94-2,62 (м, 4H), 2,13 (дкв, J = 4,3, 12,7 Гц, 2H), 1,78-1,69 (м, 2H), 1,32 (д, J = 6,8 Гц, 3H).
Смесь боргидрида натрия (0,10 г, 2,64 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(4-бензилокси-3-метоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1- ил)-пропан-1-он (1,13 г, 2,54 ммоль в 25 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь охлаждали водой и концентрировали при пониженном давлении и 40oC. Остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 1,16 г сырого продукта, который представлял собой 5:1 смесь (1R*,2R*) и (1R*,2S*) изомеров. Смесь перекристаллизовывали из этанол/эфир/гексана, а затем перекристаллизовывали из этанол/эфира с получением 0,47 г (41%) (1R*,2R*)-1-(4-бензилокси-3-метоксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ола с т.пл. 131-132oC. Анализ, рассчитано для C28H33NO4: C 75,14, H 7,43, N 3,13. Найдено: C 75,50, H 7,33, N 3,25.
Смесь продукта описанной выше реакции (0,40 г, 0,89 ммоль) и 10% палладия на угле (0,080 г) в метаноле (25 мл) и уксусной кислоты (0,5 мл) гидрогенизировали при 50 psi (начальное давление) в течение 5,5 часов при комнатной температуре, а затем фильтровали через диатомовую землю. Прокладку промывали метанолом. Фильтрат концентрировали, а остаток распределяли между этилацетатом и насыщенным водным бикарбонатом. Фазы разделяли, а органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Светло-желтую пену перекристаллизовывали из этанола с получением 0,195 г (61%) (1R*,2R*)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества с т. пл. 187,5-188oC. Анализ, рассчитано для C21H27NO4: C 70,56, H 7,61, N 3,92. Найдено: C 70,44, H 8,00, N 3,78.
ПРИМЕР 22
(1R*, 2R*)-1-(3,4-дигидроксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-пропан-1-ол
Смесь 2-бром-1-(2,2-дифенилбензо(1,3)диоксол-5-ил)-пропан-1-она (соединения по приготовлению 43, 2,00 г, 4,89 ммоль), 4-гидрокси-4-фенилпиперидина (0,90 г, 5,08 ммоль) и триэтиламина (1,40 мл, 10,04 ммоль) в этаноле (50 мл) нагревали с обратным холодильником в течение ночи. Растворитель удаляли при пониженном давлении, а остаток распределяли между эфиром и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (2 х 5 дюймов в гексане) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (500 мл), невзвешенный отгон, 50% этилацетат/гексан (500 мл), 1,76 (71%) 1-(2,2-дифенилбензо-(1,3)диоксол-5-ил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-она в виде светлой желтовато-коричневой пены, удобной для использования без дополнительной очистки, у которой: ЯМР δ 7,81 (дд, J = 1,7, 8,3 Гц, 1H), 7,70 (д, J = 1,6 Гц, 1H), 7,64-7,13 (м, 15H), 6,92 (д, J = 8,2 Гц, 1H), 4,07 (кв, J = 7,0 Гц, 1H), 3,39-3,27 (м, 1H), 2,94-2,59 (м, 3H), 2,30-2,04 (м, 2H), 1,74 (брт, J = 13,2 Гц, 2H), 1,30 (д, J = 6,8 Гц, 3H).
Смесь боргидрида натрия (0,15 г, 3,97 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(2,2-дифенилбензо(1,3)диоксол-5-ил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-пропан-1-он (1,70 г, 3,36 ммоль в 20 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение уикенда. Белый осадок собирали, промывали этанолом и эфиром и высушивали воздухом с получением 1,35 г сырого продукта. Этот продукт перекристаллизовывали из этанол/эфир/гексана, а затем перекристаллизовывали из этанол/этилацетат/метиленхлорида с получением 1,05 г (61%) (1R*,2R*)-1-(2,2-дифенилбензо(1,3)диоксол-5-ил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ола с т.пл. 224-224,5oC. Анализ, рассчитано для C33H33NO4: C 78,08, H 6,55, N 2,76. Найдено: C 78,16, H 6,46, N 2,72.
Смесь продукта описанной выше реакции (1,00 г, 1,97 ммоль) и 10% палладия на угле (0,175 г) в метаноле (50 мл) и уксусной кислоты (1,0 мл) гидрогенизировали при 50 psi (начальное давление) в течение 5 часов при комнатной температуре. Дополнительно добавляли катализатор (0,18 г) и гидрогенизацию продолжали в течение ночи. Реакционную смесь фильтровали через диатомовую землю, а прокладку фильтра промывали метанолом. Фильтрат концентрировали, а остаток распределяли между этилацетатом и насыщенным водным бикарбонатом и интенсивно перемешивали в течение 1 часа. Фазы разделяли и водный слой экстрагировали этилацетатом (2 х). Объединенный органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (500 мл), ноль, 10% метанол/этилацетат (250 мл), 20% метанол/этилацетат (250 мл) и 50% метанол/этилацетат (250 мл), 0,51 г (75%) светлого желто-зеленого твердого вещества. Это твердое вещество перекристаллизовывали из этанола с получением (1R*,2R*)-1-(3,4-дигидроксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества с т.пл. 167-168oC. Анализ, рассчитано для C20H25NO4•0,5C2H6O: C 68,83, H 7,70, N 3,82. Найдено: C 68,78, H 8,05, N 3,70.
ПРИМЕР 23
(1R*, 2R*)-1-(3-фтор-4-гидроксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3-фтор-4-триизопропилсилилокси- α -бромпропиофенона (2,0 г, 4,96 ммоль), 4- гидрокси-4-фенилпиперидина (1,1 г, 6,2 ммоль) и триэтиламина (0,9 мл, 6,5 ммоль) в этаноле (25 мл) нагревали с обратным холодильником в течение 6,5 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 6 дюймов в гексане). Продукт элюировали 15% этилацетат/гексаном с получением 1,82 г (73%) 1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-пропан-1-она в виде желтого масла, у которого: ЯМР δ 7,91 (дд, J = 2, 12 Гц, 1H), 7,84 (дд, J = 2,5, 8,5 Гц, 1H), 7,51-7,47 (м, 2H), 7,39-7,26 (м, 3H), 6,98 (т, J = 8,5 Гц, 1H), 4,07 (кв, J = 7 Гц, 1H), 2,92-2,84 (м, 2H), 2,69-2,64 (м, 2H), 2,23-1,95 (м, 2H), 1,82-1,70 (м, 2H), 1,38-1,22 (м, 6H), 1,12 (д, J = 7 Гц, 18H).
Смесь боргидрида натрия (0,12 г, 3,17 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-пропан-1-он (1,41 г, 2,82 ммоль в 25 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Выпавший белый осадок собирали фильтрованием с получением 0,14 г (10%) (1R*,2R*)-1-(3-фтор-4-триизопропилсилилоксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ола с т. пл. 140-141oC. Анализ, рассчитано для C29H44FNO3Si: C 69,42, H 8,84, N 2,79. Найдено: C 69,30, H 9,06, N 2,84. Фильтрат охлаждали водой и перемешивали в течение ночи. Выпавший осадок собирали, промывали водой и высушивали воздухом (1,5 г). Этот материал перекристаллизовывали из этанола с получением 0,72 г дополнительного продукта, в результате чего общее его количество составило 0,86 г (61%).
Продукт описанной выше реакции (0,72 г, 1,43 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли фторид тетрабутиламмония (1,45 мл, 1,45 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали в течение ночи при комнатной температуре, а затем концентрировали. К остатку добавляли эфир и воду и после интенсивного перемешивания собирали белое твердое вещество, которое высушивали на воздухе с получением 0,5 г свободного основания. Этот материал помещали в этанол и добавляли метансульфокислоту (0,093 мл, 1,43 ммоль). Смесь концентрировали и перекристаллизовывали из этанола с получением 0,476 г (75%) (1R*,2R*)-1-(3-фтор-4- гидроксифенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ол мезилата с т.пл. 198,5-199,5oC. Анализ, рассчитано для C20H24FNO3•CH4SO3: C 57,13, H 6,39, N 3,17. Найдено: C 57,02, H 6,45, N 3,33.
ПРИМЕР 24
(1R*, 2R*)-1-(3,5-дифтор-4-гидроксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ол мезилат
Смесь 3,5-дифтор-4-триизопропилокси- α -бромпропиофенона (2,46 г, 5,84 ммоль), 4-гидрокси-4-фенилпиперидина (155 г, 8,74 ммоль) и триэтиламина (1,6 мл, 11,5 ммоль) в этаноле (50 мл) нагревали с обратным холодильником в течение ночи. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 5 дюймов в гексане) и элюировали следующим образом: 10% этилацетат/гексан (250 мл), ноль, 10% этилацетат/гексан (250 мл) и 20% этилацетат/гексан (250 мл), 1,41 г (47%) 1-(3,5-дифтор-4-триизопропилсилилоксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-пропан-1-она в виде оранжевого масла, у которого: ЯМР δ 7,73 (длительно сопряженный д, J = 9 Гц, 2H), 7,46 (д, J = 8,5 Гц, 1H), 7,33 (т, J = 7,5 Гц, 2H), 7,27-7,21 (м, 1H), 4,00 (кв, J = 6,7 Гц, 1H), 2,91 (дт, J = 2,5, 13 Гц, 1H), 2,79-2,76 (м, 1H), 2,69-2,60 (м, 2H), 2,19-1,93 (м, 3H), 1,80-1,67 (м, 3H), 1,39-1,27 (м, 6H), 1,10 (д, J = 7 Гц, 18H).
Смесь боргидрида натрия (0,16 г, 4,23 ммоль) и этанола (5 мл) перемешивали 10 минут, а затем добавляли 1-(3,5-дифтор-4-триизопропилсилилоксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-пропан-1-он (1,40 г, 2,86 ммоль в 20 мл этанола). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь охлаждали водой и перемешивали в течение 4 часов. Выпавший белый осадок собирали фильтрованием и перекристаллизовывали из этанола с получением 0,46 г (32%) (1R*,2R*)-1-(3,5-дифтор-4-триизопропилсилилоксифенил)-2-
(4-гидрокси-4-фенилпиперидин-1-ил)- пропан-1-ола, у которого: ЯМР δ 7,54 (д, J = 7,5 Гц, 2H), 7,40 (т, J = 7,5 Гц, J = 8,5 Гц 2H), 7,31 (д, J = 7 Гц, 1H), 6,89 (д, J = 9 Гц, 2H), 5,28 (с, 1H), 4,18 (д, J = 9,5 Гц, 1H), 3,10 (дт, J = 2,2, 11,7 Гц, 1H), 2,73-2,69 (м, 2H), 2,62-2,51 (м, 2H), 2,30-2,06 (м, 2H), 1,90-1,83 (м, 2H), 1,36-1,20 (м, 3H), 1,10 (д, J = 7 Гц, 18H), 0,85 (д, J = 6,7 Гц, 3H). Анализ, рассчитано для C29H43F2NO3Si: C 67,02, H 8,34, N 2,69. Найдено: C 66,77, H 8,58, N 2,71.
Продукт описанной выше реакции (0,398 г, 0,81 ммоль) растворяли в тетрагидрофуране (13 мл) и добавляли фторид тетрабутиламмония (0,89 мл, 0,89 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 2 часа при комнатной температуре, а затем концентрировали. Добавляли несколько капель насыщенного водного раствора хлорида аммония и растворитель удаляли в атмосфере азота. Остаток перемешивали с насыщенным водным раствором бикарбоната и этилацетатом, а белый твердый осадок собирали и промывали водой и этилацетатом, а затем высушивали с получением 0,185 г свободного основания. Это свободное основание (0,150 г) суспендировали в метаноле и добавляли метансульфокислоту (0,027 мл, 0,417 ммоль). Смесь фильтровали, затем концентрировали при кипении до 0,5 мл с добавлением этилацетата (2 мл). Охлаждение и растирание давало белые кристаллы, которые собирали с получением 0,173 г (91%) (1R*,2R*)-1-(3,5-дифтор-4-гидроксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ол мезилата с т.пл. 216-218oC. Анализ, рассчитано для C20H23F2NO3•CH4SO3: C 54,89, H 5,92, N 3,05. Найдено: C 54,70, H 5,90, N 2,91.
ПРИМЕР 25
(1R*, 2R*)-1-(3-метил-4-гидроксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ол мезилат
Смесь 4-бензилокси-3-метил- α -бромпропиофенона (2,48 г, 7,45 ммоль), 4-гидрокси-4-фенилпиперидина (1,1 г, 6,21 ммоль) и триэтиламина (2,08 мл, 14,9 ммоль) в этаноле (17 мл) нагревали с обратным холодильником в течение 6 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между метиленхлоридом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в 10% этилацетат/гексан) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (500 мл), ноль, 20% этилацетат/гексан (250 мл), ноль, 50% этилацетат/гексан (400 мл), 2,14 г сырого продукта. Этот продукт перекристаллизовывали из эфир/гексана с получением 1,41 г (53%) 1-(4-бензилокси-3-метилфенил)-2-(4-гидрокси-4-фенилпиперидин-1- ил)-пропан-1-она в виде твердого вещества с т.пл. 98-99oC, ЯМР δ 8,02 (дд, J = 2, 8,5 Гц, 1H), 7,97 (д, J = 1,5 Гц, 1H), 7,53-7,20 (м, 10H), 6,92 (д, J = 8,5 Гц, 1H), 5,17 (с, 2H), 4,14 (кв, J = 7 Гц, 1H), 2,95-2,75 (м, 3H), 2,64 (дт, J = 2,5, 12 Гц, 1H), 2,33 (с, 3H), 2,22-2,02 (м, 2H), 1,82-1,70 (м, 2H), 1,55 (брс, 1H), 1,33 (д, J = 7 Гц, 3H).
Смесь гидрида литий-алюминия (0,246 г, 6,48 ммоль) и тетрагидрофурана (45 мл) охлаждали до 0oC и добавляли 1-(4-бензилокси-3-метилфенил)-2-(4-гидрокси-4-фенилпиперидин-1-ил)- пропан-1-он (1,39 г, 3,24 ммоль) весь сразу, в виде твердого вещества. Реакционная смесь достигала комнатной температуры и подвергалась перемешиванию в течение ночи. Реакционную смесь осторожно охлаждали водой (0,467 мл) и перемешивали 4 часа. Суспензию высушивали сульфатом натрия, фильтровали через диатомовую землю и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3 дюйма в 20% этилацетат/гексане) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (150 мл), ноль, 30% этилацетат/гексан (250 мл) и 40% этилацетат/гексан (250 мл), 0,701 г (50%) (1R*,2R*)-1-(4-бензилокси-3- метилфенил)-2-(4-гидрокси-4-фенилпиперидин)-1-ил)-пропан-1-ола в виде белого твердого вещества с т.пл. 162-163oC. ЯМР δ 7,53-7,26 (м, 10H), 7,17 (брд, 1H), 7,11 (брд, J = 8,5 Гц, 1H), 6,83 (д, J = 8,5 Гц, 2H), 5,23 (с, 1H), 5,07 (с, 2H), 4,21 (д, J = 9,5 Гц, 1H), 3,08 (сим м, 1H), 2,83-2,56 (м, 4H), 2,28 (м, 3H), 2,28-2,05 (м, 2H), 1,84 (брд, J = 13,5 Гц, 2H), 1,54 (с, 1H), 0,82 (д, J = 6,5 Гц, 3H).
Продукт описанной выше реакции (0,69 г, 1,6 ммоль) растворяли в тетрагидрофуране (30 мл) и метаноле (30 мл) и добавляли формиат аммония (1,0 г, 16 ммоль) и 10% палладий на угле (0,15 г). Реакционную смесь перемешивали 2 часа при комнатной температуре, а затем фильтровали через диатомовую землю. Прокладку фильтра промывали этанолом и водой. Фильтрат концентрировали, а остаток перемешивали с этилацетатом и насыщенным водным бикарбонатом. Твердый осадок собирали, промывали эфиром и высушивали воздухом с получением 0,611 г (100%) (1R*,2R*)-1-(4-гидрокси-3-метилфенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества. Это твердое вещество подвергали флэш-хроматографии на силикагеле (1 х 3 дюйма в 50% этилацетат/гексане) с градиентом элюирования от 50% этилацетат/гексана до 2% метанол/этилацетата. Фракции продукта объединяли, концентрировали и перекристаллизовывали из нитрометана до получения 0,063 г (11,5%) чистого свободного основания. Анализ, рассчитано для C21H27NO3: C 73,87, H 7,97, N 4,10. Найдено: C 73,60, H 8,21, N 4,22. Этот продукт превращали в его соль мезилат. Его суспендировали в метаноле (в нескольких каплях) и добавляли метансульфокислоту (0,010 мл, 0,152 ммоль). Смесь разводили изопропанолом (1 мл) и концентрировали до приблизительно 0,25 мл при кипении. Образовавшиеся при охлаждении кристаллы собирали и получали мезилат в виде кристаллического белого твердого вещества (0,053 г) с т.пл. 196-197oC.
ПРИМЕР 26
(1R*, 2R*)-1-(3,5-диметил-4-гидроксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ол мезилат
Смесь 4-бензилокси-3,5-диметил- α -бромпропиофенона (2,59 г, 7,45 ммоль), 4-гидрокси-4-фенилпиперидина (1,1 г, 6,21 ммоль) и триэтиламина (2,08 мл, 14,9 ммоль) в этаноле (15 мл) нагревали с обратным холодильником в течение 6 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между метиленхлоридом и водой. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1,5 х 3,5 дюйма в 10% этилацетат/гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (500 мл), ноль, 20% этилацетат/гексан (250 мл), ноль, 50% этилацетат/гексан (400 мл), 2,16 г (79%) 1-(4-бензилокси-3,5-диметилфенил)-2-(4-гидрокси-4-фенилпиперидин-1- ил)-пропан-1-она в виде оранжевой пены, у которой: ЯМР δ 7,82 (с, 2H), 7,55-7,21 (м, 10H), 4,87 (с, 2H), 4,17 (кв, J = 7 Гц, 1H), 2,93-2,78 (м, 3H), 2,66 (дт, J = 3, 12 Гц, 1H), 2,35 (с, 6H), 2,26-2,04 (м, 2H), 1,95-1,69 (м, 3H), 1,34 (д, J = 7 Гц, 3H). Этот материал имел около 15% неидентифицированных примесей, но был удобен для использования без дополнительной очистки.
Смесь гидрида литий-алюминия (0,257 г, 6,77 ммоль) и тетрагидрофурана (45 мл) охлаждали до 0oC и добавляли 1-(4-бензилокси-3,5-диметилфенил)-2-(4-гидрокси-4-фенилпиперидин-1- ил)-пропан-1-он (1,50 г, 3,38 ммоль) весь сразу. Реакционная смесь достигала комнатной температуры, после чего ее перемешивали в течение ночи. Реакционную смесь осторожно охлаждали водой (0,487 мл) и перемешивали 4 часа. Суспензию высушивали сульфатом натрия, фильтровали через диатомовую землю и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 3 дюйма в смеси 20% этилацетата/гексана) с элюированием, проводимым следующим образом: 20% этилацетат/гексан (100 мл), ноль, 20% этилацетат/гексан (100 мл) и 30% этилацетат/гексан (250 мл), 1,32 г (66%) (1R*,2R*)-1-(4-бензилокси-3,5-диметилфенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ола в виде желтого твердого вещества с т.пл. 133-135oC, ЯМР δ 7,54-7,48 (м, 4H), 7,47-7,25 (м, 6H), 7,02 (с, 2H), 5,23 (с, 1H), 4,79 (с, 2H), 4,19 (д, J = 9,5 Гц, 1H), 3,09 (сим м, 1H), 2,81-2,59 (м, 4H), 2,29 (с, 6H), 2,30-2,25 (м, 2H), 1,85 (брд, J = 13,5 Гц, 2H), 1,54 (с, 1H), 0,84 (д, J = 6,5 Гц, 3H).
Продукт описанной выше реакции (1,30 г, 2,92 ммоль) растворяли в тетрагидрофуране (50 мл) и метаноле (50 мл) и добавляли формиат аммония (1,8 г, 29 ммоль) и 10% палладий на угле (0,3 г). Реакционную смесь перемешивали 2 часа при комнатной температуре, а затем фильтровали через диатомовую землю. Прокладку фильтра промывали этанолом и водой. Фильтрат концентрировали, а остаток распределяли между хлороформом, насыщенным водным бикарбонатом и небольшим количеством ацетона. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали и концентрировали с получением 0,886 г (86%) (1R*,2R*)-1-(3,5-диметил-4-гидрокси-фенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества. Этот продукт превращали в его соль мезилат. Продукт суспендировали в метаноле (нескольких мл) и добавляли метансульфокислоту (0,163 мл, 2,52 ммоль). Смесь концентрировали, а остаток стирали с эфиром. Оставшееся твердое вещество перекристаллизовывали из изопропанола с получением 0,273 г (24%) соли мезилата с т.пл. 203-204oC. Анализ, рассчитано для C22H29NO3•CH4SO3•0,5H2O: C 59,98, H 7,44, N 3,04. Найдено: C 60,10, H 7,63, N 3,13.
ПРИМЕР 27
(1R*, 2R*)-1-(4-гидрокси-3-метоксифенил)-2-(4- гидрокси-4-фенилпиперидин-1-ил)-пропан-1-ол и (1S,2S)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1- ил)-пропан-1-ол.
Продукт по примеру 21 растворяли в этаноле и разделяли на два энантиомера с помощью ЖХВД со следующими хроматографическими условиями: колонка, Chiralcel OD, мобильная фаза, 25% этанол/75% гексан, температура комнатная (приблизительно 22oC), обнаружение, УФ на 215 нм. При этих условиях (1R, 2R)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4-фенилпиперидин-1- ил)-пропан-1-ол элюировали со временем задержки приблизительно 9,12 минут, а (1S, 2S)-1-(4-гидрокси-3-метоксифенил)-2-(4-гидрокси-4- фенилпиперидин-1-ил)-пропан-1-ол элюировали со временем задержки приблизительно 16,26 минут.
ПРИГОТОВЛЕНИЕ 1. 4-пропионил-2-метилфенол
Смесь 2-метилфенола (10,48 г, 96,91 ммоль), пропионовой кислоты (5,23 мл, 97,88 ммоль) и трифторметансульфокислоты (50 г) нагревали до 80oC и поддерживали эту температуру в течение 30 минут. Реакционную смесь охлаждали, выливали в лед и экстрагировали хлороформом. Органическую фазу отделяли и промывали водным бикарбонатом и насыщенным солевым раствором. Затем ее высушивали, фильтровали и концентрировали с получением коричневого твердого вещества. Этот остаток дистиллировали при 1,5 мм рт.ст. с получением двух фракций: 25-150oC (сброшенный отгон), 160oC (5,58 г, 35%) 4-пропионил-2-метилфенола в виде белого кристаллического вещества с т.пл. 83-85oC, ЯМР δ 7,81 (д, J = 1,5 Гц, 1H), 7,76 (дд, J = 2, 8,5 Гц, 1H), 6,88 (д, J = 8,5 Гц, 1H), 6,61 (с, 1H), 2,98 (кв, J = 7,5 Гц, 2H), 2,30 (с, 3H), 1,22 (т, J = 7,5 Гц, 3H).
ПРИГОТОВЛЕНИЕ 2. 4-пропионил-2-метилфенол
К смеси хлорида алюминия (51,8 г, 0,388 моль) и метиленхлорида (140 мл) добавляли пропионилхлорид (11,25 мл, 0,129 моль), затем - 2-метилфенол (7,0 г, 64,73 ммоль в метиленхлориде (25 мл с 10 мл ополаскиванием)). Смесь перемешивали 2 часа при комнатной температуре, а затем выливали ее в лед. Фазы разделяли и органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором. Органическую фазу высушивали над сульфатом кальция и концентрировали на силикагеле. Этот материал подвергали флэш-хроматографии на силикагеле (3,5 х 3 дюйма) с элюированием, проводимым следующим образом: гексан (200 мл), ноль, 4% этилацетат/гексан (1000 мл), ноль, 8% этилацетат/гексан (2000 мл), 8,17 г 4-пропионил-2-метилфенилпропионата в виде светло-желтого масла, у которого: ЯМР δ 7,86 (с, 1H), 7,82 (дд, J = 2, 8,5 Гц, 1H), 2,98 (кв, J = 7 Гц, 2H), 2,65 (кв, J = 7,5 Гц, 2H), 2,23 (с, 3H), 1,30 (т, J = 7,5 Гц, 3H), 1,22 (т, J = 7,5 Гц, 3H).
Продукт описанной выше реакции (7,61 г, 34,57 ммоль) добавляли к смеси метанола (100 мл), воды (100 мл) и гидроксида калия (3,88 г, 68,14 ммоль) и нагревали с обратным холодильником в течение 1,5 часов. Метанол удаляли при пониженном давлении, а остаток подкисляли 6 Н HCl. Водную фазу экстрагировали этилацетатом. Этот органический слой промывали водным бикарбонатом и водой, затем его высушивали и концентрировали с получением 5,29 г (93%) 4-пропионил-2-метилфенола в виде белого кристаллического твердого вещества, которое было идентично материалу, полученному в приготовлении 1.
ПРИГОТОВЛЕНИЕ 3. 4-триизопропилсилилокси-3-метилпропиофенон
К смеси 4-пропионил-2-метилфенола (соединения по приготовлениям 1 и, 2, 5,19 г, 31,63 ммоль) и имидазола (4,31 г, 63,26 ммоль) в диметилформамиде (35 мл) добавляли триизопропилсилилхлорид (7,44 мл, 34,79 ммоль в 15 мл диметилформамида с 2 мл ополаскиванием). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем выливали ее в смесь льда и 1 Н водного раствора хлорида лития. Смесь экстрагировали этилацетатом (3 х). Объединенную органическую фазу промывали 1 Н хлоридом лития и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали с получением 9,61 г (95%) 4-триизопропилсилилокси-3-метил-пропиофенона в виде желтого масла, содержавшего небольшую примесь силила, определенную при ЯМР, но удобного для использования без дополнительной очистки. ЯМР этого продукта: δ 7,80 (д, J = 2 Гц, 1H), 7,71 (дд, J = 2,5, 8,5 Гц, 1H), 6,80 (д, J = 8,5 Гц, 1H), 2,94 (кв, J = 7,5 Гц, 2H), 2,28 (с, 3H), 1,33 (сим м, 3H), 1,20 (т, J = 7,5 Гц, 3H), 1,12 (д, J = 7 Гц, 18H).
ПРИГОТОВЛЕНИЕ 4. 4-бензилокси-3-метилпропиофенон
Смесь бензилбромида (4,44 мл, 37,35 ммоль), карбоната калия (9,38 г, 67,88 ммоль) и 4-пропионил-2-метилфенола (соединения по приготовлению 1 и 2, 5,57 г, 33,94 ммоль) в ацетоне (100 мл) перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли при пониженном давлении, а остаток распределяли между водой и метиленхлоридом. Фазы разделяли, а органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Этот остаток подвергали флэш-хроматографии на силикагеле (2 х 4 дюйма) с элюированием, проводимым следующим образом: 5% этилацетат/гексан (300 мл), сброшенный отгон, 5% этилацетат/гексан (500 мл) и 10% этилацетат/гексан (600 мл), 8,03 г (93%) 4-бензилокси-3-метилпропиофенона в виде белого кристаллического твердого вещества, у которого: ЯМР δ 7,83-7,78 (м, 2H), 7,48-7,31 (м, 5H), 6,89 (д, J = 9 Гц, 1H),
5,14 (с, 2H), 2,94 (кв, J = 7,5 Гц, 2H), 2,31 (с, 3H), 1,20 (т, J = 7,5 Гц, 3H). Этот материал был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 5. 4-бензилокси-3-метил-α-бромпропиофенон
К смеси 4-бензилокси-3-метилпропиофенона (соединения по приготовлению 4, 7,89 г, 31,06 ммоль) и четыреххлористого углерода (80 мл) добавляли по каплям бром (1,63 мл, 31,68 ммоль в 20 мл четыреххлористого углерода с 5 мл ополаскиванием), цвет которого почти исчезал при контакте с реакционным раствором. Реакционную смесь перемешивали 15 минут при комнатной температуре, а затем добавляли водный раствор бисульфита натрия. Смесь перемешивали еще 30 минут. Фазы разделяли, а органический слой промывали водным бикарбонатом и насыщенным солевым раствором. Органический слой высушивали над сульфатом кальция и концентрировали с получением 10,29 г (99,5%) продукта, указанного в заглавии, в виде светлого желтовато-коричневого твердого вещества с т. пл. 88,5-89,5oC, удобного для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 6. 4-триизопропилсилилокси-3-метил-бромпропиофенон
К раствору 4-триизопропилсилилокси-3-метилпропиофенона (соединения по приготовлению 3, 9,35 г, 29,19 ммоль) в четыреххлористом углероде (100 мл) добавляли по каплям бром (1,53 мл, 29,77 ммоль в 20 мл четыреххлористого углерода), цвет которого почти исчезал при контакте с реакционным раствором. Реакционную смесь перемешивали 15 минут, затем добавляли водный бисульфит и смесь перемешивали еще 15 минут. Фазы разделяли, а органический слой промывали водным бикарбонатом и насыщенным солевым раствором. Органический слой высушивали над сульфатом кальция и концентрировали с получением 11,65 г (100%) 4-триизопропилсилилокси-3-метил- α -бромпропиофенона в виде светло-желтого масла, у которого: ЯМР δ 7,86 (д, J = 2 Гц, 1H), 7,78 (дд, J = 2,5, 8,5 Гц, 1H), 6,82 (д, J = 8,5 Гц, 1H), 5,27 (кв, J = 6,5 Гц, 1H), 2,29 (с, 3H), 1,88 (д, J = 6,5 Гц, 3H), 1,42-1,27 (м, 3H), 1,13 (д, J = 7 Гц, 18H). Этот материал был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 7. 4-пропионил-2-фторфенол
К смеси хлорида алюминия (45,8 г, 0,343 моль) и метиленхлорида (140 мл) добавляли пропионилхлорид (10,85 мл, 124,9 ммоль) весь сразу, а затем добавляли 2-фторфенол (5,57 мл, 62,44 ммоль в 25 мл метиленхлорида с 10 мл ополаскиванием).
Смесь осторожно нагревали с обратным холодильником в течение 5 часов, охлаждали до комнатной температуры и выливали в лед. Фазы разделяли, а водный слой экстрагировали метиленхлоридом. Объединенный органический слой промывали водным бикарбонатом и насыщенным солевым раствором. Органический слой высушивали над сульфатом кальция и концентрировали с получением 11,43 г (82%) 4-пропионил-2-фторфенилпропионата в виде прозрачного желтовато-коричневого масла, которое использовали без характеристики.
Продукт описанной выше реакции (10,76 г, 48,01 ммоль) добавляли к смеси метанола (125 мл), воды (125 мл) и гидроксида калия (5,39 г, 56,11 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 2 часов, охлаждали и метанол удаляли при пониженном давлении. Остаток подкисляли 6 Н HCl и экстрагировали этилацетатом. Органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали до желтовато-коричневого твердого вещества. Этот желтовато-коричневый остаток подвергали флэш-хроматографии на силикагеле (2 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: 5% этилацетат/гексан (800 мл), сброшенный отгон, 10% этилацетат/гексан (500 мл), ноль, 25% этилацетат/гексан (1000 мл), 5,56 г (69%) 4-пропионил-2-фторфенола в виде белого твердого вещества с т.пл. 104-106oC, ЯМР δ 7,74 (дд, J = 2, 9,5 Гц, 1H), 7,71-7.68 (м, 1H), 7,05 (т, J = 8,5 Гц, 1H), 5,82 (брс, 1H), 2,93 (кв, J = 7,5 Гц, 2H), 1,20 (т, J = 7 Гц, 3H).
ПРИГОТОВЛЕНИЕ 8. 4-пропионил-2-фторфенил
Смесь 4-бром-2-фторфенола (1,0 г, 5,24 ммоль) и тетрагидрофурана (15 мл) охлаждали до -78oC и быстро, по каплям добавляли бутиллитий (4,6 мл, 11,5 ммоль, 2,5 М раствор). Реакционную смесь перемешивали 12 минут и добавляли N-метил-N-метоксипропионамид (соединение по приготовлению 9, 0,735 г, 6,28 ммоль в 1 мл тетрагидрофурана с 1 мл ополаскиванием). Реакционную смесь перемешивали 5 минут при -78oC, а затем она нагревалась до комнатной температуры. Добавляли несколько капель воды, затем растворитель удаляли при пониженном давлении. Остаток помещали в метиленхлорид и промывали водным хлоридом аммония и насыщенным солевым раствором. Органический слой высушивали и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 2,5 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (250 мл), сброшенный отгон, 20% этилацетат/гексан (250 мл), 0,186 г желтого кристаллического твердого вещества, ЯМР которого был аналогичен таковому в приготовлении 7.
ПРИГОТОВЛЕНИЕ 9. N-метил-N-метоксипропионамид
Смесь N, O-диметилгидроксиламин гидрохлорида (4,43 г, 45,39 ммоль) и триэтиламина (6,93 мл, 49,71 ммоль) в метиленхлориде (150 мл) охлаждали до 0oC и по каплям добавляли пропионилхлорид (3,76 мл, 43,23 ммоль в 25 мл метиленхлорида с 25 мл ополаскиванием). Смесь оставляли стоять до приобретения ею комнатной температуры и перемешивали в течение уикенда. Реакционную смесь экстрагировали водой и насыщенным солевым раствором, высушивали и концентрировали с получением 3,08 г (61%) N-метил-N-метоксипропионамида в виде желтого масла, у которого: ЯМР δ 3,66 (с, 3H), 3,16 (с, 3H), 2,42 (кв, J = 7,5 Гц, 2H), 1,12 (т, J = 7,5 Гц, 3H). Этот материал был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 10. 4-триизопропилсилилокси-3-фторпропиофенон
Смесь 4-пропионил-2-фторфенола (соединения приготовления 7, 5,44 г, 32,37 ммоль), имидазола (4,41 г, 64,74 ммоль) и триизопропилсилилхлорида (7,62 мл, 35,60 ммоль) в диметилформамиде (55 мл) перемешивали 15 часов при комнатной температуре. Реакционную смесь выливали в смесь льда и 1 Н водного раствора хлорида лития. Смесь экстрагировали этилацетатом (3 х). Объединенную органическую фазу промывали 1 Н хлоридом лития и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали с получением 10,5 г (100%) 4-триизопропилсилилокси-3-фторпропиофенона в виде желтого масла, у которого: ЯМР δ 7,75-7,60 (м, 2H), 6,95 (т, J = 8 Гц, 1H), 2,92 (кв, J = 7 Гц, 2H), 1,25 (сим м, 3H), 1,19 (т, J = 7,5 Гц, 3H), 1,09 (д, J = 7 Гц, 18H). Этот материал был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 11. 4-триизопропилсилилокси-3-фтор- α -бромпропиофенон
К раствору 4-триизопропилсилилокси-3-фторпропиофенона (соединения по приготовлению 10, 10,27 г, 31,67 ммоль) в четыреххлористом углероде (110 мл) добавляли по каплям бром (1,66 мл, 32,3 ммоль в 20 мл четыреххлористого углерода). (Следует заметить, что после добавления первых нескольких капель раствора брома его цвет не исчезал. Для инициирования реакции добавляли одну каплю 48% HBr и смесь перемешивали 5 минут до исчезновения цвета. Затем по каплям добавляли оставшийся раствор брома). Смесь перемешивали 15 минут, затем добавляли водный бисульфит и реакционную смесь перемешивали еще 15 минут. Фазы разделяли и органический слой промывали водным бикарбонатом и насыщенным солевым раствором. Органический слой высушивали над сульфатом кальция и концентрировали с получением 11,68 г (91%) 4-триизопропилсилилокси-3-фтор- α -бромпропиофенона в виде желтого масла, у которого: ЯМР δ 7,80-7,69 (м, 2H), 6,99 (т, J = 8,5 Гц, 1H), 5,20 (кв, J = 6,5 Гц, lH), 1,89 (д, J = 6,5 Гц, 3H), 1,28 (сим м, 3H), 1,12 (д, J = 7 Гц, 18H). Этот продукт был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 12. 2,6-дихлор-4-пропионилфенол
Смесь 2,6-дихлорфенола (10,10 г, 61,96 ммоль) и пропионовой кислоты (3,34 мл, 62,58 ммоль) в трифторметансульфокислоте (50 г) нагревали до 80oC и поддерживали эту температуру в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры, выливали в лед и экстрагировали хлороформом (3 х). Объединенный органический слой промывали водным бикарбонатом и насыщенным солевым раствором, высушивали и концентрировали с получением 8,90 г (66%) 2,6-дихлор-4-пропионилфенола в виде желтовато-коричневого твердого вещества с т. пл. 50-52oC, ЯМР δ 7,89 (с, 2H), 6,29 (с, 1H), 2,91 (кв, J = 7 Гц, 2H), 1,20 (т, J = 7 Гц, 3H). Этот материал был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 13. 3,5-дихлор-4-триизопропилсилилоксипропиофенон
Смесь 2,6-дихлор-4-пропионилфенола (соединения по приготовлению 12, 8,67 г, 39,59 ммоль), имидазола (5,39 г, 79,18 ммоль) и триизопропилсилилхлорида (9,32 мл, 43,56 ммоль) в диметилформамиде (90 мл) перемешивали 15 часов при комнатной температуре. Реакционную смесь выливали в смесь льда и 1 Н раствора хлорида лития. Смесь экстрагировали этилацетатом (3 х). Объединенную органическую фазу промывали 1 Н хлоридом лития и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (3 х 3 дюйма в гексане) с элюированием, проводимым следующим образом: гексан (200 мл), ноль, 2% этилацетат/гексан (400 мл), ноль, 5% этилацетат/гексан (400 мл), ноль, 5% этилацетат/гексан (500 мл) и 8% этилацетат/гексан (200 мл), 5,72 г белого твердого вещества. Этот материал дистиллировали по способу Kugelrohr при 1,5 мм рт. ст. и собирали следующие фракции: 70oC (температура в сосуде), сброшенный отгон, 130oC, сброшенный отгон, 150-170oC, 3,84 г (26%) 3,5-дихлор-4-трииизопропилсилилоксипропиофенона в виде белого твердого вещества, содержавшего небольшую примесь по ЯМР, но удобного для использования без дополнительной очистки. Образец, вновь дистиллированный по способу Kugelrohr имел т. пл. 74-76oC. ЯМР δ 7,88 (с, 2H), 2,92 (кв, J = 7 Гц, 2H), 1,45 (сим м, 3H), 1,21 (т, J = 7 Гц, 3H), 1,14 (д, J = 7,5 Гц, 18H).
ПРИГОТОВЛЕНИЕ 14. 3,5-дихлор-4-триизопропилсилилокси- α -бромпропиофенон
К раствору 3,5-дихлор-4-триизопропилсилилоксипропиофенона (соединения по приготовлению 13, 3,84 г, 10,23 ммоль) в четыреххлористом углероде (45 мл) добавляли по каплям бром (0,54 мл, 10,48 ммоль в 5 мл четыреххлористого углерода). После того как добавляли первые несколько капель раствора брома, добавление приостанавливали до инициации реакции, которую определяли по исчезновению красного цвета раствора. Затем добавление раствора брома доводили до конца (общее время добавления составляло 20 минут). Реакционную смесь перемешивали 1 час, затем добавляли водный бисульфит и смесь перемешивали еще 1,5 часа. Слои разделяли и органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором. Органический слой высушивали над сульфатом магния и концентрировали с получением 4,88 г (100%) 3,5-дихлор-4-триизопропилсилилокси- α -бромпропиофенона в виде бледно-желтого масла, у которого: ЯМР δ 7,95 (с, 2H), 5,15 (кв, J = 6,7 Гц, 1H), 1,89 (д, J = 6,7 Гц, 3H), 1,53-1,42 (м, 3H), 1,15 (д, J = 7,4 Гц, 18H). Спектр ЯМР также показывал некоторые небольшие примеси, но продукт был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 15. 2,6-диметил-4-пропионилфенол
К смеси хлорида алюминия (32,0 г, 24 ммоль) и метиленхлорида (100 мл) добавляли пропионилхлорид (3,56 мл, 40,95 ммоль) весь сразу, а затем добавляли 2,6-диметилфенол (5,0 г, 40,93 ммоль в 25 мл метиленхлорида) в течение 5 минут. После перемешивания в течение 1 часа при комнатной температуре добавляли второй эквивалент пропионилхлорида (3,56 мл). Реакционную смесь перемешивали 2 часа, а затем осторожно охлаждали водой. Смесь экстрагировали эфиром (3 х) и объединенную органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором, затем ее высушивали над сульфатом магния и концентрировали с получением 8,18 г (85%) 2,6-диметил-4-пропионилфенилпропионата в виде воскоподобного желтовато-коричневого твердого вещества, у которого: ЯМР δ 7,68 (с, 2H), 2,95 (кв, J = 7,3 Гц, 2H), 2,65 (кв, J = 7,5 Гц, 2H), 2,19 (с, 6H), 1,32 (т, J = 7,6 Гц, 3H), 1,20 (т, J = 7,3 Гц, 3H). Этот продукт также содержал небольшое количество примесей по спектру ЯМР, но был удобен для использования без дополнительной очистки.
Продукт описанной выше реакции (8,18 г, 34,91 ммоль) добавляли к смеси метанола (100 мл), воды (100 мл) и гидроксида калия (3,9 г, 69,5 ммоль) и реакционную смесь нагревали с обратным холодильником в течение 1 часа. Метанол удаляли при пониженном давлении, а остаток подкисляли до pH 4 с помощью 6 Н HCl. Эту водную фазу экстрагировали эфиром. Органический слой промывали водным бикарбонатом (2 х), высушивали над сульфатом магния и концентрировали до получения 5,4 г (87%) 2,6-диметил-4-пропионилфенола в виде воскоподобного желтовато-коричневого твердого вещества, у которого: ЯМР δ 7,65 (с, 2H), 5,47 (с, 1H), 2,94 (кв, J = 7,3 Гц, 2H), 2,30 (с, 6H), 1,21 (т, J = 7,3 Гц, 3H).
ПРИГОТОВЛЕНИЕ 16. 2,6-диметил-4-пропионилфенол
Смесь 2,6-диметилфенола (10,5 г, 85,95 ммоль), пропионовой кислоты (4,64 мл, 86,81 ммоль) и трифторметансульфокислоты (59 г) нагревали до 80oC и поддерживали эту температуру в течение 48 часов, затем выливали смесь в лед. Смесь экстрагировали хлороформом и эту органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором. Органический слой высушивали и концентрировали до темно-оранжевого маслянистого твердого вещества. Остаток дистиллировали по способу Kugelrohr при 1,5 мм рт.ст. и собирали следующие фракции: 23-105oC (температура в сосуде), сброшенный отгон, 105-135oC, 11,2 (73%) 2,6-диметил-4-пропионилфенола в виде желтовато-белого твердого вещества, спектр ЯМР которого был идентичен таковому в приготовлении 15.
ПРИГОТОВЛЕНИЕ 17. 3,5-диметил-4-триизопропилсилилоксипропиофенон
Смесь 2,6-диметил-4-пропионилфенола (соединения по приготовлению 15 и 16, 3,0 г, 16,83 ммоль), имидазола (2,3 г, 33,8 ммоль) и триизопропилсилилхлорида (4,0 мл, 18,7 ммоль) в диметилформамиде (30 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в лед и экстрагировали эфиром. Органическую фазу промывали 1 Н хлоридом лития (2 х), водой и насыщенным солевым раствором, затем ее высушивали над сульфатом магния и концентрировали с получением 5,62 г (100%) 3,5-диметил-4-триизопропилсилил-оксипропиофенона в виде желтого твердого вещества с т.пл. 87-88,5oC. ЯМР δ 7,62 (с, 2H), 2,94 (кв, J = 7,2 Гц, 2H), 2,30 (с, 6H), 1,37-1,28 (м, 3H), 1,20 (т, J = 7,2 Гц, 3H), 1,12 (д, J = 7,1 Гц, 18H).
ПРИГОТОВЛЕНИЕ 18. 3,5-диметил-4-триизопропилсилилокси- α -бромпропиофенон
К раствору 3,5-диметил-4-триизопропилсилилоксипропиофенона (соединения по приготовлению 17, 5,50 г, 16,44 ммоль) в четыреххлористом углероде (60 мл) добавляли по каплям бром (0,87 г, 16,89 ммоль в 15 мл четыреххлористого углерода). После добавления первых нескольких капель добавление приостанавливали до инициирования реакции, что определяли по исчезновению красного цвета раствора. Затем добавление раствора брома доводили до конца (общее время добавления составляло 20 минут). Реакционную смесь перемешивали 30 минут, затем добавляли водный бисульфит и смесь перемешивали еще 1 час. Слои разделяли и органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором. Органический слой высушивали над сульфатом магния и концентрировали с получением 7,0 г (100%) 3,5-диметил-4-триизопропилсилилокси- α -бромпропиофенона в виде оранжевого твердого вещества, у которого: ЯМР δ 7,68 (с, 2H), 5,28 (кв, J = 6,6 Гц, 1H), 2,31 (с, 6H), 1,88 (д, J = 6,6 Гц, 3H), 1,38-1,27 (м, 3H), 1,13 (д, J = 7,2 Гц, 18H). 13C ЯМР δ 192,66, 158,87, 132,77, 130,18, 128,61, 126,88, 41,52, 20,40, 18,07, 17,94, 17,70, 14,26, 12,29.
ПРИГОТОВЛЕНИЕ 19. 3,5-дифтор-4-триизопропилсилилоксипропиофенон
Смесь 2,6-дифтор-4-пропионилфенола (Indofine Chemicals, Company, Inc., P. O. Box 473, Somerville, New Jersey, 08876, U.S.A., 1,69 г, 9,08 миоль), имидазола (1,24 г, 18,2 ммоль) и триизопропилсилилхлорида (2,14 мл, 10,0 ммоль) в диметилформамиде (20 мл) перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду и экстрагировали эфиром (3 х). Объединенный органический слой промывали 1 Н хлоридом лития (2 х), водой и насыщенным солевым раствором, затем его высушивали над сульфатом магния и концентрировали с получением 3,06 г (98%) 3,5-дифтор-4-триизопропилсилилоксипропиофенона в виде светлого желтовато-коричневого масла, у которого: ЯМР δ 7,51 (длительно сопряженный д, J = 8,5 Гц, 2H), 2,92 (кв, J = 7,2 Гц, 2H), 1,35-1,19 (м, 6H), 1,10 (д, J = 7,1 гц, 18H).
ПРИГОТОВЛЕНИЕ 20. 3,5-дифтор-4-триизопропилсилилокси- α -бромпропиофенон
К смеси 3,5-дифтор-4-триизопропилсилилоксипропиофенона (соединения по приготовлению 19, 3,0 г, 8,76 ммоль) в четыреххлористом углероде (35 мл) добавляли по каплям бром (0,46 мл, 8,93 ммоль в 5 мл четыреххлористого углерода). После добавления первых нескольких капель раствора брома добавление приостанавливали в ожидании начала бромирования. Спустя 5 минут добавляли 1 каплю 48% HBr. Поскольку цвет брома не исчезал спустя еще 5 минут, смесь нагревали до приблизительно 50oC. Спустя 10 минут реакция начиналась, и остававшийся раствор брома добавляли по каплям в течение 20 минут. Реакционную смесь перемешивали 15 минут, а затем добавляли водный бисульфит и перемешивали еще 30 минут. Фазы разделяли и органический слой промывали водой, водным бикарбонатом и насыщенным солевым раствором. Органический слой высушивали над сульфатом кальция и концентрировали с получением 3,26 г (88%) 3,5-дифтор-4-триизопропилсилилокси- α -бромпропиофенона в виде белого твердого вещества, у которого: ЯМР δ 7,58 (длительно сопряженные дд, J = 1,3, 7,3 Гц, 2H), 5,14 (кв, J = 6,7 Гц, 1H), 1,89 (д, J = 6,5 Гц, 3H), 1,36-1,20 (м, 3H), 1,11 (д, J = 7,4 Гц, 18H), 13C ЯМР δ 190,50, 156,16, 156,09, 152,88, 152,81, 125,99, 113,05, 112,91, 112,82, 112,71, 40,75, 20,05, 17,16, 12,90.
ПРИГОТОВЛЕНИЕ 21. 3,5-дифтор-4-бензилоксипропиофенон
Смесь 2,6-дифтор-4-пропионилфенола (Indofine Chemicals, Company, Inc., P. O. Box 473, Somerville, New Jersey, 08876, U.S.A., 2,5 г, 13,43 ммоль), карбоната калия (3,7 г, 26,8 ммоль) и бензилбромида (1,75 мл, 14,71 ммоль) в ацетоне (40 мл) перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении, а остаток распределяли между эфиром и водой. Фазы разделяли и органическую фазу промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением маслянистого твердого вещества. Этот остаток стирали с гексаном до получения 1,40 г продукта. Маточные растворы подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма) с элюированием, проводимым следующим образом: гексан (200 мл), невзвешенный бензилбромид, 20% этилацетат/гексан (150 мл), 0,38 г продукта. Таким способом получали 1,78 г (48%) 3,5-дифтор-4-бензилоксипропиофенона в виде белого твердого вещества, у которого: ЯМР δ 7,56-7,32 (м, 7H), 5,30 (с, 2H), 2,91 (кв, J = 7,2 Гц, 2H), 1,21 (т, J = 7,1 Гц, 3H).
ПРИГОТОВЛЕНИЕ 22. 3,5-дифтор-4-бензилокси- α -бромпропиофенон
К раствору 3,5-дифтор-4-бензилоксипропиофенона (соединения по приготовлению 21, 1,78 г, 6,44 ммоль) в четыреххлористом углероде (25 мл) добавляли по каплям бром (0,34 мл, 6,60 ммоль в 5 мл четыреххлористого углерода). После добавления первых нескольких капель раствора брома добавление приостанавливали до инициирования реакции, что определяли по исчезновению красного цвета раствора. Затем добавление раствора брома доводили до конца (общее время добавления составляло 15 минут). Реакционную смесь перемешивали 1 час, затем концентрировали в атмосфере азота. Остаток помещали в эфир и промывали водным бисульфитом, водным бикарбонатом и насыщенным солевым раствором, затем его высушивали и концентрировали с получением 2,16 г (94%) 3,5-дифтор-4-бензилокси- α -бромпропиофенона в виде масла соломенного цвета, у которого: ЯМР δ 7,58 (д, J = 9 Гц, 2H), 7,47-7,32 (м, 5H), 5,33 (с, 2H), 5,11 (кв, J = 6,6 Гц, 1H), 1,88 (д, J = 6,6 Гц, 3H). В этом случае также наблюдалось небольшое количество исходного материала по спектру ЯМР, но продукт был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 23. 2-ацетокси-2,6-диметил-3,4,5,6-циклогексадиен-1-он
К суспензии тетраацетата свинца (20,0 г, 45,1 ммоль) в уксусной кислоте (33 мл) добавляли 2,6-диметилфенол (5,00 г, 40,93 ммоль в 27 мл уксусной кислоты) по каплям в течение 15 минут. Реакционную смесь перемешивали при комнатной температуре 2 часа, в течение которых она постепенно приобретала равномерно желтый цвет. Смесь разводили водой и экстрагировали хлороформом (3 х). Объединенный органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали с получением желтого масла. Остаток дистиллировали по способу Kugelrohr при 0,4 мм рт.ст. Материал, который получали дистиллированием при температуре в сосуде 75-85oC (5,69 г), собирали в виде желтого масла. Образец весом 3,2 г подвергали дальнейшей очистке флэш-хроматографией на силикагеле (1 х 5 дюймов) с элюированием, проводимым следующим образом: гексан (500 мл), ноль, 5% эфир/гексан (500 мл) и 10% эфир/гексан (250 мл), 1,89 г 2-ацетокси-2,6-диметил-3,4,5,6-циклогексадиен-1-она в виде светло-желтого воскоподобного твердого вещества, у которого: ЯМР δ 6,80-6,76 (м, 2H), 6,19-6,09 (м, 4H), 2,05 (с, 3H), 1,92 (д, J = 0,8 Гц, 3H), 1,36 (с, 3H).
ПРИГОТОВЛЕНИЕ 24. 1,3-диацетокси-2,4-диметилбензол
К смеси 2-ацетокси-2,6-диметил-3,4,5,6-циклогексадиен-1-она (соединения по приготовлению 23, 0,5 г, 2,77 ммоль) в уксусном ангидриде (1 мл), охлажденной до 0oC, добавляли медленно по стенке колбы этерат трифторида бора (0,075 мл). Реакционную смесь перемешивали 15 минут при 0oC, затем она достигала комнатной температуры и перемешивалась еще 1 час. Добавляли водный бикарбонат и смесь перемешивали интенсивно в течение 30 минут. Реакционную смесь экстрагировали эфиром. Органическую фазу промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 0,59 г (97%) 1,3-диацетокси-2,4-диметилбензола в виде светло-желтого масла, у которого: ЯМР δ 7,08 (д, J = 8,2 Гц, 1H), 6,87 (д, J = 8,2 Гц, 1H), 2,35 (с, 3H), 2,32 (с, 3H), 2,15 (с, 3H), 1,98 (с, 3H).
ПРИГОТОВЛЕНИЕ 25. 1,3-дигидрокси-2,4-диметилбензол
К суспензии гидрида литий-алюминия (0,56 г, 14,76 ммоль) в эфире (35 мл) добавляли 1,3-диацетокси-2,4-диметилбензол (соединение по приготовлению 24, 1,65 г, 7,42 ммоль в 40 мл эфира) с помощью шприца. Смесь перемешивали в течение ночи, затем ее осторожно охлаждали избытком декагидрата сульфата натрия. Смесь высушивали безводным сульфатом натрия, фильтровали и концентрировали с получением 0,62 г (62%) 1,3-дигидрокси-2,4-диметилбензола в виде воскоподобного светло-желтого твердого вещества, у которого: ЯМР δ 6,83 (д, J = 8 Гц, 1H), 6,34 (д, J = 8,1 Гц, 1H), 4,71 (с, 2H), 2,19 (с, 3H), 2,17 (с, 3H). Также наблюдалось небольшое количество примеси по спектру ЯМР, но продукт был удобен для использования без дополнительной очистки. Этот продукт был до некоторой степени чувствителен к воздуху и использовался в тот же день, когда его синтезировали.
ПРИГОТОВЛЕНИЕ 26. 6,8-диметил-7-гидроксихроман-4-он
Смесь 1,3-дигидрокси-2,4-диметилбензола (соединения по приготовлению 25, 0,62 г, 4,49 ммоль), 3-хлорпропионовой кислоты (0,49 г, 4,52 ммоль) и трифторметансульфокислоты (2 мл) нагревали до 80oC и поддерживали эту температуру в течение 2,25 часов. Реакционную смесь охлаждали и выливали в хлороформ. Эту смесь экстрагировали водой и эту водную фазу повторно экстрагировали эфиром. Объединенную органическую фазу промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 2,4-дигидрокси-3,5-диметил-β-хлорпропиофенона в виде красного масла.
Продукт описанной выше реакции добавляли к 50 мл 2 Н гидроксида натрия, предварительно охлажденного до 0oC. Смесь перемешивали 2 часа, затем подкисляли до pH 1-2 с помощью 6 Н HCl и экстрагировали этилацетатом (3 х). Объединенную органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 0,55 г (64% в два этапа), 6,8-диметил-7-гидроксихроман-4-она в виде оранжевого твердого вещества, у которого: ЯМР δ 7,59 (с, 1H), 5,45 (с, 1H), 4,52 (т, J = 6,4 Гц, 2H), 2,74 (т, J = 6,4 Гц, 2H), 2,22 (с, 3H), 2,13 (с, 3H).
ПРИГОТОВЛЕНИЕ 27. 6,8-диметил-7-триизопропилсилилоксихроман-4-он
Смесь 6,8-диметил-7-гидроксихроман-4-она (соединения по приготовлению 26, 0,50 г, 2,60 ммоль), имидазола (0,35 г, 5,14 ммоль) и триизопропилсилилхлорида (0,61 мл, 2,85 ммоль) в диметилформамиде (10 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь разводили водой и экстрагировали эфиром (2 х). Объединенную органическую фазу промывали 1 Н хлоридом лития (2 х) и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 5% этилацетат/гексан (100 мл), ноль, 5% этилацетат/гексан (100 мл) и 10% этилацетат/гексан (150 мл), 0,529 г (58%) 6,8-диметил-7-триизопропилсилилоксихроман-4-она в виде воскоподобного лимонно-желтого твердого вещества, у которого: ЯМР δ 7,57 (с, 1H), 4,52 (т, J = 6,4 Гц, 2H), 2,74 (т, J = 6,4 Гц, 2H), 2,21 (с, 3H), 2,11 (с, 3H), 1,40-1,26 (м, 3H), 1,13 (д, J = 7,2 Гц, 18H), 13C ЯМР δ 191,60, 160,25, 159,93, 125,91, 122,55, 116,15, 115,29, 67,24, 37,52, 17,91, 17,69, 17,38, 14,24, 12,28, 9.97. Наблюдалась небольшая примесь силила в протонном ЯМР = 1,06 миллионных долей, однако материал был удобен для дальнейших превращений без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 28. 3,3-дибром-6,8-диметил-7-триизопропилсилилоксихроман-4-он
К раствору 6,8-диметил-7-триизопропилсилилоксихроман-4-она (соединения по приготовлению 27, 0,50 г, 1,43 ммоль) в четыреххлористом углероде (10 мл) добавляли по каплям бром (0,16 мл, 3,11 ммоль в 5 мл четыреххлористого углерода). Смесь перемешивали 1 час при комнатной температуре, затем добавляли водный бисульфит и смесь перемешивали еще 30 минут. Фазы разделяли и органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 0,64 г (89%) 3,3-дибром-6,8-диметил-7-триизопропилсилилоксихроман-4-она в виде оранжевого твердого вещества, у которого: ЯМР δ 7,64 (с, 1H), 4,68 (с, 2H), 2,22 (с, 3H), 2,13 (с, 3H), 1,38-1,15 (м, 3H), 1,11 (д, J = 7,3 Гц, 18H). В спектре ЯМР отмечали некоторые небольшие примеси. Однако материал был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 29. 6-фтор-7-гидроксихроман-4-он
Смесь 1,3-диметоксибензола (3,80 мл, 29,0 ммоль) и N-фтордибензолсульфонамида (4,21 г, 29,21 ммоль) нагревали до 60oC и поддерживали эту температуру в течение ночи. Смесь охлаждали и подвергали флэш-хроматографии на силикагеле (2 х 5 дюймов в гексане) с элюированием, проводимым следующим образом: 3% этилацетат/гексан (1000 мл), сброшенный отгон, 3% этилацетат/гексан (1000 мл), 2,69 г 2:1 смеси 2,4- диметоксифторбензола и исходного материала, которую переносили непосредственно на следующий этап.
Продукт описанной выше реакции объединяли с уксусной кислотой (11 мл) и 48% HBr (11 мл) и нагревали с обратным холодильником в течение 3 часов. Реакционную смесь концентрировали и подвергали флэш-хроматографии на силикагеле (2 х 5 дюймов в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (2000 мл), 0,95 г (43%) 2,4-дигидроксифторбензола в виде воскоподобного белого твердого вещества, которое использовали без очистки.
Смесь 2,4-дигидроксифторбензола (0,15 г, 1,17 ммоль), 3-хлорпропионовой кислоты (0,13 г, 1,20 ммоль) и трифторметансульфокислоты (1 мл) нагревали до 80oC и поддерживали эту температуру в течение 3 часов.
Реакционную смесь выливали в воду и экстрагировали эфиром (3 х). Объединенную органическую фазу промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 2,4-дигидрокси-5-фтор- β -хлорпропиофенона в виде красного твердого вещества, у которого: ЯМР δ 7,37 (д, J = 10,8 Гц, 1H), 6,54 (д, J = 7,7 Гц, 1H), 3,87 (т, J = 6,8 Гц, 2H), 3,33 (т, J = 6,8 Гц, 2H). Этот продукт все еще содержал некоторое остаточное количество 3-хлорпропионовой кислоты, но был удобен для использования в следующей реакции.
Продукт описанной выше реакции объединяли с 2 Н гидроксидом натрия (15 мл) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь подкисляли до pH 1-2 с помощью 1 Н HCl и экстрагировали этилацетатом (3 х). Объединенную органическую фазу промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 25% этилацетат/гексан (300 мл), ноль, 25% этилацетат/гексан (200 мл), 0,11 г (52% в два этапа) 6-фтор-7-гидроксихроман-4-она в виде белого твердого вещества с т.пл. 222-223oC, ЯМР δ 7,61 (д, J = 10,3 Гц, 1H), 6,58 (д, J = 7,2 Гц, 1H), 5,70-5,58 (м, 1H), 4,51 (т, J = 6,5 Гц, 2H), 2,77 (т, J = 6,4 Гц, 2H).
ПРИГОТОВЛЕНИЕ 30. 6-фтор-7-бензилоксихроман-4-он
Смесь 6-фтор-7-гидроксихроман-4-она (соединения по приготовлению 29, 0,93 г, 5,11 ммоль), бензилбромида (0,61 мл, 5,13 ммоль), карбоната калия (1,41 г, 10,2 ммоль) в ацетоне (100 мл) перемешивали при комнатной температуре в течение ночи. Смесь охлаждали, фильтровали и концентрировали до желтого твердого вещества. Остаток перекристаллизовывали из этилацетат/эфира с получением 1,02 г (73%) 6-фтор-7-бензилоксихроман-4-она за два сбора в виде кремово-белых кристаллов с т.пл. 155-156oC, ЯМР δ 7,58 (д, J = 11 Гц, 1H), 7,46-7,33 (м, 5H), 6,54 (д, J = 6,7 Гц, 1H), 5,16 (д, 2H), 4,50 (т, J = 6,4 Гц, 2H), 2,75 (т, J = 6,4 Гц, 2H). Анализ, рассчитано для C16H13FO2: C 70,58, H 4,81. Найдено: C 70,45, H 4,80.
ПРИГОТОВЛЕНИЕ 31. 3,3-дибром-6-фтор-7-бензилоксихроман-4-он
К смеси 6-фтор-7-бензилоксихроман-4-она (соединения по приготовлению 30, 0,99 г, 3,64 ммоль) и четыреххлористого углерода (45 мл) добавляли по каплям бром (0,37 мл, 7,18 ммоль в 5 мл четыреххлористого углерода). Реакционную смесь оставляли стоять на ночь при комнатной температуре. К реакционной смеси добавляли воду и фильтрованием собирали 87 мг неидентифицированного розового твердого вещества, которое удаляли. Фазы разделяли из фильтрата и органический слой промывали водой, водным бикарбонатом и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением чувствительного к действию воздуха масла, которое представляло собой смесь бромированных продуктов и исходного материала (0,93 г). Этот материал объединяли с этилацетатом (100 мл) и бромидом меди (0,6 г, 2,69 ммоль) и нагревали с обратным холодильником в течение 4 часов. Добавляли бромид меди (0,3 г, 1,35 ммоль) и реакционную смесь нагревали с обратным холодильником в течение ночи. В третий раз добавляли бромид меди (0,6 г, 2,69 ммоль) и продолжали нагревание смеси с обратным холодильником в течение ночи. Смесь охлаждали, фильтровали и концентрировали. Остаток помещали в этилацетат и промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 0,91 г смеси 3,3-дибром-6-фтор-7-бензилоксихроман-4-она и перебромированных продуктов. Главными признаками спектра ЯМР были сигналы при δ 7,69 (т, J = 9,3 Гц), 7,63-7,32 (м), 6, 62 (пара д, 3 = 5,7 и 7 Гц), 5,19 (с), 4,70 (с). Этот материал использовался сырым для экспериментов по сопряжению.
ПРИГОТОВЛЕНИЕ 32. 6-метил-7-гидроксихроман-4-он
Смесь 1,3-дигидрокси-4-метилбензола (5,0 г, 40,3 ммоль), 3-хлорпропионовой кислоты (4,38 г, 40,36 ммоль) и трифторметансульфокислоты (20 г) нагревали до 80oC и поддерживали при этой температуре в течение ночи. Реакционную смесь выливали в воду и экстрагировали 1:1 эфир/этилацетатом (2 х). Объединенную органическую фазу промывали водой (2 х) и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением оранжевой смолы (7,6 г.).
Смолу из описанной выше реакции объединяли с 2 Н гидроксидом натрия (200 мл) и нагревали с обратным холодильником в течение ночи. Смесь подкисляли до pH 1-2 с помощью 6 Н HCl и экстрагировали этилацетатом (3 х). Объединенную органическую фазу промывали водой, водным бикарбонатом (2 х) и насыщенным солевым раствором, затем ее высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1,5 х 4 дюйма в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (500 мл), ноль, 20% этилацетат/гексан (500 мл), ноль, 30% этилацетат/гексан (500 мл), 3,1 г желтого твердого вещества. Этот материал перекристаллизовывали из этилацетата с получением 1,66 г (23% в течение двух последовательных этапов) 6-метил-7-гидроксихроман-4-она в виде светло-розового твердого вещества с т. пл. 185-186oC, ЯМР δ 7,66 (с, 1H), 6,90 (брс, 1H), 6,38 (с, 1H), 4,45 (т, J = 6,4 Гц, 2H), 2,73 (т, J = 6,4 Гц, 2H), 2,17 (с, 3H).
ПРИГОТОВЛЕНИЕ 33. 6-метил-7-триизопропилсилилокси-хроман-4-он
Смесь 6-метил-7-гидроксихроман-4-она (соединения по приготовлению 32, 1,50 г, 8,42 ммоль), имидазола (1,15 г, 16,9 ммоль) и триизопропилсилилхлорида (2,0 мл, 9,2 ммоль) в диметилформамиде (30 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в воду и экстрагировали эфиром (2 х). Объединенный органический слой промывали 1 Н хлоридом лития (2 х), высушивали над сульфатом магния и концентрировали с получением 3,01 г (100%) 6-метил-7-триизопропилсилилоксихроман-4-она в виде тусклого желтого масла, у которого: ЯМР δ 7,64 (с, 1H), 6,30 (с, 1H), 4,45 (т, J = 6,4 Гц, 2H), 2,70 (т, J = 6,4 Гц, 2H), 2,14 (с, 3H), 1,40-1,25 (м, 3H), 1,09 (д, J = 7,3 Гц, 18H). Этот продукт также имел небольшую примесь силила и остаточное количество диметилформамида, но был удобен для использования в последующей реакции.
ПРИГОТОВЛЕНИЕ 34. 3,3-дибром-6-метил-7-триизопропилсилилоксихроман-4-он и 6-метил-3,3,5-трибромтриизопропилсилилоксихроман-4-он
К смеси 6-метил-7-триизопропилсилилоксихроман-4-она (соединения по приготовлению 33, 3,0 г, 8,97 ммоль) и четыреххлористового углерода (70 мл) добавляли по каплям в течение 15 минут бром (0,93 мл, 18,05 ммоль в 20 мл четыреххлористого углерода). Реакционную смесь перемешивали 1 час при комнатной температуре и добавляли водный бисульфит. Реакционную смесь перемешивали 15 минут, затем фазы разделяли и органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением оранжевого масла. Этот остаток подвергали флэш-хроматографии на силикагеле (2 х 4 дюйма) с элюированием, проводимым следующим образом: гексан (250 мл), ноль, 3% эфир/гексан (500 мл), ноль, 3% эфир/гексан (300 мл), 2,61 г смеси 3,3-дибром-6-метил-7-триизопропилсилилокси-хроман-4-она и 6-метил-3,3,5-трибром-7-триизопропилсилилокси-хроман-4-она в виде бесцветного масла (соотношение продуктов составляло приблизительно 2,5: 1). Основными признаками спектра ЯМР для 3,3-дибром-6-метил-7-триизопропилсилилоксихроман-4-она были сигналы: δ 7,77 (H при C-5), 6,38 (H при C-8), 4,68 (C-2 метилен), 2,19 (C-6 метил).
ПРИГОТОВЛЕНИЕ 35. 3-трифторметансульфонилокси-8-метил-8- азабицикло(3.2.1)октан
Тропин (14,2 г, 0,10 моль) растворяли в метиленхлориде (210 мл) и добавляли триэтиламин (23 мл, 0,16 моль). Трифторметансульфонилхлорид (9,3 мл, 0,12 моль) добавляли по каплям с такой скоростью, чтобы метиленхлорид слегка кипел. Реакционную смесь перемешивали при комнатной температуре 1 час, затем ее промывали холодным 0,5 Н гидроксидом натрия, водой и насыщенным солевым раствором. Органическую фазу высушивали и концентрировали с получением желтого твердого вещества, у которого: ЯМР δ 4,88 (т, 1H), 3,10-3,05 (м, 2H), 2,94 (с, 3H), 2,22 (с, 3H), 2,20-2,10 (м, 2H), 2,02-1,88 (м, 6H).
ПРИГОТОВЛЕНИЕ 36. 3-(4-фторфенилсульфанил)-8-метил-8-азабицикло(3.2.1)октан
В колбе с круглым дном и тремя насадками, приспособленной для поверхностного механического перемешивания, гидрид натрия (2,02 г, 50,47 ммоль, 60% дисперсия в масле) промывали для удаления масла гексаном (2 отмывания) и добавляли тетрагидрофуран (225 мл), а затем добавляли 4-фтортиофенол (4,89 мл, 45,89 ммоль в 30 мл тетрагидрофурана с 20 мл ополаскиванием). Из реакционной смеси свободно выделялся водород. После прекращения выделения водорода аккуратно добавляли в виде твердого вещества, весь сразу, с промывкой 50 мл тетрагидрофурана, 3-трифторметансульфонилокси-8-метил-8-азабицикло(3.2.1)октан (соединение по приготовлению 35, 9,42 г, 45,89 ммоль). Реакционную смесь нагревали с обратным холодильником в течение ночи, затем охлаждали. Растворитель удаляли при пониженном давлении и остаток помещали в этилацетат. Органическую фазу промывали водой и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали с получением 8,28 г (72%) 3-(4-фторфенилсульфанил)-8-метил-8-азабицикло(3.2.1)октана в виде желтовато-коричневого масла, у которого: ЯМР δ 7,38 (дд, J = 5,9 Гц, 2H), 6,96 (длительно сопряженный т, J = 9 Гц, 2H), 3,22-3,08 (м, 3H), 2,23 (с, 3H), 2,02-1,94 (м, 2H), 1,83-1,64 (м, 4H), 1,50 (ABкв: ΔV1-3 = 14,5 Гц, J = 6,5 Гц, 2H).
ПРИГОТОВЛЕНИЕ 37. 3-(4-фторфенилсульфанил)-8-(2,2,2- трихлорэтоксикарбонил)-8-азабицикло-(3.2.1)октан
Смесь 3-(4-фторфенилсульфанил)-8-метил-8-азабицикло(3.2.1)октана (соединения по приготовлению 36, 8,20 г, 32,65 ммоль), 2,2,2-трихлорэтилхлорформата (4,94 мл, 35,92 ммоль) и карбоната калия (4,96 г, 35,92 ммоль) в бензоле (140 мл) нагревали с обратным холодильником в течение ночи. Растворитель удаляли при пониженном давлении, а остаток помещали в этилацетат. Органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (3 х 4 дюйма) с элюированием, проводимым следующим образом: гексан (350 мл), ноль, 10% этилацетат/гексан (400 мл), сброшенный отгон, 10% этилацетат/гексан (600 мл), 20% этилацетат/гексан (500 мл) и 30% этилацетат/гексан (250 мл), 10,32 г (77%) 3-(4-фторфенилсульфанил)-8-(2,2,2-трихлорэтоксикарбонил)-8- азабицикло(3.2.1)октана в виде не чисто белого твердого вещества с т.пл. 65-67oC. ЯМР δ 7,40 (дд, J = 5,5, 9 Гц, 2H), 6,98 (длительно сопряженный т, J = 8,5 Гц, 2H), 4,72 (ABкв, ΔV1-3 = 60 Гц, J = 12 Гц, 2H), 4,35 (сим м, 2H), 3,32 (септет, J = 6 Гц, 1H), 2,10-1,94 (м, 2H), 1,94-1,58 (м, 6H).
ПРИГОТОВЛЕНИЕ 38. 3-(4-фторфенилсульфанил)-8-азабицикло-(3.2.1)октан
Смесь 3-(4-фторфенилсульфанил)-8-(2,2,2-трихлорэтоксикарбонил)-8- азабицикло(3.2.1)октана (соединения по приготовлению 37, 10,28 г, 24,92 ммоль), 48% HBr (20 мл) и уксусной кислоты (80 мл) нагревали до 110oC и поддерживали эту температуру в течение 78 часов. pH реакционной смеси доводили до 11 добавлением 4 Н гидроксида натрия и экстрагировали метиленхлоридом. Органическую фазу фильтровали через диатомовую землю, промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток дистиллировали по способу Kugelrohr (110oC (температура в сосуде), 1,5 мм рт.ст. ) с получением 3,30 г 3-(4-фторфенилсульфанил)-8-азабицикло(3.2.1)октана в виде желтого масла, у которого: ЯМР δ 7,41 (дд, J = 5,5, 9 Гц, 2H), 7,00 (длительно сопряженный т, J = 8,5 Гц, 2H), 4,11 (с, примесь), 3,55 (брт, J = 3,5 Гц, 2H), 3,24 (сим м, 1H), 2,58 (брс, 2H, обмен с D2O должен интегрировать с 1H), 1,90-1,77 (м, 4H), 1,70-1,51 (м, 4H). Этот продукт был удобен для использования без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 39. 3-(4-хлорфенилсульфанил)-8-метил-8-азабицикло(3.2.1)октан
В колбе с круглым дном и тремя насадками, приспособленной для поверхностного механического перемешивания, гидрид натрия (2,03 г, 50,91 ммоль, 60% дисперсия в масле) промывали для удаления масла гексаном (2 отмывания) и добавляли тетрагидрофуран (200 мл), а затем добавляли 4-хлортиофенол (6,69 г, 46,28 ммоль в 20 мл тетрагидрофурана). Из реакционной смеси свободно выделялся водород. После прекращения выделения водорода добавляли 3-трифторметансульфонилокси-8-метил-8-азабицикло(3.2.1)октан (9,5 г, 46,28 ммоль в 70 мл тетрагидрофурана). Реакционную смесь нагревали с обратным холодильником в течение ночи, затем охлаждали и фильтровали через диатомовую землю (с ополаскиванием эфиром). Фильтрат концентрировали при пониженном давлении и остаток помещали в эфир. Органическую фазу промывали водой и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали с получением 8,08 г (65%) 3-(4-хлорфенилсульфанил)-8-метил-8-азабицикло(3.2.1)октана в виде желтовато-коричневого масла, у которого: ЯМР δ 7,42-7,23 (м, 4H), 3,22 (сим м, 1H), 3,20-3,11 (м, 2H), 2,24 (с, 3H), 2,03-1,97 (м, 2H), 1,82-1,66 (м, 4H), 1,53 (ABкв ΔV1-3 = 14,5 Гц, J = 6,5 Гц, 2H).
ПРИГОТОВЛЕНИЕ 40. 3-(4-хлорфенилсульфанил)-8-(2,2,2-трихлорэтоксикарбонил)-8- азабицикло(3.2.1)октан
Смесь 3-(4-хлорфенилсульфанил)-8-метил-8-азабицикло(3.2.1)октана (8,06 г, 30,12 ммоль), 2,2,2-трихлорэтилхлорформиата (4,56 мл, 33,13 ммоль) и карбоната калия (4,58 г, 33,13 ммоль) в бензоле (150 мл) нагревали с обратным холодильником в течение ночи. Растворитель удаляли при пониженном давлении, а остаток помещали в этилацетат. Органическую фазу промывали водным бикарбонатом и насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (3 х 4 дюйма) с элюированием, проводимым следующим образом: гексан (350 мл), ноль, 10% этилацетат/гексан (500 мл), сброшенный отгон, 10% этилацетат/гексан (500 мл), 20% этилацетат/гексан (500 мл) и 30% этилацетат/гексан (250 мл), 9,26 г (72%) 3-(4-хлорфенилсульфанил)-8-(2,2,2-трихлорэтоксикарбонил)-8- азабицикло(3.2.1) октана в виде желтого твердого вещества с т.пл. 70-71,5oC. ЯМР  7,33 (длительно сопряженный д, J = 8,5 Гц, 2H), 7,26 (длительно сопряженный δ , J = 8,5 Гц, 2H), 4,73 (ABкв ΔV1-3 = 58 Гц, J = 12 Гц, 2H), 4,43-4,30 (м, 2H), 3,40 (септет, J = 6 Гц, 1H), 2,10-1,56 (м, 8H).
7,33 (длительно сопряженный д, J = 8,5 Гц, 2H), 7,26 (длительно сопряженный δ , J = 8,5 Гц, 2H), 4,73 (ABкв ΔV1-3 = 58 Гц, J = 12 Гц, 2H), 4,43-4,30 (м, 2H), 3,40 (септет, J = 6 Гц, 1H), 2,10-1,56 (м, 8H).
ПРИГОТОВЛЕНИЕ 41. 3-(4-хлорфенилсульфанил)-8-азабицикло(3.2.1)октан
Смесь 3-(4-хлорфенилсульфанил)-8-(2,2,2-трихлорэтоксикарбонил)-8- азабицикло(3.2.1)октана (8,70 г, 20,28 ммоль), 48% HB (17 мл) и уксусной кислоты (68 мл) нагревали до 110oC и поддерживали эту температуру в течение 78 часов. pH реакционной смеси доводили до 11 добавлением 4 Н гидроксида натрия и экстрагировали метиленхлоридом. Органическую фазу фильтровали через диатомовую землю, промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали с получением 3-(4-хлорфенилсульфанил)-8-азабицикло(3.2.1)октана в виде желтого масла. Этот продукт дистиллировали по способу Kugelrohr (110-130oC, (температура в сосуде), 1,5 мм рт.ст.) с получением 4,1 г (79%) 3-(4-хлорфенилсульфанил)-8-азабицикло(3.2.1)октана в виде почти бесцветного масла, которое отвердевало и имело: ЯМР δ 7,30 (м, 4H), 3,54 (брт, J = 3,5 Гц, 2H), 3,32 (сим м, 1H), 1,97-1,72 (м, 5H), 1,71-1,52 (м, 4H).
ПРИГОТОВЛЕНИЕ 42. 1-(2,2-дифенил-бензо(1,3)диоксол-5- ил)-пропан-1-он
Смесь 3,4-дигидроксипропиофенона (ICN, Biomedicals, Inc., 3300 Hyland Ave., Costa Mesa, California, 92626, U.S.A., 5,0 г, 30 ммоль) и дихлордифенилметана (10,0 мл, 52,1 ммоль) нагревали до 170oC и поддерживали эту температуру в течение 7 минут. Реакционную смесь охлаждали и выливали в 1 Н раствор гидроксида натрия. Смесь экстрагировали эфиром (2 х), а экстракты объединяли и промывали водой и насыщенным солевым раствором. Органический слой высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (2 х 5 дюймов в гексане) с элюированием, проводимым следующим образом: 2% эфир/гексан (500 мл), 0,84 г белого твердого вещества, экспериментально идентифицированного как 2-хлор-1-(2,2-дифенил-бензо(1,3) диоксол-5-ил)-пропан-1-он, 5% эфир/гексан (250 мл), 1,9 г неидентифицированного оранжевого масла, 5% эфир/гексан (250 мл), 2,18 г регенерированного дихлордифенилметана, 10% эфир/гексан (500 мл), 4,82 г (48%) 1-(2,2-дифенил-бензо(1,3)диоксол-5-ил)-пропан-1-она в виде оранжевого масла, которое отвердевало при стоянии. Этот продукт имел т.пл. 69-70,5oC. Анализ, рассчитано для C22H17ClO3: C 79,98, H 5,49. Найдено: C 80,05, H 5,34.
ПРИГОТОВЛЕНИЕ 43. 2-бром-1-(2,2-дифенил-бензо(1,3)-диоксол-5-ил)- пропан-1-он
1-(2,2-дифенил-бензо(1,3)диоксол-5-ил)-пропан-1-он (соединение приготовления 42, 4,70 г, 14,23 ммоль) растворяли в четыреххлористом углероде (60 мл) и по каплям добавляли бром (0,74 мл, 14,36 ммоль в 10 мл четыреххлористого углерода). Реакционную смесь перемешивали при комнатной температуре 30 минут, а затем экстрагировали насыщенным водным раствором бикарбоната. Органическую фазу промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 5,58 г (96%) 2-бром-1-(2,2-дифенил-бензо(1,3)диоксол-5-ил)-пропан-1-она в виде темно-оранжевого масла, у которого: ЯМР δ 7,68-7,37 (м, 12H), 6,95 (д, J = 8,3 Гц, 1H), 5,21 (кв, J = 6,7 Гц, 1H), 1,88 (д, J = 6,6 Гц, 3H).
ПРИГОТОВЛЕНИЕ 44. 4-бензилокси-3-гидроксипропиофенон
Смесь 3,4-дигидроксипропиофенона (ICN Biomedicals, Inc., 3300 Hyland Ave., Costa Meja., California, 92626, USA, 2,00 г, 12,0 ммоль), бензилбромида (1,43 мл, 12,0 ммоль) и карбоната калия (3,33 г, 24,1 ммоль) в ацетоне (100 мл) нагревали с обратным холодильником в течение 24 часов. Реакционную смесь охлаждали и фильтровали. Фильтрат концентрировали, а остаток распределяли между этилацетатом и 0,25 Н хлористоводородной кислотой. Фазы разделяли и органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1 х 5 дюймов в гексане) с элюированием, проводимым следующим образом: 10% этилацетат/гексан (500 мл), ноль, 30% этилацетат/гексан (1000 мл), 0,88 г (28%) 4-бензилокси-3-гидроксипропиофенона в виде белого твердого вещества, у которого: ЯМР δ 7,58-7,52 (м, 2H), 7,44-7,36 (м, 5H), 6,96 (д, J = 8,2 Гц, 1H), 5,72 (с, 1H), 5,19 (с, 2H), 2,94 (кв, J = 7,2 Гц, 2H), 1,21 (т, J = 7,3 Гц, 3H).
ПРИГОТОВЛЕНИЕ 45. 4-бензилокси-3-метоксипропиофенон
Смесь 4-бензилокси-3-гидроксипропиофенона (соединения по приготовлению 44, 0,88 г, 3,43 ммоль), карбоната калия (0,95 г, 6,87 ммоль) и метилиодида (0,50 мл, 8,0 ммоль) в ацетоне (50 мл) нагревали с обратным холодильником 2 часа и оставляли перемешиваться при комнатной температуре в течение уикенда. Реакционную смесь фильтровали, а фильтрат концентрировали. Остаток распределяли между эфиром и водой. Фазы разделяли и органическую фазу промывали водой и
насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением 0,88 г (95%) 4-бензилокси-3-метоксипропиофенона в виде белого твердого вещества, у которого: ЯМР δ 7,55 (д, J = 2 Гц, 1H), 7,50 (дд, J = 2, 8,4 Гц, 1H), 7,44-7,28 (м, 5H), 6,87 (д, J = 8,4 Гц, 1H), 5,22 (с, 2H), 3,93 (с, 3H), 2,93 (кв, J = 7,3 Гц, 2H), 1,20 (т, J = 7,3 Гц, 3H).
ПРИГОТОВЛЕНИЕ 46. 4-бензилокси- α -бром-3-метоксипропиофенон
4-бензилокси-3-метоксипропиофенона (соединения по приготовлению 45, 0,84 г, 3,11 моль) растворяли в четыреххлористом углероде (20 мл) и по каплям в течение 10 минут добавляли бром (0,16 мл, 3,11 ммоль в 5 мл четыреххлористого углерода). Реакционную смесь перемешивали 30 минут при комнатной температуре. Реакционную смесь выливали в насыщенный водный бикарбонат и фазы разделяли. Органический слой промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток помещали в эфир и концентрировали и этот процесс повторяли для удаления остаточного четыреххлористого углерода из продукта. Этим способом получали 1,12 г (100%) 4-бензилокси- α -бром-3-метоксипропиофенона в виде воскоподобного светло-оранжевого твердого вещества, у которого: ЯМР δ 7,58-7,54 (м, 3H), 7,42-7,23 (м, 4H), 6,89 (д, J = 8,3 Гц, 1H), 5,25-5,21 (м, 3H), 3,93 (с, 3H), 1,85 (д, J = 6,6 Гц, 3H).
ПРИГОТОВЛЕНИЕ 47. 4-(3,5-дибромфенил)-4-гидроксипиперидин гидрохлорид
Раствор 1,3,5-трибромбензола (15,75 г, 50,0 ммоль) в эфире (500 мл) охлаждали до -78oC и по каплям в течение 30 минут добавляли бутиллитий (20,8 мл, 50,0 ммоль, 2,4 М в гексане). Реакционную смесь перемешивали 30 минут, а затем по каплям в течение 30 минут добавляли 1-трет-бутилоксикарбонилпиперидин-4-он (5,0 г, 25 ммоль в 100 мл эфира) с ополаскиванием 20 мл эфира. Реакционную смесь перемешивали 2 часа при -78oC, затем реакционную смесь охлаждали водой и оставляли стоять до достижения ею комнатной температуры. Фазы разделяли и органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (4 х 4 дюйма) с элюированием, проводимым следующим образом: 1% этилацетат/гексан (1000 мл), ноль, 5% этилацетат/гексан (1000 мл) и 10% этилацетат/гексан (1000 мл), невзвешенная смесь исходного трибромида и 1,3- дибромбензола, 10% этилацетат/гексан (1000 мл), ноль, 15% этилацетат/гексан (2000 мл), ноль, 20% этилацетат/гексан (2000 мл), 6,76 (62%) 4-(3,5-дибромфенил)-4-гидрокси-1-трет-бутилоксикарбонилпиперидина в виде светло-желтой пены, у которой: ЯМР δ 7,56 (м, 3H), 4,06 (брд, J = 13 Гц, 2H), 3,21 (т, J = 13 Гц, 2H), 1,93 (дт, J = 4,5, 13 Гц, 2H), 1,80 (с, 1H), 1,68 (д, J = 13 Гц, 2H), 1,48 (с, 9H). Было установлено, что продукт имел 88% чистоту и был загрязнен 12% 1-трет-бутилоксикарбонилпиперидин-4-она (триплеты ЯМР при δ 3,71 и 2,44). Этот материал был удобен для использования без дополнительной очистки.
Продукт описанной выше реакции (6,76 г, 15,5 ммоль) растворяли в эфире (150 мл) и добавляли диоксан, насыщенный HCl (15 мл). Смесь перемешивали 30 минут при комнатной температуре, затем охлаждали до 0oC и через раствор в течение 3 минут продували газообразный HCl. Реакционную смесь оставляли стоять до достижения ею комнатной температуры и перемешивали в течение ночи. Через смесь продували азот для удаления газообразного НС1 и осадок отфильтровывали с получением 3,27 г твердого вещества кремового цвета. Фильтрат вновь насыщали газообразным HCl и перемешивали 6 часов. Вновь смесь продували азотом, а осадок собирали (1,63 г). HCl гидролиз повторяли в третий раз с получением 0,45 г дополнительного продукта. Таким образом получали 5,45 г (94%) 4-(3,5-дибромфенил)-4-гидроксипиперидин гидрохлорида в виде твердого вещества кремового цвета. Этот материал использовали без дополнительной очистки.
ПРИГОТОВЛЕНИЕ 48. (1R*, 2R*)-1-(4-гидроксифенил)-2-(4-(3,5- дибромфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ол
Смесь 4-триизопропилсилилокси- α -бромпропиофенона (3,0 г, 7,79 ммоль), 4-(3,5-дибромфенил)-4-гидроксипиперидин гидрохлорида (соединения по приготовлению 47, 2,89 г, 7,79 ммоль) и триэтиламина (3,26 мл, 23,4 ммоль) в этаноле (200 мл) нагревали с обратным холодильником в течение ночи. Растворитель удаляли при пониженном давлении, а остаток распределяли между этилацетатом и водой. Фазы разделяли и органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (2 х 4 дюйма) с элюированием, проводимым следующим образом: 1% этилацетат/гексан (500 мл), ноль, 5% этилацетат/гексан (300 мл), невзвешенный исходный кетон, 5% этилацетат/гексан (700 мл) и 15% этилацетат/гексан (300 мл), ноль, 15% этилацетат/гексан (1200 мл), 3,55 г (71%) 1-(4-триизопропилсилилоксифенил)-2-(4-(3,5-дибромфенил)-4- гидроксипиперидин-1-ил)-пропан-1-она в виде хрустящей не чисто белой пены, у которой: ЯМР δ 8,03 (д, J = 9 Гц, 2H), 7,57-7,53 (м, 3H), 6,92 (д, J = 8,5 Гц, 2H), 4,14 (кв, J = 7 Гц, 1H), 2,85 (дд, J = 2, 9,5 Гц, 2H), 2,77-2,70 (м, 1H), 2,60 (дт, J = 2,5, 11,5 Гц, 1H), 2,13-1,92 (м, 2H), 1,74-1,55 (м, 3H), 1,32 (д, J = 7 Гц, 3H), 1,36-1,18 (м, 3H), 1,12 (д, J = 7 Гц, 18H).
Смесь ледяной холодности боргидрида натрия (0,21 г, 5,56 ммоль) и этанола (50 мл) перемешивали 10 минут, затем по каплям в течение 15 минут добавляли 1-(4-триизопропилсилилоксифенил)-2-(4-(3,5- дибромфенил)-4-гидроксипиперидин-1-ил)-пропан-1-он (3,55 г, 5,56 ммоль в 50 мл этанола). Реакционную смесь оставляли стоять до приобретения ею комнатной температуры и перемешивали в течение ночи. Дополнительно добавляли боргидрид натрия (0,10 г) и реакционную смесь перемешивали еще 6 часов. Белый осадок собирали и промывали этанолом (0,84 г). Фильтрат обрабатывали боргидридом натрия (0,10 г) и перемешивали в течение ночи. Белый осадок собирали и промывали этанолом (2,56 г). Объединенный осадок (3,40 г) перекристаллизовывали из этанола с получением 3,0 г (84%) (1R*,2R*)-1-(4-триизопропилсилилоксифенил)-2-(4-(3,5- дибромфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола в виде пушистых белых игольчатых кристаллов с т.пл. 235-236,5oC. Анализ, рассчитано для C29H43Br2NO3Si: C 54,29, H 6,76, N 2,18. Найдено: C 54,17, H 6,50, N 2,35.
Продукт описанной выше реакции (0,53 г, 0,827 ммоль) растворяли в тетрагидрофуране (20 мл) и добавляли фторид тетрабутиламмония (1,25 мл, 1,25 ммоль, 1 М раствор тетрагидрофурана). Реакционную смесь перемешивали 1 час при комнатной температуре, а затем концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (1,5 х 3 дюйма) с элюированием, проводимым следующим образом: 25% этилацетат/гексан (600 мл), невзвешенный отгон, 25% этилацетат/гексан (200 мл), ноль, 25% этилацетат/гексан (200 мл) и 50% этилацетат/гексан (800 мл), 0,20 г (50%) (1R*,2R*)-1-(4-гидроксифенил)-2-(4-(3,5- дибромфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола в виде белого твердого вещества с т.пл. 232-234oC. Анализ, рассчитано для C20H23Br2NO3: C 49,51, H 4,78, N 2,89. Найдено: C 49,77, H 4,58, N 2,76.
ПРИГОТОВЛЕНИЕ 49. (1R*,2R*)-1-(4-гидроксифенил)-2-(4-(3,5- дитритиофенил)-4-гидроксипиперидин-ил)-пропан-1-ол
К раствору (1R*, 2R*)-1-(4-гидроксифенил)-2-(4-(3,5- дибромфенил)-4-гидроксипиперидин-1-ил)-пропан-1-ола (соединения по приготовлению 48, 0,015 г, 0,031 ммоль) в диоксане (3 мл) добавляли 10% палладий на угле (0,013 г) и триэтиламин (0,015 мл). Реакционную смесь дегазировали путем замораживания-оттаивания три раза, затем подвергали воздействию газообразного трития (15 кюри) в течение б часов при комнатной температуре. Реакционную смесь фильтровали через диатомовую землю и фильтр тщательно промывали метанолом (3 мл). Фильтрат концентрировали. Остаток разводили метанолом (1 мл) и концентрировали для удаления всех лабильных примесей трития. Этот процесс разведения/выпаривания повторяли три раза. Остаток растворяли в этаноле (20 мл) и фильтровали через тефлоновый шприцевой фильтр до получения активности 913 мКи. Целиком партию очищали хроматографией на силикагеле (2,5 х 8 см) с элюированием этилацетатом с получением 156 мКи (1R*,2RR*)-1-(4-гидроксифенил)-2-(4-(3,5- дитритиофенил)-4-гидроксипиперидин-ил)-пропан-1-ола с радиохимической чистотой более 98% и специфической активностью 42,8 Ки/ммоль.
ПРИГОТОВЛЕНИЕ 50. 3,5-диметил-4-гидроксипропиофенон
Смесь 2,6-диметилфенола (10,5 г, 85,9 ммоль), пропионовой кислоты (4,64 мл, 86,8 ммоль) и трифторметансульфокислоты (59 г) нагревали до 80oC и поддерживали эту температуру в течение 48 часов. Реакционную смесь охлаждали, выливали в лед и экстрагировали хлороформом. Органические экстракты промывали насыщенным водным бикарбонатом и насыщенным солевым раствором, высушивали и концентрировали до темного маслянистого твердого вещества. Этот материал дистиллировали по способу Kugelrohr при 105-135oC (1,5 мм рт.ст., температура сосуда) с получением 11,2 г (73%) 3,5-диметил-4-гидроксипропиофенона в виде твердого вещества, у которого: ЯМР δ 7,63 (с, 2H), 5,30 (с, 1H), 2,92 (кв, J = 7,5 Гц, 2H), 2,27 (с, 6H), 1,18 (т, J = 7,5 Гц, 3H).
ПРИГОТОВЛЕНИЕ 51. 4-бензилокси-3,5-диметилпропиофенон
Смесь 3,5-диметил-4-гидроксипропиофенона (11,2 г, 62,9 ммоль), бензилбромида (8,23 мл, 69,2 ммоль) и карбоната калия (17,4 г, 125,8 ммоль) в ацетоне (200 мл) перемешивали в течение ночи. Смесь фильтровали и растворитель удаляли. Остаток распределяли между этилацетатом и водой. Фазы разделяли и органический слой промывали насыщенным солевым раствором, высушивали над сульфатом кальция и концентрировали. Остаток подвергали флэш-хроматографии на силикагеле (2,5 х 3,5 дюйма в гексане) и элюировали следующим образом: 5% этилацетат/гексан (700 мл), ноль, 7% этилацетат/гексан (400 мл) и 10% этилацетат/гексан (1500 мл), 15,33 г (91%) 4-бензилокси-3,5-диметилпропиофенона в виде светло-желтого твердого вещества с т.пл. 67-68,5oC, ЯМР δ 7,66 (с, 2H), 7,47-7,32 (м, 5H), 4,83 (с, 2H), 2,95 (кв, J = 7,5 Гц, 2H), 2,32 (с, 6H), 1,20 (т, J = 7,5 Гц, 3H).
ПРИГОТОВЛЕНИЕ 52. 4-бензилокси- α -бром-3,5-диметилпропиофенон
К раствору 4-бензилокси-3,5-диметилпропиофенона (15,19 г, 56,6 ммоль) в четыреххлористом углероде (160 мл) добавляли по каплям бром (2,98 мл, 57,8 ммоль в 40 мл четыреххлористого углерода). Реакционную смесь перемешивали 15 минут после завершения добавления, а затем добавляли водный сульфит натрия и смесь перемешивали еще 30 минут. Фазы разделяли и органический слой промывали насыщенным водным бикарбонатом и насыщенным солевым раствором, высушивали и концентрировали с получением 19,55 г (99%) 4-бензилокси- α -бром-3,5-диметилпропиофенона в виде желтого твердого вещества, удобного для использования без дополнительной очистки и у которого: ЯМР δ 7,72 (с, 2H), 7,52-7,30 (м, 5H), 5,27 (кв, J = 6,5 Гц, 1H), 4,85 (с, 2H), 2,33 (с, 6H), 1,88 (д, J = 6,5 Гц, 3H).
Изобретение относится к медицине, а именно к неврологии для лечения болезни Паркинсона. Вводят млекопитающему эффективное для лечения болезни Паркинсона количество синергически эффективного сочетания двух терапевтических агентов: селективный в отношении переднего мозга антагонист М-метил-D-аспартата (NMDA), и агент, усиливающий возбуждающую обратную связь между вентральным латеральным ядром таламуса и корой головного мозга. Количество каждого из указанных агентов в указанном сочетании достаточно для достижения синергического эффекта. Агенты могут быть введены по отдельности или объединены в фармацевтической композиции. Изобретение позволяет повысить эффективность лечения. 3 с. и 39 з.п. ф-лы.

или его фармацевтически приемлемой солью присоединения кислоты,
где (a) R2 и R5 берутся раздельно, и R1, R2, R3 и R4, каждый, независимо, представляют водород, (C1-C6)алкил, галоген, СFз, ОН или OR7, и R5 представляет собой метил или этил, или (b) R2 и R5 берутся вместе и представляют собой ,
,
образуя хроман-4-оловое кольцо, и R1, R3 и R4, каждый, независимо, представляют водород, (Ci-Cg) алкил, галоген, СFз, ОН или OR7,
R6 представляет собой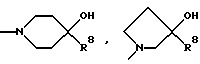
или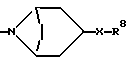
R7 представляет собой метил, этил, изопропил или н-пропил;
R8 представляет собой фенил, необязательно замещенный до трех заместителями, независимо выбранными из группы, содержащей (C1-C6) алкил, галоген и СF3;
Х представляет собой О, S или (СН2)n;
n = 0, 1, 2 или 3.
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства | |||
| -М.: Медицина, 1993, 1, с | |||
| Приспособление, заменяющее сигнальную веревку | 1921 |
|
SU168A1 |
| Способ лечения паркинсонизма | 1978 |
|
SU799723A1 |
| RU 2000114 C1, 22.07.1993. | |||

