Изобретение относится к биотехнологии, в частности к генетической инженерии, и позволяет встраивать гетерологичную ДНК в геном аденовируса птиц CELO без нарушения инфекционных и репродуктивных свойств аденовируса, реконструкцию генома аденовируса в системе in vitro, трансфекцию куриных эмбрионов рекомбинантной ДНК, получение рекомбинантного вектора-аденовируса.
Неудачные попытки использовать бактериальные и даже дрожжевые системы для экспрессии биологически активных эукариотических белков привели к необходимости создания эукариотических векторных систем. Наиболее перспективным оказался путь использования в качестве векторов различных вирусов человека и животных. Однако жесткие требования, которые предъявляются к векторным системам, резко сужают круг вирусов-кандидатов. Из уже испытываемых вирусов-векторов, наиболее обнадеживающие результаты удалось получить с аденовирусами.
Во-первых, аденовирусы полностью удовлетворяют классическим требованиям, предъявляемым к векторным системам- у них хорошо изучены циклы репродукции, транскрипции и трансляции, составлены карты рестрикции, определена несущественная область, имеется широкий набор мутантов и т.д.
Во-вторых, они обладают рядом преимуществ. Размножение вируса в клетке сопровождается мощным ингибированием синтеза клеточных белков - более 90% вновь синтезированных белков являются вирусными. Синтез вирусных белков характеризуется высоким уровнем экспрессии - до нескольких мг с 1 л суспензионной культуры клеток.
Аденовирусы способны размножаться в суспензионной культуре клеток, в частности HeLa, что позволяет достаточно просто и быстро наработать большие количества вирусной массы. Размножение вируса в культуре клеток идет очень быстро и достигает максимума уже через 24-48 ч.
Аденовирусы обладают цитопатическим эффектом, что облегчает выход вирусных белков в культуральную жидкость, а значит упрощает и очистку этих белков. Определены и локализованы на геноме аденовируса участки, ответственные за высокий уровень синтеза вирусных белков, что позволяет их использовать для экспрессии чужеродных генов.
И, наконец, в геном аденовирусов можно вставить, не нарушая процесс сборки полноценных вирионов, значительные по размеру (3-6 тыс. нуклеотидных пар) фрагменты чужеродной ДНК, а используя специализированные клетки-293 и вирус-помощник, размер фрагмента чужеродной ДНК можно увеличить до 38 тыс. нуклеотидных пар.
В настоящее время уже получены положительные результаты по конструированию рекомбинантных аденовирусов. Все они касаются аденовирусов человека 2 и 5 типов. Были сконструированы рекомбинантные аденовирусы, индуцирующие синтез вирусных трансформирующих белков, так называемых Т-антигенов. В работах [1-3], использующих генно-инженерную технику конструирования прокариотических векторов, авторы воссоединили в одной конструкции ген Т-антигена вируса SV-40, а в качестве промотора - большой поздний промотор аденовируса. Данная конструкция либо вводилась в несущественную область генома аденовируса, либо соединялась с аденовектором по удобным сайтам рестрикции, либо получали нужный вектор рекомбинацией in vitro и in vivo. Отбор рекомбинантов вели, используя обезьяньи клетки, где преимущественно размножается рекомбинантный вирус. Затем рекомбинантный вирус размножали на клетках HeLa. Рекомбинантный вирус составлял 5-10% от всей вирусной популяции. Продукция Т-антигена вируса SV-40 достигала 2 мг с 1 л культуры клеток HeLa. Был также сконструирован рекомбинантный аденовирус, содержащий Т-антиген вируса полиомы, индуцирующий синтез чужеродного вирусного белка [4,8].
Аденовирусы в качестве векторов использовались для экпрессии гена тимидин-киназы вируса простого герпеса 1-го типа [5], поверхностного белка оболочки вируса гепатита В [6,7], l-субъединицы человеческого хорионного гонадотропина [5], поверхностного белка оболочки вируса иммунодефицита человека [9].
Если в процессе создания рекомбинантного аденовируса получался дефектный вирус, то для репродукции использовали клетки-293, в которых дефектный аденовирион способен размножаться, либо использовали вирус-помощник, восстанавливающий недостающую функцию. Наиболее эффективными векторами считаются аденовирусы, способные к размножению в животной клетке без вируса-помощника, так как при совместном заражении клеток выход рекомбинантного вируса по сравнению с вирусом-помощником часто не превышает 10%. В результате синтез белков, кодиpуемых рекомбинантным вирусом, значительно ниже уровня синтеза белков вируса-помощника.
Типичная схема конструирования рекомбинантных аденовирусов без помощника (способа встраивания ДНК в геном аденовируса) приведена в работе [7].
На первом этапе авторами была получена рекомбинантная плазмида, содержащая участок вируса Ad5 левого фланга генома (0-1; 9,1-17,0); и гетерологичную часть: ген поверхностного антигена вируса гепатита Б (HBsAg) под контролем регуляторных зон вируса Ad5 и SV-40. Получив ее линейную форму, плазмиду смешивают с основной частью генома вируса Ad5, полученной расщеплением рестриктазой Xbal, и трансфицируют трансформированные (Ad5) клетки человека-293.
В результате рекомбинации по гомологии in vivo в клетках образуются рекомбинантные вирусы (по схеме, см. табл.1).
Выход аденовируса при инфекции клеток достигает 10 БОЕ/мл, а вирусспецифических белков не превышает 2 мг/л культуральной жидкости.
Недостатком изложенного способа является сложность, так как в нем для встраивания ДНК в геном необходимо предварительное дополнительное конструирование рекомбинантной бактериальной плазмиды, а для трансфекции в культуре клеток требуется специальное оборудование, специальные профессиональные навыки и дорогостоящие среды.
Более простая схема сборки генома аденовируса без клонирования фрагментов ДНК аденовирусов в прокариотических системах предложена ранее (авторское свидетельство N 1490962). С целью упрощения способа встраивания ДНК в геном аденовируса проводят гидролиз рестриктазой Xbal ДНК генома аденовируса CELO и плазмидной ДНК и неполный гидролиз рестриктазой Bgl II ДНК генома аденовируса CELO, затем смешивают Bgl II фрагменты ДНК, содержащие 39% левой части генома аденовируса и два Xbal фрагмента аденовируса CELO, содержащие 33-100% генома аденовируса CELO, которые предварительно последовательно лигированы в Xbal сайте на 94% генома с линейной плазмидной ДНК pUC19 смесью, указанных фрагментов проводят трансфекцию клеток аллантоисной оболочки куриных эмбрионов. В результате рекомбинации in vivo двух перекрывающихся фрагментов ДНК CELO образуются рекомбинантные аденовирионы (по схеме, см. табл.2).
Недостатком метода можно считать сложность получения 39% левой части генома Ad CELO путем недогидролиза ДНК CELO рестриктазой Bgl II, а также неконтролируемость процессов рекомбинации in vivo при трансфекции двумя перекрывающимися фрагментами ДНК Ad CELO.
Целью изобретения является определение размеров и точная локализация несущественной области в геноме Ad CELO, что позволяет встраивать большие фрагменты гетерологичной ДНК (от 1,5 до 5,0 т.п.н.) и использовать для этого большое количество сайтов рестрикции, а также простая схема сборки генома аденовируса CELO в контролируемой системе in vitro. Упрощение достигается за счет следующих свойств аденовируса CELO: интеграцию гетерологичной ДНК можно проводить в правом конце генома от 92 до 99,8% карты генома, не нарушая жизненно важных функций вируса; генно-инженерным конструированием in vitro возможно реконструировать полноценный геном. Обнаруженно, что в результате гидролиза ДНК CELO рестриктазами Notl и Xbal можно получить фрагменты, которые в системе in vitro методом лигазной сшивки позволяют реконструировать геном Ad CELO. В результате трансфекции куриных эмбрионов реконструированной рекомбинантной ДНК возможно получение аденовирионов несущих в несущественной области гетерологичную ДНК, т.е. рекомбинантного вектора - рекомбинантного аденовируса птиц CELO.
Способ осуществляют следующим образом.
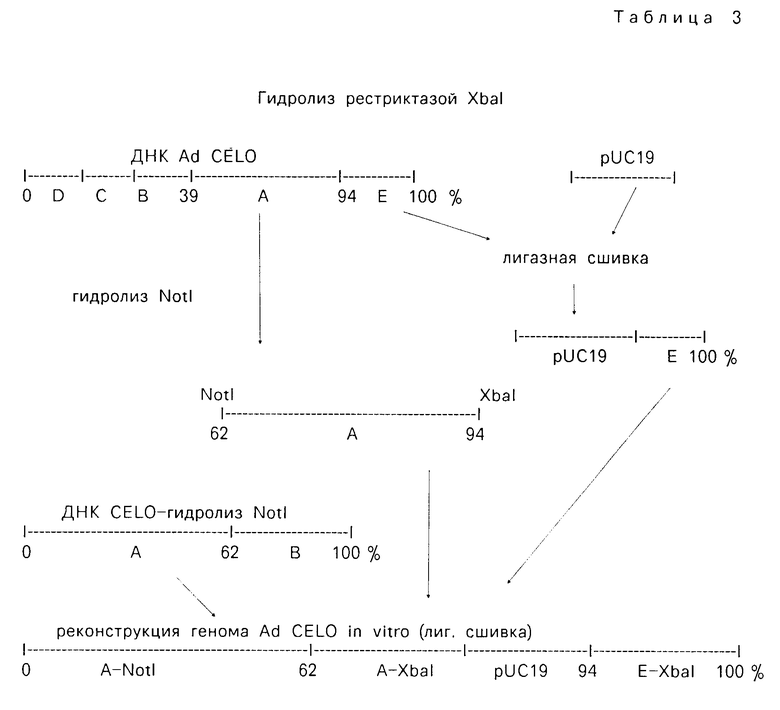
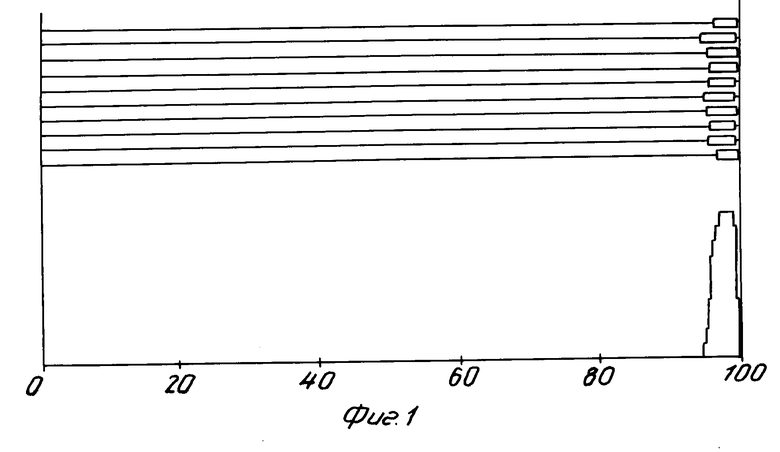
Аденовирус кур типа I (gal1, Chick embryo lethal orphan virus-CELO [1]) размножают в 9-дневных куриных эмбрионах, концентрируют и очищают последовательным центрифугированием в градиенте плотности CsCl. Очищенные аденовирионы подвергают депротеинизации обработкой протеиназой К и последующим экстрагированием фенолом. Вирионную ДНК осаждают этанолом. Методом гетеродуплексного анализа определяют локализацию несущественной области. Схема распределения спонтанных делений по геному представлена на фиг.1-2. Несущественная область локализуется в правом конце генома Ad CELO между 92-99,8% карты генома Ad CELO.
На фиг. 1 показана схема статистического анализа расположения петель 1-го типа по ДНК CELO и локализация их на карте генома Ad СELO; на фиг.2 - результаты статистического анализа расположения петель 2-го типа по ДНК CELO и локализация их на карте генома Ad CELO.
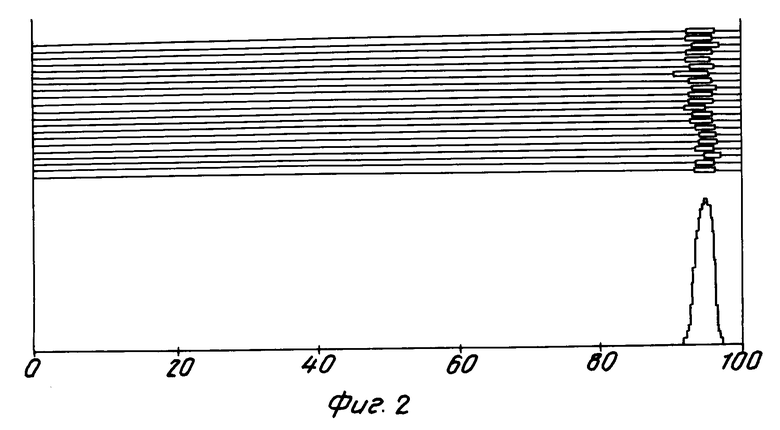
Конструирование вектора проводят по следующей схеме (см. табл.3).
После полного гидролиза ДНК Ad CELO рестриктазой Xbal и разделения фрагментов электрофорезом в 1% агаразном геле вырезают из геля и элюируют концевой фрагмент Е и затем его лигируют с предварительно разрезанной по Xbal сайту плазмидой pUC19. Фрагмент XbalE + pUC19 отделяют от непрореагировавших продуктов электрофорезом, выделяют из геля и проводят полную реконструкцию генома Ad CELO в системе in vitro путем лигазной сшивки следующих фрагментов генома Ad CELO: фрагмент NotI-A, полученный после гидролиза ДНК CELO рестриктазой NotI и выделенный после электрофореза продуктов гидролиза в 0,8% агарозном геле, фрагмента XbalA-NоtI, полученного путем гидролиза фрагмента XbalA рестриктазой NotI и выделенного из агарового геля после электрофореза продуктов гидролиза в 1% агарозном геле и фрагмента XbalE + pUC19, полученного как указано.
Сконструированный in vitro с помощью лигазной сшивки геном Ad CELO вводят в аллантоисную полость 9-дневных куриных эмбрионов. После трех слепых пассажей стандартным методом выделяют рекомбинантный аденовирус CELO.
П р и м е р. Очищенные и сконцентрированные аденовирусы CELO диализуют против 100 объемов 0,01 х SSC; 1 мм ЭДТА, ночь при +4оС. Затем добавляют до 0,5% SDS и до 1 мг/мл протеиназу К (Merck), инкубируют при 37оС 5 ч. Экстрагируют равным объемом водонасыщенного фенола (рН 7,5) дважды, затем экстракцию производят равным объемом фенол: хлороформ (1:1) и хлороформом. ДНК осаждают 2 объемами этанола и оставляют на ночь на -20оС. Осадок собирают центрифугированием на центрифуге К-23, 5000/10 мин, +5оС. Осадок подсушивают, растворяют в 1 мл 0,01 х SSC и вновь осаждают этанолом как указано. ДНК CELO растворяют в 0,01 х SSC и вновь осаждают этанолом как указано. ДНК CELO растворяют в 0,01 х SSC в концентрации 300 мкг/мл.
Гидролиз ДНК CELO рестриктазой Xbal. ДНК CELO 60 мкл (20 мкг) инкубируют с 40 ед. рестриктазы Xbal в присутствии 50 мм NaCl; 6 мМ Трис-Cl, рН 7,9; 6 мМ MgCl2; 100 мкг/мл желатины; 10 мМ 2-МЕ; при +37оС, сутки, общий объем инкубационной смеси был равен 200 мкл. Полноту гидролиза контролируют путем отбора аликвот из инкубационной смеси и проведением мини-гель-электрофореза. Продукты полного гидролиза ДНК CELO наносят на 0,8% горизонтальной агарозный гель и проводят электрофорез в Трис-боратном буфере (0,1 М Трис-Cl; 0,1 М борной кислоты; 2,4 мМ ЭДТА), ночь при напряжении 20 В. Гель окрашивают этидиум-бромидом (0,5 мкг/мг) 15 мин в темноте при +4оС. Из геля вырезают фрагменты А и Е, замораживают жидким азотом и растирают в ступке до порошкообразной массы. К порошкообразной массе добавляют 5 объемов 1,0 М NaCl, интенсивно экстрагируют 5 мин на центрифуге "Эпендорф" при 10000 об/мин. Супернатант экстрагируют фенол-хлороформ добавляют РНК до концентрации 200 мкг/мл и осаждают 3 объемами этанола при -20оС ночь. Осадок собирают центрифугированием 10000/5 мин. Осадок растворяют и 100 мкл 0,01 х SSC и переосаждают повторно этанолом как указано. Растворяют в 50 мкл 0,01 х SSC. Полученный фрагмент Xbal-А гидролизуют рестриктазой NotI. Гидролиз проводят в следующих условиях: 10 мкг (50 мкл) фрагмента Xbal-A инкубируют с 20 ед. рестриктазы NotI в присутствии 100 мМ KCl, 10 мМ MgCl2, 1 мМ DTT, 10 мМ трис-HCl, рН 8,5 при 37оС в объеме 100 мкл 3 ч. Затем отбирают аликвоту (5 мкл) и анализируют гель-электрофорезом полноту гидролиза. Продукты полного гидролиза наносят на 1% агарозный гель и проводят электрофорез в трис-боратном буфере ночь при напряжении 20 В. Гель окрашивают этидиум бромидом 15 мин и в темноте под УФ-лампой вырезают фрагмент XbalA-NotI. Кусочек геля замораживают жидким азотом и растирают в ступке до порошкообразной массы. Дальнейшее выделение и очистку фрагмента проводят как указано для фрагментов XbalA и XbalE.
Гидролиз ДНК CELO рестриктазой NotI проводят в следующих условиях: ДНК CELO 60 мкл (20 мкг) инкубируют с 60 ед. рестриктазы NotI в присутствии 100 мМ KCl, 10 мМ MgCl2, 1 мМ DTT, 10 мМ трис-Cl, рН 8,5 при 37оС в объеме 100 мкл 5 ч. Полноту гидролиза контролировали путем отбора аликвот из инкубационной смеси и проведением мини-гель-электрофореза. Продукты полного гидролиза ДНК CELO наносят на 0,8% горизонтальный агарозный гель и проводят электрофорез в трис-боратном буфере ночь при напряжении 20 В. Дальнейшее выделение и очистку фрагмента NotI-А проводили так же, как и фрагмента Xbal-A.
Фрагмент Xbal-Е используют для лигирования с плазмидой pUC-19.
Гидролиз ДНК pUC-19 рестриктазой Xbal проводят в следующих условиях. 10 мкг (20 мкл) ДНК pUC-19 инкубируют с 20 ед. (20 мкл) рестриктазы Xbal в присутствии 50 мМ NaCl; 6 мМ трис-Cl, рН 7,9; 6 мМ MgCl2; 100 мкг/мл желатины; 10 мМ 2-МЕ в объеме 100 мкл при 37оС, 5 ч. Затем отбирают аликвоту (5 мкл) и анализируют гель-электрофорезом полноту гидролиза. Продукты полного гидролиза экстрагируют равным объемом фенол-хлороформ (1:1) и осаждают 3 объемами этанола + 0,3 М Na ацетата. Осадок собирают центрифугированием при 10000/15 мин и растворяют в 20 мкл Н2О. Обработку гидролизованной Xbal ДНК pUC19 щелочной фосфатазой из Е.coli проводят следующим образом: к 20 мкл ДНК pUC19 добавляют 0,1 ед. (5 мкл) щелочной фосфатазы E.coli; 0,05 М трис-Cl; 1 мМ MgCl2; 0,1 мМ ZnCl2 и 1 мМ спермидина. Инкубацию проводят в 50 мкл при +65оС, 1 ч. Затем пробу разводят до 100 мкл Н2О, трижды экстрагируют равным объемом фенола, а затем проводят гель-хроматографию через колонку (0,5 х 3 см) с сефадексом Г-50, уравновешенную 0,01 х SSC. Пиковые фракции объединяют, добавляют до 200 мкг/мл тРНК и осаждают 3 объемами этанола при -20оС, ночь.
Лигирование обработанной щелочной фосфатазой ДНК pUC19 с фрагментом Xbal-Е проводят следующим образом. Фрагмент Xbal-E 50 мкг (10 мкл) и 10 мкг (15 мкл) ДНК pUC19 инкубировали с 60 ед. (3 мкл) ДНК-лигазы в присутствии 10 мМ АТФ, 50 мМ трис-Cl, рН 7,8, 10 мМ MgCl2 20 мМ дитиотриэтол, 100 мкг/мл БСА в общем объеме 100 мкл при +8оС, сутки. Экстрагируют равным объемом фенол: хлороформом (1:1) и осаждают 3 объемами этанола при -20оС, ночь. Осадок отбирают центрифугированием при 10000/15 мин. Растворяют в 50 мкл 0,01 х SSC и продукты лигирования разделяют электрофорезом в 1% агарозном геле в трис-боратном буфере, ночь при напряжении 20 В. После окрашивания геля этидиум-бромидом (0,5 мкг/мл) под ультрафиолетом вырезают фрагмент Xbal-E, лигировавший с pUC19. Kусочек геля замораживают в жидком азоте и растирают в ступке до порошкообразного состояния. К этой массе добавляют 5 объемов 1 М NaCl, интенсивно экстрагируют, центрифугируют при 10000/5 мин, отбирают супернатант, добавляют до 200 мкг/мл тРНК и осаждают 3 объемами этанола при -20оС, ночь. Осадок собирают центрифугированием при 10000/15 мин.
Реконструкцию генома Ad CELO проводили in vitro путем лигазной сшивки фрагментов ДНК CELO XbalA -Notl; NotIA и ДНК XbalE + pUC19. Инкубационная смесь содержала 60 ед. ДНК-лигазы, 10 мМ АИФ, 50 мМ трис-Cl, рН 7,8, 10 мМ MgCl2, 20 мМ дитиотриэтола, 100 мкг/мл желатины в объеме 150 мкл. Инкубация продолжалась ночь при +8оС. Лигазную смесь затем осаждали 3 объемами этанола, пелетировали центрифугированием и растворяли в 200 мкл стерильного физраствора. Трансфекцию куриных эмбрионов слигировавшими фрагментами Ad CELO проводят следующим образом. Описанную лигазную смесь, растворенную в 200 мкл стерильного физраствора, с помощью стерильного стеклянного капилляра вводят в аллантоисную полость двух 9-дневных куриных эмбрионов (по 100 мкл стерильного физраствора в каждый эмбрион). Куриные эмбрионы инкубируют 72 ч при 37оС. Затем эмбрион охлаждают ночь при +4оС. Стерильно отбирают аллантоисную жидкость и заражают по 0,2 мл пять 9-дневных куриных эмбрионов, инкубируют их также 72 ч при 37оС. Охлаждают и отбирают аллантоисную жидкость как указано, повторяют процедуру заражения еще 3 раза. Затем собирают аллантоисную жидкость от 5 эмбрионов и заражают 30 9-дневных куриных эмбрионов. После инкубирования 72 ч при температуре 37оС эмбрионы охлаждают ночь при +4оС. Затем осторожно и стерильно отсасывают аллантоисную жидкость, наносят на подушку CsCl с плотностью 1,345 г/см и центрифугируют в роторе SW-28 25000/15 ч, +5оС. Отбирают подушку CsCl и наслаивают ее на градиент CsCl (рефрактивный индекс 1,355 - 1,375) и центрифугируют в роторе SW-28.1 26000/20 ч при +5оС. Раскапывают на 30 фракций и исследуют их на наличие рекомбинантного CELO иммуно-дот анализом. Проводят это следующим образом: 5 мкл с каждой фракции наносят на нитроцеллюлозный фильтр (НЦ) ф-мы Millipor, высушивают 30 мин при комнатной температуре, затем инкубируют в буфере (трис-Cl, рН 7,5, 140 мМ NaCl, 0,5% NAN3 0,05% Твин-20, 8% БСА) 2 ч при 37оС. Затем буфер сливают и НЦ инкубируют в таком же буфере, дополнительно содержащем 20 мкг/мл IgG антител к вирусу CELO в течение 2 ч при 37оС. Промывают 3 раза по 15 мин буфером без антител к вирусу CELO и инкубируют в том же буфере, но содержащем 125I-меченый протеин А из расчета 1х106 имп/мин/мл. Инкубируют 4 ч при 37оС. НЦ промывают 5 раз по 15 мин стандартным буфером, подсушивают и ставят на авторадиографию на ночь на -70оС. Фракци, давшие положительный сигнал, объединяют, наносят на градиент плотности CsCl (рефрактивный индекс 1355 - 1,375) и центрифугируют в роторе SW-55 при 40000/20 ч при +5оС. Раскапывают на 20 фракций и анализируют иммунологически как указано. Фракции, содержащие вирус CELO, объединяют и диализуют против 100 объемов 0,01хSSC, 1й мМ ЭДТА ночь при +4оС. Отдиализованный материал исследуют электронно-микроскопически на наличие аденовирионов CELO. Электронно-микроскопический анализ проводят на угольно-формваровой подложке, обработанной в течение 1 мин в тлеющем электрическом разряде с контрастированием уранилацетатом. Изучение вирионов ведут на электронном микроскопе JEM-100В при увеличении 12х104 и ускоряющем напряжении 80 кВ. Были обнаружены аденовирионы с нечетко выраженной морфологией.
Из вируссодержащего материала выделяют вирионную ДНК стандартным методом, т.е. обработкой протеиназой К с последующей экстракцией фенолом. Рекомбинантную вирионную ДНК исследуют методом блот-гибридизации на идентичность с геномом Ad CELO и на содержание нуклеотидных последовательностей в геноме рекомбинанта плазмиды pUC19.
Рекомбинантную ДНК метят методом ник-трансляции следующим образом: 0,1-0,5 мкг рекомбинантной ДНК инкубируют с 1 ед. ДНК-полимеразы Е.coli в присутствии 2 нмоль немеченых дНТФ и 50-100 мкСi 32Р-меченого дНТФ, 50 нмоль ДНК-азыI, 500 мкг/мл БСА, 50 мМ трис-Cl, рН 7,2, 10 мМ MgCl2, 0,1 мМ дитиотриэтола в объеме 50 мкл в течение 2 ч при 16-18оС. Затем добавляют 5 мкл 0,25 М ЭДТА, разводят до 100 мкл Н2О, добавляют 0,4 объема 5 М уксусно-кислого аммония, 200 мкг/мл тРНК и 3 объема этанола и осаждают ночь при -20оС. Осадок собирают центрифугированием при 10000/15 мин. Подсушивают, растворяют в 100 мкл Н2О и вновь переосаждают этанолом. После второго переосаждения растворяют в 500 мкл Н2О, измеряют удельную активность меченой ДНК. Она составляет 4-10х107 имп/мин/мкг. Аналогично метят и ДНК CELO. Рекомбинантную меченую ДНК CELO гибридизуют с ДНК CELO, рекомбинантной ДНК CELO, ДНК плазмиды pUC19, ДНК фага Т5 методом блот-гибридизации. Перед блот-гибридизацией ДНК CELO, ДНК pUC19 гидролизуют рестриктазой EсoRI, ДНК фага Т5 рестриктазой HindIII. Гидролиз ДНК CELO и ДНК pUC19 проводят следующим образом: 1 мкг ДНК инкубируют с 2 ед. рестриктазы в присутствии соответствующего рестриктазного буфера в объеме 20 мкл в течение 1 ч при 37оС. Аналогичным образом гидролизуют ДНК фага Т5 рестриктазой HindIII. Продукты гидролиза наносят на 1% агарозный гель и ведут электрофорез в трис-боратном буфере ночь при напряжении 40 В. Обработку геля и перенос продуктов гидролиза на нитроцеллюлозную подложку (ф-мы Millipor) проводили согласно методике, рекомендованной фирмой Шлейхер и Шуль. Гибридизацию проводят следующим образом: нитроцеллюлозный фильтр погружают в предгибридизационный буфер (6хSSC, 5хДенхардит, 200 мкг/мл дрожжевой РНК) и инкубируют при 65оС 4ч. Затем предгибридизационный буфер сливают и заливают гибридизационным буфером (6хSSC, 2хДенхардт, 200 мкг/мл дрожжевой РНК и предварительно прогретая на кипящей водяной бане 5 мин меченая рекомбинантная ДНК CELO 1-2х106 имп/мин/мл гибридизационного буфера) и инкубируют в течение ночи при +65оС. затем гибридизационный буфер сливают и ведут отмывку фильтра, инкубируя его с 2хSSC 3 раза по 15 мин. Затем фильтр подсушивают, заворачивают в полиэтиленовую пленку и ведут авторадиографию, используя рентгеновскую пленку РМ-1, ночь или 3 - 6 дней при -70оС. На авторадиограммах видно, что рекомбинантная ДНК CELO гибридизуется с ДНК CELO и с ДHК pUC19, но не гибридизуется с ДНК фага Т5. В то же время меченая ДНК CELO гибридизуется с ДНК CELO, но не гибридизуется с ДНК pUC19 и с ДНК фага Т5. Таким образом, полученные данные показывают, что рекомбинантная ДНК CELO несет в своем составе нуклеотидные последовательности ДНК pUC19.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФРАГМЕНТ ДНК, КОДИРУЮЩИЙ СИНТЕЗ ГЛИКОПРОТЕИНА G ВИРУСА БЕШЕНСТВА, РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК PVG18-1, КОДИРУЮЩАЯ ГЛИКОПРОТЕИН G ВИРУСА БЕШЕНСТВА, ШТАММ БАКТЕРИЙ ESCHERICHIA COLI - ПРОДУЦЕНТ ГЛИКОПРОТЕИНА G ВИРУСА БЕШЕНСТВА | 1991 |
|
RU2008355C1 |
| СПОСОБ СОЗДАНИЯ РЕКОМБИНАНТНОГО АДЕНОВИРУСА ПТИЦ ДЛЯ ВАКЦИНАЦИИ ПРОТИВ ВИРУСА ГРИППА ПТИЦ Н5N1 | 2006 |
|
RU2326943C1 |
| НЕИНФЕКЦИОННЫЙ ДЛЯ ЧЕЛОВЕКА АДЕНОВИРУС КАК ВЕКТОР ДЛЯ ЗАМЕСТИТЕЛЬНОЙ ГЕННОЙ ТЕРАПИИ НАРУШЕНИЙ АНГИОГЕНЕЗА, ОБЕСПЕЧИВАЮЩИЙ ЭФФЕКТИВНЫЙ СИНТЕЗ АНГИОГЕНИНА ЧЕЛОВЕКА В ТРАНСФЕЦИРОВАННЫХ КЛЕТКАХ МЛЕКОПИТАЮЩИХ, СПОСОБ ИНДУКЦИИ АНГИОГЕНЕЗА, СПОСОБ ЛЕЧЕНИЯ ИШЕМИЧЕСКОЙ БОЛЕЗНИ, КОМПОЗИЦИЯ ДЛЯ ИНДУКЦИИ АНГИОГЕНЕЗА И ЛЕЧЕНИЯ ИШЕМИЧЕСКОЙ БОЛЕЗНИ | 2005 |
|
RU2321631C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРОРАЛЬНОЙ ВАКЦИНЫ ПРОТИВ ВИРУСА БЕШЕНСТВА | 2010 |
|
RU2432963C1 |
| СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ЛАКТОФЕРРИНА ЧЕЛОВЕКА | 2007 |
|
RU2340674C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pVR6-II, ПРЕДНАЗНАЧЕННАЯ ДЛЯ СПЕЦИФИЧЕСКОГО ВЫЯВЛЕНИЯ РОТАВИРУСОВ II СУБГРУППЫ, ШТАММ БАКТЕРИЙ ESCHERICHIA COLI - НОСИТЕЛЬ РЕКОМБИНАНТНОЙ ПЛАЗМИДНОЙ ДНК dVR6-II, ПРЕДНАЗНАЧЕННОЙ ДЛЯ ВЫЯВЛЕНИЯ РОТАВИРУСОВ II СУБГРУППЫ | 1991 |
|
RU2031949C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК, КОДИРУЮЩАЯ СИНТЕЗ HBS AG ВИРУСА ГЕПАТИТА B, И ШТАММ ВИРУСА ОСПОВАКЦИНЫ - ПРОДУЦЕНТ HBS AG ВИРУСА ГЕПАТИТА B | 1988 |
|
SU1515698A1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК РС VА ДЛЯ ЭКСПРЕССИИ РИБОЗИМА В ЭУКАРИОТИЧЕСКОЙ КЛЕТКЕ | 1995 |
|
RU2097429C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pUR 290 S ΔE, КОДИРУЮЩАЯ ГИБРИДНЫЙ ПОЛИПЕПТИД β -ГАЛАКТОЗИДАЗА-NS 1-БЕЛОК ВИРУСА КЛЕЩЕВОГО ЭНЦЕФАЛИТА, И СПОСОБ ЕЕ КОНСТРУИРОВАНИЯ | 1989 |
|
RU1679798C |
| ШТАММ РЕКОМБИНАНТНОГО ВИРУСА ОСПОВАКЦИНЫ, ЭКСРЕССИРУЮЩИЙ СТРУКТУРНЫЕ БЕЛКИ ВИРУСА ВЕНЕСУЭЛЬСКОГО ЭНЦЕФАЛОМИЕЛИТА ЛОШАДЕЙ И ПРИГОДНЫЙ ДЛЯ ПРОИЗВОДСТВА ИММУНОБИОЛОГИЧЕСКИХ ПРЕПАРАТОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2091489C1 |

Изобретение относится к биотехнологии, в частности к генетической инженерии, и позволяет встраивать гетерологичную дезоксинуклеиновую кислоту в геном аденовируса птиц CELO без нарушения инфекционных и репродуктивных свойств аденовируса, реконструкцию генома аденовируса в системе invitro, трансфекцию куриных эмбрионов рекомбинантной ДНК, получение рекомбинантного вектора-адденовируса. Сущность изобретения состоит в том, что встраивание ДНК в геном аденовируса проводят путем гидролиза рестриктазой Xba1 ДНК геном аденовируса CELO и ДНК прокариотического вектора pUC19, а также гидролиза ДНК аденовируса CELO рестриктазой Not1 и гидролиза рестриктазой Not1 фрагмента ДНК CELO / Xba 1. Затем в системе invitro методом лигазной сшивки последовательно реконструируют геном аденовируса CELO из фрагментов ДНК ad CELO, таких, как Not1 - A,Xba1A - Not1 и Xba1E + ДНК pUC19Xba1. Реконструированной в системе invitro рекомбинантной ДНК CELO проводят трансфекцию клеток хорионаллантоисной оболочки куриных эмбрионов. В результате трансфекции рекомбинантной ДНК CELO образуются полноценные вирионы птичьего аденовируса CELO, содержащие в несущественной области генома гетерологичную ДНК - ДНК прокариотического вектора pUC19. 2 с.п. ф-лы, 2 ил.
СПОСОБ ВСТРАИВАНИЯ ГЕТЕРОЛОГИЧНОЙ ДНК В ГЕНОМ АДЕНОВИРУСА CELO И РЕКОМБИНАНТНЫЙ АДЕНОВИРУСНЫЙ ВЕКТОР CELO/pUC 19.
| Авторское свидетельство СССР N 1490962, кл | |||
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |

