Изобретение относится к окислению алифатических углеводородов, таких как моноолефины и алканы, в целях получения более высоконенасыщенных алифатических углеводородов.
Ненасыщенные алифатические углеводороды, такие как моноолефины и диолефины, используются в качестве мономеров или сомономеров при получении пластиков на основе полиолефина.
Известен способ окислительного дегидрирования углеводородов парафинового ряда, таких как бутан, в присутствии катализатора, содержащего окси-соединения молибдена и магния, и носителя на основе оксида алюминия, а также может содержать ванадий и/или кремний. Процесс проводится при 500-650оС, давлении 7-210 кПа и других условиях, обеспечивающих получение реакционной смеси, содержащей 1,3-бутадиен с избирательностью не менее 40 М Если бутан взаимодействует с катализатором, то продукты содержат бутены и бутадиены. Однако избирательность и объемный выход бутадиена остается ниже желательного уровня. Кроме того, исходное содержание углеводорода и кислорода является нежелательным из соображений безопасности. И наконец, носитель на основе окиси магния не обладает достаточной ударной вязкостью и прочностью на истирание, которые необходимы для использования его в реакторах с псевдоожиженным слоем или реакторах с подвижным слоем.
В одном варианте изобретение относится к способу получения ненасыщенных алифатических углеводородов, который включает в себя взаимодействие алифатического углеводорода, имеющего по крайней мере три атома углерода, с катализатором изобретения, который будет описан ниже. При реакционных условиях предлагаемого способа более ненасыщенные алифатические углеводороды, такие как диолефины, образуются с избирательностью по крайней мере около 40 М%
Преимущественно алифатические углеводороды могут быть подвергнуты непосредственному окислению способом изобретения в целях получения более высоконенасыщенных алифатических углеводородов. Предлагаемый способ показал высокую избирательность и высокую продуктивность более высоконенасыщенных алифатических углеводородов, в частности диолефинов. Предлагаемый способ показал также низкую избирательность и низкий выход нежелательных продуктов глубокого окисления, таких как моноокись и двуокись углерода. Кроме того, с помощью предлагаемого способа бутадиен может быть получен непосредственно из бутана с высокой степенью селективности и продуктивности и при этом с низкой степенью селективности для продуктов глубокого окисления. В целях изобретения "продуктивность" определяют как граммы целевого продукта, полученного на 1 г катализатора за 1 ч.
Ненасыщенные алифатические углеводороды, такие как моноолефины или диолефины, используются в качестве мономеров или сомономеров при получении полиолефинов. Бутадиен может быть также использован как промежуточное соединение при получении стирола.
В другом варианте изобретение относится к твердой гетерогенной каталитической композиции, содержащей активный атом кислорода, причем указанный катализатор может быть использован в указанном способе получения ненасыщенных алифатических углеводородов. Этот катализатор состоит в основном из окиси магния, окиси молибдена и активатора, содержащего щелочной металл в концентрации, приблизительно 0,1-5 мас. по массе объединенных окисей магния и молибдена. Катализатор может содержать окись ванадия.
В еще одном варианте изобретение относится к каталитической композиции молибдата, содержащей реакционноспособный кислород. Указанная композиция содержит носитель, включающий в себя окись магния и по меньшей мере одну окись алюминия, выбранную из группы, содержащей Al2O3и алюминат магния (MgAl2O4). Указанный носитель имеет массовое отношение MgO/Al2O3 в пределах 0,30-4,0 и поверхностную площадь по крайней мере около 25 м2/г. Эта каталитическая композиция также содержит каталитический компонент, состоящий в основном из окиси молибдена, окиси магния и активирующее количество промотора из щелочного металла. Катализатор может содержать окись ванадия. При предпочтительном осуществлении изобретения показатель прочности на истирание, определенный ниже, составляет менее 5 мас. за 1 ч.
Каталитическая композиция изобретения используется в указанном способе окисления алифатических углеводородов в более ненасыщенные алифатические углеводороды. В предпочтительном своем исполнении каталитическая композиция обладает повышенной прочностью на истирание по сравнению с катализаторами прототипов. Таким образом, катализатор изобретения может быть использован в реакторах с псевдоожиженным слоем и реакторах с подвижным слоем, таких как реактор с восходящим слоем катализатора.
Алифатические углеводороды, которые могут быть использованы в предлагаемом способе, включают в себя алканы и олефины, которые имеют три или более атомов углерода.
Альтернативно алканы могут быть охарактеризованы как углеводороды парафинового ряда. Указанные соединения известны специалистам как насыщенные углеводороды. Как было указано, алканы содержат по крайней мере три атома углерода и могут иметь структуру с прямой или разветвленной цепью. Обычно алканы содержат до 20 атомов углерода. Примерами подходящих алканов могут служить н-бутан, н-пентан, н-гексан, н-гептан, н-октан, н-нонан, н-декан, н-додекан и более насыщенные гомологи, а также изобутан, изопентан, неопентан и, кроме того, разветвленные гексаны, гептаны, октаны, нонаны, деканы, додеканы и более разветвленные гомологи. Некоторые алициклические углеводороды являются подходящими реагентами и поэтому также используются в предлагаемом способе. Примерами алифатических углеводородов могут служить циклобутан, циклопентан, циклогексан, циклогептан, циклооктан, метилциклопентан, метилциклогексан, и другие алкил-замещенные циклоалканы. Предпочтительно, если алкан является нормальным или линейным.
Олефины, кроме того, могут быть охарактеризованы как алифатические углеводороды, содержащие по крайней мере одну ненасыщенную двойную связь. Как указывалось выше, олефины также содержат по крайней мере три атома углерода, а обычно до 20 атомов углерода. Местоположение двойной связи не является критическим; поэтому она может находиться на конце или внутри углеродной цепи. Предпочтительно, однако, чтобы олефин имел нормальную или линейную, а не разветвленную структуру. Например, 1-бутен является предпочтительней, чем изобутилен. Примерами подходящих олефинов могут служить: 1-бутен, 2-бутен, 1-пентен, 2-пентен, 3-пентен, 1-гексен, 2-гексен, 3-гексен и, кроме того, 1-гептен, 1-октен, 1-ионен, 1-децен и их изомеры, где ненасыщенность имеет место в любом другом положении углеродной цепи.
В предлагаемом способе могут быть также использованы олефины, содержащие более одной двойной связи, такие как 1,3-гексадиен и изопрен, которые превращаются в более ненасыщенные углеводороды. Некоторые алифатические олефины, такие как циклогексен и винилциклогексан, также являются подходящими исходными материалами и поэтому также могут быть включены в изобретение. Предпочтительным олефином является моноолефин. Более предпочтительным является 1- или 2-бутен. Алкины, однако, не являются подходящими реагентами в предлагаемом способе.
Приведенные выше примеры алифатических углеводородов представляют наиболее типичные из подходящих для предлагаемого способа углеводородов, однако, эти примеры не должны ограничивать другие возможные варианты. Подходящими для предлагаемого способа могут оказаться и другие алифатические углеводороды, известные специалистам.
Предпочтительными алканами являются нормальные парафины, которые могут быть представлены следующей общей формулой:
СН3-(СН2)n-СН3, где n является целым числом от 1 до 8. Более предпочтительно, если n является целым числом от 2 до 6. Наиболее предпочтительно, если n является 2, а алканом является н-бутан.
Необязательно алифатический углеводородный реагент может быть разбавлен нереакционноспособным газом, таким как азот, гелий, аргон, метан, двуокись углерода, или паром. Так как тип разбавителя выбирается в основном из соображений экономического порядка, то предпочтительным разбавителем можно считать азот. Количество разбавителя (если он используется) может варьироваться в широких пределах, в зависимости от строения реактора и реакционной способности твердого окислителя. Содержание углеводорода в смеси углеводород-разбавитель обычно составляет 1-100 М Предпочтительным содержанием углеводорода в смеси является 10-100 М% а наиболее предпочтительным 40-100 М
Каталитическая композиция изобретения представляет собой твердую гетерогенную окись, по крайней мере часть кислорода которой является активной. Это значит, что в катализаторе присутствует активная форма кислорода и что эта активная форма кислорода способна окислять алифатический углеводород. Таким образом, в одном из вариантов изобретения катализатором является твердый окислитель. После того как активный кислород удаляется из реактора, катализатор истощается. Кроме того, катализатор может еще образовывать некоторое время углеродные остатки на своей поверхности. Истощенный и отравленный катализатор может быть восстановлен с помощью источника газообразного кислорода. Таким образом, для осуществления каталитического процесса изобретения необходимо к алифатическому углеводороду добавлять кислород.
Кислород обычно поставляется от газового источника, снабженного непрерывной подачей кислорода. В рассматриваемом способе может быть использован любой источник кислорода, такой как чистый элементный кислород, воздух или закись азота. Предпочтительным источником кислорода является воздух (газ). Необязательно газообразный элементный кислород может быть разбавлен нереакционноспособным газом, таким как азот, гелий, аргон или двуокись углерода. Предпочтительным разбавителем является азот. Если используется нереакционноспособный разбавитель, то предпочтительно, чтобы содержание кислорода в смеси не превышало приблизительно 50 М% Более предпочтительно, если содержание кислорода в смеси находится в пределах 0,5-30 М% Наиболее предпочтительным содержанием кислорода в смеси является 1-20 М%
Количество кислорода, используемого в предлагаемом каталитическом способе, должно быть достаточным для полного окисления твердого гетерогенного катализатора и достаточным для удаления углеродных остатков с поверхности катализатора. Предпочтительно, если регенерацию катализатора осуществляют отдельно от окисления алифатического углеводорода.
Альтернативно можно осуществлять подачу алифатического углеводорода вместе с небольшим количеством газообразного элементного кислорода. Целью такой подачи является удаление углеродных остатков с поверхности катализатора, восстановление до некоторой степени реакционноспособного кислорода катализатора и удаление любого водорода, образующегося в процессе реакции. Концентрация кислорода в алифатическом углеводороде и при подаче диктуется лишь соображениями взрывоопасности указанной смеси. Предпочтительно, если концентрация кислорода находится ниже предела детонации.
Твердый гетерогенный катализатор, используемый в изобретении, содержит в основном окись магния, окись молибдена и щелочно-металлический промотор. В качестве окиси магния может быть использован любой источник, однако, предпочтительным является MgO. Аналогично в качестве окиси молибдена может быть использован любой приемлемый источник, например MoO3, (NH4)6Mo7O24˙4H2O и (NH4)2˙MoO4. Окись молибдена может быть также получена из предшествующего молибденового соединения, такого как карбонил молибдена, например, Mo(CO)6. Предпочтительным источником смеси молибдена является (NH4)6Mo7O24˙4H2O. Щелочно-металлический промотор является металлическим соединением (металл группы IA), имеющим основность, достаточную для образования более ненасыщенных соединений в предлагаемом способе. В катализаторе могут также присутствовать небольшие количества других элементов при условии, что указанные элементы не вносят существенных изменений в производительность катализатора.
Техника изготовления катализатора, содержащего смешанные окиси магния и молибдена, а также промотор из щелочного металла, очень проста. Обычно нужное количество окиси молибдена или ее предшественника, такого как гептамолибдат аммония или карбонил молибдена, растворяют в растворителе для получения раствора. Предпочтительным молибденовым соединением является гептамолибдат аммония, а предпочтительным растворителем вода. Полученный раствор выливают поверх окиси магния для образования шлама, который затем подвергают осушке для удаления растворителя. Если раствор является водным, то осушку проводят в печи при температуре порядка 70-120оС. Высушенную композицию подвергают кальцинированию для получения каталитически активной смеси на основе окиси магния и окиси молибдена. Кальцинирование обычно проводят при 550-650оС в течение 1-24 ч. Предпочтительно, если кальцинирование проводят приблизительно при 600оС и по меньшей мере в течение 2 ч. Альтернативно осушенная композиция, полученная как описано выше, может быть использована непосредственно в каталитическом способе изобретения, т.е. без предварительного кальцинирования. Поскольку молибденовое соединение может быть конвертировано в окись молибдена при 300оС или около того, а каталитический слой нагревают до температуры выше чем 300оС, то осушенная композиция будет in situ превращаться в каталитически активную смесь из окисей магния и молибдена.
Смешанная оксидная каталитическая композиция может показать аморфность при рентгенографии, или может показать дифракционные пики, характерные для молибдата магния. Элементный анализ кальцинированной твердой композиции дает следующий состав, мас. МоО3 6-50; MgO 94-50. Предпочтительным составом является следующий состав композиции, мас. МоО3 1-30; MgO 90-70, а наиболее предпочтительным является состав, мас. МоО3 15-25; MgO 85-75.
При предпочтительном осуществлении изобретения молибденовая каталитическая композиция содержит компонент носителя и каталитический компонент. Носитель содержит окись магния и по крайней мере одну окись алюминия, выбранную из группы, состоящей из окиси алюминия (Al2O3) и алюмомагнезиальной шпинели (MgAl2O4). Носитель имеет весовое MgO/Al2O3 в пределах 0,30-4,0 и поверхностную площадь по крайней мере около 25 м2/г. Каталитический компонент в основном состоит из окиси молибдена, окиси магния и активирующего количества промотора на основе щелочного металла. Необязательно катализатор может содержать окись ванадия.
Окись алюминия сообщает частицам катализатора главным образом твердость и прочность на истирание, так чтобы их можно было использовать в реакторах с псевдоожиженным слоем и с передвижным слоем. Для данных целей может быть использован любой источник окиси алюминия, например, α-β- и γ-формы глинозема, такие как бемский глинозем, водный коллоидальный глинозем, стехиометрический Al(OH)3 и алкоксиды алюминия, как будет показано ниже. Подходящими источниками окиси алюминия являются также алюминат магния и гидроокиси алюмината магния. В предлагаемом способе окись магния играет двойную роль: во-первых, она выступает в роли носителя для каталитических компонента, который нейтрализует кислотность окиси алюминия и другие остаточные кислотные центры. Крайне желательно, чтобы катализатор был основным, так как основность усиливает десорбцию олефиновых продуктов. В качестве окиси магния может быть использован любой приемлемый источник, однако предпочтительным является MgO. Окись молибдена вносит значительный вклад и активность катализатора, особенно если она используется в сочетании с окисью магния в виде молибдата магния. Предпочтительно, если молибден находится в степени окисления +6. В качестве окиси молибдена может быть использован любой приемлемый источник, например MoO3, (NH4)6Mo7O24˙ 4H2O и (NH4)2˙MoO4. Окись молибдена может быть также получена из молибденового предшественника, такого как карбонил молибдена, например Мо(СО)6. Предпочтительным источником окиси молибдена является гептамолибдат аммония, представленный формулой (NH4)6Mo7O24˙4H2O. Промотор из щелочного металла предназначен для повышения основности катализатора и увеличения тем самым избирательности более высоконенасыщенных соединений изобретения. В катализаторе может присутствовать небольшое количество других элементов при условии, что указанные элементы не оказывают существенного влияния на производительность катализатора.
Как отмечалось, компонент носителя может содержать фазу шпинели (MgAl2O4). Содержание шпинели в носителе может колебаться в пределах 0-100 мас.
Обычно изготовление катализатора начинают с получения смеси компонентов окиси магния и окиси алюминия в целях образования носителя для других каталитических компонентов. Для объединения указанных компонентов может быть использован любой метод; однако предпочтительными являются три способа. Первый способ заключается в том, что предварительно полученную заготовку шпинели (MgAl2O4) пропитывают раствором, содержащим растворимую соль магния, такую как нитрат магния, хлорид магния, сульфат магния, ацетат магния или т. п. при условии, что указанная соль может быть конвертирована в окись магния при кальцинировании; а затем пропитанную шпинель кальцинируют. Массовое отношение окиси магния к алюминату магния может быть выражено как массовое отношение окиси магния к окиси алюминия. Указанное отношение является критическим для производительности каталитической композиции и подробно обсуждается ниже. Температура кальци- нирования обычно колеблется в пределах 400-1200оС, предпочтительно в пределах 450-900оС, а наиболее предпочтительно в пределах 500-700оС. Кальцинирование проводят в течение периода времени, достаточного для образования сплавленной композиции, которая играет роль носителя для каталитических компонентов, и составляющего по крайней мере около 0,5 ч.
Второй способ заключается в том, что предварительно полученную заготовку глинозема пропитывают раствором, содержащим растворимую соль магния. Типичными примерами заранее заготовленных окисей алюминия являются безводные или гидратные твердые глиноземы, например α-,β- и γ-глиноземы и бемский глинозем. Массовое отношение окиси магния к окиси алюминия (MgO/Al2O3) является критическим параметром и более подробно описывается ниже. Температура кальцинирования обычно варьируется в пределах 400-1200оС, предпочтительно в пределах 450-900оС, а наиболее предпочтительно в пределах 500-700оС. Кальцинирование проводят в течение периода времени, достаточного для образования сплавленного и отвержденного композиционного материала, который играет роль носителя для каталитических компонентов. Обычно кальцинирование проводят по крайней мере около 0,5 ч. Во время кальцинации часть окиси алюминия и окиси магния могут химически смешиваться, образуя фазу шпинели (MgAl2O4), которая представляет собой однородную смесь между доменами окиси алюминия и окиси магния.
Третий способ получения носителя заключается в том, что к окиси магния добавляют коллоидный глинозем и полученную смесь высушивают при условиях, достаточных для получения носителя из окиси магния и окиси алюминия. Коллоидный глинозем представляет собой подкисленную водную суспензию гидратной окиси алюминия, где поверхностная площадь частиц является настолько больше их объема, что указанные частицы не осаждаются под действием силы тяжести. Количество указанной коллоидной суспензии окиси алюминия, добавляемое к окиси магния, должно быть таким, чтобы конечное массовое отношение окиси магния к окиси алюминия находилось в определенных пределах, которые будут определены ниже. Значение рН смеси коллоидной окиси алюминия и окиси магния составляет около 9. Затем смесь высушивают, используя один из стандартных методов, например выдерживание и выпаривание, сушка распылением, сушка в потоке горячего воздуха, сушка на барабане и т.п. Одним из предпочтительных методов является выдерживание и выпаривание смеси над горячей плитой или эквивалентным средством для нагревания в целях получения более густого геля и в итоге твердой массы, которую затем размалывают и просеивают до получения частиц желательных размеров. Температура выдерживания и выпаривания может быть любой температурой, являющейся совместимой с системой растворителей. Поскольку предпочтительным растворителем является вода, то указанная температура находится в пределах 30-100оС. Предпочтительной температурой является температура 50-90оС, а наиболее предпочтительной 60-80оС. Время, необходимое для выдерживания, зависит от количества геля и оно должно быть достаточным для получения твердой тугоплавкой массы.
В условиях промышленного производства описанную выше смесь окиси магния и коллоидной окиси алюминия предпочтительно осушивать путем распыления. В этих целях может быть использовано любое устройство для осушки распылением, которое обычно применяется в реакторах с псевдоожиженным слоем. В качестве примера может служить устройство для распыления Niro Atomizer S-12, 5-R/N. Указанное устройство имеет средство для регулирования температуры на входе и выходе. Обычно измельченные частицы, полученные при осушке распылением, имеют сферическую форму с диаметром приблизительно от 10-250 мкм и хорошие показатели текучести.
Порошок, полученный после осушки путем выдерживания распыления, кальцинируют в целях получения композиционного носителя, состоящего в основном из окисей магния и алюминия, необязательно из шпинельной фазы алюмината магния. Кальцинирование проводят в условиях, достаточных для сплавления окиси алюминия и окиси магния в отвержденную массу. Обычно кальцинирование проводят при 400-1200оС. Более предпочтительно проводить кальцинирование при 350-900оС, а самой предпочтительной является температура 500-700оС. Обычно период кальцинирования зависит от количества кальцинируемого материала и составляет по крайней мере около 0,5 ч.
Компонент носителя изобретения может иметь любое массовое отношение окиси магния к окиси алюминия при условии, что полученный в результате носитель имеет достаточную твердость и основность. Следует отметить, что хотя шпинельная фаза присутствует как отдельная композиция MgAl2O4, однако массовое отношение MgO/Al2O3 может быть рассчитано. В основном массовое отношение MgO/Al2O3 находится в пределах 0,1-9,0; при этом предпочтительным является массовое отношение 0,3-4,0, а более предпочтительным 0,3-2,0. Наиболее предпочтительное отношение составляет 0,38-0,80. Если значение массового отношения ниже указанного нижнего предпочтительного предела, то это означает слишком низкое содержание окиси магния, и поэтому катализатор может иметь слишком большую кислотность. Если значение массового отношения выше указанного верхнего предпочтительного предела, то это означает слишком большое содержание окиси магния, и поэтому катализатор может иметь слишком низкую ударную вязкость и прочность на истирание.
Кроме того, компонент носителя изобретения характеризуется своей поверхностной площадью, Обычно поверхностная площадь составляет по меньшей мере около 25 м2/г. Предпочтительное ее значение составляет по меньшей мере около 35 м2/г, а более предпочтительное по меньшей мере 50 м2/г. Еще более предпочтительно, если поверхностная площадь колеблется в пределах 50-250 м2/г, а наиболее предпочтительно 80-170 м2/г. Специалистам хорошо известно, что слишком малая поверхностная площадь соответствует низкой каталитической активности, тогда как большая поверхностная площадь обычно соответствует высокой каталитической активности. Каталитическая композиция изобретения имеет большую поверхность площади и высокую каталитическую активность.
После получения носителя на него наносят каталитические элементы окиси молибдена, щелочно-металлического промотора и необязательно окись ванадия. Если отношение MgO/Al2O3 находится в пределах допустимых значений, указанных выше, то в дополнительном добавлении окиси магния нет необходимости. Обычно необходимое количество окиси молибдена или молибденового соединения-предшественника, такого как гептамолибдат аммония или карбонил аммония, растворяют в растворителе для получения раствора. Предпочтительным молибденовым соединением является гептамолибдат аммония, а предпочтительным растворителем вода. Затем раствор подвергают взаимодействию с композитом-носителем, изготовленным как описано выше, и полученную в результате суспензию высушивают в целях удаления растворителя. Если указанный раствор является водным, то осушку проводят в печи при 70-120оС. Затем осушенную суспензию подвергают кальцинированию для образования каталитически активной композиции, содержащей окись алюминия, окись магния и окись молибдена. В основном кальцинирование проводят при 300-900оС в течение 0,5-24 ч. Предпочтительная температура кальцинации составляет 500-800оС, а более предпочтительная 550-650оС. Альтернативно осушенную суспензию, описанную выше, можно, не подвергая предварительному кальцинированию, непосредственно использовать в каталитическом процессе изобретения. Так как молибденовый предшественник может быть превращен в окись молибдена при 300оС или около того, а каталитический слой нагревают до температуры, превышающей 300оС, то осушенная композиция будет превращаться in situ в каталитически активную смесь из окисей магния и молибдена.
Смешанная оксидная каталитическая композиция, как правило, показывает один или несколько дифракционных пиков при рентгенографии, относящихся к следующим компонентам: окиси магния, молибдата магния, алюмомагнезиальной шпинели и окиси алюминия. Элементный анализ кальцинированной твердой композиции обнаруживает следующий состав, мас. МоО3 3-50; MgO 90-10, уравновешенные оксидом алюминия. Предпочтительным составом является следующий состав, мас. MoO3 10-30; MgO 60-20, а более предпочтительным МоО3 12-25; MgO 40-25.
К описанному выше катализатору на носителе необходимо добавить активирующее количество, по крайней мере одного промотора из щелочного металла. Промотор служит для повышения избирательности и продуктивности ненасыщенных продуктов, например диолефинов, в предлагаемом способе. Указанным промотором обычно являются соединения лития, натрия, калия, рубидия, цезия или фракции, имеющие достаточную основность для повышения избирательности для более высоконенасыщенных соединений изобретения. Подходящим для этой цели промоторными соединениями являются щелочные окислы, гидроокиси и карбонаты. Подходящими являются также соединения, которые при нагревании разлагаются на окиси, например, такие как ацетаты и оксалаты щелочных металлов. Также могут быть использованы соли щелочных металлов, однако галиды и силикаты щелочных металлов использовать нежелательно вследствие их низкой основности. Предпочтительными промоторами являются: окись, гидроокись, карбонат, ацетат или оксалат щелочного металла. Из них более предпочтительными являются окись или гидроокись калия или цезия. Наиболее предпочтительным промотором является окись или гидроокись калия.
Количество щелочно-металлического промотора является критическим параметром для производительности катализатора. Обычно приемлемым является любое количество указанного промотора, достаточное для увеличения избирательности и объемного выхода ненасыщенных продуктов, таких как диолефины, в предлагаемом способе. Обычно количество щелочно-металлического промотора колеблется в пределах 0,1-5 мас. по отношению к полной массе окисей магния и молибдена. Предпочтительное количество указанного промотора, такого как гидроокись щелочного металла, составляет 0,2-2 мас. по массе окисей магния и молибдена, а более предпочтительное количество составляет 0,5-1,5 мас. Если количество указанного промотора ниже нижнего предела предпочтительных значений, то избирательность для диолефинов будет более низкой, так как в этом случае повышается избирательность для продуктов глубокого окисления. Если количество промотора превышает верхний предел предпочтительных значений, то в этом случае также наблюдается снижение избирательности для диолефинов.
Обычно, количество щелочно-металлического промотора, рассчитанное на основе гидроокиси щелочного металла, составляет 0,05-5 мас. от общей массы окисей алюминия, магния и молибдена. Предпочтительное количество указанного промотора, рассчитанное на основе гидроокиси щелочного металла, составляет 0,1-2 мас. от общей массы окисей алюминия, магния и молибдена, а более предпочтительное количество 0,3-1,5 мас. Если количество указанного промотора ниже нижнего предела предпочтительных значений, то при этом наблюдается более низкая избирательность для диолефинов, а избирательность для продуктов глубокого окисления возрастает. Если количество указанного промотора превышает верхний предел предпочтительных значений, то в этом случае также наблюдается снижение избирательности и продуктивности для диолефинов.
Щелочно-металлический промотор может быть добавлен к катализатору на основе молибдата с помощью стандартных способов. Например, промотор может быть нанесен с помощью хорошо известной специалистам техники пропитки, описанной например Charles N. Satterfield в работе Heterogeneous Catalysis in Practice, McGraw-Hill Book Company, New York, 1980, pp. 82-83, которая вводится в настоящее описание посредством ссылки. В указанной технике, молибден-пропитанный носитель погружают в раствор щелочно-металлического промотора, например метаноловый раствор окиси или гидроокиси щелочного металла. Затем избыток раствора, пропитанного щелочью носителя, сливают, а остаток подвергают осушке в печи для удаления остаточного растворителя и кальцинируют при 550-650оС. Альтернативно указанный носитель пропитывают щелочно-металлическим промотором с помощью увлажняющей техники так, чтобы поры были заполнены раствором окиси щелочного металла или гидроокиси щелочного металла, но при этом раствор не должен быть в избытке. Полученный таким образом пропитанный носитель также высушивают в печи для удаления растворителя. В еще одном альтернативном варианте молибденовое соединение может быть пропитано тем же раствором, что и соединение щелочного металла.
Необязательно катализатор из молибдата изобретения может содержать активатор, который способствует повышению активности катализатора при любой данной температуре. Предпочтительно, если активатор при этом не снижает значительно избирательность к диолефинам и моноолефинам. Предпочтительно также, чтобы активатор давал возможность проводить реакцию при более низкой температуре, сохраняя при этом высокую избирательность и высокую продуктивность диолефинов. Подходящими активаторами для введения в катализатор могут служить окиси ванадия, предпочтительно V2O5. В катализатор может быть добавлено любое количество окиси ванадия при условии, что при этом активность катализатора возрастает и избирательность для алканов, включая моно- и диолефины, не слишком снижается. Обычно концентрация активатора (если он используется) колеблется в пределах 0,05-10 мас. по отношению к полной массе катализатора. Предпочтительной концентрацией активатора является концентрация от 0,1-5 мас. а более предпочтительной 0,15-1,5 мас. Указанный активатор может быть введен в носитель и суспензию окиси молибдена до кальцинации или он может быть нанесен на кальцинированные окиси алюминия-магния-молибдена с помощью техники пропитки, описанной выше.
Предпочтительным промышленным реактором, используемым в изобретении, является реактор с передвижным слоем, например с восходящим слоем катализатора. В таких реакторах каталитические частицы подвергаются постоянным столкновениям с другими каталитическими частицами и со стенками реактора. Указанные силы постепенно снижают размеры катализатора до мелких пылинок, которые теряются в реакционных продуктах; таким образом, срок службы катализатора является весьма ограниченным. Поэтому катализатор необходимо изготовлять в том виде, который мог бы противостоять ударным силам и эрозии. Катализатор для окисления бутана, содержащий молибдат магния, нанесенный на магнезию, не обладает достаточной прочностью на истирание, необходимой для промышленного производства. Напротив катализатор изобретения на основе молибдата магния, активированный щелочным металлом и нанесенный на предпочтительный композиционный материал, описанный выше, обладает достаточной прочностью на истирание, необходимой для использования его в промышленном производстве.
Очевидно, что изнашиваемость катализатора, содержащего молибдат магния, нанесенный на магнезию, непосредственно связана с высокой тепмпературой спекания окиси магния и сильными межмолекулярными связями. Температура спекания настолько превышает нормальную температуру кальцинации и нормальные рабочие температуры предлагаемого способа, что частицы не имеют возможности сплавляться и связываться одна с другой. Один из способов упрочнения катализатора, содержащего молибдат магния, заключается во введении в катализатор несущего компонента. Предпочтительно, чтобы указанный компонент имел большую поверхностную площадь и высокую прочность на истирание и представлял собой композиционный материал, содержащий окись магния и окись алюминия и необязательно фазу алюмомагнезиальной шпинели, как описано выше. Очевидно, что окись алюминия наделяет катализатор твердостью, тогда как окись магния и/или алюминат магния снижают естественную кислотность окиси алюминия. Однако приведенная выше теория не должна ограничивать раскрытие изобретения. Соответствующие способы испытаний прочности на истирание приведены ниже в примерах.
Другим важным свойством катализатора изобретения является размер его частиц. В прототипах обычно рассматривается использование катализатора со сфероидальными частицами небольшого размера, который предназначен для реакторов с неподвижным слоем и перемещающимся псевдоожиженным слоем. Эти частицы обычно имеют диаметр 20-200 мкм, а предпочтительно 80-120 мкм. Автором изобретения было обнаружено, что сфероидальные (т.е. имеющие форму, приближающуюся к сферической) частицы размером 200-1700 мкм дают лучшую производительность в реакторах с подвижным слоем. Предпочтительно, если частицы имеют размер 500-1200 мкм, а более предпочтительно 600-1000 мкм. Более крупные частицы изобретения обнаруживают меньшее "слеживание" и поэтому дают более плавный низкоскоростной поток. Таким образом, более крупные частицы дают меньший перепад давления в секциях реактора с плотным слоем и менее интенсивное столкновение частиц со стенками и обладают способностью к лучшему дифференцированию времени пребывания газа и времени пребывания катализатора в реакторе.
Предлагаемый способ может быть осуществлен в любом подходящем реакторе, например в реакторе периодического действия, в реакторе непрерывного действия с неподвижным слоем катализатора, в шламовом реакторе, в реакторе с псевдоожиженным слоем и в реакторе с восходящим уплотненным слоем катализатора. Предпочтительным является реактор непрерывного действия, такой как реактор непрерывного действия с неподвижным слоем катализатора или реактор с восходящим уплотненным слоем катализатора описанного ниже типа.
Реактор с восходящим уплотненным слоем катализатора содержит вертикальный сосуд с относительно низким отношением диаметра к длине. Катализатор непрерывно загружается в нижнюю часть реактора с восходящим слоем. Кроме того, в нижнюю часть указанного реактора одновременно подается поток алифатического углеводорода в виде паровой фазы или жидкой фазы. Предпочтительно, если алкан подается в виде паровой фазы, заранее смешанной с инертным газовым разбавителем, и необязательно с небольшим количеством кислорода. Подаваемый поток движется через реактор вверх, реагируя при этом с катализатором. При взаимодействии с катализатором исходное сырье превращается в смесь продуктов, например моноолефинов, диолефинов более высоконенасыщенных олефинов, крекинг-продуктов, продуктов глубокого окисления, таких как моноокись и двуокись углерода, и продуктов глубокого крекинга, таких как бензол и фуран, в том случае, если исходным сырьем был бутан. Поток продуктов, присутствующих в реакторе с восходящим слоем, разделяют стандартными способами, такими как перегонка, в целях получения желаемого продукта, обычно диолефина. Непрореагировавшие алканы возвращаются в реактор для дальнейшего окисления.
Технология, используемая в реакторах с восходящим слоем катализатора, является подходящей для изобретения, так как при этом устраняется опасность использования смеси алкана и/или олефина и элементного кислорода и повышается избирательность для диолефинов, особенно при высоких температурах, использующихся в данном способе. Напротив при использовании сырья, содержащего алкан и кислород, при высокой температуре и высоком молярном отношении кислород/алкан имеет место тенденция к образованию продуктов глубокого окисления, таких как моноокись и двуокись углерода. Кроме того, возрастает опасность реакции с быстро возрастающей скоростью.
Работу реактора с восходящим слоем катализатора можно смоделировать, используя метод чередующихся импульсов. Так, например, импульс потока углеводородсодержащего сырья пропускают через слой катализатора, где это сырье окисляется, образуя целевые продукты олефинов. Затем через слой катализатора пропускают импульс потока инертного газа в целях очистки слоя от остаточных алканов и алкенов. После очистки через слой катализатора пропускают импульс потока кислородсодержащего сырья в целях регенерации катализатора. И наконец, через слой катализатора пропускают второй импульс инертного газа в целях очистки слоя от кислорода, после чего цикл повторяют. Указанная процедура используется в примерах осуществления изобретения.
Алифатический углеводород взаимодействует с катализатором при любой рабочей температуре, которая способствует активации процесса окисления изобретения и повышению выхода целевых ненасыщенных продуктов. Такой температурой обычно является температура 400-700оС. Предпочтительная температура составляет 500-650оС, а более предпочтительная температура 530-600оС. Если температура ниже указанного нижнего предела предпочтительного диапазона, то конверсия реагента может снизиться. Если температура превышает верхнее значение указанного диапазона, то при этом наблюдается снижение избирательности и продуктивности диолефиновых продуктов.
Аналогично алифатический углеводород взаимодействует с катализатором при любом рабочем давлении, которое способствует активации процесса изобретения и повышению выхода целевых ненасыщенных продуктов. Обычно парциальное давление реагента корректируют так, чтобы указанный реагент находился в газообразном состоянии при данной рабочей температуре. Предпочтительно, если парциальное давление алифатического углеводорода находится в пределах диапазона: от приблизительно ниже атмосферного давления до около 100 фунтов/кв. дюйм (690 кПа). Более предпочтительно, если парциальное давление находится в пределах 1-30 фунтов/кв.дюйм (7-207 кПа). И наконец, наиболее предпочтительным парциальным давлением является давление в пределах 3-15 фунтов/кв.дюйм (21-104 кПа).
Если способ изобретения осуществляют в реакторе для непрерывного процесса, описанного ниже, то скорость потока реагентов может варьироваться. В основном в предлагаемом способе алифатический углеводород подают в реактор при любой рабочей скорости потока, которая способствует активации процесса окисления изобретения и повышению выхода и избирательности для ненасыщенных продуктов. Скорость потока выражается как объемный часовой расход газа (GHSV) и приводится в единицах объема газообразного исходного сырья, содержащего алифатический углеводород, на полный объем реактора за 1 ч или просто ч-1. Обычно эти значения колеблются в диапазоне 100-20000 ч-1. Следует отметить, что объемная скорость регулирует время пребывания реагентов в реакторе. Например, в реакторе с восходящим слоем катализатора, предпочтительное время пребывания газа в реакторе составляет менее чем 10 с, более предпочтительно менее чем 5 с и наиболее предпочтительно менее чем 1 с.
В случае использования реактора с восходящим слоем катализатора после взаимодействия катализатора с алифатическим углеводородом отработанный катализатор удаляют из верхней части реактора и переносят во второй реактор для регенерации. Регенерацию осуществляют при помощи реакции взаимодействия с кислородом. Обычно в нижнюю часть второго реактора подают заранее нагретый кислородный источник, аналогичный описанному выше. Отработанный катализатор взаимодействует с указанным источником кислорода при любых температуре, давлении и скорости потока кислородного источника, которые являются достаточными для регенерации катализатора. Параметры указанного процесса должны быть, однако, регулируемыми, так чтобы исключить возникновения неуправляемой реакции или чрезмерного разогрева. Предпочтительно, если температура колеблется в пределах 500-700оС, а более предпочтительно в пределах 550-650оС. Предпочтительное давление колеблется в пределах от давления ниже атмосферного и до 100 фунтов/кв.дюйм (690 КПа), а более предпочтительное 2-50 фунтов/кв.дюйм (14-345 кПа). Скорость потока источника кислорода зависит от теплообменных свойств конкретного реактора. Например, при слишком высоких скоростях потока может произойти чрезмерное повышение температуры, что в результате приведет к неконтролируемой реакции.
При взаимодействии алифатического углеводорода с катализатором изобретения окисление алифатического углеводорода происходит в результате потери по меньшей мере двух атомов водорода из углеводородного реагента с образованием воды как побочного продукта. Образующиеся при этом органические продукты являются в основном ненасыщенными углеводородами, таким как моноолефины и диолефины. Эти ненасыщенные продукты обычно содержат такое же число атомов углерода, как и реагентный алифатический углеводород. Поэтому указанные продукты не являются продуктами крекинга, поскольку последние содержат меньшее число атомов углерода, чем исходный углеводород. В основном ненасыщенные продукты имеют более высокую степень ненасыщенности, чем реагентный углеводород. Например алканы, такие как бутан, могут терять два атома водорода, образуя моноолефины, такие как 1-бутен, транс-2-бутен, и цис-2-бутен. В свою очередь моноолефины, такие как указанные выше бутены, могут терять два атома водорода, образуя 1,3-бутадиен.
Предпочтительные диолефиновые продукты могут быть представлены общей формулой:
СН2= СН-СН= СН-(СН2)m-Н, где m является целым числом от 0 до 6. Предпочтительно, если m от 0 до 2. Более предпочтительное значение m 0, а более предпочтительным ненасыщенным продуктом является 1,3-бутадиен. Могут быть также образованы изомеры приведенной выше формулы, где ненасыщенность может возникать в любом другом положении углеродной цепи. Предпочтительно, если ненасыщенность образуется посредством сопряженных двойных связей. Даже более ненасыщенные варианты общей формулы могут быть образованы при дальнейшем окислении, которое дает более чем две этиленовые двойные связи. Однако алканы не образуются в значительных количествах.
Наряду с алкенами продуктовый поток может содержать различные побочные продукты. Например, в случае, если насыщенным алканом является н-бутан, могут образовываться небольшие количества продукт крекинга, таких как пропилен и этилен, а также тяжелые фракции, такие как бензол и фуран, и кроме того, продукты глубокого окисления, такие как моноокись и двуокись углерода. Однако неожиданно было обнаружено, что эти побочные продукты, особенно продукты глубокого окисления, при предлагаемом способе изобретения образуются в гораздо меньших количествах.
В целях изобретения "конверсия" определяется как процентное содержание (в молях) реагирующего алифатического углеводорода, потерянного из исходного потока в результате реакции. Конверсия может широко варьироваться в зависимости от реагентов, формы катализатора и реакционных условий, таких как температура, давление, скорость потока и время пребывания катализатора в реакторе. В пределах предпочтительного диапазона температур при возрастании температуры конверсия обычно возрастает. В пределах предпочтительного диапазона часового объемного расхода газа, при увеличении объемного расхода конверсия обычно снижается. Как правило, конверсия алифатического углеводорода составляет по меньшей мере около 10 М% Предпочтительно, если конверсия составляет по крайней мере около 20 М% более предпочтительно по крайней мере около 40 М% и наиболее предпочтительно по крайней мере около 50 М%
Аналогично в целях изобретения "избирательность" определяется как процентное содержание (в молях) конвертированного углерода, который образует конкретный продукт. Обычно избирательность широко варьируется в зависимости от реагентов, формы катализатора и реакционных условий. Как правило, в предлагаемом способе достигается высокая избирательность к диолефинам. В пределах предпочтительного диапазона температур, при возрастании температуры избирательность обычно снижается. В пределах предпочтительного диапазона объемного расхода, при возрастании объемного расхода избирательность для алканов обычно возрастает. Предпочтительно, если суммарная избирательность ко всем алкенам составляет по крайней мере около 50М% более предпочтительно по крайней мере 60 М% еще более предпочтительно по крайней мере 70 М% и наиболее предпочтительно по крайней мере около 80 М% Как правило, избирательность для диолефинов составляет по крайней мере около 40 М% Предпочтительно, если избирательность для диолефинов составляет по крайней мере около 50 М% более предпочтительно по крайней мере около 60 М% наиболее предпочтительно по крайней мере около 70 М%
Понятие одновременно высокой конверсии и высокой избирательности может быть определено как выход. В целях изобретения термин "выход" означает количество продукта, образующегося в результате конверсии и избирательности при однократном пропускании через реактор. Например, в осуществлении предлагаемого способа при конверсии 0,65 или 65 М% и избирательности к диолефинам 0,75 или 75 М% выход диолефинов составляет 0,49 или 49 М% Как правило, в предлагаемом способе достигается выход диолефинов по крайней мере около 8 М% Предпочтительно, если выход диолефина составляет по меньшей мере около 18 М% более предпочтительно по крайней мере около 28 М% а наиболее предпочтительно по крайней мере 35 М% Как правило, при окислении бутана выход полных С4-олефинов составляет по меньшей мере около 25 М% Предпочтительно, если при окислении бутана выход полных С4-олефинов составляет по меньшей мере около 30 М% более предпочтительно по крайней мере около 35 М% и наиболее предпочтительно по меньшей мере около 40 М%
Скорость, при которой в предлагаемом способе образуется целевой продукт, может быть определена понятием объемного выхода. В изобретении понятие "объемный выход" определяется как выход целевого продукта (выраженный в М% за час (час-1)) и является численным выражением продукта конверсии при однократном пропускании с соответствующей избирательностью, часовым объемным расходом газа и концентрацией алифатического углеводорода в исходном сырье, где конверсия, избирательность и концентрация выражаются в десятичленных долях. Предпочтительно, если объемный выход диолефина составляет по меньшей мере около 30% за 1 ч, более предпочтительно по крайней мере около 120% за 1 ч, а более предпочтительно по меньшей мере около 200% за 1 ч.
Другим измерением скорости, при которой продуцируется целевой продукт, является "продуктивность", определяемая как граммы образованного целевого продукта на грамм катализатора за один час (г/г кат.-ч). В предлагаемом способе предпочтительная продуктивность бутадиена составляет по крайней мере около 0,2 г/г кат.-ч, более предпочтительная продуктивность составляет по меньшей мере около 0,4 г/г кат.-ч, а наиболее предпочтительная продуктивность составляет по крайней мере около 0,5 г/г кат.-ч. Предпочтительная продуктивность полных С-олефинов в предлагаемом способе по крайней мере составляет около 0,3 г/г кат.-ч; более предпочтительная продуктивность составляет по крайней мере около 0,4 г/г кат.-ч; а наиболее предпочтительная продуктивность составляет по крайней мере около 0,9 г/г кат.-ч.
Испытание прочности на истирание катализатора требует большого количества образцов катализатора. Было бы желательно разработать простую процедуру испытания прочности на истирание для небольших образцов катализатора. Такой процедурой является испытание прочности на раздавливание, поскольку повышенная прочность на раздавливание является показателем повышенной прочности на истирание. Прочность на раздавливание может быть определена с помощью любой стандартной установки, которые обычно используются в этих целях, например Instron Model 1125. Обычно в данном случае используется катализатор, который сначала просеивают для отбора гранул размером 8-меш (2,36 мм), а затем эти гранулы кальцинируют при 600оС в течение 2 ч, после чего определяют прочность на раздавливание. Прочность на раздавливание предпочтительных катализаторов изобретения составляет по меньшей мере около 5 фунтов (2270 г), предпочтительно по меньшей мере около 10 фунтов (4540 г); а более предпочтительно по крайней мере около 15 фунтов (6810 г).
Фактическая прочность на истирание предпочтительного катализатора изобретения может быть определена с помощью стандартной установки, котоpая обычно используется в этих целях. Подходящие пpиборы для испытания содержат вертикальную трубу из нержавеющей стали диаметром 1/2 дюйма (13 мм) и длиной около 30 дюймов (760 мм), которая соединяется посредством I-клапана с вертикальной колонной аналогичного диаметра и длиной около 52 дюймов (1320 мм). Циклон из нержавеющей стали диаметром 3 дюйма (76 мм) подсоединяется к указанной вертикальной колонне у входного отверстия циклона и к указанной вертикальной трубе у выходного отверстия циклона, образуя тем самым циркуляционный контур, в котором осуществляется разделение газа и твердой фазы, после чего твердые вещества возвращаются в вертикальную трубу. Обычно испытуемый материал загружают в систему и подвергают псевдоожижению путем введения газа в I-клапан со скоростью потока 0,11-1,01 с-1. Захваченный газом порошок движется в направленном вверх газовом потоке и после сепарации под действием силы тяжести возвращается в циклонную регенерационную систему. Обычно полное время испытания образцов занимает около 15 ч. Затем определяют число мелких частиц, образующихся за единицу времени и показатель истирания рассчитывают по сравнению с начальной крупнозернистой фракцией. Предпочтительный показатель истирания катализатора изобретения составляет приблизительно менее 5 мас. за 1 ч (мас.˙ч-1); а более предпочтительно приблизительно менее 1 мас.˙ч-1.
Изобретение подробно иллюстрируется конкретными примерами, которые, однако, не ограничивают возможные варианты его осуществления. Все процентные содержания приводятся в М% углерода, если это не оговорено особо.
П р и м е р 1 (а-b). Изготовление катализатора. а) Порошок окиси магния (17 г, Magox Premium Grade MgO) диспергировали в деионизованной воде (96 г) с помощью диспергатора с высоким сдвигающим усилием. К полученной суспензии постепенно добавляли 20 мас. коллоидальной окиси алюминия (200 г, Nyacol) в целях получения вязкой смеси, содержащей, мас. MgO 30; Al2O3 70. Затем добавляли еще 52 г воды для получения контролируемых реологических свойств. Полученную смесь выдерживали и желатинировали на горячей пластине, перемешивая при 70оС в течение 2 ч до получения твердого белого вещества. Это вещество измельчали, а затем нагревали в течение 4 ч при 600оС и кальцинировали еще 4 ч также при 600оС, в результате чего получали композиционный носитель на основе шпинели, имеющий массовое отношение MgO/Al2O3 0,43 и поверхностную площадь 169 м2/г. Рентгенографический спектр носителя дает дифракционную картину кристаллического MgO, а также шпинели и окиси алюминия. Носитель подвергали испытанию на приборе Instron Model 1125 и определяли среднюю прочность на раздавливание, которая составляла 15 фунтов (6810) г.
b) Шпинельный композиционный носитель изготавливали в соответствии с процедурой примера 1а за исключением того, что использовали 44 г порошка окиси магния, 280 г коллоидной окиси алюминия и 249 г деионизованной воды. Полученный в результате композиционный носитель имел массовое отношение MoO/Al2O3 0,78 и поверхностную площадь 162 м2/г. Рентгенографический спектр носителя показал отражательную картину, которая может соответствовать кристаллическому MgO, а также шпинели и окиси алюминия. Шпинельный композиционный носитель (20 г) пропитывали до начальной стадии влажности с помощью 20,44 г раствора, содержащего, мас. гептамолибдат аммония 35,25; гидроокись цезия 1,85. Пропитанный раствор высушивали в течение 2 ч при 125оС нагревали при 600оС в течение 4 ч и кальцинировали при 600оС еще 3 ч. Полученный катализатор (EI) содержал, мас. MoO3 22,4; Cs2O 1,20, и имел поверхностную площадь 122 м2/г.
П р и м е р 2. Окисление бутана. Катализатор (EI), полученный в соответствии с описанием в примере Iб, использовали для окисления бутана следующим образом: приблизительно 15 см3 катализатора загружали в камеру (18 мм наруж. диаметр х 7,6 см длина) реактора Vycor. Температуру реактора определяли с помощью кармана термопары из нержавеющей стали (1/8 дюймов (3,2 мм) внеш. диаметр), заложенного в образец катализатора. Поток исходного сырья, содержащего бутан 10-20 об. и гелий 90-80 об. пропускали через катализатор приблизительно в течение 10-30 с. Затем поток исходного сырья останавливали и через катализатор в течение 1 мин и с той же скоростью пропускали поток для очистки, содержащий чистый гелий. Затем поток для очистки прекращали и через катализатор в течение 1 мин. и с той же скоростью пропускали поток кислорода (20 об. ) в гелии, после чего пропускали другой поток гелия для очистки в течение 1 мин. Этот цикл повторяли и объединенные продукты собирали для анализа и поливинилиденхлоридную пластиковую камеру Saran® Анализ осуществляли на газовом хроматографе Carle, предназначенном для анализа С1-С5-алканов, алкенов и алкадиенов, а также постоянных газов, таких как N2, O2, CO, CO2, H2 и тяжелых продуктов, включая фуран, бензол и С6-соединения. Изобутан смешивали с исходным сырьем или с продуктами в качестве стандарта. "Неизвестные" были получены из разности между углеродным балансом и 100% Результаты и условия процесса представлены в табл.1.
Как видно из табл.1, что активированный цезием катализатор, содержащий молибдат магния и нанесенный на алюмомагнезиальный шпинельный композит, катализирует окисление бутана в бутадиен и бутен с высокой избирательностью.
П р и м е р 3. Изготовление катализатора. Алюмомагнезиальный шпинельный композиционный носитель изготавливали в соответствии с описанием примера 1б. Шпинельный композит (20 г) пропитывали до начальной влажности с помощью 19,49 г раствора, содержащего, мас. гидроокись цезия 1,66; ванадата аммония 1,39; гептамолибдат аммония 27,22. Пропитанный носитель высушивали при 125оС в течение 2 ч, нагревали до 600оС в течение 4 ч и кальцинировали при 600оС еще 3 ч. Полученный в результате катализатор (ЕЗ) содержал, мас. MoO3 17,44; V2O5 0,85; Cs2O 1,43, и имел поверхностную площадь после кальцинирования 122 м2/г.
П р и м е р 4 (а-b). Окисление бутана. Катализатор, полученный в соответствии с описанием в примере 3 (ЕЗ), использовали при окислении бутана, проводившемся в соответствии с описанием в примере 2. Условия процесса и результаты представлены в табл.1. Из табл.1 видно, что активированный цезием катализатор, содержащий молибдат магния и окиси ванадия и нанесенный на алюмомагнезиальный шпинельный композит, катализировал окисление бутана в бутадиен и бутен с высокой степенью избирательности.
П р и м е р 5. Изготовление носителя. Порошок окиси магния (600 г) диспергировали в деионизованной воде (5733 г) с помощью диспергатора с высоким сдвигающим усилием. К полученной суспензии при низком сдвигающем усилии постепенно добавляли коллоидный оксид алюминия (7000 г, 20 мас.). Полученную таким образом смесь высушивали распылением, используя распылитель с форсункой, имеющей отверстие диаметром 2 мм, под давлением 40 фунтов/кв.дюйм (276 кПа). Входная температура форсунки составляла 300оС, а выходная 120оС. Полученный в результате осушки распылением порошок белого цвета обладал прекрасными характеристиками текучести. Затем указанный порошок кальцинировали при 600оС в течение 2 ч, в результате чего получали композиционный носитель, имеющий поверхностную площадь 184,3 м2/г и массовое отношение MgO/Al2O3 0,43. Рентгенографический спектр носителя показал отражательную картину, которая может соответствовать кристаллическому MgO, а также шпинели и окиси алюминия. Кальцинирование части порошка в течение 4 ч при 800оС лишь слегка снижало поверхностную площадь до 143,2 м2/г. Носитель, полученный путем кальцинирования порошка при 600оС, имел средний размер частиц около 60 мкм. Исследование частиц с помощью сканирующего электронного микроскопа показало наличие сфероидальных частиц, имеющих прекрасные характеристики текучести. Кроме того, носитель подвергали испытанию на истирание в циркуляционном контуре, как было описано, в результате чего показатель прочности на истирание получали равным 0,28 мас.˙ч-1. Из сравнения видно, что этот носитель явно превосходит коммерческий флюидизированный катализатор для крекинга (FCC) на основе окиси алюминия, который обычно используется в реакторах с передвижным слоем и показатель прочности на истирание которого составляет 0,99 мас. ˙ч-1.
Примеры катализаторов (b-d). Носители на основе алюмомагнезиальной шпинели были получены в соответствии с приведенным описанием в п.(а), за исключением того, что эти носители содержали достаточное количество MgO для получения состава с 40, 44 и 30 мас. MgO; и кроме того они не были подвергнуты кальцинированию. После чего эти носители подвергали прессованию при 5 килофунтов/кв. дюйм (35 мПа) в изостатическом прессе с измельчением до 20-80 меш (850-180 мкм) и кальцинированию при 600оС в течение 5 ч. К 50 г каждого носителя добавляли раствор, содержащий, мас. гептамолибдат аммония 33,23; К2СO3 0,62; перекись водорода 2,5 от 30 мас. раствора пероксида, нейтрализованный до рН 9 с помощью гидроокиси аммония. Пропитанные носители (Е-5-b-d) осушали в течение 18 ч при 110оС и кальцинировали в течение 3 ч при 600оС, в результате чего получали катализаторы. Количество раствора, добавленного к каждому носителю, приведено в табл.2.
П р и м е р 6. Окисление бутана. Катализаторы, полученные в соответствии с описанием в примере 5 (Е-5b-E-5d), использовали для окисления бутана способом, описанным в примере 2. Условия способа и результаты приведены в табл. 2. Как видно из табл.2, активированные калием катализаторы, содержащие молибдат магния и нанесенные на композит из алюмомагнезиальной шпинели, катализировали окисление бутана в бутадиен и бутен с высокой степенью избирательности.
П р и м е р 7. Изготовление катализатора. Предшественник шпинели получали стандартным способом совместной преципитации гидроокисей магния и алюминия (при рН 9) из водного раствора солей магния и алюминия посредством добавления NaOH и Na2CO3. Предшественник шпинели кальцинировали при 600оС в течение 3 ч, в результате чего получали алюмомагнезиальную шпинель (MgAl2O4). Полученную шпинель пропитывали до начальной стадии влажности с помощью 1,75 М водного раствора ацетаттетрагидрата магния (37 мас.).Пропитанную шпинель кальцинировали в течение 3 ч при 600оС, в результате чего получали шпинельный композит, содержащий, кроме того, 17,1 мас. MgO и имеющий массовое отношение MgO/Al2O3 0,69. Затем композит (16,3 г) пропитывали до начальной стадии влажности с помощью водного раствора (17,53 г) гептамолибдата аммония (26,5 мас.), а затем кальцинировали в течение 4 ч при 600оС. Затем этот материал пропитывали метанольным раствором (8,5 г) гидроокиси цезия (0,63 мас.) и снова кальцинировали в течение 3 ч при 600оС. Полученный в результате катализатор (Е7) содержал, мас. МоО3 18,8; Cs2O 0,25, и имел поверхностную площадь 115 м2/г.
П р и м е р 8 (a-b). Окисление бутана. Катализатор примера 7(Е7) использовали при окислении бутана, проведенном в соответствии с описанием в примере 2. Результаты представлены в табл.3.
Как видно из табл.3, активированный цезием катализатор, содержащий молибдат магния, и нанесенный на шпинельный композит, катализировал окисление бутана в бутадиен и бутен с высокой степенью избирательности.
Сравнительный пример 1 (a-b). В соответствии с описанием в примере 7 получали композицию с тем лишь исключением, что катализатор не пропитывали гидроокисью цезия. Полученную композицию использовали в окислении бутана, проведенным в соответствии с описанием в примере 2. Результаты представлены в табл. 3. Из сравнения примера 8 и сравнительного примера 1 видно, что катализатор изобретения, содержащий небольшое количество цезиевого промотора, обладает большей избирательностью к бутадиену и бутену и меньшей избирательностью к продуктам глубокого окисления, чем аналогичный катализатор, не содержащий цезиевого промотора.
П р и м е р 9 (a-e). Изготовление катализатора. Водный раствор (1184 г), содержащий 71,5 мас. гексагидрата азотнокислого магния, добавляли к пористым глиноземным шарикам (713,3 г; VOP, 700 мкм). Полученную в результате суспензию осушали при 150оС в течение 18 ч и кальцинировали путем нагревания до 460оС в течение 4 ч, выдерживания при 460оС в течение 2 ч, еще двухчасового нагревания до 540оС и выдерживания при 540оС в течение 3 ч в потоке воздуха. Затем к осушенным глиноземным шарикам добавляли еще 1186,9 г раствора нитрата магния, после чего повторяли процедуры осушивания и кальцинирования. Полученный композиционный носитель содержал 27,7 мас. MgO и имел массовое отношение MgO/Al2O3 0,38 и поверхностную площадь 101 м2/г. Образцы (35 г) композита пропитывали раствором, содержащим гептамолибдат аммония и карбонат калия, как показано в табл.4. Концентрация гептамолибдата аммония в каждом растворе была разной, тогда как концентрация карбоната калия была приблизительно одинаковой. Пропитанные композиты кальцинировали при 600оС в течение 4 ч, в результате чего получали молибдатные активированные калием катализаторы (Е9 а-е), нанесенные на алюмомагнезиальный шпинельный композит и охарактеризованные в табл.4.
П р и м е р 10 (а-е). Окисление бутана. Катализаторы (Е9 а-е), полученные в примере 9, использовали при окислении бутана в соответствии с описанием, приведенным в примере 2, результаты которого представлены в табл.4. Из табл. 4 видно, что активированный калием молибдатный катализатор, нанесенный на алюмомагнезиальный шпинельный композит, катализировал окисление бутана в бутены и бутадиен с высокой степенью избирательности. Кроме того, было обнаружено, что избирательность к С4-олефинам снижается, если концентрация молибдата падает ниже 11 мас.
П р и м е р 11. Изготовление катализатора. Шарики из окиси алюминия (4,09 г; UOP-SAB16), имеющие номинальный диаметр 1700 мкм, три раза пропитывали до начальной стадии влажности с помощью 10 см3 каждого раствора, содержащего 1,15 г MgO в виде гексагидрата азотнокислого магния.
Между каждым добавлением раствора, пропитанную окись алюминия кальцинировали в течение 2 ч при 600оС, в результате чего получали композиционный носитель, имеющий массовое отношение MgO/Al2O3 0,75. К образцу (6,42 г) носителя добавляли раствор (9,2 мл), содержащий гептамолибдат аммония (11,8 г) К2СО3 (0,228 г) и NH4VO3 (0,60 г) на 100 мл раствора, после чего носитель кальцинировали и получали катализатор (Е-11), содержащий, мас. МоО3 12,03; V2O5 0,58; К2О 0,16, и имеющий поверхностную площадь 34 м2/г.
П р и м е р 12. Окисление бутана. Катализатор (5,65 г) примера 11 (Е-11) загружали в реактор с неподвижным слоем, как описано в примере 2. Затем при 600оС и GНSV 1200 ч-1 через реактор пропускали импульсные потоки: смеси гелия и 20 об. бутана в течение 5 с, а затем в течение 60 с соответственно гелия, воздуха и гелия. Затем цикл повторяли и получали следующие результаты: конверсия бутана 45,2% избирательность 58,3% для бутадиента, 20,6% для бутена; 78,9% и 78,9% для суммарных бутенов; 6,4% для продуктов глубокого окисления (СО2 и СО); 8,0% для крекинг-продуктов и 6,8% для неизвестных продуктов. Продуктивность составляла 0,34 г C4/г кат.-ч. и 0,25 г бутадиена/г кат. -ч. Очевидно, что продуктивность С4-углеродов и бутадиена больше, чем величина продуктивности, принятая для стандартного способа 0,2 г/г кат.-ч.
П р и м е р 13. Получение катализатора. Глиноземные шарики (713,3 г; UOP-MS-P-27) с частицами диаметром 500-1000 мкм использовали в получении композиционного носителя, содержащего алюмомагнезиальную шпинель, способом, описанным в примере 2. Полученный композиционный носитель имел массовое отношение MgO/Al2O3 0,38 и поверхностную площадь 106 м2/г. Образец (686 г) композита пропитывали 728 г раствора, содержащего, мас. гептамолибдат аммония 22,5; карбонат калия 0,66. Пропитанный композит кальцинировали в течение 4 ч при 600оС, в результате чего получали каталитическую композицию (Е-13), содержащую, мас. МоО3 18,4; К2О 0,45.
П р и м е р 14. Окисление бутана. Катализатор, полученный в соответствии с описанием примера 13, использовали в лабораторном реакторе с восходящим слоем катализатора. Сырьевой бак для псевдоожиженного слоя катализатора (емкостью 3500 мл) соединен с его нижней частью посредством длинной трубки, называемой "каталитической трубой". Указанная вертикальная труба связана посредством клапана "J" с вертикальной камерой реактора с длиной 2 м и диаметром 6 мм, которая связана с первым из трех циклонных сепараторов, последовательно соединенных друг с другом. Катализатор, восстановленный в циклонах, возвращался в верхнюю часть бака для подачи катализатора. Газообразный сырьевой поток, содержащий, об. азот 10; бутан 6,9 и балансный гелий, предварительно нагревали в нагревательной камере, имеющей лишь 4 мм в диаметре, что позволяет минимизировать время пребывания, а следовательно, и термический крекинг бутана. Камера для предварительного нагревания соединялась с системой у соединения "каталитической трубы" и клапана "J", а газообразный сырьевой поток подпивался со скоростью 500 см3/мин. Затем катализатор нагревали до 650оС в сырьевой камере, подавали в камеру реактора со скоростью 200 см3/мин. Температура камеры реактора составляла около 580оС. Время пребывания газа в реакторе составляло 1,6 с. В результате описанной процедуры получали следующие результаты: конверсия бутана 23,3% избирательность, бутадиен 61,9; бутены 22,1; продукты глубокого окисления (СО2 и СО) 5,1; С2-3-крекинг-продукты 7,7; метан 1,0; неизвестные 2,1. Продуктивность составляла около 0,43 г C4/г кат.-ч и 0,32 г бутадиена г кат.-ч. Из приведенных выше результатов видно, что продуктивность С4-углерода и бутадиена превышает величину продуктивности, принятую для стандартного способа (0,2 г/г кат.-ч).
П р и м е р 15. Окисление бутана. Катализатор примера 13 использовался в реакторе с восходящим слоем катализатора, описанном в примере 14, вместе с газообразным сырьевым потоком, содержащим 67 об. бутана и балансным азотом и гелием. Скорость подачи газа составляла 1000 см3/мин, а время пребывания газа в реакторе составляло около 0,82 с. Катализатор подавали со скоростью около 455 см3/мин, при этом время пребывания катализатора в реакторе составляло около 3,1 с. Средняя температура в вертикальной трубе составляла 584оС. В результате получали следующие данные: конверсия бутана 17,1; избирательность, бутадиен 39,1; бутаны 2,4; продукты глубокого окисления (СО и СО2) 2,5; крекинг-продукты, включая С2-С3-продукты 10,6; метан 1,6; изобутан 0,5; неизвестные продукты 1,8. Продуктивность объединенных бутадиеновых и бутеновых продуктов составляла 0,94 г C4/г кат.-ч, а продуктивность бутадиена составляла 0,49 г бутадиена/г кат.-ч. Из указанных результатов видно, что продуктивность С-углеродов и бутадиена превышает величину продуктивности, принятую для стандартного способа (0,2 г/г кат.-ч).
П р и м е р ы 16-20 (a-b).
а) Изготовление катализаторов, активированных цезием 16-20. Получали серии молибдатных катализаторов, легированных цезием, в соответствии с описанной ниже процедурой, причем количество (г) моногидрата гидроокиси цезия на 40 г метанольного раствора варьировалось следующим образом, г: катализатор 1 0,16; катализатор 2 0,23; катализатор 3 0,29; катализатор 4 0,53; и катализатор 5 0,76.
(NH4)6Mo7O24˙4H2O (72 г, 0,0583 М) растворяли в 175 мл воды. Полученный в результате раствор смешивали с гидроокисью магния 300 г Alfa Research Chemcal Materials) для получения суспензии. Суспензию осушали при 70оС в течение 18 ч и при 130оС в течение 2 ч в целях получения твердой смеси. Полученное твердое вещество измельчали и пропускали через сито для получения частиц, имеющих размер 20-120 меш. (850-122 мкм). Просеянные частицы кальцинировали путем медленного нагревания в течение 5 ч до 600оС, а затем выдерживали при этой температуре еще 2 ч. Кальцинированный твердый продукт охлаждали до комнатной температуры и получали молибдат магния, содержащий, мас. МоО3 22; MgO 78. Метаноловый раствор получали путем добавления соответствующего количества гидроокиси цезия как указано выше, к метанолу, в результате чего получали 40 г метанолового раствора. Молибдат магния (20,6 г) погружали на 15 мин в 40 г метанолового раствора. Полученную смесь фильтровали и отфильтрованное твердое вещество осушали в воздухе при комнатной температуре (около 22оС) в течение 4 ч, а затем осушали в печи при 120оС в течение 16 ч. Осушенный твердый продукт кальцинировали путем нагревания в течение 5 ч до температуры 600оС с последующим выдерживанием при этой температуре еще 2 ч, в результате чего получали активированный цезием катализатор, содержащий молибдат магния. Процентное содержание цезия в катализаторах 16-20 указано в табл.5.
b) Окисление бутана с использованием катализаторов 16-20. Полученные выше катализаторы 16-20 подвергали испытанию путем использования их в окислении бутана в соответствии со следующей процедурой. Катализатор (15 см3) загружали в стеклянную камеру реактора Vycor (7,6 см длиной х x18 мм внеш. диам. ). В своей верхней части указанная стеклянная камера соединена с впускной трубой для потока сырья, а в своей нижней части с выпускной трубой для сбора продуктов. Температура в реакторе, измеряемая из кармана термопары из нержавеющей стали с внеш. диам. 3,2 мм (1/8"), введенной в слой катализатора, понималась до 550оС. Сырьевой поток, содержащий, об. бутан 10; азот 50; гелий 40, пропускали через слой катализатора со скоростью 84 мл/мин (GHSV 336 ч-1) в течение 30 с. Затем сырьевой поток останавливали и через слой катализатора пропускали поток гелия для очистки со скоростью 84 мл/мин в течение 1 мин. Затем поток для очистки останавливали и через слой катализатора пропускали с аналогичной скоростью в течение 1 мин кислородный поток, содержащий 10 об. кислорода в гелии. Затем кислородный поток останавливали и через слой катализатора пропускали в течение 1 мин с той же скоростью поток гелия для очистки. После чего полный цикл повторяли в течение 30 мин и объединенные продукты собирали в пластиковый бак, содержащий поливинилиденхлорид (Saran® ). Полученные продукты анализировали с помощью газовой хроматографии, используя многоколоночный газовый хроматограф Carle, снабженный колонками диаметром 1/8" (3,2 мм), имеющими следующие наполнение: (1) 2,70% карбовакса (Carbowax 1540) на Поразиле Porasil (21" (530 мм), 80/100 меш. (180/150 мкм)); Тмакс 175оС (2) 3,0% Carbowax 1540 на Porasil (4'(1220 мм), 60/80 меш. (250/180 мкм)), Тмакс 150оС; (3) 27,5% 2(ХЕ) А на Chromosorb PAW (17", (5180 мм) 45/60 меш. (328/250 мкм), Тмакс 150оС; (4) Porapak Q 9'(2745 мм), 50/80 меш. (292/180 мкм), Тмакс 250оС; (5) молекулярное сито 13х (9'(2745 мм), 45/60 (328/60 мкм) меш. Тмакс 300оС; (6) молекулярное сито 5А (3'(914 мм), 80/100 меш. (180/150 мкм)), Тмакс 300оС; (7) 28%DC 200/500 на Chromosorb PAW 3,5' (1068 мм), 60/80 меш. (250/180 мкм)), Тмакс 175оС. Полученные результаты представлены в табл. 5. Из этих данных видно, что в присутствии легированного цезием катализатора, содержащего молибдат магния, бутан окисляется преимущественно в бутадиен и бутены.
С р а в н и т е л ь н ы й п р и м е р 2. Изготавливали композицию способом, описанным выше для изготовления катализаторов 16-20, за исключением того, что стадию легирования цезием не осуществляли. Полученная в результате композиция содержала, мас. MoO3 22; MgO 78. Как уже указано, эта композиция не содержала цезия. Затем указанную композицию использовали в качестве катализатора в окислении бутана, способом, описанным в примерах 16-20 (b). Полученные результаты представлены в табл. 6.
Из сравнения результатов сравнительного примера 2 (GHSV 336 ч-1 и результатов примеров 16-20 видно, что присутствие цезия в катализаторе способствует значительному улучшению избирательности тяжелым продуктам, таким как фуран и бензол, и особенно к продуктам глубокого окисления, таким как окиси углерода, соответственно уменьшается. Однако сравнение результатов сравнительного примера 2 (GHSV 336 ч-1) с результатами примеров 16-20 показало, что присутствие цезия в катализаторе приводит к снижению конверсии бутана.
П р и м е р ы 21-25. а) Изготовление катализаторов 21-25, активированных щелочью. Серии легированных щелочью катализаторов, содержащих молибдат магния, получали в соответствии с описанной ниже процедурой, причем количество гидроокиси щелочного металла на 40 г метанолового раствора варьировалось следующим образом: катализатор 6, КОН 0,049 г; катализатор 7, КОН 0,098 г; катализатор 8, КОН 0,133 г; катализатор 9, КОН 0,320 г; и катализатор 10, NaOH 0,073 г.
(NH4)6Mo7O24 ˙4H2O (72 г; 0,0583 М), растворяли в 345 мл воды, и полученный раствор добавляли к магнезии (300 г; СЕ Basic Industries, Madox Premium magnesia для получения суспензии. Суспензию осушали в течение 18 ч при 70оС и в течение 2 ч при ±30оС в целях получения твердой смеси. Полученную смесь измельчали и пропускали через сито для того, чтобы получить частицы размером 20-120 меш. (850-122 мкм). Просеянные частицы кальцинировали путем медленного нагревания в течение 5 ч до температуры 600оС, а затем выдерживали при этой температуре еще 2 ч. Кальцинированный твердый продукт охлаждали до комнатной температуры и получали молибдат магния, содержащий, мас. МоО3 22; MgO 78. Метаноловый раствор получали путем добавления соответствующего количества гидроокиси щелочного металла, как указано выше, к 40 г метанолового раствора. Молибдат магния на 15 мин погружали в метаноловый раствор. Полученную смесь фильтровали и отфильтрованное твердое вещество осушали в воздухе при комнатной температуре в течение 4 ч, а затем в печи при 120оС в течение 16 ч. Осушенный твердый продукт кальцинировали путем нагревания в течение 5 ч до 600оС, а затем выдерживали при этой температуре еще 2 ч, в результате чего получали активированный щелочным металлом катализатор, содержащий молибдат магния. Процентное содержание щелочного металла в катализаторах 21-25 указано в табл.7.
b) Окисление бутана с использованием катализаторов 21-25. Полученные выше катализаторы 21-25, легированные калием и натрием, были использованы в окислении бутана способом, описанным в примерах 16-20 b. Результаты представлены в табл.7. Полученные данные показали, что в присутствии катализатора, содержащего молибдат магния и легированного ионами калия и натрия, бутан преимущественно окисляется в бутадиен и бутаны. Кроме того, из сравнения результатов примеров 21-25 и результатов сравнительного примера 2 (GHSV 336 ч-1) видно, что при использовании катализаторов, легированных калием и натрием (катализаторы 21-25), достигается значительно более высокая избирательность к бутадиену и бутанам, чем при использовании нелегированных катализа- торов, содержащих молибдат магния. При этом избирательность к продуктам глубокого окисления при использовании легированных щелочью катализаторов значительно снижается.
П р и м е р ы 26 и 27. а) Изготовление активированной калием катализаторов 26 и 27. Получали два раствора, содержащие (NH4)6Mo7O24˙H2O (35 г), и карбонат калия в 125 г водного раствора. Первый раствор содержал 0,63 г (катализатор 26) карбоната калия, а второй раствор содержал 1,44 г (катализатор 27) карбоната калия. Затем каждый раствор добавляли, перемешивая, к окиси магния (100 г; CE Basic Industries, Madox Premium Grade magnesia и полученные смеси осушали и кальцинировали, как описано в примерах получения катализаторов 16-20, в результате чего получали легированные калием катализаторы, содержащие молибдат магния.
b) Окисление бутана с использованием катализаторов 26 и 27. Катализаторы 26 и 27 использовали в окислении бутана (примеры 26 и 27 соответственно) согласно процедуре, описанной в примерах 16-20. Результаты, полученные при различных условиях способа, представлены в табл.8. Данные табл.8 показывают, что в присутствии катализатора, содержащего молибдат магния и легированного карбонатом калия, бутан преимущественно окисляется в бутадиен и бутаны. Указанные данные, кроме того, показывают, что при увеличении реакционной температуры от 550 до 570оС конверсия бутана увеличивается, тогда как избирательность к бутенам и бутадиену уменьшается. Кроме того, из результатов видно, что при снижении объемного расхода газа от 600 до 336 ч-1 конверсия бутана увеличивается, а избирательность к бутанам и бутадиену падает. Из сравнения примеров 26 и 27 видно, что катализатор, содержащий большее количество (мас. ) калия, дает более низкую конверсию бутана и более высокую избирательность к бутенам и бутадиенам. Из сравнения примеров 27(а) и 27(с) со сравнительными примерами 2(b) и 2(а) видно, что катализатор, содержащий ионы калия, дает значительно более высокую избирательность к бутадиенам и более низкую избирательность к тяжелым продуктам и к продуктам глубокого окисления, чем катализатор, не содержащий ионов щелочных металлов.
П р и м е р 28. а) Изготовление легированного калием Катализатора 28. Ванадат аммония NH4VO3 (2,55 г; 0,022 М) добавляли к 100 мл воды и полученную смесь нагревали до 60оС в целях ускорения растворения, после чего температуру раствора поднимали приблизительно до 97оС и размешивая, добавляли (NH4)6Mo7O24˙H2O (18,5 г; 0,015 М). Полученный раствор кипятили до тех пор, пока его объем не уменьшится до 70 мл, после чего добавляли магнезию (34 г; 0,85 М) и получали густую пасту. Затем к этой густой пасте добавляли еще 50 мл воды и получали однородную кремнеобразную смесь. Эту смесь выливали в кварцевый тигель, в течение ночи осушали в воздухе, затем в течение 2 ч, осушали при 110оС и кальцинировали при 600оС в течение 2 ч, в результате чего получали кальцинированный твердый материал, содержащий, мас. V2O5 4; MoO3 30; MgO 65. Затем 8,2 мл метанолового раствора, содержащего гидроокись натрия (1 г КОН/100 мл раствора), разбавляли добавочным количеством метанола до полного объема 16 мл. После чего разбавленный раствор по капле добавляли к полученному выше кальцинированному твердому материалу (7,8 г). Пропитанное гидроокисью калия твердое вещество осушали воздухом в течение 30 мин, а затем в течение 1 ч осушали в печи при 100оС. Полученное в результате твердое вещество содержало окись ванадия, окись магния, окись молибдена и около 1 мас. калия, рассчитанное как гидроокись калия.
b) Окисление бутана с использованием катализатора 28. Катализатор 28, полученный выше, использовали в окислении бутана, проводившемся в соответствии с процедурой, описанной в примерах 16-20. Результаты представлены в табл.9.
Из результатов видно, что в присутствии катализатора, содержащего молибдат магния, окись ванадия и ионы калия, бутан окисляется преимущественно в бутадиен и бутены.
С р а в н и т е л ь н ы й п р и м е р 3. Материал для сравнительного примера получали в соответствии с описанием в примере 28 за исключением того, что катализатор не пропитывали гидроокисью калия. Полученный материал содержал, мас. V2O5 4; MoO 30; MgO 66. Этот материал затем использовали в окислении бутана в соответствии с процедурой, описанной в примерах 16-20. Полученные результаты представлены в табл.9. Из сравнения сравнительного примера 3 с примером 28 видно, что присутствие в катализаторе калиевого промотора способствует значительному увеличению избирательности для бутадиена и бутанов и уменьшению избирательности для продуктов глубокого окисления.
П р и м е р 29. а) Изготовление активированного калием катализатора 29. (NH4)6Mo7O24˙4H2O (13,5 г; 0,11 М) растворяли в 120 мл воды, и полученную смесь нагревали до 60оС для получения раствора. Затем постепенно и интенсивно перемешивая, в течение 5 мин к раствору добавляли окись магния (39 г), в результате чего получали однородную суспензию. Эту суспензию в течение ночи осушали при 110оС с целью получения спекшегося материала, который затем кальцинировали в воздухе при 600оС в течение 2 ч и в результате получали смесь окиси магния и окиси молибдена. Эту порошкообразную окисную смесь уплотняли путем прессования в прессе Carver с силой 20000 фунтов (9000 кг) и получали 1 таблетку в 1/8" 28,6 мм. Эту таблетку дробили на несколько кусочков и пропитывали метаноловым раствором гидроокиси калия, содержащим 1 г КОН на 100 мл метанола. Дозировка гидроокиси калия достигала 0,5 мас. Пропитанный гидроокисью калия катализатор, содержащий молибдат магния, осушали в воздухе в течение 2 ч, а затем еще 2 ч в печи при 110оС.
b) Окисление бутана с использованием катализатора 29. Пропитанный гидроокисью калия катализатор, содержащий молибдат магния (катализатор 29; 7,15 г), полученный описанным выше способом, загружали в трубчатый реактор, работающий в импульсном режиме, как описано в примерах 16-20. Условия окисления бутана и его результаты представлены в табл.10.
Результаты показали, что в присутствии легированного калием катализатора, содержащего молибдат магния, бутан окисляется преимущественно в бутадиен и бутены. Кроме того, было обнаружено, что при постоянной температуре и объемном часовом расходе газа объемный расход бутадиена значительно увеличивается с увеличением процентного содержания бутана в сырье. При этих же условиях, конверсия бутана и избирательность к бутадиену поддерживаются на высоком уровне. Аналогичные заключения могут быть сделаны также относительно объемного расхода и избирательности суммы С4-продуктов (бутадиена и бутенов).

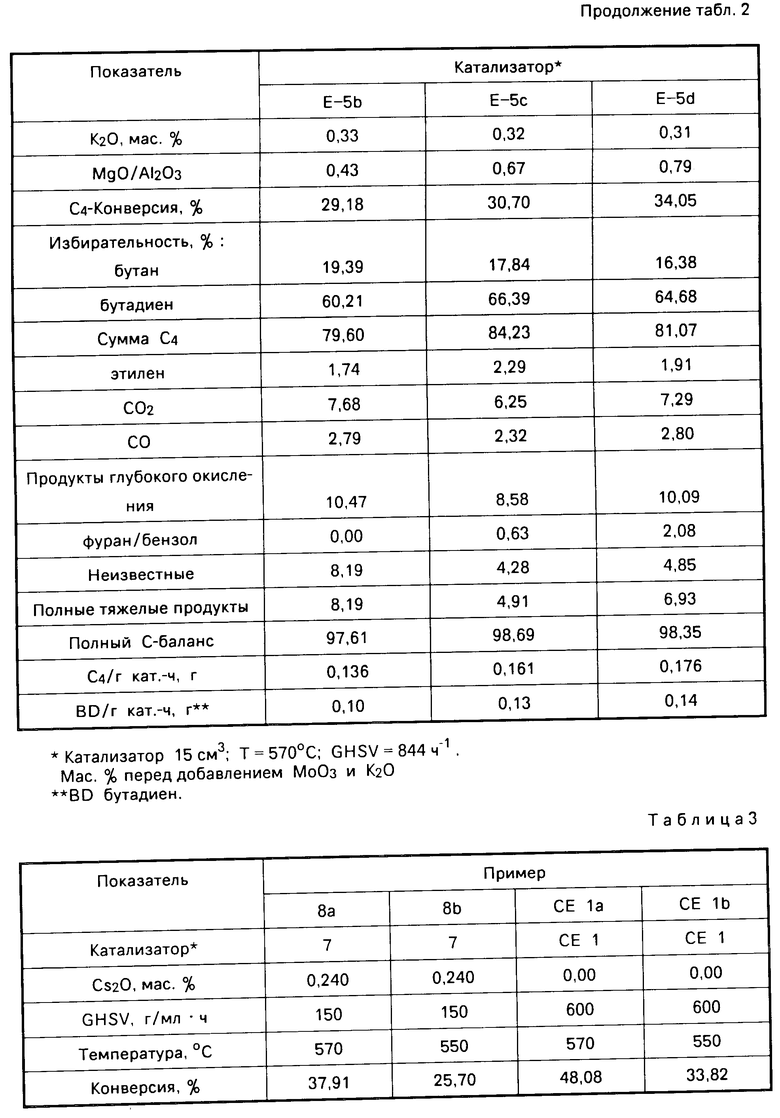
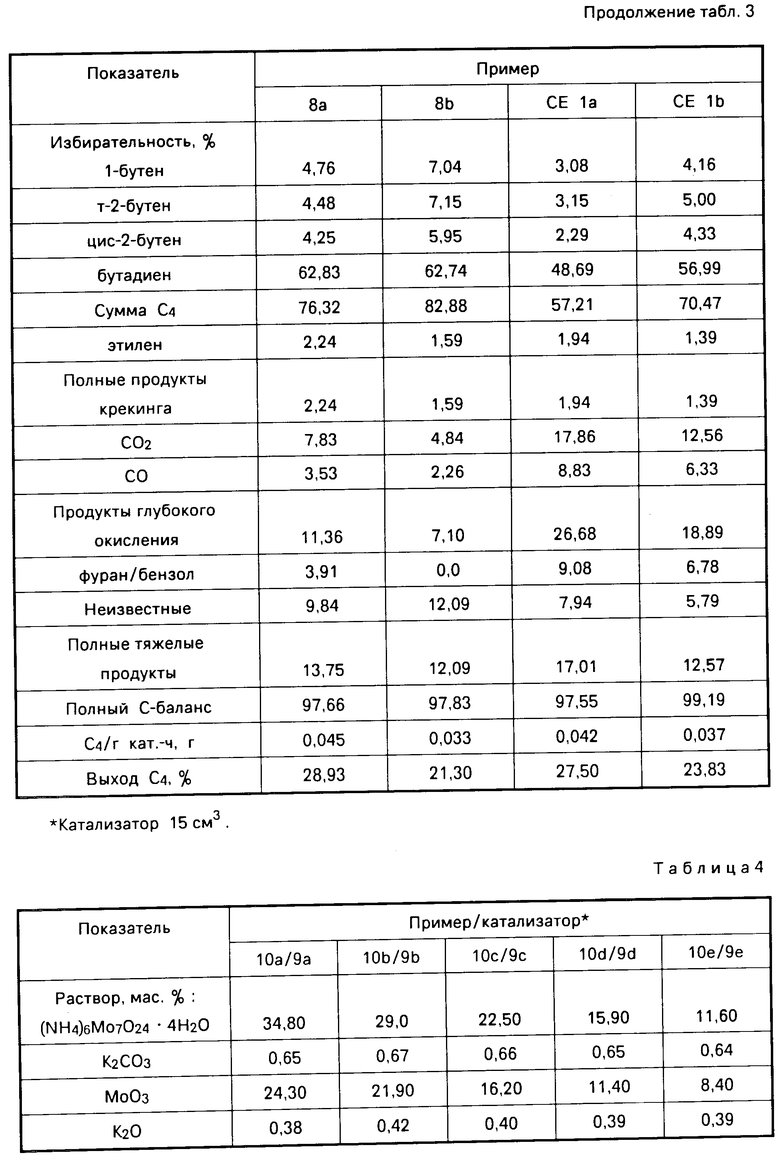
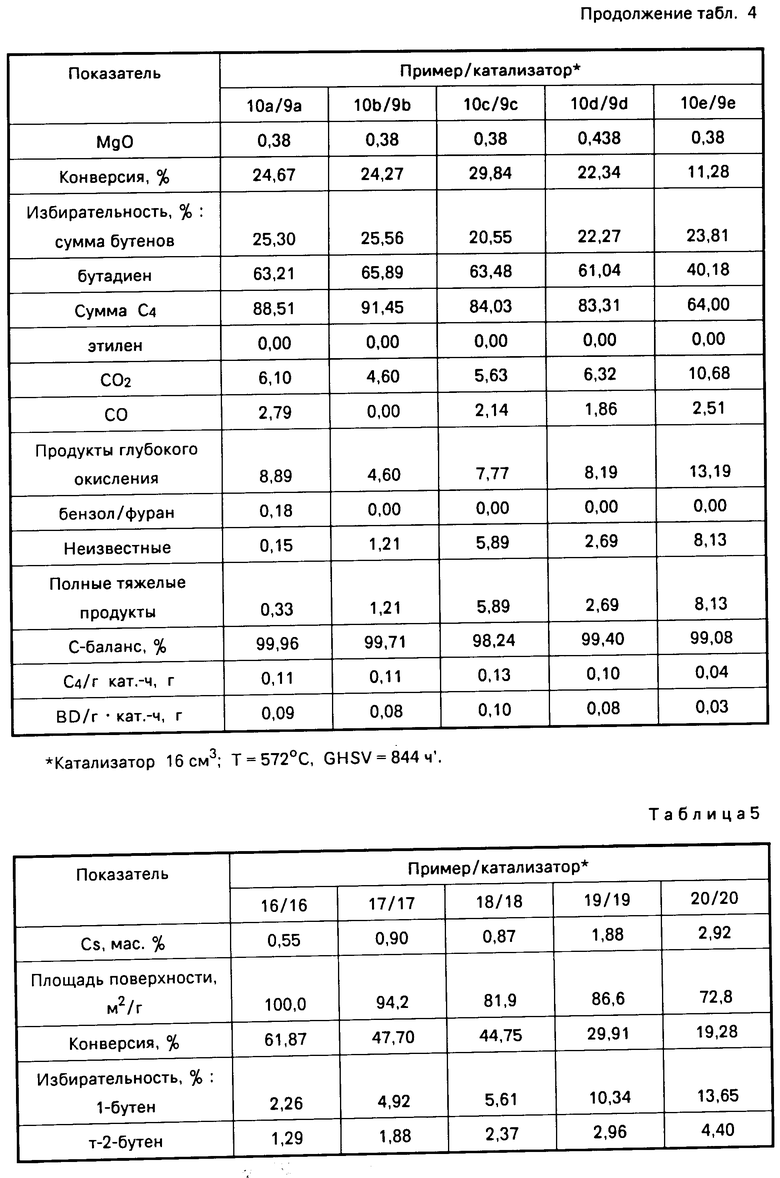
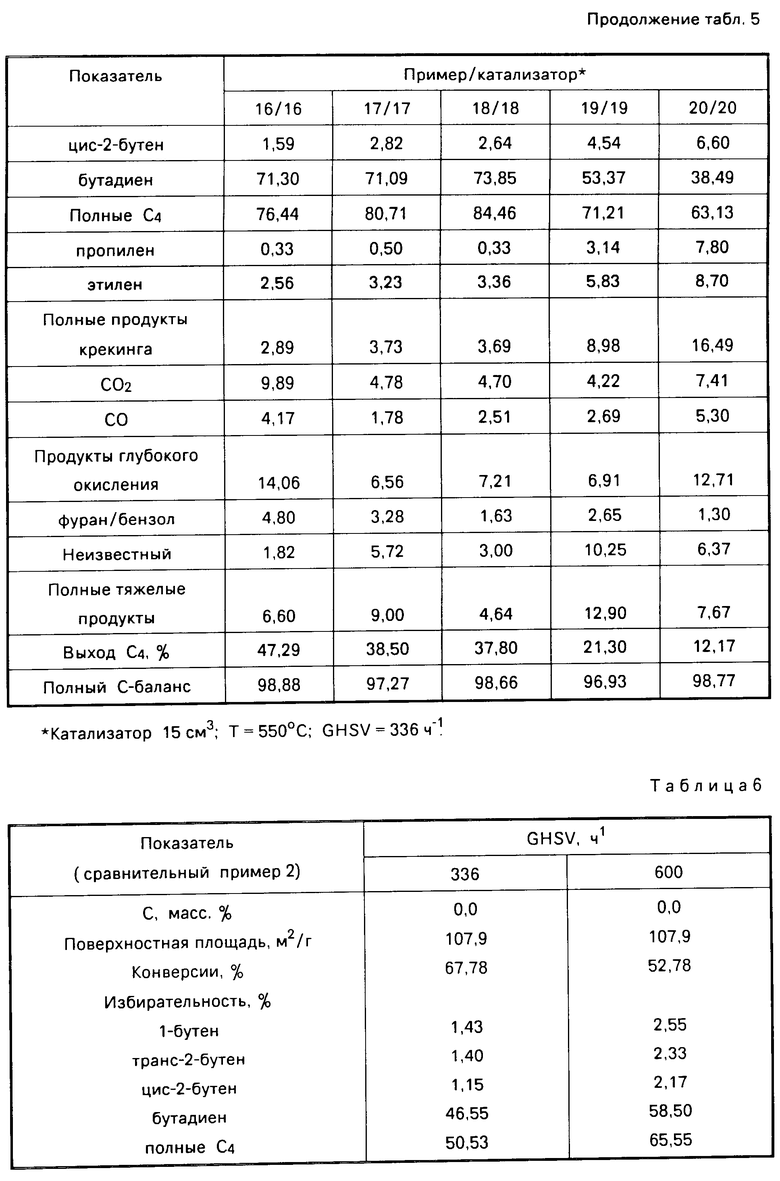
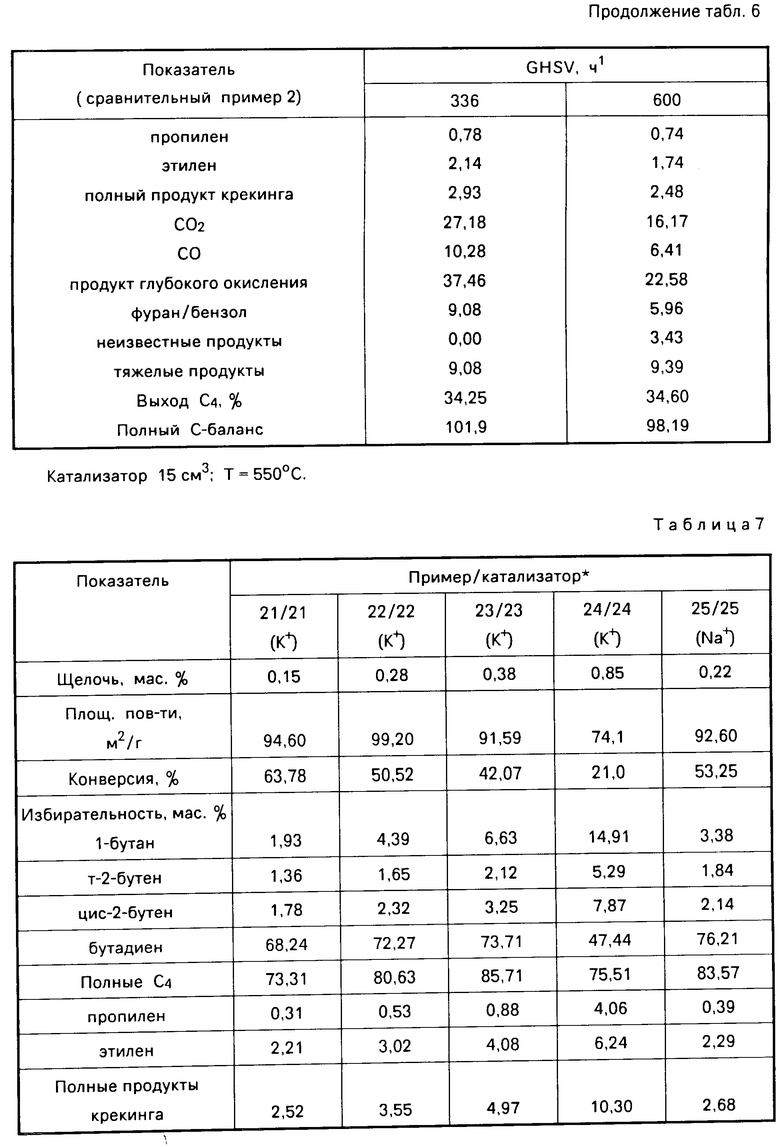
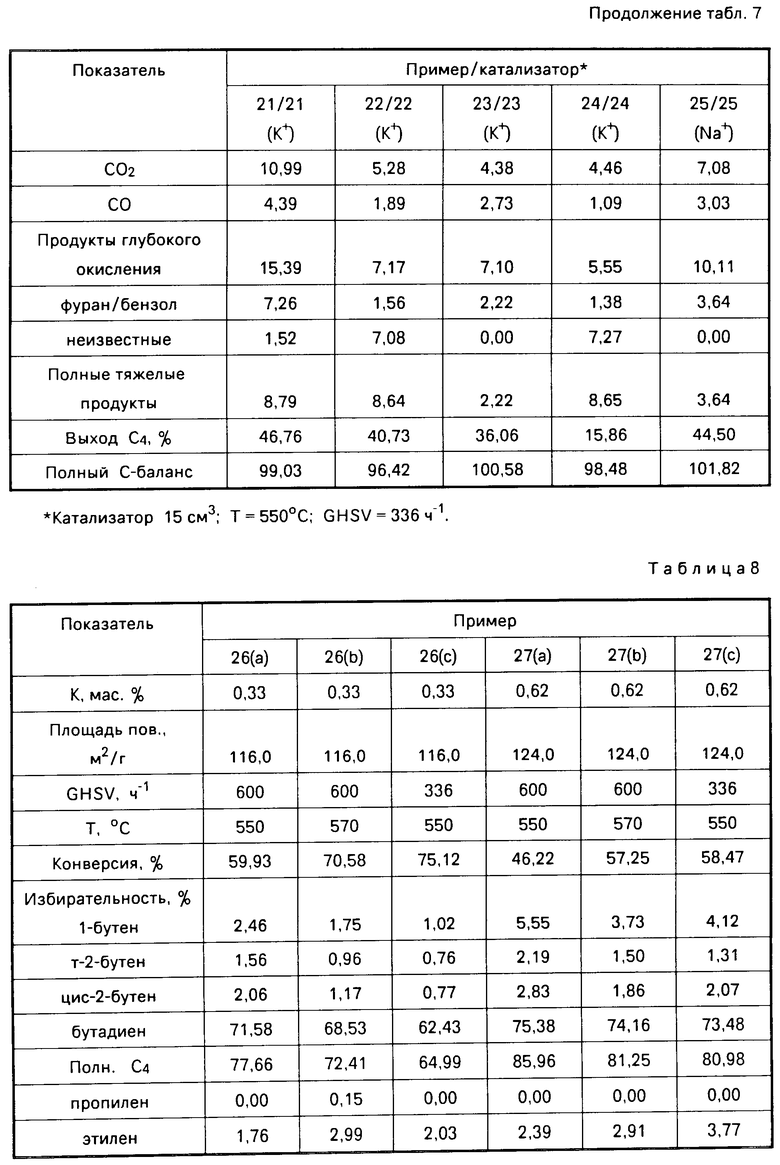
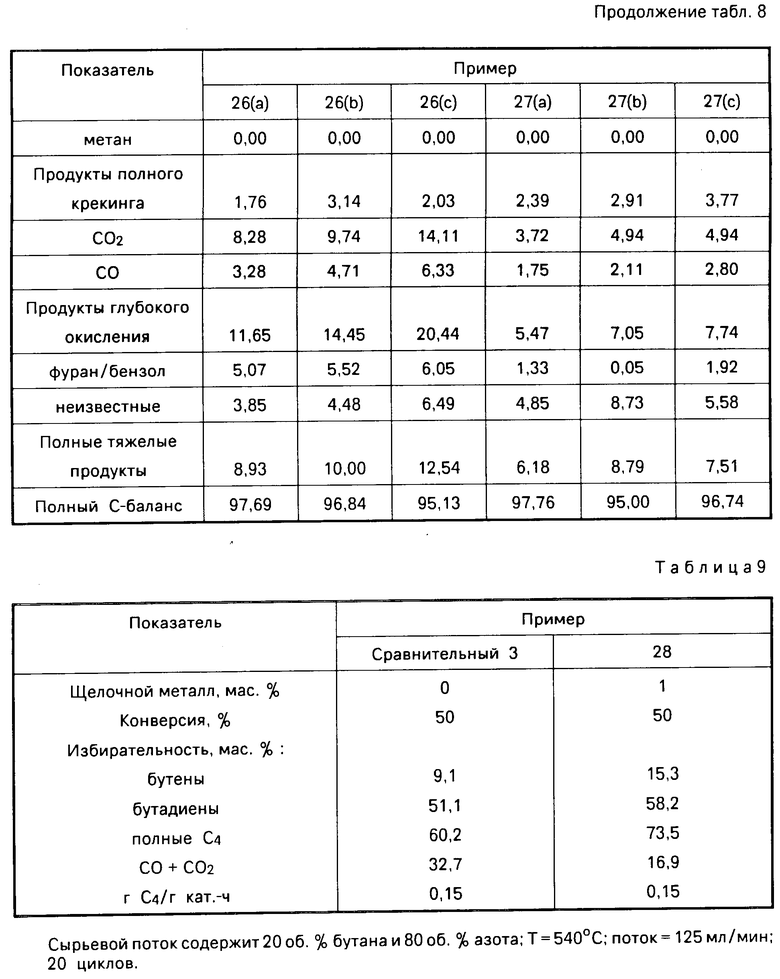
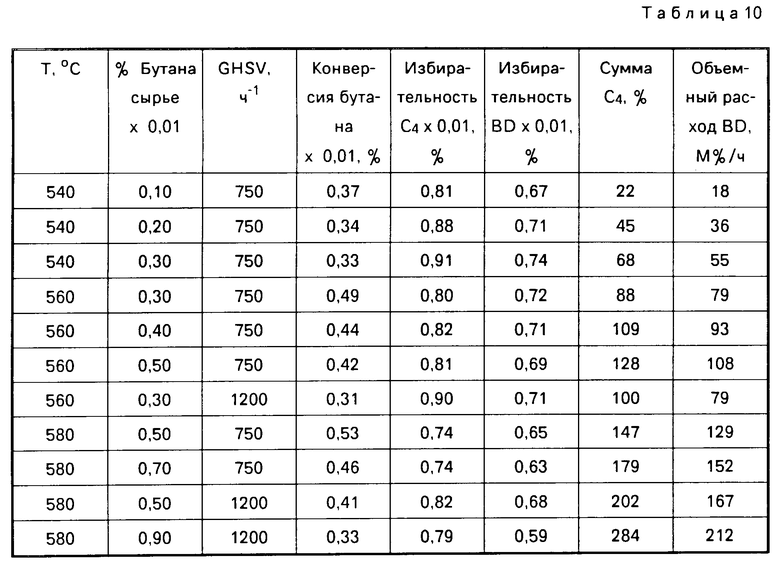
Использование: нефтехимическая промышленность. Сущность изобретения: продукт-1,3-бутадиен (БД) и катализатор для его получения с реакционноспособным кислородом. Он содержит, мас. %: оксид молибдена 8,4 - 24,30; оксид щелочного металла 0,16 - 1,43; носитель - остальное. Носитель состоит из MgO, Al2O3 и/или алюмомагниевой шпинели, массовое соотношение MgO/Al2O3 0,38 - 0,79, площадь поверхности 25-184,3 м2/г. Катализатор дополнительно содержит 0,58 - 0,85 мас.%. Условия окислительного дегидрирования бутана: 550 - 650°С, 7 - 210 КПа и другие условия, обеспечивающие избирательность по БД не менее 40 мас.%. 2 с. и 2 з.п. ф-лы, 10 табл.
Оксид молибдена 8,40 24,30
Оксид щелочного металла 0,16 1,43
Носитель Остальное
2. Способ по п.1, отличающийся тем, что используют катализатор, дополнительно содержащий 0,58 0,85 мас. пентоксида ванадия и процесс проводят при 500 650oС, 10,5 210,0 КПа, объемной скорости подачи бутана 400 4000 ч-1 и других условиях, обеспечивающих получение реакционной смеси, содержащей бутадиен с избирательностью не менее 50 мол.
Оксид молибдена 8,4 24,3
Оксид щелочного металла 0,16 1,43
Носитель Остальное
4. Катализатор по п.3, отличающийся тем, что, с целью повышения активности катализатора, он дополнительно содержит 0,58 0,85 мас. пентоксида ванадия.
| Патент США N 3862256, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

