Изобретение относится к способу извлечения сложных диэфиров дистенноксана из реакционных смесей, содержащих эти соединения. В предпочтительном варианте осуществления изобретения извлеченный сложный диэфир дистанноксана рециркулируют для последующей реакции.
Молекула сахарозы содержит три первичные (основные) гидроксильные группы и пять вторичных гидроксильных групп. Когда необходимо получить производные сахарозы, включающие реакцию гидроксильных групп, основной проблемой синтеза может оказаться направление реакции только на определенные гидроксильные группы. Например, искусственный подслаживающий агент 4,1', 6'-трихлор-4,1', 6'-тридеоксигалактосахарозу (сукралозу) получают из сахарозы при помощи замены гидроксилов в 4, 1' и 6' позициях хлором (в процессе получения подслащивающего агента стереоконфигурация в 4 позиции обращается, следовательно, это соединение является галактосахарозой). Это соединение и способы синтеза известны из патенов США NN 4343934, 4362869, 4380476 и 4435440. Направление атомов хлора только в необходимые позиции является основной проблемой синтеза, в частности, так как гидроксилы, которые при этом замещаются, обладают различной химической активностью (два являются первичными, один вторичным; кроме того, синтез усложняется тем фактом, что первичный гидроксил в 6 позиции является незамещенным в конечном продукте). Получение этого подслащивающего агента является только одной иллюстрацией синтеза производных сахарозы, в которой желательно либо модифицировать некоторые специальные гидроксильные группы и только такие гидроксильные группы, либо модифицировать только определенное количество гидроксилов. В последнем случае можно не обращать внимания на то, какие конкретно гидроксилы подвергают модификации. Получение поверхностно-активных агентов, сложных моноэфиров на основе сахарозы является промышленным примером монозамещения на молекуле сахарозы.
Известны способы получения сложного сахароза-6-эфира с использованием в качестве промежуточных соединений некоторых соединений олова. Например, получение сложных сахароза-6-эфиров на основе дистенноксана раскрыт в способе синтеза производных сахарозы при помощи региоселективной реакции (патент США N 4950746). По этому способу соответствующие материалы на основе ди(гидрокарбил) олова, такие как окись дибутилолова, окись диоктилолова, диметилат дибутилолова и т.п. могут быть соединены с содержащим гидроксильные группы соединением, например одноатомный спирт или простой фенол, так, чтобы получить химически активное промежуточное соединение дистанноксана, а именно, 1,3-ди-(гидрокарбилокси)-1,1,3,3-тетра(гидрокарбил)-дистанн- оксан, которые далее могут взаимодействовать с сахарозой, чтобы получить 1,3-ди-(6-0-сахароза)-1,1,3,3-тетра(гидрокарбил)дис- танноксан. Предложен также простой способ получения сложных сахароза-6-эфиров при помощи обработки этих аддуктов органоолова-сахарозы соответствующим ацилирующим агентом в подходящем растворителе или смеси растворителей.
Другой способ с промежуточным соединением олова для получения сложных сахароза-6-эфиров (патент США N 5023329) включает взаимодействие сахарозы с окисью ди(гидрокарбил) олова в инертном органическом носителе в течение времени и при температуре, достаточных для получения 1,3-ди(6,0-сахароза)-1,1,3,3-тетра(гидрокарбил)дистанноксана. В предпочти- тельном варианте осуществления способа, 1,3-ди-(6-0-сахароза)-1,1,3,3-тетрагидро(ги- дрокарбил) дистаноксан, полученный таким образом, взаимодействует с ацилирующим агентом при температуре и в течение времени, достаточных для получения сложного сахароза-6-эфира.
Еще один способ получения сложного сахароза-6-эфира с промежуточным соединением олова (патент США N 5089608) включает следующие стадии:
а) взаимодействие окиси ди(гидрокарбил)олова, например окись диалкилолова, с двухатомным спиртом, алканоламином или энолизируемым α -окиси кетоном, т.е. α -оксикетоном, который способен к энолизации в двухатомный спирт с одной двойной связью, в инертном органическом носителе реакции с удалением воды при помощи азеотропной дистилляции и при температуре и в течение времени, достаточных для получения циклического аддукта указанной окиси диалкилолова и двухатомного спирта, алканоламина или энолизируемого α -окиси кетона;
б) взаимодействие указанного продукта, циклического аддукта, со стадии а) с сахарозой в инертном органическом носителе реакции, в котором сахароза обладает соответствующей степенью растворимости, таком как диполярная апротонная жидкость, при температуре и в течение времени, достаточных для получения 6-0-(дигидрокарбил)окси- или амино- или оксогидрокарбил)станноксил)сахарозы;
в) взаимодействие продукта со стадии б) с ацилирующим агентом, чтобы получить сложный сахароза-6-эфир.
По этим способам реакционная смесь, содержащая сложный сахароза-6-эфир (S-6-Е), содержит также в качестве побочного продукта 1,3-диацилокси-1,1,3,3-тетра(гидрокарбил)дистанноксан или сложный диэфир дистанноксана (СЭДС).
Предлагаемый способ включает экстрагирование СЭДС из смеси, содержащей СЭДС, сложный сахароза-6-эфир и полярный апротонный растворитель, который содержит стадии:
а) контактирование указанной смеси в присутствии небольшого количества воды с органическим растворителем, который является существенно несмешивающимся с водой, чтобы получить при этом смесь экстрагирования, в которой используемое количество воды должно быть достаточным для эффективного распределения СЭДС из первой фазы, содержащей полярный апротонный растворитель, во вторую фазу, содеражащую органический растворитель;
б) перемешивание смеси экстрагирования в течение времени и при температуре, достаточных для образования двухфазной смеси, в которой преобладающее количество СЭДС, содержащегося в смеси экстрагирования, находится во второй фазе и по существу весь S-6-Е, содержащийся в смеси для экстрагирования, присутствует в первой фазе;
в) отделение первой фазы от второй фазы.
Присутствие небольшого количества воды в смеси экстрагирования служит для того, чтобы позволить или существенно ускорить распределение СЭДС в органическом растворителе в то время, как S-6-Е остается растворенным в полярном апротонном растворителе, в результате СЭДС может быть экстрагирован почти количественно при помощи органического растворителя, а сложный сахароза-6-эфир остается в растворе в полярном апротонном растворителе.
Известно ацилирование (региоселективное) в 6-позиции с промежуточным соединением органоолова сахарозы с тем, чтобы получить сложные сахароза-6-эфиры. Использование сложных сахароза-6-эфиров в способах получения искусственных подслащивающих агентов, например 4,1',6'-трихлор-4,1',6'-тридеоксигалактосахарозы описано в патентной заявке США N 382147. Распределение побочного продукта СЭДС, получаемого на месте после ацилирования сахарозы между двумя фазами органического растворителя, причем указанное распределение промотируется добавлением небольшого количества воды, не было известно ранее.
В соответствии с настоящим изобретением предлагается способ экстрагирования сложного диэфира дистанноксана из смеси, содержащей это соединение вместе со сложным сахароза-6-эфиром и полярным апротонным растворителем, при помощи контактирования указанной смеси с органическим растворителем, который является по существу несмешивающимся с водой и в котором СЭДС растворим, в присутствии небольшого количества воды. Смесь, содержащая СЭДС, S-6-Е и полярный апротонный растворитель, может быть получена по любому известному способу.
В предлагаемом способе в качестве исходной смеси используют продукт способа, по которому аддукт селективной сахарозы-олова ацилируют при помощи соответствующего ацилирующего агента (например, ангидрида кислоты) в полярной апротонной среде, такой как N,N-диметилформамид (ДМФ), диметилсульфооксид (ДМСО), N-метилпирролидинон (НМП), N,N-диметилацетамид (ДМА), гексаметилфосфорамид (ГМФА), и других полярных апротонных растворителях, в которых сахароза растворима (предпочтительным полярным апротонным растворителем является ДМФА), чтобы получить сложный сахароза-6-эфир и ацилированную форму реагента олова, который обладает существенной растворимостью в известных органических растворителях. В соответствии с настоящим изобретением полученный таким образом побочный продукт органоолова (СЭДС) отделяют от реакционной среды сложного сахароза-6-эфира при помощи обработки небольшим количеством воды с последующим экстрагированием соответствующим органическим растворителем.
Эффективность этого экстрагирования значительно и существенно увеличивается при добавлении небольшого количества воды в реакционную среду перед экстрагированием. Экстрактивное удаление олова в высшей степени эффективно (>99% удаление органоолова экстрагированием при оптимальных условиях всего за две стадии экстрагирования). Рафинат, т.е. жидкость, оставшаяся после того, как соединение олова было экстрагировано, образовавшийся после удаления экстрактивного олова, содержит полярный апротонный растворитель, ацилированные производные сахарозы, главным образом S-6-Е, остаточную воду и остаточный экстрагент, полученный в результате прекрестной растворимости. Удаление воды, которое должно быть осуществлено, когда сложный сахароза-6-эфир должен быть хлорирован в сложный сукралоза-6-эфир, и полученные в результате летучие материалы при помощи приемов дистилляции способствуют образованию раствора ацилированной сахарозы в полярном апротонном растворителе.
Когда используемым полярным апротонным растворителем является ДМФ, "нижние" составляющие этой дистилляции пригодны для непосредственного использования в реакции хлорирования без последующей обработки или изоляции сложного сахароза-6-эфира. Если S-6-Е получают по известному способу, удаление этерифицированного диаола, аминоспирта или энола может быть осуществлено перед хлорированием. Основным продуктом реакции хлорирования является сложный сукралоза-6-эфир, который после гидролиза с целью удаления ациловой группы дает высокоинтенсивный подслащивающий агент сукралозу.
Таким образом, в соответствии с настоящим изобретением предлагается экстрактивный прием для предпочтительного и эффективного отделения побочного продукта органоолова, полученного из реакций 6-ацилирования с промежуточным соеди- нением олова сахарозы из ацилированных производных углеводов, при этом имется возможность использовать неочищенный, полученный на месте углеводородный продукт этой реакции непосредственно на последующей стадии хлорирования без применения изоляции промежуточного S-6-Е. Отделение может быть осуществлено лишь при помощи небольших количеств воды, добавляемой для этого, и является важным отличительным свойством настоящего изобретения, так как вода должна быть удалена из раствора сложного сахароза-6-эфира перед последующей обработкой с целью получения сукралоза, а стоимость удаления этой воды пропорциональна ее содержанию. Полученный по способу разделения СЭДС может быть рециркулирован в последовательность реакций сукралозы при помощи удаления растворителя экстрагирования и либо обработки эквивалентным количеством алкоголята, чтобы получить химически активный диалкоголят дистанноксана, либо реакции с небольшим избытком водного каустического раствора, чтобы восстановить окись ди(гидрокарбил)олова для повторного использования по любому из трех способов получения сложного сахароза-6-эфира.
В результате того, что удается избежать изоляции твердого продукта, увеличивается общий выход для сложного сахароза-6-эфира. Например, получают выход 90-93% для сахароза-6-бензоата (S-6-Е) и 88-91% для сахароза-6-ацетата (S-6-А) в соответствии с предлагаемым способом в отличие от выходов в приблизительно 80% для твердого S-6-В и 65% для твердого S-6-А, когда используют кристаллизацию для выделения продукта. Дополнительными преимуществами являются снижение числа используемых растворителей, например для кристаллизации, в которой используют ацетон или метанол, исключение рецикла маточных жидкостей кристаллизации и выпаривания полярного апротонного растворителя, такого как ДМФ, и снижение общего количества оборудования, необходимого для осуществления способа (например, оборудования для выделения и сушки S-6-Е).
Растворители, используемые при осуществлении экстрактивного удаления СЭДС, включают алифатические и ароматические углевороды, простые эфиры, хлорированные углеводороды, кетоны и сложные эфиры, которые обладают низкой перекрестной растворимостью с водой. Под термином "низкая перекрестная растворимость с водой" подразумевается, что растворитель экстрагивания растворяет менее примерно 1 мас. воды, а вода растворяет менее примерно 1 мас. растворителя экстрагирования, причем обе растворимости определяют при температурах ниже примерно 20оС. Хотя эти растворители часто не смешиваются с ДМФ или другими полярными апротонными растворителями, присутствие сложного сахароза-6-эфира промотирует разделение смеси экстрагирования на две фазы, в то время как добавление небольшого количества воды обеспечивает эффективное разделение СЭДС, осуществляя экстрагирование. Растворители, которые можно использовать для этого, включают гексан, циклогексан, гептан и др. алифатические углеводороды, бензол, толуол, ксилолы, кумол и др. ароматические углеводороды, диэтиловый, метиловый, третичн. бутиловый, диизопропиловый и др. простые эфиры, дихлорметан, хлороформ, треххлористый углерод, ди-, трех- и четыреххлорированные этаны, полихлорированные алифатические и ароматические углеводороды, хлорбензол и др. хлорированные углеводороды, метилизобутилкетон и другие несмешивающиеся с водой кетоны и несмешивающийся с водой сложный эфир, такой как метил бензоат, изопропил ацетат и этил валерат. Предпочтительными растворителями являются по крайней мере полярные; алифатические углеводороды предпочтительны, так как они обладают более низкой перекрестной растворимостью с ДМФ. Чтобы облегчить удаление растворителя дистилляцией, в предпочтительном варианте температура точки кипения при атмосферном давлении находится в интервале 60-100оС, в наиболее предпочтительном варианте используют растворители, кипящие при температуре примерно 75-85оС при атмосферном давлении.
Растворитель экстрагирования используют в количестве, достаточном для осуществления эффективного экстрагирования содержащегося СЭДС, например, в количестве не менее примерно 1 мл растворителя экстрагирования на 1 г СЭДС, в предпочтительном варианте не менее примерно 1,5 мл растворителя экстрагирования на 1 г СЭДС, а в более предпочтительном варианте не менее примерно 2 мл растворителя экстрагирования на 1 г СЭДС, содержащегося в смеси экстрагирования. Указаные пропорции указаны для первой стадии экстрагирования. При практической реализации в общем случае используют две или три стадии экстрагирования. Несмотря на то что на второй и третьей стадиях можно использовать гораздо меньше растворителя экстрагирования, пропорции растворителя экстрагирования, используемые на последующих стадиях экстрагирования, будут больше в общем случае, чем указанные, ввиду того, что количество оставшегося СЭДС в рафинате становится все меньше и меньше с каждым экстрагированием, в некоторое минимальное количество растворителя экстрагирования необходимо использовать для того, чтобы упростить реализацию. В общей ситуации примерно от одной трети до половины количества растворителя, используемого на первой стадии экстрагирования, может быть использовано на второй, третьей и, если необходимо, на последующих стадиях экстрагирования. Примеры, которые приведены ниже, иллюстрируют порядок значений пропорций растворителя экстрагирования, которые могут быть использованы. Верхний предел используемого растворителя экстрагирования диктуется причинами экономического характера. Однако получают неудовлетворительные результаты, если более 5 мл растворителя экстрагирования используют на 1 г СЭДС на первой стадии экстрагирования.
Количество воды, используемое для облегчения распределения, зависит частично от природы используемого растворителя экстрагирования, возрастающей полярности растворителя, требующей увеличения количества воды. Так как предпочтительная цель в осуществлении экстрагирования заключается в том, чтобы получить раствор сложного сахароза-6-эфира в растворителе, таком как ДМФ, который пригоден для непосредственного хлорирования по известному способу, в соответствии с которым раствор должен быть безводным, важно по экономическим причинам минимизировать количество используемой воды. В табл.1 приведены данные экстрагирования соединения олова, диацетата дистанноксана (ДАДС) из реакционной смеси сахароза-6-ацетата с использованием 100 г сахарозы и различных количеств циклогексана и воды. Используют одну стадию экстрагирования. При промышленном осуществлении, видимо, исопльзовали бы больше, чем одну стадию экстрагирования (например две или три).
При отсутствии воды экстрагирование неэффективно, но добавление лишь небольшого количества воды достаточно для того, чтобы обеспечить оптимальное и эффективное распределение соединений олова в углеводородной фазе. Общий раствор для экстрагирования, т.е. ДМФ-раствор S-6-А и ДАДС, не включая циклогексан, содержит 540 г раствора, который содержал примерно 92,6 г (0,154 моль) ДАДС. Предпочтительными растворителями экстрагирования являются углеводородные растворители. Расход воды колеблется от примерно 3 до 20 моль воды на моль СЭДС, содержащегося в смеси для экстрагирования. Предпочтительными растворителями экстрагирования являются гексан, циклогексан и гептан; предпочтителен расход воды от примерно 5 до 10 моль воды на моль СЭДС.
Эффективное распределение СЭДС в органической фазе требует добавления небольших количеств воды (на молярной основе СЭДС) в полярную апротонную фазу. Эффективность воды при получении благоприятного распределения СЭДС может быть вызвана частично возможным разрывом связывающих взаимодействий между СЭДС и содержащимися различными углеводными материалами. Соединения четырехвалентного органолова обладают предрасположенностью к образованию пяти- и шестикоординатных соединений, если присутствуют группы со свойствами лигандов, такие как гидроксилы. После добавления воды СЭДС экстрагируют из полярной апротонной фазы в относительно неполярную органическую фазу и выделяют в форме моногидрата, при этом добавленная вода разрывает СЭДС-углеводные ассоциации и дает в результате легко экстрагируемые СЭДС, Н2О-материалы.
П р и м е р 1. Получение сахароза-6-бензоата по известному способу с экстрактивным удалением ДБДС при помощи циклогексана и воды.
В 500 мл четырехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром, холодильником и ловушкой Дина-Старка, загружали 5,16 г (анализ 87,4% 80,4 ммоль) гидрата окиси калия, 100 мл н-бутилового спирта и 35 мл гексана, смесь нагревали до дефлегмации. Воду (примерно 2,5 мл) собирали в ловушку Дина-Старка, как и азеотропную гексановую смесь, по мере ее образования в течение 45 мин. Затем в содержимое реактора добавляли раствор 32,2 г (44,5 ммоль) дибензоата дистанноксана (ДБДС), полученного, как это описано в примере 6а, в 50 мл горячего гексана. Смесь поддерживали при дефлегмации в течение 30 мин, при этом одновременно гексан испарялся. Горячую реакционную смесь фильтровали под вакуумом непосредственно в другую 500 мл колбу с круглым дном, оснащенную, как это было описано выше, заботясь о том, чтобы не проникла атмосферная влажность. Фильтровальную лепешку промывали дважды (всего 100 мл) смесью в отношении 1:1 бутанола и гексана. Твердый бензоат калия (12,4 г сухой вес, 96,6% от теоретического) сбрасывали.
В мутный фильтрат, содержащий 1,3-дибутокси-1,1,3,3-тетрабутилдистанноксан (ДБДС) в бутаноле, добавляли 25 г (73,1 ммоль) сахарозы, растворенной в 150 мл горячего (примерно 90оС) ДМФ. Реактор снаряжали для вакуумной дистилляции и дистиллят собирали при температуре на выходе 65-66оС (температура реактора 70-73оС) под вакуумом, создаваемым отсасыванием воды. После удаления примерно 200 мл дистиллята в реактор добавляли еще 50 мл ДМФ. Дистилляцию продолжали до тех пор, пока не получали всего 312 мл дистиллята. Остаток после дистилляции разбавляли 50 мл ДМФ, чтобы получить прозрачный серо-желтый раствор 1,3-ди-(6-0-сахароза)-1,1,3,3- тетрабутилдистанноксана (ДБСС) в ДМФ (объем примерно 130 мл). Этот раствор охлаждали до 18-20оС и обрабатывали одной порцией 18,2 г (806 ммоль) бензойного ангидрида. После перемешивания в течение ночи реакционную смесь переносли на делительную воронку и обрабатывали 50 мл циклогексана и 5 мл (278 ммоль, 6,24 эквивалента в пересчете на ДБДС) воды. Содержимое сепаратора слегка перемешивали и полученным в результате фазам давали возможность разделиться. Верхнюю фазу удаляли, а нижнюю (ДМФ) фазу экстрагировали дополнительным циклогексаном (5 х 50 мл). Концентрация соединенных экстрактов давала 35 г выделенного ДБДС, содержащего остатки растворителя, в виде вязкого желтого масла, которое отверждалось при выдерживании. Нижнюю фазу концентрировали под вакуумом при 50оС в липкий желтый сироп (46,1 г), который, как устанавливали при помощи ЖХВД-анализа, содержит 59,5 мас. сахароза-6-бензоата (27,4 г, 61,4 ммоль, выход 84,1%), 6,4 мас. дибензоатов сахарозы и 0,7 мас. остаточной сахарозы. Оcтаточное олово, как устанавливали при помощи спектрофотометрии поглощения атомов, выраженное в процентах ДБДС, составило 0,9 мас.
П р и м е р 2. Экстрактивное удаление ДБДС из сырой смеси сахароза-6-бензоата с использованием метил третичн.-бутилового простого эфира с добавленной водой.
В последовательности, идентичной использованной в примере 1, сырую реакционную смесь, содержащую ДБДС и S-6-В, обрабатывали при помощи 5 мл (278 ммоль, 6,24 эквивалента в пересчете на ДБДС) воды, а затем экстрагировали метил третичн. бутиловым простым эфиром (МТБЭ, 6 х 100 мл). Соединенные экстракты, которые, как было установлено при помощи ТСХ на силикагеле (Rf примерно 0,5, 15:10:2, СНСl3-СН3ОН-Н2О, которую распыляли при помощи 5%-ного этанолового раствора серной кислоты и обугливали), содержали некоторое количество S-6-В, промывали обратной струей с использованием 50 мл воды; водную фазу соединяли с рафинатом с предыдущих экстракций. Концентрация соединенных МТБЭ-экстрактов давала 33,4 г выделенного ДБДС, содержащего остаточный растворитель, в виде липкого сиропа, который отверждался при выдерживании. Соединенные нижние фазы концентрировали под вакуумом при 50оС, чтобы получить 46,6 г серо-желтого сиропа, который, как устанавливали при помощи ЖХВД-анализа, содержит 58,2 мас. cахароза-6-бензоата (27,1 г, 60,8 ммоль, 83,3% выход) и 0,3 мас. остаточной сахарозы. S-6-В рассматривали как состоящий на 100% из монобензоатов сахарозы и на 87,4% из всех видов бензоилированных углеводов. Остаточное олово в пробе, как было установлено при помощи спектрометрии атомного поглощения, составляло 0,14 мас. выраженное в процентах в пересчете на ДБДС.
П р и м е р 3. Экстрактивное удаление ДБДС из сырой смеси сахароза-6-бензоата с использованием кумола с добавленной водой.
В соответствии с процедурой, осуществляемой точно так же, как в предыдущих примерах, получали сырую реакционную смесь сахароза-6-бензоата и ДБДС (объем примерно 170 мл) и обрабатывали при помощи 100 мл кумола (изопропил бензол) и 10 мл (556 ммоль, 12,5 эквив. в пересчете на ДБДС). Мутную двухфазную систему тщательно перемешивали и затем отстаивали для того, чтобы получить две прозрачные жидкие фазы. Их разделяли и рафинат экстрагировали дополнительным кумолом (3 х 100 мл). Рафинат концентрировали при 50оС под вакуумом, чтобы получить 46,5 г сиропа, который, как было установлено при помощи ЖХВД-анализа, содержал 50,6 мас. S-6-В (23,5 г, 52,7 ммоль, 72,1% выход), 3,6 мас. остаточной сахарозы и 6,0 мас. дибензоатов сахарозы. Сахароза-6-бензоат содержал 100% монобензоатов и 91,2% всей фракции бензоилированных углеводов. Остаточное олово, выраженное в ДБДС, составило 0,04% (анализ на поглощение атомов).
П р и м е р 4. Получение сахароза-6-бензоата при помощи экстрактивного удаления ДБДС с использованием метил третичн.-бутилового простого эфира циклогексана и добавляемой воды.
В последовательности, идентичной той, что использовали в примере 1, сырую реакционную смесь, содержащую ДБДС и сахароза-6-бензоат, обрабатывали 5 мл (278 ммоль, 6,24 эквив. в пересчете на ДБДС) воды, а затем экстрагировали смесью (1: 1) МТБЭ и циклогексана (6 х 50 мл). Соединенные экстракты давали после выпаривания 35,0 г ДБДС, содержащего остаточный растворитель. Рафинат концентрировали под вакуумом при 50оС в сироп (46,12), который как устанавливали при помощи ЖХВД-анализа, содержал 59,5 мас. S-6-В (27,4 г, 61,4 ммоль, 84,1% выход). Остаточный ДБДС в сиропе составлял 0,4 г.
П р и м е р 5. Эксперименты с рециклом и использованием выделенного экстрагированием ДБДС.
В табл.2 собраны результаты серии экспериментов, связанных с получением сахароза-6-бензота, с использованием выделенного экстрагированием ДБДС. Способ аналогичен тому, что описан в примере 1, но его осуществляли в масштабе, большем в 2 раза. Исходную порцию ДБДС получали при помощи процедуры из примера 6А, используя бензойный ангидрид и изолируя при помощи кристаллизации из 5%-ного водного раствора ацетонитрила, а остаточные порции ДБДС получали с использованием рециркулированного ДБДС из предыдущего цикла.
Оловянный реагент, выделенный за 6 циклов, в среднем составлял 97,7% недостаток объясняется потерями в фильтровальной лепешке бензоата кальция и механическими потерями. Выходы сахароза-6-бензоата в среднем составили 87,7% в пересчете на исходную загрузку сахарозы.
Этот пример служит иллюстрацией того, что продукт экстрагирования сложного диэфира дистанноксана сначала взаимодействует со спиртовой щелочью, чтобы образовать 1,3-ди(гидрокарбилокси)-1,1,3,3-тетра(гидрокарбил)-дистанноксан, который затем используют по известному способу, чтобы получить 1,3-ди-(6-0-сахароза)-1,1,3,3-тетра(гидрокарбил)-дистаннок- сан, который далее взаимодействует с ацилирующим агентом, чтобы образовать сложный сахароза-6-эфир и сложный диэфир дистанноксана. Этот способ может быть затем повторен при промышленной реализации. В этом эксперименте получаемым 1,3-ди(гидрокарбилокси)-1,1,3,3-тетра(гид-рокарбил)дистанноксаном является 1,3-диалкокси-1,1,3,3-тетра(алкил)дистанноксан, в частности 1,3-дибутокси-1,1,3,3-тетрабутилдистанноксан (см. табл.2).
П р и м е р 6. Получение сложных эфиров дистанноксана (СЭДС).
А. Из карбоновой кислоты и окиси дибутилолова.
Окись дибутилолова (ОДБО, 100 г, 0,40 моль) подвергали дефлегмации с уксусной или бензовой кислотами (24,1 или 49,1 г, 0,40 моль) в толуоле или гексане (200 мл); воду реакции отделяли с использованием ловушки Дина-Старка. Удаление воды занимало примерно 2 ч. СЭДС можно было бы использовать в растворе или кристаллизовать и изолировать при помощи удаления растворителя и растворения либо в 200 мл 5%-ного водного раствора ацетонитрила (ДБДС), либо в 100 мл 5%-ного водного ДМФ (ДАДС). Эти два сложных эфира дистанноксана кристаллизовали в форме моногидратов со следующими свойствами: Выход, г ДАДС ДБДС т.т.п. oC 107 126 Анализ: Найдено 57-57 -----
С 38,37 47,26
Н 6,83 6,24 Расч. для С20Н42О5Sn2 ˙ H2O 39,39 -----
6,83 ----- Расч. для C30H46O5Sn2 ˙ H2O ----- 48,56
----- 6,20
Б. Из ангидрида и окиси дибутилолова.
Из ОДБО (100 г, 0,40 моль) приготавливали шлам в циклогексане (200 мл) при 60оС и добавляли уксусный или бензойный ангидрид (20,4 или 45,2 г, 0,20 моль). Перемешивание продолжали 2 ч при 60оС, после чего ОДБО полностью растворялся. СЭДС либо использовали в растворе, либо изолировали с использованием процедур, кратко описанных в методе А, чтобы получить продукты с тем же выходом, обладающих теми же свойствами, что были описаны выше.
П р и м е р 7. Получение сахароза-6-ацетата из окиси дибутилолова по известному способу.
ОДБО (114 г, 460 ммоль) подвергали дефлегмации в течение 2 ч в н-бутаноле (220 мл) и циклогексане (50 мл), одновременно собирая 4 мл воды в ловушке Дина-Старка. Затем всего 230 мл смешанных растворителей удаляли при помощи вакуумной дистилляции, чтобы получить серо-коричневое, слегка мутное масло, которое состояло из ДБДС, растворенного в н-бутаноле.
Сахарозу (150 г, 483 ммоль) растворяли в ДМФ (450 мл) при 110оС, раствор охлаждали до 90оС и добавляли в полученнное масло. Снова применяли вакуум и продолжали дистилляцию, собирая 200 мл дистиллята при реакционной температуре в сосуде 80-85оС. Дистилляцию продолжали в течение примерно 30 мин. Добавляли ДМФ (100 мл, 80оС) и вакуумную дистилляцию продолжали, собирая дополнительно 150 мл дистиллята. Все это повторяли с использованием 150 мл ДМФ, собирая дополнительно 100 мл дистиллята. Остаток, который состоял из ДБСС в ДМФ, охлаждали до температуры ниже 20оС.
Уксусный ангидрид (49,2 г, 482 ммоль) добавляли по каплям со скоростью, достаточной для того, чтобы поддержать температуру 10-20оС, используя внешнее охлаждение, чтобы контролировать экзотермию. Добавление продолжали 40 мин. Раствор перемешивали при 20-25оС еще в течение 0,5 ч, затем экстрагировали циклогексаном (3х250 мл), добавляя воду (15 мл, 833 ммоль, 3,62 эквив, в пересчете на ДАДС) в каждую из первых двух экстракций. Соединенные экстракты, содержащие ДАДС, хранили для рецикла, при этом рафинат концетрировали под вакуумом до 30% от исходной массы, чтобы удалить воду, затем разбавляли ДМФ (100 мл) и хранили для хлорирования в сахароза-6-ацетат. Раствор (288 г) содержал 98,2 г (256 ммоль, 58,4% выход) сахароза-6-ацетата, что устанавливали при помощи ЖХВД-анализа. При помощи спектрофотометрии атомного поглощения определяли, что раствор содержал 0,07% олова, что эквивалентно 0,4 г ОДБО.
П р и м е р 8. Экстрактивное извлечение органоолова с превращением в окись дибутилолова с целью рецикла, используя известный способ.
В 1000 мл четырехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром, воронкой (60 мл) для добавлений и средствами дистилляции верхнего потока, содержащими короткую колонну Вигре, приемник дистиллята Дина-Старка и дефлегматор, загружали 76,4 г (0,307 моль) ОДБО, 100 Г (0,292 моль) сахарозы и 350 мл ДМФ. Смесь нагревали до 100-110оС, чтобы растворить сахарозу, затем охлаждали до 85-90оС, а циклогексан (100 мл) добавляли через капельную воронку (слабая дефлегмация). Смесь нагревали при энергичном перемешивании (реакционная температура 90-95оС) в течение 4,5 ч, при этом одновременно собирали нижнюю водную фазу в приемнике Дина-Старка. Если это необходимо, чтобы поддержать температуру ниже 95оС, добавляли дополнительное количество циклогексана.
Полученый в результате чувствительный к влажности прозрачный коричневый раствор, который содержал ДБСС в смеси ДМФ и циклогексана, охлаждали до 0оС и обрабатывали по каплям 32,8 г (0,321 моль) уксусного ангидрида, одновременно поддерживая температуру ниже 10оС охлаждением в ледяной ванне. Далее смеси давали возможность нагреватьcя до 20-25оС и перемешивали в течение 1 ч.
Добавляли воду (25 мл, 1,39 моль, 9,05 эквивалента в пересчете на ДАДС) и дополнительный циклогексан (250 мл, общее содержание циклогексана примерно 350 мл), смесь энергично перемешивали 10 мин при 20-25оС, а затем переносили в 1000 мл делительную воронку. Фазы разделяли и нижнюю фазу обрабатывали дополнительным количеством воды (25 мл, 1,39 моль, 9,05 эквивалента в пересчете на ДАДС), а затем экстрагировали циклогексаном (2 х 150 мл). При экстрагировании удаляли 99,5% от общего содержания олова. Рафинат на основе ДМФ, содержащий S-6-А, воду и сохранившийся циклогексан, затем подвергали фракционной дистилляции под вакуумом 50 мм рт.ст. при 70оС (максимум), чтобы удалить воду и циклогексан перед ЖХВД-анализов (96,1 г, 0,250 моль, 85,7% выход S-6-А).
Циклогексановые экстракты, содержащие ДАДС, соединяли и концентрировали в масло, которое добавляли горячим (выше 70оС) в форме слабого потока в течение 5 мин в очень энергично перемешиваемый раствор 1,1 н. водного гидрата окиси натрия (300 мл) при 95оС. Быстро образовывались гранулы ОДРО. Шлам ОДБО энергично перемешивали при 90-95оС в течение 10-15 мин, затем охлаждали до 30оС и фильтровали. Лепешку ОДБО тщательно промывали водой (3 х 100 мл) и сушили (потери при сушке 25-33%).
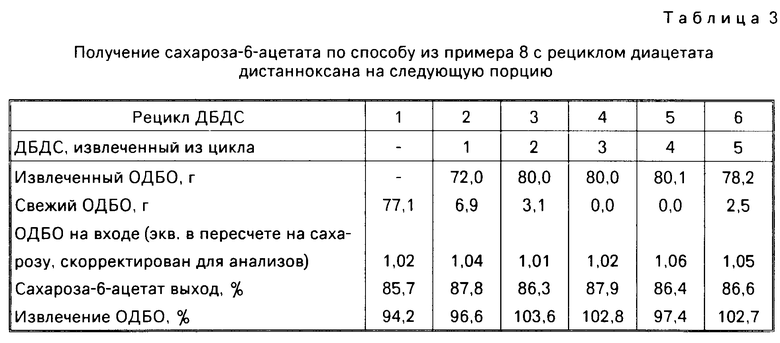
Извлеченный ОДБО использовали для получения S-6-А при помощи указанной процедуры, с ДАДС, извлеченным и экстрагированным в форме ОДБО, а затем снова рециркулировали, чтобы получить S-6-А по известному способу. Данные о извлечении окиси дибутилолова, скорректированные с учетом чистоты, выходе сахароза-6-ацетата и составе на входе ОДБО для повторяемых экспериментов приведены в табл.3 (ОДБО изолировали в форме полугидрата).
Предыдущий пример служит иллюстрацией того, что выделенный СЭДС обрабатывали водной щелочью и выделяли в форме окиси ди(гидрокарбил)олова (в данном случае), окиси диалкилолова, а более конкретно окиси дибутилолова, которую затем рециркулировали, чтобы получить сложный сахароза-6-эфир. Выделенную окись олова можно было также использовать для повторного использования по известным способам (см. табл.3).
П р и м е р 9. Хлорирование сырой смеси сахароза-6-бенозата (ДМФ) после экстрактивного извлечения оловянного реагента.
Настоящий пример иллюстрирует прямое хлорирование рафинада, полученого после экстрагирования ДБДС (в соответствии с осуществлением настоящего изобретения), в качестве промежуточной стадии в способе получения высокоинтенсивного непищевого подслащивающего агента, сукралозы.
В 500 мл четырехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром, холодильником, аргоновой оболочкой и воронкой для добавлений, загружали 16,8 г сахароза-6-бензоата (10,0 г, 22,4 ммоль) в виде сиропа, полученного как это поисано в примере 1, растворенного в 90 мл сухого ДМФ. В полученный прозрачный серо-желтый раствор, охлажденный до -38оС, по каплям добавляли всего 35,3 мл фосгена (491 ммоль), растворенного в 24 мл толуола. Добавление вызывало экзотермию с подъемом внутренней температуры от -38 до 14оС в течение добавления.
Полученный в результате шлам нагревали до 65оС, после этого прозрачный раствор нагревали до 82-83оС 1 ч, а затем до 112-113оС в течение 3 ч. Реакционную смесь охлаждали до 8оС и обрабатывали достаточно охлажденным (<5оС) 4 н. NaOH, чтобы поднять рН до 9-10 (116 мл). Это добавление давало сильную экзотермию с подъемом температуры от 8 до 51оС. Смесь перемешивали при рН 9-10 в течение 3 мин, а затем подвергали нейтрализации покапельным добавлением уксусной кислоты.
Смесь экстрагировали этилацетатом (4 х 200 мл) и соединенные экстракты промывали 150 мл воды и обесцвечивали при 45оС использованием 4 г древесного угля. Желтый фильтрат концнетрировали под вакуумом при 50оС в остаточный оранжевый сироп, который обрабатывали 50 мл воды и 50 мл МТБЭ, и нагревали до 50оС. В двухфазную смесь вводили затравку, тщательно перемешивали и охлаждали до окружающей температуры, при этом сахароза-6-бензоат кристаллизовался. Твердое вещество собирали при помощи отсасывания фильтрацией, дважды промывали 50 мл МТБЭ и сушили под вакуумом при 50оС, чтобы получить 6,17 г не совсем белого твердого вещества. При помощи ЖХВД-анализа устанавливали, что оно содержит 91,5 мас. сукралоза-6-бензоата (5,65 г, 11,3 ммоль, выход 50,2% ). Маточные жидкости после кристаллизации концентрировали, чтобы получить 7,28 г сиропа, который, как было установлено при помощи ЖХВД-анализа, содержит 28,8 мас. продукта (выход 18,6%).
П р и м е р 10. Хлорирование неочищенного сиропа сахароза-6-ацетата (ДМФ) после экстрактивного удаления олова.
Этот пример является иллюстрацией прямого хлорирования рафината после экстрагирования ДАДС (в соответствии с настоящим изобретением) в качестве промежуточной стадии при получении сукралозы.
В 1000 мл четырехгорлую колбу с круглым дном, снабженную подвешенной мешалкой, термометром, входным отверстием для аргона и средством для вакуумной дистилляции, загружали 105 г сиропа сахароза-6-ацетата (39,5 г, 103 ммоль, полученного и освобожденного от ДАДС, как в примере 8) и 272 г ДМФ. Полученный в результате раствор подвергали вакуумной дистилляции (3-5 мм рт.ст.) при 35оС, чтобы удалить загрязняющие влагу и остаточные летучие материалы. Собирали всего пример 70,5 г дистиллята.
Остаток (298 г) охлаждали до примерно 0оС и обрабатывали порциями при перемешивании 121 г (943 ммоль) производимого промышленностью хлорида хлорметилен (диметиламмония) при температуре от 0 до 32оС (экзотермия). Смесь нагревали до 114оС в течение примерно 30 мин и поддерживали при этой температуре 3 ч. Реакцию охлаждали до 0оС и обрабатывали одной порцией при помощи 238 г холодного (0-5оС водного 16 мас.) раствора гидрата окиси натрия (финальное рН 9-10). Тепло реакции повышало температуру до 54оС. Смесь нейтрализовали до рН 7 при помощи концентрированной хлористоводородной кислоты, обрабатывали 14 г обесцвечивающего углерода и фильтровали через слой фильтровального вспомогательного средства.
ДМФ удаляли из смеси продуктов при помощи дистилляции паром. Водный раствор, содержащий сукралоза-6-ацетат, концентрировали при 50оС при пониженном давлении, обрабатывали 10 г обесцвечивающего углерода и фильтровали через фильтрующее вспомогательное средство. Фильтрат экстрагировали этилацетатом (2 х 500 мл, затем 1 х 300 мл). Соединенные экстракты в этилацетате промывали 150 мл воды и концентрировали в сироп (61,1 г), который самопроизвольно начинал кристаллизовываться.
Полутвердый остаток растирали с МТВЭ (100 мл). Полученное в результате твердое вещество собирали при помощи вакуумной фильтрации и сушили под вакуумом при окружающей температуре, чтобы получить 26,8 г сукралоза-6-ацетата (70,6 мас. 18,9 г, 43,0 ммоль, 41,7% выход), содержащего 15 мас. сорбированного растворителя. В маточных жидкостях дополнительно содержалось 2,2% продукта.
П р и м е р 11. Превращение сукралоза-6-бензоата после рекристаллизации в сукралозу.
В 2000 мл четырехгорлую колбу с круглым дном, снабженную подвешенной мешалкой, термометром, сушильной трубкой и пробкой, загружали 207 г (91,4%) сукралоза-6-бензоата (189 г, 378 ммоль, полученного, как это описано в примере 9) и 1000 мл метанола. Смесь нагревали, чтобы растворить твердые вещества, добавляли, 25 мл 0,84 М раствора гидроокиси калия (21 ммоль) в метаноле одной порцией и перемешивание продолжали при комнатной температуре 5 ч.
Реакционную смесь нейтрализовали при помощи добавления слабокислой ионообменной смолы (кислая форма). Раствор фильтровали, а смолу промывали метанолом (2 х 250 г). Соединенные фильтраты выпаривали до воздушной пены (245 г), которую растворяли в 1000 мл воды и экстрагировали этилацетатом (3 х 250 мл), чтобы удалить метилбензоат, непрореагировавший сукралоза-6-бензоат и другие неполярные примеси. Водный слой концентрировали до вязкого желто-коричневого раствора (487 г, 29,0 мас. сукралозы, 94% сырой выход), который обесцвечивали при помощи древесного угля. Раствор концентрировали под вакуумом до 181 г при 70оС и кристаллизацию завершали, давая возможность раствору сначала охладиться постепенно до 40оС в течение 3,5-4 ч, затем до 10оС в течение 1,5 ч. Продукт выделяли при помощи вакумной фильтрации и сушили при 45-50оС под вакуумом, чтобы получить 112 г (282 ммоль, 74,5% выход) сукралозы (температура плавления 119-120оС, разлож. [α]20о/Д + 87,1о/С, 1,23 Н2О). Бесцветный кристаллический продукт имел чистоту (ЖХВД) 99,6 мас.
П р и м е р 12. Деацетилирование сукралоза-6-ацетата.
Неочищенный кристаллический сукралоза-6-ацетат (114 г, 258 ммоль, полученный, как в примере 10), растворяли в 400 мл метанола в 1000 мл четырехгорлой колбе, снабженной подвешенной мешалкой, термометром, дефлегматором и пробкой. Раствор нагревали до 55-60оС при помощи масляной ванны и добавляли 3,5 мл (30 мас.) КОН в метаноле. ТСХ (80:17:3, СНСl3-СН3ОН-НОАС, распыленный с 5%-ным этаноловым раствором серной кислоты и обугленный) спустя 15 мин показывали, что реакция по существу закончена. Еще спустя 15 мин смесь нейтрализовали при помощи 40 г промытой метанолом слабокислой ионообменной смолы (кислотная форма).
Смолу удаляли фильтрацией и промывали двумя порциями по 100 мл метанола. Соединенные фильтрат и промывочные жидкости выпаривали до липкого сиропа, который разбавляли водой, а затем концентрировали, чтобы удалить остатки метанола. Остаток обесцвечивали углеродом и концентрировали под вакуумом при 60оС. Кристаллизацию продукта осуществляли, давая возможность перемешиваемому сиропу с затравкой постепенно охлаждаться до окружающей температуры в течение ночи. Извлеченный материал массой 80,5 г (сухая масса) состоял на 96,6% из чистой сукралозы (77,8 г, 195 ммоль, выход 75,6%). Дополнительно 21 г (52,8 ммоль, выход 15,9%) сукралозы содержалось в маточной жидкости.
Известный способ включает взаимодействие 1,3-ди(гидрокарбилокси)-1,1,3,3-тетра(гидрокарбил)дистанноксана, который далее именуется ди(гидрокарбилокси)-ди-станноксан, с сахарозой с образованием 1,3-ди(6-0-сахароза)-1,1,3,3-тетра(гидрокар- бил)дистанноксана, (ди(гидрокарбил)станноксилсахароза), который далее взаимодействует с ацилирующим агентом с образованием сложного сахароза-6-эфира. Побочным продуктом этой реакции является 1,3-диацилокси-1,1,3,3-тетра(гидрокарбил)дистанноксан или сложный диэфир дистанноксана. Эти две ракции иллюстрируются следующей общей экспериментальной процедурой, в которой используют ОДБО для того, чтобы получить ди(гидрокарбилокси)дистанноксан на месте, а бензойный ангидрид используют в качестве ацилирующего агента.
Метанол (100 мл), сахарозу (5 г) и окись дибутилолова (3,64 г, 1,00 мол. эквив. в пересчете на сахарозу) загружали в соответствующий реакционный сосуд. Содержимое реакции дефлегмировали в течение примерно 2 ч и метанол выпаривали. Продуктом этой реакции является 1,3-ди(6-0-сахароза)-1,1,3,3-тетрабутилдистанноксан. Белое твердое вещество переносили в ДМФ (100 мл) и добавляли 3,64 г бензойного ингидрида (примерно 1,10 мол. эквив. в пересчете на сахарозу). Содержимое реакционного сосуда перемешивали при комнатной температуре в течение ночи. Продуктом является сложный сахароза-6-эфир (в данном случае S-6-В), а СЭДС (в данном случае ДБДС) продуцировался в форме побочного продукта.
Реагенты сахарозу и ди(гидрокарбилокси)дистанноксан использовали в таких пропорциях, чтобы получить целевой 1,3-ди-(6-0-сахароза)-1,1,3,3-тетра(гидрокарбил) дистанноксан. В предпочтительном варианте осуществления известного способа, в соответствии с которым ди(гидрокарбилокси)дистанноксан получают на месте в результате взаимодействия окиси ди(гидрокарбил)олова (ОДГО) с низшим алканолом, таким как метанол, ОДГО и сахарозу используют в стехиометрических отношениях примерно 1: 1. Это объясняется тем, что использование избытка сахарозы приводит к загрязнению S-6-Е сахарозой и нежелательными сложными эфирами сахарозы, а использование избыточного количества ОДГО приводит к загрязнению продукта S-6-Е сложными диэфирами сахарозы. Наиболее предпочтительное стехиометрическое отношение использует ОДГО в очень небольшом (1-3%) молярном избытке (в пересчете на сахарозу) для того, чтобы гарантировать почти полное отсутствие сахарозы в продукте.
Вместо ОДБО можно использовать другие окислы ди(гидрокарбил)олова, в которых гидрокарбильными группами, связанными с оловом, могут быть независимо друг от друга алкил, циклоалкил, арил или арилалкил, например метил, этил, пропил, бутил, октил, бензил, фенэтил, фенил, нафтил, циклогексил и замещенный фенил. Предпочтительными гидрокарбильными группами являются алкильные, содержащие до восьми атомов углерода. Вместо окиси олова можно использовать диалкоголят, дигалид, диацилат, ди(гидрокарбил)олова или другие органические соединения олова при условии, что они образуют ди(гидрокарбилокси)дистанноксан на месте.
Реакцию осуществляли в органической жидкой реакционной среде, которая является растворителем для сахарозы и ди(гидрокарбилокси)дистанноксана. Если ди(гидрокарбилокси)дистаноксан образуется на месте, то реакционная среда является в предпочтительном варианте также растворителем для соединений, используемых для образования ди(гидрокарбилокси)дистанноксана. В более предпочтительном варианте реакционная среда является также одним из реагентов, которые используют для образования ди(гидрокарбилокси)дистанноксана на месте. В качестве реакционной среды можно использовать самые разнообразные алифатические и циклоалифатические спирты или фенолы. Часто наиболее эффективна с экономической точки зрения реакция между ОДГО (или эквивалентным реагентом) и спиртом или фенолом при атмосерных условиях дефлегмации. Для этой цели предпочтительны первичные спирты низших алкилов. Таким образом, предпочтительными реакционными средами являются первичные низшие алканолы, такие как метанол, этанол, н-пропанол, н-бутанол, н-пентанол и н-гексанол. Дополнительные спирты и фенолы, которые можно использовать в качестве реагента (реакционной среды), включают фенол, замещенные фенолы, такие как низший алкилзамещенные фенолы, циклогексанол и замещенные циклогексанолы, например низший алкилзамещенные циклогексанолы. Инертные органические жидкости, такие как толуол, ксилол и другие углеводороды, можно использовать в качестве разбавителей в реакции, если это необходимо.
Реакция между сахарозой и ди(гидрокарбилокси)дистанноксаном осуществляют при температуре и в течение времени, достаточных для образования ди(гидрокарбил)станноксилсахарозы. Реакционные температуры могут находиться в интервале от 50 до 100оС, в течение от 1 до 24 ч. Ди(гидрокарбил)станноксилсахарозу извлекают при помощи выпаривания реакционной среды, которое может быть осуществлено при пониженном давлении, если это необходимо. Ди(гидрокарбил)станноксилсахарозный продукт выпаривания используют непосредственно без дальнейшей очистки в реакции ацилирования.
В предпочтительном варианте используют и больше (1-5%), чем один молярный эквивалент, ацилирующего агента (в пересчете на сахарозу). Выбор конкретного ацилирующего агента, используемого в реакции ацилирования, диктуется частично использованием получаемого ацилированного продукта. Например, если ациловая группа используется в качестве блокирующей группы, как это могло быть при получении искусственного подслащивающего агента, можно использовать такой ацилирующий агент, как бензойный или уксусный ангидрид, ввиду их низкой стоимости, причем ациловая группа легко удаляется на соответствующей стадии синтеза и она стабильна относительно реакций, которым должно быть подвергнуто ацилированное соединение перед удалением ациловой группы. Если S-6-Е должен быть конечным продуктом синтеза, то используемым ацилирующим агентом является агент, который образует целевую ациловую группу для сложного эфира.
Среди ацилирующих агентов, которые могут быть использованы, имеются различные ангидриды бензойной и замещенной бензойной кислоты, например 4-нитробензойной кислоты, 3,5-динитробензойной кислоты и т.п. алкановых кислот, таких как уксусная кислота, пропионовая кислота, масляная кислота, циклогексанкарбоновая кислота, жирных кислот с длинными цепями, как насыщенных, так и ненасыщенных, таких как стеариновая кислота, олеиновая кислота, линолевая кислота и т. п. содержащие, например, до 28 атомов углерода, ненасыщенных кислот, таких как акриловая кислота и метакриловая кислота, замещенных кислот, таких как хлоруксусная кислота, цианоуксусная кислота, феноксиуксусная кислота и т. п. и насыщенных и ненасыщенных дикарбоновых кислот, таких как фталевая кислота, малеиновая кислота, глютаровая кислота и т.п.
Реакцию ацилирования осуществляют в инертном органическом реакционном носителе, например ДМФ, или другом полярном апротонном растворителе, таком как ДМСО, НМП, ДМФ, ГМФА и др. в которых сахароза растворима. ДМФ является предпочтительным полярным апротонным растворителем ввиду его низкой стоимости, относительно низкой температуры точки плавления и эффективности в качестве растворителя на последующих стадиях способа получения сукралозы. Реакцию ацилирования осуществляют при температуре и в течение времени, достаточных для получения S-6-Е продукта.
Если ангидридом является жидкость, ее можно добавлять неразбавленной в аддукт сахарозы-органоолова или ее можно разбавить инертным совместным растворителем. Если ангидрид является твердым, его можно добавлять в твердой форме или в виде раствора в соответствующем инертном растворителе. Ангидрид можно добавлять сразу весь или медленно в течение некоторого времени.
Стехиометрия ангидрида является важным аспектом успешного осуществления настоящего изобретения. Использование слишком малого количества ангидрида приводит к S-6-Е-продукту, загрязненному остаточной сахарозой. Применение слишком большого количества ангидрида вызывает загрязнение сложным диэфиром сахарозы. Наиболее предпочтительное стехиометрическое отношение использует ангидрид в небольшом (5-10%) молярном избытке (в пересчете на сахарозу) с тем, чтобы гарантировать почти полное отсутствие сахарозы в продукте.
Температуры ацилирования изменялись от 0 до примерно 30оС в течение проведенных экспериментов. Верхний предел допустимых температур ацилирования диктуется течением термически активированных нерегиоселективных реакций ацилирования, которые способствуют образованию нежелательных сложных моно- и диэфиров сахарозы. С практической точки зрения этот температурный предел является функцией химической активности ангидрида кислоты. Например, ввиду того что уксусный ангидрид является относительно химически активным материалом, реакции ацилирования с ним в общем случае осуществляют при температуре ниже примерно 20оС. Бензойный ангидрид, с другой стороны, будучи несколько менее активным допускает ацилирование при комнатной температуре или несколько более высокой.
Реакции ацилирования являются слабо экзотермическими. В зависимости от начальной реакционной температуры и скорости добавления ангидрида и аддукт ди(гидрокарбил)олова-сахарозы может потребоваться внешнее охлаждение реакции ацилирования для того, чтобы термически активированная нерегиоселективная реакция ацилирования была минимизирована.
Время, необходимое для ацилирования аддуктов сахарозы до конца, зависит от концентрации реагентов, так как ацилирование является реакцией кратного порядка, химической активности ацилирующего агента и температуры реакционной смеси. Хотя в лаборатории использовали время от часа до нескольких дней, нет каких-либо преимуществ при расширении реакционного периода сверх времени, которое необходимо для расхода ацилирующего агента. Она в общем случае завершается в течение от примерно 1 до примерно 5 ч при общих условиях.
Известный способ (патент США N 5023329) кратко описан ниже.
Этот способ осуществляют при помощи взаимодействия сахарозы с окисью ди(гидрокарбил)олова в инертном органическом носителе. ОДГО, которые можно использовать, это те ОДГО, которые уже были описаны.
ОДГО и сахароза могут быть использованы в широкой области стехиометрических отношений. Однако предпочтительны стехиометрические отношения примерно 1:1, поскольку использование избытка сахарозы приводит к загрязнению S-6-E cахарозой и нежелательными сложными эфирами сахарозы в то время, как использование избыточного ОДГО приводит к загрязнению S-6-E сложными диэфирами сахарозы. Наиболее предпочтительное стехиометрическое отношение использует ОДГО в очень небольшом молярном избытке (1-3% в пересчете на сахарозу) для того, чтобы гарантировать почти полное отсутствие сахарозы в продукте.
Способ осуществляют в инертном органическом реакционном носителе. Под термином "инертный" подразумевают, что реакционный носитель свободен от каких-либо органических функциональных групп, которые могут взаимодействовать либо с сахарозой, либо с ОДГО. Во многих случаях, для того чтобы добиться целей настоящего изобретения, инертный органический реакционный носитель может представлять собой смешанную систему растворителей, содержащую полярный апротонный растворитель и совместный растворитель. Полярный апротонный растворитель используют с целью растворения сахарозы, а совместный растворитель используют для удаления при помощи совместной дистилляции всей воды, которая образуется в результате реакции сахарозы с ОДГО, а также промотирования растворимости ОДГО. Полярные апротонные растворители, которые можно использовать, включают те, которые уже были описаны. ДМФ является предпочтительным полярным апротонным растворителем.
К совместным растворителям, способным к совместной дистилляции с целью удаления воды конденсации, относятся хлорированные углеводороды, такие как хлороформ, самые разнообразные насыщенные и ароматические углеводороды, такие как гексан, гептан, октан, циклогексан, бензол и толуол, кетоны, такие как метилэтилкетон и метилизобутилкетон, ациклические и циклические простые эфиры, такие как тетрагидрофуран, и другие инертные органические жидкости. Самые разнообразные органические жидкости пригодны для использования в качестве совместных раствоpителей и соответствии с настоящим изобретением. Совместный растворитель дает смесь с полярным апротонным растворителем, ОДГО и сахарозой, которая дефлегмирует при атмосферном давлении с внутренней реакционной температурой от примерно 75 до примерно 125оС, он участвует в совместной дистилляции воды, полученной в результате конденсации ОДГО и сахарозы, при этом облегчается удаление воды во время реакции, и он промотирует растворимость ОДГО в реакционной смеси, так как ОДГО в общем случае не растворимы в какой-либо существенной степени в полярных апротонных растворителях, и при этом увеличивается скорость реакции ОДГО с сахарозой.
Совместные растворители, которые не смешиваются с водой и которые образуют азеотропную смесь постоянного состава с минимальным кипением с водой, предпочтительны, но, как было установлено в результате экспериментов, реакционные системы, использующиеся такие совместные растворители, дефлегмируют при температурах, которые существенно выше, чем либо температура точки кипения воды-ацеотропа, либо температуры точки кипения чистого растворителя. Имеются также данные, показывающие, что композиции воды совместного растворителя дистиллятов, образующиеся из этих систем, не являются постоянными в течение периода конденсации ОДГО-сахарозы.
Предпочтительными совместными растворителями по причинам химической стабильности, эффективности при удалении воды, дешевизны и температуры точки кипения являются циклогексан, н-гептан и изооктан (2,2,4-триметилпентан).
Реакцию между сахарозой и ОДГО осуществляют при температуре от примерно 75 до примерно 125оС. При температуре ниже 75оС реакция становится малоэффективной и медленной, а при температуре выше 125оС существует тенденция к разложению углевода. Предпочтительная реакционная температура изменяется от примерно 80 до примерно 100оС, а в более предпочтительном варианте от 85 до 90оС.
Продуктом реакции сахарозы и ОДГО является ди(гидрокарбил)-станноксилсахароза. Этот продукт подвергали ацилированию с той же смесью продуктов, т. е. сложным сахароза-6-эфиром и побочным продуктом СЭДС.
Способ по патенту США N 5089608.
Первая стадия этого способа включает реакцию ОДГО с двухатомным спиртом, алканоламином или энолизируемым α-окси-кетоном и в инертном органическом носителе, таком как жидкий углеводород, с удалением воды при температуре и в течение времени, достаточных для того, чтобы получить циклический аддукт упомянутого двухатомного спирта, алканоламина или α-оксикетона. Используемым инертным органическим носителем является в предпочтительном варианте носитель, который способен к удалению воды в результате азеотропной дистилляции. В предпочтительном варианте используют углеводороды, имеющие точку кипения от примерно 80 до 145оС. Специальными иллюстрирующими примерами таких инертных органических носителей являются циклогексан, бензол, толуол, любой из ксилолов или их смеси.
Окислы ди(гидрокарбил)олова были уже описаны. ОДГО взаимодействует с двухатомным спиртом, алканоламином или α-окси-кетоном. Специальными иллюстрирующими примерами двухатомных спиртов являются алкандиолы, такие как этилен гликоль, 2,3-пропандиол, 2,3-бутандиол, 1,3-бутандиол, 1,4-бутандиол, 1,3-пропандиол, 1,2-пентандиол, 1,2-гександиол и другие алкандиолы, которые содержат, например, до примерно 8 атомов углерода, и циклоалкан диолы, такие как 1,2-циклогександиол, 1,2-циклопентандиол и т.п. В предпочтительном варианте гидроксильных групп на двухатомном спирте не более четырех атомов углерода удалены друг от друга на углеродной цепи, с которой они связаны. Алканоламинами могут быть этаноламин, 2-амино-1-пропанол и 1-амино-2-пропанол. В предпочтительном варианте амино- и гидроксильных групп на алканоламине не более 4 атомов углерода удалены друг от друга на углеродной цепи, с которой они связаны. Специальными иллюстрирующими примерами α-оксикетонов, которые способы к энолизации в двухатомные спирты с одной двойной связью в цепи, являются бензоин (2-окси-2-фенилацетофенон) и ацетоин (3-окси-2-бутанон). Предпочтительными соединениями для использования при взаимодействии с ОДГО являются алкандиолы, в частности этилен гликоль, так как он дает исключительно высокие выходы и является дешевым материалом.
ОДГО, которые в общем случае не растворимы в инертном органическом реакционном носителе, может быть суспендирован в этот носитель. Затем добавляют используемые диол, алканоламин или α-оксикетон (в небольшом стехиометрическом избытке) для образования аддукта, и смесь нагревают до дефлегмации, которая в общем случае изменяется от примерно 80 до примерно 145оС. Воду удаляли азеотропно, так как она образуется в результате конденсации между окисью ди(гидрокарбил)олова и диолом, алканоламином или α-оксикетоном, чтобы получить гомогенные бесцветные растворы циклических аддуктов. Время реакции от 2 до примерно 4 ч является наиболее общим для этой стадии.
Эти промежуточные соединения затем могут быть изолированы при помощи концентрации и кристаллизации. Более удобно выпаривать растворитель, чтобы получить твердый или полутвердый аддукт ди(гидрокарбил) олова, который затем диспергируют в ДМФ или другом растворителе. В последнем сахароза имеет соответствующую степень растворимости, который используют в качестве реакционной среды для стадии б способа настоящего изобретения. К таким растворителям относятся ДМФ, ДМСО, ДМА и т.п. и другие полярные апротонные растворители, в которых сахароза растворима.
На стадии (б) сахарозу добавляют в реакционную смесь, которая содержит аддукт со стадии а и инертный органический реакционный носитель, такой как ДМФ. Полученную в результате суспензию перемешивают при окружающей температуре в течение времени, достаточного для образования промежуточного соединения 6-0-[дигидрокарбил(окси-, или амино-, или оксигидрокарбил)-станноксил] сахарозы, которое изменяется от примерно 12 до примерно 24 ч. В качестве альтернативы применяют нагревание для того, чтобы ускорить растворение сахарозы (например, до примерно 85оС), при этом время реакции сокращается до примерно 60 мин.
На стадии б мутные смеси, которые содержат промежуточное соединение, химически активную 6-0-[дигидрокарбил(окси-, или амино-, или оксогидрокарбил)станноксил] сахарозу и продукт стадии б, затем обрабатывают 2 мол. экв. ацилирующего агента, такого как ангидрид карбоновой кислоты, при окружающей температуре. Эти смеси перемешивают и анализируют при помощи ТСХ до тех пор, пока не убеждаются, что ацилирование закончено (в общем случае от примерно 2 до примерно 7 ч). Эти мутные смеси в общем случае становятся кристально прозрачными в течение этой фазы способа.
Смеси быстро охлаждают при помощи добавления воды или метанола, фильтруют, если необходимо уделить какие-либо посторонние твердые материалы, экстрагируют, если желательно удалять побочные продукты ди(гидрокарбил)олова, концентрируют в остаточное масло или смолу при слабом нагревании при пониженном давлении, а затем продолжают обработку и анализируют, если это необходимо (функция ациловой группы) перед дальнейшей обработкой, такой как хлорирование, если необходимо использовать S-6-E при получении сукралозы.

Сущность изобретения: экстрагирование 1,3-диацилокси-1,1,3,3-тетра (гидрокарбил) дистанноксана из смеси, содержащей 1,3-диацилокси-1,1,3,3-тетра (гидрокарбил) дистанноксана, сложный сахароза-6-эфир и полярный апротонный растворитель, который содержит стадии: а) контактирование указанной смеси в присутствии небольшого количества воды с органическим растворителем, который является существенно несмешивающимся с водой, чтобы образовать при этом смесь для экстрагирования, в которой количество используемой воды достаточно для того, чтобы вызвать эффективное распределение 1,3-диацилокси-1,1,3,3-тетра (гидрокарбил) дистанноксана из первой фазы, содержащей полярный апротонный растворитель, во вторую фазу, содержащую органический растворитель; б) перемешивание смеси для экстрагирования в течение периода времени и при температуре, достаточных для того, чтобы получить двухфазную смесь, в которой преобладающее количество 1,3-диацилокси-1,1,3,3-тетра (гидрокарбил) дистанноксана в смеси экстрагирования содержится во второй фазе, по существу весь сложный сахароза-6-эфир в смеси для эктрагирования содержится в первой фазе; в) отделение первой фазы от второй фазы. 5 з.п. ф-лы, 3 табл.
| Tetrahedron, т | |||
| Механический грохот | 1922 |
|
SU41A1 |
| J.Chem.soc.Dalton Trans, 197, 1986. | |||
