Эта заявка является частичным продолжением одновременно рассматриваемой заявки сер. N 08/323954, поданной 17 октября 1994 г.
Данное изобретение относится к способу получения сукралозы без промежуточного выделения сукралоза-6-эфира.
Синтетический подслащивающий агент 4,1',6'-трихлор-4,1',6'-тридеоксигалактосахарозу ("сукралозу") получают из сахарозы замещением гидроксилов в 4, 1', и 6' положениях хлором. В процессе получения соединения происходит обращение стереоконфигурации в 4 положении. Поэтому сукралоза представляет собой галакто-сахарозу, имеющую следующую молекулярную структуру: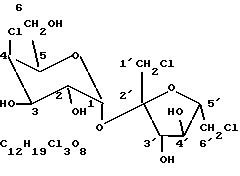
Основной проблемой синтеза является направление атомов хлора только в требуемые положения, потому что гидроксилы, которые замещаются, имеют различную реакционноспособность, два являются первичными и один - вторичным. Кроме того, синтез осложнен тем фактом, что первичный гидроксил в 6 положении не замещен в конечном продукте.
Известен целый ряд различных способов получения сукралозы, в которых реакционноспособный гидроксил в 6 положении сначала, до хлорирования гидроксилов в 4, 1', и 6' положениях, блокируют, например, в виде сложноэфирной группы, а затем гидролируют, чтобы удалить эфирный заместитель, получая сукралозу. Некоторые из таких путей синтеза включают медиированные синтезы сахароза-6-эфиров. Примерами таких медиированных оловом синтезов являются синтезы, раскрытые Navia (патент США N 4950746), Neiditch et.al. (патент США N 5023329), Walkup et al. (патент США N 5089608 - Walkup et al. - I), Vernon et al. (патент США N 5034551), и Sankey et al., заявка на патент США сер. N 08/237947, поданная 2 мая 1994, и относящиеся к решению того же самого вопроса, что и эта заявка.
Сахароза-6-эфиры, получаемые по способом, представленным в названной заявке, обычно хлорируют по способу патента США N 4980463 (Walkup et al. -II). Данный способ хлорирования дает в виде продукта сукралоза-6-эфир, такой как 4,1', 6'-трихлор-4,1',6'-тридезоксигалактосахароза-6-ацетат ("TGS-6-Ac" ("ТГС-6-Ац"), в том числе когда сукралоза-6-эфир представляет собой уксуснокислый эфир, или, в более общем виде, "TCC-6-ester") "ТГС-6-эфир" в растворе третичного амида обычно N,N-диметилформамида (ДМФ), плюс соли (получаемые в результате нейтрализации хлорирующего агента после завершения реакции хлорирования), и примеси. Одним из аспектов данного изобретения является способ выделения ТГС-6-эфира из раствора третичного амида, который является продуктом способа хлорирования по Walkup et al. - II.
В ранее известных способах, таких как способы, раскрытые Walkup et al. - II, и способе, раскрытом Navia et al., "RECOVERY OF SUCRALOSE INTERMEDIATES", заявка на патент США сер. N 08/198744, поданной 18 февраля 1994 - (и повторно поданной 1 февраля 1995, как заявка на патент США сер. N 08/368466), и относящиеся к тому же самому вопросу, что и эта заявка, сукралозу получают из реакционной смеси хлорирования по Walkup et al. - II, по следующей методике:
a) после стадии нейтрализации реакционную среду - третичный амид для реакции хлорирования удаляют, например, путем дистилляции паром (раскрыто Navia et al.), в результате чего образуется водная смесь, содержащая соли, ТГС-6-эфир и примеси,
b) затем ТГС-6-эфир выделяют из водной смеси экстракцией, используя подходящий органический растворитель, такой как этилацетат,
c) затем неочищенный ТГС-6-эфир деацилируют с образованием сукралозы, и
d) сукралозу выделяют путем противоточной экстракции и очищают путем кристаллизации.
Настоящее изобретение обеспечивает способ, при котором ТГС-6-эфир деацилируют прямо, получая водный раствор сукралозы плюс соли и примеси, из которого выделяют сукралозу, например, путем экстракции органическим растворителем и, предпочтительно, сукралозу затем очищают с помощью противоточной экстракции, кристаллизации или путем комбинации обеих методик.
Способ этого изобретения имеет несколько потенциальных экономических преимуществ по сравнению со способами получения сукралозы, описанными ранее, например, способом, раскрытым Navia et al., сер. N 08/368466. К этим преимуществам относятся следующие:
1. Уменьшение стадией манипулирования с твердыми веществами на протяжении всего процесса, в котором ТГС-6-Ац отдельно не выделяют. Это эффективно снижает необходимость в оборудовании (например, меньше потребности в центрифугах для отделения твердых веществ от жидкостей).
2. Вероятное уменьшение в необходимости повторного сбора маточных растворов (т. е. меньше стадии перекристаллизаций). Это вероятно потому, что кристаллизация сукралозы происходит легче, чем кристаллизация сукралоза-6-ацетата, и
3. Суммарные выходы кажутся незначительно увеличенными. Это может быть обусловлено конверсией диацетатов, которые имеют правильное замещение при хлорировании сукралозы. Эти диацетаты должны были бы потеряться в способе Navia et al. из-за жесткой очистки ТГС-6-Ац по Navia et al.
Данное изобретение обеспечивает способ получения сукралозы из смеси сырья (а) 6-O-ацил-4,1',6'-трихлор-4,1',6'-тридеоаксигалактосахарозы ("ТГС-6-эфир"), (b) соли, включающей хлорид щелочного металла или щелочноземельного металла, (c) воды и (d) других побочных продуктов хлорированной сахарозы, в реакционной среде, включающей третичный амид, и указанный способ включает:
(i) деацилирование 6-O-ацил-4,1'6'-трихлор-4,1',6'-тридеоксигалактосахарозы с получением водного раствора, содержащего сукралозу, (b) соль, включающую хлорид щелочного металла или щелочноземельного металла, и (d) другие побочные продукты хлорированной сахарозы, и
(ii) выделение сукралозы из продукта стадии (i) путем экстракции с последующей кристаллизацией или с помощью только методик экстракции.
Детальное описание изобретения
В одном аспекте изобретение обеспечивает способ получения сукралозы из смеси сырья (a) 6-O-ацил-4,1',6'-трихлор-4,1',6'-тридеоксигалактосахарозы, (b) соли, включающей хлорид щелочного металла или щелочноземельного металла, (c) воды, и (d) других побочных продуктов хлорирования сахарозы, в реакционной среде, включающей третичный амид, и указанный способ включает:
(i) деацилирование 6-O-ацил-4,1', 6'-трихлор-4,1',6'-тридеоксигалактосахарозы путем повышения pH водного раствора (a), (b), (c) и (d) до около 11 (±1) при температуре и в течение периода времени, достаточных для осуществления указанного деацилирования, чтобы получить водный раствор, содержащий сукралозу, соль, включающую хлорид щелочного металла или щелочноземельного металла, и другие побочные продукты хлорирования сахарозы, в реакционной среде, содержащей третичный амид,
(ii) удаление указанного третичного амида, например, дистилляцией паром или экстракцией, и
(iii) выделение сукралозы из продукта стадии (ii), например, экстракцией с последующей кристаллизацией или с помощью только методик экстракции.
В другом аспекте процесс изобретения проводят удалением третичного амида перед деацилированием, например, с помощью следующих стадий:
(i) удаление указанного третичного амида, например, дистилляцией паром, чтобы получить водный раствор (a), (b) и (d), из которого основная часть третичного амида в указанной смеси сырья удалена,
(ii) деацилирование 6-O-ацил-4,1',6'-трихлор-4,1',6'-тридеоксигалактосахарозы, например, путем повышения pH водного раствора продукта стадии (i) до pH около 11 (±1) при температуре и в течение периода времени, достаточных для осуществления указанного деацилирования, чтобы получить водный раствор, содержащий сукралозу, соль, включающую хлорид щелочного металла или щелочноземельного металла, и другие побочные продукты хлорированной сахарозы, и
(iii) выделение сукралозы из продукта стадии (ii), например, экстракцией с последующей кристаллизацией, или с помощью только методик экстракции.
В способе данного изобретения применяют в качестве сырьевой смеси композицию, содержащую 6-O-ацил-4,1',6'-трихлор-4,1',6'-тридеоксигалактосахарозу в реакционной среде третичного амида (предпочтительно ДМФ), такую как нейтрализованный (погашенный) продукт реакции хлорирования, описанный Walkup et al. -II, упомянутый выше. Предпочтительными эфирами 6-O-ацил-4,1',6'-трихлор-4,1', 6'-тридеоксигалактосахарозы являются 6-O-ацетил-4,1',6'-трихлор-4,1',6'-тридеоксигалактосахароза и 6-O-бензоил-4,1',6'-трихлор-4,1',6'-тридезоксигалактосахароза.
В лабораторном объеме неочищенный продукт хлорирования может быть погашен в периодическом процессе путем добавления (одной порцией) одного молярного эквивалента (в расчете на фосген) охлажденных льдом водных растворов или суспензией гидроксидов щелочного или щелочноземельного металла, согласно методикам Walkup et al. -II. К предпочтительным щелочным агентам относятся гидроксиды натрия, калия или кальция. Предпочтительны более разбавленные водные щелочные растворы, такие, как например, 3-4N гидроксид натрия. Можно использовать более широкие пределы концентраций (такие как, например, 2-8N гидроксид натрия). При более низких концентрациях осаждение солей уменьшается или предотвращается, что существенно уменьшает количество твердых веществ, которые должны входить в состав потока способа. Однако когда концентрация становится слишком низкой (например, ниже приблизительно 2N), поток продукта становится разбавленным до такой степени, что это может оказывать вредное воздействие на эффективность способа.
В предпочтительном способе проведения такого способа гашения холодный водный раствор щелочи добавляют при энергичном перемешивании настолько быстро, насколько это возможно, в количестве, достаточном, чтобы повысить pH до 8-10. После перемешивания в течение нескольких минут при этом умеренно повышенном pH, охлажденный раствор нейтрализуют до pH 5-7 путем добавления кислоты, такой, как, например, концентрированная водная хлористоводородная кислота или ледяная уксусная кислота. Быстрая обработка погашенной реакционной смеси хлорирования при pH 8-10 оказывает благотворное действие, заключающееся в том, что все гидроксильные группы сахароза-6-эфира, которые не были замещены атомами хлора, возвращаются в свою исходную форму в виде гидроксильной группы.
Альтернативно можно добавить достаточное количество водной щелочи, чтобы достичь pH 11 (±1), и поддерживать в течение времени, достаточного для удаления 6-ацильной функции и получения непосредственно сукралозы, в присутствии всех солей, остаточного третичного амида (ДМФ) и т.д. Это осуществляется с потерей некоторого количества ДМФ, который теряют при щелочном гидролизе до диметиламина и формиата натрия. По этой причине, и это объясняется более детально ниже, деацилирование до удаления ДМФ менее предпочтительно, поскольку желательно выделить весь ДМФ для рециркуляции и повторного использования.
Периодический способ гашения неочищенной смеси хлорированного продукта обладает недостатком, связанным с ограничением объема, обусловленным неэффективностью переноса тепла и массы. Улучшенный способ, известный как "dual-stream" ("двойной-поток") способ или способ "concurrent addition" ("одновременное добавление") включает смешение потоков водной щелочи и охлажденного (до около комнатной температуры) неочищенного хлорированного продукта вместе при тщательно измеряемых скоростях при энергичном перемешивании в условиях контролирования pH и температуры. Основные преимущества dual-stream способа гашения заключается в том, что он обеспечивает полный контроль pH, температуры и скорости смешения в ходе резкого охлаждения. Поэтому побочные реакции, приводящие к потере продукта, минимальны. Еще одно преимущество "dual-stream" ("двойной-поток") способа гашения заключается в том, что его можно проводить непрерывно путем использования сосуда для гашения, снабженного либо отверстием для выхода на дне, либо насосом. Осуществляя "dual-stream" ("двойной-поток") способ гашения в непрерывном режиме работы, относительно большое количество неочищенного хлорированного продукта можно подвергнуть обработке, используя сосуд для гашения небольшого размера. Этот непрерывный процесс представляет собой грубое приближение in-Jine способа смешения, который может быть применен для гашения в коммерческом процессе.
Применяя 1500 мл сосуд с кожухом для охлаждения, установлено, что смеси неочищенного сукралоза-6-эфирного продукта можно эффективно гасить, используя постоянную скорость подачи хлорированной смеси около 10 мл в минуту, температуру смеси закаливания около 15oC (температура охлаждающего агента 5oC), четырехлопастную пропеллерного типа мешалку 24 при скорости перемешивания, достаточной для того, чтобы обеспечить хорошее перемешивание, и установку для контроля pH для pH 8,5 на pH контролируемом насосе. Эти результаты получены с 3N или 4N NaOH в качестве щелочного агента и стартовой загрузке около 100 мл смеси от 3:1 до 1:3 ДМФ-H2O в сосуде для гашения (для того, чтобы иметь достаточный объем раствора для точного измерения pH на начальных стадиях гашения).
Удаление ДМФ
При использовании гидроксида натрия на стадии гашения и ДМФ в качестве третичного амида, соли, которые образуются на стадии гашения, включают хлорид натрия, диметиламин гидрохлорид и небольшое количество формиата натрия. Если гашение должно сопровождаться деацилированием путем повышения pH, достаточном для эффективного деацилирования, то экстрация сукралозы из погашенной и таким образом деацилированной смеси продукта должна быть осложнена присутствием ДМФ (или другого третичного амида) и склонностью сукралозы распределяться как между органической, так и водной фазами на стадии экстракции, которая должны была бы быть логически следующей стадией в цепи последовательных стадий способа для получения сукралозы. Третичный амид может растворять сукралозу в обеих фазах и также может иметь тенденцию растворять другие вещества, присутствующие в обеих фазах, из-за чего выделение сукралозы с хорошим выходом становится трудным и/или дорогостоящим. Кроме того, присутствие ДМФ или другого третичного амида является помехой для эффективной очистки сукралозы путем кристаллизации из растворителя для экстракции. Еще одно возможное осложнение может быть вызвано разложением третичного амида, катализируемого основанием. По всем этим причинам третичный амид, такой как ДМФ, следует удалять до выделения и очистки сукралозы. Кроме того, предпочтительно удалить ДМФ до стадии деацилирования.
Операцию отгонки с помощью водяного пара осуществляют таким образом, чтобы удалить основную часть ДМФ (или другого третичного амида) из погашенной смеси сырья (предпочтительный способ) или из погашенной и деацилированной реакционной смеси. Желательно удалить, по крайней мере, 95%, и предпочтительно, от около 98 до 99,9% ДМФ, для того чтобы предотвратить нежелаемые последствия, описанные в предыдущем абзаце.
При удалении ДМФ (или другого третичного амида) путем отгонки с помощью водяного пара, ДМФ эффективно замещается водой в потоке способа, и ДМФ может затем быть выделен из водных верхних погонов путем дистилляции и затем рециркулирован.
Примером лабораторного устройства для дистилляции паром типа насадочной колонки с ниспадающей пленкой, предназначенного для отгонки с помощью водяного пара ДМФ из закаленных продуктов хлорирования сукралозы-6-эфира, может служить дистилляционная колонка с вакуумным кожухом диаметром 5 см и длиной 90 см, снабженная 5 мм кольцами Рашега или другой приемлемой насадкой. Альтернативно можно использовать Oldershaw колонку с 15 тарелками, снабженную кожухом. Погашенный продукт, обычно предварительно нагретый, вводят в верхнюю часть колонки со скоростью около 5,0-5,5 г в минуту. Пар подают в колонку через боковое ответвление, расположенное в нижней части колонки. Поскольку требуется пар, свободный от конденсата, пар проходит через "пребойлер", который улавливает любой конденсат, поступающий сверху. В лаборатории этот пребойлер обычно представляет собой небольшую колбу с большим числом трубок, снабженную обогревающим кожухом. Скорости подачи водяного пара находятся в диапазоне 39-47 г в минуту (рассчитанные путем суммирования весов продуктов, отбираемых сверху и снизу колонны, и затем вычитания веса хлорированного сырья), что соответствует отношению водяной пар:сырье в пределах от 4: 1 до 12:1, причем отношения между 7,5:1 и 9:1 являются обычными для установки колонки с насадкой. В предпочтительном варианте воплощения изобретения следует использовать большее количество тарелок с более низким отношением пар:сырье, например, 15 тарелок с отношением пар: сырье около 4: 1.
Предварительное нагревание погашенного хлорированного сырья до его введения в верхнюю часть колонки проводят для того, чтобы повысить эффективность операции отгонки с водяным паром. Предварительное нагревание обычно проводят в лаборатории путем пропускания сырья через заключенный в кожух стеклянный змеевик, нагреваемый вторичным источником пара. Сырье обычно нагревают до около 90-95oC. Эффективность удаления ДМФ можно также повысить путем использования "ребойлера" (т. е. нагревания остатков от разгонки продукта так, чтобы возвратить их обратно в колонку отгонки с водяным паром).
Температуру преимущественно измеряют в двух местах аппарата, используя термопары. Помимо описанной выше температуры погашенного хлорированного сырья, также определяют температуру паров, проходящих через верхнюю часть дистилляционной колонны. Температура верхней части колонны обычно находится в пределах от около 99oC до около 104oC.
Обычно погашенный продукт хлорирования сахароза-6-ацетата содержит около 1,5-5 вес.% сукралоза-6-эфира, около 35-45 вес.% ДМФ, около 35-45 вес.% воды и около 12-18 вес.% солей. После прохождения такого продукта через аппарат отгонки с водяным паром лабораторного типа, продукты, отбираемые с нижней части колонны, обычно состоят из приблизительно 1-3 вес.% сукралоза-6-эфира, около 0,1-0,5 вес. % ДМФ, около 80-90 вес.% воды и около 8-12 вес.% солей (определенных как NaCl, исходя из анализа на натрий и хлорид).
В обычных лабораторных условиях, при которых время нахождения в колонне 7-10 минут, разложения сукралоза-6-ацетата не обнаруживается при условии, что pH погашенного хлорированного сырья является нейтральным или слабокислым (pH 5,0 - 7,0).
Аналогичные условия можно использовать для отгонки с водяным паром ДМФ из погашенной и деацилированной реакционной смеси.
Деацилирование сукралоза-6-эфира
В предпочтительном варианте способа изобретения после удаления третичного амида сукралоза-6-эфир деацилируют путем повышения pH реакционной смеси до около 11 (±1) при температуре и в течение периода времени, достаточных для осуществления деацилирования. Эту стадию обычно проводят путем добавления достаточного количества гидроксида щелочного металла, такого как гидроксид натрия, при перемешивании, до повышения pH до требуемого уровня. Установлено, что время реакции в интервале от около 30 минут до 2 часов и температура в диапазоне 15-35oC являются полезными. По окончании деацилирования присутствующее основание обычно должно быть нейтрализовано, например, путем добавления хлористоводородной кислоты до pH около 5-7. После нейтрализации водная реакционная смесь содержит сукралозу, соли (как например, вышеупомянутые, плюс соль, получаемую в результате описанной только что стадии нейтрализации) и другие побочные продукты хлорированной сахарозы.
Экстракция сукралозы
После деацилирования сукралоза может быть выделена путем экстракции водного соляного раствора целым рядом органических растворителей. К таким растворителям относятся метилацетат, этилацетат, метилэтилкетон, метил-изобутилкетон, метил-изоамилкетон, метиленхлорид, хлороформ, диэтиловый эфир, метилтретбутиловый эфир, и т.п. По причине селективности экстракции, легкости рецикла и токсикологической безопасности, предпочтительным растворителем является этилацетат.
В лаборатории выделение сукралозы обычно проводят сначала путем частичного выпаривания неочищенного нейтрализованного продукта реакции деацилирования. Около половины присутствующей воды можно необязательно удалить, получая раствор, содержащий около 2-5 мас.% углеродов и около 15-25 мас.% солей. Выделение обычно проводят путем выполнения трех последовательных экстракций этилацетатом или другим соответствующим растворителем. Экстракты объединяют, и они необязательно могут быть промыты водой (чтобы частично удалить остаточный ДМФ и производные дихлородидезоксисахарозы, которые в некоторой степени распределены в органической фазе).
Помимо методики ступенчатой экстракции, описанной выше в общих чертах, экстракцию можно также выполнять непрерывно в разбавленном (не концентрированном с помощью испарения) потоке в системе экстракции с противоточным смесителем-отстойником. Преимущество заключается в том, что не требуется предварительной стадии испарение-концентрирование. Методика такой противоточной экстракции известна в данной области техники.
В том случае, когда неочищенную сукралозу выделяют из водного соляного раствора в виде раствора в соответствующем органическом растворителе, ее раствор концентрируют и продукт может быть очищен путем кристаллизации и перекристаллизации из того же самого растворителя до требуемой чистоты. Альтернативно, чтобы достичь требуемого уровня чистоты, сукралозу можно перекристаллизовать из смешанного растворителя, такого как металон-этилацетат, или из воды. Последовательное распределение сукралозы в смесях растворитель-вода в противоточном способе также позволяет достичь очистки и кроме того открывает возможность способа прямой жидкой загрузки (т.е. не требуется никакого выделения вещества, причем финальный поток способа, имеющий требуемые технические условия, может быть непосредственно упакован для использования).
Другим заслуживающим внимания аспектом описанного выше способа очистка/выделение (т. е. экстракция с последующей кристаллизацией) является то, что для экстракции и для стадии очистки можно использовать тот же самый растворитель. Обычно (т.е. с другими химическими веществами) редко бывает, чтобы химический продукт, подлежащий очистке, перекристаллизовывали бы из того же растворителя, который используют для его экстракции. В данном случае, однако, комбинация разбавления и относительно низкие уровни примесей позволяют сукралозе оставаться в растворе во время экстракции, и затем раствор, содержащий экстрагированную сукралозу, концентрируют, и сукралозный продукт может быть кристаллизован из того же самого растворителя.
Эксперимент
Хлорирование сазароза-6-ацетата
Раствор неочищенного сахароза-6-ацетата в ДМФ (1,447 кг), содержащий 416,94 г (1,084 моля) сахароза-6-ацетата, разбавляют 2,51 кг свежего ДМФ. Раствор охлаждают до -2oC (баня со смесью сухой лед/ацетон/вода) и энергично перемешивают, в то время как добавляют фосген (1,125 кг, 99%, 11,26 молей) со скоростью от 5,4 до 6,7 г/мин. Температуру смеси поддерживают при 5-12oC во время большей части добавления.
Реакционную смесь оставляют при перемешивании при температуре окружающей среды в течение 30 минут, затем нагревают до 115oC на протяжении периода 2-3 часов, затем выдерживают при 115±1oC в течение 1,75 часов, затем охлаждают до 35oC на протяжении 30 минут. Конечную массу, 4,34 кг, подают на стадию щелочного гашения в двойном потоке и дальнейшей обработки.
Гашение в двойном потоке
Хлорированную смесь (обычно около 3,5-4,5 кг) перекачивают FMI Lab насосом (модель FR-G20) со скоростью 10 мл/мин в снабженный кожухом 2 л пластмассовый раствор (без верхней части) с запорным краном на дне, содержащий 200 мл 1:1 смеси ДМФ-вода. Водный NaOH (3,12%). 5 кг, вводят в то же время с помощью pH-контролируемого насоса с pH, установленным в точке 9,0, и ходом поршня насоса, установленным на 25%. Пропорциональная ширина полосы была установлена при максимальном отклонении (±1 pH единицы), чтобы свести к минимуму любое отклонение pH. Температуру кожуха охлаждаемого сосуда контролировали с помощью Forma Sciеntific циркуляционной ванны. Температуру кожуха поддерживали при 5oC. Температура смеси, подвергаемой гашению, составляла первоначально 6oC и затем поднималась до 20oC в течение первых 10 минут. После этого температуру стабилизировали при около 17oC на протяжении всего периода гашения. Во время гашения pH в реакторе колебалось в интервале 8,0-8,5. Смесь энергично перемешивают с помощью лабораторной мешалки, предназначенной для работы в тяжелых условиях. Закаленную смесь удаляют из реактора частями, по мере того как сосуд для гашения достигает своей мощности. Каждую порцию гасят в течение приблизительно 6 часов при вышеуказанных условиях. Для большинства порций получают приблизительно 9 кг погашенной смеси. 4,1', 6'-трихлор-4,1', 6'-тридеоксигелактосахароза-6-ацетат ["TGS-6-Ac", "ТГС-6-Ац") присутствует в смеси в количестве 2% вес. Условия и параметры оптимизируют, чтобы достичь менее чем 2 мол.% деацилирования во время гашения.
Все погашенные порции фильтруют под вакуумом для удаления нерастворимых макрочастиц материала, используя либо вакуумную фильтрацию через пористую стеклянную воронку Бюхнера, либо центрифугирование. Фильтрат отбирают на анализ и проводят отгонку с водяным паром.
Отгонка с водяным паром.
1. Лабораторный масштаб.
Погашенные смеси отгоняют с водяным паром порциями. Преследуют две цели при отгонке с водяным паром: 1) удаление ДМФ до легкого экстрагирования, 2) удаление смолистого, полимерного растворимого вещества, обнаруживаемого в погашенных смесях. Отгонку с водяным паром осуществляют в хорошо изолированной стеклянной колонке высотой 4 фута (122 см) с вн. диаметром 4 дюйма (10,16 см). Условия оптимизируют так, чтобы получить менее чем 1% ДМФ в кубе. Колонку упаковывают кольцами Рашига размером 1/4'' дюйма (0,64 см). Соотношение пар:сырье поддерживают в диапазоне 6:8. После каждых трех отгонок с водяным паром колонку очищают 1N щелочным раствором, который удаляет смолистые вещества с насадки и поверхности колонки. Обычно прохождение отгонки с паром завершается через 6 часов. На каждую порцию из 9 кг сырья получают приблизительно 13 кг куба после перегонки с водяным паром при концентрации TGS-6-Ac (ТГС-6-Ас) около 1,5% вес.
Обычная методика ведения процесса состоит в перекачивании погашенной, отфильтрованной смеси FMI Lab насосом (RP-C20) через предварительный нагреватель, представляющий собой 4 "Graham-type" конденсатор с водяным паром в кожухе, затем непосредственно в центр верхней части колонки. Водяной пар проходит через ребойлер (3-горлый, 500 мл сосуд с магнитным датчиком низкого давления и обогревающим кожухом), чтобы удалить конденсат до входа в колонку в нижней части насадки. Давление в колонке поддерживают при 0-3 дюйма (0-76,2 мм) воды на всем протяжении ведения процесса. Скорость подачи определяют первоначально путем замера времени скорости перекачивания из градуированного резервуара. Скорость сбора кубов измеряют при отборе в градуированный резервуар. Скорость дистилляции измеряют, конденсируя элюент из верхней части колонки, в мл/мин. Скорость водяного пара определяют по разнице (ПАР-ВЕРХНЯЯ ЧАСТЬ + КУБЫ - ПОДАЧА)...
2. Отгонка с водяным паром в больших масштабах.
Хлорированные и погашенные потоки способа, состава, аналогичного предшествующим примерам лабораторного масштаба, подают сверху в верхнюю тарелку колонны диаметром 10 дюймов (25,4 см), содержащей 20 сетчатых тарелок, в то время как пар подают непосредственно снизу колонны. Устанавливают соотношение пар/сырье, приблизительно равное 3, для достижения требуемого удаления ДМФ из потока отстоя (удаление > 99,2%, в расчете на определенное анализом количество ДМФ в сырье, загружаемом в колонну). Предварительный нагрев потока сырья до 80-90oC оказывает благотворное влияние на эффективность отгонки с водяным паром в колонне. Поток ДМФ/вода отгоняют с верха колонны методом противоточного потока. Кубы колонны, содержащие TGS-6-Ac (ТГС-6-Ас), соли и воду, поступают в следующую зону технологической линии для очистки. Отбираемые с верха колонны погоны направляют в другую колонну для выделения ДМФ (типичный состав 12% ДМФ, 88% воды). Таким образом, погашенное сырье, содержащее 1,8% TGS-6-Ac (ТГС-6-Ас), 8,5% солей, 54,6% воды, и 30,4% ДМФ, отгоняют с водяным паром, получая кубы, содержащие 1,6% TGS-6-Ac (ТГС-6-Ас), 9,8% солей, 84,9% воды и 0,1% остаточного ДМФ (удаляется 99,6% ДМФ). Отношение массы сырья, подаваемого на отгонку с водяным паром, к массе кубов составляло около 1,22.
Деацилирование кубов после отгонки с водяным паром
Неочищенный соляной раствор TGS-6-Ac (ТГС-6-Ас) (15,4 кг), полученный после удаления ДМФ с помощью дистилляции водяным паром, по описанной выше методике, подвергают деацилированию путем повышения pH раствора до 11,5. Это осуществляют путем добавления 50% вес./вес. гидроксида натрия в быстро перемешиваемый раствор, используя дозирующий насос при контроле pH. К раствору добавляют щелочь в количестве, достаточном для того, чтобы поднять pH до 11,5 и поддерживать его на этом уровне в течение около 2 часов при температуре окружающей среды. После того как деацитилирование посчитают завершенным, раствор нейтрализуют концентрированной хлористоводородной кислотой.
Выделение сукралозы из деацилированной смеси
Неочищенную деацилированную смесь экстрагируют непрерывно этилацетатом, используя ROBATEL противоточный экстрактор. Водную фазу (сырье) и этилацетат (экстрагирующий растворитель) оставляют в ROBATEL с помощью двух MASTER-FLEX DIGISTAT пульсирующих насосов в соотношении 4:1 (экстраген:сырье).
Раствор этилацетата, содержащий требуемый продукт, упаривают до густого сиропа, который растворяют в воде. Этот водный раствор обрабатывают углеродом для обесцвечивания, затем снова выпаривают до густого сиропа. Сироп разбавляют свежим этилацетатом. В раствор вводят в виде затравки кристаллы сукралозы, оставляют раствор стоять и кристаллизоваться при температуре окружающей среды на протяжении нескольких дней. Сукралозу получают в виде белого кристаллического твердого вещества (24 г, 92,7% вес./вес.). Дополнительные количества получают путем повторного упаривания растворителя и повторного растворения сиропа в свежем этилацетате. В целом, 33,5 г (40,6%) сукралозы выделяют в виде твердого продукта с чистотой, аналогичной прежде выделенному продукту. Остальное количество продолжает более медленно кристаллизоваться в маточном растворе на протяжении нескольких дней. Дополнительное вещество можно получить в последующих сборах или путем рецикла маточного раствора в последующих кристаллизациях.
Выделение кристаллической сукралозы из воды.
Этилацетатный раствор, полученный, как описано ранее, концентрируют до густого темного сиропа (62 г, 95-99% сукралозы). Сироп разбавляют водой в количестве, достаточном для того, чтобы получить 20% раствор сукралозы, раствор обрабатывают 5 г углерода для обесцвечивания и фильтруют, получая раствор светло-соломенного цвета, Раствор концентрируют до около 65% содержания сукралозы, охлаждают до температуры окружающей среды, вводят в него для затравки кристаллы сукралозы и дают возможность ему кристаллизоваться при перемешивании на протяжении 5 дней. Кристаллическую суспензию слегка концентрируют при помощи вакуумной дистилляции воды до около 70-75% содержания сукралозы и продолжают кристаллизацию на протяжении еще 24 часов. Кристаллический продукт выделяют фильтрацией и сушат на воздухе, получая 20,2 г продукта.
Хлорирование с помощью ARNOLD реагента/получение сукралозы
Сахароза-6-ацетат (18,93 г) и ДМФ (172 г) загружают в 500 мл 4-горлую круглодонную колбу, снабженную мешалкой, термометром и вакуумной установкой для дистилляции. Смесь дистиллируют под вакуумом до тех пор, пока не соберут 50 мл дистиллята (54,8 г теряется). (Стадия сушки для удаления остаточной влаги).
Остаток охлаждают до 0-5oC, дистилляционный аппарат заменяют на водный конденсатор и трубку для сушки, добавляют хлорид хлорметилендиметиламмония (ARNOLD Реагент, 46,8 г, 347,34 ммоль), большими порциями, поддерживая температуру реакционной смеси ниже 30oC. Затем смесь нагревают до 65oC на протяжении 20 минут, выдерживают при этой температуре в течение 5 минут, нагревают до 110-115oC на протяжении 25 минут и выдерживают в этом диапазоне в течение 3,75 часа. Затем реакционную смесь охлаждают до 0-5oC и делают ее основной путем добавления 189 г 3-молярного водного раствора гидроксида натрия, который предварительно охлаждают до 0oC. После 45 минут деацетилирование с получением неочищенной сукралозы завершается (контроль с помощью ТСХ), и смесь нейтрализуют путем добавления 17,8 г ледяной уксусной кислоты. Анализ неочищенной смеси с помощью ВЭЖХ указывал на то, что смесь содержала 11,35 г (65% выход) сукралозы.
Отгонку с водяным паром осуществляют для того, чтобы удалить ДМФ, по методикам, описанным ранее. Результаты хлорирования по методу Arnold и хлорирования, проведенного прямым введением фосгена в растворы сахароза-6-ацетата в ДМФ, указывают на то, что можно выделить ДМФ в количестве 80-90% от теоретической величины.
Неочищенный продукт сукралозы отделяют от кубов, обедненных ДМФ путем перегонки с водяным паром, с помощью экстракции подходящим органическим растворителем, не смешивающимся с водным соляным раствором (примерами которого являются дихлорметан, хлороформ, 2-бутанон, циклогексанон, этилацетат, причем последние два являются особенно эффективными и желательными для способа и по токсикологическим причинам). Водные органические экстракты снова экстрагируют водой, чтобы перевести сукралозу в водную фазу. Водный раствор обеспечивают углеродом, концентрируют и сукралозу кристаллизуют с чистотой > 91% вес. /вес. , в расчете на углевод. Маточные растворы можно использовать для рецикла, чтобы получить дополнительное количество вещества. Альтернативно, сукралозу можно очистить путем дополнительной кристаллизации из органической экстракционной среды (особенно этилацетат), которая может облегчить отделение требуемого продукта от менее полярных вещества и окрашивающих тел.
Следующий пример демонстрирует использование аналога сахароза-6-бензоата для осуществления такого же превращения, т.е. дебензоилирование непосредственно после стадии хлорирования с выделением неочищенной сукралозы. В этом случае ДМФ не удаляют при помощи отгонки с водяным паром, а применяют другой подход - экстракционную очистку. Этот аспект изобретения (т.е. экстракционная очистка в сравнении с дистилляцией водяным паром ДМФ), во время ведения процесса, не является предпочтительным, поскольку ДМФ уменьшает выход, мешая выделению неочищенной сукралозы, например, остаточный ДМФ, который уносится при помощи экстракции, служит помехой кристаллизации сукралозы.
Хлорирование сахароза-6-бензоата
Прямое превращение в неочищенную сукралозу
В 100 мл 3-горлую круглодонную колбу, снабженную магнитной мешалкой, термометром, капельной воронкой, охлаждаемым воздухом холодильником и входом для аргона, загружают 18 мл ДМФ. Колбу охлаждают до -5oC, в нее добавляют по каплям 6,8 мл (99% чистоты, 71,7 ммоль) хлорангидрида фосфорной кислоты при такой скорости, чтобы поддерживать температуру внутри колбы ниже или равной 10oC. После завершения этого добавления, смесь охлаждают до -10oC, и в нее добавляют по каплям раствор, содержащий 5,0 г сахароза-6-бензоата (91,2% чистоты, 10,21 ммоль), растворенного в 9 мл сухого ДМФ при такой скорости, чтобы температура реакционной смеси не превышала 6oC. После завершения этого добавления, прозрачную реакционную смесь нагревают до 60oC на протяжении 20 минут. Затем нагрев повышают на протяжении периода 15 минут, чтобы достигнуть температуры реакционной смеси 85oC, и при этой температуре смесь выдерживают в течение 1 часа. Образовавшийся золотисто-желтый раствор затем нагревают до 115oC и выдерживают в течение 3 часов. Полученную красно-коричневую смесь затем охлаждают до около 50oC и в нее добавляют, одной порцией, 145 мл 4N водного раствора гидроксида натрия (580 ммоль), который предварительно охлаждают вместе с 35 г льда. Щелочную смесь перемешивают при температуре окружающей среды в течение 45 минут, после чего анализ смеси с помощью ТСХ указывает на то, что конверсия сукралозы-6-бензоата в сукралозу завершена полностью. Смесь экстрагируют дважды толуолом (по 150 мл каждый раз) для удаления неполярных примесей и затем повторно экстрагируют 100 мл аликвотой 2-бутанола. Органические экстракты сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, получая 3,42 г красноватого сиропа, который содержал 36,9% вес./вес. сукралозы (31,1% выход, для двух стадий). Водная фаза содержала дополнительное количество сукралозы, которое можно выделить с помощью повторяемой экстракции 2-бутанолом.
Получение сукралозы
Сиропный раствор (20 г) сахароза-6-ацетата (43,6 ммоль) в ДМФ разбавляют 180 мл ДМФ и дегидратируют дистилляцией 51 мл ДМФ. Раствор охлаждают до от -10 до -20oC и медленно добавляют фосген (30 мл, 420 мммоль). Смеси позволяют нагреться до 20oC, затем нагревают до 65oC в течение 15 минут, выдерживают при 65oC в течение 20 минут, нагревают до 115oC в течение 20 минут, затем выдерживают при 112-114oC в течение 3,5-4 часов. Смесь охлаждают до 0oC и гасят 130 мл 4 М NaOH, добавленной порциями для контроля экзотермы нейтрализации. Для завершения деацилирования, как определено ТСХ, требуется добавление 13,15 г NaOH (гранулы). Избыточную щелочь нейтрализуют добавлением 24 мл уксусной кислоты. В этот момент смесь может быть направлена на стадию отгонки паром.
Альтернативно ДМФ может быть в значительной степени удален экстракцией и выпариванием. Неочищенную смесь экстрагируют 5 порциями 2-бутанона. Объединенные органические фазы обеспечивают 11 г угля, затем выпаривают до сиропа (21,9 г). Сироп растворяют в 50 мл этилацетата и максимально экстрагируют водой. Водную фазу максимально экстрагируют этилацетатом. Органическую фазу выпаривают до сиропа (14,5 г), который разбавляют водой с получением 72,5 г неочищенного водного раствора, содержащего сукралозу и другие хлорированные примеси, как показано ВЭЖХ.
Данные анализа сукралозы и других хлорированных примесей, образующихся на стадии хлорирования.
Сукралоза 11,5%
4,1'-дихлоргалактосахароза 0,02%
3',6'-ангидро-4,1'-дихлоргалактосахароза 0,64%
4,6'-дихлоргалактосахароза 0,26%
1',6'-дихлорсахароза 0,07%
6,1',6'-трихлорсахароза 0,47%
сукралоза-6-ацетат 0,02%
4,1',4',6'-тетрахлоргалакто-сорбо-сахароза 0,45%
4,6,1',6'-тетрахлоргалактосахароза 0,19%
Растворители:
ДМФ 6,0%
Вода (по разнице) 80,38%
Условия высокоэффективной жидкостной хроматографии (HPLC) для анализа неочищенных реакционных смесей сукралозы.
Содержание сукралозы определяют высокоэффективной жидкостной хроматографией с использованием колонки с обратной фазой C18 (Supelcosil LC18-DB, 5 мкм насадка, 15 см х 4,6 мм, Supelco # 5-8348M) и внешнего стандарта. Обнаружение достигалось с помощью детектора испарительного светорассеяния типа Varex. Подвижная фаза представляет собой ацетонитрил/водный градиент, 10-15% ацетонитрил в течение 18,3 мин. Инжектор представляет собой Waters WISP 712, градиент HPLC системы - Waters модель 680. Время пробега - 25 минут, объем инжекции - 20 мкл. Все компоненты элюируют в течение 18 мин.
Приготовление образца: соответствующее количество образца взвешивают и разбавляют до объема 10 мл деоинизированной водой и фильтруют через 0,45 мкм шприцевой фильтр (мембрана Nylon-66 или Teflon). Для определения весового процента сукралозы используют следующие расчеты:
Время удерживания компонентов смеси см. после фрмулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЭКСТРАКЦИИ 1,3-ДИАЦИЛОКСИ-1,1,3,3-ТЕТРА(ГИДРОКАРБИЛ)ДИСТАННОКСАНА ИЗ СМЕСИ | 1991 |
|
RU2036197C1 |
| СПОСОБ ВОДНОГО ДЕАЦИЛИРОВАНИЯ, СТАБИЛИЗИРОВАННОГО БУФЕРОМ | 2003 |
|
RU2325394C2 |
| СПОСОБ ПОВЫШЕНИЯ ЧИСТОТЫ И ВЫХОДА СУКРАЛОЗЫ | 2003 |
|
RU2324407C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ СЛОЖНОГО ЭФИРА САХАРОЗЫ ПО ПОЛОЖЕНИЮ 6 | 1996 |
|
RU2159773C2 |
| Способ получения 4,1,6-трихлор-4,1,6-тридеоксигалактосахарозы | 1981 |
|
SU1431680A3 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНОГО САХАРОЗА-6-ЭФИРА | 1991 |
|
RU2049791C1 |
| Способ получения 6-бензоата или 6-ацетата 6 @ ,4,1 @ -трихлорсахарозы | 1990 |
|
SU1836377A3 |
| СПОСОБ ПОЛУЧЕНИЯ СУКРАЛОЗЫ | 2016 |
|
RU2633698C1 |
| Способ получения сукралозы | 1986 |
|
SU1635905A3 |
| ФАРМАЦЕВТИЧЕСКИЕ СУСПЕНЗИИ, НЕ СОДЕРЖАЩИЕ КРАСИТЕЛЕЙ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2395275C2 |

Описывается способ получения сукралозы, включающий деацилирование 6-эфира сукралозы, отличающийся тем, что смесь сырья (а) 6-О-ацил-4,1',6'-трихлор-4,1', 6'-тридеоксигалактосахарозы, (в) соли, включающей хлорид щелочного или щелочноземельного металла, (с) воды и (d) других побочных продуктов хлорирования сахарозы обрабатывают в реакционной среде, содержащей третичный амид, и указанная обработка включает: (i) деацилирование 6-О-ацил-4,1', 6'-трихлор-4, 1',6'-тридеоксигалактосахарозы повышением рН водного раствора (а), (b), (с) и (d) до около 11 (±1) при температуре и в течение периода времени, достаточных для осуществления указанного деацилирования с получением водного раствора, содержащего сукралозу, соль, включающую хлорид щелочного или щелочноземельного металла, и другие побочные продукты хлорирования сахарозы, в реакционной среде, содержащей третичный амид, (ii) удаление указанного третичного амида и (iii) выделение сукралозы из продукта стадии (ii). Технический результат -упрощение процесса. 2 с. и 13 з.п. ф-лы.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| SU 1301316 A3, 30.03.1987 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 4980463 A, 25.12.1990 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 4380476 A, 10.04.1983 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| US 4783526 A, 08.11.1988. | |||

