Изобретение относится к способу получения производных сахарозы, который содержит взаимодействие сахарозы непосредственно с окисью ди(гидрокарбил)олова, в результате которого получают соединение 6-0-сахароза дистанноксана, которое используют в качестве промежуточного соединения при получении, среди прочего, искусственного подслащивающего агента, сукралозы.
Молекула сахарозы содержит три основные гидроксильные группы и пять вторичных гидроксильных групп. Следовательно, когда желательно получить производные сахарозы, используя реакцию гидроксильных групп, основная проблема синтеза может заключаться в том, чтобы направить реакцию только на целевые гидроксильные группы. Например, искусственный подслащивающий агент 4,1', 6'-трихлор-4,1', 6'-тридеоксигалактоса- хароза ("сукралоза") получают из сахарозы при помощи замены гидроксилов в 4,1' и 6' позициях хлором. (В процессе получения подслащивающего агента стерео-конфигурация в 4-позиции обращается поэтому соединение становится галактосахарозой). Это соединение и способы синтеза предложены в патентах США NN 4343934, 4362869, 4380476 и 4435440. Направление атомов хлора только в определенные позиции является основной проблемой синтеза, особенно по той причине, что гидроксилы, которые замещаются, обладают другой химической активностью (два являются основными и один вторичным; кроме того, синтез усложняется благодаря тому, что основной гидроксил в 6-позиции является незамещенным в конечном продукте). Получение этого подслащивающего агента является только одной иллюстрацией синтеза производных сахарозы, в котором желательно либо модифицировать некоторые специальные гидроксильные группы и только такие гидроксильные группы, либо модифицировать только определенное число гидроксилов, причем в этом последнем случае, может быть, не принимая во внимание, какие конкретно гидроксилы подвергают модификации. Получение поверхностно-активных агентов сложного моноэфира на основе сахарозы является известным примером монозамещения на молекуле сахарозы.
В соответствии с настоящим изобретением предлагается улучшенный и более эффективный способ синтеза соединений сахарозы таких, как производные 6-замещенной сахарозы, который является высоко региоселективным, как относительно направления реакции строго в 6-позицию, так и относительно получения монозамещенных производных только. Термин "региоселективный" относится к реакции, которая благоприятна только для одного основного продукта. (См. Хасснер, "Региоспецифичность. Полезная терминология в реакциях присоединения и исключения", J. Org. Chem. 33, N 7, с.2684-6, июль 1968).
Получение сложного сахароза-6-эфира на основе дистанноксана было впервые описано у Навиа. Навиа указывает, что соответствующие материалы на основе ди(гидрокарбил)олова такие, как окись дибутилолова, окись диоктилолова, диметилат дибутилолова и т.п. могут быть соединены с соединением, содержащим гидроксильную группу, таким, как одноатомный спирт или простой фенол таким образом, чтобы получить химически активный дистанноксан в качестве промежуточного соединения, а именно 1,3-ди(гидрокарбилокси)-1,1,3,3-тетра(гид- рокарбил)дистанноксан, который затем может взаимодействовать с сахарозой, чтобы получить 1,3-ди(6-0-сахароза)-1,1,3,3-тетра(гидрокарбил)дистанноксан] Навиа также указывал, на простое получение сложных сахароза-6-эфиров при помощи обработки этих аддуктов органоолова-сахарозы подходящим ацилирующим агентом в соответствующем растворителе или смеси растворителей. Кроме того, Навиа описывает простые ацетаты и бензоаты сложных эфиров, полученные из их альдегидов, в качестве защищающих групп при производстве сукралозы, благодаря своей низкой стоимости и токсикологическим свойствам, и простое их последующее удаление. Способ, предложенный Навиа для получения сложного сахароза-6-эфира ("S-6-Е") с промежуточным дистанноксаном, таким образом, состоит из трех различных стадий, которые приведены ниже (используя окись дибутилолова и н-бутанол в качестве конкретных реагентов):
(1) Реакция окиси дибутилолова ("ОДБО") с большим стехиометрическим избытком н-бутанола при азеотропном удалении воды, чтобы получить 1,3-дибутокси-1,1,3,3-тетрабутилдистанноксан (ДБДС), который как было установлено существует в форме моногидрата;
(2) Реакция ДБДС с сахарозой в N,N-диметилформамиде ("ДМФ") с удалением воды и н-бутанола, чтобы получить 1,3-ди-(6-0-сахароза)-1,1,3,3-тетрабутилдис- танноксан, более известный под наименованием дибутилстанноксилсахарозы ("ДБСС"). Ввиду того, что реакцию ацилирования на следующей стадии необходимо осуществлять в среде, свободной от гидроксилов, с целью оптимального выхода продукта, сложного эфира сазарозы, весь н-бутанол и воду необходимо удалить в течение этой стадии и заменить ДМФ;
(3) Реакция ДБСС с небольшим стехиометрическим избытком ацилирующего агента такого, как уксусный ангидрид, чтобы получить сахароза-6-акрилат такой, как сахароза-6-ацетат ("S-6-А").
Следуя этой реакционной последовательности, S-6-А в общем случае получают с хорошим выходом с только минимальными примесями в виде остатков сахарозы, диацетов сахарозы и других моацетатов сахарозы.
Описанный выше трехстадийный способ обладает несколькими недостатками при промышленной реализации. Эти недостатки являются особенно серьезными, если промышленная реализация основана на принципе периодического действия. Первый недостаток заключается в том, что необходима замена растворителем ДМФ н-бутанола в процессе образования ДБСС. Ввиду чувствительности к температуре ДБСС в этой матрице растворителя (разложение начинается при температуре примерно 90оС), такая замена растворителя должна быть осуществлена как часть вакуумной дистилляции, требующей всегда увеличения вакуума, когда содержание н-бутанола в смеси снижается. Недостаточность при удалении н-бутанола приводит к снижению эффективности в реакции ацилирования на стадии. Кроме того, рецикл дистиллированного н-бутанола усложняется, ввиду его загрязнения ДМФ и водой. (Рецикл н-бутанола необходим по экономическим причинам).
Второй недостаток трехстадийного способа состоит в чувствительности к влаге ДБДС (и связанных с ним продуктов конденсации окиси олова со спиртами или фенолами). Даже хотя ДБДС очевидно существует в форме моногидрата, контакт с атмосферной влажностью приводит к его быстрому превращению в ОДБО и н-бутанол. Поэтому с ДБДС необходимо работать при условиях, которые строго исключают атмосферную влажность. В процессе производства условия, которые бы приводили к осаждению ОДБО на поверхностях используемого оборудования, потребовали бы дополнительной дорогостоящей стадии очистки ввиду того, что ОДБО является полимерным твердым веществом, которое совершенно нерастворимо в большинстве растворителей.
Третий недостаток заключается в рецикле конечного продукта органоолова, диацетата дистанноксана ("ДАДС"). ДАДС снова превращается в ДБДС при помощи экстрагирования с последующей обработкой либо бутилатом кальция, либо бутилатом натрия. Побочные продукты этих превращений, ацетат либо калия, либо натрия очень трудно отфильтровать. Такие трудности при фильтрации приводят к потерям ДБДС и, как можно ожидать, это влияет неблагоприятно на затраты при производстве S-6-А. Кроме того, как указывалось выше, ДБДС должен быть защищен от воздействия влаги.
При осуществлении способа настоящего изобретения можно избежать этих трех недостатков и, кроме того, предлагаемый способ является более простым, более экономически эффективным и менее опасным для здоровья при производстве сложных сахароза-6-эфиров. Этот способ является особенно пригодным для использования в соответствии с периодическим принципом осуществления. Установлено, что сахароза может непосредственно взаимодействовать с окислами ди(гидрокарбил) олова такими, как ОДБО в полярном апротонном растворителе, таком, как ДМФ в присутствии совместного растворителя, способного как промотировать растворение ОДБО, так и осуществить совместную отгонку с целью удаления всей воды, образующейся в реакции окиси олова и сахарозы, с тем, чтобы получить при этом аддукт органоолова-сахарозы. Этот аддукт, как было установлено при помощи ЯМР, является дистанноксаном со структурой, идентичной полученной по способу Навиа с промежуточным спиртом (например, ДБСС). По способу Навиа ДБСС можно легко подвергнуть ацилированию на месте, чтобы получить хорошие выходы S-6-F.
Способ, являющийся предметом настоящего изобретения, является улучшением способа Навиа с промежуточным спиртом по следующим причинам:
(а) один реагент (а именно, спирт такой, как бутанол) исключен;
(b) промежуточное соединение, чувствительное к влаге (например, ДБДС) исключено;
(с) исключена сложная замена растворителя вакуумной дистилляции, вместе с необходимостью извлекать н-бутанол (или аналогичный гидроксильный реагент) из смесей, содержащих ДМФ и воду;
(d) упрощена процедура рецикла органоолова, включающая легко фильтруемую окись ди(гидрокарбил)олова (такую, как СДБО), а не трудно фильтруемую ацетатную соль и чувствительное к влаге производное органоолова (эта процедура рецикла описана в одновременно находящейся в стадии рассмотрения патентной заявке США N INOR 9 "Способ извлечения сложных эфиров органоолова из реакционных смесей, содержащих их, и повторное использование извлеченных соединений органоолова, поданной в тот же день, что и настоящая патентная заявка, М.Н. Верноном и Р.Е. Уолкапом (Вернон и др.) и подписанной тем же заявителем, что и настоящую патентную заявку); и
(е) сложные сахароза-6-эфиры такие, как S-6-А или сахароза-6-бензоат ("S-6-B") получены с более высоким выходом и с более высокой чистотой (очевидно, результат исключения одной переходной стадии из последовательности стадий способа).
Предлагается способ, который содержит взаимодействие сахарозы с окисью ди(гидрокарбил)олова в инертном органическом носителе в течение времени и при температуре достаточных для того, чтобы получить 1,3-ди-(6-0-сахароза)-1,1,3,3-тетра-(гидрокарбил)дистанноксан. В предпочтительном варианте настоящего изобретения этот 1,3-ди-(6-0-сахароза)-1,1,3,3-тетра-(гидрокарбил)дистанноксан затем взаимодействует с ацилирующим агентом при температуре и в течение времени, достаточных для получения сложного сахароза-6-эфира.
В обзорной статье "Органоселективное использование гидроксильных групп через производные органоолова", Tetrahedron т.41, N 4, с.643-663 (1985), описывается реакция соединений олова с соединениями, содержащими гидроксильные группы, с целью получения соединений станноксила, которые затем могут быть подвергнуты алкилированию или ацилированию, чтобы получить простые эфиры или сложные эфиры.
Описана также реакция окиси бис(трибутилолова) с различными углеводами (включая сахарозу) с последующим ацилированием, чтобы получить смесь сложных эфиров различных степеней насыщения. Применение окиси дибутилолова в реакции с углеводами также описано в этой статье. Описывается получение двух производных углеводов диалкилстаннилена, производного 2,3-0-дибутилстаннилена метил 4,6-0-бензилиден- α -D-глюкопиранозида и 5,6-0-бензилиден-2,3-0-дибутилстаннилен-α D-маннопиранозида.
Вагнер и др. J. Org. Chem. т.39. с.24 (1974) предлагают получение производных дибутилстаннилена нуклеозидов при помощи взаимодействия окиси дибутилолова с нуклеозидами в дефлегмирующем метаноле. После отгонки метанола производное станнилена подвергают ацилированию при помощи реакции с равномолярными количествами хлорида кислоты и триэтиламина.
Холцапфель и др. в статье "Производные сахарозы и селективное бензоилирование вторичных гидроксильных групп 6,1',6'-три-0-тритилсахарозы", S. Afr. Tydsrk. Chem. 1984, 37 (3), стр.57-61, описывают реакцию окиси дибутилолова с 6,1',6'-три-0-тритилсахарозой, с последующей реакцией с бензоил хлоридом, чтобы получить 72% выход 3'-0-бензоил-6,1',6'-три-0-тритилсахарозы и 9% производного 2-0-бензоата, и небольшие количества производного 2,3'-дибензоата.
Основные приемы известных способов (как это представлено в приведенных выше ссылках) основаны на том, что химическая активность гидроксильной группы увеличивается при образовании связи с оловом, но в полигидроксилированных соединениях таких, как сахара, нельзя предсказать, какая гидроксильная группа будет активирована (см. стр.646-7 статьи Дэвида и др. в разделе, озаглавленном "стереоэлектронные следствия связи Sn-O нуклеофильное усиление атома кислорода".
Факт, что сахароза будет взаимодействовать непосредственно с окисью ди(гидрокарбил)олова, чтобы дать высокий выход описанного производного дистанноксана, способного к дальнейшему химическому использованию, является новым и не мог быть предсказан специалистом в этой области техники. Термин "будет взаимодействовать непосредственно" означает, что сахароза взаимодействует с окисью олова без использования каких-либо промежуточных реагентов или реакций так, как в первой реакции окиси олова со спиртом или фенолом, как это делается по способу Навиа. Эта непосредственная реакция сахарозы с окисью олова является важным отличительным свойством является то, что описанный химический материал имеет структуру, в которой атом кислорода сахароза-6-гидроксила ковалентно связан с оловом, и, следовательно, нуклеофильно усиливается. Можно бы предсказать структуру, содержащую только один атом олова, такую, как станнилен в качестве наиболее вероятного реакционного продукта. (Станнилен может быть определен как производное углевода, содержащее внутримолекулярную связывающую последовательность C-O-Sn-O-C). Такие материалы видимо были бы не способны чисто превратиться в сложный сахароза-6-эфир. Все ранее описанные превращения, включающие непосредственную реакцию углеводов с окислами ди(гидрокарбил)олова, были классифицированы, как дающие продукты станнилена, причем те продукты, которые обладают пятичленными кольцами, являются предпочтительными.
Образование станнилена было описано для случаев 6,1',6'-три-0-тритилсахарозы, различных дисахаридов нуклеозидов и самых разнообразных моносахаридов. Структура станнилена некоторых из этих материалов была подтверждена при помощи рентгеновской кристаллографии и ЯМР-спектроскопии.
Аналоги отсутствуют для непосредственной реакции углеводов с окисью ди(гидрокарбил)олова, чтобы получить 1,3-углеводзамещенный дистанноксан. Этот неожиданный результат может оказаться уникальным для молекулы сахарозы.
Способ настоящего изобретения осуществляют при помощи взаимодействия сахарозы с окисью ди(гидрокарбил)олова (ОДГО) в инертном органическом носителе. К ОДГО, которые можно использовать, относятся окислы ди(гидрокарбил)олова, в которых гидрокарбиловые группы, связанные с оловом, могут быть, независимо друг от друга, алкилом, циклоалкилом, арилом или арилалкилом такими, как, например, метил, этил, пропил, бутил, октил, бензил, фенэтил, фенил, нафтил, циклогексил и замещенный фенил. Предпочтительными гидрокарбиловыми группами являются алкил, содержащий до восьми атомов углерода. Вместо окиси олова можно использовать диалкоголят, дигалид или диацилат ди(гидрокарбил)олова. Окись дибутилолова и окись диоктилолова являются особенно предпочтительными, а окись дибутилолова является наиболее предпочтительной окисью органоолова при использовании в соответствии с настоящим изобретением.
ОДГО и сахарозу можно использовать в очень широкой области стехиометрических отношений. Однако стехиометрическое отношение примерно один к одному является предпочтительным. Это объясняется тем, что использование избытка сахарозы приводит к загрязнению S-6-Е сахарозой и нежелательными сложными эфирами сахарозы в то время, как использование избытка ОДГО вызывает загрязнение S-6-Е продукта сложными диэфирами сахарозы.
Наиболее предпочтительное стехиометрическое отношение использует ОДГО в очень небольшом (1-3% ) молярном избытке (в пересчете на сахарозу) с тем, чтобы гарантировать почти полное отсутствие сахарозы в продукте.
Способ настоящего изобретения осуществляют в инертном органическом носителе реакции. Под термином "инертный" подразумевается, что реакционный носитель не содержит никаких органических функциональных групп, которые будут взаимодействовать либо с сахарозой, либо с ОДГО. Например, следует избегать таких функциональных групп, как гидроксилы спиртов или фенолов, которые могут взаимодействовать с окисью ди(гидрокарбил)олова с тем, чтобы образовать 1,3-ди(гидрокарбилокси)-1,1,3,3-тетра(гидрокарбил)дистанноксан по способу Навиа. Во многих случаях для того, чтобы достигнуть целей настоящего изобретения, инертный органический носитель реакции можно смешать с другим растворителем, образуя систему растворителей, содержащую полярный апротонный растворитель и совместный растворитель. Полярный апротонный растворитель используют с целью растворения сахарозы, а совместный растворитель используют с целью совместной перегонки, чтобы удалить воду,образующуюся в результате реакции сахарозы с ОДГО, а также для промотирования растворимости ОДГО.
К полярным апротонным растворителям, которые можно при этом использовать, относятся ДМФ, диметил сульфоокись (ДМСО), N-метилпирролидинон (НМП), N,N-диметилацетамид (ДМА), гексаметилфосфорамид (ГМФА) и другие полярные апротонные растворители, в которых сахароза растворима. ДМФ является предпочтительным полярным апротонным растворителем, благодаря своей низкой стоимости, своей относительно низкой температуре кипения и своей пригодности в качестве растворителя для последующих стадий способа получения сукралозы.
К совместным растворителям для совместной перегонки с целью удаления воды конденсации относятся хлорированные углеводороды такие, как хлороформ, самые разнообразные насыщенные и ароматические углеводороды такие, как гексан, гептан, октан, циклогексан, бензол и толуол, кетоны такие, как метил этил кетон, и метил изобутилкетон, ациклические и циклические простые эфиры такие, как метил третичн.-бутиловый простой эфир и тетрагидрофуран, и другие инертные органические жидкости, которые удовлетворяют критериям, перечисленным здесь. Самые разнообразные органические жидкости пригодны для использования в качестве совместных растворителей в соответствии с настоящим изобретением. Основными критериями для совместных растворителей являются (1) то, что он дает смесь с полярным апротонным растворителем, ОДГО и сахарозой, которая дефлегмирует при атмсферном давлении с внутренней реакционной температурой в области от примерно 75оС до примерно 125оС (2) то, что он участвует в совместной перегонке воды, образующейся в результате конденсации ОДГО и сахарозы, при этом облегчается удаление воды во время реакции, и (3) то, что он промотирует растворимость ОДГО в реакционной смеси (так как ОДГО в общем случае не растворим в какой-либо существенной степени в полярных органических растворителях) и при этом увеличивается скорость реакции ОДГО с сахарозой.
Под термином "промотирует растворимость ОДГО" подразумевается то, что совместный растворитель, по крайней мере частично сольюбилизирует ОДГО при условиях, реализуемых по способу настоящего изобретения.
Совместный растворитель не должен обладать способностью образовывать постоянно кипящий азеотроп постоянного состава с водой, чтобы быть эффективным совместным растворителем, нет также необходимости, чтобы совместный растворитель был смешивающимся с водой. Необходимо только, чтобы совместный растворитель был способен к совместной перегонке с водой конденсации из реакционной среды.
В предпочтительном варианте используют совместные растворители, которые не смешиваются с водой и которые образуют азеотроп с минимальным кипением постоянного состава с водой, но как можно заметить после изучения примеров, помещенных ниже, реакционные системы, использующие такие совместные растворители, в общем случае, дефлегмируют при температурах существенно более высоких, чем либо температура точки кипения воды-азеотропа, либо температура точки кипения чистого растворителя. Имеются также данные, показывающие, что композиции вода-растворитель дистиллятов, возникающих из этих систем не являются постоянными в течение периода конденсации ОДГО-сахарозы.
К предпочтительным совместным растворителям по причинам химической стабильности, эффективности удаления воды, стоимости и температуры точки кипения относятся циклогексан, н-гептан и изооктан (2,2,4-триметилпентан).
Реакцию между сахарозой и ОДГО осуществляют при температуре в области от примерно 75оС до примерно 125оС. При температуре ниже 75оС реакция становится слишком медленной, а выше 125оС имеется тенденция к разложению углеводов. Предпочтительной реакционной температурой является температура в области от примерно 80оС до примерно 100оС, в более предпочтительном варианте от примерно 85оС до примерно 90оС.
Реакционные температуры в общем случае регулируют эмпирическим образом при помощи регулирования отношения полярного апротонного растворителя к совместному растворителю с более низкой температурой кипения.
Отношения растворителя к совместному растворителю не являются критическим моментом настоящего изобретения. Например, отношения растворителя к совместному растворителю (об/об) от примерно одного к одному до примерно десяти к одному реализуют при осуществлении настоящего изобретения, причем отношения от примерно восьми к пяти до примерно восьми к одному были подтверждены в лаборатории. Отношения совместного растворителя к растворителю ограничиваются чисто практическими соображениями. Слишком большое количество совместного растворителя будет снижать растворимость сахарозы и может давать смесь с температурой точки кипения, слишком низкой для разумного времени превращения. Слишком малое количество совместного растворителя может оказывать неблагоприятный эффект на скорость образования аддукта ди(гидрокарбил) олова-сахарозы из-за снижения растворимости ОДГО и ограничения скорости, с которой вода может участвовать в совместной перегонке из реакционной смеси. Кроме того, использование слишком малого количества совместного растворителя может приводить к реакционным температурам, достаточно высоким, чтобы вызвать термическое разложение углеводных материалов.
При осуществлении настоящего изобретения можно использовать широкую область отношений твердых материалов (ОДГО и сахарозы) к растворителям (полярному апротонному растворителю и совместному растворителю). Но это не является решающим фактором для настоящего изобретения при условии, что имеется достаточное количество полярного апротонного растворителя, чтобы обеспечить растворение сахарозы, и достаточное количество совместного растворителя, чтобы обеспечить удаление воды, и обеспечить необходимую реакционную температуру. В экспериментах использовали отношения твердые материалы: растворители (масса/объем) от примерно одного к двум до примерно одного к шести. Более концентрированные системы предпочтительны по причинам затрат и практической реализуемости.
Время дефлегмации, необходимое для полного образования аддукта дистанноксана-сахарозы, является функцией эффективности удаления всей воды конденсации (плюс любой воды, которая присутствует в результате использования влажных реагентов или растворителей) из системы при помощи совместной перегонки. (Один эквивалент воды продуцируется на каждый эквивалент окиси олова). Эффективность удаления воды из реакционной системы является функцией числа взаимодействующих переменных. Эти переменные, которые в значительной степени могут быть экспериментально регулируемы, включают: (а) внутреннюю реакционную температуру; (b) температуру точки кипения совместного растворителя; (с) содержание воды при совместной перегонке; (d) скорость тепловыделения в систему; (е) эффективность перемешивания; и (f) конфигурацию используемого реактора.
Твердые полимерные ОДБО содержат воду гидратации, которая составляет половину эквивалента воды на один эквивалент окиси олова. (Эту воду гидратации измеряют при помощи нескольких приемов, наиболее эффективным из которых является анализ на воду Карла Фишера ОДГО, растворенного в ледяной уксусной кислоте). Реакция конденсации между сахарозой и ОДБО высвобождает эту воду гидратации. Таким образом, вода, продуцируемая по способу настоящего изобретения, для удаления в результате совместной перегонки представляет собой сумму воды конденсации и высвобожденной воды гидратации. Стехиометрия этой процедуры, таким образом, представляет один моль воды, продуцированной для совместной перегонки, на моль окиси олова. Термин "вода конденсации" используется для обозначения общего количества воды реакции (т.е. сумма воды двух типов) в соответствии со стехиометрией один моль-один моль.
Образование аддукта сахарозы-органоолова продолжается в течение от примерно двух часов до примерно двадцати четырех часов. Период дефлегмации завершается, когда совместно перегоняется теоретическое количество воды из системы. Такое определение в общем случае осуществляют при помощи анализа на воду с использованием метода Карла Фишера. Удаление воды в общем случае составляет от примерно 101 до 110% от теории. Избыток воды является результатом случайного присутствия влаги в растворителе, совместном растворителе и сахарозе. При помощи соответствующего изменения переменных, описанных выше, во время экспериментов достигали общего необходимого времени дефлегмации от трех до пяти часов.
После завершения удаления воды, в общем случае, двухфазные реакционные смеси (но не содержащие твердых частиц) охлаждают до комнатной температуры или ниже и подвергают ацилированию. Ангидриды кислот являются предпочтительными ацилирующими агентами. Выбор конкретного ацилирующего агента, используемого здесь, диктуется частично использованием, для которого предназначен ацилированный продукт. Например, если ациловая группа предназначена для использования в качестве блокирующей группы, как это было бы при производстве искусственных подслащивающих агентов, как это было указано выше, то можно было бы использовать такой ацилирующий агент, как бензойный или уксусный ангидрид, так как он является дешевым материалом, ациловая группа легко удаляется на соответствующей стадии синтеза и она стабильна относительно реакций, которым ацилированное соединение должно подвергаться перед удалением ациловой группы. Если сложный сахароза-6-эфир является конечным продуктом синтеза, тогда используемым ацилирующим агентом является агент, который будет давать целевую ациловую группу для продукта сложного эфира. Имея в виду эти принципы, среди ацилирующих агентов, которые можно использовать, находятся различные ангидриды и галиды кислот, бензойной или замещенной бензойной кислоты (например, 4-нитробензойная кислоты, 3,5-динитро-бензойной кислоты и т.п.), алкановой кислоты такой, как уксусная кислота, пропионовая кислота, масляная кислота, циклогексанкарбоновая кислота, жирных кислот с длинными цепями, как насыщенных, так и ненасыщенных таких, как стеариновая кислота, олеиновая кислота, линолевая кислота и т.п. содержащих, например, до 28 атомов углерода, ненасыщенных кислот таких, как акриловая кислота и метакриловая кислота, замещенных кислот таких, как хлоруксусная кислота, цианоуксусная кислота, феноксиуксусная кислота и т.п. и насыщенных и ненасыщенных дикарбоновых кислот таких, как фталевая кислота, малеиновая кислота, глютаровая кислота и т.п.
Если ангидрид является жидким, то его можно добавлять неразбавленным в реакционный продукт смеси реакции конденсации сахарозы/окиси олова, или он может быть разбавленным инертным совместным растворителем. Если ангидрид является твердым, то он может быть добавлен в твердой форме или добавлен в виде раствора в соответствующем инертном растворителе. Ангидрид может быть добавлен сразу весь или он может быть добавлен медленно в течение некоторого периода времени.
Стехиометрия ангидрида является важным аспектом успешной реализации настоящего изобретения. Использование слишком малого количества ангидрида будет приводить к загрязнению S-6-E-продукта остатками сахарозы. Использование слишком большого количества ангидрида будет приводить к загрязнению сложным диэфиром сахарозы. В наиболее предпочтительном стехиометрическом отношении используют ангидрид в небольшом (5-10%) молярном избытке (в пересчете на сахарозу) с тем, чтобы гарантировать почти полное отсутствие сахарозы в продукте.
В экспериментах использовали температуры ацилирования в области от ниже 0оС до примерно 30оС. Верхний предел приемлемых температур ацилирования диктуется началом термически активированных перегиселективных реакций ацилирования, которые будут приводить к образованию нежелательных сложных моно- и диэфиров сахарозы. С практической точки зрения такой температурный предел является функцией химической активности ангидрида кислоты. Например, ввиду того, что уксусный ангидрид является относительно химически активным материалом, ацилирование с его применением в общем случае осуществляют при температуре ниже примерно 20оС. С другой стороны, бензойный ангидрид, будучи несколько менее химически активным, допускает ацилирование при комнатной температуре или несколько выше.
Реакция ацилирования является слабо экзотермической. В зависимости от реакционной температуры и скорости добавления ангидрида в аддукт ди(гидрокарбоил)олова-сахарозы, внешнее охлаждение реакции ацилирования может потребоваться с тем, чтобы минимизировать термически активированное перегиоселективное ацилирование.
Времена, необходимые для ацилирования аддуктов сахарозы до завершения, зависят от концентрации реагентов (так как ацилирование является реакцией кратного порядка), химической активности ацилирующего агента и температуры реакционной смеси. Хотя в лаборатории использовали времена от одного часа до нескольких дней, никаких преимуществ не было получено при расширении реакционного периода до времен, необходимых для расхода ацилирующего агента. Реакция в общем случае завершалась в течение от примерно одного до примерно пяти часов при общих условиях.
Когда для получения сукралозы необходимо использовать сложный сахароза-6-эфир, реакционные смеси после ацилирования содержат S-6-E-, полярный апротонный растворитель, совместный растворитель и 1,3-ди(гидрокарбокси)-1,1,3,3-тетра(гидрокарбил)-дистанноксан или сложный диэфир дистанноксана (СЭДС), который является содержащим олово конечным продуктом последовательности реакций, S-6-E-продукты могут быть извлечены из смеси при помощи самых разнообразных приемов. Например, летучие растворители могут быть удалены выпариванием и/или при помощи выпаривания под вакуумом, чтобы получить сироп или смолу, состоящую главным образом из S-6-E и СЭДС. Производное сахарозы затем можно изолировать при помощи осаждения или кристаллизации из растворителя, в котором он нерастворим, но в котором СЭДС растворим. В качестве альтернативы, относительно летучий совместный растворитель может быть удален при помощи выпаривания, а СЭДС экстрагируют (для рецикла) из полярного апротонного растворителя при помощи соответствующего несмешивающегося растворителя так, как это описано у Вернона и др. см.выше. Выпаривание полярного апротонного растворителя будет давать сироп или смолу, состоящую главным образом из S-6-E- и остаточного полярного апротонного растворителя. Твердый S-6-E может быть выделен при помощи осаждения или кристаллизации.
Предпочтительный вариант осуществления настоящего изобретения включает получение описанного выше, не содержащего СЭДС, сиропа, содержащего примерно одну или две части ДМФ на одну часть S-6-E-E (мас/мас). Этот сироп непосредственно пригоден для получения сложных сукралоза-6-эфиров и сукралозы при помощи хлорирования (такого, как процедура хлорирования, описанная в поданной одновременно патентной заявке США N 382 147, "Улучшенный способ хлорирования сложного сахароза-6-эфира", поданной 18 июля 1989 г. Р.Е. Уолкапом, Н.М. Верноном и Дж.Л.Навиа).
При осуществлении настоящего изобретения получают S-6-E- с выходами от примерно 70-ти с чем то до примерно 90-ти с чем то. Некристаллизованные продукты в общем случае содержат следовые количества остаточной сахарозы и несколько большие количества сложных диэфиров сахарозы. Сложные моноэфиры сахарозы, имеющие перемежающие точки присоединения, в общем случае не продуцируются по этому способу. Например, нормальные S-6-E- выходы для ацетата и бензоата в общем случае изменяются от 86 до 94% Сложные диэфиры сахарозы в общем случае составляют от примерно 3% до примерно 10% от исходной сахарозы, и от примерно 0,5% до примерно 1,5% от исходной сахарозы извлекают в непрореагировавшем виде.
Подробное описание способа, являющегося предметом настоящего изобретения, продолжается для специального случая, включающего ОДБО в качестве окиси ди(гидрокарбил)олова, ДМФ в качестве полярного апротонного растворителя, н-гептан в качестве совместного растворителя и бензойный ангидрид в качестве ацилирующего агента.
Сахарозу (1,00 мол. эквивалент) и ОДБО (1,05 мол.эквивалент) суспендировали в ДМФ (примерно 6 мл на 1 г сахарозы) и н-гептан (примерно 3 мл на грамм сахарозы), и смесь энергично дефлегмировали и перемешивали в течение 3 ч. Смесь освобождали от твердых частиц спустя примерно 30 мин, а реакционная температура составляла 98оС. Воду удаляли из реакционной смеси при помощи совместной перегонки с использованием сепаратора воды Дина-Старка и анализировали при помощи процедуры Карла Фишера. Она соответствовала 104% от теоретического (в пересчете на: одна вода на одну окись олова). Раствор ДБСС охлаждали в ледяной ванне, обрабатывали по каплям раствором бензойного ангидрида (1,10 мол.эквив.) в ДМФ, а затем перемешивали несколько часов, сначала, при температуре ледяной ванны, а затем при комнатной температуре. Неочищенную смесь продуктов, содержащую главным образом сахарозу-6-бензоат, дибензоат дистанноксан (ДБДС), ДМФ и н-гептан, далее обрабатывали водой и экстрагировали циклогексаном (примерно 15 мл на 1 г сахарозы), чтобы удалить ДБДС в соответствии с приемами Вернона и др. см.выше. Затем раствор ДМФ подвергали роторному испарению под высоким вакуумом, чтобы удалить оставшийся н-гептан, воду и часть ДМФ, чтобы получить сироп, который, как было установлено при помощи ЖХВД, содержит с 95,9% выходом S-6-B.
ЖХВД-анализ также показал, что сироп содержит необнаруживаемые другие типы монобензоата сахарозы, 3,57% выход дибензоатов сахарозы и 0,46% извлечение сахарозы. Содержание олова в сиропе, как было установлено, составляло 0,1% (при помощи АП-спектрофотометра (АП атомное поглощение (пер.)). Этот сироп пригоден для хлорирования, чтобы получить сукралозу-6-бензоат.
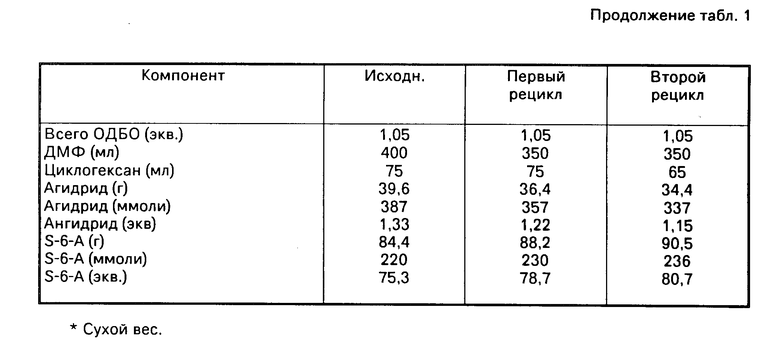
П р и м е р 1. Получение сукролоза-6-бензоата с использованием окиси диоктилолова, диметилформамида и гептана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка, на котором установлен дефлегматор, загружали 68,5 г (200 ммолей) сахарозы, 75,8 г (210 ммолей) окиси диоктилолова, 400 мл ДМФ и 200 мл н-гептана. Суспензию нагревали до дефлегмации (реакционная температура 98оС) и полученный в результате прозрачный раствор подвергали дефлегмации в течение 3 ч. Содержимое сепаратора воды удаляли, растворяли в безводном изопропаноле и анализировали на воду по методу Карла Фишера (4,08 г, 224 ммолей, 107% от теоретического).
Раствор охлаждали до примерно 5оС, по каплям обрабатывали при помощи 49,8 г (220 ммолей) бензойного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ, и перемешивали еще в течение 60 мин при примерно 5оС. Образование S-6-B (Rf 0,5) и исчезновение сахарозы (Rf 0,2) наблюдали при помощи ТСХ SiO2 (15: 10:2, CHCl3-CH3OH-H2O, распыленный с использованием 5% этаноловой серной кислоты и обугленный).
После перемешивания в течение ночи при комнатной температуре в аргоне реакционную смесь обрабатывали водой (50 мл), экстрагировали циклогексаном (2х500 мл), чтобы удалить побочные продукты олова, а ДМФ выпаривали) (роторный испаритель, вакуум при помощи механического насоса, водная ванна 30оС), чтобы получить светло-коричневое вязкое масло, которое как было установлено при помощи ЖХВД-анализа, содержит 80,4 г (180 ммолей, выход 90,1%) сахароза-6-бензоата. Это масло, как было установлено при помощи АП-спектрофотометрии, содержит 0,1% олова.
П р и м е р 2. Получение сахароза-6-ацетата с использованием окиси диоктилолова, диметилформамида и изооктана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 молей) сахарозы, 75,8 г (210 ммолей) окиси диоктил олова, 400 мл ДМФ и 200 мл изооктана (2,2,4-триметилпентана). Суспензию нагревали до дефлегмации (реакционная температура 101оС) и полученный в результате прозрачный раствор дефлегмировали 3 ч. Содержимое сепаратора воды удаляли, растворяли в безводном изопропаноле и анализировали (Карл Фишер) на воду (3,99 г, 222 моля, 106% от теории).
Раствор охлаждали до примерно 5оС, обрабатывали по каплям с использованием 22,5 г (220 ммолей) уксусного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ, и перемешивали еще в течение 3 ч при температуре примерно 50оС. Образование сахароза-6-ацетата (Rf 0,4) наблюдали при помощи системы ТСХ, описанной в примере 1.
После перемешивания в течение ночи при комнатной температуре в атмосфере аргона реакционную смесь обрабатывали водой (50 мл), экстрагировали циклогексаном (2х500 мл) и ДМФ выпаривали (роторный испаритель, вакуум при помощи механического насоса, водная ванна 30оС), чтобы получить коричневатое вязкое масло, которое, как было установлено при помощи ЖХВД, содержало 60,3 г (157 ммолей, выход 78,5%) сахароза-6-ацетата. AN-спектрофотометрия давала содержание олова в этом масле в 0,26.
П р и м е р 3. Получение сахароза-6-бензоата с использованием окиси дибутилолова, диметилформамида и циклогексана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутилолова, 400 мл ДМФ и 100 мл циклогексана. Суспензию нагревали до температуры дефлегмации (реакционная температура 93оС) и полученный в результате прозрачный раствор подвергали дефлегмации 4 ч. Содержимое сепаратора воды удаляли, растворяли в безводном изопропаноле и анализировали на воду, используя процедуру Карла Фишера (3,97 г, 221 ммолей, 105% от теоретического).
Раствор охлаждали до примерно 5оС, обрабатывали по каплям при помощи 49,8 г (220 ммолей) бензойного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ, и перемешивали в течение дополнительных 30 мин при температуре примерно 5оС. После перемешивания в течение ночи при комнатной температуре в атмосфере аргона реакционную смесь обрабатывали с использованием воды, экстрагировали при помощи 1000 мг циклогексана, а ДМФ выпаривали (роторный испаритель, вакуум создавали механическим насосом, водяная ванна 30оС), чтобы получить светло-рыжевато-коричневое вязкое масло, которое, как устанавливали при помощи ЖХВД, содержит 84,2 г (188 ммолей, выход 94,4%) сахароза-6-бензоата.
П р и м е р 4. Получение сахароза-6-ацетата с использованием окиси дибутилолова, диметилформамида и бензола.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутилолова, 400 мл ДМФ и 200 мл бензола. Суспензию нагревали до дефлегмации (реакционная температура 106оС) и полученный в результате прозрачный раствор дефлегмировали 2 часа.
Смесь охлаждали до примерно 5оС, обрабатывали по каплям с использованием 22,5 г (220 ммолей) уксусного ангидрида в 50 мл охлажденного льдом ДМФ и перемешивали еще в течение 30 мин при примерно 5оС. После перемешивания в течение 3,5 дней при комнатной температуре в атмосфере аргона реакционную смесь обрабатывали 50 мл воды, экстрагировали при помощи 1000 мл циклогексана, чтобы удалить побочные продукты олова, а ДМФ выпаривали (роторный испаритель, вакуум с использованием механического насоса, водная ванна 30оС), чтобы получить светло-рыжевато-коричневое масло, которое, как устанавливали при помощи ЖХВД. Содержит 64,5 г (168 молей, 83,9% выход) сахароза-6-ацетата.
П р и м е р 5. Получение сахароза-6-ацетата с использованием окиси дибутилолова, диметилформамида и гексана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 молей) сахарозы, 52,3 г (210 молей) окиси дибутилолова, 400 мл ДМФ и 100 мл н-гексана. Суспензию нагревали до дефлегмации (реакционная температура 77оС) и выдерживали при этой температуре 24 ч.
Смесь охлаждали до примерно 5оС, обрабатывали по каплям 22,5 г (220 молей) уксусного ангидрида в 50 мл охлажденного льдом ДМФ и перемешивали еще 60 мин при примерно 5оС. После перемешивания в течение 90 мин при окружающей температуре в атмосфере аргона, реакционную смесь фильтровали (сила тяжести, рифленая бумага), обрабатывали водой (50 мл), экстрагировали циклогексаном (1х1000 мл), а ДМФ выпаривали (роторный испаритель, вакуум создавали механическим насосом, водная ванна 30оС), чтобы получить коричневатое вязкое масло, которое, как было установлено при помощи ЖХВД-анализа, содержало 58,7 г (153 моля, 75,4% выход) сахароза-6-ацетата.
П р и м е р 6. Получение сахароза-6-ацетата с использованием окиси дибутилолова, диметилформамида и гептана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутилолова, 400 мл ДМФ и 200 мл н-гептана. Суспензию нагревали до дефлегмации (реакционная температура 98оС) и полученный в результате прозрачный раствор дефлегмировали в течение 3 ч. Содержимое сепаратора воды удаляли, растворяли в безводном изопропаноле и анализировали (Карл Фишер) на воду (3,81 г, 212 ммолей, 101% от теоретического). Раствор охлаждали до примерно 5оС, обрабатывали по каплям 22,5 г (220 ммолей) уксусного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ и перемешивали еще в течение 60 мин при примерно 5оС. После перемешивания в течение ночи при комнатной температуре в аргоне реакционную смесь обрабатывали водой (50 мл), экстрагировали циклогексаном 1х1000 мл), чтобы удалить побочные продукты на основе олова, а ДМФ выпаривали (роторный испаритель, вакуум создавали механическим насосом, водная ванна 30оС), чтобы получить серо-желтое вязкое масло, которое, как было установлено, при помощи ЖХВД-анализа, содержит 65,5 г (171 ммолей, выход 85,3%) сахароза-6-ацетата.
П р и м е р 7. Получение сахароза-6-бензоата с использованием окиси дибутилолова, диметилформамида и гептана
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутиолова, 400 мл ДМФ и 200 мл н-гептана.
Суспензию нагревали до дефлегмации (реакционная температура 98оС) и полученный в результате прозрачный раствор дефлегмировали 3 часа. Содержимое сепаратора воды удаляли, растворяли в безводном изопропаноле и подвергали анализу на воду Карла Фишера (3,93 г 218 ммолей, 104% от теоретического).
Раствор охлаждали в ледяной ванне, обрабатывали по каплям 49,8 г (220 ммолей) бензойного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ и перемешивали еще в течение 2 часов при температуре ледяной ванны. После перемешивания в течение ночи при комнатной температуре в атмосфере аргона реакционную смесь обрабатывали водой (50 мл), экстрагировали циклогексаном (1х1000 мл), а ДМФ выпаривали (роторный испаритель, вакуум создавали механическим насосом, водная ванна 30оС), чтобы получить светло-желтое вязкое масло, которое, как было установлено при помощи ЖХВД-анализа, содержит 85,6 г (192 ммоля, выход 95,9%) сахароза-6-бензоата. Масло, как было установлено при помощи АП-спектрофотометрии, содержит 0,1% олова.
П р и м е р 8. Получение сахароза-6-ацетата с использованием окиси дибутил олова, диметилформамида и метилэтилкетона.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутил олова, 400 мл ДМФ и 200 мл метилэтил кетона. Суспензию нагревали до дефлегмации (реакционная температура 110оС) и полученный в результате прозрачный раствор дефлегмировали 3 ч.
Отдельный водный слой не образуется в сепараторе воды. Спустя 1 ч и 2 ч после начала дефлегмации содержимое сепаратора воды удаляли и в реакционную смесь одновременно добавляли достаточное количество метилэтилкетона (примерно 25 мл), чтобы поддержать 108-112оС. Спустя 3 ч после начала дефлегмации содержимое сепаратора удаляли и соединяли с более ранними двумя образцами. При помощи процедуры Карла Фишера устанавливали содержание 3,30 г (183 ммолей, 87,3% от теоретического) воды.
Раствор охлаждали в ледяной ванне, обрабатывали по каплям 22,5 г (220 ммолей) уксусного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ, и перемешивали еще в течение 30 мин при температуре ледяной ванны. После перемешивания в течение ночи при комнатной температуре в атмосфере аргона, реакционную смесь подвергали роторному испарению (вакуум создавали при помощи отсасывания воды, температура ванны 60оС), чтобы удалить метилэтилкетон, обрабатывали 100 мл ДМФ и 50 мл воды, затем экстрагировали циклогексаном (1х1000 мл), чтобы удалить побочные продукты органоолова. В результате выпаривания ДМФ (роторный испаритель, вакуум при помощи механического насоса, водная ванна 30оС) получали темное вязкое масло, которое, как устанавливали при помощи ЖХВД-анализа, содержит 56,2 г (146 ммолей, выход 73,1%) сахароза-6-ацетата и 4,68 г (13,7 ммоля, извлечение 6,84%) сахарозы.
П р и м е р 9. Получение сахароза-6-ацетата с использованием окиси дибутилолова, диметилформамида и изооктана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутилолова, 400 мл ДМФ и 200 мл изооктана. Суспензию нагревали до дефлегмации (реакционная температура 100оС) и полученный в результате прозрачный раствор дефлегмировали 3 ч. Содержимое сепаратора воды удаляли, растворяли в безводном изопропаноле и подвергали анализу на воду Карла Фишера (4,20 г, 233 ммоля, 111% от теоретического).
Раствор охлаждали в ледяной ванне, обрабатывали по каплям 22,5 г (220 ммолей) уксусного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ, и перемешивали еще в течение 60 мин в атмосфере аргона при температуре ледяной ванны. Система ТСУ, описанная в примере 1, показывала, что превращение завершается в этот момент.
Реакционную смесь обрабатывали 50 мл воды, экстрагировали 1000 мл циклогексана и ДМФ удаляли (роторный испаритель, вакуум при помощи механического насоса, водная ванна 30оС), чтобы получить серо-желтое вязкое масло, которое, как было установлено при помощи ЖХВД-анализа, содержит 60,6 г (158 ммолей, выход 78,8%) сахароза-6-ацетата.
П р и м е р 10. Получение сахароза-6-ацетата с использованием окиси дибутилолова, диметилформамида и циклогексана.
В 2000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 200 г (0,584 моля) сахарозы, 153 г (0,613 моля) окиси дибутилолова, 700 мл ДМФ и 200 мл циклогексана. Суспензию нагревали до дефлегмации (реакционная температура 88оС) и полученный в результате прозрачный раствор дефлегмировали в течение 5 ч. Сепаратор воды осушали по мере необходимости с одновременным добавлением 25 мл порций циклогексана в реакционную смесь, чтобы поддержать температуру 92-93оС. Соединенное содержимое сепаратора воды растворяли в безводном изопропаноле и подвергали анализу на воду Карла Фишера (12,2 г, 0,676 моля, 110% от теоретического).
Раствор охлаждали до примерно 5оС, обрабатывали по каплям в течение 10 мин 65,6 г (0,643 моля) уксусного ангидрида (максимальная температура 10оС) и перемешивали еще в течение 60 минут при 5-10оС в атмосфере аргона. Реакция завершалась в конце этого времени, что устанавливали с использованием ТСХ-системы, описанной в примере 1.
Реакционную смесь обрабатывали 50 мл воды и экстрагировали циклогексаном (500 мл). Слои разделяли и слой циклогексана сливали. Далее слой ДМФ обрабатывали дополнительными 50 мл воды и 250 мл ДМФ, а затем экстрагировали циклогексаном (3х500 мл). Слои циклогексана сливали, а слой ДМФ выпаривали (роторный испаритель, вакуум при помощи механического насоса, водная ванна 30оС), чтобы получить серо-желтое вязкое масло, которое, как было установлено при помощи ЖХВД-анализа, содержит 198 г (0,517 моля выход 88,4%) сахароза-6-ацетата. Как было установлено при помощи AN-спектрофотометрии, это масло содержало 0,08% олова.
П р и м е р 11. Получение сахароза-6-ацетата с использованием окиси дибутилолова, N-метил-2-пирролидона и циклогексана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром, сепаратором Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутилолова, 400 мл N-метил-2-пирролидона и 200 мл циклогексана. Смесь подвергали дефлегмации 5,5 часа (реакционная температура 90оС). Суспензия становилась однородной спустя примерно 4 ч. Содержимое сепаратора воды растворяли в безводном изопропаноле и анализировали на воду при помощи процедуры Карла Фишера (3,63 г, 201 ммоль, 95,9% от теоретического).
Раствор охлаждали до примерно 5оС, по каплям обрабатывали в течение 15 мин с использованием 22,5 г (220 ммолей) уксусного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ, и перемешивали еще в течение 30 мин при 3-5оС. После перемешивания в течение ночи при комнатной температуре в атмосфере аргона реакционную смесь обрабатывали водой (50 мл), экстрагировали циклогексаном (2х500 мл) и слой метилпирролидона выпаривали (роторный испаритель, вакуум при помощи механического насоса, водная ванна 45оС), чтобы получить коричневатое вязкое масло, которое, как было установлено при помощи ЖХВД-анализа, содержит 64,1 г (167 ммолей, выход 83,4%) сахароза-6-ацетата. Это масло (AN-спектрофотометрия) содержит 0,1 мас. олова.
П р и м е р 12. Получение твердого сахароза-6-бензоата с использованием окиси дибутилолова, диметилформамида и бензола.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром, сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутилолова, 400 мл ДМФ и 200 мл бензола. Суспензию нагревали до дефлегмации (реакционная температура 107оС) и полученный в результате прозрачный раствор подвергали дефлегмации в течение 2 ч. Содержимое сепаратора воды растворяли в безводном изопропаноле и анализировали на содержание воды (метод Карла Фишера) (3,66 г, 203 ммоля, 96,9% от теоретического).
Раствор охлаждали в ледяной ванне, по каплям обрабатывали в течение 30 мин при помощи 49,8 г (220 ммолей) бензойного ангидрида, растворенного в 50 мл охлажденного льдом ДМФ, а затем перемешивали еще в течение 30 мин при температуре ледяной ванны. После перемешивания в течение ночи при комнатной температуре в атмосфере аргона два растворителя удаляли в роторном испарителе (вакуум при помощи отсасывания воды, ванна при 50оС, затем вакуум при помощи механического насоса, ванна 30оС), чтобы получить вязкое масло, которое обрабатывали в роторном испарителе при помощи 250 мл ацетона. В результате нагревания до примерно 50оС получали прозрачный раствор, из которого сахароза-6-бензоат легко кристаллизовали при охлаждении до комнатной температуры.
Продукт фильтровали на грубо фриттованном, спекшемся стекле в качестве фильтра, промывали ацетоном (2х100 мл) и сушили под вакуумом (50оС) 0,5 мм рт. ст. (16 ч), чтобы получить 70,0 г не совсем белого твердого вещества, которое, как было установлено при помощи ЖХВД-анализа, состоит на 98,1% из сахарозы-6-бензоата (68,7 г. 154 ммоля, выход 76,9%). Этот твердый материал, как было установлено при помощи AN-спектрофотометрии, содержит 0,43% олова.
П р и м е р 13. Получение сахароза-6-ацетата с использованием окиси дибутилолова, диметилформамида и хлороформа.
В 1000 мл трехгорлую колбу с круглым дном, снабжженную механической мешалкой, термометром и сепаратором Контес Глассвеа растворителя воды, тяжелее, чем вода (каталог 535800-0000) с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутилолова, 400 мл ДМФ и 250 мл хлороформа. Суспензию нагревали до дефлегмации (реакционная температура 103оС, температура пара сепаратора 87оС) и полученный в результате прозрачный раствор дефлегмировали 3 ч.
При окружающей температуре раствор обрабатывали одной порцией при помощи 22,5 г (220 ммолей) уксусного ангидрида. Медленная экзотермия приводила к подъему температуры от 23 до 30оС в течение примерно 15 мин. После перемешивания в течение ночи при комнатной температуре в атмосфере аргона реакционную смесь обрабатывали водой (50 мл), экстрагировали циклогексаном (2х1000 мл), чтобы удалить побочные продукты олова, а слой ДМФ выпаривали, чтобы получить черновато-коричневое вязкое масло, которое, как было установлено при помощи ЖХВД-анализа, содержит 56,0 г (146 ммоля, выход 72,9%) сахароза-6-ацетата. Это масло, как было установлено с использованием AN-спектрофотометрии, содержит 0,1% олова.
П р и м е р 14. Получение твердого сахароза-6-бензоата с использованием окиси дибутилолова, диметилформамида и гептана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммоль) окиси дибутилолова, 400 мл ДМФ и 200 мл н-гептана. Суспензию нагревали до дефлегмации (реакционная температура 98оС) и полученный в результате прозрачный раствор дефлегмировали 3 ч. Содержание сепаратора воды растворяли в безводном изопропаноле и анализировали на содержание воды при помощи процедуры Карла Фишера (3,39 г, 168 ммолей, 89,9% от теоретического).
Раствор обрабатывали одной порцией при комнатной температуре с использованием 49,8 г (220 ммолей) бензойного ангидрида, растворенного в 50 мл ДМФ. Медленная экзотермия поднимала реакционную температуру от 26 до 30оС в течение примерно 20 мин. После перемешивания в течение ночи при комнатной температуре в атмосфере аргона два растворителя удаляли в роторном испарителе (вакуум при помощи отсасывания воды, ванна в 40оС, затем вакуум при помощи механического насоса, ванна 30оС), чтобы получить вязкое масло, которое обрабатывали на роторном испарителе при помощи 250 мл ацетона. В результате нагревания этой смеси до примерно 50оС получали прозрачный раствор, из которого сахароза-6-бензоат легко кристаллизовали при охлаждении до комнатной температуры.
Этот продукт фильтровали на грубо фриттированном, агломерированном стеклянном фильтре, промывали ацетоном (2 х 100 мл) и сушили под вакуумом (50оС) 0,5 мм рт.ст. (14 ч), чтобы получить 69,3 г белого твердого вещества, которое, как было установлено при помощи ЖХВД-анализа, состоит на 97,0% из сахароза-6-бензоата (67,2 г, 151 ммолей, выход 75,3%). Это твердое вещество, как устанавливали при помощи AN-спектрофотометрии, содержит 0,40% олова.
П р и м е р 15. Получение твердого сахароза-6-ацетата с использованием окиси дибутилолова, диметилформамида и циклогексана
В 2000 мл четырехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром, капельной воронкой и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 200 г (0,584 моля) сахарозы, 153 г (0,613 моля) окиси дибутилолова, 700 мл ДМФ и 100 мл циклогексана. Суспензию нагревали до дефлегмации (реакционная температура 100оС) и полученный в результате прозрачный раствор дефлегмировали 5 ч. Нижний слой в сепараторе воды удаляли по мере необходимости при одновременном добавлении циклогексана (всего 25 мл) с тем, чтобы поддержать температуру 100 ± 1оС. Соединенные порции сепаратора воды анализировали на воду при помощи процедуры Карла Фишера (11,7 г, 0,651 моль, 106% от теоретического).
Смесь охлаждали до примерно 4оС, используя ванну сухой лед-ацетон, обрабатывали по каплям в течение 40 мин при помощи 64,4 г (0,631 моля) уксусного ангидрида (максимальная температура 1оС) и перемешивали еще в течение 20 мин при температуре примерно 2оС. Оказалось, что реакция завершалась в этот момент, что устанавливали при помощи ТСХ-системы, описанной в примере 1.
После перемешивания в течение ночи при комнатной температуре реакционную смесь обрабатывали 20 мл воды и экстрагировали циклогексаном (1 х 500 мл, затем 1 х 250 мл). Слои циклогексана сливали. Слой ДМФ затем обрабатывали дополнительно 20 мл воды, а затем экстрагировали циклогексаном (2 х 250 мл). Слои циклогексана сливали, а слой ДМФ выпаривали (роторный испаритель, вакуум при помощи механического насоса, водная ванна 30оС), чтобы получить темно-рыжевато-коричневое вязкое масло, которое, как было установлено при помощи ЖХВД-анализа, содержит 187 г (0,513 моля, выход 87,9%) сахароза-6-ацетата. Масло, как было установлено при помощи AN-спектрометрии, содержит 0,1% олова.
Сироп, полученный выше, соединяли с аналогичным образцом, содержащим 101 г (0,263 моля) сахароза-6-ацетата, и удаляли столько остаточного ДМФ, сколько это возможно (роторный испаритель, вакуум при помощи механического насоса, водная ванна 50оС). Остаток растворяли в горячем метаноле (300 мл), охлаждали до примерно 5оС, помещали в качестве затравки кристаллы сахароза-6-ацетата, выдерживали при 5оС в течение ночи, и фильтровали с использованием грубо фриттиванного, агломерированного стеклянного фильтра. Фильтровальную лепешку снова превращали в шлам в 200 мл метанола, фильтровали, а фильтровальную лепешку промывали 100 мл метанола. После сушки под вакуумом (25оС) 0,5 мм рт.ст. 18 ч), получали 246 г не совсем белого твердого вещества, которое, как устанавливали при помощи ЖХВД-анализа, состоит на 82,4% из сахароза-6-ацетата (203 г, 0,528 моля, извлечение 68,0%), на 1,3% из сахарозы и на 2,5% из диацетатов сахарозы. Газовый хроматографический анализ и процедура Карла Фишера показали, что это твердое вещество содержит также существенные количества метанола (5,8%), ДМФ (3,3%) и воды (1,1).
Пробу в 100 г неочищенного твердого вещества (82,4 г. 0,215 моля) S-6-A обрабатывали 5 г активированного углерода в 550 мл метанола при дефлегмации в течение 10 мин. Углерод удаляли фильтрацией и углеродную лепешку промывали 150 мл горячего метанола. Фильтрат и промывочные жидкости соединяли, концентрировали до объема примерно 500 мл, охлаждали до примерно 10оС и вводили затравку.
Продукт фильтровали, сразу же снова растворяли в 550 мл дефлегмирующего метанола, концентрировали до объема примерно 500 мл, охлаждали до примерно 10оС и выдерживали в течение ночи. Полученное таким образом белое вещество фильтровали, промывали холодным метанолом (100 мл) и сушили под вакуумом (50оС) 0,5 мм рт.ст. (16 ч), чтобы получить продукт (74,4 г), который, как определяли при помощи комбинации анализов, состоит на 90,2% из сахарозы-6-ацетата (67,1 г, 0,175 моля, извлечение 81,3% в пересчете на неочищенное твердое вещество и извлечение в 55,3% в пересчете на экстрагированный ДМФ сироп), на 0,6% из сахарозы, 1,7% из диацетатов сахарозы, 6,8% метанола и 0,2% воды. Установлено, что содержание метанола в твердом сахарозе-6-ацетате, полученном таким образом, не может быть снижено при помощи удлинения вакуумной сушки.
П р и м е р 16. Получение твердого сахароза-6-бензоата с использованием окиси дибутилолова, диметилформамида и толуола
В 2000 мл одногорлую колбу с круглым дном, снабженную магнитной мешалкой и сепаратором воды Дина-Старка с установленным на нем дефлегматором, загружали 100 г (292 ммоля) сахарозы и 400 мг ДМФ. Эту смесь нагревали и перемешивали при температуре 90оС (ванна) до однородности (приблизительно 10 минут).
Раствор обрабатывали при помощи 73,6 г (296 ммолей) окиси дибутилолова и 50 мл толуола, и полученную таким образом суспензию нагревали до 110оС (ванна) на 1,5 ч, затем дополнительно нагревали до 125оС (ванна) на 1,5 ч. В течение этого периода нагревания применяли слабый вакуум к системе для того, чтобы получить дефлегмацию. Содержимое сепаратора воды удаляли по мере необходимости при одновременном добавлении толуола (всего 30 мл), чтобы гарантировать адекватную дефлегмацию.
Полученный таким образом темный раствор охлаждали до комнатной температуры и обрабатывали одной порцией при помощи 69,4 г (307 ммолей) бензойного ангидрида. После перемешивания в течение ночи при комнатной температуре растворители удаляли в роторном испарителе (вакуум при помощи механического насоса, водная ванна 50оС), чтобы получить сироп, который обрабатывали на роторном испарителе при помощи 500 мл ацетона. В результате нагревания до примерно 50оС получали раствор, из которого инициировали кристаллизацию сахароза-6-бензоата при охлаждении до комнатной температуры и введения затравки.
Шлам разбавляли 250 мл ацетона, перемешивали в течение 2 ч при температуре 0-5оС и фильтровали на грубо фриттированном, агломерированном стеклянном фильтре. Из продукта дважды приготавливали шлам в 100 мл ацетона, снова фильтровали и фильтровальную лепешку промывали 50 мл ацетона. В результате вакуумной сушки (25оС) 0,5 мм рт.ст. (14 ч) получали 103 г белого твердого вещества, которое, как было установлено при помощи ЖХВД-анализа, состоит на 92,3% сахароза-6-бензоата (95,0 г, 213 ммоля, выход 72,9%).
П р и м е р 17. Получение сахароза-6-бензоата с использованием окиси дибутилолова, диметилформамида и тетрагидрофурана.
В 1000 мл трехгорлую колбу с круглым дном, снабженную механической мешалкой, термометром, 125 мл капельной воронкой без выравнивания давления и сепаратором воды-растворителя Контес Глассвеа с установленным на нем дефлегматором, загружали 68,5 г (200 ммолей) сахарозы, 52,3 г (210 ммолей) окиси дибутилолова, 400 мл ДМФ и 200 мл тетрагидрофурана (ТГФ). Суспензию нагревали до дефлегмации и полученный в результате прозрачный раствор подвергали дефлегмации в течение 7,5 ч.
Отдельный слой воды не образовывался в сепараторе воды, через приблизительно 30-минутные интервалы содержимое сепаратора воды извлекали и одновременно добавляли достаточное количество ТГФ (всего 375 мл), чтобы поддержать реакционную температуру на уровне 100 ± 2оС, а температуру пара в сепараторе воды анализировали при помощи процедуры Карла Фишера (3,54 г, 197 ммолей, 93,7% от теоретического).
Раствор обрабатывали при комнатной температуре в течение 20 мин при помощи 49,8 г (220 ммолей) бензойного ангидрида, растворенного в 50 мл ДМФ. Медленная экзотермия поднимала температуру реакции с 19 до 25оС. После перемешивания в течение ночи при комнатной температуре в атмосфере аргона реакционную смесь обрабатывали водой (50 мл), экстрагировали циклогексаном (1 х 1000 мл), а ДМФ выпаривали (роторный испаритель, вакуум при помощи механического насоса, водная ванна 30оС), чтобы получить темно-коричневое вязкое масло, которое, как было установлено при помощи ЖХВД-анализа, содержит 69,9 г (157 ммолей, 78,4% выход) сахароза-6-бензоата.
П р и м е р 18. Получение сахароза-6-ацетата с использованием окиси дибутилолова, диметилформамида и циклогексана с демонстрацией рецикла окиси дибутилолова
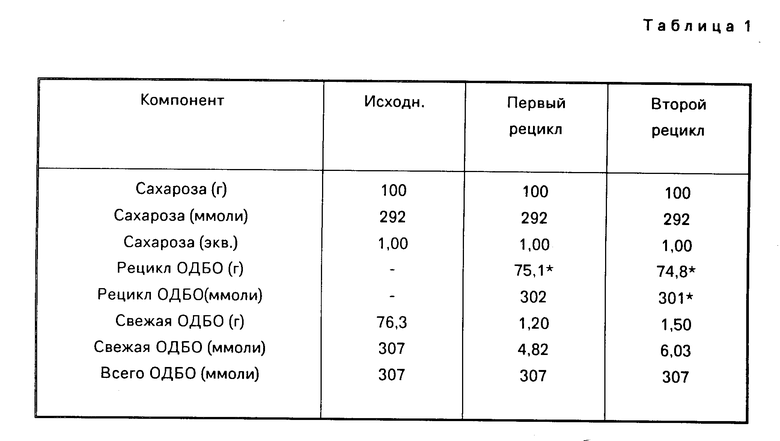
Сахарозу (100 г, 292 ммоля) обрабатывали при помощи 76,3 г (307 ммоля) окиси дибутилолова и превращали в сахароза-6-ацетат (84,4 г, 220 ммолей, выход 75,3% ) по существу также, как это описано в примере 10 за тем исключением, что соединенные и выпаренные циклогексановые экстракты, которые содержали 1,3-диацетил-1,1,3,3-тетрабутилдистаноксан или диацетат дистанноксана ("ДАДС"), добавляли при 60оС в 13,0 г (325 ммолей) гидрата окиси натрия в 250 мл воды.
После удаления остаточного циклогексана при помощи отгонки при атмосферном давлении полученный таким образом шлам охлаждали до 30оС, фильтровали (корзиночная центрифуга) и выделенную окись дибутилолова ("ОДБО") промывали водой (3 х 100 мл). Влажный вес извлеченного твердого вещества составил 81,8 г.
Извлеченную ОДБО использовали для того, чтобы получить вторую порцию сахароза-6-ацетата и ОДБО извлекали еще раз. Затем получали третью порцию сахароза-6-ацетата при помощи дважды выделенной ОДБО. Полные данные для этой серии из трех последовательных реакций приведены в табл.1.
В приведенной ниже табл.2 приведены экспериментальные подробности и выходы для примеров 1-17:
Использование: в качестве промежуточного продукта при получении искусственного подслащивающего агента сукралозы. Сущность изобретения: продукт-сложный сахароза-6-эфир. Реагент 1: сахароза. Реагент 2: оксид дибутилолова. Условия реакции: температура 75 125°С с одновременным удалением воды в среде смеси полярного апротонного растворителя и органической жидкости с образованием 1,3-ди-(6-о-сахароза) -1,1,3,3-тетра(гидрокарбил) дистанноксана. Реагент 3: ангидрид карбоновой кислоты. Условия реакции: ацилирование с выделением целевого продукта. 2 з. п. ф-лы, 2 табл.
| 0 |
|
SU220641A1 |
